 Ariel Laub
Ariel Laub Nathalia Rodrigues de Almeida
Nathalia Rodrigues de Almeida Shouxiong Huang
Shouxiong Huang- 1Host-Pathogen Interactions Program, Texas Biomedical Research Program, San Antonio, TX, United States
- 2Department of Molecular Microbiology and Immunology, University of Texas at San Antonio, San Antonio, TX, United States
Unlike conventional T cells that detect peptide antigens loaded to major histocompatibility complex (MHC) molecules, unconventional T cells respond to non-peptidic metabolite antigens presented by MHC class I-like proteins, such as CD1 and MHC-related protein 1 (MR1). Semi-invariant mucosal-associated invariant T (MAIT) cells, γδ T cells, and invariant natural killer T (iNKT) cells, together with other CD1- or MR1-restricted T cell subsets expressing diverse T cell receptors (TCR), elicit an innate-like response independent of diverse MHC genetics. In contrast to an overall enhanced response to bacterial-derived riboflavin precursor metabolites in infections, MAIT cells often exhibit an immunosuppressive or exhausted phenotype in glioblastoma, lung cancer, colorectal cancer, and various hematological malignancies. Whereas some tumor cells can activate MAIT cells, the structures and functions of tumor-derived MR1 ligands remain largely unknown. Novel discoveries of mammalian-derived agonists and antagonists binding to MR1 protein are our knowledge of MR1 ligand structures and functions from MAIT cell activation in healthy conditions to anti-cancer immunity. Recent findings reveal that nucleoside and nucleobase analogs, as self-metabolites to activate MR1-restricted T cells, are regulated in the tumor microenvironment. Likewise, iNKT cells exhibit a dynamic role in cancer, capable of both protumor and antitumor immunity. Similarly, γδ T cells have also demonstrated both protective and tumor-promoting roles, via recognizing stress-induced protein and metabolite ligands. This review further depicts the distinct kinetics of responses, highlighting a rapid activation of unconventional T cells in solid versus hematological cancers. Emerging therapeutic strategies, including antigen-loaded MR1 and CD1, adoptive T cell transfer, chimeric antigen receptor-T (CAR-T) cells, T cell receptor-T (TCR-T) cells, and combination treatments with immune checkpoint inhibitors, yet remain challenging, hold promise in overcoming tumor-induced immunosuppression and genetic restriction of conventional T cell therapies. By addressing critical gaps, such as novel structures and functions of cancer metabolite antigens, unconventional T cells offer unique advantages in anti-cancer immunotherapy.
Introduction
Antitumor T cell immunity against malignancy has been generally focused on studying conventional T cell activation, which relies on recognizing tumor peptide antigens presented by polymorphic major histocompatibility complex (MHC) or human leukocyte antigen (HLA) class I and II molecules in various human populations (1, 2). Conventional cytotoxic CD8+ T lymphocytes (CTLs) recognize peptide antigens presented by MHC class I, and CD4+ T cells engage with peptide-MHC class II complexes (3), driving crucial anticancer immune responses and framing cancer immunotherapies (4, 5). These adaptive T cells, particularly CD8+ CTLs, mediate tumor cell killing through antigen-specific recognition of tumor-associated antigens (TAAs) or neoantigens, eliciting potent cytotoxic molecular mediators capable of direct tumor lysis. However, tumor cells often evade this response by downregulating MHC class I expression or inducing an immunosuppressive tumor microenvironment (TME), limiting the effectiveness of conventional T cells and leading to immune escape for cancer progression (5). In contrast, unconventional T cells rely on recognizing polar or lipid metabolite antigens presented by non-classical MHC class I or MHC class Ib molecules with limited polymorphisms. These include lipids by the Cluster of Differentiation 1 (CD1) proteins for CD1-restricted T cells and polar metabolites by MHC-related protein 1 (MR1) for MR1-restricted T cells (6–13). Notably, the non-classical antigen presentation mechanisms allow unconventional T cells to bypass MHC restriction, enabling rapid and individual-unrestricted immune activation that does not rely on genetically diverse classical HLA proteins in various human populations (6, 12, 14).
Unconventional T cells are generally first classified based on antigen presentation mechanisms for activation. CD1-restricted T cells can recognize bacterial and mammalian lipids to exert various immune regulation and effector responses (10, 15). Particularly, CD1d-restricted invariant natural killer T (iNKT) cells are well-characterized to robustly produce cytokines that enhance both pro-inflammatory and regulatory immune pathways (7). MR1-restricted T cells recognize polar metabolites with bacterial sources mostly from vitamin B biosynthetic pathways, particularly microbial riboflavin precursors (16, 17), and with currently known mammalian ligands mostly from nucleoside metabolism (18–20). γδ T cells are known to detect phosphoantigens through members of the butyrophilin (BTN) family, which form a receptor complex to engage γδ TCR for activation. The γδ T cells also detect CD1-presented lipids and MR1-presented polar metabolites. The capacity to sense cellular stress and metabolite compounds enables unconventional T cells to play key roles in both microbial defense and tumor surveillance (21, 22). Further classification of CD1- or MR1-restricted T cell subsets usually relies on the invariant or diverse TCR sequences. CD1d-restricted T cells are typically divided into invariant NKT cells (iNKT or type I NKT cells) expressing an invariant TCRα chain (human TRAV10 or mouse TRAV11) and diverse NKT cells (dNKT or type II NKT cells) expressing variable TCRα chains (15, 23). Similar to NKT cells, MR1-restricted T cells (MR1T) can be divided into mucosal-associated invariant T (MAIT) cells expressing invariant TCRα chains (human TRAV1-2 or mouse TRAV1) (24) and diverse MR1-restricted T cells expressing diverse TCRs (diverse MR1T, dMR1T, TRAV1-2- MR1T, or Vα7.2- MR1T in humans). The γδ T cells are typically classified into three primary subsets based on their δ chain usage: Vδ1+, Vδ2+, and Vδ3+ (22, 25). This semi-invariant TCR expression and interaction with metabolite antigens presented by limited polymorphic MHC class I-like proteins overall define the innate-like nature of unconventional T cell responses.
In cancer immunity, unconventional T cells generally play dual roles, either promoting tumor clearance through cytotoxicity and cytokine production or, conversely, contributing to tumor progression when exposed to chronic immunosuppressive signaling in the TME and leading to undesirable exhausted phenotypes (26). This review will focus on MR1-restricted MAIT cells and diverse MR1T cells in cancer immunity, comparing their roles to other unconventional T cell subsets, including iNKT cells and γδ T cells. By analyzing their dual nature of immune responses in different tumor settings, we center on the structures and functions of metabolite antigens for unconventional T cell activation in the cancer context and discuss their potential as targets for cancer immunotherapy. Additionally, we highlight emerging therapeutic strategies, primarily by activating semi-invariant and diverse T cells through MR1- and CD1-mediated antigen presentation, harnessing these T cells for adoptive T cell transfer, and combining multiple immune therapies such as checkpoint blockade. Given their ability to bypass MHC restrictions and their innate-like rapid responses, MAIT cells and other unconventional T cells are expected to provide promising opportunities for improving immune-based cancer treatments.
Self-metabolite antigen presentation
Binary and tertiary structures of antigen-presentation complex containing an MHC class I-like protein and a self-metabolite antigen with or without a TCR provide critical knowledge on the topology, interacting sites, and affinity binding of metabolite antigens with proteins. Different from peptide antigens that generally use multiple hydrogen bonds to interact with classical MHC class I proteins, metabolite antigens bind a smaller number of amino acid residues via polar and hydrophobic interactions, and even covalent bonds to interact with CD1 and MR1 proteins (Figure 1A) (27–30). Specifically, polar metabolite antigens such as nucleoside derivatives or riboflavin intermediate metabolites are small in size and reasonably interact with a small number of MR1 residues. Some mammalian cell-derived polar metabolites, such as the carbonyl adduct of adenine (18) and 5-formyl-deoxyuridine (30), form a Schiff’s base with lysine 43 (K43) of human MR1, similarly to the bacterial riboflavin precursor metabolites 5-[2-oxopropylideneamino]-6-D-ribitylaminouracil (5-OP-RU) (16). Lipid metabolites generally consist of hydrophilic head groups to form polar interactions with CD1 proteins and TCR chains, while hydrophobic fatty acyl or sphingosine chains interact with the ligand-binding clefts of CD1 proteins through hydrophobic interactions (Figure 1A) (29). Binding affinity of mammalian cell metabolite antigens to MR1 or CD1 proteins is detected by Kd, a dissociation constant reflecting the concentration of ligand binding to 50% of receptor molecules, generally at a micromolar range for self-metabolite antigens (Figure 1A), different from an overall nanomolar range of Kd for non-self-metabolite antigens such as human CD1d binding to marine sponge-derived α-galactosylceramide (α-GalCer) (31) and human MR1 binding to a bacterial metabolite derivative 5-OP-RU (32). By examining the polar interactions between metabolite ligand and invariant TCR, it appears that invariant TCRα chains form major contacts, while TCRβ chains point to different positions away from the center of metabolite antigens, unlike conventional TCRα and TCRβ chains generally center on α1 and α2 domains of the HLA-A2 protein (Figures 1A, B) and other classical MHC class I proteins (33). A comparison of these tertiary antigen-presentation complexes depicts an unconventional interacting pattern utilizing less polar interactions and uncentered engagement with TCR at a low affinity for unconventional T cell activation in cancers. Various identified mammalian-derived metabolite compounds for unconventional T cell activation are detailed in the following sections.
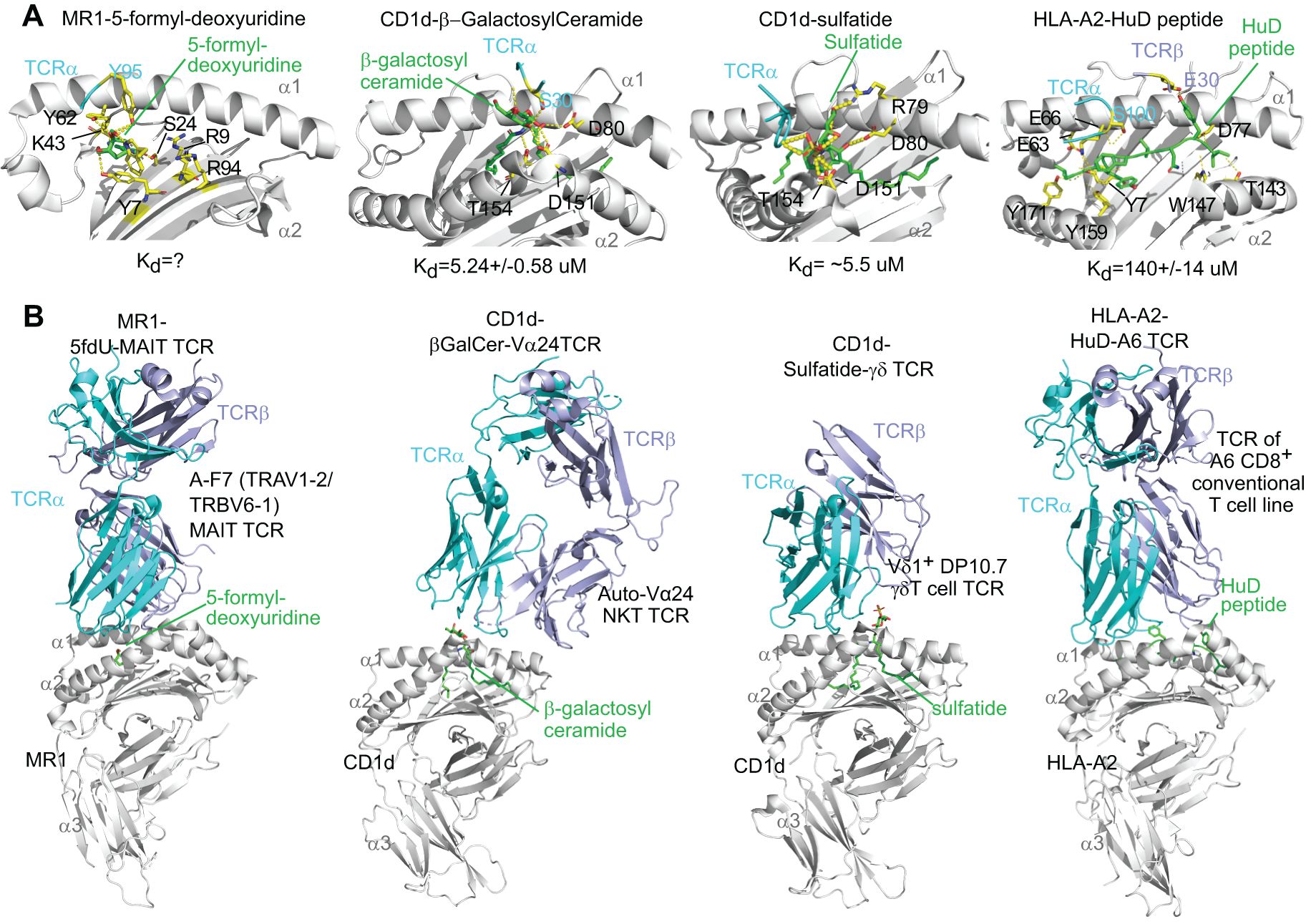
Figure 1. Re-analyses of tertiary crystal structures of MR1 or CD1, mammalian metabolite antigens, and invariant T cell receptors, in parallel with an HLA-A2-auto peptide-TCR complex. The reported crystal structures for human MR1, 5-formyl-deoxyuridine, and A-F7 MAIT cell TCR (9EK7 from the PDB database), human CD1d, β-galactosylceramide, and autoreactive Vα24 TCR (3SDX), human CD1d, sulfatide, and DP10.7 γδ TCR (4MNG), and HLA-A2, glioblastoma peptide HuD, and A6 conventional CD8+ T cell TCR (3PWP) were compared via Pymol for metabolite antigen interaction with MR1, CD1, and TCR chains. (A) Metabolite antigen binding to MR1, CD1, and TCR chains is shown through polar interactions (yellow dots), particularly hydrogen bonding, but hydrophobic interaction is not shown. Interacting residues were annotated. α1, α2, and α3 label α1, α2, and α3 domains with α2 domains partially removed to show protein-ligand interactions. Kd, the dissociation constant, represents the concentration of ligand binding to 50% of receptor molecules. (B) Metabolite antigen binding to MHC class I-like proteins differentially shapes the orientation of TCRα and TCRβ chains.
Polar metabolites as MR1 ligands
MR1 is an MHC class I-like antigen-presentation molecule for presenting polar metabolite antigens to MR1-restricted T cells (6, 34–36). Although surface expression is generally low, MR1 expresses broadly across tissues at RNA and protein levels (11, 37), and activates MAIT cells upon bacterial (38) or cancer metabolite stimulation (20, 39). Similar to classical MHC molecules, MR1 remains largely retained intracellular until it binds a ligand, at which point it is transported to the cell surface for antigen presentation (11, 35–37). This ligand-regulated expression mechanism allows MR1 to serve as a metabolic checkpoint, particularly in recognizing small molecule metabolites derived from microbial riboflavin (16) or mammalian metabolite biosynthesis (18, 20) (Figure 2). It was well known that the semi-invariant conserved MAIT TCR predominantly utilizes TRAV1-2 (Vα7.2 in humans) conjuncted with TRAJ33 (Jα33) (40), or with TRAJ12 or TRAJ20 (41) for TCRα chains and a limitedly diverse TCRβ chains (TRBV6 or Vβ13.2~13.5, Vβ6.5~6.8, TRBV20 or Vβ2.1, 2.3) in humans (40). In mice, MAIT cells express Vα19-Jα33 TCRα, mainly paired with Vβ8 and Vβ6 segments (11, 12, 34, 35). Diverse MR1T cells, which do not express TRAV1-2 segment (TRAV1-2-) but respond to various metabolites in infection and cancer, were also reported recently (Figure 2) (13, 16–19, 32, 42–44).
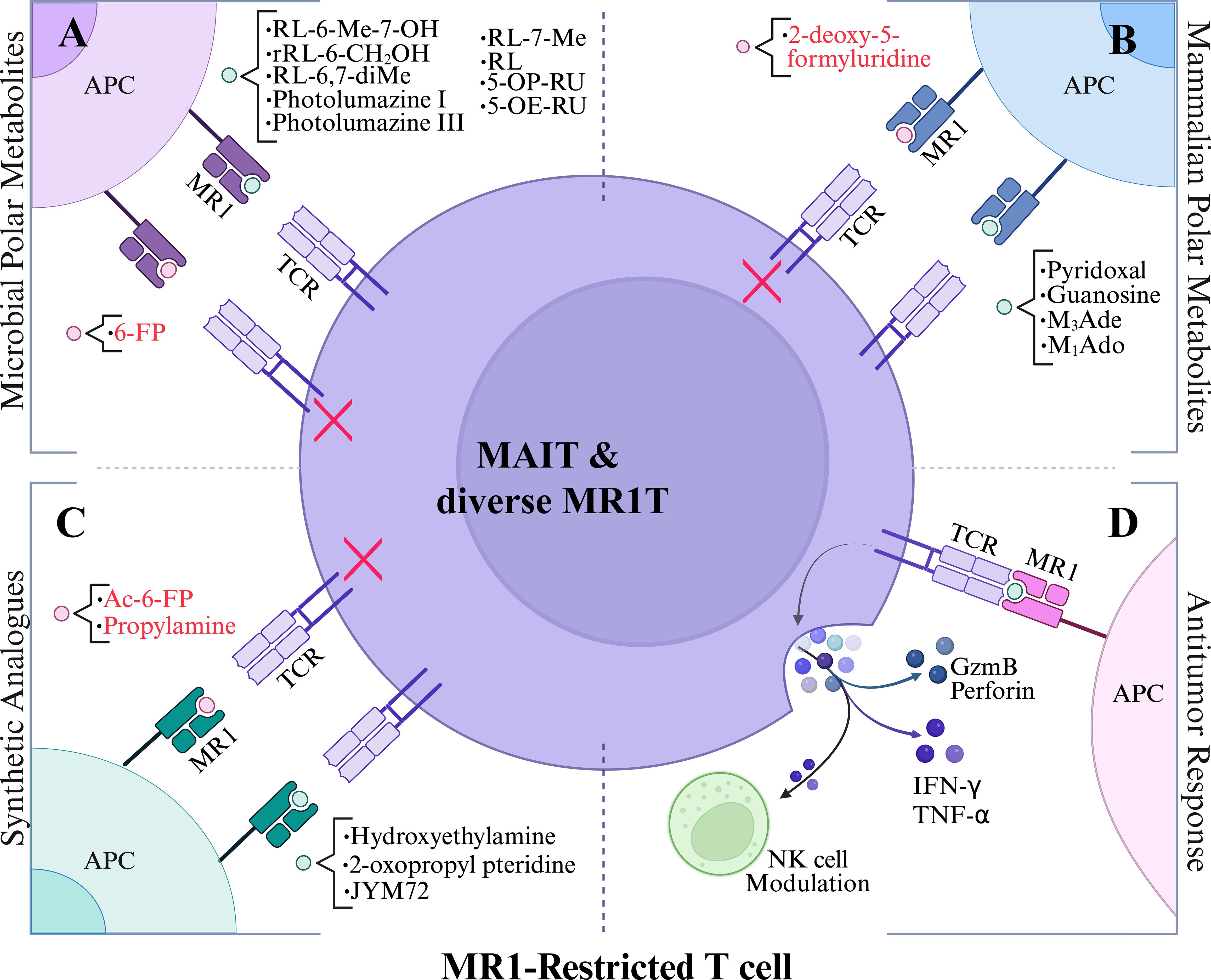
Figure 2. Polar metabolite antigen presentation to MR1-restricted T cells. (A) Vitamin B1, B6, and B9 precursors as MR1 ligands. (B) tumor-associated mammalian-derived metabolites as MR1 ligands. (C) synthetic analogues as MR1 ligands. (D) reported antitumor responses upon stimulation with polar metabolites. Black lettering denotes agonists and red lettering depicts antagonists. Red X indicates inhibited MR1T cell response. Abbreviations are as follows: 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU), 5-(2-oxoethylideneamino)-6-D-ribitylaminouracil (5-OE-RU), ribityl lumazine (RL), 7-methyl-ribityllumazine (RL-7-Me), 6,7-dimethyl-8-ribityllumazine (RL-6,7-diMe), 6-methyl-7-hydroxy-ribityllumazine (RL-6-Me-7-OH), 3-(2-deoxy-β-D-erythro-pentofuranosyl)-6-(hydroxymethyl)-8-oxo-9H-purine-2-carbaldehyde (M1Ado), 6-(hydroxymethyl)-8-oxo-9H-purine-2-carbaldehyde (M3Ade), and acetyl-6-formylpterin (Ac-6-FP).
Polar microbial metabolites
Most identified MAIT cell antigens may not be directly associated with cancer but have been used for MAIT cell activation in cancer cell killing assays. The first identified MR1 ligand, 6-formylpterin (6-FP), was reported in 2012 from the refolded MR1 protein potentially binding with nutrient metabolites from culture media, and it was a photodegradation product of folic acid (vitamin B9) (17). 6-FP and its synthetic analog acetyl-6-formylpterin (Ac-6-FP) bind to MR1 and potently upregulate the cell surface expression of MR1 on human lymphoid C1R cells, but generally inhibit MAIT cells activation (17, 41). Both 6-FP and Ac-6-FP function as competitive inhibitors and stain MAIT or diverse MR1T cells, indicating possible roles in immune modulation rather than robust effector function. Further, MAIT cells are known to recognize derivatives of the bacterial riboflavin (Vitamin B2) intermediate 5-Amino-6-(D-ribitylamino) uracil (5-A-RU), such as 5-(2-oxoethylideneamino)-6-D-ribitylaminouracil (5-OE-RU) and 5-[2-oxopropylideneamino]-6-D-ribitylaminouracil (5-OP-RU), as potent MAIT cell activators, formed by the non-enzymatic condensation of 5-A-RU with small carbonyl metabolites such as glyoxal and methylglyoxal, respectively (16, 41) (Table 1; Figure 3). Among these, 5-OP-RU remains the most potent activator of human and mouse MAIT cells. It induces robust cytokine production and has demonstrated pronounced antitumor activity in murine models of liver, lung, and subcutaneous tumors. Notably, pre-pulsing B16F10 melanoma cells with 5-OP-RU enhances MAIT cell-mediated tumor control, partly through modulation of NK cell responses (45, 46), although, in a different context, MR1-expressing B16F10 cells can suppress NK cell frequency via MAIT activation (47). In contrast, 5-OE-RU is formed with a condensation reaction with glyoxal rather than methylglyoxal and also activates MAIT TCR-expressing Jurkat cells, but with reduced potency (16).
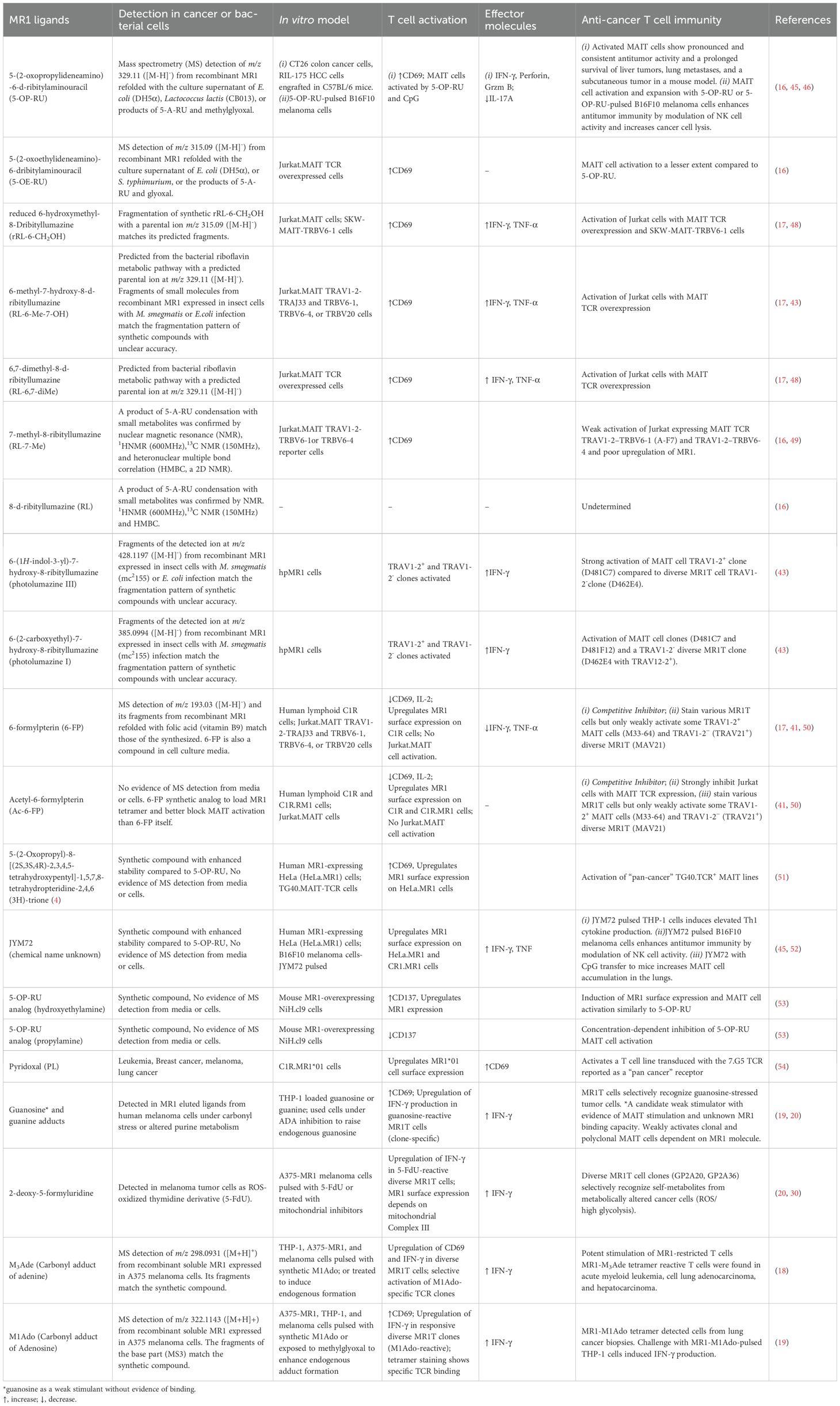
Table 1. Mammalian and bacterial polar metabolites regulating MAIT cell responses.
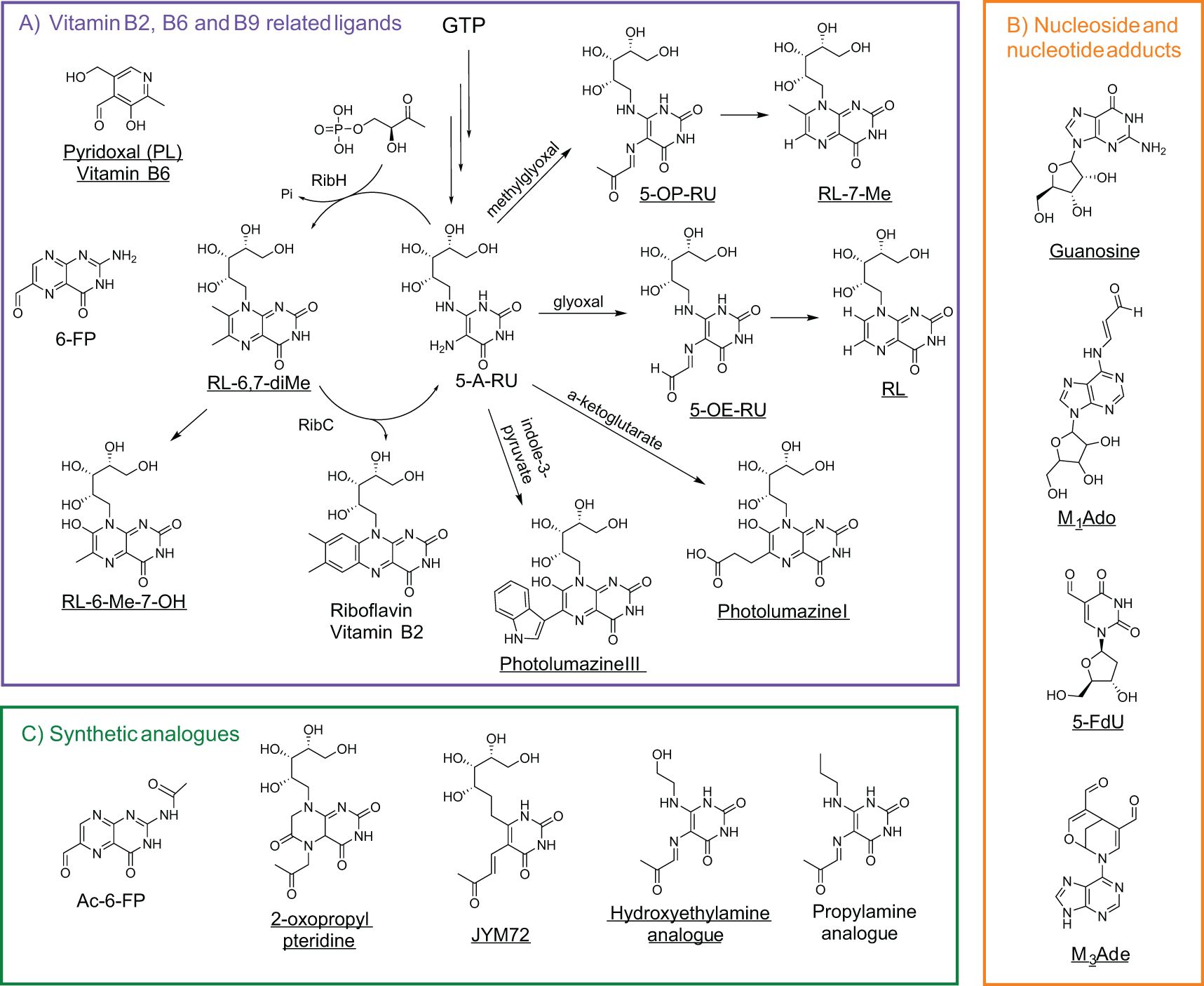
Figure 3. Polar metabolites as MR1 ligands. (A) Vitamin B1, B6, and B9 precursors or related metabolites shown in relation to their metabolic pathways (GTP for riboflavin biosynthesis), with reactants labeled above arrows indicating product formation. RibH (lumazine synthase) and RibC (riboflavin synthase) enzymatically convert early vitamin B2 precursors into riboflavin derivatives. (B) Nucleoside and nucleoside adducts but with unknown MR1-binding capacity for guanosine and 2-deoxy-5-formyluridine. (C) Synthetic analogues associated with vitamin precursor metabolites. Ac-6-FP is derived from vitamin B9 precursors, while 2-oxopropyl pteridine, JYM72 (chemical name unknown), hydroxyethylamine, and propylamine are linked to vitamin B2 derivatives. MR1 agonists or MAIT stimulators are underlined. Abbreviations are as follows: guanosine triphosphate (GTP), 5-amino-6-(D-ribitylamino)uracil (5-A-RU), 5-(2-oxopropylideneamino)-6-D-ribitylaminouracil (5-OP-RU), 5-(2-oxoethylideneamino)-6-D-ribitylaminouracil (5-OE-RU), ribityl lumazine (RL), 6,7-dimethyl-8-ribityllumazine (RL-6,7-diMe), 6-methyl-7-hydroxy-ribityllumazine (RL-6-Me-7-OH), pyridoxal (PL), 2-deoxy-5-formyluridine (fdU), 3-(2-deoxy-β-D-erythro-pentofuranosyl)-6-(hydroxymethyl)-8-oxo-9H-purine-2-carbaldehyde (M1Ado), 6-(hydroxymethyl)-8-oxo-9H-purine-2-carbaldehyde (M3Ade), and acetyl-6-formylpterin (Ac-6-FP).
These ribityl-pyrimidines are very unstable in aqueous acidic conditions and lead to the formation of the ribityl lumazine (RL) and 7-methyl-ribityllumazine (RL-7-Me), which present weak stimulatory activity and poorly upregulate MR1 surface expression. MAIT cells also recognize intermediate metabolites from the bacterial riboflavin pathway, such as 6,7-dimethyl-8-ribityllumazine (RL-6,7-diMe, a natural chromophore in lumazine protein) and 6-methyl-7-hydroxyl-ribityllumazine (RL-6-Me-7-OH), inducing weak stimulation of Jurkat cells with MAIT TCR overexpression and human MAIT cells from peripheral blood (17, 41, 48). These predicted and further synthesized ribityllumazine compounds match the detected mass-to-charge (m/z) unit of 329.11 from the Salmonella typhimurium culture media (17). As in Table 1 and Figure 3, more ribityllumazine compounds, such as photolumazine I and photolumazine III, were detected from the recombinant MR1 protein expressed in Mycobacterium smegmatis-infected insect cells through matching collided fragment patterns with those of synthetic compounds (43). Both metabolites activated MAIT cell clones (e.g., D481C7 and D481F12 clones) and TRAV1-2- MR1T clones (e.g., D462E4 with TRAV12-2) (43). More recently, pyridoxal (PL), another vitamin-related ligand, was identified in leukemia, breast cancer, melanoma, and lung cancer using mass spectrometry. Pyridoxal and pyridoxal phosphate have been reported to activate T cell lines transduced with the 7.G5 TCR, a receptor recently characterized for its ability to recognize MR1-presented ligands across a broad range of tumor types, including both hematologic and solid malignancies, thus demonstrating functional “pan-cancer” reactivity (54).
Synthetic analogues
To evaluate the impact of the ribityl chain on MAIT cell activity, several 5-OP-RU analogs varying the 6-alkylamino substituents on the uracil were designed and synthesized (Table 1; Figure 2; Figure 3) (53). From these analogs, hydroxyethylamine induces MR1 surface expression and activates mouse MAIT cell line 6C2 cells comparable to 5-OP-RU stimulation, while the propylamine analog shows inhibition of 5-OP-RU-activated MAIT cells, similarly to AC-6-FP (53). To enhance MR1-ligand binding stability compared to 5-OP-RU and maintain MAIT cell activity, some vitamin-related synthetic analogs have been designed and synthesized, for example, the 2-oxopropyl pteridine (51) and JYM72 with an unknown chemical name (52) that display stimulatory activity for a murine T hybridoma cell TG40 and a human T cell line Jurkat expressing MAIT TCRs, respectively. Interestingly, B16F10 melanoma cells pre-pulsed with the MAIT cell antigen JYM72 have shown an enhanced antitumor immunity via an MAIT cell-modulated NK cell response (45) (Table 1; Figure 3). This is consistent with earlier findings that MAIT cells can promote NK cell activation and cytotoxicity within the tumor microenvironment, highlighting the potential for MAIT-NK crosstalk as a mechanism of antitumor immunity (45, 46) and the need for an appropriate stimulating strategy to induce protection (47).
Polar self-metabolites
Cancer cells or mammalian cells-derived polar metabolites as agonists or antagonists for self-reactive MAIT and diverse MR1T cell activation can be closely linked to altered metabolic pathways in cancer, bridging cancer cell metabolism with immune regulation and surveillance. Recent discoveries indicate that physiologically relevant nucleobase and nucleoside compounds, as critical precursors for RNA synthesis and metabolism in mammalian systems, stimulate diverse MR1T cells via MR1-mediated antigen presentation (18–20). One such compound, 5-formyl-deoxyuridine (5-fdU), a modified nucleoside formed by oxidative damage to pyrimidine generated during cellular stress, was recently identified as an MR1 antigen capable of activating diverse MR1T cells (20, 30, 73). This result suggests the existence of diverse mammalian-derived polar metabolites that can modulate diverse MR1T cell responses, emphasizing the need to understand metabolite production and function within distinct metabolic pathways. Scientists from Switzerland recently demonstrated that carbonyl-nucleobase adducts (Table 1; Figure 2; Figure 3), including carbonyl adduct formation with adenosine (M1Ado), deoxyadenosine (M1dA), adenine (M1Ade), and guanine (M1Gua) with various activities for diverse MR1T cell activation, which may be accredited to upregulated metabolic pathways in cancer cells that generate high amounts of these identified carbonyl adducts (19). A subsequent study complemented these findings by additionally identifying a carbonyl nucleobase adduct of adenine (M3Ade), MR1-M3Ade loaded tetramers recognized heterogeneous MR1-reactive T-cells in healthy donors and patients with acute myeloid leukemia, and tumor-infiltrating lymphocytes from non-small cell lung adenocarcinoma and hepatocarcinoma ex vivo (18, 74, 75). These findings suggest that while the small molecules themselves are not unique to cancer cells, their elevated production under cancer-associated metabolic dysregulation may create a window for immune recognition. In culmination, these intriguing developments give rise to potential tumor-targeting strategies by using novel self-antigens, which occur in different metabolic pathways and are regulated by oxidative or carbonyl stress, to harness future TCR-based cancer immunotherapies and deepen the understanding of unconventional T cells in tumor immunity.
To comprehend metabolic pathways and metabolite profiles altered in cancer, research has indicated that cancer cell metabolism undergoes various transformations, such as a shift from oxidative phosphorylation to aerobic glycolysis (Warburg effect), or higher pressure of oxygen or carbonyl species, leading to changes in competitive nutrient utilization and metabolite prevalence compared to non-malignant cells (76, 77). Metabolic reprogramming throughout tumor progression stages is expected to be different between tumor regression and tumor progression stages. An early immune surveillance stage may be attributed to an effective anti-tumorigenic inflammatory microenvironment mediated by local immune cells and factors to inhibit tumor progression. However, a later cancer progression stage likely fosters an immunosuppressive landscape with a pro-tumorigenic microenvironment. In this context, the immune-dysregulated cancer microenvironment inherently impacts the availability and specificity of polar metabolites for MR1, which in turn critically influences MAIT cell activation downstream. Specifically, this metabolic shift may impact the production and loading of MR1 ligands, likely further regulating MR1 expression and effector function in the cancer microenvironment.
MR1-restricted T cells in cancer
Self-reactive MAIT cells expressing an invariant Vα19 TCRα (TRAV1) chain were initially cloned and tested for their dependence on MR1-mediated antigen presentation in mice (11, 12, 34, 35, 40). In humans, MR1-restricted T cells exhibit high clonal diversity, characterized by variant TCR chains, the recognition of cancer cells, and the potential ability to kill cancer cells. Specifically, these MR1-restricted self-reactive human T cells from blood samples of healthy donors or cancer patients mostly express diverse TCRs (diverse MR1T or TRAV1-2- MR1T) (13, 18, 44). A TRAV1-2- MR1T clone (MC.7.G5), as an early example of antitumor diverse MR1T cells, is generated from the blood of healthy donors and is capable of cancer cell killing, via responding to a cancer instead of a bacterial metabolite (44). This diverse MR1T clone exhibits pan-cancer cytotoxicity to kill various types of cancer cells (44), but not a pan-population effect in humans, due to its restriction by an unusual MR1 allomorph with an R9H mutation in humans (78, 79). More diverse MR1T clones respond to cancer cells, dependent on the dominant MR1 allomorph in humans (13, 30). Many of these diverse MR1T clones are later shown to recognize nucleobase adducts with a carbonyl or aldehyde group (18, 19), which is potentially related to mitochondrial metabolic reprogramming with oxidative stress in melanoma and leukemia cell lines (30). The activation of diverse MR1T cells with self-metabolite stimulation induces the differentiation of memory subsets (18), demanding further understanding the dynamics and transcriptional programs of these diverse MR1T cells, compared with MAIT and conventional T cells.
In later years, MAIT and diverse MR1T cell responses in cancer immunity have been suggested to be highly dependent on context and influenced by the tumor microenvironment (TME), metabolic constraints, and immune-suppressive factors (13, 80). It is known that MAIT cells are enriched in mucosal tissues, including the lungs, liver, and intestines, and have been detected in tumor-infiltrating lymphocytes (TILs) across cancer types (81). Clinical research has reported that MR1 expression in tumors varies significantly, with some cancers upregulating MR1 as a potential immune evasion strategy, correlating with poor prognosis (72), while others exhibit reduced MR1 expression, potentially limiting MAIT and diverse MR1T cell activation (26, 82). Overall, MR1-restricted T cells display functional plasticity with tumor-suppressive or tumor-promoting responses across various malignancies, reflecting the dynamic influence of the tumor microenvironment (Table 2). Local factors, including cytokine signals, metabolic composition, and cellular interactions, influence MAIT cell activity, which supports either tumor control or tumor growth. This duality arises from their ability to produce inflammatory cytokines such as IFN-γ and TNF-α, contributing to tumor cell lysis, while also potentially promoting immune suppression through IL-17, IL-10, and regulatory interactions within the TME (83). These divergent outcomes depend on tissue context, disease stage, and ligand availability, and the mechanisms governing this dichotomy have been recently reviewed in greater detail (26).
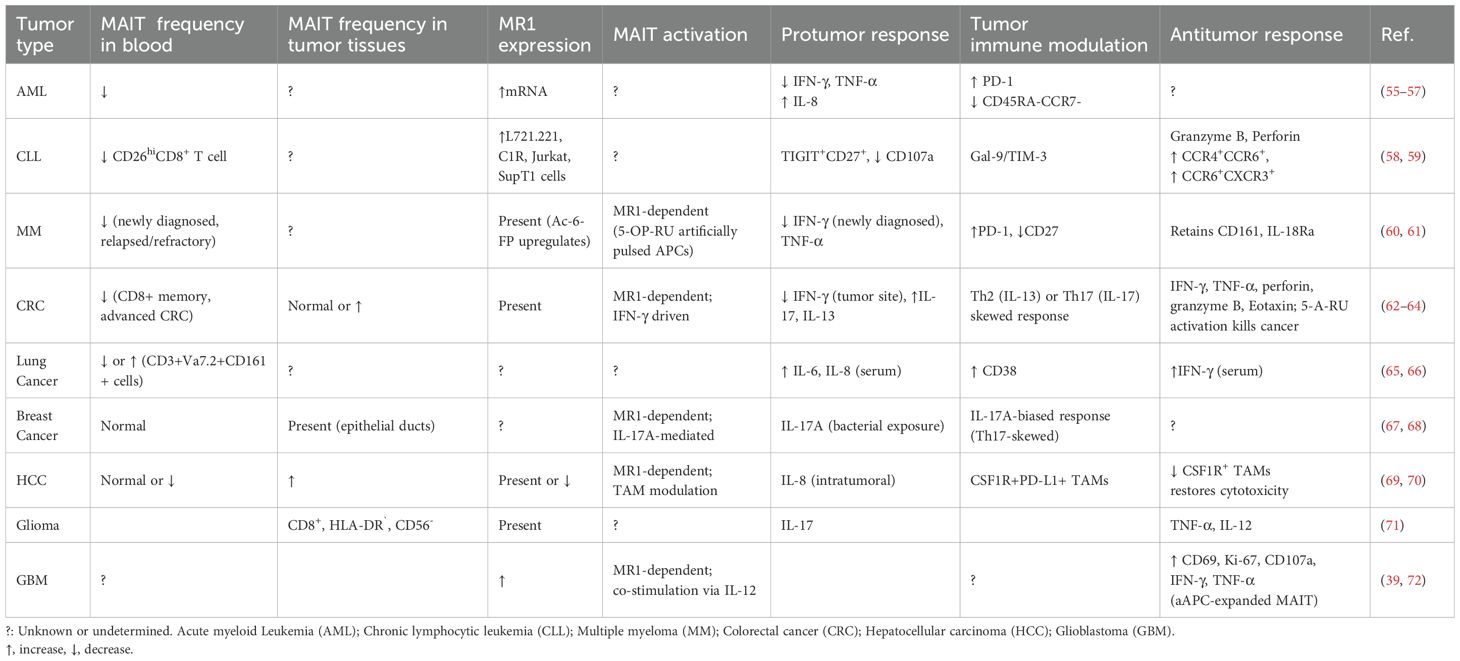
Table 2. MAIT cell frequencies, responses, and functions in tumors.
However, whether the availability and identities of MR1 ligands regulate the levels of MR1 expression and shape the impact of MAIT cells across different cancers remains largely unknown, making their role in tumor immunity an area of active investigation. In hematological malignancies, such as multiple myeloma and leukemias, the role of MAIT cells remains largely underexplored, with limited clarity on whether MAIT cells contribute to tumor suppression or progression (Table 2; Figure 4). In solid tumors, MAIT cells exhibit more complex and variable functions, influenced by the tumor microenvironment, cancer metabolic program, local immune cell response, and immune checkpoint regulation (Table 2; Figure 4).
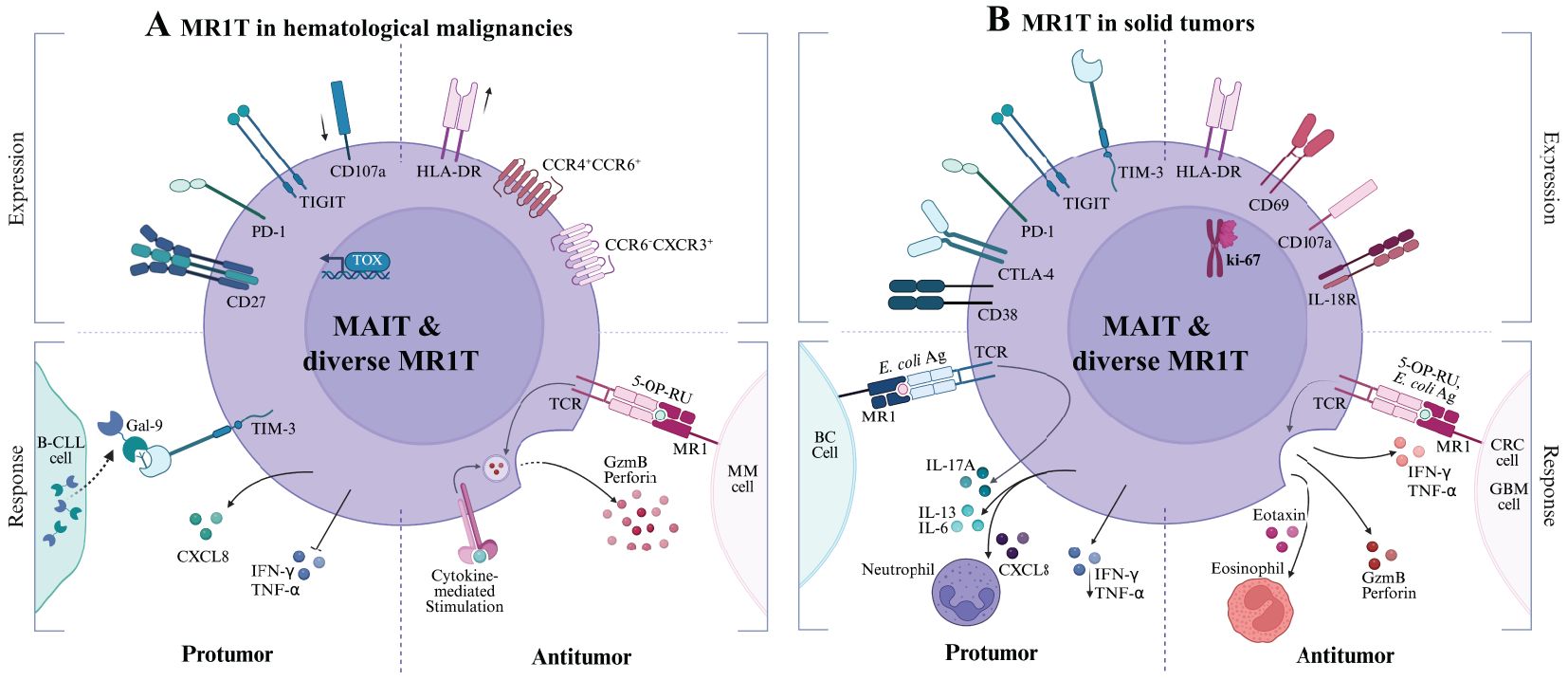
Figure 4. MR1T cell responses in Hematological Malignancies (A) and Solid Tumors (B). Each panel is divided into quadrants to illustrate the relationship between MR1T cell phenotype (top, labeled “Expression”) and functional outcome (bottom, labeled “Response”). Top left quadrant: pro-tumor immunosuppressive or exhausted phenotypes. Bottom left quadrant: tumor-promoting cytokine profiles and dysfunctional responses. Top right quadrant: antitumor effector and activation phenotypes. Bottom right quadrant: tumor-controlling protective responses. Cancer cells depicted include (A) malignant B cell-chronic lymphocytic leukemia (B-CLL), multiple myeloma (MM); (B) Breast cancer (BC), colorectal cancer (CRC), glioblastoma (GBM).
Hematological malignancies
In acute myelogenous leukemia (AML), a disease marked by the uncontrolled proliferation of undifferentiated myeloid cells, MAIT cells show significant reduction in circulation among newly diagnosed patients, coinciding with elevated HLA-DR expression on MAIT cells, suggesting a recent activation state (56). Upon chemotherapy for 15 days in these AML patients (n=25), MAIT cell frequency is dramatically reduced to an average frequency of 3% of that prior to chemotherapy (56). A similar study analyzing a cohort of AML patients reveals similar alterations in MAIT cell frequency and function, correlating with disease burden and progression status. MAIT cells exhibit an upregulated PD-1 expression, a downregulated CD45RA-CCR7- effector memory subset, impaired IFN-γ and TNF-α production, and begin favoring a cytokine profile (IL-8) linked to tumor progression (55). Notably, a lower MAIT cell frequency is independently associated with poorer patient prognosis, contrasting conclusions seen in other tumor types (56). To date, clinical data underscore the probable roles of MAIT cells adopting dysfunctional responses, potentially impacted by poorly understood metabolic and immune regulatory factors occurring in AML cells. For example, Tet methylcytosine dioxygenases 2 mutation promotes leukemogenesis likely through stabilizing methyl-5-cytosine for epigenetic regulation (84, 85) and affects nucleoside modification (84, 86).
A similar pattern of MAIT cell depletion is observed in chronic lymphocytic leukemia (CLL), where MAIT cell frequency is markedly reduced, particularly within the CD26hi T cell population previously reported (58). Findings indicate that the CML microenvironment employs immune evasion modulation that promotes the apoptosis of CD8+CD26hi T cells through the galectin-9 (Gal-9)/TIM-3 axis, leading to a pronounced depletion of this subset occupying the majority of MAIT cells (58). Interestingly, CD8+CD26hi T cells enriched with MAIT cells also significantly reduce in CLL patients, and show a propensity to highly express cytotoxic molecules when stimulated with cytokines rather than CD3/CD28-dependent stimulation, indicating MR1 recognizing CLL-derived ligand is remiss but potentially important for MAIT cell activation in this cancer context.
In multiple myeloma (MM), a plasma cell malignancy thriving within an immunosuppressive bone marrow niche, MAIT cells are again depleted in circulation, along with a diminished frequency present in the BM (60), suggestive of depleted cell numbers not attributable to redistribution but rather dysregulation of MAIT cell differentiation and function within TME. Reduced frequencies are especially prominent in newly diagnosed and relapsed/refractory patients in recent observations (61). A defining feature of MAIT cells in MM is their upregulation of PD-1+ and CD27+, indicative of T cell exhaustion. Ex vivo and in vivo experiments demonstrate that blocking PD-1 signaling combined with α-GalCer-stimulated iNKT cells partially restores MAIT cell cytokine production (60), highlighting the potential for checkpoint blockade therapies in MAIT cell reactivation. Other research reveals MM cell lines exhibit detectable basal MR1 surface expression upon exposure to the folate-derived ligand 6-FP from the vitamin B9 pathway. These findings suggest that MM cells possess a reservoir of ER-resident MR1 capable of rapidly trafficking to the cell surface, where MR1 can bind or replace ligands for MAIT cell activation or inhibition. Additionally, MAIT cell-mediated cancer killing was apparent in MM cell lines pulsed with the potent 5-OP-RU agonist (61). This approach highlights the possibility that selective MR1 agonists can enrich MAIT cell-mediated tumor immunity in MM and may also prove relevant in other tumors; however, in vivo models are essential to examine the stimulatory molecules to induce an anti-cancer effect of MAIT cells.
Solid tumors
Malignant gliomas, a type of gliogenic brain tumors, show evidence of MAIT cell tumor infiltration that displays an exhausted phenotype characterized by high expression of PD-1+, TIM-3+, and LAG-3+. Although MAIT cells in glioma tumors have been shown to express CD8+HLA-DR+, demonstrating a favorable activated phenotype, they lack CD56, a NK marker associated with enhanced MAIT cell responsiveness to Th1 cytokine stimulation (71). TRAV1-2+ TRAJ12/33 MAIT cells have been detected within brain tumor lesions, with MR1 expression identified in some cancerous glial cells. In glioblastoma (GBM), one of the most aggressive glioma malignancies, higher MR1 expression has been correlated with poor prognosis (72). While the mechanism remains unclear, this correlation raises the possibility that MR1-restricted MAIT cells may play a tumor-promoting role in GBM, or that elevated MR1 expression does not necessarily reflect increased presentation of stimulatory ligands capable of inducing protective MAIT responses. However, when expanded ex vivo and activated using artificial antigen-presenting cells, MAIT cells demonstrate strong cytotoxic potential against GBM cells via CD107a degranulation and lactate dehydrogenase detection. Upon activation, flow cytometric results illustrate elevated levels of IFN-γ and TNF-α, along with enhanced expression of CD69+ and Ki-67 activation and proliferation markers (39). MAIT cells have also demonstrated the ability to effectively lyse GBM cells in an MR1-dependent manner at higher effector-to-target ratios, underscoring their potential to target gliomas via MR1-antigen recognition and highlighting new avenues for glioma immunotherapy.
A similarly complex pattern emerges in lung cancer (LC), where MAIT cells exhibit various functions depending on disease stages and tumor microenvironmental factors. A clinical study investigating MAIT cell relevance in mucosal-localized tumors, inclusive of lung cancer, found reduced circulating MAIT cells, in contrast to elevated frequencies within tumor tissue. Strikingly, MAIT cells retain normal cytokine profiles with capacities for IFN-γ, IL-17, and TNF-α production (65). Conversely, another clinical study has observed CD3+Va7.2+CD161+ MAIT cells in circulation are significantly elevated in lung cancer patients, showing an activated pro-inflammatory state by CD38+CD8+ expression. Serum cytokine profile from this patient cohort reveals elevated IFN-γ, IL-6, and IL-8 production, suggesting that MAIT cells may contribute to shaping the inflammatory milieu (66). Further analysis shows that the IL-6 expression level correlated positively with tumor-associated MAIT cells frequently expressing CD38, leading to a possible immunosuppressive role. LC patients with an overall higher MAIT cell level, particularly CD38+CD8+ expression, are associated with worse progression-free survival, highlighting a detrimental role in lung cancer progression (66).
In colorectal cancer (CRC), MAIT cells exhibit both antitumor and protumor roles, with their function heavily influenced by the local inflammatory environment (64). Tumor-controlling potential of MAIT cells is generally associated with the increased tumor-infiltrating MAIT cells exhibiting a Th1 phenotype that secreted granzyme B and, to a lesser extent, perforin, suggesting a protective antitumor role in colon adenocarcinomas (87). Supporting this, RAG-/- mice bearing the murine colon adenocarcinoma cell line MC38-derived tumors demonstrate significant tumor growth inhibition when MAIT cells are injected peritumorally. Tumors treated with MAIT cells display elevated levels of pro-inflammatory cytokines (IFN-γ, IL-17, GM-CSF), and eosinophil-attracting chemokines (eotaxin-1), alongside increased caspase 3/7 activity, indicative of enhanced tumor cell death (62). Complementary in vitro experiments with human MAIT cells stimulated by 5-A-RU further demonstrate their capacity to kill COLO 205 cancer cells, enhance cytokine production, and promote eosinophil activation and recruitment, as evidenced by upregulated CD69+ and granzyme A expression (62). However, MAIT cells, characterized by impaired Th1 cytokine production or increased IL-17 secretion, adopt an immunosuppressive phenotype within the TME, which sustains chronic inflammation and tumor growth, or enhance IL-13 expression, fostering a Th2-skewed and protumor microenvironment (63, 64). This shift dampens effective cytotoxic responses while promoting tumor-associated inflammation and myeloid cell recruitment. Notably, IL-17-driven inflammation has been strongly linked to tumor progression, correlating with worsened CRC prognosis (63). Furthermore, decreased circulating MAIT cells, particularly within the CD8+ memory subset, have been associated with advanced-stage CRC. Whereas paradoxically, a higher tumor-infiltration of MAIT cells, detected in CRC tissues compared with that of non-tumor tissues (63), may modulate anti-cancer immunity and affect patient survival, potentially dependent on Th1 or Th17-like phenotype of the infiltrated MAIT cells. This duality underscores the need for further research into the regulatory mechanisms that dictate MAIT cell functional polarization in CRC. Understanding cancer cell metabolism, in particular cancer metabolite profiles for MAIT cell activation or inhibition, may reveal new therapeutic strategies aimed at modulating MAIT cells to enhance antitumor immunity while limiting their protumor activities.
MAIT cells in hepatocellular carcinoma (HCC) underline their functional plasticity, with studies presenting conflicting evidence regarding their impact on prognosis. Research suggests that a higher abundance of MAIT cell infiltrates correlates with improved patient outcomes (88), while other clinical studies indicate that elevated intratumoral MAIT cells are associated with poor prognosis (70). This study further demonstrates that intratumoral MAIT cells from HCC patients upregulate the expression of PD-1+, CTLA-4+, and TIM-3+ inhibitor or exhaustion markers along with diminished cytotoxic molecules, including IFN-γ, granzyme B, and perforin, in comparison to MAIT cells from peritumor regions. Within a detrimental TME, infiltrating MAIT cells display an exhausted phenotype, largely driven by tumor-associated macrophages (TAMs), which induce dysfunction through increased expression of CSF1R+ (colony stimulating factor 1 receptor), PD-L1+ (programmed cell death ligand 1), and CD69+. This direct cell-cell interaction suppresses MAIT cell activity, leading to exhaustion and the loss of cytotoxic function (69). Via paracrine regulation, the presence of IL-8 within the HCC microenvironment indicates MAIT cell dysfunction via inhibiting IFN-γ production (70). More recent findings reveal that HCC patients exhibit a significant reduction in circulating MAIT cells alongside limited infiltration into liver tumors. Despite these immunosuppressive mechanisms, lower levels of MAIT cell infiltration have been linked to a worse prognosis, suggesting that MAIT cells may contribute to tumor control if their functional integrity and protectivity can be maintained, and they are not pushed into a chronically exhausted state. Notably, murine HCC models have demonstrated that depletion of CSF1R+ TAMs improves MAIT cell infiltration and restores cytotoxic function (69), highlighting the potential of targeting the MAIT cell-TAM axis as a promising strategy to enhance immunotherapy responses in HCC. Current findings emphasize the need for further investigation into strategies that can preserve MAIT cell functionality while preventing their immunosuppressive conversion within a cancerous microenvironment of the liver.
In the breast cancer context, MAIT cells are primarily retained in circulation but are also detectable within the epithelial ducts of human breast tissue. Gene transcriptomic analysis has identified MAIT cell-specific markers, including TRAV1-2+ TCR, CD161, PLZF, and IL-18Rα, within the epithelial ducts, suggesting their presence and potential functional role in breast TME (67). Notably, these tumor-associated MAIT cells exhibit a Th17-skewed functional profile, characterized by an enrichment of IL-17A-producing cells, the MAIT17 subset. This IL-17A bias is particularly significant, as IL-17A-mediated inflammation has been implicated in promoting tumor progression through the recruitment of protumor immune cells and the establishment of a chronic inflammatory milieu. Further in vitro experiments demonstrate that when MAIT cells are activated by E. coli in an MR1-dependent manner and co-cultured with breast cancer cell lines, they predominantly produce IL-17A while exhibiting a diminished Th1 or cytotoxic response (67). This shift away from IFN-γ and TNF-α production suggests that MAIT cells in the breast cancer microenvironment may contribute to tumor-promoting inflammation rather than effective antitumor immunity. Additionally, research has shown that MR1-restricted TRAV1-2+ and TRAV26-1+ TCRs identified from the tumor-infiltrating T cells of breast cancer patients specifically respond to some breast cancer cell lines but not to other cancer types (68). This observation implies a degree of antigen-specific recognition of breast cancer cells, highlighting the potential for tumor-selective immune interactions mediated by MR1. However, which breast cancer cell-derived metabolites bind to MR1 for MAIT cell activation and whether this selective recognition can be leveraged for therapeutic intervention or if it predominantly contributes to tumor immune evasion remain open questions, necessitating further exploration into the functional dynamics of MAIT cells in breast cancer.
Overall, the function of MAIT cells in cancer is shaped by the interplay between cancer metabolism, immune checkpoint regulation, and local cytokine signaling. In some settings, MAIT cells exhibit potent tumor control, whereas in others, they are co-opted into tumor-promoting activities. Understanding the mechanisms governing this functional plasticity is critical for designing effective immunotherapies that harness their tumor-killing potential capacities while mitigating their protumor advancements. Strategies aimed at reactivating exhausted MAIT cells, modulating MR1 antigen presentation, minimizing protumor MAIT cell subsets, and leveraging cytotoxic MAIT subsets or capabilities represent promising avenues for future cancer treatment.
CD1-restricted T cells in cancer
CD1 molecules are nonpolymorphic in humans and present lipid-based antigens to unconventional T cells. Unlike classical MHC molecules using shallow hydrophilic ligand-binding grooves for peptide antigen presentation (Figure 1) (1, 2), CD1 molecules possess hydrophobic antigen-binding clefts and present lipid-biased ligands, providing a distinct mechanism for immune recognition (7, 14, 89). Studies demonstrated that CD1 molecules present mammalian cell-derived and tumor-associated lipids, such as gangliosides, phospholipids, and sphingolipids. CD1-mediated lipid antigen presentation permits CD1-restricted T cells to become lipid metabolite sensors, detect lipid metabolic alteration in cancer cells, and induce cancer immune surveillance and metabolic modulation. Translationally, lipid antigen presentation through CD1 may serve as a valuable target for immunotherapeutic strategies aimed at harnessing the antitumor potential of CD1-restricted T cells. CD1 proteins with lipid antigen-presentation functions consist of four isoforms in humans: CD1a, CD1b, and CD1c as group 1 CD1 proteins, and CD1d as a group 2 CD1 protein (8, 89, 90). Tumor cells manifest an altered lipid metabolism, leading to the overrepresentation of specific lipid antigens that may be loaded to CD1 proteins for the recognition of CD1-restricted T cells. Advanced by mass spectrometry-based lipid profiling, recent discoveries demonstrated that CD1 proteins bind various classes of heterogeneous mammalian cell-derived lipids by groups I and II human CD1 proteins (20, 89, 91–94). CD1a binds skin-derived lipids (95), implying a potential involvement in cutaneous malignancies. CD1b has been shown to bind to tumor-derived phospholipids in T-cell lymphoma (96). CD1c recognizes methyl-lysophosphatidic acid (97), a lipid abundantly expressed in leukemic cells. Perhaps due to the first characterized invariant αβ T cell population called invariant natural killer T (iNKT, type I NKT) cells and the availability of lipid-preloaded CD1d tetramers for staining iNKT cells, CD1d has been particularly well studied for its role in presenting lipid antigens to iNKT cells (23, 98) and later shown to activate diverse NKT (dNKT, type II NKT) cells as well (99, 100).
The iNKT cells express a highly conserved TCR, comprising TRAV10 or TRAJ18 α-chain with limited variable β-chain such as TRBV24 usage (8), which recognizes α-GalCer purified originally from the marine sponge, Agelas mauritianus. Type I iNKT cells are known for their rapid cytokine responses upon activation, producing IFN-γ and TNF-α, which enhance antitumor immunity by recruiting and activating dendritic cells (DCs), NK cells, and CTLs. The iNKT cells also possess the capacity to exert direct cytotoxic effects on tumor cells via perforin, granzyme B, and FasL-mediated apoptosis (101). Given their pivotal role in modulating immune responses, CD1-restricted T cells have emerged as attractive candidates for therapeutic intervention. Several strategies have been explored to harness iNKT cells in cancer therapy (102), including lipid-based vaccines such as α-GalCer, which potently activate iNKT cells and induce robust Th1-skewed antitumor responses (103). Modified α-GalCer derivatives and CD1d-binding glycolipid agonists have been developed to improve cytokine bias and overcome iNKT cell anergy (104, 105). Advances in cellular therapies include chimeric antigen receptor (CAR)-NKT cells, which combine innate tumor-homing capabilities with engineered antigen specificity and have demonstrated enhanced cytotoxicity and persistence in preclinical lymphoma and melanoma models (106–108). Additionally, dendritic cell-based lipid vaccines have shown promise in expanding functional iNKT populations and boosting antitumor immunity (109). The balance between Type I and Type II iNKT cell activity influences the overall immune response within the TME. Type II iNKT cells exhibit TCR diversity and respond to a broader range of lipid antigens (8). While Type I iNKT cells largely promote antitumor immunity, Type II iNKT cells can occupy an immunosuppressive functionality, modulating immune responses through the production of IL-13 and TGF-β, which promote regulatory T cell expansion and limit effective immune responses. In multiple myeloma (110), Type II iNKT cells have been implicated in suppressing effective immune responses by inducing myeloid-derived suppressor cells (MDSCs) that modulate immune cells toward a suppressed, regulatory state (23). As the roles of CD1-restricted T cells in cancer immunity and therapeutic trials based on lipid antigens have been comprehensively reviewed (90, 111, 112), we focus on discussing the structures and functions of lipid metabolites as CD1 ligands in cancer immunity.
Self lipid metabolites as CD1 ligands
As CD1-loaded lipids likely bridge cancer cell lipid metabolism with T cell sensing of lipids, cellular lipids loaded to CD1 proteins are particularly interesting to be identified using tandem mass spectrometry in multiple studies. Two major structural categories of tumor-associated lipids, phospholipids and sphingolipids with or without glycosylated modification (Table 3; Figure 5), are sampled by human CD1d protein and function as agonists or antagonists for NKT cell responses (15, 20, 89, 91–94). Multiple classes of CD1-sampled lipids, including cardiolipin, sphingomyelin, sulfatide, and ganglioside, have been detected in tumor tissues by MS imaging from human glioblastoma (113).
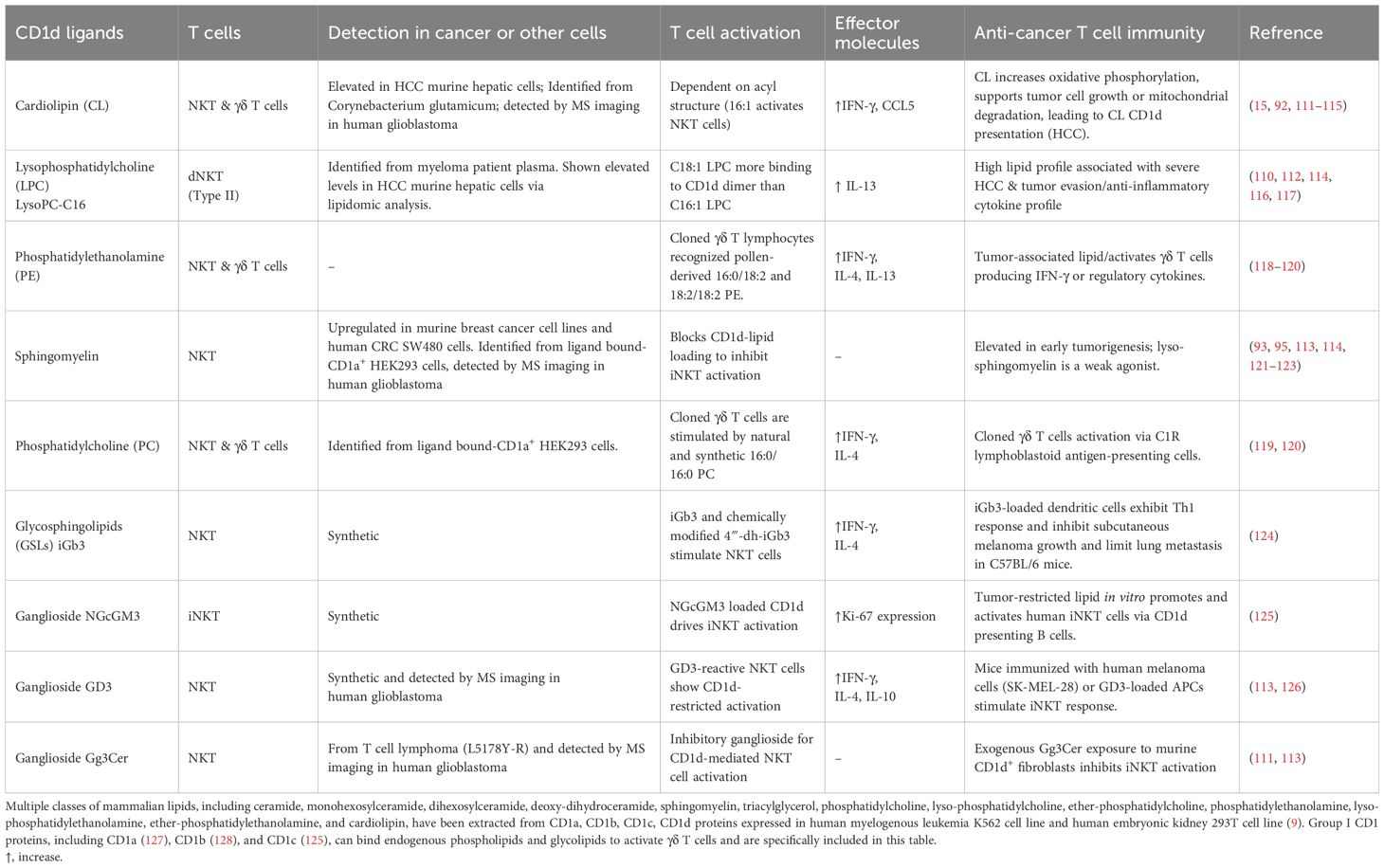
Table 3. CD1d ligands detected from cancer cells activate or block CD1d-restricted T cell responses.
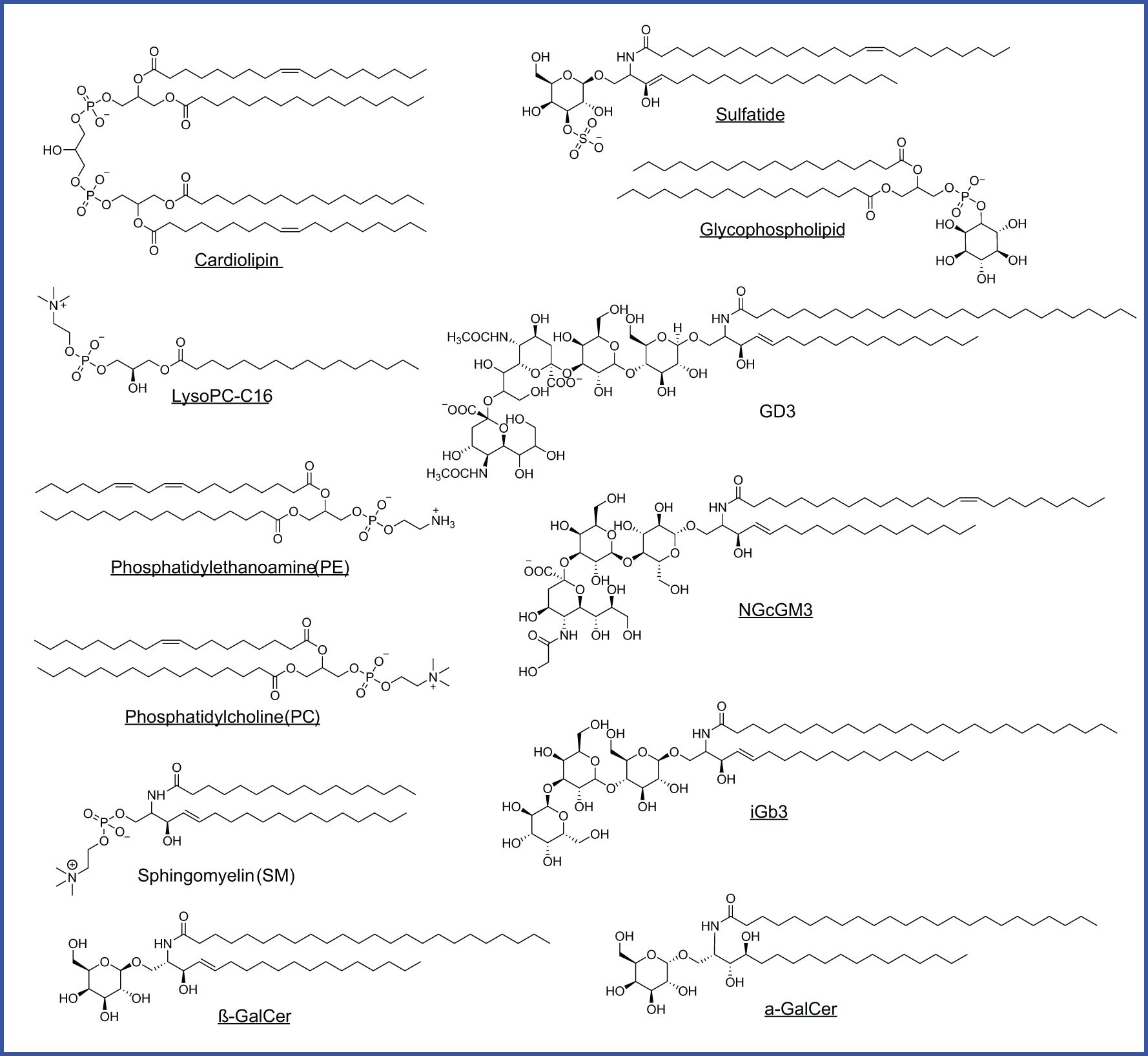
Figure 5. CD1d-bound lipids detected in different cancers. Agonists are underlined. Glycophospholipid is identified from HLA class I deficient human lymphoblastoid, MS detection with m/z 861.8 of [M-H]-, but with an unknown hexosyl group.
Phospholipids
Phospholipids, as generally defined by a glycerol backbone linked to fatty acids and a phosphate-containing head group, are key components of cellular membranes and signaling pathways. Within this class, cardiolipin (CL), overexpressed early in hepatocellular carcinoma (HCC) in mice, is released during mitochondrial degradation and is associated with CD1d lipid presentation to iNKT and γδ T cells, promoting tumor growth by supporting mitochondrial function (114, 115, 120). Lysophosphatidylcholine (LPC), elevated in mouse HCC and multiple myeloma patients, fosters a tumorigenic lipid profile and immune evasion by activating anti-inflammatory NKT cells (110, 112, 116). Similarly, phosphatidylethanolamine (PE) and phosphatidylcholine (PC) (Table 3; Figure 5), both abundant membrane phospholipids, are presented by CD1d and CD1c to activate iNKT and γδ T cells, promoting either regulatory or Th1 cytokine responses (118, 119).
Sphingolipids
Sphingolipids, in contrast, are built on a sphingosine base and often incorporate ceramide backbones, playing essential roles in membrane stability and signal transduction. Tumor-elevated sphingomyelin (Table 3; Figure 5), a sphingolipid enriched in the mammalian plasma membrane, has been shown to weakly stimulate iNKT cells through a mono-acylated derivative, whereas the di-acylated form is predominantly reported to act as an inhibitor when presented on CD1d (92, 93). Sphingomyelin also displays as an iNKT antagonist and contributes functionally to immune dysregulation across mouse breast cancer cells alongside human colon and B cell cancers (120–122). Glycosylated sphingolipids (GSLs), particularly isoglobotrihexosylceramide (iGb3), are a class of glycolipids containing amino alcohol sphingosine that have been shown to promote antitumor Th1 responses by activating iNKT cells to produce IFN-γ (124). Gangliosides, such as GD3, NGcGM3, Gg3Cer (Table 3; Figure 5), and salic-acid containing GSLs enriched in certain malignancies, can either inhibit or promote iNKT activation in a context-dependent manner, acting as tumor-specific antigens or immune modulators (125, 126, 129, 130). Although identifying CD1-loaded endogenous lipids (Table 3; Figure 5) has offered a critical example and framework for understanding intracellular metabolite loading for unconventional T cell activation, only a small list of endogenous ligands has been discovered, and their functional roles in cancer remain largely unexplored.
The γδ T cells in cancer
The γδ T cells represent a distinct arm of the T cell lineage, defined by their TCR expression of γ and δ chains, as opposed to the αβ TCRs found on other invariant T cell subsets and conventional T cells. Although γδ T cells can be activated by nonclassical MHC molecules and metabolite antigen complexes, γδ T cells can also recognize other metabolites or protein ligands, such as phosphoantigens. Briefly, Vδ1+ cells are more prevalent in mucosal and epithelial tissues; Vδ2+ γδ T cells, frequently paired with the Vγ9 chain, are the most abundant subset in peripheral blood; while Vδ3+ cells are less common and enriched primarily in the liver and gut (25). In TME, γδ T cells exhibit dual tumor responses, depending on their subsets, cytokine profiles, and tissue microenvironments. Vδ2+ cells generally contribute to antitumor immunity by secreting IFN-γ and TNF-α and inducing tumor cell lysis through secreting perforin, granzyme B, and TRAIL. Their expression of NKG2D enables direct recognition and killing of stressed or transformed cells expressing MICA/B and ULBPs (25). Studies have shown an increased infiltration of Vδ2+ TILs correlating with improved patient survival in malignancies such as malignant melanoma, AML, and ALL (145, 146). Other research suggests Vδ5+ or TRDV5+ γδ T cells that recognize EPCR demonstrate potent tumoricidal activity (147). On the other hand, Vδ1+ γδ T cells have demonstrated a more complex role. While capable of effective cytotoxicity and IFN-γ production, certain Vδ1+ populations secrete IL-17A, a cytokine associated with angiogenesis, neutrophil recruitment, and the promotion of MDSCs (134), all of which contribute to tumor progression. In rectal cancer, patients with increased Vδ1+ cell infiltration indicated higher tumor burden, whereas Vδ2+ cell presence exhibited a negative association with tumor size (135). Beyond this basic outline of important subsets of γδ T cells, we will briefly describe the roles of metabolite and protein ligands in anti-cancer γδ T cell responses (Figures 5, 6; Table 4), as immune responses and therapeutic effects of γδ T cells have been reviewed recently (22, 25, 134).
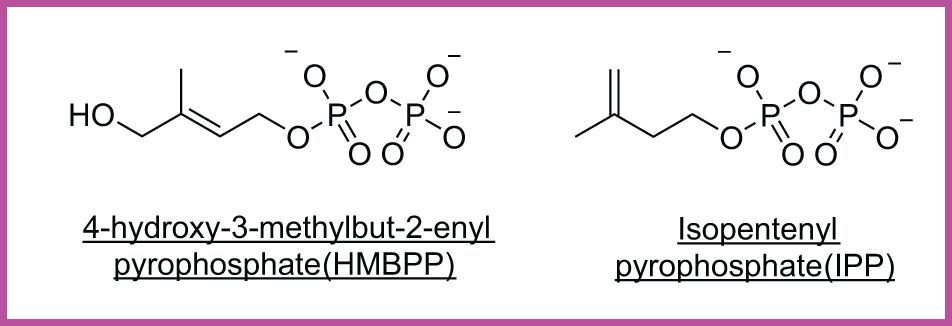
Figure 6. Cancer-associated phosphoantigens (pAgs). HMBPP, a microbial metabolite, and IPP, a host-derived mevalonate intermediate, are presented by butyrophilin proteins to activate Vγ9Vδ2 γδ T cells.
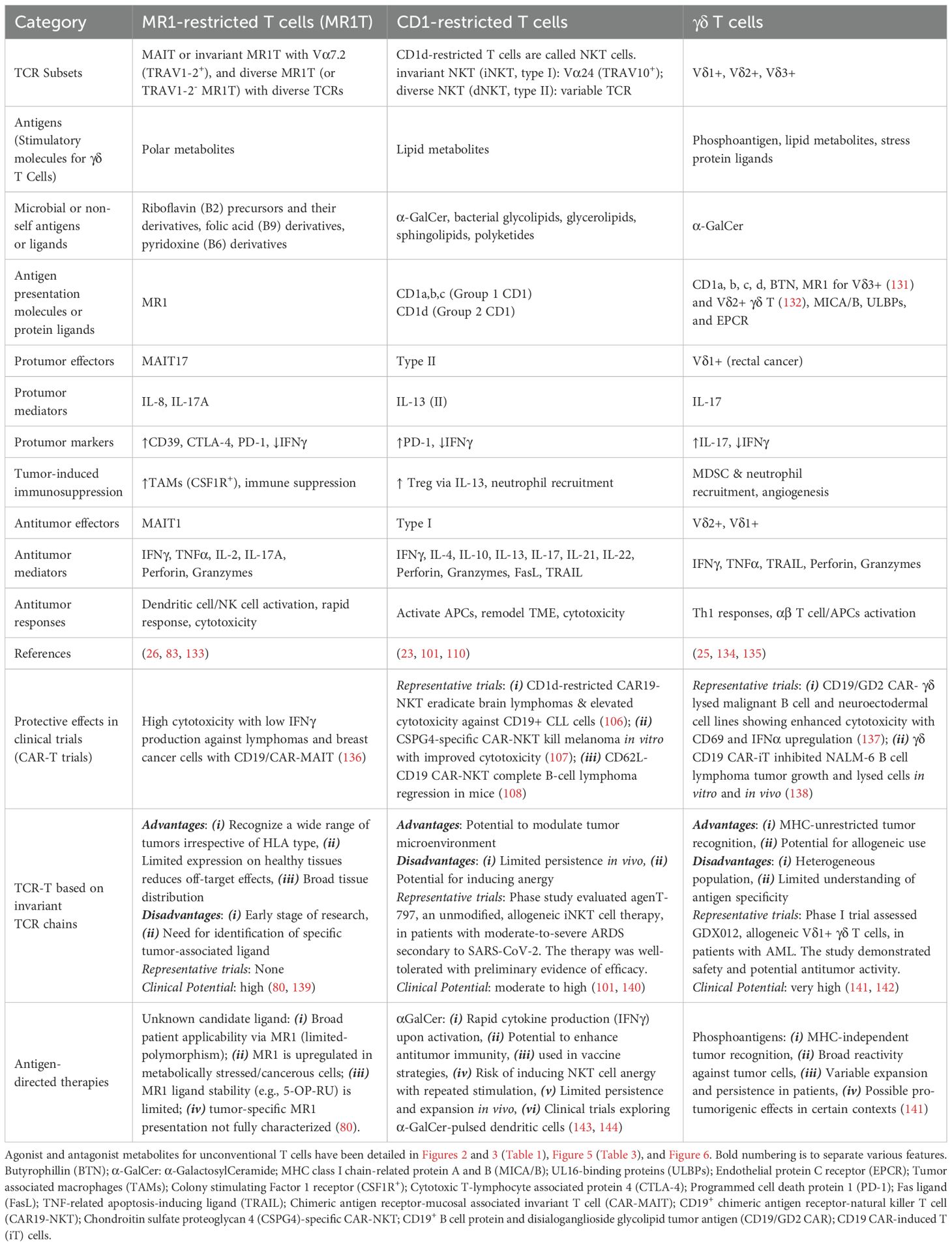
Table 4. Unconventional T cells in cancer immunity.
Stress-induced metabolites for γδ T cell activation
The γδ T subsets interact with protein ligands or metabolite antigens and shape their roles in immune surveillance and tumor immunity. A key feature of γδ T cells is their ability to recognize a broad array of stress-induced metabolites and tumor-associated metabolites not necessarily via antigen processing or presentation of MHC-like proteins (22, 25), which can be classified based on different Vδ chain expression. Different Vδ1+ T cell clones have demonstrated the ability to recognize respective lipid antigens presented by human CD1 proteins, including various endogenous phospholipids and glycolipids presented by CD1a (127), CD1b (128), and CD1c (125), as well as CD1d loaded with α-GalCer and sulfatide (28) (Table 4; Figure 5). Vδ1+ T cells can also be sorted using MR1 tetramers loaded with 5-OP-RU (148) and can differentiate with the stimulation of HLA-A2 from hematopoietic stem or progenitor cells (149). In other cases, Vδ1+ T cells interact with protein stress ligands such as non-classical MHC class I proteins, MICA/B and ULBPs, via Vδ1+ T cell surface NKG2D receptor, and annexin A2, a molecule linked to tumor cell stress and apoptosis (150, 151). Vδ1+ cells also engage ligands like heat shock proteins (HSPs) and the endothelial protein C receptor (EPCR) (147), which are upregulated under cellular stress. Functionally, Vδ1+ cells are shown to be dysregulated in malignancies such as colorectal cancer (152), or a candidate target of immunotherapy against neuroblastoma (153).
Vδ2+ T cells, represented by Vγ9Vδ2 T cells, the dominant circulating subset in humans, are activated by non-peptidic phosphorylated intermediates of the mevalonate pathway, collectively known as phosphoantigens (pAgs) (Figure 6), which are often upregulated in transformed or infected cells (25, 154). These cells require engagement of the butyrophilin family members BTN3A1 and BTN2A1, which cooperatively mediate γδ TCR activation (155). Recent structural studies have revealed that BTN2A1 binds to the lateral surface of the γδ TCR, leaving the apical region accessible for co-engagement by a second ligand in a BTN3A1-dependent manner. BTN2A1 and BTN3A1 also directly interact in cis to form multimeric W-shaped complexes, and this coordinated interaction is critical for full γδ TCR activation (156). Vδ2+ cells have also been shown to respond to human MutS homologue 2 (hMSH2) (157), a DNA repair protein aberrantly expressed on tumor cells, as well as bacterial superantigens and certain microbial-associated proteins (21, 158). Recent work has further expanded the functional scope of this subset by identifying a population of MR1-autoreactive Vγ9Vδ2 T cells that recognize MR1-self-antigen complexes in a butyrophilin-independent, CDR3δ-dependent manner, revealing a novel avenue for γδ T cell involvement in antigen-specific immunity (132).
Differently, a Vδ3+ T cell clone has shown ability to sense cell stress factors and recognize annexin A2 that binds to cell surface lipids (151) on tumor cells responding to stress and depending on the induction of reactive oxygen species. Additionally, processed insulin and Igλ light chains of host origin have been reported to stimulate γδ T cells in multiple myeloma and insulinoma (159, 160). These diverse recognition mechanisms underscore the capacity of γδ T cells to serve as versatile sentinels in tumor detection.
Recent studies have revealed variable roles for γδ T cell subsets across different cancer types. In human breast cancers, tumor-infiltrating Vδ1+ T cells have been associated with improved prognosis and are enriched in the tumor epithelium, where they exert cytotoxic capacity against cancer cell lines (161). Conversely, in glioblastoma and certain lung cancers, γδ T cells have been shown to skew toward an IL-17-producing phenotype, which may promote tumor progression by enhancing angiogenesis and recruiting myeloid-derived suppressor cells (162, 163). In hematological malignancies such as leukemia and lymphoma, circulating Vγ9Vδ2+ T cells often exhibit cytotoxic activity and are being investigated for adoptive immunotherapy (134). Clinical trials are underway evaluating the safety and efficacy of expanded or engineered γδ T cells, though challenges remain in ensuring their persistence, tumor infiltration, and avoidance of exhaustion within the tumor microenvironment. For a more comprehensive overview of γδ T cell antitumor effector responses and therapeutic trials, please refer to the recent reviews by Arias-Badia et al., Hayday et al, and Schoünefeldt et al. (22, 25, 134), as we focus on discussing the activation of γδ T cells and its association with cancer metabolism.
Overall, γδ T cells demonstrate remarkable diversity to small molecules and protein ligands, and are not constrained by antigen-presenting molecules or MHC class I-like proteins. This grants γδ T cells enhanced flexibility in recognizing a variety of tumor-derived and stress-induced signals. γδ T cells, like MAIT cells, often retain cytotoxic potential in immunosuppressive environments and can exhibit partial resistance to exhaustion when phenotypically expressing PD-1+CTLA-4+TIGIT+, making them promising candidates for cancer immunotherapy. However, the duality of their roles, particularly the context-dependent effects of IL-17-producing γδ subsets, warrants careful consideration in therapeutic applications. Ongoing research is needed to elucidate the precise factors governing γδ T cell polarization and function within tumors, including their interactions with other immune cells, metabolic conditions, and cytokine milieu.
Therapeutic potential of unconventional T cells in cancer
Invariant T cell-based immune therapies are expected to overcome the restriction of the extensive polymorphism of HLA genes in individual patients and the dependence on HLA haplotype-matching for prolonging antigen stimulation. These restricting elements make conventional T cell-based immune therapies costly, time-consuming, and sometimes harmful or toxic in immunopathology. Inaccurate or partial HLA crossmatching can diminish efficacy or lead to adverse events, including graft-versus-host disease. These limitations are evident in recent therapeutic advances involving conventional T cells. For instance, Lifileucel, a tumor-infiltrating lymphocyte (TIL)-based therapy, received FDA approval for treatment-resistant metastatic melanoma after showing durable responses in roughly one-third of patients (164). Another example came with afamitresgene autoleucel, a TCR-engineered T cell therapy developed for HLA-A*02:01-positive synovial sarcoma, which achieved a 40% response rate (165). However, the treatment’s efficacy was constrained by its reliance on HLA restriction, confining eligibility to patients expressing HLA-A2 subtypes and still posing a risk of ‘on-target, off-tumor effects’ toxicities, where healthy tissues are inadvertently targeted, leading to severe or fatal side effects (166).
In this context, unconventional T cells offer an exciting alternative platform for extending the benefits of T cell-based immunotherapies to a wider patient base. Invariant T cells express conserved TCR domains that enable population-wide responses with minimal variability. Unlike the highly diverse αβ TCR repertoire of conventional T cells, which exhibits vast signatures up to 1011 variations within a single individual (167), invariant TCRs confer broad antigen recognition with limited specificity, allowing a rapid and innate-like activation. It is expected that responses in early tumor surveillance will lead to early detection of malignancies and early defense against emerging malignant cells by the existing high baseline frequency of unconventional T cells in tissues. This rapid responding kinetics of unconventional T cells occurs prior to the prolonged cancer antigen-stimulation and clonal expansion of conventional T cells, which often take place alongside the establishment of TME-mediated suppression and exhaustion under chronic stimulation (168). Unconventional T cells, while not associated with traditional memory, exhibit greater resistance to exhaustion than conventional αβ T cells (169), retain cytotoxic potential in certain immune-suppressive environments, bridge innate and adaptive responses, making them valuable in immunotherapy contexts. Their ability to recognize metabolite ligands through semi-invariant TCRs independent of peptide antigen processing and HLA restriction could bypass one of the key bottlenecks in current adoptive T cell approaches. Concepts like TIL therapy and TCR-engineered strategies, already proven in conventional T cells, could be adapted for invariant T cells by exploiting their unique recognition pathways.
NKT cell therapies
Most importantly, three different invariant T cell populations respond to distinct metabolite compounds. CD1-restricted T cells, mainly CD1d-restricted NKT cells, have been intensively tested in numerous pre-clinical studies (90, 111), as briefly summarized in Table 4. These studies generally apply α-GalCer in mouse models, stimulate dendritic cells with α-GalCer, transfer ex vivo-expanded NKT cells, expand NKT cells with α-GalCer-loaded CD1d protein, or generate invariant NKT cells with chimeric antigen receptor (CAR) expression, mostly leading to a prolonged survival or inhibited tumor growth in animals bearing metastatic tumor cells (108, 170–173). The α-GalCer-loaded APCs and synthetic glycolipid agonists can expand and activate iNKT cells, skewing them toward Th1-like subsets that favor antitumor responses. Modified α-GalCer analogs and nanoparticle delivery systems may potentiate iNKT-mediated anti-tumor responses (174, 175). Strategies to overcome iNKT cell anergy, such as anti-PD-1 checkpoint blockade and cytokine supplementation with IL-2 or G-CSF, are promising approaches. In one study, a blockade of PD-1/PD-L interactions during α-GalCer treatment preserved iNKT cell responsiveness and enhanced anti-metastatic activity by restoring IFN-γ production and NK cell cytotoxicity (104). Both autologous and allogeneic iNKT cells have shown safety and early signs of efficacy in clinical trials, while monoclonal antibody-based approaches have demonstrated the ability to either expand iNKT cells to enhance antitumor responses or deplete them in settings where they may promote tumor growth (176). To test the anti-cancer efficacy of NKT cells in humans, CAR-NKT cell therapies and recombinant TCRs (rTCRs) are under active investigation in clinical trials, including using direct α-GalCer injection (177), α-GalCer-pulsed monocyte-derived dendritic cells (178), adoptive transfer of autologous ex vivo-expanded NKT cells (179), and some trials with combined therapies (180), with overall outcomes of stable diseases or effectiveness in a small percentage of patients. It also appears that combined therapies may increase the efficacy of therapy in patients with head and neck squamous cell carcinoma using respective injection of expanded NKT cells and α-GalCer-pulsed blood cells to observe objective tumor regression in 50% of patients (180, 181). Expanding IFNγ-producing NKT cells in tumor tissues likely contributes to an enhanced protection of NKT cell-based anti-cancer immune therapies in multiple clinical trials.
γδ T cell therapies
Through responding to various stress-induced metabolite or protein ligands, an adoptive transfer of activated γδ T cells is promising as a candidate for immunotherapy. These innate-like T cells have shown greater resistance to exhaustion compared to αβ T cells and retained cytotoxicity in checkpoint-inhibited environments. These findings may indicate γδ T cells are ideal for adoptive T cell therapies, particularly in tumors resistant to conventional strategies (182). Approaches proposed or under investigation include the expansion of γδ T cells using phosphoantigen stimulation, followed by reinfusion in combination with cytokines such as IL-2 or IL-15 to enhance persistence and proliferation. Allogeneic γδ T cell therapies are also being discussed to create “off-the-shelf” products, circumventing the need for patient-specific cell manufacturing (183–185). CAR-γδ T cells are in development to enhance tumor specificity, and bispecific antibody platforms are being designed to simultaneously activate both γδ T cells and NK cells. These approaches show particular promise in hematologic malignancies but may also extend to solid tumors with improved cytokine engineering and tumor-homing strategies.
MR1T cell therapies
Through the activation of polar metabolites from bacterial and mammalian cells, MR1T, including MAIT and diverse MR1T cells, could potentially serve as the foundation for novel cancer vaccines or cell therapies using tumor-derived or synthetic MR1 ligands to drive antitumor responses. Although preclinical or clinical studies remain missing, targeting the MR1-TCR axis with synthetic MR1 ligands is expected to effectively enhance MR1T-mediated cytotoxicity or suppress MR1T cell function when dysregulation contributes to tumor growth. Combination strategies involving PD-1/PD-L1 checkpoint blockade have also been proposed to restore MR1T function in tumors where exhaustion markers are highly expressed, while cytokine support with IL-12 or IL-18 may synergistically boost effector activity (186).
Current challenges and future perspectives
One conceptual challenge in studying anti-cancer unconventional T cells is a poor understanding of the antigen structures, antigenic specificity, and TCR of unconventional T cells that induce efficient cancer cell killing in vitro and inhibit cancer growth in vivo. Due to non-peptidic nature of metabolite antigens, robust high-throughput methods are yet missing to identify and validate endogenous tumor-associated ligands presented by MR1 and CD1 via profiling a pool of differential metabolites in cancer vs. normal cells. Although some candidate ligands (e.g., 5-OP-RU or tumor-elevated phospholipids) have been proposed to induce antitumor reactivity, the spectrum of ligands driving optimal antitumor or regulatory responses remains poorly defined. As precise cancer metabolic conditions can shape cancer antigen landscape and specify unconventional T cell responses, knowledge is currently missing to link metabolic pathways and products in cancer cells with unconventional T cell activation, warranting further investigation of cancer cell-derived metabolite antigens.
A further conceptual challenge is to define tumor-specific and tumor-associated antigens, which have been used to differentiate peptide antigens that are solely expressed or just enriched in cancer cells. For polar metabolite antigens, little is known about their expression in cancer versus normal cells, because the definition of tumor-specific or associated metabolite antigens has to be validated by mass spectrometry or NMR detection of metabolite compounds in cells or from MHC class I-like proteins between various cancer cells and normal cells, comparatively. For the newly reported nucleoside or nucleobase analogs from a preprint or peer-reviewed articles for MAIT cell or diverse MR1T activation (18–20), their expression levels in cancer vs. normal cells remain unknown. For lipid metabolites that activate iNKT cells, determining the expression levels of tumor-associated CD1-presented lipid antigens is equally crucial. Thus, a full range of tumor-associated or specific metabolite antigens and their in vivo relevance remain elusive. Further, whether effector MAIT cells or NKT cells rely on differential expression levels of metabolite antigens to differentiate cancer cells from normal cells is also an imperative question in future studies.
Technical challenges also hamper the generation and delivery of unconventional T cell-targeted reagents for therapies. Major efforts are needed to cultivate and engineer unconventional T cells for expressing cancer tissue-targeting molecules, such as CAR-MAIT targeting the Her2 protein in breast cancer, CD19-targeted CAR-T against B cell malignancy, or tissue-homing chemotaxis receptors, allowing unconventional T cells to better reach cancer tissues. Although preclinical models show promise (Table 4), it remains unclear from a clinical standpoint whether the adoptively transferred unconventional T cells can efficiently migrate to and be detected in cancer tissues for cancer cell killing. Technological barriers to clinical translation also include the need for improved ligand delivery systems that enable stable, tumor-specific MR1 or CD1 ligand loading in vivo. Metabolite antigens are different from peptide antigens in chemical nature, therefore, the chemical formulations designed for delivering peptide antigens are likely not optimal for delivering polar and lipid metabolites. Early studies for delivering α-GalCer and other glycolipids, such as PEGylated (polyethylene glycol-formulated), lipid nanocarriers (187), liposome formulations with glycolipids or peptides (188–190), and codelivery with mRNA (191), are valuable to be further tested in lipid and polar metabolite antigen delivery.
Challenges in translational applications are also multifold. Long-term therapeutic efficacy usually depends on maintaining effector memory phenotypes and minimizing T cell exhaustion within the immunosuppressive tumor microenvironment (TME), involving strategies to enhance T cell infiltration and retention within solid tumors. This may require the development of metabolic reprogramming approaches, cytokine support systems (e.g., IL-7 or IL-15 co-administration), or engineering of resistance to TME-associated inhibitory pathways. Thus, a central challenge for the clinical success of unconventional T cell immunotherapies is to understand how MR1- or CD1-restricted T cells can be sustainably maintained with durable responses, while resisting cancer-driven immunosuppression and T cell exhaustion within tumor environments. Similar to the importance of correct peptide antigens in inducing sustainable conventional T cell responses for protection, the identification of metabolite antigens to induce durable unconventional T cell protection against cancer is a feasible method for designing novel anti-cancer therapies. However, different from the protective peptide antigens specifically restricted by individual HLA alleles, a protective metabolite antigen, if identified, can be applied to all patients as a universal treatment, regardless of their diverse HLA genetics.
A further relevant critical gap is the lack of clinically validated methods to monitor unconventional T cell trafficking and activity in real time. Emerging technologies such as non-invasive imaging using labeled TCR ligands, single-cell transcriptomics of tumor-infiltrating lymphocytes, and circulating T cell receptor repertoire profiling, and antigen loading on antigen-presenting cells for antitumor T cell activation offer promising avenues to track dynamic T cell response during therapy. Furthermore, the development of preclinical models that accurately recapitulate invariant T cell function in human tumors is needed to accelerate clinical translation. Humanized mouse models considering their comparative differences (192) offer promising platforms to evaluate the therapeutic potential of unconventional T cells in diverse cancer settings.
Together, a better understanding of antigen presentation dynamics, TCR repertoire diversity, and intratumoral localization of these T cells is needed to inform therapeutic design. Ultimately, bridging ligand discovery, engineering strategies for T cell persistence, and real-time monitoring tools will be key to unlocking the full clinical potential of these unconventional T cell populations in cancer immunotherapy (136). These efforts underscore the expanding frontier of T cell-based immunotherapies, where invariant T cell subsets may soon complement or enhance the current landscape shaped by conventional T cells.
Author contributions
AL: Conceptualization, Validation, Writing – review & editing, Writing – original draft. NA: Writing – original draft, Validation, Conceptualization, Writing – review & editing. SH: Writing – original draft, Funding acquisition, Validation, Conceptualization, Writing – review & editing, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. We thank funding support from Cancer Prevention and Research Institute of Texas (CPRIT) for grant RP240605 and National Institute of Allergy and Infectious Diseases (NIAID) for grant 1R01AI173245.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Rudolph MG, Stanfield RL, and Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. (2006) 24:419–66. doi: 10.1146/annurev.immunol.23.021704.115658
2. Roche PA and Cresswell P. Antigen processing and presentation mechanisms in myeloid cells. Microbiol Spectr. (2016) 4:1–14. doi: 10.1128/microbiolspec.MCHD-0008-2015
3. Koh C-H, Lee S, Kwak M, Kim B-S, and Chung Y. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp Mol Med. (2023) 55:2287–99. doi: 10.1038/s12276-023-01105-x
4. Giles JR, Globig AM, Kaech SM, and Wherry EJ. CD8(+) T cells in the cancer-immunity cycle. Immunity. (2023) 56:2231–53. doi: 10.1016/j.immuni.2023.09.005
5. Raskov H, Orhan A, Christensen JP, and Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer. (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
6. Hansen TH, Huang S, Arnold PL, and Fremont DH. Patterns of nonclassical MHC antigen presentation. Nat Immunol. (2007) 8:563–8. doi: 10.1038/ni1475
7. Mori L, Lepore M, and De Libero G. The immunology of CD1- and MR1-restricted T cells. Annu Rev Immunol. (2016) 34:479–510. doi: 10.1146/annurev-immunol-032414-112008
8. Godfrey DI, Le Nours J, Andrews DM, Uldrich AP, and Rossjohn J. Unconventional T cell targets for cancer immunotherapy. Immunity. (2018) 48:453–73. doi: 10.1016/j.immuni.2018.03.009
9. Huang S, Shahine A, Cheng TY, Chen YL, Ng SW, Balaji GR, et al. CD1 lipidomes reveal lipid-binding motifs and size-based antigen-display mechanisms. Cell. (2023) 186:4583–96.e13. doi: 10.1016/j.cell.2023.08.022
10. Moody DB, Zajonc DM, and Wilson IA. Anatomy of CD1-lipid antigen complexes. Nat Rev Immunol. (2005) 5:387–99. doi: 10.1038/nri1605
11. Huang S, Gilfillan S, Kim S, Thompson B, Wang X, Sant AJ, et al. MR1 uses an endocytic pathway to activate mucosal-associated invariant T cells. J Exp Med. (2008) 205:1201–11. doi: 10.1084/jem.20072579
12. Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, et al. Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature. (2003) 422:164–9. doi: 10.1038/nature01433
13. Lepore M, Kalinichenko A, Calogero S, Kumar P, Paleja B, Schmaler M, et al. Functionally diverse human T cells recognize non-microbial antigens presented by MR1. Elife. (2017) 6:e24476. doi: 10.7554/eLife.24476
14. Huang S and Moody DB. Donor-unrestricted T cells in the human CD1 system. Immunogenetics. (2016) 68:577–96. doi: 10.1007/s00251-016-0942-x
15. Tatituri RV, Watts GF, Bhowruth V, Barton N, Rothchild A, Hsu FF, et al. Recognition of microbial and mammalian phospholipid antigens by NKT cells with diverse TCRs. Proc Natl Acad Sci U S A. (2013) 110:1827–32. doi: 10.1073/pnas.1220601110
16. Corbett AJ, Eckle SBG, Birkinshaw RW, Liu L, Patel O, Mahony J, et al. T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature. (2014) 509:361–5. doi: 10.1038/nature13160
17. Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature. (2012) 491:717–23. doi: 10.1038/nature11605
18. Chancellor A, Constantin D, Berloffa G, Yang Q, Nosi V, Loureiro JP, et al. The carbonyl nucleobase adduct M3Ade is a potent antigen for adaptive polyclonal MR1-restricted T cells. Immunity. (2025) 58:431–47.e10. doi: 10.1016/j.immuni.2024.11.019
19. Vacchini A, Chancellor A, Yang Q, Colombo R, Spagnuolo J, Berloffa G, et al. Nucleobase adducts bind MR1 and stimulate MR1-restricted T cells. Sci Immunol. (2024) 9:eadn0126. doi: 10.1126/sciimmunol.adn0126
20. Huang S, Sharma M, Sallans L, Li C, Kh Z, Choubey D, et al. Metabolomics reveals nucleoside analogs for regulating mucosal-associated invariant T cell responses. bioRxiv. (2023). doi: 10.1101/2023.01.30.526332
21. Deseke M and Prinz I. Ligand recognition by the γδ TCR and discrimination between homeostasis and stress conditions. Cell Mol Immunol. (2020) 17:914–24. doi: 10.1038/s41423-020-0503-y
22. Hayday A, Dechanet-Merville J, Rossjohn J, and Silva-Santos B. Cancer immunotherapy by γδ T cells. Science. (2024) 386:eabq7248. doi: 10.1126/science.abq7248
23. Dhodapkar MV and Kumar V. Type II NKT cells and their emerging role in health and disease. J Immunol. (2017) 198:1015–21. doi: 10.4049/jimmunol.1601399
24. Boudinot P, Mondot S, Jouneau L, Teyton L, Lefranc MP, and Lantz O. Restricting nonclassical MHC genes coevolve with TRAV genes used by innate-like T cells in mammals. Proc Natl Acad Sci U S A. (2016) 113:E2983–92. doi: 10.1073/pnas.1600674113
25. Arias-Badia M, Chang R, and Fong L. γδ T cells as critical anti-tumor immune effectors. Nat Cancer. (2024) 5:1145–57. doi: 10.1038/s43018-024-00798-x
26. Yigit M, Basoglu OF, and Unutmaz D. Mucosal-associated invariant T cells in cancer: dual roles, complex interactions and therapeutic potential. Front Immunol. (2024) 15:1369236. doi: 10.3389/fimmu.2024.1369236
27. Borbulevych OY, Piepenbrink KH, and Baker BM. Conformational melding permits a conserved binding geometry in TCR recognition of foreign and self molecular mimics. J Immunol. (2011) 186:2950–8. doi: 10.4049/jimmunol.1003150
28. Luoma AM, Castro CD, Mayassi T, Bembinster LA, Bai L, Picard D, et al. Crystal structure of Vδ1 T cell receptor in complex with CD1d-sulfatide shows MHC-like recognition of a self-lipid by human γδ T cells. Immunity. (2013) 39:1032–42. doi: 10.1016/j.immuni.2013.11.001
29. Pellicci DG, Clarke AJ, Patel O, Mallevaey T, Beddoe T, Le Nours J, et al. Recognition of beta-linked self glycolipids mediated by natural killer T cell antigen receptors. Nat Immunol. (2011) 12:827–33. doi: 10.1038/ni.2076
30. Prota G, Berloffa G, Awad W, Vacchini A, Chancellor A, Schaefer V, et al. Mitochondria regulate MR1 protein expression and produce self-metabolites that activate MR1-restricted T cells. Proc Natl Acad Sci U S A. (2025) 122:e2418525122. doi: 10.1073/pnas.2418525122
31. Pellicci DG, Patel O, Kjer-Nielsen L, Pang SS, Sullivan LC, Kyparissoudis K, et al. Differential recognition of CD1d-alpha-galactosyl ceramide by the V beta 8.2 and V beta 7 semi-invariant NKT T cell receptors. Immunity. (2009) 31:47–59. doi: 10.1016/j.immuni.2009.04.018
32. Chancellor A, Alan Simmons R, Khanolkar RC, Nosi V, Beshirova A, Berloffa G, et al. Promiscuous recognition of MR1 drives self-reactive mucosal-associated invariant T cell responses. J Exp Med. (2023) 220:e20221939. doi: 10.1084/jem.20221939
33. Rossjohn J, Gras S, Miles JJ, Turner SJ, Godfrey DI, and McCluskey J. T cell antigen receptor recognition of antigen-presenting molecules. Annu Rev Immunol. (2015) 33:169–200. doi: 10.1146/annurev-immunol-032414-112334
34. Huang S, Martin E, Kim S, Yu L, Soudais C, Fremont DH, et al. MR1 antigen presentation to mucosal-associated invariant T cells was highly conserved in evolution. Proc Natl Acad Sci U S A. (2009) 106:8290–5. doi: 10.1073/pnas.0903196106
35. Huang S, Gilfillan S, Cella M, Miley MJ, Lantz O, Lybarger L, et al. Evidence for MR1 antigen presentation to mucosal-associated invariant T cells. J Biol Chem. (2005) 280:21183–93. doi: 10.1074/jbc.M501087200
36. McWilliam HEG, Eckle SBG, Theodossis A, Liu L, Chen Z, Wubben JM, et al. The intracellular pathway for the presentation of vitamin B–related antigens by the antigen-presenting molecule MR1. Nat Immunol. (2016) 17:531–7. doi: 10.1038/ni.3416
37. Riegert P, Wanner V, and Bahram S. Genomics, isoforms, expression, and phylogeny of the MHC class I-related MR1 gene. J Immunol. (1998) 161:4066–77. doi: 10.4049/jimmunol.161.8.4066
38. Leeansyah E, Svärd J, Dias J, Buggert M, Nyström J, Quigley MF, et al. Arming of MAIT cell cytolytic antimicrobial activity is induced by IL-7 and defective in HIV-1 infection. PloS Pathog. (2015) 11:e1005072. doi: 10.1371/journal.ppat.1005072
39. Priya R and Brutkiewicz RR. MR1 tetramer-based artificial APCs expand MAIT cells from human peripheral blood that effectively kill glioblastoma cells. Immunohorizons. (2021) 5:500–11. doi: 10.4049/immunohorizons.2100003
40. Tilloy F, Treiner E, Park SH, Garcia C, Lemonnier F, de la Salle H, et al. An invariant T cell receptor alpha chain defines a novel TAP-independent major histocompatibility complex class Ib-restricted alpha/beta T cell subpopulation in mammals. J Exp Med. (1999) 189:1907–21. doi: 10.1084/jem.189.12.1907
41. Eckle SB, Birkinshaw RW, Kostenko L, Corbett AJ, McWilliam HE, Reantragoon R, et al. A molecular basis underpinning the T cell receptor heterogeneity of mucosal-associated invariant T cells. J Exp Med. (2014) 211:1585–600. doi: 10.1084/jem.20140484
42. Jin H, Ladd NA, Peev AM, Swarbrick GM, Cansler M, Null M, et al. Deaza-modification of MR1 ligands modulates recognition by MR1-restricted T cells. Sci Rep. (2022) 12:22539. doi: 10.1038/s41598-022-26259-y
43. Harriff MJ, McMurtrey C, Froyd CA, Jin H, Cansler M, Null M, et al. MR1 displays the microbial metabolome driving selective MR1-restricted T cell receptor usage. Sci Immunol. (2018) 3:eaao2556. doi: 10.1126/sciimmunol.aao2556
44. Crowther MD, Dolton G, Legut M, Caillaud ME, Lloyd A, Attaf M, et al. Genome-wide CRISPR-Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nat Immunol. (2020) 21:178–85. doi: 10.1038/s41590-019-0578-8
45. Petley EV, Koay H-F, Henderson MA, Sek K, Todd KL, Keam SP, et al. MAIT cells regulate NK cell-mediated tumor immunity. Nat Commun. (2021) 12:4746. doi: 10.1038/s41467-021-25009-4
46. Ruf B, Catania VV, Wabitsch S, Ma C, Diggs LP, Zhang Q, et al. Activating mucosal-associated invariant T cells induces a broad antitumor response. Cancer Immunol Res. (2021) 9:1024–34. doi: 10.1158/2326-6066.Cir-20-0925
47. Yan J, Allen S, McDonald E, Das I, Mak JYW, Liu L, et al. MAIT cells promote tumor initiation, growth, and metastases via tumor MR1. Cancer Discov. (2020) 10:124–41. doi: 10.1158/2159-8290.CD-19-0569
48. Patel O, Kjer-Nielsen L, Le Nours J, Eckle SBG, Birkinshaw R, Beddoe T, et al. Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat Commun. (2013) 4:2142. doi: 10.1038/ncomms3142
49. Awad W, Ler GJM, Xu W, Keller AN, Mak JYW, Lim XY, et al. The molecular basis underpinning the potency and specificity of MAIT cell antigens. Nat Immunol. (2020) 21:400–11. doi: 10.1038/s41590-020-0616-6
50. Gherardin Nicholas A, Keller Andrew N, Woolley Rachel E, Le Nours J, Ritchie David S, Neeson Paul J, et al. Diversity of T cells restricted by the MHC class I-related molecule MR1 facilitates differential antigen recognition. Immunity. (2016) 44:32–45. doi: 10.1016/j.immuni.2015.12.005
51. Takasaki R, Nagae M, Takahashi Y, Ito E, Matsuoka T, Yasue W, et al. Development of ribityllumazine analogue as mucosal-associated invariant T cell ligands. Am Chem Soc. (2024) 146:29964–76. doi: 10.1021/jacs.4c12997
52. Mak JYW, Xu W, Reid RC, Corbett AJ, Meehan BS, Wang H, et al. Stabilizing short-lived Schiff base derivatives of 5-aminouracils that activate mucosal-associated invariant T cells. Nat Commun. (2017) 8:14599. doi: 10.1038/ncomms14599
53. Braganza CD, Motozono C, Sonoda K-H, Yamasaki S, Shibata K, Timmer MSM, et al. Agonistic or antagonistic mucosal-associated invariant T (MAIT) cell activity is determined by the 6-alkylamino substituent on uracil MR1 ligands. Chem Commun. (2020) 56:5291–4. doi: 10.1039/D0CC00247J
54. McInerney MP, Awad W, Souter MNT, Kang Y, Wang CJH, Chan Yew Poa K, et al. MR1 presents vitamin B6-related compounds for recognition by MR1-reactive T cells. Proc Natl Acad Sci U S A. (2024) 121:e2414792121. doi: 10.1073/pnas.2414792121
55. Peng Q, Huang R, Wang H, Xiao H, Wang Y, Zhai Z, et al. Immune characteristics and prognostic implications of mucosal-associated invariant T cells in acute myeloid leukemia. Cancer Immunol Immunother. (2023) 72:4399–414. doi: 10.1007/s00262-023-03574-5
56. Comont T, Nicolau-Travers ML, Bertoli S, Recher C, Vergez F, and Treiner E. MAIT cells numbers and frequencies in patients with acute myeloid leukemia at diagnosis: association with cytogenetic profile and gene mutations. Cancer Immunol Immunother. (2022) 71:875–87. doi: 10.1007/s00262-021-03037-9
57. Cornel AM, van der Sman L, van Dinter JT, Arrabito M, Dunnebach E, van Hoesel M, et al. Targeting pediatric cancers via T-cell recognition of the monomorphic MHC class I-related protein MR1. J Immunother Cancer. (2024) 12:e007538. doi: 10.1136/jitc-2023-007538
58. Bozorgmehr N, Hnatiuk M, Peters AC, and Elahi S. Depletion of polyfunctional CD26highCD8+ T cells repertoire in chronic lymphocytic leukemia. Exp Hematol Oncol. (2023) 12:13. doi: 10.1186/s40164-023-00375-5
59. Abos B, Gomez Del Moral M, Gozalbo-Lopez B, Lopez-Relano J, Viana V, and Martinez-Naves E. Human MR1 expression on the cell surface is acid sensitive, proteasome independent and increases after culturing at 26 degrees C. Biochem Biophys Res Commun. (2011) 411:632–6. doi: 10.1016/j.bbrc.2011.07.007
60. Mérédis F, Koen V, Sylvia F, Ken M, Kim De V, Elke De B, et al. Both mucosal-associated invariant and natural killer T-cell deficiency in multiple myeloma can be countered by PD-1 inhibition. Haematologica. (2017) 102:e266–e70. doi: 10.3324/haematol.2017.163758
61. Gherardin NA, Loh L, Admojo L, Davenport AJ, Richardson K, Rogers A, et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci Rep. (2018) 8:4159. doi: 10.1038/s41598-018-22130-1
62. Cheng OJ, Lebish EJ, Jensen O, Jacenik D, Trivedi S, Cacioppo J, et al. Mucosal-associated invariant T cells modulate innate immune cells and inhibit colon cancer growth. Scand J Immunol. (2024) 100:e13391. doi: 10.1111/sji.13391
63. Ling L, Lin Y, Zheng W, Hong S, Tang X, Zhao P, et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci Rep. (2016) 6:20358. doi: 10.1038/srep20358
64. Berzins SP, Wallace ME, Kannourakis G, and Kelly J. A role for MAIT cells in colorectal cancer. Front Immunol. (2020) 11:949. doi: 10.3389/fimmu.2020.00949
65. Won EJ, Ju JK, Cho YN, Jin HM, Park KJ, Kim TJ, et al. Clinical relevance of circulating mucosal-associated invariant T cell levels and their anti-cancer activity in patients with mucosal-associated cancer. Oncotarget. (2016) 7:76274–90. doi: 10.18632/oncotarget.11187
66. Zhang Q, Li P, Zhou W, and Fang S. Wang J. Participation of increased circulating MAIT cells in lung cancer: a pilot study. J Cancer. (2022) 13:1623–9. doi: 10.7150/jca.69415
67. Zumwalde NA, Haag JD, Gould MN, and Gumperz JE. Mucosal associated invariant T cells from human breast ducts mediate a Th17-skewed response to bacterially exposed breast carcinoma cells. Breast Cancer Res. (2018) 20:111. doi: 10.1186/s13058-018-1036-5
68. Hayee A, Kobayashi E, Motozono C, Hamana H, My HTV, Okada T, et al. Characterization of tumor-infiltrating lymphocyte-derived atypical TCRs recognizing breast cancer in an MR1-dependent manner. Cells. (2024) 13:1711. doi: 10.3390/cells13201711
69. Ruf B, Bruhns M, Babaei S, Kedei N, Ma L, Revsine M, et al. Tumor-associated macrophages trigger MAIT cell dysfunction at the HCC invasive margin. Cell. (2023) 186:3686–705.e32. doi: 10.1016/j.cell.2023.07.026
70. Duan M, Goswami S, Shi JY, Wu LJ, Wang XY, Ma JQ, et al. Activated and exhausted MAIT cells foster disease progression and indicate poor outcome in hepatocellular carcinoma. Clin Cancer Res. (2019) 25:3304–16. doi: 10.1158/1078-0432.Ccr-18-3040
71. Peterfalvi A, Gomori E, Magyarlaki T, Pal J, Banati M, Javorhazy A, et al. Invariant Valpha7.2-Jalpha33 TCR is expressed in human kidney and brain tumors indicating infiltration by mucosal-associated invariant T (MAIT) cells. Int Immunol. (2008) 20:1517–25. doi: 10.1093/intimm/dxn111
72. Kubica P, Lara-Velazquez M, Bam M, Siraj S, Ong I, Liu P, et al. MR1 overexpression correlates with poor clinical prognosis in glioma patients. Neurooncol Adv. (2021) 3:vdab034. doi: 10.1093/noajnl/vdab034
73. Runtsch LS, Stadlmeier M, Schon A, Muller M, and Carell T. Comparative nucleosomal reactivity of 5-formyl-uridine and 5-formyl-cytidine. Chemistry. (2021) 27:12747–52. doi: 10.1002/chem.202102159
74. Moldogazieva NT, Zavadskiy SP, Astakhov DV, and Terentiev AA. Lipid peroxidation: Reactive carbonyl species, protein/DNA adducts, and signaling switches in oxidative stress and cancer. Biochem Biophys Res Commun. (2023) 687:e149167. doi: 10.1016/j.bbrc.2023.149167
75. Hayes JD, Dinkova-Kostova AT, and Tew KD. Oxidative stress in cancer. Cancer Cell. (2020) 38:167–97. doi: 10.1016/j.ccell.2020.06.001
76. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. (2012) 22:547–60. doi: 10.1016/j.ccr.2012.08.014
77. Ricci JE. Tumor-induced metabolic immunosuppression: Mechanisms and therapeutic targets. Cell Rep. (2025) 44:115206. doi: 10.1016/j.celrep.2024.115206
78. Cornforth TV, Moyo N, Cole S, Lam EPS, Lobry T, Wolchinsky R, et al. Conserved allomorphs of MR1 drive the specificity of MR1-restricted TCRs. Front Oncol. (2024) 14:1419528. doi: 10.3389/fonc.2024.1419528
79. Suckling RJ, Pamukcu C, Simmons RA, Fonseca D, Grant E, Harrison R, et al. Molecular basis underpinning MR1 allomorph recognition by an MR1-restricted T cell receptor. Front Immunol. (2025) 16:1547664. doi: 10.3389/fimmu.2025.1547664
80. Dolton G, Thomas H, Tan LR, Rius Rafael C, Doetsch S, Ionescu GA, et al. MHC-related protein 1-restricted recognition of cancer via a semi-invariant TCR-alpha chain. J Clin Invest. (2025) 135:e181895. doi: 10.1172/JCI181895
81. Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science. (2021) 374:abe6474. doi: 10.1126/science.abe6474
82. Godfrey DI, Koay HF, McCluskey J, and Gherardin NA. The biology and functional importance of MAIT cells. Nat Immunol. (2019) 20:1110–28. doi: 10.1038/s41590-019-0444-8
83. Vacchini A, Chancellor A, Spagnuolo J, Mori L, and De Libero G. MR1-restricted T cells are unprecedented cancer fighters. Front Immunol. (2020) 11:751. doi: 10.3389/fimmu.2020.00751
84. Zou Z, Dou X, Li Y, Zhang Z, Wang J, Gao B, et al. RNA m(5)C oxidation by TET2 regulates chromatin state and leukaemogenesis. Nature. (2024) 634:986–94. doi: 10.1038/s41586-024-07969-x
85. Li Y, Xue M, Deng X, Dong L, Nguyen LXT, Ren L, et al. TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal. Cell Stem Cell. (2023) 30:1072–90.e10. doi: 10.1016/j.stem.2023.07.001
86. Gao Q, Shen K, and Xiao M. TET2 mutation in acute myeloid leukemia: biology, clinical significance, and therapeutic insights. Clin Epigenetics. (2024) 16:155. doi: 10.1186/s13148-024-01771-2
87. Sundström P, Szeponik L, Ahlmanner F, Sundquist M, Wong JSB, Lindskog EB, et al. Tumor-infiltrating mucosal-associated invariant T (MAIT) cells retain expression of cytotoxic effector molecules. Oncotarget. (2019) 10:2810–23. doi: 10.18632/oncotarget.26866
88. Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. (2017) 169:1342–56.e16. doi: 10.1016/j.cell.2017.05.035
89. Van Rhijn I, Godfrey DI, Rossjohn J, and Moody DB. Lipid and small-molecule display by CD1 and MR1. Nat Rev Immunol. (2015) 15:643–54. doi: 10.1038/nri3889
90. Exley MA, Dellabona P, and Casorati G. Exploiting CD1-restricted T cells for clinical benefit. Mol Immunol. (2021) 132:126–31. doi: 10.1016/j.molimm.2020.12.015
91. Huang S, Cheng TY, Young DC, Layre E, Madigan CA, Shires J, et al. Discovery of deoxyceramides and diacylglycerols as CD1b scaffold lipids among diverse groove-blocking lipids of the human CD1 system. Proc Natl Acad Sci U S A. (2011) 108:19335–40. doi: 10.1073/pnas.1112969108
92. Cox D, Fox L, Tian R, Bardet W, Skaley M, Mojsilovic D, et al. Determination of cellular lipids bound to human CD1d molecules. PloS One. (2009) 4:e5325. doi: 10.1371/journal.pone.0005325
93. Fox LM, Cox DG, Lockridge JL, Wang X, Chen X, Scharf L, et al. Recognition of lyso-phospholipids by human natural killer T lymphocytes. PloS Biol. (2009) 7:e1000228. doi: 10.1371/journal.pbio.1000228
94. Yuan W, Kang SJ, Evans JE, and Cresswell P. Natural lipid ligands associated with human CD1d targeted to different subcellular compartments. J Immunol. (2009) 182:4784–91. doi: 10.4049/jimmunol.0803981
95. de Jong A, Cheng T-Y, Huang S, Gras S, Birkinshaw RW, Kasmar AG, et al. CD1a-autoreactive T cells recognize natural skin oils that function as headless antigens. Nat Immunol. (2014) 15:177–85. doi: 10.1038/ni.2790
96. Bagchi S, He Y, Zhang H, Cao L, Van Rhijn I, Moody DB, et al. CD1b-autoreactive T cells contribute to hyperlipidemia-induced skin inflammation in mice. J Clin Invest. (2017) 127:2339–52. doi: 10.1172/jci92217
97. Lepore M, de Lalla C, Gundimeda SR, Gsellinger H, Consonni M, Garavaglia C, et al. A novel self-lipid antigen targets human T cells against CD1c(+) leukemias. J Exp Med. (2014) 211:1363–77. doi: 10.1084/jem.20140410
98. Porcelli S, Yockey CE, Brenner MB, and Balk SP. Analysis of T cell antigen receptor (TCR) expression by human peripheral blood CD4-8- alpha/beta T cells demonstrates preferential use of several V beta genes and an invariant TCR alpha chain. J Exp Med. (1993) 178:1–16. doi: 10.1084/jem.178.1.1
99. Jahng A, Maricic I, Aguilera C, Cardell S, Halder RC, and Kumar V. Prevention of autoimmunity by targeting a distinct, noninvariant CD1d-reactive T cell population reactive to sulfatide. J Exp Med. (2004) 199:947–57. doi: 10.1084/jem.20031389
100. Exley MA, Tahir SM, Cheng O, Shaulov A, Joyce R, Avigan D, et al. A major fraction of human bone marrow lymphocytes are Th2-like CD1d-reactive T cells that can suppress mixed lymphocyte responses. J Immunol. (2001) 167:5531–4. doi: 10.4049/jimmunol.167.10.5531
101. Cui G, Abe S, Kato R, and Ikuta K. Insights into the heterogeneity of iNKT cells: tissue-resident and circulating subsets shaped by local microenvironmental cues. Front Immunol. (2024) 15:1349184. doi: 10.3389/fimmu.2024.1349184
102. Speir M, Hermans IF, and Weinkove R. Engaging natural killer T cells as ‘Universal helpers’ for vaccination. Drugs. (2017) 77:1–15. doi: 10.1007/s40265-016-0675-z
103. Praveena T and Le Nours J. State of play in the molecular presentation and recognition of anti-tumor lipid-based analogues. Front Immunol. (2024) 15:1479382. doi: 10.3389/fimmu.2024.1479382
104. Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, et al. PD-1/PD-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant NKT cells. J Immunol. (2009) 182:2816–26. doi: 10.4049/jimmunol.0803648
105. Kerzerho J, Yu ED, Barra CM, Alari-Pahissa E, Girardi E, Harrak Y, et al. Structural and functional characterization of a novel nonglycosidic type I NKT agonist with immunomodulatory properties. J Immunol. (2012) 188:2254–65. doi: 10.4049/jimmunol.1103049
106. Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced anti-lymphoma activity of CAR19-iNKT cells underpinned by dual CD19 and CD1d targeting. Cancer Cell. (2018) 34:596–610.e11. doi: 10.1016/j.ccell.2018.08.017
107. Simon B, Wiesinger M, März J, Wistuba-Hamprecht K, Weide B, Schuler-Thurner B, et al. The generation of CAR-transfected natural killer T cells for the immunotherapy of melanoma. Int J Mol Sci. (2018) 19:2365. doi: 10.3390/ijms19082365
108. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity. vivo. J Clin Invest. (2016) 126:2341–55. doi: 10.1172/jci83476
109. Petersen TR, Sika-Paotonu D, Knight DA, Simkins HM, and Hermans IF. Exploiting the role of endogenous lymphoid-resident dendritic cells in the priming of NKT cells and CD8+ T cells to dendritic cell-based vaccines. PloS One. (2011) 6:e17657. doi: 10.1371/journal.pone.0017657
110. Chang DH, Deng H, Matthews P, Krasovsky J, Ragupathi G, Spisek R, et al. Inflammation-associated lysophospholipids as ligands for CD1d-restricted T cells in human cancer. Blood. (2008) 112:1308–16. doi: 10.1182/blood-2008-04-149831
111. Lee MS and Webb TJ. Novel lipid antigens for NKT cells in cancer. Front Immunol. (2023) 14:1173375. doi: 10.3389/fimmu.2023.1173375
112. Terabe M and Berzofsky JA. The immunoregulatory role of type I and type II NKT cells in cancer and other diseases. Cancer Immunology Immunother. (2014) 63:199–213. doi: 10.1007/s00262-013-1509-4
113. O’Neill KC, Liapis E, Harris BT, Perlin DS, and Carter CL. Mass spectrometry imaging discriminates glioblastoma tumor cell subpopulations and different microvascular formations based on their lipid profiles. Sci Rep. (2022) 12:17069. doi: 10.1038/s41598-022-22093-4
114. Guri Y, Colombi M, Dazert E, Hindupur SK, Roszik J, Moes S, et al. mTORC2 promotes tumorigenesis via lipid synthesis. Cancer Cell. (2017) 32:807–23.e12. doi: 10.1016/j.ccell.2017.11.011
115. Nielson JR and Rutter JP. Lipid-mediated signals that regulate mitochondrial biology. J Biol Chem. (2018) 293:7517–21. doi: 10.1074/jbc.R117.001655
116. Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, and Cravatt BF. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell. (2010) 140:49–61. doi: 10.1016/j.cell.2009.11.027
117. Neparidze N and Dhodapkar MV. Harnessing CD1d-restricted T cells toward antitumor immunity in humans. Ann N Y Acad Sci. (2009) 1174:61–7. doi: 10.1111/j.1749-6632.2009.04931.x
118. Russano AM, Agea E, Corazzi L, Postle AD, De Libero G, Porcelli S, et al. Recognition of pollen-derived phosphatidyl-ethanolamine by human CD1d-restricted gammadelta+ T cells. J Allergy Clin Immunol. (2006) 117:1178–84. doi: 10.1016/j.jaci.2006.01.001
119. Russano AM, Bassotti G, Agea E, Bistoni O, Mazzocchi A, Morelli A, et al. CD1-restricted recognition of exogenous and self-lipid antigens by duodenal gammadelta+ T lymphocytes. J Immunol. (2007) 178:3620–6. doi: 10.4049/jimmunol.178.6.3620
120. Lee MS, Sun W, and Webb TJ. Sphingosine kinase blockade leads to increased natural killer T cell responses to mantle cell lymphoma. Cells. (2020) 9:1030. doi: 10.3390/cells9041030
121. Fhaner CJ, Liu S, Ji H, Simpson RJ, and Reid GE. Comprehensive lipidome profiling of isogenic primary and metastatic colon adenocarcinoma cell lines. Anal Chem. (2012) 84:8917–26. doi: 10.1021/ac302154g
122. Dória ML, Cotrim CZ, Simões C, Macedo B, Domingues P, Domingues MR, et al. Lipidomic analysis of phospholipids from human mammary epithelial and breast cancer cell lines. J Cell Physiol. (2013) 228:457–68. doi: 10.1002/jcp.24152
123. Melum E, Jiang X, Baker KD, Macedo MF, Fritsch J, Dowds CM, et al. Control of CD1d-restricted antigen presentation and inflammation by sphingomyelin. Nat Immunol. (2019) 20:1644–55. doi: 10.1038/s41590-019-0504-0
124. Zhou Z, Zhang C, Xia C, Chen W, Zhu H, Shang P, et al. Enhanced antitumor effects by chemical modified IGb3 analogues. Mol Cancer Ther. (2011) 10:1375–84. doi: 10.1158/1535-7163.Mct-11-0030
125. Gentilini MV, Pérez ME, Fernández PM, Fainboim L, and Arana E. The tumor antigen N-glycolyl-GM3 is a human CD1d ligand capable of mediating B cell and natural killer T cell interaction. Cancer Immunol Immunother. (2016) 65:551–62. doi: 10.1007/s00262-016-1812-y
126. Wu DY, Segal NH, Sidobre S, Kronenberg M, and Chapman PB. Cross-presentation of disialoganglioside GD3 to natural killer T cells. J Exp Med. (2003) 198:173–81. doi: 10.1084/jem.20030446
127. Wegrecki M, Ocampo TA, Gunasinghe SD, von Borstel A, Tin SY, Reijneveld JF, et al. Atypical sideways recognition of CD1a by autoreactive γδ T cell receptors. Nat Commun. (2022) 13:3872. doi: 10.1038/s41467-022-31443-9
128. Reijneveld JF, Ocampo TA, Shahine A, Gully BS, Vantourout P, Hayday AC, et al. Human γδ T cells recognize CD1b by two distinct mechanisms. Proc Natl Acad Sci U S A. (2020) 117:22944–52. doi: 10.1073/pnas.2010545117
129. Roberts TJ, Sriram V, Spence PM, Gui M, Hayakawa K, Bacik I, et al. Recycling CD1d1 molecules present endogenous antigens processed in an endocytic compartment to NKT cells. J Immunol. (2002) 168:5409–14. doi: 10.4049/jimmunol.168.11.5409
130. Bunney PE, Zink AN, Holm AA, Billington CJ, and Kotz CM. Orexin activation counteracts decreases in nonexercise activity thermogenesis (NEAT) caused by high-fat diet. Physiol Behav. (2017) 176:139–48. doi: 10.1016/j.physbeh.2017.03.040
131. Rice MT, von Borstel A, Chevour P, Awad W, Howson LJ, Littler DR, et al. Recognition of the antigen-presenting molecule MR1 by a Vδ3(+) γδ T cell receptor. Proc Natl Acad Sci U S A. (2021) 118:e2110288118. doi: 10.1073/pnas.2110288118
132. Loureiro JP, Vacchini A, Berloffa G, Devan J, Schaefer V, Nosi V, et al. Recognition of MR1-antigen complexes by TCR vgamma9Vdelta2. Front Immunol. (2025) 16:1519128. doi: 10.3389/fimmu.2025.1519128
133. Treiner E. Mucosal-associated invariant T cells in hematological Malignancies: Current knowledge, pending questions. Front Immunol. (2023) 14:1160943. doi: 10.3389/fimmu.2023.1160943
134. Schönefeldt S, Wais T, Herling M, Mustjoki S, Bekiaris V, Moriggl R, et al. The diverse roles of γδ T cells in cancer: from rapid immunity to aggressive lymphoma. Cancers. (2021) 13:6212. doi: 10.3390/cancers13246212
135. Rong L, Li K, Li R, Liu HM, Sun R, and Liu XY. Analysis of tumor-infiltrating gamma delta T cells in rectal cancer. World J Gastroenterol. (2016) 22:3573–80. doi: 10.3748/wjg.v22.i13.3573
136. Dogan M, Karhan E, Kozhaya L, Placek L, Chen X, Yigit M, et al. Engineering human MAIT cells with chimeric antigen receptors for cancer immunotherapy. J Immunol. (2022) 209:1523–31. doi: 10.4049/jimmunol.2100856
137. Rischer M, Pscherer S, Duwe S, Vormoor J, Jürgens H, and Rossig C. Human gammadelta T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br J Haematol. (2004) 126:583–92. doi: 10.1111/j.1365-2141.2004.05077.x
138. Wallet MA, Nishimura T, Del Casale C, Lebid A, Salantes B, Santostefano K, et al. Induced pluripotent stem cell-derived gamma delta CAR-T cells for cancer immunotherapy. Blood. (2021) 138:2771. doi: 10.1182/blood-2021-149095
139. Wang Z, Wang M, Chen J, Zhang L, Zhang L, and Yu L. MR1-restricted T cells: the new dawn of cancer immunotherapy. Biosci Rep. (2020) 40:BSR20202962. doi: 10.1042/BSR20202962
140. Hammond TC, Purbhoo MA, Kadel S, Ritz J, Nikiforow S, Daley H, et al. A phase 1/2 clinical trial of invariant natural killer T cell therapy in moderate-severe acute respiratory distress syndrome. Nat Commun. (2024) 15:974. doi: 10.1038/s41467-024-44905-z
141. Legut M, Cole DK, and Sewell AK. The promise of gammadelta T cells and the gammadelta T cell receptor for cancer immunotherapy. Cell Mol Immunol. (2015) 12:656–68. doi: 10.1038/cmi.2015.28
142. Rao A, Agrawal A, Borthakur G, Battula VL, and Maiti A. Gamma delta T cells in acute myeloid leukemia: biology and emerging therapeutic strategies. J Immunother Cancer. (2024) 12:e007981. doi: 10.1136/jitc-2023-007981
143. Escriba-Garcia L, Alvarez-Fernandez C, Tellez-Gabriel M, Sierra J, and Briones J. Dendritic cells combined with tumor cells and alpha-galactosylceramide induce a potent, therapeutic and NK-cell dependent antitumor immunity in B cell lymphoma. J Transl Med. (2017) 15:115. doi: 10.1186/s12967-017-1219-3
144. Parekh VV, Wilson MT, Olivares-Villagomez D, Singh AK, Wu L, Wang CR, et al. Glycolipid antigen induces long-term natural killer T cell anergy in mice. J Clin Invest. (2005) 115:2572–83. doi: 10.1172/JCI24762
145. Cordova A, Toia F, La Mendola C, Orlando V, Meraviglia S, Rinaldi G, et al. Characterization of human γδ T lymphocytes infiltrating primary Malignant melanomas. PloS One. (2012) 7:e49878. doi: 10.1371/journal.pone.0049878
146. Nussbaumer O and Koslowski M. The emerging role of γδ T cells in cancer immunotherapy. Immuno-Oncology Technol. (2019) 1:3–10. doi: 10.1016/j.iotech.2019.06.002
147. Willcox CR, Pitard V, Netzer S, Couzi L, Salim M, Silberzahn T, et al. Cytomegalovirus and tumor stress surveillance by binding of a human γδ T cell antigen receptor to endothelial protein C receptor. Nat Immunol. (2012) 13:872–9. doi: 10.1038/ni.2394
148. Le Nours J, Gherardin NA, Ramarathinam SH, Awad W, Wiede F, Gully BS, et al. A class of γδ T cell receptors recognize the underside of the antigen-presenting molecule MR1. Science. (2019) 366:1522–7. doi: 10.1126/science.aav3900
149. Benveniste PM, Roy S, Nakatsugawa M, Chen ELY, Nguyen L, Millar DG, et al. Generation and molecular recognition of melanoma-associated antigen-specific human γδ T cells. Sci Immunol. (2018) 3:eaav4036. doi: 10.1126/sciimmunol.aav4036
150. Wrobel P, Shojaei H, Schittek B, Gieseler F, Wollenberg B, Kalthoff H, et al. Lysis of a Broad Range of Epithelial Tumour Cells by Human γδ T Cells: Involvement of NKG2D ligands and T-cell Receptor- versus NKG2D-dependent Recognition. Scandinavian J Immunol. (2007) 66:320–8. doi: 10.1111/j.1365-3083.2007.01963.x
151. Marlin R, Pappalardo A, Kaminski H, Willcox CR, Pitard V, Netzer S, et al. Sensing of cell stress by human γδ TCR-dependent recognition of annexin A2. Proc Natl Acad Sci. (2017) 114:3163–8. doi: 10.1073/pnas.1621052114
152. Stary V, Pandey RV, List J, Kleissl L, Deckert F, Kabiljo J, et al. Dysfunctional tumor-infiltrating Vdelta1 + T lymphocytes in microsatellite-stable colorectal cancer. Nat Commun. (2024) 15:6949. doi: 10.1038/s41467-024-51025-1
153. Schilbach K, Frommer K, Meier S, Handgretinger R, and Eyrich M. Immune response of human propagated gammadelta-T-cells to neuroblastoma recommend the Vdelta1+ subset for gammadelta-T-cell-based immunotherapy. J Immunother. (2008) 31:896–905. doi: 10.1097/CJI.0b013e31818955ad
154. Fournié JJ and Bonneville M. Stimulation of γδ T cells by phosphoantigens. Res Immunol. (1996) 147:338–47. doi: 10.1016/0923-2494(96)89648-9
155. Rigau M, Ostrouska S, Fulford TS, Johnson DN, Woods K, Ruan Z, et al. Butyrophilin 2A1 is essential for phosphoantigen reactivity by γδ T cells. Science. (2020) 367:eaay5516. doi: 10.1126/science.aay5516
156. Fulford TS, Soliman C, Castle RG, Rigau M, Ruan Z, Dolezal O, et al. Vgamma9Vdelta2 T cells recognize butyrophilin 2A1 and 3A1 heteromers. Nat Immunol. (2024) 25:1355–66. doi: 10.1038/s41590-024-01892-z
157. Dai Y, Chen H, Mo C, Cui L, and He W. Ectopically expressed human tumor biomarker MutS homologue 2 is a novel endogenous ligand that is recognized by human γδ T cells to induce innate anti-tumor/virus immunity. J Biol Chem. (2012) 287:16812–9. doi: 10.1074/jbc.M111.327650
158. Xi X, Han X, Li L, and Zhao Z. Identification of a new tuberculosis antigen recognized by γδ T cell receptor. Clin Vaccine Immunol. (2013) 20:530–9. doi: 10.1128/CVI.00584-12
159. Kim HT, Nelson EL, Clayberger C, Sanjanwala M, Sklar J, and Krensky AM. Gamma delta T cell recognition of tumor Ig peptide. J Immunol. (1995) 154:1614–23. doi: 10.4049/jimmunol.154.4.1614
160. de Sousa TR and Victor JR. Natural self-ligand gamma delta T cell receptors (γδTCRs) insight: the potential of induced igG. Vaccines (Basel). (2020) 8:436. doi: 10.3390/vaccines8030436
161. Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, et al. An innate-like Vdelta1(+) gammadelta T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Transl Med. (2019) 11:eaax9364. doi: 10.1126/scitranslmed.aax9364
162. Kang I, Kim Y, and Lee HK. gammadelta T cells as a potential therapeutic agent for glioblastoma. Front Immunol. (2023) 14:1273986. doi: 10.3389/fimmu.2023.1273986
163. Jin C, Lagoudas GK, Zhao C, Bullman S, Bhutkar A, Hu B, et al. Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell. (2019) 176:998–1013 e16. doi: 10.1016/j.cell.2018.12.040
164. Sarnaik AA, Hamid O, Khushalani NI, Lewis KD, Medina T, Kluger HM, et al. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J Clin Oncol. (2021) 39:2656–66. doi: 10.1200/JCO.21.00612
165. Mullard A. FDA approves first TCR-engineered T cell therapy, for rare soft-tissue cancer. Nat Rev Drug Discov. (2024) 23:731. doi: 10.1038/d41573-024-00134-z
166. Kishton RJ and Restifo NP. T cells lead the charge against solid tumors. Nat Cancer. (2024) 5:1762–4. doi: 10.1038/s43018-024-00860-8
167. Weng N-p. Numbers and odds: TCR repertoire size and its age changes impacting on T cell functions. Semin Immunol. (2023) 69:101810. doi: 10.1016/j.smim.2023.101810
168. Zakeri N, Hall A, Swadling L, Pallett LJ, Schmidt NM, Diniz MO, et al. Characterisation and induction of tissue-resident gamma delta T-cells to target hepatocellular carcinoma. Nat Commun. (2022) 13:1372. doi: 10.1038/s41467-022-29012-1
169. Katsnelson EN, Spengler A, Domenico J, Couts KL, Loh L, Gapin L, et al. Dysfunctional states of unconventional T-cell subsets in cancer. J Leukoc Biol. (2024) 115:36–46. doi: 10.1093/jleuko/qiad129
170. Cui J, Shin T, Kawano T, Sato H, Kondo E, Toura I, et al. Requirement for Valpha14 NKT cells in IL-12-mediated rejection of tumors. Science. (1997) 278:1623–6. doi: 10.1126/science.278.5343.1623
171. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Sato H, et al. Natural killer-like nonspecific tumor cell lysis mediated by specific ligand-activated Valpha14 NKT cells. Proc Natl Acad Sci U S A. (1998) 95:5690–3. doi: 10.1073/pnas.95.10.5690
172. Silk JD, Hermans IF, Gileadi U, Chong TW, Shepherd D, Salio M, et al. Utilizing the adjuvant properties of CD1d-dependent NK T cells in T cell-mediated immunotherapy. J Clin Invest. (2004) 114:1800–11. doi: 10.1172/jci22046
173. Liu K, Idoyaga J, Charalambous A, Fujii S, Bonito A, Mordoh J, et al. Innate NKT lymphocytes confer superior adaptive immunity via tumor-capturing dendritic cells. J Exp Med. (2005) 202:1507–16. doi: 10.1084/jem.20050956
174. Zhang L and Donda A. Alpha-galactosylceramide/CD1d-antibody fusion proteins redirect invariant natural killer T cell immunity to solid tumors and promote prolonged therapeutic responses. Front Immunol. (2017) 8:1417. doi: 10.3389/fimmu.2017.01417
175. Macho-Fernandez E, Cruz LJ, Ghinnagow R, Fontaine J, Bialecki E, Frisch B, et al. Targeted delivery of α-galactosylceramide to CD8α+ dendritic cells optimizes type I NKT cell-based antitumor responses. J Immunol. (2014) 193:961–9. doi: 10.4049/jimmunol.1303029
176. Boonchalermvichian C, Yan H, Gupta B, Rubin A, Baker J, and Negrin RS. invariant Natural Killer T cell therapy as a novel therapeutic approach in hematological Malignancies. Front Transplant. (2024) 3:1353803. doi: 10.3389/frtra.2024.1353803
177. Giaccone G, Punt CJ, Ando Y, Ruijter R, Nishi N, Peters M, et al. A phase I study of the natural killer T-cell ligand alpha-galactosylceramide (KRN7000) in patients with solid tumors. Clin Cancer Res. (2002) 8:3702–9.
178. Chang DH, Osman K, Connolly J, Kukreja A, Krasovsky J, Pack M, et al. Sustained expansion of NKT cells and antigen-specific T cells after injection of alpha-galactosyl-ceramide loaded mature dendritic cells in cancer patients. J Exp Med. (2005) 201:1503–17. doi: 10.1084/jem.20042592
179. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. (2006) 12:6079–86. doi: 10.1158/1078-0432.Ccr-06-0114
180. Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin Immunol. (2011) 138:255–65. doi: 10.1016/j.clim.2010.11.014
181. Nair S and Dhodapkar MV. Natural killer T cells in cancer immunotherapy. Front Immunol. (2017) 8:1178. doi: 10.3389/fimmu.2017.01178
182. Liu J, Wu M, Yang Y, Wang Z, He S, Tian X, et al. γδ T cells and the PD-1/PD-L1 axis: a love–hate relationship in the tumor microenvironment. J Trans Med. (2024) 22:553. doi: 10.1186/s12967-024-05327-z
183. Makkouk A, Yang XC, Barca T, Lucas A, Turkoz M, Wong JTS, et al. Off-the-shelf Vδ1 gamma delta T cells engineered with glypican-3 (GPC-3)-specific chimeric antigen receptor (CAR) and soluble IL-15 display robust antitumor efficacy against hepatocellular carcinoma. J Immunother Cancer. (2021) 9:e003441. doi: 10.1136/jitc-2021-003441
184. Wilhelm M, Kunzmann V, Eckstein S, Reimer P, Weissinger F, Ruediger T, et al. Gammadelta T cells for immune therapy of patients with lymphoid Malignancies. Blood. (2003) 102:200–6. doi: 10.1182/blood-2002-12-3665
185. Kunzmann V, Smetak M, Kimmel B, Weigang-Koehler K, Goebeler M, Birkmann J, et al. Tumor-promoting versus tumor-antagonizing roles of γδ T cells in cancer immunotherapy: results from a prospective phase I/II trial. J Immunother. (2012) 35:205–13. doi: 10.1097/CJI.0b013e318245bb1e
186. Sundstrom P, Dutta N, Rodin W, Hallqvist A, Raghavan S, and Quiding Jarbrink M. Immune checkpoint blockade improves the activation and function of circulating mucosal-associated invariant T (MAIT) cells in patients with non-small cell lung cancer. Oncoimmunology. (2024) 13:2312631. doi: 10.1080/2162402X.2024.2312631
187. Hung JT, Chiou SP, Tang YH, Huang JR, Lo FY, and Yu AL. Bioactivities and anti-cancer activities of NKT-stimulatory phenyl-glycolipid formulated with a PEGylated lipid nanocarrier. Drug Des Devel Ther. (2024) 18:5323–32. doi: 10.2147/DDDT.S484130
188. Romano C, Jiang H, Tahvili S, Wei P, Keiding UB, Clergeaud G, et al. Chemical synthesis and immunological evaluation of cancer vaccines based on ganglioside antigens and alpha-galactosylceramide. RSC Med Chem. (2024) 15:2718–28. doi: 10.1039/d4md00387j
189. de Haas AM, Stolk DA, Schetters STT, Goossens-Kruijssen L, Keuning E, Ambrosini M, et al. Vaccination with DC-SIGN-targeting alphaGC liposomes leads to tumor control, irrespective of suboptimally activated T-cells. Pharmaceutics. (2024) 16:1–14. doi: 10.3390/pharmaceutics16050581
190. Broecker F, Gotze S, Hudon J, Rathwell DCK, Pereira CL, Stallforth P, et al. Synthesis, liposomal formulation, and immunological evaluation of a minimalistic carbohydrate-alpha-galCer vaccine candidate. J Med Chem. (2018) 61:4918–27. doi: 10.1021/acs.jmedchem.8b00312
191. Guevara ML, Jilesen Z, Stojdl D, and Persano S. Codelivery of mRNA with alpha-Galactosylceramide Using a New Lipopolyplex Formulation Induces a Strong Antitumor Response upon Intravenous Administration. ACS Omega. (2019) 4:13015–26. doi: 10.1021/acsomega.9b00489
Keywords: unconventional T cells, polar metabolites, lipids, cancer, immunotherapy, MHC class I-related protein 1 (MR1), CD1
Citation: Laub A, Rodrigues de Almeida N and Huang S (2025) Unconventional T cells in anti-cancer immunity. Front. Immunol. 16:1618393. doi: 10.3389/fimmu.2025.1618393
Received: 26 April 2025; Accepted: 23 June 2025;
Published: 17 July 2025.
Edited by:
Luc Van Kaer, Vanderbilt University Medical Center, United StatesReviewed by:
Aquib Ehtram, La Jolla Institute for Immunology (LJI), United StatesAgnieszka Bojarska-Junak, Medical University of Lublin, Poland
Alessandro Vacchini, University Hospital of Basel, Switzerland
Copyright © 2025 Laub, Rodrigues de Almeida and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shouxiong Huang, c2h1YW5nQHR4YmlvbWVkLm9yZw==