 Vijay Kumar
Vijay Kumar John H. Stewart IV
John H. Stewart IV- Department of Surgery, Laboratory of Tumor Immunology and Immunotherapy, Medical Education Building-C, Morehouse School of Medicine, Atlanta, GA, United States
Human pregnancy is a complex condition that poses significant challenges for women due to the necessity of a uterus for key processes such as fertilization, embryo implantation, fetal development, and childbirth. These processes are governed by immunological factors and accompanied by various physiological changes. For a successful pregnancy, maternal immune reprogramming is crucial because the developing embryo is considered a semi-allograft. Any immunological alteration during pregnancy induces recurrent pregnancy loss and other fetal–maternal health issues, including preeclampsia. However, despite advances in reproductive immunology, the exact immunopathogenesis of preeclampsia remains unclear. The complement system (CS) is an evolutionarily ancient and critical innate immune component that plays a significant role in maintaining immune homeostasis. The current article discusses the critical role of the CS in human pregnancy and how its dysregulation predisposes pregnant women to preeclampsia. The article introduces the concept of the Th1 to Th2 immunological shift as a prerequisite for a successful pregnancy and the evolution of decidualization via transposable elements, which recruit genes responsible for the process in the endometrium. The immune system plays a critical role in decidualization. The second section discusses the CS signaling pathway, its negative regulators, and the roles of the C3a/C3aR and C5a/C5aR1/C5aR2 or C5L2 axis in immune homeostasis. The third section elaborates on the role of the CS in the establishment of human pregnancy, such as fertilization, implantation, and fetal development. The fourth section describes maternal CS signaling alteration during successful human pregnancy. The fifth section describes the role of CS signaling in preeclampsia, including its systemic and local (placental) alterations and the responsible mechanisms. The article closes with future perspectives and a summary that describes important complement-based approaches for diagnosing and treating preeclampsia.
1 Introduction
Human pregnancy occurs in the very specialized organ, the uterus, which protects the developing embryo and fetus through its mucosal lining or decidua, making human pregnancy a unique immune challenge that further develops trained immunity with subsequent pregnancies (1–4). The maternal–fetal interaction during human pregnancy is an example of fetal allograft acceptance by the pregnant female as indicated by the shift from a pro-inflammatory Th1 immune response to an anti-inflammatory Th2 immune response (Figure 1) (5, 6). Furthermore, the maternal innate immune system plays a critical role in the successful outcome of human pregnancy. For example, uterine natural killer (uNK) cells are critical for the early embryonic establishment and spiral artery formation (1, 7). Along with the local uterine immune microenvironment, systemic factors, such as hormonal status and cytokine (pro- and anti-inflammatory) levels governing the systemic and local immunological status, determine pregnancy success (1). The details of fetal–maternal immune interactions during human pregnancy have been discussed elsewhere (4, 8–10).
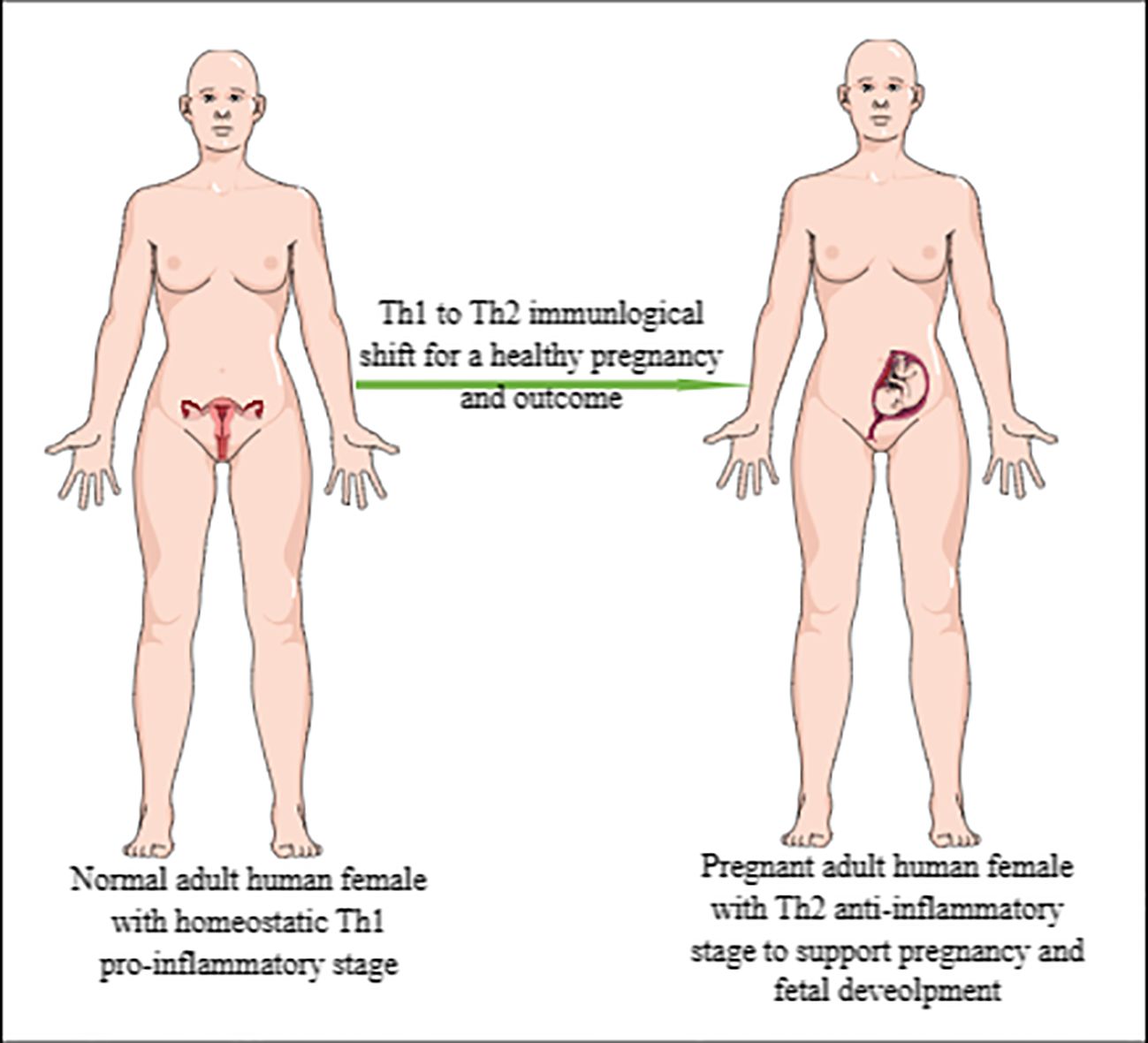
Figure 1. Representation of Th1 to Th2 immunological shift during pregnancy. Normal/healthy non-pregnant adult woman exhibiting pro-inflammatory Th1 immune response to maintain immune homeostasis and fight against invading pathogens and other foreign particles. However, during pregnancy, this pro-inflammatory Th1 immune response shifts to anti-inflammatory Th2 immune response to support pregnancy or developing embryo/fetus, which is an allograft for a pregnant woman.
The complement system (CS) is a component of the innate immune system. It is composed of more than 50 humoral components (fluid-phase proteins present in the blood, saliva, lymph, and interstitial fluids), which recognize pathogens and interact with antibodies (Abs)/immunoglobulins (IgG and IgM) and their cognate receptors expressed on different immune cells to maintain immune homeostasis (11, 12). Evolutionarily, the CS is one of the most ancient and primitive components of innate immunity (12, 13). For example, the complement component C3 and factor B genes comprising the central components of the CS originated at least 1,000 million (one billion) years ago (MYA) (13). Furthermore, developmental evolution studies focusing on the origin of pregnancy indicate that the recruitment of genes ancestrally expressed in other organ and tissue systems into endometrial expression transmitted new functions to the uterine endometrium, such as immune regulation and fetal–maternal signaling for a healthy pregnancy (14). The transposable elements (TEs) evolved/amplified prior to the divergence of eutherian mammals were critical for recruiting these genes to the endometrium to induce the development of decidualization, as indicated by the deposition of binding sites for master transcriptional regulators of endometrial stromal cell type identity and progesterone responsiveness to numerous genes across the genome (14). For example, the progesterone receptor (PGR) is the principal transcriptional effector of progesterone signaling and decidualization (14). Thus, decidualization in mammalian pregnancy has also evolved from acquiring genes from other organs required to maintain immune balance for normal functioning. The CS is one of the most ancient components of the innate immune system; therefore, it is critical to understand its role in human pregnancy and preeclampsia.
2 CS as a critical component of the immune system
The CS is composed of circulating or humoral components and its receptors called complement receptors (CRs), such as C1q, which is a pattern recognition receptor (PRR) of the complement component C1 (C1 is composed of C1r, C1q, and C1s) and mediates the complement recognition of surface-bound immunoglobulin (Ig) G and IgM, CR1 (CD35), CR2 (CD21), CR3 (CD11b/CD18 or Mac1), CR4 (CD11c/CD18), CRIg (VSIG4, expressed on Kupffer cells and several other tissue-resident macrophages), C3aR, C5aR1 (CD88), and C5aR2 or C5L2 (12, 15, 16). The liver is a major producer of circulating CS components (17, 18). However, epithelial, endothelial, and immune cells, such as neutrophils, monocytes and macrophages, dendritic cells (DCs), mast cells, B cells, and T cells, also produce different CS components or proteins (19). CS activation is a rapid innate immune response against invading pathogens, including microbe/pathogen-associated molecular patterns (MAMPs/PAMPs) and death/damage-associated molecular patterns (DAMPs), aimed at containing the infection and inflammation.
The CS activation further activates innate immune cells by promoting phagocytosis by producing opsonins, which induce opsonization, and the stimulation of different CRs (C3aR, C5aR1, and C5aR2) expressed on innate and adaptive immune cells, further activating both (innate and adaptive) arms of the immune system to maintain immune homeostasis. The CS activation pathway diverges mainly into three pathways: 1) classical CS activation, 2) lectin or mannan-binding lectin (MBL) pathway, and 3) alternative CS activation (Figure 2). It is critical to note that the alternative CS signaling pathway is evolutionarily older and that the classical CS signaling pathway evolved from it (20, 21). Complement component C3 activation is common to all three CS pathways, or all these pathways converge at C3 to form the end product, called the membrane attack complex (MAC), which is composed of C5bC6-9 (Figure 2). A brief description of all three CS signaling pathways forming the MAC has been discussed below and is shown in Figure 2.
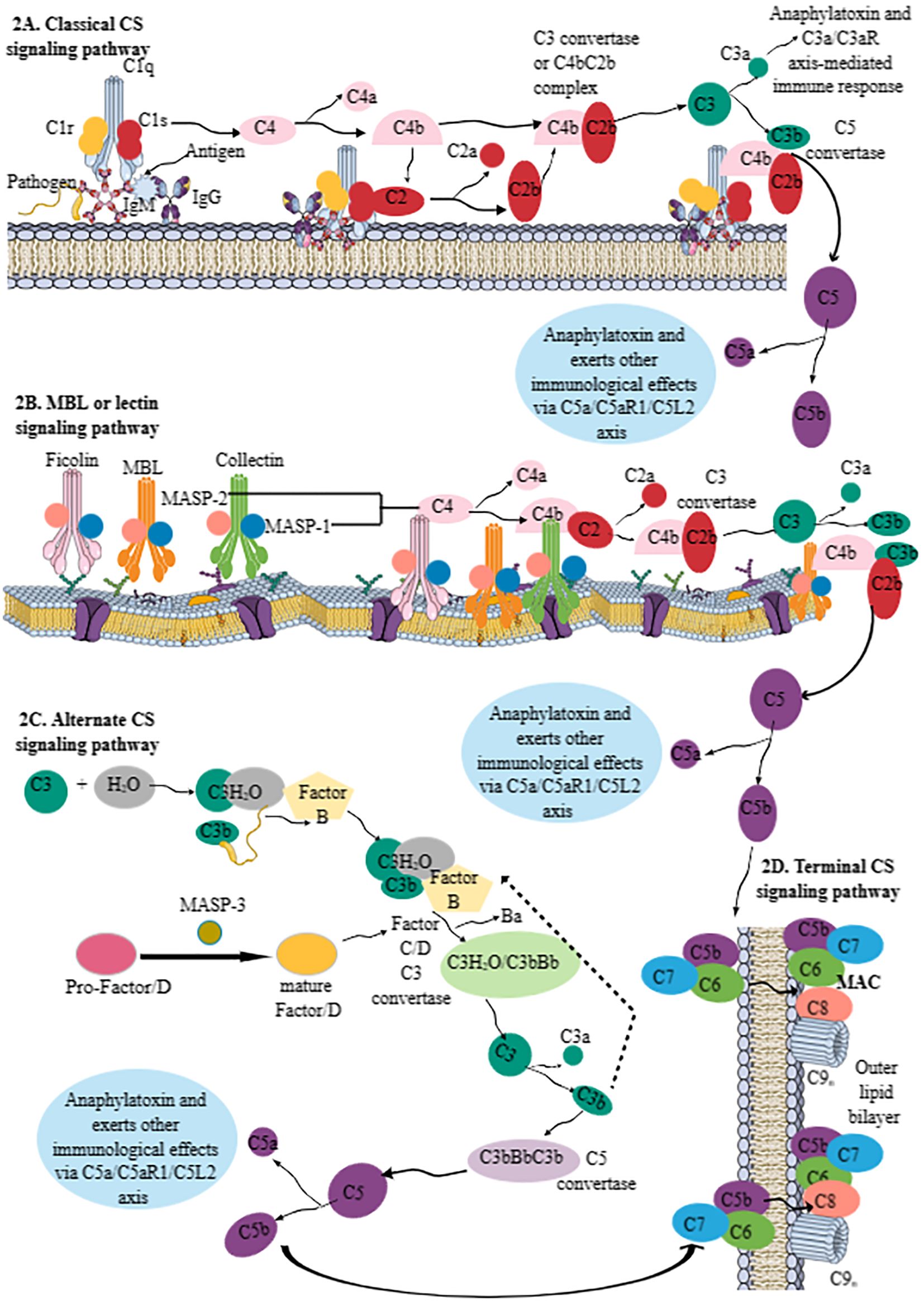
Figure 2. Schematic representation of the CS signaling pathways. (A) Classical CS signaling pathway. The C1 component [comprising C1q (serves as a PRR) and 2C1r and 2C1s components, which are serine proteases] initiates the classical CS signaling pathway by recognizing IgG and/or IgM bound to the pathogen, cell surfaces, or other immune complexes. The C1q binding to the pathogen/antigen/IC-IgM/IgG complex activates two serine proteases (C1r and C1s). The activated C1s recognizes C4 and generates C4a and C4b components. C4b recruits C2 and C1s to generate C2a and C2b. The C4bC2b complex serves as C3 convertase of the classical CS signaling pathway. C3 convertase cleaves C3 into C3a (an anaphylatoxin; alters immune response via C3a/C3aR axis on different immune cells) and C3b (opsonin). The remaining C3b attached to C4bC2b forms C4bC2bC3b complex called C5 convertase, which cleaves C5 into C5a (an anaphylatoxin; alters immune response via C3a/C3aR axis on different immune cells) and C5b, which forms MAC by activating the terminal pathway. (B) The lectin or MBL pathway. The MBL pathway does not require C1 but instead depends on ficolin-, MBL-, and collectin-mediated pathogen recognition. MASP-1 and MASP-2 of these molecules upon pathogen recognition become active. For example, MASP-1 activation stimulates MASP-2 enzymatic activity for C4 and C2 molecules to generate C4bC2b or lectin pathway C3 convertase to generate C3a and C3b. This pathway also generates C5a and C5b, like classical CS signaling pathway, to generate MAC. (C) The alternative CS signaling pathway. The alternative CS signaling pathway involves hydroxylation of C3 to form C3(H2O) complex, which recognizes circulating pathogens. The bound C3b on the pathogen surface is recognized by FB. The MASP-3 of the MBL pathway cleaves pro-factor D to mature factor D that serves as a serine protease to cleave factor B (FB) and generate the C3 convertases C3(H2O)Bb and C3bBb. Thus, the C3(H2O)/C3bFB complex generates C3(H2O)/C3bBb as a C3 convertase of the alternative CS signaling pathway that cleaves C3 into C3a and C3b. The fast production of C3bBbC3b (C4b2b3b) serves as a C5 convertase of the alternative CS signaling pathway to generate C5a and C5b. (D) The terminal pathway of the CS. C5b, generated due to the activation of all three CS pathways, forms a complex with C6, C7, C8, and C9 components called MAC. MAC kills invading pathogens by forming pores on their cell membranes. Kindly see the text for details. CS, complement system; PRR, pattern recognition receptor; MAC, membrane attack complex; MBL, mannan-binding lectin; FB, factor B.
Classical CS activation starts from the C1 component, which is composed of three components: C1q, C1r, and C1s (Figure 2A). C1q serves as a pattern recognition molecule (PRM) or PRR. It recognizes structural changes induced by IgM and/or IgG1, IgG2, and IgG3 Abs binding to pathogens, cell surfaces, or immune complexes (ICs) (Figure 2A). This recognition, or the C1q–pathogen/antigen/IC-IgM/IgG complex, activates two serine proteases (C1r and C1s) of C1. The enzymatically activated C1s recognizes complement component C4 and cleaves it into C4a (smaller fraction) and C4b (larger fraction) (Figure 2A). The biological role of C4a is not yet clear, whereas C4b within or near the Ig–C1 complex recruits fluid phase C2 and C1s, which process C2 to C2a (biological function unknown) and C2b (which is an active serine protease) (Figure 2A). The C4bC2b complex serves as a classical CS pathway C3 convertase, a central player for all three CS pathways (Figure 2A). C3 convertase promotes C3 activation, a central component of CS signaling pathways. C3 cleavage produces C3a (serves as an anaphylatoxin and induces C3a–C3aR interaction-mediated immune response) (Figure 2A), and C3b serves as an opsonin to aid in phagocytosis through its exposed thioester group that recognizes amino or hydroxyl groups on the target (15, 22, 23). The remaining C3b within C4bC2b forms a complex called C4bC2bC3b or C5 convertase (Figure 2A). The C2b component of C5 convertase cleaves C5 into C5a (serves as anaphylatoxin and inflammogen and induces C5aR1- and C5aR2-mediated immune functions) and C5b (mediates the activation of terminal pathway or MAC formation) (Figure 2A).
The lectin or MBL pathway is independent of Abs and shares many characteristics of classical and alternative CS signaling pathways (Figure 2B). The MBL or lectin pathway does not require C1 for its activation; instead, it depends on the recognition of PAMPs by MBL, three ficolins (Ficolins 1–3), and two collectins (Collectin-10 and Collectin-11), which have serine protease activity (Figure 2B). MBL and collectins (Collectin-10 and Collectin-11), due to their carbohydrate recognition domain, are part of the superfamily of fibrinogen-like proteins, whereas ficolins have a fibrinogen-like recognition domain and belong to the superfamily of fibrinogen-like proteins. Thus, MBL, ficolins, and collectins can recognize different carbohydrate entities, such as mannose of bacterial pathogens by MBL, N-acetylglucosamine (GlcNAc) of injured and dying cells by MBL and ficolins, and altered l-fucose and D-galactose patterns of cells under severe stress by ficolins and Collectin-10 and Collectin-11. Furthermore, the lectin pathway can also recognize the host DNA exposed on apoptotic cells, and Collectin-12 may activate the alternative CS signaling pathway in its soluble form in conjunction with properdin. MBL-associated serine protease-1 (MASP-1), MASP-2, and MASP-3 are MBL or lectin pathway serine proteases. MASP-1 and MASP-2 are associated with common collagen regions within MBL, ficolins, and collectins (Figure 2B), whereas MASP-3 activity is connected to the alternative CS pathway activation via the proteolytic cleavage of pro-Factor D (FD) to enzymatically active mature FD (Figure 2C). FD is a serine protease that is critical for activating the alternative CS signaling pathway by cleaving factor B (FB) and generating the C3 convertases C3(H2O)Bb and C3bBb (discussed later in the alternative CS signaling pathway section) (24, 25). Adipocytes are the main producers of circulating FD (24, 25). The MASP-3-mediated cleavage of FD to its mature form prepares it for its initiation and amplification function of the alternative CS signaling pathway (25). The details of FD in the CS signaling pathway and complement-mediated inflammatory diseases have been discussed elsewhere (24, 25).
The MBL or lectin pathway recognizes the target molecule and autoactivates MASP-1, cleaving MASP-2 for its enzymatic activity toward C4 and C2 bound to initiating MBL, ficolins, and collectin–MASP-1/2 complexes. This is followed by classical CS signaling, such as forming a C4bC2b complex and a C3 convertase, activating C3 and C5 (Figure 2B). The C3 and C5 convertases of the lectin pathway are also known as lectin pathway convertases.
The alternative CS signaling pathway has been considered a separate CS activation pathway (Figure 2C). However, it can activate the CS itself and may account for approximately 80%–90% of the total complement activation, even in conditions triggered by classical or lectin/MBL pathways (26). For example, it idles in the serum constantly at low levels of activation, as 3%–5% of circulating C3 constantly exists in a hydrolyzed [C3(H2O)] form, and its exposed thioester can spontaneously interact with complement targets, such as microbes in the peripheral circulation (blood, lymph, and interstitial fluids). Thus, C3(H2O) is bound to the circulating pathogen, and C3b is deposited on the target surface during the classical or lectin/MBL pathway, recognized by the inactive serine protease, FB (Figure 2C). The C3(H2O)/C3bFB complex interacts with another serine protease, FC, that cleaves FB into Ba and Bb (enzymatically active) units (Figure 2C). The smaller Ba subunit detaches itself from the C3(H2O)/C3bFB complex, generating C3(H2O)/C3bBb, the C3 convertase of the alternative CS signaling pathway (Figure 2C). The Bb component of the alternative CS signaling pathway’s C3 convertase cleaves C3 into C3a and C3b, as occurs during classical or lectin/MBL pathways (Figure 2C). The rapid production of C3bBbC3b (C4b2b3b) complexes during the alternative CS signaling pathway forms its C5 convertase, cleaving C5 into C5a and C5b (Figure 2C) (27). Properdin, a complement protein, along with stabilizing the C3 convertase complexes (C3bBbP) of the alternative CS signaling pathway, may also serve as an initiator or focus point for the subsequent C3b deposition during different conditions, such as apoptotic immune cell death as seen during acute infections, such as sepsis (27, 28). For example, properdin targets specific proteoglycans of apoptotic immune cells, serving as DAMPs and microbial PAMPs, and recruits C3b, which promotes phagocytic clearance of these pathogens and apoptotic cells (27).
The terminal pathway of CS signaling involves the convergence of all three CS signaling pathways that form the final and lytic component of the CS called the MAC, which forms pores in the targeted microbial surface (Figure 2D) (27). The MAC is composed C5b, C6, C7, C8, and C9 components of the CS (Figure 2D) (27, 29, 30). The complement components C6 to C9 are members of the MAC/perforin/cholesterol-dependent cytolysin (MACPF/CDC) protein superfamily. Thus, the MAC is the CS signaling end product that kills the target by forming pores. However, continuous CS activation is governed by several circulating or surface-bound regulators to prevent host damage, as described in Table 1.
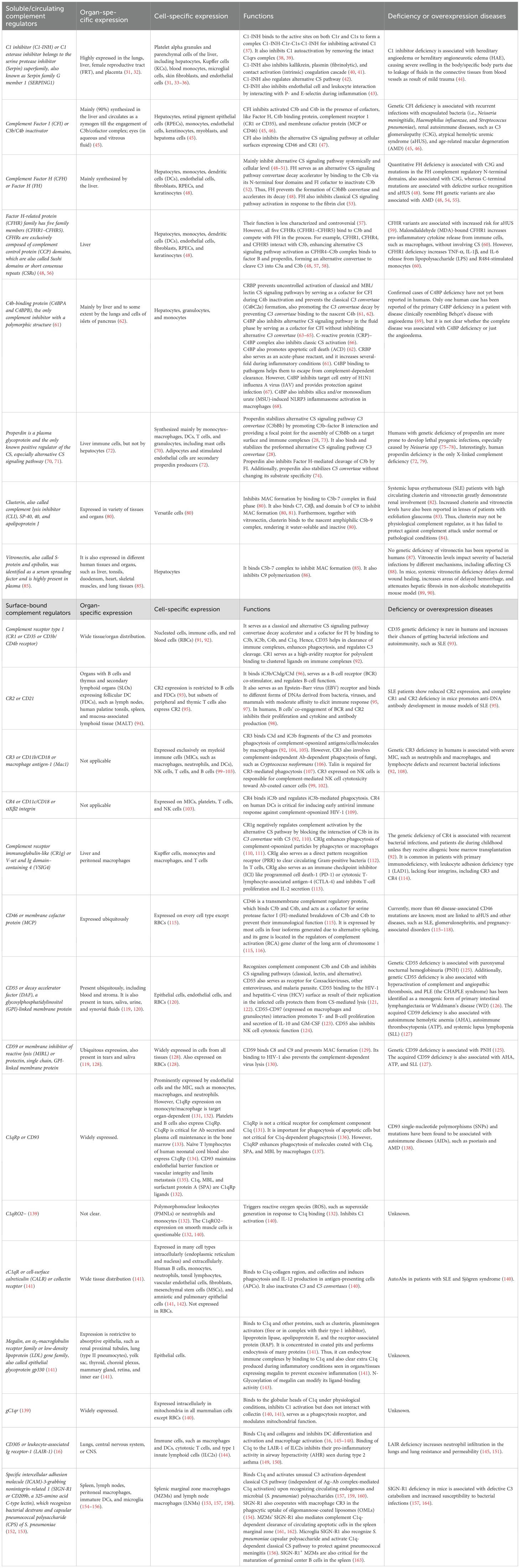
Table 1. Circulating and cell surface/membrane-bound complement regulators.
2.1 CS in immunoregulation and immune homeostasis
The CS was first described as a critical antimicrobial defense component of innate immunity in circulation between 1888 and 1894 (165, 166). As discussed earlier, the activation of three components of the CS in response to different stimuli, such as Ag–Ab complexes, pathogens (PAMPs/MAMPs), and DAMPs, induces the release of different complement proteins, such as C3a, C3b, C5a, and C5b, and the formation of terminal MAC to clear the exo- or endogenous threat. C3a and C5a are critical immunomodulators that, in addition to serving as anaphylatoxins, also function as potent immunomodulatory agents through their cognate receptors (C3aR, C5aR1, and C5aR2 or C5L2), which are expressed on various innate and adaptive immune cells. Therefore, this section discusses the impact of C3a and C5a on immune cells expressing their cognate receptors.
2.1.1 Impact of C3a on various immune cells via direct interaction with C3aR
In addition to the CS signaling events, C3a can also be generated by different systemic proteases, such as thrombin and immune cell-derived cathepsin G and L (a lysosomal protease) (167–169). However, the cathepsin L (CTSL)-mediated cleavage of the C3 into C3a and C3b has been reported intracellularly in T cells, which maintains their survival through intracellular C3a–C3aR interaction-mediated downstream mammalian target of rapamycin complex 1 (mTORC1), Raptor, and p56 signaling, and extracellular C3a–C3aR promotes the generation of pro-inflammatory Th1 cells as indicated by the generation of pro-inflammatory cytokines, such as interferon-γ (IFN-γ) (170, 171). Resting T-cell lysosomes and endosomes contain C3, and its cleavage by the CTSL to generate “tonic” intracellular C3a is critical in maintaining homeostatic T-cell survival. The transfer of this intracellular C3a to the T-cell surface induces the autocrine pro-inflammatory cytokine production of the Th1 phenotype, which has been seen in T cells isolated from patients with autoimmune arthritis (170). The T cells of patients with autoimmune arthritis exhibit overactivated intracellular CS and IFN-γ production that can be blocked by targeting the intracellular CTSL. Furthermore, intracellular C3–C3aR signaling in intestinal Paneth cells [intestinal secretory epithelial cells with innate immune functions, such as the production and secretion of antimicrobial peptides (AMPs) and other immunomodulatory molecules] also regulates their mTORC1 signaling to enhance their intestinal protective function by supporting the expansion of intestinal stem cells (ISCs) in the intestinal crypts during acute inflammatory intestinal injury (172–174).
Interestingly, in contrast to the extracellular/secreted C3, the intracellular C3 generated via alternative translation in the cytosol is non-glycosylated and present in the reduced state. Intracellular non-glycosylated C3 is turned over by the ubiquitin–proteasome system (UPS) (175). Furthermore, C3 can also be retranslocated from the endoplasmic reticulum (ER) to the cytosol and structurally resembles extracellular/secreted C3. Notably, cytosolic C3 also exerts antimicrobial action in epithelial cells by opsonizing invasive pathogens, such as Staphylococcus aureus, decreasing the vacuolar escape, and impacting the bacterial survival by presenting the pathogen to phagocytes, such as macrophages (175). Furthermore, the cytosolic C3 in the β cells of the pancreas protects them from IL-1β-induced inflammatory cell death by interacting with and inhibiting the downstream Fyn-related kinase (FRK) (176, 177). Another study has indicated that the C3-mediated protective effect on pancreatic islet β cells involves AKT activation and c-Jun N-terminal kinase (JNK) inhibition upon treatment with pro-inflammatory cytokines, such as IL-1β and IFN-γ (178). Thus, in different cell types, such as T cells, Paneth cells, and pancreatic β cells, the cytosolic C3 supports their survival and division. Additionally, C3 present in the breast milk protects suckling mouse pups from Citrobacter rodentium-mediated enteric infection by shaping the evolving pup gut microbiota (killing of commensal Gram-positive Staphylococcus lentus B3) but without affecting the production of secretory antibodies in the breast milk (179, 180). A more detailed review of C3a–C3aR interactions on different immune cells would be too cumbersome for the main text and is summarized in Table 2.
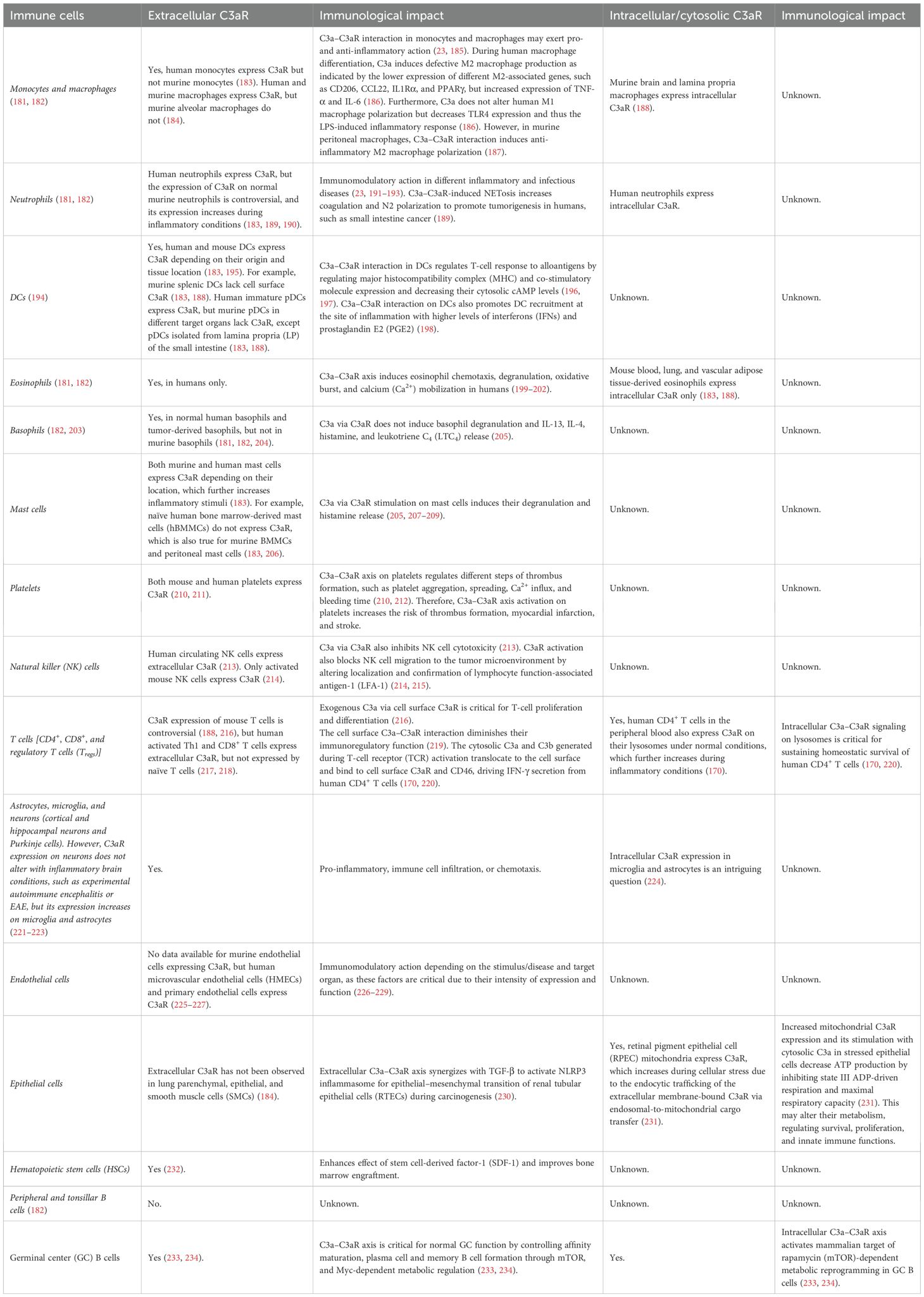
Table 2. C3aR expression (cell surface and cytosolic) on different immune cells and the impact of C3 fragments on their immune functions.
2.1.2 Impact of C5a on various immune cells via direct interaction with C5aRs (C5aR1 and C5aR2 or C5L2)
The C5a generated during CS pathways due to the breakdown of C5 into C5a and C5b exerts immunomodulatory actions by interacting with C5aR1 (CD88) and C5aR2/C5L2. For example, C5a–C5aR1 interaction mediates potent leukocyte chemoattraction at the site of inflammation. It induces pro-inflammatory phenotype and functions on different immune cells during sterile and infectious inflammatory conditions, such as autoimmune diseases [rheumatoid arthritis (RA) and Crohn’s disease (CD)], allergies (asthma), ischemia–reperfusion injuries, and sepsis (235–239). However, the discovery of the second C5a receptor called C5L2 [a seven-transmembrane domain G protein-coupled receptor (GPCR)] or C5aR2 in 2000 has generated controversies in the previously established pro-inflammatory functions of C5a, as C5L2 has now been considered an anti-inflammatory C5aR (240, 241). C5L2 is also considered an active metabolic receptor, and its ligand C5a and C5adesArg or acylation-stimulating protein (ASP) [the degraded/desarginated C5a fragment generated via enzymatic (carboxypeptidase) degradation-dependent endocytosis], is time-, clathrin-, and cholesterol-dependent (241, 242).
Circulating ASP levels increase in people with obesity, insulin resistance or Type 2 Diabetes Mellitus (T2DM), and metabolic syndrome, which increases monocyte chemoattractant protein-1 (MCP-1) and keratinocyte-derived chemokine (KC or IL-8) from their adipocytes through C5L2 or C5aR2 interaction without impacting IL-6 and adiponectin production (243, 244). The MCP-1 and KC production from adipocytes in response to ASP/C5L2 interaction involves phosphatidylinositol-3 kinase (PI3K) and nuclear factor-kappa B (NF-κB) activation (244). However, ASP via C5L2 interaction in macrophages does not induce MCP-1 and KC production. In contrast, the adipocyte-mediated production of these cytokines in the adipose tissue (AT) increases monocyte/macrophage chemotaxis and their inflammatory function (243). For example, C5adesArg-induced C5L2 activation in adipocytes induces triglyceride synthesis and glucose and fatty acid (FA) uptake, which is absent in the adipocytes of C5L2 knockout mice (243–245).
C5L2 ligand binding induces their internalization and degradation, which decreases their extracellular expression level, which is a step to decrease the generation of profound or tissue-damaging complement-mediated inflammatory immune response (241). In contrast, C5aR1 internalizes ligands at a slow rate, which are further expelled back into the extracellular environment without undergoing degradation, which further aggravates the inflammatory cascade (241). Furthermore, Thr196Asn mutations in the C5L2 gene were associated with hyperlipidemia and retinitis pigmentosa (RP) in a Chinese family (246).
In humans, the GPR77 gene on chromosome 19, q13.33-13.34, is located downstream of the C5aR1 gene encoding C5aR2 or C5L2 (247). Interestingly, C5L2, belonging to C3a, C5a, and formyl Met-Leu-Ph (fMLP) receptors related to the chemokine receptor family, also binds to C3a with a moderate affinity (247, 248). However, the C3a binding to C5L2 can be easily displaced by C4a, indicating that C3a has a lower affinity to C5L2 than C5a. Moreover, the binding of anaphylatoxins (C3a and C5a) to C5L2 only increases the immune cell degranulation potential upon cross-linking high-affinity immunoglobulin E (IgE) receptor by a pertussis toxin-sensitive mechanism. C5L2 binding affinity to C5a is similar to that of C5aR1. C5L2 binds to C5adesArg with a higher affinity than C5aR1 (242, 247). The C5L2 transcripts have been widely expressed in different organs, such as the spleen, testis, brain (frontal cortex, hippocampus, and hypothalamus), heart, lung, liver, kidney, adrenal gland, thyroid gland, spinal cord, ovary, and colon, and in immune and non-immune cells, like granulocytes, immature DCs, adipocytes, and skin fibroblasts, but not in monocyte-derived macrophages (MDMs) (248–253). Table 3 shows the impact of C5a and C5aR1/C5L2 or C5aR2 interaction on different immune cells.
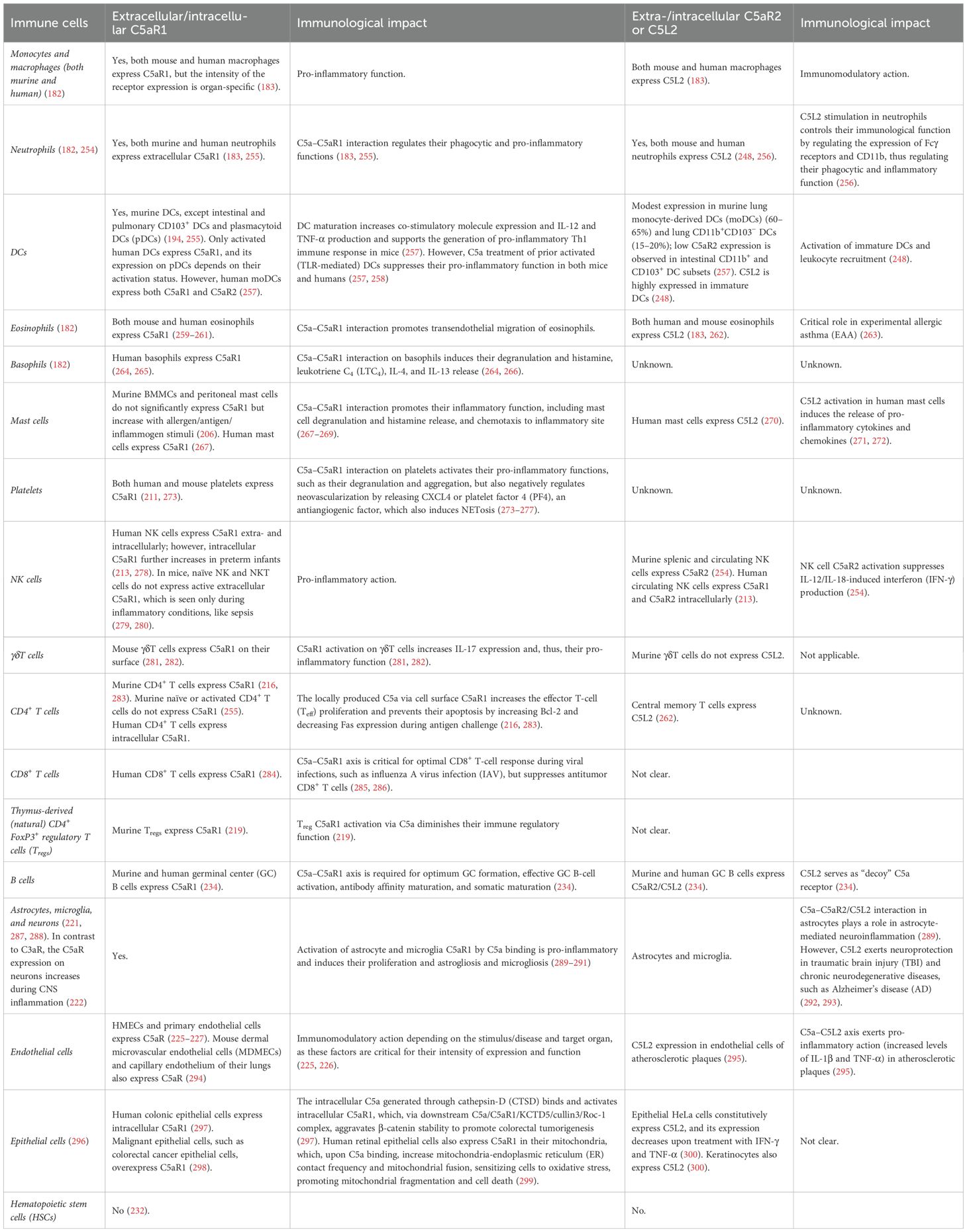
Table 3. C5aR1 and C5L2 expression (cell surface and cytosolic) on different immune cells and the impact of C5a on their immune functions.
3 CS in human pregnancy
3.1 CS in human fertilization
The CS is critical for women’s reproductive health and successful pregnancy (fertilization to childbirth). For example, the human female reproductive tract (FRT), comprising the ovaries, fallopian tubes, uterine endometrium, myometrium, and cervix, expresses complement regulatory proteins (CRPs), such as CD55 and CD59 (protectin), and CD46 [membrane cofactor protein (MCP)] (Figure 3) (301). The CRPs (CD55, CD59, and CD46) are overexpressed in stressed endometrial cells, indicating that their endometrial cells develop a complement-mediated lysis process, modifying their inflammatory outcome in different immune-mediated inflammatory diseases (IMIDs) (302). However, all CS components/proteins of classical and alternative pathways are present in the uterine, tubal, and follicular fluids (303).
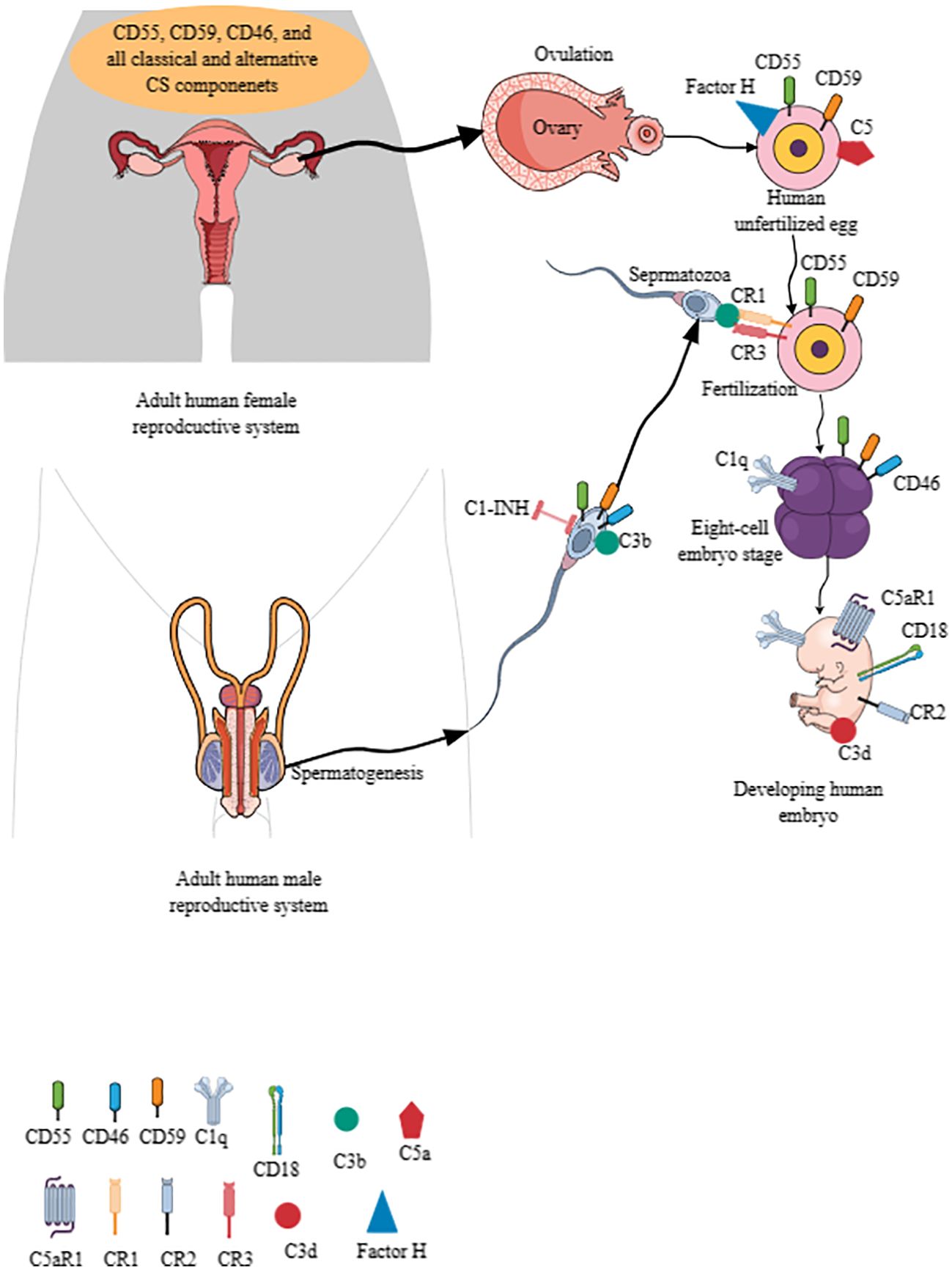
Figure 3. The CS signaling pathway in establishing human pregnancy. The complement components are present in human male and female reproductive tracts, and their gametes (eggs and sperms) also express different CS components. For example, CD55, C59, and C46 are present in the human uterus, ovaries, fallopian tubes, and cervix. Furthermore, the uterine, tubal, and follicular fluids have all the components of classical and alternative CS signaling pathways. Human unfertilized eggs also express CD55, CD59, factor H, C5, CR1, and CR3. Human sperms also express CD55, CD59, CD46, C3b, and C1-INH. The C3b interaction with CR1 and CR3 between sperm and ovum is critical for fertilization. Different complement components are expressed on developing embryo depending on the developmental stage. For example, CD46 appears at six-to-eight embryonic stage, which also expresses C1q. C5aR1, CD18, CR2, and C3d are also expressed by the developing embryo to check the complement activity for healthy pregnancy. Kindly see the text for details. CS, complement system.
Moreover, human unfertilized eggs (plasma membrane and zona pellucida) and pre-implantation embryos express CD55/DAF and CD59, preventing them from complement attack (Figure 3) (304–306). Unfertilized oocytes do not express CD46, but it appears at the six-to-eight cell embryonic stage on their cell membrane as the first embryonic human gene expression begins (Figure 3) (304, 305). Interestingly, oocytes and pre-implantation embryos do not express complement receptor 1 (CR1 or CD35)/C3b/C4b receptor and major histocompatibility complex 1 (MHC-1) proteins (303, 304). Of note, mRNA transcripts of soluble complement inhibitors, including C4b-binding protein (C4BP beta chain), factor I, and clusterin, are present in oocytes through their eight-cell stage embryo blastomeres (306).
The mRNA transcripts of central complement activating components, such as C3 and C5, along with their activators (factor B and D, C3 activators in the alternative pathway) and other complement cascade proteins (C1s and C2) of the classical and MBL CS pathways are also present in oocytes and embryos, indicating that their transcripts remain after fertilization (306). Human embryos also express C5aR1, CR2, and CD18 (Figure 3). CD18/CD11b serves as CR3, and CD18/CD11c comprises CR4. C1q is present at all embryonic developmental stages (Figure 3) (306). The C3b/iC3b complex on the cell surface of different early embryonic developmental stages indicates targeting of the embryo by the activated complement. The inactivated C3, known as C3d, is present on the cell surface of the developing embryo, indicating that a complement activation check is crucial for embryo development (Figure 3) (306). However, C5 is expressed on the zona pellucida surface of the oocyte but not on the surface of blastomeres (Figure 3). Interestingly, oocytes and blastomeres do not have intracellular C3 and C5 but express C4bp and factor H on their cell surface (306). Factor H is also present in the zona pellucida of oocytes (Figure 3).
In addition to the expression of complement proteins in the human female ovum, the sperm also expresses different complement components. For example, human sperms express C1 inhibitor (C1-INH), C1qR (cC1qR and gC1qR/p33), CD46, CD55, and CD59, which may be critical for their survival and motility (Figure 3) (307–309). CD46, CD55, and CD59 are expressed on the inner acrosomal membrane of a human sperm, which are critical for the fertilization process but do not protect sperm from anti-sperm antibodies and complement-mediated immune attack (310). Furthermore, C1q promotes the agglutination of capacitated sperms, and C1qR (cC1qR and gC1qR) expression increases in this process (311, 312). The cleaved complement component C3b on sperm acrosome (formed during acrosome reaction) binds with CR1 and CR3 of the oocyte to facilitate the fertilization process (Figure 3) (313). Factor H, but not CD46, serves as a cofactor in C3b cleavage, which contributes to the fertilization process (314). Human sperm CD46 also contributes to the survival of acrosome-reacted spermatozoa in the FRT by modulating CS activation (308). The complement activation critical for the fertilization process is initiated by the C-reactive protein (CRP) and is dependent on other complement components, such as C1q, C2, and factor B (314). Hence, complement components (expressed in ovum and sperm) are critical for mammalian/human fertilization to create a new life.
3.2 CS in embryo implantation and spiral artery formation
The trophoblast is a local producer of complement components in the maternal decidua, serving as a primary source of complement components C3 and C4 at the maternal–fetal interface (315). Local IFN-γ at the maternal–fetal interface positively impacts trophoblast C3 and C4 production. Uterine or decidual NK (dNK) cells comprise the majority of immune cells at the time of blastocyst implantation, are the primary source of local IFN-γ during the early stages of human pregnancy, and are critical mediators of spiral artery formation (1, 316–319). Human circulating NK cells express C3aR, which migrate to the uterine microenvironment during pregnancy to serve as dNK or uNK cells, comprising 70% of the decidual lymphocyte population. The C3a–C3aR interaction on NK cells inhibits their cytotoxic action (320, 321). Additionally, C3b deposition on target cells also inhibits NK cell cytotoxicity (322). It is well known that human dNK cells exhibit lower cytotoxic activity and secrete high levels of immunoregulatory cytokines and molecules to support a healthy pregnancy (323–325). Therefore, it would be interesting to explore how homeostatic complement activation during embryo implantation influences dNK cell functions (decreasing cytotoxic function but increasing their secretory function to release immunoregulatory cytokines) to support their pregnancy functions.
Human decidual stroma widely expresses and secretes complement component C1q, which interacts with proteins expressed on decidual extracellular matrix (DEM) and promotes trophoblast adhesion and migration by activating ERK1/2 mitogen-activated protein kinases (MAPKs) to promote trophoblast invasion of decidua and placental development (326, 327). Furthermore, epithelial–mesenchymal transition (EMT) is critical for trophoblast differentiation and maternal–fetal interface establishment, as indicated by the differentiation of trophoblast [extravillous trophoblast (EVT)] cells from proximal epithelial phenotype to a distal invasive mesenchymal phenotype called interstitial trophoblast penetrating the maternal decidua basalis and into the maternal myometrium (328). Complement components, such as the C3a–C3aR axis, promote EMT during fibrosis and cancer metastasis (230, 329–331). Trophoblast cells also exhibit characteristics of cancer cells and pseudotumorigenesis to nourish the developing embryo (332–334). Thus, CS components are critical for embryo implantation and placental development to maintain a healthy human pregnancy.
4 Impact of pregnancy on maternal circulating CS components
The circulating levels of C3a, C4a, and C5a increase in normal pregnant women and remain elevated throughout pregnancy, from 20 weeks post-gestation to the newborn’s delivery (335–337). However, some studies have indicated a decrease in circulating C5a with no alteration in C3a and C4a levels in women with healthy pregnancies than non-pregnant women (338). Moreover, an early (first trimester of pregnancy) increase in circulating C3a levels is associated with adverse pregnancy outcomes (336, 339). Nevertheless, a study from China has indicated that the circulating C1q, C5a, and C5b-9 (MAC) levels in the first and second trimesters are similar to those of non-pregnant healthy women (340). In contrast, increased levels of C3, properdin, C1q, factors H and B, C4, and adipsin and decreased levels of circulating C2 and C5a have been associated with successful implantation as indicated by a study comprising Middle Eastern (Qatar) women with obesity undergoing in vitro fertilization (IVF)-assisted conception (341). The maternal circulating C1-INH level decreases during this period (336). Thus, in a healthy pregnancy, maternal circulating complement components, such as C3a, adipsin (FD), and C5a, increase above baseline during the second and third trimesters and remain stable afterward (340, 342). However, women with preeclampsia develop higher circulating adipsin levels later in their pregnancy (343). In addition, catalyzing the rate-limiting step of alternative CS signaling pathway activation, adipsin (FD) is also involved in MAC formation and C3a and C5a anaphylatoxin generation (27, 344, 345). Thus, increased circulating adipsin levels during pregnancy and preeclampsia indicate associated metabolic and cardiovascular changes, as circulating adipsin levels are directly associated with the metabolic and cardiovascular health of an individual (346–348).
The MBL–MASP2 activity also increases during normal pregnancy (349). Any alteration in circulating complement components beyond their regulatory/protective function at the early stages of pregnancy (first trimester) causes an abnormal pregnancy outcome (337, 350–352). Preeclampsia is one of the conditions that affect the fetus and mother, which is discussed in the following section, specifically in the context of the complement system to maintain the article’s specificity. Moreover, preeclampsia predisposes surviving women to develop hypertension, cardiovascular diseases (CVDs), and metabolic syndrome later in life (353, 354).
5 CS in preeclampsia
Clinically, preeclampsia is characterized by the new onset of hypertension and proteinuria in pregnant women or any other maternal signs of maternal vascular dysfunction, such as edema or dysfunction of any other organ (liver, kidney, pulmonary, cerebral, or visual) or restricted fetal growth after 20 weeks of gestation (355, 356). A comparative preeclampsia study based on American College of Obstetricians and Gynecologists (ACOG) and International Society for the Study of Hypertension in Pregnancy (ISSHP) definitions of preeclampsia at term gestational age (≥37 0/7 weeks) to identify adverse maternal and perinatal outcomes has indicated the inclusion of broad definition for preeclampsia, as it can better identify women and babies at risk of adverse outcomes (357). For example, the more inclusive ISSHP definition of maternal end-organ dysfunction seemed to be more sensitive in identifying adverse maternal and perinatal outcomes associated with preeclampsia than following the less inclusive ACOG definition (357). Moreover, the inclusion of uteroplacental dysfunction (particularly when angiogenic factors are included) to diagnose preeclampsia on its broad definition optimizes preeclampsia identification in pregnant women and babies at risk (357). In contrast, eclampsia represents severe convulsions/seizures in women with gestational hypertension or preeclampsia (355, 358). The etiology of preeclampsia, including placentation’s impact, has been described elsewhere and will not be discussed in the current article (359, 360).
It is interesting to note that preeclampsia was described in the early 20th century, while eclampsia was recognized much earlier, with descriptions dating back thousands of years (355, 361). Immunology, including the study of the innate immune system, is also a new branch of modern medicine. For example, the field of the innate immune system was revolutionized after the discovery of macrophages/phagocytosis by Elia Metchnikoff in 1882 (362). Furthermore, the functional studies associated with the CS were first described between 1888 and 1894, although evolutionarily, the CS is the most ancient component of the innate immune system, as a complement component called C3-like protein has existed a billion years ago (BYA) (12, 13, 166). Although immunological advances in preeclampsia immunopathogenesis have been made, information about the CS in preeclampsia is scarce (363–369). Furthermore, inflammation (local and systemic) also plays a critical role in preeclampsia immunopathogenesis, and the CS is a key mediator of the inflammatory process (370–373). Therefore, understanding the role of this (CS) evolutionarily ancient innate immune component in preeclampsia pathogenesis is critical to understanding its immunology.
5.1 Maternal circulating CS components during preeclampsia
Recent studies have indicated the alteration of CS (classical, lectin, and alternative complement activation pathways) proteins in maternal and fetal circulation and placental tissues (342, 374–376). Nevertheless, data are not equivocal for some maternal circulating complement proteins, which may be due to race, ethnicity, comorbidity, and geographical locations, as race and ethnicity also play a crucial role in the origin, pathophysiology, and outcomes of preeclampsia (377). For example, women with preeclampsia have significantly lower circulating properdin and C4 levels but higher factor B (Ba and Bb) than women with normal pregnancy, which starts to increase during early pregnancy (first trimester) (350, 374, 375, 378, 379). The decreased circulating C1q and factor H levels in patients with early- and late-onset severe preeclampsia have also been observed (379). However, another study has indicated a significant increase in circulating C1q and C4d levels in late-onset severe preeclampsia (LOSPE). C3a, C5a, and MAC levels also increase in maternal circulation with early-onset severe preeclampsia (EOSPE) and LOSPE (380–382). The placentae of pregnant women with preeclampsia express lower C3aR and C5aR levels than those of women with normal pregnancy (381, 383). Nevertheless, a positive correlation between higher serum C3a and C5a levels in women with preeclampsia with circulating angiotensin II type 1 (AT1) receptor agonistic autoantibody (AT1-AA) has been observed (381). AT1-AA is one of several mediators of hypertension during pregnancy, along with increasing soluble fms-like tyrosine kinase-1 (sFlt1) and soluble endoglin (CD105) (sEng), and endothelin-1, which are elevated in women with preeclampsia (384–386). Increased sEng and sFlt1 in EOSPE directly correlate with MAC levels and, inversely, with circulating C3a levels (382). Thus, increased AT1-AA in circulation during pregnancy may activate the CS signaling pathway to generate potent anaphylatoxins, such as C3a and C5a, to induce EOSPE. Increased circulating sFlt1 during pregnancy induces hypertension, proteinuria, and glomerular endotheliosis, which are associated with preeclampsia (387). In addition to the circulating complement component alteration during preeclampsia, the cell surface component, such as CD93 (C1qRp or C1qR1 expressed on endothelial cells), level decreases in the circulation, whereas its level increases in the serum during the first trimester of normal pregnancy (388). Therefore, further studies are needed in this direction.
A study of women with singleton pregnancies in Colombia has indicated the association between decreased maternal circulating factor H in the first trimester and spontaneous preterm birth (389). Recently, a genome-wide association study (GWAS) in Finland has identified five rare heterozygous factor H variants (L3V, R127H, R166Q, C1077S, and N1176K) only in women with severe preeclampsia (390, 391). Of these five factor H variants in women with severe preeclampsia, variants R127H and C1077S are associated with normal factor H synthesis without its release in the circulation. In contrast, variants R166Q and N1176K are associated with normal factor H secretion with reduced binding to C3b, causing dysregulated CS activation associated with severe preeclampsia (390). However, the authors could not find any defect in patients with severe preeclampsia exhibiting the L3V factor H variant. Thus, CS-associated genetic mutations can also determine women’s susceptibility or resistance to developing preeclampsia during pregnancy. The decreased maternal circulating factor H levels have also been associated with preeclampsia in women from European countries, such as the Netherlands, Finland, Norway, Italy, and the United Kingdom, without any increase in circulating anti-factor H autoantibodies (392, 393). Furthermore, the placentae [decidual stromal cells (DSCs), decidual endothelial cells (DECs), and EVTs] of women with preeclampsia express lower factor H levels (mRNA and protein) than those of women with normal pregnancy (393). In addition to factor H, other C3b regulators, such as MCP and Complement Factor I (CFI) genetic mutants [typically associated with atypical hemolytic uremic syndrome (aHUS)], in pregnant women with systemic lupus erythematosus (SLE) or antiphospholipid antibodies (APL Ab) have been identified, indicating their higher susceptibility to develop preeclampsia than normal women (394). Moreover, normal pregnant women (without SLE and APL Ab) with hypomorphic MCP and CFI genetic variants are more susceptible to developing preeclampsia than those without these variants.
Nevertheless, despite inconsistencies in different circulating CS components, a systematic meta-analysis of selected 41 studies out of a total of 456 studies has retrieved results consistently reporting reduction of C4, C3, and factor H and increase of C4d, Bb, factor D, C3a, C5a, and MAC or C5b-9 in maternal circulation during preeclampsia than in women with normal pregnancies (395). In addition to altered circulating CS components/proteins, the CR variants, such as CR3 (CD11b/18, Mac-1, or integrin αMβ2) variant M441K, display a trend toward an increased adhesion to iC3b, which is most significantly associated with preeclampsia in the Finnish Genetics of Pre-eclampsia Consortium (FINNPEC) cohort (396). The CR4 variant A251T increases C4 adhesion to iC3b, and the W48R CR4 variant decreases CR4 binding to iC3b, which may have functional consequences on the CS signaling pathway to impact preeclampsia susceptibility/resistance and severity in the population (396, 397). Even 14 variants within nine genes coding for components of the MAC or C5b-9 have a strong association with preeclampsia (398). For example, two missense variants (rs200674959 and rs147430470) of C5 are strongly associated with preeclampsia predisposition among pregnant women. Moreover, the C6 variant rs41271067 predisposes women to preeclampsia, whereas its rs114609505 variant protects against preeclampsia (398).
In addition to the classical and alternative CS components, the circulating MBL pathway components are critical for pregnancy and preeclampsia (399); for example, H-ficolin and MASP-3 of the MBL pathway of the CS decrease in women with preeclampsia (399). Ficolin-2 and ficolin-3 are also lower in pregnant women with preeclampsia than in those with normal pregnancy (400). The decreased plasma ficolin-2 level of women with preeclampsia positively correlates with circulating placental growth factor (PIGF) and inversely correlates with circulating sFlt1. However, pregnant women with preeclampsia have higher plasma MBL concentration than women with normal pregnancy (376). Women with MBL codon 54 gene polymorphism are protected from preeclampsia development (401). The protective effect of the MBL codon 54 gene against preeclampsia may be due to low MBL production, as low-MBL production genotypes are considered disease (preeclampsia) modifiers (402). Therefore, further studies are needed to establish preeclampsia’s genetic and immunological mechanisms with MBL pathway dysregulation.
Additionally, second-trimester amniotic fluids of pregnant (singleton) women have shown upregulated C3a and factor Bb before the onset of preeclampsia, indicating that CS activation during early pregnancy is associated with early-onset preeclampsia (403). Elevated C5a levels in the amniotic fluid of pregnant women developing preeclampsia have also been observed (404). Women with preeclampsia exhibit urinary excretion of the MAC because of an antiangiogenic state (high circulating sFlt1 and low PIGF and VEGF levels) associated with severe preeclampsia (405–407). Hence, altered maternal complement components during pregnancy are critical for the onset and severity of preeclampsia.
5.2 Placental CS components and preeclampsia
The complement proteins expressed on placental tissues are also critical for a healthy pregnancy, and their alteration plays a critical role in inducing preeclampsia (408). For example, a term placenta obtained after a healthy delivery expresses complement inhibitor C4b-binding protein (C4bBP) on its outer layer (syncytial knots of syncytiotrophoblast) that facilitates material exchange between the mother and the developing fetus (409). C4d is rarely present in the placentae of normal pregnant women, but its expression increases in syncytiotrophoblasts of women with preeclampsia (408, 410). In contrast, factor H is abundant in the placental tissue stroma of normal pregnancy, which is decreased in the placentae obtained from women with preeclampsia (409). EVTs express CD46, CD55, and CD59 in all three trimesters of normal pregnancy (411, 412). The placentae obtained from women with preeclampsia overexpress CD55 and CD59 (408). The C1q, MBL, and properdin expression in the placenta do not change between a healthy pregnancy and preeclampsia (408). However, despite no difference between control and preeclampsia in control and EOSPE patients, C1q expression decreases in LOSPE patients, which needs further investigation (408, 409). Moreover, the placental macrophages of women with preeclampsia overexpress C5a, and their trophoblasts overexpress C5aR (413). The C5a–C5aR interaction on trophoblast cells polarizes them toward an anti-angiogenic phenotype by balancing their angiogenic factors, such as sFlt1 or soluble vascular endothelial growth factor receptor-1 (sVEGFR-1) and PIGF (410, 413). Another study has indicated an increase in placental sFlt1 and PIGF in women with preeclampsia, which increases maternal circulating sFlt1 and falls post-delivery (387). The upregulated C4d, sFlt1, and MAC in the placentae of women with preeclampsia correlate well, indicating the aberrant CS activation. C5a–C5aR axis inhibition has prevented aberrant placental development by decreasing sFlt1 levels and rescued pregnancies (413, 414). Furthermore, C3a also induces the upregulation of cellular sFlt1 in human syncytiotrophoblast cells, and upregulated MAC induces its release (415). Increased sFlt1 and decreased PIGF-mediated angiogenic imbalance suppress the expression and secretion of factor H from placental endothelial cells, further activating the CS to cause endothelial cell damage and systemic vascular dysfunction, hypertension, and proteinuria during preeclampsia (416). Fetal cord blood factor B levels do not vary during healthy pregnancy and preeclampsia, and other complement components (C1q, C3, C4, and C3d) are much lower than those in healthy maternal circulation (375, 417). Nevertheless, C3d levels increase in fetal cord blood with the degree of placental inflammation, indicating their increase during preeclampsia (418).
It is interesting to mention that the placentae of women with SLE and APL syndrome show higher classical CS pathway activation, including higher C4d (a most important classical CS pathway activation marker) expression at the feto-maternal interface, leading to fetal loss and preeclampsia development, than normal healthy pregnant women (408, 419, 420). However, a very recent case–control study from Finland comprising pregnancies from 2000 to 2018 has indicated no statistical difference between pregnant women with SLE and normal pregnant women in the occurrence of preeclampsia or any other congenital malformation despite a significantly shorter duration of pregnancies and a more urgent need for cesarean section among pregnant women with SLE (421). A retrospective study comprising all SLE pregnancies during a period of 10 years (2006–2015) from a single center in Malaysia has indicated the development of complicated pregnancies, including preeclampsia, fetal losses, and the need for cesarean deliveries (422). Another retrospective study by a different group in Malaysia comprising pregnant SLE women for the period January 2008 to 2020 indicated the development of complicated pregnancies, such as SLE flares, preeclampsia, and eclampsia (423). Another retrospective study from Beijing, China, comprising 105 SLE-associated pregnancies for the period from January 1990 to December 2008, has also indicated complicated pregnancies, fetal loss, and preeclampsia development in active SLE pregnant women (424). A retrospective cohort study of 149 pregnancies in 98 women with SLE conducted over 10 years in Oman has also indicated the development of preeclampsia and eclampsia in these women along with an increase in SLE-associated pathologies, such as lupus nephritis and flares (425). The data from four retrospective studies performed in Sub-Saharan African pregnant women with SLE (137 pregnancies in 102 women) over a 26-year period have indicated a higher incidence of preeclampsia and aggravation of SLE symptoms, such as lupus nephritis and SLE flares, which further increased adverse pregnancy outcomes, including preeclampsia (426). This difference [geographical and ethnic origin (Europe, Asia, and Africa)] indicates the genetic and environmental impact on SLE and other autoimmune conditions affecting pregnancy outcomes, including preeclampsia, which must be explored. SLE and APL syndrome-mediated immune alteration, including CS pathway association with pregnancy loss and preeclampsia discussion, is beyond the scope of the current article and has been discussed elsewhere (427–429).
5.3 Mechanisms of CS pathway activation during preeclampsia
We have discussed earlier that altered CS signaling pathways are critical players in the onset and severity of preeclampsia. However, we do not know the triggers activating the CS pathway to induce inflammatory consequences that support and aggravate preeclampsia pathogenesis and outcome. For example, maternal hypertension and proteinuria (endothelial dysfunction) after 20 weeks of gestation are significant characteristics of preeclampsia, along with increased platelet aggregation and the hyperactivation of the coagulation system (430, 431). The pathogenesis of preeclampsia varies in nulliparous women compared with women with pre-existing vascular diseases, metabolic syndrome, multifetal gestation, and/or previous preeclampsia. However, some pregnant women with HELLP (hemolysis, elevated liver enzymes, or low platelet counts) syndrome (10%–15%) or eclampsia (38%) may not exhibit hypertension or proteinuria, which are associated with higher rates of maternal and fetal morbidities than in mild preeclampsia (432, 433). Interestingly, HELLP syndrome exhibits elevated maternal CS pathway activation as indicated by the increased systemic levels of different complement components, such as C5a, MAC, CFH, and CFH-related 1 and 3 (CFHR1 and 3), which are comparable to preeclampsia systemic values of complement components (434–437).
Furthermore, HELLP syndrome patients with complement gene variants exhibit poorer clinical outcomes than those with no complement gene variants. These patients have higher complement mutation frequency, including rare germline mutations in the alternative CS pathway (CFHR1, CFHR3, CFI, CFB, and CD46) than women with preeclampsia, having similar rates of variants as the control population (438, 439). Thus, pregnant women with complement gene variants are more likely to progress from preeclampsia to HELLP syndrome, where gene variants and pregnancy provide first and second hits, like other complement disorders, such as aHUS and paroxysmal nocturnal hemoglobinuria (PNH). Furthermore, a clinical trial with eculizumab, a long-acting human monoclonal antibody targeting C5 to block its cleavage to C5a and C5b, has shown positive results in phase 1 clinical trials of pregnant women with early-onset HELLP syndrome (440). Hence, CS activation is critical for the pathogenesis and severity of preeclampsia and HELLP syndrome. Therefore, it is critical to identify factors that dysregulate the CS activation during preeclampsia and its severe forms.
5.3.1 Maternal factors associated with increased risk of preeclampsia and their association with complement dysregulation
Women facing infertility associated with polycystic ovary syndrome (PCOS) and recurrent pregnancy loss (RPL; defined as ≥3 consecutive embryonic losses before 10 weeks’ gestation or ≥2 fetal losses after 10 weeks’ gestation) are more prone to develop preeclampsia (441). The immune system plays a critical role in the pathogenesis of PCOS and RPL, and the CS is a critical component of the immune system. Studies have shown that high maternal circulating C3 and C4 levels via CS signaling pathway activation before conception are associated with RPL independent of MBL CS pathways and other components of immunity, such as B cells and antibodies (442–445). Furthermore, women with specific mutations in C4BP carrying C4BP gene polymorphism R120H also suffer from RPL due to decreased C3b degradation that elevates their circulating C3b level (446). Another study has indicated several C3 gene variants associated with defective secretion/function of C3 in European women (n = 192) who suffered from RPL (447). Thus, dysregulated CS activation, such as the alternative CS signaling pathway in women who suffered RPL previously, predisposes them to develop preeclampsia during a successful pregnancy due to their altered CS signaling pathway, even in the presence of healthy placental development. Furthermore, fasting circulating complement components, such as C3 and C3a (desArg), are higher in PCOS women with insulin resistance, which increases to a similar extent in the control and PCOS groups (448). However, higher factor H levels are present in the circulation in PCOS women with obesity (448, 449).
Non-obese, non-insulin-resistant women with PCOS have higher systemic alternative and classical CS signaling pathway components, such as C3, iC3b, properdin, and C4 levels (450). Further study has indicated that upregulated alternative CS pathway components, such as C3, properdin, factor B, and factor I, are elevated in non-obese patients with PCOS, which further increases with obesity (449, 450). Systemic C5a levels are also increased in normal-weight women and women with obesity suffering from PCOS (449). Hence, activation and terminal CS pathway components are altered in PCOS women, which increases their propensity to develop RPL. Thus, we can speculate that infertile women (due to RPL and PCOS) with otherwise normal sexual function and immune components may have altered CS components, which increases their propensity to develop preeclampsia.
Elevated body mass index (BMI) and obesity are other definite preeclampsia risk factors (431, 451–454). AT comprises adipocytes and stromal vascular fraction (SVF), containing different cell types, such as preadipocytes, fibroblasts, and immune cells (macrophages and T cells). Adipocytes are the primary source of FD, a critical player in the alternative CS signaling pathway activation (455). Along with FD, other alternative CS components, such as C3, factor B, factor H, factor I, and properdin, are overexpressed in ATs, which increase with BMI and obesity status (455). Furthermore, adipocytes express C3aR and C5aRs (C5aR1 and C5aR2), which, via their corresponding ligands (C3a and C5a), increase the local and systemic inflammation along with increasing insulin resistance (244, 456, 457). Women with obesity with elevated circulating Bb (active protease, generated during the alternative CS signaling pathway) and C3a levels compared with the control group are more likely to develop preeclampsia (458, 459). Thus, women with obesity, high BMI, and insulin resistance or glucose tolerance have hyperactivated alternative CS signaling, which predisposes them to develop preeclampsia during their pregnancy. Hence, pre-pregnancy CS component dysregulation due to the above-mentioned factors in women increases their chances of facing preeclampsia during their pregnancy.
6 Future perspective and conclusion
Preeclampsia is a disease that specifically occurs during pregnancy; therefore, the placenta and dysregulated maternal immune response are key factors for its pathogenesis. However, immunological advances in pregnancy and preeclampsia have now clarified that poor placentation is not the only driving force behind preeclampsia pathogenesis but rather serves as a critical factor for preeclampsia predisposition (431). The degree of maternal physiological reaction, including the immune response severity, determines the predisposition to preeclampsia and its severity. For example, the immune system governs the effective allotransplantation of the fetus in the uterus of the pregnant woman by modifying the maternal local (uterine) and systemic immune response, vascular, and coagulatory functions, which are further governed by the hormonal and psychogenic changes taking place in a pregnant woman (460–465). Hence, any pre-pregnancy immune dysfunction can be lethal to the future mother and developing fetus, as the CS is the first and rapid immune component of innate immunity; therefore, its homeostatic levels during pregnancy are critical for a healthy outcome.
Hypertension development during pregnancy is a critical factor in the development of preeclampsia. A study has indicated systemic elevation of the clusterin (a complement regulatory protein) in pregnant women during pre-hypertension disorder of pregnancy (pre-HDP) development, which proved to be the critical factor for HDP development (466). Further studies have indicated increased clusterin systemic levels before the onset of preeclampsia clinical symptoms in pregnant women that increase with preeclampsia severity (467–469). Moreover, clusterin plays a critical role in the decidualization process by interacting with the triggering receptor expressed on myeloid cells 2 (TREM-2) receptor expressed on decidual cells, and its alteration may impact placental development, including trophoblast invasion (467, 470). The higher clusterin levels in the placenta inhibit MAC formation; therefore, it will be interesting to investigate the systemic clusterin and MAC levels during normal human pregnancy and preeclampsia.
Furthermore, aberrant and persistent CS activation (local and systemic) elevates systemic MAC or C5b-9 and C1q, causing systemic vasculitis or thromboinflammation that impairs endothelial function, which may cause hypertension (471–476). Thus, aberrant CS activation in pregnant women and their placentae may induce endothelial damage that may cause hypertension and elevate circulating endothelial cells, further aggravating the CS, predisposing them to preeclampsia, and increasing its severity (476, 477). For example, fetal endothelial cell damage and CS dysregulation (elevated MAC and C3a levels but decreased factor H and Bb) have been observed in pregnancies complicated by preeclampsia (478).
C3 is a critical component in hypertension pathogenesis due to its maintenance effect on undifferentiated mesenchymal stem cells (MSCs), and maternal and placental C3a levels are upregulated in women with preeclampsia, indicating aberrant CS activation (479). Furthermore, increased maternal circulating C5a in women with preeclampsia is positively correlated with maternal blood pressure and arterial stiffness (413). Targeting the CS during preeclampsia may prevent associated organ damage, such as renal manifestations, as the kidneys are among the most affected organs in preeclampsia (480). Hence, CS proteins must be checked for a healthy pregnancy.
Women with preeclampsia also exhibit a hypercoagulable state than women with normal pregnancy at an early stage of the disease (481). Women with preeclampsia have elevated circulating levels of factor VIII, von Willebrand factor (vWF; due to endothelial cell damage/inflammation), the thrombin–antithrombin complex (TAT), D dimers, soluble fibrin, and thrombomodulin levels than women with normal pregnancy (481–483). Increased fibrin deposition in women with preeclampsia occurs in the glomerulus sub-endothelium, spiral arteries, decidual components, and occlusive lesions of placental vasculature (484, 485). Fibrin deposition activates the classical CS signaling pathway by interacting with C1q by covalent interaction mediated by FXIIIa (53). However, this fibrin–C1q interaction is antagonized by factor H, which is downregulated in women with preeclampsia.
The activated CS signaling pathway in patients with preeclampsia can stimulate the extrinsic coagulation system pathway to form thrombin by increasing the tissue factor (TF) activity on different cells, such as endothelial cells, as evolutionarily the CS and coagulation system have a common origin and interact to maintain homeostasis and hemostasis (486–489). Moreover, overproduced plasmin, a protease generated in response to thrombin production and fibrin deposition, also serves as C5 convertase and cleaves C5 into C5a and C5b to induce the inflammatory cascade and the assembly of procoagulant C5b-9 or the MAC (490). In addition to C5 cleavage, plasmin also cleaves C3 into C3a, which is upregulated in women with preeclampsia (491). Furthermore, inflammatory events, including organ injuries, complement (increase in C5a levels), coagulation (thrombin–antithrombin complexes), activation, and cross-talk, are very early events, which have also been reported in patients with preeclampsia (491). Several other coagulation pathway components, such as Factor Xa, thrombin, FIXa, and FXIa, also cleave C3 and C5 into biologically active C3a and C5a capable of exerting their pro-inflammatory effects (491, 492). Further maternal proteomics-based study has indicated the increased deposition of C5b-9 or the MAC and vWF in the endothelial cells of women with early-onset severe preeclampsia, indicating that the complement and coagulation systems are the critical pathways for early-onset severe preeclampsia (493). Thus, aberrant complement activation not only dysregulates immune homeostasis but also affects the coagulation system, hypertension, and metabolism to initiate and increase the severity of preeclampsia.
Moreover, increased circulating C3a levels have been observed in women with depression; therefore, it may be interesting to investigate whether increased circulating C3a levels predispose women to develop preeclampsia upon getting pregnant (494). Increased circulating C3a levels during pregnancy are a critical biomarker not only for preeclampsia prediction but also for depression during pregnancy, as the Edinburgh postnatal depression scale (EPDS) alone is not perfectly sufficient to detect major depressive disorder during pregnancy (495). Interestingly, more than 10% of pregnant women in high-income countries, such as the United States, have depression during pregnancy (496).
6.1 Conclusion
Measuring different circulating complement proteins in pregnant women may serve as a biomarker for early preeclampsia detection. Targeting the CS in pregnant women with preeclampsia will complement normal pregnancy and associated organ damage. Understanding CS signaling during preeclampsia will further help to track future maternal health issues, such as metabolic, cardiovascular, and neurologic disorders in survivors. Complementing helps in a healthy pregnancy, but decomplementing will equip us to fight against preeclampsia and other future health issues in preeclampsia survivors. Therefore, future studies are warranted to understand the CS signaling pathways’ alteration and their mechanism of action in human pregnancy and preeclampsia.
Author contributions
VK: Writing – original draft, Conceptualization, Software, Writing – review & editing. JS: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kumar V and Medhi B. Emerging role of uterine natural killer cells in establishing pregnancy. Iranian J Immunol. (2008) 5:71–81. doi: 10.22034/iji.2008.48555
2. Feyaerts D, Joosten I, and van der Molen RG. A pregnancy to remember: trained immunity of the uterine mucosae. Mucosal Immunol. (2021) 14:539–41. doi: 10.1038/s41385-020-00362-7
3. Zhou JZ, Way SS, and Chen K. Immunology of uterine and vaginal mucosae: (Trends in immunology 39, 302-314, 2018). Trends Immunol. (2018) 39:355. doi: 10.1016/j.it.2018.02.006
4. Solano ME. Decidual immune cells: Guardians of human pregnancies. Best Pract Res Clin Obstetrics Gynaecology. (2019) 60:3–16. doi: 10.1016/j.bpobgyn.2019.05.009
5. Wegmann TG, Lin H, Guilbert L, and Mosmann TR. Bidirectional cytokine interactions in the maternal-fetal relationship: is successful pregnancy a TH2 phenomenon? Immunol Today. (1993) 14:353–6. doi: 10.1016/0167-5699(93)90235-D
6. Vince GS and Johnson PM. Is there a Th2 bias in human pregnancy? J Reprod Immunol. (1996) 32:101–4. doi: 10.1016/S0165-0378(96)00995-3
7. Male V and Moffett A. Natural killer cells in the human uterine mucosa. Annu Rev Immunol. (2023) 41:127–51. doi: 10.1146/annurev-immunol-102119-075119
8. Sacks G, Sargent I, and Redman C. An innate view of human pregnancy. Immunol Today. (1999) 20:114–8. doi: 10.1016/S0167-5699(98)01393-0
9. Colamatteo A, Fusco C, Micillo T, D’Hooghe T, de Candia P, Alviggi C, et al. Immunobiology of pregnancy: from basic science to translational medicine. Trends Mol Med. (2023) 29:711–25. doi: 10.1016/j.molmed.2023.05.009
10. Orefice R. Immunology and the immunological response in pregnancy. Best Pract Res Clin Obstetrics Gynaecology. (2021) 76:3–12. doi: 10.1016/j.bpobgyn.2020.07.013
11. Kolev M, Le Friec G, and Kemper C. Complement–tapping into new sites and effector systems. Nat Rev Immunol. (2014) 14:811–20. doi: 10.1038/nri3761
12. Kumar V. The complement system, toll-like receptors and inflammasomes in host defense: three musketeers’ one target. Int Rev Immunol. (2019) 38:131–56. doi: 10.1080/08830185.2019.1609962
13. Nonaka M and Kimura A. Genomic view of the evolution of the complement system. Immunogenetics. (2006) 58:701–13. doi: 10.1007/s00251-006-0142-1
14. Lynch VJ, Nnamani MC, Kapusta A, Brayer K, Plaza SL, Mazur EC, et al. Ancient transposable elements transformed the uterine regulatory landscape and transcriptome during the evolution of mammalian pregnancy. Cell Rep. (2015) 10:551–61. doi: 10.1016/j.celrep.2014.12.052
15. Mastellos DC, Hajishengallis G, and Lambris JD. A guide to complement biology, pathology and therapeutic opportunity. Nat Rev Immunol. (2024) 24:118–41. doi: 10.1038/s41577-023-00926-1
16. Son M. Understanding the contextual functions of C1q and LAIR-1 and their applications. Exp Mol Med. (2022) 54:567–72. doi: 10.1038/s12276-022-00774-4
18. Thorgersen EB, Barratt-Due A, Haugaa H, Harboe M, Pischke SE, Nilsson PH, et al. The role of complement in liver injury, regeneration, and transplantation. Hepatology. (2019) 70:725–36. doi: 10.1002/hep.30508
19. Lubbers R, van Essen MF, van Kooten C, and Trouw LA. Production of complement components by cells of the immune system. Clin Exp Immunol. (2017) 188:183–94. doi: 10.1111/cei.12952
20. Farries TC, Steuer KL, and Atkinson JP. Evolutionary implications of a new bypass activation pathway of the complement system. Immunol Today. (1990) 11:78–80. doi: 10.1016/0167-5699(90)90031-4
21. Farries TC and Atkinson JP. Evolution of the complement system. Immunol Today. (1991) 12:295–300. doi: 10.1016/0167-5699(91)90002-B
22. Ricklin D, Reis ES, Mastellos DC, Gros P, and Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. (2016) 274:33–58. doi: 10.1111/imr.12500
23. Coulthard LG and Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Immunol. (2015) 194:3542–8. doi: 10.4049/jimmunol.1403068
24. Sekine H, Machida T, and Fujita T. Factor D. Immunol Rev. (2023) 313:15–24. doi: 10.1111/imr.13155
25. Barratt J and Weitz I. Complement factor D as a strategic target for regulating the alternative complement pathway. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.712572
26. Harboe M and Mollnes TE. The alternative complement pathway revisited. J Cell Mol Med. (2008) 12:1074–84. doi: 10.1111/j.1582-4934.2008.00350.x
27. Ricklin D, Hajishengallis G, Yang K, and Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785–97. doi: 10.1038/ni.1923
28. Fearon DT and Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med. (1975) 142:856–63. doi: 10.1084/jem.142.4.856
29. Müller-Eberhard HJ. The killer molecule of complement. J Invest Dermatol. (1985) 85:47s–52s. doi: 10.1111/1523-1747.ep12275445
30. Podack ER and Tschopp J. Membrane attack by complement. Mol Immunol. (1984) 21:589–603. doi: 10.1016/0161-5890(84)90044-0
31. Prada AE, Zahedi K, and Davis AE. 3rd, Regulation of C1 inhibitor synthesis. Immunobiology. (1998) 199:377–88. doi: 10.1016/S0171-2985(98)80042-9
32. Gitlin D and Biasucci A. Development of γG, γA, γM, β 1C/β 1A, C′ 1 esterase inhibitor, ceruloplasmin, transferrin, hemopexin, haptoglobin, fibrinogen, plasminogen, α 1-antitrypsin, orosomucoid, β-lipoprotein, α 2-macroglobulin, and prealbumin in the human conceptus. J Clin Invest. (1969) 48:1433–46. doi: 10.1172/JCI106109
33. Schmaier AH, Smith PM, and Colman RW. Platelet C1- inhibitor. A secreted alpha-granule protein. J Clin Invest. (1985) 75:242–50. doi: 10.1172/JCI111680
34. Schmaier AH, Amenta S, Xiong T, Heda GD, and Gewirtz AM. Expression of platelet C1 inhibitor. Blood. (1993) 82:465–74. doi: 10.1182/blood.V82.2.465.465
35. Armbrust T, Schwögler S, Zöhrens G, and Ramadori G. C1 esterase inhibitor gene expression in rat Kupffer cells, peritoneal macrophages and blood monocytes: modulation by interferon gamma. J Exp Med. (1993) 178:373–80. doi: 10.1084/jem.178.2.373
36. Katz Y and Strunk RC. Synthesis and regulation of C1 inhibitor in human skin fibroblasts. J Immunol. (1989) 142:2041–5. doi: 10.4049/jimmunol.142.6.2041
37. Harpel PC and Cooper NR. Studies on human plasma C1 inactivator-enzyme interactions. I. Mechanisms of interaction with C1s, plasmin, and trypsin. J Clin Invest. (1975) 55:593–604. doi: 10.1172/JCI107967
38. Chen CH and Boackle RJ. A newly discovered function for C1 inhibitor, removal of the entire C1qr2s2 complex from immobilized human IgG subclasses. Clin Immunol Immunopathol. (1998) 87:68–74. doi: 10.1006/clin.1997.4515
39. Chen CH, Lam CF, and Boackle RJ. C1 inhibitor removes the entire C1qr2s2 complex from anti-C1Q monoclonal antibodies with low binding affinities. Immunology. (1998) 95:648–54. doi: 10.1046/j.1365-2567.1998.00635.x
40. Schreiber AD, Kaplan AP, and Austen KF. Inhibition by C1INH of Hagemann factor fragment activation of coagulation, fibrinolysis, and kinin generation. J Clin Invest. (1973) 52:1402–9. doi: 10.1172/JCI107313
41. Ratnoff OD, Pensky J, Ogston D, and Naff GB. The inhibition of plasmin, plasma kallikrein, plasma permeability factor, and the C’1r subcomponent of the first component of complement by serum C’1 esterase inhibitor. J Exp Med. (1969) 129:315–31. doi: 10.1084/jem.129.2.315
42. Jiang H, Wagner E, Zhang H, and Frank MM. Complement 1 inhibitor is a regulator of the alternative complement pathway. J Exp Med. (2001) 194:1609–16. doi: 10.1084/jem.194.11.1609
43. Cai S and Davis AE. III, complement regulatory protein C1 inhibitor binds to selectins and interferes with endothelial-leukocyte adhesion 1. J Immunol. (2003) 171:4786–91. doi: 10.4049/jimmunol.171.9.4786
44. Gompels MM, Lock RJ, Abinun M, Bethune CA, Davies G, Grattan C, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol. (2005) 139:379–94. doi: 10.1111/j.1365-2249.2005.02726.x
45. Hallam TM, Sharp SJ, Andreadi A, and Kavanagh D. Complement factor I: Regulatory nexus, driver of immunopathology, and therapeutic. Immunobiology. (2023) 228:152410. doi: 10.1016/j.imbio.2023.152410
46. Nilsson SC, Sim RB, Lea SM, Fremeaux-Bacchi V, and Blom AM. Complement factor I in health and disease. Mol Immunol. (2011) 48:1611–20. doi: 10.1016/j.molimm.2011.04.004
47. Cole JL, Housley GA Jr., Dykman TR, MacDermott RP, and Atkinson JP. Identification of an additional class of C3-binding membrane proteins of human peripheral blood leukocytes and cell lines. Proc Natl Acad Sci U.S.A. (1985) 82:859–63.
48. Cserhalmi M, Papp A, Brandus B, Uzonyi B, and Józsi M. Regulation of regulators: Role of the complement factor H-related proteins. Semin Immunol. (2019) 45:101341. doi: 10.1016/j.smim.2019.101341
49. Kiss MG, Papac-Miličević N, Porsch F, Tsiantoulas D, Hendrikx T, Takaoka M, et al. Cell-autonomous regulation of complement C3 by factor H limits macrophage efferocytosis and exacerbates atherosclerosis. Immunity. (2023) 56:1809–1824.e1810. doi: 10.1016/j.immuni.2023.06.026
50. Pouw RB, Brouwer MC, de Gast M, van Beek AE, van den Heuvel LP, Schmidt CQ, et al. Potentiation of complement regulator factor H protects human endothelial cells from complement attack in aHUS sera. Blood Adv. (2019) 3:621–32. doi: 10.1182/bloodadvances.2018025692
51. Mahajan S, Jacob A, Kelkar A, Chang A, McSkimming D, Neelamegham S, et al. Local complement factor H protects kidney endothelial cell structure and function. Kidney Int. (2021) 100:824–36. doi: 10.1016/j.kint.2021.05.033
52. Wu J, Wu Y-Q, Ricklin D, Janssen BJC, Lambris JD, and Gros P. Structure of complement fragment C3b–factor H and implications for host protection by complement regulators. Nat Immunol. (2009) 10:728–33. doi: 10.1038/ni.1755
53. Kang YH, Varghese PM, Aiyan AA, Pondman K, Kishore U, and Sim RB. Complement-Coagulation Cross-talk: Factor H-mediated regulation of the Complement Classical Pathway activation by fibrin clots. Front Immunol. (2024) 15:1368852. doi: 10.3389/fimmu.2024.1368852
54. Cipriani V, Tierney A, Griffiths JR, Zuber V, Sergouniotis PI, Yates JRW, et al. Beyond factor H: The impact of genetic-risk variants for age-related macular degeneration on circulating factor-H-like 1 and factor-H-related protein concentrations. Am J Hum Genet. (2021) 108:1385–400. doi: 10.1016/j.ajhg.2021.05.015
55. Lorés-Motta L, van Beek AE, Willems E, Zandstra J, van Mierlo G, Einhaus A, et al. Common haplotypes at the CFH locus and low-frequency variants in CFHR2 and CFHR5 associate with systemic FHR concentrations and age-related macular degeneration. Am J Hum Genet. (2021) 108:1367–84. doi: 10.1016/j.ajhg.2021.06.002
56. Sándor N, Schneider AE, Matola AT, Barbai VH, Bencze D, Hammad HH, et al. The human factor H protein family – an update. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1135490
57. Józsi M, Tortajada A, Uzonyi B, Goicoechea de Jorge E, and Rodríguez de Córdoba S. Factor H-related proteins determine complement-activating surfaces. Trends Immunol. (2015) 36:374–84. doi: 10.1016/j.it.2015.04.008
58. Hebecker M and Józsi M. Factor H-related protein 4 activates complement by serving as a platform for the assembly of alternative pathway C3 convertase via its interaction with C3b protein. J Biol Chem. (2012) 287:19528–36. doi: 10.1074/jbc.M112.364471
59. Bernabéu-Herrero ME, Jiménez-Alcázar M, Anter J, Pinto S, Sánchez Chinchilla D, Garrido S, et al. FHR-3 and FHR-1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. (2015) 67:276–86. doi: 10.1016/j.molimm.2015.06.021
60. Li X, Zong J, and Si S. Complement Factor H related protein 1 and immune inflammatory disorders. Mol Immunol. (2022) 145:43–9. doi: 10.1016/j.molimm.2022.03.117
61. Blom AM, Villoutreix BO, and Dahlbäck B. Complement inhibitor C4b-binding protein—friend or foe in the innate immune system? Mol Immunol. (2004) 40:1333–46. doi: 10.1016/j.molimm.2003.12.002
62. Werner LM and Criss AK. Diverse functions of C4b-binding protein in health and disease. J Immunol. (2023) 211:1443–9. doi: 10.4049/jimmunol.2300333
63. Seya T, Holers VM, and Atkinson JP. Purification and functional analysis of the polymorphic variants of the C3b/C4b receptor (CR1) and comparison with H, C4b-binding protein (C4bp), and decay accelerating factor (DAF). J Immunol (Baltimore Md.: 1950). (1985) 135:2661–7. doi: 10.4049/jimmunol.135.4.2661
64. Seya T, Nakamura K, Masaki T, Ichihara-Itoh C, Matsumoto M, Nagasawa S, et al. and C4b-binding protein serve as factor I-cofactors both encompassing inactivation of C3b and C4b. Mol Immunol. (1995) 32:355–60. doi: 10.1016/0161-5890(94)00157-V
65. Blom AM, Kask L, and Dahlbäck B. Structural requirements for the complement regulatory activities of C4BP*. J Biol Chem. (2001) 276:27136–44. doi: 10.1074/jbc.M102445200
66. Sjöberg AP, Trouw LA, McGrath FDG, Hack CE, and Blom AM. Regulation of complement activation by C-reactive protein: targeting of the inhibitory activity of C4b-binding protein1. J Immunol. (2006) 176:7612–20. doi: 10.4049/jimmunol.176.12.7612
67. Varghese PM, Murugaiah V, Beirag N, Temperton N, Khan HA, Alrokayan SH, et al. C4b binding protein acts as an innate immune effector against influenza A virus. Front Immunol. (2021) 11. doi: 10.3389/fimmu.2020.585361
68. Bierschenk D, Papac-Milicevic N, Bresch IP, Kovacic V, Bettoni S, Dziedzic M, et al. C4b-binding protein inhibits particulate- and crystalline-induced NLRP3 inflammasome activation. Front Immunol. (2023) 14:1149822. doi: 10.3389/fimmu.2023.1149822
69. Trapp RG, Fletcher M, Forristal J, and West CD. C4 binding protein deficiency in a patient with atypical Behçet’s disease. J Rheumatol. (1987) 14:135–8.
70. Schwaeble WJ and Reid KBM. Does properdin crosslink the cellular and the humoral immune response? Immunol Today. (1999) 20:17–21. doi: 10.1016/s0167-5699(98)01376-0
71. Pillemer L, Blum L, Lepow IH, Ross OA, Todd EW, and Wardlaw AC. The properdin system and immunity: I. Demonstration and isolation of a new serum protein, properdin, and its role in immune phenomena. Science. (1954) 120:279–85. doi: 10.1126/science.120.3112.279
72. Ferreira VP. Chapter 27 - properdin. In: Barnum S and Schein T, editors. The Complement FactsBook, 2nd ed. USA: Academic Press (2018). p. pp 283–293.
73. Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement*. J Biol Chem. (2006) 281:2128–32. doi: 10.1074/jbc.M508928200
74. Medicus RG, Götze O, and Müller-Eberhard HJ. Alternative pathway of complement: recruitment of precursor properdin by the labile C3/C5 convertase and the potentiation of the pathway. J Exp Med. (1976) 144:1076–93. doi: 10.1084/jem.144.4.1076
75. Densen P, Weiler JM, Griffiss JM, and Hoffmann LG. Familial properdin deficiency and fatal meningococcemia. Correction of the bactericidal defect by vaccination. N Engl J Med. (1987) 316:922–6. doi: 10.1056/NEJM198704093161506
76. Schlesinger M, Nave Z, Levy Y, Slater PE, and Fishelson Z. Prevalence of hereditary properdin, C7 and C8 deficiencies in patients with meningococcal infections. Clin Exp Immunol. (1990) 81:423–7. doi: 10.1111/j.1365-2249.1990.tb05350.x
77. Linton SM and Morgan BP. Properdin deficiency and meningococcal disease–identifying those most at risk. Clin Exp Immunol. (1999) 118:189–91. doi: 10.1046/j.1365-2249.1999.01057.x
78. Fijen CA, van den Bogaard R, Schipper M, Mannens M, Schlesinger M, Nordin FG, et al. Properdin deficiency: molecular basis and disease association. Mol Immunol. (1999) 36:863–7. doi: 10.1016/S0161-5890(99)00107-8
79. Sullivan KE. Chapter 81 - inherited complement deficiencies. In: Rimoin D, Pyeritz R, and Korf B, editors. Emery and Rimoin’s Principles and Practice of Medical Genetics, Sixth Edition. Academic Press, Oxford (2013). p. pp 1–13. doi: 10.1016/B978-0-12-383834-6.00085-9
80. Tschopp J, Chonn A, Hertig S, and French LE. Clusterin, the human apolipoprotein and complement inhibitor, binds to complement C7, C8 beta, and the b domain of C9. J Immunol. (1993) 151:2159–65. doi: 10.4049/jimmunol.151.4.2159
81. Massri M, Toonen EJM, Sarg B, Kremser L, Grasse M, Fleischer V, et al. Complement C7 and clusterin form a complex in circulation. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1330095
82. Chauhan AK and Moore TL. Presence of plasma complement regulatory proteins clusterin (Apo J) and vitronectin (S40) on circulating immune complexes (CIC). Clin Exp Immunol. (2006) 145:398–406. doi: 10.1111/j.1365-2249.2006.03135.x
83. Doudevski I, Rostagno A, Cowman M, Liebmann J, Ritch R, and Ghiso J. Clusterin and complement activation in exfoliation glaucoma. Invest Ophthalmol Visual Sci. (2014) 55:2491–9. doi: 10.1167/iovs.13-12941
84. Hochgrebe TT, Humphreys D, Wilson MR, and Easterbrook-Smith SB. A reexamination of the role of clusterin as a complement regulator. Exp Cell Res. (1999) 249:13–21. doi: 10.1006/excr.1999.4459
85. Schvartz I, Seger D, and Shaltiel S. Vitronectin. Int J Biochem Cell Biol. (1999) 31:539–44. doi: 10.1016/S1357-2725(99)00005-9
86. Milis L, Morris CA, Sheehan MC, Charlesworth JA, and Pussell BA. Vitronectin-mediated inhibition of complement: evidence for different binding sites for C5b-7 and C9. Clin Exp Immunol. (1993) 92:114–9. doi: 10.1111/j.1365-2249.1993.tb05956.x
87. Zheng X, Saunders TL, Camper SA, Samuelson LC, and Ginsburg D. Vitronectin is not essential for normal mammalian development and fertility. Proc Natl Acad Sci U.S.A. (1995) 92:12426–30. doi: 10.1073/pnas.92.26.12426
88. Singh B, Su Y-C, and Riesbeck K. Vitronectin in bacterial pathogenesis: a host protein used in complement escape and cellular invasion. Mol Microbiol. (2010) 78:545–60. doi: 10.1111/j.1365-2958.2010.07373.x
89. Jang Y-C, Tsou R, Gibran NS, and Isik FF. Vitronectin deficiency is associated with increased wound fibrinolysis and decreased microvascular angiogenesis in mice. Surgery. (2000) 127:696–704. doi: 10.1067/msy.2000.105858
90. Hayashida M, Hashimoto K, Ishikawa T, and Miyamoto Y. Vitronectin deficiency attenuates hepatic fibrosis in a non-alcoholic steatohepatitis-induced mouse model. Int J Exp Pathol. (2019) 100:72–82. doi: 10.1111/iep.12306
91. Fearon DT. Identification of the membrane glycoprotein that is the C3b receptor of the human erythrocyte, polymorphonuclear leukocyte, B lymphocyte, and monocyte. J Exp Med. (1980) 152:20–30. doi: 10.1084/jem.152.1.20
92. Dustin ML. Complement receptors in myeloid cell adhesion and phagocytosis. Microbiol Spectr. (2016) 4. doi: 10.1128/microbiolspec.mchd-0034-2016
93. Kazatchkine MD and Fearon DT. Deficiencies of human C3 complement receptors type 1 (CR1, CD35) and type 2 (CR2, CD21). Immunodefic Rev. (1990) 2:17–41.
94. Steiniger B, Trabandt M, and Barth PJ. The follicular dendritic cell network in secondary follicles of human palatine tonsils and spleens. Histochem Cell Biol. (2011) 135:327–36. doi: 10.1007/s00418-011-0799-x
95. Asokan R, Banda NK, Szakonyi G, Chen XS, and Holers VM. Human complement receptor 2 (CR2/CD21) as a receptor for DNA: implications for its roles in the immune response and the pathogenesis of systemic lupus erythematosus (SLE). Mol Immunol. (2013) 53:99–110. doi: 10.1016/j.molimm.2012.07.002
96. Iida K, Nadler L, and Nussenzweig V. Identification of the membrane receptor for the complement fragment C3d by means of a monoclonal antibody. J Exp Med. (1983) 158:1021–33. doi: 10.1084/jem.158.4.1021
97. Wagner C, Ochmann C, Schoels M, Giese T, Stegmaier S, Richter R, et al. The complement receptor 1, CR1 (CD35), mediates inhibitory signals in human T-lymphocytes. Mol Immunol. (2006) 43:643–51. doi: 10.1016/j.molimm.2005.04.006
98. Kovács KG, Mácsik-Valent B, Matkó J, Bajtay Z, and Erdei A. Revisiting the coreceptor function of complement receptor type 2 (CR2, CD21); coengagement with the B-cell receptor inhibits the activation, proliferation, and antibody production of human B cells. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.620427
99. Vorup-Jensen T and Jensen RK. Structural immunology of complement receptors 3 and 4. Front Immunol. (2018) 9:2716. doi: 10.3389/fimmu.2018.02716
100. Sándor N, Kristóf K, Paréj K, Pap D, Erdei A, and Bajtay Z. CR3 is the dominant phagocytotic complement receptor on human dendritic cells. Immunobiology. (2013) 218:652–63. doi: 10.1016/j.imbio.2012.07.031
101. Wagner C, Hänsch GM, Stegmaier S, Denefleh B, Hug F, and Schoels M. The complement receptor 3, CR3 (CD11b/CD18), on T lymphocytes: activation-dependent up-regulation and regulatory function. Eur J Immunol. (2001) 31:1173–80. doi: 10.1002/1521-4141(200104)31:4<1173::AID-IMMU1173>3.0.CO;2-9
102. Klein E, Di Renzo L, and Yefenof E. Contribution of CR3, CD11b/CD18 to cytolysis by human NK cells. Mol Immunol. (1990) 27:1343–7. doi: 10.1016/0161-5890(90)90041-W
103. Vik DP and Fearon DT. Cellular distribution of complement receptor type 4 (CR4): expression on human platelets. J Immunol. (1987) 138:254–8. doi: 10.4049/jimmunol.138.1.254
104. Walbaum S, Ambrosy B, Schütz P, Bachg AC, Horsthemke M, Leusen JHW, et al. Complement receptor 3 mediates both sinking phagocytosis and phagocytic cup formation via distinct mechanisms. J Biol Chem. (2021) 296. doi: 10.1016/j.jbc.2021.100256
105. Lukácsi S, Nagy-Baló Z, Erdei A, Sándor N, and Bajtay Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol Lett. (2017) 189:64–72. doi: 10.1016/j.imlet.2017.05.014
106. Taborda CP and Casadevall A. CR3 (CD11b/CD18) and CR4 (CD11c/CD18) are involved in complement-independent antibody-mediated phagocytosis of Cryptococcus neoformans. Immunity. (2002) 16:791–802. doi: 10.1016/S1074-7613(02)00328-X
107. Lim J, Wiedemann A, Tzircotis G, Monkley SJ, Critchley DR, and Caron E. An essential role for talin during αMβ2-mediated phagocytosis. Mol Biol Cell. (2007) 18:976–85. doi: 10.1091/mbc.e06-09-0813
108. Kazatchkine MD, Jouvin MH, Wilson JG, Fischer E, and Fischer A. Human diseases associated with C3 receptor deficiencies. Immunol Lett. (1987) 14:191–5. doi: 10.1016/0165-2478(87)90100-3
109. Bermejo-Jambrina M, Blatzer M, Jauregui-Onieva P, Yordanov TE, Hörtnagl P, Valovka T, et al. CR4 signaling contributes to a DC-driven enhanced immune response against complement-opsonized HIV-1. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.02010
110. Wiesmann C, Katschke KJ, Yin J, Helmy KY, Steffek M, Fairbrother WJ, et al. Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature. (2006) 444:217–20. doi: 10.1038/nature05263
111. Gorgani NN, He JQ, Katschke KJ, Helmy KY, Xi H, Steffek M, et al. Complement receptor of the Ig superfamily enhances complement-mediated phagocytosis in a subpopulation of tissue resident macrophages. J Immunol. (2008) 181:7902–8. doi: 10.4049/jimmunol.181.11.7902
112. Zeng Z, Surewaard BGJ, Wong CHY, Geoghegan JA, Jenne CN, and Kubes P. CRIg functions as a macrophage pattern recognition receptor to directly bind and capture blood-borne gram-positive bacteria. Cell Host Microbe. (2016) 20:99–106. doi: 10.1016/j.chom.2016.06.002
113. Vogt L, Schmitz N, Kurrer MO, Bauer M, Hinton HI, Behnke S, et al. VSIG4, a B7 family–related protein, is a negative regulator of T cell activation. J Clin Invest. (2006) 116:2817–26. doi: 10.1172/JCI25673
114. Anderson DC and Springer TA. Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p 150, 95 glycoproteins. Annu Rev Med. (1987) 38:175–94. doi: 10.1146/annurev.me.38.020187.001135
115. Liszewski MK and Atkinson JP. Membrane cofactor protein (MCP; CD46): deficiency states and pathogen connections. Curr Opin Immunol. (2021) 72:126–34. doi: 10.1016/j.coi.2021.04.005
116. Liszewski MK and Kemper C. Complement in motion: the evolution of CD46 from a complement regulator to an orchestrator of normal cell physiology. J Immunol. (2019) 203:3–5. doi: 10.4049/jimmunol.1900527
117. Liszewski MK and Atkinson JP. Complement regulator CD46: genetic variants and disease associations. Hum Genomics. (2015) 9:7. doi: 10.1186/s40246-015-0029-z
118. Richards A, Kemp EJ, Liszewski MK, Goodship JA, Lampe AK, Decorte R, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci. (2003) 100:12966–71. doi: 10.1073/pnas.2135497100
119. Cocuzzi E, Szczotka LB, Brodbeck WG, Bardenstein DS, Wei T, and Medof ME. Tears contain the complement regulator CD59 as well as decay-accelerating factor (DAF). Clin Exp Immunol. (2001) 123:188–95. doi: 10.1046/j.1365-2249.2001.01408.x
120. Dho SH, Lim JC, and Kim LK. Beyond the role of CD55 as a complement component. Immune Netw. (2018) 18:e11. doi: 10.4110/in.2018.18.e11
121. Marschang P, Sodroski J, Würzner R, and Dierich MP. Decay-accelerating factor (CD55) protects human immunodeficiency virus type 1 from inactivation by human complement. Eur J Immunol. (1995) 25:285–90. doi: 10.1002/eji.1830250147
122. Kwon Y-C, Kim H, Meyer K, Di Bisceglie AM, and Ray R. Distinct CD55 isoform synthesis and inhibition of complement-dependent cytolysis by hepatitis C virus. J Immunol. (2016) 197:1127–36. doi: 10.4049/jimmunol.1600631
123. Capasso M, Durrant LG, Stacey M, Gordon S, Ramage J, and Spendlove I. Costimulation via CD55 on human CD4+ T cells mediated by CD97. J Immunol. (2006) 177:1070–7. doi: 10.4049/jimmunol.177.2.1070
124. Finberg RW, White W, and Nicholson-Weller A. Decay-accelerating factor expression on either effector or target cells inhibits cytotoxicity by human natural killer cells. J Immunol (Baltimore Md.: 1950). (1992) 149:2055–60. doi: 10.4049/jimmunol.149.6.2055
125. Parker C, Omine M, Richards S, Nishimura J-I, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. (2005) 106:3699–709. doi: 10.1182/blood-2005-04-1717
126. Ozen A. CHAPLE syndrome uncovers the primary role of complement in a familial form of Waldmann’s disease. Immunol Rev. (2019) 287:20–32. doi: 10.1111/imr.12715
127. Ruiz-Argüelles A and Llorente L. The role of complement regulatory proteins (CD55 and CD59) in the pathogenesis of autoimmune hemocytopenias. Autoimmun Rev. (2007) 6:155–61. doi: 10.1016/j.autrev.2006.09.008
128. Meri S, Waldmann H, and Lachmann PJ. Distribution of protectin (CD59), a complement membrane attack inhibitor, in normal human tissues. Lab Invest. (1991) 65:532–7.
129. Couves EC, Gardner S, Voisin TB, Bickel JK, Stansfeld PJ, Tate EW, et al. Structural basis for membrane attack complex inhibition by CD59. Nat Commun. (2023) 14:890. doi: 10.1038/s41467-023-36441-z
130. Spear GT, Lurain NS, Parker CJ, Ghassemi M, Payne GH, and Saifuddin M. Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV). J Immunol (Baltimore Md.: 1950). (1995) 155:4376–81. doi: 10.4049/jimmunol.155.9.4376
131. McGreal EP, Ikewaki N, Akatsu H, Morgan BP, and Gasque P. Human C1qRp is identical with CD93 and the mNI-11 antigen but does not bind C1q. J Immunol. (2002) 168:5222–32. doi: 10.4049/jimmunol.168.10.5222
132. Tenner AJ. C1q receptors: regulating specific functions of phagocytic cells. Immunobiology. (1998) 199:250–64. doi: 10.1016/S0171-2985(98)80031-4
133. Chevrier S, Genton C, Kallies A, Karnowski A, Otten LA, Malissen B, et al. CD93 is required for maintenance of antibody secretion and persistence of plasma cells in the bone marrow niche. Proc Natl Acad Sci U.S.A. (2009) 106:3895–900. doi: 10.1073/pnas.0809736106
134. Ikewaki N, Yamao H, Kulski JK, and Inoko H. Flow cytometric identification of CD93 expression on naive T lymphocytes (CD4(+)CD45RA (+) cells) in human neonatal umbilical cord blood. J Clin Immunol. (2010) 30:723–33. doi: 10.1007/s10875-010-9426-1
135. Vemuri K, de Alves Pereira B, Fuenzalida P, Subashi Y, Barbera S, van Hooren L, et al. CD93 maintains endothelial barrier function and limits metastatic dissemination. JCI Insight. (2024) 9. doi: 10.1172/jci.insight.169830
136. Norsworthy PJ, Fossati-Jimack L, Cortes-Hernandez J, Taylor PR, Bygrave AE, Thompson RD, et al. Murine CD93 (C1qRp) contributes to the removal of apoptotic cells in vivo but is not required for C1q-mediated enhancement of phagocytosis. J Immunol. (2004) 172:3406–14. doi: 10.4049/jimmunol.172.6.3406
137. Nepomuceno RR, Ruiz S, Park M, and Tenner AJ. C1qRP is a heavily O-glycosylated cell surface protein involved in the regulation of phagocytic activity. J Immunol. (1999) 162:3583–9. doi: 10.4049/jimmunol.162.6.3583
138. Tossetta G, Piani F, Borghi C, and Marzioni D. Role of CD93 in health and disease. Cells. (2023) 12. doi: 10.3390/cells12131778
139. Kishore U and Reid KB. C1q: structure, function, and receptors. Immunopharmacology. (2000) 49:159–70. doi: 10.1016/S0162-3109(00)80301-X
140. Eggleton P, Tenner AJ, and Reid KB. C1q receptors. Clin Exp Immunol. (2000) 120:406–12. doi: 10.1046/j.1365-2249.2000.01218.x
141. Sim RB, Moestrup SK, Stuart GR, Lynch NJ, Lu J, Schwaeble WJ, et al. Interaction of C1q and the collectins with the potential receptors calreticulin (cClqR/collectin receptor) and megalin. Immunobiology. (1998) 199:208–24. doi: 10.1016/S0171-2985(98)80028-4
142. Qiu Y, Marquez-Curtis LA, and Janowska-Wieczorek A. Mesenchymal stem cells express the surface receptor calreticulin (cC1qR) and complement C1q chemoattracts them. Blood. (2010) 116:3855–5. doi: 10.1182/blood.V116.21.3855.3855
143. Hirano M, Totani K, Fukuda T, Gu J, and Suzuki A. N-Glycoform-dependent interactions of megalin with its ligands. Biochim Biophys Acta (BBA) - Gen Subj. (2017) 1861:3106–18. doi: 10.1016/j.bbagen.2016.10.015
144. Hammad R, Aglan RB, Mohammed SA, Awad EA, Elsaid MA, Bedair HM, et al. Cytotoxic T cell expression of leukocyte-associated immunoglobulin-like receptor-1 (LAIR-1) in viral hepatitis C-mediated hepatocellular carcinoma. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232012541
145. Helou DG, Quach C, Hurrell BP, Li X, Li M, Akbari A, et al. LAIR-1 limits macrophage activation in acute inflammatory lung injury. Mucosal Immunol. (2023) 16:788–800. doi: 10.1016/j.mucimm.2023.08.003
146. Son M, Santiago-Schwarz F, Al-Abed Y, and Diamond B. C1q limits dendritic cell differentiation and activation by engaging LAIR-1. Proc Natl Acad Sci U.S.A. (2012) 109:E3160–3167. doi: 10.1073/pnas.1212753109
147. Son M, Diamond B, Volpe BT, Aranow CB, Mackay MC, and Santiago-Schwarz F. Evidence for C1q-mediated crosslinking of CD33/LAIR-1 inhibitory immunoreceptors and biological control of CD33/LAIR-1 expression. Sci Rep. (2017) 7:270. doi: 10.1038/s41598-017-00290-w
148. Carvalheiro T, Garcia S, Pascoal Ramos MI, Giovannone B, Radstake T, Marut W, et al. Leukocyte associated immunoglobulin like receptor 1 regulation and function on monocytes and dendritic cells during inflammation. Front Immunol. (2020) 11:1793. doi: 10.3389/fimmu.2020.01793
149. Helou DG, Shafiei-Jahani P, Hurrell BP, Painter JD, Quach C, Howard E, et al. LAIR-1 acts as an immune checkpoint on activated ILC2s and regulates the induction of airway hyperreactivity. J Allergy Clin Immunol. (2022) 149:223–236.e226. doi: 10.1016/j.jaci.2021.05.042
150. Morita H, Saito H, and Matsumoto K. Immune checkpoint molecules on ILC2s as potential therapeutic targets for allergic diseases. J Allergy Clin Immunol. (2022) 149:60–2. doi: 10.1016/j.jaci.2021.10.021
151. Kumawat K, Geerdink RJ, Hennus MP, Roda MA, van Ark I, Leusink-Muis T, et al. LAIR-1 limits neutrophilic airway inflammation. Front Immunol. (2019) 10:842. doi: 10.3389/fimmu.2019.00842
152. Park CG, Takahara K, Umemoto E, Yashima Y, Matsubara K, Matsuda Y, et al. Five mouse homologues of the human dendritic cell C-type lectin, DC-SIGN. Int Immunol. (2001) 13:1283–90. doi: 10.1093/intimm/13.10.1283
153. Kang Y-S, Kim JY, Bruening SA, Pack M, Charalambous A, Pritsker A, et al. The C-type lectin SIGN-R1 mediates uptake of the capsular polysaccharide of Streptococcus pneumoniae in the marginal zone of mouse spleen. Proc Natl Acad Sci. (2004) 101:215–20. doi: 10.1073/pnas.0307124101
154. Takagi H, Numazaki M, Kajiwara T, Abe Y, Ishii M, Kato C, et al. Cooperation of specific ICAM-3 grabbing nonintegrin-related 1 (SIGNR1) and complement receptor type 3 (CR3) in the uptake of oligomannose-coated liposomes by macrophages. Glycobiology. (2009) 19:258–66. doi: 10.1093/glycob/cwn128
155. Kawauchi Y, Igarashi M, and Kojima N. C-type lectin receptor SIGNR1 expressed on peritoneal phagocytic cells with an immature dendritic cell-like phenotype is involved in uptake of oligomannose-coated liposomes and subsequent cell maturation. Cell Immunol. (2014) 287:121–8. doi: 10.1016/j.cellimm.2014.01.004
156. Park JY, Choi HJ, Prabagar MG, Choi WS, Kim SJ, Cheong C, et al. The C-type lectin CD209b is expressed on microglia and it mediates the uptake of capsular polysaccharides of Streptococcus pneumoniae. Neurosci Lett. (2009) 450:246–51. doi: 10.1016/j.neulet.2008.11.070
157. Kang Y-S, Do Y, Lee H-K, Park SH, Cheong C, Lynch RM, et al. A dominant complement fixation pathway for pneumococcal polysaccharides initiated by SIGN-R1 interacting with C1q. Cell. (2006) 125:47–58. doi: 10.1016/j.cell.2006.01.046
158. Geijtenbeek TB, Groot PC, Nolte MA, van Vliet SJ, Gangaram-Panday ST, van Duijnhoven GC, et al. Marginal zone macrophages express a murine homologue of DC-SIGN that captures blood-borne antigens. vivo. Blood. (2002) 100:2908–16. doi: 10.1182/blood-2002-04-1044
159. Roozendaal R and Carroll MC. Emerging patterns in complement-mediated pathogen recognition. Cell. (2006) 125:29–32. doi: 10.1016/j.cell.2006.03.018
160. Silva-Martín N, Bartual SG, Ramírez-Aportela E, Chacón P, Park CG, and Hermoso JA. Structural basis for selective recognition of endogenous and microbial polysaccharides by macrophage receptor SIGN-R1. Structure. (2014) 22:1595–606. doi: 10.1016/j.str.2014.09.001
161. Prabagar MG, Do Y, Ryu S, Park JY, Choi HJ, Choi WS, et al. SIGN-R1, a C-type lectin, enhances apoptotic cell clearance through the complement deposition pathway by interacting with C1q in the spleen. Cell Death Differentiation. (2013) 20:535–45. doi: 10.1038/cdd.2012.160
162. Park JY, Loh S, Cho EH, Choi HJ, Na TY, Nemeno JG, et al. SIGN-R1 and complement factors are involved in the systemic clearance of radiation-induced apoptotic cells in whole-body irradiated mice. Biochem Biophys Res Commun. (2015) 463:1064–70. doi: 10.1016/j.bbrc.2015.06.059
163. Pirgova G, Chauveau A, MacLean AJ, Cyster JG, and Arnon TI. Marginal zone SIGN-R1+ macrophages are essential for the maturation of germinal center B cells in the spleen. Proc Natl Acad Sci. (2020) 117:12295–305. doi: 10.1073/pnas.1921673117
164. Koppel EA, Litjens M, van den Berg VC, van Kooyk Y, and Geijtenbeek TB. Interaction of SIGNR1 expressed by marginal zone macrophages with marginal zone B cells is essential to early IgM responses against Streptococcus pneumoniae. Mol Immunol. (2008) 45:2881–7. doi: 10.1016/j.molimm.2008.01.032
165. Figueroa JE and Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. (1991) 4:359–95. doi: 10.1128/CMR.4.3.359
166. Ross GD. Introduction and history of complement research. In: Ross GD, editor. Immunobiology of the Complement System. USA: Academic Press (1986). p. pp 1–19.
167. Markiewski MM, Nilsson B, Nilsson Ekdahl K, Mollnes TE, and Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. (2007) 28:184–92. doi: 10.1016/j.it.2007.02.006
168. Maison CM, Villiers CL, and Colomb MG. Proteolysis of C3 on U937 cell plasma membranes. Purification cathepsin G. J Immunol. (1991) 147:921–6. doi: 10.4049/jimmunol.147.3.921
169. Matsuda T, Nagasawa S, Koide T, and Koyama J. Limited proteolysis of a chemically modified third component of human complement, C3, by cathepsin G of human leukocytes. J Biochem. (1985) 98:229–36. doi: 10.1093/oxfordjournals.jbchem.a135262
170. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. (2013) 39:1143–57. doi: 10.1016/j.immuni.2013.10.018
171. Lajoie S and Wills-Karp M. New twist on an ancient innate immune pathway. Immunity. (2013) 39:1000–2. doi: 10.1016/j.immuni.2013.11.015
172. Ye J, Yuan K, Dai W, Sun K, Li G, Tan M, et al. The mTORC1 signaling modulated by intracellular C3 activation in Paneth cells promotes intestinal epithelial regeneration during acute injury. Int Immunopharmacol. (2019) 67:54–61. doi: 10.1016/j.intimp.2018.12.002
173. Lueschow SR and McElroy SJ. The paneth cell: the curator and defender of the immature small intestine. Front Immunol. (2020) 11:587. doi: 10.3389/fimmu.2020.00587
174. Ouellette AJ. Paneth cell alpha-defensins: peptide mediators of innate immunity in the small intestine. Springer Semin Immunopathol. (2005) 27:133–46. doi: 10.1007/s00281-005-0202-x
175. Kremlitzka M, Colineau L, Nowacka AA, Mohlin FC, Wozniak K, Blom AM, et al. Alternative translation and retrotranslocation of cytosolic C3 that detects cytoinvasive bacteria. Cell Mol Life Sci. (2022) 79:291. doi: 10.1007/s00018-022-04308-z
176. Kulak K, Kuska K, Colineau L, McKay M, Maziarz K, Slaby J, et al. Intracellular C3 protects β-cells from IL-1β-driven cytotoxicity via interaction with Fyn-related kinase. Proc Natl Acad Sci U.S.A. (2024) 121:e2312621121. doi: 10.1073/pnas.2312621121
177. Welsh M, Welsh C, Ekman M, Dixelius J, Hägerkvist R, Annerén C, et al. The tyrosine kinase FRK/RAK participates in cytokine-induced islet cell cytotoxicity. Biochem J. (2004) 382:261–8. doi: 10.1042/BJ20040285
178. Dos Santos RS, Marroqui L, Grieco FA, Marselli L, Suleiman M, Henz SR, et al. Protective role of complement C3 against cytokine-mediated β-cell apoptosis. Endocrinology. (2017) 158:2503–21. doi: 10.1210/en.2017-00104
179. Xu D, Zhou S, Liu Y, Scott AL, Yang J, and Wan F. Complement in breast milk modifies offspring gut microbiota to promote infant health. Cell. (2024) 187:750–763.e720. doi: 10.1016/j.cell.2023.12.019
180. Papatriantafyllou M. Complement in maternal milk shapes the infant microbiome. Nat Rev Immunol. (2024) 24:159–9. doi: 10.1038/s41577-024-01006-8
181. Martin U, Bock D, Arseniev L, Tornetta MA, Ames RS, Bautsch W, et al. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J Exp Med. (1997) 186:199–207. doi: 10.1084/jem.186.2.199
182. Zwirner J, Götze O, Begemann G, Kapp A, Kirchhoff K, and Werfel T. Evaluation of C3a receptor expression on human leucocytes by the use of novel monoclonal antibodies. Immunology. (1999) 97:166–72. doi: 10.1046/j.1365-2567.1999.00764.x
183. Laumonnier Y, Karsten CM, and Köhl J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Mol Immunol. (2017) 89:44–58. doi: 10.1016/j.molimm.2017.05.019
184. Tschernig T, Kiafard Z, Dibbert C, Neumann D, and Zwirner J. Use of monoclonal antibodies to assess expression of anaphylatoxin receptors in rat and murine models of lung inflammation. Exp Toxicologic Pathol. (2007) 58:419–25. doi: 10.1016/j.etp.2007.03.004
185. Asgari E, Le Friec G, Yamamoto H, Perucha E, Sacks SS, Köhl J, et al. C3a modulates IL-1β secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. (2013) 122:3473–81. doi: 10.1182/blood-2013-05-502229
186. Mogilenko DA, Danko K, Larionova EE, Shavva VS, Kudriavtsev IV, Nekrasova EV, et al. Differentiation of human macrophages with anaphylatoxin C3a impairs alternative M2 polarization and decreases lipopolysaccharide-induced cytokine secretion. Immunol Cell Biol. (2022) 100:186–204. doi: 10.1111/imcb.12534
187. Wei L-L, Ma N, Wu K-Y, Wang J-X, Diao T-Y, Zhao S-J, et al. Protective role of C3aR (C3a anaphylatoxin receptor) against atherosclerosis in atherosclerosis-prone mice. Arteriosclerosis Thrombosis Vasc Biol. (2020) 40:2070–83. doi: 10.1161/ATVBAHA.120.314150
188. Quell KM, Karsten CM, Kordowski A, Almeida LN, Briukhovetska D, Wiese AV, et al. Monitoring C3aR expression using a floxed tdTomato-C3aR reporter knock-in mouse. J Immunol. (2017) 199:688–706. doi: 10.4049/jimmunol.1700318
189. Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. (2016) 7:11037. doi: 10.1038/ncomms11037
190. Gasque P, Singhrao SK, Neal JW, Wang P, Sayah S, Fontaine M, et al. The receptor for complement anaphylatoxin C3a is expressed by myeloid cells and nonmyeloid cells in inflamed human central nervous system: analysis in multiple sclerosis and bacterial meningitis. J Immunol. (1998) 160:3543–54. doi: 10.4049/jimmunol.160.7.3543
191. Corcoran JA and Napier BA. C3aR plays both sides in regulating resistance to bacterial infections. PLoS Pathog. (2022) 18:e1010657. doi: 10.1371/journal.ppat.1010657
192. Brennan FH, Jogia T, Gillespie ER, Blomster LV, Li XX, Nowlan B, et al. Complement receptor C3aR1 controls neutrophil mobilization following spinal cord injury through physiological antagonism of CXCR2. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.98254
193. Wu MCL, Brennan FH, Lynch JPL, Mantovani S, Phipps S, Wetsel RA, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci. (2013) 110:9439–44. doi: 10.1073/pnas.1218815110
194. Kirchhoff K, Weinmann O, Zwirner J, Begemann G, Götze O, Kapp A, et al. Detection of anaphylatoxin receptors on CD83+ dendritic cells derived from human skin. Immunology. (2001) 103:210–7. doi: 10.1046/j.1365-2567.2001.01197.x
195. Li K, Fazekasova H, Wang N, Sagoo P, Peng Q, Khamri W, et al. Expression of complement components, receptors and regulators by human dendritic cells. Mol Immunol. (2011) 48:1121–7. doi: 10.1016/j.molimm.2011.02.003
196. Peng Q, Li K, Anderson K, Farrar CA, Lu B, Smith RAG, et al. Local production and activation of complement up-regulates the allostimulatory function of dendritic cells through C3a–C3aR interaction. Blood. (2008) 111:2452–61. doi: 10.1182/blood-2007-06-095018
197. Li K, Anderson KJ, Peng Q, Noble A, Lu B, Kelly AP, et al. Cyclic AMP plays a critical role in C3a-receptor–mediated regulation of dendritic cells in antigen uptake and T-cell stimulation. Blood. (2008) 112:5084–94. doi: 10.1182/blood-2008-05-156646
198. Gutzmer R, Lisewski M, Zwirner J, Mommert S, Diesel C, Wittmann M, et al. Human monocyte-derived dendritic cells are chemoattracted to C3a after up-regulation of the C3a receptor with interferons. Immunology. (2004) 111:435–43. doi: 10.1111/j.1365-2567.2004.01829.x
199. Petering H, Köhl JR, Weyergraf A, Dulkys Y, Kimmig D, Smolarski R, et al. Characterization of synthetic C3a analog peptides on human eosinophils in comparison to the native complement component C3a1. J Immunol. (2000) 164:3783–9. doi: 10.4049/jimmunol.164.7.3783
200. Takafuji S, Tadokoro K, Ito K, and Dahinden CA. Degranulation from human eosinophils stimulated with C3a and C5a. Int Arch Allergy Immunol. (1994) 104 Suppl 1:27–9. doi: 10.1159/000236743
201. Daffern PJ, Pfeifer PH, Ember JA, and Hugli TE. C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activation. J Exp Med. (1995) 181:2119–27. doi: 10.1084/jem.181.6.2119
202. Elsner J, Oppermann M, Czech W, Dobos G, Schöpf E, Norgauer J, et al. C3a activates reactive oxygen radical species production and intracellular calcium transients in human eosinophils. Eur J Immunol. (1994) 24:518–22. doi: 10.1002/eji.1830240304
203. Dahinden CA, Bischoff SC, Brunner T, Krieger M, Takafuji S, and de Weck AL. Regulation of mediator release by human basophils: importance of the sequence and time of addition in the combined action of different agonists. Int Arch Allergy Appl Immunol. (1991) 94:161–4. doi: 10.1159/000235350
204. Kretzschmar T, Jeromin A, Gietz C, Bautsch W, Klos A, Köhl J, et al. Chronic myelogenous leukemia-derived basophilic granulocytes express a functional active receptor for the anaphylatoxin C3a. Eur J Immunol. (1993) 23:558–61. doi: 10.1002/eji.1830230239
205. Ali H. Regulation of human mast cell and basophil function by anaphylatoxins C3a and C5a. Immunol Lett. (2010) 128:36–45. doi: 10.1016/j.imlet.2009.10.007
206. Soruri A, Grigat J, Kiafard Z, and Zwirner J. Mast cell activation is characterized by upregulation of a functional anaphylatoxin C5a receptor. BMC Immunol. (2008) 9:29. doi: 10.1186/1471-2172-9-29
207. Guo Q, Subramanian H, Gupta K, and Ali H. Regulation of C3a receptor signaling in human mast cells by G protein coupled receptor kinases. PLoS One. (2011) 6:e22559. doi: 10.1371/journal.pone.0022559
208. Elieh Ali Komi D, Shafaghat F, Kovanen PT, and Meri S. Mast cells and complement system: Ancient interactions between components of innate immunity. Allergy. (2020) 75:2818–28. doi: 10.1111/all.14413
209. Lohman R-J, Hamidon JK, Reid RC, Rowley JA, Yau M-K, Halili MA, et al. Exploiting a novel conformational switch to control innate immunity mediated by complement protein C3a. Nat Commun. (2017) 8:351. doi: 10.1038/s41467-017-00414-w
210. Sauter RJ, Sauter M, Reis ES, Emschermann FN, Nording H, Ebenhöch S, et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation. (2018) 138:1720–35. doi: 10.1161/CIRCULATIONAHA.118.034600
211. Patzelt J, Mueller KAL, Breuning S, Karathanos A, Schleicher R, Seizer P, et al. Expression of anaphylatoxin receptors on platelets in patients with coronary heart disease. Atherosclerosis. (2015) 238:289–95. doi: 10.1016/j.atherosclerosis.2014.12.002
212. Polley MJ and Nachman RL. Human platelet activation by C3a and C3a des-arg. J Exp Med. (1983) 158:603–15. doi: 10.1084/jem.158.2.603
213. Min X, Liu C, Wei Y, Wang N, Yuan G, Liu D, et al. Expression and regulation of complement receptors by human natural killer cells. Immunobiology. (2014) 219:671–9. doi: 10.1016/j.imbio.2014.03.018
214. Nandagopal S, Li CG, Xu Y, Sodji QH, Graves EE, and Giaccia AJ. C3aR signaling inhibits NK-cell infiltration into the tumor microenvironment in mouse models. Cancer Immunol Res. (2022) 10:245–58. doi: 10.1158/2326-6066.CIR-21-0435
215. Sodji QH, Nambiar DK, Viswanathan V, von Eyben R, Colburg D, Binkley MS, et al. The combination of radiotherapy and complement C3a inhibition potentiates natural killer cell functions against pancreatic cancer. Cancer Res Commun. (2022) 2:725–38. doi: 10.1158/2767-9764.CRC-22-0069
216. Strainic MG, Liu J, Huang D, An F, Lalli PN, Muqim N, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. (2008) 28:425–35. doi: 10.1016/j.immuni.2008.02.001
217. Ghannam A, Fauquert J-L, Thomas C, Kemper C, and Drouet C. Human complement C3 deficiency: Th1 induction requires T cell-derived complement C3a and CD46 activation. Mol Immunol. (2014) 58:98–107. doi: 10.1016/j.molimm.2013.11.010
218. Werfel T, Kirchhoff K, Wittmann M, Begemann G, Kapp A, Heidenreich F, et al. Activated human T lymphocytes express a functional C3a receptor1. J Immunol. (2000) 165:6599–605. doi: 10.4049/jimmunol.165.11.6599
219. Kwan WH, van der Touw W, Paz-Artal E, Li MO, and Heeger PS. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J Exp Med. (2013) 210:257–68. doi: 10.1084/jem.20121525
220. Hess C and Kemper C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity. (2016) 45:240–54. doi: 10.1016/j.immuni.2016.08.003
221. Nataf S, Stahel PF, Davoust N, and Barnum SR. Complement anaphylatoxin receptors on neurons: new tricks for old receptors? Trends Neurosci. (1999) 22:397–402. doi: 10.1016/s0166-2236(98)01390-3
222. Davoust N, Jones J, Stahel PF, Ames RS, and Barnum SR. Receptor for the C3a anaphylatoxin is expressed by neurons and glial cells. Glia. (1999) 26:201–11. doi: 10.1002/(SICI)1098-1136(199905)26:3<201::AID-GLIA2>3.0.CO;2-M
223. Ischenko A, Sayah S, Patte C, Andreev S, Gasque P, Schouft MT, et al. Expression of a functional anaphylatoxin C3a receptor by astrocytes. J Neurochem. (1998) 71:2487–96. doi: 10.1046/j.1471-4159.1998.71062487.x
224. Gedam M and Zheng H. Complement C3aR signaling: Immune and metabolic modulation and its impact on Alzheimer’s disease. Eur J Immunol. (2024) 54:2350815. doi: 10.1002/eji.202350815
225. Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, and DiScipio R. Complement C3a and C5a induce different signal transduction cascades in endothelial cells1. J Immunol. (2002) 169:2102–10. doi: 10.4049/jimmunol.169.4.2102
226. Sartain SE, Turner NA, and Moake JL. Brain microvascular endothelial cells exhibit lower activation of the alternative complement pathway than glomerular microvascular endothelial cells. J Biol Chem. (2018) 293:7195–208. doi: 10.1074/jbc.RA118.002639
227. Shivshankar P, Li Y-D, Mueller-Ortiz SL, and Wetsel RA. In response to complement anaphylatoxin peptides C3a and C5a, human vascular endothelial cells migrate and mediate the activation of B-cells and polarization of T-cells. FASEB J. (2020) 34:7540–60. doi: 10.1096/fj.201902397R
228. Propson NE, Roy ER, Litvinchuk A, Köhl J, and Zheng H. Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J Clin Invest. (2021) 131. doi: 10.1172/JCI140966
229. Wolf HN, Guempelein L, Schikora J, and Pauly D. C3a Mediates Endothelial Barrier Disruption in Brain-Derived, but Not Retinal, Human Endothelial Cells. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms252011240
230. You D, Nie K, Wu X, Weng M, Yang L, Chen Y, et al. C3a/C3aR synergies with TGF-β to promote epithelial-mesenchymal transition of renal tubular epithelial cells via the activation of the NLRP3 inflammasome. J Transl Med. (2023) 21:904. doi: 10.1186/s12967-023-04764-6
231. Ishii M, Beeson G, Beeson C, and Rohrer B. Mitochondrial C3a receptor activation in oxidatively stressed epithelial cells reduces mitochondrial respiration and metabolism. Front Immunol. (2021) 12:628062. doi: 10.3389/fimmu.2021.628062
232. Schraufstatter IU, Khaldoyanidi SK, and DiScipio RG. Complement activation in the context of stem cells and tissue repair. World J Stem Cells. (2015) 7:1090–108. doi: 10.4252/wjsc.v7.i8.1090
233. Robinson MJ, Quast I, and Tarlinton DM. Complement-in’ the germinal center response. Nat Immunol. (2021) 22:673–4. doi: 10.1038/s41590-021-00946-w
234. Cumpelik A, Heja D, Hu Y, Varano G, Ordikhani F, Roberto MP, et al. Dynamic regulation of B cell complement signaling is integral to germinal center responses. Nat Immunol. (2021) 22:757–68. doi: 10.1038/s41590-021-00926-0
235. Schanzenbacher J, Hendrika Kähler K, Mesler E, Kleingarn M, Marcel Karsten C, and Leonard Seiler D. The role of C5a receptors in autoimmunity. Immunobiology. (2023) 228:152413. doi: 10.1016/j.imbio.2023.152413
236. Zhang K, Li G-Q, He Q-H, Li Y, Tang M, Zheng Q-Y, et al. C5a/C5aR pathway accelerates renal ischemia-reperfusion injury by downregulating PGRN expression. Int Immunopharmacol. (2017) 53:17–23. doi: 10.1016/j.intimp.2017.10.006
237. Gao H and Yan C. New insights for C5a and C5a receptors in sepsis. Front Immunol. (2012) 3. doi: 10.3389/fimmu.2012.00368
238. Riedemann NC, Guo RF, and Ward PA. A key role of C5a/C5aR activation for the development of sepsis. J Leukoc Biol. (2003) 74:966–70. doi: 10.1189/jlb.0403137
239. Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol. (2004) 4:133–42. doi: 10.1038/nri1269
240. Li R, Coulthard LG, Wu MC, Taylor SM, and Woodruff TM. C5L2: a controversial receptor of complement anaphylatoxin, C5a. FASEB J. (2013) 27:855–64. doi: 10.1096/fj.12-220509
241. Scola AM, Johswich KO, Morgan BP, Klos A, and Monk PN. The human complement fragment receptor, C5L2, is a recycling decoy receptor. Mol Immunol. (2009) 46:1149–62. doi: 10.1016/j.molimm.2008.11.001
242. Cui W, Simaan M, Laporte S, Lodge R, and Cianflone K. C5a- and ASP-mediated C5L2 activation, endocytosis and recycling are lost in S323I-C5L2 mutation. Mol Immunol. (2009) 46:3086–98. doi: 10.1016/j.molimm.2009.06.007
243. Tom FQ, Gauvreau D, Lapointe M, Lu H, Poursharifi P, Luo XP, et al. Differential chemoattractant response in adipocytes and macrophages to the action of acylation stimulating protein. Eur J Cell Biol. (2013) 92:61–9. doi: 10.1016/j.ejcb.2012.10.005
244. Poursharifi P, Lapointe M, Fisette A, Lu H, Roy C, Munkonda MN, et al. C5aR and C5L2 act in concert to balance immunometabolism in adipose tissue. Mol Cell Endocrinol. (2014) 382:325–33. doi: 10.1016/j.mce.2013.10.019
245. Cui W, Lapointe M, Gauvreau D, Kalant D, and Cianflone K. Recombinant C3adesArg/acylation stimulating protein (ASP) is highly bioactive: a critical evaluation of C5L2 binding and 3T3-L1 adipocyte activation. Mol Immunol. (2009) 46:3207–17. doi: 10.1016/j.molimm.2009.08.013
246. Qu LH, Jin X, Li LM, Li SY, and Xie HP. A novel mutation in C5L2 gene was associated with hyperlipidemia and retinitis pigmentosa in a Chinese family. Lipids Health Dis. (2014) 13:75. doi: 10.1186/1476-511X-13-75
247. Cain SA and Monk PN. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-arg74*. J Biol Chem. (2002) 277:7165–9. doi: 10.1074/jbc.C100714200
248. Ohno M, Hirata T, Enomoto M, Araki T, Ishimaru H, and Takahashi TA. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Mol Immunol. (2000) 37:407–12. doi: 10.1016/S0161-5890(00)00067-5
249. Lee DK, George SR, Cheng R, Nguyen T, Liu Y, Brown M, et al. Identification of four novel human G protein-coupled receptors expressed in the brain. Brain Res Mol Brain Res. (2001) 86:13–22. doi: 10.1016/S0169-328X(00)00242-4
250. Gao H, Neff TA, Guo RF, Speyer CL, Sarma JV, Tomlins S, et al. Evidence for a functional role of the second C5a receptor C5L2. FASEB J. (2005) 19:1003–5. doi: 10.1096/fj.04-3424fje
251. Chen NJ, Mirtsos C, Suh D, Lu YC, Lin WJ, McKerlie C, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature. (2007) 446:203–7. doi: 10.1038/nature05559
252. Kalant D, MacLaren R, Cui W, Samanta R, Monk PN, Laporte SA, et al. C5L2 is a functional receptor for acylation-stimulating protein. J Biol Chem. (2005) 280:23936–44. doi: 10.1074/jbc.M406921200
253. Lee H, Whitfeld PL, and Mackay CR. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol. (2008) 86:153–60. doi: 10.1038/sj.icb.7100166
254. Karsten CM, Wiese AV, Mey F, Figge J, Woodruff TM, Reuter T, et al. Monitoring C5aR2 expression using a floxed tdTomato-C5aR2 knock-in mouse. J Immunol. (2017) 199:3234–48. doi: 10.4049/jimmunol.1700710
255. Karsten CM, Laumonnier Y, Eurich B, Ender F, Bröker K, Roy S, et al. Monitoring and cell-specific deletion of C5aR1 using a novel floxed GFP-C5aR1 reporter knock-in mouse. J Immunol. (2015) 194:1841–55. doi: 10.4049/jimmunol.1401401
256. Seiler DL, Kähler KH, Kleingarn M, Sadik CD, Bieber K, Köhl J, et al. The complement receptor C5aR2 regulates neutrophil activation and function contributing to neutrophil-driven epidermolysis bullosa acquisita. Front Immunol. (2023) 14:1197709. doi: 10.3389/fimmu.2023.1197709
257. Zaal A, van Ham SM, and Ten Brinke A. Differential effects of anaphylatoxin C5a on antigen presenting cells, roles for C5aR1 and C5aR2. Immunol Lett. (2019) 209:45–52. doi: 10.1016/j.imlet.2019.03.014
258. Zaal A, Dieker M, Oudenampsen M, Turksma AW, Lissenberg-Thunnissen SN, Wouters D, et al. Anaphylatoxin C5a regulates 6-sulfo-lacNAc dendritic cell function in human through crosstalk with toll-like receptor-induced CREB signaling. Front Immunol. (2017) 8. doi: 10.3389/fimmu.2017.00818
259. Gerard NP, Hodges MK, Drazen JM, Weller PF, and Gerard C. Characterization of a receptor for C5a anaphylatoxin on human eosinophils. J Biol Chem. (1989) 264:1760–6. doi: 10.1016/S0021-9258(18)94252-3
260. DiScipio RG, Daffern PJ, Jagels MA, Broide DH, and Sriramarao P. A comparison of C3a and C5a-mediated stable adhesion of rolling eosinophils in postcapillary venules and transendothelial migration in vitro and in vivo1. J Immunol. (1999) 162:1127–36. doi: 10.4049/jimmunol.162.2.1127
261. Elsner J, Oppermann M, and Kapp A. Detection of C5a receptors on human eosinophils and inhibition of eosinophil effector functions by anti-C5a receptor (CD88) antibodies. Eur J Immunol. (1996) 26:1560–4. doi: 10.1002/eji.1830260723
262. Bamberg CE, Mackay CR, Lee H, Zahra D, Jackson J, Lim YS, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction*. J Biol Chem. (2010) 285:7633–44. doi: 10.1074/jbc.M109.092106
263. Zhang X, Schmudde I, Laumonnier Y, Pandey MK, Clark JR, König P, et al. A critical role for C5L2 in the pathogenesis of experimental allergic asthma. J Immunol. (2010) 185:6741–52. doi: 10.4049/jimmunol.1000892
264. Füreder W, Agis H, Willheim M, Bankl HC, Maier U, Kishi K, et al. Differential expression of complement receptors on human basophils and mast cells. Evidence for mast cell heterogeneity and CD88/C5aR expression on skin mast cells. J Immunol. (1995) 155:3152–60. doi: 10.4049/jimmunol.155.6.3152
265. Schulman ES, Post TJ, Henson PM, and Giclas PC. Differential effects of the complement peptides, C5a and C5a des Arg on human basophil and lung mast cell histamine release. J Clin Invest. (1988) 81:918–23. doi: 10.1172/JCI113403
266. Eglite S, Plüss K, and Dahinden CA. Requirements for C5a receptor-mediated IL-4 and IL-13 production and leukotriene C4 generation in human basophils. J Immunol. (2000) 165:2183–9. doi: 10.4049/jimmunol.165.4.2183
267. Nilsson G, Johnell M, Hammer CH, Tiffany HL, Nilsson K, Metcalfe DD, et al. C3a and C5a are chemotaxins for human mast cells and act through distinct receptors via a pertussis toxin-sensitive signal transduction pathway. J Immunol. (1996) 157:1693–8. doi: 10.4049/jimmunol.157.4.1693
268. Hartmann K, Henz BM, Krüger-Krasagakes S, Köhl J, Burger R, Guhl S, et al. C3a and C5a stimulate chemotaxis of human mast cells. Blood. (1997) 89:2863–70. doi: 10.1182/blood.V89.8.2863
269. el-Lati SG, Dahinden CA, and Church MK. Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J Invest Dermatol. (1994) 102:803–6. doi: 10.1111/1523-1747.ep12378589
270. Pundir P, MacDonald CA, and Kulka M. The novel receptor C5aR2 is required for C5a-mediated human mast cell adhesion, migration, and proinflammatory mediator production. J Immunol. (2015) 195:2774–87. doi: 10.4049/jimmunol.1401348
271. Pundir P and Kulka M. The seven-transmembrane receptor, C5L2, is a stimulatory receptor on human mast cells. Allergy Asthma Clin Immunol. (2010) 6:P25. doi: 10.1186/1710-1492-6-S2-P25
272. Pundir P and Kulka M. Functional expression of the novel C5a receptor C5L2 in human mast cells. J Allergy Clin Immunol. (2013) 131:AB239.
273. Nording H, Baron L, Haberthür D, Emschermann F, Mezger M, Sauter M, et al. The C5a/C5a receptor 1 axis controls tissue neovascularization through CXCL4 release from platelets. Nat Commun. (2021) 12:3352. doi: 10.1038/s41467-021-23499-w
274. Meuer S, Ecker U, Hadding U, and Bitter-Suermann D. Platelet-serotonin release by C3a and C5a: two independent pathways of activation. J Immunol. (1981) 126:1506–9. doi: 10.4049/jimmunol.126.4.1506
275. Matsumoto K, Yasuoka H, Yoshimoto K, Suzuki K, and Takeuchi T. Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci Rep. (2021) 11:222. doi: 10.1038/s41598-020-80685-4
276. Apostolidis SA, Sarkar A, Giannini HM, Goel RR, Mathew D, Suzuki A, et al. Signaling through fcγRIIA and the C5a-C5aR pathway mediate platelet hyperactivation in COVID-19. Front Immunol. (2022) 13:834988. doi: 10.3389/fimmu.2022.834988
277. Aiello S, Gastoldi S, Galbusera M, Ruggenenti P, Portalupi V, Rota S, et al. C5a and C5aR1 are key drivers of microvascular platelet aggregation in clinical entities spanning from aHUS to COVID-19. Blood Adv. (2022) 6:866–81. doi: 10.1182/bloodadvances.2021005246
278. Boeckel H, Karsten CM, Göpel W, Herting E, Rupp J, Härtel C, et al. Increased expression of anaphylatoxin C5a-receptor-1 in neutrophils and natural killer cells of preterm infants. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241210321
279. Fusakio ME, Mohammed JP, Laumonnier Y, Hoebe K, Köhl J, and Mattner J. C5a regulates NKT and NK cell functions in sepsis. J Immunol. (2011) 187:5805–12. doi: 10.4049/jimmunol.1100338
280. Qing X, Koo GC, and Salmon JE. Complement regulates conventional DC-mediated NK-cell activation by inducing TGF-β1 in Gr-1+ myeloid cells. Eur J Immunol. (2012) 42:1723–34. doi: 10.1002/eji.201142290
281. Han G, Geng S, Li Y, Chen G, Wang R, Li X, et al. γδT-cell function in sepsis is modulated by C5a receptor signalling. Immunology. (2011) 133:340–9. doi: 10.1111/j.1365-2567.2011.03445.x
282. Zheng QY, Xu F, Yang Y, Sun DD, Zhong Y, Wu S, et al. C5a/C5aR1 mediates IMQ-induced psoriasiform skin inflammation by promoting IL-17A production from γδ-T cells. FASEB J. (2020) 34:10590–604. doi: 10.1096/fj.202000384R
283. Lalli PN, Strainic MG, Yang M, Lin F, Medof ME, and Heeger PS. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. (2008) 112:1759–66. doi: 10.1182/blood-2008-04-151068
284. Nataf S, Davoust N, Ames RS, and Barnum SR. Human T cells express the C5a receptor and are chemoattracted to C5a. J Immunol. (1999) 162:4018–23. doi: 10.4049/jimmunol.162.7.4018
285. Kim AHJ, Dimitriou ID, Holland MCH, Mastellos D, Mueller YM, Altman JD, et al. Complement C5a receptor is essential for the optimal generation of antiviral CD8+ T cell responses1. J Immunol. (2004) 173:2524–9. doi: 10.4049/jimmunol.173.4.2524
286. Luan X, Lei T, Fang J, Liu X, Fu H, Li Y, et al. Blockade of C5a receptor unleashes tumor-associated macrophage antitumor response and enhances CXCL9-dependent CD8+ T cell activity. Mol Ther. (2024) 32:469–89. doi: 10.1016/j.ymthe.2023.12.010
287. Ager RR, Fonseca MI, Chu SH, Sanderson SD, Taylor SM, Woodruff TM, et al. Microglial C5aR (CD88) expression correlates with amyloid-beta deposition in murine models of Alzheimer’s disease. J Neurochem. (2010) 113:389–401. doi: 10.1111/j.1471-4159.2010.06595.x
288. Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, et al. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J Neurosci. (2007) 27:8699–708. doi: 10.1523/JNEUROSCI.2018-07.2007
289. Pekna M and Pekny M. The complement system: A powerful modulator and effector of astrocyte function in the healthy and diseased central nervous system. Cells. (2021) 10. doi: 10.3390/cells10071812
290. Brennan FH, Gordon R, Lao HW, Biggins PJ, Taylor SM, Franklin RJ, et al. The complement receptor C5aR controls acute inflammation and astrogliosis following spinal cord injury. J Neurosci. (2015) 35:6517–31. doi: 10.1523/JNEUROSCI.5218-14.2015
291. Schartz ND, Liang HY, Carvalho K, Chu S-H, Mendoza-Arvilla A, Petrisko TJ, et al. C5aR1 antagonism suppresses inflammatory glial responses and alters cellular signaling in an Alzheimer’s disease mouse model. Nat Commun. (2024) 15:7028. doi: 10.1038/s41467-024-51163-6
292. Biggins PJC, Brennan FH, Taylor SM, Woodruff TM, and Ruitenberg MJ. The alternative receptor for complement component 5a, C5aR2, conveys neuroprotection in traumatic spinal cord injury. J Neurotrauma. (2017) 34:2075–85. doi: 10.1089/neu.2016.4701
293. Carvalho K, Schartz ND, Balderrama-Gutierrez G, Liang HY, Chu SH, Selvan P, et al. Modulation of C5a-C5aR1 signaling alters the dynamics of AD progression. J Neuroinflamm. (2022) 19:178. doi: 10.1186/s12974-022-02539-2
294. Laudes IJ, Chu JC, Huber-Lang M, Guo RF, Riedemann NC, Sarma JV, et al. Expression and function of C5a receptor in mouse microvascular endothelial cells. J Immunol. (2002) 169:5962–70. doi: 10.4049/jimmunol.169.10.5962
295. Vijayan S, Asare Y, Grommes J, Soehnlein O, Lutgens E, Shagdarsuren G, et al. High expression of C5L2 correlates with high proinflammatory cytokine expression in advanced human atherosclerotic plaques. Am J Pathol. (2014) 184:2123–33. doi: 10.1016/j.ajpath.2014.04.004
296. Monk PN, Scola AM, Madala P, and Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol. (2007) 152:429–48. doi: 10.1038/sj.bjp.0707332
297. Ding P, Xu Y, Li L, Lv X, Li L, Chen J, et al. Intracellular complement C5a/C5aR1 stabilizes β-catenin to promote colorectal tumorigenesis. Cell Rep. (2022) 39:110851. doi: 10.1016/j.celrep.2022.110851
298. Beach C, MacLean D, Majorova D, Melemenidis S, Nambiar DK, Kim RK, et al. Innate immune receptor C5aR1 regulates cancer cell fate and can be targeted to improve radiotherapy in tumours with immunosuppressive microenvironments. bioRxiv. (2023). doi: 10.1101/2023.01.10.521547
299. Ishii M and Rohrer B. Anaphylatoxin C5a receptor signaling induces mitochondrial fusion and sensitizes retinal pigment epithelial cells to oxidative stress. Biochim Biophys Acta (BBA) - Gen Subj. (2023) 1867:130374. doi: 10.1016/j.bbagen.2023.130374
300. Johswich K, Martin M, Thalmann J, Rheinheimer C, Monk PN, and Klos A. Ligand specificity of the anaphylatoxin C5L2 receptor and its regulation on myeloid and epithelial cell lines. J Biol Chem. (2006) 281:39088–95. doi: 10.1074/jbc.M609734200
301. Oglesby TJ, Longwith JE, and Huettner PC. Human complement regulator expression by the normal female reproductive tract. Anat Rec. (1996) 246:78–86. doi: 10.1002/(SICI)1097-0185(199609)246:1<78::AID-AR9>3.0.CO;2-B
302. Iborra A, Mayorga M, Llobet N, and Martínez P. Expression of complement regulatory proteins [membrane cofactor protein (CD46), decay accelerating factor (CD55), and protectin (CD59)] in endometrial stressed cells. Cell Immunol. (2003) 223:46–51. doi: 10.1016/S0008-8749(03)00127-8
303. Fénichel P, Cervoni F, Donzeau M, and Hsi BL. Expression and role of complement regulatory proteins on human gametes and pre-implantation embryos. Contracept Fertil Sex. (1995) 23:576–80.
304. Fenichel P, Donzeau M, Cervoni F, Menezo Y, and Hsi BL. Expression of complement regulatory proteins on human eggs and preimplantation embryos. Am J Reprod Immunol. (1995) 33:155–64. doi: 10.1111/j.1600-0897.1995.tb00879.x
305. Taylor CT and Johnson PM. Complement-binding proteins are strongly expressed by human preimplantation blastocysts and cumulus cells as well as gametes. Mol Hum Reprod. (1996) 2:52–9. doi: 10.1093/molehr/2.1.52
306. Reichhardt MP, Lundin K, Lokki AI, Recher G, Vuoristo S, Katayama S, et al. Complement in human pre-implantation embryos: attack and defense. Front Immunol. (2019) 10:2234. doi: 10.3389/fimmu.2019.02234
307. Jiang H and Pillai S. Complement regulatory proteins on the sperm surface: relevance to sperm motility. Am J Reprod Immunol. (1998) 39:243–8. doi: 10.1111/j.1600-0897.1998.tb00360.x
308. Cervoni F, Oglesby TJ, Adams EM, Milesifluet C, Nickells M, Fenichel P, et al. Identification and characterization of membrane cofactor protein of human spermatozoa. J Immunol. (1992) 148:1431–7. doi: 10.4049/jimmunol.148.5.1431
309. Cummerson JA, Flanagan BF, Spiller DG, and Johnson PM. The complement regulatory proteins CD55 (decay accelerating factor) and CD59 are expressed on the inner acrosomal membrane of human spermatozoa as well as CD46 (membrane cofactor protein). Immunology. (2006) 118:333–42. doi: 10.1111/j.1365-2567.2006.02374.x
310. D’Cruz OJ and Haas GG Jr. The expression of the complement regulators CD46, CD55, and CD59 by human sperm does not protect them from antisperm antibody- and complement-mediated immune injury. Fertil Steril. (1993) 59:876–84. doi: 10.1016/S0015-0282(16)55875-0
311. Bronson R, Bronson S, Oula L, Zhang W, and Ghebrehiwet B. Detection of complement C1q receptors on human spermatozoa. J Reprod Immunol. (1998) 38:1–14. doi: 10.1016/S0165-0378(98)00006-0
312. Grace KS, Bronson RA, and Ghebrehiwet B. Surface Expression of Complement Receptor gC1q-R/p33 Is Increased on the Plasma Membrane of Human Spermatozoa after Capacitation1. Biol Reprod. (2002) 66:823–9. doi: 10.1095/biolreprod66.3.823
313. Anderson DJ, Abbott AF, and Jack RM. The role of complement component C3b and its receptors in sperm-oocyte interaction. Proc Natl Acad Sci U.S.A. (1993) 90:10051–5. doi: 10.1073/pnas.90.21.10051
314. Riley-Vargas RC, Lanzendorf S, and Atkinson JP. Targeted and restricted complement activation on acrosome-reacted spermatozoa. J Clin Invest. (2005) 115:1241–9. doi: 10.1172/JCI23213
315. Bulla R, Bossi F, Agostinis C, Radillo O, Colombo F, De Seta F, et al. Complement production by trophoblast cells at the feto-maternal interface. J Reprod Immunol. (2009) 82:119–25. doi: 10.1016/j.jri.2009.06.124
316. Hanna J, Wald O, Goldman-Wohl D, Prus D, Markel G, Gazit R, et al. CXCL12 expression by invasive trophoblasts induces the specific migration of CD16- human natural killer cells. Blood. (2003) 102:1569–77. doi: 10.1182/blood-2003-02-0517
317. Drake PM, Gunn MD, Charo IF, Tsou CL, Zhou Y, Huang L, et al. Human placental cytotrophoblasts attract monocytes and CD56(bright) natural killer cells via the actions of monocyte inflammatory protein 1alpha. J Exp Med. (2001) 193:1199–212. doi: 10.1084/jem.193.10.1199
318. Koopman LA, Kopcow HD, Rybalov B, Boyson JE, Orange JS, Schatz F, et al. Human decidual natural killer cells are a unique NK cell subset with immunomodulatory potential. J Exp Med. (2003) 198:1201–12. doi: 10.1084/jem.20030305
319. Pijnenborg R, Vercruysse L, and Hanssens M. The uterine spiral arteries in human pregnancy: facts and controversies. Placenta. (2006) 27:939–58. doi: 10.1016/j.placenta.2005.12.006
320. Charriaut C, Senik A, Kolb JP, Barel M, and Frade R. Inhibition of in vitro natural killer activity by the third component of complement: role for the C3a fragment. Proc Natl Acad Sci U.S.A. (1982) 79:6003–7.
321. Wang SY, Veeramani S, Racila E, Cagley J, Fritzinger DC, Vogel CW, et al. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood. (2009) 114:5322–30. doi: 10.1182/blood-2009-01-200469
322. Wang SY, Racila E, Taylor RP, and Weiner GJ. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood. (2008) 111:1456–63. doi: 10.1182/blood-2007-02-074716
323. Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. (2006) 12:1065–74. doi: 10.1038/nm1452
324. Karimi K and Arck PC. Natural Killer cells: keepers of pregnancy in the turnstile of the environment. Brain Behav Immun. (2010) 24:339–47. doi: 10.1016/j.bbi.2009.09.015
325. Kopcow HD, Allan DS, Chen X, Rybalov B, Andzelm MM, Ge B, et al. Human decidual NK cells form immature activating synapses and are not cytotoxic. Proc Natl Acad Sci U.S.A. (2005) 102:15563–8. doi: 10.1073/pnas.0507835102
326. Agostinis C, Bulla R, Tripodo C, Gismondi A, Stabile H, Bossi F, et al. An alternative role of C1q in cell migration and tissue remodeling: contribution to trophoblast invasion and placental development. J Immunol. (2010) 185:4420–9. doi: 10.4049/jimmunol.0903215
327. Agostinis C, Mangogna A, Balduit A, Kishore U, and Bulla R. A non-redundant role of complement protein C1q in normal and adverse pregnancy. Explor Immunol. (2022) 2:622–36. doi: 10.37349/ei
328. E Davies J, Pollheimer J, Yong HE, Kokkinos MI, Kalionis B, Knöfler M, et al. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adh Migr. (2016) 10:310–21. doi: 10.1080/19336918.2016.1170258
329. Tang Z, Lu B, Hatch E, Sacks SH, and Sheerin NS. C3a mediates epithelial-to-mesenchymal transition in proteinuric nephropathy. J Am Soc Nephrol. (2009) 20:593–603. doi: 10.1681/ASN.2008040434
330. Otsuki T, Fukuda N, Chen L, Tsunemi A, and Abe M. Twist-related protein 1 induces epithelial-mesenchymal transition and renal fibrosis through the upregulation of complement 3. PLoS One. (2022) 17:e0272917. doi: 10.1371/journal.pone.0272917
331. Cho MS, Rupaimoole R, Choi H-J, Noh K, Chen J, Hu Q, et al. Complement component 3 is regulated by TWIST1 and mediates epithelial–mesenchymal transition. J Immunol. (2016) 196:1412–8. doi: 10.4049/jimmunol.1501886
332. Soundararajan R and Rao AJ. Trophoblast ‘pseudo-tumorigenesis’: significance and contributory factors. Reprod Biol Endocrinol. (2004) 2:15. doi: 10.1186/1477-7827-2-15
333. Carvajal L, Gutiérrez J, Morselli E, and Leiva A. Autophagy process in trophoblast cells invasion and differentiation: similitude and differences with cancer cells. Front Oncol. (2021) 11. doi: 10.3389/fonc.2021.637594
334. Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, and Bellet D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update. (2006) 13:121–41. doi: 10.1093/humupd/dml048
335. Richani K, Soto E, Romero R, Espinoza J, Chaiworapongsa T, Nien JK, et al. Normal pregnancy is characterized by systemic activation of the complement system. J Matern Fetal Neonatal Med. (2005) 17:239–45. doi: 10.1080/14767050500072722
336. Derzsy Z, Prohászka Z, Rigó J Jr., Füst G, and Molvarec A. Activation of the complement system in normal pregnancy and preeclampsia. Mol Immunol. (2010) 47:1500–6. doi: 10.1016/j.molimm.2010.01.021
337. Abramson SB and Buyon JP. Activation of the complement pathway: comparison of normal pregnancy, preeclampsia, and systemic lupus erythematosus during pregnancy. Am J Reprod Immunol. (1992) 28:183–7. doi: 10.1111/j.1600-0897.1992.tb00787.x
338. Soto E, Romero R, Richani K, Espinoza J, Nien JK, Chaiworapongsa T, et al. Anaphylatoxins in preterm and term labor. J Perinat Med. (2005) 33:306–13. doi: 10.1515/JPM.2005.051
339. Lynch AM, Gibbs RS, Murphy JR, Giclas PC, Salmon JE, and Holers VM. Early elevations of the complement activation fragment C3a and adverse pregnancy outcomes. Obstet Gynecol. (2011) 117:75–83. doi: 10.1097/AOG.0b013e3181fc3afa
340. He YD, Xu BN, Song D, Wang YQ, Yu F, Chen Q, et al. Normal range of complement components during pregnancy: A prospective study. Am J Reprod Immunol. (2020) 83:e13202. doi: 10.1111/aji.13202
341. Ramanjaneya M, Diboun I, Rizwana N, Dajani Y, Ahmed L, Butler AE, et al. Elevated adipsin and reduced C5a levels in the maternal serum and follicular fluid during implantation are associated with successful pregnancy in obese women. Front Endocrinol (Lausanne). (2022) 13:918320. doi: 10.3389/fendo.2022.918320
342. Liu M, Luo X, Xu Q, Yu H, Gao L, Zhou R, et al. Adipsin of the alternative complement pathway is a potential predictor for preeclampsia in early pregnancy. Front Immunol. (2021) 12:702385. doi: 10.3389/fimmu.2021.702385
343. Poveda NE, Garcés MF, Ruiz-Linares CE, Varón D, Valderrama S, Sanchez E, et al. Serum adipsin levels throughout normal pregnancy and preeclampsia. Sci Rep. (2016) 6:20073. doi: 10.1038/srep20073
344. Rosen BS, Cook KS, Yaglom J, Groves DL, Volanakis JE, Damm D, et al. Adipsin and complement factor D activity: an immune-related defect in obesity. Science. (1989) 244:1483–7. doi: 10.1126/science.2734615
345. Xu Y, Ma M, Ippolito GC, Schroeder HW Jr., Carroll MC, and Volanakis JE. Complement activation in factor D-deficient mice. Proc Natl Acad Sci U.S.A. (2001) 98:14577–82. doi: 10.1073/pnas.261428398
346. Milek M, Moulla Y, Kern M, Stroh C, Dietrich A, Schön MR, et al. Adipsin serum concentrations and adipose tissue expression in people with obesity and type 2 diabetes. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23042222
347. Lo JC, Ljubicic S, Leibiger B, Kern M, Leibiger IB, Moede T, et al. Adipsin is an adipokine that improves β cell function in diabetes. Cell. (2014) 158:41–53. doi: 10.1016/j.cell.2014.06.005
348. Dare A and Chen S-Y. Adipsin in the pathogenesis of cardiovascular diseases. Vasc Pharmacol. (2024) 154:107270. doi: 10.1016/j.vph.2023.107270
349. Csuka D, Molvarec A, Derzsy Z, Varga L, Füst G, Rigó J Jr., et al. Functional analysis of the mannose-binding lectin complement pathway in normal pregnancy and preeclampsia. J Reprod Immunol. (2010) 87:90–6. doi: 10.1016/j.jri.2010.07.004
350. Lynch AM, Wagner BD, Giclas PC, West NA, Gibbs RS, and Holers VM. The relationship of longitudinal levels of complement bb during pregnancy with preeclampsia. Am J Reprod Immunol. (2016) 75:104–11. doi: 10.1111/aji.12439
351. Regal JF, Gilbert JS, and Burwick RM. The complement system and adverse pregnancy outcomes. Mol Immunol. (2015) 67:56–70. doi: 10.1016/j.molimm.2015.02.030
352. Denny KJ, Woodruff TM, Taylor SM, and Callaway LK. Complement in pregnancy: a delicate balance. Am J Reprod Immunol. (2013) 69:3–11. doi: 10.1111/aji.12000
353. Alonso-Ventura V, Li Y, Pasupuleti V, Roman YM, Hernandez AV, and Pérez-López FR. Effects of preeclampsia and eclampsia on maternal metabolic and biochemical outcomes in later life: a systematic review and meta-analysis. Metabolism. (2020) 102:154012. doi: 10.1016/j.metabol.2019.154012
354. Perry H, Khalil A, and Thilaganathan B. Preeclampsia and the cardiovascular system: An update. Trends Cardiovasc Med. (2018) 28:505–13. doi: 10.1016/j.tcm.2018.04.009
355. Robillard PY, Dekker G, Chaouat G, Scioscia M, Iacobelli S, and Hulsey TC. Historical evolution of ideas on eclampsia/preeclampsia: A proposed optimistic view of preeclampsia. J Reprod Immunol. (2017) 123:72–7. doi: 10.1016/j.jri.2017.09.006
356. Garrido-Gómez T, Castillo-Marco N, Cordero T, and Simón C. Decidualization resistance in the origin of preeclampsia. Am J Obstet Gynecol. (2022) 226:S886–s894. doi: 10.1016/j.ajog.2020.09.039
357. Lai J, Syngelaki A, Nicolaides KH, von Dadelszen P, and Magee LA. Impact of new definitions of preeclampsia at term on identification of adverse maternal and perinatal outcomes. Am J Obstetrics Gynecology. (2021) 224:518.e511–518.e511. doi: 10.1016/j.ajog.2020.11.004
358. Ghulmiyyah L and Sibai B. Maternal mortality from preeclampsia/eclampsia. Semin Perinatol. (2012) 36:56–9. doi: 10.1053/j.semperi.2011.09.011
359. Jung E, Romero R, Yeo L, Gomez-Lopez N, Chaemsaithong P, Jaovisidha A, et al. The etiology of preeclampsia. Am J Obstet Gynecol. (2022) 226:S844–s866. doi: 10.1016/j.ajog.2021.11.1356
360. Fisher SJ. Why is placentation abnormal in preeclampsia? Am J Obstetrics Gynecology. (2015) 213:S115–22. doi: 10.1016/j.ajog.2015.08.042
361. Erez O, Romero R, Jung E, Chaemsaithong P, Bosco M, Suksai M, et al. Preeclampsia and eclampsia: the conceptual evolution of a syndrome. Am J Obstet Gynecol. (2022) 226:S786–s803. doi: 10.1016/j.ajog.2021.12.001
362. Kumar V. Introductory chapter: macrophages – more than sentinel innate immune cells. In: Kumar V, editor. Macrophages - Celebrating 140 Years of Discovery. IntechOpen, Rijeka (2022). doi: 10.5772/intechopen.109647
363. Dekker GA and Sibai BM. The immunology of preeclampsia. Semin Perinatol. (1999) 23:24–33. doi: 10.1016/S0146-0005(99)80057-3
364. Redman CW and Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol. (2010) 63:534–43. doi: 10.1111/j.1600-0897.2010.00831.x
365. Collier AY, Modest AM, Aguayo RA, Bondzie EA, Patel S, Hacker MR, et al. Altered cytokine production in human intervillous blood T cells in preeclampsia. Reprod Sci. (2023) 30:2655–64. doi: 10.1007/s43032-023-01165-4
366. Darmochwal-Kolarz D, Kludka-Sternik M, Tabarkiewicz J, Kolarz B, Rolinski J, Leszczynska-Gorzelak B, et al. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J Reprod Immunol. (2012) 93:75–81. doi: 10.1016/j.jri.2012.01.006
367. Stefańska K, Kurkowiak M, Piekarska K, Chruściel E, Zamkowska D, Jassem-Bobowicz J, et al. High maternal-fetal HLA eplet compatibility is associated with severe manifestation of preeclampsia. Front Immunol. (2023) 14:1272021. doi: 10.3389/fimmu.2023.1272021
368. van Bentem K, Bos M, van der Keur C, Brand-Schaaf SH, Haasnoot GW, Roelen DL, et al. The development of preeclampsia in oocyte donation pregnancies is related to the number of fetal-maternal HLA class II mismatches. J Reprod Immunol. (2020) 137:103074. doi: 10.1016/j.jri.2019.103074
369. Boulanger H, Bounan S, Mahdhi A, Drouin D, Ahriz-Saksi S, Guimiot F, et al. Immunologic aspects of preeclampsia. AJOG Global Rep. (2024) 4:100321. doi: 10.1016/j.xagr.2024.100321
370. Borzychowski AM, Sargent IL, and Redman CW. Inflammation and pre-eclampsia. Semin Fetal Neonatal Med. (2006) 11:309–16. doi: 10.1016/j.siny.2006.04.001
371. Ramma W and Ahmed A. Is inflammation the cause of pre-eclampsia? Biochem Soc Trans. (2011) 39:1619–27. doi: 10.1042/BST20110672
372. Markiewski MM and Lambris JD. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am J Pathol. (2007) 171:715–27. doi: 10.2353/ajpath.2007.070166
373. Ruan C-C and Gao P-J. Role of complement-related inflammation and vascular dysfunction in hypertension. Hypertension. (2019) 73:965–71. doi: 10.1161/HYPERTENSIONAHA.118.11210
374. Blakey H, Sun R, Xie L, Russell R, Sarween N, Hodson J, et al. Pre-eclampsia is associated with complement pathway activation in the maternal and fetal circulation, and placental tissue. Pregnancy Hypertension. (2023) 32:43–9. doi: 10.1016/j.preghy.2023.04.001
375. Velickovic I, Dalloul M, Wong KA, Bakare O, Schweis F, Garala M, et al. Complement factor B activation in patients with preeclampsia. J Reprod Immunol. (2015) 109:94–100. doi: 10.1016/j.jri.2014.12.002
376. Than NG, Romero R, Erez O, Kusanovic JP, Tarca AL, Edwin SS, et al. A role for mannose-binding lectin, a component of the innate immune system in pre-eclampsia. Am J Reprod Immunol. (2008) 60:333–45. doi: 10.1111/j.1600-0897.2008.00631.x
377. Johnson JD and Louis JM. Does race or ethnicity play a role in the origin, pathophysiology, and outcomes of preeclampsia? An expert review of the literature. Am J Obstetrics Gynecology. (2022) 226:S876–85. doi: 10.1016/j.ajog.2020.07.038
378. Lynch AM, Murphy JR, Byers T, Gibbs RS, Neville MC, Giclas PC, et al. Alternative complement pathway activation fragment Bb in early pregnancy as a predictor of preeclampsia. Am J Obstet Gynecol. (2008) 198:385.e381–389. doi: 10.1016/j.ajog.2007.10.793
379. Jia K, Ma L, Wu S, and Yang W. Serum levels of complement factors C1q, bb, and H in normal pregnancy and severe pre-eclampsia. Med Sci Monit. (2019) 25:7087–93. doi: 10.12659/MSM.915777
380. He Y, Xu B, Song D, Yu F, Chen Q, and Zhao M. Expression of the complement system’s activation factors in plasma of patients with early/late-onset severe pre-eclampsia. Am J Reprod Immunol. (2016) 76:205–11. doi: 10.1111/aji.12541
381. Ye Y, Kong Y, and Zhang Y. Complement split products C3a/C5a and receptors: are they regulated by circulating angiotensin II type 1 receptor autoantibody in severe preeclampsia? Gynecol Obstet Invest. (2016) 81:28–33. doi: 10.1159/000440651
382. He Y, Xu B, Song D, Yu F, Chen Q, and Zhao M. Correlations between complement system’s activation factors and anti-angiogenesis factors in plasma of patients with early/late-onset severe preeclampsia. Hypertens Pregnancy. (2016) 35:499–509. doi: 10.1080/10641955.2016.1190845
383. Lim R and Lappas M. Decreased expression of complement 3a receptor (C3aR) in human placentas from severe preeclamptic pregnancies. Eur J Obstet Gynecol Reprod Biol. (2012) 165:194–8. doi: 10.1016/j.ejogrb.2012.08.003
384. LaMarca B, Wallace K, and Granger J. Role of angiotensin II type I receptor agonistic autoantibodies (AT1-AA) in preeclampsia. Curr Opin Pharmacol. (2011) 11:175–9. doi: 10.1016/j.coph.2011.01.003
385. Herse F and LaMarca B. Angiotensin II type 1 receptor autoantibody (AT1-AA)-mediated pregnancy hypertension. Am J Reprod Immunol. (2013) 69:413–8. doi: 10.1111/aji.12072
386. Palmer KR, Tong S, and Kaitu’u-Lino TJ. Placental-specific sFLT-1: role in pre-eclamptic pathophysiology and its translational possibilities for clinical prediction and diagnosis. Mol Hum Reprod. (2017) 23:69–78. doi: 10.1093/molehr/gaw077
387. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. (2003) 111:649–58. doi: 10.1172/JCI17189
388. Piani F, Tossetta G, Fantone S, Agostinis C, Di Simone N, Mandalà M, et al. First trimester CD93 as a novel marker of preeclampsia and its complications: A pilot study. High Blood Pressure Cardiovasc Prev. (2023) 30:591–4. doi: 10.1007/s40292-023-00608-y
389. Becerra-Mojica CH, Mora-Guevara E, Parra-Saavedra MA, Martínez-Vega RA, Díaz-Martínez LA, and Rincón-Orozco B. Low levels of complement factor H in the first trimester of pregnancy are associated with spontaneous preterm birth. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms251910549
390. Lokki AI, Ren Z, Triebwasser M, Daly E, Perola M, Auro K, et al. Identification of complement factor H variants that predispose to pre-eclampsia: A genetic and functional study. Bjog. (2023) 130:1473–82. doi: 10.1111/1471-0528.17529
391. Jääskeläinen T, Heinonen S, Kajantie E, Kere J, Kivinen K, Pouta A, et al. Cohort profile: the finnish genetics of pre-eclampsia consortium (FINNPEC). BMJ Open. (2016) 6:e013148. doi: 10.1136/bmjopen-2016-013148
392. Dijkstra DJ, Lokki AI, Gierman LM, Borggreven NV, van der Keur C, Eikmans M, et al. Circulating levels of anti-C1q and anti-factor H autoantibodies and their targets in normal pregnancy and preeclampsia. Front Immunol. (2022) 13. doi: 10.3389/fimmu.2022.842451
393. Yasmin H, Agostinis C, Toffoli M, Roy T, Pegoraro S, Balduit A, et al. Protective role of complement factor H against the development of preeclampsia. Front Immunol. (2024) 15. doi: 10.3389/fimmu.2024.1351898
394. Salmon JE, Heuser C, Triebwasser M, Liszewski MK, Kavanagh D, Roumenina L, et al. Mutations in complement regulatory proteins predispose to preeclampsia: a genetic analysis of the PROMISSE cohort. PLoS Med. (2011) 8:e1001013. doi: 10.1371/journal.pmed.1001013
395. Balduit A, Agostinis C, Mangogna A, Zito G, Stampalija T, Ricci G, et al. Systematic review of the complement components as potential biomarkers of pre-eclampsia: pitfalls and opportunities. Front Immunol. (2024) 15:1419540. doi: 10.3389/fimmu.2024.1419540
396. Lokki AI, Teirilä L, Triebwasser M, Daly E, Bhattacharjee A, Uotila L, et al. Dysfunction of complement receptors CR3 (CD11b/18) and CR4 (CD11c/18) in pre-eclampsia: a genetic and functional study. Bjog. (2021) 128:1282–91. doi: 10.1111/1471-0528.16660
397. Burwick RM. Complement receptors in pre-eclampsia: cleaning up placental debris. Bjog. (2021) 128:1292. doi: 10.1111/1471-0528.16664
398. Lokki AI, Triebwasser M, Daly E, Kurki MI, Perola M, Auro K, et al. Understanding rare genetic variants within the terminal pathway of complement system in preeclampsia. Genes Immun. (2025) 26:22–6. doi: 10.1038/s41435-024-00310-6
399. Larsen JB, Andersen AS, Hvas CL, Thiel S, Lassen MR, Hvas AM, et al. Lectin pathway proteins of the complement system in normotensive pregnancy and pre-eclampsia. Am J Reprod Immunol. (2019) 81:e13092. doi: 10.1111/aji.13092
400. Halmos A, Rigó J Jr., Szijártó J, Füst G, Prohászka Z, and Molvarec A. Circulating ficolin-2 and ficolin-3 in normal pregnancy and pre-eclampsia. Clin Exp Immunol. (2012) 169:49–56. doi: 10.1111/j.1365-2249.2012.04590.x
401. Sziller I, Babula O, Hupuczi P, Nagy B, Rigó B, Szabó G, et al. Mannose-binding lectin (MBL) codon 54 gene polymorphism protects against development of pre-eclampsia, HELLP syndrome and pre-eclampsia-associated intrauterine growth restriction. Mol Hum Reprod. (2007) 13:281–5. doi: 10.1093/molehr/gam003
402. van de Geijn FE, Dolhain RJ, van Rijs W, Hazes JM, and de Groot CJ. Mannose-binding lectin genotypes and pre-eclampsia: a case-control study. Hum Immunol. (2007) 68:888–93. doi: 10.1016/j.humimm.2007.10.002
403. Banadakoppa M, Vidaeff AC, Yallampalli U, Ramin SM, Belfort MA, and Yallampalli C. Complement split products in amniotic fluid in pregnancies subsequently developing early-onset preeclampsia. Dis Markers. (2015) 2015:263109. doi: 10.1155/2015/263109
404. Haeger M, Bengtson A, Karlsson K, and Heideman M. Complement activation and anaphylatoxin (C3a and C5a) formation in preeclampsia and by amniotic fluid. Obstet Gynecol. (1989) 73:551–6.
405. Guseh SH, Feinberg BB, Dawood HY, Yamamoto HS, Fichorova RN, and Burwick RM. Urinary excretion of C5b-9 is associated with the anti-angiogenic state in severe preeclampsia. Am J Reprod Immunol. (2015) 73:437–44. doi: 10.1111/aji.12349
406. Burwick RM, Fichorova RN, Dawood HY, Yamamoto HS, and Feinberg BB. Urinary excretion of C5b-9 in severe preeclampsia: tipping the balance of complement activation in pregnancy. Hypertension. (2013) 62:1040–5. doi: 10.1161/HYPERTENSIONAHA.113.01420
407. Burwick RM, Velásquez JA, Valencia CM, Gutiérrez-Marín J, Edna-Estrada F, Silva JL, et al. Terminal complement activation in preeclampsia. Obstet Gynecol. (2018) 132:1477–85. doi: 10.1097/AOG.0000000000002980
408. Buurma A, Cohen D, Veraar K, Schonkeren D, Claas FH, Bruijn JA, et al. Preeclampsia is characterized by placental complement dysregulation. Hypertension. (2012) 60:1332–7. doi: 10.1161/HYPERTENSIONAHA.112.194324
409. Lokki AI, Heikkinen-Eloranta J, Jarva H, Saisto T, Lokki ML, Laivuori H, et al. Complement activation and regulation in preeclamptic placenta. Front Immunol. (2014) 5:312. doi: 10.3389/fimmu.2014.00312
410. Yonekura Collier AR, Zsengeller Z, Pernicone E, Salahuddin S, Khankin EV, and Karumanchi SA. Placental sFLT1 is associated with complement activation and syncytiotrophoblast damage in preeclampsia. Hypertens Pregnancy. (2019) 38:193–9. doi: 10.1080/10641955.2019.1640725
411. Holmes CH, Simpson KL, Wainwright SD, Tate CG, Houlihan JM, Sawyer IH, et al. Preferential expression of the complement regulatory protein decay accelerating factor at the fetomaternal interface during human pregnancy. J Immunol. (1990) 144:3099–105. doi: 10.4049/jimmunol.144.8.3099
412. Holmes CH, Simpson KL, Okada H, Okada N, Wainwright SD, Purcell DF, et al. Complement regulatory proteins at the feto-maternal interface during human placental development: distribution of CD59 by comparison with membrane cofactor protein (CD46) and decay accelerating factor (CD55). Eur J Immunol. (1992) 22:1579–85. doi: 10.1002/eji.1830220635
413. Ma Y, Kong LR, Ge Q, Lu YY, Hong MN, Zhang Y, et al. Complement 5a-mediated trophoblasts dysfunction is involved in the development of pre-eclampsia. J Cell Mol Med. (2018) 22:1034–46. doi: 10.1111/jcmm.13466
414. Girardi G, Yarilin D, Thurman JM, Holers VM, and Salmon JE. Complement activation induces dysregulation of angiogenic factors and causes fetal rejection and growth restriction. J Exp Med. (2006) 203:2165–75. doi: 10.1084/jem.20061022
415. Banadakoppa M, Balakrishnan M, and Yallampalli C. Upregulation and release of soluble fms-like tyrosine kinase receptor 1 mediated by complement activation in human syncytiotrophoblast cells. Am J Reprod Immunol. (2018) 80:e13033. doi: 10.1111/aji.13033
416. Matsuyama T, Tomimatsu T, Mimura K, Yagi K, Kawanishi Y, Kakigano A, et al. Complement activation by an angiogenic imbalance leads to systemic vascular endothelial dysfunction: A new proposal for the pathophysiology of preeclampsia. J Reprod Immunol. (2021) 145:103322. doi: 10.1016/j.jri.2021.103322
417. Saleh M, Compagno M, Pihl S, Strevens H, Persson B, Wetterö J, et al. Variation of complement protein levels in maternal plasma and umbilical cord blood during normal pregnancy: an observational study. J Clin Med. (2022) 11. doi: 10.3390/jcm11133611
418. Miyano A, Nakayama M, Fujita T, Kitajima H, Imai S, and Shimizu A. Complement activation in fetuses: assessment by the levels of complement components and split products in cord blood. Diagn Clin Immunol. (1987) 5:86–90.
419. Holers VM, Girardi G, Mo L, Guthridge JM, Molina H, Pierangeli SS, et al. Complement C3 activation is required for antiphospholipid antibody-induced fetal loss. J Exp Med. (2002) 195:211–20. doi: 10.1084/jem.200116116
420. Cohen D, Buurma A, Goemaere NN, Girardi G, le Cessie S, Scherjon S, et al. Classical complement activation as a footprint for murine and human antiphospholipid antibody-induced fetal loss. J Pathol. (2011) 225:502–11. doi: 10.1002/path.2893
421. Elfving P, Kariniemi S, Kautiainen H, Rantalaiho V, Virta LJ, Puolakka K, et al. Pregnancies in patients with systemic lupus erythematosus during 2000–2018 in Finland: a case-control study. Rheumatol Int. (2024) 44:1101–9. doi: 10.1007/s00296-024-05564-x
422. Teh CL, Wan SA, Cheong YK, and Ling GR. Systemic lupus erythematosus pregnancies: ten-year data from a single centre in Malaysia. Lupus. (2017) 26:218–23. doi: 10.1177/0961203316664996
423. Ong SG and Ding HJ. Predictors of adverse pregnancy outcome in a cohort of women with systemic lupus erythematosus in Malaysia. Med J Malaysia. (2021) 76:466–73.
424. Liu J, Zhao Y, Song Y, Zhang W, Bian X, Yang J, et al. Pregnancy in women with systemic lupus erythematosus: a retrospective study of 111 pregnancies in Chinese women. J Matern Fetal Neonatal Med. (2012) 25:261–6. doi: 10.3109/14767058.2011.572310
425. Al-Riyami N, Salman B, Al-Rashdi A, Al-Dughaishi T, Al-Haddabi R, and Hassan B. Pregnancy Outcomes in Systemic Lupus Erythematosus Women: A single tertiary centre experience. Sultan Qaboos Univ Med J. (2021) 21:e244–52. doi: 10.18295/squmj.2021.21.02.013
426. Essouma M, Nkeck JR, Motolouze K, Bigna JJ, Tchaptchet P, Nkoro GA, et al. Outcomes of pregnancy and associated factors in sub-Saharan African women with systemic lupus erythematosus: a scoping review. Lupus Sci Med. (2020) 7. doi: 10.1136/lupus-2020-000400
427. Salmon JE, Girardi G, and Holers VM. Activation of complement mediates antiphospholipid antibody-induced pregnancy loss. Lupus. (2003) 12:535–8. doi: 10.1191/0961203303lu397oa
428. Castellanos Gutierrez AS, Figueras F, Morales-Prieto DM, Schleußner E, Espinosa G, and Baños N. Placental damage in pregnancies with systemic lupus erythematosus: A narrative review. Front Immunol. (2022) 13:941586. doi: 10.3389/fimmu.2022.941586
429. Ostensen M and Clowse M. Pathogenesis of pregnancy complications in systemic lupus erythematosus. Curr Opin Rheumatol. (2013) 25:591–6. doi: 10.1097/BOR.0b013e328363ebf7
430. Steegers EA, von Dadelszen P, Duvekot JJ, and Pijnenborg R. Pre-eclampsia. Lancet. (2010) 376:631–44. doi: 10.1016/S0140-6736(10)60279-6
431. Sibai B, Dekker G, and Kupferminc M. Pre-eclampsia. Lancet. (2005) 365:785–99. doi: 10.1016/S0140-6736(05)17987-2
432. Sibai BM. Diagnosis, controversies, and management of the syndrome of hemolysis, elevated liver enzymes, and low platelet count. Obstet Gynecol. (2004) 103:981–91. doi: 10.1097/01.AOG.0000126245.35811.2a
433. Douglas KA and Redman CW. Eclampsia in the United Kingdom. Bmj. (1994) 309:1395–400. doi: 10.1136/bmj.309.6966.1395
434. Vaught AJ, Gavriilaki E, Hueppchen N, Blakemore K, Yuan X, Seifert SM, et al. Direct evidence of complement activation in HELLP syndrome: A link to atypical hemolytic uremic syndrome. Exp Hematol. (2016) 44:390–8. doi: 10.1016/j.exphem.2016.01.005
435. Vaught AJ, Braunstein E, Chaturvedi S, Blakemore K, and Brodsky RA. A review of the alternative pathway of complement and its relation to HELLP syndrome: is it time to consider HELLP syndrome a disease of the alternative pathway. J Matern Fetal Neonatal Med. (2022) 35:1392–400. doi: 10.1080/14767058.2020.1755650
436. Burwick RM, Java A, Gray KJ, Combs D, Rincon M, Roberts VH, et al. HELLP syndrome is characterized by a unique pattern of complement protein biomarkers. Am J Obstetrics Gynecology. (2022) 226:S484–5. doi: 10.1016/j.ajog.2021.11.802
437. Chen S, Zheng L, Yingdong H, and Chen Q. Dysregulation of complement system in HELLP syndrome. Hypertension Pregnancy. (2021) 40:303–11. doi: 10.1080/10641955.2021.1983593
438. Shah H, Boyer T, Chen H, Chaturvedi S, Vaught A, and Braunstein EM. Complement activation drives progression of pre-eclampsia to HELLP syndrome. Blood. (2021) 138:772–2. doi: 10.1182/blood-2021-148162
439. Vaught AJ, Braunstein EM, Jasem J, Yuan X, Makhlin I, Eloundou S, et al. Germline mutations in the alternative pathway of complement predispose to HELLP syndrome. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.99128
440. Gerber GF, Brodsky RA, and Vaught AJ. Complement C5 Inhibition in early onset HELLP Syndrome. Blood Adv. (2025) 9(13):3304–7. doi: 10.1182/bloodadvances.2024014746
441. Jivraj S, Anstie B, Cheong Y-C, Fairlie FM, Laird SM, and Li TC. Obstetric and neonatal outcome in women with a history of recurrent miscarriage: a cohort study. Hum Reprod. (2001) 16:102–6. doi: 10.1093/humrep/16.1.102
442. Mao D, Wu X, Deppong C, Friend LD, Dolecki G, Nelson DM, et al. Negligible role of antibodies and C5 in pregnancy loss associated exclusively with C3-dependent mechanisms through complement alternative pathway. Immunity. (2003) 19:813–22. doi: 10.1016/S1074-7613(03)00321-2
443. Zhou Z, Xie H, Liu M, Li R, Jiang W, Zheng Y, et al. Expression and correlation of complement C3 and C4 in serum and maternal-fetal interface in patients with unexplained recurrent spontaneous abortion: A prospective cohort study. CEOG. (2023) 50. doi: 10.31083/j.ceog5011252
444. Sugiura-Ogasawara M, Nozawa K, Nakanishi T, Hattori Y, and Ozaki Y. Complement as a predictor of further miscarriage in couples with recurrent miscarriages. Hum Reprod. (2006) 21:2711–4. doi: 10.1093/humrep/del229
445. Krog MC, Flachs EM, Kolte AM, de Jager W, Meyaard L, Christiansen OB, et al. Angiogenic factors and the lectin pathway of complement in women with secondary recurrent pregnancy loss. J Reprod Immunol. (2024) 163:104221. doi: 10.1016/j.jri.2024.104221
446. Mohlin FC, Mercier E, Fremeaux-Bacchi V, Liszewski MK, Atkinson JP, Gris JC, et al. Analysis of genes coding for CD46, CD55, and C4b-binding protein in patients with idiopathic, recurrent, spontaneous pregnancy loss. Eur J Immunol. (2013) 43:1617–29. doi: 10.1002/eji.201243196
447. Mohlin FC, Gros P, Mercier E, Gris JR, and Blom AM. Analysis of C3 gene variants in patients with idiopathic recurrent spontaneous pregnancy loss. Front Immunol. (2018) 9:1813. doi: 10.3389/fimmu.2018.01813
448. Lewis RD, Narayanaswamy AK, Farewell D, and Rees DA. Complement activation in polycystic ovary syndrome occurs in the postprandial and fasted state and is influenced by obesity and insulin sensitivity. Clin Endocrinol (Oxf). (2021) 94:74–84. doi: 10.1111/cen.14322
449. Butler AE, Moin ASM, Sathyapalan T, and Atkin SL. Complement dysregulation in obese versus nonobese polycystic ovary syndrome patients. Cells. (2023) 12. doi: 10.3390/cells12152002
450. Moin ASM, Sathyapalan T, Butler AE, and Atkin SL. Classical and alternate complement factor overexpression in non-obese weight matched women with polycystic ovary syndrome does not correlate with vitamin D. Front Endocrinol (Lausanne). (2022) 13:935750. doi: 10.3389/fendo.2022.935750
451. O’Brien TE, Ray JG, and Chan WS. Maternal body mass index and the risk of preeclampsia: a systematic overview. Epidemiology. (2003) 14:368–74. doi: 10.1097/01.EDE.0000059921.71494.D1
452. Canto-Cetina T, Coral-Vázquez RM, Rojano-Mejía D, Pérez Godoy S, Coronel A, and Canto P. Higher prepregnancy body mass index is a risk factor for developing preeclampsia in Maya-Mestizo women: a cohort study. Ethn Health. (2018) 23:682–90. doi: 10.1080/13557858.2017.1315367
453. Vinturache A, Moledina N, McDonald S, Slater D, and Tough S. Pre-pregnancy Body Mass Index (BMI) and delivery outcomes in a Canadian population. BMC Pregnancy Childbirth. (2014) 14:422. doi: 10.1186/s12884-014-0422-y
454. Schiavone MJ, Pérez MP, Aquieri A, Nosetto D, Pronotti MV, Mazzei M, et al. The role of obesity in the development of preeclampsia. Curr Hypertens Rep. (2024) 26:247–58. doi: 10.1007/s11906-024-01299-z
455. Shim K, Begum R, Yang C, and Wang H. Complement activation in obesity, insulin resistance, and type 2 diabetes mellitus. World J Diabetes. (2020) 11:1–12. doi: 10.4239/wjd.v11.i1.1
456. Mamane Y, Chung Chan C, Lavallee G, Morin N, Xu LJ, Huang J, et al. The C3a anaphylatoxin receptor is a key mediator of insulin resistance and functions by modulating adipose tissue macrophage infiltration and activation. Diabetes. (2009) 58:2006–17. doi: 10.2337/db09-0323
457. Phieler J, Chung KJ, Chatzigeorgiou A, Klotzsche-von Ameln A, Garcia-Martin R, Sprott D, et al. The complement anaphylatoxin C5a receptor contributes to obese adipose tissue inflammation and insulin resistance. J Immunol. (2013) 191:4367–74. doi: 10.4049/jimmunol.1300038
458. Lynch AM, Eckel RH, Murphy JR, Gibbs RS, West NA, Giclas PC, et al. Prepregnancy obesity and complement system activation in early pregnancy and the subsequent development of preeclampsia. Am J Obstet Gynecol. (2012) 206:428.e421–428. doi: 10.1016/j.ajog.2012.02.035
459. Olson KN, Redman LM, and Sones JL. Obesity “complements” preeclampsia. Physiol Genomics. (2019) 51:73–6. doi: 10.1152/physiolgenomics.00102.2018
460. Robinson DP and Klein SL. Pregnancy and pregnancy-associated hormones alter immune responses and disease pathogenesis. Horm Behav. (2012) 62:263–71. doi: 10.1016/j.yhbeh.2012.02.023
461. Abu-Raya B, Michalski C, Sadarangani M, and Lavoie PM. Maternal immunological adaptation during normal pregnancy. Front Immunol. (2020) 11. doi: 10.3389/fimmu.2020.575197
462. Sherer ML, Posillico CK, and Schwarz JM. An examination of changes in maternal neuroimmune function during pregnancy and the postpartum period. Brain Behavior Immun. (2017) 66:201–9. doi: 10.1016/j.bbi.2017.06.016
463. Brunton PJ. Neuroactive steroids and stress axis regulation: Pregnancy and beyond. J Steroid Biochem Mol Biol. (2016) 160:160–8. doi: 10.1016/j.jsbmb.2015.08.003
464. Kopaliani I, Elsaid B, Speier S, and Deussen A. Immune and metabolic mechanisms of endothelial dysfunction. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms252413337
465. Rizzoni D, De Ciuceis C, Szczepaniak P, Paradis P, Schiffrin EL, and Guzik TJ. Immune system and microvascular remodeling in humans. Hypertension. (2022) 79:691–705. doi: 10.1161/HYPERTENSIONAHA.121.17955
466. Zeng S, Han M, Jiang M, Liu F, Hu Y, Long Y, et al. Serum complement proteomics reveal biomarkers for hypertension disorder of pregnancy and the potential role of Clusterin. Reprod Biol Endocrinol. (2021) 19:56. doi: 10.1186/s12958-021-00742-z
467. Zeng S, Pan Y, Liu F, Yin J, Jiang M, Long Y, et al. Role of clusterin in the regulation of trophoblast development and preeclampsia. Biochem Biophys Res Commun. (2021) 583:128–34. doi: 10.1016/j.bbrc.2021.10.064
468. Watanabe H, Hamada H, Yamada N, Sohda S, Yamakawa-Kobayashi K, Yoshikawa H, et al. Proteome analysis reveals elevated serum levels of clusterin in patients with preeclampsia. PROTEOMICS. (2004) 4:537–43. doi: 10.1002/pmic.200300565
469. Oztas E, Ozler S, Ersoy AO, Iskender CT, Sucak A, Ergin M, et al. Increased levels of serum clusterin is associated with intrauterine growth restriction and adverse pregnancy outcomes in preeclampsia. J Perinatal Med. (2016) 44:269–75. doi: 10.1515/jpm-2015-0120
470. Yao S, Chen Y, Cao R, Lu L, Yang J, Lei W, et al. Clusterin from endometrial glands plays a critical role in decidualization via Trem2. BMC Genomics. (2024) 25:969. doi: 10.1186/s12864-024-10827-9
471. Dolgyras P, Anyfanti P, Lazaridis A, Gavriilaki E, Koletsos N, Triantafyllou A, et al. Endothelial dysfunction and complement activation are independently associated with disease duration in patients with systemic vasculitis. Microvasc Res. (2024) 154:104692. doi: 10.1016/j.mvr.2024.104692
472. Konukoglu D and Uzun H. Endothelial dysfunction and hypertension. Adv Exp Med Biol. (2017) 956:511–40. doi: 10.1007/5584_2016_90
473. Wang Z, Zhang Z, Li Y, Zhang Y, Wei M, Li H, et al. Endothelial-derived complement factor D contributes to endothelial dysfunction in Malignant nephrosclerosis via local complement activation. Hypertension Res. (2023) 46:1759–70. doi: 10.1038/s41440-023-01300-3
474. Cervia-Hasler C, Brüningk SC, Hoch T, Fan B, Muzio G, Thompson RC, et al. Persistent complement dysregulation with signs of thromboinflammation in active Long Covid. Science. (2024) 383:eadg7942. doi: 10.1126/science.adg7942
475. Baillie K, Davies HE, Keat SBK, Ladell K, Miners KL, Jones SA, et al. Complement dysregulation is a prevalent and therapeutically amenable feature of long COVID. Med. (2024) 5:239–253.e235. doi: 10.1016/j.medj.2024.01.011
476. Wenzel UO, Kemper C, and Bode M. The role of complement in arterial hypertension and hypertensive end organ damage. Br J Pharmacol. (2021) 178:2849–62. doi: 10.1111/bph.15171
477. Karthikeyan VJ and Lip GY. Endothelial damage/dysfunction and hypertension in pregnancy. Front Biosci (Elite Ed). (2011) 3:1100–8. doi: 10.2741/314
478. Ramos A, Youssef L, Molina P, Martinez-Sanchez J, Moreno-Castaño AB, Blasco M, et al. Endothelial damage and complement dysregulation in fetuses from pregnancies complicated by preeclampsia. Acta Obstetricia Gynecologica Scandinavica. (2025) 104(5):829–38. doi: 10.1111/aogs.15072
479. Chen L, Fukuda N, Matsumoto T, and Abe M. Role of complement 3 in the pathogenesis of hypertension. Hypertension Res. (2020) 43:255–62. doi: 10.1038/s41440-019-0371-y
480. Penning M, Chua JS, van Kooten C, Zandbergen M, Buurma A, Schutte J, et al. Classical complement pathway activation in the kidneys of women with preeclampsia. Hypertension. (2015) 66:117–25. doi: 10.1161/HYPERTENSIONAHA.115.05484
481. Dusse LM, Rios DRA, Pinheiro MB, Cooper AJ, and Lwaleed BA. Pre-eclampsia: Relationship between coagulation, fibrinolysis and inflammation. Clinica Chimica Acta. (2011) 412:17–21. doi: 10.1016/j.cca.2010.09.030
482. Drury-Stewart DN, Hoppe KK, Lannert KW, Chung DW, Gammill HS, and Johnsen JM. Pregnancies complicated by severe preeclampsia exhibit perturbed von willebrand factor (VWF)-associated parameters. Blood. (2013) 122:3525. doi: 10.1182/blood.V122.21.3525.3525
483. Soleimani Samarkhazan H, Khaksari MN, Rahmati A, Esfahani ML, Solouki A, and Aghaei M. Von Willebrand disease (VWD) and pregnancy: a comprehensive overview. Thromb J. (2025) 23:41. doi: 10.1186/s12959-025-00727-7
484. MCKAY DG. Hematologic evidence of disseminated intravascular coagulation in eclampsia. Obstetrical Gynecological Survey. (1972) 27:399–417. doi: 10.1097/00006254-197206000-00001
485. He S, Bremme K, and Blombäck M. Acquired deficiency of antithrombin in association with a hypercoagulable state and impaired function of liver and/or kidney in preeclampsia. Blood coagulation fibrinolysis. (1997) 8:232–8. doi: 10.1097/00001721-199706000-00004
486. Carson SD and Johnson DR. Consecutive enzyme cascades: complement activation at the cell surface triggers increased tissue factor activity. Blood. (1990) 76:361–7. doi: 10.1182/blood.V76.2.361.361
487. Heurich M and McCluskey G. Complement and coagulation crosstalk – Factor H in the spotlight. Immunobiology. (2023) 228:152707. doi: 10.1016/j.imbio.2023.152707
488. Muhlfelder TW, Niemetz J, Kreutzer D, Beebe D, Ward PA, and Rosenfeld SI. C5 chemotactic fragment induces leukocyte production of tissue factor activity: a link between complement and coagulation. J Clin Invest. (1979) 63:147–50. doi: 10.1172/JCI109269
489. Ikeda K, Nagasawa K, Horiuchi T, Tsuru T, Nishizaka H, and Niho Y. C5a induces tissue factor activity on endothelial cells. Thromb Haemost. (1997) 77:394–8.
490. Foley JH, Walton BL, Aleman MM, O’Byrne AM, Lei V, Harrasser M, et al. Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine. (2016) 5:175–82. doi: 10.1016/j.ebiom.2016.02.011
491. Amara U, Flierl MA, Rittirsch D, Klos A, Chen H, Acker B, et al. Molecular intercommunication between the complement and coagulation systems. J Immunol. (2010) 185:5628–36. doi: 10.4049/jimmunol.0903678
492. Amara U, Rittirsch D, Flierl M, Bruckner U, Klos A, Gebhard F, et al. Interaction between the coagulation and complement system. Adv Exp Med Biol. (2008) 632:71–9. doi: 10.1007/978-0-387-78952-1_6
493. Youssef L, Miranda J, Blasco M, Paules C, Crovetto F, Palomo M, et al. Complement and coagulation cascades activation is the main pathophysiological pathway in early-onset severe preeclampsia revealed by maternal proteomics. Sci Rep. (2021) 11:3048. doi: 10.1038/s41598-021-82733-z
494. Quintanilla B, Greenstein D, Tripathi A, Bartosh A, Yuan P, Zarate CA, et al. Assessment of complement cascade components in patients with major depressive disorder. Brain Behavior Immun. (2025) 127:229–37. doi: 10.1016/j.bbi.2025.03.009
495. Rondung E, Massoudi P, Nieminen K, Wickberg B, Peira N, Silverstein R, et al. Identification of depression and anxiety during pregnancy: A systematic review and meta-analysis of test accuracy. Acta Obstetricia Gynecologica Scandinavica. (2024) 103:423–36. doi: 10.1111/aogs.14734
Keywords: human pregnancy, preeclampsia, CS, placenta, immunoregulation, immune homeostasis
Citation: Kumar V and Stewart JH IV (2025) The complement system in human pregnancy and preeclampsia. Front. Immunol. 16:1617140. doi: 10.3389/fimmu.2025.1617140
Received: 23 April 2025; Accepted: 23 July 2025;
Published: 19 August 2025.
Edited by:
Paola Triggianese, University of Rome Tor Vergata, ItalyReviewed by:
Guilherme Ramires De Jesús, Rio de Janeiro State University, BrazilKonstantine Halkidis, University of Kansas Medical Center, United States
Copyright © 2025 Kumar and Stewart. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vijay Kumar, dmlqa3VtYXJAbXNtLmVkdQ==; dmlqX3RveEB5YWhvby5jb20=
†ORCID: Vijay Kumar, orcid.org/0000-0001-9741-3597