 Karen Helene Bronken Martinsen1,2*
Karen Helene Bronken Martinsen1,2* Torstein Øverland1
Torstein Øverland1 Asbjørg Stray-Pedersen3
Asbjørg Stray-Pedersen3 Tore G. Abrahamsen2Børre Fevang2,4Hans Christian Erichsen Landsverk1,2
Tore G. Abrahamsen2Børre Fevang2,4Hans Christian Erichsen Landsverk1,2- 1Division of Pediatric and Adolescent Medicine, Oslo University Hospital, Oslo, Norway
- 2Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway
- 3Norwegian National Unit for Newborn Screening, Division of Pediatric and Adolescent Medicine, Oslo University Hospital Rikshospitalet, Oslo, Norway
- 4Section of Clinical Immunology and Infectious Diseases, Division of Specialized Medicine And Surgery, Oslo University Hospital, Oslo, Norway
Purpose: Inborn errors of immunity (IEIs) caused by mutations in STAT1 are associated with a broad range of clinical manifestations, ranging from relatively mild to life-threatening. Our aim was to give a clinical and molecular description of a Norwegian cohort with STAT1-related disease.
Methods: This is a descriptive epidemiological study.
Results: We present 23 patients with heterozygous STAT1 mutations, from 12 unrelated Norwegian families. Eighteen individuals had STAT1 gain-of-function (GOF) variants. Chronic mucocutaneous candidiasis (CMC) was the most common manifestation, observed in 94% of patients. Herpesviruses caused morbidity in one-third of patients, with severe complications such as varicella meningitis, varicella retinitis and ulcerative CMV esophagitis seen in 17%. Autoimmune hypertriglyceridemia with GPIHBP1 autoantibodies was diagnosed in one patient, adding a new entity to STAT1 GOF. Fifty percent of patients suffered chronic ophthalmologic manifestations. Severe gastrointestinal manifestations were observed in 22% of patients. Five of the 23 patients had STAT1 loss-of-function (LOF) variants. Mendelian susceptibility to mycobacterial disease (MSMD) was detected in three patients. Malignancy and autoimmunity were observed in two patients, both were heterozygous for the p.Ala246Thr variant, which is likely associated with a more complex phenotype. Significant viral infections were also observed. Presently, our cohort represents the largest nationwide study on STAT1-related disease.
Conclusion: We report novel clinical manifestations in STAT1 GOF, and suggest that heterozygous STAT1 LOF might be a more complex condition than originally presumed.
1 Introduction
Mutations in Signal transducer and activator of transcription 1 (STAT1) lead to various inborn errors of immunity (IEIs). More than 100 different mutations in STAT1 located in all domains of the protein have been reported (1). Mutations can be transmitted in an autosomal dominant (AD) or autosomal recessive (AR) manner, and exert either a gain-of-function (GOF) or loss-of-function (LOF) effect (1). GOF variants are believed to result in enhanced STAT1-dependent responses to interferons (IFNs) I and II and interleukin-27 (IL-27) (2–4). LOF variants cause impaired STAT1-mediated signaling after stimulation with IFNs and ILs (5).
Chronic mucocutaneous candidiasis (CMC) has been known for almost 100 years (6) and is characterized by the persistent or recurrent presence of candida infections affecting the skin, nails and mucous membranes (7, 8). In 2011, AD STAT1 GOF was found to be the underlying genetic cause in approximately 50% of cases of AD CMC (8). Currently, more than 400 patients with STAT1 GOF have been described worldwide (3). STAT1 GOF has a broad range of phenotypes, ranging from relatively mild to life-threatening. The range of severity varies even within families with identical mutations. CMC is the hallmark symptom, described in 98% of cases. Patients also show increased rates of bacterial, viral and invasive fungal infections. More than one-third have autoimmune disease, most frequently affecting the thyroid gland. It is also associated with an increased prevalence of malignancy and vascular aneurysms. The presence of malignancy, invasive infections or aneurysms are considered negative prognostic factors, which decrease the cumulative survival of patients aged 60 years from 87% to 31% (7). Hematopoietic stem cell transplantation (HSCT) was previously associated with poor outcome and a mortality rate around 50% (9). The outcome after HSCT has however improved dramatically after the introduction of JAK (Janus kinase) inhibitors (JAKi) as a bridge-to-transplant treatment, with survival rates of around 90% according to recent reports (10).
STAT1 LOF is rarer than STAT1 GOF, with approximately 60 patients described (11). The phenotype depends on the mode of inheritance, with AR cases usually being more severe. AR STAT1 LOF can result in partial or complete STAT1 deficiency, with the complete form presenting at an earlier age and with more severe symptoms. Mendelian susceptibility to mycobacterial disease (MSMD) is the most frequent manifestation. A vulnerability to viruses belonging to the Herpesviridae is also seen, as well as bacterial infections and secondary hemophagocytic syndrome (12). AD STAT1 LOF is described as isolated MSMD (5, 11, 13).
2 Methods and materials
To date, the Norwegian patient population with a genetic STAT1 diagnosis has not been described. The primary aim of this study was to provide a detailed description of this cohort. Patients carrying a disease-causing, suspected or likely disease-causing STAT1 variant were included in the study from January 2023 to April 2024.
Eligible patients were identified with help from members of the Norwegian network for primary immunodeficiency, a nationwide network for healthcare professionals involved in the treatment of patients with IEIs. We also collaborated with the departments of medical genetics with STAT1 included in their targeted gene sequencing panels (Oslo University Hospital, Haukeland University Hospital and Telemark Hospital Trust) to identify patients.
Oslo University Hospital is a national competence center for patients with IEIs in Norway, and patients were invited to participate in the study during their regular follow-up appointment. Patients who did not have a follow-up in Oslo, were recruited via their local hospital.
Patients included in the study were asked to complete a detailed questionnaire. This was a non-standardized questionnaire based on previously described symptoms and manifestations of STAT1-related disease. The questionnaire was also designed to identify other affected family members.
As part of routine follow-up, blood samples which included various immunological parameters were drawn from the participants. In cases where a new blood sample was not feasible, previous results were evaluated.
The patients’ electronic records at Oslo University Hospital were analyzed. The records included documents sent from the patient’s local hospital or general practitioner.
The patients were considered to have STAT1 GOF or LOF based on a comprehensive evaluation of the genetic variant identified, clinical manifestations, family history, laboratory parameters, and currently available relevant literature. For all patients, clinical manifestations were assessed to determine whether they indicated GOF or LOF. Patients with known pathogenic variants in STAT1 were classified accordingly. In cases where functional analysis of variants has been reported in the literature, the methodology of these studies was evaluated. If the methodology were found to be satisfactory, we used these results to interpret the functional effect of our variants. In case of previously unreported variants, blood samples for functional analysis were sent abroad as part of clinical investigations, and an evaluation of the patient’s symptoms was conducted to determine whether the variant indicated GOF or LOF. Samples for functional analysis were sent to the French National Institute for Health and Medical Research (Inserm) in Paris and the Advanced diagnostic unit at Freiburg University clinic.
Informed written consent was obtained from all patients or their legal guardians.
The study was approved by the Regional Committee for Health and Research Ethics in Norway.
3 Results
3.1 Patient population
We identified 24 patients from 12 nonrelated families, one of whom died at age 71, possibly due to cancer. All 23 surviving patients consented to participate in the study and are described here in detail. There was a male predominance, with 14 male patients.
Eighteen patients from nine families were classified as having STAT1 GOF. The patients were aged 5–57 years. Eleven patients (61%) were male. Debut of symptoms was early, with 67% (12/18) reporting symptoms within their first year of life.
Five patients from three families were classified as having AD STAT1 LOF. Three patients (60%) were male. The patients ranged in age from 8–54 years.
3.2 Genetic features
Whole exome sequencing with targeted gene sequencing panels identified eleven different variants in STAT1. The variants were located in different domains of STAT1, with the coiled-coil domain (CCD) most frequently affected (45%) (Figure 1).
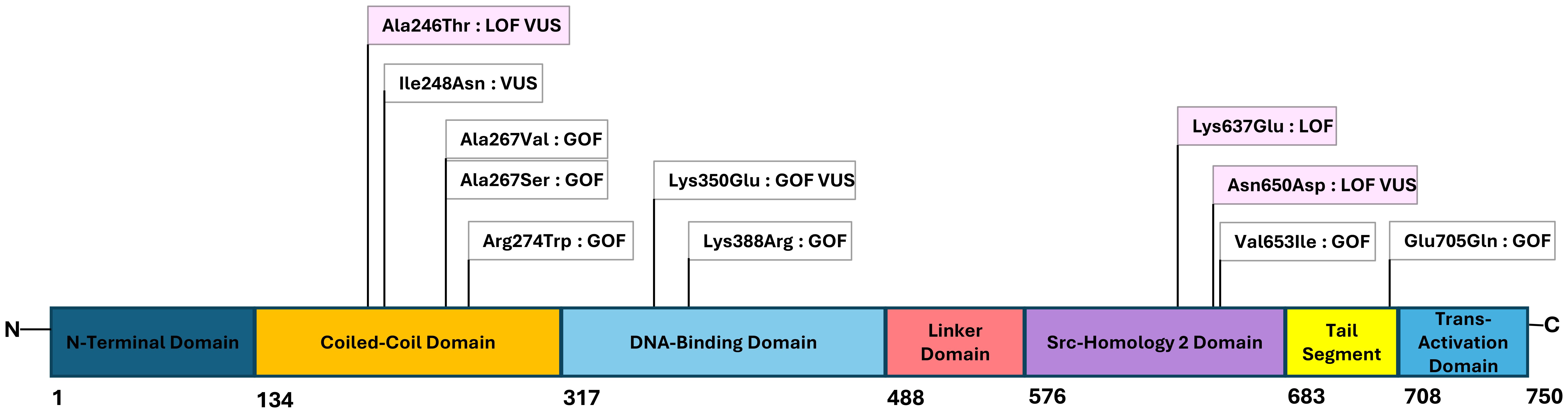
Figure 1. Figure with STAT1-mutations and their locations within the functional domains of the gene. The numbers below the figure indicating the amino acid number in the protein. Gain-of-function (GOF) and loss-of-function (LOF) variants are marked white and pink, respectively, and the novel variants of uncertain significance (VUS) noted.
Eight variants were considered GOF and three were considered LOF. Individuals within the same family had identical genetic variants. The variant NM_007315.4(STAT1):c.820C>T, p.Arg274Trp was shared by two families (Family 2 and Family 12). Seven variants were regarded as pathogenic or likely pathogenic, and four missense variants were variants of uncertain significance (VUSs). Functional testing of these variants indicated either GOF or LOF, as shown in Table 1.
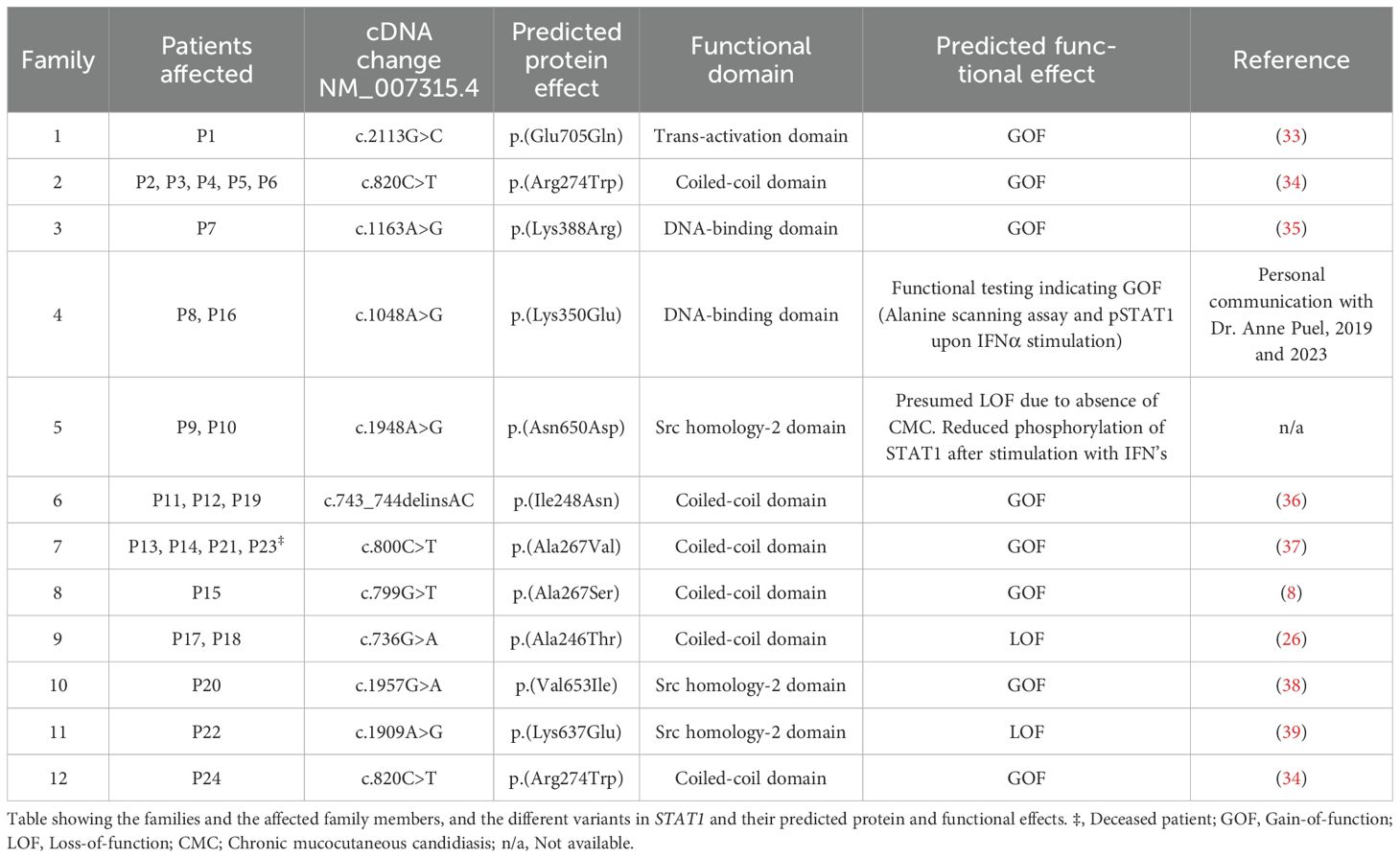
Table 1. The Norwegian families’ STAT1 variants with predicted functional effects.
Table 1 and Figure 1 show an overview of the demographics and the molecular findings. Figure 2 shows family pedigrees of the families with several affected family members.
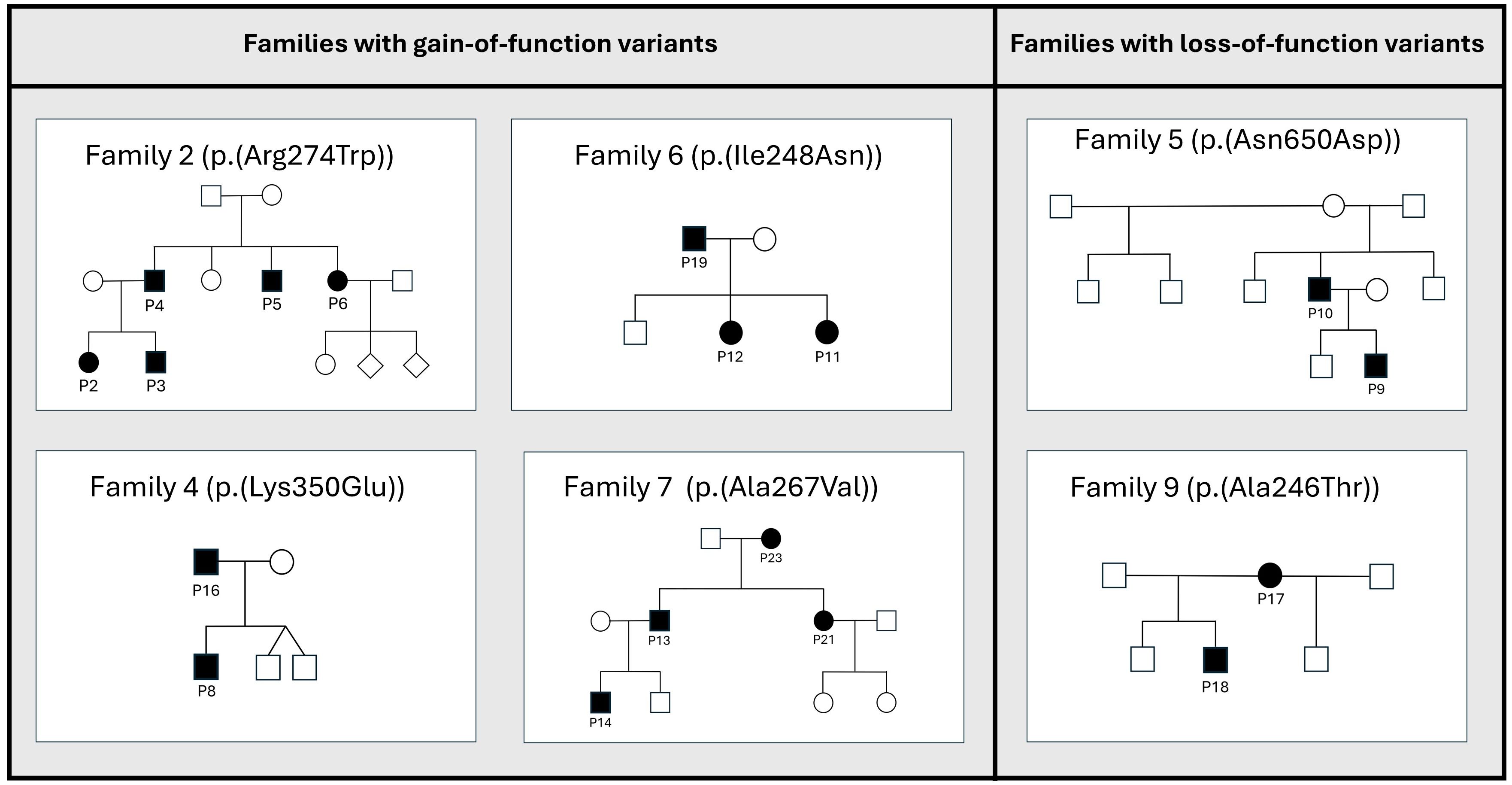
Figure 2. Pedigrees of the STAT1-families with several affected family members. Families with de novo variants, and only one affected member(i.e.families 1, 3, 8, 10, 11 and 12) are not shown in the figure. Affected members shown as black circles (females) or squares (males), unaffected members shown as white circles/squares. Diamond shape indicating unknown gender.
3.3 Clinical manifestations in STAT1 GOF
3.3.1 Infections
The patients had an increased infectious susceptibility, with CMC and recurrent respiratory tract infections being the most common manifestations. Infections in the eye and periorbital region affected 50% (9/18) of the patients. Bacterial skin infections were detected in 50% (9/18) of the patients.
Table 2 summarizes the pathogens detected in the patients.
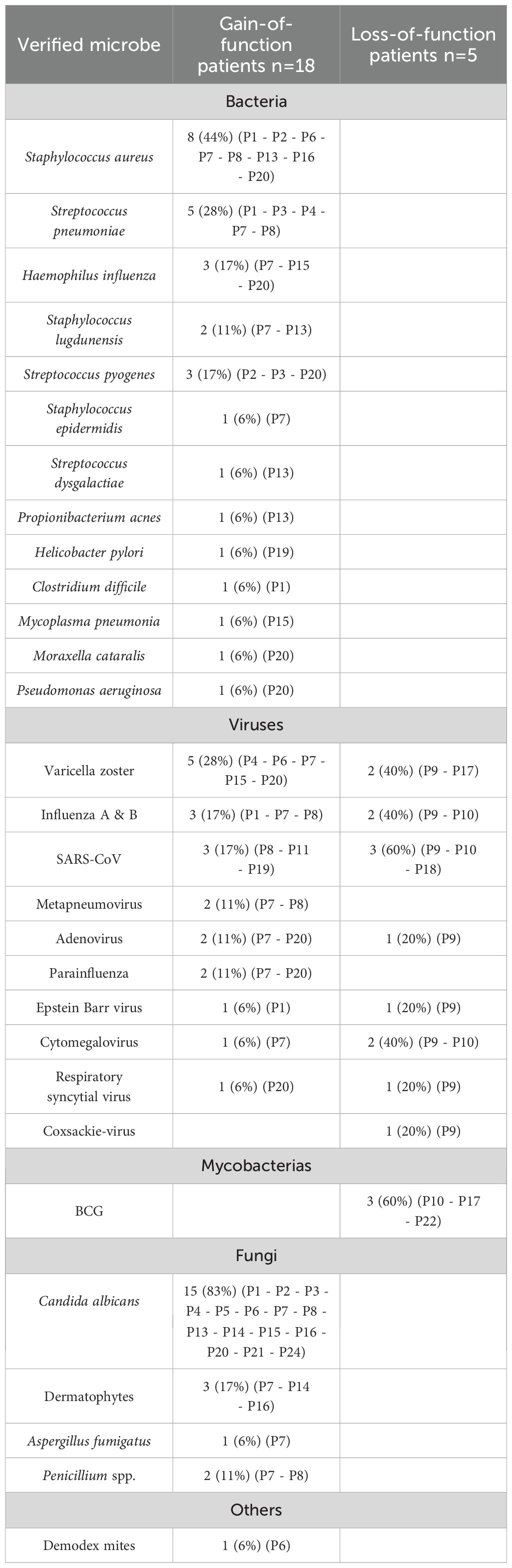
Table 2. Table showing the various pathogens detected in the STAT1-patients, with the specific patients in whom the pathogens were detected listed in brackets.
3.3.1.1 Fungal infections
CMC was the most common infectious manifestation, observed in 94% (17/18). Debut of CMC was early, with 61% reporting fungal infections within their first year of life. Candida albicans was identified in 83% (15/17), and dermatophytes were identified in 18% (3/17). The oral cavity was affected in all of the patients suffering CMC (17/17), the esophagus and skin were affected in 59% (10/17). Onychomycosis affected 35% (6/17) and fungal scalp infections affected 12% (2/17) of the patients. Five out of seven female patients (71%) suffered recurrent vulvovaginal candidiasis, with debut in childhood/adolescence. Among the male patients, 36% (4/11) had genital fungal infections.
Two patients were treated for systemic fungal infections, caused by Candida albicans and Aspergillus fumigatus. Penicillium species were identified in two patients.
3.3.1.2 Bacterial infections
Staphylococcal infections were most frequently identified (n=11), with Staphylococcus aureus being responsible for most documented infections (n=8). Streptococcal infections were documented in eight patients, with Streptococcus pneumoniae most commonly identified (n=5). One patient suffered severe necrotizing pneumonia caused by S.pneumoniae. Another patient contracted vision-threatening endophthalmitis due to Propionibacterium acnes.
3.3.1.3 Viral infections
Herpesviruses were associated with morbidity, particularly due to reactivation of Varicella zoster virus (VZV). Herpes zoster was documented in 28% (5/18) of patients, with recurrent episodes in one patient. One patient had VZV retinitis complicated with acute retinal necrosis, and another experienced VZV meningitis. Notably, none of the patients had severe or prolonged course of primary VZV-infection. One patient suffered severe, treatment-resistant ulcerative cytomegalovirus (CMV) esophagitis. Epstein-Barr virus (EBV) was found in biopsies from the gastrointestinal tract on several occasions in one patient.
Two pediatric patients underwent reactive viral polyarthritis, one of which were secondary to adenovirus.
Warts were detected in 35%, and 12% of patients had molluscum contagiosum.
3.3.1.4 Other infectious manifestations
One patient had a pustular skin infection with multiple Demodex mites within the pustules.
3.3.2 Autoimmunity and inflammatory manifestations
Autoimmune and inflammatory manifestations were observed in 78% (14/18) of the patients, 93% of whom had multiple manifestations.
3.3.2.1 Autoimmunity
Hypothyroidism was observed in 39% (7/18) of patients, and one patient had subclinical/latent hypothyroidism. Anti-thyroid antibodies were detected in 29% (2/7) of patients. Type 1 diabetes with positive anti-GAD antibodies was diagnosed in 6% (1/18), 11% (2/18) had DAT-positive autoimmune hemolytic anemia (AIHA), 6% (1/18) had seronegative rheumatoid arthritis, and 6% (1/18) had primary hyperparathyroidism.
One patient suffered from extreme hypertriglyceridemia. High titers of autoantibodies against GPIHBP1 (glycosylphosphatidylinositol-anchored high density lipoprotein binding protein-1) were found to be the underlying cause.
3.3.2.2 Organ specific inflammatory manifestations
3.3.2.2.1 Ophthalmologic manifestations
Chronic ophthalmologic manifestations were observed in 50% (9/18) of the patients; these manifestations were a combination of infectious and inflammatory. The patients suffered from chronic conjunctivitis, keratitis, blepharitis, chalazion, hordeolum and meibomian gland dysfunction. Vision-threatening manifestations were observed in 1/3 of the patients.
3.3.2.2.2 Gastrointestinal manifestations
Gastrointestinal manifestations were observed in 72% (13/18) of patients, with abdominal pain being the most prominent symptom. Complicated manifestations such as substantial esophageal strictures and stenosis, ulcerative inflammatory bowel disease and eosinophilic esophagitis were observed in 22% (4/18) of patients.
3.3.2.2.3 Respiratory tract manifestations
Chronic respiratory symptoms were observed in 56% (10/18) of patients, with chronic mucous production being the most common complaint (80%), followed by chronic cough (60%) and dyspnea (50%). Two patients were diagnosed with asthma. Bronchiectasis was found in 11% (2/18) of patients.
3.3.2.2.4 Dermal manifestations
Chronic dermal manifestations were described in 83% (15/18), with eczema seen most frequently (33%, 6/18). Patients were also diagnosed with psoriasis, rosacea, urticarial rash and severe acne vulgaris.
3.3.2.2.5 Vascular manifestations
Cerebral imaging was performed in 22% (4/18) of patients as a screening for cerebral aneurysms. No aneurysms were identified. Calcifications in the abdominal and thoracic aorta were described in one patient, and another had vasculitic histology changes in a biopsy from the genital area.
3.3.3 Malignancy/premalignant manifestations
None of our patients were diagnosed with cancer. Premalignant conditions were seen in 39% (7/18), mainly affecting the gastrointestinal tract (in 6 out of 7), but also affecting the genital tract and hematological system.
Table 3 provides a comprehensive overview of the clinical manifestations.
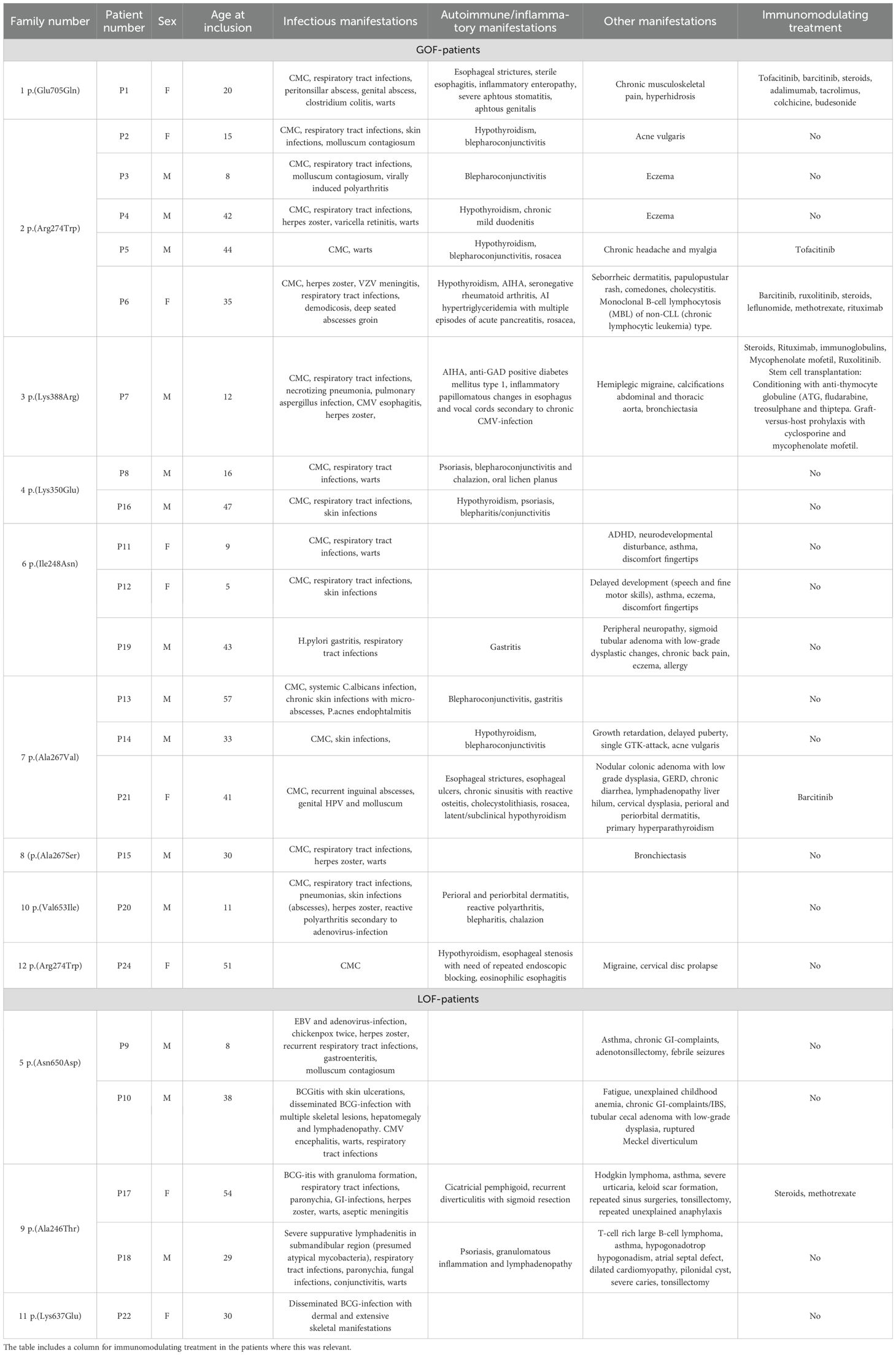
Table 3. Table showing clinical manifestations in each of the STAT1-patients.
3.4 Clinical manifestations in STAT1 LOF
3.4.1 Infections
Increased rate of infections was reported by 80% (4/5), with debut at an early age. The patients suffered frequent respiratory tract infections (80%, 4/5), mycobacterial infections (60%, 3/5), gastrointestinal tract infections (40%, 2/5) and paronychia (40%, 2/5).
One patient had MSMD only, the remaining patients had complex phenotypes.
3.4.1.1 Fungal infections
Onychomycosis since early childhood was reported in one patient. The same patient had substantial mucous membrane candidiasis under ongoing chemotherapy, which was more pronounced than normally seen.
3.4.1.2 Viral infections
Morbidity due to viruses, mainly herpesviruses, was seen. This included CMV encephalitis in one patient and disseminated herpes zoster in another. One patient reported having chickenpox twice, followed by herpes zoster infection. This patient was also admitted to the hospital with primary EBV-infection and adenovirus.
Warts were reported by 60% (3/5), and 20% (1/5) had molluscum contagiosum.
3.4.1.3 Mycobacterial infections
Complications secondary to Bacillus Calmette-Guerin (BCG) vaccination were observed in 60% (3/5). Two patients had disseminated BCG infection with extensive skeletal manifestations, and one patient developed a large granuloma. The patients were successfully treated with long-term tuberculostatica. The remaining two LOF patients were never BCG vaccinated.
One patient suffered severe suppurative lymphadenitis at five years of age which was suspected to be mycobacterial.
3.4.2 Autoimmunity and inflammatory manifestations
Cicatricial pemphigoid affecting the oral cavity, conjunctiva and skin was documented in one patient. The same patient additionally had chronically elevated inflammatory markers, recurrent episodes of diverticulitis and keloid scar formation.
Another patient had recurrent episodes of granulomatous inflammation and lymphadenopathy of unknown cause.
3.4.3 Malignancy
Hematological malignancy was documented in 40% (2/5) of patients. One patient was diagnosed with histiocyte/T-cell rich large B-cell lymphoma at the age of 26 years. The other patient was diagnosed with nodular lymphocyte rich Hodgkin lymphoma at the age of 30 years, and suffered a late recurrence of the malignancy at 55 years of age. The two patients shared the same genetic variant (p.Ala246Thr).
3.4.4 Other manifestations
One patient was diagnosed with atrial septum defect and later developed dilated cardiomyopathy. One patient had a ruptured Meckel diverticulum.
Table 3 provides a comprehensive overview of the manifestations in the patients.
3.5 Immunological investigations
All patients underwent extensive immunological investigations. A blood sample was obtained at the time of inclusion in 78% (18/23) of the patients. Previous laboratory values of interest were also evaluated. The patients had from one to more than 20 lymphocyte subset quantifications performed, with a median of six measurements per patient for STAT1 GOF and a median of two measurements per patient for the LOF patients. T- and B-cell subpopulations were analyzed in all but one patient, with a median of two measurements per patient (range 0–10 for T cells and 0–9 for B cells).
The results of the immunological investigations are summarized in Table 4 and Supplementary Tables 1-4.
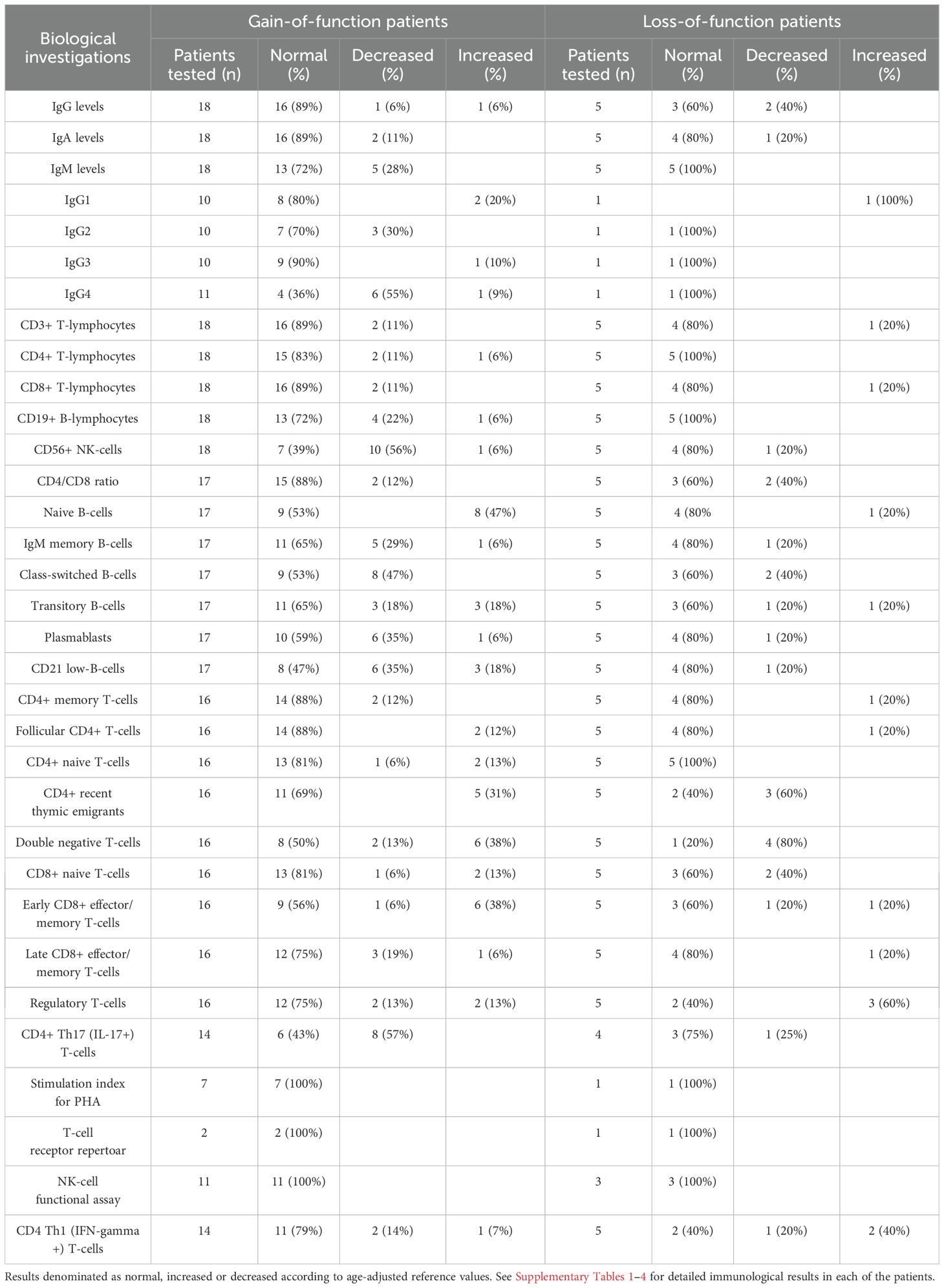
Table 4. Immunological investigations for STAT1 GOF and LOF patients summarized.
3.5.1 Immunological findings in GOF patients (n=18)
Immunoglobulins (IgG, IgA and IgM) were within the reference range in 61% (11/18) of patients. One patient had decreased levels of all immunoglobulin classes and received immunoglobulin substitution treatment. IgG4 was reduced in 55% (6/11) and IgG2 reduced in 30% (3/10) of patients.
NK-cell lymphopenia was found in 56% (10/18) and B-cell lymphopenia in 22% (4/18) of patients. One patient had reduced numbers of all lymphocyte subsets.
Deviating B-cell subpopulations were observed in 88% (15/17) of the patients, with an increased proportion of naïve B cells and a decreased proportion of class-switched B cells observed in 47% (8/17). Among the T cell subpopulations, 38% (6/16) had increased percentages of CD4-CD8- double-negative T cells (DNTs).
The percentage of Th17 cells was reduced on one or more measurements in 57% (8/14) of the patients. In patients with serial measurements (n=9), the values fluctuated from below the normal range to within the normal range in 33% of patients. The percentage of Th17 cells increased after the initiation of treatment with JAKi (n=5).
A systematic functional NK-cell degranulation assay was performed in 11 patients, with quantitative flow cytometric analysis of changes in surface-expression of CD107a after exposure to K562 cells. The assay results were normal in all patients tested.
Lymphocyte proliferation upon stimulation with phytohemagglutinin (PHA) was normal in all tested patients (n=6).
3.5.2 Immunological findings in LOF patients (n=5)
Reduced levels of IgG were observed in 40% (2/5) of the patients, none of whom received immunoglobulin substitution treatment.
Lymphocyte quantification was normal in 60% (3/5) of patients. The CD4+/CD8+ T cell ratio was reduced in 40% (2/5) of patients.
Lymphocyte proliferation upon stimulation with PHA was normal in the one patient tested.
3.6 Treatment
3.6.1 Treatment in STAT1 GOF patients
3.6.1.1 Antifungals
All the patients used antifungals, either on-demand or long-term prophylactic treatment (reported in 100% and 41%, respectively). Fluconazole was the antifungal most commonly used, in 94% (17/18) of patients. Fluconazole resistance was detected in one patient, but the patient responded well to voriconazole. Amphotericin B for suspected Aspergillus infection was used in one patient.
3.6.1.2 JAK inhibitors
JAKi were used in 28% (5/18) of STAT1 GOF patients, including one child. Barcitinib was used in three, and tofacitinib and ruxolitinib were used in two patients. Two patients were sequentially treated with two different JAKi due to the unsatisfactory effect of the first treatment.
The indications for starting JAKi were CMC, autoimmunity and gastrointestinal problems (i.e. esophageal inflammation, strictures and ulcers, inflammatory enteropathy). JAKi was used as a bridge-to-transplant in one patient with severe immune dysregulation and combined immunodeficiency. Decisions on initiating JAKi treatment were done on an individual basis. Clinical improvement was observed in 80% (4/5) of patients. One patient had no response to treatment and another had a declining response over time.
The daily dose administered of Tofacitinib was 10 mg (corresponding to 4,8 and 6,1 mg/m2/d), while the daily dose for Barcitinib was 4mg (corresponding to 2,3 and 2,4mg/m2/d). The pediatric patient was treated with Ruxolitinib at a dose of 10mg/m2/d. The daily dose of Tofacitinib and Ruxolitinib was divided in two equal doses, while Barcitinib was administered once daily.
Reported side effects were peritonsillar abscess, fatigue and weight gain.
3.6.1.3 Other immunosuppressants
Rituximab was effective in treating AIHA and autoimmune hypertriglyceridemia. Intravenous immunoglobulins and steroids were not effective in treating AIHA in our cohort.
Other immunosuppressants used were colchicine, adalimumab, steroids, tacrolimus and mycophenolate.
3.6.1.4 Stem cell transplantation
HSCT was successfully performed in one patient at the age of 9.5 years, with a 10/10 matched sibling donor. The patient’s conditioning regimen consisted of anti-thymocyte globuline (ATG), fludarabine, treosulphane and thiotepa. Graft-versus-host prophylaxis consisted of cyclosporine and mycophenolate mofetil. Two years post-SCT the patient was healthy and with good immune-reconstitution. Prior to SCT, the patient had diabetes mellitus and reduced pulmonary function, both of which persisted after SCT.
3.6.2 Treatment in STAT1 LOF patients
3.6.2.1 Antituberculosis treatment
Disseminated BCG infections were managed according to current national guidelines and resistance pattern related to the BCG-strain used in Norway.
3.6.2.2 Antifungals
In the LOF group, 40% (2/5) of the patients used antifungals on demand.
3.6.2.3 Immunosuppressive treatment
Methotrexate was successfully administered for mucocutaneous pemphigoid.
4 Discussion
In the present study 23 Norwegian patients with STAT1-related disease are presented, making this the largest nationwide study on STAT1 patients to date. To our best knowledge, all patients with a molecularly confirmed STAT1-related disease in Norway were identified. This was enabled by a well-established national immunological network and extended collaboration with the genetic departments performing STAT1 sequencing. This provides us with a unique opportunity to estimate the prevalence of STAT1-related disease in Norway. Norway has a population of approximately 5.56 million people, giving an estimated prevalence of STAT1 related disease in Norway of 1:240 000. However, due to the broad phenotype and unawareness of the diagnosis among clinicians not familiar with IEIs, there is probably many undiagnosed patients. Thus, the true prevalence is probably higher. Nonetheless, our findings indicate that STAT1-related disease is one of the more common IEIs (14).
4.1 STAT1 GOF patients
CMC was the most consistent clinical feature in the STAT1 GOF patients (94%), as was increased susceptibility to infections in general, consistent with previous reports (7).
Increased morbidity due to herpesviruses was observed, consistent with previous findings (9). Severe ulcerative esophagitis due to CMV was observed in one patient and necessitated parenteral nutrition. Reactivation of VZV caused severe disease in 11% of patients, including VZV meningitis and retinitis, and around 30% suffered herpes zoster. As a measure of the immune system’s capacity to handle herpesviruses patients are routinely asked about the clinical course of their primary varicella infection. Importantly, our findings show that a mild primary infection does not exclude the possibility of severe morbidity from herpesvirus later in life.
Autoimmune phenomena are well documented in STAT1 GOF and were seen in >50% of our patients. As reported by Strøm (15), one of our patients adds a new form of autoimmunity to STAT1 GOF, namely autoimmune hypertriglyceridemia due to GPIHBP1 autoantibodies. Autoimmune hypertriglyceridemia has been found in a limited number of patients (16). It seems reasonable to screen for hyperlipidemia in the routine follow-up of STAT1 patients and to consider autoimmune hypertriglyceridemia in cases of an unexplained hypertriglyceridemia.
Severe ophthalmic manifestations were seen in 50% of our STAT1 GOF patients. Ophthalmologic complications are sparsely documented in STAT1 GOF (7), ophthalmic health is in fact not mentioned at all in some newer review papers (17, 18). Our findings suggest that ophthalmic complications are underreported in STAT1 GOF, but are of major impact for the affected patients. When following STAT1 patients, it is important to ask about eye health in the routine anamnesis. To prevent chronic eye damage, these patients require regular ophthalmologic follow-up.
Inflammatory dermal and ophthalmic manifestations due to Demodex mites are increasingly being reported in IEIs like STAT1 GOF (19–22). Demodex mites were found to be the causative agent for a pustular skin infection in one of our patient. Systematic search for Demodex were not undertaken in the other patients suffering dermal and ophthalmic manifestations. There is a well-established association between Demodex and rosacea (23), and Demodex are also believed to play a role in ocular rosacea (24). In our patient population, three patients were diagnosed with rosacea, of which two of them also had ophthalmic manifestations. Although not formally diagnosed, the patients with blepharoconjunctivitis and chalazion have symptoms compatible with ocular rosacea. Our findings add more data to the association between rosacea-like demodicosis and STAT1 GOF (20). In patients with persistent dermatological and ophthalmologic manifestations, rosacea and Demodex should be considered possible etiologies, as this may influence the choice of treatment. Rosacea and demodicosis might be underrecognized in STAT1 GOF.
4.2 STAT1 LOF patients
AD STAT1 LOF is associated with MSMD, but has not previously been associated with an increased susceptibility to autoimmunity, inflammation and malignancy (25). Our cohort comprise five patients with AD STAT1 LOF, of which only one of them presented with a phenotype of MSMD only. Disseminated BCG infection was diagnosed in two more patients, but these had additional heterogeneous disease manifestations.
The STAT1 LOF patients not suffering BCG infection was not vaccinated and thus were never exposed to the bacillus. STAT1 GOF have also been linked to a predisposition to mycobacterial infections (7, 9). Of note, complications secondary to BCG vaccination were not seen in any of our STAT1 GOF patients. This was despite the fact that more than 50% of the patients were vaccinated as part of the routine Childhood Immunization Program.
The authors question whether the p.Ala246Thr variant might be associated with a more severe and multifaceted phenotype. Our two patients had autoimmune, inflammatory and oncological manifestations, which up until now have not been associated with STAT1 LOF (9, 12, 25). Both patients had hematological malignancies, which was also reported by Chen et al. in one patient with the same mutation (26).
One of our patients had congenital heart disease, similarly to what has been previously reported in another STAT1 LOF patient (26). Our findings add more data to the speculation of whether there is an association between STAT1 LOF and congenital malformations.
4.3 Immunological findings
Broad immunological investigations were performed in our patient cohort, with various immunological deviations observed. However, there are no pathognomonic tests for STAT1, and genetic testing is necessary to diagnose STAT1-related disease.
The most consistent immunological abnormality previously reported in STAT1 GOF is a reduced frequency of Th17-cells (9). This can, however, also be found in other conditions associated with CMC and is not pathognomonic for STAT1 GOF (27). Reduced levels of Th17 cells were found in 57% of STAT1 GOF patients. However, the values fluctuated over time, from below to within the normal range. The measurement of Th17 cells cannot be considered a robust assay to screen for STAT1 GOF, and a normal Th17 value cannot be used to exclude the possibility of STAT1 GOF.
The immunological background for the increased autoimmunity in STAT1 GOF is not fully understood but is believed to be caused by increased responses to type I IFN signaling (3, 7). Approximately 40% of our patients had increased levels of DNTs. Decreased percentages of regulatory T cells were seen in 13% (2/16) of the patients, one of whom had an IPEX-like (Immune dysregulation, polyendocrinopathy, enteropathy X-linked) phenotype. Deviations in DNTs and regulatory T cells have previously not been associated with STAT1 GOF (28, 29).
Altered humoral immunity has been reported in STAT1 GOF (7, 30, 31). The present study add additional data to this matter, as 50% of our patients had deviations in B-cell subpopulations and 22% had B cell lymphopenia.
NK-cell lymphopenia is well documented in STAT1 GOF (2, 7, 9). Compared to previous findings, a greater proportion of our patients (60%) had decreased numbers of NK-cells. However, none of our patients had pathologic NK-cell functional assays, contrary to previous findings (32).
4.4 Summary
The present study of STAT1-related disease in Norway adds novel clinical characteristics to STAT1 GOF, making an already broad phenotype even broader. The surprising finding of troublesome ophthalmic manifestations in 50% of patients should be considered, as this warrants specialized treatment and follow-up.
Our findings strongly support the assumption that STAT1 LOF may be a more complex condition than originally thought (26). A multidisciplinary approach is warranted for both STAT1 GOF and LOF patients, and one must be aware of the possibility of malignancies in STAT1 LOF.
Data availability statement
The datasets presented in this article are not readily available because according to Norwegian GDP-rules, clinical data and lab results cannot be made publicly available. Immunological assays were done as part of routine follow-up of patients, and are part of electronic patient records. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Regional Committee for Health and Research Ethics in Norway. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
KM: Methodology, Investigation, Writing – review & editing, Conceptualization, Writing – original draft, Formal Analysis, Project administration. TØ: Writing – review & editing, Methodology, Supervision. AS: Writing – review & editing. TA: Supervision, Writing – review & editing. BF: Writing – review & editing. HL: Supervision, Project administration, Conceptualization, Writing – review & editing, Methodology.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
The authors thank the patients and their relatives for participating in the study. We also thank the following doctors for help with identifying and recruiting patients and for valuable help with the completion of the manuscript; Stina Jordal, Anders Aune Tveita, Jørgen Østensjø, Dag Seeger Halvorsen, Mette Engan, Franziskus Johannes Bosse, Kristian Tveten and Andreas Benneche; Emma Maria Haapaniemi; Per Kristian Knudsen; and Liv Osnes for invaluable help with immunological assays.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1620291/full#supplementary-material
References
1. Mizoguchi Y and Okada S. Inborn errors of STAT1 immunity. Curr Opin Immunol. (2021) 72:59–64. doi: 10.1016/j.coi.2021.02.009
2. Chen X, Xu Q, Li X, Wang L, Yang L, Chen Z, et al. Molecular and phenotypic characterization of nine patients with STAT1 GOF mutations in China. J Clin Immunol. (2020) 40:82–95. doi: 10.1007/s10875-019-00688-3
3. Okada S, Asano T, Moriya K, Boisson-Dupuis S, Kobayashi M, Casanova JL, et al. Human STAT1 gain-of-function heterozygous mutations: chronic mucocutaneous candidiasis and type I interferonopathy. J Clin Immunol. (2020) 40:1065–81. doi: 10.1007/s10875-020-00847-x
4. Olbrich P and Freeman AF. STAT1 and STAT3 mutations: important lessons for clinical immunologists. Expert Rev Clin Immunol J Translated Name Expert Rev Clin Immunol Keyword Heading chronic mucocutaneous candidiasis Gain Funct (GOF) STAT1 gain Funct (GOF) STAT3 hyper IgE syndrome Job’s syndrom. (2018) 14:1029–41. doi: 10.1080/1744666X.2018.1531704
5. Ott N, Faletti L, Heeg M, Andreani V, and Grimbacher B. JAKs and STATs from a clinical perspective: loss-of-function mutations, gain-of-function mutations, and their multidimensional consequences. J Clin Immunol. (2023) 43:1326–59. doi: 10.1007/s10875-023-01483-x
6. Thorpe JR ES and Handley HE. CHRONIC TETANY AND CHRONIC MYCELIAL STOMATITIS IN A CHILD AGED FOUR AND ONE-HALF YEARS. Am J Dis Children. (1929) 38:328–38. doi: 10.1001/archpedi.1929.01930080104011
7. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. (2016) 127:3154–64. doi: 10.1182/blood-2015-11-679902
8. van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, et al. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N Engl J Med. (2011) 365:54–61. doi: 10.1056/NEJMoa1100102
9. Zhang W, Chen X, Gao G, Xing S, Zhou L, Tang X, et al. Clinical relevance of gain- and loss-of-function germline mutations in STAT1: A systematic review. Front Immunol. (2021) 12:654406. doi: 10.3389/fimmu.2021.654406
10. Fischer M, Olbrich P, Hadjadj J, Aumann V, Bakhtiar S, Barlogis V, et al. JAK inhibitor treatment for inborn errors of JAK/STAT signaling: An ESID/EBMT-IEWP retrospective study. J Allergy Clin Immunol. (2024) 153:275–86 e18. doi: 10.1016/j.jaci.2023.10.018
11. Ye F, Zhang W, Dong J, Peng M, Fan C, Deng W, et al. A novel STAT1 loss-of-function mutation associated with Mendelian susceptibility to mycobacterial disease. Front Cell Infect Microbiol. (2022) 12:1002140. doi: 10.3389/fcimb.2022.1002140
12. Le Voyer T, Sakata S, Tsumura M, Khan T, Esteve-Sole A, Al-Saud BK, et al. Genetic, immunological, and clinical features of 32 patients with autosomal recessive STAT1 deficiency. J Immunol. (2021) 207:133–52. doi: 10.4049/jimmunol.2001451
13. Bustamante J. Mendelian susceptibility to mycobacterial disease: recent discoveries. Hum Genet. (2020) 139:993–1000. doi: 10.1007/s00439-020-02120-y
14. Lopes JP and Cunningham-Rundles C. The importance of primary immune deficiency registries: the United States immunodeficiency network registry. Immunol Allergy Clin North Am. (2020) 40:385–402. doi: 10.1016/j.iac.2020.03.002
15. Strom TB, Tveita AA, Bogsrud MP, and Leren TP. Molecular genetic testing and measurement of levels of GPIHBP1 autoantibodies in patients with severe hypertriglyceridemia: The importance of identifying the underlying cause of hypertriglyceridemia. J Clin Lipidol. (2023) 18:e80-e89. doi: 10.1016/j.jacl.2023.11.002
16. Miyashita K, Lutz J, Hudgins LC, Toib D, Ashraf AP, Song W, et al. Chylomicronemia from GPIHBP1 autoantibodies. J Lipid Res. (2020) 61:1365–76. doi: 10.1194/jlr.R120001116
17. Olbrich P and Freeman AF. STAT1 and STAT3 gain of function: clinically heterogenous immune regulatory disorders. Curr Opin Allergy Clin Immunol. (2024) 24:440–7. doi: 10.1097/ACI.0000000000001039
18. Asano T, Noma K, Mizoguchi Y, Karakawa S, and Okada S. Human STAT1 gain of function with chronic mucocutaneous candidiasis: A comprehensive review for strengthening the connection between bedside observations and laboratory research. Immunol Rev. (2024) 322:81–97. doi: 10.1111/imr.v322.1
19. Baghad B, El Fatoiki FZ, Benhsaien I, Bousfiha AA, Puel A, Migaud M, et al. Pediatric demodicosis associated with gain-of-function variant in STAT1 presenting as rosacea-type rash. J Clin Immunol. (2021) 41:698–700. doi: 10.1007/s10875-020-00942-z
20. Martinot M, Korganow AS, Wald M, Second J, Birckel E, Mahe A, et al. Case report: A new gain-of-function mutation of STAT1 identified in a patient with chronic mucocutaneous candidiasis and rosacea-like demodicosis: an emerging association. Front Immunol. (2021) 12:760019. doi: 10.3389/fimmu.2021.760019
21. Molho-Pessach V, Meltser A, Kamshov A, Ramot Y, and Zlotogorski A. STAT1 gain-of-function and chronic demodicosis. Pediatr Dermatol. (2020) 37:153–5. doi: 10.1111/pde.14011
22. Jacob S, VanDaele MA, and Brown JN. Treatment of Demodex-associated inflammatory skin conditions: A systematic review. Dermatol Ther. (2019) 32:e13103. doi: 10.1111/dth.13103
23. Zhao YE, Wu LP, Peng Y, and Cheng H. Retrospective analysis of the association between Demodex infestation and rosacea. Arch Dermatol. (2010) 146:896–902. doi: 10.1001/archdermatol.2010.196
24. Mohamed-Noriega K, Loya-Garcia D, Vera-Duarte GR, Morales-Wong F, Ortiz-Morales G, Navas A, et al. Ocular rosacea: an updated review. Cornea. (2025) 44:525–37. doi: 10.1097/ICO.0000000000003785
25. Asano T, Utsumi T, Kagawa R, Karakawa S, and Okada S. Inborn errors of immunity with loss- and gain-of-function germline mutations in STAT1. Clin Exp Immunol. (2023) 212:96–106. doi: 10.1093/cei/uxac106
26. Chen X, Chen J, Chen R, Mou H, Sun G, Yang L, et al. Genetic and functional identifying of novel STAT1 loss-of-function mutations in patients with diverse clinical phenotypes. J Clin Immunol. (2022) 42:1778–94. doi: 10.1007/s10875-022-01339-w
27. Tangye SG and Puel A. The th17/IL-17 axis and host defense against fungal infections. J Allergy Clin Immunol Pract. (2023) 11:1624–34. doi: 10.1016/j.jaip.2023.04.015
28. Largent AD, Lambert K, Chiang K, Shumlak N, Liggitt D, Oukka M, et al. Dysregulated IFN-gamma signals promote autoimmunity in STAT1 gain-of-function syndrome. Sci Transl Med. (2023) 15:eade7028. doi: 10.1126/scitranslmed.ade7028
29. Uzel G, Sampaio EP, Lawrence MG, Hsu AP, Hackett M, Dorsey MJ, et al. Dominant gain-of-function STAT1 mutations in FOXP3 wild-type immune dysregulation-polyendocrinopathy-enteropathy-X-linked-like syndrome. J Allergy Clin Immunol. (2013) 131:1611–23. doi: 10.1016/j.jaci.2012.11.054
30. Depner M, Fuchs S, Raabe J, Frede N, Glocker C, Doffinger R, et al. The extended clinical phenotype of 26 patients with chronic mucocutaneous candidiasis due to gain-of-function mutations in STAT1. J Clin Immunol. (2016) 36:73–84. doi: 10.1007/s10875-015-0214-9
31. Romberg N, Morbach H, Lawrence MG, Kim S, Kang I, Holland SM, et al. Gain-of-function STAT1 mutations are associated with PD-L1 overexpression and a defect in B-cell survival. J Allergy Clin Immunol. (2013) 131:1691–3. doi: 10.1016/j.jaci.2013.01.004
32. Tabellini G, Vairo D, Scomodon O, Tamassia N, Ferraro RM, Patrizi O, et al. Impaired natural killer cell functions in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. (2017) 140:553–64 e4. doi: 10.1016/j.jaci.2016.10.051
33. Al Shehri T, Gilmour K, Gothe F, Loughlin S, Bibi S, Rowan AD, et al. Novel gain-of-function mutation in stat1 sumoylation site leads to CMC/CID phenotype responsive to ruxolitinib. J Clin Immunol. (2019) 39:776–85. doi: 10.1007/s10875-019-00687-4
34. Fujiki R, Hijikata A, Shirai T, Okada S, Kobayashi M, and Ohara O. Molecular mechanism and structural basis of gain-of-function of STAT1 caused by pathogenic R274Q mutation. J Biol Chem. (2017) 292:6240–54. doi: 10.1074/jbc.M116.753848
35. Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. (2017) 139:232–45. doi: 10.1016/j.jaci.2016.05.042
36. Kagawa R, Fujiki R, Tsumura M, Sakata S, Nishimura S, Itan Y, et al. Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J Allergy Clin Immunol. (2017) 140:232–41. doi: 10.1016/j.jaci.2016.09.035
37. Mizoguchi Y, Tsumura M, Okada S, Hirata O, Minegishi S, Imai K, et al. Simple diagnosis of STAT1 gain-of-function alleles in patients with chronic mucocutaneous candidiasis. J Leukoc Biol. (2014) 95:667–76. doi: 10.1189/jlb.0513250
38. Meesilpavikkai K, Dik WA, Schrijver B, Nagtzaam NM, van Rijswijk A, Driessen GJ, et al. A novel heterozygous mutation in the STAT1 SH2 domain causes chronic mucocutaneous candidiasis, atypically diverse infections, autoimmunity, and impaired cytokine regulation. Front Immunol. (2017) 8:274. doi: 10.3389/fimmu.2017.00274
Keywords: STAT1 GOF, STAT1 LOF, chronic mucocutaneous candidiasis (CMC), autoimmune hypertriglyceridemia, malignancy, ophthalmologic
Citation: Martinsen KHB, Øverland T, Stray-Pedersen A, Abrahamsen TG, Fevang B and Landsverk HCE (2025) A Norwegian cohort with STAT1-related disease – further expanding the clinical phenotype. Front. Immunol. 16:1620291. doi: 10.3389/fimmu.2025.1620291
Received: 29 April 2025; Accepted: 02 June 2025;
Published: 13 August 2025.
Edited by:
Stephanie Boisson-Dupuis, The Rockefeller University, United StatesReviewed by:
Satoshi Okada, Hiroshima University, JapanEyad Gadour, King Abdulaziz National Guard Hospital, Saudi Arabia
Copyright © 2025 Martinsen, Øverland, Stray-Pedersen, Abrahamsen, Fevang and Landsverk. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karen Helene Bronken Martinsen, YjM0NDcxQG91cy1oZi5ubw==