 Alessandro Costa1,2
Alessandro Costa1,2 Olga Mulas1,2*Angela Maria Mereu1Mercede Schintu1Marianna Greco2
Olga Mulas1,2*Angela Maria Mereu1Mercede Schintu1Marianna Greco2 Giovanni Caocci1,2
Giovanni Caocci1,2- 1Department of Medical Sciences and Public Health, University of Cagliari, Cagliari, Italy
- 2Hematology Unit, Businco Hospital, ARNAS Brotzu, Cagliari, Italy
In recent years, the pathophysiologic framework of autoimmune hemolytic anemias (AIHAs) has evolved considerably, extending beyond the simplistic paradigm of antibody-mediated red blood cell (RBC) destruction, which is now recognized as a downstream consequence of a broader immune dysregulation. AIHA is fundamentally orchestrated by a complex interplay between innate and adaptive immune components, including autoreactive B and T lymphocytes, macrophages, and the reticuloendothelial system. Central to disease pathogenesis are two interrelated mechanisms: clonal B-cell expansion with autoantibody production and complement activation. These immunologic processes support the heterogeneity of AIHA, delineating distinct clinical entities such as warm AIHA, cold agglutinin disease/syndrome (CAD/CAS), and atypical variants, each characterized by specific therapeutic susceptibilities. Glucocorticoids remain the standard first-line therapy for warm AIHA; in contrast, CAD/CAS is increasingly managed with agents targeting B-cell function or complement activation, including rituximab and sutimlimab. However, therapeutic algorithms are rapidly shifting, particularly in the context of treatment-refractory disease. Emerging therapeutics targeting the classical complement pathway include novel anti-C1s monoclonal antibodies such as riliprubart, which exhibits an extended half-life due to enhanced affinity for the neonatal Fc receptor. Parallel strategies aim to disrupt B-cell receptor (BCR) signaling cascades, employing Bruton tyrosine kinase (BTK) inhibitors such as ibrutinib, spleen tyrosine kinase (SYK) inhibitors such as fostamatinib and sovleplenib, and phosphoinositide 3-kinase (PI3K) inhibitors such as parsaclisib. Collectively, these advances are reshaping the therapeutic landscape of AIHA toward a precision medicine model guided by mechanistic insights into disease biology. In this review, we delineate the evolving immunopathogenesis of AIHAs and examine emerging therapeutic strategies, integrating their underlying rationale, clinical data, and implications for future treatment paradigms.
1 Introduction
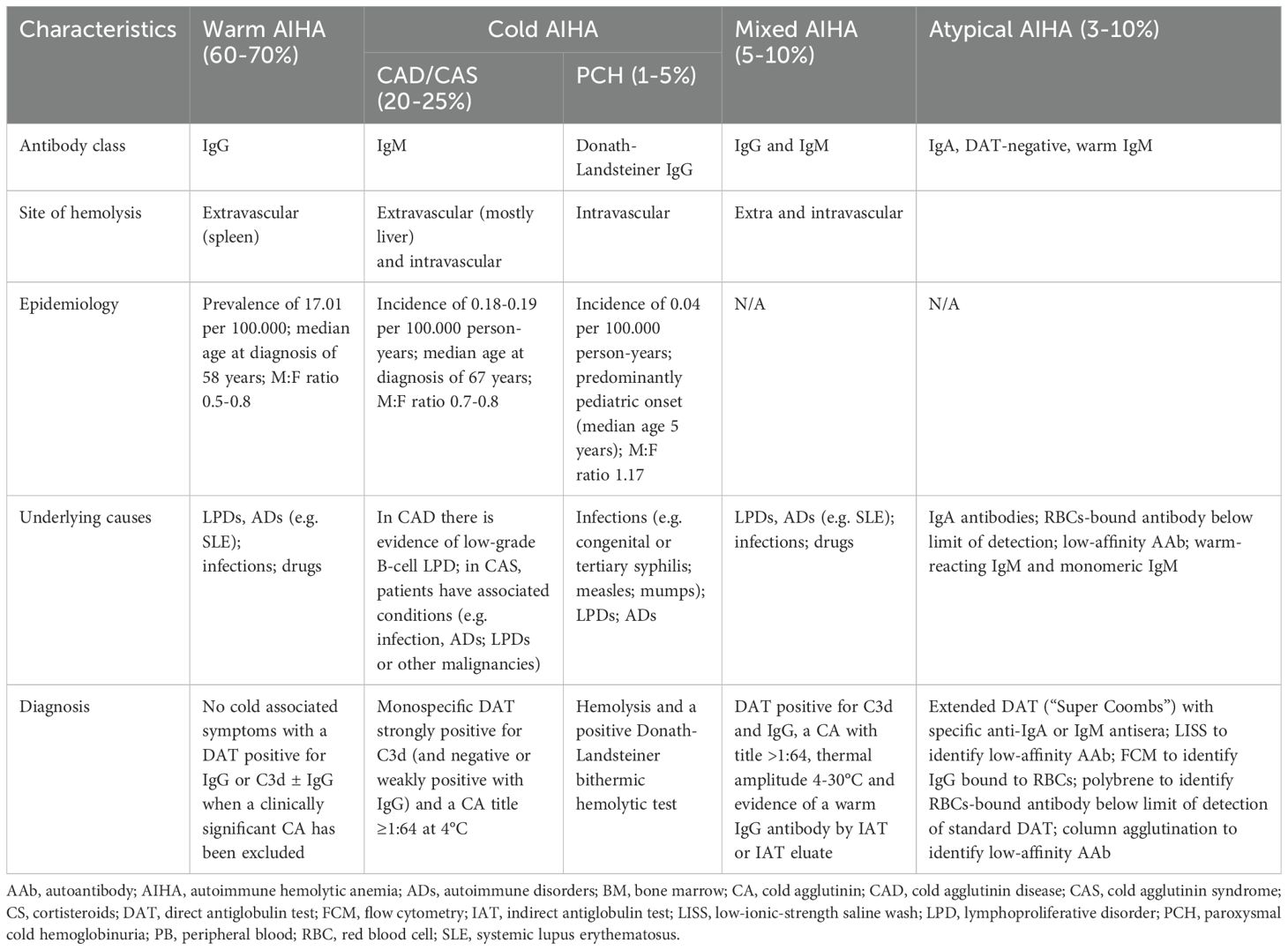
Autoimmune hemolytic anemias (AIHAs) are now recognized as complex immune-mediated disorders that extend beyond the traditional concept of isolated autoimmune erythrocyte destruction (1). The modern framework acknowledges this complexity, emphasizing the critical roles of both the innate and adaptive immune systems. Based on the isotype and thermal amplitude of the pathogenic autoantibody, these disorders are categorized into warm, cold, and mixed AIHA. However, the recognition of atypical forms, such as those with non-standard antibodies, negative direct antiglobulin test (DAT), or warm IgM involvement, has added complexity to diagnosis and classification (Table 1) (2). Thus, AIHAs figure as highly heterogeneous diseases, ranging from mild, stable cases to severe, relapsing, and potentially fatal ones (4). This heterogeneity is further illustrated by the variety of causes that can contribute to its onset. Genetic defects, alterations in immune response, neoplastic processes, infections, or drug exposure are commonly cited as pathogenic or predisposing factors (5).
Two interconnected immunopathogenic mechanisms are central to AIHA development. The first involves an autoreactive B-cell clone, sustained by a dysfunctional T-cell regulatory network, which produces autoantibodies that mediate primarily extravascular hemolysis via the reticuloendothelial system (2). The second crucial mechanism is complement activation, which exacerbates hemolysis, promotes cellular injury and sustains immune dysregulation. The resulting inflammation extends beyond red blood cell (RBC) destruction, affecting innate and adaptive immunity and increasing the risk of comorbidities such as vascular injury and thrombotic complications (3).
Emerging treatments targeting complement and B-cell clones are changing the AIHA treatment paradigm. As our understanding of AIHA deepens, it’s clear that a more comprehensive investigation of the connections between hemolysis, immune dysregulation, and inflammation is essential for improving patient outcomes and refining treatment strategies. This review examines the roles of innate and adaptive immunity and emerging therapies in AIHAs, aiming to enhance diagnosis, optimize treatment, and improve outcomes in these complex diseases.
2 Beyond the surface: divergent hemolytic pathways in warm and cold AIHA and the intricate cross-talk between complement and coagulation
2.1 Hemolytic mechanisms in warm AIHA
Warm AIHA, accounting for 60-70% of adult cases and approximately 50% of pediatric cases, is characterized by IgG autoantibodies with optimal binding at physiological temperature (∼37°C) (Table 1) (6). In the affected individuals, DAT is typically positive for IgG, and in most cases, the autoantibodies are polyclonal and directed against a range of erythrocyte antigens. The principal target is band 3 (SCL4A1), a major transmembrane anion exchanger. However, reactivity is frequently observed against other antigens, including Rh, Jk, Kell, Gerbich, Wright, glycophorin A and D, and Landsteiner-Wiener (7).
Hemolysis in warm AIHA predominantly occurs via extravascular mechanisms localized to the spleen and, to a lesser extent, the liver. Three principal immune effector pathways contribute to erythrocyte destruction (Figure 1): (1) antibody-dependent cellular phagocytosis (ADCP), (2) antibody-dependent cellular cytotoxicity (ADCC), and (3) complement-dependent cytotoxicity (CDC) (8). In the first two processes, the spleen serves as the primary site of extravascular hemolysis and the generation of autoreactive plasma cells, which sustain autoantibody production and contribute to hemolysis (9). Opsonized RBCs are recognized and phagocytized by red pulp macrophages via Fcγ receptors (FcγRs) IIa and IIIa (10). Among the IgG subclasses, FcγRs exhibit the highest affinity for IgG1 and IgG3 compared to IgG2 and IgG4 (11). Notably, splenic macrophages also express FcγRIIb, an inhibitory receptor whose activation may underlie the limited efficacy of intravenous immunoglobulin (IVIG) therapy in AIHA, as IVIG is thought to exert its effects, at least in part, through interactions with FcγRIIb (12). Natural killer (NK) cells and neutrophils, which express Fcγ receptors, can also mediate ADCC (13).
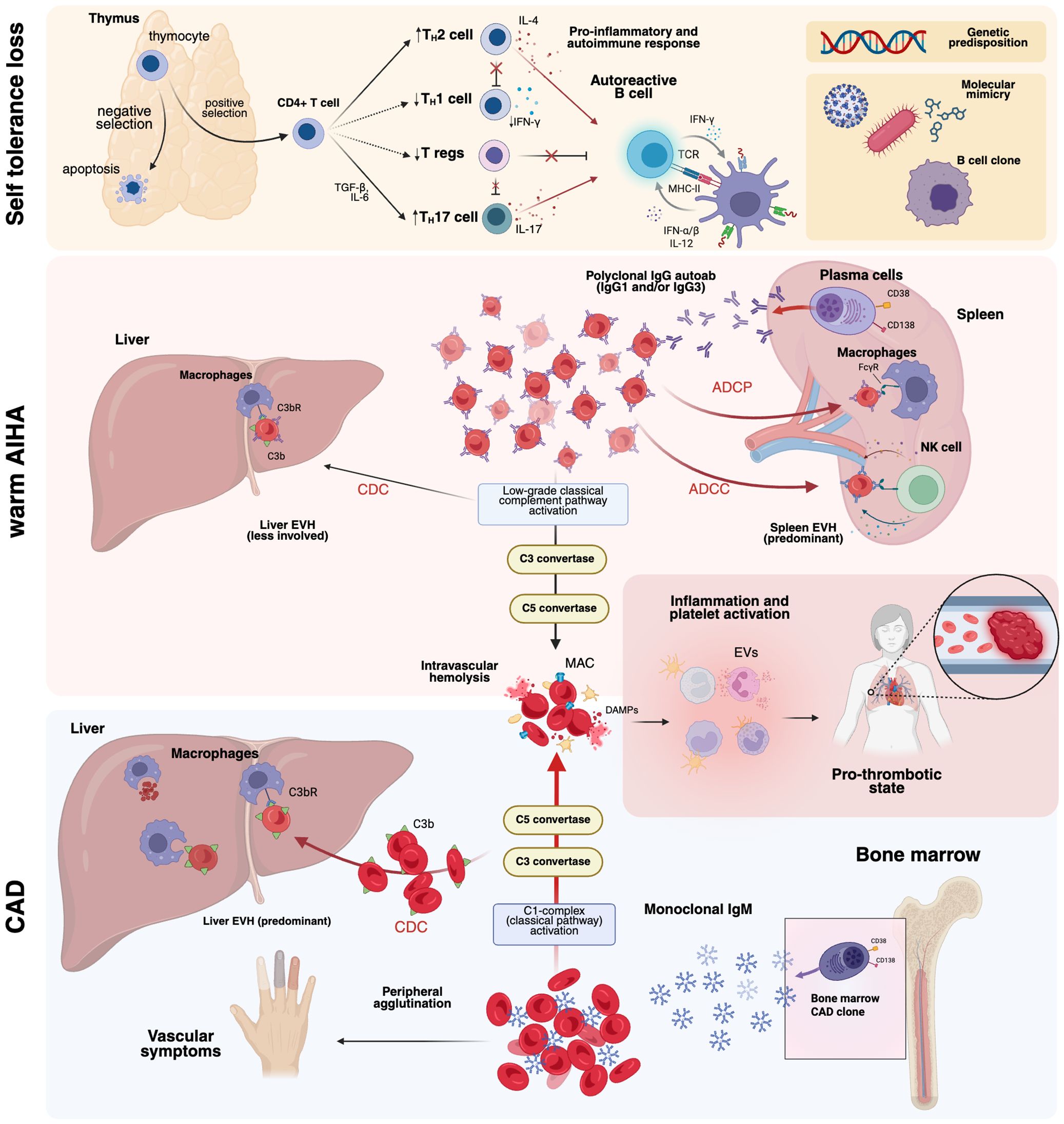
Figure 1. Overview of the pathogenesis of autoimmune hemolytic anemias (AIHAs). AIHAs result from a failure of immune tolerance, wherein defects in negative thymic selection permit the survival of autoreactive T lymphocytes. Dysregulation of TH1/TH2 homeostasis, coupled with genetic predisposition, contribute to aberrant B-cell activation. Molecular mimicry, often initiated by viral or bacterial infections, may further drive the clonal expansion of autoreactive B cells. In warm AIHA, autoantibodies primarily mediate EVH through splenic clearance via ADCP and ADCC, with a minor contribution from CDC in the liver. In contrast, in CAD, IgM autoantibodies bind erythrocytes at low temperatures, activating the classical complement pathway and leading to both IVH and EVH, primarily in the liver. ADCC, antibody-dependent cellular cytotoxicity; ADCP, antibody-dependent cellular phagocytosis; CAD, cold agglutinin disease; CDC, complement-dependent cytotoxicity. C3bR, C3b receptor; EVs, extracellular vesicles; EVH, extravascular hemolysis; IVH, intravascular hemolysis; MAC, membrane attack complex; TCR, T-cell receptor. Created in BioRender. Costa, A. (2025) https://BioRender.com/l08cmw5.
Although complement activation is generally less prominent in warm AIHA than in IgM-mediated cold AIHAs, IgG1 and IgG3 autoantibodies retain the capacity to activate the classical complement cascade through C1q binding. Following C1q engagement, conformational changes in the C1 complex (C1q, C1r, and C1s) initiate sequential proteolytic cleavage of C4 and C2 into C4a, C4b, C2a, and C2b, culminating in C3 convertase formation and downstream activation of the terminal complement cascade. This may result in variable degrees of intravascular hemolysis, although it typically remains a minor component compared to extravascular mechanism (14).
2.2 Hemolytic mechanisms in cold AIHA
Cold AIHA accounts for 20–25% of cases and is primarily driven by complement-mediated hemolysis. In these cases, DAT positivity is restricted to C3, and high-titer cold agglutinins exhibit optimal reactivity at low temperatures (typically 4–10°C) (Table 1) (15). A key distinction exists between cold agglutinin syndromes (CAS) associated with an underlying disorder and primary cold agglutinin disease (CAD), recently redefined as a clonal B-cell lymphoproliferative disorder in the 5th edition of the World Health Organization (WHO) classification (16). Specifically, CAD occurs in a clinically significant monoclonal gammopathy, where a clonal B-cell population produces auto-reactive IgM antibody. In contrast, in infection-associated CAS, IgM autoantibodies are usually polyclonal and less pathogenic than monoclonal ones (17).
In CAD/CAS, IgM autoantibodies primarily target the I antigen, which emerges on the erythrocyte membrane in adults following the enzymatic conversion of the fetal i antigen by β1,6-N-acetylglucosaminyltransferase (18). The pentameric nature of IgM promotes red cell agglutination, causing RBC aggregates that impair microcirculation in acral regions, where temperatures may fall within the thermal range permissive for agglutination even in the absence of environmental cold exposure (Figure 1) (19). Indeed, disease severity is primarily determined by the thermal amplitude of IgM rather than its serum titer, with a high thermal amplitude (>28°C) allowing for effective antibody binding even at normal body temperature (19). Notably, IgMκ binding to the erythrocyte surface in CAD induces agglutination and hemolysis by activating the classical complement pathway. Opsonization of RBCs by C3b promotes their extravascular clearance, primarily mediated by hepatic macrophages (20). However, terminal complement activation with membrane attack complex (MAC) C5b-C9 formation is generally absent during stable clinical phases and occurs predominantly during acute exacerbations or in severe disease (20, 21).
2.3 Hemolytic mechanisms in paroxysmal cold hemoglobinuria
Paroxysmal cold hemoglobinuria (PCH) is a rare and challenging entity within the spectrum of AIHA, accounting for less than 5% of cases and with an estimated annual incidence of 0.04 per 100.00 individuals (22). PCH is mediated by the Donath-Landsteiner antibody, a biphasic IgG that targets the RBC surface P antigen. Specifically, the Donath-Landsteiner IgG binds to its antigen at low temperatures and fixes early complement components C1, C4, and C2; upon warming to 37°C, the classical complement pathway is further activated, resulting in C3 cleavage and progression to terminal complement activation, which leads to acute intravascular hemolysis (22, 23). This pathogenic mechanism is unique among AIHA subtypes and contrasts with the primarily extravascular hemolysis observed in warm AIHA and CAD/CAS.
Historically, PCH was described as a chronic or recurrent complication of congenital or tertiary syphilis. The widespread use of penicillin led to a dramatic decline in syphilitic PCH, and the condition has since been reported predominantly in children following upper respiratory tract infections (24). However, recent epidemiological trends indicating a resurgence of syphilis underscore the need to reintegrate syphilis-associated PCH into the differential diagnosis of AIHA, including in adult patients (24).
2.4 Pro-thrombotic state in AIHA
Thrombosis is a major contributor to morbidity and mortality in AIHA, with both arterial and venous events occurring in 10–20% of cases (25–29). Retrospective data from 378 AIHA patients reported a 15% prevalence of venous thromboembolism (VTE), with warm AIHA and atypical variants carrying the highest risk (26). Notably, thrombotic rates appear consistent across different etiologies (30). Both a decade-long retrospective analysis and a Danish population-based study revealed a twofold increase in VTE risk in AIHA patients compared to non-AIHA controls (27, 28). Key risk factors include active hemolysis (hemoglobin ≤8 g/dL, elevated LDH), anemia severity, and prior splenectomy, emphasizing the role of hemolysis intensity in driving thrombosis (25, 29).
The classical coagulation model as a linear proteolytic cascade has evolved into a more integrated framework, incorporating the interplay between hemostasis, innate immunity, and the complement system (31). This revised paradigm is particularly relevant to AIHA, where intravascular hemolysis, complement activation, and inflammatory signaling converge to establish a prothrombotic state. While the mechanisms of hemolysis are well-characterized (32), emerging evidence provides novel insights. Mannes et al. (33) demonstrated that adenosine diphosphate (ADP), released upon MAC-dependent erythrocyte lysis, contributes to platelet activation and thrombotic propagation, in line with the pro-inflammatory role of damage-associated molecular patterns (DAMPs), such as heme (Figure 1) (31). Erythrocyte-derived extracellular vesicles (EVs), enriched in phosphatidylserine and tissue factor, have been implicated in AIHA-associated hypercoagulability. Barcellini et al. (34) found that EV levels were elevated in hemolytic patients, showing a correlation with the severity of anemia. Notably, patients with AIHA had much higher EV counts than healthy controls. Beyond hemolysis and erythrocyte-derived DAMPs, complement activation emerges as a key driver of the prothrombotic environment. A bidirectional interaction between complement and coagulation has been described (35), where complement activation enhances coagulation and vice versa through multiple mechanisms. C3 directly binds to fibrin, stabilizing clots and increasing resistance to fibrinolysis (36). Moreover, MAC activation on platelets at sublytic levels induces the externalization of procoagulant phospholipids, amplifying thrombin generation (37). Complement activation also drives the release of C3a and C5a, potent anaphylatoxins that recruit inflammatory cells and sustain thrombogenic inflammation (38). Conversely, thrombin cleaves C5 and C5b into non-canonical fragments that promote highly lytic MAC formation, linking coagulation and complement in a self-perpetuating cycle (35). These findings suggest that thrombotic risk in AIHA is not solely driven by hemolysis but reflects complex interactions between complement, platelets, and inflammation. This crosstalk underscores the rationale for complement-targeted therapies to mitigate thrombosis and improve clinical outcomes in AIHA.
3 Beneath the surface: self-tolerance breakdown and hidden drivers of hemolysis in AIHA
3.1 Mechanisms of self-tolerance loss
Historically, autoimmunity has been attributed to the interplay between genetic predisposition and environmental triggers, including infections, medications, or concomitant diseases, which together drive the emergence and clonal expansion of autoreactive lymphocytes (Figure 1) (39). In AIHAs, a known mechanism is molecular mimicry, where foreign antigens share structural or functional similarities with self-proteins, triggering cross-reactive immune responses. Notably, Herpesviridae (e.g., EBV, CMV, Varicella-Zoster virus) and Poxviridae exhibit a significantly higher degree of linear peptide homology with human proteins than other viral families, supporting their role in immune dysregulation (40). This concept gained attraction during the COVID-19 pandemic, as autoimmune cytopenias emerged in SARS-CoV-2-infected patients (41). Angileri et al. (42) identified an immunogenic epitope (LLLQY) shared between the RBC cytoskeletal protein ANK-1 and the SARS-CoV-2 spike glycoprotein, providing direct evidence of viral-induced AIHA (43). Beyond mimicry, viruses may further drive autoimmunity by exposing cryptic intracellular antigens upon cell lysis (44).
A similar mechanism may underlie drug-induced immune hemolytic anemia (DIIHA), where drug-dependent autoantibody formation occurs through immune-allergic hypersensitivity or non-immunologic protein adsorption (45). Among drugs commonly associated with AIHA, furosemide, antibiotics (e.g., amoxicillin, ceftriaxone, cefixime, cefpodoxime, ciprofloxacin, amphotericin B, sulfamethoxazole/trimethoprim, norfloxacin), azathioprine, NSAIDs (e.g., ibuprofen), and paracetamol have been implicated, with azathioprine carrying the highest associated risk (46).
While T-cell dysregulation has been a dominant focus, recent attention has shifted back to B-cell involvement, particularly the concept of a “forbidden clone”, an autoreactive B-cell population that escapes immune tolerance checkpoints, fueling persistent autoantibody production (47). Whether autoimmunity precedes or follows B-cell clonal expansion remains debated. Epidemiologic data show an increased risk of Non Hodgkin lymphomas (NHL) in patients with preexisting autoimmune diseases such as celiac disease and rheumatoid arthritis, supporting a two-hit model in which loss of immune tolerance precedes oncogenesis through chronic inflammation, persistent antigenic stimulation, and genetic susceptibility (48). Conversely, B-cell malignancies can drive autoimmunity via humoral and cellular dysregulation, as seen in hypogammaglobulinemia and altered regulatory T-cell subsets, underscoring the bidirectional link between lymphoproliferation and immune dysregulation (49). This is exemplified in AIHA associated with chronic lymphocytic leukemia (CLL), characterized by autoreactive polyclonal B cells alongside neoplastic monoclonal B cells (49).
The involvement of autoreactive lymphocyte clones is well established in warm AIHA and CAS; however, recent insights have refined the pathogenic understanding of CAD. Although overt lymphoid malignancy is typically absent, most CAD patients harbor a monoclonal IgMκ B-cell population, indicative of an underlying lymphoproliferative disorder (Figure 1). Randen et al. (50) delineated a clinically and immunophenotypically homogeneous entity in a cohort of 54 CAD patients. Bone marrow examinations revealed nodular infiltrates of mature B cells, lacking distinct morphological features, negative for the MYD88 L265P mutation, and not fulfilling diagnostic criteria for lymphoplasmacytic lymphoma (50). This evolving paradigm redefines CAD as a distinct disorder, separate from other lymphoproliferative neoplasms, thereby sustaining its pathogenic framework and inclusion in the latest WHO classification.
3.2 Genetic background
Susceptibility to autoimmune diseases, including AIHA, is influenced by genetic variants that modulate immune responses. Given the critical role of the human leukocyte antigen (HLA) system, studies have focused on polymorphisms in specific HLA loci that alter the selection and activation of autoreactive T lymphocytes. Notably, HLA-B8 and BW6 alleles have been associated with an increased risk of AIHA, suggesting their involvement in the persistence of autoreactive lymphocyte clones (51). Additionally, specific immunoglobulin heavy-chain variable (IGHV) gene configurations appear to promote the selection of autoreactive B-cell clones (52, 53). For instance, IGHV4-34, IGHV4-31, IGHV3-23, and IGK3–20 gene rearrangements have been found in CAD patients (54–56). Moreover, genetic variants such as the G polymorphism in CTLA4 and the AG configuration in the lymphotoxin-α gene have been reported at higher frequencies in AIHA patients (57, 58).
In CAD, next-generation sequencing (NGS) has revealed gene mutations affecting B- and T-cell function. Notably, mutations in KMT2D, which encodes a histone methyltransferase involved in B-cell survival, differentiation, and homing, are present in 69% of cases. In contrast, mutations in CARD11, a key regulator of the NF-κB signaling pathway, are detected in 31% (59). Emerging candidate genes include IGLL5, whose precise role in B-cell development remains under investigation, and CXCR4, which plays a critical role in B-cell migration and trafficking (60). Additionally, decreased expression of CR1, a negative regulator of B-cell activation and differentiation, has been reported in CAD (56). The genetic contribution is even more pronounced in pediatric Evans syndrome (pES), a condition characterized by AIHA, immune thrombocytopenia, or autoimmune neutropenia (61). In a cohort of 80 pES patients, 40% were found to harbor mutations in immune-regulatory genes such as TNFRSF6, CTLA4, STAT3, PIK3CD, CBL, ADAR1, LRBA, RAG1, and KRAS (62).
Moreover, the role of polymorphisms in inflammatory cytokines has gained recent attention, further elucidating the complex genetic and immune dysregulation underlying AIHA. Specifically, Zaninoni et al. (63) identified single-nucleotide polymorphisms (SNPs) in genes encoding TNF-α, TGF-β1, IL-10, IL-6, and interferon γ (IFN-α), showing associations with disease severity and treatment response. In line with Pavkovic et al. (64), AIHA patients exhibited a lower frequency of the TNF-α -308 G/A polymorphism compared to controls, as well as a reduced genotypic frequency of TNF-α -308 G/A and TGF-β codon 25G/C. The genetic link with cytokines was further emphasized in pES, where PTPN2 haploinsufficiency has been identified, contributing to heightened sensitivity to cytokine signaling through the JAK/STAT pathway (65).
3.3 T cell dysregulation and polarization
Self-tolerance is a fundamental principle of immune homeostasis, essential for preventing aberrant autoreactive responses (Figure 1). In the thymus, central tolerance serves as the primary checkpoint for T lymphocyte selection and is coordinated by medullary thymic epithelial cells, cortical thymic epithelial cells, dendritic cells, and thymic B cells (66). These cells mediate antigen presentation and the clonal deletion of autoreactive T lymphocytes. Thymocytes expressing T cell receptors (TCRs) that fail to engage self-peptides presented via major histocompatibility complex (MHC) molecules undergo death by neglect due to insufficient survival signaling (67). Despite these mechanisms, up to 40% of autoreactive T cells and a comparable fraction of autoreactive B cells evade central tolerance (68, 69). Peripheral regulatory networks act as additional safeguards to limit their activation and expansion. These include T cell anergy, mediated by inhibitory receptors such as CTLA-4, clonal deletion via Fas-Fas ligand interactions, and suppression by CD4+/CD25+/Foxp3+ regulatory T cells (Tregs), which exert immunomodulatory effects through cytokines such as interleukin 10 (IL-10) and TGF-β (66, 70, 71).
T cells play a pivotal role in the pathogenesis of AIHA, contributing to the breakdown of immune tolerance and to the production of anti-erythrocyte autoantibodies. Unlike other self-reactive T cells, those targeting RBC antigens escape thymic negative selection. Instead, CD4+ recent thymic emigrants (RTEs) encounter RBCs in the periphery, where they modulate inhibitory receptor expression, particularly programmed death cell 1 (PD-1), and transcription factors associated with anergy, functional exhaustion, and regulatory differentiation (72). This dynamic suggests that tolerance to RBC autoantigens is primarily maintained through peripheral rather than central mechanisms.
In murine models, an imbalance between T helper 1 (TH1) and T helper 2 (TH2) subsets has been described, with a predominant TH1 response characterized by elevated IFN-γ production. Disease improvement in these models occurred in response to the TH2 cytokine IL-4 (73, 74). Conversely, human warm AIHA has traditionally been classified as a TH2-driven disorder, as indicated by elevated IL-4 levels and reduced IFN-γ secretion (75). Moreover, like other autoimmune diseases, AIHA seems to be marked by an imbalance between effector T cells and Tregs (76). The pathogenic role of T helper 17 (TH17) cells has been well documented in multiple inflammatory disorders (77), and an elevated presence of TH17 cells has been observed in AIHA patients, strongly correlating with disease activity (78). In murine models, dysfunction in CD4+/CD25+ Tregs impedes autoimmunity suppression, facilitating the production of erythrocyte autoantibodies (79). Recently, Ciudad et al. (80) observed a shift favoring TH17 polarization within T effector cells, associated with elevated serum IL-17 levels, aligning with findings by Xu et al. (78). Additionally, a reduction in circulating Tregs and decreased Foxp3 expression, a key transcription factor for Treg stability and suppressive function, were noted (80). Consistent with these observations, interference with RTE tolerance mechanisms using immune checkpoint inhibitors led to a concurrent decline in Foxp3+ Tregs and an increase in TH17 cells (81).
CD39 single-positive CD4+ T cells emerged as a predictive marker for AIHA development, suggesting their direct involvement in tolerance breakdown. CD39, an ectonucleotidase that hydrolyzes ATP and other extracellular nucleotides, modulates the immune microenvironment. Its dysregulation may contribute to the expansion of proinflammatory T cells, particularly TH17, while concurrently reducing regulatory populations, thereby promoting an autoimmune response against erythrocytes (81).
Increasing attention is given to the roles of follicular helper T cells (THF) and follicular regulatory T cells (TFR) in autoimmune pathogenesis and erythrocyte hemolysis. THF cells play a central role in complex regulatory events within the germinal center, including germinal center formation, B-cell support, isotype switching, high-affinity antibody production, and memory cell differentiation (82). In this setting, IL-6 and IL-21 contribute to THF differentiation and function by upregulating the transcriptional repressor BCL6. In AIHA, a murine study reported increased levels of both THF and TFR cells alongside elevated IL-21 and IL-6 levels. The pathogenic involvement of these subsets was further confirmed by the adoptive transfer of purified CD4+/CXCR5+/CD25− THF cells from immunized mice, which induced autoantibody production in an AIHA murine model (83). Similarly, 24 patients with pES exhibited increased circulating THF cells, heightened T-cell activation, and a reduction in naïve CD4+ T cells compared to healthy controls and patients with chronic immune thrombocytopenia (84). Post-activation exhaustion features were also noted, with upregulation of canonical checkpoint inhibitors, and a higher degree of β-chain oligoclonality in the TCR was observed in THF cells compared to healthy controls.
3.4 Long-lived plasma cells and treatment failure
Following B-cell activation and differentiation within secondary lymphoid organs and inflamed tissues, a subset of plasma cells migrates to bone marrow niches, where they persist as long-lived plasma cells (LLPCs), comprising approximately 25% of the total bone marrow plasma cell pool (85). These CD38+/CD138+/CD20- cells are responsible for sustained long-term antibody production, a function that can persist for decades (86). Their survival is regulated by stromal and immunoregulatory signals from dendritic cells, stromal cells, and regulatory T cells, which, through molecules such as CD80/86, CXCL12, and B-cell activating factor (BAFF), promote their persistence (86). At the molecular level, LLPCs overexpress anti-apoptotic genes (MCL1, BCL2, BCL-XL), making them highly resistant to immune-mediated depletion (87).
In warm AIHA, persistent autoantibody production is driven by autoreactive plasmablasts and plasma cells, with circulating plasmablasts indicating ongoing B-cell activation (9). Once settled in the spleen and bone marrow, LLPCs become refractory primarily to conventional therapies, including corticosteroids, rituximab, and other immunosuppressive agents targeting CD20-positive B cells. This limitation in therapeutic efficacy is particularly relevant in steroid-refractory or relapsing AIHAs, where ongoing autoantibody production by LLPCs sustains hemolysis despite the effective depletion of peripheral B cells (9). Moreover, it has been observed that B-cell depletion-induced alterations in the splenic microenvironment create a dependency of plasma cells on BAFF and CD4+ T cells. This suggests that the expression profile of LLPCs is influenced by signals derived from the splenic microenvironment (88).
4 Diagnostic approach and challenges in AIHA
4.1 Role and pitfalls of DAT
Precise classification of AIHA subtypes is essential to tailor treatment strategies and predict clinical outcomes. AIHA should be suspected in patients presenting with anemia and laboratory evidence of hemolysis, with the DAT serving as the primary diagnostic tool to confirm the immune-mediated nature of hemolysis (Table 1) (89, 90). The DAT detects immunoglobulin and/or complement bound to RBCs by using reagents that bind these components and induce visible agglutination in vitro (91).
The DAT-tube has traditionally been performed using polyspecific antiglobulin reagents, which yield semi-quantitative results without differentiating antibody isotypes or thermal amplitude. The composition of these reagents can vary significantly in the proportion of anti-IgG, anti-IgM, anti-IgA, and anti-complement components, often favoring anti-IgG (92). This approach can lead to false-negative results in cases involving IgA antibodies, low-affinity autoantibodies, or when the number of IgG molecules bound to RBCs falls below the assay’s detection threshold. The use of monospecific DAT enables accurate identification of specific autoantibody classes, thereby enhancing differential diagnosis and informing more precise therapeutic strategies (2, 15, 93) Additionally, employing low ionic strength solutions (LISS) or cold washes can overcome DAT negativity. Although less specific, more sensitive methods such as microcolumn and solid-phase antiglobulin tests allow for detection of low levels of IgG coating RBCs (93).
Among the most diagnostically challenging variants of AIHA, PCH warrants particular attention. The diagnostic process in PCH is often complex and far from straightforward (22). The DAT usually reveals isolated C3d positivity, which may also be observed in other complement-mediated forms of AIHA (22). Definitive diagnosis relies on the Donath-Landsteiner test, a technically demanding assay that is not routinely available outside of specialized laboratories (94). Therefore, clinicians should consider PCH in patients presenting with acute hemolysis and hemoglobinuria, especially when typical warm or cold autoantibody patterns are absent (22, 23).
4.2 Clinical assessment and diagnostic work-up
A detailed clinical history is essential to exclude alternative causes of hemolysis or confounding factors, such as recent transfusion reactions, drug-induced hemolysis, or glucose-6-phosphate dehydrogenase (G6PD) deficiency, which is more prevalent in specific populations (95). Evaluation should also include assessment for underlying disorders, particularly systemic autoimmune diseases (e.g., systemic lupus erythematosus) and lymphoproliferative malignancies like CLL or indolent NHL (90). Patient age can inform diagnostic considerations, with younger individuals more likely to present with underlying infections or autoimmune diseases, while older patients have an increased likelihood of harboring an active neoplastic disorder. Notably, infection screening is warranted when patients present with recent fever or respiratory symptoms; testing for EBV infection is particularly relevant in children and young adults (3). Signs such as weight loss, lymphadenopathy, hepatosplenomegaly, lymphocytosis, or cytopenias should prompt investigation for lymphoproliferative disorders.
Beyond standard hemolytic markers, extended immunohematologic and biochemical workup is essential to distinguish primary from secondary forms of AIHA (90, 96). Although this distinction is not immediately critical, since first-line treatment generally follows the same approach as for primary AIHA, it becomes clinically relevant when an underlying disorder requires targeted management (97). Autoantibody panels that include antinuclear antibodies (ANA), extractable nuclear antigens (ENA), anti-DNA, and antithyroid antibodies, should be considered in patients with suggestive clinical features such as arthralgias, serositis, or thyroid dysfunction. These tests may uncover an underlying connective tissue disease, guiding the use of immunosuppressants or targeted biologics (90). Moreover, in patients with a history of thrombotic events or recurrent abortion, antiphospholipid antibody testing is advisable. Viral serologies are also recommended (98). Serum protein electrophoresis, immunofixation, and quantitative immunoglobulin assays can help identify cases associated with plasma cell dyscrasias, such as Waldenström macroglobulinemia (99), or with primary immunodeficiencies (100). Bone marrow evaluation, including morphologic, histopathologic, immunophenotypic, and cytogenetic analysis, is essential for diagnosing AIHA associated with lymphoproliferative disorders, myelodysplastic syndromes, or marrow failure syndromes (90). This is particularly important at diagnosis in patients with CAD and in relapsed warm AIHA patients who are corticosteroid-refractory (90). Additionally, total-body computed tomography (CT) scanning is fundamental for staging and identifying lymphadenopathy or splenomegaly, which may indicate underlying lymphoid malignancies.
5 From biology to current and future therapeutic solutions in AIHA
Over the past decades, substantial advances in understanding AIHA pathophysiology have laid for a more tailored, subtype-specific therapeutic approach. However, despite these biological insights, current treatments still present important limitations. In warm AIHA, corticosteroids remain the mainstay of first-line treatment, with initial response rates of approximately 80% (97). Yet, sustained remission after tapering is achieved in only 30–40% of cases at one year (2, 4, 97). In CAD, corticosteroids are largely ineffective and should be avoided due to poor efficacy and frequent relapses after withdrawal. This reflects their inability to suppress autoantibody production in CAD. Likewise, splenectomy has no role in CAD due to hepatic rather than splenic clearance of erythrocytes (2, 4). Rituximab, targeting CD20+ B cells, has become the preferred second-line option in wAIHA and is increasingly used as a first-line treatment in CAD (97). Nonetheless, responses are often transient, and the efficacy of retreatment remains unclear. Other immunosuppressive agents, once used more broadly before the rituximab era, now play a limited role, often confined to later-line, steroid-sparing strategies, and lack strong prospective evidence (101, 102).
These limitations underscore a critical unmet need. The identification of B-cell dysregulation and complement activation as core pathogenic pathways has led to the development of novel targeted therapies. This progress reinforces the importance of precise disease classification, which now plays a central role in therapeutic planning and sequencing, marking a shift toward increasingly personalized and tailored management.
5.1 B cell-directed therapeutic strategies
Several therapeutic strategies that directly target B cells involved in autoantibody production (Figure 2) have been explored. These approaches aim to counteract relapse and treatment resistance, often driven by the persistence and escape mechanisms of autoreactive B cells at different stages of maturation. Among B-cell-directed therapies, targeting the B-cell receptor (BCR) signaling pathway is particularly relevant, given its central role in lymphoproliferative disorders and autoimmune diseases. The BCR regulates B-cell survival, proliferation, and activation through a complex intracellular signaling network involving Bruton tyrosine kinase (BTK), phosphoinositide 3-kinase (PI3K), and spleen tyrosine kinase (SYK) (103). The following sections explore these therapeutic strategies, evaluating their mechanisms of action, clinical efficacy, and limitations in the context of B cell-mediated autoimmunity.
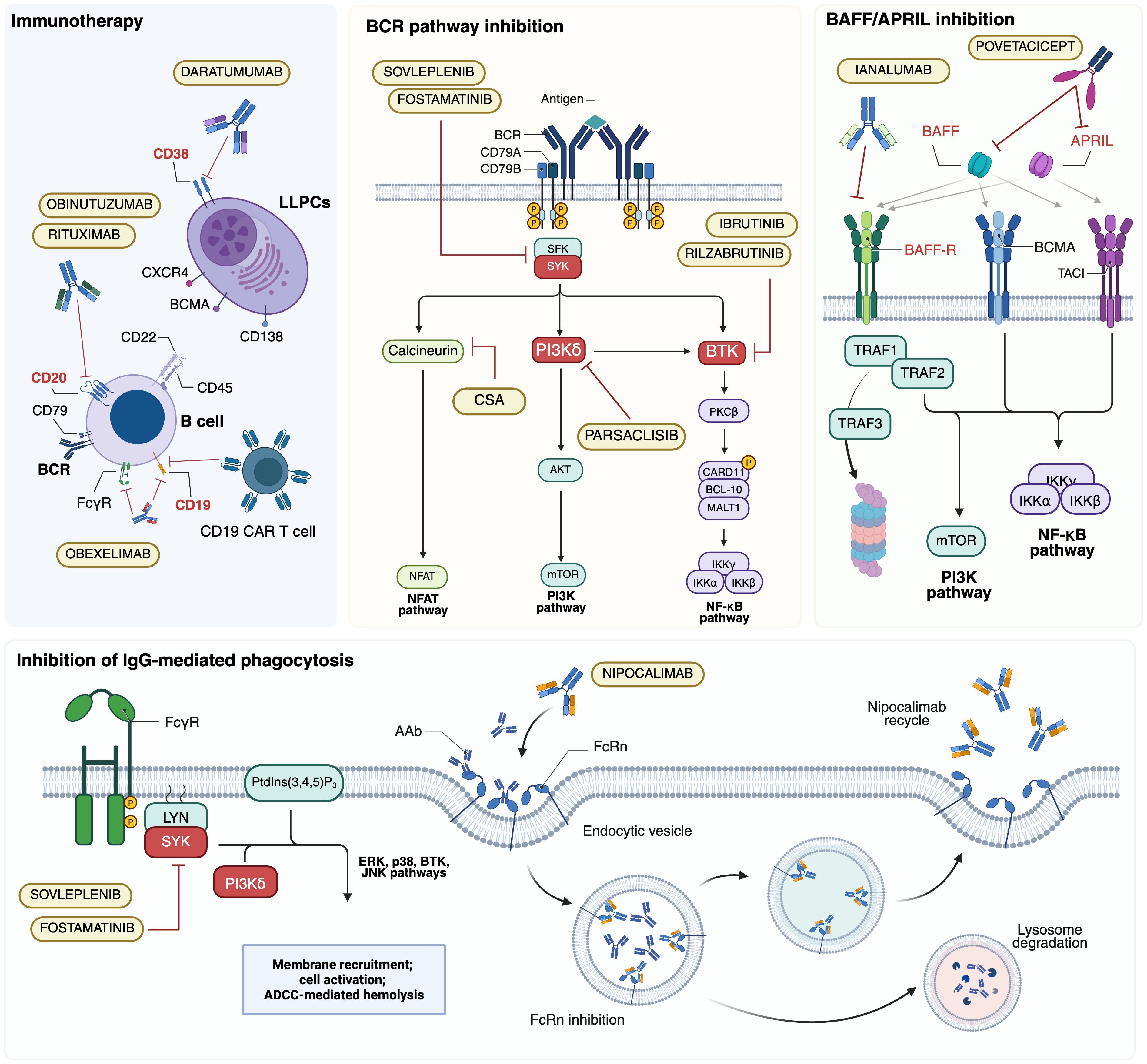
Figure 2. Major B-cell targeted therapy approaches investigated in autoimmune hemolytic anemias (AIHAs). AAb, autoantibody; ADCC, antibody-dependent cellular cytotoxicity; APRIL, a proliferation-inducing ligand; BAFF, B cell activating factor; BAFF-R, BAFF receptor; BCMA, B cell maturation antigen; BCR, B-cell receptor; BTK; Bruton tyrosine kinase; CAR T, chimeric antigen receptor T; CSA, ciclosporin A; FcγR; Fc γ receptor; FcRn, neonatal Fc receptor; LLPCs, long-lived plasma cells; PI3K, phosphoinositol 3-kinase; PtdIns(3,4,5)P3, phosphatidylinositol-3,4,5-trisphosphate; SYK, splenic tyrosine kinase; TACI, transmembrane activator and CAML Interactor. Created in BioRender. Costa, A. (2025) https://BioRender.com/kl39pwf.
5.1.1 Anti-CD20 and anti-CD38 immunotherapies
Autoreactive CD20+ B lymphocytes play a central role in the pathogenesis of AIHAs, making them a crucial therapeutic target. Rituximab, a human IgG1κ MoAb originally developed for B-cell malignancies, was the first MoAb introduced in this setting (Table 2) (110). By binding to CD20 on the lymphocyte surface, rituximab induces apoptosis through ADCC and CDC mechanisms while modulating immune homeostasis by increasing Tregs and rebalancing the TH1/TH2 axis (104). In CAD, rituximab remains the preferred first-line therapy, inducing responses in approximately 50% of patients at one year, though durability remains a concern (111). Adding fludarabine enhances response rates but at the cost of increased toxicity, whereas bendamustine appears safer and potentially more effective, with response rates exceeding 70% (112, 113). In warm AIHA, rituximab is the preferred second-line treatment for steroid-refractory cases, achieving 70–80% response rates within 3–6 weeks (105, 114).
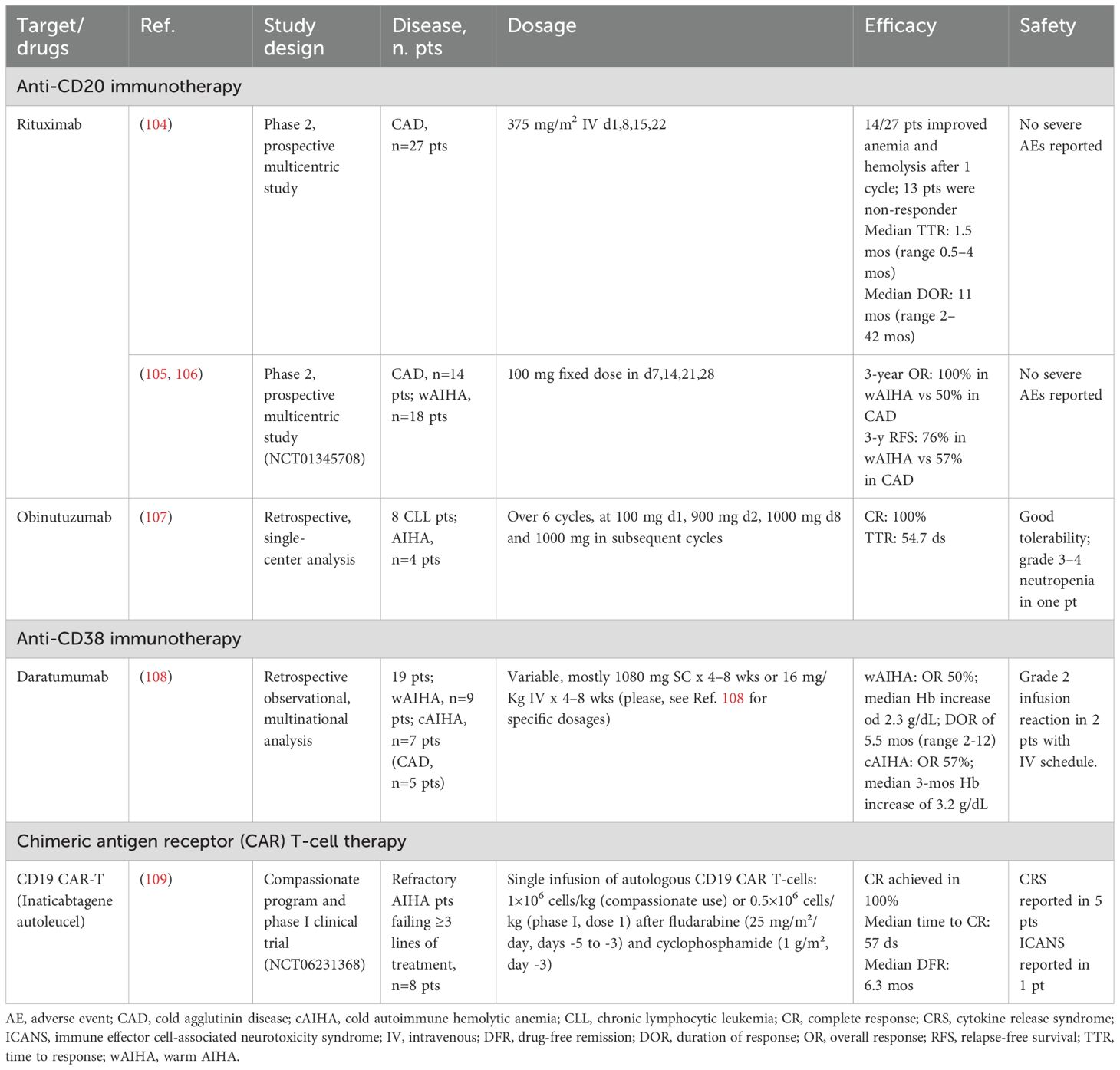
Table 2. Immunotherapeutic target and strategies in AIHA.
Recognizing the lower B-cell burden in autoimmune diseases compared to lymphoproliferative disorders, reduced-dose rituximab regimens have been explored. Studies investigating low-dose schedules (100 mg weekly for four weeks) reported response rates of 90%, with warm AIHA patients exhibiting superior responses and longer disease-free survival than cold forms (106, 115). Further insights come from a recent phase 2 pilot study assessing ultra-low rituximab doses (5 mg/m² every three weeks, 20 mg every four weeks, 50 mg every three months, and 100 mg every three months) in relation to CD20+ cell suppression (116). While nearly all patients achieved ≥95% CD20+ clearance after the first infusion, sustained depletion was not maintained. Instead, plasma rituximab levels above 0.4 μg/ml were required for complete peripheral CD20+ clearance.
Obinutuzumab, a second-generation anti-CD20 MoAb with enhanced direct cytotoxicity and ADCC has also been evaluated in AIHA, although data remains scarce. The most extensive analysis to date is a retrospective study of eight patients with CLL/SLL treated with obinutuzumab monotherapy for AIHA (n=4) or immune thrombocytopenia (ITP, n=4) (107). All AIHA patients achieved complete response, maintained at a median follow-up of 15 months. Ofatumumab, another anti-CD20 MoAb, has been used in isolated cases (117, 118). However, further studies are needed to establish the efficacy of anti-CD20 MoAbs beyond rituximab.
Considering the persistent autoantibody production by LLPCs, targeting CD38+ cells offers a novel therapeutic approach. Daratumumab, an IgGκ MoAb developed initially for multiple myeloma, was first reported by Scheutz et al. (108) in two cases of life-threatening warm AIHA post-stem cell transplantation, both successfully treated. At the same time, a third patient experienced a fatal relapse. Recent studies report a 50% response rate in warm AIHA with a median duration of 5.5 months, whereas in CAD, 57% of patients showed hemoglobin improvement, with clinical benefits such as reduced acrocyanosis (119). Additionally, due to CD38 expression on T lymphocytes, daratumumab may exert an immunomodulatory effect. A prospective analysis of two warm AIHA patients revealed a complete depletion of CD38+ T cells, impairing their activation and proliferation. Notably, in one patient, disease relapse coincided with the reappearance of CD38+ T cells, suggesting a link between therapeutic response and T cell repopulation.
Another anti-CD38 antibody of interest is isatuximab, which has a higher affinity for CD38 than daratumumab. A phase 1b/2 trial investigating its use in warm AIHA was initiated but was prematurely discontinued due to sponsor-driven strategic decisions (120).
Finally, obexelimab, a bifunctional, non-depleting humanized monoclonal antibody targeting CD19 and FcγRII, represents a promising therapeutic option for AIHA. Currently under investigation in a Phase 2 study (NCT05786573), its safety and efficacy are being evaluated in patients with warm AIHA. Interim results from six patients show a notable improvement in hemoglobin levels from baseline. Adverse events were reported in four patients, with three of 18 events considered drug-related. Further data are awaited to confirm these findings (121).
5.1.2 BTK inhibitors
The BTK inhibitors have significantly improved treatment outcomes for patients with CLL/SLL and other related conditions (122). These agents are also being explored in autoimmune, allergic, and inflammatory disorders, including CAD. Notably, safety and efficacy data have emerged from a multinational retrospective analysis of 15 cold AIHA patients, 4 diagnosed with CAD and 11 with CAS, all receiving ibrutinib (123). Hemoglobin levels improved in all patients, with 12 achieving complete responses and one partial response, alongside transfusion independence and improved acrocyanosis. Adverse events were consistent with the known safety profile of ibrutinib. Rilzabrutinib, a reversible covalent BTK inhibitor, has been evaluated in a multicentric open-label phase 2b trial (Table 3). Preliminary results indicate a 64% response rate, with durable effects in 41% of patients (124). It is important to note that 86% of patients experienced adverse events, 18% of which were severe, with 45.5% deemed treatment-related. Further data is necessary to assess its safety and efficacy fully. Lastly, zanubrutinib, a next-generation, irreversible BTK inhibitor, is under investigation in a phase 2 trial (NCT05922839) for patients with warm AIHA and in a separate phase 2 trial involving CAD (NCT06067048).
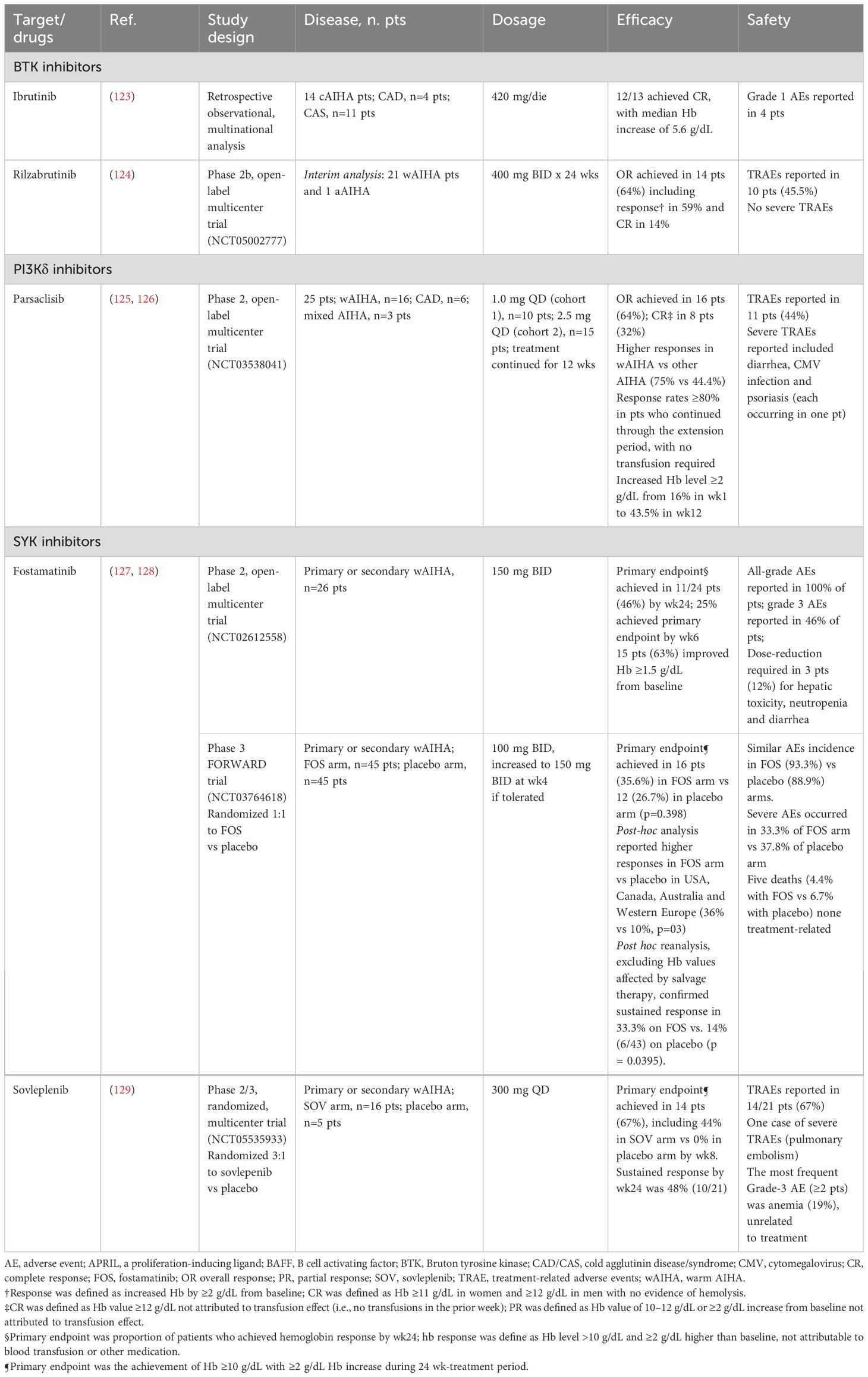
Table 3. B cell receptor (BCR) pathway directed therapies in autoimmune hemolytic anemias (AIHAs).
5.1.3 PI3Kδ inhibitors
Another key target downstream of the BCR is PI3K, which plays an essential role in B-cell differentiation and survival (130). Parsaclisib, a selective and potent PI3Kδ inhibitor, has shown considerable efficacy in treating B-cell malignancies. Preclinical studies have demonstrated its ability to suppress B-cell proliferation, modulate regulatory T-cell homeostasis, inhibit CD8+ T-cell maturation, and influence immune compartment dynamics (125). In AIHA, preliminary findings from a phase 2 study of primary AIHA patients showed good tolerance and normalization of hemoglobin levels after 12 weeks of treatment with parsaclisib (126). Recent long-term data from 25 patients revealed early hemoglobin responses within the first week for half of the patients, with sustained improvements in the following weeks, leading to a 64% rate of partial or complete responses (131). Notably, higher responses were seen in patients with warm AIHA than those with CAD or mixed types. The drug was well tolerated, with 44% of patients experiencing treatment-related adverse events. A phase 3 randomized placebo-controlled trial is currently underway to evaluate its safety and efficacy in patients with primary warm AIHA (132).
5.1.4 Inhibition of IgG-mediated phagocytosis
SYK is a key player in Fcγ-mediated phagocytosis of opsonized erythrocytes and B-cell activation/differentiation, making it an attractive therapeutic target. Its inhibition offers a twofold strategy: reducing autoantibody production and erythrocyte destruction. Fostamatinib, an oral SYK inhibitor, has received regulatory approval for ITP and has shown potential in warm AIHA. Based on preclinical efficacy in murine models (133), a Phase 2 study (NCT02612558) in warm AIHA reported a hemoglobin response in 46% of patients (Table 3) (127). The FORWARD trial further assessed fostamatinib in 90 patients with warm AIHA, randomized 1:1 to either placebo or fostamatinib (100 mg twice daily, escalating to 150 mg BID if tolerated) (128). Notably, two-thirds of patients had received at least one prior therapy, nearly all had been exposed to steroids, and half had been treated with rituximab. Although a greater proportion of fostamatinib-treated patients achieved hemoglobin improvement versus placebo, statistical significance was not reached. Regional differences were noted, with a more pronounced response in North American, Australian, and Western European patients, likely attributable to protocol deviations (including salvage steroid therapy during screening and two placebo-treated cases later determined unlikely to have warm AIHA) rather than true geographic differences (128). The drug was well tolerated, with diarrhea, hypertension, and fatigue as the most frequent adverse events.
A next-generation oral SYK inhibitor, sovleplenib, was evaluated in a Chinese Phase 2/3 study (NCT05535933) in 16 patients, with five receiving a placebo (129). By week 24, 67% of sovleplenib-treated patients achieved a hemoglobin response. Adverse events occurred in all patients, with 67% of them deemed drug-related. Further data are awaited to define its clinical role better.
FcRn inhibition, aimed at blocking IgG recycling and lowering circulating pathogenic antibodies, is also under investigation. Nipocalimab (M281), an anti-FcRn monoclonal antibody, has demonstrated promising preliminary results in a placebo-controlled Phase 1 trial on healthy volunteers (134). It is currently being evaluated in the Phase 2/3 ENERGY trial (NCT04119050), a randomized placebo-controlled study as an adjunct to standard therapy (135). Orilanolimab (SYNT001) and batoclimab (RVT-1401) are also under investigation for warm AIHA, though available data remain limited, necessitating further studies to assess their therapeutic potential (136).
5.1.5 Inhibition of BAFF/APRIL signaling
BAFF (also known as BLyS, B lymphocyte stimulator) and APRIL (a proliferation-inducing ligand) are key TNF family members involved in B-cell survival, maturation, and activation (Figure 2). As previously highlighted, BAFF plays a role in B-cell repopulation following rituximab-induced depletion in warm AIHA, making it a target to prevent relapse via BAFF modulation (88).
Ianalumab (VAY736), an IgG1κ monoclonal antibody targeting BAFF-R, promotes B-cell depletion via ADCC and apoptosis. Preliminary data suggest that its depleting capacity may exceed that of anti-CD20 monoclonal antibodies. Based on these findings, the Phase 3 VAYHIA trial evaluates ianalumab in warm AIHA (137).
Povetacicept (ALPN-303) is a next-generation fusion protein designed to inhibit both BAFF and APRIL, acting through multiple receptors, including BAFF-R, TACI (transmembrane activator and calcium-modulating cyclophilin ligand interactor), and BCMA (B-cell maturation antigen). Preclinical models demonstrated that povetacicept reduces anti-RBC autoantibody production and antibody-secreting cells, depleting the spleen’s THF cells, B cells, and plasma cells. These effects were associated with increased hematocrit and a mild elevation in serum LDH (138). Further insights are expected from an ongoing Phase 1b open-label study in patients with autoimmune cytopenias, including warm AIHA and CAD (NCT05757570) (138).
5.1.6 CD19 CAR T in AIHA
Chimeric antigen receptor (CAR) T cells have revolutionized lymphoproliferative malignancies management are now being explored in other hematologic and solid tumors. Emerging evidence also suggests efficacy in autoimmune diseases, including lupus, idiopathic inflammatory myopathies, and systemic sclerosis (139). Preliminary data from a median 6.8-month follow-up were recently reported in five patients treated with compassionate-use CD19 CAR-T therapy and three enrolled in a Phase 1 trial (NCT06231368) for refractory AIHA (109). Toxicity was manageable, with five patients experiencing grade 1 cytokine release syndrome (CRS) and one developing grade 1 immune effector cell-associated neurotoxicity syndrome (ICANS). Notably, all patients achieved complete responses.
5.2 Complement inhibition
Complement activation is a central driver of hemolysis in AIHA, particularly in CAD, where it mediates intravascular destruction and extravascular clearance of erythrocytes (2). Beyond hemolysis, excessive complement activation contributes to a procoagulant and inflammatory environment, exacerbating disease severity and increasing thrombotic risk. As shown in Figure 3, these pathogenic effects highlight the classical complement pathway as a critical therapeutic target, with emerging inhibitors showing promise in mitigating hemolysis and its systemic complications.
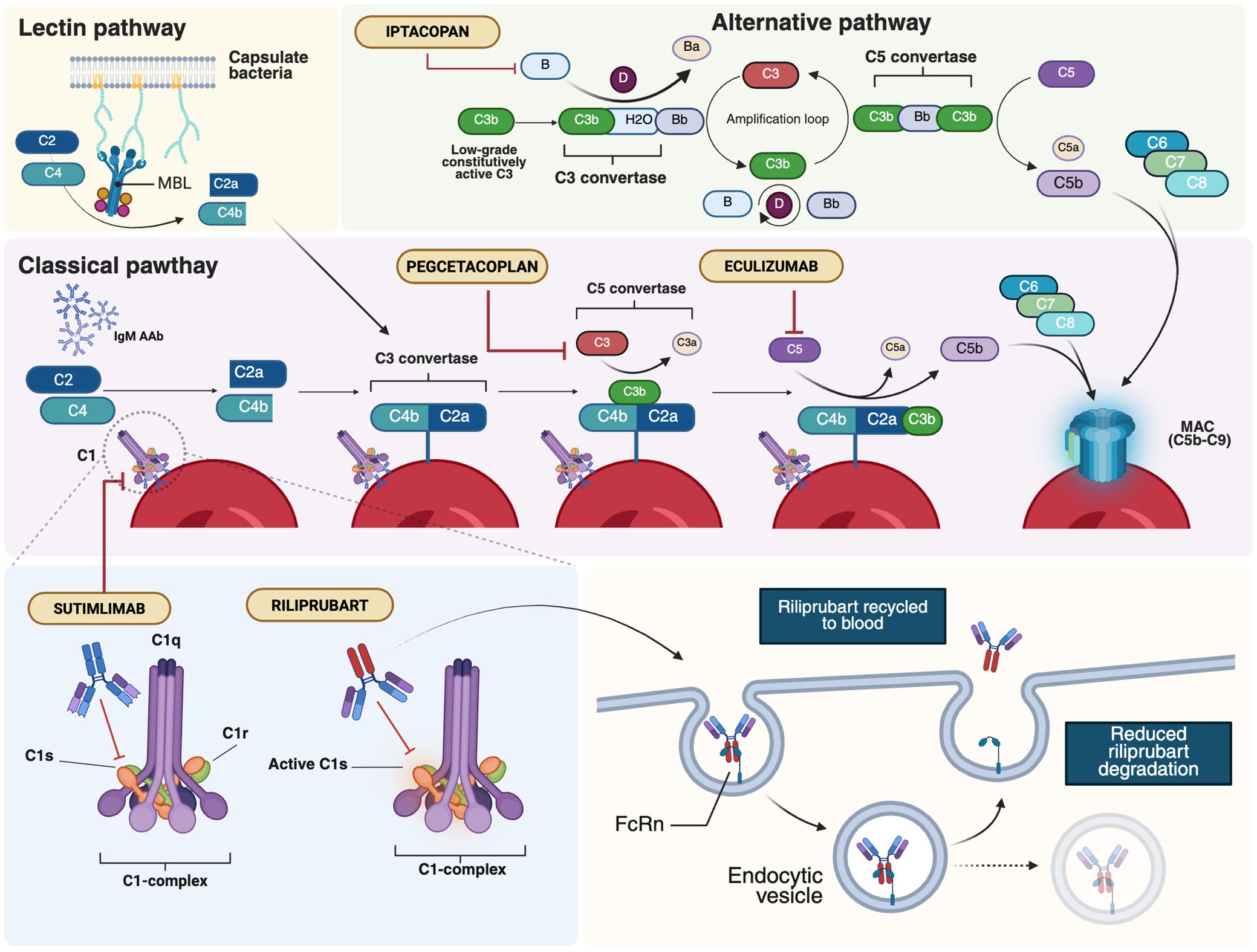
Figure 3. Major therapeutic targets within the complement system in AIHAs. AIHAs, autoimmune hemolytic anemias; AAb, autoantibodies; FcRn, neonatal Fc Receptor; MAC, membrane attack complex; MBL, mannan-binding lectin. Created in BioRender. Costa, A. (2025) https://BioRender.com/lj5h9fv.
5.2.1 C1 inhibitors
Given the central role of the classical complement pathway in CAD pathogenesis and the impact of C1 activation on extravascular hemolysis, selective inhibition of C1 has emerged as a promising strategy to modulate the disease while preserving other complement pathways.
Sutimlimab, a humanized IgG4 monoclonal antibody (MoAb) targeting C1s, is the first selective inhibitor of the classical complement pathway to receive approval for CAD (Figure 3) (140). By targeting an early step in the complement cascade, sutimlimab prevents C3b deposition on erythrocytes while sparing the alternative and lectin pathways. In vitro studies have demonstrated its ability to block C3 deposition on erythrocytes and inhibit the generation of anaphylatoxins, including C3a, C4a, and C5a, laying the foundation for clinical evaluation (20). In a phase 1b study with 64 healthy volunteers, sutimlimab infusions were well tolerated and showed a rapid concentration-effect relationship (141). The drug inhibited over 90% of classical complement activity within one hour of infusion, with doses ≥60 mg/kg every 14 days required to maintain stable inhibition. A phase 1b study in CAD patients revealed a significant increase in hemoglobin levels, although anemia recurred 3–4 weeks after discontinuation (Table 4) (141, 142). An extension of the follow-up to the study conducted by Jäger et al. (142) evaluated seven patients who had previously responded to the drug and were subsequently treated under a named patient protocol program. All patients exhibited a positive response to the treatment and maintained a transfusion-free status during the administration of sutimlimab (143). The safety and efficacy of sutimlimab were further assessed in the phase 2/3 CARDINAL trial, with the primary endpoint being the normalization of hemoglobin levels to ≥12 g/dL or an increase of ≥2 g/dL from baseline, without the need for RBC transfusions or the use of prohibited medications (144). A total of 24 CAD patients received sutimlimab (6.5 g for patients <75 kg, 7.5 g for those ≥75 kg) on days 0 and 7, followed by biweekly infusions for 26 weeks. Of these, 54% met the primary endpoint, and 83% achieved a stable increase of ≥1 g/dL in hemoglobin, with 71% not requiring transfusions between weeks 5 and 26. The most common adverse events included respiratory infections, diarrhea, and arthralgia, with no treatment discontinuations. No meningococcal events were reported. While reductions in D-dimer and thrombin-antithrombin complex levels were observed, conclusive evidence regarding the effect of complement inhibition on thrombotic prevention remains lacking. The two-year extension phase involving 22 patients from the part A study showed persistent improvements in hemolysis and anemia and positive effects on patient well-being (145, 146). Crucial findings also emerged from the phase 3 CADENZA trial, a randomized, placebo-controlled study in 42 CAD patients without a recent transfusion history, hemoglobin ≤10 g/dL, and at least one CAD-related symptom (147). Primary endpoints were the increase in hemoglobin level from baseline of ≥ 1.5 g/dL at treatment assessment timepoint (mean value from weeks 23, 25 and 26), absence of blood transfusions from week 5 to 26, avoidance of protocol-prohibited CAD medications from week 5 to week 26 (Table 4). Notably, 73% of the 22 patients receiving sutimlimab achieved the primary endpoint, compared to 10% of the 20 patients receiving placebo. Significant improvements in average hemoglobin levels and quality-of-life scores were observed in the sutimlimab cohort relative to the placebo group. As with the CARDINAL trial, the 1-year extension confirmed further improvements in hemoglobin levels from baseline (148). However, discontinuation of the drug was associated with a recurrence of hemolytic symptoms. A post-hoc analysis of combined phase 3 trial data stratified by baseline anemia severity included 24 patients from CARDINAL and 22 from the sutimlimab arm of CADENZA (153). Hemoglobin increases were proportional to baseline anemia severity, though differences between subgroups were not statistically significant. Real-world data remain sparse, and results from the multicenter, prospective CADENCE registry in CAD/CAS patients are eagerly awaited (154).
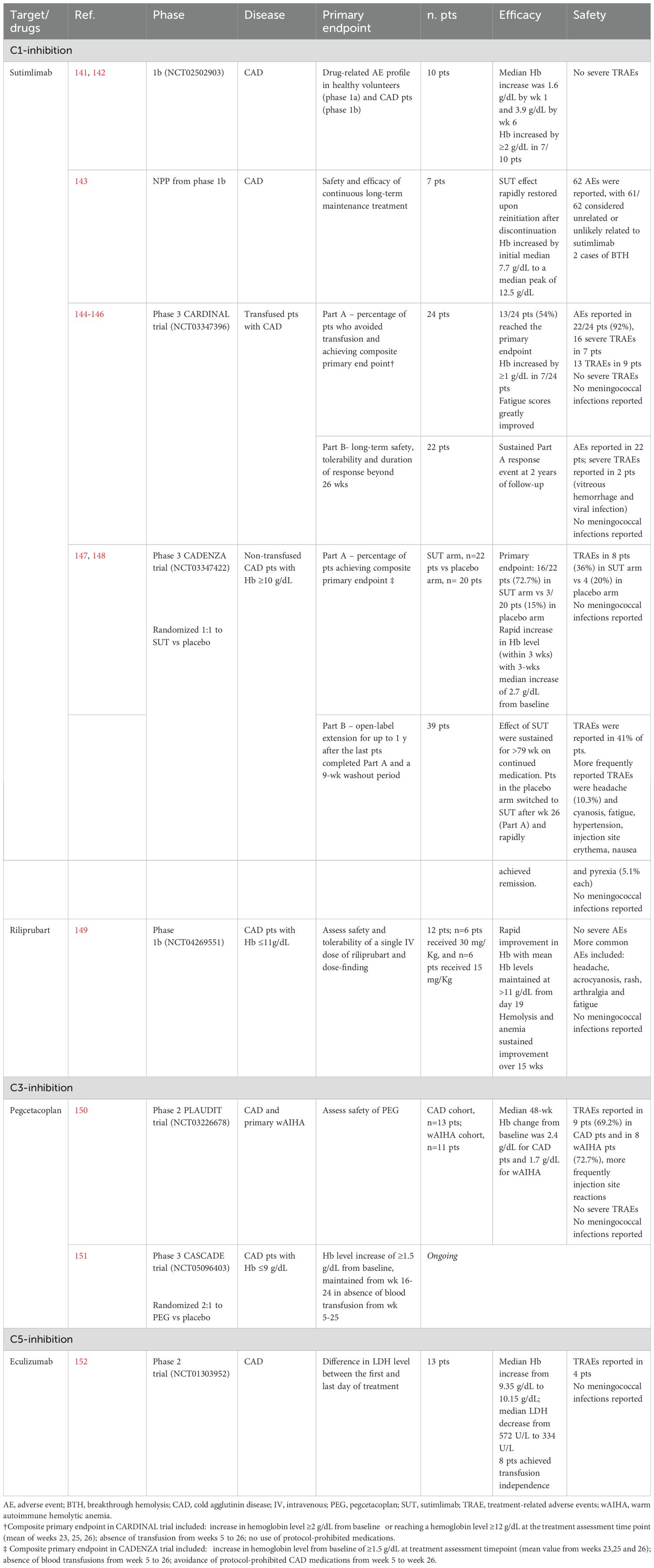
Table 4. Registered clinical trials exploring complement-inhibitions in AIHA.
The second-generation C1 inhibitor riliprubart (SAR445088, BIVV020) is a humanized IgG4 MoAb that selectively inhibits the activated form of C1s, in contrast to sutimlimab, which targets both the active and inactive forms of C1s (Figure 2) (155, 156). Additionally, riliprubart is engineered with mutations that increase its binding affinity to the neonatal Fc receptor (FcRn), preventing lysosomal degradation and allowing for the recycling of the antibody within the system, thereby prolonging its half-life (157). A randomized, double-blind, placebo-controlled phase 1 trial was conducted to assess the safety, tolerability, and pharmacokinetics of riliprubart in 93 healthy subjects. The drug was administered either subcutaneously or intravenously in escalating doses, with both formulations demonstrating an acceptable safety profile. The subcutaneous formulation exhibited slow absorption and a prolonged half-life of 8 to 15 weeks (155). Based on these favorable pharmacokinetic properties, riliprubart was further evaluated in a phase 1b study (NCT04269551) involving 12 patients with CAD (Table 4) (149). Patients were administered a single intravenous dose of either 30 mg/kg or 15 mg/kg on day 1, followed by a 15-week monitoring period. The drug demonstrated a good safety profile, with no treatment-related adverse events leading to death or permanent study discontinuation. Furthermore, rapid improvements in hemoglobin levels and hemolysis biomarkers were observed in both dose groups within the first 30 days, with continued suppression of hemolysis throughout the 15-week study duration. An ongoing phase 1b extension study (NCT04802057) is evaluating the long-term safety and efficacy of riliprubart. Patients who completed the Phase 1a study and Part 1 of the LTS16637 study (receiving 600 mg subcutaneously every 4 weeks) transitioned to Part 2, where they received an intravenous regimen of 3.5 g every 12 weeks, with an additional loading dose on day 29. Preliminary data from four patients indicate sustained control of hemolysis and anemia, with hemoglobin and bilirubin levels maintained within normal ranges. The safety profile remains manageable, with no new or unexpected toxicities reported. Pharmacokinetic and pharmacodynamic analyses confirm predictable systemic drug exposure and sustained inhibition of the classical complement pathway, supporting the potential clinical applicability of the intravenous regimen (158).
Another investigated strategy involves administering a C1 inhibitor (C1-INH). Given its role as an endogenous complement regulator, C1-INH was evaluated in a phase 2 study (EudraCT2012-003710-13) in AIHA patients requiring transfusion support (159). Four intravenous doses were administered at 12-hour intervals. Hemoglobin levels temporarily increased post-transfusion but returned to baseline within 48 hours, suggesting the limited efficacy of this approach.
Finally, considering that over one-third of patients with warm AIHA exhibit complement activation, complement inhibitors may offer a promising therapeutic strategy, particularly in mixed forms with IgG and C3d positivity (2). In this regard, results are awaited from a phase 2 study (NCT04691570) evaluating subjects with warm AIHA showing evidence of complement activation or mixed forms, treated with ANX005, a recombinant anti-C1q IgG4 humanized MoAb.
5.2.2 C3 inhibitors
Further downstream in the complement cascade, C3 inhibition has gained increasing interest due to its key role in CAD-related hemolysis. Pegcetacoplan, a polyethylene glycol-conjugated C3 inhibitor designed to prolong its half-life, exhibits high-affinity binding to both C3 and its activated fragment, C3b (Figure 3). The drug has already demonstrated robust efficacy and safety in paroxysmal nocturnal hemoglobinuria (PNH), securing Food and Drug Administration (FDA) and European Medicine Agency (EMA) approval (160). More recently, it has been evaluated in CAD in the phase 2 PLAUDIT trial (NCT03226678), which, unlike other studies, also included patients with relapsed, refractory, or treatment-intolerant warm AIHA (Table 4) (150). Overall, 13 CAD and 11 warm AIHA patients were enrolled and randomized to receive pegcetacoplan at either 270 mg/d (CAD, n=7; warm AIHA, n=5) or 360 mg/d (CAD, n=6; warm AIHA, n=6). In terms of efficacy, pegcetacoplan increased hemoglobin levels, with a median rise from baseline at 48 weeks of 2.4 g/dL in CAD and 1.7 g/dL in warm AIHA. In the warm AIHA cohort, hemoglobin levels increased until week 8, then plateaued. The drug was well tolerated, with diarrhea, headache, hypertension, nausea, and vitamin B12 deficiency being the most frequently reported adverse events. Injection-site reactions occurred in 46.2% of patients, and seven developed infections. One AIHA-related death was reported, attributed not to the drug but to the patient’s complex clinical management due to other complications. Although ten patients experienced at least one serious adverse event, none were considered treatment-related. The phase 3 CASCADE trial, a randomized, placebo-controlled study (NCT05096403), is ongoing, with eagerly awaited results expected to further define this agent’s efficacy and safety profile (151).
5.2.3 Other complement target and inhibition strategies
Another explored inhibitor is eculizumab, an anti-C5 MoAb known for revolutionizing the therapeutic landscape of PNH, another form of complement-mediated, DAT-negative hemolytic anemia (Figure 3) (161). Building on promising off-label reports from individual case studies (162–164), the phase 2 DECADE trial, conducted in 13 CAD patients, demonstrated a reduction in transfusion requirements, although the median hemoglobin increase remained modest, likely due to persistent C3b-mediated opsonization and ongoing extravascular hemolysis (152). Eculizumab has also shown clinical efficacy in a pediatric case of PCH in a 4-year-old child who was refractory to corticosteroids but experienced marked clinical improvement following a single dose of the anti-C5 antibody (23).
More recently, factor B inhibition has garnered interest, mirroring therapeutic advancements in PNH. Iptacopan, an oral selective factor B inhibitor, has shown efficacy in PNH and other complement-mediated disorders (Figure 3) (165). The drug is currently under evaluation in CAD (NCT05086744), aiming for a hemoglobin increase of at least 1.5 g/dL from baseline. An interim analysis of 10 enrolled CAD patients reported that the primary endpoint was met in 50% of cases, with a mean hemoglobin increase of 1.8 g/dL and positive trends in other biomarkers and FACIT-fatigue scores (166). The drug was well tolerated, with no reported adverse events deemed treatment-related.
6 Conclusion and open questions
The landscape of AIHA has evolved, revealing a complex interplay between innate and adaptive immune mechanisms. While recent advances have led to more targeted therapeutic strategies, uncertainty persists, particularly in patients with refractory or relapsing disease. Moving forward, key challenges include refining the management of refractory cases, identifying reliable biomarkers to guide treatment selection, and addressing the underlying mechanisms of therapeutic resistance and autoantibody persistence. A deeper understanding of these factors is crucial, not only to enhance treatment effectiveness but also to prevent chronicity. The future of AIHA lies in integrating novel therapeutic approaches with personalized care to address ongoing clinical challenges and improve patient outcomes.
Author contributions
AC: Writing – review & editing, Writing – original draft, Visualization, Methodology, Conceptualization. OM: Writing – original draft, Writing – review & editing, Methodology, Visualization, Conceptualization. AM: Writing – original draft, Writing – review & editing. MS: Writing – original draft, Writing – review & editing. MG: Writing – review & editing, Writing – original draft. GC: Writing – original draft, Methodology, Conceptualization, Writing – review & editing, Visualization.
Funding
The authors declare that financial support was received for the research and/or publication of this article. The research leading to these results has received funding from the European Union -NextGenerationEU through the Italian Ministry of University and Research under PNRR-M4C2-I1.3 Project PE_00000019 “HEAL ITALIA” to Giovanni Caocci CUP F53C22000750006 University of Cagliari. The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them.
Acknowledgments
The authors wish to thank Professor Andrea Perra (Oncology and Molecular Pathology Unit, Department of Biomedical Sciences, University of Cagliari, Cagliari, Italy) for his valuable support and commitment to this project.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bozza MT and Jeney V. Pro-inflammatory actions of heme and other hemoglobin-derived DAMPs. Front Immunol. (2020) 11:1323. doi: 10.3389/fimmu.2020.01323
2. Barcellini W, Zaninoni A, Giannotta JA, and Fattizzo B. New insights in autoimmune hemolytic anemia: from pathogenesis to therapy stage 1. J Clin Med. (2020) 9:3859. doi: 10.3390/jcm9123859
3. Fattizzo B and Barcellini W. Autoimmune hemolytic anemia: causes and consequences. Expert Rev Clin Immunol. (2022) 18:731–45. doi: 10.1080/1744666X.2022.2089115
4. Mulder FVM, Evers D, de Haas M, Cruijsen MJ, Bernelot Moens SJ, Barcellini W, et al. Severe autoimmune hemolytic anemia; epidemiology, clinical management, outcomes and knowledge gaps. Front Immunol. (2023) 14:1228142. doi: 10.3389/fimmu.2023.1228142
5. Tefferi A. Anemia in adults: a contemporary approach to diagnosis. Mayo Clin Proc. (2003) 78:1274–80. doi: 10.4065/78.10.1274
6. Kalfa TA. Warm antibody autoimmune hemolytic anemia. Hematol Am Soc Hematol Educ Program. (2016) 2016:690–7. doi: 10.1182/asheducation-2016.1.690
7. Branch DR. Warm autoimmune hemolytic anemia: new insights and hypotheses. Curr Opin Hematol. (2023) 30:203–9. doi: 10.1097/MOH.0000000000000779
8. Howie HL and Hudson KE. Murine models of autoimmune hemolytic anemia. Curr Opin Hematol. (2018) 25:473–81. doi: 10.1097/MOH.0000000000000459
9. Mahévas M, Michel M, Vingert B, Moroch J, Boutboul D, Audia S, et al. Emergence of long-lived autoreactive plasma cells in the spleen of primary warm auto-immune hemolytic anemia patients treated with rituximab. J Autoimmun. (2015) 62:22–30. doi: 10.1016/j.jaut.2015.05.006
10. Nagelkerke SQ, Bruggeman CW, den Haan JMM, Mul EPJ, van den Berg TK, van Bruggen R, et al. Red pulp macrophages in the human spleen are a distinct cell population with a unique expression of Fc-γ receptors. Blood Adv. (2018) 2:941–53. doi: 10.1182/bloodadvances.2017015008
11. Vidarsson G, Dekkers G, and Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. (2014) 5:520. doi: 10.3389/fimmu.2014.00520
12. Thiagarajan P, Parker CJ, and Prchal JT. How do red blood cells die? Front Physiol. (2021) 12:655393. doi: 10.3389/fphys.2021.655393
13. Wang Y and Jönsson F. Expression, role, and regulation of neutrophil fcγ Receptors. Front Immunol. (2019) 10:1958. doi: 10.3389/fimmu.2019.01958
14. Frischauf N, Strasser J, Borg EGF, Labrijn AF, Beurskens FJ, and Preiner J. Complement activation by IgG subclasses is governed by their ability to oligomerize upon antigen binding. Proc Natl Acad Sci U S A. (2024) 121:e2406192121. doi: 10.1073/pnas.2406192121
15. Jäger U, Barcellini W, Broome CM, Gertz MA, Hill A, Hill QA, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: Recommendations from the First International Consensus Meeting. Blood Rev. (2020) 41:100648. doi: 10.1016/j.blre.2019.100648
16. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. (2022) 36:1720–48. doi: 10.1038/s41375-022-01620-2
17. Berentsen S, D’Sa S, Randen U, Małecka A, and Vos JMI. Cold agglutinin disease: improved understanding of pathogenesis helps define targets for therapy. Hemato. (2022) 3:574–94. doi: 10.3390/hemato3040040
18. Daniels G. I and i Antigens, and Cold Agglutination. Daniels G, editor. In: Human blood groups. 3rd ed. New York (NY): Wiley. (2013). doi: 10.1002/9781118493595.ch25
19. Berentsen S, Barcellini W, D'Sa S, Randen U, Tvedt THA, Fattizzo B, et al. Cold agglutinin disease revisited: a multinational, observational study of 232 patients. Blood. (2020) 136:480–8. doi: 10.1182/blood.2020005674
20. Shi J, Rose EL, Singh A, Hussain S, Stagliano NE, Parry GC, et al. TNT003, an inhibitor of the serine protease C1s, prevents complement activation induced by cold agglutinins. Blood. (2014) 123:4015–22. doi: 10.1182/blood-2014-02-556027
21. Wouters D and Zeerleder S. Complement inhibitors to treat IgM-mediated autoimmune hemolysis. Haematologica. (2015) 100:1388–95. doi: 10.3324/haematol.2015.128538
22. Jacobs JW, Figueroa Villalba CA, Booth GS, Woo JS, Stephens LD, Adkins BD, et al. Clinical and epidemiological features of paroxysmal cold hemoglobinuria: a systematic review. Blood Adv. (2023) 7(11):2520–7. doi: 10.1182/bloodadvances.2022009516
23. Lau-Braunhut SA, Stone H, Collins G, Berentsen S, Braun BS, and Zinter MS. Paroxysmal cold hemoglobinuria successfully treated with complement inhibition. Blood Adv. (2019) 3:3575–8. doi: 10.1182/bloodadvances.2019000897
24. Jacobs JW, Booth GS, Woo JS, Adkins BD, and Stephens LD. How the United States syphilis epidemic may portend a resurgence of an unusual hematologic condition: The connection between syphilis and paroxysmal cold hemoglobinuria. Am J Hematol. (2024) 99:484–5. doi: 10.1002/ajh.27190
25. Barcellini W, Fattizzo B, Zaninoni A, Radice T, Nichele I, Di Bona E, et al. Clinical heterogeneity and predictors of outcome in primary autoimmune hemolytic anemia: a GIMEMA study of 308 patients. Blood. (2014) 124:2930–6. doi: 10.1182/blood-2014-06-583021
26. Barcellini W, Zaninoni A, Fattizzo B, Giannotta JA, Lunghi M, Ferrari A, et al. Predictors of refractoriness to therapy and healthcare resource utilization in 378 patients with primary autoimmune hemolytic anemia from eight Italian reference centers. Am J Hematol. (2018) 93:E243–6. doi: 10.1002/ajh.25212
27. Bylsma LC, Gulbech Ording A, Rosenthal A, Öztürk B, Fryzek JP, Arias JM, et al. Occurrence, thromboembolic risk, and mortality in Danish patients with cold agglutinin disease. Blood Adv. (2019) 3:2980–5. doi: 10.1182/bloodadvances.2019000476
28. Broome CM, Cunningham JM, Mullins M, Jiang X, Bylsma LC, Fryzek JP, et al. Increased risk of thrombotic events in cold agglutinin disease: A 10-year retrospective analysis. Res Pract Thromb Haemost. (2020) 4:628–35. doi: 10.1002/rth2.12333
29. Fattizzo B, Bortolotti M, Giannotta JA, Zaninoni A, Consonni D, and Barcellini W. Intravascular hemolysis and multitreatment predict thrombosis in patients with autoimmune hemolytic anemia. J Thromb Haemost. (2022) 20:1852–8. doi: 10.1111/jth.15757
30. Eltaher B, Mahmoud A, and Shah P. Risk of venous thromboembolism in hospitalized patients with chronic lymphocytic leukemia complicated with autoimmune hemolytic anemia. Blood. (2024) 144:6789. doi: 10.1182/blood-2024-212067
31. Yong J and Toh CH. Rethinking coagulation: from enzymatic cascade and cell-based reactions to a convergent model involving innate immune activation. Blood. (2023) 142:2133–45. doi: 10.1182/blood.2023021166
32. Dimitrov JD, Roumenina LT, Perrella G, and Rayes J. Basic mechanisms of hemolysis-associated thrombo-inflammation and immune dysregulation. Arterioscler Thromb Vasc Biol. (2023) 43:1349–61. doi: 10.1161/ATVBAHA.123.318780
33. Mannes M, Pechtl V, Hafner S, Dopler A, Eriksson O, Manivel VA, et al. Complement and platelets: prothrombotic cell activation requires membrane attack complex-induced release of danger signals. Blood Adv. (2023) 7:6367–80. doi: 10.1182/bloodadvances.2023010817
34. Barcellini W, Zaninoni A, Giannotta JA, Merati G, Capecchi M, Fattizzo B, et al. Circulating extracellular vesicles and cytokines in congenital and acquired hemolytic anemias. Am J Hematol. (2021) 96:E129–32. doi: 10.1002/ajh.26108
35. Pryzdial ELG, Leatherdale A, and Conway EM. Coagulation and complement: Key innate defense participants in a seamless web. Front Immunol. (2022) 13:918775. doi: 10.3389/fimmu.2022.918775
36. Gushiken FC, Han H, Li J, Rumbaut RE, and Afshar-Kharghan V. Abnormal platelet function in C3-deficient mice. J Thromb Haemost. (2009) 7:865–70. doi: 10.1111/j.1538-7836.2009.03334.x
37. Wiedmer T, Esmon C, and Sims P. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood. (1986) 68:875–80. doi: 10.1182/blood.V68.4.875.875
38. Shivshankar P, Li YD, Mueller-Ortiz SL, and Wetsel RA. In response to complement anaphylatoxin peptides C3a and C5a, human vascular endothelial cells migrate and mediate the activation of B-cells and polarization of T-cells. FASEB J. (2020) 34:7540–60. doi: 10.1096/fj.201902397R
39. Michalak SS, Olewicz-Gawlik A, Rupa-Matysek J, Wolny-Rokicka E, Nowakowska E, and Gil L. Autoimmune hemolytic anemia: current knowledge and perspectives. Immun Ageing. (2020) 17:38. doi: 10.1186/s12979-020-00208-7
40. Maguire C, Wang C, Ramasamy A, Fonken C, Morse B, Lopez N, et al. Molecular mimicry as a mechanism of viral immune evasion and autoimmunity. Nat Commun. (2024) 15:9403. doi: 10.1038/s41467-024-53658-8
41. Taherifard E, Taherifard E, Movahed H, and Mousavi MR. Hematologic autoimmune disorders in the course of COVID-19: a systematic review of reported cases. Hematology. (2021) 26:225–39. doi: 10.1080/16078454.2021.1881225
42. Angileri F, Légaré S, Marino Gammazza A, Conway de Macario E, Macario AJL, and Cappello F. Is molecular mimicry the culprit in the autoimmune haemolytic anaemia affecting patients with COVID-19? Br J Haematol. (2020) 190:e92–3. doi: 10.1111/bjh.16883
43. Xia X, Liu S, and Zhou ZH. Structure, dynamics and assembly of the ankyrin complex on human red blood cell membrane. Nat Struct Mol Biol. (2022) 29:698–705. doi: 10.1038/s41594-022-00779-7
44. Root-Bernstein R and Fairweather D. Complexities in the relationship between infection and autoimmunity. Curr Allergy Asthma Rep. (2014) 14:407. doi: 10.1007/s11882-013-0407-3
45. Garratty G. Immune hemolytic anemia associated with drug therapy. Blood Rev. (2010) 24:143–50. doi: 10.1016/j.blre.2010.06.004
46. Maquet J, Lafaurie M, Michel M, Lapeyre-Mestre M, and Moulis G. Drug-induced immune hemolytic anemia: detection of new signals and risk assessment in a nationwide cohort study. Blood Adv. (2024) 8:817–26. doi: 10.1182/bloodadvances.2023009801
47. McQueen F. A B cell explanation for autoimmune disease: the forbidden clone returns. Postgrad Med J. (2012) 88:226–33. doi: 10.1136/postgradmedj-2011-130364
48. Shi X, Wallach JD, Ma X, and Rogne T. Autoimmune diseases and risk of non-hodgkin lymphoma: A mendelian randomisation study. Cancer Med. (2024) 13:e70327. doi: 10.1002/cam4.70327
49. Autore F, Pasquale R, Innocenti I, Fresa A, Sora' F, and Laurenti L. Autoimmune hemolytic anemia in chronic lymphocytic leukemia: A comprehensive review. Cancers (Basel). (2021) 13:5804. doi: 10.3390/cancers13225804
50. Randen U, Trøen G, Tierens A, Steen C, Warsame A, Beiske K, et al. Primary cold agglutinin-associated lymphoproliferative disease: a B-cell lymphoma of the bone marrow distinct from lymphoplasmacytic lymphoma. Haematologica. (2014) 99:497–504. doi: 10.3324/haematol.2013.091702
51. Abdel-Khalik A, Paton L, White AG, and Urbaniak SJ. Human leucocyte antigens A, B, C, and DRW in idiopathic "warm" autoimmune haemolytic anaemia. Br Med J. (1980) 280:760–1. doi: 10.1136/bmj.280.6216.760
52. Silberstein LE, Jefferies LC, Goldman J, Friedman D, Moore JS, Nowell PC, et al. ariable region gene analysis of pathologic human autoantibodies to the related i and I red blood cell antigens. Blood. (1991) 78:2372–86.
53. Jefferies LC, Carchidi CM, and Silberstein LE. Naturally occurring anti-i/I cold agglutinins may be encoded by different VH3 genes as well as the VH4. 21 Gene segment J Clin Invest. (1993) 92:2821–33. doi: 10.1172/JCI116902
54. Małecka A, Trøen G, Tierens A, Østlie I, Małecki J, Randen U, et al. Immunoglobulin heavy and light chain gene features are correlated with primary cold agglutinin disease onset and activity. Haematologica. (2016) 101:e361–4. doi: 10.3324/haematol.2016.146126
55. Zhang R, Zhuang J, and Han B. Exploration of B and T cell receptor repertoires reveals distinct mechanisms in pure red cell aplasia, autoimmune hemolytic anemia, and aplastic anemia. Blood. (2023) 142:5196. doi: 10.1182/blood-2023-174862
56. Małecka A, Østlie I, Trøen G, Małecki J, Delabie J, Tierens A, et al. Gene expression analysis revealed downregulation of complement receptor 1 in clonal B cells in cold agglutinin disease. Clin Exp Immunol. (2024) 216:45–54. doi: 10.1093/cei/uxad135
57. D'Abronzo LS, Barros MM, Bordin JO, and Figueiredo MS. Analysis of polymorphisms of TNF-α, LT-α, IL-10, IL-12 and CTLA-4 in patients with warm autoimmune haemolytic anaemia. Int J Lab Hematol. (2012) 34:356–61. doi: 10.1111/j.1751-553X.2012.01400.x
58. Pavkovic M, Georgievski B, Cevreska L, Spiroski M, and Efremov DG. CTLA-4 exon 1 polymorphism in patients with autoimmune blood disorders. Am J Hematol. (2003) 72:147–9. doi: 10.1002/ajh.10278
59. Małecka A, Trøen G, Tierens A, Østlie I, Małecki J, Randen U, et al. Frequent somatic mutations of KMT2D (MLL2) and CARD11 genes in primary cold agglutinin disease. Br J Haematol. (2018) 183:838–42. doi: 10.1111/bjh.15063
60. Małecka A, Trøen G, Delabie J, Ostlie I, Tierens A, Randen U, et al. The mutational landscape of cold agglutinin disease. Blood. (2020) 136:14–5. doi: 10.1182/blood-2020-133760
61. Fattizzo B, Marchetti M, Michel M, Cantoni S, Frederiksen H, Giordano G, et al. Diagnosis and management of Evans syndrome in adults: first consensus recommendations. Lancet Haematol. (2024) 11:e617–28. doi: 10.1016/S2352-3026(24)00144-3
62. Hadjadj J, Aladjidi N, Fernandes H, Leverger G, Magérus-Chatinet A, Mazerolles F, et al. Pediatric Evans syndrome is associated with a high frequency of potentially damaging variants in immune genes. Blood. (2019) 134:9–21. doi: 10.1182/blood-2018-11-887141
63. Zaninoni A, Fattizzo B, Pettine L, Vercellati C, Marcello AP, and Barcellini W. Cytokine polymorphisms in patients with autoimmune hemolytic anemia. Front Immunol. (2023) 14:1221582. doi: 10.3389/fimmu.2023.1221582
64. Pavkovic M, Petlichkovski A, Angelovic R, Genadieva-Stavric S, Cevreska L, and Stojanovic A. Tumor necrosis factor gene polymorphisms in adult patients with autoimmune hemolytic anemia. Int J Lab Hematol. (2017) 39:e74–6. doi: 10.1111/ijlh.12624
65. Jeanpierre M, Cognard J, Tusseau M, Riller Q, Bui LC, Berthelet J, et al. Haploinsufficiency in PTPN2 leads to early-onset systemic autoimmunity from Evans syndrome to lupus. J Exp Med. (2024) 221:e20232337. doi: 10.1084/jem.20232337
66. Shirafkan F, Hensel L, and Rattay K. Immune tolerance and the prevention of autoimmune diseases essentially depend on thymic tissue homeostasis. Front Immunol. (2024) 15:1339714. doi: 10.3389/fimmu.2024.1339714
67. Kenison JE, Stevens NA, and Quintana FJ. Therapeutic induction of antigen-specific immune tolerance. Nat Rev Immunol. (2024) 24:338–57. doi: 10.1038/s41577-023-00970-x
68. Bouneaud C, Kourilsky P, and Bousso P. Impact of negative selection on the T cell repertoire reactive to a self-peptide: a large fraction of T cell clones escapes clonal deletion. Immunity. (2000) 13:829–40. doi: 10.1016/s1074-7613(00)00080-7
69. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. (2003) 301:1374–7. doi: 10.1126/science.1086907
70. Song Y, Li J, and Wu Y. Evolving understanding of autoimmune mechanisms and new therapeutic strategies of autoimmune disorders. Signal Transduct Target Ther. (2024) 9:1–40. doi: 10.1038/s41392-024-01952-8
71. Hasegawa H and Matsumoto T. Mechanisms of tolerance induction by dendritic cells in vivo. Front Immunol. (2018) 9:350. doi: 10.3389/fimmu.2018.00350
72. Wong ASL, Gruber DR, Richards AL, Sheldon K, Qiu A, Hay A, et al. Tolerization of recent thymic emigrants is required to prevent RBC-specific autoimmunity. J Autoimmun. (2020) 114:102489. doi: 10.1016/j.jaut.2020.102489
73. Shen CR, Mazza G, Perry FE, Thompson SJ, Corato A, Newton S, et al. T-helper 1 dominated responses to erythrocyte Band 3 in NZB mice. Immunology. (1996) 89:195–9. doi: 10.1046/j.1365-2567.1996.d01-731.x
74. Youssef AR, Shen CR, Lin CL, Barker RN, and Elson CJ. IL-4 and IL-10 modulate autoimmune haemolytic anaemia in NZB mice. Clin Exp Immunol. (2005) 139:84–9. doi: 10.1111/j.1365-2249.2005.02663.x
75. Fagiolo E and Toriani-Terenzi C. Th1 and Th2 cytokine modulation by IL-10/IL-12 imbalance in autoimmune haemolytic anaemia (AIHA). Autoimmunity. (2002) 35:39–44. doi: 10.1080/08916930290005891
76. Hall AM, Ward FJ, Vickers MA, Stott LM, Urbaniak SJ, and Barker RN. Interleukin-10-mediated regulatory T-cell responses to epitopes on a human red blood cell autoantigen. Blood. (2002) 100:4529–36. doi: 10.1182/blood-2002-05-1383
77. Stockinger B and Omenetti S. The dichotomous nature of T helper 17 cells. Nat Rev Immunol. (2017) 17:535–44. doi: 10.1038/nri.2017.50
78. Xu L, Zhang T, Liu Z, Li Q, Xu Z, and Ren T. Critical role of Th17 cells in development of autoimmune hemolytic anemia. Exp Hematol. (2012) 40:994–1004.e4. doi: 10.1016/j.exphem.2012.08.008
79. Mqadmi A, Zheng X, and Yazdanbakhsh K. CD4+CD25+ regulatory T cells control induction of autoimmune hemolytic anemia. Blood. (2005) 105:3746–8. doi: 10.1182/blood-2004-12-4692
80. Ciudad M, Ouandji S, Lamarthée B, Cladière C, Ghesquière T, Nivet M, et al. Regulatory T-cell dysfunctions are associated with increase in tumor necrosis factor α in autoimmune hemolytic anemia and participate in Th17 polarization. Haematologica. (2024) 109:444–57. doi: 10.3324/haematol.2023.282859
81. Dei Zotti F, Qiu A, D'Agati VD, Jagnarine S, Kyritsis E, Miller A, et al. Mitigation of checkpoint inhibitor-induced autoimmune hemolytic anemia through modulation of purinergic signaling. Blood. (2024) 144:1581–94. doi: 10.1182/blood.2024024230
82. Qi J, Liu C, Bai Z, Li X, and Yao G. T follicular helper cells and T follicular regulatory cells in autoimmune diseases. Front Immunol. (2023) 14:1178792. doi: 10.3389/fimmu.2023.1178792
83. Gao Y, Jin H, Nan D, Yu W, Zhang J, Yang Y, et al. The role of T follicular helper cells and T follicular regulatory cells in the pathogenesis of autoimmune hemolytic anemia. Sci Rep. (2019) 9:19767. doi: 10.1038/s41598-019-56365-3
84. Kumar D, Prince C, Bennett CM, Briones M, Lucas L, Russell A, et al. T-follicular helper cell expansion and chronic T-cell activation are characteristic immune anomalies in Evans syndrome. Blood. (2022) 139:369–83. doi: 10.1182/blood.2021012924
85. Liu X, Yao J, Zhao Y, Wang J, and Qi H. Heterogeneous plasma cells and long-lived subsets in response to immunization, autoantigen and microbiota. Nat Immunol. (2022) 24(11):1564–76. doi: 10.1038/s41590-022-01345-5
86. Lightman SM, Utley A, and Lee KP. Survival of long-lived plasma cells (LLPC): piecing together the puzzle. Front Immunol. (2019) 10:965. doi: 10.3389/fimmu.2019.00965
87. Joyner CJ, Ley AM, Nguyen DC, Ali M, Corrado A, Tipton C, et al. Generation of human long-lived plasma cells by developmentally regulated epigenetic imprinting. Life Sci Alliance. (2021) 5:e202101285. doi: 10.26508/lsa.202101285
88. Thai LH, Le Gallou S, Robbins A, Crickx E, Fadeev T, Zhou Z, et al. BAFF and CD4+ T cells are major survival factors for long-lived splenic plasma cells in a B-cell-depletion context. Blood. (2018) 131:1545–55. doi: 10.1182/blood-2017-06-789578
89. Scheckel CJ and Go RS. Autoimmune hemolytic anemia: diagnosis and differential diagnosis. Hematol Oncol Clin North Am. (2022) 36:315–24. doi: 10.1016/j.hoc.2021.12.001
90. Barcellini W and Fattizzo B. Strategies to overcome the diagnostic challenges of autoimmune hemolytic anemias. Expert Rev Hematol. (2023) 16:515–24. doi: 10.1080/17474086.2023.2216930
91. Zantek ND, Koepsell SA, Tharp DR Jr, and Cohn CS. The direct antiglobulin test: a critical step in the evaluation of hemolysis. Am J Hematol. (2012) 87:707–9. doi: 10.1002/ajh.23218
92. Parker V and Tormey CA. The direct antiglobulin test: indications, interpretation, and pitfalls. Arch Pathol Lab Med. (2017) 141:305–10. doi: 10.5858/arpa.2015-0444-RS
93. Barcellini W. Pitfalls in the diagnosis of autoimmune haemolytic anaemia. Blood Transfus. (2015) 13:3–5. doi: 10.2450/2014.0252-14
94. Zeller MP, Arnold DM, Al Habsi K, Cserti-Gazdewich C, Delage G, Lebrun A, et al. Paroxysmal cold hemoglobinuria: a difficult diagnosis in adult patients. Transfusion. (2017) 57:137–43. doi: 10.1111/trf.13888
95. Luzzatto L, Ally M, and Notaro R. Glucose-6-phosphate dehydrogenase deficiency. Blood. (2020) 136:1225–40. doi: 10.1182/blood.2019000944
96. Barcellini W, Giannotta J, and Fattizzo B. Autoimmune hemolytic anemia in adults: primary risk factors and diagnostic procedures. Expert Rev Hematol. (2020) 13:585–97. doi: 10.1080/17474086.2020.1754791
97. Berentsen S, Fattizzo B, and Barcellini W. The choice of new treatments in autoimmune hemolytic anemia: how to pick from the basket? Front Immunol. (2023) 14:1180509. doi: 10.3389/fimmu.2023.1180509
98. Giannotta JA, Fattizzo B, Cavallaro F, and Barcellini W. Infectious complications in autoimmune hemolytic anemia. J Clin Med. (2021) 10:164. doi: 10.3390/jcm10010164
99. Bockorny B, Atienza JA, and Dasanu CA. Autoimmune manifestations in patients with Waldenström macroglobulinemia. Clin Lymphoma Myeloma Leuk. (2014) 14:456–9. doi: 10.1016/j.clml.2014.04.009
100. Notarangelo LD. Primary immunodeficiencies (PIDs) presenting with cytopenias. Hematol Am Soc Hematol Educ Program. (2009) (1): 139–43. doi: 10.1182/asheducation-2009.1.139
101. Fattizzo B, Cantoni S, Giannotta JA, Bandiera L, Zavaglia R, Bortolotti M, et al. Efficacy and safety of cyclosporine A treatment in autoimmune cytopenias: the experience of two Italian reference centers. Ther Adv Hematol. (2022) 13:20406207221097780. doi: 10.1177/20406207221097780
102. Kotb R, Pinganaud C, Trichet C, Lambotte O, Dreyfus M, Delfraissy JF, et al. Efficacy of mycophenolate mofetil in adult refractory auto-immune cytopenias: a single center preliminary study. Eur J Haematol. (2005) 75:60–4. doi: 10.1111/j.1600-0609.2005.00437.x
103. Burger JA and Wiestner A. Targeting B cell receptor signalling in cancer: preclinical and clinical advances. Nat Rev Cancer. (2018) 18:148–67. doi: 10.1038/nrc.2017.121
104. Stasi R, Del Poeta G, Stipa E, Evangelista ML, Trawinska MM, Cooper N, et al. Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood. (2007) 110:2924–30. doi: 10.1182/blood-2007-02-068999
105. Birgens H, Frederiksen H, Hasselbalch HC, Rasmussen IH, Nielsen OJ, Kjeldsen L, et al. A phase III randomized trial comparing glucocorticoid monotherapy versus glucocorticoid and rituximab in patients with autoimmune haemolytic anaemia. Br J Haematol. (2013) 163:393–9. doi: 10.1111/bjh.12541
106. Barcellini W, Zaja F, Zaninoni A, Imperiali FG, Di Bona E, Fattizzo B, et al. Sustained response to low-dose rituximab in idiopathic autoimmune hemolytic anemia. Eur J Haematol. (2013) 91:546–51. doi: 10.1111/ejh.12199
107. Herishanu Y, Levi S, Kamdjou T, Bornstein Y, Ram R, Benyamini N, et al. Obinutuzumab in the treatment of autoimmune haemolytic anaemia and immune thrombocytopenia in patients with chronic lymphocytic leukaemia/small lymphocytic lymphoma. Br J Haematol. (2021) 192:e1–4. doi: 10.1111/bjh.17105
108. Schuetz C, Hoenig M, Moshous D, Weinstock C, Castelle M, Bendavid M, et al. Daratumumab in life-threatening autoimmune hemolytic anemia following hematopoietic stem cell transplantation. Blood Adv. (2018) 2:2550–3. doi: 10.1182/bloodadvances.2018020883
109. Li R, Zhang L, Pan H, Li W, Ma J, Tian L, et al. CD19 CAR T-cell therapy in refractory autoimmune hemolytic anemia. Blood. (2024) 144:682. doi: 10.1182/blood-2024-202980
110. Barcellini W and Zanella A. Rituximab therapy for autoimmune haematological diseases. Eur J Intern Med. (2011) 22:220–9. doi: 10.1016/j.ejim.2010.12.016
111. Berentsen S, Ulvestad E, Gjertsen BT, Hjorth-Hansen H, Langholm R, Knutsen H, et al. Rituximab for primary chronic cold agglutinin disease: a prospective study of 37 courses of therapy in 27 patients. Blood. (2004) 103:2925–8. doi: 10.1182/blood-2003-10-3597
112. Berentsen S, Randen U, Oksman M, Birgens H, Tvedt THA, Dalgaard J, et al. Bendamustine plus rituximab for chronic cold agglutinin disease: results of a Nordic prospective multicenter trial. Blood. (2017) 130:537–41. doi: 10.1182/blood-2017-04-778175
113. Berentsen S, Randen U, Vågan AM, Hjorth-Hansen H, Vik A, Dalgaard J, et al. High response rate and durable remissions following fludarabine and rituximab combination therapy for chronic cold agglutinin disease. Blood. (2010) 116:3180–4. doi: 10.1182/blood-2010-06-288647
114. Michel M, Terriou L, Roudot-Thoraval F, Hamidou M, Ebbo M, Le Guenno G, et al. A randomized and double-blind controlled trial evaluating the safety and efficacy of rituximab for warm auto-immune hemolytic anemia in adults (the RAIHA study). Am J Hematol. (2017) 92:23–7. doi: 10.1002/ajh.24570
115. Barcellini W, Zaja F, Zaninoni A, Imperiali FG, Battista ML, Di Bona E, et al. Low-dose rituximab in adult patients with idiopathic autoimmune hemolytic anemia: clinical efficacy and biologic studies. Blood. (2012) 119:3691–7. doi: 10.1182/blood-2011-06-363556
116. Moser MM, Thalhammer R, Sillaber C, Derhaschnig U, Firbas C, Jäger U, et al. Very low doses of rituximab in autoimmune hemolytic anemia-an open-label, phase II pilot trial. Front Med (Lausanne). (2024) 11:1481333. doi: 10.3389/fmed.2024.1481333
117. Karageorgas T, Zomas A, Kazakou P, Katsimbri P, Mantzourani M, and Boumpas D. Successful treatment of life-threatening autoimmune haemolytic anaemia with ofatumumab in a patient with systemic lupus erythematosus. Rheumatol (Oxford). (2016) 55:2085–7. doi: 10.1093/rheumatology/kew267
118. Pidala J, Kim J, Betts BC, Alsina M, Ayala E, Fernandez HF, et al. Ofatumumab in combination with glucocorticoids for primary therapy of chronic graft-versus-host disease: phase I trial results. Biol Blood Marrow Transplant. (2015) 21:1074–82. doi: 10.1016/j.bbmt.2015.03.014
119. Jalink M, Jacobs CF, Khwaja J, Evers D, Bruggeman C, Fattizzo B, et al. Daratumumab monotherapy in refractory warm autoimmune hemolytic anemia and cold agglutinin disease. Blood Adv. (2024) 8:2622–34. doi: 10.1182/bloodadvances.2024012585
120. Safety, Pharmacokinetics, and Efficacy of Subcutaneous Isatuximab in Adults With Warm Autoimmune Hemolytic Anemia (wAIHA) - NCI . Available online at: https://www.cancer.gov/about-cancer/treatment/clinical-trials/search/v?id=NCI-2022-08954 (Accessed March 21, 2025).
121. Fattizzo B, Evans G, Wells A, Poma A, Quinn S, Zhang D, et al. Evaluation of obexelimab in patients with warm autoimmune hemolytic anemia: preliminary results from an open label phase 2 study. Blood. (2024) 144:5212.
122. Neys SFH, Rip J, Hendriks RW, and Corneth OBJ. Bruton's tyrosine kinase inhibition as an emerging therapy in systemic autoimmune disease. Drugs. (2021) 81:1605–26. doi: 10.1007/s40265-021-01592-0
123. Jalink M, Berentsen S, Castillo JJ, Treon SP, Cruijsen M, Fattizzo B, et al. Effect of ibrutinib treatment on hemolytic anemia and acrocyanosis in cold agglutinin disease/cold agglutinin syndrome. Blood. (2021) 138:2002–5. doi: 10.1182/blood.2021012039
124. Cooper N, Kuter DJ, Frederiksen H, Han B, Chen F, Barcellini W, et al. Part a efficacy and safety of oral bruton tyrosine kinase inhibitor (BTKi) rilzabrutinib in patients with warm autoimmune hemolytic anemia (wAIHA): multicenter, open-label, phase 2b study. Blood. (2024) 144:3836. doi: 10.1182/blood-2024-200325
125. Shin N, Stubbs M, Koblish H, Yue EW, Soloviev M, Douty B, et al. Parsaclisib is a next-generation phosphoinositide 3-kinase δ Inhibitor with reduced hepatotoxicity and potent antitumor and immunomodulatory activities in models of B-cell Malignancy. J Pharmacol Exp Ther. (2020) 374:211–22. doi: 10.1124/jpet.120.265538
126. Barcellini W, Murakhovskaya I, Terriou L, Pane F, Patriarca A, Butler K, et al. S286: Long-term efficacy and safety results from an ongoing open-label phase 2 study of parsaclisib for the treatment of autoimmune hemolytic anemia (AIHA). HemaSphere. (2022) 6:187. doi: 10.1097/01.HS9.0000844036.02998.15
127. Kuter DJ, Rogers KA, Boxer MA, Choi M, Agajanian R, Arnold DM, et al. Fostamatinib for the treatment of warm antibody autoimmune hemolytic anemia: Phase 2, multicenter, open-label study. Am J Hematol. (2022) 97:691–9. doi: 10.1002/ajh.26508
128. Kuter DJ, Piatek C, Röth A, Siddiqui A, Numerof RP, Dummer W, et al. Fostamatinib for warm antibody autoimmune hemolytic anemia: Phase 3, randomized, double-blind, placebo-controlled, global study (FORWARD). Am J Hematol. (2024) 99:79–87. doi: 10.1002/ajh.27144
129. Zhao X, Sun J, Zhang Z, Chen M, Gong T, He G, et al. Sovleplenib in patients with primary or secondary warm autoimmune haemolytic anaemia: results from phase 2 of a randomised, double-blind, placebo-controlled, phase 2/3 study. Lancet Haematol. (2025) 12:e97–e108. doi: 10.1016/S2352-3026(24)00344-2
130. Abdelrasoul H, Werner M, Setz CS, Okkenhaug K, and Jumaa H. PI3K induces B-cell development and regulates B cell identity. Sci Rep. (2018) 8:1327. doi: 10.1038/s41598-018-19460-5
131. Barcellini W, Pane F, Patriarca A, Murakhovskaya I, Terriou L, DeSancho MT, et al. Parsaclisib for the treatment of primary autoimmune hemolytic anemia: Results from a phase 2, open-label study. Am J Hematol. (2024) 99:2313–20. doi: 10.1002/ajh.27493
132. Incyte Corporation. A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of the Efficacy and Safety of Parsaclisib in Participants With Primary Warm Autoimmune Hemolytic Anemia (2024). Available online at: https://clinicaltrials.gov/study/NCT05073458 (Accessed March 24, 2025).
133. Podolanczuk A, Lazarus AH, Crow AR, Grossbard E, and Bussel JB. Of mice and men: an open-label pilot study for treatment of immune thrombocytopenic purpura by an inhibitor of Syk. Blood. (2009) 113:3154–60. doi: 10.1182/blood-2008-07-166439
134. Leu JH, Vermeulen A, Abbes C, Arroyo S, Denney WS, and Ling LE. Pharmacokinetics and pharmacodynamics across infusion rates of intravenously administered nipocalimab: results of a phase 1, placebo-controlled study. Front Neurosci. (2024) 18:1302714. doi: 10.3389/fnins.2024.1302714
135. Murakhovskaya I, Fattizzo B, Ebrahim T, Sweet K, and Shu C. Energy trial in warm autoimmune hemolytic anemia (wAIHA): design of a phase 2/3 randomized, double-blind, placebo-controlled study to assess the efficacy and safety of nipocalimab, an fcRn blocker. Blood. (2022) 140:2443–4. doi: 10.1182/blood-2022-165585
136. Jacobs JW, Booth GS, Raza S, Clark LM, Fasano RM, Gavriilaki E, et al. Current state and potential applications of neonatal Fc receptor (FcRn) inhibitors in hematologic conditions. Am J Hematol. (2024) 99:2351–66. doi: 10.1002/ajh.27487
137. Barcellini W, Michel M, Cuker A, Cooper N, Ghanima W, Wong R, et al. Pb2552: vayhia: A randomized, double-blind, phase iii trial to assess the efficacy and safety of ianalumab vs placebo in patients with warm autoimmune hemolytic anemia that failed ≥1 line of treatment. Hemasphere. (2023) 7:e28542f2. doi: 10.1097/01.HS9.0000976904.28542.f2
138. Lewis KE, Griffin L, Maria Z, Enstrom A, Bankhead AR, Peng SL, et al. Povetacicept (ALPN-303), a potent dual BAFF/APRIL antagonist in development for the treatment of autoimmune cytopenias, suppresses disease in a mouse model of autoimmune hemolytic anemia. Blood. (2023) 142:6462. doi: 10.1182/blood-2023-177786
139. Müller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Völkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - A case series with follow-up. N Engl J Med. (2024) 390:687–700. doi: 10.1056/NEJMoa2308917
140. Berentsen S, Barcellini W, D'Sa S, and Jilma B. Sutimlimab for treatment of cold agglutinin disease: why, how and for whom? Immunotherapy. (2022) 14:1191–204. doi: 10.2217/imt-2022-0085
141. Bartko J, Schoergenhofer C, Schwameis M, Firbas C, Beliveau M, Chang C, et al. Randomized, first-in-human, healthy volunteer trial of sutimlimab, a humanized antibody for the specific inhibition of the classical complement pathway. Clin Pharmacol Ther. (2018) 104:655–63. doi: 10.1002/cpt.1111
142. Jäger U, D'Sa S, Schörgenhofer C, Bartko J, Derhaschnig U, Sillaber C, et al. Inhibition of complement C1s improves severe hemolytic anemia in cold agglutinin disease: a first-in-human trial. Blood. (2019) 133:893–901. doi: 10.1182/blood-2018-06-856930
143. Gelbenegger G, Schoergenhofer C, Derhaschnig U, Buchtele N, Sillaber C, Fillitz M, et al. Inhibition of complement C1s in patients with cold agglutinin disease: lessons learned from a named patient program. Blood Adv. (2020) 4:997–1005. doi: 10.1182/bloodadvances.2019001321
144. Röth A, Barcellini W, D'Sa S, Miyakawa Y, Broome CM, Michel M, et al. Sutimlimab in cold agglutinin disease. N Engl J Med. (2021) 384:1323–34. doi: 10.1056/NEJMoa2027760
145. Röth A, Broome CM, Barcellini W, Tvedt THA, Miyakawa Y, D'Sa S, et al. Long-term sutimlimab improves quality of life for patients with cold agglutinin disease: CARDINAL 2-year follow-up. Blood Adv. (2024) 8:2650. doi: 10.1182/bloodadvances.2024013366
146. Röth A, Barcellini W, D'Sa S, Miyakawa Y, Broome CM, Michel M, et al. Sustained inhibition of complement C1s with sutimlimab over 2 years in patients with cold agglutinin disease. Am J Hematol. (2023) 98:1246–53. doi: 10.1002/ajh.26965
147. Röth A, Berentsen S, Barcellini W, D'Sa S, Jilma B, Michel M, et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood. (2022) 140:980–91. doi: 10.1182/blood.2021014955
148. Röth A, Berentsen S, Barcellini W, D'Sa S, Jilma B, Michel M, et al. Long-term efficacy and safety of continued complement C1s inhibition with sutimlimab in cold agglutinin disease: CADENZA study Part B. EClinicalMedicine. (2024) 74:102733. doi: 10.1016/j.eclinm.2024.102733
149. D'Sa S, Vos JMI, Barcellini W, Wardęcki M, Perrin L, Barker G, et al. Safety, tolerability, and activity of the active C1s antibody riliprubart in cold agglutinin disease: a phase 1b study. Blood. (2024) 143:713–20. doi: 10.1182/blood.2023022153
150. Roman E, Fattizzo B, Shum M, Hanna W, Lentz SR, Araujo SSS, et al. Safety and efficacy of pegcetacoplan treatment for cold agglutinin disease and warm antibody autoimmune hemolytic anemia. Blood. (2025) 145:397–408. doi: 10.1182/blood.2023022549
151. Jilma B, Roman E, Ueda Y, Mappa S, Szamosi J, Savage J, et al. Trial in progress: randomized, double-blind, placebo-controlled multicenter phase 3 study to evaluate the efficacy and safety of pegcetacoplan in patients with cold agglutinin disease (CASCADE). Blood. (2022) 140:5326–7. doi: 10.1182/blood-2022-155989
152. Röth A, Bommer M, Hüttmann A, Herich-Terhürne D, Kuklik N, Rekowski J, et al. Eculizumab in cold agglutinin disease (DECADE): an open-label, prospective, bicentric, nonrandomized phase 2 trial. Blood Adv. (2018) 2:2543–9. doi: 10.1182/bloodadvances.2018024190
153. Röth A, Broome CM, Barcellini W, Michel M, Vos J, Khan U, et al. Efficacy of sutimlimab stratified by baseline hemoglobin in patients with cold agglutinin disease: a post hoc analysis from CARDINAL and CADENZA. HemaSphere. (2024) 8:2857–8.
154. Michel M, Barcellini W, Broome CM, Jaeger U, Ueda Y, Hill QA, et al. Baseline characteristics from the first inter analysis of CADENCE, the cold agglutinin disease (CAD) /Cold agglutinin syndrome (CAS) registry. HemaSphere. (2024) 8:2855–6.
155. Chow T, Shamszad P, Vinnard C, Yoon E, Belinski J, Karpenko I, et al. First-in-human study with SAR445088: A novel selective classical complement pathway inhibitor. Clin Transl Sci. (2023) 16:673–85. doi: 10.1111/cts.13481
156. Simmons KT, Chan J, Hussain S, Rose EL, Markham K, Byun TS, et al. Anti-C1s humanized monoclonal antibody SAR445088: A classical pathway complement inhibitor specific for the active form of C1s. Clin Immunol. (2023) 251:109629. doi: 10.1016/j.clim.2023.109629
157. Mackness BC, Jaworski JA, Boudanova E, Park A, Valente D, Mauriac C, et al. Antibody Fc engineering for enhanced neonatal Fc receptor binding and prolonged circulation half-life. MAbs. (2019) 11:1276–88. doi: 10.1080/19420862.2019.1633883
158. Röth A, Vos J, Barcellini W, Storek M, Chow T, Wardęcki M, et al. Riliprubart (SAR445088) in cold agglutinin disease: interim analysis from part 2 of a long-term phase 1B trial confirms acceptable efficacy and safety with 12-weekly IV administration. HemaSphere. (2024) 8:2853–4.
159. de Boer ECW, Jalink M, Delvasto-Nuñez L, Meulenbroek EM, Baas I, Janssen SR, et al. C1-inhibitor treatment in patients with severe complement-mediated autoimmune hemolytic anemia. Blood Adv. (2023) 7:3128–39. doi: 10.1182/bloodadvances.2022009402
160. Hillmen P, Szer J, Weitz I, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. (2024) 390:1060. doi: 10.1056/NEJMx240003
161. Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N Engl J Med. (2006) 355:1233–43. doi: 10.1056/NEJMoa061648
162. Röth A, Hüttmann A, Rother RP, Dührsen U, and Philipp T. Long-term efficacy of the complement inhibitor eculizumab in cold agglutinin disease. Blood. (2009) 113:3885–6. doi: 10.1182/blood-2009-01-196329
163. Gupta N and Wang ES. Long-term response of refractory primary cold agglutinin disease to eculizumab therapy. Ann Hematol. (2014) 93:343–4. doi: 10.1007/s00277-013-1800-7
164. Shapiro R, Chin-Yee I, and Lam S. Eculizumab as a bridge to immunosuppressive therapy in severe cold agglutinin disease of anti-Pr specificity. Clin Case Rep. (2015) 3:942–4. doi: 10.1002/ccr3.399
165. Peffault de Latour R, Röth A, Kulasekararaj AG, Han B, Scheinberg P, Maciejewski JP, et al. Oral iptacopan monotherapy in paroxysmal nocturnal hemoglobinuria. N Engl J Med. (2024) 390:994–1008. doi: 10.1056/NEJMoa2308695
Keywords: autoimmune hemolytic anemias, cold agglutinin disease, warm autoimmune hemolytic anemia, pathogenesis, strategy, target therapy, complement system, immunotherapy
Citation: Costa A, Mulas O, Mereu AM, Schintu M, Greco M and Caocci G (2025) Beneath the surface in autoimmune hemolytic anemia: pathogenetic networks, therapeutic advancements and open questions. Front. Immunol. 16:1624667. doi: 10.3389/fimmu.2025.1624667
Received: 07 May 2025; Accepted: 03 July 2025;
Published: 31 July 2025.
Edited by:
Alenka Gagro, Children’s Hospital Zagreb, CroatiaReviewed by:
Emanuele Bizzi, Vita-Salute San Raffaele University, ItalySigbjørn Berentsen, Fonna Hospital Trust, Norway
Copyright © 2025 Costa, Mulas, Mereu, Schintu, Greco and Caocci. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Olga Mulas, bXVsYXNvbGdhQHVuaWNhLml0