 Xiufeng Wang
Xiufeng Wang Cong Luo
Cong Luo Danying Zhang
Danying Zhang- 1Department of Orthopedic Trauma, Zhuji People’s Hospital of Zhejiang Province, Shaoxing, China
- 2Department of Emergency and Critical Care, Shanghai Changzheng Hospital, Naval Medical University, Shanghai, China
Cancer-associated fibroblasts (CAFs) are pivotal in shaping the immunosuppressive and chemoresistant tumor microenvironment (TME) of osteosarcoma (OS). This review explores how CAFs drive OS progression through paracrine signaling (e.g., TGF-β, IL-6), extracellular matrix (ECM) remodeling, exosome-mediated crosstalk, and metabolic reprogramming. We highlight CAF heterogeneity (e.g., myCAFs, iCAFs) and their roles in therapy resistance, emphasizing emerging strategies such as FAP inhibitors, TGF-β blockers, and CXCR4 antagonists. Combining these approaches with immunotherapy or chemotherapy offers promise for overcoming chemoresistance. Challenges like CAF plasticity and biomarker development are discussed, alongside future directions for precision targeting in OS.
1 Introduction
Osteosarcoma (OS) is a highly aggressive and easily metastasizing malignant bone tumor originating from mesenchymal cells in the bone marrow cavity or bone surface (1, 2). OS predominantly affects children and adolescents, with a peak incidence between 10 and 19 years of age (3). This age correlation stems from rapid skeletal growth during this period, where increased bone cell proliferation and differentiation elevate the risk of malignant transformation (4). OS cells exhibit marked cellular atypia and possess the unique ability to directly produce osteoid matrix or immature bone tissue, which serves as the pathological hallmark for diagnosis (5, 6). The treatment of OS typically employs multi-agent chemotherapy regimens to enhance therapeutic efficacy. Standard chemotherapeutic agents include methotrexate, cisplatin, doxorubicin and so on (7). However, the clinical utility of these drugs is significantly constrained by the development of chemoresistance and severe adverse effects (8–10). Indeed, overcoming drug resistance remains a key challenge in OS research.
The tumor microenvironment (TME) refers to the local milieu surrounding tumor cells, encompassing not only the malignant cells themselves but also adjacent stromal cells, extracellular matrix (ECM), cytokines, chemokines and metabolic byproducts (11–13). Cancer-associated fibroblasts (CAFs) represent a predominant cellular component of the TME, exhibiting remarkable functional and molecular heterogeneity (14). Through the secretion of cytokines, chemokines, growth factors and extracellular matrix components, CAFs significantly promote tumor progression and confer treatment resistance (15). For example, periostin, a CAF-secreted protein, promoted platinum drug resistance in ovarian cancer cells through activation of the PI3K/Akt signaling pathway (16). More importantly, a recent study demonstrated that CAFs promoted the occurrence of OS through the MIF-CD74 signalling axis, and their abundance was strongly correlated with the prognosis of OS patients (17). Recent single-cell RNA sequencing studies have revealed that CAFs are not a uniform population but rather consist of multiple functionally distinct subtypes that differentially influence OS progression and therapy resistance (18). This heterogeneity manifests through diverse secretory profiles, metabolic programs, and interactions with tumor cells, which collectively shape the immunosuppressive and chemoresistant TME (19). For instance, inflammatory CAFs (iCAFs) and myofibroblastic CAFs (myCAFs) exhibit opposing roles in OS metastasis, with the former promoting immune evasion via IL-6/STAT3 signaling and the latter driving ECM remodeling to impede drug delivery (20). Understanding these subsets is critical for developing precision therapies targeting CAF-specific vulnerabilities. Therefore, targeting CAFs has emerged as a promising strategy for OS treatment (21).
In this review, we comprehensively analyze the contributions of CAFs to OS chemoresistance, elucidating the underlying molecular mechanisms by which CAFs regulate chemotherapy sensitivity in OS, thereby underscoring their potential as innovative therapeutic targets.
2 Communication mechanisms between CAFs and OS cells
CAFs are a predominant component of the tumor microenvironment that modulate tumor cell proliferation, therapy resistance and immune evasion through diverse mechanisms. The interaction between CAFs and OS cells is summarized below (Figure 1).
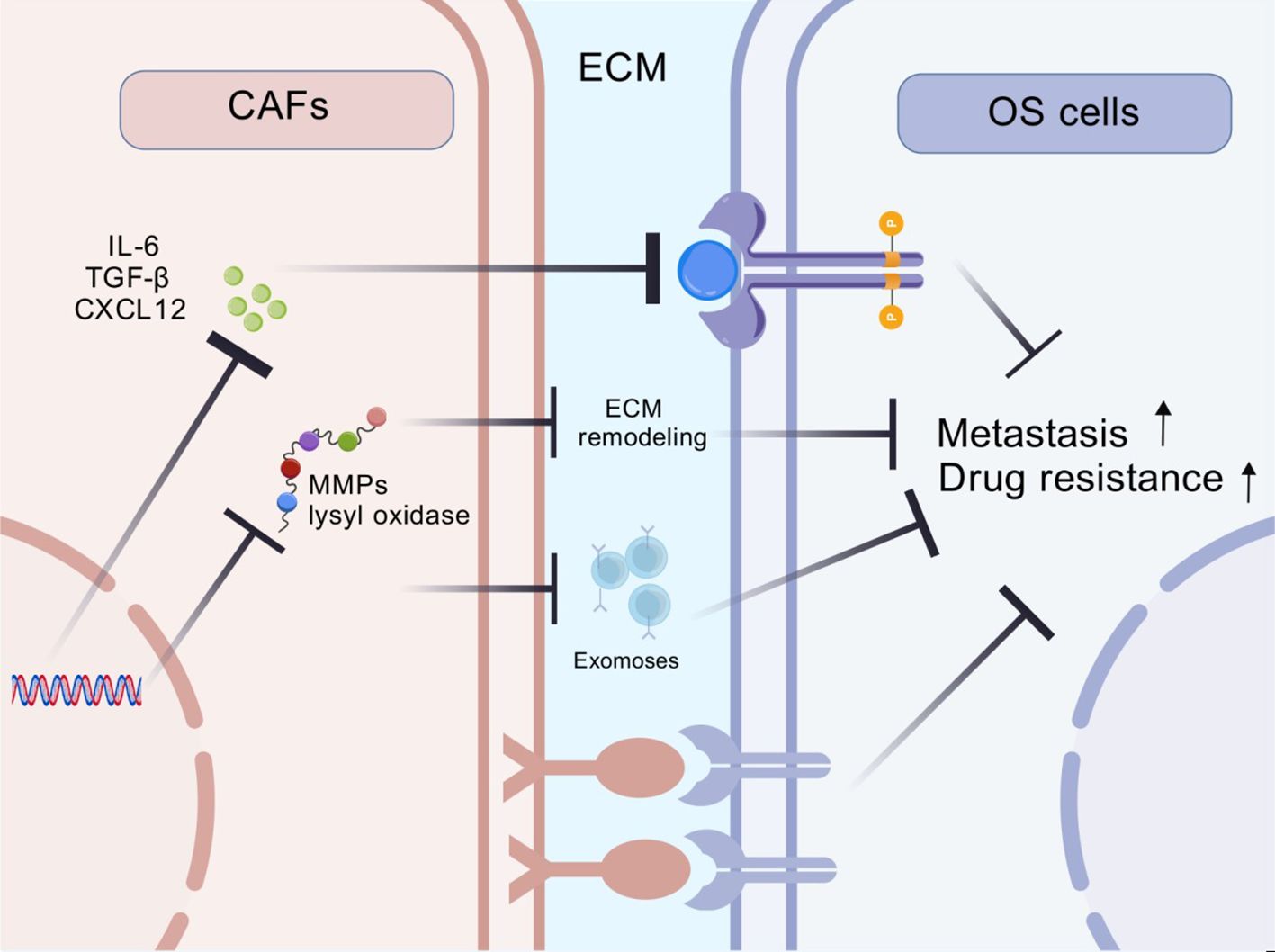
Figure 1. The key mechanisms of CAFs-mediated drug resistance and malignant progression in OS.
2.1 Paracrine signaling
CAFs secrete diverse cytokines and chemokines that regulate OS progression, immune responses and drug resistance. These secretory factors not only shape the composition and function of immune cells within the tumor microenvironment but also profoundly influence malignant biological behaviors of cancer cells.
CAFs are one of the primary sources of TGF-β (22). Studies demonstrate that TGF-β influence cellular behavior through both SMAD-dependent and SMAD-independent pathways, inducing epithelial-mesenchymal transition (EMT) and thereby enhancing tumor cell migration and invasion (23). In OS, elevated TGF-β expression is closely associated with tumor metastasis and recurrence (24). TGF-β also plays a critical role in the development of tumor drug resistance. It enables a wide range of tumour cells to resist chemotherapy and radiotherapy by modulating different signalling pathways (25).
TGF-β derived from CAFs orchestrates a multifaceted resistance network: (1) Tumor-intrinsically, it activates SMAD4-dependent transcription of ABCB1 drug efflux pumps while suppressing pro-apoptotic Bim via HIF1α stabilization (26); (2) Immunologically, TGF-β polarizes macrophages to M2 phenotypes through SMAD3/IL-10 signaling and recruits Tregs via CCL22/CCR4 axis activation (27); (3) Stromally, it induces LOXL2-mediated collagen cross-linking that physically impedes drug penetration (28). This tripartite mechanism creates a chemoprotective niche, as demonstrated by a 68% reduction in cisplatin uptake when OS cells are co-cultured with TGF-β-secreting CAFs (24).
Furthermore, TGF-β establishes an immunosuppressive axis through: (1) Upregulating PD-L1 on CAFs via p38 MAPK/STAT3 signaling, enabling T-cell exhaustion through PD-1 ligation (29); (2) Inducing FAS ligand expression that triggers apoptosis of tumor-infiltrating CD8+ T cells (30); (3) Activating IDO1/kynurenine pathway in dendritic cells, which expands myeloid-derived suppressor cells (31). This network is clinically relevant, as OS patients with high TGF-β activity show 3.2-fold fewer infiltrating cytotoxic lymphocytes than low-TGF-β counterparts (32).
This comprehensive network of TGF-β-mediated effects underscores its central role in both immune evasion and chemotherapy resistance in osteosarcoma, highlighting the importance of targeting TGF-β signaling pathways in therapeutic strategies.
CAFs also secrete IL-6, which promotes tumor cells proliferation and invasion through multiple mechanisms (33). Studies showed that IL-6 activates the STAT3 signaling pathway, thereby enhancing OS cell proliferation and metastasis (34). In U2OS and MG-63 OS cells, IL-6 promoted cancer stemness and tumorigenicity by activating the OPN-STAT3 pathway (35). Besides, irisin reversed IL-6-induced EMT process in OS cells via the STAT3/Snail signaling pathway, consequently suppressing cancer cell migration and invasion (36). Additionally, the inhibition of IL-6 increased cisplatin resistance in human OS cells (37). In summary, IL-6 played a pivotal role in the initiation, progression, and drug resistance of OS.
CXCL12 is another important chemokine secreted by CAFs (38). The interaction between CXCL12 and its receptor CXCR4 promoted the proliferation and invasion of pancreatic cancer cells (39). In neuroblastoma, SOX17 inhibited cancer cell proliferation and invasion through the CXCL12/CXCR4 signaling axis (40). In OS, CXCL12 expression was epigenetically regulated. Studies demonstrated that CXCL12 expression was downregulated in OS cells via DNA methyltransferase 1, thereby regulating the metastasis and immune response in OS (41).
2.2 ECM remodeling
CAFs play a pivotal role in the TME of solid tumors such as OS. CAFs interact with tumor cells through ECM remodeling, influencing tumor growth, metastasis and therapeutic response (42). The ECM is a complex network composed of various macromolecules, including collagen, fibronectin, laminin, hyaluronic acid and others, which provide structural support and biochemical signals for cells (43). In the tumor microenvironment, CAFs are the primary producers and significantly alter composition and physical properties of ECM (44).
CAFs secrete enzymes such as lysyl oxidase to promote collagen cross-linking, thereby increasing ECM stiffness and density (28). These alterations not only provide a physical barrier for OS cells but also activate intracellular signaling pathways that enhance tumor cell invasion and metastasis (44). Additionally, CAFs produce matrix metalloproteinases (MMPs) to degrade ECM components, creating space for tumor cell migration (45). Meanwhile, CAFs regulate the deposition and organization of ECM constituents to guide directional tumor cell movement, further facilitating invasion and metastasis (42). Moreover, CAFs modify ECM architecture and mechanical properties through cellular contractile forces, influencing OS cells behavior (46).
2.3 Exosome-mediated communication
Exosomes are small extracellular vesicles secreted by cells that serve as critical mediators of intercellular communication between CAFs and tumor cells. CAFs utilize exosomes to transfer various bioactive molecules—including miRNAs, lncRNAs, proteins, and metabolites—to tumor cells (47). These molecules regulate gene expression in tumor cells, promoting proliferation, migration, and invasion (48, 49). CAF-derived exosomes induce EMT process, enhancing tumor cell metastatic potential (50). Exosomes derived from CAFs modulate other cells in the tumor microenvironment, such as immune cells and vascular endothelial cells, thereby promoting angiogenesis and facilitating tumor growth through inhibiting the activation of immune cells (51). In addition, exosomes produced by CAFs reduce tumor cell sensitivity to drugs and support tumor cell survival (50). Interestingly, tumor cells can also modulate CAFs properties through exosomes. For example, tumor-derived exosomes activate normal fibroblasts and induce the transformation from normal fibroblasts to CAFs, thereby further promoting tumor progression (52).
2.4 Direct cell-cell contact
The interaction between CAFs and OS cells includes direct cell-to-cell contact. Direct cell-to-cell contact facilitate membrane surface molecular engagements, such as receptor-ligand binding, which subsequently activate intracellular signaling pathways (53). This signal transduction critically influences OS cells proliferation, migration and invasive capabilities. The physical contact between CAFs and OS cells may induce cytoskeletal remodeling, thereby altering cellular morphology and motility. For instance, CAFs secrete small extracellular vesicles that mediate collagen cross-linking and promote EMT process through the p-FAK/p-paxillin/YAP signaling axis, ultimately enhancing OS cells invasion and metastasis (54). CAFs transfer metabolic substrates (lactate, pyruvate and ketone bodies) to OS cells via direct contact, supporting tumor cell growth and survival (55). This metabolic coupling enables tumor cells to better adapt to the nutrient-deprived and hypoxic conditions within the tumor microenvironment. Additionally, CAFs modulate immune cell function through direct contact. For example, CAFs directly suppresses T-cell activation through expressing immune checkpoint molecules PD-L1 (56).
2.5 CAFs heterogeneity in osteosarcoma
CAFs exhibit significant functional and molecular heterogeneity, which plays a crucial role in shaping the TME and influencing OS progression and therapy resistance. This heterogeneity arises from diverse cellular origins, spatial distribution within tumors, and dynamic interactions with other TME components, leading to distinct CAF subpopulations with varying pro-tumorigenic functions (57)
2.5.1 Origins and subtypes of CAFs in OS
CAFs in OS can originate from multiple precursor cells, including resident fibroblasts, mesenchymal stem cells (MSCs), endothelial cells undergoing endothelial-to-mesenchymal transition (EndMT), and even transdifferentiated osteoblasts (58). Single-cell RNA sequencing studies have identified at least three major CAF subtypes in OS: Myofibroblastic CAFs (myCAFs): Characterized by high expression of α-SMA (ACTA2) and TGF-β signaling markers, these CAFs are typically located near tumor cells and contribute to extracellular matrix (ECM) remodeling and mechanical stiffness (59); Inflammatory CAFs (iCAFs): Enriched in cytokine secretion (e.g., IL-6, CXCL12) and JAK/STAT signaling, iCAFs promote immune suppression and angiogenesis (60);Antigen-presenting CAFs (apCAFs): Express MHC class II molecules and co-stimulatory proteins, potentially modulating T-cell responses (59).
2.5.2 Functional implications of CAF heterogeneity
The spatial distribution of CAF subtypes correlates with distinct pathological features of OS. For example, myCAFs are predominantly found in the tumor core, where they drive collagen cross-linking and create a physical barrier to drug penetration, while iCAFs localize to the invasive front, facilitating metastasis through immune evasion (59). Metabolically, CAF subpopulations exhibit divergent behaviors. Lactate-secreting CAFs (marked by MCT4 overexpression) fuel OS cell glycolysis, while lipid-rich CAFs promote chemoresistance by transferring fatty acids to tumor cells via direct contact or exosomes (61). This metabolic coupling is further regulated by hypoxia, with peri-necrotic CAFs showing upregulated HIF-1α signaling and enhanced secretion of pro-angiogenic factors like VEGF (62). Single-cell studies identify COL11A1+ CAFs as a chemoresistance-driving subtype in OS, activating IGF-1R/Akt signaling to promote cancer stemness (42). Conversely, CD10+ CAFs recruit tumor-associated neutrophils (TANs) via CCL2 secretion, accelerating lung metastasis (63). These findings underscore the need for subtype-specific targeting, such as COL11A1-neutralizing antibodies.
CAFs directly modulate immune cell function through multiple mechanisms. For instance, PD-L1 overexpression on CAFs inhibits CD8+ T cell activation by binding to PD-1, facilitating immune evasion (56). Additionally, CAF-secreted IL-6 and TGF-β polarize macrophages toward an M2 phenotype, which further suppresses antitumor immunity (35). Single-cell RNA sequencing reveals that COL11A1+ CAFs correlate with T cell exhaustion markers in OS, suggesting subtype-specific immunosuppressive roles (57). Single-cell studies also identify COL11A1+ CAFs as a chemoresistance-driving subtype in OS, activating IGF-1R/Akt signaling to promote cancer stemness (58). Conversely, CD10+ CAFs recruit tumor-associated neutrophils (TANs) via CCL2 secretion, accelerating lung metastasis (18). These findings underscore the need for subtype-specific targeting, such as COL11A1-neutralizing antibodies.
3 Therapeutic strategies targeting CAFs to overcome OS resistance
Targeting CAFs has emerged as a crucial strategy to overcome OS drug resistance. Below are the primary CAF-targeting approaches and their research advancements (Figure 2).
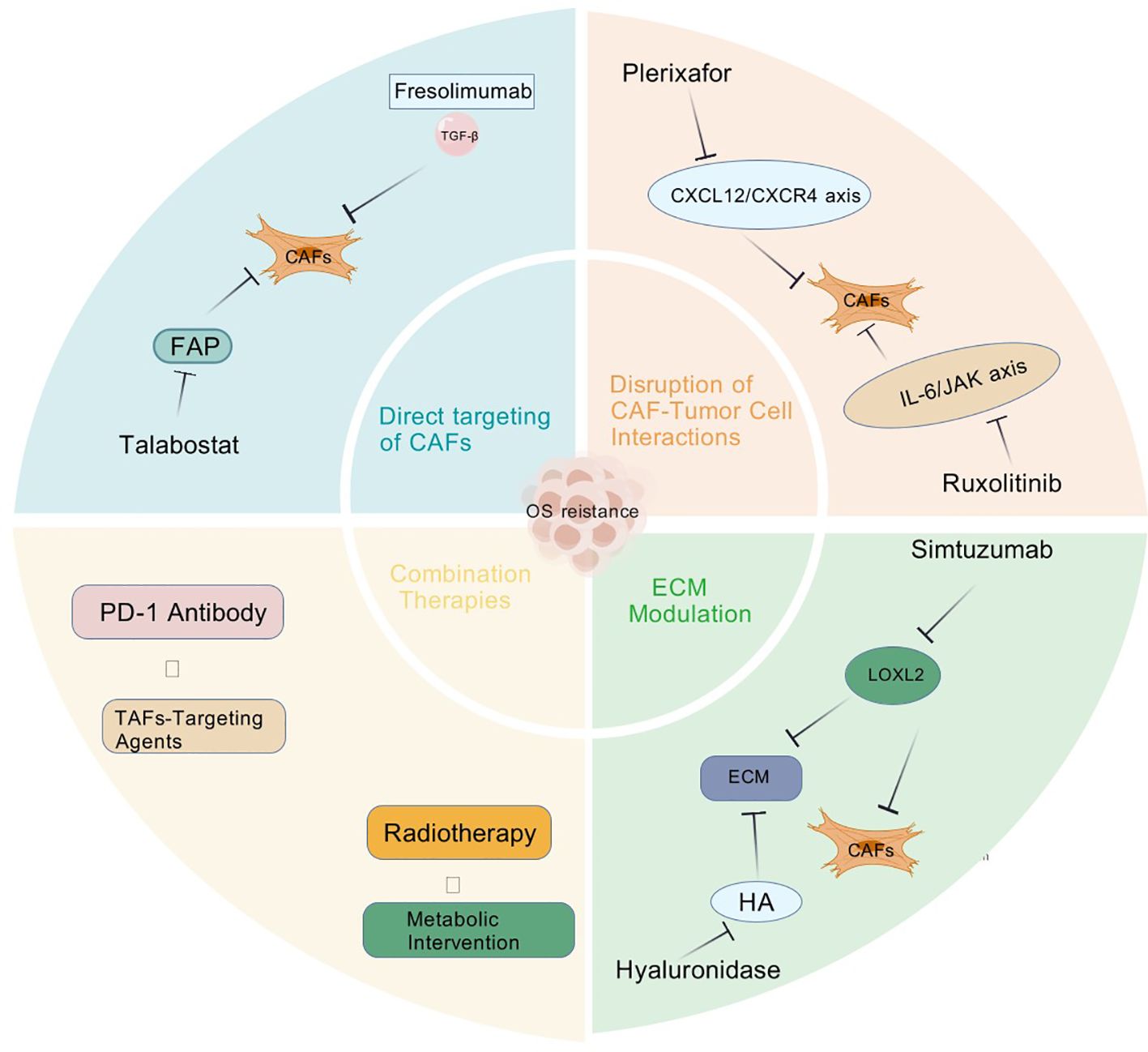
Figure 2. Several therapeutic strategies targeting CAFs to overcome OS resistance via: (1) Direct inhibition (FAP/TGF-β blockers); (2) Disrupting crosstalk (CXCR4/IL-6 inhibitors); (3) ECM modulation (LOXL2/hyaluronidase).
3.1 Direct targeting of CAFs
3.1.1 Fibroblast activation protein inhibitors
FAP is a specific marker on the surface of CAFs, and its high expression in CAFs makes it an ideal target for targeting these cells (64). FAP inhibitors work by suppressing the activity of FAP, thereby reducing the pro-tumor effects of CAFs and enhancing the sensitivity of tumors to chemotherapy and immunotherapy (65). For example, studies have shown that Talabostat (an oral FAP inhibitor) significantly reduces the activity of CAFs, thereby inhibiting OS growth and invasion (66). Moreover, Talabostat exhibits synergistic effects when combined with other anticancer agents. A phase II clinical trial evaluated the antitumor activity of Talabostat in combination with pembrolizumab (an anti-PD-1 antibody) in patients with advanced solid tumors, and the results revealed that the development of tumor was notably suppressed (67). Recent phase I trials have shown promising safety and preliminary efficacy of FAP-targeted therapies in solid tumors. For example, FAP-2286, a novel radioligand therapy, demonstrated favorable tumor uptake in a phase I study (NCT04939610) for advanced solid tumors (68, 69). However, OS-specific clinical data remain limited, partly due to heterogeneous FAP expression across CAF subtypes in osteosarcoma, potentially leading to variable responses. Additionally, on-target/off-tumor effects in normal tissues (e.g., healing wounds, fibrotic lesions) necessitate careful monitoring. Ongoing trials combining FAP inhibitors with immune checkpoint blockade (e.g., NCT05552703) may offer insights for OS treatment strategies (68).
3.1.2 TGF-β neutralizing antibodies
TGF-β is a multifunctional cytokine, playing a crucial role in various biological processes, including cell growth, differentiation, and immune regulation. In the context of cancer, TGF-β has a dual role: it can act as a tumor suppressor in the early stages of tumorigenesis but often promotes tumor progression, metastasis, and immune evasion in advanced stages (31). The TGF-β signaling pathway is a crucial regulator of CAF phenotype and function. Blocking TGF-β can inhibit the transformation of normal fibroblasts into CAFs, reduce the expression of markers such as α-SMA, and thereby diminish the tumor-promoting activity of CAFs (70). Additionally, CAFs promote collagen cross-linking and EMT process through the release of sEVs, and inhibiting TGF-β affects this process (54). CAFs can influence the recruitment and differentiation of immune cells through various pathways, thereby impacting tumor immune evasion. The inhibition of TGF-β can alter the immunomodulatory function of CAFs, promoting immune cell infiltration and antitumor immune responses (30). Fresolimumab is a humanized monoclonal antibody that exerts its therapeutic effects by specifically neutralizing TGF-β. It displays significant potential in modulating the TME and regulating CAFs activity (71). In mouse models of breast cancer and pancreatic cancer, Fresolimumab treatment significantly reduced CAF activation and decreased ECM deposition, thereby inhibiting the formation of physical barriers and promoting drug delivery and immune cell infiltration (72). Fresolimumab treatment also improved the immune microenvironment of the TME, increasing the infiltration of cytotoxic T cells and reducing the levels of immunosuppressive cells such as regulatory T cells and myeloid-derived suppressor cells, thereby alleviating immune suppression and enhancing the antitumor activity of cytotoxic T cells (73, 74). Although Fresolimumab has potential, it is important to note that TGF-β may play a dual role in different tumor types and disease stages. In some cases, TGF-β may suppress early tumor development, while in advanced stages, it may promote tumor progression. Therefore, the use of TGF-β inhibitors such as Fresolimumab requires careful evaluation and should be combined with personalized treatment based on the specific conditions of the patient. Fresolimumab, a TGF-β neutralizing antibody, has shown mixed results in clinical trials. As of September 2023, TGF-β neutralizing antibodies are under clinical investigation for osteosarcoma, often combined with immunotherapy. For example, TQB2858, an anti-PD-L1/TGF-β bispecific antibody, is being explored in clinical settings (75). Preclinical studies indicate that TGF-β promotes chemoresistance and tumor progression, prompting trials testing its blockade to overcome resistance. Early approaches, such as combining anti-TGF-β with dendritic cell therapy, show antitumor potential. Given the dismal <30% 5-year survival rate in metastatic osteosarcoma (76), targeting TGF-β—a key mediator of bone metastasis—represents a promising strategy to improve outcomes.
3.2 Disruption of CAFs-tumor cells interactions
CAFs play a critical role in tumor progression and drug resistance through their interactions with tumor cells. CAFs support tumor cell growth, invasion, and drug resistance via multiple mechanisms, including the secretion of cytokines, chemokines, growth factors, and ECM components. Therefore, disrupting CAF-tumor cell interactions has become an important strategy to overcome tumor drug resistance.
3.2.1 CXCR4 antagonists
CXCR4 (C-X-C chemokine receptor type 4) is a G protein-coupled receptor (GPCR), and its ligand CXCL12 (also known as stromal cell-derived factor-1, SDF-1) plays important roles in various physiological and pathological processes. The CXCL12/CXCR4 axis is involved in multiple critical processes, including cell proliferation, survival, migration, invasion and metastasis, and is associated with more than 20 different types of cancer (77). Upon binding of CXCL12 to CXCR4, multiple downstream signaling pathways are activated, including G proteins, PI3K/AKT, MAPK and RhoA/ROCK2 pathways (78). After G protein activation, it further regulates adenylate cyclase, phospholipase C, and others, generating second messengers such as cAMP, IP3, and DAG, thereby influencing cellular functions (79). The CXCL12/CXCR4 axis also plays a crucial role in guiding cell migration, particularly in immune cell homing, hematopoietic stem cell mobilization and tumor metastasis (80). The high expression of CXCL12 in the bone marrow directs leukemia stem cells expressing CXCR4 to localize within the bone marrow microenvironment, maintaining LSC quiescence and protecting them from chemotherapy (80). Tumor cells, by expressing CXCR4, respond to CXCL12 secreted by metastatic target organs (such as lymph nodes, lungs, liver and bone marrow), thereby promoting directional migration of tumor cells (81). The CXCL12/CXCR4 axis is involved in various inflammatory and immune responses. In a rat model of vascular dementia, inhibition of the CXCL12/CXCR4 axis alleviates neuroinflammation and cognitive dysfunction (82). Plerixafor was initially developed as an anti-HIV drug and later identified as a potent CXCR4 antagonist, subsequently approved for hematopoietic stem cell mobilization in autologous stem cell transplantation (83). Currently, plerixafor has demonstrated promising antitumor effects in various tumor models, particularly in overcoming drug resistance, with applications in both hematologic malignancies and solid tumors (84). The mechanism of action of plerixafor primarily revolves around the inhibition of the CXCL12/CXCR4 axis, exerting multiple effects in cancer therapy (85). In preclinical models of breast cancer and pancreatic cancer, plerixafor combined with chemotherapeutic agents (such as paclitaxel) significantly reduces tumor burden and improves survival rates (86). Additionally, plerixafor has been explored for enhancing the efficacy of immunotherapy by improving immune cell infiltration and augmenting antitumor immune responses. In p53-related therapies, CXCR4 can serve as a target in combination with anti-PD1 therapy (87). Although plerixafor has shown potential in both preclinical and clinical studies, its application in cancer treatment still faces challenges. For instance, some studies indicate that in Ewing sarcoma cell lines, plerixafor may instead promote cell proliferation and activate receptor tyrosine kinase signaling (88). Overall, as a CXCR4 antagonist, plerixafor influences tumor cells and the tumor microenvironment through multifaceted mechanisms, holding broad clinical prospects in OS applications.
In p53-mutant OS, CXCR4 inhibition has emerged as a promising strategy to enhance immunotherapeutic responses, especially when combined with anti-PD1 therapy. This approach leverages the role of CXCR4 in shaping the TME and modulating immune cell infiltration. Inhibiting CXCR4 can reduce tumor growth and potentially restore or enhance the efficacy of immune checkpoint inhibitors. This strategy aligns with broader efforts to convert “cold” tumors into “hot” ones by enhancing tumor immunogenicity and improving responses to immune checkpoint blockade (ICB) (89). For instance, in pancreatic cancer, inhibiting tumor-associated neutrophils (TANs) enhances the effectiveness of anti-PD-1 therapy (90). Similar strategies could be applied to OS, considering the immunosuppressive features associated with p53 mutations. Mechanistically, p53 mutations often lead to immune evasion and resistance to apoptosis, which can be countered by strategies that induce ferroptosis or modulate immune cell infiltration (91, 92).
3.2.2 IL-6/JAK inhibitors
IL-6 is a pleiotropic cytokine that plays a critical role in various physiological and pathological processes, particularly in the tumor microenvironment, where it promotes tumorigenesis and progression by activating the JAK/STAT3 signaling pathway (93). Upon activation of the IL-6/JAK/STAT3 signaling pathway, the expression of genes such as Cyclin D1 and Bcl-2 is upregulated, promoting tumor cell cycle progression and inhibiting apoptosis, thereby enhancing tumor cell proliferation and survival (94). Activated STAT3 can induce the expression of EMT-related transcription factors, including Snail, ZEB1, and Twist, downregulate E-cadherin expression, and upregulate N-cadherin expression, thereby promoting the EMT process in tumor cells and enhancing their invasive and metastatic capabilities (94, 95). The activation of the IL-6/STAT3 pathway is associated with resistance to multiple chemotherapeutic drugs. STAT3 activation reduce tumor cell sensitivity to chemotherapy by regulating the expression of drug transporters, enhancing DNA repair capacity, or influencing apoptosis pathways (96, 97). Ruxolitinib is a JAK1/2 inhibitor that exerts anti-inflammatory and anti-tumor effects by inhibiting the JAK-STAT signaling pathway. Numerous studies have demonstrated its potential therapeutic value in various cancers, as it can influence tumor cell biology through multiple mechanisms. In renal cell carcinoma, Ruxolitinib suppresses tumor cell proliferation and survival by inhibiting the IL-6/JAK/STAT signaling pathway and downregulating PIM1 expression (98). In head and neck squamous cell carcinoma, Ruxolitinib overcomes EGFR-TKI resistance by blocking IL-6/STAT3 signaling, thereby improving therapeutic efficacy (99). Similarly, in NSCLC cells, Ruxolitinib reverses cisplatin resistance by inhibiting the JAK/STAT pathway (100). Additionally, Ruxolitinib suppresses pancreatic cancer progression by attenuating the pro-tumor effects of tumor-associated macrophages through inhibition of the STAT3 signaling pathway (101). In glioma cells, Ruxolitinib exhibits a dose-dependent inhibitory effect on interferon γ-dependent JAK/STAT signaling, thereby impairing tumor cell invasion and tumorigenesis (102). Recent research has shown that targeting the IL-6/JAK/STAT3 signaling pathway can significantly inhibit osteosarcoma growth and metastasis by reducing tumor self-seeding and enhancing antitumor immunity (103). For instance, a study demonstrated that the STAT3 inhibitor cryptotanshinone effectively reduced tumor progression and improved survival rates in osteosarcoma models by inhibiting IL-6 signaling (103). Additionally, JAK inhibitors are being explored in combination with other therapies to enhance treatment efficacy, as seen in preclinical and early-phase clinical trials (104). These findings suggest that IL-6/JAK inhibitors hold promise for improving outcomes in osteosarcoma patients, although further clinical trials are needed to confirm their safety and efficacy (105).
3.3 ECM modulation
ECM is a critical component of the tumor microenvironment, providing structural support and regulating cellular behavior (106). Abnormal ECM remodeling can contribute to tumor progression, metastasis, and drug resistance. Modulating ECM components disrupt these supportive interactions and enhance the efficacy of cancer therapies.
3.3.1 LOXL2 inhibitors
Lysyl oxidase-like 2 (LOXL2) is an enzyme that catalyzes the cross-linking of collagen fibers, contributing to the stiffness and rigidity of the ECM (107). Elevated LOXL2 activity in the tumor microenvironment can promote tumor cell invasion and resistance to therapy (108). Inhibiting LOXL2 can reduce ECM stiffness and disrupt the pro-tumor effects of CAFs. Simtuzumab is a humanized monoclonal antibody that specifically targets LOXL2. By binding to LOXL2, Simtuzumab inhibits its enzymatic activity, thereby reducing collagen cross-linking and ECM stiffness. Preclinical studies have shown that Simtuzumab can decrease tumor-associated fibrosis and improve the efficacy of chemotherapy and immunotherapy (109). In clinical trials, Simtuzumab has demonstrated promising results in reducing tumor stiffness and enhancing drug delivery to tumor cells (109).
3.3.2 Hyaluronidase
Hyaluronic acid (HA) is a major component of the ECM, contributing to its viscoelastic properties and influencing cellular behavior. High levels of HA in the tumor microenvironment can create a dense and impenetrable ECM, limiting drug delivery and promoting tumor progression (108). Hyaluronidase is an enzyme that degrades HA, thereby reducing ECM density and enhancing drug penetration. Hyaluronidase can be administered systemically or locally to degrade HA in the tumor microenvironment. By reducing HA levels, hyaluronidase can enhance the permeability of the ECM, allowing better penetration of chemotherapy drugs and immune cells. Preclinical studies have shown that combining hyaluronidase with chemotherapy or immunotherapy can significantly improve treatment outcomes (110). For example, PEGPH20 (a pegylated form of hyaluronidase) has been shown to reduce tumor interstitial pressure and enhance drug delivery in various cancer models (111).
3.4 Combination strategies
Combination therapies are becoming increasingly important in the treatment of cancer, as they leverage the synergistic effects of multiple treatment modalities to enhance efficacy and overcome resistance mechanisms. By integrating different therapeutic approaches, combination therapies can target multiple pathways involved in tumor progression and resistance, leading to improved patient outcomes.
3.4.1 Chemotherapy + immune checkpoint inhibitors
Combining chemotherapy with immune checkpoint inhibitors has emerged as a powerful strategy to enhance anti-tumor effects by leveraging the cytotoxic effects of chemotherapy and the immune-boosting effects of checkpoint inhibitors (112). Nivolumab is a humanized monoclonal antibody that targets the PD-1 receptor, thereby blocking the inhibitory signals that cancer cells use to evade the immune system (113). By combining Nivolumab with CAF-targeting agents, such as TGF-β inhibitors or FAP inhibitors, the therapy can simultaneously reduce the immunosuppressive effects of CAFs and enhance the immune response against tumor cells (114). Preclinical studies have shown that this combination can significantly increase the number of tumor-infiltrating lymphocytes (TILs) and improve overall survival rates in various cancer models (115).
3.4.2 Metabolic intervention + radiotherapy
Metabolic interventions aim to disrupt the altered metabolism of cancer cells, making them more susceptible to other treatments. Combining metabolic interventions with radiotherapy can enhance the efficacy of radiation by targeting metabolic pathways that contribute to radioresistance. Monocarboxylate transporter 4 is involved in the transport of lactate and other monocarboxylates, contributing to the acidic tumor microenvironment and promoting radioresistance (116). AZD3965 is a selective MCT4 inhibitor that blocks the transport of lactate, thereby reducing the acidic environment and enhancing the sensitivity of tumor cells to radiation (117). Studies have shown that AZD3965 can significantly enhance the efficacy of radiotherapy by normalizing the tumor microenvironment and reducing hypoxia (118). This combination has demonstrated promising results in improving local control and survival rates in various cancer models, including those resistant to conventional radiotherapy.
4 Challenges and future perspectives
CAFs significantly contribute to drug resistance in OS, a highly aggressive bone malignancy. These cells foster tumor progression and resilience to treatment through diverse mechanisms, such as secreting cytokines, chemokines, and extracellular matrix components. However, targeting CAFs to surmount drug resistance in OS faces several challenges. The heterogeneity of CAFs, with their varied phenotypes and functions, complicates the identification of specific targets and the development of effective therapies. Moreover, CAFs’ complex interactions with other cells in the tumor microenvironment, including tumor cells, immune cells, and endothelial cells, require a comprehensive understanding of the involved signaling pathways and potential compensatory mechanisms. Additionally, resistance to CAF-targeting therapies can emerge through genetic mutations, epigenetic changes, and adaptive responses within the tumor microenvironment. The lack of well-defined biomarkers to predict response to CAF-targeting therapies further hampers the development and application of personalized treatment strategies.
Despite these challenges, the future outlook for targeting CAFs in osteosarcoma is promising. Developing targeted therapies that specifically inhibit CAFs’ pro-tumor effects, such as TGF-β inhibitors, FAP inhibitors, and CXCR4 antagonists, holds potential for overcoming drug resistance. These therapies can be used alone or in combination with existing treatments to enhance efficacy. Combining CAF-targeting agents with chemotherapy, immunotherapy, or targeted therapies may provide synergistic effects, overcome multiple resistance mechanisms and improving patient outcomes. Advances in genomics, proteomics, and imaging technologies offer the potential for personalized medicine approaches. Identifying patients who are most likely to benefit from CAF-targeting therapies based on their tumor and CAF characteristics can improve treatment outcomes and reduce unnecessary side effects. Given CAFs’ immunosuppressive effects, combining CAF-targeting therapies with immunomodulatory agents may enhance anti-tumor immune responses, potentially overcome immune evasion mechanisms and improve the efficacy of immunotherapy in OS.
Long-term preclinical and clinical studies are essential to understand the chronic effects of CAF-targeting therapies and to identify potential late-onset resistance mechanisms. Future research should focus on elucidating the molecular mechanisms of CAF heterogeneity, developing biomarkers for CAF-targeted therapies, investigating the role of CAFs in immune modulation, and optimizing combination therapies. Specifically, further studies are needed to uncover the molecular pathways that drive the differentiation and functional diversity of CAFs in OS. This could involve single-cell sequencing technologies to identify novel CAF subtypes and their unique signaling pathways. Identifying reliable biomarkers that predict response to CAF-targeted therapies is crucial, and this could involve exploring the expression of specific proteins, miRNAs, or metabolic signatures in CAFs that correlate with treatment response. Understanding how CAFs interact with immune cells in the TME and identifying strategies to enhance immune surveillance by targeting these interactions could lead to more effective combination therapies. Preclinical studies should focus on optimizing the sequence and timing of CAF-targeted therapies with other treatments, such as chemotherapy or immunotherapy, to maximize therapeutic efficacy and minimize resistance. Conducting translational studies that incorporate patient-derived models and clinical trials will be essential to validate the efficacy of CAF-targeted therapies in OS and to identify potential challenges in clinical application.
In summary, while targeting CAFs in OS presents significant challenges, ongoing research and future studies hold promise for developing novel therapeutic strategies that can overcome drug resistance and improve patient outcomes. A multidisciplinary approach that integrates basic research, translational studies, and clinical trials will be critical in advancing this field. By addressing the heterogeneity of CAFs, understanding their complex interactions, and developing personalized combination therapies, we can pave the way for more effective treatments for osteosarcoma patients.
Author contributions
XW: Writing – original draft. CL: Writing – original draft. DZ: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hu T, Wang G, Wang D, Deng Y, and Wang W. m6A methylation modification: Potential pathways to suppress osteosarcoma metastasis. Int Immunopharmacol. (2025) 145:113759. doi: 10.1016/j.intimp.2024.113759
2. Zhang Y, Xu Y, Bao Y, Luo Y, Qiu G, He M, et al. N6-methyladenosine (m6A) modification in osteosarcoma: expression, function and interaction with noncoding RNAs - an updated review. Epigenetics. (2023) 18:2260213. doi: 10.1080/15592294.2023.2260213
3. Nie Z and Peng H. Osteosarcoma in patients below 25 years of age: An observational study of incidence, metastasis, treatment and outcomes. Oncol Lett. (2018) 16:6502–14. doi: 10.3892/ol.2018.9453
4. Żybowska-Męczyńska M, Pawłowski B, Sienkiewicz M, Zatłoka-Mazur D, Klimas F, Rusiński K, et al. Bone tumors in children and adolescents in pediatric practice. Qual Sport. (2025) 38:58257. doi: 10.12775/QS.2025.38.58257
5. Dry SM. Dedifferentiation in bone and soft tissue sarcomas: How do we define it? What is prognostically relevant? Hum Pathol. (2024) 147:139–47. doi: 10.1016/j.humpath.2024.02.001
6. Picci P. Osteosarcoma (osteogenic sarcoma). Orphanet J Rare Dis. (2007) 2:6. doi: 10.1186/1750-1172-2-6
7. Sellers GS, Poirier MA, Mayberry TG, Cowan BC, Wakefield MR, and Fang Y. From conventional to cutting edge: an exploration of osteosarcoma treatments. Med Oncol. (2025) 42:81. doi: 10.1007/s12032-025-02629-0
8. Postovsky S and Ben Arush MW. Acral erythema caused by high-dose methotrexate therapy in patients with osteogenic sarcoma. Pediatr Hematol Oncol. (2005) 22:167–73. doi: 10.1080/08880010590907320
9. Pan B, Li Y, Han H, Zhang L, Hu X, Pan Y, et al. FoxG1/BNIP3 axis promotes mitophagy and blunts cisplatin resistance in osteosarcoma. Cancer Sci. (2024) 115:2565–77. doi: 10.1111/cas.16242
10. Liu Y, Raina DB, Sebastian S, Nagesh H, Isaksson H, Engellau J, et al. Sustained and controlled delivery of doxorubicin from an in-situ setting biphasic hydroxyapatite carrier for local treatment of a highly proliferative human osteosarcoma. Acta Biomater. (2021) 131:555–71. doi: 10.1016/j.actbio.2021.07.016
11. Harris MA, Savas P, Virassamy B, O’Malley MMR, Kay J, Mueller SN, et al. Towards targeting the breast cancer immune microenvironment. Nat Rev Cancer. (2024) 24:554–77. doi: 10.1038/s41568-024-00714-6
12. Tiwari S, Siddiqui B, Singh S, and Praveen A. Tumor microenvironment: a therapeutic aid in cancer. Indian J Surgery. (2024) 86:57–63. doi: 10.1007/s12262-023-03828-7
13. Meng W, Huang L, Guo J, Xin Q, Liu J, and Hu Y. Innovative nanomedicine delivery: targeting tumor microenvironment to defeat drug resistance. Pharmaceutics. (2024) 16:1549. doi: 10.3390/pharmaceutics16121549
14. Han C, Liu T, and Yin R. Biomarkers for cancer-associated fibroblasts. biomark Res. (2020) 8:64. doi: 10.1186/s40364-020-00245-w
15. Rizzolio S, Giordano S, and Corso S. The importance of being CAFs (in cancer resistance to targeted therapies). J Exp Clin Cancer Res. (2022) 41:319. doi: 10.1186/s13046-022-02524-w
16. Chu L, Wang F, Zhang W, Li HF, Xu J, and Tong XW. Periostin secreted by carcinoma-associated fibroblasts promotes ovarian cancer cell platinum resistance through the PI3K/akt signaling pathway. Technol Cancer Res Treat. (2020) 19:1533033820977535. doi: 10.1177/1533033820977535
17. Adamaki M and Zoumpourlis V. Prostate Cancer Biomarkers: From diagnosis to prognosis and precision-guided therapeutics. Pharmacol Ther. (2021) 228:107932. doi: 10.1016/j.pharmthera.2021.107932
18. Chen X and Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
19. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
20. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
21. Hsu C-Y, Alkhathami AG, Ahmed TA, Chandra M, Mohammed JS, Malathi H, et al. Decoding the function of cancer-associated fibroblasts in osteosarcoma: Molecular pathways, therapeutic approaches and prognostic significance. Exp Cell Res. (2025) 450:114612. doi: 10.1016/j.yexcr.2025.114612
22. Itatani Y, Kawada K, and Sakai Y. Transforming growth factor-β Signaling pathway in colorectal cancer and its tumor microenvironment. Int J Mol Sci. (2019) 20:5822. doi: 10.3390/ijms20235822
23. Wang G, Zhou X, Guo Z, Huang N, Li J, Lv Y, et al. The Anti-fibrosis drug Pirfenidone modifies the immunosuppressive tumor microenvironment and prevents the progression of renal cell carcinoma by inhibiting tumor autocrine TGF-β. Cancer Biol Ther. (2022) 23:150–62. doi: 10.1080/15384047.2022.2035629
24. Ma K, Zhang C, and Li W. TGF-β is associated with poor prognosis and promotes osteosarcoma progression via PI3K/Akt pathway activation. Cell Cycle. (2020) 19:2327–39. doi: 10.1080/15384101.2020.1805552
25. Zhang M, Zhang YY, Chen Y, Wang J, Wang Q, and Lu H. TGF-β Signaling and resistance to cancer therapy. Front Cell Dev Biol. (2021) 9:786728. doi: 10.3389/fcell.2021.786728
26. Xu Y, Li Y, Chen X, Xiang F, Deng Y, Li Z, et al. TGF-β protects osteosarcoma cells from chemotherapeutic cytotoxicity in a SDH/HIF1α dependent manner. BMC Cancer. (2021) 21:1200. doi: 10.1186/s12885-021-08954-7
27. van den Bulk J, de Miranda N, and Ten Dijke P. Therapeutic targeting of TGF-β in cancer: hacking a master switch of immune suppression. Clin Sci (Lond). (2021) 135:35–52. doi: 10.1042/CS20201236
28. Cuesta C, Alcón-Pérez M, Zheng J, Prócel N, Ramírez-Cota R, Galparsoro DF, et al. RAS-PI3K pathway in CAFs shapes physicochemical properties of tumor ECM to impact tumor progression. bioRxiv. (2025). doi: 10.1101/2025.01.20.633776
29. Konen JM, Wu H, and Gibbons DL. Immune checkpoint blockade resistance in lung cancer: emerging mechanisms and therapeutic opportunities. Trends Pharmacol Sci. (2024) 45:520–36. doi: 10.1016/j.tips.2024.04.006
30. Dauer P, Zhao X, Gupta VK, Sharma N, Kesh K, Gnamlin P, et al. Inactivation of cancer-associated-fibroblasts disrupts oncogenic signaling in pancreatic cancer cells and promotes its regression. Cancer Res. (2018) 78:1321–33. doi: 10.1158/0008-5472.CAN-17-2320
31. Mojsilovic S, Mojsilovic SS, Bjelica S, and Santibanez JF. Transforming growth factor-beta1 and myeloid-derived suppressor cells: A cancerous partnership. Dev Dyn. (2022) 251:105–24. doi: 10.1002/dvdy.339
32. Kawano M, Itonaga I, Iwasaki T, Tsuchiya H, and Tsumura H. Anti-TGF-β antibody combined with dendritic cells produce antitumor effects in osteosarcoma. Clin Orthop Relat Res. (2012) 470:2288–94. doi: 10.1007/s11999-012-2299-2
33. Rašková M, Lacina L, Kejík Z, Venhauerová A, Skaličková M, Kolář M, et al. The role of IL-6 in cancer cell invasiveness and metastasis-overview and therapeutic opportunities. Cells. (2022) 11:3698. doi: 10.3390/cells11223698
34. Lu M, Xie K, Lu X, Lu L, Shi Y, and Tang Y. Notoginsenoside R1 counteracts mesenchymal stem cell-evoked oncogenesis and doxorubicin resistance in osteosarcoma cells by blocking IL-6 secretion-induced JAK2/STAT3 signaling. Invest New Drugs. (2021) 39:416–25. doi: 10.1007/s10637-020-01027-9
35. Zhang C, Ma K, and Li WY. IL-6 promotes cancer stemness and oncogenicity in U2OS and MG-63 osteosarcoma cells by upregulating the OPN-STAT3 pathway. J Cancer. (2019) 10:6511–25. doi: 10.7150/jca.29931
36. Kong G, Jiang Y, Sun X, Cao Z, Zhang G, Zhao Z, et al. Irisin reverses the IL-6 induced epithelial-mesenchymal transition in osteosarcoma cell migration and invasion through the STAT3/Snail signaling pathway. Oncol Rep. (2017) 38:2647–56. doi: 10.3892/or.2017.5973
37. Han XG, Mo HM, Liu XQ, Li Y, Du L, Qiao H, et al. TIMP3 overexpression improves the sensitivity of osteosarcoma to cisplatin by reducing IL-6 production. Front Genet. (2018) 9:135. doi: 10.3389/fgene.2018.00135
38. Aronovich A, Moyal L, Gorovitz B, Amitay-Laish I, Naveh HP, Forer Y, et al. Cancer-associated fibroblasts in mycosis fungoides promote tumor cell migration and drug resistance through CXCL12/CXCR4. J Invest Dermatol. (2021) 141:619–27.e2. doi: 10.1016/j.jid.2020.06.034
39. Shen B, Zheng MQ, Lu JW, Jiang Q, Wang TH, and Huang XE. CXCL12-CXCR4 promotes proliferation and invasion of pancreatic cancer cells. Asian Pac J Cancer Prev. (2013) 14:5403–8. doi: 10.7314/APJCP.2013.14.9.5403
40. Wang XH, Zhang SF, Wu HY, Gao J, Wang XH, and Gao TH. SOX17 inhibits proliferation and invasion of neuroblastoma through CXCL12/CXCR4 signaling axis. Cell Signal. (2021) 87:110093. doi: 10.1016/j.cellsig.2021.110093
41. Li B, Wang Z, Wu H, Xue M, Lin P, Wang S, et al. Epigenetic regulation of CXCL12 plays a critical role in mediating tumor progression and the immune response in osteosarcoma. Cancer Res. (2018) 78:3938–53. doi: 10.1158/0008-5472.CAN-17-3801
42. Zhang J, Lu S, Lu T, Han D, Zhang K, Gan L, et al. Single-cell analysis reveals the COL11A1(+) fibroblasts are cancer-specific fibroblasts that promote tumor progression. Front Pharmacol. (2023) 14:1121586. doi: 10.3389/fphar.2023.1121586
43. Li X, Zhou J, Wang X, Li C, Ma Z, Wan Q, et al. Pancreatic cancer and fibrosis: Targeting metabolic reprogramming and crosstalk of cancer-associated fibroblasts in the tumor microenvironment. Front Immunol. (2023) 14:1152312. doi: 10.3389/fimmu.2023.1152312
44. Arpinati L, Carradori G, and Scherz-Shouval R. CAF-induced physical constraints controlling T cell state and localization in solid tumours. Nat Rev Cancer. (2024) 24:676–93. doi: 10.1038/s41568-024-00740-4
45. Piwocka O, Piotrowski I, Suchorska WM, and Kulcenty K. Dynamic interactions in the tumor niche: how the cross-talk between CAFs and the tumor microenvironment impacts resistance to therapy. Front Mol Biosci. (2024) 11:1343523. doi: 10.3389/fmolb.2024.1343523
46. Yu S, Wang S, Wang X, and Xu X. The axis of tumor-associated macrophages, extracellular matrix proteins, and cancer-associated fibroblasts in oncogenesis. Cancer Cell Int. (2024) 24:335. doi: 10.1186/s12935-024-03518-8
47. Yang X, Li Y, Zou L, and Zhu Z. Role of exosomes in crosstalk between cancer-associated fibroblasts and cancer cells. Front Oncol. (2019) 9:356. doi: 10.3389/fonc.2019.00356
48. Ye F, Liang Y, Wang Y, Le Yang R, Luo D, Li Y, et al. Cancer-associated fibroblasts facilitate breast cancer progression through exosomal circTBPL1-mediated intercellular communication. Cell Death Dis. (2023) 14:471. doi: 10.1038/s41419-023-05986-8
49. Wu HJ, Hao M, Yeo SK, and Guan JL. FAK signaling in cancer-associated fibroblasts promotes breast cancer cell migration and metastasis by exosomal miRNAs-mediated intercellular communication. Oncogene. (2020) 39:2539–49. doi: 10.1038/s41388-020-1162-2
50. Zhang Y, Yin C, Wei C, Xia S, Qiao Z, Zhang XW, et al. Exosomal miR-625-3p secreted by cancer-associated fibroblasts in colorectal cancer promotes EMT and chemotherapeutic resistance by blocking the CELF2/WWOX pathway. Pharmacol Res. (2022) 186:106534. doi: 10.1016/j.phrs.2022.106534
51. Zheng J and Hao H. The importance of cancer-associated fibroblasts in targeted therapies and drug resistance in breast cancer. Front Oncol. (2023) 13:1333839. doi: 10.3389/fonc.2023.1333839
52. Ringuette Goulet C, Bernard G, Tremblay S, Chabaud S, Bolduc S, and Pouliot F. Exosomes Induce Fibroblast Differentiation into Cancer-Associated Fibroblasts through TGFβ Signaling. Mol Cancer Res. (2018) 16:1196–204. doi: 10.1158/1541-7786.MCR-17-0784
53. Dhungel N and Dragoi AM. Exploring the multifaceted role of direct interaction between cancer cells and fibroblasts in cancer progression. Front Mol Biosci. (2024) 11:1379971. doi: 10.3389/fmolb.2024.1379971
54. Liu X, Li J, Yang X, Li X, Kong J, Qi D, et al. Carcinoma-associated fibroblast-derived lysyl oxidase-rich extracellular vesicles mediate collagen crosslinking and promote epithelial-mesenchymal transition via p-FAK/p-paxillin/YAP signaling. Int J Oral Sci. (2023) 15:32. doi: 10.1038/s41368-023-00236-1
55. Guil-Luna S, Sanchez-Montero MT, and Rodríguez-Ariza A. S-Nitrosylation at the intersection of metabolism and autophagy: Implications for cancer. Biochim Biophys Acta Rev Cancer. (2023) 1878:189012. doi: 10.1016/j.bbcan.2023.189012
56. Li X, González-Maroto C, and Tavassoli M. Crosstalk between CAFs and tumour cells in head and neck cancer. Cell Death Discov. (2024) 10:303. doi: 10.1038/s41420-024-02053-9
57. Zhou Y, Yang D, Yang Q, Lv X, Huang W, Zhou Z, et al. Single-cell RNA landscape of intratumoral heterogeneity and immunosuppressive microenvironment in advanced osteosarcoma. Nat Commun. (2020) 11:6322. doi: 10.1038/s41467-020-20059-6
58. Feng L, Chen Y, and Jin W. Research progress on cancer-associated fibroblasts in osteosarcoma. Oncol Res. (2025) 33:1091–103. doi: 10.32604/or.2024.054207
59. Zhang T, Ren Y, Yang P, Wang J, and Zhou H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. (2022) 13:897. doi: 10.1038/s41419-022-05351-1
60. Tsoumakidou M. The advent of immune stimulating CAFs in cancer. Nat Rev Cancer. (2023) 23:258–69. doi: 10.1038/s41568-023-00549-7
61. Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. (2020) 17:557–88. doi: 10.1038/s41575-020-0310-z
62. Kugeratski FG, Atkinson SJ, Neilson LJ, Lilla S, Knight JRP, Serneels J, et al. Hypoxic cancer-associated fibroblasts increase NCBP2-AS2/HIAR to promote endothelial sprouting through enhanced VEGF signaling. Sci Signal. (2019) 12:8247. doi: 10.1126/scisignal.aan8247
63. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. (2018) 172:841–56.e16. doi: 10.1016/j.cell.2018.01.009
64. Bughda R, Dimou P, D’Souza RR, and Klampatsa A. Fibroblast activation protein (FAP)-targeted CAR-T cells: launching an attack on tumor stroma. Immunotargets Ther. (2021) 10:313–23. doi: 10.2147/ITT.S291767
65. Wu Z, Hua Y, Shen Q, and Yu C. Research progress on the role of fibroblast activation protein in diagnosis and treatment of cancer. Nucl Med Commun. (2022) 43:746–55. doi: 10.1097/MNM.0000000000001565
66. Kim SY, Cohen SA, Epps J, Smedley J, Mendoza A, Khanna C, et al. Reduction of murine osteosarcoma lung metastases using the dipeptidyl peptidase inhibitor talabostat. J Clin Oncol. (2006) 24:9009. doi: 10.1200/jco.2006.24.18_suppl.9009
67. Ahmed J, Janku F, Karp DD, Piha-Paul SA, Tsimberidou AM, Yap TA, et al. A phase 2 basket study of talabostat, a small-molecule inhibitor of dipeptidyl peptidases, administered in combination with pembrolizumab in patients with advanced solid cancers. Cancer. (2025) 131:e35728. doi: 10.1002/cncr.35728
68. Privé BM, Boussihmad MA, Timmermans B, van Gemert WA, Peters SMB, Derks YHW, et al. Fibroblast activation protein-targeted radionuclide therapy: background, opportunities, and challenges of first (pre)clinical studies. Eur J Nucl Med Mol Imaging. (2023) 50:1906–18. doi: 10.1007/s00259-023-06144-0
69. Singaravelu I, Spitz H, Mahoney M, Dong Z, and Kotagiri N. Antiandrogen therapy radiosensitizes androgen receptor-positive cancers to (18)F-FDG. J Nucl Med. (2022) 63:1177–83. doi: 10.2967/jnumed.121.262958
70. Shangguan L, Ti X, Krause U, Hai B, Zhao Y, Yang Z, et al. Inhibition of TGF-β/Smad signaling by BAMBI blocks differentiation of human mesenchymal stem cells to carcinoma-associated fibroblasts and abolishes their protumor effects. Stem Cells. (2012) 30:2810–9. doi: 10.1002/stem.1251
71. den Hollander MW, Bensch F, Glaudemans AW, Oude Munnink TH, Enting RH, den Dunnen WF, et al. TGF-β Antibody uptake in recurrent high-grade glioma imaged with 89Zr-fresolimumab PET. J Nucl Med. (2015) 56:1310–4. doi: 10.2967/jnumed.115.154401
72. Chakkera M, Foote JB, Farran B, and Nagaraju GP. Breaking the stromal barrier in pancreatic cancer: Advances and challenges. Biochim Biophys Acta Rev Cancer. (2024) 1879:189065. doi: 10.1016/j.bbcan.2023.189065
73. Hanahan D, Michielin O, and Pittet MJ. Convergent inducers and effectors of T cell paralysis in the tumour microenvironment. Nat Rev Cancer. (2025) 25:41–58. doi: 10.1038/s41568-024-00761-z
74. Linke JA, Munn LL, and Jain RK. Compressive stresses in cancer: characterization and implications for tumour progression and treatment. Nat Rev Cancer. (2024) 24:768–91. doi: 10.1038/s41568-024-00745-z
75. Xie L, Liang X, Xu J, Sun X, Liu K, Sun K, et al. Exploratory study of an anti-PD-L1/TGF-β antibody, TQB2858, in patients with refractory or recurrent osteosarcoma and alveolar soft part sarcoma: a report from Chinese sarcoma study group (TQB2858-Ib-02). BMC Cancer. (2023) 23:868. doi: 10.1186/s12885-023-11390-4
76. Harrison DJ, Geller DS, Gill JD, Lewis VO, and Gorlick R. Current and future therapeutic approaches for osteosarcoma. Expert Rev Anticancer Ther. (2018) 18:39–50. doi: 10.1080/14737140.2018.1413939
77. Wang L, Cheng M, Wang Y, Chen J, Xie F, Huang LH, et al. Fasting-activated ventrolateral medulla neurons regulate T cell homing and suppress autoimmune disease in mice. Nat Neurosci. (2024) 27:462–70. doi: 10.1038/s41593-023-01543-w
78. Bao S, Darvishi M, HA A, Al-Haideri MT, Patra I, Kashikova K, et al. CXC chemokine receptor 4 (CXCR4) blockade in cancer treatment. J Cancer Res Clin Oncol. (2023) 149:7945–68. doi: 10.1007/s00432-022-04444-w
79. Sudarsa IW and Yasa IWPS. C-X-C RECEPTOR 4 {CXCR4} METASTASIS KANKER PAYUDARA. INDONESIAN J OF Clin Pathol AND Med LABORATORY. (2018) 19:126–31. doi: 10.24293/ijcpml.v19i2.1068
80. Pansy K, Feichtinger J, Ehall B, Uhl B, Sedej M, Roula D, et al. The CXCR4-CXCL12-axis is of prognostic relevance in DLBCL and its antagonists exert pro-apoptotic effects in vitro. Int J Mol Sci. (2019) 20:4740. doi: 10.3390/ijms20194740
81. Ma K-T, Wu Y-J, Yang Y-X, Wu T, Chen C, Peng F, et al. A novel phthalein component ameliorates neuroinflammation and cognitive dysfunction by suppressing the CXCL12/CXCR4 axis in rats with vascular dementia. J Ethnopharmacology. (2024) 328:118117. doi: 10.1016/j.jep.2024.118117
82. Parab S, Setten E, Astanina E, Bussolino F, and Doronzo G. The tissue-specific transcriptional landscape underlines the involvement of endothelial cells in health and disease. Pharmacol Ther. (2023) 246:108418. doi: 10.1016/j.pharmthera.2023.108418
83. Erik De C. Recent advances on the use of the CXCR4 antagonist plerixafor (AMD3100, Mozobil™) and potential of other CXCR4 antagonists as stem cell mobilizers. Pharmacol Ther. (2010) 128:509–18. doi: 10.1016/j.pharmthera.2010.08.009
84. Jane L. Plerixafor: potential role in acute leukemia therapy. Expert Opin Orphan Drugs. (2015) 3:467–75. doi: 10.1517/21678707.2015.1020297
85. Liu Z, Wang J, and Chen H. CXCR4 antagonist AMD3100 (Plerixafor) modulates immune responses in the tumor microenvironment. Int J Cancer Clin Res. (2021) 8:566–81. doi: 10.23937/2378-3419/1410144
86. Chengu N, Jing Z, and Patrick IO. Harnessing plant flavonoids to fight pancreatic cancer. Curr Nutr Rep. (2024) 13:566–81. doi: 10.1007/s13668-024-00545-9
87. Shivi C, Shivani J, Vibhuti J, Bhavana S, Sujata B, Manoj G, et al. Potential role of p53 deregulation in modulating immune responses in human Malignancies: A paradigm to develop immunotherapy. Cancer Letters. (2024) 588:216766. doi: 10.1016/j.canlet.2024.216766
88. Philipp B, Christiane S, Dagmar C, Eberhard K, Uta D, and Jenny P. The CXCR4 antagonist plerixafor (AMD3100) promotes proliferation of Ewing sarcoma cell lines in vitro and activates receptor tyrosine kinase signaling. Cell Communication Signaling. (2018) 16:21. doi: 10.1186/s12964-018-0233-2
89. Zheng J, Wang S, Xia L, Sun Z, Chan KM, Bernards R, et al. Hepatocellular carcinoma: signaling pathways and therapeutic advances. Signal Transduct Target Ther. (2025) 10:35. doi: 10.1038/s41392-024-02075-w
90. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. (2016) 29:832–45. doi: 10.1016/j.ccell.2016.04.014
91. Burger JA and Peled A. CXCR4 antagonists: targeting the microenvironment in leukemia and other cancers. Leukemia. (2009) 23:43–52. doi: 10.1038/leu.2008.299
92. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
93. Sara GM, Daniel LD, Grace LW, and Hui-Wen L. IL-6/JAK/STAT3 signaling in breast cancer metastasis: biology and treatment. Front Oncol. (2022) 12. doi: 10.3389/fonc.2022.866014
94. Rajalaxmi P, Subarno P, Sushree Subhadra A, Saptarshi S, Somya Ranjan D, and Chanakya Nath K. Nano formulated Resveratrol inhibits PD-L1 in oral cancer cells by deregulating the association between tumor associated macrophages and cancer associated fibroblasts through IL-6/JAK2/STAT3 signaling axis. J Nutr Biochem. (2024) 125:109568. doi: 10.1016/j.jnutbio.2024.109568
95. Eskiler GG, Bezdegumeli E, Ozman Z, Ozkan AD, Bilir C, Kucukakca BN, et al. IL-6 mediated JAK/STAT3 signaling pathway in cancer patients with cachexia. Bratislava Med J. (2019) 120:819–26. doi: 10.4149/BLL_2019_136
96. Kang-Ning W, Kan Z, Nian-Nian Z, Lei-Ming C, Zi-Zhan L, Yao X, et al. Enhancing cancer therapy: The role of drug delivery systems in STAT3 inhibitor efficacy and safety. Life Sci. (2024) 346:122635. doi: 10.1016/j.lfs.2024.122635
97. Yongtao B, Lianjie N, Lihua S, Guoliang D, Wenzhou Z, Baoxia H, et al. Uncovering the effect and mechanism of Jiawei Xiaoyao Wan in treating breast cancer complicated with depression based on network pharmacology and experimental analysis. Phytomedicine. (2024) 128:155427. doi: 10.1016/j.phymed.2024.155427
98. Kimberly Stephanie M and Sheldon LH. Targeting an IL-6/JAK/STAT/PIM1 pathway in renal cell carcinoma. J Clin Oncol. (2022) 40:376–. doi: 10.1200/JCO.2022.40.6_suppl.376
99. Kimberly SM, Kimberly S, and Sheldon LH. Abstract 627: IL-6 signaling via JAK/STAT axis influences PIM1 expression in renal cell carcinoma. Cancer Res. (2024) 84:627–. doi: 10.1158/1538-7445.AM2024-627
100. Emre D, Oguzhan D, Zeynep Banu D, and Ozlem D. Inhibition of the invasion of human glioblastoma u87 cell line by ruxolitinib: a molecular player of mir-17 and mir-20a regulating jak/stat pathway. Turkish Neurosurg. (2019) 30(2):182–9. doi: 10.5137/1019-5149.JTN.26122-19.1
101. Emilee S, Julie H, Caroline A, Joao TB, Wenqing L, and Scott D. The JAK inhibitor ruxolitinib is effective in treating T cell acute lymphoblastic leukemia with gain of function mutations in IL-7R alpha. Blood. (2015) 126:1330–. doi: 10.1182/blood.V126.23.1330.1330
102. Olaf N, Pilar Blanco L, Paloma Guisado H, Isabel VilLaoslada C, Beatriz de Felipe C, Carmen C, et al. Ex vivo effect of JAK inhibition on JAK-STAT1 pathway hyperactivation in patients with dominant negative STAT3 mutations. (New York, United States: Springer). (2022).
103. Zhang Y, Ma Q, Liu T, Guan G, Zhang K, Chen J, et al. Interleukin-6 suppression reduces tumour self-seeding by circulating tumour cells in a human osteosarcoma nude mouse model. Oncotarget. (2016) 7:446–58. doi: 10.18632/oncotarget.6371
104. Barrett LE, Gardner HL, Barber LG, Sadowski A, and London CA. Safety and toxicity of combined oclacitinib and carboplatin or doxorubicin in dogs with solid tumors: a pilot study. BMC Vet Res. (2019) 15:291. doi: 10.1186/s12917-019-2032-4
105. El-Sherbiny M, El-Sayed RM, Helal MA, Ibrahiem AT, Elmahdi HS, Eladl MA, et al. Nifuroxazide mitigates angiogenesis in ehlrich’s solid carcinoma: molecular docking, bioinformatic and experimental studies on inhibition of il-6/jak2/stat3 signaling. Molecules. (2021) 26:6858. doi: 10.3390/molecules26226858
106. Kellen W, Thuc L, Matthew K, Andras C, and Sufi Mary T. Cancer-associated fibroblasts: master tumor microenvironment modifiers. Cancers. (2023) 15:1899. doi: 10.3390/cancers15061899
107. Alan P, Marta Martinez Y, Huilei W, and Lakshmi S. Lysyl oxidase like-2 in fibrosis and cardiovascular disease. Am J Physiology-Cell Physiol. (2023) 325:C694–707. doi: 10.1152/ajpcell.00176.2023
108. Julia AL, Lance LM, and Rakesh KJ. Compressive stresses in cancer: characterization and implications for tumour progression and treatment. Nat Rev Cancer. (2024) 24:768–91. doi: 10.1038/s41568-024-00745-z
109. Naoki I, Zhen-Wei P, Kahini AV, Susan BL, Shuhei Y, Deanna YS, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. (2017) 66:1697–708. doi: 10.1136/gutjnl-2016-312473
110. Ge S, Ying W, Jiekai L, Mingjie Y, Hang X, Yiping L, et al. Quercetin liposomes conjugated with hyaluronidase: An efficient drug delivery system to block pancreatic cancer. J Controlled Release. (2025) 382:113642. doi: 10.1016/j.jconrel.2025.113642
111. Sophie G, Kairbaan H-D, and Gabriele B. Targeting the tumour vasculature: from vessel destruction to promotion. Nat Rev Cancer. (2024) 24:655–75. doi: 10.1038/s41568-024-00736-0
112. Meng W, Pan L, Huang L, Li Q, and Sun Y. Applications of image-guided locoregional transarterial chemotherapy in patients with inoperable colorectal cancer: a review. Front Oncol. (2024) 14:1464242. doi: 10.3389/fonc.2024.1464242
113. Bahman Abedi K, Arash A, Nadia Ghasemi D, Nasim A, Arsalan M, Yalda Y, et al. Combination therapy with nivolumab (anti-PD-1 monoclonal antibody): A new era in tumor immunotherapy. Int Immunopharmacology. (2022) 113:109365. doi: 10.1016/j.intimp.2022.109365
114. Hongyun W and He R. Precision treatment of pancreatic ductal adenocarcinoma. Cancer Letters. (2024) 585:216636. doi: 10.1016/j.canlet.2024.216636
115. Meng Q, Fei Z, Xinyu L, Tao J, Haowei W, Xuefei L, et al. Targeting focal adhesion kinase boosts immune response in KRAS/LKB1 co-mutated lung adenocarcinoma via remodeling the tumor microenvironment. Exp Hematol Oncol. (2024) 13:11. doi: 10.1186/s40164-023-00471-6
116. Qinghua Z, Guizhen P, Lu Z, Yidan X, and Jiqing H. The predictive value of monocarboxylate transporter 4 (MCT4) on lung adenocarcinoma patients treated with PD-1 inhibitors. J Inflammation Res. (2024), 10515–31. doi: 10.2147/JIR.S493632
117. Anna Maria R, Dominik G, Ali G, Michael B, and Nicolai S. MCT4 promotes tumor Malignancy in F98 glioma cells. J Oncol. (2021) 2021:1–20. doi: 10.1155/2021/6655529
118. Athina M, Tzanikou E, Kallergi G, Pantazaka E, Georgoulias V, Kotsakis A, et al. Evaluation of monocarboxylate transporter 4 (MCT4) expression and its prognostic significance in circulating tumor cells from patients with early stage non-small-cell lung cancer. Front Cell Dev Biol. (2021) 9. doi: 10.3389/fcell.2021.641978
Keywords: osteosarcoma, cancer-associated fibroblasts, chemoresistance, extracellular matrix, tumor microenvironment
Citation: Wang X, Luo C and Zhang D (2025) The malignant dialogue between cancer-associated fibroblasts and osteosarcoma cells: microenvironment-mediated drug resistance and therapeutic targets. Front. Immunol. 16:1621521. doi: 10.3389/fimmu.2025.1621521
Received: 01 May 2025; Accepted: 31 July 2025;
Published: 20 August 2025.
Edited by:
Qiong Lu, Central South University, ChinaReviewed by:
Qiang Huang, Fudan University, ChinaCopyright © 2025 Wang, Luo and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Danying Zhang, emhhbmdkYW55aW5nMTEyNkAxMjYuY29t