 Santhosh Mudipalli Elavarasu
Santhosh Mudipalli Elavarasu Sasikumar K
Sasikumar K- 1Department of Integrative Biology, School of Biosciences and Technology, Vellore Institute of Technology (VIT), Vellore, Tamil Nadu, India
- 2Department of Sensor and Biomedical Technology, School of Electronics Engineering, Vellore Institute of Technology (VIT), Vellore, Tamil Nadu, India
Introduction: As a highly adaptable opportunistic pathogen, Pseudomonas aeruginosa presents a significant threat to people with weakened immune systems. This is because it naturally resists antibiotics and can form biofilms. These factors complicate treatment and underscore the urgent need for innovative therapeutic strategies, such as vaccines, to combat this pathogen.
Methods: A pangenome analysis of P. aeruginosa genomes was performed to identify conserved core genes critical for bacterial survival and virulence. LptF, an outer membrane protein, was prioritized as a target for vaccine development. B-cell and T-cell epitopes were predicted from LptF using immunoinformatics tools, and a multi-epitope peptide vaccine was designed. The interaction between the vaccine candidate and Toll-like receptors (TLRs) was investigated through molecular docking and molecular dynamics simulations. Codon optimization and in-silico cloning were carried out to validate the vaccine’s expression potential in E. coli. Immune response simulations evaluated the vaccine’s immunogenicity.
Results: Our pangenome analysis identified highly conserved core genes, including LptF, which proved crucial for bacterial virulence. A multi-epitope peptide vaccine was designed using the most immunogenic B-cell and T-cell epitopes derived from LptF. Studies using molecular docking and dynamic simulation have shown stable interactions between the vaccine and TLRs, with the POA_V_RS09 construct exhibiting the highest stability. Codon optimization indicated high expression efficiency in E. coli. Immune simulations revealed robust adaptive immune responses, including sustained IgG production, the formation of memory B cells, and the activation of T-cell responses.
Discussion: The POA_V_RS09 vaccine candidate exhibited excellent stability, immunogenic potential, and expression efficiency, making it a promising candidate for combating P. aeruginosa infections. This study provides a strong foundation for developing effective therapeutic strategies to address the growing issue of antimicrobial resistance in P. aeruginosa. More experimental validation is needed to verify its effectiveness in preclinical and clinical environments.
Graphical Abstract.
1 Introduction
Pseudomonas aeruginosa (P. aeruginosa), a highly adaptable opportunistic pathogen, is a significant cause of multidrug-resistant (MDR) infections, including diabetic foot infections, ventilator-associated pneumonia, wound infections, septicemia, and catheter-associated urinary tract infections (1). It poses a significant threat, particularly to immunocompromised individuals, due to its intrinsic resistance to antibiotics and its ability to thrive in diverse environments. Furthermore, P. aeruginosa can spread through medical equipment, increasing the risk of cross-contamination between patients and complicating infection control in healthcare settings (2). According to the World Health Organization (WHO), antimicrobial resistance (AMR) is expected to cause 10 million deaths annually by 2050, highlighting its severe impact as a global health threat (3). Hospital-acquired infections caused by ESKAPE pathogens, Enterobacter species, P. aeruginosa, Staphylococcus aureus, Acinetobacter baumannii, Klebsiella pneumoniae, and Enterococcus faecium are particularly concerning as they employ diverse mechanisms to resist antibiotics, making treatment increasingly challenging (4). Addressing P. aeruginosa’s virulence and its role as a key contributor to AMR highlights the urgent need for new therapeutic strategies, such as vaccines, to mitigate its impact (5, 6).
According to the WHO’s 2024 list of critical diseases, P. aeruginosa is a high-burden resistant bacterium resistant to last-resort antibiotics (7). Factors contributing to its pathogenicity include secretion systems, biofilm formation, and toxin production. Biofilms protect bacteria from host immune responses and medications, promoting the formation of multidrug-resistant persister cells that cause recurrent infections, particularly in patients with cystic fibrosis (8). P. aeruginosa employs its Type III secretion system to inject effector proteins into the host cells, interfering with cellular processes and facilitating immune evasion (9). The bacterium exhibits three primary resistance mechanisms: intrinsic resistance (efflux pumps, antibiotic-inactivating enzymes, limited outer membrane permeability), acquired resistance (mutations or horizontal gene transfer leading to resistance to aminoglycosides, quinolones, and β-lactams), and adaptive resistance (driven by persister cells and biofilm formation) (10). Clinical outcomes of P. aeruginosa infections are generally worse than those caused by other bacteria (11–13). During the COVID-19 pandemic, despite a decrease in the overall number of isolates, the percentage of MDR P. aeruginosa isolates increased significantly, from 23.8% in 2019 to 38.8% in 2020 (14). This trend was influenced by longer hospital stays, increased ICU admissions, and a greater reliance on empirical antibiotics, primarily due to the severity of cases and the extensive use of mechanical ventilation. This highlights how AMR is exacerbated in healthcare settings during pandemics (15). With the overuse of antibiotics, slow development of new drugs, and increasing complexity of healthcare, AMR is expected to worsen, leading to higher mortality rates and a greater burden on healthcare systems globally. Traditional antibiotics are becoming ineffective against MDR and extensively drug-resistant (XDR) strains, which no longer respond to standard treatments (16). The limited efficacy of last-resort drugs, such as colistin, coupled with their toxicity risks, makes managing resistant infections even more challenging (17). The lack of specific, targeted therapies for resistant infections leaves healthcare providers with limited options, underscoring the need for novel treatments and more effective alternatives to combat AMR (18). Among vaccine development studies for P. aeruginosa, outer membrane proteins such as Porin F (OprF) and Lipoprotein I (OprI) have been extensively explored as potential antigen targets (19).
Vaccines are crucial for preventing infections and reducing antibiotic use in low- and middle-income countries, significantly contributing to the fight against AMR. By lowering the incidence of infectious diseases, vaccines help minimize antibiotic misuse and overuse, particularly in populations with limited access to healthcare (20). Vaccines hold significant promise in addressing AMR by preventing infections, reducing antibiotic dependency, and curbing the spread of resistant strains (21). However, designing a vaccine for P. aeruginosa has been challenging due to its complex genetic diversity, biofilm formation, and immune evasion capabilities (22). Recent advancements in genomics and immunoinformatics offer new opportunities to overcome these obstacles. Computational tools for identifying novel vaccine candidates pave the way for developing targeted vaccines that can address the diversity of P. aeruginosa strains and enhance immune protection (23). In this study, we employed a high-resolution pan-genomic analysis of complete P. aeruginosa genomes from the NCBI RefSeq database to identify core, virulence-associated proteins. Among the prioritized candidates, LptF, a component of the LPS transport system, has been classified as a lipotoxin (LPT) due to its ability to trigger strong pro-inflammatory responses via TLR2 activation, particularly in cystic fibrosis. LptF is a pro-inflammatory lipotoxin involved in the excessive induction of IL-8 in cystic fibrosis and remains underexplored as a vaccine target (24). Our pan-genome analysis has identified LptF as a key membrane-associated protein that interacts with virulence factors, such as OprI and LptE, which supports its potential as a new therapeutic candidate (25). Our pipeline integrates reverse vaccinology, structural modeling, and molecular dynamics simulations to design a multi-epitope subunit vaccine construct. Unlike previous studies that relied on reference strains, metabolic enzymes, or limited proteome screening, our approach emphasizes strain-wide conservation, immune accessibility, and functional relevance. This integrative, pathogen-focused design offers a rational and potentially effective strategy for developing a broad-coverage vaccine against MDR P. aeruginosa. Using linkers, these epitopes can be linked to effective adjuvants to develop vaccines.
2 Materials and methods
2.1 Genome data retrieval
A comprehensive dataset of P. aeruginosa genomes, all at the “complete” assembly level, was obtained from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/) using the NCBI Genome Download Toolkit (26). To ensure comprehensive genomic representation, this dataset included a variety of strains, encompassing both clinical isolates and reference strains.
2.2 Pangenome construction and analysis
P. aeruginosa strains underwent pangenome analysis using the Roary tool (Version 3.13.0) (27). A diverse set of strains was initially selected to capture extensive genetic variability by collecting whole genomes from the NCBI RefSeq database. These genomes were annotated using Prokka (Version 1.14.6), which converted raw sequences into functional gene and protein data (28). Prokka is used to annotate essential genetic elements such as transfer RNA (tRNA), ribosomal RNA (rRNA), and coding sequences (CDS) for each genome, ensuring consistent annotation across all strains. Roary identifies the core and the accessory genes, revealing the conserved and variable genomic regions among P. aeruginosa strains. Core genes from all genomes were extracted from the Roary output for further detailed analysis, providing insights into essential genomic elements and potential targets for vaccine or therapeutic development. This pangenome analysis elucidated the genetic composition of the species and identified potential targets for further therapeutic advancements.
2.3 Prediction of subcellular localization
Following the identification of core genes, we employed the PSORTb tool (version 3.0.3) to predict their subcellular localization (29). PSORTb, a robust tool for prokaryotic genome analysis, categorized the core genes based on their predicted cellular locations, including cytoplasmic, periplasmic, and outer membrane regions. This study primarily focused on outer membrane proteins due to their accessibility on the bacterial surface, making them ideal targets for vaccine development. To confirm that the selected outer membrane-associated genes did not show homology with the human proteins, we conducted a comparison against the human proteome using BLASTP analysis (E-value 0.0001) (30). This step was essential to avoid potential cross-reactivity and enhance the specificity of vaccine candidate selection.
2.4 Analysis using the virulence factor database
The identified outer membrane proteins were analyzed by comparing them to the Virulence Factor Database (VFDB) using BLASTP [E-value = 0.0001, protein sequences from the VFDB full dataset (set B)] (31). This comparative analysis aimed to determine whether the selected protein candidates possess virulence potential, thereby assessing their suitability as targets for therapeutic or vaccine development. By aligning these outer membrane proteins with known virulence factors, we identified candidates with established roles in pathogenicity, enhancing the selection of proteins with significant implications in host-pathogen interactions. The selected target underwent an additional BLASTP search against the P. aeruginosa database for further validation (32). This analysis provided insights into the protein’s potential role, supporting its relevance for subsequent stages of the study.
2.5 Immunogenic potential and structural characterization of vaccine candidate
We evaluated the selected sequence’s antigenic potential using the VaxiJen v2.0 (https://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) server (33) to determine its suitability as an antigenic candidate. The sequence was analyzed with Allertop v2.0 (34) to assess allergenic risk, ensuring it lacked properties that could trigger allergic reactions. We used the ProtParam tool (https://web.expasy.org/protparam/) to determine the physicochemical properties, including molecular weight, instability index, grand average of hydropathicity (GRAVY), and hydrophobicity (35). These analyses provided essential insights into the protein’s suitability for vaccine development by assessing its immunogenicity, safety, and stability.
2.6 Signal peptide prediction analysis
SignalP 6.0 (https://services.healthtech.dtu.dk/services/SignalP-6.0/) is a sophisticated bioinformatics tool designed to detect signal peptides in protein sequences and pinpoint their cleavage sites (36). Utilizing protein language models (LMs), it analyzes the N-terminal region of proteins. Based on the predicted pathway and cleavage mechanism, SignalP classifies signal peptides into various types, such as Sec/SPI and Tat/SPI. The tool provides crucial scores, including the S-score for signal peptide probability and the C-score for predicting cleavage sites. This is essential for developing vaccines, as it helps identify secreted or surface-exposed proteins that could serve as potential immunogenic targets.
2.7 Prediction of linear B-cell epitopes
For the prediction of linear B-cell epitopes, we utilized BepiPred 2.0, which relies on the Immune Epitope Database (IEDB) Analysis resource (https://www.iedb.org/) (37, 38). This tool analyses amino acid propensity scores and identifies patterns typical of B-cell epitopes, using propensity scales to identify regions likely to consist of these epitopes. Improved accuracy of predictions is achieved by training on known antigen-antibody complexes, with the Random Forest method refining the results. The antigenic potential of the predicted epitopes was further assessed using VaxiJen v2.0 to determine their ability to stimulate an immune response. In this study, it served as an additional screening tool to prioritize epitopes (B and T Cell epitopes) with higher intrinsic antigenic potential before subjecting them to downstream immunoinformatics and structural analyses. Allertop v2.0 assessed allergenicity, ensuring the epitopes would not trigger allergic reactions. Additionally, the toxicity profiles of the selected epitopes were evaluated using the ToxinPred server (39), making sure they had a low risk of allergic reactions was a key step in designing the vaccine.
2.8 Prediction of T-cell epitopes (MHC Class I and II)
Epitope prediction for helper (HTL) and cytotoxic (CTL) T lymphocytes was performed using the NetMHCpan 4.1 algorithm provided by the Immune Epitope Database (IEDB) Analysis Resource (40). The focus was on non-structural (NS) proteins, which are conserved across various strains of P. aeruginosa and serve as key targets for immune responses. A human-specific approach was employed for CTL epitopes, identifying 10-mer peptides (ten amino acids long) that included 27 common HLA alleles as a reference panel. These epitopes were chosen for their ability to bind to MHC class I molecules and activate cytotoxic T cells, which is essential for targeting and eliminating infected cells. We selected T-cell epitopes based on recommendations from the IEDB for binding predictions. Specifically, we selected epitopes with a percentile rank of ≤ 1% for MHC class I, and a median percentile rank of ≤20% for MHC class II. These thresholds represent high and moderate affinity binders, and we mapped them to our scoring scale (≥ 0.60 for class I and ≥ 0.75 for class II) to include biologically relevant epitopes (41). For HTL epitopes, 15-mer peptides likely to stimulate helper T cells were identified using the IEDB-recommended method. These epitopes were designed to bind to MHC class II molecules, thereby activating B cells and initiating the humoral immune response. The input included antigenic, non-allergenic, and NS proteins from P. aeruginosa to ensure the predicted epitope’s efficacy and safety for vaccine development.
2.9 Prediction of interferon-γ inducing MHC-II epitopes
In this study, the IFNepitope server was used to predict MHC-II epitopes capable of inducing Interferon-gamma (IFN-γ) responses. This web-based tool leverages a comprehensive dataset from the IEDB, comprising 6,728 non-inducing epitopes and 3,705 confirmed IFN-γ-inducing epitopes (42). Utilizing the Support Vector Machine (SVM) technique, the server combines sequence analysis with predictive algorithms to identify epitopes with a high potential to stimulate IFN-γ production. We also analyzed the IL-4 prediction web server (43), the IL-6 prediction web server (44), the IL-10 prediction web server (45), and the IL-13 prediction web server (46). Epitopes were selected for vaccine development based on their prior assessment for antigenicity and non-allergenicity. This tool also prioritizes safe and immunologically relevant epitopes, which boosts the chances of a successful immune response.
2.10 Analysis of population coverage
The finalized T-cell epitopes and their associated HLA binding data were evaluated using the IEDB’s Population Coverage module to determine their global distribution across diverse populations (47). This analysis provided valuable insights into the epitope’s coverage across different demographics and regions, enhancing our understanding of their potential immunological effectiveness. By examining the global distribution of these epitopes, the study highlighted their relevance to diverse demographic groups. This crucial step demonstrated the epitope’s ability to address global healthcare needs, ensuring the vaccine candidate’s potential to protect a wide range of populations, thereby increasing its worldwide applicability and efficacy.
2.11 Vaccine design and construction
This study enhanced the vaccine design by incorporating carefully selected adjuvants, linkers, and epitopes to amplify the immune response. Two adjuvants were selected for their immune-boosting properties: RS-09 (APPHALS), a short peptide mimicking bacterial lipopolysaccharide, and Beta-defensin, a potent antimicrobial peptide known for its strong immunological activation (48, 49). Four linkers were used to achieve the best positioning and presentation of the epitopes. The EAAAK linker connected the adjuvants to the epitopes. This rigid helical linker promotes spatial separation between the adjuvant and the epitope region, thereby minimizing potential structural interference and enhancing adjuvant-mediated immune activation. The Alanine-Alanine-Tyrosine (AAY) linker was employed between MHC-I epitopes to enhance processing and presentation by MHC class I molecules. The MHC-II epitopes were separated using the Glycine-Proline-Glycine-Proline-Glycine (GPGPG) linker, which is a flexible and hydrophilic linker that aids in preserving epitope integrity and enhances recognition by helper T cells. Finally, the KK (Lysine-Lysine) linker was used to connect B-cell epitopes, ensuring adequate exposure for B-cell activation while maintaining their conformational flexibility and immunogenicity (50). These strategic additions of adjuvants and linkers were designed to optimize the vaccine’s ability to elicit strong and targeted immune responses, effectively combating the intended disease.
2.12 Analysis of the physicochemical properties of the formulated vaccines
The ProtParam server was utilized to conduct a physicochemical analysis of the developed vaccine candidates, assessing their stability and suitability for development (51). We analyzed the amino acid sequences to identify key structural and functional features. We calculated the molecular weight to estimate the proteins’ size, solubility, and potential antigenicity. To assess their biochemical behavior under physiological conditions, we determined the theoretical isoelectric point (pI), which indicated their net charge and acid–base characteristics. We also computed the instability index to predict the likelihood of protein degradation. However, the aliphatic index was evaluated to determine temperature stability based on the contribution of aliphatic amino acids. The GRAVY index was also evaluated to determine the vaccine’s overall hydrophobic or hydrophilic nature, aiding in understanding its solubility and stability.
2.13 Secondary structure analysis and prediction
The secondary structure of the developed vaccine was predicted using the PSIPRED tool (52), a widely used online resource for protein structure annotation and prediction. PSIPRED offers comprehensive protein analysis tools (53), with a focus on structural feature prediction. This analysis yielded valuable insights into how the vaccine might interact, its stability, and its functional properties. After entering the amino acid sequence of the final vaccine construct, the PSIPRED server analyzed the sequence and predicted the secondary structure, identifying coil, β-sheet, and α-helical regions. These predictions provide crucial insights about the overall structure and organization of the vaccine’s protein backbone.
2.14 Prediction and computational refinement of tertiary structure
To predict the three-dimensional (3D) structure of the developed vaccine and facilitate docking analysis, the ROBETTA server and AlphaFold (54, 55), which employ deep-learning techniques for accurate protein modeling, were utilized. The complete amino acid sequence of the vaccine was entered into both platforms, resulting in the prediction of multiple 3D structures in PDB format. These structures were enhanced in quality and accuracy using the GalaxyRefine tool (56). This tool refines the models by correcting structural errors, optimizing energy levels, and minimizing steric clashes. A comparative analysis of the refined models was conducted, and the best-performing structure, as determined by structural validation using a Ramachandran plot and other quality metrics, was selected for further docking studies.
2.15 Molecular docking and interaction studies
We employed molecular docking analysis to examine the interactions between the vaccine construct and Toll-like receptors TLR2 and TLR4, which are critical mediators of innate immune responses to infection. TLR2 was selected for its ability to detect various pathogen-associated molecular patterns and initiate immune responses (57). RS09 is a synthetic TLR4 agonist peptide that stimulates innate immunity. The TLR4 receptor recognizes a TLR4 agonist linked to the N-terminus of the vaccine construct. When TLR4 is activated, it triggers an intracellular signaling process via the NF-kB pathway, resulting in the production of inflammatory cytokines (58, 59). We retrieved the 3D structures of TLR2 and TLR4 from the RCSB PDB database, using IDs 2Z7X and 3FXI for TLR2 and TLR4, respectively, for further analysis (60, 61). Before docking, we thoroughly prepared the receptor structures by removing heteroatoms, water molecules, and bound ligands to ensure accurate analysis. This step was vital to prevent any interference during the docking process. Docking simulations were performed using the HDOCK web server (http://hdock.phys.hust.edu.cn/) (62). It employs a hybrid docking algorithm that combines template-based and free docking methods. In this study, we did blind docking to allow unbiased prediction of potential interaction sites between the vaccine construct and immune receptors. HDOCK, which is known for its intuitive interface and robust protein-protein docking capabilities, facilitated the simulation process by leveraging the refined 3D structure of the vaccine and the immune receptor models of TLR2 and TLR4. The docking affinity scores, indicating the strength of interaction between the vaccine and the receptors, were used to evaluate the results. Additionally, key residues involved in binding interactions were identified, providing insights into how these immune receptors recognize the vaccine. This study helps elucidate how the vaccine may effectively interact with TLR2 and TLR4, key components of the innate immune system, to trigger an immune response.
2.16 Molecular dynamics simulation analysis
To conduct molecular dynamics (MD) simulations for 1000ns, we utilized the CHARMM-GUI server’s Solution Builder protocol, applying the CHARMM36 force field to generate the necessary input files (63). The TIP3P water model was used to solvate the protein-protein complexes, creating a realistic simulation environment by enclosing the system in a periodic cubic box extending 10 Å from the protein atoms in all directions (64). Counter ions were added to neutralize the system, ensuring overall charge balance. The Verlet cutoff method was employed with a 10 Å cutoff distance, striking a balance between computational efficiency and accuracy to calculate electrostatic and van der Waals interactions. Bond constraints were applied using the LINCS algorithm to stabilize the simulation by maintaining fixed bond lengths. The Particle Mesh Ewald (PME) method was used to precisely calculate long-range electrostatic interactions, enhancing simulation accuracy in systems with periodic boundary conditions (65). To remove undesirable interactions and stabilize the system, the solvated system was subjected to energy minimization using the steepest descent technique (66). Two equilibration phases followed: the first in the NVT ensemble (constant Number of particles, Volume, and Temperature) to stabilize temperature, and the second in the NPT ensemble (constant number of particles, Pressure, and Temperature) to stabilize pressure. Proper thermostat and barostat techniques maintained constant temperature and pressure levels. This dual equilibration ensured system stability before the production run. The simulation recorded coordinates every 1 ps with a time step of 2 fs, striking a balance between computational efficiency and accuracy. CHARMM-GUI provided Python scripts to convert topology (top) and parameter (itp) files into GROMACS-compatible formats, simplifying input file preparation (67). Following the post-production run, we performed thorough trajectory analyses, including calculating Root Mean Square Deviation (RMSD) for structural stability, Root Mean Square Fluctuation (RMSF) for flexibility, hydrogen bond analysis (HBOND) for molecular interactions, Principal Component Analysis (PCA) for dominant motion patterns, Buried Surface Area (BSA) for evaluating binding stability, and Free Energy Landscape (FEL) analysis for the conformational states of the protein-protein complexes. Free energy calculations were performed for the interaction between TLR complexes and the vaccine construct (POA_V_RS09, POA_V_BDEF) using the MM-PBSA method with a Poisson-Boltzmann approach (68, 69). These approaches account for various energy components, including bonded interactions, van der Waals forces, electrostatic effects, and both polar and non-polar solvation energies. Here in MM-PBSA, the polar solvation energy is derived from the Poisson–Boltzmann equation, utilizing the molecular dynamics (MD) trajectory to compute interaction energies throughout the simulation. These analyses provided valuable insights into structural stability, flexibility, interaction dynamics, and potential conformational changes, enhancing our understanding of protein-protein interactions over time (70–72).
2.17 In silico cloning and expression analysis
To ensure optimal expression in the desired host, the gene of interest was first subjected to codon optimization using the GenScript program (www.genscript.com), aligning the gene sequence with the host’s preferred codon usage (73). Using SnapGene software (https://www.snapgene.com/), we cloned the vaccine construct via in silico. The result showed that the gene of interest and the pET-28a(+) plasmid did not share any restriction sites. This was addressed by adding specific nucleotide sequences to the gene’s N-terminal and C-terminal ends, which matched the restriction sites XhoI and NdeI, thereby aiding in cloning. These sequences provided suitable restriction sites for the accurate insertion of the gene into the plasmid. The appropriate recombinant plasmid construct was then produced by cloning the codon-optimized gene into the pET-28a(+) plasmid in silico using additional restriction sequences (74).
2.18 C-IMMSIM-based immune simulation
The C-IMMSIM server (https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php) (75), a widely used tool for simulating immune responses, was employed to evaluate the in-silico immunological response of the developed vaccine. This server utilizes a simulation-based framework to replicate the function of the human immune system and its organs, with a particular emphasis on primary lymphoid tissues. It uses a position-specific scoring matrix, enhanced by machine learning algorithms, to predict immune reactions. To achieve a total simulation period of 1050 steps, the vaccine and adjuvant were given in three doses: an initial dose, a second dose administered 84 days later, and a third dose administered 1050 days later, spaced eight hours apart. The adjuvant concentration was set to 100, and the injected antigen amount was 1000, following the server’s default parameters. This setup enabled a comprehensive evaluation of the immune response triggered by the vaccine or the adjuvant.
3 Results
3.1 NCBI data retrieval
A diverse array of 864 complete P. aeruginosa genomes, encompassing strains such as PAO1, PA14, PAK, LESB58, and CF39S, was sourced from the NCBI Assembly database utilizing the NCBI-genome-download toolkit. Supplementary Table S1 contains detailed information on all included genomes, ensuring a comprehensive genomic representation for subsequent analyses.
3.2 Pangenome analysis
A thorough pangenome analysis was performed on 864 complete genomes of P. aeruginosa sourced from the NCBI Assembly database. Genome annotation was executed using Prokka, followed by pangenome analysis with Roary, which identified a total of 63,239 genes. Of these, 3,325 were classified as core genes. Within this core set, 296 genes were consistently present across all genomes, with 79 hypothetical genes excluded from further analysis. Additionally, 3,149 accessory genes were identified in 15–95% of the genomes. The significant genomic diversity revealed by this open pangenome analysis highlights the extensive variability within P. aeruginosa (Figure 1). The figures were generated using R. This variability provides crucial insights into strain-specific adaptations, pathogenicity, and antibiotic resistance. Furthermore, identifying universally conserved targets among the core genes points to promising candidates for vaccine development applicable across diverse P. aeruginosa strains. These findings are pivotal in guiding future research and therapeutic strategies.
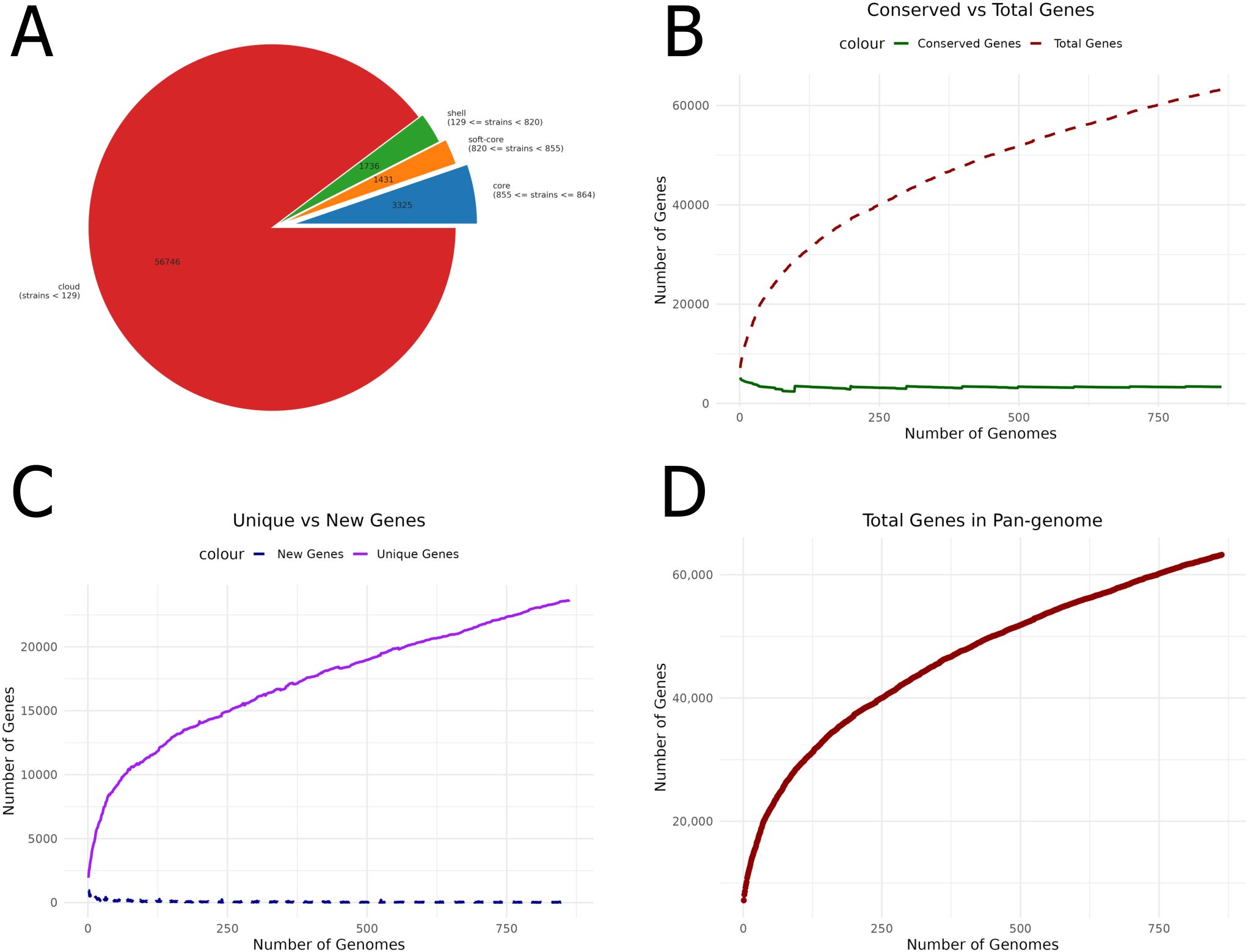
Figure 1. Pangenome analysis of P. aeruginosa genomes. (A) Pie chart showing the distribution of core, accessory, and unique genes. (B) Comparison of conserved genes with the total number of genes, highlighting genetic conservation across genomes. (C) Unique versus new gene ratio, emphasizing genome variability. (D) The number of genes identified within the pangenome provides insights into overall genomic diversity and its potential impact on vaccine development.
3.3 Subcellular localization and virulence prediction
PSORTb analysis identified three outer membrane proteins, while the remaining proteins were classified as cytoplasmic or belonging to other categories (Table 1). Subsequent BLASTP analysis against the human proteome in NCBI showed no homologous hits for the outer membrane proteins, ensuring their specificity and minimizing the risk of cross-reactivity in vaccine development. BLASTP analysis against the VFDB revealed that only the PAL_1 protein matched known virulence factors, confirming its potential as a relevant target for further therapeutic or vaccine development. Further analysis of PAL_1 against the P. aeruginosa database identified the protein as LptF, with an e-value of 0. To validate the conservation of the selected vaccine target LptF across diverse P. aeruginosa strains, a multiple sequence alignment was performed using LptF sequences from 864 genomes using Python (76). The conservation analysis revealed that over 98% of the amino acid positions were fully conserved (with 100% identity), and a pairwise sequence identity of greater than 99% was observed among all strains. A corresponding heatmap of the pairwise identity matrix further confirmed the uniform conservation pattern (Supplementary Figure S1). These results underscore the evolutionary stability of LptF and support its candidacy as a universal target for vaccine or therapeutic development.
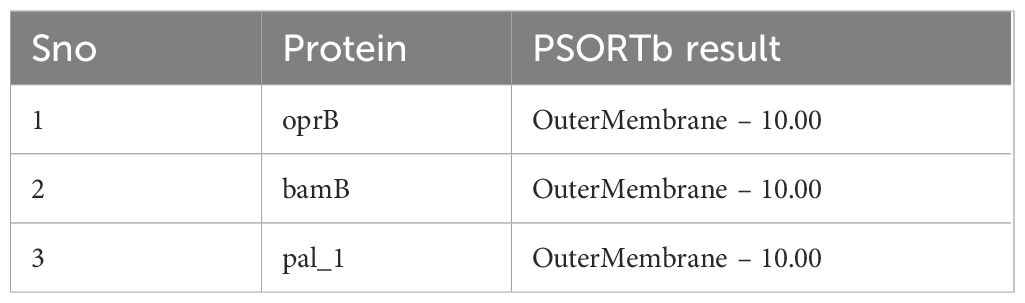
Table 1. Subcellular localization predictions for selected proteins based on PsortB analysis.
3.4 Analysis of immunogenic and physicochemical characteristics
The ProtParam tool was used to predict the physicochemical characteristics of the LptF protein. It has a molecular weight of 28.5 kDa and displays slight instability under standard laboratory conditions, with an instability index of 42.30. The GRAVY index of -0.574 indicates its hydrophilic nature. With an aliphatic index of 80.15, which reflects the protein’s thermostability, LptF is considered a strong candidate for vaccine development due to its stability at physiological temperatures. Its potential as an immunogenic candidate is further supported by an antigenicity score of 0.6442 (classified as likely antigenic with a threshold of 0.4) and its classification as non-allergenic by AllerTOP.
3.5 Signal peptide prediction
The analysis identified a Sec/SPII cleavage site at position 20 of the protein sequence, indicating the presence of a signal peptide that is likely cleaved during the maturation process via the Sec-dependent secretion pathway or the Sec/SPII system (Figure 2). With the signal peptide removed, the mature protein sequence begins at position 20. The signal peptide was excluded from further analysis, and the mature protein sequence was used in subsequent bioinformatics analyses. This sequence underwent secondary structure prediction, functional annotation, and potential epitope mapping, all of which are essential for understanding the protein’s biological function and its potential use in vaccine design. This approach ensures that only the biologically relevant mature protein is considered for downstream analyses.
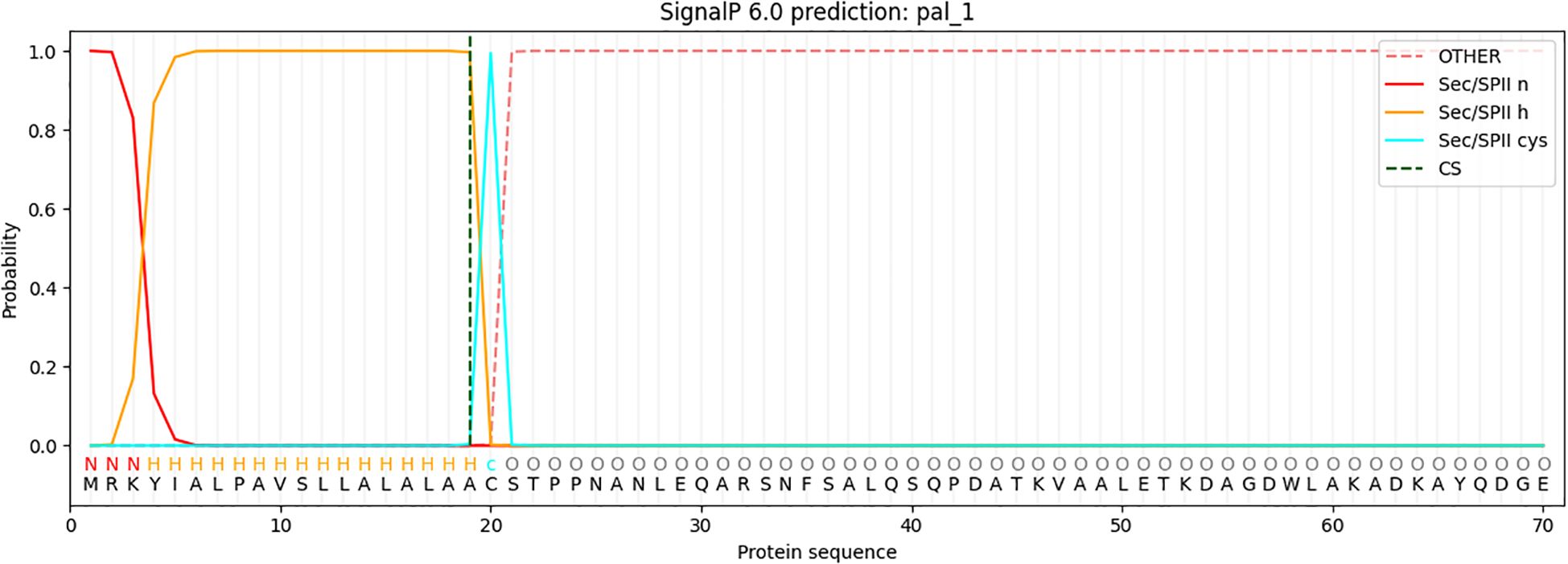
Figure 2. Signal peptide prediction for the LptF protein sequence was performed using SignalP.
3.6 Prediction of B-cell epitope
The BepiPred Linear Epitope Prediction 2.0 tool was initially used to predict B-cell epitopes, identifying nine epitopes for the LptF protein. One of these epitopes, a 72-mer, was re-analyzed to ensure no potential epitopes were missed. This re-evaluation revealed eight additional epitopes (Figure 3), with figures generated in R (77). They were carefully selected based on several critical factors to confirm the suitability of the identified epitopes for vaccine development. VaxiJen v2.0 predicted high antigenicity scores for these epitopes, indicating their potential to trigger a robust immune response. Additionally, their non-toxic nature was confirmed using ToxinPred, ensuring they would not cause adverse effects. The non-allergenic properties of the epitopes were verified using AllerTOP v2.0, further ensuring their safety. As shown in Table 2, the selected epitopes were chosen for further research after careful consideration of these factors. Supplementary Table S2 provides a detailed analysis of the epitope’s suitability for inclusion in potential vaccine formulations, including their toxicity, allergenicity, and antigenicity profiles.
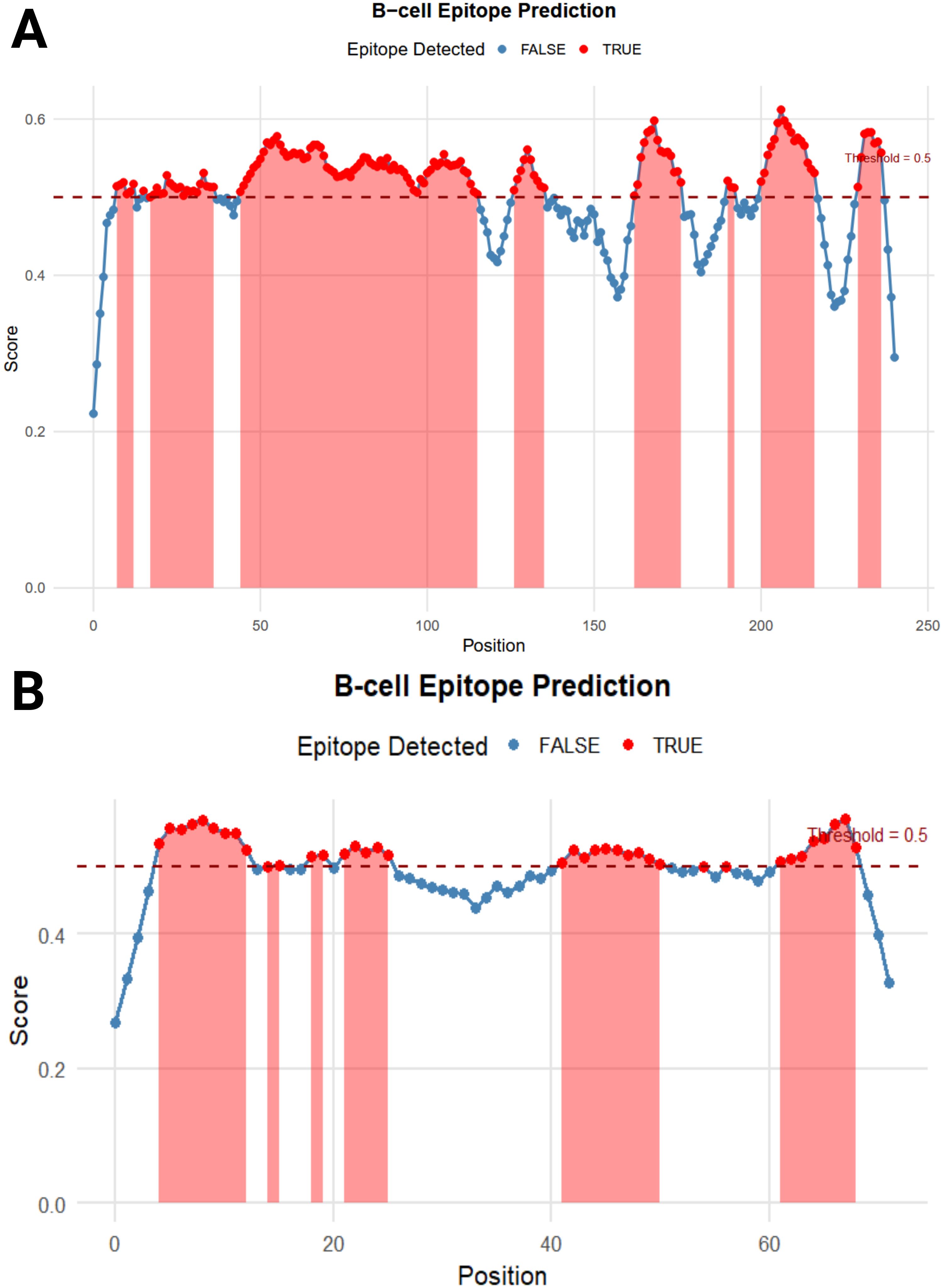
Figure 3. B-cell Epitope Prediction. (A) depicts the prediction of epitopes from the entire protein sequence, while (B) highlights the re-analyzed 72-mer epitope to ensure no potential epitope is overlooked.
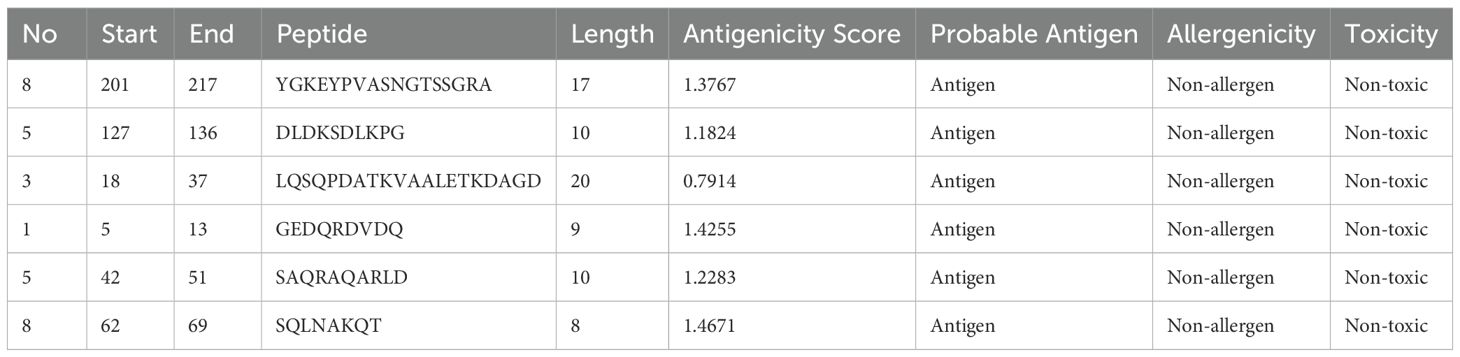
Table 2. Predicted B-cell epitopes for the LptF protein, identified as potential targets for vaccine development.
3.7 Prediction of T-cell epitope (MHC-I and MHC-II)
The MHC-I and MHC-II epitopes were predicted for the LptF protein sequence using NetMHCpan 4.1 from IEDB. The finalized epitopes are presented in Tables 3, 4, with detailed T-cell epitope analyses in Supplementary Tables S3, S4. While VaxiJen v2.0 is primarily designed for complete proteins, it was utilized here as an additional tool to assess the antigenicity of both MHC class I and II T cell epitopes, supporting selection alongside MHC binding, immunogenicity, and toxicity criteria. Initially, 6,265 MHC-I epitopes were predicted and filtered based on a rank cutoff of 0.5 and a core score cutoff of 0.60. Similarly, 6,130 MHC-II epitopes were filtered using a rank cutoff of 2 and a score of 0.75. These thresholds were chosen because lower rank and score values indicate a higher binding affinity to MHC alleles, which is crucial for identifying effective immunogenic candidates. The finalized epitopes were further assessed for toxicity, antigenicity, and allergenicity to confirm their immunogenic potential while minimizing the risk of adverse reactions. All selected epitopes were predicted to be IL-10 inducers, suggesting their potential to regulate immune responses and prevent excessive inflammation. Notably, epitope 3 exhibited balanced induction of IL-4, IL-6, IL-10, and IL-13, making it a strong vaccine candidate. Epitopes 2 and 4 also induced IL-6 alongside IL-10, supporting a mixed pro-inflammatory and regulatory profile (Supplementary Table S5). Interferon-γ scores were computed for MHC-II epitopes to rank those that could elicit a strong immunological response. The chosen MHC-I and MHC-II epitopes, identified according to these criteria, are presented in Table 4.
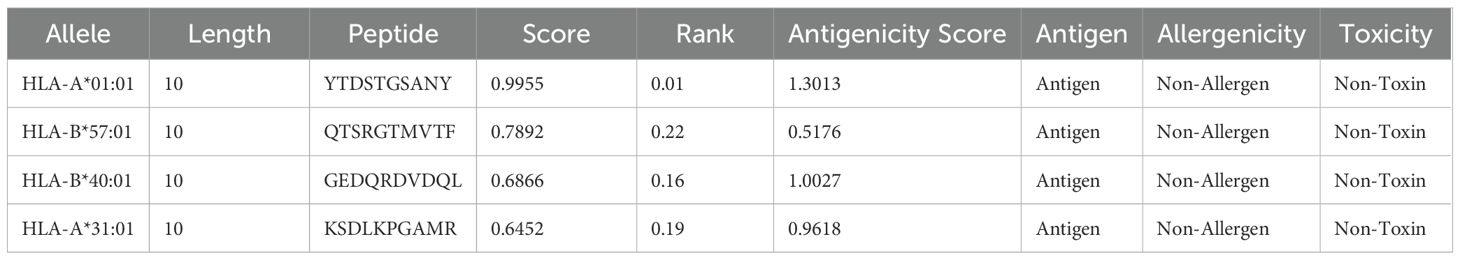
Table 3. Finalized MHC-I epitopes identified for the LptF protein.
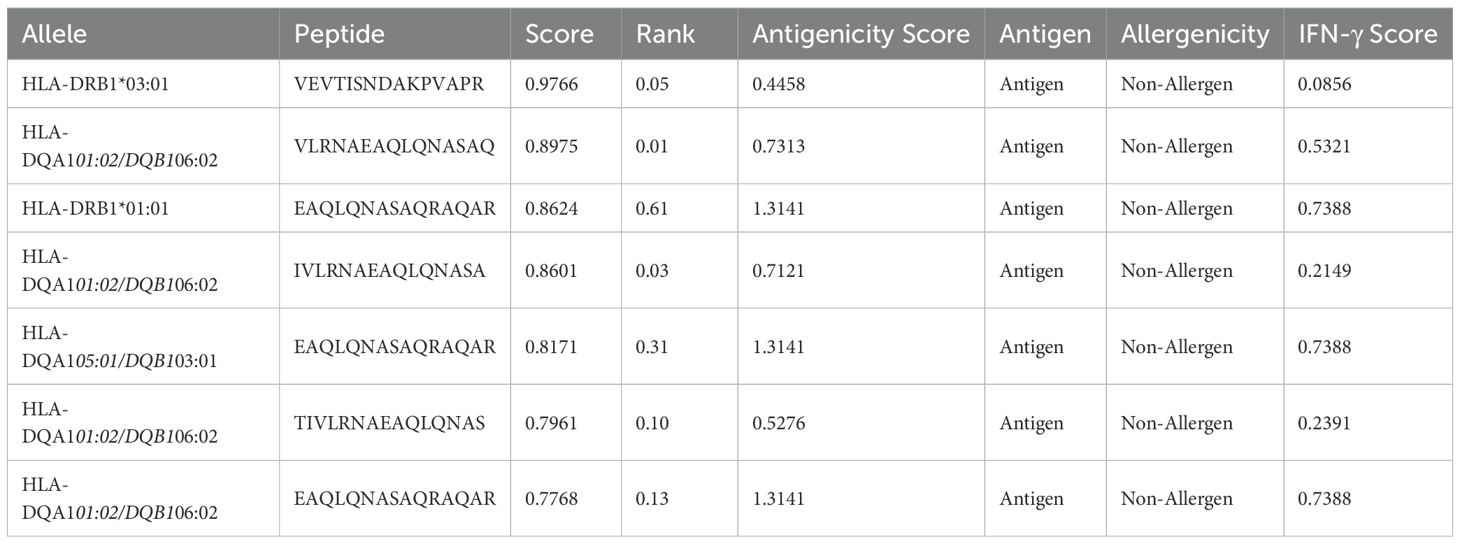
Table 4. Finalized MHC-II epitopes identified for the LptF protein, optimized for vaccine design.
3.8 Vaccine design and conservancy evaluation
Two vaccine constructs were developed, incorporating adjuvants such as RS-09 and Beta-defensin and with the predicted epitopes from the LptF protein. Each vaccine included the selected epitopes, comprising five MHC-II, four MHC-I, and five B-cell epitopes. Fifteen epitopes were incorporated into the final vaccine constructs (Figure 4). The sequences and corresponding lengths of both constructions are described in Table 5, and the proposed vaccines ranged from 248 to 283 amino acids. The combined term for these constructs was POA_V. The presence of the chosen epitopes in P. aeruginosa was verified using a BLASTP analysis. The results demonstrated 100% sequence similarity across P. aeruginosa strains, indicating that the chosen epitopes are conserved and present in all strains.
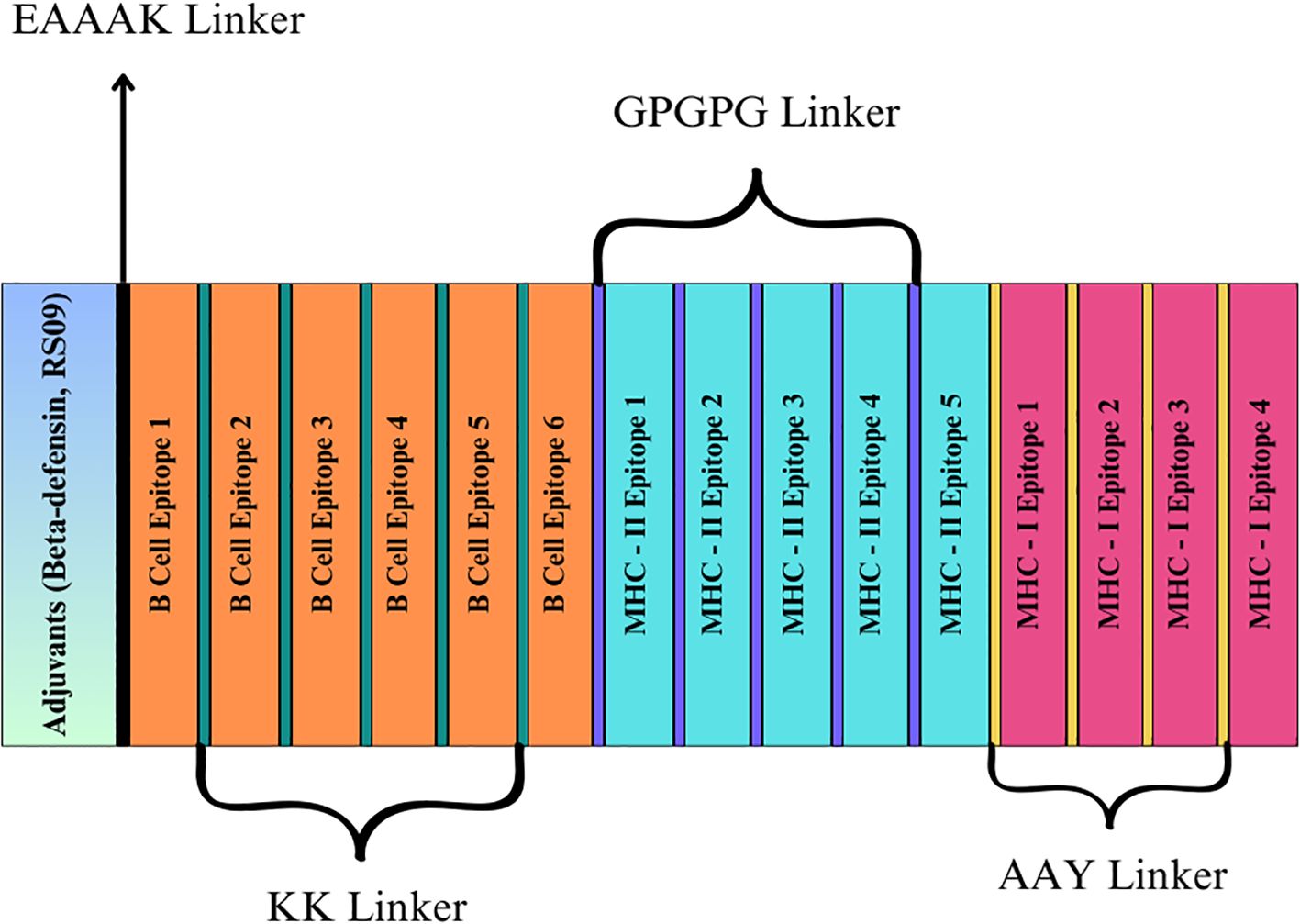
Figure 4. Graphical representation showcasing the formulation of the designed vaccine.

Table 5. Amino acid sequences and sequence lengths of the finalized vaccine constructs.
3.9 Analysis of population coverage
Based on estimated population coverage, the vaccine could potentially reach 87.35% of the global population. Tables 3, 4 comprehensively analyse the epitope distribution, demonstrating its adaptability across different regions and demographic groups. Additionally, Figure 5 visually represents the global coverage, underscoring the vaccine’s potential for widespread impact (Supplementary Table S6, Supplementary Figure S2). Notably, regions such as the United States (98.33%), Kenya (98.58%), Germany (98.26%), Brazil (97.93%), France (98.04%), and Canada (95.58%) showed high predicted population coverage, emphasizing the vaccine’s potential effectiveness across diverse geographic and genetic backgrounds. Moderate coverage was observed in countries such as India (85.56%), Japan (87.60%), and China (89.81%), further confirming the vaccine’s adaptability in densely populated and genetically diverse regions. On the other hand, lower coverage was observed in regions such as the United Kingdom (56.38%), Hong Kong (56.64%), and American Samoa (56.40%), which may be attributed to regional HLA allele distribution patterns. Overall, the population coverage analysis strongly supports the broad usability and potential of the designed vaccine to fight the targeted pathogen worldwide.
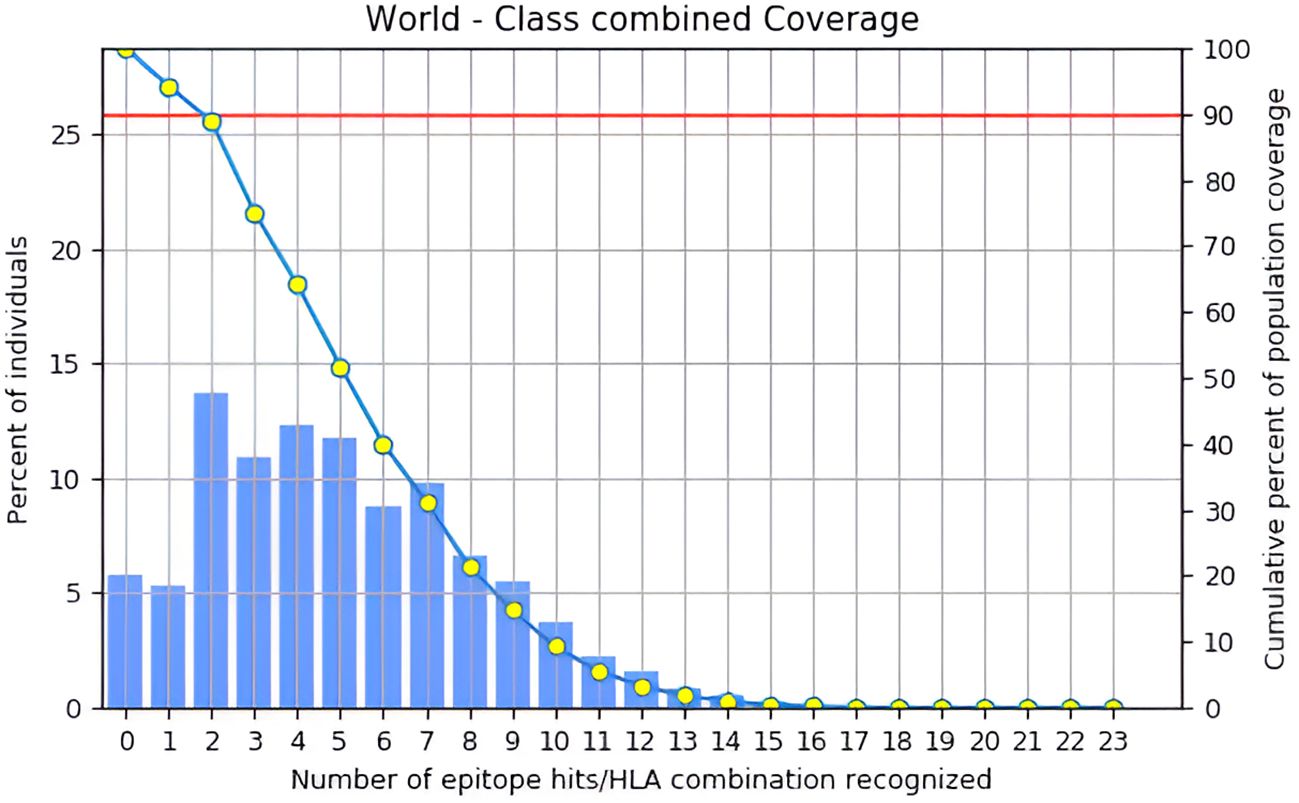
Figure 5. Global population coverage of the designed vaccine was analyzed using the IEDB tool, considering HLA allele frequencies across regions.
3.10 Physicochemical property analysis
The physicochemical evaluation of the vaccine candidates POA_V_RS09 and POA_V_BDEF underscores their potential viability. POA_V_RS09, with a molecular weight of 25,734.39 Da comprising 248 amino acids, has an isoelectric point (pI) of 9.43. It exhibits hydrophilicity, as indicated by a GRAVY score of -0.856, and is considered stable with an instability index of 23.29. Similarly, POA_V_BDEF has a molecular weight of 29,763.15 Da, a pI of 9.53, and consists of 283 amino acids. Its instability index, 26.34, also suggests stability, and the GRAVY score of -0.761, which confirms its hydrophilic nature. These favorable stability and solubility properties render both candidates promising for further validation as vaccine prospects.
3.11 Secondary structure, tertiary structure, and refinement
The secondary structure of the vaccines was estimated using the PSIPRED approach, concentrating on the ratios of coils, β-sheets, and α-helices due to their immunogenic potential. PSIPRED’s analysis of the vaccine candidates POA_V_RS09 and POA_V_BDEF revealed distinct structural features. POA_V_RS09 comprised 58.87% alpha helices, 4.03% beta strands, and 37.10% random coils. In contrast, POA_V_BDEF consisted of 50.53% alpha helices, 7.07% beta strands, and 42.40% random coils (Supplementary Figure S3). These findings indicate a predominance of alpha helices and a significant presence of coils in both candidates, with a relatively low content of beta strands. This structural profile suggests a balance between stability and flexibility, which is beneficial for antigenic presentation in vaccine design. Using ROBETTA and AlphaFold, we modelled the vaccine’s 3D structures. Following structure generation, we refined all the models using GalaxyRefine to improve stereochemical accuracy. Among the generated models, Model 1 demonstrated superior performance for both vaccines, with RMSD values ranging from 0.9744 to 0.9889. Further validation was conducted using QMEAN4 scores and Ramachandran plot analysis to evaluate the structural quality at both global and local levels. For the POA_V_BDEF construct, the ROBETTA model yielded a QMEAN4 score of –0.72, while the AlphaFold model scored –2.67. Similarly, for the POA_V_RS09 construct, the ROBETTA model scored –0.19, compared to –2.24 for the AlphaFold prediction. QMEAN4 integrates four structural descriptors and is widely used to evaluate model quality in the absence of a native structure. These results indicate that the ROBETTA-generated models exhibit superior reliability and structural accuracy for both constructs. Further structural assessment using Ramachandran plot analysis (Table 6) revealed that ROBETTA models have over 96% of residues in favored regions, with only 0.71–0.81% falling in the disallowed areas. In contrast, AlphaFold models had a higher percentage of disallowed residues (up to 2.44%), particularly in functionally important loops and epitope-accessible regions. While AlphaFold has shown remarkable success in protein structure prediction and has been used in several recent vaccine design studies with promising results (78), we opted for ROBETTA-refined models in our research. This decision was based on comparative structural validation, where ROBETTA constructs exhibited fewer steric clashes and better Ramachandran statistics. Therefore, the ROBETTA-generated models were chosen for both POA_V_RS09 and POA_V_BDEF constructs and used in all downstream docking and immunological simulations to ensure structural reliability and predictive robustness. The Ramachandran plot of POA_V_RS09 and POA_V_BDEF shows that the structural value exceeds 90% of residues in favored regions, indicating a good overall geometry (Supplementary Figure S4). For the POA_V_BDEF construct, residues like Ser-26 and Gly-134 were located in disallowed areas, while for the POA_V_RS09 construct, residues Pro-100 and Gly-141 were also found in similar disallowed areas. The vaccine models are detailed in Supplementary Figure S5.
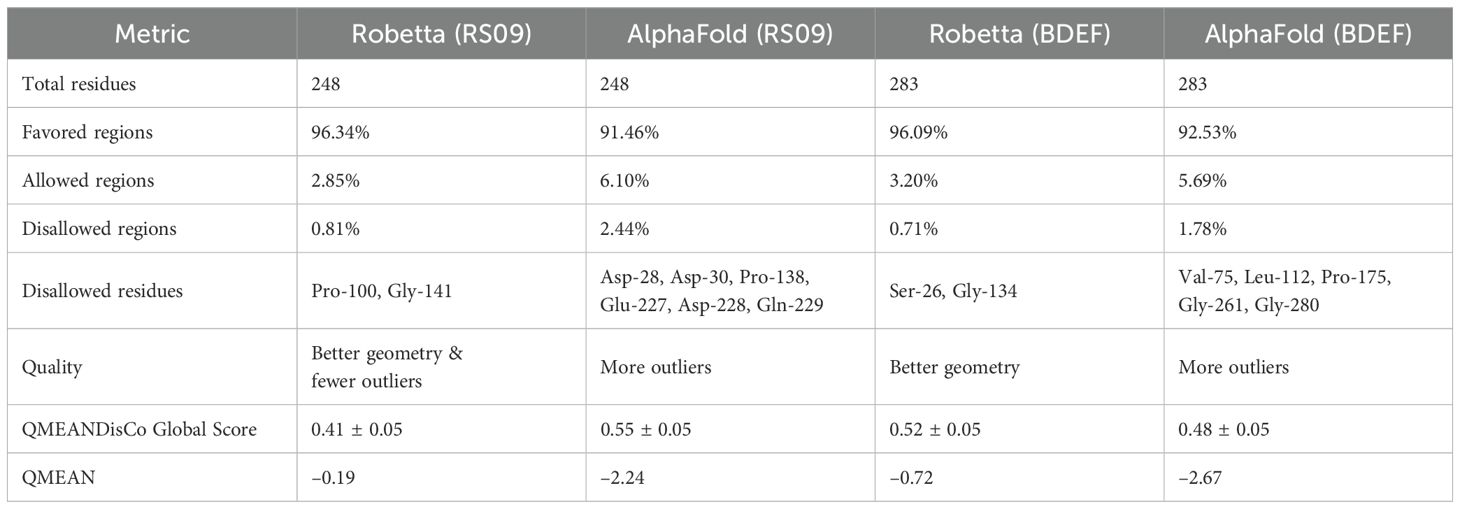
Table 6. Structural validation of POA_V_RS09 and POA_V_BDEF-based vaccine models generated using Robetta and AlphaFold.
3.12 Molecular docking analysis
The HDOCK server performed docking tests to evaluate the interactions between the suggested vaccine candidates POA_V_RS09 and POA_V_BDEF and the immunological receptors TLR2 and TLR4, respectively. These receptors are vital in recognizing pathogen-associated molecular patterns (PAMPs) and triggering immune responses, such as cytokine production and the recruitment of immune cells. The results indicated strong binding affinities for all complexes, with POA_V_RS09 achieving the highest docking scores of -310.2 (kcal/mol) for TLR4 and -286.76 (kcal/mol) for TLR2 (Table 7, Figure 6). The MD simulations were conducted to further validate the interactions by examining the stability and conformational behavior of the docked complexes under physiological conditions. Both vaccine candidates exhibited stable interactions, with minimal fluctuations at the receptor-binding interface, suggesting their ability to engage immune receptors and potentially elicit robust immune responses effectively. These findings highlight the promising immunogenic potential of the designed vaccines.
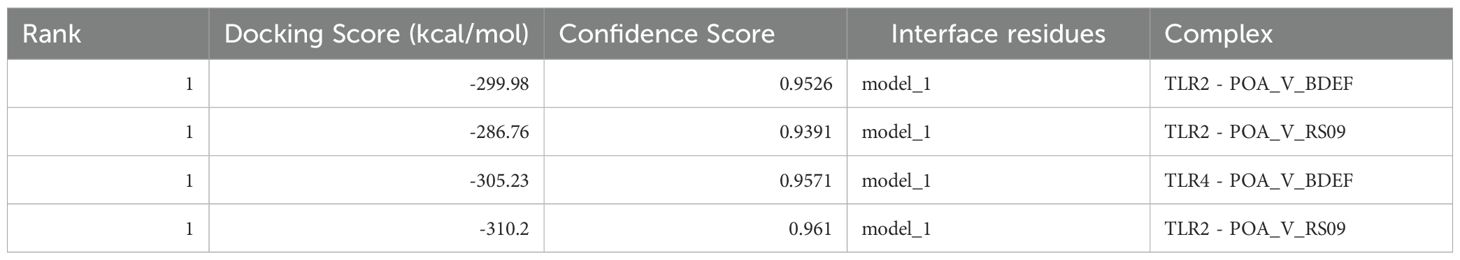
Table 7. Molecular docking scores of POA_V_BDEF and POA_V_RS09 with TLR2 and TLR4, showing binding affinities.
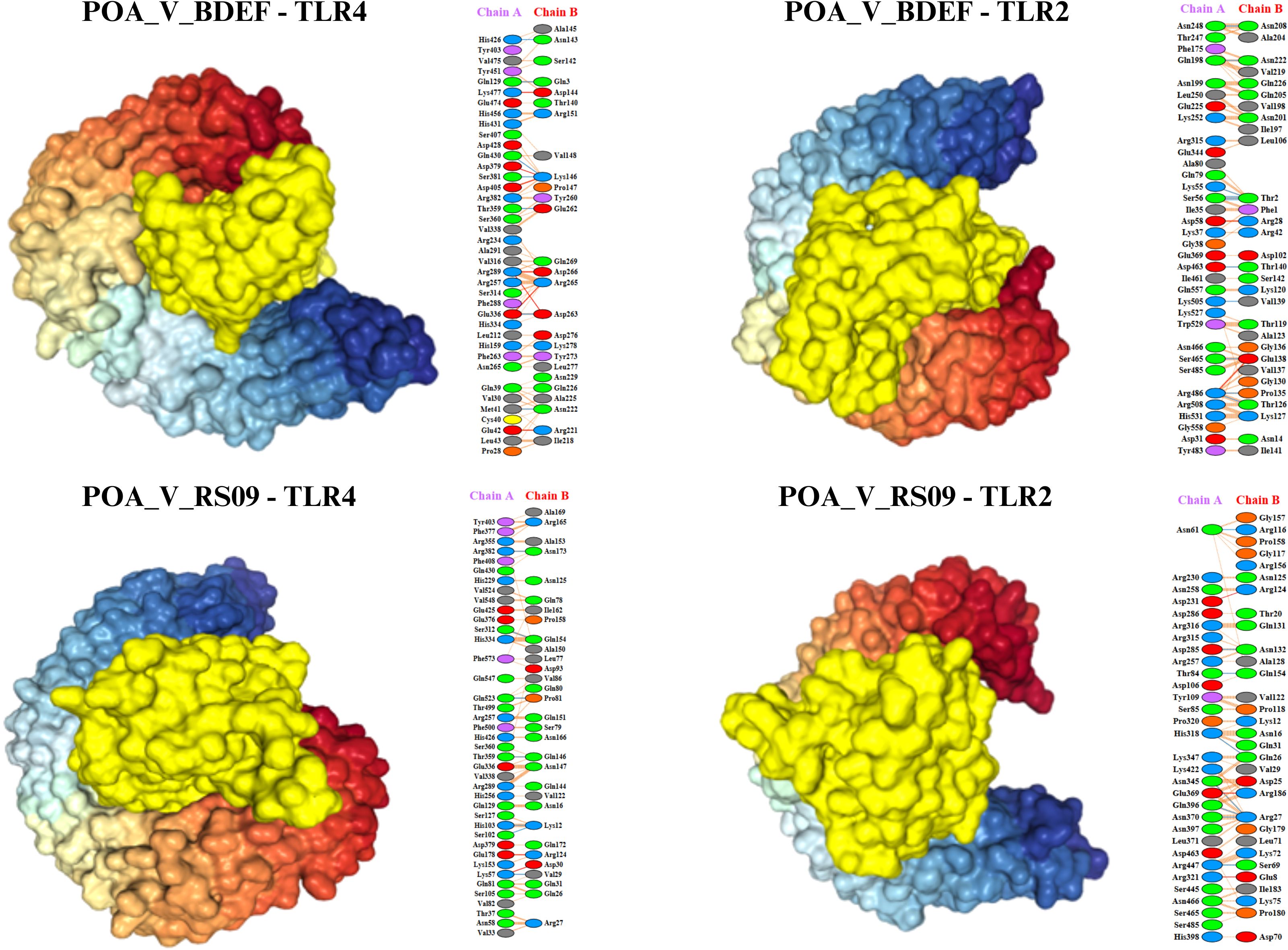
Figure 6. Illustrates the docking models of the vaccine constructs with the receptor, focusing on the lowest binding energy conformations. The identified interaction residues reveal strong binding affinities, highlighting critical contacts that contribute to the complex’s stability and potential efficacy.
3.13 Molecular dynamics simulation analysis
The MD simulations for the vaccine complexes (POA_V_RS09 and POA_V_BDEF) with TLR2 and TLR4 were conducted over 1000 ns and revealed notable differences in stability and interaction properties (Table 8). The RMSD (backbone) value indicated that the POA_V_RS09 vaccine complex was the most stable, with the TLR4_POA_V_RS09 complex showing the lowest RMSD (0.57 ± 0.06 nm), followed by TLR2_POA_V_RS09 (0.80 ± 0.19) nm. These complexes remained stable throughout the 1000-ns MD simulation. In contrast, the POA_V_BDEF-based vaccine complexes had higher RMSD values, with TLR2_POA_V_BDEF (1.08 ± 0.14 nm) and TLR4_POA_V_BDEF (1.03 ± 0.10 nm), indicating more significant structural deviations and less stable interactions (Figure 7A). These complexes slightly fluctuated at the beginning of the MD simulation (0-200ns), and later they equilibrated at 1 nm. To investigate the observed fluctuations, we analyzed the backbone RMSD and Calpha RMSF of TLR2 and TLR4. Both receptors exhibited considerable structural stability, with average RMSD values of TLR2 in POA_V_BDEF at (0.33 ± 0.03 nm), TLR2 in POA_V_RS09 at (0.40 ± 0.07 nm), TLR4 in POA_V_BDEF at (0.26 ± 0.03 nm), and TLR4 in POA_V_RS09 at (0.23 ± 0.03 nm). The RMSF profiles also indicated stable conformations across all complexes, TLR2 in POA_V_BDEF at (0.15 ± 0.08 nm), TLR2 in POA_V_RS09 at (0.16 ± 0.14 nm), TLR4 in POA_V_BDEF at (0.15 ± 0.07 nm), and TLR4 in POA_V_RS09 at (0.14 ± 0.07 nm), as illustrated in Supplementary Figure S6. The predicted POA_V_BDEF complex displayed enhanced flexibility, primarily attributed to the presence of less structured epitope and linker regions Supplementary Figure S7. This inherent structural looseness likely accounts for the comparatively elevated average RMSD observed across its associated complexes. When coming to the vaccine stability in residue wise, RMSF (Calpha) analysis showed that the RS09 vaccine complexes were more rigid, with the TLR4_POA_V_RS09 complex showing the lowest RMSF (0.21 ± 0.07 nm) and the TLR2_POA_V_RS09 complex showing (0.32 ± 0.18 nm), indicating minimal flexibility at the interaction interface and that all the residues were around 0.5nm. Conversely, the POA_V_BDEF-based vaccine complexes had higher RMSF values, with TLR2_POA_V_BDEF (0.64 ± 0.23 nm) and TLR4_POA_V_BDEF (0.47 ± 0.25 nm), suggesting increased flexibility and dynamic behaviour (Figure 7B). In contrast, the POA_V_BDEF vaccine construct exhibited pronounced fluctuations, particularly in regions interacting with TLR4 and TLR2. For the TLR4_POA_V_BDEF complex, notable flexibility was observed in the N-terminal linker region (residues 53–57), as well as in combined epitope and linker segments spanning residues 50–85, 165–180, and 207–216, in addition to the C-terminal end. Similarly, the TLR2_POA_V_BDEF complex showed continuous fluctuation across the linker (55–60), the epitope region (73–83), the extended linker–epitope stretch (109–150), and residues 173–180 and 195–220, along with the C-terminal region. In contrast to the POA_V_BDEF construct, the POA_V_RS09-based vaccine formulation demonstrated notably greater structural stability.
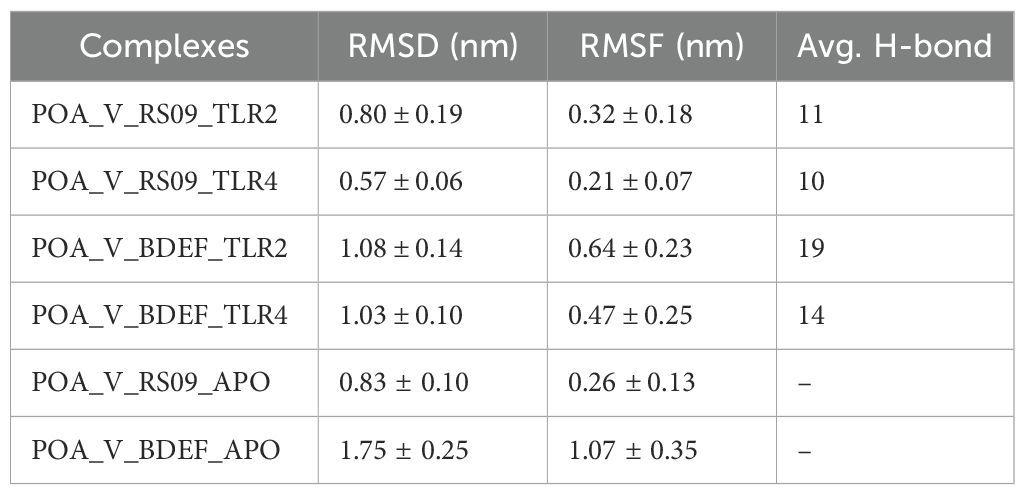
Table 8. Post-MD analysis averages for protein-protein complexes, including RMSD, RMSF, and H-bond values, reflecting structural stability and interactions.
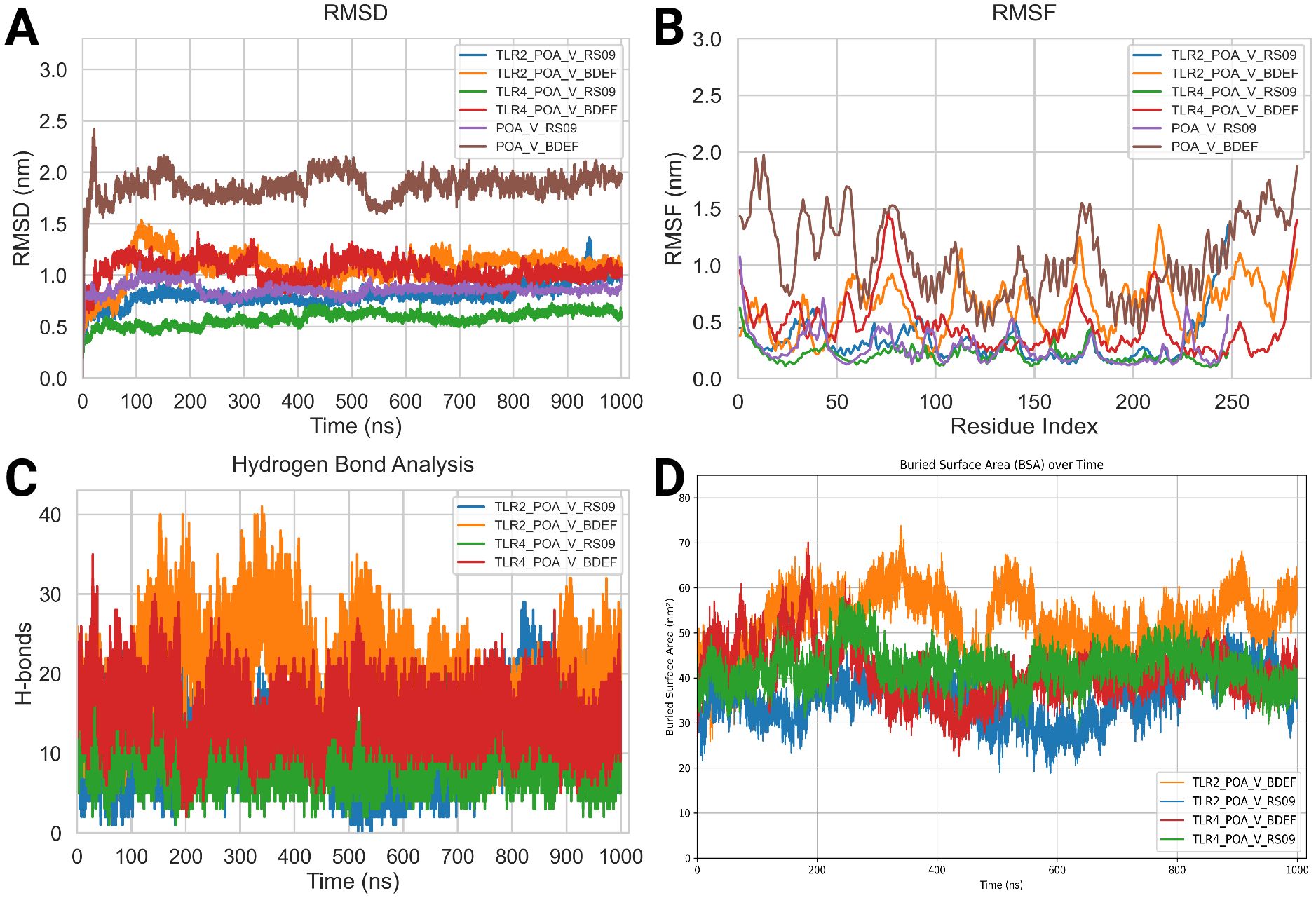
Figure 7. Molecular dynamics (MD) analysis results. (A) Root mean square deviation (RMSD) Backbone analysis. (B) Root mean square fluctuation (RMSF) Calpha Analysis. (C) Hydrogen bond (HBOND) analysis. (D) Buried surface analysis (BSA).
PCA was performed on the vaccine constructs extracted from their respective TLR2 and TLR4 complexes to evaluate their conformational dynamics. The POA_V_RS09 construct, when analyzed post-interaction with both TLR2 and TLR4, exhibited compact PCA clusters, indicating limited conformational fluctuations and stable structural behavior throughout the 1000 ns simulation. In contrast, the POA_V_BDEF construct displayed broader dispersions in PCA space, suggesting greater structural flexibility and reduced conformational stability. This trend remained consistent when the standalone vaccine models were analyzed, where POA_V_RS09 continued to show tight clustering and structural integrity, while POA_V_BDEF exhibited higher variability. These results align with earlier RMSD and RMSF analyses, collectively highlighting POA_V_RS09 as the more stable and potentially immunogenic vaccine candidate. (Supplementary Figure S8). Hydrogen bond (HBOND) analysis was conducted over the 1000 ns molecular dynamics simulation using GROMACS. The default criteria were used, which include a donor–acceptor distance cutoff of 0.35 nm and a hydrogen–donor–acceptor angle cutoff of ≥150° (i.e., ≤30° deviation from linearity), consistent with established definitions for biologically relevant hydrogen bonds. The analysis focused specifically on the intermolecular hydrogen bonds formed between the vaccine constructs and the TLR receptors. The POA_V_BDEF vaccine complexes exhibited a higher average number of hydrogen bonds (19 with TLR2 and 14 with TLR4) compared to the POA_V_RS09 complexes (11 with TLR2 and 10 with TLR4). However, the relatively higher RMSD and RMSF values observed in the POA_V_BDEF complexes suggest that these additional hydrogen bonds may be less stable or more transient (Figure 7C). The buried surface area (BSA) during the 1000 ns simulation at the interface of the TLR4_POA_V_RS09 complex was 42.09 nm² ± 3.98, indicating stable interactions and low variability. This was closely followed by TLR4_POA_V_BDEF, with a BSA of 41.12 nm² ± 5.83, showing a similar interaction pattern. In contrast, TLR2_POA_V_BDEF had a higher BSA of 52.37 nm² ± 6.60, while TLR2_POA_V_RS09 showed a BSA of 35.70 nm² ± 5.19, both with higher standard deviations, suggesting relatively fewer stable interactions (Figure 7D). Analysis of the apo forms revealed that POA_V_BDEF exhibited the highest RMSD (1.75 ± 0.25 nm) and RMSF (1.07 ± 0.35 nm), indicating significant conformational flexibility in the absence of receptor binding. In contrast, POA_V_RS09 exhibited lower deviation (0.83 ± 0.10 nm RMSD and 0.26 ± 0.13 nm RMSF), suggesting it remains relatively stable even when unbound. The FEL analysis effectively showed us the structural stability and flexibility of the vaccine-protein complexes. All complexes exhibited energy basins, indicating the presence of metastable states. However, notable differences were observed in the shape and depth of these energy wells. Complexes involving TLR4 exhibited more compact and deeper energy minima compared to those involving TLR2, suggesting a higher degree of structural stability. In particular, the TLR4_POA_V_RS09 complex exhibited a well-defined global minimum, indicating a stable and energetically favorable conformation throughout the simulation. Although the TLR4_POA_V_BDEF complex also reached stable conformations, it showed slightly more conformational variability. Conversely, the TLR2 complexes exhibited broader and more scattered low-energy regions, indicating increased conformational flexibility and less stable interaction patterns. Among them, the TLR2_POA_V_RS09 complex exhibited relatively smoother energy transitions compared to TLR2_POA_V_BDEF, which displayed more rugged features in its energy landscape (Supplementary Figure S9).
We calculated the binding free energy using MMPBSA, which revealed notable differences between the POA_V_RS09 and POA_V_BDEF-based vaccine constructs in complex with TLR2 and TLR4 receptors. The TLR2_POA_V_RS09 showed a more favorable binding energy (–1483.14 kJ/mol) than TLR2_POA_V_BDEF (–1335.16 kJ/mol), indicating that RS09 forms a more stable and energy-efficient complex with TLR2, compared to POA_V_BDEF. In contrast, TLR4_POA_V_BDEF exhibited a significantly stronger binding energy (–4600.83 kJ/mol) than TLR4_RS09 (–2682.66 kJ/mol), likely due to its extended area, which enables an increased contact surface. However, prior dynamic and structural analyses, such as RMSF and FEL plots, indicate that POA_V_BDEF is more flexible, particularly at the linker and epitope regions. This flexibility may contribute to reduced structural stability, especially in the TLR2 complex compared to POA_V_RS09.
3.14 In silico codon adaptation, cloning, and immune simulation
Codon optimization was performed using GenScript to enhance the expression of the POA_V_RS09 vaccine sequence in E. coli K-12. With a GC content of 60.22% and a total length of 744 base pairs, the optimized sequence falls within the ideal range (30–70%) for effective expression in E. coli. This balanced GC content ensures efficient transcription and translation, making the sequence suitable for high-level expression in the host. The optimized vaccine sequence was then used for in-silico cloning with SnapGene software, successfully inserting the gene into the pET-28a(+) expression plasmid (Figure 8). The immune response dynamics elicited by POA_V_RS09 are shown in Figure 9. Figure 9A illustrates the antigen (Ag) and antibody responses over a 350-day period, where an early antigen peak, followed by a sharp decline, indicates effective recognition and clearance by the host immune system. This is accompanied by a strong humoral response, characterized by an initial surge in IgM, typical of a primary response, followed by a sustained increase in IgG1 and IgG2, which shows class switching and maturation of the immune response. The dominance of IgG subclasses over time reflects the development of long-term protective immunity. Notably, IgG1 and IgG2 are associated with Th1-type immune responses, which are essential for combating pathogens such as viruses and certain bacteria. Figure 9B illustrates cytokine dynamics, where high levels of Interleukin-2 (IL-2) and IFN-γ early on indicate strong T-cell activation and a Th1-biased immune response, which is particularly important for combating pathogens. Their gradual decline over time suggests immune regulation and resolution of inflammation, highlighting the vaccine’s safety profile. Figure 9C illustrates the dynamics of the B-cell population, exhibiting an increasing trend in memory B cells and a shift in isotype expression from IgM to IgG, which further validates class switching and the generation of long-lasting humoral memory. Figure 9D focuses on B-cell states, showing that active and antigen-internalizing B cells peak early, while anergic cells remain relatively constant, suggesting efficient antigen processing and presentation. The low level of anergic B cells suggests that immune tolerance is not induced, further supporting the vaccine’s immunogenicity. Figure 9E presents helper T-cell (TH) populations, with an initial spike in total TH cells, followed by the emergence of memory TH cells, supporting long-term immunity. Finally, Figure 9F depicts cytotoxic T-cell (TC) states, showing early activity and duplication followed by a steady increase in resting memory TC cells, which are critical for sustained immune surveillance. This can contribute to the direct killing of bacteria or infected host cells in bacterial infections that evade extracellular immune mechanisms. These outcomes underscore the successful initiation of both humoral and cellular immune responses, supporting the potential for the development of long-lasting immunological memory. Moreover, the observed immune signatures align with the protective responses typically seen in effective bacterial vaccines, validating the rational design of POA_V_RS09, which incorporates TLR agonists, multi-epitope constructs, and immune-enhancing linkers to induce broad, durable immunity against bacterial pathogens.
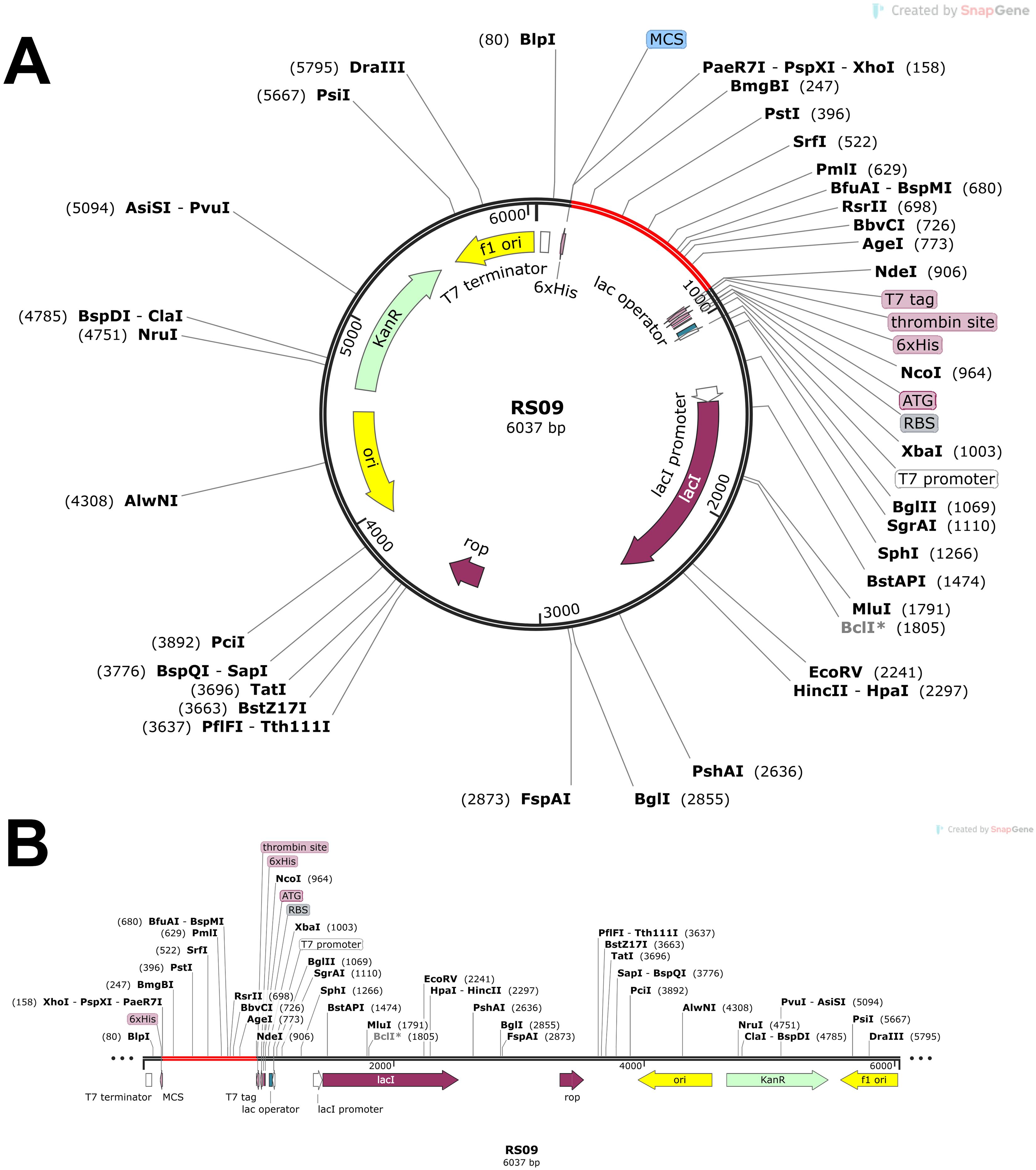
Figure 8. In silico cloning of POA_V_RS09. (A) Cloning of RS09 (which is POA_V_RS09) into the pET-28a(+) vector. (B) final vaccination design with additional restriction sites.
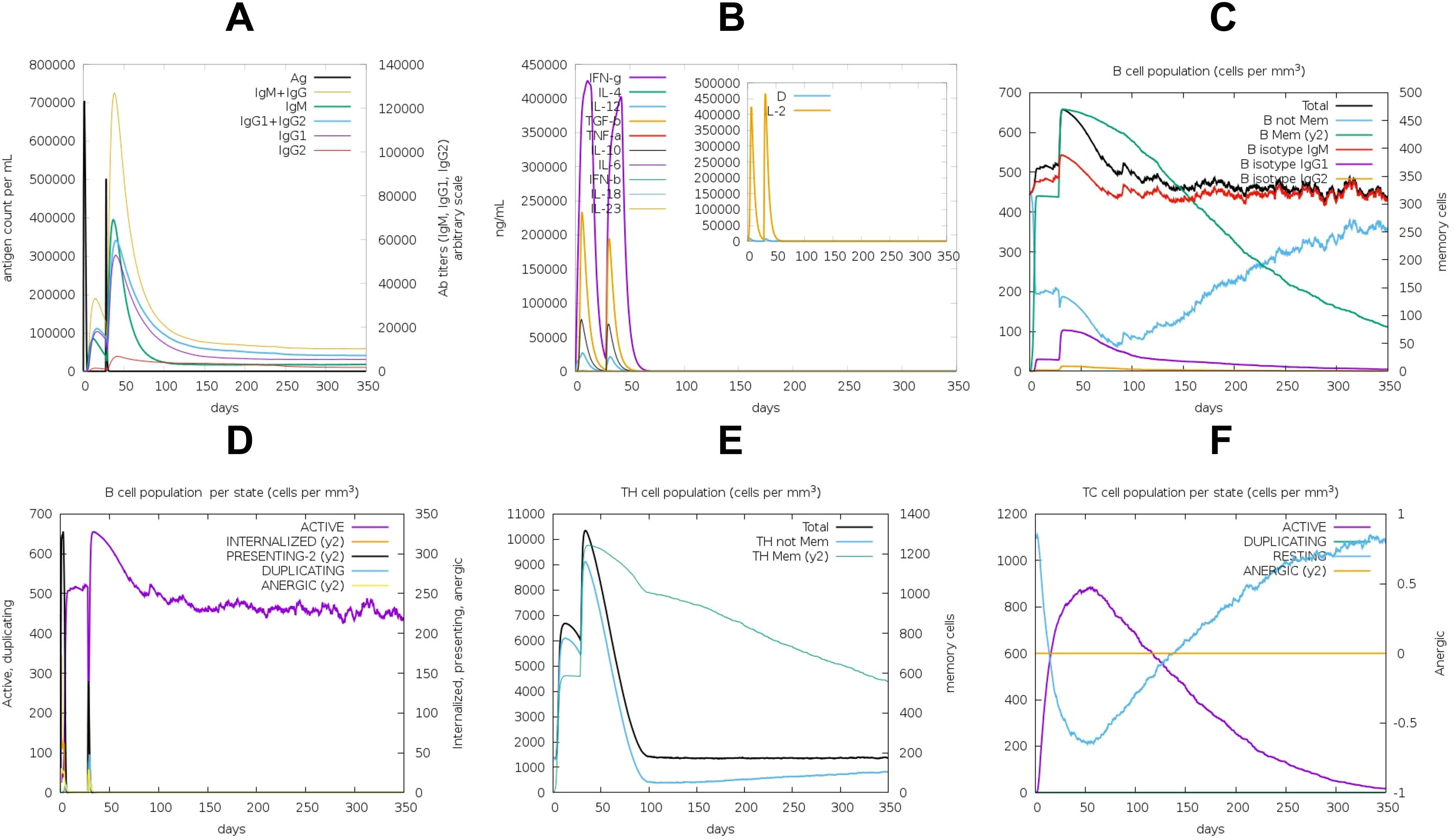
Figure 9. Immune Response Induced by the POA_V_RS09 Vaccine: (A) antibody response. (B) Cytokine response. (C) B-Cell population (cells/mm³). (D) B-Cell population by state. (E) TH-cell population by state. (F) TC-cell population by state.
4 Discussion
The opportunistic pathogen P. aeruginosa is a significant cause of hospital-acquired infections worldwide. It presents a serious threat to human health, especially in immunocompromised individuals, due to its innate antibiotic resistance and ability to develop biofilms (79, 80). The increasing prevalence of drug-resistant strains has complicated treatment options, highlighting the urgent need for alternative therapeutic strategies (81). Despite progress in antimicrobial therapies, no licensed vaccine exists for P. aeruginosa, revealing a critical gap in combating this pathogen (82). Immunoinformatics has emerged as a powerful tool in vaccine development, facilitating the rational design of in silico vaccines, as demonstrated in the development of vaccines against pathogens such as the Ebola virus, SARS-CoV-2, and Mycobacterium tuberculosis (83–85). These approaches hold significant promise for addressing the challenges posed by P. aeruginosa.
Previous immunoinformatics-based vaccine studies against P. aeruginosa often relied on single-strain datasets or focused on a narrow range of targets. Some selected cytoplasmic proteins have limited surface accessibility, while others used previously known antigens without assessing their conservation across diverse strains. Additionally, several studies selected targets which is completely based on literature without genome-wide screening (86–89). Other broader approaches involving multiple pathogens have also identified shared virulence or essential gene-derived epitopes while filtering for self-tolerance (90). However, such strategies typically lack species-specific optimization, structural validation, and comprehensive strain-level genomic coverage—critical aspects that our study addresses. In this study, we conducted a comprehensive pangenome analysis (91) of 864 P. aeruginosa genomes. This extensive dataset enabled robust pangenome analysis and the identification of conserved, surface-exposed, and virulence-associated targets, distinguishing our study from previous investigations. Here we identified 63,239 genes, including 3,325 core genes and 3,149 accessory genes. We focused on conserved core genes essential for bacterial survival and pathogenicity to ensure broad-spectrum coverage. The Pal_1 protein was identified as a potential vaccine candidate, which is classified as an outer membrane protein via PSORTb, and confirmed as a virulence-associated factor through BLASTP analysis against the VFDB. Its sequence was validated against the P. aeruginosa database, where it was identified with 100% confidence and an E-value of 0, and it is known as LptF (lipotoxin F). Sequence comparison with the human proteome confirmed the absence of homologous hits, minimizing the risk of adverse cross-reactivity. LptF, an OmpA-like outer membrane protein, plays a crucial role in P. aeruginosa’s survival, particularly in stressful environments such as lung colonization in cystic fibrosis, and may serve as an important target for therapeutic strategies (92). LptF remains an underexplored target. Its classification as a lipotoxin, along with evidence from structural proteomics revealing interactions with key membrane proteins like OprI and LptE, further highlights its relevance as a promising vaccine candidate against P. aeruginosa. Due to the increasing antibiotic resistance of P. aeruginosa, an effective vaccine is urgently needed, and lipotoxins have been identified as potential targets in studies (24). The LptF protein exhibited favorable physicochemical and immunogenic properties, with a molecular weight of 28.5 kDa, thermostability indicated by an aliphatic index of 80.15, and a hydrophilic nature reflected in a GRAVY index of -0.574. Immunogenic analysis revealed its suitability as a vaccine target, with an antigenicity score of 0.6442 and classification as a non-allergen. SignalP analysis showed that the protein has a signal peptide, which is likely removed during maturation. This was accounted for in later analyses that focused on the mature protein sequence. We identified B-cell epitopes and chose high-affinity T-cell epitopes based on their binding affinities to MHC-I and MHC-II molecules, with additional refinement based on their potential to stimulate interferon-γ production. Ultimately, 15 epitopes were incorporated into the vaccine design, comprising four MHC-I epitopes, five MHC-II epitopes, and six B-cell epitopes. Additionally, the cytokine prediction analysis revealed that all selected epitopes possess IL-10-inducing potential, a cytokine shown to be critical in controlling inflammation and enhancing bacterial clearance during P. aeruginosa infection (93). The vaccine constructs POA_V_RS09 and POA_V_BDEF were designed, incorporating RS-09 and Beta-defensin as adjuvants. We selected RS09 as it functions as a TLR4 agonist, effectively stimulating the innate immune response. β-defensin was chosen for its dual role in activating both innate and adaptive immunity. Incorporating these adjuvants aimed to enhance the immunogenic potential of the constructs. This strategic design allowed us to evaluate and compare their impact on vaccine performance. These constructs exhibited broad global population coverage (87.35%) and strong immunogenic potential. Secondary structure analysis revealed that both vaccines predominantly consisted of α-helices and random coils, enhancing antigenic presentation. Structural validation through Ramachandran plot analysis ensured the reliability of the tertiary structures. We selected the Robetta server over AlphaFold for tertiary structure prediction because Robetta offers more reliable modeling for synthetic, chimeric constructs involving multiple domains, such as adjuvants, epitopes, and linkers. Unlike AlphaFold, which is optimized for natural protein sequences, Robetta’s de novo prediction approach is better suited for handling novel vaccine constructs. For the BDEF-based construct, serine-26 (Ser-26) and glycine-134 (Gly-234) residues were found in disallowed regions, while the RS09-based construct showed proline-100 (Pro-100) and glycine-141 (Gly-141) residues in disallowed regions. These residues were primarily located in loop and linker regions and were not associated with key epitope or adjuvant domains, suggesting that they are unlikely to compromise the overall structural integrity of the protein. Therefore, the refined and validated vaccine structures were subsequently used for molecular docking studies to assess receptor interactions. Molecular docking analyses revealed high-affinity interactions between the vaccine constructs and Toll-like receptors TLR2 and TLR4, which play pivotal roles in initiating innate immune responses. POA_V_RS09 showed superior docking scores, with -310.2 (kcal/mol) for TLR4 and -286.76 (kcal/mol) for TLR2, while MD simulations confirmed the stability of these interactions under physiological conditions. We conducted extensive 1000-ns molecular dynamics simulations to analyze the long-term structural stability and interaction dynamics of the vaccine-receptor complexes. This extended simulation duration exceeds the standard practice in similar studies, providing deeper insights into conformational behavior, particularly in flexible regions such as linkers and epitopes. It enhances the structural validation of our vaccine constructs and reinforces the reliability of our results. The TLR4_POA_V_RS09 complex demonstrated minimal structural fluctuations, with the lowest RMSD and RMSF values, indicating stable interactions compared to POA_V_BDEF. Although POA_V_BDEF demonstrated good structural quality based on validation metrics, MD simulations revealed considerable flexibility, even in its apo form (without receptor binding). This inherent flexibility, especially in the epitope-linker regions, might weaken stable receptor binding and influence immune activation. While some mobility facilitates epitope presentation, too much fluctuation can reduce vaccine effectiveness. These findings underscore the importance of dynamic assessment in conjunction with static validation when evaluating multi-epitope vaccine designs. Further analysis confirmed POA_V_RS09’s stable conformational states, with tighter cluster dispersion and distinct energy minima. MMPBSA analysis showed that although POA_V_BDEF has a strong binding affinity with TLR4, POA_V_RS09 exhibits a more balanced and consistent interaction profile with both TLR2 and TLR4, along with fewer structural fluctuations. These qualities make POA_V_RS09 a robust and dependable adjuvant candidate for the design of multi-epitope vaccines. Codon optimization for POA_V_RS09 allowed efficient expression in E. coli (K-12), and in silico cloning into pET-28a(+) validated its expression potential. Immune simulations demonstrated robust adaptive immune responses, characterized by sustained IgG production, memory B-cell formation, and effective cytokine engagement, rendering POA_V_RS09 a promising vaccine candidate for long-term immunity. The POA_V_RS09 vaccine demonstrated strong stability, optimal expression potential, and robust immune activation, positioning it as an ideal candidate for further development. By targeting P. aeruginosa, a highly resistant pathogen, the POA_V_RS09 vaccine could offer a valuable strategy for preventing infections and addressing the global threat of antimicrobial resistance, ultimately improving patient outcomes. In this study, we designed two separate vaccine constructs using RS09 and β-defensin adjuvants to independently evaluate their immunostimulatory potential. This separation allows for comparative assessment of construct stability, population coverage, and immunogenicity. However, future studies could explore the integration of both adjuvants into a single construct, as combinatorial adjuvants have been shown to enhance immune responses more effectively than individual components (78). One limitation of this study is that it lacks experimental validation. While our computational approach provides a cost-effective and time-efficient method for epitope screening, future in vitro and in vivo studies (e.g., ELISA, ELISPOT) are essential to confirm immunogenicity and support vaccine development of the POA_V_RS09 vaccine candidate against P. aeruginosa. However, we have thoroughly examined the structural and immunological characteristics of the vaccine candidate through in silico methods, including 1000 ns molecular dynamics simulations, epitope mapping, TLR docking, population coverage analysis, and immunogenicity prediction. Long-timescale MD simulations allow for the capture of biologically relevant conformational changes, showing that microsecond to millisecond scale simulations can uncover protein folding pathways and slow structural transitions. This supports the use of 1000 ns MD to study dynamic molecular interactions (94). Several previous studies have demonstrated that immunoinformatics-based vaccine designs can reliably predict antigenic determinants and immune interactions, often correlating well with experimental outcomes (95–97). These findings support the translational relevance of computational predictions in the early stages of vaccine design. Further validation using comprehensive in vitro assays is also necessary to evaluate the safety profile and immunogenic potential of the POA_V_RS09-based vaccine, including its ability to induce pro-inflammatory cytokines, activate T cells, and generate specific antibody responses. Such investigations will provide valuable insights into the clinical feasibility of POA_V_RS09 as a vaccine candidate for P. aeruginosa infections.
5 Conclusion
This study utilized an integrated pangenome and immunoinformatics approach to develop an epitope-based peptide vaccine targeting P. aeruginosa. Through pangenome analysis, we identified LptF as a promising and underexplored vaccine target, specifically. From LptF, we predicted potential epitopes. The resulting vaccine candidate, POA_V_RS09, demonstrated promising immune response outcomes and strong binding affinity to immunological receptors (TLRs). Notably, the 1000-ns molecular dynamics simulation provided valuable insights into the structural stability of the vaccine–receptor complexes over an extended timescale, reinforcing the robustness of the construct under physiological conditions. This computational strategy holds significant potential for addressing the escalating issue of antimicrobial resistance, particularly in resource-limited settings and low-income countries. This strategy provides a comprehensive and practical approach to combating infections by targeting conserved NS proteins, identifying high-affinity B-cell and T-cell epitopes, and utilizing suitable adjuvants. Future studies should assess the vaccine’s safety, effectiveness, and scalability through in vitro investigations, animal model testing, and ensuing clinical trials. To transform this computational framework into a valuable tool for combating P. aeruginosa resistance to multiple drugs, these steps are crucial.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Author contributions
SE: Validation, Writing – original draft, Formal analysis, Investigation, Visualization, Data curation, Methodology, Conceptualization, Writing – review & editing. SK: Visualization, Writing – review & editing, Conceptualization, Validation, Supervision, Methodology.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
The authors express deep gratitude to the management of VIT University for all the support, assistance, and constant encouragement to carry out this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1617251/full#supplementary-material
Abbreviations
WHO, World Health Organization; AMR, Antimicrobial Resistance; P. aeruginosa, Pseudomonas aeruginosa; NCBI, National Center for Biotechnology Information; VFDB, Virulence Factor Database; MD, Molecular dynamics; RMSD, Root Mean Square Deviation; RMSF, Root Mean Square Fluctuation; PCA, Principal Component Analysis; FEL, Free Energy Landscape; HBOND, Hydrogen bond analysis.
References
1. Qin S, Xiao W, Zhou C, Pu Q, Deng X, Lan L, et al. Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics. Signal Transduct Target Ther. (2022) 7:199. doi: 10.1038/s41392-022-01056-1
2. Menon ND, Somanath P, Jossart J, Vijayakumar G, Shetty K, Baswe M, et al. Comparative molecular profiling of multidrug-resistant Pseudomonas aeruginosa identifies novel mutations in regional clinical isolates from South India. JAC Antimicrob Resist. (2023) 6. doi: 10.1093/jacamr/dlae001
3. De Oliveira DMP, Forde BM, Kidd TJ, Harris PNA, Schembri MA, Beatson SA, et al. Antimicrobial resistance in ESKAPE pathogens. Clinical Microbiology Reviews (2020) 33. doi: 10.1128/cmr.00181-19
4. Kalpana S, Lin W-Y, Wang Y-C, Fu Y, Lakshmi A, and Wang H-Y. Antibiotic resistance diagnosis in ESKAPE pathogens—A review on proteomic perspective. Diagnostics. (2023) 13:1014. doi: 10.3390/diagnostics13061014
5. Teney C, Poupelin J-C, Briot T, Le Bouar M, Fevre C, Brosset S, et al. Phage therapy in a burn patient colonized with extensively drug-resistant pseudomonas aeruginosa responsible for relapsing ventilator-associated pneumonia and bacteriemia. Viruses. (2024) 16:1080. doi: 10.3390/v16071080
6. Pham TM, Büchler AC, Voor in ‘t holt AF, Severin JA, Bootsma MCJ, Gommers D, et al. Routes of transmission of VIM-positive Pseudomonas aeruginosa in the adult intensive care unit-analysis of 9 years of surveillance at a university hospital using a mathematical model. Antimicrob Resist Infect Control. (2022) 11:55. doi: 10.1186/s13756-022-01095-x
7. Organization WH. WHO bacterial priority pathogens list, 2024: Bacterial pathogens of public health importance to guide research, development and strategies to prevent and control antimicrobial resistance. (2024). Available online at: https://www.who.int/publications/i/item/9789240093461 (Accessed August 19, 2025).
8. Liao C, Huang X, Wang Q, Yao D, and Lu W. Virulence factors of pseudomonas aeruginosa and antivirulence strategies to combat its drug resistance. Front Cell Infect Microbiol. (2022) 12:926758. doi: 10.3389/fcimb.2022.926758
9. Horna G and Ruiz J. Type 3 secretion system of Pseudomonas aeruginosa. Microbiol Res. (2021) 246:126719. doi: 10.1016/j.micres.2021.126719
10. Giovagnorio F, De Vito A, Madeddu G, Parisi SG, and Geremia N. Resistance in pseudomonas aeruginosa: A narrative review of antibiogram interpretation and emerging treatments. Antibiotics. (2023) 12:1621. doi: 10.3390/antibiotics12111621
11. Kang C-I, Kim S-H, Park WB, Lee K-D, Kim H-B, Kim E-C, et al. Bloodstream infections caused by antibiotic-resistant gram-negative bacilli: risk factors for mortality and impact of inappropriate initial antimicrobial therapy on outcome. Antimicrob Agents Chemother. (2005) 49:760–6. doi: 10.1128/AAC.49.2.760-766.2005
12. Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, and Edmond MB. Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis. (2004) 39:309–17. doi: 10.1086/421946
13. Li J, Wang Y, Liu P, Zhang Y, Yang Y, Zhao S, et al. Trends and implications of antimicrobial resistance in Pseudomonas aeruginosa: Insights from a 19-year study in Zhejiang Province. Medicine. (2024) 103:e40606. doi: 10.1097/MD.0000000000040606
14. Yang AF, Huang V, Samaroo-Campbell J, and Augenbraun M. Multi-drug resistant Pseudomonas aeruginosa: a 2019–2020 single center retrospective case control study. Infect Prev Pract. (2023) 5:100296. doi: 10.1016/j.infpip.2023.100296
15. Almutairy B. Extensively and multidrug-resistant bacterial strains: case studies of antibiotics resistance. Front Microbiol. (2024) 15:1381511. doi: 10.3389/fmicb.2024.1381511
16. Muteeb G, Rehman MT, Shahwan M, and Aatif M. Origin of antibiotics and antibiotic resistance, and their impacts on drug development: A narrative review. Pharmaceuticals. (2023) 16:1615. doi: 10.3390/ph16111615
17. Mondal AH, Khare K, Saxena P, Debnath P, Mukhopadhyay K, and Yadav D. A review on colistin resistance: an antibiotic of last resort. Microorganisms. (2024) 12:772. doi: 10.3390/microorganisms12040772
18. Singha B, Singh V, and Soni V. Alternative therapeutics to control antimicrobial resistance: a general perspective. Front Drug Discov. (2024) 4:1385460. doi: 10.3389/fddsv.2024.1385460
19. Hassan R, El-Naggar W, Abd El-Aziz AM, Shaaban M, Kenawy HI, and Ali YM. Immunization with outer membrane proteins (OprF and OprI) and flagellin B protects mice from pulmonary infection with mucoid and nonmucoid Pseudomonas aeruginosa. J Microbiol Immunol Infect. (2018) 51:312–20. doi: 10.1016/j.jmii.2016.08.014
20. Hasso-Agopsowicz M, Sparrow E, Cameron AM, Sati H, Srikantiah P, Gottlieb S, et al. The role of vaccines in reducing antimicrobial resistance: A review of potential impact of vaccines on AMR and insights across 16 vaccines and pathogens. Vaccine. (2024) 42:S1–8. doi: 10.1016/j.vaccine.2024.06.017
21. Costanzo V and Roviello GN. The potential role of vaccines in preventing antimicrobial resistance (AMR): an update and future perspectives. Vaccines (Basel). (2023) 11:333. doi: 10.3390/vaccines11020333
22. Aroca Molina KJ, Gutiérrez SJ, Benítez-Campo N, and Correa A. Genomic differences associated with resistance and virulence in pseudomonas aeruginosa isolates from clinical and environmental sites. Microorganisms. (2024) 12:1116. doi: 10.3390/microorganisms12061116
23. Sethi G, Varghese RP, Lakra AK, Nayak SS, Krishna R, and Hwang JH. Immunoinformatics and structural aided approach to develop multi-epitope based subunit vaccine against Mycobacterium tuberculosis. Sci Rep. (2024) 14:15923. doi: 10.1038/s41598-024-66858-5
24. Firoved AM, Ornatowski W, and Deretic V. Microarray analysis reveals induction of lipoprotein genes in mucoidPseudomonas aeruginosa: implications for inflammation in cystic fibrosis. Infect Immun. (2004) 72:5012–8. doi: 10.1128/iai.72.9.5012-5018.2004
25. Adila Nazli, Qiu J, Tang Z, and He Y. Recent advances and techniques for identifying novel antibacterial targets. Curr Medicinal Chem. (2023) 31:464–501. doi: 10.2174/0929867330666230123143458
27. Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. (2015) 31:3691–3. doi: 10.1093/bioinformatics/btv421
28. Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. (2014) 30:2068–9. doi: 10.1093/bioinformatics/btu153
29. Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. (2010) 26:1608–15. doi: 10.1093/bioinformatics/btq249
30. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinf. (2009) 10:421. doi: 10.1186/1471-2105-10-421
31. Chen L. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. (2004) 33:D325–8. doi: 10.1093/nar/gki008
32. Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, and Brinkman FSL. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res. (2016) 44:D646–53. doi: 10.1093/nar/gkv1227
33. Doytchinova IA and Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf. (2007) 8:4. doi: 10.1186/1471-2105-8-4
34. Dimitrov I, Bangov I, Flower DR, and Doytchinova I. AllerTOP v.2—a server for in silico prediction of allergens. J Mol Model. (2014) 20:2278. doi: 10.1007/s00894-014-2278-5
35. Kyte J and Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology. (1982) 157:105–32. doi: 10.1016/0022-2836(82)90515-0
36. Teufel F, Almagro Armenteros JJ, Johansen AR, Gíslason MH, Pihl SI, Tsirigos KD, et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol. (2022) 40:1023–5. doi: 10.1038/s41587-021-01156-3
37. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. (2019) 47:D339–43. doi: 10.1093/nar/gky1006
38. Jespersen MC, Peters B, Nielsen M, and Marcatili P. BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. (2017) 45:W24–9. doi: 10.1093/nar/gkx346
39. Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, and Raghava GPS. In silico approach for predicting toxicity of peptides and proteins. PloS One. (2013) 8:e73957. doi: 10.1371/journal.pone.0073957
40. Reynisson B, Alvarez B, Paul S, Peters B, and Nielsen M. NetMHCpan-4.1 and NetMHCIIpan-4.0: improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. (2020) 48:W449–54. doi: 10.1093/nar/gkaa379
41. Fleri W, Paul S, Dhanda SK, Mahajan S, Xu X, Peters B, et al. The immune epitope database and analysis resource in epitope discovery and synthetic vaccine design. Front Immunol. (2017) 8:278. doi: 10.3389/fimmu.2017.00278
42. Dhanda SK, Vir P, and Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biol Direct. (2013) 8:30. doi: 10.1186/1745-6150-8-30
43. Dhanda SK, Gupta S, Vir P, and Raghava GPS. Prediction of IL4 inducing peptides. Clin Dev Immunol. (2013) 2013:1–9. doi: 10.1155/2013/263952
44. Dhall A, Patiyal S, Sharma N, Usmani SS, and Raghava GPS. Computer-aided prediction and design of IL-6 inducing peptides: IL-6 plays a crucial role in COVID-19. Briefings Bioinf. (2020) 22. doi: 10.1093/bib/bbaa259
45. Nagpal G, Usmani SS, Dhanda SK, Kaur H, Singh S, Sharma M, et al. Computer-aided designing of immunosuppressive peptides based on IL-10 inducing potential. Sci Rep. (2017) 7. doi: 10.1038/srep42851
46. Jain S, Dhall A, Patiyal S, and Raghava GPS. IL13Pred: A method for predicting immunoregulatory cytokine IL-13 inducing peptides. Comput Biol Med. (2022) 143:105297–7. doi: 10.1016/j.compbiomed.2022.105297
47. Soltan MA, Elbassiouny N, Gamal H, Elkaeed EB, Eid RA, Eldeen MA, et al. In Silico Prediction of a Multitope Vaccine against Moraxella catarrhalis: Reverse Vaccinology and Immunoinformatics. Vaccines (Basel). (2021) 9:669. doi: 10.3390/vaccines9060669
48. Li X, Wang X, Du J, Bu X, Peng C, Duan X, et al. Applications of β-defensins against infectious pathogenic microorganisms. Expert Rev Anti Infect Ther. (2024) 22:501–10. doi: 10.1080/14787210.2024.2377677
49. Ruaro-Moreno M, Monterrubio-López GP, Reyes-Gastellou A, Castelán-Vega JA, Jiménez-Alberto A, Aparicio-Ozores G, et al. Design of a Multi-Epitope Vaccine against Tuberculosis from Mycobacterium tuberculosis PE_PGRS49 and PE_PGRS56 Proteins by Reverse Vaccinology. Microorganisms. (2023) 11:1647. doi: 10.3390/microorganisms11071647
50. Khan S, Rizwan M, Zeb A, Eldeen MA, Hassan S, Ur Rehman A, et al. Identification of a Potential Vaccine against Treponema pallidum Using Subtractive Proteomics and Reverse-Vaccinology Approaches. Vaccines. (2023) 11:72. doi: 10.3390/vaccines11010072
51. Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, et al. Protein identification and analysis tools on the ExPASy server. In: The Proteomics Protocols Handbook. Humana Press, Totowa, NJ (2005). p. 571–607. doi: 10.1385/1-59259-890-0:571
52. McGuffin LJ, Bryson K, and Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. (2000) 16:404–5. doi: 10.1093/bioinformatics/16.4.404
53. Buchan DWA and Jones DT. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res. (2019) 47:W402–7. doi: 10.1093/nar/gkz297
54. Kim DE, Chivian D, and Baker D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. (2004). doi: 10.1093/nar/gkh468
55. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–9. doi: 10.1038/s41586-021-03819-2
56. Heo L, Park H, and Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Res. (2013) 41:W384–8. doi: 10.1093/nar/gkt458
57. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
58. Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, and Tiwari RK. Synthetic toll like receptor-4 (TLR-4) agonist peptides as a novel class of adjuvants. PloS One. (2012) 7:e30839. doi: 10.1371/journal.pone.0030839
59. Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, et al. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. (1999) 11:443–51. doi: 10.1016/S1074-7613(00)80119-3
60. Park BS, Song DH, Kim HM, Choi B-S, Lee H, and Lee J-O. The structural basis of lipopolysaccharide recognition by the TLR4–MD-2 complex. Nature. (2009) 458:1191–5. doi: 10.1038/nature07830
61. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik S-G, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. (2007) 130:1071–82. doi: 10.1016/j.cell.2007.09.008
62. Yan Y, Tao H, He J, and Huang S-Y. The HDOCK server for integrated protein–protein docking. Nat Protoc. (2020) 15:1829–52. doi: 10.1038/s41596-020-0312-x
63. Jo S, Kim T, Iyer VG, and Im W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J Comput Chem. (2008) 29:1859–65. doi: 10.1002/jcc.20945
64. Ong EES and Liow J-L. The temperature-dependent structure, hydrogen bonding and other related dynamic properties of the standard TIP3P and CHARMM-modified TIP3P water models. Fluid Phase Equilib. (2019) 481:55–65. doi: 10.1016/j.fluid.2018.10.016
65. Petersen HG. Accuracy and efficiency of the particle mesh Ewald method. J Chem Phys. (1995) 103:3668 3679. doi: 10.1063/1.470043
66. Haug EJ, Arora JS, and Matsui K. A steepest-descent method for optimization of mechanical systems. J Optim Theory Appl. (1976) 19:401 424. doi: 10.1007/BF00941484
67. Nesabi A, Kalayan J, Al-Rawashdeh S, Ghattas MA, and Bryce RA. Molecular dynamics simulations as a guide for modulating small molecule aggregation. J Comput Aided Mol Des. (2024) 38:11. doi: 10.1007/s10822-024-00557-1
68. Kumari R, Kumar R, and Lynn A. g_mmpbsa—A GROMACS tool for highThroughput MMPBSA calculations. J Chem Inf Model. (2014) 54:1951–62. doi: 10.1021/ci500020m
69. Baker NA, Sept D, Joseph S, Holst MJ, and McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci United States America. (2001) 98:10037–41. doi: 10.1073/pnas.181342398
70. Sharma AD, Samuel J, Singh H, Cuspoca F, Cavallo L, and Chawla M. Immunoinformatics-driven design of a multi-epitope vaccine targeting neonatal rotavirus with focus on outer capsid proteins VP4 and VP7 and non structural proteins NSP2 and NSP5. Sci Rep. (2025) 15. doi: 10.1038/s41598-025-95256-8
71. Kaushik V, G SK, Gupta LR, Kalra U, Shaikh AR, Cavallo L, et al. Immunoinformatics aided design and in-vivo validation of a cross-reactive peptide based multi-epitope vaccine targeting multiple serotypes of dengue virus. Front Immunol. (2022) 13:3389/fimmu.2022.865180. doi: 10.3389/fimmu.2022.865180
72. Kaushik V, Jain P, Akhtar N, Joshi A, Gupta LR, Grewal RK, et al. ImmunoinformaticsAided design and in vivo validation of a peptideBased multi-epitope vaccine targeting canine circovirus. ACS Pharmacol Transl Sci. (2022) 5:679–91. doi: 10.1021/acsptsci.2c00130
73. GenScript. Make Research Easy - The leader in molecular cloning and gene synthesis, peptide synthesis, protein and antibody engineering. Available online at: https://www.genscript.com/.
74. Kamens J. The Addgene repository: an international nonprofit plasmid and data resource. Nucleic Acids Res. (2015) 43:D1152–7. doi: 10.1093/nar/gku893
75. Rapin N, Lund O, Bernaschi M, and Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PloS One. (2010) 5:e9862. doi: 10.1371/journal.pone.0009862
76. Sanner MF. Python: a programming language for software integration and development. J Mol Graph Model. (1999) 17:57–61.
77. Tuimala J and Kallio A. R, programming language. In: Encyclopedia of Systems Biology. Springer New York, New York, NY (2013). p. 1809–11. doi: 10.1007/978-1-4419-9863-7_619
78. Chawla M, Cuspoca AF, Akthar N, Magdaleno JSL, Rattanabunyong S, Suwattanasophon C, et al. Immunoinformatics-aided rational design of a multi-epitope vaccine targeting feline infectious peritonitis virus. Front Vet Sci. (2023) 10:1280273. doi: 10.3389/fvets.2023.1280273
79. Elfadadny A, Ragab RF, AlHarbi M, Badshah F, Ibáñez-Arancibia E, Farag A, et al. Antimicrobial resistance of Pseudomonas aeruginosa: navigating clinical impacts, current resistance trends, and innovations in breaking therapies. Front Microbiol. (2024) 15:1374466. doi: 10.3389/fmicb.2024.1374466
80. Assefa M and Amare A. Biofilm-associated multi-drug resistance in hospital-acquired infections: A review. Infect Drug Resist. (2022) 15:5061–8. doi: 10.2147/IDR.S379502
81. Sharma D, Misba L, and Khan AU. Antibiotics versus biofilm: an emerging battleground in microbial communities. Antimicrob Resist Infect Control. (2019) 8:76. doi: 10.1186/s13756-019-0533-3
82. Lopes JA, Rghei AD, Thompson B, Susta L, Khursigara CM, and Wootton SK. Overcoming barriers to preventing and treating P. aeruginosa infections using AAV vectored immunoprophylaxis. Biomedicines. (2022) 10:3162. doi: 10.3390/biomedicines10123162
83. Sharma R, Rajput VS, Jamal S, Grover A, and Grover S. An immunoinformatics approach to design a multi-epitope vaccine against Mycobacterium tuberculosis exploiting secreted exosome proteins. Sci Rep. (2021) 11:13836. doi: 10.1038/s41598-021-93266-w
84. Alizadeh M, Amini-Khoei H, Tahmasebian S, Ghatrehsamani M, Ghatreh Samani K, Edalatpanah Y, et al. Designing a novel multi epitope vaccine against Ebola virus using reverse vaccinology approach. Sci Rep. (2022) 12:7757. doi: 10.1038/s41598-022-11851-z
85. Sarkar B, Ullah M, Johora FT, Taniya MA, and Araf Y. Immunoinformatics-guided designing of epitope-based subunit vaccines against the SARS Coronavirus-2 (SARS-CoV-2). Immunobiology. (2020) 225:151955. doi: 10.1016/j.imbio.2020.151955
86. Elhag M, Ruaa Mohamed Alaagib, Ahmed NM, Abubaker M, Haroun EM, Sahar, et al. Design of Epitope-Based Peptide Vaccine against Pseudomonas aeruginosa Fructose Bisphosphate Aldolase Protein Using Immunoinformatics. J Immunol Res. (2020) 2020:1–11. doi: 10.1155/2020/9475058
87. Zhu F, Qin R, Ma S, Zhou Z, Tan C, Yang H, et al. Designing a multi-epitope vaccine against Pseudomonas aeruginosa via integrating reverse vaccinology with immunoinformatics approaches. Sci Rep. (2025) 15. doi: 10.1038/s41598-025-90226-6
88. Solanki V, Tiwari M, and Tiwari V. Prioritization of potential vaccine targets using comparative proteomics and designing of the chimeric multi-epitope vaccine against Pseudomonas aeruginosa. Sci Rep. (2019) 9. doi: 10.1038/s41598-019-41496-4
89. Roy SK, Biswas MS, Raman MF, Hasan R, Zahidur Rahmann, and Moyen M. A computational approach to developing a multi-epitope vaccine for combating Pseudomonas aeruginosa–induced pneumonia and sepsis. Briefings Bioinf. (2024) 25. doi: 10.1093/bib/bbae401
90. Nagpal G, Usmani SS, and Raghava GPS. A web resource for designing subunit vaccine against major pathogenic species of bacteria. Front Immunol. (2018) 9:3389/fimmu.2018.02280. doi: 10.3389/fimmu.2018.02280
91. Naz K, Naz A, Ashraf ST, Rizwan M, Ahmad J, Baumbach J, et al. PanRV: Pangenome-reverse vaccinology approach for identifications of potential vaccine candidates in microbial pangenome. BMC Bioinf. (2019) 20:123. doi: 10.1186/s12859-019-2713-9
92. Damron FH, Napper J, Teter MA, and Yu HD. Lipotoxin F of Pseudomonas aeruginosa is an AlgU-dependent and alginate-independent outer membrane protein involved in resistance to oxidative stress and adhesion to A549 human lung epithelia. Microbiol (N Y). (2009) 155:1028–38. doi: 10.1099/mic.0.025833-0
93. Belo VA, Pereira JA, Souza SFD, Tana FL, Pereira BP, Lopes D de O, et al. The role of IL-10 in immune responses against Pseudomonas aeruginosa during acute lung infection. Cell Tissue Res. (2020) 383:1123–33. doi: 10.1007/s00441-020-03308-4
94. Shaw DE, Maragakis P, Lindorff-Larsen K, Piana S, Dror RO, Eastwood MP, et al. Atomic-level characterization of the structural dynamics of proteins. Science. (2010) 330:341–6. doi: 10.1126/science.1187409
95. Cheng X, Chen Z, Gao C, Zhang Y, Yang L, Wan J, et al. Structural and biological insights into outer membrane protein lipotoxin F of Pseudomonas aeruginosa: Implications for vaccine application. Int J Biol Macromol. (2023) 253:127634. doi: 10.1016/j.ijbiomac.2023.127634
96. Sharma S, Yadav PD, and Cherian S. Comprehensive immunoinformatics and bioinformatics strategies for designing a multi-epitope based vaccine targeting structural proteins of Nipah virus. Front Immunol. (2025) 16:1535322. doi: 10.3389/fimmu.2025.1535322
Keywords: Pseudomonas aeruginosa, pangenome analysis, immunoinformatics, epitope-based vaccine, molecular docking, immune simulation
Citation: Mudipalli Elavarasu S and K S (2025) Rational design of an epitope-centric vaccine against Pseudomonas aeruginosa using pangenomic insights and immunoinformatics approach. Front. Immunol. 16:1617251. doi: 10.3389/fimmu.2025.1617251
Received: 24 April 2025; Accepted: 06 August 2025;
Published: 01 September 2025.
Edited by:
Gurudeeban Selvaraj, Aarupadai Veedu Medical College and Hospital, IndiaReviewed by:
Salman Sadullah Usmani, Albert Einstein College of Medicine, United StatesXiao-Yan Yang, Zhuhai Campus of Zunyi Medical University, China
Mohit Chawla, King Abdullah University of Science and Technology, Saudi Arabia
Copyright © 2025 Mudipalli Elavarasu and K. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sasikumar K, c2FzaWt1bWFyLmtAdml0LmFjLmlu