 Arij Fouzat Hassan
Arij Fouzat Hassan Hadeel Kheraldine
Hadeel Kheraldine Lama Abujamous
Lama Abujamous Hamda Al-Thawadi
Hamda Al-Thawadi Abdelbary Elhissi1
Abdelbary Elhissi1- 1College of Pharmacy, Department of Pharmaceutical Sciences, QU Health, Qatar University, Doha, Qatar
- 2College of Medicine, Department of Basic Medical Science, QU Health, Qatar University, Doha, Qatar
Triple-negative breast cancer (TNBC) is an aggressive and clinically challenging subtype of breast cancer characterized by the absence of hormone receptors and HER2 amplification. This molecular profile limits the effectiveness of targeted therapies, leaving chemotherapy as the mainstay of treatment a strategy often met with limited success due to rapid disease progression and high recurrence rates. Increasing evidence underscores the pivotal role of the tumor microenvironment (TME) in driving TNBC pathogenesis, particularly through chronic inflammation and cytokine dysregulation. Inflammatory cytokines such as TNF-α, TGF-β, IL-6, and IL-10 orchestrate a complex network of cellular interactions that remodel the TME into an immunosuppressive niche. This inflammatory landscape not only promotes tumor cell proliferation and metastasis but also compromises antitumor immune responses and contributes to therapeutic resistance. Recent preclinical and clinical studies have explored the therapeutic potential of targeting cytokine signaling to disrupt this inflammatory axis and overcome resistance. In this review, we critically examine the multifaceted interplay between cytokines, inflammation, and the TME in TNBC, with a focus on mechanisms of resistance. We further evaluate current and emerging therapeutic approaches targeting the inflammatory axis, highlighting both the promise and the complexities of this evolving landscape.
1 Introduction
Triple-negative breast cancer (TNBC) is an aggressive subtype lacking the expression of estrogen receptor (ER), progesterone receptor (PR), and epidermal growth factor receptor type 2 (HER2) (1, 2). It accounts for approximately 15–20% of all breast cancer cases globally (~170,000 annually) (3). It disproportionately affects premenopausal women, with higher prevalence among African American women and those carrying pathogenic breast cancer susceptibility genes, such as BRCA mutations (4).TNBC is characterized by rapid tumor growth, early metastasis (often to the lungs, liver, and brain), and a higher likelihood of recurrence, particularly within the first three to five years after treatment, where the survival rate is less than 10% (1, 2). Its clinical behavior varies due to underlying molecular heterogeneity, influencing therapeutic response and outcomes at both early and advanced stages (5). The absence of targetable receptors makes TNBC unresponsive to hormonal therapies such as tamoxifen and aromatase inhibitors, as well as HER2-targeted treatments like trastuzumab (Figure 1). As a result, chemotherapy remains the primary systemic treatment option, highlighting the need for a deeper understanding of TNBC’s unique biology and tumor microenvironment (TME) (6, 7).
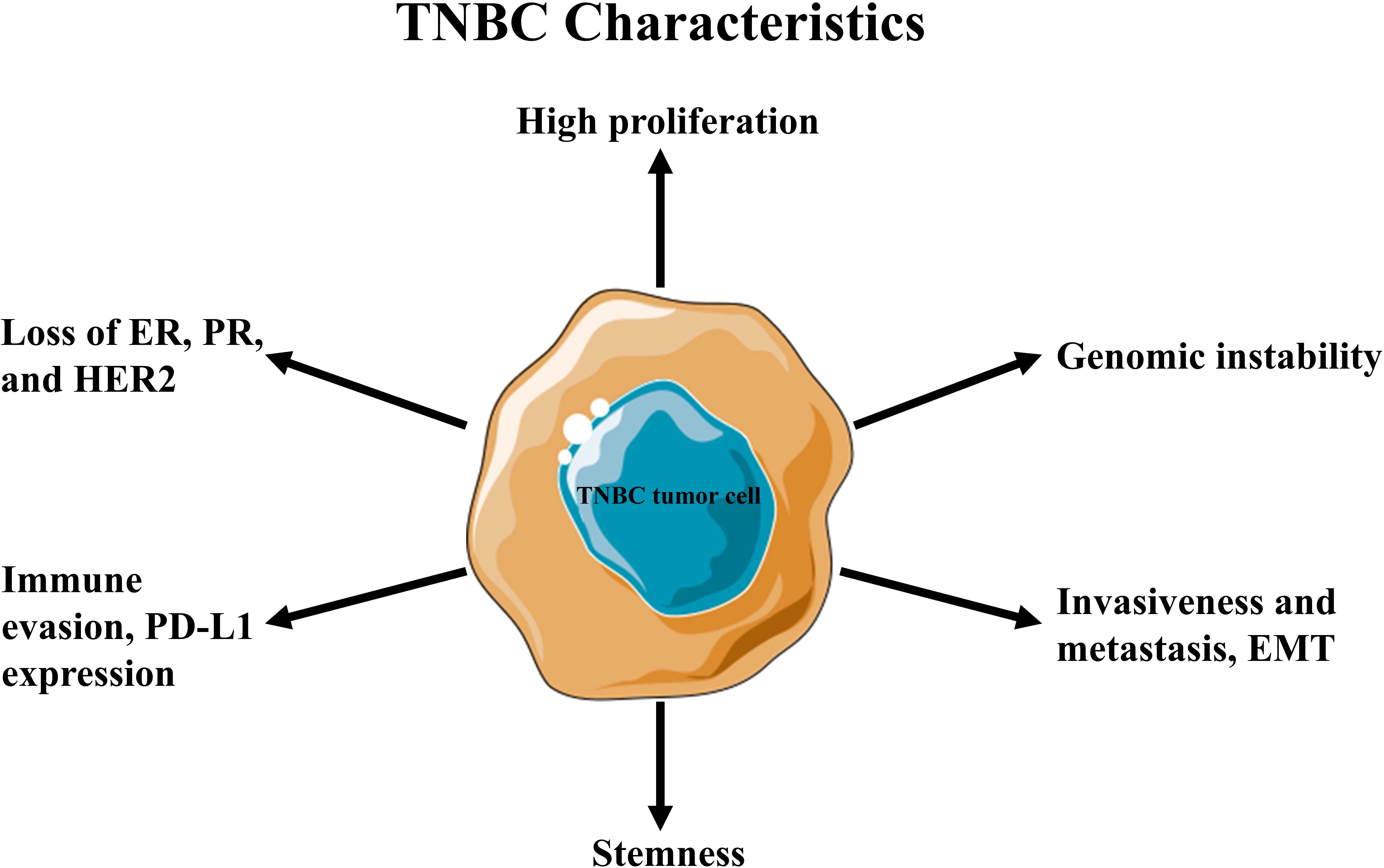
Figure 1. Hallmark characteristics of triple-negative breast cancer (TNBC).
A key contributor to TNBC progression and resistance is the TME a complex and dynamic network of stromal cells, immune cells, extracellular matrix, and signaling molecules (8). Cytokines within the TME drive inflammation, immune evasion, angiogenesis, and epithelial-mesenchymal transition (EMT) in TNBC, supporting tumor survival and metastasis and making them attractive candidates for therapeutic intervention (9, 10). Moreover, immunosuppressive components such as tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs) exacerbate resistance by shielding tumor cells from immune attack (11, 12).
Recent studies have highlighted the therapeutic potential of targeting cytokine signaling and the inflammatory TME to restore treatment sensitivity in TNBC. Strategies such as cytokine inhibitors, immune checkpoint blockade, and agents that reprogram the TME have shown promise in preclinical and clinical settings, offering hope for more effective and durable treatments (13, 14).
This review explores the interplay between inflammatory cytokines, the TME, and drug resistance in TNBC, with a focus on mechanisms that drive progression and immune suppression. We also evaluate emerging interventions aimed at disrupting this axis to enhance therapeutic efficacy and overcome resistance.
1.1 Tumor microenvironment in TNBC
TME refers to the complex and dynamic milieu of cells, extracellular matrix, and signaling molecules that surround and interact with tumor cells (15). Since TME is necessary for the growth and invasiveness of cancer cells, it has emerged as a promising target for the development of novel cancer therapeutics (16). The TME is comprised of a variety of cell types, including cancer cells, immune cells, endothelial cells, pericytes, and cancer-associated fibroblasts (CAFs) (17). These cells can both support and impede the growth and spread of tumors (18). The endothelial cells, as well as CAFs, secrete cytokines to regulate EMT (19). EMT-triggered tumor cells produce immunosuppressive cytokines, leading to an immunosuppressive TME (19).
To maintain their own survival and growth, tumor cells regularly actively change their microenvironment, resulting in a dynamic and complicated interplay between neoplastic cells and the TME (20). TME can be divided into immunosuppressive and immunoreactive categories based on its contribution to the immune response (21). The predominant immune cells within the tumor microenvironment are lymphocytes, commonly referred to as tumor-infiltrating lymphocytes (TILs), which are frequently observed in solid tumors. TILs consist of various subtypes, with CD3+ T cells being the most abundant in solid tumors. While CD20+ B cells may also be present, their infiltration is relatively uncommon. Indeed, these various TIL subtypes play diverse roles in the management of breast cancer and contribute to immunomodulation through distinct pathways (22).
In the early stages of tumor development, immune cells can recognize and eliminate highly immunogenic cancer cells (23). However, under conditions such as cellular stress, infection, or inflammation, cancer cells can evade immune surveillance, allowing tumor progression to occur unchecked (24, 25). Increasing evidence has highlighted inflammation as a key factor in shaping the TME and promoting cancer development.
1.2 Inflammation in TME of TNBC
Inflammation significantly contributes to the emergence and development of TNBC, particularly when chronic inflammation permeates the TME. Chronic inflammation can affect different elements of TNBC etiology and is characterized by a protracted and dysregulated immune response. Recent studies strongly imply that chronic inflammation is associated with aberrant metabolism, which is most likely to alter the TME and promote cancer-associated signaling cascades (26–28). The immune system mounts a response to harmful agents, such as pathogens, irradiation, or tissue injury, resulting in inflammation (29).
In many instances, the development and spread of cancer are correlated with inflammation. Since cells responsible for inflammation-induced cancers are often stable and resistant to current anticancer therapies, targeting inflammation has emerged as a promising therapeutic strategy. Inflammation that arises within tumors is associated with several risk factors, including pathogenic infections, autoimmune diseases, lifestyle factors, smoking, and excessive alcohol consumption. Additionally, cancer-intrinsic or cancer-induced inflammation can originate from cancer-initiating mutations, which recruit and activate inflammatory cells, ultimately supporting tumor growth (30).
The primary goal of the inflammatory response is to eliminate the foreign substance that interferes with tissue homeostasis. During acute inflammation, cellular and molecular interactions restore the homeostatic condition in the typical physiological environment. On the other hand, when inflammation is not resolved adequately, it leads to chronic inflammation (31). Chronic inflammation can increase the risk of cancer by supplying bioactive molecules from invasive cells into TME, such as cytokines, to prevent proangiogenic factors, and ECM-modifying enzymes like metalloproteinases that promote EMT and facilitate other carcinogenesis programs (32).
In TNBC, pro-inflammatory pathways, tissue damage, infection, and other factors can contribute to chronic inflammation. Cytokines are pivotal mediators in chronic inflammation, orchestrating the communication between immune cells and influencing cellular behaviors in TME (33). TNBC has been associated with many cytokines, including those necessary for tumor growth, such as TGF-β, TNF-α, IL-6, and IL-10 (34, 35). Understanding the mechanisms behind the interactions between TNBC cells and their microenvironment is therefore essential for the development of effective cancer therapies.
For the purpose of developing effective therapeutic strategies that specifically target these pathways to improve outcomes for TNBC patients, it is crucial to understand the role of cytokines in TNBC-associated immunosuppression and drug resistance. This review explores the mechanisms by which cytokines act within the TME, highlighting the inflammatory responses that contribute not only to immune evasion but also to the promotion of therapeutic resistance in TNBC.
2 Cytokines in TNBC
Cytokines are small secreted proteins of size less than 40kDa produced by cells involved in the immune response (36). Although different types of cells produce cytokines, helper T cells and macrophages are the chief producers. Schwann cells, mast cells, endothelial cells, and resident and recruited macrophages may produce cytokines in and via peripheral nerve tissue during normal and pathological processes (37). The release of pro-inflammatory cytokines stimulates and activates immune cells, as well as induces the release of further cytokines (36).
Cytokines are generated in response to a wide variety of cellular stressors, such as inflammation, infection, and damage brought on by carcinogens. These conditions trigger cytokines to activate a host response targeted at reducing cellular damage and managing cellular stress. Effective damage containment encourages tissue repair, but failure to treat the damage might result in cytokine production that persists and worsens tissue loss. As a result, host responses to cellular stress may influence various phases of cancer development and progression (38). Cytokines enable paracrine and autocrine immune system cell communication over short distances in the innate and adaptive immune systems. Since the immune system is capable of identifying and eliminating cancer cells, research has focused on using cytokines to treat cancer during the past few decades (39). Cytokines play an important role in cancer by regulating cell differentiation and proliferation, tumor development, progression, and immune response (40). Generally, cytokines can be categorized into pro-inflammatory and anti-inflammatory based on their function and the type of immune responses they regulate (41, 42). (see Table 1; Figure 2).
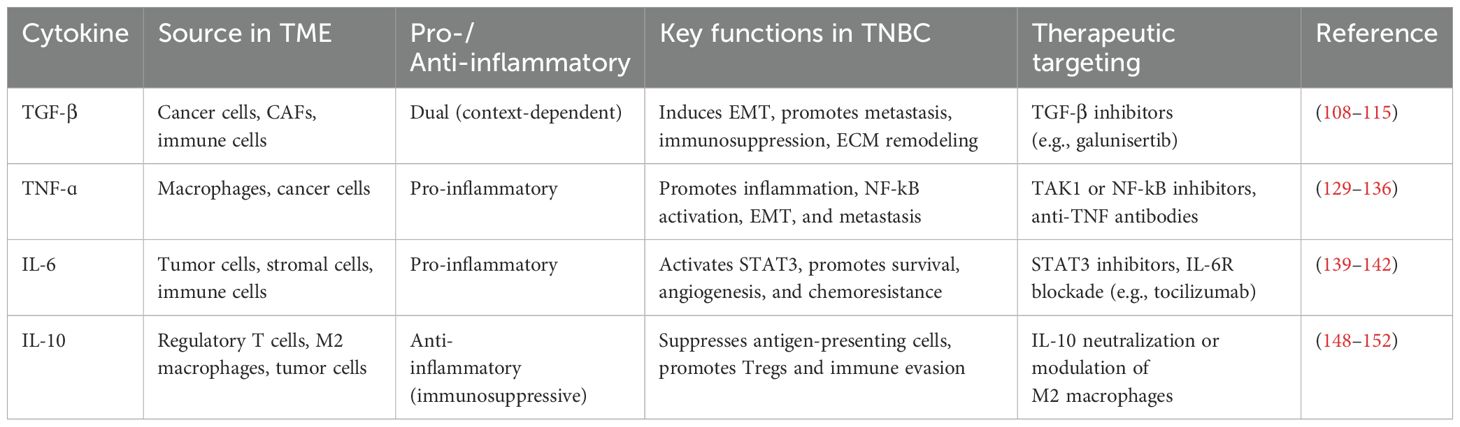
Table 1. Key cytokines in triple-negative breast cancer (TNBC) tumor microenvironment (TME).
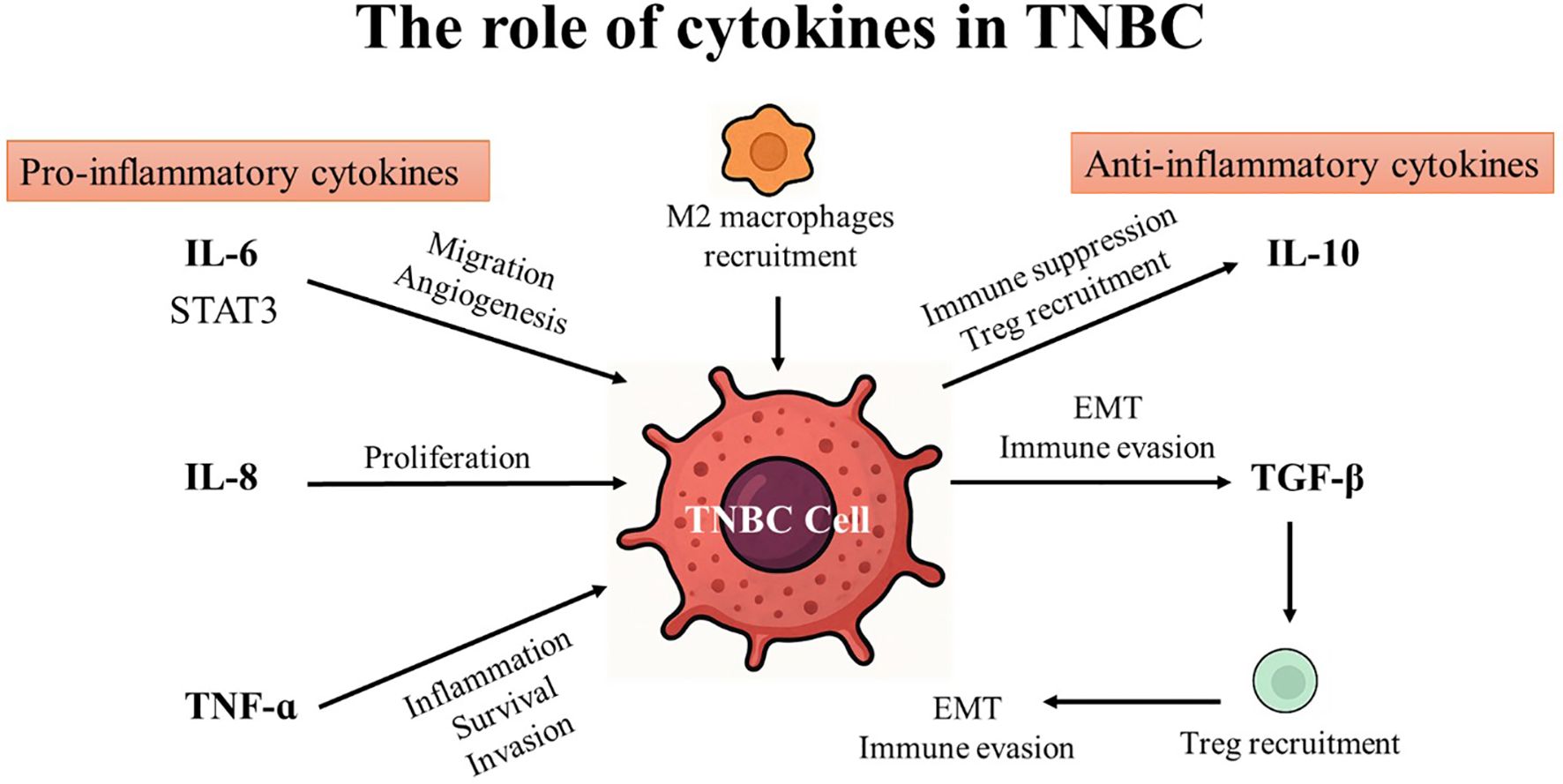
Figure 2. The role of pro- and anti-inflammatory cytokines in triple-negative breast cancer (TNBC).
For instance, Th1 cells, CD4+ cells, macrophages, and dendritic cells release pro-inflammatory cytokines including Interleukins (IL), interferon (IFN), and tumor necrosis factor (TNF). The three primary cytokines that encourage inflammation are IL-6, IL-10, and TNF. These cytokines communicate their signals through type I cytokine receptors (CCR1), which are physically distinct from other cytokine receptor types. They are crucial for modulating the immune system and for coordinating cell-mediated immune responses. Pro-inflammatory cytokines typically affect immune cell proliferation, activation, differentiation, and homing to the sites of infection to control and eradicate intracellular pathogens (99).
2.1 Pro-inflammatory cytokines in TNBC
Pro-inflammatory cytokines have a significant impact on the development of TME in TNBC. Numerous stromal cells and immunological cells produce these cytokines within the TME, and their presence contributes to chronic inflammation within the tumor milieu. Pro-inflammatory cytokines act as key signaling molecules, orchestrating complex interactions between malignant and immune cells, and stromal components. Through their actions, they can modulate tumor cell survival, proliferation, angiogenesis, and immune evasion mechanisms (100, 101). Pro-inflammatory cytokines also affect immune cell recruitment, activation, and performance inside the TME, affecting the anti-tumor immune response. Their ability to shape the TME creates a favorable environment for tumor growth, invasion, and metastasis. It is crucial to comprehend how pro-inflammatory cytokines interact with the TME to create targeted therapeutics that disrupt tumor-promoting pathways and enhance clinical outcomes (102). TGF-β, TNF-α, IL-6, and IL-10 are a few pro-inflammatory cytokines that regulate TME in TNBC and promote cancer progression and invasiveness (103).
2.2 Anti-inflammatory cytokines in TNBC
Anti-inflammatory cytokines play a critical role in maintaining immune homeostasis and regulating inflammation. Key cytokines in this group include interleukin-10 (IL-10), transforming growth factor-beta (TGF-β), and interleukin-35 (IL-35) (104). These cytokines suppress excessive immune activation, protect tissues from damage during inflammation, and promote wound healing (105). However, in the context of cancer, their immunosuppressive properties can paradoxically facilitate tumor progression. For example, TGF-β suppresses cytotoxic T cell and natural killer (NK) cell activity while promoting regulatory T cell (Treg) expansion, contributing to immune evasion by cancer cells. TGF-β is also involved in EMT, a key process in cancer metastasis (106). Further, IL-10 inhibits the activation of antigen-presenting cells and effector T cells, thereby impairing anti-tumor immune responses and creating an immunosuppressive TME (105). Similarly, IL-35, though less studied, has also been shown to enhance Treg function and inhibit anti-tumor immunity (107). Collectively, anti-inflammatory cytokines contribute to the establishment of a tolerogenic TME that favors tumor growth, metastasis, and resistance to therapies. Targeting these cytokines or their signaling pathways holds promise for reversing immunosuppression and enhancing anti-cancer immune responses (Figure 3).
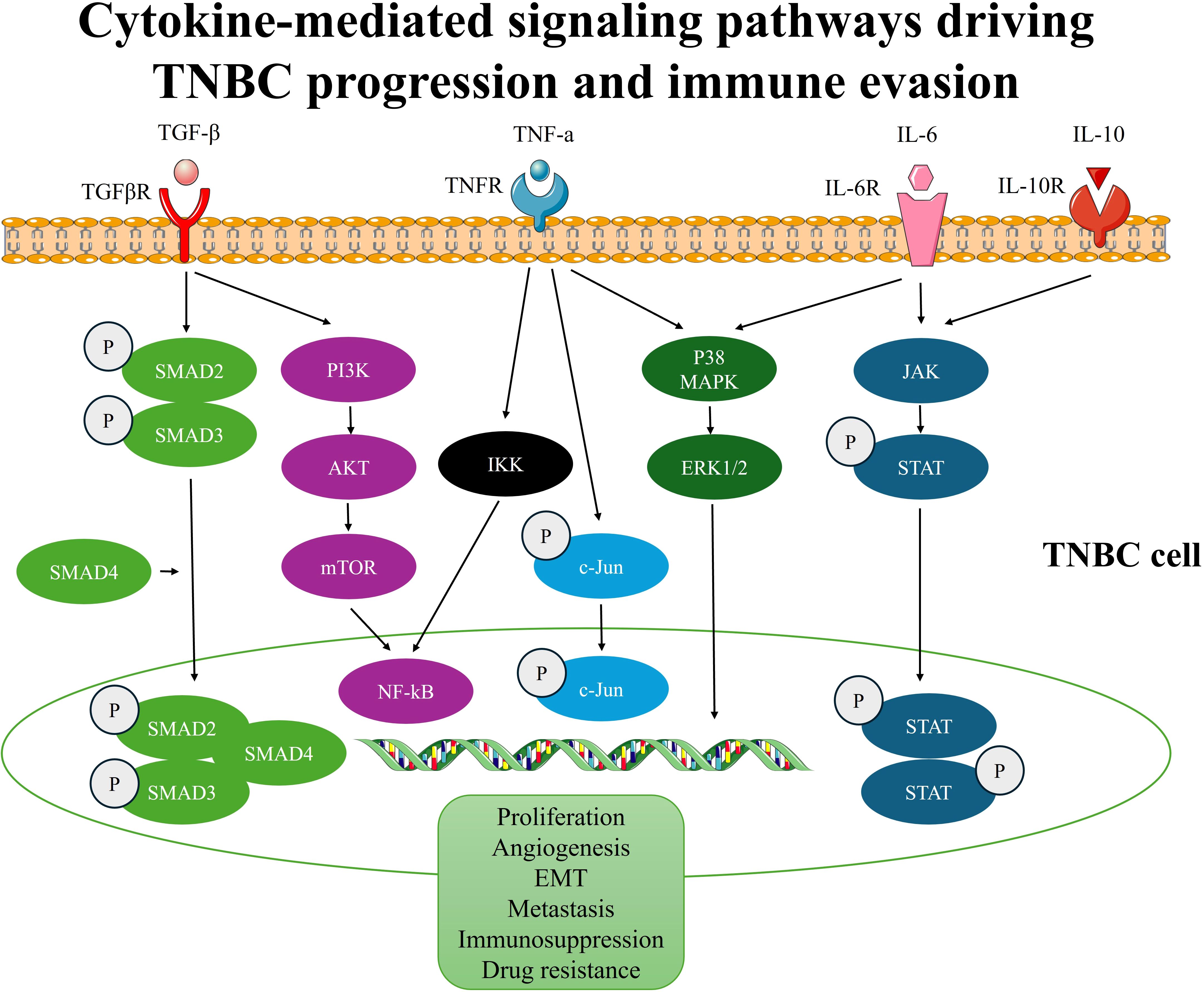
Figure 3. Cytokine-mediated signaling pathways driving TNBC progression and immune evasion.
2.3 Transforming growth factor-β
TGF-β is a versatile cytokine essential for many physiological and pathological processes. It is a member of the TGF-β superfamily of cytokines and includes various isoforms such as TGF-β1, TGF-β2, and TGF-β3. It is a powerful regulator of cell development, differentiation, apoptosis, immune responses, tissue development, and homeostasis. TGF-β is produced by many cell types, including immune cells, fibroblasts, and epithelial cells. It exists as an inactive precursor that needs to be activated to have a biological impact. Upon activation, TGF-β binds to its receptors on the cell surface, initiating downstream signaling pathways that regulate gene expression and cellular responses. TGF-β has a wide range of functions based on the cell type, microenvironment, and the disease (108, 109).
TGF-β functions as a tumor suppressor under normal physiological circumstances by preventing cell growth, causing cell cycle arrest, and encouraging apoptosis. It is also involved in tissue repair and immune regulation, contributing to wound healing and immunosuppressive effects. TGF-β signaling dysregulation, on the other hand, is linked to the onset and development of several illnesses, including cancer (110). TGF-β displays a dual role; while it suppresses tumor growth in the early stages of tumorigenesis, it promotes tumor development and metastasis in advanced stages. Moreover, it has the ability to trigger EMT, an important event driving tumor cell invasion and metastasis. In addition, TGF-β also contributes to immunosuppression by inhibiting immune cell function and promoting regulatory T-cell differentiation (111, 112).
In other words, a crucial TME regulator, TGF-β, is involved in the complex interactions between stromal cells and tumors. Malignant cells and cells that exist in TME can produce TGF-β, leading to autocrine and paracrine signaling. TGF-β signaling influences multiple aspects of the TME, including immune cell infiltration, angiogenesis, extracellular matrix remodeling, and EMT. One of the critical functions of TGF-β in the TME is its immunomodulatory role. TGF-β suppresses the activity of immune cells, such as cytotoxic T cells, NK cells, and dendritic cells, impairing their anti-tumor functions. It promotes the differentiation and activity of immunosuppressive Tregs and myeloid-derived suppressor cells (MDSCs). This immunosuppressive environment allows tumors to evade immune surveillance and promotes immune tolerance (113, 114).
In TNBC, the TGF-β cascade is often dysregulated, resulting in aberrant activation and an imbalance in its effects within the TME. TGF-β signaling in TNBC exerts diverse effects on different components of the TME, influencing tumor progression and therapeutic resistance (115). In highly complex diseases like TNBC, the most important factor that controls carcinogenesis and treatment response is the signaling cascades between tumors and adjacent cell populations, such as ECM and CAFs. Despite the lack of knowledge regarding TGF-β’s role in TNBC, CAF in axillary lymph nodes causes BC metastases via comparable processes, including EMT that utilizes the TGF-β pathway. The aggressive BC phenotype is stimulated by the interplay of TGF-β pathways, specifically the downregulation of TGF-β receptors, which creates an irrational association with patient outcomes (116). According to a recent study, intratumorally high levels of TGF-β1 were observed in more than 50% of TNBC as opposed to non-TNBC, suggesting a possible function for TGF signaling in the biology of TNBC (117). Additionally, an in-vitro study showed that TGF-β1 was discovered to increase MDA-MB-231 cells’ propensity to form tumors by triggering the SMAD2 and P38 signaling pathways (118). Collectively, several investigations show a connection between TGF-β and TNBC’s aggressiveness, poor disease-free survival, and metastatic status (115, 119, 120). The TGF-β signaling cascade is a well-known oncogenic pathway in TNBC that promotes tumor stemness and EMT-mediated cancer plasticity. The stimulation of TGF-β signaling results in the production of EMT-activating transcription factors (EMT-ATFs), which function as molecular switches for the EMT and stemness nuclear reprogramming (121, 122). TGF-β typically acts as a tumor promoter in late-stage cancers by promoting EMT (123). In addition, the TGF-β signaling pathway is initiated when TGF-β ligands bind to type II receptors (TGFBR2) on the cell surface. This binding leads to the recruitment and activation of type I receptors (TGFBR1), resulting in phosphorylation and activation of downstream signaling molecules called SMADS (124). The TGF-β signaling pathway can be canonical or non-canonical. During the canonical TGF-β signaling, TGF-β is initiated through the activation of its receptor and the recruitment of phosphorylated SMADs (SMAD2/SMAD3), which form complexes with SMAD4 and translocate to the nucleus (124). Once within the nucleus, these complexes control the transcription of target genes involved in a variety of cellular activities, including EMT by interacting with transcription factors and co-regulators (125). However, through a non-canonical TGF-β axis, it can also control physiological and pathological responses. The non-canonical cascade, for instance, also includes the extracellular signal-regulated kinases (ERK), p38, and c-Jun N-terminal kinase (JNK), as well as the cell survival mediator’s protein kinase B (AKT), mammalian target of rapamycin (mTOR), and nuclear factor kappa-B (NF-kB) (126). The development of cancer-related disease is significantly influenced by the dysregulation of these non-canonical TGF-β signaling pathways. Further, overexpression of the c-Myc gene, along with activation of the p15, p21, and SMADs, eliminates the antitumor impact of TGF-β on cell development, resulting in the evolution of tumors (127). Furthermore, TGF-β signaling in TNBC is involved in angiogenesis and the remodeling of the ECM. Increased tumor vascularization results from TGF-β’s stimulation of the synthesis of pro-angiogenic substances, including vascular endothelial growth factor (VEGF), and enhancement of endothelial cell migration and tube formation. Moreover, TGF-β stimulates the recruitment of ECM proteins, including fibronectin and collagen, and modulates the activity of matrix metalloproteinases (MMPs), which contribute to the breakdown of the ECM and the infiltration of malignant cells (128).
2.4 Tumor necrosis factor
TNF-α is a cytokine that is primarily generated by immune cells like macrophages upon activation. TNF-α participates in different physiological and pathological processes, and its dysregulation is implicated in several diseases. TNF-α is a cytokine that promotes inflammation and is essential for the initiation and progression of the inflammatory response (129). TNF-α stimulates the recruitment of other inflammatory molecules, such as interleukins and chemokines, and promotes the migration of immune cells to the site of inflammation. TNF-α production can cause chronic inflammation and tissue damage, even though it is necessary for immune responses and host defense. Abnormal TNF-α signaling has been associated with various autoimmune diseases. TNF-α encourages the activation of immune cells, the release of inflammatory mediators, and the death of healthy tissues under abnormal circumstances (129).
In cancer, TNF-α has an essential role, exhibiting both pro-tumor and anti-tumor effects depending on the context and stage of cancer development (130). It can induce apoptotic proteins in certain malignant cells, particularly in the early stages of tumor development. It can also promote inflammation and immune responses that can aid in the elimination of cancer cells. Nevertheless, TNF-α can exert pro-tumor effects by promoting tumor cell proliferation, survival, and angiogenesis. It can increase the production of cytokines and growth factors that encourage the development and invasion of tumor cells (130). TNF-α may also stimulate the growth of a microenvironment that is supportive of tumors by luring immune cells that inhibit anti-tumor immune responses and fasten tumor development. The fact that TNF-α may affect TME further complicates the dual role that it plays in cancer. TNF-α may affect the recruitment and activation of immune cells within the tumor, resulting in a dynamic interaction between immune responses that promote and suppress tumor growth. The relative importance of these conflicting effects can define TNF-α ‘s overall influence on tumorigenesis (131).
In BC, patients’ biopsies revealed high TNFα-mRNA and protein expression, particularly in those with a worse prognosis (132). Recently, the pro-metastatic function of TNF-α and its involvement in the EMT for tumor cell migration to initiate metastasis were reported (133). In fact, it is considered one of the inflammatory cytokines produced by TME and is linked to metastasis and aggressiveness in several malignancies, including TNBC (134). The role of TNF-α in survival and proliferation has produced contradictory outcomes. Depending on the cell type and cellular milieu in which it is detected, TNF-α can cause apoptosis and hasten the formation of tumors. TNF-α, on the other hand, can stimulate c-Jun, which in turn activates pro-apoptotic pathways and downregulates anti-apoptotic genes, resulting in apoptosis. However, the same study showed that TNF-α may encourage tumor growth (135, 136). During tumorigenesis, and through the activation of the NF-kB and p38/MAPK pathways, TNF-α activates the signal transducer and activator of transcription 3 (STAT3), a well-known transcription factor classified as an oncogene. This cascade creates a positive feedback loop, as when STAT3 is activated, HBXIP (Hepatitis B Virus X-Interacting Protein) is expressed more abundantly, which increases the level of TNFR1. As a result, TNFR1 binds to TNF-α, maintaining the activation of the p38/MAPK and NF-kB pathways. Additionally, the membrane glycoprotein MUC4 can promote cell proliferation in TNBC cell lines by upregulating cyclin D1 expression and mediating β-catenin (137). Another in-vitro study showed that TNF-α induces cancer metastasis by the generation of MMP, which can change the ECM during metastasis. It is important to highlight that the TGF-β-activated kinase 1 (TAK1) complex is essential for TNF-α-mediated MMP9 synthesis in this procedure (138).
2.5 Interleukin-6
IL-6 is a glycosylated protein also known as B cell differentiation factor (BSF-2), which stimulates B-cell maturation into antibody-producing cells. It has important functions in maintaining hematopoietic progenitor cells, the cardiovascular and nervous systems (139). Additionally, IL-6 contributes to both acute and chronic inflammation. At the location of tissue injury, IL-6 is released during an immunological response and aids in directing immune defense. It increases the liver’s ability to produce acute-phase proteins, including C-reactive protein (CRP), which aids in the identification and removal of infections. Additionally, IL-6 encourages immune cell activation and differentiation, including B and T cell differentiation, and modifies the balance between pro-inflammatory and anti-inflammatory responses (140).
In the TME of cancer, IL-6 is important. It is produced by a variety of cell types in TME, including stromal cells, immune cells, and cancer cells themselves. The growth, development, and immunological responses of tumors can be significantly impacted by the presence of IL-6 in TME (141). Essentially, in TME, the signaling pathway of IL-6 is considered a malevolent player, due to its function in cancer development and progression. Based on animal studies, chronic IL-6 signaling is highly associated with carcinogenesis. Through a variety of downstream mediators, IL-6 exerts an intrinsic effect on tumor cells that promotes cancer cell survival, proliferation, and metastasis. Furthermore, IL-6 can exert extrinsic effects on other cells in the complex TME to maintain a pro-tumor milieu by increasing angiogenesis and tumor immunity evasion (142).
IL-6 was discovered to be overexpressed in BC patients’ blood and tumor sites, which is often associated with poor prognosis and decreased survival. IL-6 can influence every stage of the BC process by regulating proliferation, apoptosis, metabolism, survival, angiogenesis, and metastasis (143). In the context of TNBC, IL-6 is expressed in 50% of TNBC cases (144). IL-6, one of the main mediators of the inflammatory response, has a substantial impact on both the defense mechanisms of the host immune system and the control of cellular development. The transmission of IL-6 signals triggers STAT3 and involves both the cell-surface IL-6 receptor (IL-6R) and the soluble IL-6 receptor (sIL-6R). In TNBC patients, STAT3 activation is associated with lower survival rates and chemoresistance (145). According to a new study, the STAT3 pathway, which is controlled by IL-6 under genotoxic stress, is stimulated by the stimulator of interferon genes (STING), which circumvents immune suppression by raising PD-L1 in TNBC. The activation of STING pathways in tumor cells and immune cells recruited in the TME is of significant interest since it may help to alter tumor intrinsic cell survival and death pathways. STING induces the production of IL-6 in TNBC cells under genotoxic stress, which binds to its specific receptor and promotes phosphorylation of the tyrosine (705) residue of STAT3. This suggests that the IL-6/STAT3 pathway is triggered via the STING pathway (146). Another study illustrated that TNBC cells activate the IL-6-STAT3 signaling pathway, which increases levels of the chemokines, VEGF, and chemokine (C-C motif) ligand 5 (CCL5) and encourages lymph node angiogenesis and lung extravasation of tumor cells (147).
2.6 Interleukin-10
Interleukin-10 (IL-10) is an immunoregulatory cytokine that plays a critical role in modulating immune responses and maintaining immune homeostasis. IL-10 acts on multiple cells types and exerts both anti-inflammatory and immunosuppressive effects. One of the key functions of IL-10 is its ability to dampen immune responses and limit inflammation. TNF-α and IL-6 are two examples of pro-inflammatory cytokines whose synthesis and activity are inhibited by IL-10. Additionally, it prevents immune cells like T cells, NK cells, and antigen-presenting cells from activating and functioning. By reducing the inflammatory response, IL-10 helps prevent tissue damage caused by excessive or prolonged immune activation. IL-10 is particularly important in regulating immune responses at mucosal surfaces, such as the gastrointestinal tract. It helps maintain immune tolerance to commensal microorganisms and food antigens while still allowing for effective immune responses against pathogens. Dysregulation of IL-10 signaling can contribute to chronic inflammation and diseases, including cancer (148, 149). It is well recognized that IL-10 has dual roles as anti-tumor and pro-tumor. Tumor regression activity is demonstrated by IL-10. The anti-tumor effect of IL-10 depends on CD8+ or CD4+ T cell activity, contrary to some who have suggested that the anti-tumor action of IL-10 is caused by increased NK cell activity (150). The tumor can thwart the host immune system’s ability to eliminate it by using IL-10 production at the tumor location. It is essential for local cancer immunotherapy that IL-10 has the capacity to stop an immune response from growing at the tumor site (151). In fact, cancer cell proliferation and metastasis are enhanced by IL-10 through the regulation of antitumor immunity (152).
Although little is known about how IL-10 impacts TNBC, there is mounting evidence that it plays a role in the growth of cancer. However, it was shown that TNBC samples had a significant expression of IL-10 (153). IL-10 exerts both direct and indirect effects on TNBC cells and TME. One mechanism by which IL-10 may influence TNBC is through IgG4. It was reported that IgG4+B cells are linked to higher tumor recurrence and unfavorable patient prognosis in TME of TNBC. IgG4+B cell presence correlates with IL-10 tumor expression. These findings imply that, due to the presence of IL-10 overexpression, TNBC may offer a TME that supports class change to the IgG4 subtype. Furthermore, hormone-receptor negative tumors, higher-grade tumors, and locally advanced cancer are more prevalent in TNBC patients with IL-10 expression in their malignancies (154). Another mechanism responsible for IL-10 overexpression is through the production of proinflammatory cytokines, M1 and M2-type macrophages. M1-macrophages are linked to an inflammatory response and trigger a Th1 immune response. M2-type macrophages, which are linked to tumor progression, secrete IL-10 and suppress the Th1 immune response, further inducing tumor invasion and metastasis. Additionally, angiogenic factors like VEGF can be secreted by M2-type macrophages to support tumor angiogenesis and supply nutrients and channels for metastasis. Additionally, TNBC cells also release IL-10 and M-CSF to promote the polarization of M1-type macrophages into M2-type states (155).
Collectively, these findings suggest that cytokines in the TME of TNBC function within a tightly regulated yet dynamic signaling network that promotes survival, plasticity, and immune evasion (156). We propose a mechanistic model in which chronic cytokine signaling—particularly via IL-6, TGF-β, and TNF-α—drives a feedforward loop that reinforces tumor aggressiveness. Through activation of key pathways such as STAT3, NF-κB, and SMAD, these cytokines promote EMT, reprogram immune cell behavior (e.g., M2 macrophage polarization), and sustain transcriptional plasticity in tumor cells (157–159). Notably, TGF-β may initiate early transcriptional reprogramming, while IL-6–mediated STAT3 activation supports long-term drug resistance by maintaining survival and angiogenic signaling (43). These feedback circuits, often enhanced by stress responses such as STING or hypoxia-induced HIF-1α, create a self-sustaining cytokine–TME axis that contributes to therapeutic failure (44, 45). Understanding these interconnected signaling events may offer new avenues for combination therapies targeting multiple cytokine-driven nodes simultaneously.
2.7 Metastatic potential of TNBC and cytokines involvement
TNBC is notably aggressive, with a high propensity to metastasize to the bone, lungs, liver, and brain (46, 47). This metastatic potential largely determines patient prognosis and therapeutic failure (48). Metastasis is a complex, multi-step process that includes local invasion, intravasation, survival in circulation, extravasation, and colonization at distant sites. Each step is governed by dynamic interactions between TNBC cells, the TME, and host immune responses (13, 49).
Early in metastasis, hypoxia, acidosis, and mechanical stress in the primary tumor promote the secretion of cytokines and recruitment of bone marrow-derived immune cells (50, 51). These events facilitate extracellular matrix (ECM) remodeling, angiogenesis, and EMT. Key cytokines such as TGF-β, IL-6, and TNF-α drive EMT by activating SMAD, STAT3, and NF-κB pathways, loosening cell–cell adhesion and promoting a migratory phenotype (52–54). Additionally, cytokines such as CXCL12, VEGF, and MMP-9 contribute to the formation of pre-metastatic niches, conditioning distant organs and recruiting supportive stromal and immune cells (55, 56).
Organ-specific metastasis (organotropism) in TNBC involves both tumor-intrinsic traits and microenvironmental cues (57). For instance, bone metastasis involves integrins (αvβ3, α5β1), TGF-β, HIF-1α, and MMP-mediated signaling (58, 59). Brain colonization requires breaching the blood-brain barrier via CXCR4, VEGF, and COX-2, aided by astrocyte-derived cytokines and WNT pathway activation (60, 61). Lung and liver metastases are facilitated by metabolic reprogramming and cytokine-driven ECM modulation, including pyruvate carboxylase, LOX, and β-catenin-independent WNT signaling (62, 63).
Emerging regulators such as non-coding RNAs (miRNAs and lncRNAs) and ferroptosis-related genes are also implicated in metastasis (64). Notably, miRNAs like miR-125b suppress EMT and metastatic behavior, while ferroptosis, an iron-dependent form of cell death, interacts with cytokine signaling and redox balance, representing a novel axis of metastatic regulation in TNBC (65, 66).
3 TME targeted therapeutics in TNBC
Compared to other BC subtypes, TNBC typically manifests as a high-grade invasive ductal carcinoma with a higher rate of early recurrence, frequent distant metastases, and is associated with worse prognosis. Although there is progress in understanding tumor biology, clinical outcomes for TNBC are, regrettably, still not sufficient. This fact underscores how urgently better therapeutic interventions must be developed for TNBC patients (67). Further, the lack of targetable receptors renders TNBC unresponsive to hormonal therapies such as tamoxifen and aromatase inhibitors, as well as HER2-targeted therapies like trastuzumab. This limitation leaves chemotherapy as the main systemic treatment option and underscores the need for a deeper understanding of its distinct biology and TME (Table 2).
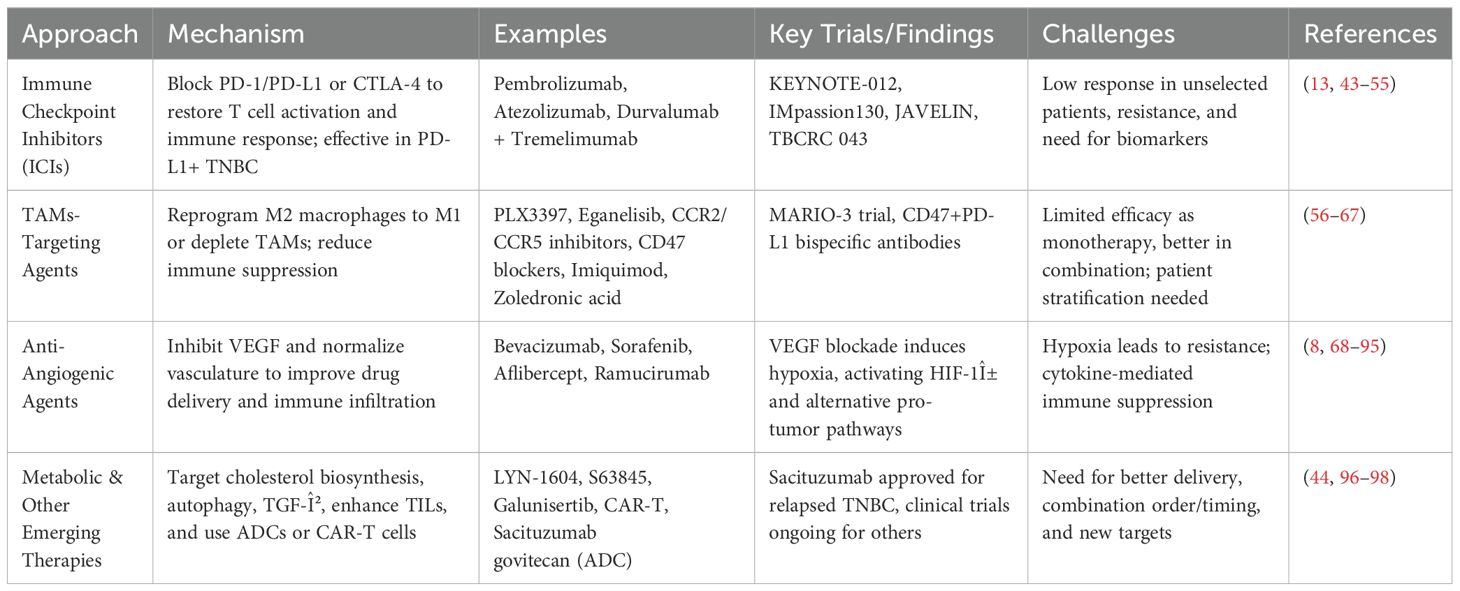
Table 2. Summary of therapeutic strategies targeting the tumor microenvironment (TME) in triple-negative breast cancer (TNBC).
3.1 Immune checkpoint inhibitors in TNBC
Several targeted therapeutic approaches have shown promise in addressing the TME of TNBC, such as immune checkpoint inhibitors (ICIs), which aim to unleash the anti-tumor immune response and transform the TME from an “immune cold” to an “immune hot” state (8). Ongoing research and clinical trials continue to expand the landscape of targeted therapeutics, offering hope for improved outcomes in TNBC patients (68, 69). The two most widely studied ICIs function by blocking the PD-1/PD-L1 axis or CTLA-4. PD-L1 was shown to be present in 20-30% of TNBC cases, which made PD-1/PD-L1 inhibitors effective against TNBC. ICIs such as pembrolizumab and atezolizumab block the interaction between PD-1 and its ligand, restoring T cell activation and promoting an effective immune response (70).
Clinical trials, including the KEYNOTE-012 study, demonstrated that pembrolizumab showed manageable safety and promising antitumor activity in patients with PD-L1-positive advanced TNBC (71). Further, the phase I trial of JS001, an anti-PD-1 monoclonal antibody, showed encouraging safety and preliminary efficacy in patients with advanced TNBC who had undergone multiple lines of systemic therapy, suggesting its potential as a treatment option for this heavily pretreated population (72). Further, the JAVELIN study evaluated the outcome of avelumab, an anti-PD-L1 monoclonal antibody, in patients with metastatic TNBC. It was found that while avelumab demonstrated manageable safety, its overall efficacy as monotherapy was limited (73). These clinical trials highlighted the need for combination approaches or better patient selection strategies, particularly in PD-L1-positive.
CTLA-4 inhibitors, though less explored in TNBC, have shown promise in preclinical studies and early-phase trials. By targeting CTLA-4, these ICIs enhance T cell priming and activation, boosting antitumor immune responses. Dual blockade of PD-1/PD-L1 and CTLA-4 pathways is being investigated as a strategy to achieve synergistic effects, particularly in TNBC cases with cold or poorly immunogenic tumors (74). For instance, a pilot study of durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) in metastatic TNBC demonstrated manageable safety and provided insights into immunogenomic dynamics, showing potential for combination immune checkpoint blockade in specific patient subgroups (75). Despite their potential, ICIs face challenges in TNBC, including low response rates in unselected patient populations and the development of resistance. Ongoing research aims to identify biomarkers such as PD-L1 expression to better predict response to ICIs.
Interestingly, the combination of ICIs with chemotherapy or radiotherapy can make the TME more conducive to immune activation, improving the clinical outcomes (76). For instance, the IMpassion130 study has demonstrated the efficacy of atezolizumab in combination with nab-paclitaxel, showing improved progression-free survival (PFS) in PD-L1-positive TNBC patients (77). Another multicenter phase II trial investigated the efficacy and safety of combining camrelizumab; an anti-PD-1 antibody, apatinib; a VEGFR2 tyrosine kinase inhibitor, and eribulin; a chemotherapy in patients with heavily pretreated advanced TNBC. This trial revealed that the combination therapy demonstrated promising efficacy with a manageable safety profile in this group of patients (78). Most recently, the TBCRC 043, a phase II randomized clinical trial, evaluated the efficacy of combining atezolizumab with carboplatin chemotherapy in patients with metastatic TNBC. Interestingly, the benefits of adding atezolizumab were observed regardless of PD-L1 status. Patients with high tumor-infiltrating lymphocytes (TILs), higher mutation burden, obesity, and uncontrolled blood glucose levels experienced greater benefits from the combination therapy. Notably, all TNBC subtypes benefited from the addition of atezolizumab, except for the luminal androgen receptor subtype (79). These trials have demonstrated improved progression-free survival, overall survival, and objective response rates, especially in patients with PD-L1-positive tumors, while also highlighting the potential for durable responses and manageable safety profiles, making this approach a promising treatment option in both early-stage and metastatic TNBC settings.
The Food and Drug Administration (FDA) of the United States has approved the following PD-1/PD-L1 inhibitors for the treatment of TNBC: pembrolizumab for high-risk early-stage triple-negative breast cancer, pembrolizumab for locally recurrent, unresectable, or metastatic TNBC, and atezolizumab for PD-L1-positive positive unresectable, locally advanced, or metastatic TNBC (80). Overall, ICIs represent a promising advancement in the treatment of TNBC, offering hope for improved outcomes in this difficult-to-treat cancer subtype. However, further studies are needed to optimize their use and broaden their benefits to more patients.
3.2 TAMs-targeting agents
Another immunological approach to target TNBC focuses on targeting tumor-associated macrophages (TAMs), in order to reprogram their tumor-promoting (M2-like) phenotype into a tumor-suppressing (M1-like) state or deplete TAMs to reduce their protumorigenic effects (81–83). It was stated that high densities of TAMs, particularly those expressing the CD163 marker, are associated with decreased overall survival and progression-free survival in breast cancer patients, with the prognostic impact varying by breast cancer subtype (84). In TNBC, the role of TAMs in tumor progression has been widely investigated, suggesting a potential benefit of targeting them. For example, a study showed that the combination of a chemotherapy agent, cyclophosphamide, with macrophage inhibition using either PLX3397 (a CSF1R inhibitor) or an anti-CSF1R antibody, led to increased infiltration of T and B cells into the tumor microenvironment and resulted in durable tumor regression in TNBC models (85). Moreover, a study revealed the combination of eganelisib, an oral immunomodulatory PI3K-γ inhibitor, with ICIs and chemotherapy reprogrammed tumor-associated macrophages to a cancer-fighting state, enhanced immune system activation, and remodeled the tumor environment to make it less supportive of cancer growth in TNBC. The results of this study are based on translational data derived from the MARIO-3 clinical trial (86). Another approach is using the chemokine receptors CCR2/CCR5 antagonists, which mediate the recruitment of monocytes to tumors where they differentiate into TAMs. For instance, a study suggested that inhibiting CCR2 can effectively reduce MCP-1-driven invasiveness and metastasis in TNBC by blocking the recruitment of pro-tumoral monocytes and macrophages to the tumor microenvironment (87). Further, inhibiting CCR5 through a novel antagonist targeting the CCL5/CCR5 axis suppresses tumor growth and metastasis in TNBC by regulating the CCR5-YAP1 signaling pathway, thereby disrupting pro-tumoral mechanisms (88). On the other hand, CD47-SIRPα pathway blockers were found to enhance the ability of macrophages to attack tumor cells, as it is stated that CD47 is overexpressed on cancer cells and inhibits macrophage-mediated phagocytosis (89). TNBC cells often express high levels of CD47, making this an attractive target. For example, PPAB001, a bispecific antibody targeting CD47 and CD24, enhances anti-PD-L1 efficacy in TNBC by reprogramming tumor-associated macrophages to an anti-tumoral M1 phenotype (90). Further, dual-targeting fusion protein against PD-L1 and CD47 inhibits TNBC by enhancing anti-tumor immunity and disrupting immune evasion mechanisms (91). Other approaches, like TLR agonists, can repolarize TAMs from an M2-like to an M1-like phenotype. An example of TLR agonists is Imiquimod (R837), which acts as a key immune activator in the nanoformulation, driving M1 macrophage polarization and synergizing with photothermal therapy to enhance the therapeutic efficacy against TNBC (92). While, bisphosphonates, such as zoledronic acid, were stated to effectively target TAMs in TNBC, reducing their pro-tumoral activity and inhibiting tumor growth and metastasis. Overall, targeting TAMs in TNBC is a promising strategy to reprogram the tumor microenvironment, reduce immune suppression, and enhance the efficacy of existing therapies against this aggressive cancer subtype. However, the effectiveness of TAM inhibitors as standalone treatments is limited, while combination therapies (e.g., chemotherapy, immunotherapy, or radiation) show greater potential. Further biomarker development is critical to identify patients who will benefit most from TAM-targeting therapies.
3.3 Anti-angiogenic agents
Angiogenesis is considered one of the main hallmarks of cancer, including TNBC (93). In tumors, the blood vessels are unevenly spread and disorganized. As a result, poor blood flow limits the delivery of chemotherapy and immunotherapy drugs to the inside of the tumor. It also prevents the removal of immune-suppressing cells from the TME (94). However, vascular endothelial cells lining the tumor express proteins like PD-L1, which activate regulatory T-cells and block the activity of cytotoxic T cells. Thus, the immunosuppressive environment of the tumor will be supported (95). In TNBC, the promotion of blood vessel growth in tumors is regulated by several factors, including VEGF, angiopoietin-2, placental growth factor, and TGF-β. All were reported to contribute to the tumor’s immunosuppressive environment in TNBC (96).
The anti-angiogenic agents targeting different angiogenic factors aim to inhibit tumor angiogenesis, normalize abnormal blood vessels, and improve the delivery of chemotherapy in the hypoxic TNBC microenvironment. These strategies aim to improve the immune system’s ability to fight TNBC (97). VEGF is the main driver of new blood vessel formation in cancer (98). Treatments that block VEGF pathways include monoclonal antibodies against VEGF, like bevacizumab (160), small-molecule tyrosine kinase inhibitors such as sorafenib (161), VEGF traps or decoy receptors like aflibercept (162), and VEGFR2 inhibitors such as ramucirumab (163, 164).
Anti-angiogenic therapy, primarily targeting VEGF signaling, represents a cornerstone approach for impairing tumor vascularization and inhibiting tumor growth (165). However, paradoxically, the blockade of VEGF pathways and subsequent disruption of tumor-associated vasculature often induce a hypoxic microenvironment within the tumor tissue in TNBC (56, 166). Upon inhibition of VEGF-mediated angiogenesis, tumors often experience a state of hypoxia due to the impaired formation of new blood vessels and the consequent reduction in oxygen supply. Hypoxia acts as a potent stressor in the TME, leading to the stabilization and activation of hypoxia-inducible factor-1α (HIF-1α), a key transcription factor that orchestrates cellular responses to low oxygen conditions (167). Activated HIF-1α induces the transcription of a variety of genes associated with survival, metabolism, angiogenesis, and immune modulation, including a range of pro-inflammatory and pro-angiogenic cytokines such as VEGF, interleukin-8 (IL-8), stromal cell-derived factor-1 (CXCL12), and transforming growth factor-β (TGF-β) (168–171).
The resultant cytokine surge within the TME facilitates tumor adaptation through several mechanisms (172). These cytokines activate alternative angiogenic pathways independent of VEGF, allowing the tumor to circumvent the blockade and restore a blood supply essential for its survival and proliferation (173). Furthermore, elevated cytokine levels modulate the immune microenvironment, fostering a more immunosuppressive and inflammatory milieu that favors tumor progression (174). Following anti-angiogenic therapy, these cytokine-driven changes also critically contribute to the development of drug resistance (94, 175, 176). Specifically, signaling pathways downstream of cytokine receptors, such as STAT3 (177), NF-κB (178), and PI3K/AKT (179), become persistently activated.
Collectively, these processes render tumors progressively less sensitive to both chemotherapeutic agents and continued anti-angiogenic therapy, ultimately culminating in multidrug resistance. Thus, while anti-angiogenic therapies initially restrict tumor growth, the induced hypoxic and inflammatory responses mediated via cytokines significantly undermine their long-term efficacy and highlight the need for combinatorial strategies to overcome resistance, the induced hypoxic and inflammatory responses mediated via cytokines significantly undermine their long-term efficacy and highlight the need for combinatorial strategies to overcome resistance (180, 181).
3.4 Other approaches to target TNBC’s TME
Since the cholesterol biosynthesis pathway is associated with TME responses and activities, there is a need to develop innovative inhibitors with low toxicity to target this metabolic pathway. Such novel inhibitors would offer a fresh therapeutic approach for treating the tumor microenvironment. The autophagy initiator LYN-1604 and myeloid cell leukemia-1 inhibitor S63845 are new targeted small-molecule medications that have been developed to cause cancer cell death (182, 183). Galunisertib, a TGF-β inhibitor, is now being studied in a clinical trial to determine the adverse effects and best dosage for treating patients with metastatic androgen receptor-negative TNBC (184). Moreover, tumor-infiltrating lymphocytes (TILs) are the major element in tumor cell immune infiltration in TNBC. TILs engage in interactions with tumor cells, alter the tumor immune microenvironment, and play a role in either immune suppression or attack by T1-cells. Therefore, therapeutic strategies like chimeric antigen receptor T (CAR-T) that encourage immune cell activation and penetration into tumor tissue show great potential. Another promising drugs for TNBC is antibody–drug conjugates (ADCs). The ability to target the treatment to tumor tissue rather than normal tissue is made possible by the identification between tumor cell antigens and antibodies. Due to certain characteristics of the extracellular or intracellular microenvironment (such as a low pH in the high metabolic TME), the entire ADC molecule is destroyed after recognizing the targeted antigens. ADCs provide an innovative, individualized therapeutic method with highly selective drug delivery. For patients with relapsed, refractory metastatic TNBC who have received at least two prior treatments, sacituzumab govitecan was the only ADC approved by the FDA in 2020 (68).
Despite the growing recognition of inflammation and cytokine signaling as central drivers of TNBC progression and therapy resistance, a deeper mechanistic understanding remains elusive. The dual roles of key cytokines such as TNF-α, TGF-β, IL-6, and IL-10, which act as both tumor suppressors and promoters depending on the tumor context, highlight the complexity of targeting these pathways therapeutically. While current strategies show promise, especially in preclinical settings, clinical translation remains limited by tumor heterogeneity, dynamic immune evasion, and the lack of predictive biomarkers for therapy responsiveness. Moreover, combinatorial therapies, such as immune checkpoint inhibitors with anti-angiogenic agents or TAM modulators, have yet to overcome resistance in a durable and broadly applicable manner. Future studies must move beyond observational correlations and aim to dissect temporal and spatial cytokine dynamics within the TME. Embracing integrative multi-omics and spatial transcriptomics approaches could illuminate the contextual behavior of cytokines and enable precision-targeted interventions. Ultimately, TNBC subtypes should be defined and stratified based not only on tumor-intrinsic features but also on immunologic and stromal landscapes. This will be crucial to overcoming current therapeutic limitations and improving patient outcomes.
While several TME-targeted therapies are in development, most focus on inhibiting single pathways such as PD-1/PD-L1 or STAT3. However, our proposed model suggests that TNBC resistance emerges from multi-node cytokine circuits, where immune, stromal, and tumor components reinforce each other through redundant signaling. This implies that combination strategies targeting both upstream cytokine receptors (e.g., IL-6R, TGFβR) and downstream effectors (e.g., STAT3, SMAD3, NF-κB) may be necessary to dismantle these feedback networks and reverse resistance.
3.5 Experimental models to study cytokine-driven drug resistance in TNBC
To better understand cytokine-mediated drug resistance and tumor progression, biologically relevant preclinical models are essential. Current therapies are limited by drug resistance and toxicity, underscoring the need for models that accurately reflect the TME and its complex cytokine networks (185). Preclinical models include traditional 2D cultures and advanced in vitro systems such as 3D spheroids, organoids, and co-cultures alongside in vivo models like xenografts, genetically engineered mice, and humanized systems enables more faithful modeling of cancer biology and supports the development of effective immunomodulatory therapies (186, 187).
In the context of TNBC, cytokine-mediated signaling plays a central role in shaping the TME and influencing therapeutic resistance (188). To dissect these pathways, a variety of experimental models have been developed, each offering unique advantages and limitations in modeling cytokine-driven processes. Conventional monolayer cultures of TNBC cell lines such as MDA-MB-231, BT-549 and Hs578T have been extensively used to study the effect of cytokine expression patterns and their signaling cascades on cancer hallmarks including proliferation, migration, and chemoresistance (189–192). However, these 2D models oversimplify the tumor environment, often lacking immune and stromal components crucial for cytokine crosstalk (193). To address this, researchers have turned to 3D spheroid cultures, tumor organoids, and co-culture systems that incorporate fibroblasts, macrophages, or lymphocytes (194, 195). These platforms better mimic the spatial organization and paracrine signaling of the native TME, enabling more accurate investigation of pro-inflammatory cytokines such as IL-6, IL-8, TNF-α, and TGF-β in drug resistance.
Furthermore, in vivo models offer essential insights into how cytokines modulate tumor behavior within a systemic physiological context. Orthotopic xenograft models using TNBC cell lines implanted into the mammary fat pad of immunodeficient mice allow for the study of cytokine-driven metastasis and treatment response in situ (196–198). More advanced models, such as humanized mouse models, enable the evaluation of immune–cytokine interactions in the presence of a human immune system, which is critical for testing immunotherapeutic strategies (199, 200). Similarly, patient-derived xenografts retain the cytokine expression profiles and heterogeneity of the original tumors, providing a powerful tool for translational studies (201, 202). Furthermore, innovative systems, such as zebrafish xenografts, have also emerged as dynamic models to study cytokine-mediated immune evasion and metastasis due to their optical transparency and compatibility with real-time imaging (203, 204).
These models, when combined with single-cell RNA sequencing and spatial profiling technologies, further enhance our ability to map cytokine networks and their functional impact within the evolving TNBC landscape. Overall, integrating multiple preclinical models is crucial to fully capture the complexity of cytokine-driven mechanisms in TNBC. This systems-based approach not only provides mechanistic insights but also facilitates the identification of novel targets for overcoming drug resistance and improving clinical outcomes.
4 Conclusions
The dynamic balance between pro-inflammatory and anti-inflammatory cytokines in TNBC plays a critical role in determining tumor progression. Dysregulation of cytokine signaling disrupts this balance, leading to an immunosuppressive TME that promotes immune evasion, tumor growth, and resistance to therapies. Cytokines such as TNF-α, TGF-β, IL-6, and IL-10 are pivotal players in these processes, driving inflammation, tumor progression, and immune suppression. Understanding the intricate interplay of cytokines within the TME is essential for developing targeted therapeutics that can combat immune suppression and enhance anti-tumor immune responses. Promising therapeutic strategies include inhibiting pro-inflammatory cytokines, encouraging the release of anti-tumorigenic cytokines, modulating the TME with cytokine inhibitors, and employing immune checkpoint inhibitors. These approaches hold significant potential to transform TNBC treatment by reducing inflammation, enhancing immune responses, and overcoming therapeutic resistance. However, further research is required to refine these strategies, unravel the complexities of the cytokine network, and develop personalized therapies. Harnessing the potential of cytokines as therapeutic targets may pave the way for novel treatment strategies, ultimately improving clinical outcomes and offering new hope for patients battling this aggressive breast cancer subtype. Future research should prioritize identifying key cytokine interaction hubs within the TME and developing multi-targeted interventions informed by such integrative mechanistic frameworks.
Author contributions
AH: Writing – original draft, Conceptualization, Writing – review & editing. HK: Writing – review & editing, Writing – original draft. LA: Writing – original draft. HA-T: Writing – review & editing. AE: Supervision, Conceptualization, Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The authors acknowledge the support of QU Health, Qatar University, for covering the open access publication fees of this review. The authors acknowledge the support of Qatar University for the Graduate Assistantship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. De Francesco EM, Cirillo F, Vella V, Belfiore A, Maggiolini M, and Lappano R. Triple-negative breast cancer drug resistance, durable efficacy, and cure: how advanced biological insights and emerging drug modalities could transform progress. Expert Opin Ther Targets. (2022) 26:513–35. doi: 10.1080/14728222.2022.2094762, PMID: 35761781
2. Almansour NM. Triple-negative breast cancer: A brief review about epidemiology, risk factors, signaling pathways, treatment and role of artificial intelligence. Front Mol Biosci. (2022) 9:836417. doi: 10.3389/fmolb.2022.836417, PMID: 35145999
3. Derakhshan F and Reis-Filho JS. Pathogenesis of triple-negative breast cancer. Annu Rev Pathol. (2022) 17:181–204. doi: 10.1146/annurev-pathol-042420-093238, PMID: 35073169
4. Karim AM, Eun Kwon J, Ali T, Jang J, Ullah I, Lee Y-G, et al. Triple-negative breast cancer: epidemiology, molecular mechanisms, and modern vaccine-based treatment strategies. Biochem Pharmacol. (2023) 212:115545. doi: 10.1016/j.bcp.2023.115545, PMID: 37044296
5. Leon-Ferre RA and Goetz MP. Advances in systemic therapies for triple negative breast cancer. BMJ. (2023) 381:e071674. doi: 10.1136/bmj-2022-071674, PMID: 37253507
6. Hsu JY, Chang CJ, and Cheng JS. Survival, treatment regimens and medical costs of women newly diagnosed with metastatic triple-negative breast cancer. Sci Rep. (2022) 12:729. doi: 10.1038/s41598-021-04316-2, PMID: 35031634
7. Afghahi A, Telli ML, and Kurian AW. Genetics of triple-negative breast cancer: Implications for patient care. Curr Probl Cancer. (2016) 40:130–40. doi: 10.1016/j.currproblcancer.2016.09.007, PMID: 28340968
8. Liu Y, Hu Y, Xue J, Li J, Yi J, Bu J, et al. Advances in immunotherapy for triple-negative breast cancer. Mol Cancer. (2023) 22:145. doi: 10.1186/s12943-023-01850-7, PMID: 37660039
9. Yuan J, Yang L, Zhang H, Beeraka NM, Zhang D, Wang Q, et al. Decoding tumor microenvironment: EMT modulation in breast cancer metastasis and therapeutic resistance, and implications of novel immune checkpoint blockers. Biomedicine Pharmacotherapy. (2024) 181:117714. doi: 10.1016/j.biopha.2024.117714, PMID: 39615165
10. Kajihara N, Kobayashi T, Otsuka R, Nio-Kobayashi J, Oshino T, Takahashi M, et al. Tumor-derived interleukin-34 creates an immunosuppressive and chemoresistant tumor microenvironment by modulating myeloid-derived suppressor cells in triple-negative breast cancer. Cancer Immunol Immunother. (2023) 72:851–64. doi: 10.1007/s00262-022-03293-3, PMID: 36104597
11. Mantovani A, Marchesi F, Jaillon S, Garlanda C, and Allavena P. Tumor-associated myeloid cells: diversity and therapeutic targeting. Cell Mol Immunol. (2021) 18:566–78. doi: 10.1038/s41423-020-00613-4, PMID: 33473192
12. Wang S, Wang J, Chen Z, Luo J, Guo W, Sun L, et al. Targeting M2-like tumor-associated macrophages is a potential therapeutic approach to overcome antitumor drug resistance. NPJ Precis Oncol. (2024) 8:31. doi: 10.1038/s41698-024-00522-z, PMID: 38341519
13. Deepak KGK, Vempati R, Nagaraju GP, Dasari VR, S N, Rao DN, et al. Tumor microenvironment: Challenges and opportunities in targeting metastasis of triple negative breast cancer. Pharmacological Res. (2020) 153:104683. doi: 10.1016/j.semcancer.2019.09.011, PMID: 32050092
14. Harris MA, Savas P, Virassamy B, O’Malley MMR, Kay J, Mueller SN, et al. Towards targeting the breast cancer immune microenvironment. Nat Rev Cancer. (2024) 24:554–77. doi: 10.1038/s41568-024-00714-6, PMID: 38969810
15. Peng C, Xu Y, Wu J, Wu D, Zhou L, and Xia X. TME-related biomimetic strategies against cancer. Int J Nanomedicine. (2024) 19:109–35. doi: 10.2147/IJN.S438793
16. Park M, Kim D, Ko S, Kim A, Mo K, and Yoon H. Breast cancer metastasis: mechanisms and therapeutic implications. Int J Mol Sci. (2022) 23:6806. doi: 10.3390/ijms23126806, PMID: 35743249
17. Wang Q, Shao X, Zhang Y, Zhu M, Wang FXC, Mu J, et al. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. (2023) 12:11149–65. doi: 10.1002/cam4.5698, PMID: 36807772
18. de Visser KE and Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016, PMID: 36917948
19. Gupta I, Hussein O, Sastry KS, Bougarn S, Gopinath N, Chin-Smith E, et al. Deciphering the complexities of cancer cell immune evasion: Mechanisms and therapeutic implications. Adv Cancer Biol - Metastasis. (2023) 8:100107. doi: 10.1016/j.adcanc.2023.100107
20. Mukherjee A, Bilecz AJ, and Lengyel E. The adipocyte microenvironment and cancer. Cancer Metastasis Rev. (2022) 41:575–87. doi: 10.1007/s10555-022-10059-x, PMID: 35941408
21. Kondou R, Iizuka A, Nonomura C, Miyata H, Ashizawa T, Nagashima T, et al. Classification of tumor microenvironment immune types based on immune response-associated gene expression. Int J Oncol. (2019) 54:219–28. doi: 10.3892/ijo.2018.4605, PMID: 30365153
22. Zheng H, Siddharth S, Parida S, Wu X, and Sharma D. Tumor microenvironment: key players in triple negative breast cancer immunomodulation. Cancers (Basel). (2021) 13:3357. doi: 10.3390/cancers13133357, PMID: 34283088
23. Pardoll D. Cancer and the immune system: basic concepts and targets for intervention. Seminars in Oncology (Semin Oncol). (2015) 42(4):523–38. doi: 10.1038/nrc3247, PMID: 26320058
24. Vinay DS, Ryan EP, Pawelec G, Talib WH, Stagg J, Elkord E, et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. Semin Cancer Biol. (2015) 35:S185–S98. doi: 10.1016/j.semcancer.2015.03.004, PMID: 25818339
25. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduction Targeted Ther. (2021) 6:263. doi: 10.1038/s41392-021-00658-5, PMID: 34248142
26. Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. (2004) 4:540–50. doi: 10.1038/nrc1388, PMID: 15229479
27. Jang JH, Kim DH, and Surh YJ. Dynamic roles of inflammasomes in inflammatory tumor microenvironment. NPJ Precis Oncol. (2021) 5:18. doi: 10.1038/s41698-021-00154-7, PMID: 33686176
28. McLaughlin M, Patin EC, Pedersen M, Wilkins A, Dillon MT, Melcher AA, et al. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat Rev Cancer. (2020) 20:203–17. doi: 10.1038/s41568-020-0246-1, PMID: 32161398
29. Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. (2018) 9:7204–18. doi: 10.18632/oncotarget.23208, PMID: 29467962
30. Singh N, Baby D, Rajguru JP, Patil PB, Thakkannavar SS, and Pujari VB. Inflammation and cancer. Annals African Med (Ann Afr Med). (2019) 18(3):121–6. doi: 10.1016/j.jor.2019.06.005, PMID: 31879481
31. Greten FR and Grivennikov SI. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. (2019) 51:27–41. doi: 10.1016/j.immuni.2019.06.025, PMID: 31315034
32. Lim B, Woodward WA, Wang X, Reuben JM, and Ueno NT. Inflammatory breast cancer biology: the tumour microenvironment is key. Nat Rev Cancer. (2018) 18:485–99. doi: 10.1038/s41568-018-0010-y, PMID: 29703913
33. Kuroda H, Jamiyan T, Yamaguchi R, Kakumoto A, Abe A, Harada O, et al. Tumor microenvironment in triple-negative breast cancer: the correlation of tumor-associated macrophages and tumor-infiltrating lymphocytes. Clin Transl Oncol. (2021) 23:2513–25. doi: 10.1007/s12094-021-02652-3, PMID: 34089486
34. Malone MK, Smrekar K, Park S, Blakely B, Walter A, Nasta N, et al. Cytokines secreted by stromal cells in TNBC microenvironment as potential targets for cancer therapy. Cancer Biol Ther. (2020) 21:560–9. doi: 10.1080/15384047.2020.1739484, PMID: 32213106
35. Mehraj U, Mushtaq U, Mir MA, Saleem A, Macha MA, Lone MN, et al. Chemokines in triple-negative breast cancer heterogeneity: New challenges for clinical implications. Semin Cancer Biol. (2022) 86:769–83. doi: 10.1016/j.semcancer.2022.03.008, PMID: 35278636
36. Takeuchi O and Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022, PMID: 20303872
37. Zhang JM and An J. Cytokines, inflammation, and pain. Int Anesthesiol Clin. (2007) 45:27–37. doi: 10.1097/AIA.0b013e318034194e, PMID: 17426506
38. Dranoff G. Cytokines in cancer pathogenesis and cancer therapy. Nat Rev Cancer. (2004) 4:11–22. doi: 10.1038/nrc1252, PMID: 14708024
39. Conlon KC, Miljkovic MD, and Waldmann TA. Cytokines in the treatment of cancer. J Interferon Cytokine Res. (2019) 39:6–21. doi: 10.1089/jir.2018.0019, PMID: 29889594
40. Waldmann TA. Cytokines in cancer immunotherapy. Cold Spring Harb Perspect Biol. (2018) 10:a028472. doi: 10.1101/cshperspect.a028472, PMID: 29101107
41. Hillier SG. Pro-inflammatory cytokines and steroids. In: Henry HL and Norman AW, editors. Encyclopedia of hormones. Academic Press, New York (2003). p. 257–63.
42. Marafini I, Sedda S, Dinallo V, and Monteleone G. Inflammatory cytokines: from discoveries to therapies in IBD. Expert Opin Biol Ther. (2019) 19:1207–17. doi: 10.1080/14712598.2019.1652267, PMID: 31373244
43. Jeong H, Koh J, Kim S, Yim J, Song SG, Kim H, et al. Cell-intrinsic PD-L1 signaling drives immunosuppression by myeloid-derived suppressor cells through IL-6/Jak/Stat3 in PD-L1-high lung cancer. J Immunother Cancer. (2025) 13:e010612. doi: 10.1136/jitc-2024-010612, PMID: 40050048
44. Hines JB, Kacew AJ, and Sweis RF. The development of STING agonists and emerging results as a cancer immunotherapy. Curr Oncol Rep. (2023) 25:189–99. doi: 10.1007/s11912-023-01361-0, PMID: 36705879
45. Li S, Mirlekar B, Johnson BM, Brickey WJ, Wrobel JA, Yang N, et al. STING-induced regulatory B cells compromise NK function in cancer immunity. Nature. (2022) 610:373–80. doi: 10.1038/s41586-022-05254-3, PMID: 36198789
46. Nakhjavani M, Samarasinghe RM, and Shigdar S. Triple-negative breast cancer brain metastasis: An update on druggable targets, current clinical trials, and future treatment options. Drug Discov Today. (2022) 27:1298–314. doi: 10.1016/j.drudis.2022.01.010, PMID: 35101641
47. Yang M, Wang C, Ouyang L, Zhang H, and Lin J. Establishment of prognostic model for invasive ductal carcinoma with distant metastasis within the triple-negative breast cancer: a SEER population-based study. European J Can Prevention. (2025) 34(5):392–404. doi: 10.1097/CEJ.0000000000000925, PMID: 39724567
48. Kumar H, Gupta NV, Jain R, Madhunapantula SV, Babu CS, Kesharwani SS, et al. A review of biological targets and therapeutic approaches in the management of triple-negative breast cancer. J Adv Res. (2023) 54:271–92. doi: 10.1016/j.jare.2023.02.005, PMID: 36791960
49. Gan S, Macalinao DG, Shahoei SH, Tian L, Jin X, Basnet H, et al. Distinct tumor architectures and microenvironments for the initiation of breast cancer metastasis in the brain. Cancer Cell. (2024) 42:1693–712.e24. doi: 10.1016/j.ccell.2024.08.015, PMID: 39270646
50. Deng J and Fleming JB. Inflammation and myeloid cells in cancer progression and metastasis. Front Cell Dev Biol. (2021) 9:759691. doi: 10.3389/fcell.2021.759691, PMID: 35127700
51. Lu C, Liu Y, Ali NM, Zhang B, and Cui X. The role of innate immune cells in the tumor microenvironment and research progress in anti-tumor therapy. Front Immunol. (2022) 13:1039260. doi: 10.3389/fimmu.2022.1039260, PMID: 36741415
52. Chiba N, Ochiai S, Gunji T, Kobayashi T, Sano T, Tomita K, et al. HOXB9 mediates resistance to chemotherapy and patient outcomes through the TGFβ pathway in pancreatic cancer. Oncotarget. (2022) 13:747–54. doi: 10.1038/s41467-021-21986-0, PMID: 35634239
53. Watabe T, Takahashi K, Pietras K, and Yoshimatsu Y. Roles of TGF-β signals in tumor microenvironment via regulation of the formation and plasticity of vascular system. Semin Cancer Biol. (2023) 92:130–8. doi: 10.1016/j.semcancer.2023.04.007, PMID: 37068553
54. Oh A, Pardo M, Rodriguez A, Yu C, Nguyen L, Liang O, et al. NF-kappaB signaling in neoplastic transition from epithelial to mesenchymal phenotype. Cell Commun Signal. (2023) 21:291. doi: 10.1186/s12964-023-01207-z, PMID: 37853467
55. Chen J, Feng W, Sun M, Huang W, Wang G, Chen X, et al. TGF-&x3b2;1-induced SOX18 elevation promotes hepatocellular carcinoma progression and metastasis through transcriptionally upregulating PD-L1 and CXCL12. Gastroenterology. (2024) 167:264–80. doi: 10.1053/j.gastro.2024.04.031, PMID: 38729450
56. Mahaki H, Nobari S, Tanzadehpanah H, Babaeizad A, Kazemzadeh G, Mehrabzadeh M, et al. Targeting VEGF signaling for tumor microenvironment remodeling and metastasis inhibition: Therapeutic strategies and insights. Biomedicine Pharmacotherapy. (2025) 186:118023. doi: 10.1016/j.biopha.2025.118023, PMID: 40164047
57. Wei S and Siegal GP. Metastatic organotropism: an intrinsic property of breast cancer molecular subtypes. Adv Anat Pathol. (2017) 24(2):78–81. doi: 0.1038/s41523-017-0013-3, PMID: 28098572
58. Chen M, Wu C, Fu Z, and Liu S. ICAM1 promotes bone metastasis via integrin-mediated TGF-beta/EMT signaling in triple-negative breast cancer. Cancer Sci. (2022) 113:3751–65. doi: 10.1111/cas.15532, PMID: 35969372
59. Cao J, Cao R, Liu Y, and Dai T. CPNE1 mediates glycolysis and metastasis of breast cancer through activation of PI3K/AKT/HIF-1α signaling. Pathol - Res Practice. (2023) 248:154634. doi: 10.1016/j.prp.2023.154634, PMID: 37454492
60. Liu D, Guo P, McCarthy C, Wang B, Tao Y, and Auguste D. Peptide density targets and impedes triple negative breast cancer metastasis. Nat Commun. (2018) 9:2612. doi: 10.1038/s41467-018-05035-5, PMID: 29973594
61. Benchama O, Tyukhtenko S, Malamas MS, Williams MK, Makriyannis A, and Avraham HK. Inhibition of triple negative breast cancer-associated inflammation, tumor growth and brain colonization by targeting monoacylglycerol lipase. Sci Rep. (2022) 12:5328. doi: 10.1038/s41598-022-09358-8, PMID: 35351947
62. Tan B, Zhou K, Liu W, Prince E, Qing Y, Li Y, et al. RNA N(6) -methyladenosine reader YTHDC1 is essential for TGF-beta-mediated metastasis of triple negative breast cancer. Theranostics. (2022) 12:5727–43. doi: 10.7150/thno.71872, PMID: 35966596
63. Zhao G, Li C, Liu W, Wu J, and Yang X. Understanding the molecular mechanisms of SORBS2 in TNBC lung metastasis. Biochem Biophys Res Commun. (2025) 762:151762. doi: 10.1016/j.bbrc.2025.151762, PMID: 40199126
64. Chen Z and Zhao Y. The mechanism underlying metastasis in triple-negative breast cancer: focusing on the interplay between ferroptosis, epithelial-mesenchymal transition, and non-coding RNAs. Front Pharmacol. (2024) 15:1437022. doi: 10.3389/fphar.2024.1437022, PMID: 39881868
65. Nie J, Jiang H-C, Zhou Y-C, Jiang B, He W-J, Wang Y-F, et al. MiR-125b regulates the proliferation and metastasis of triple negative breast cancer cells via the Wnt/β-catenin pathway and EMT. Bioscience Biotechnol Biochem. (2019) 83:1062–71. doi: 10.1080/09168451.2019.1584521, PMID: 30950326
66. Luo B, Zheng H, Liang G, Luo Y, Zhang Q, and Li X. HMGB3 contributes to anti-PD-1 resistance by inhibiting IFN-γ-driven ferroptosis in TNBC. Mol Carcinogenesis. (2025) 64:490–501. doi: 10.1038/s41467-023-36000-y, PMID: 39660968
67. Vagia E, Mahalingam D, and Cristofanilli M. The landscape of targeted therapies in TNBC. Cancers (Basel). (2020) 12:916. doi: 10.3390/cancers12040916, PMID: 32276534
68. Li Y, Zhang H, Merkher Y, Chen L, Liu N, Leonov S, et al. Recent advances in therapeutic strategies for triple-negative breast cancer. J Hematol Oncol. (2022) 15:121. doi: 10.1186/s13045-022-01341-0, PMID: 36038913
69. Malla RR, Deepak K, Merchant N, and Dasari VR. Breast Tumor Microenvironment: Emerging target of therapeutic phytochemicals. Phytomedicine. (2020) 70:153227. doi: 10.1016/j.phymed.2020.153227, PMID: 32339885
70. Zhu Y, Zhu X, Tang C, Guan X, and Zhang W. Progress and challenges of immunotherapy in triple-negative breast cancer. Biochim Biophys Acta Rev Cancer. (2021) 1876(2):188593. doi: 10.1038/s41571-021-00576-2
71. Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, et al. Open-label clinical trial of niraparib combined with pembrolizumab for treatment of advanced or metastatic triple-negative breast cancer. JAMA Oncol. (2019) 5:1132–40. doi: 10.1001/jamaoncol.2019.1029, PMID: 31194225
72. Bian L, Zhang H, Wang T, Zhang S, Song H, Xu M, et al. JS001, an anti-PD-1 mAb for advanced triple negative breast cancer patients after multi-line systemic therapy in a phase I trial. Annals Translational Med. (2019) 7(18):435 doi: 10.1136/jitc-2020-001240, PMID: 31700871
73. Dirix LA-O, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Research and Treatment. (2018) 167(3):671–686. doi: 10.1002/cncr.30507, PMID: 28112816
74. Li HD, Chen YQ, Li Y, Wei X, Wang SY, Cao Y, et al. Harnessing virus flexibility to selectively capture and profile rare circulating target cells for precise cancer subtyping. Nature Communications. (2024) 15(1):5849. doi: 10.1038/s41467-021-27858-4, PMID: 38992001
75. Santa-Maria CA, Kato T, Park JH, Kiyotani K, Rademaker A, Shah AN, et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget. (2018) 9(27):18985–96. doi: 10.1038/s41523-022-00399-y, PMID: 29721177
76. Haiderali A, Huang M, Pan W, Akers KG, Maciel D, and Frederickson AM. Pembrolizumab plus chemotherapy for first-line treatment of advanced triple-negative breast cancer. Future Oncol. (2024) 20:1587–600. doi: 10.2217/fon-2023-0301, PMID: 38597713
77. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. The New England Journal of Medicine. (2018) 379(22):2108–2121. doi: 10.1056/NEJMoa1809615, PMID: 30345906
78. Li YA-O, Xu CA-OX, Wang BA-O, Xu FA-O, Ma FA-O, Qu Y, et al. Proteomic characterization of gastric cancer response to chemotherapy and targeted therapy reveals new therapeutic strategies. Nature Communications. (2022) 13:5723. doi: 10.1038/s41586-022-05283-1, PMID: 36175412
79. Lehmann BD, Abramson VG, Dees EC, Shah PD, Ballinger TJ, Isaacs C, et al. Atezolizumab in combination with carboplatin and survival outcomes in patients with metastatic triple-negative breast cancer: the TBCRC 043 phase 2 randomized clinical trial. JAMA Oncol. (2024) 10:193–201. doi: 10.1001/jamaoncol.2021.0001, PMID: 38095878
80. Shah MA-O, Osgood CA-O, Amatya AA-O, Fiero MA-OX, Pierce WA-O, Nair AA-OX, et al. FDA approval summary: pembrolizumab for neoadjuvant and adjuvant treatment of patients with high-risk early-stage triple-negative breast cancer. Clinical Cancer Research. (2022) 28(24):5249–5253. doi: 10.1158/1078-0432.CCR-21-3863, PMID: 35925043
81. Patwardhan KA, RaviPrakash H, Nikolaou N, Gonzalez-García I, Salazar JD, Metcalfe P, et al. Towards a survival risk prediction model for metastatic NSCLC patients on durvalumab using whole-lung CT radiomics. Front Immunol. (2024) 15:1383644. doi: 10.3389/fimmu.2024.1383644, PMID: 38915397
82. Padzińska-Pruszyńska I, Kucharzewska P, Matejuk A, Górczak MA-O, Kubiak MA-O, Taciak BA-O, et al. Macrophages: key players in the battle against triple-negative breast cancer. International J Mol Sci (2024) 25(19):10781. doi: 10.3390/ijms251910781, PMID: 39409110
83. Li M, He L, Zhu J, Zhang P, and Liang S. Targeting tumor-associated macrophages for cancer treatment. Cell Bioscience. (2022) 12:85. doi: 10.1186/s13578-022-00823-5, PMID: 35672862
84. Allison E, Edirimanne S, Matthews J, and Fuller SA-O. Breast cancer survival outcomes and tumor-associated macrophage markers: A systematic review and meta-analysis. Oncology Therapy. (2023) 11:27–48. doi: 10.1007/s40487-022-00214-3, PMID: 36484945
85. Singh SA-OX, Lee N, Pedroza DA-O, Bado IA-O, Hamor C, Zhang LA-OX, et al. Chemotherapy coupled to macrophage inhibition induces T-cell and B-cell infiltration and durable regression in triple-negative breast cancer. Cancer Research. (2022) 82(12):2281–2297. doi: 10.1158/0008-5472.CAN-21-3714, PMID: 35442423
86. Qi CA-O, Liu D, Liu C, Wei X, Ma M, Lu X, et al. Antigen-independent activation is critical for the durable antitumor effect of GUCY2C-targeted CAR-T cells. J ImmunoTherapy Can. (2024) 12. doi: 10.1136/jitc-2024–009960, PMID: 39366753
87. Munzone EA-O, Gray KP, Fumagalli C, Guerini-Rocco E, Láng I, Ruhstaller T, et al. Mutational analysis of triple-negative breast cancers within the International Breast Cancer Study Group (IBCSG) Trial 22-00. Breast Cancer Research and Treatment. (2018) 170(2):351–360. doi: 10.1007/s10549-018-4767-1, PMID: 29589138
88. Chen L, Xu G, Song X, Zhang L, Chen C, Xiang G, et al. A novel antagonist of the CCL5/CCR5 axis suppresses the tumor growth and metastasis of triple-negative breast cancer by CCR5-YAP1 regulation. Cancer Letters. (2024) 583:216635. doi: 10.1016/j.canlet.2024.216635, PMID: 38237887
89. Si Y, Zhang Y, Guan JS, Ngo HG, Totoro A, Singh AP, et al. Anti-CD47 monoclonal antibody-drug conjugate: A targeted therapy to treat triple-negative breast cancers. Vaccines (Basel). (2021) 9(8):882. doi: 10.3390/vaccines9080882, PMID: 34452008
90. Yang Y, Li J, Zhang J, Wu H, Yang Y, Guo H, et al. PPAB001, a novel bispecific antibody against CD47 and CD24, enhances anti-PD-L1 efficacy in triple-negative breast cancer via reprogramming tumor-associated macrophages towards M1 phenotype. Molecular Therapy – Oncolytics. (2023) 31:100747. doi: 10.1016/j.intimp.2024.113740, PMID: 39622130
91. Bian Y, Lin T, Jakos T, Xiao X, and Zhu JA-O. The generation of dual-targeting fusion protein PD-L1/CD47 for the inhibition of triple-negative breast cancer. Biomedicines. (2022) 10(8):1843. doi: 10.3390/biomedicines10081843, PMID: 36009390
92. Geng S, Fang B, Wang K, Wang F, Zhou Y, Hou Y, et al. Polydopamine nanoformulations induced ICD and M1 phenotype macrophage polarization for enhanced TNBC synergistic photothermal immunotherapy. ACS Appl Materials Interfaces. (2024) 16:59814–32. doi: 10.1021/acsami.4c11594, PMID: 39450881
93. Ribatti D, Nico B, Ruggieri S, Tamma R, Simone G, and Mangia A. Angiogenesis and antiangiogenesis in triple-negative breast cancer. Transl Oncol. (2016) 9:453–7. doi: 10.1016/j.tranon.2016.07.002, PMID: 27751350
94. Chen W, Shen L, Jiang J, Zhang L, Zhang Z, Pan J, et al. Antiangiogenic therapy reverses the immunosuppressive breast cancer microenvironment. biomark Res. (2021) 9:59. doi: 10.1186/s40364-021-00312-w, PMID: 34294146
95. Wang Y, Chen Y, Liu Y, Zhao J, Wang G, Chen H, et al. Tumor vascular endothelial cells promote immune escape by upregulating PD-L1 expression via crosstalk between NF-κB and STAT3 signaling pathways in nasopharyngeal carcinoma. Cell Death Dis. (2025) 16:129. doi: 10.1038/s41419-025-07444-z, PMID: 40000620
96. Malekian S, Rahmati M, Sari S, Kazemimanesh M, Kheirbakhsh R, Muhammadnejad A, et al. Expression of diverse angiogenesis factor in different stages of the 4T1 tumor as a mouse model of triple-negative breast cancer. Adv Pharm Bull. (2020) 10:323–8. doi: 10.34172/apb.2020.039, PMID: 32373503
97. Oguntade AS, Al-Amodi F, Alrumayh A, Alobaida M, and Bwalya M. Anti-angiogenesis in cancer therapeutics: the magic bullet. J Egypt Natl Canc Inst. (2021) 33:15. doi: 10.1186/s43046-021-00072-6, PMID: 34212275
98. Yang Y and Cao Y. The impact of VEGF on cancer metastasis and systemic disease. Semin Cancer Biol. (2022) 86:251–61. doi: 10.1016/j.semcancer.2022.03.011, PMID: 35307547
99. Turner MD, Nedjai B, Hurst T, and Pennington DJ. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta. (2014) 1843:2563–82. doi: 10.1016/j.bbamcr.2014.05.014, PMID: 24892271
100. Dinarello CA. The paradox of pro-inflammatory cytokines in cancer. Cancer Metastasis Rev. (2006) 25:307–13. doi: 10.1007/s10555-006-9000-8, PMID: 17029030
101. Kabel AM. Relationship between cancer and cytokines. J Cancer Res Treat. (2014) 2:41–3. doi: 10.12691/jcrt-2-2-3
102. Mantovani A, Allavena P, Sica A, and Balkwill F. Cancer-related inflammation. Nature. (2008) 454:436–44. doi: 10.1038/nature07205, PMID: 18650914
103. Esquivel-Velazquez M, Ostoa-Saloma P, Palacios-Arreola MI, Nava-Castro KE, Castro JI, and Morales-Montor J. The role of cytokines in breast cancer development and progression. J Interferon Cytokine Res. (2015) 35:1–16. doi: 10.1089/jir.2014.0026, PMID: 25068787
104. Balkwill F and Mantovani A. Inflammation and cancer: back to Virchow?. The Lancet. (2001) 357:539–545. doi: 10.1016/S0140-6736(00)04046-0, PMID: 11229684
105. Habanjar OA-O, Bingula RA-O, Decombat CA-O, Diab-Assaf M, Caldefie-Chezet FA-O, and Delort LA-O. Crosstalk of inflammatory cytokines within the breast tumor microenvironment. International J Mol Sci. (2023) 24(4):4002. doi: 10.3390/ijms24044002, PMID: 36835413
106. Thomas DA and Massagué J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. (2005) 8:369–80. doi: 10.1016/j.ccr.2005.10.012, PMID: 16286245
107. Yazdani Z, Rafiei A, Golpour M, Zafari PA-O, Moonesi M, and Ghaffari S. IL-35, a double-edged sword in cancer. J Cell Biochemistry. (2020) 121(3):2064–2076. doi: 10.1002/jcb.27894, PMID: 31633232
108. Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X, et al. Transforming growth factor-beta in stem cells and tissue homeostasis. Bone Res. (2018) 6:2. doi: 10.1038/s41413-017-0005-4, PMID: 29423331
109. Wang H, Wang P, Xu M, Song X, Wu H, Evert M, et al. Distinct functions of transforming growth factor-beta signaling in c-MYC driven hepatocellular carcinoma initiation and progression. Cell Death Dis. (2021) 12:200. doi: 10.1038/s41419-021-03488-z, PMID: 33608500
110. Vander Ark A, Cao J, and Li X. TGF-β receptors: In and beyond TGF-β signaling. Cell Signalling. (2018) 52:112–20. doi: 10.1016/j.cellsig.2018.09.002, PMID: 30184463
111. Tauriello DVF, Sancho E, and Batlle E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat Rev Cancer. (2022) 22:25–44. doi: 10.1038/s41568-021-00413-6, PMID: 34671117
112. Gorelik L and Flavell RA. Transforming growth factor-beta in T-cell biology. Nat Rev Immunol. (2002) 2:46–53. doi: 10.1038/nri704, PMID: 11905837
113. Derynck R, Turley SJ, and Akhurst RJ. TGFbeta biology in cancer progression and immunotherapy. Nat Rev Clin Oncol. (2021) 18:9–34. doi: 10.1038/s41571-020-0403-1, PMID: 32710082
114. Smith PM, Means AL, and Beauchamp RD. Immunomodulatory effects of TGF-beta family signaling within intestinal epithelial cells and carcinomas. Gastrointest Disord (Basel). (2019) 1:290–300. doi: 10.3390/gidisord1030023, PMID: 33834163
115. Xu X, Zhang L, He X, Zhang P, Sun C, Xu X, et al. TGF-beta plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem Biophys Res Commun. (2018) 502:160–5. doi: 10.1016/j.bbrc.2018.05.139, PMID: 29792857
116. Vishnubalaji R and Alajez NM. Epigenetic regulation of triple negative breast cancer (TNBC) by TGF-β signaling. Sci Rep. (2021) 11:15410. doi: 10.1038/s41598-021-94514-9, PMID: 34326372
117. Vishnubalaji R and Alajez NM. Epigenetic regulation of triple negative breast cancer (TNBC) by TGF-beta signaling. Sci Rep. (2021) 11:15410. doi: 10.1038/s41598-021-94514-9, PMID: 34326372
118. Ding MJ, Su KE, Cui GZ, Yang WH, Chen L, Yang M, et al. Association between transforming growth factor-beta1 expression and the clinical features of triple negative breast cancer. Oncol Lett. (2016) 11:4040–4. doi: 10.3892/ol.2016.4497, PMID: 27313737
119. Xing Y, Ren ZQ, Jin R, Liu L, Pei JP, and Yu K. Therapeutic efficacy and mechanism of CD73-TGFbeta dual-blockade in a mouse model of triple-negative breast cancer. Acta Pharmacol Sin. (2022) 43:2410–8. doi: 10.1038/s41401-021-00840-z, PMID: 35082394
120. Yan X, He Y, Yang S, Zeng T, Hua Y, Bao S, et al. A positive feedback loop: RAD18-YAP-TGF-beta between triple-negative breast cancer and macrophages regulates cancer stemness and progression. Cell Death Discov. (2022) 8:196. doi: 10.1038/s41420-022-00968-9, PMID: 35413945
121. Zhang H, Wang J, Yin Y, Meng Q, and Lyu Y. The role of EMT-related lncRNA in the process of triple-negative breast cancer metastasis. Biosci Rep. (2021) 41:BSR20203121. doi: 10.1042/bsr20203121, PMID: 33443534
122. Jalalirad M, Haddad TC, Salisbury JL, Radisky D, Zhang M, Schroeder M, et al. Aurora-A kinase oncogenic signaling mediates TGF-beta-induced triple-negative breast cancer plasticity and chemoresistance. Oncogene. (2021) 40:2509–23. doi: 10.1038/s41388-021-01711-x, PMID: 33674749
123. Sundqvist A, Vasilaki E, Voytyuk O, Bai Y, Morikawa M, Moustakas A, et al. TGFbeta and EGF signaling orchestrates the AP-1- and p63 transcriptional regulation of breast cancer invasiveness. Oncogene. (2020) 39:4436–49. doi: 10.1038/s41388-020-1299-z, PMID: 32350443
124. Zhao Y, Sun H, Li X, Liu Q, Liu Y, Hou Y, et al. DGKZ promotes TGFβ signaling pathway and metastasis in triple-negative breast cancer by suppressing lipid raft-dependent endocytosis of TGFβR2. Cell Death Dis. (2022) 13:636. doi: 10.1038/s41419-022-04537-x, PMID: 35115500
125. Yang Y, Ye WL, Zhang RN, He XS, Wang JR, Liu YX, et al. The role of TGF-beta signaling pathways in cancer and its potential as a therapeutic target. Evid Based Complement Alternat Med. (2021) 2021:6675208. doi: 10.1155/2021/6675208, PMID: 34335834
126. Johnson GL and Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. (2002) 298:1911–1912. doi: 10.1126/science.1072682, PMID: 12471242
127. Khoshakhlagh M, Soleimani A, Binabaj MM, Avan A, Ferns GA, Khazaei M, et al. Therapeutic potential of pharmacological TGF-beta signaling pathway inhibitors in the pathogenesis of breast cancer. Biochem Pharmacol. (2019) 164:17–22. doi: 10.1016/j.bcp.2019.03.031, PMID: 30905655
128. Liu ZL, Chen HH, Zheng LL, Sun LP, and Shi L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct Target Ther. (2023) 8:198. doi: 10.1038/s41392-023-01460-1, PMID: 37169756
129. Jang DI, Lee AH, Shin HY, Song HR, Park JH, Kang TB, et al. The role of tumor necrosis factor alpha (TNF-alpha) in autoimmune disease and current TNF-alpha inhibitors in therapeutics. Int J Mol Sci. (2021) 22:2719. doi: 10.3390/ijms22094419, PMID: 33800290
130. Mahdavi Sharif P, Jabbari P, Razi S, Keshavarz-Fathi M, and Rezaei N. Importance of TNF-alpha and its alterations in the development of cancers. Cytokine. (2020) 130:155066. doi: 10.1016/j.cyto.2020.155066, PMID: 32208336
131. Cruceriu D, Baldasici O, Balacescu O, and Berindan-Neagoe I. The dual role of tumor necrosis factor-alpha (TNF-alpha) in breast cancer: molecular insights and therapeutic approaches. Cell Oncol (Dordr). (2020) 43:1–18. doi: 10.1007/s13402-019-00489-1, PMID: 31900901
132. Liu W, Lu X, Shi P, Yang G, Zhou Z, Li W, et al. TNF-alpha increases breast cancer stem-like cells through up-regulating TAZ expression via the non-canonical NF-kappaB pathway. Sci Rep. (2020) 10:1804. doi: 10.1038/s41598-020-58642-y, PMID: 32019974
133. Wang Y and Zhou BP. Epithelial-mesenchymal transition in breast cancer progression and metastasis. Chinese J Cancer. (2011) 30(9):603–611. doi: 10.1016/j.semcancer.2011.03.002
134. Raphael I, Nalawade S, Eagar TN, and Forsthuber TG. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine. (2015) 74(1):5–17. doi: 10.1016/j.immuni.2015.07.017, PMID: 25458968
135. Qiao Y, He H, Jonsson P, Sinha I, Zhao C, and Dahlman-Wright K. AP-1 is a key regulator of proinflammatory cytokine TNF&x3b1;-mediated triple-negative breast cancer progression. J Biol Chem. (2016) 291:18309. doi: 10.1074/jbc.a115.702571, PMID: 26792858
136. Cai X, Cao C, Li J, Chen F, Zhang S, Liu B, et al. Inflammatory factor TNF-خ▒ promotes the growth of breast cancer via the positive feedback loop of TNFR1/NF-خ╙B (and/or p38)/p-STAT3/HBXIP/TNFR1. Oncotarget. (2017) 8:58338–52. doi: 10.18632/oncotarget.16873, PMID: 28938560
137. Mercogliano MF, Bruni S, Elizalde PV, and Schillaci R. Tumor necrosis factor alpha blockade: an opportunity to tackle breast cancer. Front Oncol. (2020) 10:584. doi: 10.3389/fonc.2020.00584, PMID: 32391269
138. Sun EG, Vijayan V, Park MR, Yoo KH, Cho SH, Bae WK, et al. Suppression of triple-negative breast cancer aggressiveness by LGALS3BP via inhibition of the TNF-alpha-TAK1-MMP9 axis. Cell Death Discov. (2023) 9:122. doi: 10.1038/s41420-023-01419-9, PMID: 37041137
139. Kumari N, Dwarakanath BS, Das A, and Bhatt AN. Role of interleukin-6 in cancer progression and therapeutic resistance. Tumour Biol. (2016) 37:11553–72. doi: 10.1007/s13277-016-5098-7, PMID: 27260630
140. Kang S and Kishimoto T. Interplay between interleukin-6 signaling and the vascular endothelium in cytokine storms. Exp Mol Med. (2021) 53:1116–23. doi: 10.1038/s12276-021-00649-0, PMID: 34253862
141. Chonov DC, Ignatova MMK, Ananiev JR, and Gulubova MV. IL-6 activities in the tumour microenvironment. Part 1. Open Access Maced J Med Sci. (2019) 7:2391–8. doi: 10.3889/oamjms.2019.589, PMID: 31592285
142. Fisher DT, Appenheimer MM, and Evans SS. The two faces of IL-6 in the tumor microenvironment. Semin Immunol. (2014) 26:38–47. doi: 10.1016/j.smim.2014.01.008, PMID: 24602448
143. Masjedi A, Hashemi V, Hojjat-Farsangi M, Ghalamfarsa G, Azizi G, Yousefi M, et al. The significant role of interleukin-6 and its signaling pathway in the immunopathogenesis and treatment of breast cancer. BioMed Pharmacother. (2018) 108:1415–24. doi: 10.1016/j.biopha.2018.09.177, PMID: 30372844
144. Heo T-H, Wahler J, and Suh N. Potential therapeutic implications of IL-6/IL-6R/gp130-targeting agents in breast cancer. Oncotarget. (2016) 7:15460–73. doi: 10.18632/oncotarget.7102, PMID: 26840088
145. Vecchi L, Mota STS, Zoia MAP, Martins IC, de Souza JB, Santos TG, et al. Interleukin-6 signaling in triple negative breast cancer cells elicits the annexin A1/formyl peptide receptor 1 axis and affects the tumor microenvironment. Cells. (2022) 11:1705. doi: 10.3390/cells11101705, PMID: 35626741
146. Vasiyani H, Mane M, Rana K, Shinde A, Roy M, Singh J, et al. DNA damage induces STING mediated IL-6-STAT3 survival pathway in triple-negative breast cancer cells and decreased survival of breast cancer patients. Apoptosis. (2022) 27:961–78. doi: 10.1007/s10495-022-01763-8, PMID: 36018392
147. Jin K, Pandey NB, and Popel AS. Simultaneous blockade of IL-6 and CCL5 signaling for synergistic inhibition of triple-negative breast cancer growth and metastasis. Breast Cancer Res. (2018) 20:54. doi: 10.1186/s13058-018-0981-3, PMID: 29898755
148. Saraiva M and O’Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. (2010) 10:170–81. doi: 10.1038/nri2711, PMID: 20154735
149. Wang K and Karin M. Chapter five - tumor-elicited inflammation and colorectal cancer. In: Wang X-Y and Fisher PB, editors. Advances in Cancer Research, vol. 128 . Academic Press (2015). p. 173–96., PMID: 26216633
150. Hamidullah, Changkija B, and Konwar R. Role of interleukin-10 in breast cancer. Breast Cancer Res Treat. (2012) 133:11–21. doi: 10.1007/s10549-011-1855-x, PMID: 22057973
151. Llanes-Fernandez L, Alvarez-Goyanes RI, Arango-Prado Mdel C, Alcocer-Gonzalez JM, Mojarrieta JC, Perez XE, et al. Relationship between IL-10 and tumor markers in breast cancer patients. Breast. (2006) 15:482–9. doi: 10.1016/j.breast.2005.10.002, PMID: 16403632
152. Mirlekar B. Tumor promoting roles of IL-10, TGF-beta, IL-4, and IL-35: Its implications in cancer immunotherapy. SAGE Open Med. (2022) 10:20503121211069012. doi: 10.1177/20503121211069012, PMID: 35096390
153. Kai-Yuan C, Wei-Ching C, Jiuan-Miaw L, Chia-Wen T, Wen-Shin C, Chen-Hsien SU, et al. Contribution of interleukin-10 genotype to triple negative breast cancer risk. Anticancer Res. (2021) 41:2451. doi: 10.21873/anticanres.15020, PMID: 33952470
154. Toney NJ, Opdenaker LM, Cicek K, Frerichs L, Kennington CR, Oberly S, et al. Tumor-B-cell interactions promote isotype switching to an immunosuppressive IgG4 antibody response through upregulation of IL-10 in triple negative breast cancers. J Transl Med. (2022) 20:112. doi: 10.1186/s12967-022-03319-5, PMID: 35255925
155. Fan Y and He S. The characteristics of tumor microenvironment in triple negative breast cancer. Cancer Manag Res. (2022) 14:1–17. doi: 10.2147/CMAR.S346248
156. Guo Z, Zhu Z, Lin X, Wang S, Wen Y, Wang L, et al. Tumor microenvironment and immunotherapy for triple-negative breast cancer. Biomark Res. (2024) 12:166. doi: 10.1186/s40364-024-00714-6, PMID: 39741315
157. Vishnubalaji R and Alajez NM. Disrupted lipid metabolism, cytokine signaling, and dormancy: hallmarks of doxorubicin-resistant triple-negative breast cancer models. Cancers (Basel). (2024) 16:4273. doi: 10.3390/cancers16244273, PMID: 39766172
158. Zhou L and Yu CW. Epigenetic modulations in triple-negative breast cancer: Therapeutic implications for tumor microenvironment. Pharmacol Res. (2024) 204:107205. doi: 10.1016/j.phrs.2024.107205, PMID: 38719195
159. Palakurthi B, Fross SR, Guldner IH, Aleksandrovic E, Liu X, Martino AK, et al. Targeting CXCL16 and STAT1 augments immune checkpoint blockade therapy in triple-negative breast cancer. Nat Commun. (2023) 14:2109. doi: 10.1038/s41467-023-37727-y, PMID: 37055410
160. Ferrara N, Hillan Kj Fau - Novotny W, and Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophysical Res Communications. (2005) 333(2):328–335. doi: 10.1038/nrd1504
161. Ishihara S, Onoda N, Noda S, Asano Y, Tauchi Y, Morisaki T, et al. Sorafenib inhibits vascular endothelial cell proliferation stimulated by anaplastic thyroid cancer cells regardless of BRAF mutation status. International J Oncol. (2019) 55(5):1069–1076. doi: 10.3892/ijo.2014.2758, PMID: 31545405
162. Stewart MW. Aflibercept (VEGF-TRAP): the next anti-VEGF drug. Inflammation & Allergy – Drug Targets. (2011) 10(6):497–508. doi: 10.1038/eye.2011.81, PMID: 21999177
163. Clarke JM and Hurwitz HI. Targeted inhibition of VEGF receptor 2: an update on ramucirumab. Expert Opinion on Biological Therapy. (2013) 13(8):1187–1196. doi: 10.2217/tme-2016-0002, PMID: 23803182
164. Khanna S, Komati R, Eichenbaum DA, Hariprasad I, Ciulla TA, and Hariprasad SA-O. Current and upcoming anti-VEGF therapies and dosing strategies for the treatment of neovascular AMD: a comparative review. BMJ Open Ophthalmology. (2019) 4(1):e000398. doi: 10.2147/OPTH.S211877, PMID: 31909196
165. Wang L, Liu WQ, Broussy S, Han B, and Fang H. Recent advances of anti-angiogenic inhibitors targeting VEGF/VEGFR axis. Front Pharmacol. (2024) 14:1307860. doi: 10.1016/j.actbio.2023.07.018, PMID: 38239196
166. Ray A, Dhar S, and Ray BK. Control of VEGF expression in triple-negative breast carcinoma cells by suppression of SAF-1 transcription factor activity. Mol Cancer Res. (2011) 9:1030–41. doi: 10.1158/1541-7786.mcr-10-0598, PMID: 21665940
167. Hendriksen EM, Span PN, Schuuring J, Peters JPW, Sweep FCGJ, van der Kogel AJ, et al. Angiogenesis, hypoxia and VEGF expression during tumour growth in a human xenograft tumour model. Microvascular Res. (2009) 77:96–103. doi: 10.1016/j.mvr.2008.11.002, PMID: 19118564
168. Gómez-Ferrer M, Villanueva-Badenas EA-O, Sánchez-Sánchez R, Sánchez-López CA-O, Baquero MC, Sepúlveda PA-O, et al. HIF-1α and pro-inflammatory signaling improves the immunomodulatory activity of MSC-derived extracellular vesicles. International J Mol Sci. (2021) 22(7):3416. doi: 10.3390/ijms22073416, PMID: 33810359
169. Magar AG, Morya VA-O, Kwak MK, Oh JU, and Noh KC. A molecular perspective on HIF-1α and angiogenic stimulator networks and their role in solid tumors: an update. International J Mol Sci. (2024) 25(6):3313. doi: 10.3390/ijms25063313, PMID: 38542288
170. Ramakrishnan S, Anand V Fau - Roy S, and Roy S. Vascular endothelial growth factor signaling in hypoxia and inflammation. J Neuroimmune Pharmacol. (2014) 9(2):142–160. doi: 10.1007/s11481-014-9531-7, PMID: 24610033
171. Liu Q, Guan C, Liu C, Li H, Wu J, and Sun C. Targeting hypoxia-inducible factor-1alpha: A new strategy for triple-negative breast cancer therapy. Biomed. Pharmacotherapy. (2022) 156:113861. doi: 10.1016/j.biopha.2022.113861, PMID: 36228375
172. Pradhan R, Kundu A, and Kundu CN. The cytokines in tumor microenvironment: from cancer initiation-elongation-progression to metastatic outgrowth. Crit Rev Oncology/Hematology. (2024) 196:104311. doi: 10.1016/j.critrevonc.2024.104311, PMID: 38442808
173. Yi M, Li T, Niu M, Zhang H, Wu Y, Wu K, et al. Targeting cytokine and chemokine signaling pathways for cancer therapy. Signal Transduct Target Ther. (2024) 9:176. doi: 10.1038/s41392-024-01868-3, PMID: 39034318
174. Li L, Yu R, Cai T, Chen Z, Lan M, Zou T, et al. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int Immunopharmacology. (2020) 88:106939. doi: 10.1016/j.intimp.2020.106939, PMID: 33182039
175. Eikesdal HP and Kalluri R. Drug resistance associated with antiangiogenesis therapy. Seminars in Cancer Biology. (2009) 19(5):310–317 doi: 10.1038/nrc3624, PMID: 19524042
176. Hu H, Chen Y, Tan S, Wu S, Huang Y, Fu S, et al. The research progress of antiangiogenic therapy, immune therapy and tumor microenvironment. Front Immunol. (2022) 13:802846. doi: 10.3389/fimmu.2022.802846, PMID: 35281003
177. Qin JJ, Yan L, Zhang J, and Zhang WD. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. (2019) 38:195. doi: 10.1186/s13046-019-1206-z, PMID: 31088482
178. De La Cruz P, McAdams J, Morales Aquino M, Fernandez AI, Elliott A, Lustberg M, et al. NF-κB associated markers of prognosis in early and metastatic triple negative breast cancer. Breast Cancer Res. (2024) 26:175. doi: 10.1186/s13058-024-01925-3, PMID: 39623404
179. Khan MA, Jain VK, Rizwanullah M, Ahmad J, and Jain K. PI3K/AKT/mTOR pathway inhibitors in triple-negative breast cancer: a review on drug discovery and future challenges. Drug Discov Today. (2019) 24:2181–91. doi: 10.1016/j.drudis.2019.09.001, PMID: 31520748
180. Malkov MA-O, Lee CT, and Taylor CA-O. Regulation of the hypoxia-inducible factor (HIF) by pro-inflammatory cytokines. Cells. (2021) 10(9):2340. doi: 10.3390/cells10092340, PMID: 34571989
181. Semenza GL. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim Biophys Acta (BBA) - Mol Cell Res. (2016) 1863:382–91. doi: 10.1016/j.bbamcr.2015.05.036, PMID: 26079100
182. Cai D, Wang J, Gao B, Li J, Wu F, Zou JX, et al. RORgamma is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat Commun. (2019) 10:4621. doi: 10.1038/s41467-019-12529-3, PMID: 31604910
183. Ouyang L, Zhang L, Fu L, and Liu B. A small-molecule activator induces ULK1-modulating autophagy-associated cell death in triple negative breast cancer. Autophagy. (2017) 13:777–8. doi: 10.1080/15548627.2017.1283470, PMID: 28165887
184. Kelley RK, Gane E, Assenat E, Siebler J, Galle PR, Merle P, et al. A phase 2 study of galunisertib (TGF-beta1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin Transl Gastroenterol. (2019) 10:e00056. doi: 10.14309/ctg.0000000000000056, PMID: 31295152
185. Chaves P, Garrido M, Oliver J, Perez-Ruiz E, Barragan I, and Rueda-Dominguez A. Preclinical models in head and neck squamous cell carcinoma. Br J Cancer. (2023) 128:1819–27. doi: 10.1038/s41416-023-02186-1, PMID: 36765175
186. Miserocchi G, Bocchini M, Cortesi M, Arienti C, De Vita A, Liverani C, et al. Combining preclinical tools and models to unravel tumor complexity: Jump into the next dimension. Front Immunol. (2023) 14:1171141. doi: 10.3389/fimmu.2023.1171141, PMID: 37033986
187. Tinhofer I, Braunholz D, and Klinghammer K. Preclinical models of head and neck squamous cell carcinoma for a basic understanding of cancer biology and its translation into efficient therapies. Cancers Head Neck. (2020) 5:9. doi: 10.1186/s41199-020-00056-4, PMID: 32714605
188. Ding R, Wang Y, Fan J, Tian Z, Wang S, Qin X, et al. Identification of immunosuppressive signature subtypes and prognostic risk signatures in triple-negative breast cancer. Front Oncol. (2023) 13:1108472. doi: 10.3389/fonc.2023.1108472, PMID: 37377907
189. Pham TH, Park HM, Kim J, Hong JT, and Yoon DY. Interleukin-32theta triggers cellular senescence and reduces sensitivity to doxorubicin-mediated cytotoxicity in MDA-MB-231 cells. Int J Mol Sci. (2021) 22:6342. doi: 10.3390/ijms22041893, PMID: 34067074
190. Coker-Gurkan AA-O, Ozakaltun B, Akdeniz BS, Ergen B, Obakan-Yerlikaya P, Akkoc T, et al. Proinflammatory cytokine profile is critical in autocrine GH-triggered curcumin resistance engulf by atiprimod cotreatment in MCF-7 and MDA-MB-231 breast cancer cells. Mol Biol Reports. (2020) 47(11):8797–8808. doi: 10.1007/s11033‑020‑05928‑z, PMID: 33130987
191. Zhu Y, Zhang C, Yin Q, Xu W, Luo Y, and Ou J. FOXO4 suppresses cisplatin resistance of triple-negative breast cancer by inhibiting autophagy. Am J Med Sci. (2025) 369:252–63. doi: 10.1016/j.amjms.2024.08.012, PMID: 39154963
192. Jurj A, Pop LA, Zanoaga O, Ciocan-Cârtiţă CA, Cojocneanu R, Moldovan C, et al. New insights in gene expression alteration as effect of paclitaxel drug resistance in triple negative breast cancer cells. Journal of Experimental & Clinical Cancer Research. (2020) 39:241. doi: 10.1186/s13046-020-01736-2, PMID: 32619350
193. Abuwatfa WH, Pitt WG, and Husseini GA. Scaffold-based 3D cell culture models in cancer research. J BioMed Sci. (2024) 31:7. doi: 10.1186/s12929-024-00994-y, PMID: 38221607
194. Kumar R, Iden M, Tsaih SW, Schmidt R, Ojesina AI, and Rader JS. Deciphering the divergent transcriptomic landscapes of cervical cancer cells grown in 3D and 2D cell culture systems. Front Cell Dev Biol. (2024) 12:1413882. doi: 10.3389/fcell.2024.1413882, PMID: 39193365
195. Wang Z, McWilliams-Koeppen HP, Reza H, Ostberg JR, Chen W, Wang X, et al. 3D-organoid culture supports differentiation of human CAR(+) iPSCs into highly functional CAR T cells. Cell Stem Cell. (2022) 29:515–27.e8. doi: 10.1016/j.stem.2022.02.009, PMID: 35278370
196. Deng YW, Hao WJ, Li YW, Li YX, Zhao BC, and Lu D. Hsa-miRNA-143-3p reverses multidrug resistance of triple-negative breast cancer by inhibiting the expression of its target protein cytokine-induced apoptosis inhibitor 1 in vivo. J Breast Cancer. (2018) 21:251–8. doi: 10.4048/jbc.2018.21.e40, PMID: 30275853
197. Bao G, Wang Z, Liu L, Zhang B, Song S, Wang D, et al. Targeting CXCR4/CXCL12 axis via [(177)Lu]Lu-DOTAGA.(SA.FAPi)(2) with CXCR4 antagonist in triple-negative breast cancer. Eur J Nucl Med Mol Imaging. (2024) 51:2744–57. doi: 10.1007/s00259-024-06704-y, PMID: 38587644
198. Caldeira IDS, Giovanini G, Adorno LF, Fernandes D, Ramos CR, Cruz-Visalaya SR, et al. Antiapoptotic and prometastatic roles of cytokine FAM3B in triple-negative breast cancer. Clin Breast Cancer. (2024) 24:e633–e44.e2. doi: 10.1016/j.clbc.2024.06.008, PMID: 38997857
199. Xu Y, Shan W, Luo Q, Zhang M, Huo D, Chen Y, et al. Establishment of a humanized mouse model using steady-state peripheral blood-derived hematopoietic stem and progenitor cells facilitates screening of cancer-targeted T-cell repertoires. Cancer Innov. (2024) 3:e118. doi: 10.1002/cai2.118, PMID: 38947755
200. Lee HK, Ji HJ, Shin SK, Koo J, Kim TH, Kim CW, et al. Targeting transforming growth factor-beta2 by antisense oligodeoxynucleotide accelerates T cell-mediated tumor rejection in a humanized mouse model of triple-negative breast cancer. Can Immunol Immunother. (2022) 71:2213–26. doi: 10.1007/s00262-022-03157-w, PMID: 35099588
201. Yoshida GJ. Applications of patient-derived tumor xenograft models and tumor organoids. J Hematol Oncol. (2020) 13:4. doi: 10.1186/s13045-019-0829-z, PMID: 31910904
202. Yao F, Deng Y, Zhao Y, Mei Y, Zhang Y, Liu X, et al. A targetable LIFR-NF-kappaB-LCN2 axis controls liver tumorigenesis and vulnerability to ferroptosis. Nat Commun. (2021) 12:7333. doi: 10.1038/s41467-021-27452-9, PMID: 34921145
203. Wang S, Li J, Hong S, Wang N, Xu S, Yang B, et al. Chemotherapy-elicited extracellular vesicle CXCL1 from dying cells promotes triple-negative breast cancer metastasis by activating TAM/PD-L1 signaling. J Exp Clin Cancer Res. (2024) 43:121. doi: 10.1186/s13046-024-03050-7, PMID: 38654356
Keywords: triple-negative breast cancer (TNBC), tumor microenvironment (TME), inflammation, cytokines, drug resistance
Citation: Hassan AF, Kheraldine H, Abujamous L, Al-Thawadi H and Elhissi A (2025) Inflammation-associated drug resistance and tumor growth in TNBC. Front. Immunol. 16:1623137. doi: 10.3389/fimmu.2025.1623137
Received: 06 May 2025; Accepted: 30 June 2025;
Published: 26 August 2025.
Edited by:
Remo Castro Russo, Federal University of Minas Gerais, BrazilReviewed by:
Athanasios Armakolas, National and Kapodistrian University of Athens, GreeceGuangwen Cao, Second Military Medical University, China
Copyright © 2025 Hassan, Kheraldine, Abujamous, Al-Thawadi and Elhissi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arij Fouzat Hassan, YWgxMzA0NDE4QHF1LmVkdS5xYQ==