 Siqing Chen
Siqing Chen Zhang Qin2†
Zhang Qin2† Sainan Zhou
Sainan Zhou Yin Xu
Yin Xu- 1Department of Gastroenterology, The First Hospital of Hunan University of Chinese Medicine, Changsha, Hunan, China
- 2The Fourth Hospital of Changsha (Changsha Hospital Affiliated with Hunan Normal University), Changsha, Hunan, China
Background: Ulcerative colitis (UC) is a chronic inflammatory bowel (IBD) disease characterized by a complex pathogenesis and limited treatment options. Macrophages play a key role in the pathophysiology of UC by regulating inflammatory responses and tissue repair processes. Currently, there is no comprehensive summary of macrophage regulatory pathways in UC, either domestically or internationally.
Objective: This review aims to systematically elucidate the role of macrophages in UC and their specific regulatory mechanisms, and to identify potential therapeutic strategies and future research directions.
Methods: A comprehensive literature review was conducted, integrating recent advances from global studies to explore macrophage-related pathways and functional alterations in UC. Special attention was given to studies investigating molecular mechanisms underlying macrophage polarization and function.
Results: Evidence indicates that macrophage dysfunction is a central mechanism in the pathogenesis of UC. Major findings demonstrate that metabolic reprogramming serves as a fundamental pathway inducing phenotypic and functional alterations in macrophages. Additional mechanisms mediating these changes include epigenetic modifications, chemokine-driven recruitment, microbial metabolite induction, autophagy, and apoptosis. Multiple drugs targeting macrophages have shown effectiveness in treating UC.
Conclusion: Targeting macrophage-related pathways represents an effective therapeutic approach for UC. This review provides a theoretical foundation for developing precision treatments focused on macrophage modulation and highlights important new avenues for future research.
1 Introduction
Ulcerative colitis (UC), a chronic relapsing-remitting inflammatory bowel disease (IBD), imposes a substantial global health burden with rising incidence in industrialized nations (1). Characterized by diffuse mucosal inflammation extending from the rectum to the colon, UC manifests as bloody diarrhea, abdominal pain, and systemic complications, significantly impairing patients’ quality of life (2).It can be classified into distinct subtypes based on the anatomical extent and severity of mucosal inflammation. Clinically, UC is commonly categorized into proctitis (limited to the rectum), left-sided colitis (extending up to the splenic flexure), and pancolitis (diffuse involvement of the entire colon) (3). These classifications have significant implications for both therapeutic decision-making and long-term prognosis: localized proctitis often responds well to topical therapies, whereas pancolitis typically requires systemic immunosuppression or biologic agents. Furthermore, UC can be stratified by disease activity using validated scoring systems such as the Mayo score or the simple clinical colitis activity index, which integrate parameters including stool frequency, rectal bleeding, systemic symptoms, and endoscopic findings (4, 5). Accurate classification not only guides optimal treatment strategies but also aids in predicting complications such as steroid dependency, hospitalization risk, and the long-term development of colitis-associated cancer (6). Despite advances in anti-inflammatory and immunosuppressive therapies, some patients exhibit inadequate response to conventional treatments, highlighting the unmet need for novel therapeutic strategies targeting the core pathogenic mechanisms (7). The pathophysiology of UC involves complex interactions between genetic predisposition, epithelial barrier dysfunction, dysregulated immune responses, and environmental triggers (8, 9). Within this intricate network, macrophages emerge as pivotal orchestrators of intestinal homeostasis and inflammation (10, 11). As the most abundant population of innate immune cells in the gut mucosa, macrophages demonstrate remarkable functional plasticity, dynamically shifting between pro-inflammatory (M1-like) and tissue-reparative (M2-like) phenotypes in response to microenvironmental signals (12). Emerging evidence suggests that macrophage polarization imbalance - skewed toward pro-inflammatory states during active inflammation and insufficient resolution during remission - constitutes a critical driver of UC pathogenesis (13). This functional duality positions macrophages as both instigators of tissue damage and architects of mucosal healing, creating a therapeutic paradox that demands precise regulatory interventions.
Recent breakthroughs have unveiled multidimensional regulatory mechanisms governing macrophage behavior in UC. Metabolic reprogramming, particularly shifts in glycolysis, oxidative phosphorylation (OXPHOS), amino acid metabolism, and lipid metabolism, has been identified as a master regulator of macrophage polarization (14, 15). Concurrently, epigenetic modifications including DNA methylation, histone acetylation, and non-coding RNA regulation have been shown to establish long-term activation states in gut macrophages (16, 17). Furthermore, the chemokines modulate macrophage functional differentiation through intricate crosstalk with other immune cells (18, 19). Notably, the microbiota and its metabolites engage in bidirectional communication with macrophages via pattern recognition receptors and metabolite-sensing pathways, creating a self-perpetuating cycle of dysbiosis and immune dysregulation (20, 21). Despite these advances, current literature lacks a systematic integration of macrophage-centered regulatory networks in UC. In this review, we aim to bridge this gap by synthesizing recent findings into a cohesive framework of macrophage regulation in UC. We systematically examine five major regulatory axes:1) metabolic reprogramming driving functional polarization, 2) epigenetic modifications establishing activation thresholds, 3) chemokine networks shaping microenvironmental communication, 4) macrophage apoptosis and colitis, and 5) microbiota-macrophage crosstalk maintaining mucosal homeostasis. By critically evaluating preclinical and clinical studies, we highlight promising therapeutic strategies for UC. Finally, we propose future research directions that emphasize multi-dimensional drug formulations to precisely target macrophages for UC.
2 Fundamental biological characteristics of macrophages
Macrophages, with their diverse origins, phenotypic plasticity, and functional versatility, serve as central regulators in inflammatory diseases. Understanding their fundamental biology is essential for elucidating their mechanistic roles in pathological processes. As key components of the innate immune system, macrophages not only initiate immune and inflammatory responses against pathogens but also maintain tissue homeostasis and contribute to tissue repair and remodeling (22, 23). Monocytes, the precursors of macrophages, originate from hematopoietic stem cells in the bone marrow. These monocytes circulate in the bloodstream as resident or inflammatory monocytes and, upon migrating to peripheral tissues, differentiate into tissue-resident macrophages under specific microenvironmental cues (24). Notably, most tissue-resident macrophages are embryonically derived (25), originating from the yolk sac, fetal liver, or bone marrow, whereas macrophages recruited to tissues in response to injury or infection arise from bone marrow-derived hematopoietic stem cells later in life (26). Colonic macrophages are primarily found in the colonic tissue and represent one of the largest populations of cells in the colonic microenvironment. They play a significant role in the immune response and inflammatory processes of the colon (27). Peritoneal macrophages, which originate from embryos, are the primary immune cells found in the abdominal cavity and are essential for maintaining immune homeostasis. It is important to note that the gut is part of this abdominal environment. There are distinct differences in the distribution and functional characteristics of peritoneal and colonic macrophages. These differences enable each type of macrophage to carry out unique roles under various physiological and pathological conditions. For instance, intraperitoneal injection of mitomycin C can activate peritoneal macrophages and induce colitis in rats (28). Conversely, the administration of NF-κB inhibitors via intraperitoneal delivery has been shown to improve severe colitis by facilitating the migration of drug-carrying macrophages from the peritoneal cavity to the site of inflammation (29). These indicate that the activation and migration of peritoneal macrophages are also closely related to the occurrence and development of colitis. Macrophages are ubiquitously distributed across all tissues and organs, making them indispensable to the innate immune system (23).
Macrophages are highly plastic cells capable of adopting diverse functional phenotypes in response to microenvironmental signals (30). The classical dichotomy describes two major activation states: classically activated (M1) and alternatively activated (M2) macrophages. M1 macrophages are typically induced by pro-inflammatory cytokines and exhibit potent immunostimulatory and tumoricidal activity, often contributing to tissue damage during chronic inflammation (31–33). In contrast, M2 macrophages — sometimes referred to as “wound-healing” macrophages — promote anti-inflammatory responses, tissue repair, and resistance to parasitic infections (31–33). However, the M1/M2 dichotomy is an oversimplified model (34, 35). Macrophage phenotypes exist along a continuum, with dynamic interconversion between states under specific conditions, finely regulated by metabolic reprogramming, epigenetic modifications and other pathways (36).
Functionally, macrophages play essential roles in both innate immunity and tissue homeostasis. Upon recruitment to inflammatory sites, they migrate into affected tissues, where they phagocytose cellular debris and release bioactive mediators that modulate immune responses, inflammation, and tissue repair (37, 38). Furthermore, macrophages act as antigen-presenting cells, presenting processed antigens to T cells and thereby initiating adaptive immune responses (39). They also secrete chemokines that regulate the activation and trafficking of other immune cell populations, reinforcing the crosstalk between innate and adaptive immunity (40).
An imbalance in macrophage polarization—particularly excessive M1 activation during disease flares and impaired M2-driven resolution during remission—has been implicated in the pathogenesis of multiple inflammatory disorders (41–43). Given the reversible nature of macrophage polarization, targeting their functional state represents a promising therapeutic strategy for inflammatory diseases, notably UC (26, 36) (Figure 1).
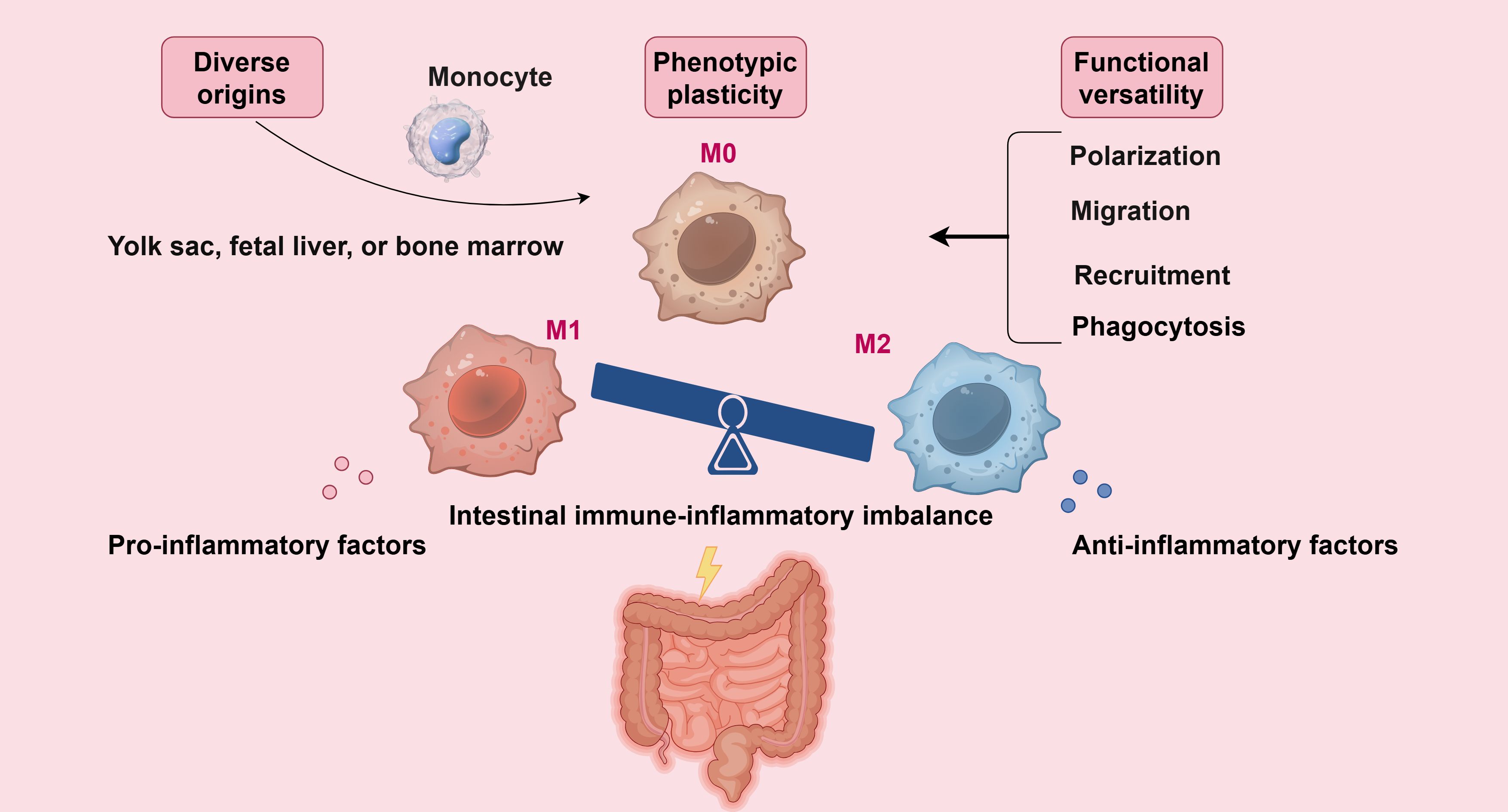
Figure 1. Macrophages originate from various sources, exhibit phenotypic plasticity, and display functional diversity, making them crucial regulators of intestinal inflammation development and progression (By Figdraw).
3 The dual role of macrophages in UC
3.1 Pro-inflammatory effects: driving intestinal inflammation and barrier disruption
Macrophages exhibit pronounced pro-inflammatory properties in the pathogenesis of UC. Upon intestinal barrier damage, pathogenic bacteria or damage-associated molecular patterns activate lamina propria macrophages via pattern recognition receptors, triggering their polarization toward the M1 phenotype. Activated M1-like macrophages (inflammatory macrophages) subsequently secrete pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF), interleukin-6 (IL-6), and interleukin-1β (IL-1β), which are critical for pathogen clearance but also exacerbate intestinal inflammation and mucosal barrier injury in UC. Spalinger et al. demonstrated that macrophage-specific PTPN2 deficiency promotes IL-6 upregulation, reducing the expression of tight junction proteins (Claudin-2, occludin) and increasing colonic permeability in mice, thereby elevating the risk of inflammatory bowel disease (IBD) (44). Intriguingly, IL-6 inhibition reversed these barrier defects and inflammatory infiltration (45), confirming the detrimental role of M1 macrophage-derived cytokines in UC progression. Further studies reveal that the toll-like receptor 2/4 (TLR2/4)-NF-κB-NOD-like receptor thermal protein domain associated protein 3 (NLRP3) axis exacerbates colitis by inducing macrophage pyroptosis and IL-1β secretion, amplifying intestinal inflammation and barrier dysfunction (46). Single-cell RNA sequencing highlights that UC patient-derived macrophages display M1-polarized signatures, with elevated CXCL9 (T-cell chemoattractant) and CD40 (T-cell activation marker), intensifying Th1/Th17-mediated immunity (47). Additionally, Tim-3–deficient macrophages recruit neutrophils and induce necroptosis, disrupting the mucosal barrier and perpetuating a vicious cycle of colitis (48).
3.2 Reparative functions: guardians of homeostasis and tissue repair
Despite their pro-inflammatory dominance in UC, macrophages also play indispensable reparative roles via M2 polarization. During inflammation resolution, macrophages transition to an M2-like phenotype (anti-inflammatory macrophages), secreting arginase-1 (Arg1) and interleukin-10 (IL-10) to promote epithelial regeneration and extracellular matrix remodeling (49–51). Notably, Arg1 from M2 macrophages provides polyamine precursors for collagen synthesis, facilitating tissue repair (52, 53). Numerous studies have demonstrated that various signaling pathways mediated by M2 macrophages reduce colonic inflammation and enhance the repair of the intestinal barrier. For instance, the activation of the M2-type macrophage/Foxo3 axis has been shown to alleviate the colonic inflammatory response and improve intestinal mucosal barrier function in mice with dextran sulfate sodium (DSS)-induced colitis (54). Additionally, the activation of the M2 macrophage/Wnt/ERK signaling pathway can significantly aid in the repair of the intestinal mucus barrier during colitis (55). Notably, the protective effects of therapeutic drugs on colitis were significantly diminished after the depletion of macrophages (56). It is believed that inhibiting macrophage apoptosis may be a crucial strategy for slowing down the progression of UC (57). Additionally, many chemokines and cytokines produced due to M2 macrophage polarization help restore the Th1/Th17 balance, which significantly alleviates colitis (58–60).
In conclusion, Macrophages in UC are a double-edged sword—both drivers of inflammation and mediators of repair. Deciphering their regulatory mechanisms unlocks therapies to amplify beneficial functions while mitigating harm, offering hope for UC treatment.
4 Regulation and operational mechanisms of macrophage function in UC
4.1 Metabolic reprogramming
4.1.1 Metabolic reprogramming and macrophage plasticity
Macrophages exhibit an extraordinary ability to adapt, known as high plasticity, which enables them to adjust their phenotypic characteristics and functions in response to subtle changes in their microenvironment. This remarkable plasticity is primarily regulated by the cell’s metabolic state (61). Metabolic reprogramming is a key process through which cells actively modify their metabolic pathways to adapt to various physiological and pathological conditions. In macrophages, metabolic reprogramming plays a crucial role in determining their activation state and effector functions; it is a core mechanism in the processes of macrophage polarization and function regulation (61). By finely regulating glucose, lipid, and amino acid metabolism, metabolic reprogramming impacts not only the energy supply of macrophages but also the efficiency of signaling and gene expression patterns (62–64). This comprehensive regulation ultimately influences the phenotypic traits and functional capabilities of macrophages. For example, an excess of classical M1 macrophages combined with a deficiency of alternative M2 macrophages can lead to severe colitis, a condition that can be significantly reversed through the modulation of metabolic reprogramming. This presents a highly innovative and practical approach for the clinical treatment of UC.
4.1.2 Role of macrophage metabolic reprogramming in colitis
Glycolysis is a metabolic pathway that converts glucose into pyruvate and lactate through a series of enzyme-mediated reactions in the cytoplasm (65). Upregulation of glycolytic rates enhances the pro-inflammatory properties of macrophages activated by pathogens (66). Numerous studies have indicated that increased glycolysis in macrophages can augment their pro-inflammatory characteristics and contribute to the development of various inflammatory diseases (67–69). The classical M1 polarization program primarily relies on glycolysis and is regulated by key glycolytic enzymes such as pyruvate kinase M2 (PKM2) and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3(PFKFB3) (66). In experiments involving colitis in mice, the expression of PFKFB3 was significantly upregulated, which coincided with an increase in glycolytic activity and a higher ratio of M1 macrophages (70). Notably, these effects were reversed and colonic inflammation was alleviated following the administration of the PFKFB3-specific inhibitor 3PO (70). HIF-1α is highly expressed in macrophages stimulated by LPS and IFN-γ, functioning as a key transcription factor that regulates the expression of glycolytic enzymes (71–73). The combined activation of LPS and IFN-γ appears to stabilize HIF-1α subunits and induce metabolic reprogramming of M1 macrophages towards glycolysis (74). Recent studies have demonstrated that the ubiquitination of HIF-1α inhibits glycolysis and promotes a balance between M1 and M2 macrophages, which could improve conditions like UC (75). PKM2 activators, such as DASA-58 and TEPP-46, can directly inhibit the expression of M1 macrophages induced by LPS while enhancing the expression of several marker proteins associated with M2 macrophages. When LPS promotes the expression of PKM2, HIF-1α, and IL-1β in macrophages, these components form a complex that inhibits the increase of glycolysis in macrophages and reduces the expression of pro-inflammatory signals (76). Further research has indicated that the dimerization of PKM2 is crucial for maintaining the stability of the HIF-1α protein; this dimerization enhances glycolysis and leads to the polarization of macrophages towards a pro-inflammatory phenotype in colitis models, which can be detrimental to recovery from colitis (77, 78). These studies have highlighted the crucial role of key enzymes in the glycolytic pathway in regulating the proportion of inflammatory macrophages and alleviating colitis. They provide a theoretical foundation for further exploration of how these enzymes impact inflammatory macrophage regulation and the management of colitis. This research not only enhances our understanding of the interactions between metabolism and immunity in the development of UC, but also suggests a potential direction for developing novel therapeutic strategies that target the glycolytic pathway. In addition, mTORC1 signaling induces macrophages to adopt a pro-inflammatory state and plays a crucial role in the development of various inflammatory diseases (79–81). Recent studies have shown that regulating glycolysis and M1 polarization facilitate the recovery from colitis by inhibiting the mTORC1/HIF-1α signaling pathway; this therapeutic effect can be counteracted by the mTORC1 agonist, l-leucine (82). IL-10 is a well-known anti-inflammatory cytokine that contributes to the treatment of many inflammatory diseases (83, 84). Research by Eddie et al. has indicated that IL-10 reduces colonic inflammation in mice with colitis by inhibiting lipopolysaccharide-induced glucose uptake and glycolysis in macrophages while promoting OXPHOS; furthermore, in macrophages from colitis mice and patients with IBD, a deficiency of IL-10 exacerbates mitochondrial damage and enhances the activation of mTORC1 signaling, leading to increased expression of NLRP3 and IL-1β (85). These studies not only highlight the role of mTORC1 signaling pathway in the process of macrophage metabolic reprogramming and polarization, but also clarify the mechanism of IL-10 as its key upstream regulator. Together, these findings create a multi-layered network that links metabolic regulation, signaling, and immune responses. This framework offers a strong theoretical foundation and significant opportunities for further research aimed at developing new therapeutic strategies for UC and other chronic inflammatory conditions. Interestingly, recent findings have revealed that M2 macrophages can also promote glycolysis, which challenges the traditional belief that glycolysis is minimal in anti-inflammatory macrophages (86). However, reducing glycolysis has not affected the activation of M2 macrophages (87). Overall, enhanced glycolysis in macrophages contributes to the development of UC. Therefore, manipulating the expression of M1 macrophages through the regulation of glycolytic reprogramming remains an important strategy for promoting recovery from colitis.
The cessation of tricarboxylic acid(TCA) cycle metabolism is another feature of M-type 1 macrophages and is closely linked to macrophage-mediated inflammatory and immune responses (88). Research has demonstrated that succinate, a byproduct of TCA cycle metabolism, accumulates at sites of inflammation and modulates immune function through signaling via the succinate receptor (SUCNR1); this receptor signaling has been shown to influence immune responses, potentially leading to intestinal inflammation and compromised intestinal barrier function (88). Patients with UC and mice with DSS-induced colitis exhibit increased expression of SUCNR1, while SUCNR1-silenced mice show significantly greater resistance to DSS-induced colitis (89). Recent studies have also found that succinate exacerbates colonic injury and the inflammatory response in DSS-induced colitis models by inhibiting the expression of Treg cells, which have anti-inflammatory properties (90). These studies reveal the important regulatory role of metabolic arrest in the TCA cycle and its key intermediate, succinate, in macrophage-mediated intestinal inflammation. When this metabolism ceases, it prevents the next steps in processing succinic acid, leading to the progression and persistence of colonic inflammation. Targeting the accumulation of succinic acid or its downstream signaling pathways offers a new strategy for treating UC.
Lipogenesis involves a series of enzymatic reactions that synthesize fatty acids and triglycerides. The metabolism of fatty acids is a significant pathway for generating pro-inflammatory effects in M1 macrophages (91). M1 macrophages synthesize fatty acids and utilize them as precursors for inflammatory mediators while primarily obtaining most of their adenosine triphosphate (ATP) from aerobic glycolysis (92). In contrast, M2 macrophages rely on a functional mitochondrial respiratory chain driven by fatty acid oxidation (FAO) to fuel the TCA cycle and subsequent OXPHOS (92). FAO is a catabolic process that breaks down fatty acids into acetyl-CoA, which is then used in the TCA cycle and OXPHOS (93). Although FAO is not exclusive to anti-inflammatory macrophages and is also present in M1 macrophages, this phenomenon does not appear to impact the pro-inflammatory effects of M1 (92, 94, 95). Numerous studies have shown that FAO in macrophages is crucial for influencing their anti-inflammatory effects (96–98). Peroxisome proliferator-activated receptors (PPARs) are a class of nuclear receptors and transcription factors that regulate gene expression; they play key roles in various processes, including lipid metabolism, energy balance, inflammatory response, and cell differentiation (99–101). In the context of FAO, the activation of PPARs affects fatty acid uptake, transport, and oxidation processes, acting as a regulatory switch in fatty acid management (102). Free fatty acid receptors (FFARs) sense changes in the concentration of intracellular and extracellular free fatty acids and regulate the rate of FAO through signaling pathways (103, 104). Research by Li et al. involving DSS-induced mice and LPS-induced RAW264.7 cells treated with the FFAR4 agonist GSK137647 found that the FFAR4-PPARα axis regulates FAO and promotes M2 macrophage polarization (105). This suggests that the FFAR4-PPARα axis may play a role in modulating M2 macrophage polarization, alleviating colitis (105). Similar studies have indicated that the symptoms of UC, colon length, and tissue damage in DSS-induced colitis mice were significantly improved through the combined intervention of the FFAR1 agonist GW9508 and the FFAR4 agonist GSK137647 (106). Moreover, the in vitro experiments of this study indicated that these improvements in colitis symptoms and histopathology have been achieved by promoting FAO, which reduces lipid accumulation and enhances M2 macrophage polarization (106). Recently, it was reported that mTORC2/PPAR-γ-mediated increases in FAO could induce M2 polarization, thereby improving colonic tissue inflammation and alleviating symptoms in colitis mice (82). These studies demonstrate the critical role of fatty acid metabolism in the polarization of macrophages and their inflammatory responses. Regulating fatty acid metabolism in macrophages can not only shift their polarization but also significantly aid in the resolution of tissue inflammation. This approach offers a promising therapeutic avenue for the treatment of UC.
The reprogramming of amino acid metabolism in macrophages also plays a significant role in the development of UC. Specifically, the regulation of amino acid metabolism directly influence the response mechanisms of macrophages; this metabolic adaptation not only supports the functional activities of macrophages but also plays a crucial role in their development, ensuring the stability of their polarization state within specific microenvironments (107). Traditionally, two metabolites of amino acids, iNOS and Arg1, are recognized as characteristic markers of anti-inflammatory and pro-inflammatory macrophages, respectively (108). This method of distinguishing macrophage phenotypes based on amino acid markers shows that the metabolic processes associated with different macrophage phenotypes are distinct, highlighting their important relationship with inflammation. Chen et al. discovered that tryptophan catabolism products significantly accumulate during the activation of M1 macrophages, as determined through metabolite set enrichment analysis (86). Tryptophan metabolism serves as a primary means of distinguishing between M1 and M2 macrophages. This study also indicated that using dual inhibitors of tryptophan catabolic enzymes enhance the expression of anti-inflammatory factors in macrophages (86). The ability to promote the expression of anti-inflammatory factors through these dual inhibitors exemplifies a typical process for reprogramming macrophages to switch their phenotype. Lu et al. demonstrated that branched-chain amino acid (BCAA) metabolite-mediated alterations in the TCA cycle modulate M2 macrophage polarization in response to specific immune responses and inflammatory conditions (109). They achieved this by inducing M2 polarization in mice using chitin and by knocking down genes for key enzymes involved in BCAA metabolism in M2 macrophages in vitro (109). Additionally, the metabolism of other amino acids, such as L-glutamine, serine, and glycine, influence the phenotype of both macrophage types by affecting the TCA cycle; this modulation subsequently impacts the inflammatory response and immune regulation of macrophages (108, 110, 111). These studies underscore the critical role of amino acid metabolic reprogramming in determining the pro- or anti-inflammatory properties of macrophages. In human primary macrophages, branched-chain aminotransferase 1 (BCAT1) is the dominant isoform of BCAT while the specific inhibitor of it is ERG240 (112). Papathanassiu et al. found that the expression of BCAT1 influences the polarization of macrophages in mice with various inflammatory diseases; by using an ERG240 inhibitor in model mice with multiple inflammatory conditions, they suggested that BCAT1 may play a role in the development of these diseases and thus regulate their progression (112). Chen et al. employed several functional enrichment algorithms to analyze tryptophan metabolism and activation patterns in different cell types in UC; they discovered that the tryptophan metabolism in macrophages plays a regulatory role in the abnormal immune response and inflammation observed in UC (21). A recent study indicated that 5-methoxytryptophan reduces M1 polarization of macrophages and alleviates colonic inflammation in mice with DSS-induced colitis (113). This findings suggest that the catabolism of 5-methoxytryptophan is crucial for transforming the macrophage phenotype in colitis mice. Hashimoto et al. reported that serum levels of D-alanine were significantly lower in patients with UC compared to healthy controls; they found that the transformation of inflammatory macrophages was inhibited, and colitis symptoms in mice were alleviated through intraperitoneal injection of D-alanine into DSS-induced colitis mice (114). The metabolic changes of D-alanine in colitis mice contribute to the remission of colitis. Zhang et al. studied macrophage-specific knockout of ring finger protein 99 (RNF99) in DSS-induced colitis mice and examined RNF99-overexpressing inflammatory macrophages in vitro; they demonstrated that RNF99 degrades TAK1-binding protein 2 (TAB2) through the lysine ubiquitination pathway, thereby regulating the inflammatory state of macrophages and promoting recovery from colitis (115). This research highlights the significance of lysine metabolic reprogramming in regulating macrophage polarization and its role in colitis development.
In summary, cellular metabolism serves as a key regulatory factor in macrophage polarization and functional modulation, and targeting these metabolic pathways may represent a promising strategy for modulating macrophages to treat colitis. Future studies should focus on optimizing such metabolic regulatory approaches and translating them into clinical applications, while maintaining a solid theoretical foundation to achieve more effective inflammation control and disease management (Figure 2).
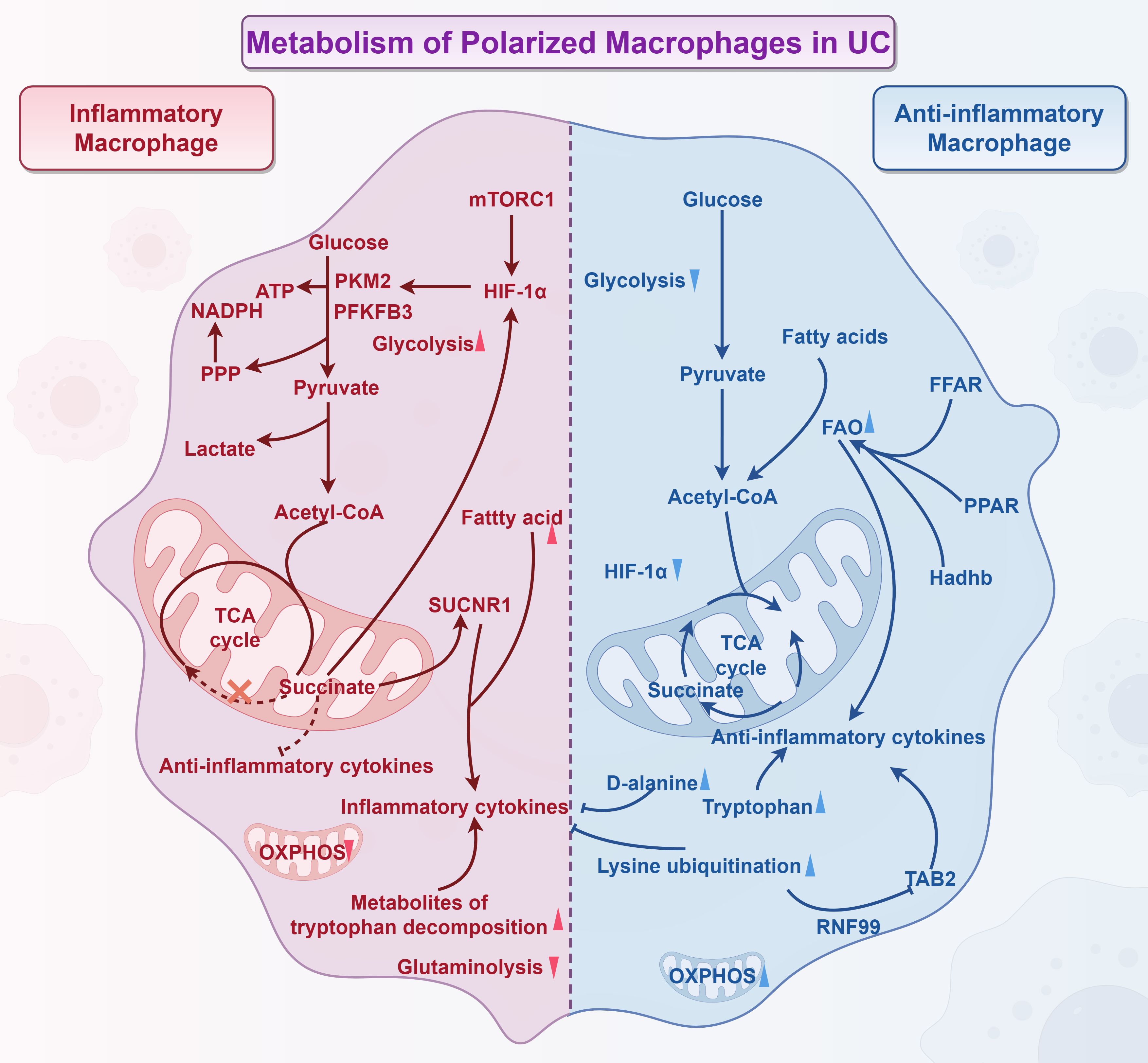
Figure 2. Metabolism of polarized macrophages in UC (By Figdraw). Inflammatory macrophage: 1) The classical polarization of inflammatory macrophages primarily relies on glycolysis, which is regulated by key enzymes such as PKM2 and PFKFB3. 2) IL-10 serves as a crucial upstream regulator of mTORC1-mediated glycolysis. The mTORC1/HIF-1α signaling pathway influences glycolysis and M1 polarization, promoting the recovery from colitis. 3) Succinate, a byproduct of TCA cycle metabolism, accumulates at sites of inflammation and regulates immune function through SUCNR1 signaling. This accumulation leads to intestinal inflammation and damages the intestinal barrier. Succinate also modulates HIF-1α expression, which indirectly affects macrophage glycolysis levels. Furthermore, succinate inhibits the expression of regulatory T cells, which have anti-inflammatory properties, thereby exacerbating intestinal inflammation. 4) Fatty acids serve as precursors for the synthesis of inflammatory mediators in M1 macrophages. 5) The accumulation of tryptophan metabolites, along with decreased L-glutamine catabolism, encourages macrophages to transition to the M1 phenotype. Anti-inflammatory macrophage: Anti-inflammatory macrophages utilize FAO to drive the functional mitochondrial respiratory chain, which powers the TCA cycle and subsequent OXPHOS. FAO plays a crucial role in enhancing the anti-inflammatory effects of M2 macrophages. The expression of proteins such as FFAR, PPAR, and Hadhb all promote FAO, contributing to the improvement of colitis. Additionally, the ubiquitination of lysine along with the accumulation of D-alanine and tryptophan regulate macrophage polarization and reduce inflammation in macrophages.
4.2 Beyond metabolism
4.2.1 Macrophage epigenetic regulation and colitis
Cellular epigenetics refers to the chemical modifications that regulate gene expression without altering the DNA sequence (116). These modifications impact gene expression by changing the structure of DNA and proteins on chromosomes, and they are both heritable and reversible (117, 118). The biological characteristics of macrophages are closely linked to their epigenetic regulatory networks. The epigenetic regulation mechanisms in macrophages involve various molecular modifications, including DNA methylation, histone post-translational modifications, and the regulation of non-coding RNAs; these epigenetic factors play a significant role in regulating macrophage phenotypic plasticity (119). Research conducted by Solier et al. identified a chemically active pool of copper ions in the mitochondria of inflammatory macrophages, which regulates the redox cycle of NAD by catalyzing the activation of hydrogen peroxide; this unique metabolic mechanism involving metal ions drives the macrophage epigenetic regulatory network toward a pro-inflammatory phenotype by maintaining intracellular NAD dynamics (120). Furthermore, studies have shown that the histone H3K9 acetylation and H3K18 lactate modifications promote a shift of M1 macrophages towards an anti-inflammatory phenotype (121, 122). It is important to note that the role of lactate—as a product of macrophage glycolysis—and glycolysis itself in the inflammatory response is complex and multifaceted. There exists a mutually reinforcing relationship between these processes, as well as a balance mechanism of mutual inhibition. This intricate interaction is essential for the body’s ability to maintain homeostasis and adaptability in response to pathophysiological challenges such as inflammation. Histone methyltransferases are a class of enzymes that catalyze the transfer of methyl groups to histones; these modifications are crucial for regulating gene expression (123). Histone H3 is a core histone in chromatin, which, along with other histones such as H2A, H2B, and H4, forms the basic structure of chromatin (124–126). SET1 is a histone methyltransferase that specifically methylates lysine 4 on histone H3 (127). Megakaryoblastic Leukemia 1 (MKL1) influences cellular behavior by recruiting SET1 to the promoter region of P65 target genes, thereby initiating pro-inflammatory transcription in macrophages (128). An et al. found that mice deficient in MKL1 were less sensitive to DSS-induced colitis; they observed that macrophages from transgenic mice expressing human MKL1 were more sensitive to DSS-induced colitis than those from normal control mice (129). In these transgenic mice, the number of macrophages in the lamina propria was decreased, and most of them exhibited a pro-inflammatory phenotype (129). The regulatory effect of MKL1 on macrophage phenotype in colitis could be attributed to epigenetic modifications mediated by MKL1, which require further experimental validation. Jumonji domain-containing 3 (JMJD3) is an important histone demethylase that regulates epigenetic modifications in macrophages, thus influencing macrophage phenotypes (130). It has been implicated in various immune-inflammatory diseases (130–132). Recent study has shown that the inhibition the JMJD3-NLRP3 signaling axis alter the trimethylation of histone H3 on lysine 27 (H3K27me3) in macrophages, which alleviates the colonic inflammatory state in DSS-induced colitis mice (133). In a study conducted by Duan et al., the JMJD3-specific inhibitor GSK-J4 was used to intervene in mice with DSS-induced colitis and in inflammatory macrophages. The researchers found that this intervention significantly improved the inflammatory state and alleviated colonic pathological changes. These enhancements resulted from the JMJD3 manipulation of H3K27me3 in macrophages, which led to a higher proportion of anti-inflammatory macrophages and a decreased expression of pro-inflammatory factors (133). Epigenetic modifications in macrophages play a crucial role in regulating their phenotypic responses, offering novel insights into the pathogenesis of UC. Targeting key enzymes involved in these epigenetic processes has been shown to influence macrophage polarization, dampen excessive inflammatory activity, and support tissue homeostasis. A deeper understanding of how these modifications shape macrophage function may guide the identification of more precise therapeutic targets. Further investigation into the underlying mechanisms could therefore contribute to the development of innovative interventions for UC.
4.2.2 Chemokine-mediated macrophage response and colitis
Chemokines, also called chemotactic cytokines, are small signaling proteins secreted by cells (134). They are named for their ability to induce directional chemotaxis, guiding the movement of nearby responding cells (135, 136). During the inflammatory response, chemokines play a crucial role by directing the migration and proliferation of inflammatory cells, including macrophages; this participation is vital for chemotaxis, cell aggregation, and the activation of various immune cells, which can significantly contribute to the development of inflammatory diseases and the rapid formation of an immune-inflammatory response (18). Chemerin is a chemotactic protein that plays an essential role in the inflammatory response by recruiting macrophages to the site of inflammation, primarily through its interaction with the macrophage-expressed chemerin receptor 1 (CMKLR1) (137). A study by Dander et al. on DSS-induced colitis in CMKLR1 knockout mice found that the deletion of this chemokine receptor increased the mice’s sensitivity to DSS, resulting in more severe colonic pathology (138). This research highlights the importance of immune cell surface chemokine activity in the development of colitis. Additionally, it has been reported that the chemokine PC3-secreted microprotein (PSMP) is expressed in the colon tissues of patients with colitis (139). Moreover, the expression of this chemokine in colon tissues was found to be up-regulated in the early stages of DSS-induced colitis (139). Researchers discovered that intervening in mice with DSS-induced colitis using adenoviral methods or PSMP-neutralizing antibodies resulted in PSMP promoting the expression of multiple inflammatory factors during the early stages of colitis, furthermore, this intervention prompted the conversion of macrophage phenotypes into pro-inflammatory macrophages (139). Xue et al. conducted a study on DSS-induced colitis in mice and LPS-induced inflammation in RAW264.7 cells, finding that reducing the migration of macrophages induced by C-C motif chemokine ligand 2 (CCL2), specifically by decreasing both migration distance and velocity, can help alleviate colitis (140). Chemokines produced by macrophages also play a significant role in influencing colitis by impacting other immune cells. Research conducted by Jones et al. on patients with IBD and mice with DSS-induced colitis demonstrated that macrophages producing the chemokines C-C motif chemokine ligand 7 and C-C motif chemokine ligand 8 recruit more monocytes from the bloodstream into colon tissue; these recruited monocytes serve as a major source of IL-1β and TNF in the inflamed tissue (19). This study reveals a harmful cycle of inflammation in colonic disorders:monocytes are attracted to the site of inflammation, where they release pro-inflammatory factors that worsen the condition; at the same time, macrophages release chemokines to draw in more monocytes, further perpetuating and intensifying the inflammation. Overall, chemokines are closely linked with macrophages as small-molecular-weight cytokines or signaling proteins. Regulating these chemokines could be a strategic approach to limit UC by promoting the migration of immune cells, including macrophages, to areas of colonic inflammation.
4.2.3 Microbial metabolism-mediated macrophage polarization and colitis
Gut microbiota and their metabolites play a crucial role in regulating the immune system (141). By understanding how microbial metabolism products affect the differentiation and function of immune cells, we can gain insights into the pathogenesis of various systemic diseases and develop new strategies for their prevention and treatment. Macrophages are crucial immune cells that sense environmental changes and play a role in regulating immunity of various diseases (142, 143). The gut microbiota influences macrophage characteristics through microbial signals, and this interaction is crucial in the development of various inflammatory conditions, particularly UC (144–146). Short-chain fatty acids (SCFAs) are organic fatty acids containing 1 to 6 carbon atoms, and they are the main end products of anaerobic bacteria in the colon (147). In patients with UC, there is a significantly low abundance of anaerobic bacteria that produce SCFAs (54). Research has demonstrated that a mixture of supernatants containing seven SCFAs promotes an anti-inflammatory macrophage phenotype in the colon by inhibiting the Janus Kinase-Forkhead box O3 signaling pathway, which leads to improved intestinal inflammation and enhanced barrier function in mice with DSS-induced colitis (54). Butyrate, one of the SCFAs produced by gut microbes during metabolism, has also been studied for its effects on macrophages. A recent study using clodronate liposomes to deplete macrophages found that butyrate increases mucus production and the proportion of mucus-secreting goblet cells in the colonic crypts in a macrophage-dependent manner (55). Researchers demonstrated both in vivo and in vitro that butyrate stimulates the polarization of macrophages toward the M2 phenotype, which, in turn, promotes goblet cell formation and aids in repairing the mucus barrier, especially following DSS-induced injury (55). Butyrate has protective effects in UC by promoting the polarization of macrophages toward an anti-inflammatory phenotype, enhancing goblet cell function, and aiding in mucus barrier repair. This finding uncovers a novel interaction mechanism between macrophages and goblet cells and suggests that butyrate is a potential therapeutic target for UC. Interestingly, individuals with gastrointestinal motility disorders are more prone to developing colonic inflammation (148). The accumulation of linoleic acid caused by intestinal flora imbalance in patients with gastrointestinal motility disorder leads to the recruitment of pro-inflammatory macrophages, which is susceptible to colitis (149). Additionally, polyamines produced by the gut microbiota, such as putrescine and spermidine, play a vital role in maintaining intestinal health (150, 151). These polyamines enhance the proliferation of colonic epithelial cells, regulate macrophage differentiation, and alleviate colitis symptoms (152). It has also been reported that putrescine produced by bacteria acts as a substrate for symbiotic metabolism and increases the number of anti-inflammatory macrophages in the colon to help alleviate colonic tissue inflammation and maintain intestinal mucosal homeostasis (152). These findings highlight the crucial role of microbial metabolism in mediating macrophage responses that support host intestinal health and offer new perspectives for treating intestinal inflammation.
4.2.4 Macrophage apoptosis and autophagy in colitis
Apoptosis refers to the process of cell autonomous and orderly death controlled by genes to maintain the stability of the internal environment (44). It has complex molecular biological mechanisms and important biological significance, it plays an important role in the growth and development of organisms, the stability of the internal environment and the resistance to external factors (153). The disorder of macrophage apoptosis process is closely related to the occurrence of a variety of inflammation-related diseases (154, 155). Therefore, it is of great significance to study the mechanism and regulation of apoptosis for the treatment and prevention of diseases. In addition to regulating macrophage polarization and chemotaxis, inhibiting macrophage apoptosis is another strategy for managing UC. Cai et al. conducted a study on mice with DSS-induced colitis and inflammatory macrophages; they found that intervention with exosomes that highly express miR-378a-5p inhibits the activation of the NLRP3 inflammasome and the secretion of IL-18 and IL-1β (57). Additionally, this intervention reduced the cleavage of Caspase-1, thereby inhibiting the occurrence of pyroptosis and alleviating DSS-induced colitis (57). This discovery offers new insights and potential drug targets for treating IBD. Liu et al. conducted a study on mice with colitis induced by DSS and on bone marrow-derived macrophage models triggered by LPS; they discovered that salidroside inhibits the activation of the NLRP3 inflammasome by blocking the triggering receptor expressed on myeloid cells 1; this inhibition leads to a reduction in macrophage pyroptosis and decreases the release of the inflammatory factor IL-1β (156). This study highlights the promising potential of drugs designed to improve colitis by regulating macrophage autophagy. Farnesoid X receptor (FXR) is a nuclear receptor, mainly expressed in the liver and intestine, involved in the regulation of bile acid metabolism, inflammatory response and intestinal barrier function, and its activation has anti-inflammatory effects (157, 158). Recently, Yang et al. found that the supernatant from saccharomyces boulardii (SB) activates FXR, thus inhibiting NLRP3 expression and preventing macrophage apoptosis in DSS-induced colitis mice, which significantly improves the symptoms of colitis. Additionally, in vitro experiments showed that the effect of SB is reversed by the FXR inhibitor Guggulsterone, which increases levels of apoptosis-related proteins (159). This underscores the importance of macrophage apoptosis in protecting against colitis. While SB is beneficial, other strains worsen UC by regulating autophagy, with the specific mechanisms still under investigation. In Shen et al.’s study, peptostreptococcus anaerobius aggravated DSS-induced colitis in mice by inducing macrophage pyroptosis and IL-1β secretion through the activation of the TLR2/4-NF-κB-NLRP3 signaling axis (46). These studies illustrate that the distorted macrophage apoptotic response is one of the intrinsic mechanisms of intestinal dysbiosis leading to colitis in patients with UC.
Overall, in UC, defective and insufficient apoptosis prolongs and amplifies the inflammatory response. Meanwhile, the role of autophagy in the pathogenesis of UC has attracted considerable attention in recent years. Impaired autophagy in macrophages compromises their ability to clear pathogens and regulate inflammation, thereby exacerbating the disease (160, 161). Autophagy is a crucial mechanism for maintaining cellular homeostasis, playing an important role in immune regulation in inflammatory bowel disease by degrading damaged organelles, misfolded proteins, and intracellular pathogens (162). In the context of UC, dysfunctional autophagy resulting from genetic polymorphisms in macrophage autophagy-related genes is closely associated with disease onset and progression (163). Altered expression of autophagy-related genes can impair the formation of autophagosomes, thereby reducing the capacity of macrophages to eliminate invasive Escherichia coli; this leads to persistent intestinal inflammation and increases susceptibility to chronic inflammatory responses (164, 165).
At the molecular level, macrophage autophagy contributes to the pathology of UC through multiple mechanisms (161). Autophagy serves as a key regulator of the NLRP3 inflammasome. An intact autophagic pathway facilitates the clearance of inflammasome activators such as damaged mitochondria, suppresses caspase-1 activation, and reduces the production of pro-inflammatory cytokines including IL-1β and IL-18, thereby preventing excessive inflammation (166). Conversely, impaired autophagy leads to hyperactivation of the NLRP3 inflammasome and aggravates intestinal inflammation. Besides, there is a dynamic interaction between macrophage autophagy and the gut microbiota (167). On one hand, the intestinal microbial community regulates macrophage autophagy activity via metabolites and pattern recognition receptor signaling; on the other hand, macrophages help maintain microbial homeostasis and eliminate invading pathogens through autophagy (168–170). In UC, this balance is disrupted, creating a vicious cycle of dysbiosis and autophagy dysfunction that drives disease progression.
Notably, macrophage autophagy is also involved in UC pathogenesis through the modulation of immune responses. It influences antigen presentation and helps maintain immune homeostasis by removing inflammatory signaling molecules, such as reactive oxygen species (ROS) and inflammasome components (171–173). Macrophages with defective autophagy exhibit increased production of pro-inflammatory cytokines and impaired anti-inflammatory functions, promoting a chronic inflammatory state in UC (161).
Abnormalities in macrophage apoptosis and autophagy silently contribute to the initiation and progression of UC. Further investigation into the specific mechanisms and targets underlying dysregulated macrophage apoptosis and autophagy represents a promising therapeutic strategy for colitis. Incorporating the regulation of macrophage apoptosis and autophagy into UC treatment research enhances conventional immunomodulatory approaches. Moreover, this exploration offers new perspectives for the future development of precision immunotherapies focused on modulating cell fate (Figure 3).
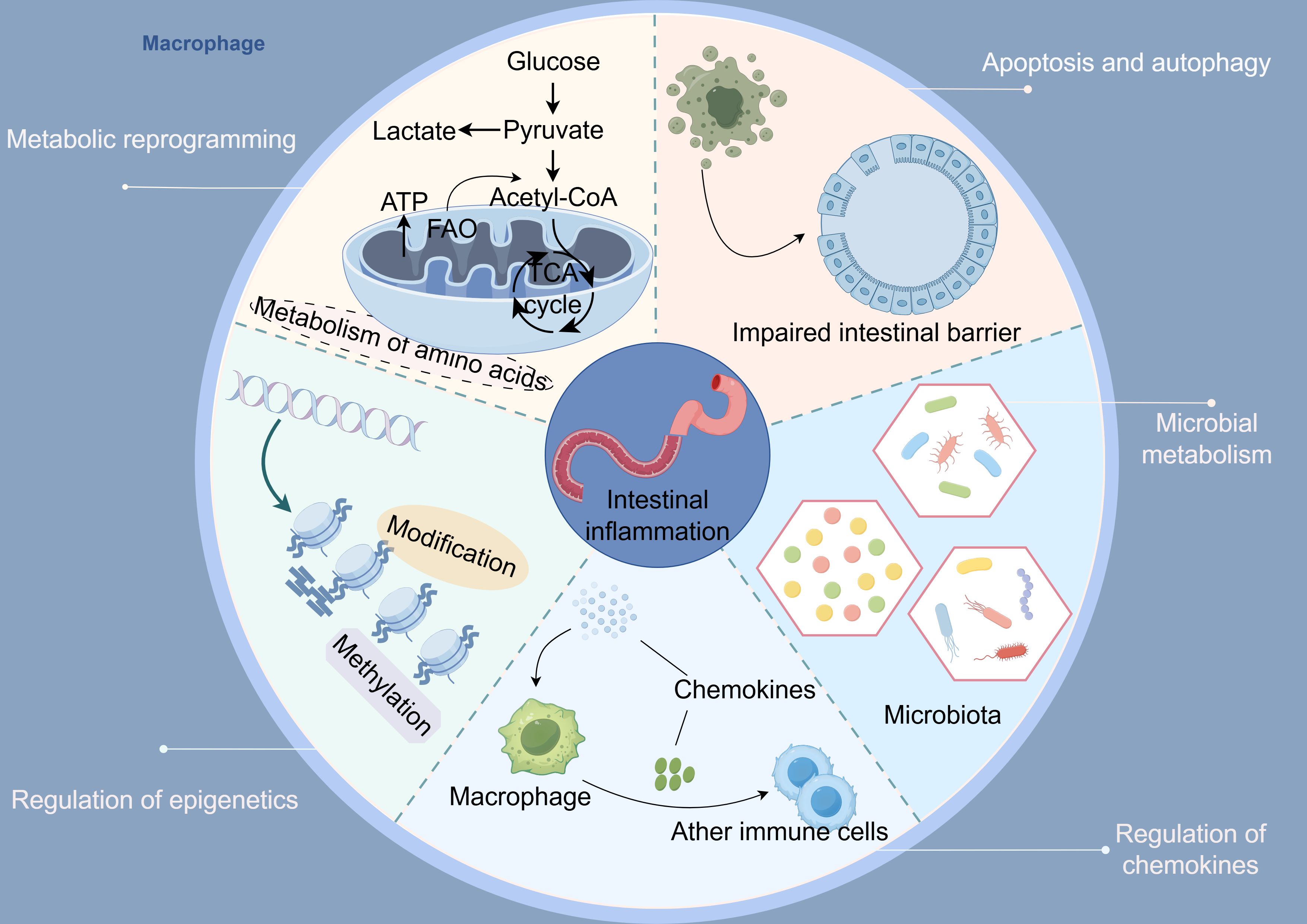
Figure 3. Macrophages regulate five key axes involved in the pathogenesis of ulcerative colitis: 1) metabolic reprogramming, 2) epigenetics, 3) chemokine networks, 4) apoptosis and autophagy, and 5) microbes along with their metabolites (By Figdraw).
5 Targeting macrophages for UC therapy
Intestinal inflammation and immune imbalance represent core pathogenic mechanisms underlying the onset and progression of UC, with macrophages serving as pivotal mediators in this complex interplay. As key innate immune cells residing in the intestinal lamina propria, macrophages exhibit remarkable functional plasticity, dynamically switching between pro-inflammatory and anti-inflammatory phenotypes in response to environmental cues. Dysregulated macrophage activation contributes significantly to mucosal damage, epithelial barrier disruption, and persistent immune activation in UC. Consequently, modulating macrophage polarization, infiltration, and cytokine production has emerged as a promising therapeutic strategy. Recent advances in immunology and pharmacology have led to the identification of multiple targeted agents that regulate macrophage function in UC.
These include:
Nanoformulations: Engineered nanoparticles capable of delivering drugs specifically to macrophages, thereby enhancing therapeutic efficacy while minimizing systemic side effects.
Natural or Synthetic Compounds: Compounds such as a-aminobutyric acid (AABA), xylan acetate (XylA), and genistein that modulate macrophage signaling to improve the balance of macrophage polarization.
Microbial Preparations:Probiotics, specifically Akkermansia muciniphila (A. muciniphila) and Lactobacillus johnsonii (L. johnsonii), have an impact on macrophage behavior through interactions between the host and the microbes.
Extracts and compounds of traditional Chinese medicine (TCM): Extracts and compounds of TCM that exert anti-inflammatory effects by regulating macrophage polarization and cytokine secretion.
These strategies offer novel insights into precision immunomodulation in UC and highlight the potential of macrophage-targeted therapies to restore immune homeostasis and promote mucosal healing (Table 1).
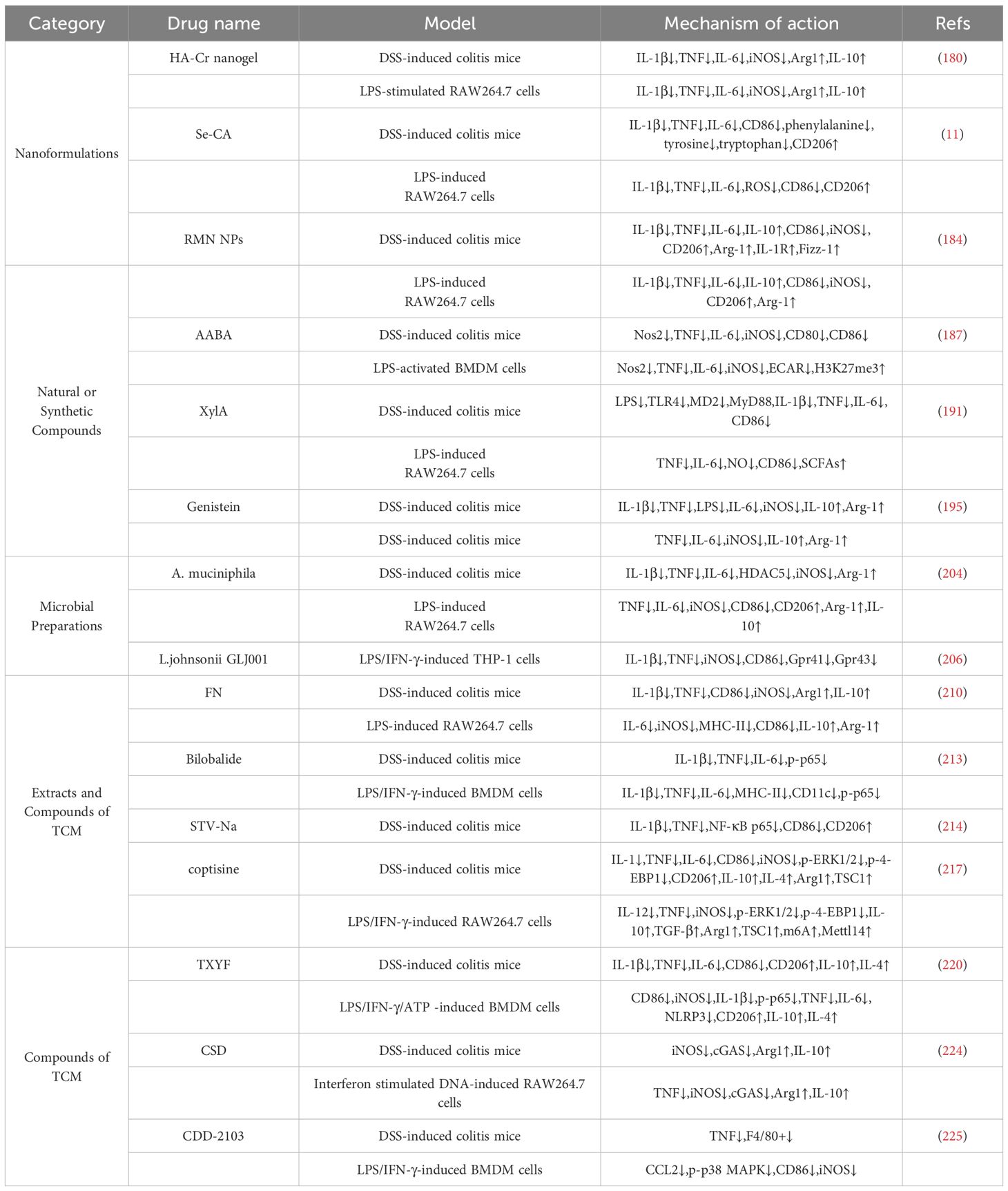
Table 1. Summary of potential targeted macrophages therapeutics for UC.
5.1 Nanoformulations
Nanoformulations have demonstrated significant potential in the treatment of UC. They utilize nanocarriers, such as liposomes and polymer nanoparticles, to achieve targeted delivery and controlled release of drugs (174). This approach enhances the local concentration of medications in the inflamed intestine, improve drug delivery efficiency, and reduce systemic exposure and side effects (174, 175). The advantages of nanoformulations include enhanced drug stability, improved bioavailability, prolonged retention time of the drug, and the ability to actively target inflammatory sites through surface modification (176–178). These features help regulate the intestinal immune-inflammatory response, making nanoformulations an effective and precise strategy for UC therapy. Creatine (Cr), a nitrogen-containing organic acid that provides energy to cells, has shown potential benefits in treating UC. Research indicates that oral administration of Cr helps prevent the development of the disease (179). Huai et al. combined Cr with nanomaterials to create a Cr-modified selenium-based hyaluronic acid nanogel, referred to as HA-Cr Nanogel; this formulation exploits the CD44 targeting ability of hyaluronic acid and its positive charge at the site of inflammation for precise delivery (180). The researchers found that HA-Cr nanogel was not only more effective than Cr alone but also more specifically targeted inflammation in mice with DSS-induced colitis and in Raw 264.7 cells (180). Recently, Li et al. used chitosan (CS) as the backbone and covalently modified L-arginine (L-Arg) onto CS through an EDC/NHS catalytic system to form a polymeric complex known as CS-L-Arg (CA); they then prepared a modified selenium nanozyme, referred to as Se-CA, through a chemical reduction method (11). In both in vitro and in vivo studies, they validated the anti-UC properties of Se-CA, finding that Se-CA significantly improves colonic pathology in colitis mice by enhancing epithelial repair, reducing inflammation, reversing macrophage type 1 responses, and decreasing reactive oxygen species (ROS) levels (11). Importantly, long-term use of Se-CA was found safe (11). Additionally, the PPAR gamma agonist Rosiglitazone (RLZ) is believed to have therapeutic effects in various inflammatory diseases by inducing macrophages to switch to the M2 phenotype and secrete multiple anti-inflammatory factors (181–183). Similar studies have utilized nanoparticles coated with macrophage membranes (from RAW264.7 cells) and loaded with Rosiglitazone (RLZ) form RMN NPs; these nanoparticles effectively modulate the M1 to M2 macrophage ratio and protect intestinal epithelial cells in vitro; in vivo, they significantly improve pathological changes in the colon, reduce levels of inflammatory factors in DSS mice, and restore the intestinal barrier function (184). Compared to nanoparticles or free drugs alone, RMN NPs specifically target inflamed intestinal tissue, producing superior therapeutic effects at the lesion site (184). In conclusion, although these nanomedicines show promise in treating UC, their potential application is still unclear. Further research and improvements are needed regarding large-scale production, long-term safety, targeting efficiency, individualized treatment, and stability. There is a new expectation for better utilization of nanomaterials to regulate macrophage function in colon tissue for the treatment of this refractory disease.
5.2 Natural or synthetic compounds
AABA is a non-protein amino acid that can be metabolized from various neutral amino acids, previously thought to be linked to metabolic sexual dysfunction (185, 186). Li et al. proposed that AABA regulates macrophage polarization and function in UC, providing a new foundation for targeting amino acid metabolism to modulate macrophage function for UC treatment (187). The researchers examined DSS-induced colitis in mice and LPS-induced inflammatory macrophages, findings that AABA reduces disease severity in colitis-affected mice through metabolic reprogramming and epigenetic modification of M1 macrophages (187). Specifically, AABA enhances the amino acid metabolism in macrophages, increases H3K27 trimethylation in the promoter region of M1 macrophage-associated inflammatory genes, and inhibits the glycolytic process of cells (187). These results suggest that AABA plays a crucial role in macrophage development, significantly reducing inflammation and excessive immune responses in the colon. Xylan and its derivatives, as insoluble polysaccharides that are difficult to digest in the stomach and duodenum after oral administration, play a crucial role in re-establishing intestinal homeostasis (188). For instance, xylan butyrate ester and wheat bran arabinoxylan are potent compounds that help regulate the balance between intestinal inflammation and immunity, promoting recovery from UC (189, 190). Interestingly, a recent study by Tang et al. demonstrated that XylA, a derivative of xylan, significantly alleviates the symptoms and pathological manifestations of UC; they found that in both DSS-induced colitis in mice and LPS-induced inflammation in RAW264 cells, XylA significantly reduces the polarization of M1 macrophages in the inflammatory colon and decreases the expression of various pro-inflammatory factors in an in vitro inflammatory environment (191). In addition, these effects are closely linked to the short-chain fatty acids (SCFAs) derived from XylA. Genistein is a significant active compound derived from soybean, known for its wide range of biological functions, including regulating the cell cycle, anti-inflammatory effects, antioxidant properties, and promoting autophagy (192). These attributes make it valuable in treating various diseases, such as diabetes, lipid disorders, and acute pancreatitis (193, 194). Recently, Jia et al. demonstrated that Genisteind enhances symptom recovery and improves colonic histomorphology in mice with DSS-induced colitis (195). It was also found to reduce the level of inflammatory cell infiltration in the affected colon (195). This therapeutic effect primarily stems from Genistein’s ability to regulate macrophage phenotypes, leading to a decrease in pro-inflammatory macrophages and an increase in anti-inflammatory macrophages in diseased mice. Consequently, Genistein is regarded as a promising macrophage-targeted therapeutic agent for UC.
5.3 Microbial preparations
Intestinal epithelial cells are constantly exposed to a complex microenvironment composed of various immune cells and immeasurable microorganisms, which has a tremendous impact on the normal function of the intestinal barrier (196). Multiple studies have shown that microbial agents and their metabolites are able to modulate beneficial effects on the host by improving gut microbial balance, enhancing intestinal barrier function, and improving local inflammatory-immune responses (197, 198). Unfortunately, in recent years, there have not been many studies on the treatment of UC using microbial agents that target macrophages. Given the importance of macrophage polarization balance in the pathogenesis of UC, it is important to continue to explore the mechanism of various microbial agents-mediated macrophage treatment of UC. A. muciniphila regulates macrophage polarization state to participate in the occurrence and development of various diseases including hepatocellular carcinoma and intestinal infectious diseases (199, 200). Histone deacetylase 5 (HDAC5) belongs to the HDAC family and is able to alter chromatin structure and function by removing acetyl groups on histone lysine residues, thereby repressing gene transcription (201). It can also interact with specific transcription factors and be recruited to specific gene promoter regions to achieve fine regulation of specific gene expression (202). Recently, it was reported that the abundance of A.muciniphila is significantly decreased in UC, which may be related to the development of UC (203). Miao et al. demonstrated through their study of DSS-induced colitis in mice and inflammatory macrophages that A. muciniphila can reduce the expression of HDAC5; this reduction prevents the H3K9 acetylation modification of the promoter for the HDAC5 deacetylation-disabled homolog 2 gene, resulting in the suppression of this gene’s expression no longer occurring (204). Consequently, this mechanism leads to a reduction in pro-inflammatory macrophage polarization in the colonic tissue. The study delves deeper into the role of A. muciniphila-mediated epigenetic modification in the inflammatory phenotype of macrophages in UC and enriches the existing targets for macrophage phenotype regulation, which has contributed to the expansion of potential macrophage-targeting drugs for UC. Jia et al. conducted a study on L. johnsonii to explore its relationship with pathogenicity in mice with DSS-induced colitis (205). Both in vivo and in vitro studies demonstrated that L. johnsonii promotes an increase in the ratio of intestinal M2 macrophages by activating the STAT3 signaling pathway; this process enhances the expression of anti-inflammatory factors, effectively controlling intestinal inflammation (205). Recently, Cai et al. demonstrated that L. johnsonii GLJ001, which becomes enriched in rat feces following intragastric administration of Pu-erh tea, plays a crucial role in restoring the macrophage-mediated inflammatory-immune balance in colitis (206). Specifically, their research, involving DSS-induced colitis in mice and inflammatory THP-1 cells, revealed that L. johnsonii GLJ001 influences the levels of SCFAs, which are closely linked to macrophage polarization, by improving gut microbiota balance in colitis-affected mice (206) Furthermore, the use of SCFAs to intervene in an in vitro inflammatory macrophage model reduces inflammation and significantly decreases the levels of GPR41 and GPR43, which play a role in immune and inflammatory responses (206). Therefore, L. johnsonii GLJ001 presents as a potential therapeutic agent to promote recovery from UC by modulating macrophage polarization through the gut microbiota/SCFAs axis.
5.4 Extracts and compounds of TCM
Formononetin (FN) is an isoflavone that plays a significant role in maintaining the balance between inflammation and immunity, and it is the primary active compound found in Pueraria montana and astragalus (207–209). A study by Xiao et al. demonstrated that FN improves symptoms, reduces colonic lesions, and decreases mortality in mice with DSS-induced colitis (209). Importantly, both in vivo and in vitro studies have shown that FN alleviates UC by promoting the transformation of inflammatory colonic tissues from pro-inflammatory to anti-inflammatory macrophages; this transformation may be crucial in the pathogenesis of UC (210). However, the efficacy of FN can be diminished by the depletion of macrophages using clodronate liposomes (210). Bilobalide is a natural active ingredient extracted from ginkgo biloba leaves and is widely researched for its potential in treating neurological disorders and inflammatory diseases (211, 212). Zhang et al. discovered that bilobalide significantly improves colon shortening and damage while reducing inflammation in mice with DSS-induced colitis (212). In vitro studies on inflammatory macrophages showed that bilobalide decreases the expression of inflammatory phenotype markers such as MHC-II and CD11c, as well as multiple inflammatory factors produced by these macrophages; this therapeutic effect is attributed to the modulation of the NF-κB pathway (213). A similar study involving mice with DSS-induced colitis highlighted isosteviol sodium (STV-Na), a terpenoid sodium salt derived from the acid hydrolysis of stevioside; this compound may also ameliorate UC by inhibiting NF-κB p65 signaling, thereby reducing M1 macrophage polarization (214). Various TCM extracts have shown promise in alleviating colonic inflammation and improving symptoms of UC through NF-κB/p65 signaling-mediated M1 macrophage polarization and this research provides valuable insights for further exploring the mechanisms of other TCM extracts that have therapeutic effects on UC. Coptisine is an active ingredient extracted from Coptis chinensis, known for its ability to treat various inflammatory diseases, including IBD and psoriasis (215, 216). A recent study has shown that Coptisine regulates the epigenetics of macrophages, which helps reduce the number of inflammatory macrophages in the colon, ultimately aiding in the treatment of UC (217). Researchers conducted experiments on mice with DSS-induced colitis and found that Coptisine improves colonic pathology and symptoms while altering the polarization balance of macrophages in the colonic tissue to enhance the anti-inflammatory response (217). Further in vivo and in vitro studies revealed that Coptisine regulates the ERK pathways by methylating TSC1, which mediates the polarization balance of macrophages and contributes to the improvement of UC (217). Epigenetics involves modifications that change the expression of target proteins without altering the gene sequence. Therefore, changing the epigenetics of macrophages is a significant method for regulating their function. In summary, the outstanding potential of TCM extracts in regulating macrophage function for UC treatment is highlighted.
5.5 Compounds of TCM
Compounds of TCM are composed of various herbs that work together in a coordinated manner, following the hierarchical roles of the sovereign, the minister, the assistant, and the courier within the theoretical framework of TCM. Each herb in the formulation interacts with the others to achieve therapeutic effects that may be difficult to attain with a single herb alone. Tongxie-yaofang (TXYF) is a safe and effective compound of TCM used for treating UC, although its precise therapeutic mechanism remains unclear (218, 219). Research conducted by Zhang et al. demonstrated that TXYF restores the polarization balance of M1 and M2 macrophages, while also improving both the pathological changes in colon tissues and the symptoms of UC in mice with colitis (220). Furthermore, their study found that TXYF reverses inflammation induced by LPS and IFN-γ in macrophages in vitro, a process linked to the NF-κB-mediated activation of NLRP3 (220). This research provides valuable insights into the therapeutic mechanism of TXYF and offers a theoretical basis for the clinical application of TXYF-targeted macrophage therapy in the treatment of UC. Compound sophorae decoction (CSD) is commonly used to treat UC, particularly in improving patients’ blood stool conditions (221). Many studies have demonstrated that CSD intervenes in the occurrence and progression of UC through multiple targets and pathways (222, 223). However, there are few studies specifically addressing its effects on macrophages. Wu et al. discovered that, in DSS-induced colitis mice, the expression of pro-inflammatory factors TNF and IL-6 was significantly down-regulated, while the level of the anti-inflammatory factor IL-10 was significantly up-regulated in colon tissue (221). These changes may result from phenotypic shifts in macrophages. They further supported this hypothesis by examining macrophage phenotype changes in the spleen and mesenteric lymph nodes of the mice. Unfortunately, they did not conclusively prove whether the changes in inflammatory factors were solely due to the shifts in macrophage phenotype. Fei et al. recently confirmed that CSD targets macrophages in the treatment of UC using a combination of in vivo and in vitro experiments along with a network pharmacology approach (224). Specifically, they identified hundreds of targets associated with CSD that have therapeutic effects on UC, including proteins such as inducible nitric oxide synthase (iNOS) and TNF, which are classic products of macrophage polarization (224). Subsequently, in vivo and in vitro experiments confirmed that CSD alleviated UC via cyclic GMP-AMP synthase (cGAS)-mediated macrophage polarization (224). This study enhances our understanding of CSD’s role in treating UC and contributes to the mechanistic research concerning its modulation of macrophage function. A similar study used the theories of traditional Chinese medicine to develop a new decoction called modified Zhenwu decoction (CDD-2103), it targets macrophage function and migration through the C-C Chemokine receptor 2/p38 mitogen-activated protein kinase (p38MAPK) signaling axis, which is involved in the regulation of macrophage migration, however, adoptive macrophage transfer experiments undermined the protective effect of this treatment on colitis (225). This further confirms that CDD-2103 primarily treats UC by targeting macrophage function and behavioral changes.
All of the above are the latest therapeutic drugs with the potential to target macrophages. Focusing on macrophages represents a promising new approach for managing UC. The role and behavior of macrophages are crucial in the onset and progression of UC. Therefore, it remains a significant topic within the digestive health community to develop methods for precisely regulating the types of macrophages that aid in UC recovery and effectively directing drug treatment to the affected areas of the colon.
6 Conclusions
Currently, there is no consensus on the pathogenesis of UC, but the primary pathological feature is the imbalance of intestinal inflammation and immune response. Metabolic reprogramming signals in macrophages and signals beyond metabolism collectively alter macrophage function and behavior, significantly influencing intestinal inflammation and modifying local inflammatory-immune dysregulation in the gut. Ultimately, this helps in the treatment of UC. Therefore, targeting the functions and behaviors of macrophages to improve intestinal inflammation is a crucial strategy in UC treatment. This review provides a theoretical basis for macrophage-targeted therapy in UC and offers important new avenues for future research.
7 Future perspectives
Future therapeutic directions can also further explore single-cell omics appge co-culture models, and nanotechnology-enabled targeted delivery systems, aiming to bridge mechanistic understanding with clinical translation for precision medicine in UC management. Despite promising anti-inflammatory effects observed with macrophage-targeted therapies in preclinical models, their successful clinical translation remains limited by several practical challenges. One of the most clinically relevant issues is the phenomenon of “loss of response (LoR)”, wherein patients who initially respond well to treatment gradually lose efficacy over time—a problem frequently encountered with biologic agents such as anti-TNF monoclonal antibodies (226). LoR is distinct from primary non-response and is often attributed to multiple interrelated mechanisms, including the development of anti-drug antibodies, activation of compensatory pro-inflammatory pathways, and microbial-driven alterations in drug metabolism or target expression (227, 228). Moreover, adaptive changes within the host immune system, such as shifts in T-cell subsets or altered macrophage polarization dynamics, may further compromise therapeutic durability (81, 229). To address these challenges, emerging strategies include therapeutic drug monitoring to guide dose optimization and combination therapy with immunomodulators, such as azathioprine, to reduce immunogenicity (230, 231). Nonetheless, more mechanistic studies and well-designed clinical trials are warranted to fully understand the underlying biology of treatment failure and to develop rational interventions that improve long-term outcomes for patients with UC.
This review primarily focuses on the regulatory roles of macrophages in the pathogenesis of UC while also exploring potential therapeutic interventions targeting these cells. It is essential to recognize the long-term oncogenic risks linked to chronic intestinal inflammation. Patients with long-term UC have an increased susceptibility to colitis-associated cancer (CAC), a complication driven by persistent immune dysregulation and epithelial damage. In this context, tumor-associated macrophages (TAMs) play a crucial role in tumor initiation and progression. They contribute to significant processes such as remodeling the immune microenvironment, promoting angiogenesis, and facilitating immune evasion (232, 233). Remarkably, TAMs exhibit considerable plasticity and undergo functional changes when transitioning from chronic inflammation to malignancy. This transition involves a shift from a pro-inflammatory M1-like phenotype to an immunosuppressive M2-like phenotype (234). Accompanying this phenotypic switch are alterations in metabolic programming—such as enhanced lipid metabolism and downregulated tryptophan metabolism—along with the activation of signaling pathways like the phosphorylation of STAT6 and protein kinase B (235–237). Targeted inhibition of these changes that promote colitis-associated cancer could theoretically enhance human health. Specifically, a deeper understanding of these dynamic changes may inform the development of novel therapeutic strategies aimed at modulating macrophage function to prevent or delay the onset of CAC. Future studies should concentrate on defining the temporal evolution of macrophage subsets and their interactions with tumor cells and other stromal components throughout the inflammatory-to-malignant transition in UC.
Author contributions
SC: Methodology, Conceptualization, Writing – original draft. ZQ: Conceptualization, Methodology, Writing – original draft. XL: Writing – original draft, Methodology, Conceptualization. SZ: Project administration, Writing – review & editing, Supervision. YX: Project administration, Writing – review & editing, Supervision. YZ: Writing – review & editing, Supervision, Project administration.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was funded by the National Natural Science Foundation of China (82374426), the Hunan Provincial Education Department (24A0271), the Changsha Natural Science Foundation (kq2403098), and the Domestic First-class Construction Discipline of Chinese Medicine in Hunan University of Chinese Medicine.
Acknowledgments
Thanks to all the authors for their contributions to this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
TNF: tumor necrosis factor
IL-6: interleukin-6
IL-1β: interleukin-1β
IBD: inflammatory bowel disease
DSS: dextran sulfate sodium
Arg1: arginase-1
IL-10: interleukin-10
IL-4: interleukin-4
PKM2: pyruvate kinase M2
PFKFB3: 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3
NLRP3: NOD-like receptor thermal protein domain associated protein 3
ROS: oxygen species
TCA: tricarboxylic acid
ATP: adenosine triphosphate
FAO: fatty acid oxidation
PPAR: speroxisome proliferator-activated receptors
FFARs: free fatty acid receptors
Hadhb: Hydroxyacyl Coenzyme A Dehydrogenase Beta
BCAA: branched-chain amino acid
BCAT1: branched-chain aminotransferase 1
RNF99: ring finger protein 99
MKL1: megakaryoblastic leukemia 1
JMJD3: jumonji domain-containing 3
H3K27me3: histone H3 on lysine 27
CMKLR1: chemerin receptor 1
PSMP: PC3-secreted microprotein
CCL2: C-C motif chemokine ligand 2
SCFAs: short-chain fatty acids
FXR: farnesoid X receptor
SB: saccharomyces boulardii
TCM: traditional Chinese medicine
Cr: creatine
CS: chitosan
L-Arg: L-arginine
CA: CS-L-Arg
RLZ: rosiglitazone
AABA: a-aminobutyric acid
XylA: xylan acetate
A. muciniphila: Akkermansia muciniphila
HDAC5: histone deacetylase 5
L. johnsonii: lactobacillus johnsonii
FN: formononetin
STV-Na: isosteviol sodium
TXYF: Tongxie-yaofang
CSD: compound sophorae decoction
iNOS: inducible nitric oxide synthase
TAB2: TAK1-binding protein 2
OXPHOS: oxidative phosphorylation
ECAR: extracellular acidification rate
IL-1R: interleukin-1R
Fizz-1: found in inflammatory zone
TLR4: toll-like receptor 4
MD2: myeloid differentiation protein 2
p-p65: phospho-NF-κB p65
p65: NF-κB p65
p-ERK1/2: phosphorylated extracellular regulated protein kinases 1/2
p-4-EBP1: phosphorylated 4E binding protein 1
TSC1: tuberous sclerosis complex 1
m6A: N6-methyladenosine
Mettl14: methyltransferase-like protein 14
cGAS: cyclic GMP-AMP synthase
p-p38 MAPK: phospho-P38 mitogen-activated proteinkinase
CAC: colitis-associated cancer
TAMs: tumor-associated macrophages
LoR: loss of response.
References
1. Du L and Ha C. Epidemiology and pathogenesis of ulcerative colitis. Gastroenterol Clin North Am. (2020) 49:643–54. doi: 10.1016/j.gtc.2020.07.005
2. Wangchuk P, Yeshi K, and Loukas A. Ulcerative colitis: clinical biomarkers, therapeutic targets, and emerging treatments. Trends Pharmacol Sci. (2024) 45:892–903. doi: 10.1016/j.tips.2024.08.003
3. Conrad K, Roggenbuck D, and Laass MW. Diagnosis and classification of ulcerative colitis. Autoimmun Rev. (2014) 13:463–6. doi: 10.1016/j.autrev.2014.01.028
4. Sandborn WJ, Sands BE, Vermeire S, Leung Y, Guo X, Modesto I, et al. Modified Mayo score versus Mayo score for evaluation of treatment efficacy in patients with ulcerative colitis: data from the tofacitinib OCTAVE program. Therap Adv Gastroenterol. (2022) 15:17562848221136331. doi: 10.1177/17562848221136331
5. Walsh A, Cao R, Wong D, Kantschuster R, Matini L, Wilson J, et al. Using item response theory (IRT) to improve the efficiency of the Simple Clinical Colitis Activity Index (SCCAI) for patients with ulcerative colitis. BMC Gastroenterol. (2021) 21:132. doi: 10.1186/s12876-021-01621-y
6. Barahona-Garrido J, Camacho-Escobedo J, García-Martínez CI, Tocay H, Cabiedes J, and Yamamoto-Furusho JK. Antinuclear antibodies: a marker associated with steroid dependence in patients with ulcerative colitis. Inflammation Bowel Dis. (2009) 15:1039–43. doi: 10.1002/ibd.20852
7. Mansouri P, Mansouri P, Behmard E, Najafipour S, Kouhpayeh A, and Farjadfar A. Novel targets for mucosal healing in inflammatory bowel disease therapy. Int Immunopharmacol. (2025) 144:113544. doi: 10.1016/j.intimp.2024.113544
8. Hu L, Wu S, Shu Y, Su K, Wang C, Wang D, et al. Impact of maternal smoking, offspring smoking, and genetic susceptibility on crohn’s disease and ulcerative colitis. J Crohns Colitis. (2024) 18:671–8. doi: 10.1093/ecco-jcc/jjad200
9. Le Berre C, Honap S, and Peyrin-Biroulet L. Ulcerative colitis. Lancet. (2023) 402:571–84. doi: 10.1016/S0140-6736(23)00966-2
10. Zhang M, Li X, Zhang Q, Yang J, and Liu G. Roles of macrophages on ulcerative colitis and colitis-associated colorectal cancer. Front Immunol. (2023) 14:1103617. doi: 10.3389/fimmu.2023.1103617
11. Li S, Chen Z, Wang M, Rao Y, Yang F, Liu M, et al. L-arginine-modified selenium nanozymes targeting M1 macrophages for oral treatment of ulcerative colitis. Small. (2025) 21:e2408205. doi: 10.1002/smll.202408205
12. Wang Y, Yao Y, Zhang Y, Yu Y, Luo J, Sweet MJ, et al. Rational design of advanced gene delivery carriers: macrophage phenotype matters. Adv Mater. (2025) 37:e2401504. doi: 10.1002/adma.202401504
13. Li P, Zhu L, Song C, Wu M, Zhu X, He S, et al. Triple-functional probiotics with intracellularly synthesized selenium nanoparticles for colitis therapy by regulating the macrophage phenotype and modulating gut microbiota. ACS Nano. (2025) 19:14213–32. doi: 10.1021/acsnano.5c00574
14. Zhang QY, Zhang HY, Feng SG, Yao MD, Ding JJ, Li XM, et al. Macrophage metabolic reprogramming ameliorates diabetes-induced microvascular dysfunction. Redox Biol. (2025) 79:103449. doi: 10.1016/j.redox.2024.103449
15. Liu L, Li M, Zhang C, Zhong Y, Liao B, Feng J, et al. Macrophage metabolic reprogramming: A trigger for cardiac damage in autoimmune diseases. Autoimmun Rev. (2025) 24:103733. doi: 10.1016/j.autrev.2024.103733
16. Chen S, Yang J, Wei Y, and Wei X. Epigenetic regulation of macrophages: from homeostasis maintenance to host defense. Cell Mol Immunol. (2020) 17:36–49. doi: 10.1038/s41423-019-0315-0
17. Mehta S, Cronkite DA, Basavappa M, Saunders TL, Adiliaghdam F, Amatullah H, et al. Maintenance of macrophage transcriptional programs and intestinal homeostasis by epigenetic reader SP140. Sci Immunol. (2017) 2:eaag3160. doi: 10.1126/sciimmunol.aag3160
18. Ruytinx P, Proost P, Van Damme J, and Struyf S. Chemokine-induced macrophage polarization in inflammatory conditions. Front Immunol. (2018) 9:1930. doi: 10.3389/fimmu.2018.01930
19. Jones GR, Bain CC, Fenton TM, Kelly A, Brown SL, Ivens AC, et al. Dynamics of colon monocyte and macrophage activation during colitis. Front Immunol. (2018) 9:2764. doi: 10.3389/fimmu.2018.02764
20. Microbial metabolism activates immune-suppressive tumor macrophages. Cancer Discov. (2022) 12:883. doi: 10.1158/2159-8290.CD-RW2022-028
21. Chen G, Qi H, Jiang L, Sun S, Zhang J, Yu J, et al. Integrating single-cell RNA-Seq and machine learning to dissect tryptophan metabolism in ulcerative colitis. J Transl Med. (2024) 22:1121. doi: 10.1186/s12967-024-05934-w
22. Laskin DL, Sunil VR, Gardner CR, and Laskin JD. Macrophages and tissue injury: agents of defense or destruction. Annu Rev Pharmacol Toxicol. (2011) 51:267–88. doi: 10.1146/annurev.pharmtox.010909.105812
23. Chen Y and Zhang X. Pivotal regulators of tissue homeostasis and cancer: macrophages. Exp Hematol Oncol. (2017) 6:23. doi: 10.1186/s40164-017-0083-4
24. Lazarov T, Juarez-Carreño S, Cox N, and Geissmann F. Physiology and diseases of tissue-resident macrophages. Nature. (2023) 618:698–707. doi: 10.1038/s41586-023-06002-x
25. Mass E, Ballesteros I, Farlik M, Halbritter F, Günther P, Crozet L, et al. Specification of tissue-resident macrophages during organogenesis. Science. (2016) 353:aaf4238. doi: 10.1126/science.aaf4238
26. Wynn TA, Chawla A, and Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. (2013) 496:445–55. doi: 10.1038/nature12034
27. Fritsch SD, Sukhbaatar N, Gonzales K, Sahu A, Tran L, Vogel A, et al. Metabolic support by macrophages sustains colonic epithelial homeostasis. Cell Metab. (2023) 35:1931–1943.e8. doi: 10.1016/j.cmet.2023.09.010
28. Keshavarzian A, Doria MI, Sedghi S, Kanofsky JR, Hecht D, Holmes EW, et al. Mitomycin C-induced colitis in rats: a new animal model of acute colonic inflammation implicating reactive oxygen species. J Lab Clin Med. (1992) 120:778–91.
29. El-Salhy M, Umezawa K, Gilja OH, Hatlebakk JG, Gundersen D, and Hausken T. Amelioration of severe TNBS induced colitis by novel AP-1 and NF- κ B inhibitors in rats. ScientificWorldJournal. (2014) 2014:813804. doi: 10.1155/2014/813804
30. Smith TD, Nagalla RR, Chen EY, and Liu WF. Harnessing macrophage plasticity for tissue regeneration. Adv Drug Delivery Rev. (2017) 114:193–205. doi: 10.1016/j.addr.2017.04.012
31. Huang J, Wu Q, Geller DA, and Yan Y. Macrophage metabolism, phenotype, function, and therapy in hepatocellular carcinoma (HCC). J Transl Med. (2023) 21:815. doi: 10.1186/s12967-023-04716-0
32. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol. (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
33. Zhang J, Zhou X, and Hao H. Macrophage phenotype-switching in cancer. Eur J Pharmacol. (2022) 931:175229. doi: 10.1016/j.ejphar.2022.175229
34. Yan L, Wang J, Cai X, Liou YC, Shen HM, Hao J, et al. Macrophage plasticity: signaling pathways, tissue repair, and regeneration. MedComm. (2020) . 2024. 5:e658. doi: 10.1002/mco2.658
35. Kumar Jha P, Aikawa M, and Aikawa E. Macrophage heterogeneity and efferocytosis: beyond the M1/M2 dichotomy. Circ Res. (2024) 134:186–8. doi: 10.1161/CIRCRESAHA.123.324011
36. Funes SC, Rios M, Escobar-Vera J, and Kalergis AM. Implications of macrophage polarization in autoimmunity. Immunology. (2018) 154:186–95. doi: 10.1111/imm.12910
37. Hassanshahi A, Moradzad M, Ghalamkari S, Fadaei M, Cowin AJ, and Hassanshahi M. Macrophage-mediated inflammation in skin wound healing. Cells. (2022) 11:2953. doi: 10.3390/cells11192953
38. Schilperoort M, Ngai D, Sukka SR, Avrampou K, Shi H, and Tabas I. The role of efferocytosis-fueled macrophage metabolism in the resolution of inflammation. Immunol Rev. (2023) 319:65–80. doi: 10.1111/imr.13214
39. Vilbois S, Xu Y, and Ho PC. Metabolic interplay: tumor macrophages and regulatory T cells. Trends Cancer. (2024) 10:242–55. doi: 10.1016/j.trecan.2023.11.007
40. Flytlie HA, Hvid M, Lindgreen E, Kofod-Olsen E, Petersen EL, Jørgensen A, et al. Expression of MDC/CCL22 and its receptor CCR4 in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Cytokine. (2010) 49:24–9. doi: 10.1016/j.cyto.2009.10.005
41. Zhang Q and Sioud M. Tumor-associated macrophage subsets: shaping polarization and targeting. Int J Mol Sci. (2023) 24:7493. doi: 10.3390/ijms24087493
42. Wang Z and Wang Z. The role of macrophages polarization in sepsis-induced acute lung injury. Front Immunol. (2023) 14:1209438. doi: 10.3389/fimmu.2023.1209438
43. Zhang L, Chen X, Cai P, Sun H, Shen S, Guo B, et al. Reprogramming mitochondrial metabolism in synovial macrophages of early osteoarthritis by a camouflaged meta-defensome. Adv Mater. (2022) 34:e2202715. doi: 10.1002/adma.202202715
44. Khanna R, Chande N, and Marshall JK. Ozanimod for the treatment of ulcerative colitis. Gastroenterology. (2022) 162:2104–6. doi: 10.1053/j.gastro.2022.01.033
45. Spalinger MR, Sayoc-Becerra A, Santos AN, Shawki A, Canale V, Krishnan M, et al. PTPN2 regulates interactions between macrophages and intestinal epithelial cells to promote intestinal barrier function. Gastroenterology. (2020) 159:1763–1777.e14. doi: 10.1053/j.gastro.2020.07.004
46. Shen XH, Guan J, Lu DP, Hong SC, Yu L, and Chen X. Peptostreptococcus Anaerobius enhances dextran sulfate sodium-induced colitis by promoting nf-κB-NLRP3-Dependent macrophage pyroptosis. Virulence. (2024) 15:2435391. doi: 10.1080/21505594.2024.2435391
47. Dharmasiri S, Garrido-Martin EM, Harris RJ, Bateman AC, Collins JE, Cummings JRF, et al. Human intestinal macrophages are involved in the pathology of both ulcerative colitis and crohn disease. Inflammation Bowel Dis. (2021) 27:1641–52. doi: 10.1093/ibd/izab029
48. Wang F, Zhou F, Peng J, Chen H, Xie J, Liu C, et al. Macrophage Tim-3 maintains intestinal homeostasis in DSS-induced colitis by suppressing neutrophil necroptosis. Redox Biol. (2024) 70:103072. doi: 10.1016/j.redox.2024.103072
49. Xiong K, Deng J, Yue T, Hu W, Zeng X, Yang T, et al. Berberine promotes M2 macrophage polarisation through the IL-4-STAT6 signalling pathway in ulcerative colitis treatment. Heliyon. (2023) 9:e14176. doi: 10.1016/j.heliyon.2023.e14176
50. Huang C, Wang J, Liu H, Huang R, Yan X, Song M, et al. Ketone body β-hydroxybutyrate ameliorates colitis by promoting M2 macrophage polarization through the STAT6-dependent signaling pathway. BMC Med. (2022) 20:148. doi: 10.1186/s12916-022-02352-x
51. Nighot M, Ganapathy AS, Saha K, Suchanec E, Castillo EF, Gregory A, et al. Matrix metalloproteinase MMP-12 promotes macrophage transmigration across intestinal epithelial tight junctions and increases severity of experimental colitis. J Crohns Colitis. (2021) 15:1751–65. doi: 10.1093/ecco-jcc/jjab064
52. Li LX, Xia YT, Sun XY, Li LR, Yao L, Ali MI, et al. CXCL-10/CXCR3 in macrophages regulates tissue repair by controlling the expression of Arg1, VEGFa and TNFα. J Biol Regul Homeost Agents. (2020) 34:987–99. doi: 10.23812/20-59-A-65
53. Liu J, Chen B, Bao J, Zhang Y, Lei L, and Yan F. Macrophage polarization in periodontal ligament stem cells enhanced periodontal regeneration. Stem Cell Res Ther. (2019) 10:320. doi: 10.1186/s13287-019-1409-4
54. Zhao H, Zhou Y, Xu J, Zhang Y, Wang H, Zhao C, et al. Short-chain fatty acid-producing bacterial strains attenuate experimental ulcerative colitis by promoting M2 macrophage polarization via JAK/STAT3/FOXO3 axis inactivation. J Transl Med. (2024) 22:369. doi: 10.1186/s12967-024-05122-w
55. Liang L, Liu L, Zhou W, Yang C, Mai G, Li H, et al. Gut microbiota-derived butyrate regulates gut mucus barrier repair by activating the macrophage/WNT/ERK signaling pathway. Clin Sci (Lond). (2022) 136:291–307. doi: 10.1042/CS20210778
56. Wang M, Pan W, Xu Y, Zhang J, Wan J, and Jiang H. Microglia-mediated neuroinflammation: A potential target for the treatment of cardiovascular diseases. J Inflammation Res. (2022) 15:3083–94. doi: 10.2147/JIR.S350109
57. Cai X, Zhang ZY, Yuan JT, Ocansey DKW, Tu Q, Zhang X, et al. hucMSC-derived exosomes attenuate colitis by regulating macrophage pyroptosis via the miR-378a-5p/NLRP3 axis. Stem Cell Res Ther. (2021) 12:416. doi: 10.1186/s13287-021-02492-6
58. Manoharan I, Shanmugam A, Ramalingam M, Patel N, Thangaraju M, Ande S, et al. The transcription factor RXRα in CD11c+ APCs regulates intestinal immune homeostasis and inflammation. J Immunol. (2023) 211:853–61. doi: 10.4049/jimmunol.2200909
59. Zou Y, Ghaderpour A, Munkhbileg B, Seo SU, and Seong SY. Taurodeoxycholate ameliorates DSS-induced colitis in mice. Int Immunopharmacol. (2023) 122:110628. doi: 10.1016/j.intimp.2023.110628
60. Yin H, Ju Z, Zhang X, Zuo W, Yang Y, Zheng M, et al. Inhibition of METTL3 in macrophages provides protection against intestinal inflammation. Cell Mol Immunol. (2024) 21:589–603. doi: 10.1038/s41423-024-01156-8
61. El Kasmi KC and Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol. (2015) 27:267–75. doi: 10.1016/j.smim.2015.09.001
62. Wang S, Liu G, Li Y, and Pan Y. Metabolic reprogramming induces macrophage polarization in the tumor microenvironment. Front Immunol. (2022) 13:840029. doi: 10.3389/fimmu.2022.840029
63. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. α-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. (2017) 18:985–94. doi: 10.1038/ni.3796
64. Liu PS, Chen YT, Li X, Hsueh PC, Tzeng SF, Chen H, et al. CD40 signal rewires fatty acid and glutamine metabolism for stimulating macrophage anti-tumorigenic functions. Nat Immunol. (2023) 24:452–62. doi: 10.1038/s41590-023-01430-3
65. Shen D, Yu X, Fan X, Liang Y, Lu D, Ke Z, et al. CDCA3-MYC positive feedback loop promotes bladder cancer progression via ENO1-mediated glycolysis. J Exp Clin Cancer Res. (2025) 44:63. doi: 10.1186/s13046-025-03325-7
66. Wu KK, Xu X, Wu M, Li X, Hoque M, Li GHY, et al. MDM2 induces pro-inflammatory and glycolytic responses in M1 macrophages by integrating iNOS-nitric oxide and HIF-1α pathways in mice. Nat Commun. (2024) 15:8624. doi: 10.1038/s41467-024-53006-w
67. Rao J, Wang H, Ni M, Wang Z, Wang Z, Wei S, et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut. (2022) 71:2539–50. doi: 10.1136/gutjnl-2021-325150
68. Feng T, Zhao X, Gu P, Yang W, Wang C, Guo Q, et al. Adipocyte-derived lactate is a signalling metabolite that potentiates adipose macrophage inflammation via targeting PHD2. Nat Commun. (2022) 13:5208. doi: 10.1038/s41467-022-32871-3
69. Ayyangar U, Karkhanis A, Tay H, Afandi AFB, Bhattacharjee O, Ks L, et al. Metabolic rewiring of macrophages by epidermal-derived lactate promotes sterile inflammation in the murine skin. EMBO J. (2024) 43:1113–34. doi: 10.1038/s44318-024-00039-y
70. Duan S, Lou X, Chen S, Jiang H, Chen D, Yin R, et al. Macrophage LMO7 deficiency facilitates inflammatory injury via metabolic-epigenetic reprogramming. Acta Pharm Sin B. (2023) 13:4785–800. doi: 10.1016/j.apsb.2023.09.012
71. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
72. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. (2014) 345:1250684. doi: 10.1126/science.1250684
73. Alexander RK, Liou YH, Knudsen NH, Starost KA, Xu C, Hyde AL, et al. Bmal1 integrates mitochondrial metabolism and macrophage activation. Elife. (2020) 9:e54090. doi: 10.7554/eLife.54090
74. Corcoran SE and O’Neill LA. HIF1α and metabolic reprogramming in inflammation. J Clin Invest. (2016) 126:3699–707. doi: 10.1172/JCI84431
75. Zhuang H, Lv Q, Zhong C, Cui Y, He L, Zhang C, et al. Tiliroside ameliorates ulcerative colitis by restoring the M1/M2 macrophage balance via the HIF-1α/glycolysis pathway. Front Immunol. (2021) 12:649463. doi: 10.3389/fimmu.2021.649463
76. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
77. Huang B, Wang Q, Jiang L, Lu S, Li C, Xu C, et al. Shikonin ameliorated mice colitis by inhibiting dimerization and tetramerization of PKM2 in macrophages. Front Pharmacol. (2022) 13:926945. doi: 10.3389/fphar.2022.926945
78. Feng YL, Xu XR, Zhu QM, Chang J, Zhang HL, Wang N, et al. Aucklandiae radix targeted PKM2 to alleviate ulcerative colitis: Insights from the photocrosslinking target fishing technique. Phytomedicine. (2024) 134:155973. doi: 10.1016/j.phymed.2024.155973
79. Shen X, Sun C, Cheng Y, Ma D, Sun Y, Lin Y, et al. cGAS Mediates Inflammation by Polarizing Macrophages to M1 Phenotype via the mTORC1 Pathway. J Immunol. (2023) 210:1098–107. doi: 10.4049/jimmunol.2200351
80. Xiao J, Wang S, Chen L, Ding X, Dang Y, Han M, et al. 25-Hydroxycholesterol regulates lysosome AMP kinase activation and metabolic reprogramming to educate immunosuppressive macrophages. Immunity. (2024) 57:1087–1104.e7. doi: 10.1016/j.immuni.2024.03.021
81. Cortés M, Brischetto A, Martínez-Campanario MC, Ninfali C, Domínguez V, Fernández S, et al. Inflammatory macrophages reprogram to immunosuppression by reducing mitochondrial translation. Nat Commun. (2023) 14:7471. doi: 10.1038/s41467-023-42277-4
82. Wu MM, Wang QM, Huáng BY, Mai CT, Wang CL, Wang TT, et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol Res. (2021) 172:105796. doi: 10.1016/j.phrs.2021.105796
83. York AG, Skadow MH, Oh J, Qu R, Zhòu QD, Hsieh WY, et al. IL-10 constrains sphingolipid metabolism to limit inflammation. Nature. (2024) 627:628–35. doi: 10.1038/s41586-024-07098-5
84. Richter FC, Friedrich M, Kampschulte N, Piletic K, Alsaleh G, Zummach R, et al. Adipocyte autophagy limits gut inflammation by controlling oxylipin and IL-10. EMBO J. (2023) 42:e112202. doi: 10.15252/embj.2022112202
85. Ip W, Hoshi N, Shouval DS, Snapper S, and Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science. (2017) 356:513–9. doi: 10.1126/science.aal3535
86. Chen HJ, Sévin DC, Griffith GR, Vappiani J, Booty LM, van Roomen C, et al. Integrated metabolic-transcriptomic network identifies immunometabolic modulations in human macrophages. Cell Rep. (2024) 43:114741. doi: 10.1016/j.celrep.2024.114741
87. Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, et al. Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab. (2018) 28:463–475.e4. doi: 10.1016/j.cmet.2018.08.012
88. Yang Y, Cui BB, Li J, Shan JJ, Xu J, Zhang CY, et al. Tricarboxylic acid cycle metabolites: new players in macrophage. Inflammation Res. (2024) 73:531–9. doi: 10.1007/s00011-024-01853-0
89. Bauset C, Lis-López L, Coll S, Gisbert-Ferrándiz L, Macias-Ceja DC, Seco-Cervera M, et al. SUCNR1 mediates the priming step of the inflammasome in intestinal epithelial cells: relevance in ulcerative colitis. Biomedicines. (2022) 10:532. doi: 10.3390/biomedicines10030532
90. Liu CS, Hu YX, Luo ZY, Qiu CW, Deng XH, and Chen FL. Xianglian pill modulates gut microbial production of succinate and induces regulatory T cells to alleviate ulcerative colitis in rats. J Ethnopharmacol. (2023) 303:116007. doi: 10.1016/j.jep.2022.116007
91. Torretta S, Scagliola A, Ricci L, Mainini F, Di Marco S, Cuccovillo I, et al. D-mannose suppresses macrophage IL-1β production. Nat Commun. (2020) 11:6343. doi: 10.1038/s41467-020-20164-6
92. Batista-Gonzalez A, Vidal R, Criollo A, and Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. (2019) 10:2993. doi: 10.3389/fimmu.2019.02993
93. Vassiliou E and Farias-Pereira R. Impact of lipid metabolism on macrophage polarization: implications for inflammation and tumor immunity. Int J Mol Sci. (2023) 24:12032. doi: 10.3390/ijms241512032
94. Huáng SC, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. (2016) 45:817–30. doi: 10.1016/j.immuni.2016.09.016
95. Korbecki J and Bajdak-Rusinek K. The effect of palmitic acid on inflammatory response in macrophages: an overview of molecular mechanisms. Inflammation Res. (2019) 68:915–32. doi: 10.1007/s00011-019-01273-5
96. Wen JH, Li DY, Liang S, Yang C, Tang JX, and Liu HF. Macrophage autophagy in macrophage polarization, chronic inflammation and organ fibrosis. Front Immunol. (2022) 13:946832. doi: 10.3389/fimmu.2022.946832
97. Soliman E, Elhassanny A, Malur A, McPeek M, Bell A, Leffler N, et al. Impaired mitochondrial function of alveolar macrophages in carbon nanotube-induced chronic pulmonary granulomatous disease. Toxicology. (2020) 445:152598. doi: 10.1016/j.tox.2020.152598
98. Malandrino MI, Fucho R, Weber M, Calderón-Dominguez M, Mir JF, Valcarcel L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab. (2015) 308:E756–69. doi: 10.1152/ajpendo.00362.2014
99. Chen H, Tan H, Wan J, Zeng Y, Wang J, Wang H, et al. PPAR-γ signaling in nonalcoholic fatty liver disease: Pathogenesis and therapeutic targets. Pharmacol Ther. (2023) 245:108391. doi: 10.1016/j.pharmthera.2023.108391
100. Qiu Y, Gan M, Wang X, Liao T, Chen Q, Lei Y, et al. The global perspective on peroxisome proliferator-activated receptor γ (PPARγ) in ectopic fat deposition: A review. Int J Biol Macromol. (2023) 253:127042. doi: 10.1016/j.ijbiomac.2023.127042
101. Bougarne N, Weyers B, Desmet SJ, Deckers J, Ray DW, Staels B, et al. Molecular actions of PPARα in lipid metabolism and inflammation. Endocr Rev. (2018) 39:760–802. doi: 10.1210/er.2018-00064
102. Liu S, Zhang H, Li Y, Zhang Y, Bian Y, Zeng Y, et al. S100A4 enhances protumor macrophage polarization by control of PPAR-γ-dependent induction of fatty acid oxidation. J Immunother Cancer. (2021) 9:e002548. doi: 10.1136/jitc-2021-002548
103. Duah M, Zhang K, Liang Y, Ayarick VA, Xu K, and Pan B. Immune regulation of poly unsaturated fatty acids and free fatty acid receptor 4. J Nutr Biochem. (2023) 112:109222. doi: 10.1016/j.jnutbio.2022.109222
104. Binienda A and Fichna J. Current understanding of free fatty acids and their receptors in colorectal cancer treatment. Nutr Res. (2024) 127:133–43. doi: 10.1016/j.nutres.2024.05.007
105. Li MY, Wu YZ, Qiu JG, Lei JX, Li MX, Xu N, et al. Huangqin Decoction ameliorates ulcerative colitis by regulating fatty acid metabolism to mediate macrophage polarization via activating FFAR4-AMPK-PPARα pathway. J Ethnopharmacol. (2023) 311:116430. doi: 10.1016/j.jep.2023.116430
106. Zhang LS, Zhang ZS, Wu YZ, Guo B, Li J, Huáng XQ, et al. Activation of free fatty acid receptors, FFAR1 and FFAR4, ameliorates ulcerative colitis by promote fatty acid metabolism and mediate macrophage polarization. Int Immunopharmacol. (2024) 130:111778. doi: 10.1016/j.intimp.2024.111778
107. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, and Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
108. Kieler M, Hofmann M, and Schabbauer G. More than just protein building blocks: how amino acids and related metabolic pathways fuel macrophage polarization. FEBS J. (2021) 288:3694–714. doi: 10.1111/febs.15715
109. Lu M, Luo D, Zhang Z, Ouyang F, Shi Y, Hu C, et al. Branched-chain amino acid catabolism promotes M2 macrophage polarization. Front Immunol. (2024) 15:1469163. doi: 10.3389/fimmu.2024.1469163
110. Jiang Q and Shi L. Coordination of the uptake and metabolism of amino acids in mycobacterium tuberculosis-infected macrophages. Front Immunol. (2021) 12:711462. doi: 10.3389/fimmu.2021.711462
111. Zhang Z, Wang Y, Xia L, and Zhang Y. Roles of critical amino acids metabolism in the interactions between intracellular bacterial infection and macrophage function. Curr Microbiol. (2024) 81:280. doi: 10.1007/s00284-024-03801-x
112. Papathanassiu AE, Ko JH, Imprialou M, Bagnati M, Srivastava PK, Vu HA, et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat Commun. (2017) 8:16040. doi: 10.1038/ncomms16040
113. Wang Y, Li J, Yang Q, Zhù Z, Cheng F, Ai X, et al. 5-methoxytryptophan alleviates dextran sulfate sodium-induced colitis by inhibiting the intestinal epithelial damage and inflammatory response. Mediators Inflamm. (2024) 2024:1484806. doi: 10.1155/2024/1484806
114. Hashimoto H, Takagi T, Asaeda K, Yasuda T, Kajiwara M, Sugaya T, et al. D-alanine inhibits murine intestinal inflammation by suppressing IL-12 and IL-23 production in macrophages. J Crohns Colitis. (2024) 18:908–19. doi: 10.1093/ecco-jcc/jjad217
115. Zhang J, Cao L, Gao A, Ren R, Yu L, Li Q, et al. E3 ligase RNF99 negatively regulates TLR-mediated inflammatory immune response via K48-linked ubiquitination of TAB2. Cell Death Differ. (2023) 30:966–78. doi: 10.1038/s41418-023-01115-2
116. Xiong H, Wang Q, Li CC, and He A. Single-cell joint profiling of multiple epigenetic proteins and gene transcription. Sci Adv. (2024) 10:eadi3664. doi: 10.1126/sciadv.adi3664
117. Du W, Shi G, Shan CM, Li Z, Zhu B, Jia S, et al. Mechanisms of chromatin-based epigenetic inheritance. Sci China Life Sci. (2022) 65:2162–90. doi: 10.1007/s11427-022-2120-1
118. Sun L, Zhang H, and Gao P. Metabolic reprogramming and epigenetic modifications on the path to cancer. Protein Cell. (2022) 13:877–919. doi: 10.1007/s13238-021-00846-7
119. Song J, Wu Y, Chen Y, Sun X, and Zhang Z. Epigenetic regulatory mechanism of macrophage polarization in diabetic wound healing (Review). Mol Med Rep. (2025) 31:2. doi: 10.3892/mmr.2024.13367
120. Solier S, Müller S, Cãneque T, Versini A, Mansart A, Sindikubwabo F, et al. A druggable copper-signalling pathway that drives inflammation. Nature. (2023) 617:386–94. doi: 10.1038/s41586-023-06017-4
121. Sun S, XU X, Liang L, Wang X, Bai X, Zhù L, et al. Lactic Acid-Producing Probiotic Saccharomyces cerevisiae Attenuates Ulcerative Colitis via Suppressing Macrophage Pyroptosis and Modulating Gut Microbiota. Front Immunol. (2021) 12:777665. doi: 10.3389/fimmu.2021.777665
122. Xu B, Liu Y, Li N, and Geng Q. Lactate and lactylation in macrophage metabolic reprogramming: current progress and outstanding issues. Front Immunol. (2024) 15:1395786. doi: 10.3389/fimmu.2024.1395786
123. Perez MF and Sarkies P. Histone methyltransferase activity affects metabolism in human cells independently of transcriptional regulation. PloS Biol. (2023) 21:e3002354. doi: 10.1371/journal.pbio.3002354
124. Li B, Xia Y, Mei S, Ye Z, Song B, Yan X, et al. Histone H3K27 methyltransferase EZH2 regulates apoptotic and inflammatory responses in sepsis-induced AKI. Theranostics. (2023) 13:1860–75. doi: 10.7150/thno.83353
125. Ray-Gallet D and Almouzni G. H3-H4 histone chaperones and cancer. Curr Opin Genet Dev. (2022) 73:101900. doi: 10.1016/j.gde.2022.101900
126. Flury V, Reverón-Gómez N, Alcaraz N, Stewart-Morgan KR, Wenger A, Klose RJ, et al. Recycling of modified H2A-H2B provides short-term memory of chromatin states. Cell. (2023) 186:1050–1065.e19. doi: 10.1016/j.cell.2023.01.007
127. Woo H, Oh J, Cho YJ, Oh GT, Kim SY, Dan K, et al. N-terminal acetylation of Set1-COMPASS fine-tunes H3K4 methylation patterns. Sci Adv. (2024) 10:eadl6280. doi: 10.1126/sciadv.adl6280
128. Yu L, Fang F, Dai X, Xu H, Qi X, Fang M, et al. MKL1 defines the H3K4Me3 landscape for NF-κB dependent inflammatory response. Sci Rep. (2017) 7:191. doi: 10.1038/s41598-017-00301-w
129. An J, Nagaishi T, Watabe T, Naruse TK, Watanabe M, and Kimura A. MKL1 expressed in macrophages contributes to the development of murine colitis. Sci Rep. (2017) 7:13650. doi: 10.1038/s41598-017-13629-0
130. Davis FM, Tsoi LC, Melvin WJ, denDekker A, Wasikowski R, Joshi AD, et al. Inhibition of macrophage histone demethylase JMJD3 protects against abdominal aortic aneurysms. J Exp Med. (2021) 218:e20201839. doi: 10.1084/jem.20201839
131. An C, Jiao B, Du H, Tran M, Song B, Wang P, et al. Jumonji domain-containing protein-3 (JMJD3) promotes myeloid fibroblast activation and macrophage polarization in kidney fibrosis. Br J Pharmacol. (2023) 180:2250–65. doi: 10.1111/bph.16096
132. Audu CO, Melvin WJ, Joshi AD, Wolf SJ, Moon JY, Davis FM, et al. Macrophage-specific inhibition of the histone demethylase JMJD3 decreases STING and pathologic inflammation in diabetic wound repair. Cell Mol Immunol. (2022) 19:1251–62. doi: 10.1038/s41423-022-00919-5
133. Huang M, Wang Q, Long F, Di Y, Wang J, Zhun ZY, et al. Jmjd3 regulates inflammasome activation and aggravates DSS-induced colitis in mice. FASEB J. (2020) 34:4107–19. doi: 10.1096/fj.201902200RR
134. Miller MC and Mayo KH. Chemokines from a structural perspective. Int J Mol Sci. (2017) 18:2088. doi: 10.3390/ijms18102088
135. Urvas L and Kellenberger E. Structural insights into molecular recognition and receptor activation in chemokine-chemokine receptor complexes. J Med Chem. (2023) 66:7070–85. doi: 10.1021/acs.jmedchem.3c00352
136. Eberle SA and Gustavsson M. A scintillation proximity assay for real-time kinetic analysis of chemokine-chemokine receptor interactions. Cells. (2022) 11:1317. doi: 10.3390/cells11081317
137. Liu A, Liu Y, Wang J, and Ye RD. Structural basis for full-length chemerin recognition and signaling through chemerin receptor 1. Commun Biol. (2024) 7:1598. doi: 10.1038/s42003-024-07228-9
138. Dander E, Vinci P, Vetrano S, Recordati C, Piazza R, Fazio G, et al. The chemerin/CMKLR1 axis regulates intestinal graft-versus-host disease. JCI Insight. (2023) 8:e154440. doi: 10.1172/jci.insight.154440
139. Pei X, Zheng D, She S, Ma J, Guo C, Mo X, et al. The PSMP-CCR2 interactions trigger monocyte/macrophage-dependent colitis. Sci Rep. (2017) 7:5107. doi: 10.1038/s41598-017-05255-7
140. Xue L, Jin X, Ji T, Li R, Zhuge X, Xu F, et al. Luteolin ameliorates DSS-induced colitis in mice via suppressing macrophage activation and chemotaxis. Int Immunopharmacol. (2023) 124:110996. doi: 10.1016/j.intimp.2023.110996
141. Takeuchi T, Nakanishi Y, and Ohno H. Microbial metabolites and gut immunology. Annu Rev Immunol. (2024) 42:153–78. doi: 10.1146/annurev-immunol-090222-102035
142. Kolypetri P and Weiner HL. Monocyte regulation by gut microbial signals. Trends Microbiol. (2023) 31:1044–57. doi: 10.1016/j.tim.2023.05.006
143. Sheu KM and Hoffmann A. Functional hallmarks of healthy macrophage responses: their regulatory basis and disease relevance. Annu Rev Immunol. (2022) 40:295–321. doi: 10.1146/annurev-immunol-101320-031555
144. Saez A, Herrero-Fernandez B, Gomez-Bris R, Sánchez-Martinez H, and Gonzalez-Granado JM. Pathophysiology of inflammatory bowel disease: innate immune system. Int J Mol Sci. (2023) 24:1526. doi: 10.3390/ijms24021526
145. Chen X, Wu R, Li L, Zeng Y, Chen J, Wei M, et al. Pregnancy-induced changes to the gut microbiota drive macrophage pyroptosis and exacerbate septic inflammation. Immunity. (2023) 56:336–352.e9. doi: 10.1016/j.immuni.2023.01.015
146. Ngo VL, Lieber CM, Kang HJ, Sakamoto K, Kuczma M, Plemper RK, et al. Intestinal microbiota programming of alveolar macrophages influences severity of respiratory viral infection. Cell Host Microbe. (2024) 32:335–348.e8. doi: 10.1016/j.chom.2024.01.002
147. Bishehsari F, Engen PA, Preite NZ, Tuncil YE, Naqib A, Shaikh M, et al. Dietary fiber treatment corrects the composition of gut microbiota, promotes SCFA production, and suppresses colon carcinogenesis. Genes (Basel). (2018) 9:102. doi: 10.3390/genes9020102
148. Bassotti G, Antonelli E, Villanacci V, Nascimbeni R, Dore MP, Pes GM, et al. Abnormal gut motility in inflammatory bowel disease: an update. Tech Coloproctol. (2020) 24:275–82. doi: 10.1007/s10151-020-02168-y
149. Zhang Y, Song F, Yang M, Chen C, Cui J, Xing M, et al. Gastrointestinal dysmotility predisposes to colitis through regulation of gut microbial composition and linoleic acid metabolism. Adv Sci (Weinh). (2024) 11:e2306297. doi: 10.1002/advs.202306297
150. Ma L, Ni Y, Wang Z, Tu W, Ni L, Zhuge F, et al. Spermidine improves gut barrier integrity and gut microbiota function in diet-induced obese mice. Gut Microbes. (2020) 12:1–19. doi: 10.1080/19490976.2020.1832857
151. Yang Y, Misra BB, Liang L, Bi D, Weng W, Wu W, et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics. (2019) 9:4101–14. doi: 10.7150/thno.35186
152. Nakamura A, Kurihara S, Takahashi D, Ohashi W, Nakamura Y, Kimura S, et al. Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nat Commun. (2021) 12:2105. doi: 10.1038/s41467-021-22212-1
153. Bertheloot D, Latz E, and Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. (2021) 18:1106–21. doi: 10.1038/s41423-020-00630-3
154. Preston JA, Bewley MA, Marriott HM, McGarry Houghton, Mohasin M, Jubrail J, et al. Alveolar macrophage apoptosis-associated bacterial killing helps prevent murine pneumonia. Am J Respir Crit Care Med. (2019) 200:84–97. doi: 10.1164/rccm.201804-0646OC
155. Sinha SK, Miikeda A, Fouladian Z, Mehrabian M, Edillor C, Shih D, et al. Local M-CSF (Macrophage colony-stimulating factor) expression regulates macrophage proliferation and apoptosis in atherosclerosis. Arterioscler Thromb Vasc Biol. (2021) 41:220–33. doi: 10.1161/ATVBAHA.120.315255
156. Liu X, Zhou M, Dai Z, Luo S, Shi Y, He Z, et al. Salidroside alleviates ulcerative colitis via inhibiting macrophage pyroptosis and repairing the dysbacteriosis-associated Th17/Treg imbalance. Phytother Res. (2023) 37:367–82. doi: 10.1002/ptr.7636
157. Adorini L and Trauner M. FXR agonists in NASH treatment. J Hepatol. (2023) 79:1317–31. doi: 10.1016/j.jhep.2023.07.034
158. Tian SY, Chen SM, Pan CX, and Li Y. FXR: structures, biology, and drug development for NASH and fibrosis diseases. Acta Pharmacol Sin. (2022) 43:1120–32. doi: 10.1038/s41401-021-00849-4
159. Yang L, Li W, Zhao Q, Mo Q, Liu T, and Cao H. Saccharomyces boulardii alleviates colitis by regulating FXR-NLRP3 mediated macrophage pyroptosis. J Inflammation Res. (2025) 18:3161–76. doi: 10.2147/JIR.S504957
160. Lv Q, Xing Y, Liu J, Dong D, Liu Y, Qiao H, et al. Lonicerin targets EZH2 to alleviate ulcerative colitis by autophagy-mediated NLRP3 inflammasome inactivation. Acta Pharm Sin B. (2021) 11:2880–99. doi: 10.1016/j.apsb.2021.03.011
161. Zheng Y, Yu Y, Chen XF, Yang SL, Tang XL, and Xiang ZG. Intestinal macrophage autophagy and its pharmacological application in inflammatory bowel disease. Front Pharmacol. (2021) 12:803686. doi: 10.3389/fphar.2021.803686
162. Chen SL, Li CM, Li W, Liu QS, Hu SY, Zhao MY, et al. How autophagy, a potential therapeutic target, regulates intestinal inflammation. Front Immunol. (2023) 14:1087677. doi: 10.3389/fimmu.2023.1087677
163. Subramanian A, Kumarasamy V, Begum MY, Sekar M, Subramaniyan V, Wong LS, et al. Exploring the connections: autophagy, gut microbiota, and inflammatory bowel disease pathogenesis. J Inflammation Res. (2024) 17:10453–70. doi: 10.2147/JIR.S483958
164. Kabi A, Nickerson KP, Homer CR, and McDonald C. Digesting the genetics of inflammatory bowel disease: insights from studies of autophagy risk genes. Inflammation Bowel Dis. (2012) 18:782–92. doi: 10.1002/ibd.21868
165. McGovern DP, Kugathasan S, and Cho JH. Genetics of inflammatory bowel diseases. Gastroenterology. (2015) 149:1163–1176.e2. doi: 10.1053/j.gastro.2015.08.001
166. Chen M, Chen Y, Fu R, Liu S, Li H, and Shen T. Atox1 regulates macrophage polarization in intestinal inflammation via ROS-NLRP3 inflammasome pathway. J Transl Med. (2024) 22:497. doi: 10.1186/s12967-024-05314-4
167. Subramanian A, Jahabardeen A, Thamaraikani T, and Vellapandian C. More on the interplay between gut microbiota, autophagy, and inflammatory bowel disease is needed. World J Gastroenterol. (2024) 30:3356–60. doi: 10.3748/wjg.v30.i27.3356
168. Larabi A, Barnich N, and Nguyen H. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy. (2020) 16:38–51. doi: 10.1080/15548627.2019.1635384
169. Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, et al. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut. (2018) 67:574–87. doi: 10.1136/gutjnl-2017-314903
170. Zhao Y, Xiang Z, Pan H, Huang X, Chen W, and Huang Z. FGL2 improves experimental colitis related to gut microbiota structure and bile acid metabolism by regulating macrophage autophagy and apoptosis. Heliyon. (2024) 10:e34349. doi: 10.1016/j.heliyon.2024.e34349
171. Wang E, Wu MY, Ren ZY, Zheng Y, Ye RD, Tan C, et al. Targeting macrophage autophagy for inflammation resolution and tissue repair in inflammatory bowel disease. Burns Trauma. (2023) 11:tkad004. doi: 10.1093/burnst/tkad004
172. Hontecillas R, Horne WT, Climent M, Guri AJ, Evans C, Zhang Y, et al. Immunoregulatory mechanisms of macrophage PPAR-γ in mice with experimental inflammatory bowel disease. Mucosal Immunol. (2011) 4:304–13. doi: 10.1038/mi.2010.75
173. Zhang S, Zhou X, Ou M, Fu X, Lin Q, Tao X, et al. Berbamine promotes macrophage autophagy to clear Mycobacterium tuberculosis by regulating the ROS/Ca(2+) axis. mBio. (2023) 14:e0027223. doi: 10.1128/mbio.00272-23
174. Zhang J, Zhao Y, Hou T, Zeng H, Kalambhe D, Wang B, et al. Macrophage-based nanotherapeutic strategies in ulcerative colitis. J Control Release. (2020) 320:363–80. doi: 10.1016/j.jconrel.2020.01.047
175. Zhuo Z, Guo K, Luo Y, Yang Q, Wu H, Zeng R, et al. Targeted modulation of intestinal epithelial regeneration and immune response in ulcerative colitis using dual-targeting bilirubin nanoparticles. Theranostics. (2024) 14:528–46. doi: 10.7150/thno.87739
176. Ferreira-Faria I, Yousefiasl S, Macário-Soares A, Pereira-Silva M, Peixoto D, Zafar H, et al. Stem cell membrane-coated abiotic nanomaterials for biomedical applications. J Control Release. (2022) 351:174–97. doi: 10.1016/j.jconrel.2022.09.012
177. Vargas López JM, Cruz Ramos JA, and Carbajal Arizaga GG. Revisiting the characteristics of nanomaterials, composites, hybrid and functionalized materials in medical microbiology. Colloids Surf B Biointerf. (2025) 250:114556. doi: 10.1016/j.colsurfb.2025.114556
178. He H, Qin Q, Xu F, Chen Y, Rao S, Wang Cc, et al. Oral polyphenol-armored nanomedicine for targeted modulation of gut microbiota-brain interactions in colitis. Sci Adv. (2023) 9:eadf3887. doi: 10.1126/sciadv.adf3887
179. Wallimann T, Hall C, Colgan SP, and Glover LE. Creatine supplementation for patients with inflammatory bowel diseases: A scientific rationale for a clinical trial. Nutrients. (2021) 13:1429. doi: 10.3390/nu13051429
180. Huai M, Pei M, Chen J, Duan X, Zhu Y, Yang F, et al. Oral creatine-modified selenium-based hyaluronic acid nanogel mediated mitochondrial energy recovery to drive the treatment of inflammatory bowel disease. J Nanobiotechnol. (2024) 22:740. doi: 10.1186/s12951-024-03007-0
181. Zhang YA, Li FW, Dong YX, Xie WJ, and Wang HB. PPAR-γ regulates the polarization of M2 macrophages to improve the microenvironment for autologous fat grafting. FASEB J. (2024) 38:e23613. doi: 10.1096/fj.202400126R
182. Hasegawa-Moriyama M, Kurimoto T, Nakama M, Godai K, Kojima M, Kuwaki T, et al. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone attenuates inflammatory pain through the induction of heme oxygenase-1 in macrophages. Pain. (2013) 154:1402–12. doi: 10.1016/j.pain.2013.04.039
183. Rohm TV, Castellani Gomes Dos Reis F, Isaac R, Murphy C, Cunha E, Rocha K, et al. Adipose tissue macrophages secrete small extracellular vesicles that mediate rosiglitazone-induced insulin sensitization. Nat Metab. (2024) 6:880–98. doi: 10.1038/s42255-024-01023-w
184. Sun T, Kwong C, Gao C, Wei J, Yue L, Zhang J, et al. Amelioration of ulcerative colitis via inflammatory regulation by macrophage-biomimetic nanomedicine. Theranostics. (2020) 10:10106–19. doi: 10.7150/thno.48448
185. Felicianna Lo, Chen C, Ismaiah MJ, Zhang F, Leung H, El-Nezami H, et al. Alpha-aminobutyric acid ameliorates diet-induced metabolic dysfunction-associated steatotic liver disease (MASLD) progression in mice via enhancing AMPK/SIRT1 pathway and modulating the gut-liver axis. J Nutr Biochem. (2025) 140:109885. doi: 10.1016/j.jnutbio.2025.109885
186. Ismaiah MJ, Lo E, Chen C, Tsui JS, Johnson-Hill WA, Felicianna , et al. Alpha-aminobutyric acid administration suppressed visceral obesity and modulated hepatic oxidized PUFA metabolism via gut microbiota modulation. Free Radic Biol Med. (2025) 232:86–96. doi: 10.1016/j.freeradbiomed.2025.02.029
187. Li F, Xia Y, Yuan S, Xie X, Li L, Luo Y, et al. α-aminobutyric acid constrains macrophage-associated inflammatory diseases through metabolic reprogramming and epigenetic modification. Int J Mol Sci. (2023) 24:10444. doi: 10.3390/ijms241310444
188. Zhang B, Zhong Y, Dong D, Zheng Z, and Hu J. Gut microbial utilization of xylan and its implication in gut homeostasis and metabolic response. Carbohydr Polym. (2022) 286:119271. doi: 10.1016/j.carbpol.2022.119271
189. Zha Z, Lv Y, Tang H, Li T, Miao Y, Cheng J, et al. An orally administered butyrate-releasing xylan derivative reduces inflammation in dextran sulphate sodium-induced murine colitis. Int J Biol Macromol. (2020) 156:1217–33. doi: 10.1016/j.ijbiomac.2019.11.159
190. Zhou L, Song W, Liu T, Yan T, He Z, He W, et al. Multi-omics insights into anti-colitis benefits of the synbiotic and postbiotic derived from wheat bran arabinoxylan and Limosilactobacillus reuteri. Int J Biol Macromol. (2024) 278:134860. doi: 10.1016/j.ijbiomac.2024.134860
191. Tang H, Li Q, Zha Z, Jiao Y, Yang B, Cheng Z, et al. Xylan acetate ester ameliorates ulcerative colitis through intestinal barrier repair and inflammation inhibition via regulation of macrophage M1 polarization. Int J Biol Macromol. (2024) 280:135551. doi: 10.1016/j.ijbiomac.2024.135551
192. Mas-Bargues C, Borrás C, and Viña J. Genistein, a tool for geroscience. Mech Ageing Dev. (2022) 204:111665. doi: 10.1016/j.mad.2022.111665
193. Sharifi-Rad J, Quispe C, Imran M, Rauf A, Nadeem M, Gondal TA, et al. Genistein: an integrative overview of its mode of action, pharmacological properties, and health benefits. Oxid Med Cell Longev. (2021) 2021:3268136. doi: 10.1155/2021/3268136
194. Siriviriyakul P, Sriko J, Somanawat K, Chayanupatkul M, Klaikeaw N, and Werawatganon D. Genistein attenuated oxidative stress, inflammation, and apoptosis in L-arginine induced acute pancreatitis in mice. BMC Complement Med Ther. (2022) 22:208. doi: 10.1186/s12906-022-03689-9
195. Jia Q, Fang S, Yang R, Ling Y, Mehmood S, Ni H, et al. Genistein alleviates dextran sulfate sodium-induced colitis in mice through modulation of intestinal microbiota and macrophage polarization. Eur J Nutr. (2024) 63:1877–88. doi: 10.1007/s00394-024-03391-1
196. Díaz-Garrido N, Badia J, and Baldomà L. Microbiota-derived extracellular vesicles in interkingdom communication in the gut. J Extracell Vesicles. (2021) 10:e12161. doi: 10.1002/jev2.12161
197. Iheozor-Ejiofor Z, Kaur L, Gordon M, Baines PA, Sinopoulou V, and Akobeng AK. Probiotics for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. (2020) 3:CD007443. doi: 10.1002/14651858.CD007443.pub2
198. Caenepeel C, Deleu S, Vazquez Castellanos JF, Arnauts K, Braekeleire S, Machiels K, et al. Rigorous donor selection for fecal microbiota transplantation in active ulcerative colitis: key lessons from a randomized controlled trial halted for futility. Clin Gastroenterol Hepatol. (2025) 23:621–631.e7. doi: 10.1016/j.cgh.2024.05.017
199. Calvo A, Pastor Y, Rosas-Val P, and Gamazo C. Unveiling the immunomodulatory effect of the novel probiotic Akkermansia muciniphila and its protective effect in vitro. Microbiol Res. (2024) 283:127677. doi: 10.1016/j.micres.2024.127677
200. Wu XQ, Ying F, Chung K, Leung C, Leung R, So K, et al. Intestinal Akkermansia muciniphila complements the efficacy of PD1 therapy in MAFLD-related hepatocellular carcinoma. Cell Rep Med. (2025) 6:101900. doi: 10.1016/j.xcrm.2024.101900
201. Sharma S, Tyagi W, Tamang R, and Das S. HDAC5 modulates SATB1 transcriptional activity to promote lung adenocarcinoma. Br J Cancer. (2023) 129:586–600. doi: 10.1038/s41416-023-02341-8
202. Gao Y, Liu Y, Zheng D, Ho C, Wen D, Sun J, et al. HDAC5-mediated Smad7 silencing through MEF2A is critical for fibroblast activation and hypertrophic scar formation. Int J Biol Sci. (2022) 18:5724–39. doi: 10.7150/ijbs.76140
203. Yilmaz O, Okullu SO, Catakci M, Elmas MA, Pinheiro Y, Arbak S, et al. Akkermansia muciniphila improves chronic colitis-induced enteric neuroinflammation in mice. Neurogastroenterol Motil. (2024) 36:e14745. doi: 10.1111/nmo.14745
204. Miao Y, Wang M, Sun H, Zhang Y, Zhou W, Yang W, et al. Akkermansia muciniphila ameliorates colonic injury in mice with DSS-induced acute colitis by blocking macrophage pro-inflammatory phenotype switching via the HDAC5/DAB2 axis. Biochim Biophys Acta Mol Cell Res. (2024) 1871:119751. doi: 10.1016/j.bbamcr.2024.119751
205. Jia DJ, Wang QW, Hu YY, He JM, Ge QW, Qi YD, et al. Lactobacillus johnsonii alleviates colitis by TLR1/2-STAT3 mediated CD206(+) macrophages(IL-10) activation. Gut Microbes. (2022) 14:2145843. doi: 10.1080/19490976.2022.2145843
206. Cai Y, Huang Y, Wang Y, Lin C, Qiu L, and Wei H. Lactobacillus johnsonii GLJ001 prevents DSS-induced colitis in mice by inhibiting M1 macrophage polarization via gut microbiota-SCFAs axis. Int Immunopharmacol. (2025) 144:113671. doi: 10.1016/j.intimp.2024.113671
207. Xu HT, Zheng Q, Tai ZG, Jiang WC, Xie SQ, Luo Y, et al. Formononetin attenuates psoriasiform inflammation by regulating interferon signaling pathway. Phytomedicine. (2024) 128:155412. doi: 10.1016/j.phymed.2024.155412
208. He H, Peng S, Song X, Jia R, Zou Y, Li Lc, et al. Protective effect of isoflavones and triterpenoid saponins from pueraria lobata on liver diseases: A review. Food Sci Nutr. (2022) 10:272–85. doi: 10.1002/fsn3.2668
209. Zhang L, Wu Q, Huang Y, Zheng J, Guo S, and He L. Formononetin ameliorates airway inflammation by suppressing ESR1/NLRP3/Caspase-1 signaling in asthma. BioMed Pharmacother. (2023) 168:115799. doi: 10.1016/j.biopha.2023.115799
210. Xiao Q, Huang J, Zhu X, Shi M, Chen LL, Chen L, et al. Formononetin ameliorates dextran sulfate sodium-induced colitis via enhancing antioxidant capacity, promoting tight junction protein expression and reshaping M1/M2 macrophage polarization balance. Int Immunopharmacol. (2024) 142:113174. doi: 10.1016/j.intimp.2024.113174
211. Lu J, Xie L, Liu K, Zhang X, Wang X, Dai X, et al. Bilobalide: A review of its pharmacology, pharmacokinetics, toxicity, and safety. Phytother Res. (2021) 35:6114–30. doi: 10.1002/ptr.7220
212. Ma T, Lv L, Yu Y, Jia L, Song X, Xu X, et al. Bilobalide exerts anti-inflammatory effects on chondrocytes through the AMPK/SIRT1/mTOR pathway to attenuate ACLT-induced post-traumatic osteoarthritis in rats. Front Pharmacol. (2022) 13:783506. doi: 10.3389/fphar.2022.783506
213. Zhang H, Cao N, Yang Z, Fang X, Yang X, Li H, et al. Bilobalide alleviated dextran sulfate sodium-induced experimental colitis by inhibiting M1 macrophage polarization through the NF-κB signaling pathway. Front Pharmacol. (2020) 11:718. doi: 10.3389/fphar.2020.00718
214. Wang S, Huang J, Tan KS, Deng L, Liu F, and Tan W. Isosteviol sodium ameliorates dextran sodium sulfate-induced chronic colitis through the regulation of metabolic profiling, macrophage polarization, and NF-κB pathway. Oxid Med Cell Longev. (2022) 2022:4636618. doi: 10.1155/2022/4636618
215. Nguyen L, Choi MJ, Shin HM, and Yang IJ. Coptisine alleviates imiquimod-induced psoriasis-like skin lesions and anxiety-like behavior in mice. Molecules. (2022) 27:1412. doi: 10.3390/molecules27041412
216. Li C, Deng L, Pu M, Ye X, and Lu Q. Coptisine alleviates colitis through modulating gut microbiota and inhibiting TXNIP/NLRP3 inflammasome. J Ethnopharmacol. (2024) 335:118680. doi: 10.1016/j.jep.2024.118680
217. Zhao M, Li P, Qiao D, Hua S, Yue Q, Dai Y, et al. N6-methyladenosine modification of TSC1 mRNA contributes to macrophage polarization regulated by Coptisine in DSS-induced ulcerative colitis. Phytomedicine. (2024) 122:155153. doi: 10.1016/j.phymed.2023.155153
218. Liu X, Ye M, He Y, Lai Q, Liu B, and Zhang L. Investigation of Tongxie-Yaofang formula in treating ulcerative colitis based on network pharmacology via regulating MAPK/AKT signaling pathway. Aging (Albany NY). (2024) 16:1911–24. doi: 10.18632/aging.205467
219. Gong SS, Fan YH, Wang SY, Han QQ, Lv B, Xu Y, et al. Mucosa repair mechanisms of Tong-Xie-Yao-Fang mediated by CRH-R2 in murine, dextran sulfate sodium-induced colitis. World J Gastroenterol. (2018) 24:1766–78. doi: 10.3748/wjg.v24.i16.1766
220. Zhang HY, Zeng HR, Wei HZ, Chu XY, Zhu HT, Zhao B, et al. Tongxie-Yaofang formula regulated macrophage polarization to ameliorate DSS-induced colitis via NF-κB/NLRP3 signaling pathway. Phytomedicine. (2022) 107:154455. doi: 10.1016/j.phymed.2022.154455
221. Wu H, Chen QY, Wang WZ, Chu S, Liu XX, Liu YJ, et al. Compound sophorae decoction enhances intestinal barrier function of dextran sodium sulfate induced colitis via regulating notch signaling pathway in mice. BioMed Pharmacother. (2021) 133:110937. doi: 10.1016/j.biopha.2020.110937
222. Hong ZC, Cai Q, Wu HZ, Yang YF, Fan H, and Duan XY. Compound Sophorae Decoction: treating ulcerative colitis by affecting multiple metabolic pathways. Chin J Nat Med. (2021) 19:267–83. doi: 10.1016/S1875-5364(21)60029-8
223. Liu Y, Deng S, Sun L, He H, Zhou Q, Fan H, et al. Compound sophorae decoction mitigates DSS-induced ulcerative colitis by activating autophagy through PI3K-AKT pathway: A integrative research combining network pharmacology and in vivo animal model validation. J Ethnopharmacol. (2025) 337:118885. doi: 10.1016/j.jep.2024.118885
224. Gao F, Deng S, Liu Y, Wu P, Huang L, Zhu F, et al. Compound sophora decoction alleviates ulcerative colitis by regulating macrophage polarization through cGAS inhibition: network pharmacology and experimental validation. Aging (Albany NY). (2024) 16:6921–36. doi: 10.18632/aging.205734
225. Mok HL, Cheng KW, Xu Y, Huang C, Lyu C, Xu J, et al. Modified Zhenwu Decoction suppresses chronic colitis via targeting macrophage CCR2/Fyn/p38 MAPK signaling axis. Phytomedicine. (2024) 129:155694. doi: 10.1016/j.phymed.2024.155694
226. Nguyen VQ, Eden K, Morrison HA, Sammons MB, Knight KK, Sorrentino S, et al. Noncanonical NF-κB signaling upregulation in inflammatory bowel disease patients is associated with loss of response to anti-TNF agents. Front Pharmacol. (2021) 12:655887. doi: 10.3389/fphar.2021.655887
227. Nencini F, Vultaggio A, Pratesi S, Cammelli D, Milla M, Fiori Gc, et al. The kinetics of antidrug antibodies, drug levels, and clinical outcomes in infliximab-exposed patients with immune-mediated disorders. J Allergy Clin Immunol Pract. (2018) 6:2065–2072.e2. doi: 10.1016/j.jaip.2018.04.007
228. Shin J, Baek GH, Cha B, Park SH, Lee JH, Kim JS, et al. Complementary therapeutic effect of fecal microbiota transplantation in ulcerative colitis after the response to anti-tumor necrosis factor alpha agent was lost: A case report. Biomedicines. (2024) 12:800. doi: 10.3390/biomedicines12040800
229. Sono K, Yamada A, Yoshimatsu Y, Takada N, and Suzuki Y. Factors associated with the loss of response to infliximab in patients with Crohn’s disease. Cytokine. (2012) 59:410–6. doi: 10.1016/j.cyto.2012.04.026
230. Khoury T and Ilan Y. Introducing patterns of variability for overcoming compensatory adaptation of the immune system to immunomodulatory agents: A novel method for improving clinical response to anti-TNF therapies. Front Immunol. (2019) 10:2726. doi: 10.3389/fimmu.2019.02726
231. Roblin X, Williet N, Boschetti G, Phelip JM, Del Tedesco, Berger AE, et al. Addition of azathioprine to the switch of anti-TNF in patients with IBD in clinical relapse with undetectable anti-TNF trough levels and antidrug antibodies: a prospective randomised trial. Gut. (2020) 69:1206–12. doi: 10.1136/gutjnl-2019-319758
232. Matusiak M, Hickey JW, van IJzendoorn, Lu G, Kidziński L, Zhu S, et al. Spatially segregated macrophage populations predict distinct outcomes in colon cancer. Cancer Discov. (2024) 14:1418–39. doi: 10.1158/2159-8290.CD-23-1300
233. Deng Y, Jia X, Liu L, He Q, and Liu L. The role of intestinal macrophage polarization in colitis-associated colon cancer. Front Immunol. (2025) 16:1537631. doi: 10.3389/fimmu.2025.1537631
234. Shu Y and Cheng P. Targeting tumor-associated macrophages for cancer immunotherapy. Biochim Biophys Acta Rev Cancer. (2020) 1874:188434. doi: 10.1016/j.bbcan.2020.188434
235. Li Y, Li Q, Yuan R, Wang Y, Guo C, and Wang L. Bifidobacterium breve-derived indole-3-lactic acid ameliorates colitis-associated tumorigenesis by directing the differentiation of immature colonic macrophages. Theranostics. (2024) 14:2719–35. doi: 10.7150/thno.92350
236. Liu T, Zhang X, Yan X, Cheng L, Yan X, Zeng F, et al. Smad4 deficiency in S100A4(+) macrophages enhances colitis-associated tumorigenesis by promoting macrophage lipid metabolism augmented M2 polarization. Int J Biol Sci. (2024) 20:6114–29. doi: 10.7150/ijbs.98529
Keywords: ulcerative colitis (UC), macrophage, immune, inflammation, treatment
Citation: Chen S, Qin Z, Lin X, Zhou S, Xu Y and Zhu Y (2025) Macrophages: emerging targets for ulcerative colitis. Front. Immunol. 16:1623491. doi: 10.3389/fimmu.2025.1623491
Received: 06 May 2025; Accepted: 27 August 2025;
Published: 15 September 2025.
Edited by:
Shuai Wang, Guangzhou University of Chinese Medicine, ChinaReviewed by:
Stanislava Stanojevic, University of Belgrade, SerbiaArunkumar Subramanian, Thanthai Roever College of Pharmacy, India
Copyright © 2025 Chen, Qin, Lin, Zhou, Xu and Zhu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Zhu, emh1eWluZzA4OUAxMjYuY29t; Yin Xu, MzExMTE4QGhudWNtLmVkdS5jbg==; Sainan Zhou, bGFuY3lpdnlAaG51Y20uZWR1LmNu
†These authors have contributed equally to this work and share first authorship