 Man Wang1†
Man Wang1† Yuanzhuo Zhao
Yuanzhuo Zhao Ke Zhang
Ke Zhang- 1Department of Nephrology, Shuyang Hospital of Traditional Chinese Medicine, Shuyang, Jiangsu, China
- 2The Second Clinical Medical College, Nanjing Medical University, Nanjing, Jiangsu, China
- 3Department of Urology, The Affiliated Suzhou Hospital of Nanjing Medical University, Suzhou Municipal Hospital, Suzhou, Jiangsu, China
Clear cell renal cell carcinoma (ccRCC) progression heavily relies on the immunosuppressive tumor microenvironment (TME). In the ccRCC TME, the cancer-associated fibroblasts (CAFs) drive a self-perpetuating cycle of immune evasion and therapeutic resistance through diverse interactions between cells and molecules. Furthermore, heterogeneous CAFs facilitate tumor growth through metabolic reprogramming and modulate immune suppression by driving the M2 polarization of tumor-associated macrophages (TAMs) and the expansion of regulatory T cells (Tregs), which promote a multilayered immunosuppressive network. In addition, CAFs reshape the mechanical properties of extracellular matrix (ECM), hinder the infiltration of cytotoxic T lymphocytes (CTLs) and further exacerbate immune escape. Moreover, CAF-derived exosomes can confer resistance to chemoradiation therapy. Interleukin-6 (IL-6) secreted by CAFs synergizes with vascular endothelial growth factor (VEGF) to facilitate adaptive resistance to targeted therapy. Emerging therapeutic strategies—including fibroblast activation protein (FAP)-targeted CAR-T cells and transforming growth factor-β (TGF-β) inhibitors—can partially reverse this immunosuppressive property. Combination therapies employing immune checkpoint inhibitors and VEGF antagonists exhibit promising synergistic effects, although the clinical translation remains hampered by CAF heterogeneity, dual functional roles, and the lack of specific biomarkers. Future studies should integrate single-cell sequencing and spatial multi-omics techniques to comprehensively analyze the spatio-temporal dynamic heterogeneity of CAF subpopulations and develop precision treatment strategies based on molecular subtyping, aiming to break the vicious cycle of “CAF-TME-resistance” in ccRCC.
1 Introduction
Renal cell carcinoma (RCC) originates from the epithelial cells of the renal tubules and is a highly heterogeneous malignancy. Clear cell renal cell carcinoma (ccRCC), the predominant histological subtype of renal cancer, is principally characterized by the inactivation of the von Hippel-Lindau (VHL) tumor suppressor gene. This genetic alteration leads to constitutive activation of the hypoxia-inducible factor (HIF) pathway, which promotes metabolic reprogramming, angiogenesis, and an immunosuppressive tumor microenvironment (TME) (1).
Although targeted therapy—particularly vascular endothelial growth factor (VEGF) inhibitors—and immune checkpoint blockade have significantly improved prognosis in cancer patients, therapeutic resistance remains as a major challenge (2). This clinical dilemma highlights an urgent need of the transition from the traditional “tumor cell-centric” paradigm to a “TME-centric regulatory” conception.
The tumor microenvironment (TME) is a dynamic ecosystem composed of diverse cell types and stromal components. Among the stromal components, cancer-associated fibroblasts (CAFs) emerge as pivotal regulators that are dynamically activated through various signals involved in extracellular matrix (ECM) remodeling. Through complex molecular and cellular interactions, CAFs foster a vicious cycle of immune suppression, therapeutic resistance, and tumor progression (3).
This review systematically analyzes the multidimensional regulatory network of CAFs within the ccRCC TME, and focused on their heterogeneous origins, functional subtype, molecular mechanisms of interaction with other cells, and evolutionary potentials of CAF-targeting strategies.
2 Biological characteristics and functional roles of CAFs in ccRCC
2.1 Cellular origins and phenotypic heterogeneity of CAFs
The heterogeneity of CAFs in ccRCC is primarily attributed to their diverse cellular origins and differentiation pathways: First, resident renal fibroblasts undergo in situ activation into α-smooth muscle actin positive (α-SMA+) myofibroblasts under the influence of transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF); Second, bone marrow-derived mesenchymal stem cells (BM-MSCs) are recruited to the TME via the CXCL12/CXCR4 axis, where they adopt CAF-like phenotypes; Third, transdifferentiation processes such as epithelial-to-mesenchymal transition (EMT) provide an additional source of CAFs (4, 5). Recent advances in single-cell RNA sequencing (scRNA-seq) have enabled a finer classification of CAF subtypes and their functional roles in ccRCC. Kieffer et al. showed in multiple solid tumor models that FAP+ CAFs are a key source of pro-inflammatory and immunomodulatory cytokines that impair anti-tumor immunity and confer resistance to immune checkpoint inhibitors (6). Although originally defined in pancreatic cancer, myofibroblastic CAFs (myCAFs) and inflammatory CAFs (iCAFs) exhibit conserved gene expression programs in ccRCC as well—myCAFs express TGF-β response genes and ECM components, while iCAFs secrete IL-6, LIF, and CXCL8, which amplify immunosuppression and tumor-promoting inflammation (7, 8). These findings highlight the functional specialization of CAF subsets and their relevance as therapeutic targets in ccRCC.
2.2 CAF-mediated tumor-promoting functions
Once activated, CAFs play an essential role in promoting tumor progression (9). Metabolically, CAFs undergo reprogramming that enhances glycolysis and facilitates the supply of metabolic intermediates to tumor cells. They secrete a variety of pro-angiogenic factors—such as CXCL12, vascular endothelial growth factor A (VEGFA), platelet-derived growth factor C (PDGFC), and osteopontin—that stimulate neovascularization. In addition, CAFs actively participate in ECM remodeling, thereby altering tissue stiffness and promoting tumor invasion and metastasis (10–12). Furthermore, CAFs also engage in direct physical and paracrine interactions with tumor cells, inducing EMT and supporting processes such as vascular mimicry (13–15). These collective actions establish a tumor-permissive microenvironment and fuel aggressive behaviors, rendering CAFs indispensable facilitators of tumor proliferation, angiogenesis, metastasis, and therapy resistance (16).
2.3 Signaling mechanisms driving CAF activation and differentiation
Normal fibroblasts can inhibit tumor cells proliferation and invasion, and inhibit epithelial tumors (17, 18). Therefore, the transformation of normal fibroblasts to cancer-promoting CAFs is an essential mechanism for the survival of malignant cells. The generation of CAFs results from the synergistic effect of paracrine signaling and mechanical stimulation. Once activated, CAFs create a self-reinforcing feedback loop within the TME that continuously activates these signaling pathways, driving CAF population expansion (19–21).
Paracrine signaling molecules, including transforming growth factor-β (TGF-β), interleukin-1α (IL-1α), and PDGF, play key regulatory roles in CAFs reprogramming. Specifically, the TGF-β axis has been shown to primarily drive the differentiation of myCAFs, while the IL-1α axis regulates the formation of iCAFs (22, 23). After activation of the above signaling pathways, downstream effects of CAFs reprogramming gradually emerge, such as a shift in metabolic pattern. Specifically, TGF-β-stimulated CAFs exhibit an activated oxidative stress program that provides energy substrates to cancer cells (24, 25). In addition, macrophage-derived factors and CAFs’ autocrine products together constitute additional signaling axes regulating CAFs differentiation (23, 26). This dynamic feedback network reinforces the malignant properties of CAFs, and contributes to the sustained immunosuppressive niche.
3 CAFs and the tumor microenvironment: a dynamic crosstalk network
3.1 Bidirectional interactions between CAFs and tumor cells
CAFs are essential modulators within the TME, interacting with tumor cells through various mechanisms, including tumor cell proliferation [20], regulating angiogenesis [21], construction of an immunosuppressive niche to evade immune surveillance [9], and promoting tumor formation and therapeutic resistance [22]. Through the secretion of diverse cytokines, chemokines (e.g., CXCL2), extracellular matrix proteins (e.g., collagen, laminin), and matrix metalloproteinases (MMPs), CAFs regulate immune cell recruitment, ECM remodeling, and tissue architecture (27, 28). These factors collectively facilitate cancer progression by enhancing tumor cell motility, promoting EMT, and contributing to vascular mimicry. Increasing evidence indicates that CAFs complement other components of the microenvironment to combat immune cells and regulate tumor immune microenvironment (TIME) (29, 30). CAFs coordinate the immunosuppressive TME through dynamic interactions with tumor-associated immune cells (17). Specifically, CAFs regulate immune cell-mediated anti-tumor responses through the following mechanisms: CAFs enhancing the recruitment, activation and immunosuppressive function of immunosuppressive cells (31). At the same time, the killing activity and cytokine secretion of effector immune cells (such as natural killer cells (NK cells) and cytotoxic T lymphocytes (CTLs) are inhibited, and the bidirectional regulation of immune response is realized (32, 33). Infiltrating immune cells enhance the activation state and functional activity of CAFs in both ways, thereby establishing a self-sustaining immunosuppressive feedback loop (34). CAFs induce T cell dysfunction by upregulating the expression of immune checkpoint molecules, including programmed cell death ligand 1 (PD-L1)/receptor 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4)/B7, on cell surface of both TME itself and adjacent cells (35). CAFs secrete fibronectin, collagen and Matrix Metalloproteinases (MMPs), signaling pathway of Focal Adhesion Kinase (FAK) is activated to reshape the ECM (36, 37). This biomechanical remodeling further solidifies the immunosuppressive state by forming a physical barrier that prevents immune cell infiltration (37, 38). By synergistically interacting with ECM-mediated survival signals, CAFs acquire resistance against apoptosis and maintain their cancer-promoting activity (34). These tumor-supportive interactions form the basis for subsequent immunomodulatory effects within the TME.
3.2 Immunomodulatory Roles of CAFs in the TIME
3.2.1 CAFs-TAM axis: co-amplification of innate immunosuppression
TAMs, a key component of TIME, make a significant contribution to maintaining immunosuppression (39, 40). CAFs promote the recruitment and polarization of monocytes into M2-type TAMs through secretion of factors such as IL-6, CCL2, and CXCL12. These M2-TAMs exhibit immunosuppressive phenotypes characterized by elevated PD-1 expression and reduced phagocytic activity, which impairs both innate and adaptive immune responses (41). High co-expression of CAF and M2-TAM markers (e.g., FAP and CD163) correlates with poor prognosis in ccRCC and other solid tumors (42, 43). In addition, CAFs can mediate the induction of immunosuppressive phenotypes in TAMs. For example, Gok et al. demonstrated through flow cytometry analysis that PD-1 expression is specifically elevated on M2-polarized TAMs, and CAF-mediated upregulation of PD-1 expression in TAMs significantly impaired their phagocytic capacity against tumor cells. Furthermore, this PD-1 overexpression establishes an immunosuppressive microenvironment by suppressing T cell infiltration and proliferation, while simultaneously compromising both innate and adaptive arms of anti-tumor immunity (44). This axis exemplifies the CAFs’ capacity to manipulate the innate immune compartment and primes them for interactions with adaptive immune cells.
3.2.2 CAFs-Tregs axis: inhibition of adaptive immune responses
Lymphocytes, which play a crucial role in regulating adaptive immune responses, consist of different functional subgroups, including Tregs, CTLs, and Helper T (Th) cells (35, 45). There is substantial evidence of dynamic interactions between CAFs and T cell populations. The interaction between CAFs and Tregs exemplifies this immunoregulatory property (46, 47). Tregs with high Foxp3 expression have been confirmed to play a key role in inhibiting anti-tumor immunity (48, 49). Kinoshita et al. reported the spatial co-localization of Tregs and CAFs in tumor tissues (50). Clinical data further showed that co-infiltration of Foxp3+ Tregs and CAFs in tumor stroma was significantly associated with poor prognosis (50, 51). These findings suggest that there is a potential interaction between CAFs and Tregs. CAFs can also actively induce the phenotypic plasticity of Tregs and amplify their immunosuppressive effects (6). Chen et al. confirmed that CAF-derived TGF-β could drive the differentiation of initial T cells into CD4+/CD25+ Tregs (52). Zhao et al. reported that CAFs facilitate Treg expansion and functional activation by secreting TGF-β and promoting chemokine-driven recruitment (e.g., via CCL22 and CCL17) (53, 54). Paradoxically, Ozdemir et al. found in the pancreatic ductal adenocarcinoma (PDAC) model that myofibroblast depletion could increase the proliferation of Foxp3+ Tregs, thereby suppressing immune surveillance (22). This counterintuitive phenomenon suggests that CAFs-Tregs interactions have a dual, context-dependent character.
3.3 Mechanical remodeling of the ECM by CAFs
Fibroblasts, the primary constructors of the ECM, play a crucial role in tissue repair and homeostasis maintenance through the synthesis and remodeling of the interstitial matrix (45). CAFs play a central role in extracellular matrix remodeling, a process that not only facilitates tumor cell invasion but also modulates immune cell infiltration and accessibility. During normal wound healing, cytokines and growth factors stimulate the recruitment of fibroblasts, which respond by increasing the mechanical stress within the microenvironment. This process drives the transdifferentiation of fibroblasts into myofibroblasts, characterized by the expression of α-SMA (55). When stimulated by TGF-β1 and mechanical cues, CAFs upregulate α-SMA expression and produce fibronectin variants, such as EDA-FN. These changes facilitate the assembly of actomyosin fibers and induce persistent tissue contractility (56–58). Notably, α-SMA+ myofibroblasts sustain ECM contraction and drive permanent tissue remodeling, unlike smooth muscle cells, which exhibit only transient contraction properties (59). Chronic tissue contraction leads to ECM stiffening, which in turn activates TGF-β1 through tension-induced release from latent complexes. This activation further amplifies fibroblast activity via a feedforward mechanism. Additionally, actomyosin-mediated contraction of fibroblasts activates the YAP/TAZ and MRTF pathways, increasing ECM protein expression through transcriptional activation, thereby linking mechanical stress to the gene transcription of myofibroblasts (60–62). In healthy tissues, myofibroblasts return to a quiescent state or undergo apoptosis following tissue repair (63–65). However, within the tumor microenvironment, sustained signaling and mechanical tension maintain the activated state of CAFs, resulting in chronic ECM stiffness and an immunosuppressive microenvironment. These biomechanical changes impede CTL infiltration and contribute to immune evasion. Thus, ECM remodeling constitutes a mechanical barrier that acts in concert with immune suppression, further enhancing CAF-mediated resistance mechanisms.
4 CAF-induced immune evasion and therapeutic resistance
4.1 CAF-mediated construction of an immunosuppressive microenvironment
Tumors employ multiple strategies to escape immune surveillance, with CAFs involved in the TME playing a particularly crucial role in reinforcing these mechanisms (66). In ccRCC, immune evasion is primarily mediated through impaired antigen presentation, overexpression of immune checkpoints, and establishment of immunosuppressive cellular networks (67). As key architects of the immunosuppressive TME, CAFs directly impair T cell activation through secretion of TGF-β and CXCL12 (68). These mediators exert dual immunosuppressive effects: CXCL12 not only induces spatial exclusion of T cells from tumor cores but also disrupts dendritic cell maturation, while TGF-β drives the differentiation of naïve T cells into regulatory T cells (Tregs). Notably, CXCL12-mediated signaling concurrently promotes tumor angiogenesis, thereby coupling immune evasion with pro-tumorigenic processes (69). At the cellular level, ccRCC exhibits characteristic downregulation of major histocompatibility complex (MHC) molecules, severely compromising T cell-mediated tumor recognition (70). This antigen presentation defect synergizes with tumor cell upregulation of immune checkpoint molecules, which systematically inhibit T cell activation through receptor-ligand interactions (71). Immune checkpoint pathway is the core mechanism of tumor immune escape, and these checkpoints inhibit T cell activation and proliferation, which enables tumor cells to evade immunosurveillance (72). Therapeutic inhibitors targeting these checkpoints, such as anti-PD-1 antibodies and anti-CTLA-4 antibodies, are applied in treating ccRCC and other malignancies, and have shown tremendous clinical benefits (73). The presence of immunosuppressive cell populations—including TAMs, Tregs, and myeloid-derived suppressor cells (MDSCs)—further reinforces the tolerogenic environment (74). CAFs orchestrate this process through chemokine-mediated recruitment and functional reprogramming of these cell populations (75). For example, TAMs excrete cytokines and growth factors which drive tumor progression (76). On the other hand, Tregs create an immunosuppressive microenvironment by inhibiting T cell activation and secreting TGF-β and interleukin-10 (IL-10), which promote tumor evasion of immunosurveillance (77). Importantly, CAFs do not only coexist with but also actively coordinate these immunosuppressive pathways, and these roles could establish an immunological framework that facilitates subsequent therapy resistance development.
4.2 Molecular mechanisms of CAF-driven therapy resistance
Theatment resistance is one of the reasons for treatment failure, and tumor recurrence in cancer patients. CAFs contribute to this resistance through both direct and indirect mechanisms, including the release of exosomes, paracrine signaling, and metabolic modulation. One of the most well-characterized mechanisms involves exosomes derived from CAFs, which are enriched in non-coding RNAs such as miR-590-3p. These exosomes are taken up by tumor cells, where they activate the PI3K/AKT signaling pathway, thereby inhibiting apoptosis and promoting radioresistance (78, 79). In colorectal cancer (CRC), similar exosomal cargo has been shown to suppress cleaved caspase-3 activity, a key mediator of programmed cell death, suggesting a conserved mechanism across tumor types (80). Beyond exosomal communication, CAFs secrete cytokines that modulate tumor cell plasticity and resistance. For example, IL-6, frequently overexpressed by CAFs, synergizes with VEGF to support tumor angiogenesis and adaptive resistance to VEGF-targeted therapies (81). CAFs also produce stromal-derived factor-1 (SDF-1/CXCL12), which has been implicated in tumor cell survival and evasion of anti-angiogenic treatments (82). Taken together, these findings underscore the centrality of CAFs in facilitating immune evasion and treatment failure. Addressing CAF-driven resistance requires a comprehensive understanding of their signaling networks, which will guide the development of combination therapies capable of disrupting this malignant crosstalk.
5 Therapeutic strategies targeting CAF–TME interactions in ccRCC
Table 1 summarizes representative ongoing clinical studies evaluating CAF-related biomarkers across solid tumors and sets the stage for subsequent therapeutic strategies (Data source: ClinicalTrials.gov: https://beta.clinicaltrials.gov/ provided by the U.S. National Library of Medicine).

Table 1. Overview of CAF-related biomarker applications in solid tumor clinical studies.
5.1 Molecular targeting of CAFs and their signaling pathways
Given the multifaceted roles of CAFs in tumor progression, immune evasion, and therapeutic resistance, direct targeting of these cells has emerged as a rational therapeutic strategy. These approaches include selective depletion, functional inhibition, or reprogramming of CAFs to attenuate their tumor-promoting effects. A primary strategy involves targeting surface biomarkers specific to CAFs (15). FAP, a serine protease overexpressed on CAFs in multiple tumor types, has become a leading candidate. A number of studies have showed FAP-directed chimeric antigen receptor (CAR) T cells demonstrated potent antitumor effects in preclinical models (83–85). However, the widespread expression of FAP in non-malignant tissues poses a significant challenge for clinical translation, particularly due to potential stromal toxicity. In addition to surface markers, CAF-derived cytokines and signaling pathways have been explored as therapeutic targets. The blockade of TGF-β signaling, a master regulatory axis governing CAF activation, using selective inhibitors like Galunisertib has demonstrated therapeutic efficacy in mitigating CAF-driven ECM fibrogenesis while concomitantly enhancing CD8+ T cell immunosurveillance (86). Moreover, CAFs influence the TME through other signaling circuits, such as IL-6/STAT3 and Hedgehog pathway, which have become attractive drug targets. Emerging compounds that inhibit these axes may indirectly mitigate CAF activity and sensitize tumors to conventional and immune therapies [80]. Epigenetic regulators, including histone deacetylase (HDAC) and Smoothened (SMO) inhibitors, also influence CAF reprogramming and stromal remodeling. These agents are under investigation for their dual activity on both tumor cells and the surrounding microenvironment. Thus, CAF-directed interventions hold promise as standalone or adjunctive therapies.
5.2 Combination strategies with immunotherapy to overcome resistance
As summarized in Table 2, several phase II/III trials—including CheckMate 214, KEYNOTE-426, and CLEAR—have established ICI-based regimens as the cornerstone of first-line therapy for advanced ccRCC. The complex crosstalk between CAFs and immune components in the TME provides a compelling rationale for combining CAF-targeting approaches with immunotherapy. Immune checkpoint inhibitors (ICIs), including those targeting PD-1/PD-L1 and CTLA-4 pathways, have revolutionized treatment paradigms for advanced ccRCC. However, their efficacy remains limited in CAF-rich tumors due to impaired T cell infiltration, sustained immune suppression, and stromal resistance mechanisms (87, 88). A clinical trial of combination therapy based on an immune checkpoint inhibitor recently confirms, combination therapy is significantly better than monotherapy in an untreated patient with metastatic ccRCC (89). A clinical trial in melanoma patients compared the efficacy of anti-CTLA4 antibody (ipilimumab) alone with the combined gp100 peptide vaccine, showing that the first two regimen significantly improved overall survival (86, 90). The CheckMate 214 study highlighted the fact that nivolumab combined with ipilimumab achieved higher objective response rates and longer overall survival compared to sunitinib in patients with untreated metastatic ccRCC. However, these effects are still compromised by the suppressive stromal environment, which attenuates immune infiltration and facilitates adaptive resistance (91). To address this challenge, combination regimens integrating CAF-targeted agents with ICIs are being actively explored. TGF-β inhibitors, such as Galunisertib and FAK inhibitors, have been shown in preclinical models to modulate the fibrotic and immunosuppressive properties of the stroma, thereby improving immune cell infiltration and sensitizing tumors to immunotherapy. These strategies not only enhance the efficacy of ICIs but also reverse the CAF-driven immune exclusion phenotype (92). Interestingly, the depletion of CAFs is not always beneficial. Some studies suggest that total ablation may lead to compensatory immunosuppressive mechanisms, such as increased Treg infiltration. Therefore, the focus has shifted toward functional reprogramming or partial inhibition to normalize CAF behavior without eliminating their structural roles. Adoptive cell therapies such as CAR-T cells targeting CAF-specific antigens like FAP have also shown potential in reversing immune resistance in solid tumors. Although promising, these approaches require further optimization to mitigate off-target toxicities and ensure safe, durable responses in patients (93). The overall response rate, overall survival rate, and objective response rate (ORR) in the nivolumab plus ipilimumab group were better. Analysis of patients after recovery showed that compared with Sunitinib, the combination regimen of nivolumab could significantly improve patients’ health status (94, 95). Moreover, the addition of CAF-targeting agents has the potential to modulate other immune checkpoints beyond PD-1 and CTLA-4, enhancing the breadth and depth of immune activation. Combination strategies are being tested in ongoing clinical trials and represent a promising path to maximize the benefit of immunotherapy in CAF-rich tumors (91, 96). Together, these approaches provide a foundation for rational combination therapies that address not only tumor-intrinsic immune escape but also the stromal impediments orchestrated by CAFs.
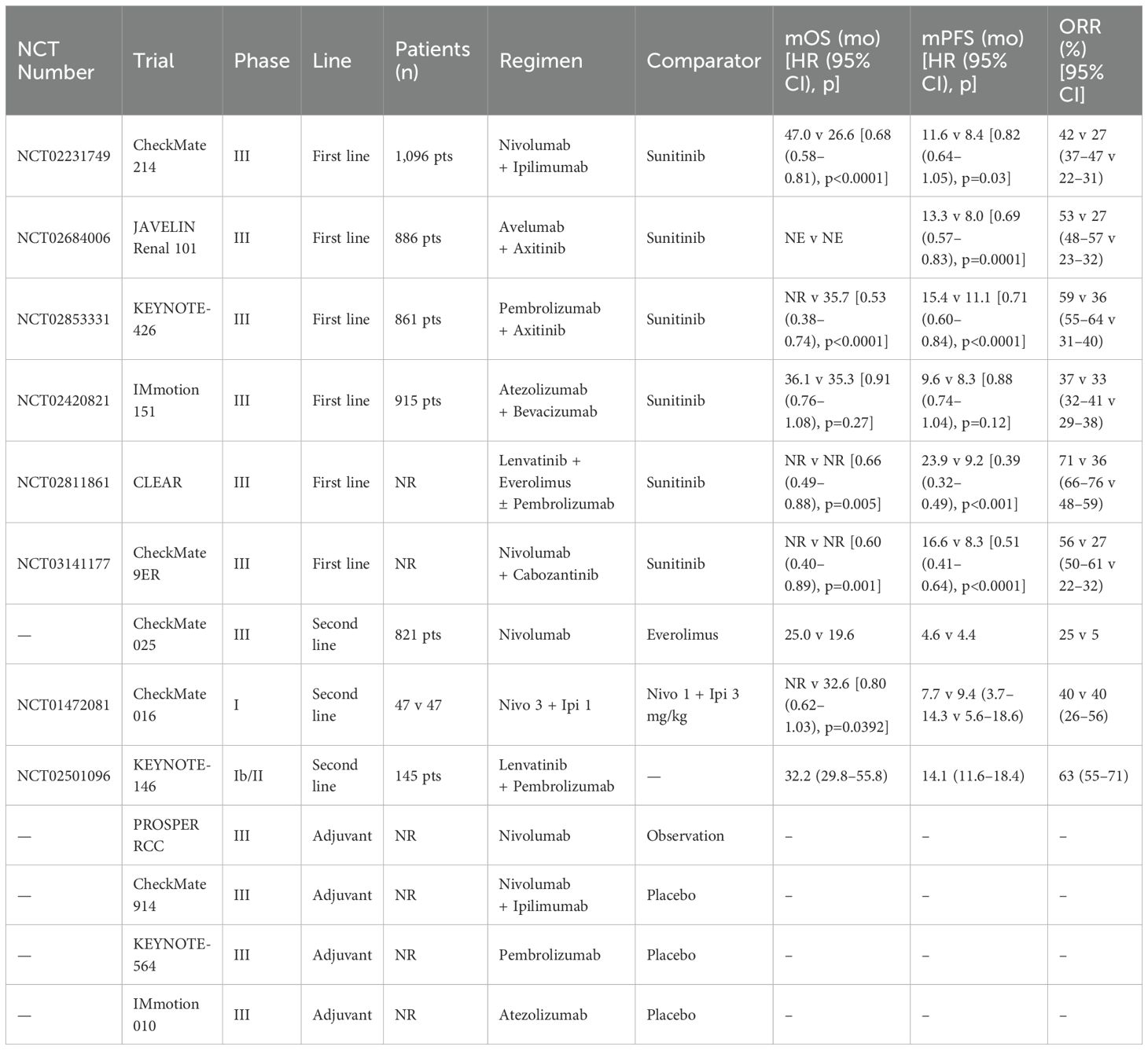
Table 2. Key clinical trials of immune checkpoint inhibitor monotherapy and combination regimens in renal cell carcinoma.
5.3 Integrative approaches combining anti-VEGF and immunotherapies
Standard-of-care treatment for metastatic ccRCC has traditionally relied on VEGF-targeted tyrosine kinase inhibitors (TKIs) (97). These agents inhibit angiogenesis, which is critical in ccRCC due to its highly vascular nature. However, monotherapies targeting VEGF pathways often lead to adaptive resistance, limited response duration, and enhanced tumor aggressiveness, partly due to immune evasion and compensatory stromal signaling. In addition to stimulating angiogenesis, VEGF not only promotes neovascularization but also contributes to immune suppression by enhancing the infiltration and function of immunosuppressive cell subsets such as Tregs and MDSCs, as well as by inhibiting dendritic cell maturation (98, 99). Consequently, combining VEGF inhibition with immunotherapies has emerged as a promising strategy to both normalize the tumor vasculature and relieve immunosuppressive constraints within the TME. Multiple clinical trials have investigated these integrative approaches. For instance, the combination of bevacizumab (anti-VEGF antibody) with interferon-alpha showed survival benefits in metastatic RCC patients. More recent data from trials evaluating ICIs plus VEGF-TKIs—such as pembrolizumab with axitinib or nivolumab with cabozantinib—demonstrated significantly improved objective response rates and progression-free survival compared to monotherapy (96, 100). From a mechanistic perspective, anti-VEGF therapy reduces vascular permeability and interstitial pressure, thereby enhancing immune cell trafficking into tumors. When combined with ICIs, this dual action fosters a reactivation of anti-tumor immunity. Importantly, VEGF blockade also indirectly modulates CAF behavior by altering paracrine signaling and ECM remodeling dynamics (101). Nevertheless, treatment optimization requires deeper insight into the temporal dynamics of VEGF-immune-CAF interactions and the identification of biomarkers predictive of response. Integrating spatial multi-omics and longitudinal immune profiling could facilitate precision stratification and enhance clinical outcomes (102). In summary, the combination therapy of anti-VEGF agents and ICIs represents a synergistic modality that addresses both vascular and immunological components of the TME. When further integrated with CAF-targeting strategies, such combinations hold the potential to overcome multidimensional resistance and transform the treatment landscape of advanced ccRCC. An integrative schematic overview of CAF-related signaling, regulation, and therapeutic implications is presented in Figure 1.
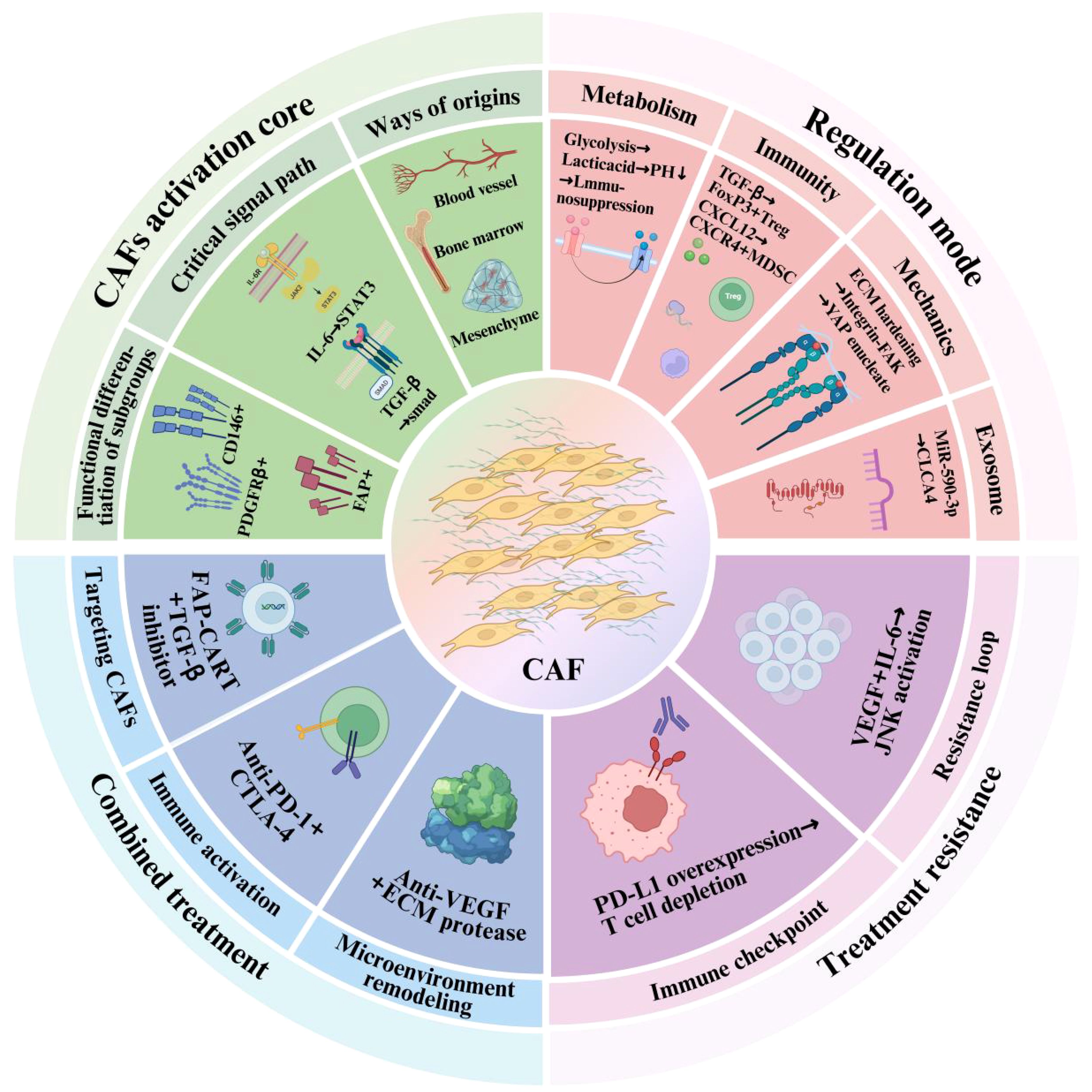
Figure 1. Functional frameworks of cancer-associated fibroblasts (CAFs) in tumor progression and therapy. The inner green sector highlights CAF activation, including their cellular origins (e.g., mesenchymal, bone marrow, endothelial), major signaling pathways (e.g., IL-6/STAT3, TGF-β), and functional subgroups (e.g., FAP+, PDGFRβ+). The pink sector shows the regulatory mechanisms by which CAFs influence the TME, including metabolic reprogramming, immunosuppression, mechanical remodeling, and exosome-mediated communication. The outer blue and purple regions demonstrate the involvement of CAFs in treatment resistance and combination therapy strategies, including immune checkpoint blockade, anti-VEGF therapy, and CAF-targeting interventions.
6 Conclusions and prospects
TME plays a pivotal role in the development, progression, and therapeutic response of ccRCC. Among these constituents within TME, CAFs have emerged as central orchestrators of immune evasion, treatment resistance, and stromal remodeling. Recent advances in single-cell sequencing and spatial transcriptomics have revealed the phenotypic and functional heterogeneity of CAFs and enabled the identification of distinct subsets with angiogenic, immunosuppressive, and ECM-remodeling properties. Therapeutic strategies targeting CAFs are advancing rapidly. Agents aimed at CAF-specific surface markers (e.g., FAP, PDGFRβ), secreted factors (e.g., TGF-β, IL-6), and key regulatory pathways (e.g., Hedgehog, STAT3, WNT) have progressed preclinical or clinical development. Functional reprogramming of CAFs, rather than complete depletion, appears to be a safer and potentially more effective approach, given their context-dependent pro- and anti-tumor functions. Furthermore, combining CAF-targeted interventions with ICIs and anti-angiogenic agents has demonstrated promising synergistic effects. These integrative strategies address not only tumor-intrinsic mechanisms but also the stromal and immune components of resistance. However, their clinical translation faces challenges from CAF heterogeneity, dynamic plasticity, off-target effects, and the lack of validated biomarkers to predict therapeutic response. To overcome these barriers, future studies should focus on the integration of single-cell and spatial multi-omics profiling to map the spatial and temporal evolution of CAF subtypes and their interactions with other TME elements. Identifying molecular signatures associated with response or resistance is crucial for guiding precision treatment. Ultimately, disrupting the vicious cycle between CAFs, the TME, and therapy resistance holds great promise for improving outcomes in patients with advanced ccRCC. As our understanding deepens, CAFs are likely to shift from therapeutic challenge to therapeutic opportunity in the evolving landscape of renal cancer treatment.
Author contributions
MW: Conceptualization, Writing – original draft, Writing – review & editing. YZ: Writing – original draft. KX: Software, Writing – original draft. CL: Software, Visualization, Writing – review & editing. HZ: Writing – original draft. YW: Writing – review & editing. KZ: Conceptualization, Visualization, Writing – review & editing. SW: Conceptualization, Formal Analysis, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wolf MM, Kimryn Rathmell W, and Beckermann KE. Modeling clear cell renal cell carcinoma and therapeutic implications. Oncogene. (2020) 39:3413–26. doi: 10.1038/s41388-020-1234-3
2. Wang Y, Suarez ER, Kastrunes G, de Campos NSP, Abbas R, Pivetta RS, et al. Evolution of cell therapy for renal cell carcinoma. Mol Cancer. (2024) 23:8. doi: 10.1186/s12943-023-01911-x
3. Monjaras-Avila CU, Lorenzo-Leal AC, Luque-Badillo AC, D’Costa N, Chavez-Muñoz C, and Bach H. The tumor immune microenvironment in clear cell renal cell carcinoma. Int J Mol Sci. (2023) 24:7946. doi: 10.3390/ijms24097946
4. Arina A, Idel C, Hyjek EM, Alegre ML, Wang Y, Bindokas VP, et al. Tumor-associated fibroblasts predominantly come from local and not circulating precursors. Proc Natl Acad Sci U S A. (2016) 113:7551–6. doi: 10.1073/pnas.1600363113
5. Ridge SM, Sullivan FJ, and Glynn SA. Mesenchymal stem cells: key players in cancer progression. Mol Cancer. (2017) 16:31. doi: 10.1186/s12943-017-0597-8
6. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. (2020) 10:1330–51. doi: 10.1158/2159-8290.Cd-19-1384
7. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
8. Peng YL, Xiong LB, Zhou ZH, Ning K, Li Z, Wu ZS, et al. Single-cell transcriptomics reveals a low cd8(+) T cell infiltrating state mediated by fibroblasts in recurrent renal cell carcinoma. J immunotherapy Cancer. (2022) 10:e004206. doi: 10.1136/jitc-2021-004206
9. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
10. De Francesco EM, Lappano R, Santolla MF, Marsico S, Caruso A, and Maggiolini M. Hif-1α/gper signaling mediates the expression of vegf induced by hypoxia in breast cancer associated fibroblasts (Cafs). Breast Cancer Res. (2013) 15:R64. doi: 10.1186/bcr3458
11. Apte RS, Chen DS, and Ferrara N. Vegf in signaling and disease: beyond discovery and development. Cell. (2019) 176:1248–64. doi: 10.1016/j.cell.2019.01.021
12. Wan X, Guan S, Hou Y, Qin Y, Zeng H, Yang L, et al. Fosl2 promotes vegf-independent angiogenesis by transcriptionnally activating wnt5a in breast cancer-associated fibroblasts. Theranostics. (2021) 11:4975–91. doi: 10.7150/thno.55074
13. De Palma M, Biziato D, and Petrova TV. Microenvironmental regulation of tumour angiogenesis. Nat Rev Cancer. (2017) 17:457–74. doi: 10.1038/nrc.2017.51
14. Chen Y, McAndrews KM, and Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. (2021) 18:792–804. doi: 10.1038/s41571-021-00546-5
15. Chen X and Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
16. Zhang H, Yue X, Chen Z, Liu C, Wu W, Zhang N, et al. Define cancer-associated fibroblasts (Cafs) in the tumor microenvironment: new opportunities in cancer immunotherapy and advances in clinical trials. Mol Cancer. (2023) 22:159. doi: 10.1186/s12943-023-01860-5
17. Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, et al. Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol. (2019) 12:86. doi: 10.1186/s13045-019-0770-1
18. Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, and Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. (2002) 3:349–63. doi: 10.1038/nrm809
19. Caligiuri G and Tuveson DA. Activated fibroblasts in cancer: perspectives and challenges. Cancer Cell. (2023) 41:434–49. doi: 10.1016/j.ccell.2023.02.015
20. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. (2016) 16:582–98. doi: 10.1038/nrc.2016.73
21. Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. (2013) 504:277–81. doi: 10.1038/nature12783
22. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
23. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. Il1-induced jak/stat signaling is antagonized by tgfβ to shape caf heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. (2019) 9:282–301. doi: 10.1158/2159-8290.Cd-18-0710
24. Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med. (2010) 2:211–30. doi: 10.1002/emmm.201000073
25. Martinez-Outschoorn UE, Sotgia F, and Lisanti MP. Metabolic asymmetry in cancer: A “Balancing act” That promotes tumor growth. Cancer Cell. (2014) 26:5–7. doi: 10.1016/j.ccr.2014.06.021
26. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
27. Ziani L, Chouaib S, and Thiery J. Alteration of the antitumor immune response by cancer-associated fibroblasts. Front Immunol. (2018) 9:414. doi: 10.3389/fimmu.2018.00414
28. Kim R, Emi M, and Tanabe K. Cancer immunosuppression and autoimmune disease: beyond immunosuppressive networks for tumour immunity. Immunology. (2006) 119:254–64. doi: 10.1111/j.1365-2567.2006.02430.x
29. Sun Q, Zhang B, Hu Q, Qin Y, Xu W, Liu W, et al. The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Theranostics. (2018) 8:5072–87. doi: 10.7150/thno.26546
30. Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H, and Takeyama H. Cancer-associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel). (2015) 7:2443–58. doi: 10.3390/cancers7040902
31. Harper J and Sainson RC. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol. (2014) 25:69–77. doi: 10.1016/j.semcancer.2013.12.005
32. Stojanovic A and Cerwenka A. Natural killer cells and solid tumors. J Innate Immun. (2011) 3:355–64. doi: 10.1159/000325465
33. Habif G, Crinier A, André P, Vivier E, and Narni-Mancinelli E. Targeting natural killer cells in solid tumors. Cell Mol Immunol. (2019) 16:415–22. doi: 10.1038/s41423-019-0224-2
34. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. (2021) 20:131. doi: 10.1186/s12943-021-01428-1
35. Lakins MA, Ghorani E, Munir H, Martins CP, and Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of cd8 (+) T cells to protect tumour cells. Nat Commun. (2018) 9:948. doi: 10.1038/s41467-018-03347-0
36. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. (2016) 22:851–60. doi: 10.1038/nm.4123
37. Serrels A, Lund T, Serrels B, Byron A, McPherson RC, von Kriegsheim A, et al. Nuclear fak controls chemokine transcription, tregs, and evasion of anti-tumor immunity. Cell. (2015) 163:160–73. doi: 10.1016/j.cell.2015.09.001
38. Bae YH, Mui KL, Hsu BY, Liu SL, Cretu A, Razinia Z, et al. A fak-cas-rac-lamellipodin signaling module transduces extracellular matrix stiffness into mechanosensitive cell cycling. Sci Signaling. (2014) 7:ra57. doi: 10.1126/scisignal.2004838
39. Hu B, Wang Z, Zeng H, Qi Y, Chen Y, Wang T, et al. Blockade of dc-sign(+) tumor-associated macrophages reactivates antitumor immunity and improves immunotherapy in muscle-invasive bladder cancer. Cancer Res. (2020) 80:1707–19. doi: 10.1158/0008-5472.Can-19-2254
40. Yugawa K, Itoh S, Yoshizumi T, Iseda N, Tomiyama T, Morinaga A, et al. Cmtm6 stabilizes pd-L1 expression and is a new prognostic impact factor in hepatocellular carcinoma. Hepatol Commun. (2021) 5:334–48. doi: 10.1002/hep4.1643
41. Tan B, Shi X, Zhang J, Qin J, Zhang N, Ren H, et al. Inhibition of rspo-lgr4 facilitates checkpoint blockade therapy by switching macrophage polarization. Cancer Res. (2018) 78:4929–42. doi: 10.1158/0008-5472.Can-18-0152
42. Herrera M, Herrera A, Domínguez G, Silva J, García V, García JM, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. (2013) 104:437–44. doi: 10.1111/cas.12096
43. Fujii N, Shomori K, Shiomi T, Nakabayashi M, Takeda C, Ryoke K, et al. Cancer-associated fibroblasts and cd163-positive macrophages in oral squamous cell carcinoma: their clinicopathological and prognostic significance. J Pathol Med. (2012) 41:444–51. doi: 10.1111/j.1600-0714.2012.01127.x
44. Gok Yavuz B, Gunaydin G, Gedik ME, Kosemehmetoglu K, Karakoc D, Ozgur F, et al. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive pd-1(+) tams. Sci Rep. (2019) 9:3172. doi: 10.1038/s41598-019-39553-z
45. Kumar BV, Connors TJ, and Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
46. Shimizu J, Yamazaki S, and Sakaguchi S. Induction of tumor immunity by removing cd25+Cd4+ T cells: A common basis between tumor immunity and autoimmunity. J Immunol. (1999) 163:5211–8. doi: 10.4049/jimmunol.163.10.5211
47. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. (2018) 33:463–79.e10. doi: 10.1016/j.ccell.2018.01.011
48. Tanaka A and Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. (2017) 27:109–18. doi: 10.1038/cr.2016.151
49. Kumar V, Donthireddy L, Marvel D, Condamine T, Wang F, Lavilla-Alonso S, et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of csf1 receptor blockade by inducing pmn-mdsc infiltration of tumors. Cancer Cell. (2017) 32:654–68.e5. doi: 10.1016/j.ccell.2017.10.005
50. Kinoshita T, Ishii G, Hiraoka N, Hirayama S, Yamauchi C, Aokage K, et al. Forkhead box P3 regulatory T cells coexisting with cancer associated fibroblasts are correlated with a poor outcome in lung adenocarcinoma. Cancer Sci. (2013) 104:409–15. doi: 10.1111/cas.12099
51. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. Tgfβ Drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
52. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral cd4+Cd25- naive T cells to cd4+Cd25+ Regulatory T cells by tgf-beta induction of transcription factor foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
53. Zhao X, Ding L, Lu Z, Huang X, Jing Y, Yang Y, et al. Diminished cd68(+) cancer-associated fibroblast subset induces regulatory T-cell (Treg) infiltration and predicts poor prognosis of oral squamous cell carcinoma patients. Am J Pathol. (2020) 190:886–99. doi: 10.1016/j.ajpath.2019.12.007
54. You W, Shang B, Sun J, Liu X, Su L, and Jiang S. Mechanistic insight of predictive biomarkers for antitumor pd−1/pd−L1 blockade: A paradigm shift towards immunome evaluation (Review). Oncol Rep. (2020) 44:424–37. doi: 10.3892/or.2020.7643
55. Bonnans C, Chou J, and Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. (2014) 15:786–801. doi: 10.1038/nrm3904
56. Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, et al. The fibronectin domain ed-a is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol. (1998) 142:873–81. doi: 10.1083/jcb.142.3.873
57. Klingberg F, Chau G, Walraven M, Boo S, Koehler A, Chow ML, et al. The fibronectin ed-a domain enhances recruitment of latent tgf-B-binding protein-1 to the fibroblast matrix. J Cell Sci. (2018) 131:jcs201293. doi: 10.1242/jcs.201293
58. Kollmannsberger P, Bidan CM, Dunlop JWC, Fratzl P, and Vogel V. Tensile forces drive a reversible fibroblast-to-myofibroblast transition during tissue growth in engineered clefts. Sci Adv. (2018) 4:eaao4881. doi: 10.1126/sciadv.aao4881
59. Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, and Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol. (2006) 172:259–68. doi: 10.1083/jcb.200506179
60. Tomasek JJ, Vaughan MB, Kropp BP, Gabbiani G, Martin MD, Haaksma CJ, et al. Contraction of myofibroblasts in granulation tissue is dependent on rho/rho kinase/myosin light chain phosphatase activity. Wound Repair Regener. (2006) 14:313–20. doi: 10.1111/j.1743-6109.2006.00126.x
61. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through yap and taz drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L344–57. doi: 10.1152/ajplung.00300.2014
62. Piersma B, de Rond S, Werker PM, Boo S, Hinz B, van Beuge MM, et al. Yap1 is a driver of myofibroblast differentiation in normal and diseased fibroblasts. Am J Pathol. (2015) 185:3326–37. doi: 10.1016/j.ajpath.2015.08.011
63. Foster CT, Gualdrini F, and Treisman R. Mutual dependence of the mrtf-srf and yap-tead pathways in cancer-associated fibroblasts is indirect and mediated by cytoskeletal dynamics. Genes Dev. (2017) 31:2361–75. doi: 10.1101/gad.304501.117
64. Darby I, Skalli O, and Gabbiani G. Alpha-smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing. Lab Invest. (1990) 63:21–9.
65. Desmoulière A, Redard M, Darby I, and Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. (1995) 146:56–66.
66. Yang D, Liu J, Qian H, and Zhuang Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp Mol Med. (2023) 55:1322–32. doi: 10.1038/s12276-023-01013-0
67. Díaz-Montero CM, Rini BI, and Finke JH. The immunology of renal cell carcinoma. Nat Rev Nephrol. (2020) 16:721–35. doi: 10.1038/s41581-020-0316-3
68. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J clinicians. (2021) 71:209–49. doi: 10.3322/caac.21660
69. Karin N. The multiple faces of cxcl12 (Sdf-1alpha) in the regulation of immunity during health and disease. J Leukoc Biol. (2010) 88:463–73. doi: 10.1189/jlb.0909602
70. Kim HL, Seligson D, Liu X, Janzen N, Bui MH, Yu H, et al. Using protein expressions to predict survival in clear cell renal carcinoma. Clin Cancer research: an Off J Am Assoc Cancer Res. (2004) 10:5464–71. doi: 10.1158/1078-0432.Ccr-04-0488
71. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. (2012) 12:252–64. doi: 10.1038/nrc3239
72. Li B, Chan HL, and Chen P. Immune checkpoint inhibitors: basics and challenges. Curr medicinal Chem. (2019) 26:3009–25. doi: 10.2174/0929867324666170804143706
73. Atkins MB and Tannir NM. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treat Rev. (2018) 70:127–37. doi: 10.1016/j.ctrv.2018.07.009
74. Jarosz-Biej M, Smolarczyk R, Cichoń T, and Kułach N. Tumor microenvironment as a “Game changer” in cancer radiotherapy. Int J Mol Sci. (2019) 20:3212. doi: 10.3390/ijms20133212
75. Barker HE, Paget JT, Khan AA, and Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. (2015) 15:409–25. doi: 10.1038/nrc3958
76. Vitale I, Manic G, Coussens LM, Kroemer G, and Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. (2019) 30:36–50. doi: 10.1016/j.cmet.2019.06.001
77. Zhou Y, Wang Y, Chen H, Xu Y, Luo Y, Deng Y, et al. Immuno-oncology: are tam receptors in glioblastoma friends or foes? Cell Commun Signal. (2021) 19:11. doi: 10.1186/s12964-020-00694-8
78. Nishishita R, Morohashi S, Seino H, Wu Y, Yoshizawa T, Haga T, et al. Expression of cancer-associated fibroblast markers in advanced colorectal cancer. Oncol Lett. (2018) 15:6195–202. doi: 10.3892/ol.2018.8097
79. Yang B, Cao L, Liu J, Xu Y, Milne G, Chan W, et al. Low expression of chloride channel accessory 1 predicts a poor prognosis in colorectal cancer. Cancer. (2015) 121:1570–80. doi: 10.1002/cncr.29235
80. Chen X, Liu Y, Zhang Q, Liu B, Cheng Y, Zhang Y, et al. Exosomal mir-590-3p derived from cancer-associated fibroblasts confers radioresistance in colorectal cancer. Mol Ther Nucleic Acids. (2021) 24:113–26. doi: 10.1016/j.omtn.2020.11.003
81. Kilvaer TK, Khanehkenari MR, Hellevik T, Al-Saad S, Paulsen EE, Bremnes RM, et al. Cancer associated fibroblasts in stage I-iiia nsclc: prognostic impact and their correlations with tumor molecular markers. PloS One. (2015) 10:e0134965. doi: 10.1371/journal.pone.0134965
82. de Araujo Farias V, O’Valle F, Serrano-Saenz S, Anderson P, Andrés E, López-Peñalver J, et al. Exosomes derived from mesenchymal stem cells enhance radiotherapy-induced cell death in tumor and metastatic tumor foci. Mol Cancer. (2018) 17:122. doi: 10.1186/s12943-018-0867-0
83. Lee J, Fassnacht M, Nair S, Boczkowski D, and Gilboa E. Tumor immunotherapy targeting fibroblast activation protein, a product expressed in tumor-associated fibroblasts. Cancer Res. (2005) 65:11156–63. doi: 10.1158/0008-5472.Can-05-2805
84. Loeffler M, Krüger JA, Niethammer AG, and Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. (2006) 116:1955–62. doi: 10.1172/jci26532
85. Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, et al. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. (2010) 101:2325–32. doi: 10.1111/j.1349-7006.2010.01695.x
86. Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal transduction targeted Ther. (2021) 6:218. doi: 10.1038/s41392-021-00641-0
87. Buonerba C, Dolce P, Iaccarino S, Scafuri L, Verde A, Costabile F, et al. Outcomes associated with first-line anti-pd-1/pd-L1 agents vs. Sunitinib in patients with sarcomatoid renal cell carcinoma: A systematic review and meta-analysis. Cancers (Basel). (2020) 12:408. doi: 10.3390/cancers12020408
88. Powles T. Treatment choices for front-line metastatic clear cell renal cancer. Eur Urol. (2020) 77:454–6. doi: 10.1016/j.eururo.2020.01.011
89. Rustum YM, Reis R, and Rustum TM. Druggable biomarkers altered in clear cell renal cell carcinoma: strategy for the development of mechanism-based combination therapy. Int J Mol Sci. (2023) 24:902. doi: 10.3390/ijms24020902
90. Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. New Engl J Med. (2011) 364:2517–26. doi: 10.1056/NEJMoa1104621
91. Yang JC, Hughes M, Kammula U, Royal R, Sherry RM, Topalian SL, et al. Ipilimumab (Anti-ctla4 antibody) causes regression of metastatic renal cell cancer associated with enteritis and hypophysitis. J Immunother. (2007) 30:825–30. doi: 10.1097/CJI.0b013e318156e47e
92. Motzer RJ, Tannir NM, McDermott DF, Arén Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. New Engl J Med. (2018) 378:1277–90. doi: 10.1056/NEJMoa1712126
93. Hammers HJ, Plimack ER, Infante JR, Rini BI, McDermott DF, Lewis LD, et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the checkmate 016 study. J Clin Oncol. (2017) 35:3851–8. doi: 10.1200/jco.2016.72.1985
94. Cella D, Grünwald V, Escudier B, Hammers HJ, George S, Nathan P, et al. Patient-reported outcomes of patients with advanced renal cell carcinoma treated with nivolumab plus ipilimumab versus sunitinib (Checkmate 214): A randomised, phase 3 trial. Lancet Oncol. (2019) 20:297–310. doi: 10.1016/s1470-2045(18)30778-2
95. Doroshow DB, Bhalla S, Beasley MB, Sholl LM, Kerr KM, Gnjatic S, et al. Pd-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat Rev Clin Oncol. (2021) 18:345–62. doi: 10.1038/s41571-021-00473-5
96. Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Archer L, et al. Phase iii trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of calgb 90206. J Clin Oncol. (2010) 28:2137–43. doi: 10.1200/jco.2009.26.5561
97. Takyar S, Diaz J, Sehgal M, Sapunar F, and Pandha H. First-line therapy for treatment-naive patients with advanced/metastatic renal cell carcinoma: A systematic review of published randomized controlled trials. Anticancer Drugs. (2016) 27:383–97. doi: 10.1097/cad.0000000000000335
98. Almand B, Clark JI, Nikitina E, van Beynen J, English NR, Knight SC, et al. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J Immunol. (2001) 166:678–89. doi: 10.4049/jimmunol.166.1.678
99. Yang J, Yan J, and Liu B. Targeting vegf/vegfr to modulate antitumor immunity. Front Immunol. (2018) 9:978. doi: 10.3389/fimmu.2018.00978
100. Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase iii trial. Lancet (London England). (2007) 370:2103–11. doi: 10.1016/s0140-6736(07)61904-7
101. Considine B and Hurwitz ME. Current status and future directions of immunotherapy in renal cell carcinoma. Curr Oncol Rep. (2019) 21:34. doi: 10.1007/s11912-019-0779-1
Keywords: clear cell renal cell carcinoma (ccRCC), cancer-associated fibroblasts (CAFs), tumor microenvironment (TME), immune evasion, therapy resistance, combination immunotherapy
Citation: Wang M, Zhao Y, Xu K, Liu C, Zhong H, Wu Y, Zhang K and Wei S (2025) Cancer-associated fibroblasts in clear cell renal cell carcinoma: functional heterogeneity, tumor microenvironment crosstalk, and therapeutic opportunities. Front. Immunol. 16:1617968. doi: 10.3389/fimmu.2025.1617968
Received: 25 April 2025; Accepted: 21 May 2025;
Published: 04 June 2025.
Edited by:
Qiong Lu, Central South University, ChinaReviewed by:
Muhammad Shamsul Alam, National Cancer Institute (NIH), United StatesCopyright © 2025 Wang, Zhao, Xu, Liu, Zhong, Wu, Zhang and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ke Zhang, emhhbmdrZW1ud2tAZm94bWFpbC5jb20=; Shanzhai Wei, YXpoYWk3NjcxQDE2My5jb20=
†These authors have contributed equally to this work