 Yuxin Shang
Yuxin Shang Yuqing Pan
Yuqing Pan Lingling Xie
Lingling Xie Yan Zhao
Yan Zhao Wei Mao
Wei Mao Tingting Chen
Tingting Chen- 1Zhejiang Key Laboratory of Integrative Chinese and Western Medicine for Diagnosis and Treatment of Circulatory Diseases, Zhejiang Hospital (Affiliated Zhejiang Hospital, Zhejiang University School of Medicine), Hangzhou, Zhejiang, China
- 2Zhejiang Engineering Research Center for Precise Diagnosis and Innovative Traditional Chinese Medicine for Cardiovascular Diseases, Zhejiang Hospital (Affiliated Zhejiang Hospital, Zhejiang University School of Medicine), Hangzhou, Zhejiang, China
- 3The First School of Clinical Medicine, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, China
- 4Cardiovascular Department, Zhejiang Hospital (Affiliated Zhejiang Hospital, Zhejiang University School of Medicine), Hangzhou, Zhejiang, China
Atherosclerosis, the leading cause of cardiovascular morbidity and mortality worldwide, is now firmly established as a chronic immune-mediated disorder rather than a purely lipid-storage disease. Accumulating evidence has uncovered a previously underappreciated dimension of atherogenesis: the dynamic and bidirectional crosstalk between the nervous and immune systems. This neuroimmune axis, involving intricate communication between autonomic neural circuits and vascular immune cells, plays a central role in regulating arterial inflammation and plaque development. In particular, neuroimmune cardiovascular interfaces (NICIs)—specialized anatomical and functional hubs—have emerged as key sites for signal integration. Here, we review recent mechanistic insights into how sympathetic and parasympathetic pathways influence immune responses in atherosclerotic vessels and hematopoietic organs. We focus on the roles of neuromodulators such as pituitary adenylate cyclase-activating polypeptide (PACAP), calcitonin gene-related peptide (CGRP), neuropeptide Y (NPY), and galanin in shaping myeloid cell behavior, vascular tone, and endothelial activation. Additionally, we examine translational advances in neuromodulatory interventions—ranging from vagus nerve stimulation (VNS) to selective α7 nicotinic acetylcholine receptor (α7nAChR) agonists—that target these pathways to mitigate vascular inflammation in experimental models. These findings suggest that spatially resolved and temporally dynamic neuroimmune interactions constitute a critical layer of regulation in atherogenesis, offering a compelling framework for novel anti-inflammatory therapies beyond traditional lipid-lowering strategies.
1 Introduction
Atherosclerosis (AS) is a chronic inflammatory disease of the arterial wall and remains the leading cause of cardiovascular morbidity and mortality worldwide (1, 2). The disease originates from subendothelial accumulation of apolipoprotein B-containing lipoproteins (e.g. LDL), which triggers innate and adaptive immune responses leading to plaque formation (3). Despite advances in lipid-lowering and anti-inflammatory therapies, a significant proportion of patients continue to experience recurrent cardiovascular events (4, 5). This highlights an urgent need for novel therapeutic strategies that address not only lipid accumulation but also the underlying immune and inflammatory mechanisms driving disease progression.
Emerging evidence suggests that AS is not merely a lipid-driven disorder but also a chronic inflammatory disease shaped by neuroimmune dysregulation (6, 7). Among the neural regulators, the autonomic nervous system (ANS), comprising sympathetic (SNS) and parasympathetic (PNS) branches, plays a pivotal role in vascular inflammation. Chronic SNS activation enhances leukocyte recruitment and proinflammatory cytokine release via β-adrenergic signaling (8–10). On the other hand, PNS activity, particularly through vagus nerve, mediated cholinergic anti-inflammatory pathways, reduces inflammation by directly modulating macrophages, as shown in recent work indicating direct β2-adrenergic receptor (β2-AR) signaling to splenic myeloid cells, independent of a T-cell relay (11). This finding contrasts with the earlier paradigm proposed by Rosas-Ballina et al. (12), relaying vagal input to α7nAChR on macrophages. Notably, ChAT+ T cells have also been observed in ATLOs adjacent to adrenergic varicosities (7), suggesting that both mechanisms may operate in atherosclerosis but in context-dependent manners, direct β2-AR signaling may predominate in diffuse inflammatory milieus, whereas a ChAT+ T-cell relay could be engaged in structured adventitial lymphoid aggregates. Reconciling these pathways will require spatially resolved functional studies in vascular neuroimmune niches. Taken together, these contrasting effects, mediated by distinct neurotransmitters such as norepinephrine and acetylcholine, highlight the possible neuroimmune crosstalk relevant to AS pathophysiology (13, 14), rather than a uniformly established mechanism.
Specialized neuroimmune cardiovascular interfaces (NICIs) in the arterial adventitia enable direct communication between sensory nerves and immune aggregates, dynamically modulating local inflammation (7). Dysregulation of the neuro-immune axis, such as elevated corticotropin-releasing hormone (CRH) and reduced vagal activity, as measured by decreased heart rate variability and diminished efferent cholinergic signaling, which reflects reduced parasympathetic signaling through the vagus nerve, can exacerbate vascular inflammation and disrupt cardiovascular homeostasis (15, 16).
Therapeutically, targeting the neuroimmune axis has shown promise in preclinical models. For example, bioelectronic vagus nerve stimulation attenuates systemic inflammation via activation of the cholinergic anti-inflammatory pathway (17). Optogenetic approaches offer promising precision control of sympathetic circuits in neural regulation, but their direct therapeutic role in atherosclerosis remains to be investigated. On the other hand, clinical translation of neuromodulatory approaches remains limited by the difficulty of selectively targeting specific neural circuits without off-target effects or systemic interference. Emerging technologies, such as single-cell transcriptomics and spatial mapping, may help identify discrete neural-immune modules, offering new avenues for precise therapeutic targeting (18, 19).
This review synthesizes recent advances in understanding neuroimmune regulation in AS, with a particular focus on adventitial interfaces and macrophage plasticity. We further evaluate the therapeutic potential of both neuromodulatory and pharmacological strategies, highlight current limitations, and discuss future directions for precise, integrative interventions that bridge neural and immune modulation in vascular disease.
2 Immune dysregulation in atherosclerosis
AS begins with endothelial dysfunction, triggered by cardiovascular risk factors such as hyperlipidemia, hypertension, and oxidative stress (20). Dysfunctional endothelium promotes the retention and oxidation of low-density lipoprotein cholesterol (LDL-C) in the subendothelial space (21, 22). Oxidized LDL (oxLDL) acts as a damage-associated molecular pattern (DAMP), activating endothelial cells and inducing the expression of adhesion molecules, such as vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) (23, 24). These molecules mediate the recruitment of circulating monocytes and lymphocytes, facilitating the recruitment of monocytes and lymphocytes (25, 26) (Figure 1).
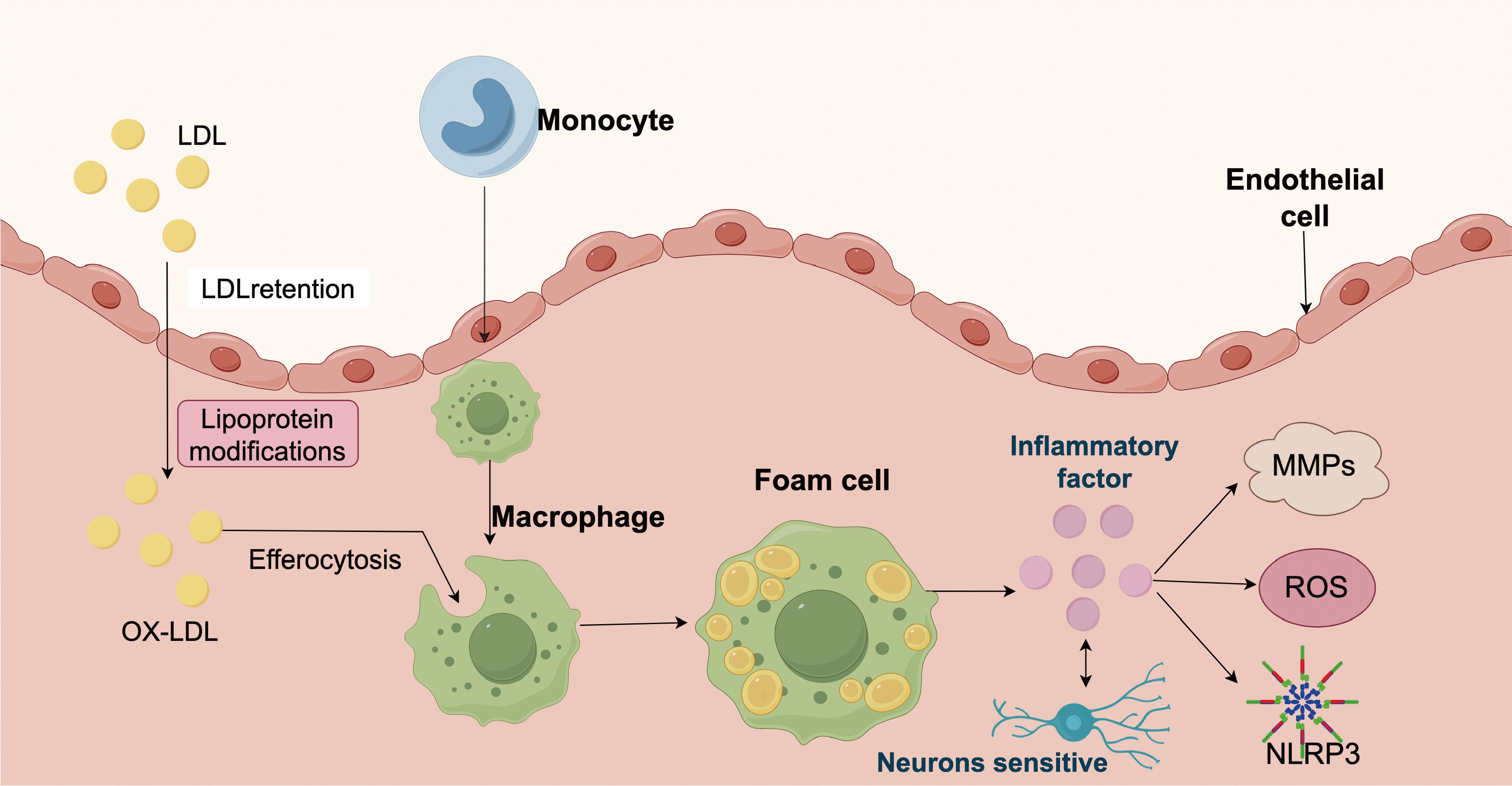
Figure 1. Schematic representation of the pathological mechanisms involved in atherosclerosis. Low-density lipoprotein (LDL) particles infiltrate the arterial intima, where they become retained and undergo various modifications, leading to the formation of oxidized LDL (ox-LDL). These modified lipoproteins trigger recruitment of circulating monocytes into the subendothelial space, where they differentiate into macrophages and engulf ox-LDL through efferocytosis. Lipid-laden macrophages transform into foam cells, a hallmark of early atherosclerotic lesions. Foam cells release inflammatory factors, which can activate endothelial cells, induce matrix metalloproteinases (MMPs), and stimulate the production of reactive oxygen species (ROS). These inflammatory mediators further amplify vascular inflammation, enhance oxidative stress, and activate the NLRP3 inflammasome. Activation of NLRP3 leads to further cytokine release, contributing to chronic inflammation and tissue remodeling. Sensory neurons in the vascular wall may also respond to inflammatory cues, suggesting a potential role in neuroimmune interactions during atherosclerosis progression.
Early innate immune activation, dominated by monocyte-derived macrophages, initiates lipid clearance but paradoxically sustains inflammation through cytokine secretion (27). Monocytes differentiate into macrophages upon entering the intima, where they engulf oxidized LDL to form foam cells, which is a hallmark of early fatty streaks (28, 29). Foam cells secrete pro-inflammatory cytokines, including tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), which amplifies local inflammation and promotes further leukocyte recruitment (30). Activated macrophages further release reactive oxygen species (ROS) and matrix metalloproteinases (MMPs), driving oxidative stress and extracellular matrix degradation, thereby weakening the fibrous cap and increase the risk of plaque rupture (31, 32).
Transition from fatty streaks to complex plaques involves macrophage phenotypic plasticity: pro-inflammatory M1 subsets exacerbate necrotic core formation via NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome activation (33). In contrast, alternatively activated M2 macrophages secrete TGF-β and promote tissue repair and fibrosis (34). Meanwhile, Adaptive immune responses are also engaged. Th1 and Th17 cells recognize modified self-antigens such as apoB-100, releasing INF-γ and IL-17 to perpetuate inflammation and impair vascular integrity (35).
3 Immunopathological crosstalk
The progression of AS is fundamentally orchestrated by a dynamic crosstalk between innate and adaptive immune cells within the arterial wall. Endothelial dysfunction triggered by oxLDL and hemodynamic stress initiates a cascade of immune recruitment and activation that dictates plaque evolution.
3.1 Innate immune orchestrators
Monocytes and macrophages are the dominant innate effectors in early atherogenesis (Figure 2). oxLDL-induced endothelial activation upregulates VCAM-1 and ICAM-1, facilitating monocyte adhesion and transendothelial transmigration (36). Intimal monocytes differentiate into macrophages that internalize oxLDL via scavenger receptors (e.g., CD36, LOX-1), forming lipid-laden foam cells, which is a process amplified by pro-inflammatory cytokines such as IL-1β and TNF-α (37, 38). This process amplifies local inflammation and recruiting additional immune cells and destabilizes the plaque microenvironment. In parallel, macrophages-derived ROS, contributes to matrix degradation and promote necrotic core formation (39).
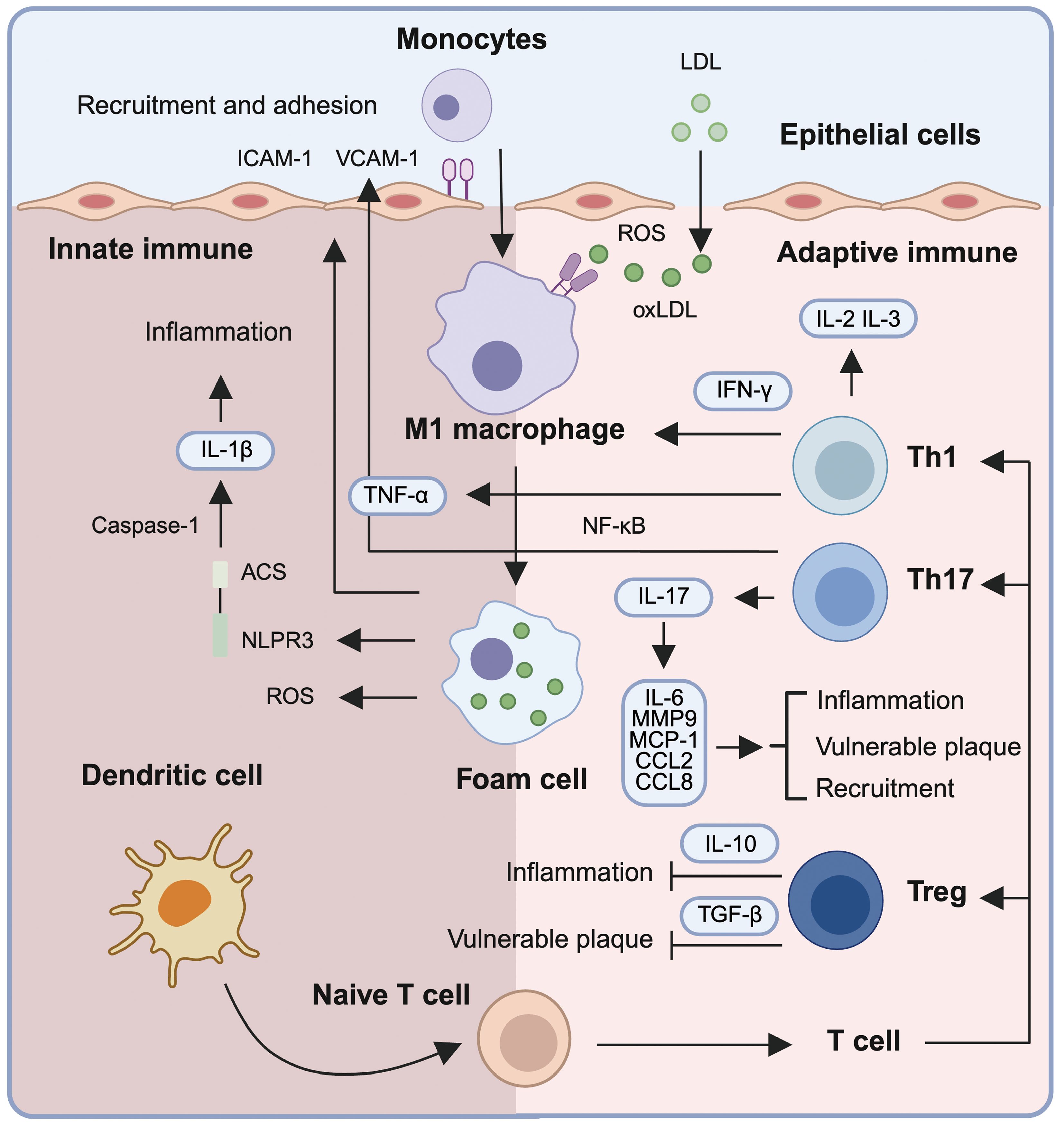
Figure 2. Immune mechanisms driving inflammation and plaque vulnerability in atherosclerosis. Low-density lipoprotein (LDL) infiltrates the subendothelial space and undergoes oxidative modification by reactive oxygen species (ROS), forming oxidized LDL (oxLDL). oxLDL triggers the upregulation of adhesion molecules (ICAM-1 and VCAM-1) on endothelial cells, facilitating monocyte recruitment and trans-endothelial migration. Within the intima, monocytes differentiate into M1 macrophages that internalize oxLDL, forming lipid-laden foam cells. These macrophages amplify inflammation via secretion of pro-inflammatory cytokines such as TNF-α and IL-1β, the latter being activated through the NLRP3 inflammasome. Foam cells and activated macrophages release mediators (e.g., IL-6, MCP-1, CCL2, CCL8, MMP9) that contribute to immune cell recruitment, plaque instability, and chronic vascular inflammation. Dendritic cells present antigens to naïve T cells, initiating adaptive immune responses. T helper (Th) subsets including Th1 and Th17 secrete IFN-γ and IL-17, respectively, further propagating inflammation and destabilizing plaques. In contrast, regulatory T cells (Tregs) produce IL-10 and TGF-β, exerting anti-inflammatory effects and promoting immune homeostasis. This coordinated interplay between innate and adaptive immune cells drives atherosclerotic progression and highlights potential targets for immunomodulatory therapy.
Dendritic cells (DCs) are another critical innate immune cell type, functioning as antigen-presenting cells (APCs) that process and present oxLDL-derived antigens to T cells (40, 41). Both conventional dendritic cells (cDCs) and plasmacytoid dendritic cells (pDCs) are involved in AS by promoting antigen-specific T cell activation, pro-inflammatory responses, and loss of tolerance (42, 43). The maturation of plaque DCs, driven by inflammatory stimuli and necrotic signals, contributes to atherogenesis through enhanced T cell activation and reduced anti-inflammatory signaling (Figure 2) (44, 45).
3.2 Adaptive immune modulators
CD4+ T cell subsets play divergent roles in atherogenesis: Th1 cells secrete IFN-γ to sustain macrophage activation, whereas Th17-derived IL-17 promotes endothelial dysfunction (Figure 2) (46, 47). Regulatory T cells (Tregs) counterbalance inflammation via the production of interleukin-10 (IL-10) and transforming growth factor-beta (TGF-β), yet their stability and function are impaired in the inflammatory plaque milieu due to oxLDL-induced endoplasmic reticulum stress and IL-6-mediated suppression of FoxP3 expression (48, 49).
B cells also contribute to plaque dynamics in a subset-specific manner. B2 cells exacerbate atherosclerosis through the production of IgG antibodies that that engage Fcγ receptors on macrophages, enhancing macrophage lipid uptake and inflammatory activation, while B1 cells produce IgM antibodies that facilitate apoptotic cell clearance and neutralize modified lipids, conferring atheroprotective effects (50, 51).
4 Neuroimmune communication in physiological states
Bidirectional communication between the nervous and immune systems is essential for maintaining homeostasis. Though a network of neurotransmitters, neuropeptides, and signaling pathways, the ANS, sensory fibers, and central neural circuits dynamically regulate immune cell development, activation, and trafficking. This section outlines key physiological mechanisms by which neural inputs influence immune function (Figure 3).
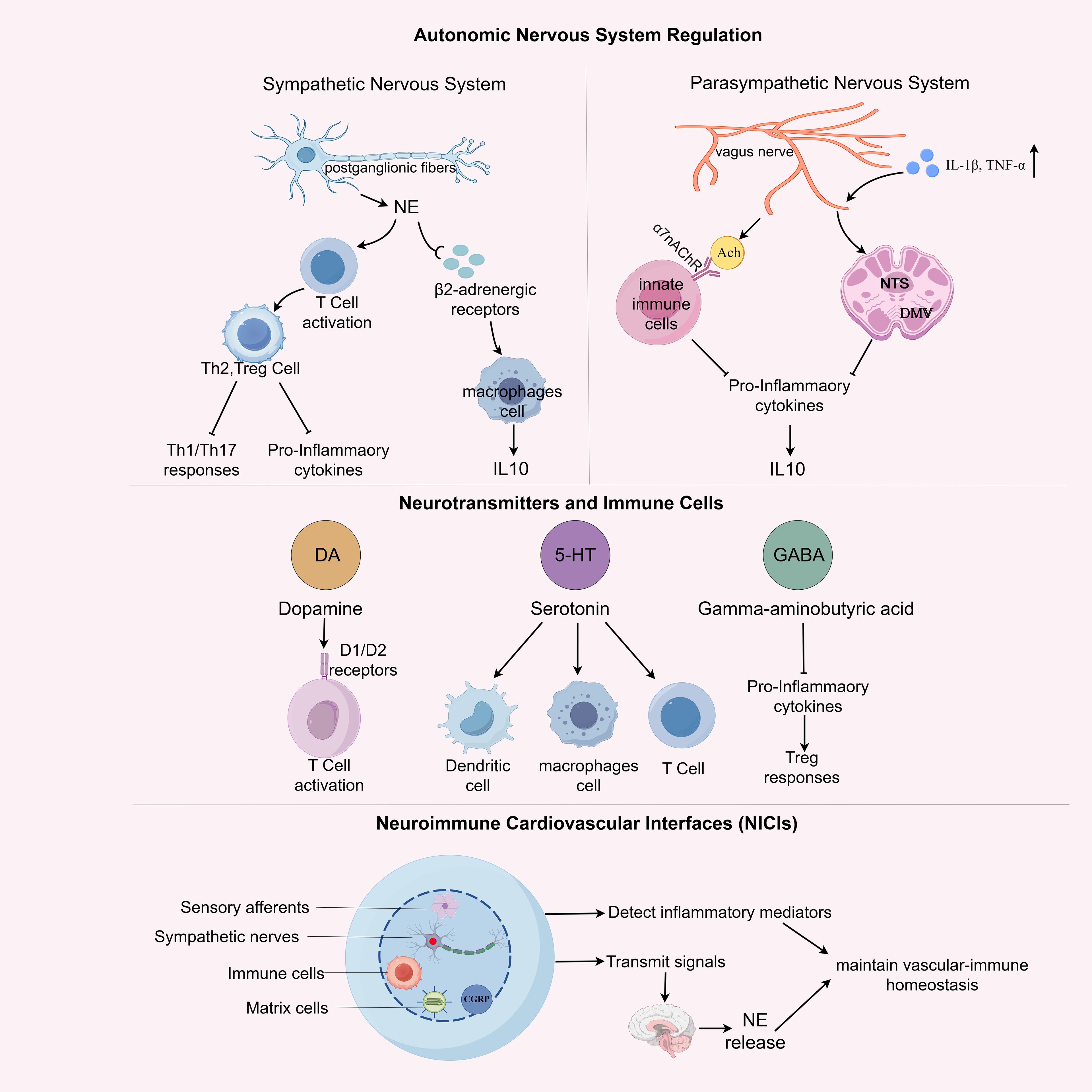
Figure 3. Neuroimmune regulation in physiological homeostasis. This schematic illustrates the coordinated regulation of immune function by the autonomic nervous system (ANS), neuromodulatory transmitters, and neuroimmune cardiovascular interfaces (NICIs) under physiological conditions. The sympathetic nervous system (SNS) exerts pro-inflammatory effects via norepinephrine (NE) and β2-adrenergic receptor (β2-AR) signaling on T cells and macrophages, favoring Th1/Th17 polarization and production of pro-inflammatory cytokines (e.g., TNF-α, IL-1β). In contrast, the parasympathetic nervous system (PNS), primarily via the vagus nerve, engages anti-inflammatory pathways through acetylcholine release and α7 nicotinic acetylcholine receptor (α7nAChR) activation on immune cells, promoting Th2/Treg responses and IL-10 secretion. Central neuromodulators such as dopamine, serotonin, and γ-aminobutyric acid (GABA) further influence immune cell activation, shaping T cell phenotypes and dendritic cell function. At the structural interface between nerves and immune cells, NICIs enable bidirectional crosstalk: sensory afferents detect local inflammatory cues, while efferent autonomic fibers modulate immune activity via neurotransmitter release into the perivascular niche. Together, these multi-layered neural mechanisms maintain vascular-immune homeostasis and prepare the groundwork for neuroimmune dysregulation in disease.
4.1 Autonomic neuro-immune regulation
The SNS and PNS represent the major autonomic branches regulating immune homeostasis.
The SNS modulates immune responses primarily through the release of norepinephrine (NE) from postganglionic fibers innervating lymphoid organs, including bone marrow, spleen, and lymph nodes. NE binds to β2-adrenergic receptors (β2-ARs) expressed on various immune cells (52). In macrophages, β2-AR activation suppresses pro-inflammatory cytokines and promotes anti-inflammatory IL-10 production, shifting them toward an M2-like phenotype (53). In CD4+ T cells, NE signaling enhances differentiation toward Th2 and Treg subsets, while dampening Th1/Th17 responses and inflammatory cytokine release (54).
Conversely, the PNS, primarily via the vagus nerve, exerts anti-inflammatory control through the cholinergic anti-inflammatory pathway. Acetylcholine (ACh), the primary neurotransmitter of the vagus nerve, binds to α7 nicotinic acetylcholine receptors (α7nAChR) on innate immune cells, particularly macrophages and dendritic cells, or potentially at upstream autonomic ganglia such as the celiac ganglion, as suggested by recent work (11). While α7nAChR signaling can downregulate TNF-α and IL-1β and enhance IL-10 production (55), the precise anatomical locus of this modulation remains incompletely defined and may vary across inflammatory contexts.
In parallel, vagal afferent fibers can detect peripheral inflammatory mediators, such as IL-1β and TNF-α, and relay these signals to the nucleus tractus solitarius (NTS) and dorsal motor nucleus of the vagus (DMV), thereby initiating a reflexive efferent anti-inflammatory output that constrains systemic immune activation (56).
Together, these autonomic circuits calibrate immune surveillance and tolerance in steady-state conditions, allowing the body to rapidly respond to threats while preventing excessive inflammation.
4.2 Neurotransmitters and immune cells
Beyond NE and ACh, a range of classical neurotransmitters and neuropeptides act as immune modulators.
Dopamine (DA) modulates both innate and adaptive immune responses. D1-like receptor signaling has been linked to enhanced T cell activation and pro-inflammatory cytokine release, whereas D2-like receptor activity promotes the generation and function of Tregs, thus maintaining immune homeostasis (57, 58). In APCs, DA regulates IL-12 and IL-23 expression in a receptor-dependent manner: activation of D1-like receptors, particularly D5R, enhances IL-12 and IL-23 production (via STAT3 inhibition), whereas absence of D5R markedly reduces these cytokines, illustrating how receptor subtype and local concentration shape APC-driven adaptive immunity (59).
Serotonin (5-HT), derived from both platelets and enterochromaffin cells, regulates dendritic cell maturation, macrophage chemotaxis, and T cell polarization. It exerts its effects through multiple 5-HT receptor subtypes, which are differentially expressed across immune cell types (60, 61). Serotonin signaling is also implicated in modulating vascular permeability and platelet aggregation, thereby linking neuroimmune signaling with early inflammatory responses (62, 63).
Gamma-aminobutyric acid (GABA), the principal inhibitory neurotransmitter in the CNS, also modulates immune responses. GABA A and GABA B receptors are expressed on T cells and macrophages (64). GABAergic signaling suppresses pro-inflammatory cytokine production, inhibits antigen presentation, and promotes regulatory T cell responses. GABA analogs and receptor agonists are currently under investigation for their potential to modulate neuroinflammation and peripheral immune responses in chronic inflammatory diseases (64–66). Together, these neurotransmitters form a regulatory axis that enables context-dependent modulation of immune function across peripheral and central compartments.
4.3 Neuroimmune cardiovascular interfaces as structural hubs
NICIs have recently been identified as specialized sites in the adventitia of large arteries, including the aorta and carotids. These interfaces consist of sensory afferents, sympathetic nerve fibers, resident immune cell clusters (e.g., macrophages, DCs), and stromal cells. NICIs function as neuroimmune synapse-like structures that permit real-time communication between vascular inflammation and central neural circuits (7).
In healthy vessels, NICIs contribute to homeostasis by regulating immune tone and facilitating vessel wall monitoring. Afferent nociceptive neurons detect inflammatory mediators (e.g., IL-1β, CCL2) and relay signals to the brainstem, particularly the nucleus tractus solitarius (NTS), thereby initiating autonomic feedback loops. Sympathetic efferents project back to the same vascular sites, releasing NE locally to modulate immune cell activation, antigen presentation, and cytokine production (7).
In addition to neurotransmitters, NICIs also house neuropeptides such as calcitonin gene-related peptide (CGRP), which has been shown to influence endothelial barrier function and shift macrophage phenotype (67, 68). In healthy arteries, functionally relevant NICIs are absent and arise de novo during disease onset, but their structural and functional plasticity makes them susceptible to extensive remodeling during pathological states such as atherosclerosis, hypertension, and autoimmune vasculitis. This potential for pathological rewiring underscores their importance as early sentinels of immune dysregulation.
5 Neuroimmune dysregulation and atherosclerosis pathophysiology
In atherosclerosis, the delicate balance between neural and immune regulation is disrupted, fueling chronic vascular inflammation and plaque progression (Figure 4).
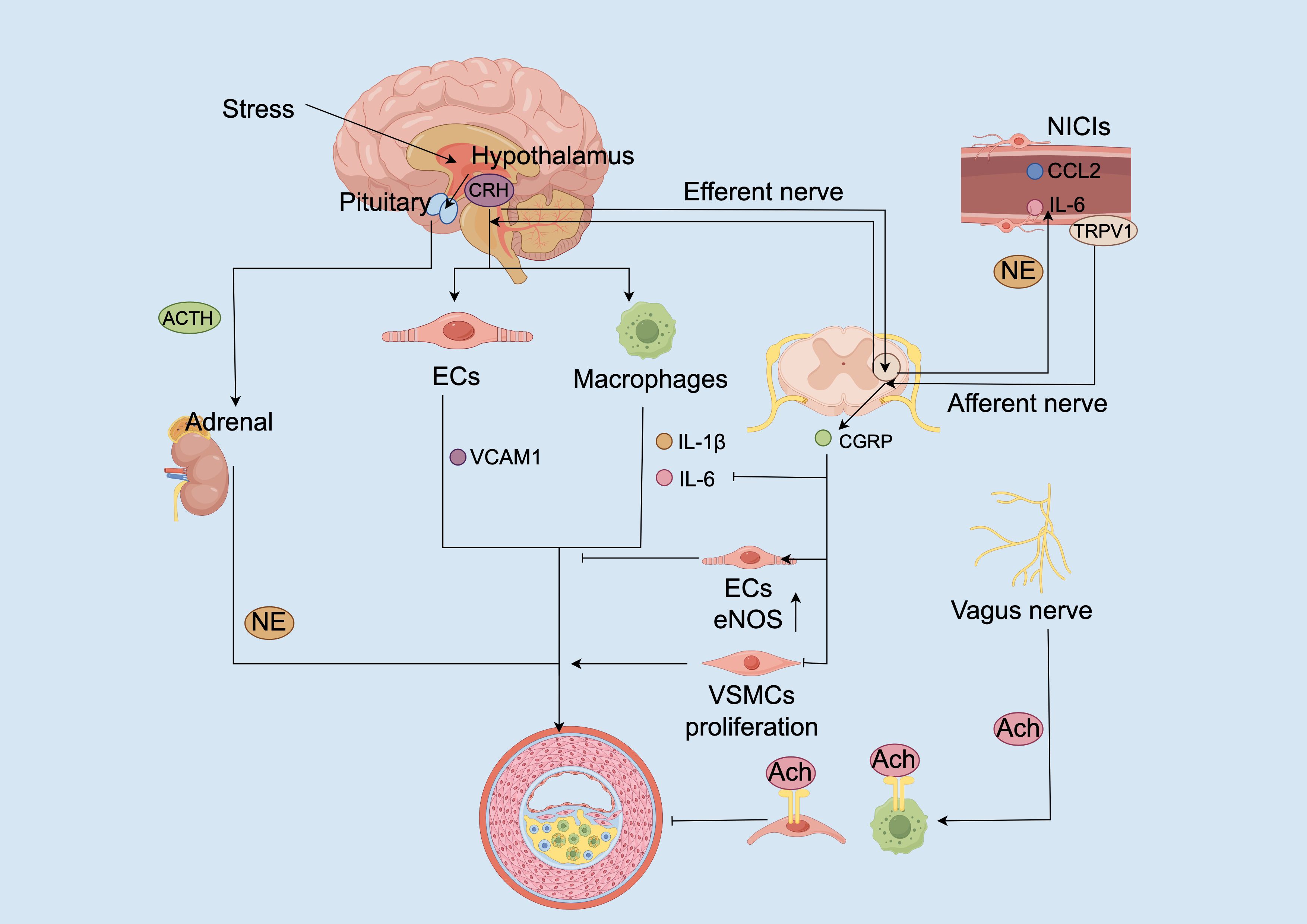
Figure 4. Neuroimmune circuit dysregulation promotes vascular inflammation and atherogenesis. Chronic stress activates the hypothalamic–pituitary–adrenal (HPA) axis, leading to corticotropin-releasing hormone (CRH) secretion from the hypothalamus. CRH acts directly on endothelial cells (ECs) and macrophages to upregulate vascular cell adhesion molecule-1 (VCAM1) and stimulate pro-inflammatory cytokine release (e.g., IL-1β, IL-6). Concurrently, CRH stimulates adrenocorticotropic hormone (ACTH) release from the pituitary, promoting adrenal norepinephrine (NE) production. NE further exacerbates immune activation through β-adrenergic signaling. In peripheral neuroimmune cardiovascular interfaces (NICIs), stress-induced norepinephrine and inflammatory cues (e.g., IL-6, CCL2) sensitize TRPV1 channels on afferent sensory terminals, leading to CGRP release. CGRP promotes vasodilation and inhibits cytokine production in macrophages and dendritic cells. Efferent vagal signaling through acetylcholine (Ach) activates α7 nicotinic acetylcholine receptors (α7nAChRs) on macrophages and ECs, suppressing inflammation and enhancing eNOS expression. Imbalances in these neuroimmune loops, characterized by sympathetic hyperactivity, reduced vagal activity, and sustained TRPV1 sensitization, converge to promote EC dysfunction, vascular smooth muscle cell (VSMC) proliferation, and plaque progression.
5.1 Dysregulation of autonomic control in atherosclerosis
Chronic psychosocial stress is increasingly recognized as a non-traditional but powerful accelerator of atherosclerosis through coordinated dysregulation of both the hypothalamic–pituitary–adrenal (HPA) axis and the autonomic nervous system. Activation of the HPA axis leads to elevated levels of CRH, which not only orchestrates systemic glucocorticoid responses but also exerts direct proinflammatory effects at the vascular level—promoting endothelial hyperpermeability and enhancing pro-inflammatory cytokine release from macrophages (69). Study has demonstrated that CRH directly promotes atherosclerosis progression by inducing endothelial inflammation. In LDLR⁻/⁻ mice, exogenous CRH administration significantly exacerbated plaque development, accompanied by increased VCAM-1 expression and activation of the NF-κB signaling pathway, which together facilitate leukocyte recruitment. These pro-atherogenic effects were receptor-dependent and effectively suppressed by pharmacological inhibition of CRH receptor 1 (CRHR1). Importantly, this CRH–CRHR1 signaling pathway acts independently of systemic lipid metabolism, indicating a local vascular mechanism contributing to atherogenesis beyond classical metabolic factors (70).
Concomitantly, stress reduces parasympathetic tone, impairing the cholinergic anti-inflammatory reflex that is essential for limiting vascular inflammation. In this pathway, acetylcholine—originating from neuronal or potentially non-neuronal sources—binds to α7nAChRs on macrophages and endothelial cells, a receptor interaction supported by evidence from in vivo and in vitro studies demonstrating suppression of pro-inflammatory cytokine production (11, 55). However, this regulatory pathway is often compromised in chronic inflammatory states. In ApoE⁻/⁻ mice, functional impairment of baroreflex control through sino-aortic denervation significantly exacerbates plaque burden, which is partially rescued by administration of selective α7nAChR agonists. This supports a critical role for vagal integrity in restraining local immune activation and endothelial dysfunction during atherogenesis (71).
Conversely, SNS overactivity enhances leukocyte recruitment and proinflammatory polarization via local NE release, primarily through β1-adrenergic receptor (β1-AR) and α-AR activation. Chronic SNS activation promotes myelopoiesis via β1-AR, increasing circulating inflammatory monocytes that infiltrate atherosclerotic plaques, effects reversed by non-selective β-adrenergic blockade (targeting both β1- and β2-AR) (72). NE also modulates neutrophil function and polarization, skewing them toward an immunosuppressive N2 phenotype while reducing their proinflammatory activity, although the precise implications of this polarization in atherosclerosis remain context-dependent (73).
Importantly, these autonomic imbalances do not occur in isolation but contribute to a pathophysiological feed−forward loop. CRH can directly enhance macrophage pro−inflammatory cytokine production, as shown by augmented TNF−α, IL−1β and IL−6 responses in LPS−stimulated macrophages (74). Meanwhile, catecholamines released from sympathetic nerve activation exhibit rhythmic, tissue-specific modulation of leukocyte adhesion to the vascular endothelium, promoting localized vascular inflammation with circadian variations (75). Notably, chronic inflammation is associated with increased sympathetic innervation density in the perivascular adventitia—suggesting neural remodeling reinforces immune activation (7). Collectively, these findings support a circuit in which autonomic imbalance, excessive sympathetic tone coupled with impaired parasympathetic anti-inflammatory signaling, facilitates vascular inflammation and plaque progression.
5.2 Neuromodulators in atherosclerosis
A growing body of experimental evidence supports the pivotal role of neuropeptides in orchestrating local immune responses in atherosclerosis. Among these, pituitary adenylate cyclase-activating polypeptide (PACAP), particularly its predominant isoform PACAP-38, has emerged as a key anti-inflammatory neuromodulator. In PACAP-deficient ApoE⁻/⁻ mice (PACAP⁻/⁻:ApoE⁻/⁻), plaques exhibit significantly increased area, macrophage infiltration, and foam cell formation, accompanied by elevated TNF-α expression, indicating that endogenous PACAP constrains proinflammatory macrophage activation (76). Activation of its high-affinity receptor PAC1 using the selective agonist Maxadilan attenuates plaque burden by over 40%, reduces apoptotic cell accumulation, and suppresses IL-1β and TNF-α in LDLR⁻/⁻ mice, without altering circulating lipids, suggesting local immunomodulatory effects (76). These findings underscore PACAP–PAC1 signaling as a vascular anti-inflammatory pathway with translational potential (77).
Beyond PACAP, other neuromodulators are increasingly recognized as crucial players in vascular immune regulation. Neuropeptide Y (NPY), highly expressed in sympathetic nerves, promotes vascular smooth muscle cell (VSMC) proliferation and M1 macrophage polarization (78). In a rat model of carotid balloon injury, slow-release NPY delivery induced occlusive atherosclerotic-like lesions containing lipid cores, macrophage infiltrates, thrombi, and neovessels, despite the absence of metabolic abnormalities or atherogenic diet. These effects were significantly attenuated by selective blockade of the Y1 receptor, with partial efficacy by Y5 receptor inhibition, implicating Y1R-mediated signaling as a key driver of NPY-induced vascular pathology (79).
Substance P (SP), via NK1 receptors, exerts dual effects depending on disease phase. In early atherogenesis, SP enhances monocyte recruitment and proinflammatory cytokine release, whereas in chronic stages it may promote tissue repair by stimulating Treg responses (80). Similarly, Secretoneurin is a pleiotropic peptide that promotes endothelial regeneration and angiogenesis but may also facilitate VSMC migration and contribute to fibrous cap destabilization (81).
Other modulators such as brain-derived neurotrophic factor (BDNF) and Galanin add further complexity. BDNF, traditionally known for its role in neuronal plasticity, is also expressed in human atherosclerotic plaques, particularly within smooth muscle cells, macrophages, and fibroblasts. Clinical imaging and immunohistochemistry studies have shown that elevated plasma BDNF levels correlate with increased macrophage infiltration in coronary plaques, suggesting a potential link to plaque vulnerability. Additionally, in vitro studies indicate that BDNF can enhance NAD(P)H oxidase activity in vascular cells, implicating a role in promoting oxidative stress within the vascular (82). Galanin is a neuropeptide that modulates autonomic tone and metabolic function and exhibits context-dependent immunoregulatory properties. In human monocytes and macrophages, galanin induces either pro-inflammatory (e.g., TNF-α, IL-1β, CXCL8) or anti-inflammatory (e.g., IL-10) cytokine expression depending on the activation state, demonstrating its bidirectional immune effects (83). Moreover, signaling through the GAL3 receptor has been shown to enhance the responsiveness of innate immune cells to inflammatory cues in specific inflammatory contexts, further supporting a potential pro-inflammatory role of galanin in chronic vascular inflammation (84).
Together, these findings suggest that atherosclerosis is modulated by a dynamic array of neuropeptides with context- and cell-specific effects. PACAP plays dominant anti-inflammatory roles, whereas NPY and SP may exacerbate inflammation depending on timing and receptor signaling. Understanding the spatial and temporal orchestration of these neuromodulators—and their interactions with neuroimmune interfaces such as NICIs, offers new opportunities for precision therapy.
5.3 Neuroimmune cardiovascular interfaces in atherosclerosis
Emerging evidence reveals that atherosclerotic inflammation induces profound structural and functional remodeling of the vascular adventitia, giving rise to specialized NICIs. In advanced lesions of both ApoE⁻/⁻ mice and human coronary arteries, high-resolution imaging has demonstrated dense networks of sympathetic and sensory nerve fibers interwoven with clusters of macrophages, T and B lymphocytes, and smooth muscle cells in the adventitial layer, forming integrated hubs of bidirectional artery–brain communication (7, 75).
NICIs play dual and dynamic roles in coordinating neural and immune signals. First, afferent sensory fibers expressing TRPV1 and CGRP detect inflammatory cues and transmit them via dorsal root and nodose ganglia to autonomic centers in the brainstem, parabrachial nucleus, and hypothalamus (7). Second, efferent sympathetic pathways descend from these regions to the vascular adventitia through spinal and celiac ganglia, where they release NE, modulating leukocyte behavior and endothelial activation (75). Ablation of the celiac ganglion in aged atherosclerotic mice disrupted NICI integrity, attenuated local immune activation, and reduced plaque progression, indicating that this peripheral neural circuit contributes to disease progression at late stages (7).
Within NICIs, the TRPV1–CGRP axis functions as a key sensory immunomodulatory pathway. In the context of atherosclerosis, inflammatory cues such as LPS/TLR4 signaling, local acidosis, and endogenous lipid mediators, including capsaisin, anandamide (AEA) and N-arachidonoyl dopamine (NADA), sensitize TRPV1 channels expressed on perivascular sensory afferents (85–87). Upon activation, TRPV1 induces the release of CGRP, a vasodilatory neuropeptide that suppresses pro-inflammatory cytokine production, notably TNF-α in macrophages and IL-12 in dendritic cells (88, 89). Although specific suppression of CXCL2 has not been directly demonstrated, these findings collectively support a broader anti-inflammatory role for CGRP in myeloid cell regulation. Moreover, CGRP enhances endothelial nitric oxide synthase (eNOS) activity and inhibits NF-κB–dependent chemokine expression, such as CCL2 and CXCL8, thus limiting monocyte adhesion and transmigration (90). Additionally, CGRP restrains smooth muscle cell proliferation and reducing neointimal hyperplasia (91). Of note, prolonged TRPV1 activation may induce receptor desensitization through calcium-dependent dephosphorylation mechanisms, potentially weakening its neuroprotective output in chronic inflammation settings (92).
Crucially, NICIs structurally integrate with adventitial tertiary lymphoid organs (ATLOs), which arise in response to chronic vascular inflammation and whose development depends on de novo innervation from sympathetic and sensory fibers, as demonstrated in recent experimental models (7, 93). Such nerve ingrowth not only provides structural linkage but also enables bidirectional communication between vascular sensory/autonomic circuits and ATLO-resident immune cells, potentially shaping local antigen presentation, cytokine milieus, and lymphoid organization during atherogenesis. In aged ApoE⁻/⁻ mice, ATLOs emerge as organized immune aggregates within the adventitia, comprising B-cell follicles, germinal centers, and high endothelial venules (93). study reported extensive nerve fiber sprouting within ATLOs, marked by GAP-43 and Synapsin, forming ultrastructural synapse-like contacts within nanometer proximity to CD45+ immune cells (7). These neuro-immune junctions function as dynamic interfaces for rapid sensing and modulation of immune activity within the vascular wall.
Collectively, NICIs and ATLOs convert the adventitia into a neuroimmune nexus—capable of integrating, amplifying, and transmitting vascular inflammatory signals through bidirectional neuroimmune communication. These findings shift the paradigm of atherosclerosis from a purely metabolic and inflammatory disease to one with defined neuroimmune architecture and functionality. An unresolved challenge is reconciling the anti-inflammatory and vasoprotective actions of CGRP with the potentially deleterious effects of de novo innervation and NICI/ATLO expansion during atherogenesis, whether these represent stage-dependent phenomena or context-specific signaling outcomes remains to be determined. Therapeutic targeting of specific NICI components, such as reinforcing CGRP-mediated vascular protection, restoring vagal α7nAChR signaling, or blocking maladaptive sympathetic outputs, may offer novel strategies for halting plaque progression and reducing cardiovascular events. Future studies should prioritize the temporal dynamics, cell-specific interactions, and plasticity of NICIs across disease stages to enable precision neuromodulation in atherosclerosis.
6 Therapeutic opportunities targeting the neuroimmune axis in atherosclerosis
Recent advances in neuroimmunology have highlighted the therapeutic potential of modulating neural circuits to restrain vascular inflammation and atherogenesis. Traditional approaches to atherosclerosis management primarily target lipid metabolism and systemic inflammation; however, these strategies often overlook the critical contribution of neuroimmune interactions in disease progression. The identification of NICIs and neuromodulatory targets such as α7nAChRs, TRPV1 channels, and neuropeptides (e.g., PACAP, CGRP) has opened new avenues for intervention. Therapeutic strategies that restore autonomic balance, modulate neuroimmune signaling, or disrupt pathogenic nerve–immune crosstalk offer a promising adjunct or alternative to conventional treatments. This section reviews emerging modalities—including bioelectronic devices, pharmacologic agonists, and anti-cytokine therapies—with a focus on their mechanistic underpinnings, preclinical efficacy, and translational challenges in the context of atherosclerosis (Table 1).
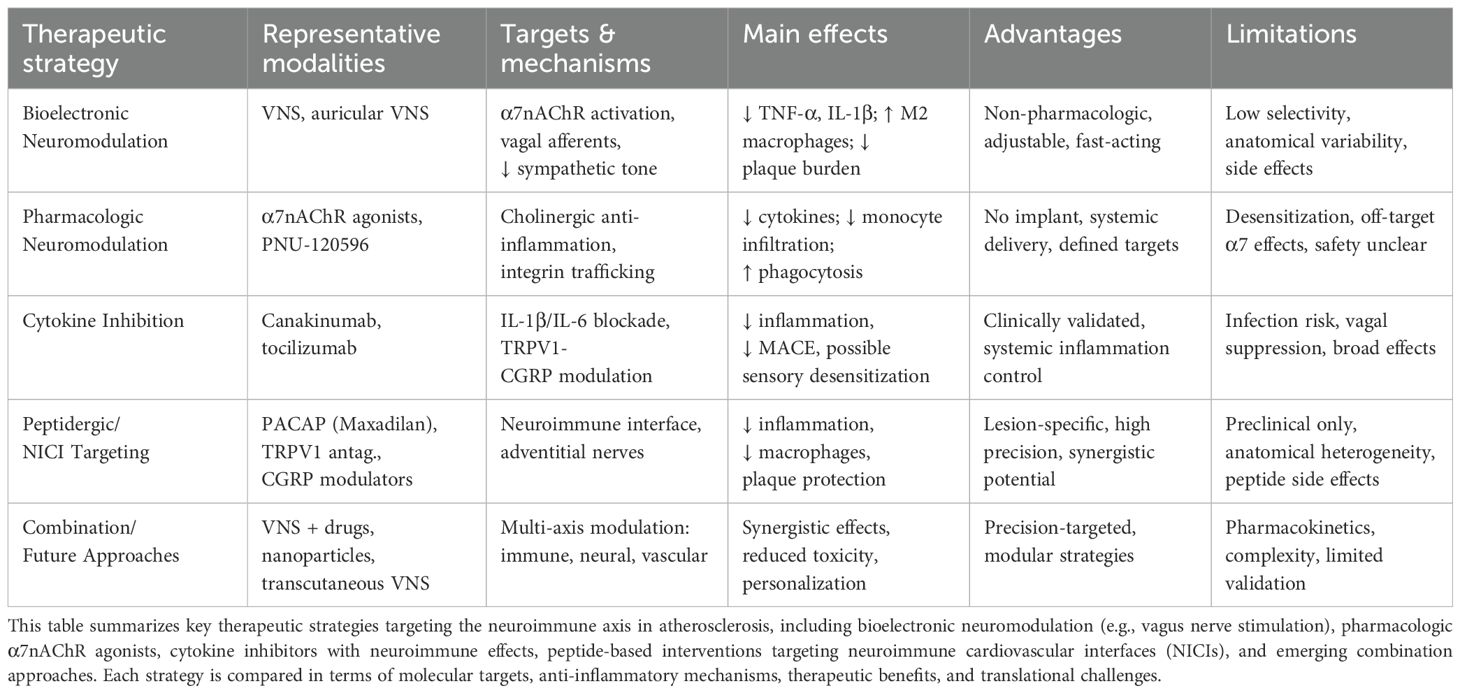
Table 1. Comparative overview of neuroimmune-targeted therapies in atherosclerosis.
Before discussing dedicated neuromodulatory strategies, it is worth noting that several current frontline atherosclerosis therapies exert incidental effects on inflammatory and autonomic pathways. For example, high-intensity statins not only lower LDL-C but also reduce vascular macrophage activation and circulating IL-6 levels, effects associated with modest improvements in heart rate variability, a surrogate of parasympathetic tone (94, 95). PCSK9 inhibitors similarly attenuate arterial wall inflammation, potentially through reduced monocyte recruitment (96). Beyond lipid-lowering agents, GLP-1 receptor agonists dampen systemic cytokine production and have been shown to decrease sympathetic nerve activity in hypertensive patients (97). Likewise, SGLT2 inhibitors can improve autonomic balance and lower plasma norepinephrine in heart failure cohorts (98). While these effects are secondary to their primary indications, they highlight that modulation of the inflammatory, autonomic axis is already occurring in clinical practice. However, emerging neuromodulatory interventions, such as vagus nerve stimulation or selective α7nAChR agonists, offer the prospect of more precise and potent targeting of pathogenic neuroimmune circuits in atherosclerosis.
6.1 Bioelectronic neuromodulation: vagus nerve stimulation
Preclinical studies support the efficacy of vagus nerve stimulation (VNS) as a targeted anti-inflammatory approach in atherosclerosis. While direct evidence in ApoE⁻/⁻ mice is limited, pharmacological activation of α7nAChRs, a key mediator of vagal signaling, significantly reduces plaque burden, promotes M2 macrophage polarization, and lowers IL-1β and TNF-α levels (99).
A recent study of a novel small-molecule agonist, AZ6983, confirmed the central role of α7nAChR: in ApoE⁻/⁻ mice AZ6983 reduced plaque burden by ~37–49 % versus control, dampened serum cytokines, and increased macrophage phagocytic capacity, recapitulating key functional endpoints attributed to VNS (100). Complementing these findings, imaging in human atherosclerotic vessels identified α7nAChR expression co-localized with macrophage-rich areas, lending translational significance to the mouse data (100).
Mechanistically, α7-mediated anti-inflammatory effects extend beyond cytokine suppression. A study demonstrated that macrophage recruitment into inflamed tissue depends on α7nAChR regulation of integrin αMβ2 expression; macrophages lacking α7 signal showed impaired migration in 3D matrices and in vivo endotoxemia models (101). These findings help explain how VNS and α7 agonism modulate plaque cellularity and stability by restricting monocyte/macrophage infiltration and favoring efferocytosis.
Despite therapeutic promise, VNS efficacy depends heavily on stimulation parameters. Low-frequency stimulation (1–10 Hz) primarily activates afferent fibers and suppresses sympathetic output, while higher frequencies (20–30 Hz) engage broader autonomic responses, including cardiac effects (102). Efforts to achieve fiber-selective targeting in VNS are challenged by significant anatomical variability and inconsistent electrode positioning in humans. The vagus nerve’s fascicular organization exhibits considerable interindividual differences, complicating reproducible and precise activation of desired fiber populations using cuff electrodes (103). Moreover, long-term clinical use of VNS can produce side effects such as bradycardia and hoarseness, which may impact patient compliance and quality of life (104); rare but serious cardiac adverse events like periodic bradycardia have also been reported, especially in pediatric cases (105). These technical and physiological limitations, combined with individual differences in disease stage and comorbidities, remain key barriers to the widespread clinical translation of fiber-selective VNS therapies.
6.2 Precision neuromodulatory pharmacology
α7nAChR agonists represent a pharmacologic route to harness the anti-inflammatory benefits of VNS without implantable devices. AZ6983 offers proof-of-concept: systemic administration in ApoE⁻/⁻ mice reduced plaque burden, suppressed TNF-α, and enhanced macrophage phagocytosis via α7 engagement (100). Translational relevance is bolstered by evidence that α7nAChR is expressed in human atherosclerotic plaques, suggesting direct targeting of lesion macrophage populations may be feasible (106).
Beyond cytokine regulation, recent in vivo model of endotoxemia have demonstrated a distinct role for α7nAChR in regulating immune cell trafficking: α7nAChR deficiency markedly impairs macrophage recruitment to inflamed organs, including lungs and liver, by downregulating key integrins such as αMβ2, thereby limiting integrin-mediated migration (101). These findings underscore α7 agonists’ potential not only to suppress pro-inflammatory cytokine production but also to prevent immune cell accumulation in atherosclerotic plaques via modulation of integrin-dependent macrophage trafficking.
While these features suggest a degree of pathway specificity, their actual spatial precision in vivo remains an open question. For example, work by Simon et al. (11), indicates that in the splenic anti-inflammatory reflex, key α7nAChR-dependent steps may occur in upstream autonomic ganglia such as the celiac ganglion, rather than solely on plaque-resident macrophages. This raises the possibility that systemic α7 agonists might influence multiple anatomical sites along the reflex arc, not all of which are disease specific.
These pharmacologic strategies offer precision over systemic anti-inflammatories. Targeting α7nAChR allows pathway-specific modulation of immune responses, minimizing off-target immunosuppression (107). Moreover, allosteric modulators like PNU-120596 significantly enhance receptor responsiveness, enabling lower dosing and increased specificity in reducing inflammatory signaling (108).
However, concerns remain: α7 agonists must avoid desensitization of receptor function and do not yet have established long-term safety profiles in vascular patients (109). Another unresolved issue is tissue specificity, α7nAChR is expressed in multiple cell types (e.g. neurons, epithelial cells), raising potential for unintended effects unless cell-targeted delivery approaches are employed (110).
Efforts are ongoing to develop next-generation α7 modulators with biased signaling properties or restricted biodistribution. Combining pharmacologic agents with localized VNS or transcutaneous auricular VNS may synergize inflammatory suppression while reducing systemic exposure. Overall, precision α7 pharmacology offers a promising, device-independent alternative to neuromodulation, but its optimal targeting strategy in atherosclerosis will require further refinement and experimental validation.
6.3 Anti-inflammatory drugs with neuroimmune effects
Study highlights that beyond conventional lipid-lowering therapies, anti-cytokine agents also exert profound modulatory effects on neuroimmune circuits involved in atherosclerosis. The landmark CANTOS trial demonstrated that inhibiting IL-1β with canakinumab reduces major adverse cardiovascular events without altering lipid profiles, supporting the immunomodulatory relevance of IL-1β signaling in plaques (4). While CANTOS itself did not directly assess neural endpoints, more recent preclinical studies indicate that IL-1β can also modulate arterial nociceptive sensory fibers and neuroimmune signaling (6), thereby providing a mechanistic link between cytokine blockade and potential effects on vascular innervation. Genetic deletion of IL-1β in ApoE−/− mice leads to a significant reduction in plaque burden, accompanied by decreased monocyte adhesion and inflammatory cytokine expression (111). Intriguingly, IL-1β has been shown to sensitize TRPV1+ sensory neurons in inflammatory skin and joint diseases, promoting neuropeptide release and local immune amplification (112). Given the established role of TRPV1–CGRP signaling within NICIs in atherosclerosis, it is plausible that IL-1β blockade might also dampen neurogenic inflammation in vascular lesions. While direct evidence for IL-1β–TRPV1 crosstalk in atherosclerosis is lacking, future investigations could reveal whether sensory neuroimmune desensitization contributes to the plaque-stabilizing effects of anti-cytokine therapy.
IL-6 receptor antagonism, using agents like tocilizumab, may provide benefits that extend beyond standard anti-inflammatory effects. Although direct evidence in ApoE⁻/⁻ mice linking IL-6R blockade to modulation of plaque-associated sympathetic fibers remains unavailable, preclinical research indicates that IL-6 signaling robustly influences sympathetic nervous system activity. For example, intracerebroventricular IL-6 administration in rats was found to significantly increase splenic sympathetic nerve discharge and elevate circulating norepinephrine levels, highlighting a central IL-6–driven sympathetoneural axis (113). Additionally, systemic IL-6 elevation is commonly associated with increased sympathetic tone in chronic inflammatory conditions, providing a mechanistic link to neuroimmune modulation in vascular disease (113, 114). These findings support a plausible dual mechanism: IL-6R blockade could suppress inflammatory signaling and indirectly attenuate sympathetic-driven neuroimmune activation in atherosclerosis. However, targeted in vivo studies are needed to confirm whether IL-6R-targeted therapies modulate sympathetic innervation or β2-adrenoceptor expression in plaque-associated nerves.
These immunomodulators differ from traditional neuromodulatory therapies. Whereas VNS and pharmacologic agonists directly target neural circuits, cytokine inhibitors reprogram neuroimmune interactions indirectly, quieting both inflammatory triggers and sensitized neural feedback loops. While no studies have directly examined the combined effects of VNS and canakinumab in LDLR⁻/⁻ mice, their complementary mechanisms of neural circuit modulation and cytokine inhibition suggest the potential for synergistic benefits in reducing plaque size and systemic inflammation. Future experimental work is needed to evaluate this possibility.
However, targeting cytokines is not without challenges. Systemic neutralization of IL-1β or IL-6 carries infection risk, as these mediators are essential for host defense. For instance, the CANTOS trial reported significantly elevated rates of serious infections and sepsis among individuals receiving canakinumab compared to placebo (4). Moreover, IL-1β antagonists can blunt fever-induced vagal reflexes, potentially shifting autonomic balance—animal studies show that subdiaphragmatic vagotomy blocks IL-1β–induced fever and behavioral changes, underscoring the role of vagal afferents in mediating inflammatory responses (115). Emerging clinical data also link sustained IL-6 suppression to transient vagal hypoactivity: heart rate variability (HRV), a key measure of parasympathetic function, inversely correlates with inflammatory markers such as IL-6 and CRP in patients with rheumatoid arthritis (116). These observations underscore the need for therapies that selectively modulate pathogenic neuroimmune pathways while preserving beneficial autonomic reflexes.
In summary, anti-cytokine agents have proven efficacy in cardiovascular trials and show emerging neuromodulatory effects by quelling both inflammatory and neural feedback. However, their non-specific nature raises safety concerns. Combinatorial approaches with electrical or pharmacologic neuromodulation may achieve greater precision and reduced side effects, which sheds light on a balanced “neuro-cytokine” therapeutic axis for atherosclerosis.
6.4 Future directions: targeting key neuroimmune pathways
The evolving understanding of plaque-centric neuroimmune systems, NICIs, creates fertile ground for novel therapies that directly target nerve–immune interfaces within the adventitia. For instance, Sensory TRPV1 signaling has been implicated in modulating vascular–sympathetic reflexes and homeostatic blood pressure regulation, suggesting that TRPV1 antagonists may offer a way to interrupt stress-induced pro-inflammatory circuits—though direct evidence in atherosclerotic plaque models remains pending (117). Separately, centrally administered gabapentin modulates blood pressure and heart rate via the NTS in hypertensive rats through NOS-dependent mechanisms, indicating its capacity to recalibrate autonomic reflexes—though synergy with α7nAChR agonists in AS has yet to be explored (118).
Within peptide neuromodulators, PACAP and CGRP have emerged as promising yet divergent targets. As mentioned, PACAP–PAC1 activation via Maxadilan inhibits macrophage TNF-α production and increases regulatory IL-10 expression in murine plaques independent of lipids (76). Conversely, CGRP antagonism, widely used in migraine, presents a cautionary tale: galcanezumab-treated ApoE⁻/⁻ mice exhibit increased plaque macrophage infiltration and vessel wall inflammation, highlighting CGRP’s protective role in vascular homeostasis (119). These findings have major translational implications—identifying CGRP inhibitors as potentially contraindicated in high-risk cardiovascular patients, while spotlighting PACAP agonists as therapeutic candidates.
Challenges loom in targeting NICIs and peptidergic systems. Anatomical heterogeneity across arterial beds may limit drug access, and long-term manipulation of sensory fibers may disrupt homeostatic neurovascular reflexes, as sensory innervation density and function vary widely by vascular territory (120). Moreover, biased agonism at PAC1 receptors may differentially modulate vascular versus neural signaling, with PAC1-mediated vascular relaxation and central neural sensitization showing distinct pharmacology—necessitating careful pharmacodynamic profiling (121).
However, the emerging clinical use of CGRP blockade in migraine and the translational potential of PAC1 agonists such as Maxadilan underscore the urgent need to evaluate cardiovascular safety and long-term immunomodulatory outcomes in relevant patient populations. Future studies should systematically map neuropeptide expression dynamics and receptor distribution across atherosclerosis stages to inform the rational design of targeted interventions.
Nevertheless, integrating structural NICI mapping with focused neuromodulation holds promise. Combined approaches, e.g., nanoparticle-mediated PACAP delivery to adventitial nerve sheaths, or localized TRPV1 blockade using targeted antibodies, could allow lesion-specific intervention, although agonist such as capsaicin should be avoided due to its side effects (122, 123). These strategies may showcase integrated mechanistic insight and therapeutic innovation. Overall, therapy aimed at disrupting pathological nerve–immune crosstalk, via PACAP agonists, CGRP modulators, and NICI-directed interventions, offers a new frontier in precision atherosclerosis management.
6.5 Challenges in clinical translation
Several translational barriers complicate the therapeutic potential of targeting neuroimmune pathways in AS application in human disease. One major obstacle lies in the anatomical and functional complexity of autonomic circuits. VNS, while potently anti-inflammatory in ApoE⁻/⁻ mouse models via activation of α7nAChR, expressing macrophages, often results in non-specific activation of adjacent neural branches when translated into larger animals or humans, due to the anatomical complexity and lack of fiber-type selectivity with conventional cuff electrodes (124). Standard cuff electrodes lack fiber-specific resolution, inadvertently activating laryngeal, leading to coughing or bradycardia (124, 125). Even in recent animal studies (adult male Sprague-Dawley rats and Merino ewes) employing closed-loop or optogenetic approaches, achieving selectivity for anti-inflammatory vagal efferents remains technically demanding and largely untested in AS-specific contexts (126).
Beyond technical hurdles, the systemic effects of neuromodulatory peptides, such as CGRP and PACAP, raise concerns about long-term safety and vascular specificity. CGRP receptor antagonists (e.g., erenumab) are now widely used for migraine prophylaxis, and large-scale post-marketing data suggest they do not increase major adverse cardiovascular events (127). However, these findings are based on low-risk populations and may not fully translate to individuals with established atherosclerosis, where CGRP plays a vasoprotective and anti-inflammatory role. Indeed, ApoE/CGRP double-knockout mice develop significantly larger plaques and enhanced macrophage activation, underscoring α-CGRP’s protective role in atherogenesis (119). Conversely, PACAP-based therapies remain at the preclinical stage: the PAC1 receptor agonist Maxadilan markedly reduced plaque incidence (from ~75 % to ~28 %) and lumen stenosis in ApoE−/− mice, lowered TNF-α+ and IL-1β+ areas, and decreased apoptosis in lesions—even under cholesterol-enriched diets (76). However, these effects are confined to murine models; no safety or efficacy data exist in non-rodent species, and translation to humans remains speculative.
Another challenge is the striking inter-individual variability in autonomic tone, inflammatory responses, and neural-immune coupling across patients. Factors such as age, metabolic syndrome, autonomic neuropathy, and even sleep quality alter neuroimmune balance, potentially modulating response to interventions. For instance, HRV, widely used as a proxy for vagal tone, is inversely associated with circulating markers of inflammation in cardiovascular disease cohorts: lower HRV correlates with elevated IL-6, CRP, and fibrinogen levels (128–130). Additionally, in a large young adult cohort (N = 2,064), reduced HRV was independently associated with higher monocyte and leukocyte counts, further suggesting HRV as a physiological biomarker of immune activation (131). These findings underscore the need for physiological stratification using HRV or related autonomic markers to identify individuals most likely to benefit from neuromodulatory therapies.
Adding further complexity, recent evidence indicates that VNS does not always act through the canonical α7nAChR pathway. In inflammatory disease models of acute lung injury, VNS-induced immunosuppression was abolished by adrenalectomy, but not by α7nAChR blockade, suggesting a vagus–adrenal–catecholamine axis rather than direct cholinergic signaling (132, 133). This distinction is crucial for AS, where peripheral nerve terminals may act via local versus systemic pathways, making target engagement and mechanistic verification essential in future trials.
Finally, chronic neuromodulation may induce neural plasticity or receptor desensitization, as seen in prolonged CGRP or PACAP exposure, potentially diminishing efficacy over time (134). Despite their promising immunomodulatory profiles, long-term studies evaluating tolerance, feedback inhibition, or compensatory sympathetic activation are largely absent in current AS models. Moreover, while bioelectronic and pharmacologic therapies hold distinct promise, their combined effects, whether synergistic or antagonistic, remain unexplored, raising critical questions for therapeutic design.
In conclusion, advancing neuroimmune-targeted therapies for atherosclerosis will require refined neuromodulation tools with enhanced circuit selectivity, rigorous safety validation of peptidergic drugs, and patient stratification approaches grounded in real-time autonomic and inflammatory biomarkers. Addressing these challenges through integrated engineering, pharmacological, and immunological frameworks is essential to realize the clinical potential of this emerging field.
7 Conclusion
Mounting evidence positions neuroimmune dysregulation as a central mechanism linking autonomic imbalance to chronic vascular inflammation and atherogenesis. Disruptions in sympathetic-parasympathetic tone, aberrant neuropeptide signaling, and the pathological remodeling of NICIs collectively shape the inflammatory milieu of the arterial wall. These neurogenic cues not only influence local immune cell activation and trafficking but also modulate hematopoietic niches and systemic cytokine output, reinforcing a feed-forward loop of vascular injury. Preclinical studies have highlighted the therapeutic potential of neuromodulatory interventions, including selective α7nAChR agonists, VNS, and neuropeptide-targeting strategies, to attenuate immune activation and stabilize atherosclerotic plaques. However, significant translational hurdles remain, particularly in achieving spatial precision, minimizing off-target autonomic effects, and tailoring therapies to interindividual variability in neural-immune architecture. Moving forward, integrating neuroimmunology with systems-level vascular biology holds promise for the rational design of precision therapies that transcend lipid-lowering paradigms. Such strategies may ultimately reshape the clinical approach to atherosclerosis, offering durable immunomodulation and improved cardiovascular outcomes.
Author contributions
YS: Writing – original draft. YP: Writing – original draft. LX: Writing – review & editing. YZ: Writing – review & editing. WM: Writing – review & editing. TC: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (grant numbers U24A20799, 2025), National Key Research and Development Program (grant numbers 2023YFC3606201, 2023), Central guidance for local scientific and technological development funding projects (grant numbers 2024ZY01016, 2024).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tellides G and Pober JS. Inflammatory and immune responses in the arterial media. Circ Res. (2015) 116:312–22. doi: 10.1161/CIRCRESAHA.116.301312
2. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. (2019) 139:e56–e528. doi: 10.1161/CIR.0000000000000659
3. Robinson JG, Williams KJ, Gidding S, Boren J, Tabas I, Fisher EA, et al. Eradicating the burden of atherosclerotic cardiovascular disease by lowering apolipoprotein B lipoproteins earlier in life. J Am Heart Assoc. (2018) 7:e009778. doi: 10.1161/JAHA.118.009778
4. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
5. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. (2019) 380:11–22. doi: 10.1056/NEJMoa1812792
6. Gerhardt T, Huynh P, and McAlpine CS. Neuroimmune circuits in the plaque and bone marrow regulate atherosclerosis. Cardiovasc Res. (2025) 120:2395–407. doi: 10.1093/cvr/cvae167
7. Mohanta SK, Peng L, Li Y, Lu S, Sun T, Carnevale L, et al. Neuroimmune cardiovascular interfaces control atherosclerosis. Nature. (2022) 605:152–9. doi: 10.1038/s41586-022-04673-6
8. Wang Y, Anesi J, Maier MC, Myers MA, Oqueli E, Sobey CG, et al. Sympathetic nervous system and atherosclerosis. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms241713132
9. Sohrabi Y, Reinecke H, and Soehnlein O. Trilateral interaction between innervation, leukocyte, and adventitia: a new driver of atherosclerotic plaque formation. Signal Transduct Target Ther. (2022) 7:249. doi: 10.1038/s41392-022-01121-9
10. de Lucia C, Eguchi A, and Koch WJ. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front Pharmacol. (2018) 9:904. doi: 10.3389/fphar.2018.00904
11. Simon T, Kirk J, Dolezalova N, Guyot M, Panzolini C, Bondue A, et al. The cholinergic anti-inflammatory pathway inhibits inflammation without lymphocyte relay. Front Neurosci. (2023) 17:1125492. doi: 10.3389/fnins.2023.1125492
12. Rosas-Ballina M, Olofsson PS, Ochani M, Valdés-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. (2011) 334:98–101. doi: 10.1126/science.1209985
13. Bellocchi C, Carandina A, Montinaro B, Targetti E, Furlan L, Rodrigues GD, et al. The interplay between autonomic nervous system and inflammation across systemic autoimmune diseases. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23052449
14. Sheng Y and Zhu L. The crosstalk between autonomic nervous system and blood vessels. Int J Physiol Pathophysiol Pharmacol. (2018) 10:17–28.
15. Cho W, Kang JL, and Park YM. Corticotropin-releasing hormone (CRH) promotes macrophage foam cell formation via reduced expression of ATP binding cassette transporter-1 (ABCA1). PloS One. (2015) 10:e0130587. doi: 10.1371/journal.pone.0130587
16. Costa MD, Redline S, Davis RB, Mittleman M, Goldberger AL, and Heckbert SR. Vagal impairment and cardiovascular risk in those with zero to low coronary artery calcification scores: the Multi-Ethnic Study of Atherosclerosis. Am J Physiol Heart Circ Physiol. (2025) 329:H258–H66. doi: 10.1152/ajpheart.00295.2025
17. Matei D, Buculei I, Luca C, Corciova CP, Andritoi D, Fuior R, et al. Impact of non-pharmacological interventions on the mechanisms of atherosclerosis. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23169097
18. Zhao Z, Qin Y, Wu R, Li W, and Dong Y. Single-cell analysis identified key macrophage subpopulations associated with atherosclerosis. Open Med (Wars). (2024) 19:20241088. doi: 10.1515/med-2024-1088
19. Lai Z, Kong D, Li Q, Wang Y, Li K, Duan X, et al. Single-cell spatial transcriptomics of tertiary lymphoid organ-like structures in human atherosclerotic plaques. Nat Cardiovasc Res. (2025) 4:547–66. doi: 10.1038/s44161-025-00639-9
20. Franco C, Sciatti E, Favero G, Bonomini F, Vizzardi E, and Rezzani R. Essential hypertension and oxidative stress: novel future perspectives. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms232214489
21. Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. (2002) 417:750–4. doi: 10.1038/nature00804
22. El-Hajjar L, Hindieh J, Andraos R, El-Sabban M, and Daher J. Myeloperoxidase-oxidized LDL activates human aortic endothelial cells through the LOX-1 scavenger receptor. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23052837
23. Peng K, Jiang P, Du Y, Zeng D, Zhao J, Li M, et al. Oxidized low-density lipoprotein accelerates the injury of endothelial cells via circ-USP36/miR-98-5p/VCAM1 axis. IUBMB Life. (2021) 73:177–87. doi: 10.1002/iub.2419
24. Obermayer G, Afonyushkin T, and Binder CJ. Oxidized low-density lipoprotein in inflammation-driven thrombosis. J Thromb Haemost. (2018) 16:418–28. doi: 10.1111/jth.13925
25. Nakashima Y, Raines EW, Plump AS, Breslow JL, and Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler Thromb Vasc Biol. (1998) 18:842–51. doi: 10.1161/01.ATV.18.5.842
26. Fu Z, Zhou E, Wang X, Tian M, Kong J, Li J, et al. Oxidized low-density lipoprotein-induced microparticles promote endothelial monocyte adhesion via intercellular adhesion molecule 1. Am J Physiol Cell Physiol. (2017) 313:C567–C74. doi: 10.1152/ajpcell.00158.2016
27. Blagov AV, Markin AM, Bogatyreva AI, Tolstik TV, Sukhorukov VN, and Orekhov AN. The role of macrophages in the pathogenesis of atherosclerosis. Cells. (2023) 12. doi: 10.3390/cells12040522
28. Groh L, Keating ST, Joosten LAB, Netea MG, and Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. (2018) 40:203–14. doi: 10.1007/s00281-017-0656-7
29. Farahi L, Sinha SK, and Lusis AJ. Roles of macrophages in atherogenesis. Front Pharmacol. (2021) 12:785220. doi: 10.3389/fphar.2021.785220
30. Persson J, Nilsson J, and Lindholm MW. Interleukin-1beta and tumour necrosis factor-alpha impede neutral lipid turnover in macrophage-derived foam cells. BMC Immunol. (2008) 9:70. doi: 10.1186/1471-2172-9-70
31. Davies MJ. Reactive oxygen species, metalloproteinases, and plaque stability. Circulation. (1998) 97:2382–3. doi: 10.1161/01.CIR.97.24.2382
32. Shah PK and Galis ZS. Matrix metalloproteinase hypothesis of plaque rupture: players keep piling up but questions remain. Circulation. (2001) 104:1878–80. doi: 10.1161/circ.104.16.1878
33. Wang X, Liu X, Wu W, Liao L, Zhou M, Wang X, et al. Hypoxia activates macrophage-NLRP3 inflammasome promoting atherosclerosis via PFKFB3-driven glycolysis. FASEB J. (2024) 38:e23854. doi: 10.1096/fj.202400283R
34. Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y, et al. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. (2011) 108:985–95. doi: 10.1161/CIRCRESAHA.110.233775
35. Wolf D, Gerhardt T, Winkels H, Michel NA, Pramod AB, Ghosheh Y, et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B(100)-reactive CD4(+) T-regulatory cells. Circulation. (2020) 142:1279–93. doi: 10.1161/CIRCULATIONAHA.119.042863
36. Huang Z, Cai X, Li S, Zhou H, Chu M, Shan P, et al. Berberine−attenuated monocyte adhesion to endothelial cells induced by oxidized low−density lipoprotein via inhibition of adhesion molecule expression. Mol Med Rep. (2013) 7:461–5. doi: 10.3892/mmr.2012.1236
37. Pirillo A, Uboldi P, Ferri N, Corsini A, Kuhn H, and Catapano AL. Upregulation of lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) expression in human endothelial cells by modified high density lipoproteins. Biochem Biophys Res Commun. (2012) 428:230–3. doi: 10.1016/j.bbrc.2012.10.020
38. Liu W, Yin Y, Zhou Z, He M, and Dai Y. OxLDL-induced IL-1 beta secretion promoting foam cells formation was mainly via CD36 mediated ROS production leading to NLRP3 inflammasome activation. Inflammation Res. (2014) 63:33–43. doi: 10.1007/s00011-013-0667-3
39. Bennett MR. Reactive oxygen species and death: oxidative DNA damage in atherosclerosis. Circ Res. (2001) 88:648–50. doi: 10.1161/hh0701.089955
40. Huang D, Gao W, Lu H, Qian JY, and Ge JB. Oxidized low-density lipoprotein stimulates dendritic cells maturation via LOX-1-mediated MAPK/NF-κB pathway. Braz J Med Biol Res. (2021) 54:e11062. doi: 10.1590/1414-431X2021e11062
41. Frostegård J, Zhang Y, Sun J, Yan K, and Liu A. Oxidized Low-Density Lipoprotein (OxLDL)-Treated Dendritic Cells Promote Activation of T Cells in Human Atherosclerotic Plaque and Blood, Which Is Repressed by Statins: microRNA let-7c Is Integral to the Effect. J Am Heart Assoc. (2016) 5:e003976. doi: 10.1161/JAHA.116.003976
42. Gautier EL, Huby T, Saint-Charles F, Ouzilleau B, Pirault J, Deswaerte V, et al. Conventional dendritic cells at the crossroads between immunity and cholesterol homeostasis in atherosclerosis. Circulation. (2009) 119:2367–75. doi: 10.1161/CIRCULATIONAHA.108.807537
43. Macritchie N, Grassia G, Sabir SR, Maddaluno M, Welsh P, Sattar N, et al. Plasmacytoid dendritic cells play a key role in promoting atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2012) 32:2569–79. doi: 10.1161/ATVBAHA.112.251314
44. Subramanian M, Thorp E, Hansson GK, and Tabas I. Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J Clin Invest. (2013) 123:179–88. doi: 10.1172/JCI64617
45. Koltsova EK, Garcia Z, Chodaczek G, Landau M, McArdle S, Scott SR, et al. Dynamic T cell-APC interactions sustain chronic inflammation in atherosclerosis. J Clin Invest. (2012) 122:3114–26. doi: 10.1172/JCI61758
46. Boshuizen MC and de Winther MP. Interferons as essential modulators of atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35:1579–88. doi: 10.1161/ATVBAHA.115.305464
47. Karbach S, Croxford AL, Oelze M, Schuler R, Minwegen D, Wegner J, et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol. (2014) 34:2658–68. doi: 10.1161/ATVBAHA.114.304108
48. He X, Liang B, and Gu N. Th17/Treg Imbalance and Atherosclerosis. Dis Markers. (2020) 2020:8821029. doi: 10.1155/2020/8821029
49. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. (2008) 105:18460–5. doi: 10.1073/pnas.0809850105
50. Hutchinson MA, Park HS, Zanotti KJ, Alvarez-Gonzalez J, Zhang J, Zhang L, et al. Auto-antibody production during experimental atherosclerosis in apoE(-/-) mice. Front Immunol. (2021) 12:695220. doi: 10.3389/fimmu.2021.695220
51. Rosenfeld SM, Perry HM, Gonen A, Prohaska TA, Srikakulapu P, Grewal S, et al. B-1b cells secrete atheroprotective igM and attenuate atherosclerosis. Circ Res. (2015) 117:e28–39. doi: 10.1161/CIRCRESAHA.117.306044
52. Felten DL, Livnat S, Felten SY, Carlson SL, Bellinger DL, and Yeh P. Sympathetic innervation of lymph nodes in mice. Brain Res Bull. (1984) 13:693–9. doi: 10.1016/0361-9230(84)90230-2
53. Ağaç D, Estrada LD, Maples R, Hooper LV, and Farrar JD. The β2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav Immun. (2018) 74:176–85. doi: 10.1016/j.bbi.2018.09.004
54. Takenaka MC, Araujo LP, Maricato JT, Nascimento VM, Guereschi MG, Rezende RM, et al. Norepinephrine Controls Effector T Cell Differentiation through beta2-Adrenergic Receptor-Mediated Inhibition of NF-kappaB and AP-1 in Dendritic Cells. J Immunol. (2016) 196:637–44. doi: 10.4049/jimmunol.1501206
55. Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. (2003) 421:384–8. doi: 10.1038/nature01339
56. Jin H, Li M, Jeong E, Castro-Martinez F, and Zuker CS. A body-brain circuit that regulates body inflammatory responses. Nature. (2024) 630:695–703. doi: 10.1038/s41586-024-07469-y
57. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell. (2015) 160:62–73. doi: 10.1016/j.cell.2014.11.047
58. Contreras F, Prado C, Gonzalez H, Franz D, Osorio-Barrios F, Osorio F, et al. Dopamine receptor D3 signaling on CD4+ T cells favors th1- and th17-mediated immunity. J Immunol. (2016) 196:4143–9. doi: 10.4049/jimmunol.1502420
59. Prado C, Gaiazzi M, Gonzalez H, Ugalde V, Figueroa A, Osorio-Barrios FJ, et al. Dopaminergic stimulation of myeloid antigen-presenting cells attenuates signal transducer and activator of transcription 3-activation favouring the development of experimental autoimmune encephalomyelitis. Front Immunol. (2018) 9:571. doi: 10.3389/fimmu.2018.00571
60. Bahr FS, Muller FE, Kasten M, Benen N, Sieve I, Scherr M, et al. Serotonin receptor 5-HT7 modulates inflammatory-associated functions of macrophages. Cell Mol Life Sci. (2025) 82:51. doi: 10.1007/s00018-024-05570-z
61. Muller T, Durk T, Blumenthal B, Grimm M, Cicko S, Panther E, et al. 5-hydroxytryptamine modulates migration, cytokine and chemokine release and T-cell priming capacity of dendritic cells in vitro and in vivo. PloS One. (2009) 4:e6453. doi: 10.1371/journal.pone.0006453
62. Retamal JS, Grace MS, Dill LK, Ramirez-Garcia P, Peng S, Gondin AB, et al. Serotonin-induced vascular permeability is mediated by transient receptor potential vanilloid 4 in the airways and upper gastrointestinal tract of mice. Lab Invest. (2021) 101:851–64. doi: 10.1038/s41374-021-00593-7
63. Duerschmied D, Suidan GL, Demers M, Herr N, Carbo C, Brill A, et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood. (2013) 121:1008–15. doi: 10.1182/blood-2012-06-437392
64. Fu J, Han Z, Wu Z, Xia Y, Yang G, Yin Y, et al. GABA regulates IL-1β production in macrophages. Cell Rep. (2022) 41:111770. doi: 10.1016/j.celrep.2022.111770
65. Bhandage AK, Jin Z, Korol SV, Shen Q, Pei Y, Deng Q, et al. GABA regulates release of inflammatory cytokines from peripheral blood mononuclear cells and CD4(+) T cells and is immunosuppressive in type 1 diabetes. EBioMedicine. (2018) 30:283–94. doi: 10.1016/j.ebiom.2018.03.019
66. Bhat R, Axtell R, Mitra A, Miranda M, Lock C, Tsien RW, et al. Inhibitory role for GABA in autoimmune inflammation. Proc Natl Acad Sci U S A. (2010) 107:2580–5. doi: 10.1073/pnas.0915139107
67. Kong Q, Gao S, Li P, Sun H, Zhang Z, Yu X, et al. Calcitonin gene-related peptide-modulated macrophage phenotypic alteration regulates angiogenesis in early bone healing. Int Immunopharmacol. (2024) 130:111766. doi: 10.1016/j.intimp.2024.111766
68. Nelson-Maney NP, Bálint L, Beeson AL, Serafin DS, Kistner BM, Douglas ES, et al. Meningeal lymphatic CGRP signaling governs pain via cerebrospinal fluid efflux and neuroinflammation in migraine models. J Clin Invest. (2024) 134. doi: 10.1172/JCI175616
69. Mu J, Que Y, Li X, Zhou F, Jin L, Li S, et al. CRH/CRHR1 modulates cerebrovascular endothelial cell permeability in association with S1PR2 and S1PR3 under oxidative stress. Vascul Pharmacol. (2022) 142:106941. doi: 10.1016/j.vph.2021.106941
70. Wu Y, Zhang R, Zhou C, Xu Y, Guan X, Hu J, et al. Enhanced expression of vascular cell adhesion molecule-1 by corticotrophin-releasing hormone contributes to progression of atherosclerosis in LDL receptor-deficient mice. Atherosclerosis. (2009) 203:360–70. doi: 10.1016/j.atherosclerosis.2008.05.059
71. Chen L, Liu DH, Zhang X, Zhang EH, Liu C, Su DF, et al. Baroreflex deficiency aggravates atherosclerosis via α7 nicotinic acetylcholine receptor in mice. Vascul Pharmacol. (2016) 87:92–9. doi: 10.1016/j.vph.2016.08.008
72. Al-Sharea A, Lee MKS, Whillas A, Michell DL, Shihata WA, Nicholls AJ, et al. Chronic sympathetic driven hypertension promotes atherosclerosis by enhancing hematopoiesis. Haematologica. (2019) 104:456–67. doi: 10.3324/haematol.2018.192898
73. Nicholls AJ, Wen SW, Hall P, Hickey MJ, and Wong CHY. Activation of the sympathetic nervous system modulates neutrophil function. J Leukoc Biol. (2018) 103:295–309. doi: 10.1002/JLB.3MA0517-194RR
74. Agelaki S, Tsatsanis C, Gravanis A, and Margioris AN. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect Immun. (2002) 70:6068–74. doi: 10.1128/IAI.70.11.6068-6074.2002
75. de Juan A, Ince LM, Pick R, Chen CS, Molica F, Zuchtriegel G, et al. Artery-associated sympathetic innervation drives rhythmic vascular inflammation of arteries and veins. Circulation. (2019) 140:1100–14. doi: 10.1161/CIRCULATIONAHA.119.040232
76. Mey L, Bonaterra GA, Hoffmann J, Schwarzbach H, Schwarz A, Eiden LE, et al. PAC1 agonist maxadilan reduces atherosclerotic lesions in hypercholesterolemic apoE-deficient mice. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms252413245
77. Perrotta M and Carnevale D. Neuroimmune modulation for targeting organ damage in hypertension and atherosclerosis. J Physiol. (2024) 602:4789–802. doi: 10.1113/JP284078
78. Choi B, Shin MK, Kim EY, Park JE, Lee H, Kim SW, et al. Elevated neuropeptide Y in endothelial dysfunction promotes macrophage infiltration and smooth muscle foam cell formation. Front Immunol. (2019) 10:1701. doi: 10.3389/fimmu.2019.01701
79. Li L, Lee EW, Ji H, and Zukowska Z. Neuropeptide Y-induced acceleration of postangioplasty occlusion of rat carotid artery. Arterioscler Thromb Vasc Biol. (2003) 23:1204–10. doi: 10.1161/01.ATV.0000071349.30914.25
80. Feickert M and Burckhardt BB. Substance P in cardiovascular diseases - A bioanalytical review. Clin Chim Acta. (2019) 495:501–6. doi: 10.1016/j.cca.2019.05.014
81. Kähler CM, Schratzberger P, and Wiedermann CJ. Response of vascular smooth muscle cells to the neuropeptide secretoneurin. A functional role for migration and proliferation in vitro. Arterioscler Thromb Vasc Biol. (1997) 17:2029–35. doi: 10.1161/01.ATV.17.10.2029
82. Amadio P, Cosentino N, Eligini S, Barbieri S, Tedesco CC, Sandrini L, et al. Potential relation between plasma BDNF levels and human coronary plaque morphology. Diagnostics (Basel). (2021) 11. doi: 10.3390/diagnostics11061010
83. Ramspacher A, Neudert M, Koller A, Schlager S, Kofler B, and Brunner SM. Influence of the regulatory peptide galanin on cytokine expression in human monocytes. Ann N Y Acad Sci. (2019) 1455:185–95. doi: 10.1111/nyas.14111
84. Kofler B, Brunner S, Koller A, Wiesmayr S, Locker F, Lang R, et al. Contribution of the galanin system to inflammation. Springerplus. (2015) 4:L57. doi: 10.1186/2193-1801-4-S1-L57
85. Munjuluri S, Wilkerson DA, Sooch G, Chen X, White FA, and Obukhov AG. Capsaicin and TRPV1 channels in the cardiovascular system: the role of inflammation. Cells. (2021) 11. doi: 10.3390/cells11010018
86. Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. (2003) 140:790–801. doi: 10.1038/sj.bjp.0705467
87. Abdalla SS, Harb AA, Almasri IM, and Bustanji YK. The interaction of TRPV1 and lipids: Insights into lipid metabolism. Front Physiol. (2022) 13:1066023. doi: 10.3389/fphys.2022.1066023
88. Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. (1999) 400:452–7. doi: 10.1038/22761
89. Assas BM, Miyan JA, and Pennock JL. Cross-talk between neural and immune receptors provides a potential mechanism of homeostatic regulation in the gut mucosa. Mucosal Immunol. (2014) 7:1283–9. doi: 10.1038/mi.2014.80
90. Wang Y, Cui L, Xu H, Liu S, Zhu F, Yan F, et al. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis. (2017) 260:13–9. doi: 10.1016/j.atherosclerosis.2017.03.016
91. Yang L, Sakurai T, Kamiyoshi A, Ichikawa-Shindo Y, Kawate H, Yoshizawa T, et al. Endogenous CGRP protects against neointimal hyperplasia following wire-induced vascular injury. J Mol Cell Cardiol. (2013) 59:55–66. doi: 10.1016/j.yjmcc.2013.02.002
92. Mandadi S, Tominaga T, Numazaki M, Murayama N, Saito N, Armati PJ, et al. Increased sensitivity of desensitized TRPV1 by PMA occurs through PKCepsilon-mediated phosphorylation at S800. Pain. (2006) 123:106–16. doi: 10.1016/j.pain.2006.02.016
93. Gräbner R, Lötzer K, Döpping S, Hildner M, Radke D, Beer M, et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE-/- mice. J Exp Med. (2009) 206:233–48. doi: 10.1084/jem.20080752
94. de Lemos JA, Blazing MA, Wiviott SD, Lewis EF, Fox KA, White HD, et al. Early intensive vs a delayed conservative simvastatin strategy in patients with acute coronary syndromes: phase Z of the A to Z trial. Jama. (2004) 292:1307–16. doi: 10.1001/jama.292.11.1307
95. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr., Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. (2008) 359:2195–207. doi: 10.1056/NEJMoa0807646
96. Ruscica M, Tokgözoğlu L, Corsini A, and Sirtori CR. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis. (2019) 288:146–55. doi: 10.1016/j.atherosclerosis.2019.07.015
97. Xu XY, Wang JX, Chen JL, Dai M, Wang YM, Chen Q, et al. GLP-1 in the hypothalamic paraventricular nucleus promotes sympathetic activation and hypertension. J Neurosci. (2024) 44. doi: 10.1523/JNEUROSCI.2032-23.2024
98. Katsurada K, Nandi SS, Sharma NM, and Patel KP. Enhanced expression and function of renal SGLT2 (Sodium-glucose cotransporter 2) in heart failure: role of renal nerves. Circ Heart Fail. (2021) 14:e008365. doi: 10.1161/CIRCHEARTFAILURE.121.008365
99. Hashimoto T, Ichiki T, Watanabe A, Hurt-Camejo E, Michaëlsson E, Ikeda J, et al. Stimulation of α7 nicotinic acetylcholine receptor by AR-R17779 suppresses atherosclerosis and aortic aneurysm formation in apolipoprotein E-deficient mice. Vascul Pharmacol. (2014) 61:49–55. doi: 10.1016/j.vph.2014.03.006
100. Ulleryd MA, Mjörnstedt F, Panagaki D, Yang LJ, Engevall K, Gutiérrez S, et al. Stimulation of alpha 7 nicotinic acetylcholine receptor (α7nAChR) inhibits atherosclerosis via immunomodulatory effects on myeloid cells. Atherosclerosis. (2019) 287:122–33. doi: 10.1016/j.atherosclerosis.2019.06.903
101. Ulleryd MA, Mjörnstedt F, Panagaki D, Yang LJ, Engevall K, Gutiérrez S, et al. Cholinergic signaling via the α7 nicotinic acetylcholine receptor regulates the migration of monocyte-derived macrophages during acute inflammation. J Neuroinflammation. (2024) 21:3. doi: 10.1186/s12974-023-03001-7
102. Huffman WJ, Musselman ED, Pelot NA, and Grill WM. Measuring and modeling the effects of vagus nerve stimulation on heart rate and laryngeal muscles. Bioelectron Med. (2023) 9:3. doi: 10.1186/s42234-023-00107-4
103. Aristovich K, Donega M, Fjordbakk C, Tarotin I, Chapman CAR, Viscasillas J, et al. Model-based geometrical optimisation and in vivo validation of a spatially selective multielectrode cuff array for vagus nerve neuromodulation. J Neurosci Methods. (2021) 352:109079. doi: 10.1016/j.jneumeth.2021.109079
104. Ben-Menachem E. Vagus nerve stimulation, side effects, and long-term safety. J Clin Neurophysiol. (2001) 18:415–8. doi: 10.1097/00004691-200109000-00005
105. Cantarín-Extremera V, Ruíz-Falcó-Rojas ML, Tamaríz-Martel-Moreno A, García-Fernández M, Duat-Rodriguez A, and Rivero-Martín B. Late-onset periodic bradycardia during vagus nerve stimulation in a pediatric patient. A new case and review of the literature. Eur J Paediatr Neurol. (2016) 20:678–83. doi: 10.1016/j.ejpn.2016.02.014
106. Johansson ME, Ulleryd MA, Bernardi A, Lundberg AM, Andersson A, Folkersen L, et al. α7 Nicotinic acetylcholine receptor is expressed in human atherosclerosis and inhibits disease in mice–brief report. Arterioscler Thromb Vasc Biol. (2014) 34:2632–6. doi: 10.1161/ATVBAHA.114.303892
107. Siniavin AE, Streltsova MA, Kudryavtsev DS, Shelukhina IV, Utkin YN, and Tsetlin VI. Activation of α7 nicotinic acetylcholine receptor upregulates HLA-DR and macrophage receptors: potential role in adaptive immunity and in preventing immunosuppression. Biomolecules. (2020) 10. doi: 10.3390/biom10040507
108. Uwada J, Nakazawa H, Mikami D, Islam MS, Muramatsu I, Taniguchi T, et al. PNU-120596, a positive allosteric modulator of α7 nicotinic acetylcholine receptor, directly inhibits p38 MAPK. Biochem Pharmacol. (2020) 182:114297. doi: 10.1016/j.bcp.2020.114297
109. Papke RL, Kem WR, Soti F, López-Hernández GY, and Horenstein NA. Activation and desensitization of nicotinic alpha7-type acetylcholine receptors by benzylidene anabaseines and nicotine. J Pharmacol Exp Ther. (2009) 329:791–807. doi: 10.1124/jpet.108.150151
110. Cordero-Erausquin M, Pons S, Faure P, and Changeux JP. Nicotine differentially activates inhibitory and excitatory neurons in the dorsal spinal cord. Pain. (2004) 109:308–18. doi: 10.1016/j.pain.2004.01.034
111. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, et al. Lack of interleukin-1beta decreases the severity of atherosclerosis in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. (2003) 23:656–60. doi: 10.1161/01.ATV.0000064374.15232.C3
112. Saraiva-Santos T, Zaninelli TH, Manchope MF, Andrade KC, Ferraz CR, Bertozzi MM, et al. Therapeutic activity of lipoxin A(4) in TiO(2)-induced arthritis in mice: NF-κB and Nrf2 in synovial fluid leukocytes and neuronal TRPV1 mechanisms. Front Immunol. (2023) 14:949407. doi: 10.3389/fimmu.2023.949407
113. Helwig BG, Craig RA, Fels RJ, Blecha F, and Kenney MJ. Central nervous system administration of interleukin-6 produces splenic sympathoexcitation. Auton Neurosci. (2008) 141:104–11. doi: 10.1016/j.autneu.2008.04.008
114. Pongratz G and Straub RH. The sympathetic nervous response in inflammation. Arthritis Res Ther. (2014) 16:504. doi: 10.1186/s13075-014-0504-2
115. Wieczorek M, Swiergiel AH, Pournajafi-Nazarloo H, and Dunn AJ. Physiological and behavioral responses to interleukin-1beta and LPS in vagotomized mice. Physiol Behav. (2005) 85:500–11. doi: 10.1016/j.physbeh.2005.05.012
116. Adlan AM, Veldhuijzen van Zanten J, Lip GYH, Paton JFR, Kitas GD, and Fisher JP. Cardiovascular autonomic regulation, inflammation and pain in rheumatoid arthritis. Auton Neurosci. (2017) 208:137–45. doi: 10.1016/j.autneu.2017.09.003
117. Zhong B, Ma S, and Wang DH. Knockout of TRPV1 Exacerbates Ischemia-reperfusion-induced Renal Inflammation and Injury in Obese Mice. In Vivo. (2020) 34:2259–68. doi: 10.21873/invivo.12036
118. Chen HH, Li YD, Cheng PW, Fang YC, Lai CC, Tseng CJ, et al. Gabapentin Reduces Blood Pressure and Heart Rate through the Nucleus Tractus Solitarii. Acta Cardiol Sin. (2019) 35:627–33. doi: 10.6515/ACS.201911_35(6).20190429B
119. Hashikawa-Hobara N, Inoue S, and Hashikawa N. Lack of alpha CGRP exacerbates the development of atherosclerosis in ApoE-knockout mice. Sci Rep. (2024) 14:18377. doi: 10.1038/s41598-024-69331-5
120. Abu Bakar H, Robert Dunn W, Daly C, and Ralevic V. Sensory innervation of perivascular adipose tissue: a crucial role in artery vasodilatation and leptin release. Cardiovasc Res. (2017) 113:962–72. doi: 10.1093/cvr/cvx062
121. Tasma Z, Siow A, Harris PWR, Brimble MA, Hay DL, and Walker CS. Characterisation of agonist signalling profiles and agonist-dependent antagonism at PACAP-responsive receptors: Implications for drug discovery. Br J Pharmacol. (2022) 179:435–53. doi: 10.1111/bph.15700
122. Gao W, Sun Y, Cai M, Zhao Y, Cao W, Liu Z, et al. Copper sulfide nanoparticles as a photothermal switch for TRPV1 signaling to attenuate atherosclerosis. Nat Commun. (2018) 9:231. doi: 10.1038/s41467-017-02657-z
123. Ho E, Li DX, Cui H, Akbar D, Siu R, Morshead CM, et al. Immunomodulation with local, sustained delivery of pituitary adenylate cyclase activating polypeptide results in improved functional recovery in stroke-injured mice. Adv Healthc Mater. (2025):e2500765. doi: 10.1002/adhm.202500765
124. Nicolai EN, Settell ML, Knudsen BE, McConico AL, Gosink BA, Trevathan JK, et al. Sources of off-target effects of vagus nerve stimulation using the helical clinical lead in domestic pigs. J Neural Eng. (2020) 17:046017. doi: 10.1088/1741-2552/ab9db8
125. De Ferrari GM, Stolen C, Tuinenburg AE, Wright DJ, Brugada J, Butter C, et al. Long-term vagal stimulation for heart failure: Eighteen month results from the NEural Cardiac TherApy foR Heart Failure (NECTAR-HF) trial. Int J Cardiol. (2017) 244:229–34. doi: 10.1016/j.ijcard.2017.06.036
126. Booth LC, Yao ST, Korsak A, Farmer DGS, Hood SG, McCormick D, et al. Selective optogenetic stimulation of efferent fibers in the vagus nerve of a large mammal. Brain Stimul. (2021) 14:88–96. doi: 10.1016/j.brs.2020.11.010
127. Yang S, Orlova Y, Park H, Smith SM, Guo Y, Chapin BA, et al. Cardiovascular safety of anti-CGRP monoclonal antibodies in older adults or adults with disability with migraine. JAMA Neurol. (2025) 82:132–41. doi: 10.1001/jamaneurol.2024.4537
128. Alen NV, Parenteau AM, Sloan RP, and Hostinar CE. Heart rate variability and circulating inflammatory markers in midlife. Brain Behav Immun Health. (2021) 15. doi: 10.1016/j.bbih.2021.100273
129. Cooper TM, McKinley PS, Seeman TE, Choo TH, Lee S, and Sloan RP. Heart rate variability predicts levels of inflammatory markers: Evidence for the vagal anti-inflammatory pathway. Brain Behav Immun. (2015) 49:94–100. doi: 10.1016/j.bbi.2014.12.017
130. von Känel R, Carney RM, Zhao S, and Whooley MA. Heart rate variability and biomarkers of systemic inflammation in patients with stable coronary heart disease: findings from the Heart and Soul Study. Clin Res Cardiol. (2011) 100:241–7. doi: 10.1007/s00392-010-0236-5
131. Aeschbacher S, Schoen T, Dörig L, Kreuzmann R, Neuhauser C, Schmidt-Trucksäss A, et al. Heart rate, heart rate variability and inflammatory biomarkers among young and healthy adults. Ann Med. (2017) 49:32–41. doi: 10.1080/07853890.2016.1226512
132. Murray K, Cremin M, Tay E, Sanchez K, Schreiber S, Lloyd E, et al. Inhibition of acute lung inflammation by a neuroimmune circuit induced by vagal nerve stimulation. Sci Adv. (2025) 11:eadw7080. doi: 10.1126/sciadv.adw7080
133. Trevizan-Baú P and McAllen RM. What is the Vagal-Adrenal Axis? J Comp Neurol. (2024) 532:e25656. doi: 10.1002/cne.25656
Keywords: atherosclerosis, immune, nervous system, mechanism, therapy
Citation: Shang Y, Pan Y, Xie L, Zhao Y, Mao W and Chen T (2025) Neuro-immune axis in atherosclerosis: mechanisms of regulation and therapeutic opportunities. Front. Immunol. 16:1619338. doi: 10.3389/fimmu.2025.1619338
Received: 28 April 2025; Accepted: 18 August 2025;
Published: 05 September 2025.
Edited by:
Francesco Gentile, Sant’Anna School of Advanced Studies, ItalyReviewed by:
Vladimir Stanislavovich Shavva, Karolinska Institutet (KI), SwedenSabrina Montuoro, Sant’Anna School of Advanced Studies, Italy
Copyright © 2025 Shang, Pan, Xie, Zhao, Mao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tingting Chen, Y3R0MzAzNEB6Y211LmVkdS5jbg==; Wei Mao, bWFvd2VpbHdAMTYzLmNvbQ==
†These authors have contributed equally to this work