 Cuiting Dong
Cuiting Dong Mingda Wu
Mingda Wu Haonan Liu1†
Haonan Liu1† Kunpeng Yang
Kunpeng Yang Yuejiao Lan
Yuejiao Lan Xiaodan Lu
Xiaodan Lu- 1Changchun University of Chinese Medicine, Changchun, Jilin, China
- 2Jilin Province People’s Hospital, Changchun, Jilin, China
Lupus nephritis (LN), a severe manifestation of systemic lupus erythematosus (SLE), is driven by immune complex deposition and complement activation, resulting in glomerular inflammation and podocyte injury. Beyond being passive targets, podocytes actively modulate renal immunity through cytokine secretion and antigen presentation. Recent advances in urinary biomarkers such as NGAL, TWEAK, and MCP-1 and composite indices like the Renal Activity Index for Lupus (RAIL) offer dynamic and noninvasive monitoring of disease activity. Immunotherapy has transitioned from nonspecific immunosuppression to targeted biologics, with agents such as belimumab and telitacicept improving outcomes by modulating B-cell function. Additionally, emerging therapies including bortezomib and daratumumab demonstrate efficacy in refractory LN through plasma cell depletion. This review summarizes current immunological insights, biomarker innovations, and immunotherapy strategy to support precision medicine and improve long-term renal prognosis in LN.
1 Introduction
Lupus nephritis (LN), one of the most common and severe manifestations of systemic lupus erythematosus (SLE), results from immune complex (IC) deposition in the kidneys (1, 2). SLE is a chronic, multisystem autoimmune disease triggered by genetic and environmental factors—including hormonal imbalances, infections, and drug exposures—that disrupt immune tolerance and promote autoantibody production (3, 4). Approximately 50% of SLE patients develop LN (5), characterized by glomerular accumulation of autoantibodies, such as anti-dsDNA, which activate the complement cascade, recruit inflammatory cells, and injure podocytes (6, 7). Rather than serving only as passive targets, podocytes actively contribute to disease by expressing pattern recognition receptors, secreting cytokines, and presenting antigens that amplify the immune responses and injury (8, 9).
Despite renal biopsy being the definitive method for LN histopathological assessment, its invasiveness limits repeated use in routine clinical management (10). Current therapies induce complete renal response in only approximately 20–40% of patients, and 20% progress to end-stage kidney disease (ESKD) within five years (11), underscoring the need for improved biomarkers and treatments. Urinary biomarkers, including NGAL and TWEAK, offer potential for disease monitoring (12), while biologics such as belimumab and telitacicept target B-cell signaling pathways with increasing success in clinical trials (13–15). This review integrates recent insights into LN pathogenesis, diagnostic biomarkers, and therapeutic innovations to support personalized care and research progress.
2 Pathogenesis of lupus nephritis
The development of LN occurs within the genetic framework of SLE, with its increased prevalence highlighting the critical influence of epigenetic modifications (16). The disease process involves a complex breakdown of immune regulation, triggered by viral infections, environmental pollutants, and drug-induced effects (17–20). This cascade begins with impaired removal of apoptotic cells, resulting in the abnormal release of nuclear antigens. Unprocessed nucleic acids and nuclear proteins stimulate B-cell activation through Toll-like receptor (TLR)-dependent pathways, promoting the production of anti-nuclear antibodies (21). These antibodies contribute to the formation of circulating immune complexes (CICs) or, via molecular mimicry, generate in situ complexes that target structural elements of the glomerular basement membrane (22, 23). While CICs accumulate in renal tissues due to hemodynamic forces, in situ complexes directly bind glomerular antigens (24, 25). Both mechanisms ultimately trigger complement activation and neutrophil recruitment (26). Concurrently, immune complexes induce glomerular endothelial and mesangial cells to release proinflammatory cytokines, including monocyte chemoattractant protein-1 (MCP-1) and tumor necrosis factor-α (TNF-α) (27, 28). These mediators intensify inflammation via NF-κB signaling, culminating in podocyte damage and impairment of the glomerular filtration barrier (29).
3 Podocyte injury in lupus nephritis
3.1 Immune complex-mediated injury and complement activation
Podocyte injury in LN displays significant pathological diversity across different disease classes (30). In proliferative LN (classes III/IV), anti-dsDNA antibodies activate complement pathways and induce endothelial inflammation, while subendothelial immune complex (IC) accumulation leads to structural alterations in podocytes (6, 31). These changes include cytoskeletal reorganization through reduced nephrin expression and inhibited VEGF-A signaling (32). Conversely, class V membranous LN primarily involves subepithelial IC deposition, which stimulates localized complement activation and functional impairment of podocytes (33). This manifests as foot process effacement without basement membrane disruption, differing mechanistically from the structural damage (podocyte detachment, basement membrane rupture) seen in proliferative forms. These distinctions may account for the more favorable long-term outcomes observed in class V disease (34). Emerging evidence indicates that the extent of glomerular endothelial damage in proliferative LN positively associates with foot process width (FPW). Furthermore, IgG antibodies promote podocyte cytoskeletal reorganization by activating β1-integrin-dependent pathways (32). Dysregulation of the VEGF-endothelin axis also contributes to filtration barrier dysfunction through increased proinflammatory mediator release and impaired podocyte–endothelial crosstalk (35). Interestingly, while VEGF-A downregulation in proliferative LN contributes to endothelial injury and proteinuria, class V membranous LN often shows preserved or even upregulated VEGF-A levels, potentially reflecting a compensatory response to subepithelial immune complex deposition (36, 37). These subtype-specific differences underscore a knowledge gap in understanding VEGF signaling dynamics and highlight the need for tailored therapeutic strategies targeting angiogenic pathways.
3.2 Cytoskeletal disruption and signaling pathways
3.2.1 Immune complex deposition
The deposition patterns of ICs in LN vary significantly based on their anatomical distribution. In proliferative LN (classes III/IV), ICs predominantly accumulate in subendothelial and mesangial regions (38). This initiates podocyte injury through cytoskeletal destabilization, which is mediated by reduced expression of nephrin and podocin, and promotes endothelial-mesenchymal transition (6). These effects occur via Fcγ receptor-dependent complement activation and NF-κB-driven release of inflammatory cytokines such as MCP-1 and TNF-α (7). Conversely, class V membranous LN is distinguished by subepithelial IC deposition, where the hydrophobic properties of the glomerular basement membrane restrict complement activation, primarily involving C5b-9 membrane attack complexes (39, 40). This results in foot process effacement and internalization of slit diaphragm proteins, leading to selective filtration defects rather than extensive inflammatory damage (7, 41). Importantly, FPW expansion correlates with clinical markers of disease severity across LN subtypes. However, the underlying mechanisms differ. In class V LN, FPW alterations reflect charge-selective barrier impairment, whereas in proliferative LN, FPW abnormalities stem from structural damage to the lamina rara interna and podocyte detachment. These distinctions support the need for histotype-specific treatment approaches (41) (Figure 1).
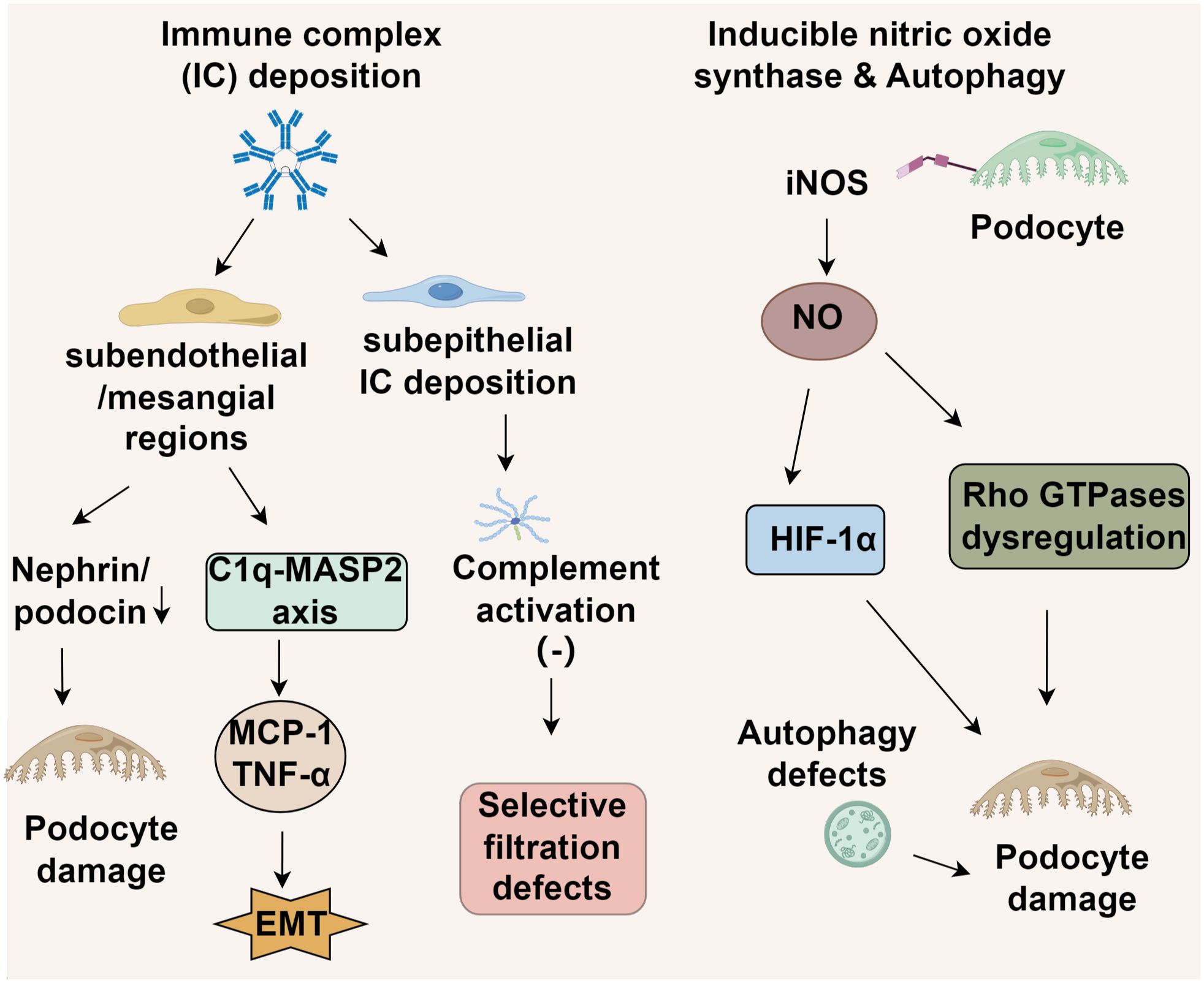
Figure 1. Molecular mechanisms of lupus nephritis podocyte injury.
Endothelial damage in LN demonstrates subtype-dependent characteristics (42). In proliferative LN (classes III/IV), excessive complement activation triggered by anti-dsDNA antibodies causes widening of the basement membrane-endothelial cleft and degradation of the endothelial glycocalyx (43). In contrast, class V LN features endothelial-mesenchymal transition (EndMT) induced by podocyte-derived TGF-β overexpression, leading to basement membrane thickening while largely sparing endothelial integrity (42). Notably, genetic deficiencies in complement regulatory proteins or autoantibodies targeting complement factors can perpetuate alternative pathway activation. This promotes podocyte dysfunction and disrupts the thrombomodulin-protein C system, potentially precipitating aHUS-like pathology and identifying a distinct molecular subset of nephrotic-range proteinuria (44).
3.2.2 iNOS-mediated podocyte injury in lupus nephritis
Emerging evidence implicates podocyte-specific upregulation of inducible nitric oxide synthase (iNOS) as a key mediator of structural damage in lupus nephritis (45), though its exact pathogenic role remains to be fully elucidated. The current paradigm suggests that excessive nitric oxide production leads to podocyte injury through hypoxia-inducible factor 1α (HIF-1α) activation and subsequent dysregulation of Rho GTPases, particularly Cdc42 and Rac1 (46). This dysregulation disrupts podocyte structural integrity, highlighting iNOS and its downstream effectors as potential therapeutic targets to mitigate NO-induced podocyte injury and slow disease progression (46). Parallel studies have identified autophagy as a critical regulator of podocyte survival in LN. Early disease stages are characterized by distinct alterations in autophagy-related pathways, which may determine podocyte fate. In vitro models demonstrate that LN-like conditions upregulate cyclooxygenase-2 (COX-2) and induce endoplasmic reticulum (ER) stress in podocytes, mechanistically linked to activation of the unfolded protein response (UPR) transcription factor ATF4 (47). Pharmacological inhibition of COX-2 attenuates autophagy induction in these models, while ATF4 knockdown abolishes LN-associated COX-2 overexpression, suggesting a causal role for the ATF4-COX-2 axis in podocyte injury (47). Multiple studies have demonstrated a strong correlation between iNOS level and the progression of disease phenotypes in several murine LN models (48, 49). Notably, the iNOS inhibitor SD-3651 significantly ameliorates both proteinuria and podocytopathy in experimental LN mice (49). Despite promising preclinical data supporting iNOS inhibition, clinical translation faces challenges, including off-target effects of pan-iNOS inhibitors and the absence of reliable biomarkers for patient stratification. Future therapeutic strategies require selective iNOS modulation combined with autophagy restoration to preserve podocyte function in LN.
3.3 Podocyte-related biomarkers in lupus nephritis
Studies on podocyte-linked biomarkers in LN highlight their dual function as both immune complex-mediated injury targets and disease progression markers. The reduced expression of podocyte-specific proteins in glomeruli emphasizes the critical role of cytoskeletal disruption in LN glomerulopathy pathogenesis (50). Furthermore, urinary sediment analysis in active LN patients detects abnormal levels of podocalyxin, synaptopodin mRNA, and immature nephrin/GLEPP1 proteins, supporting their use as noninvasive diagnostic tools for glomerular damage (51). Proteomic analyses have identified α-enolase and annexin A1 as podocyte-targeted autoantigens in LN, where their strong interaction with glomerular IgG suggests involvement in immune complex formation (52, 53). Podocyte-derived microparticles (MPs), carrying surface markers like annexin V and podocalyxin, serve as dynamic indicators of renal disease activity and histopathological changes in SLE (54). Importantly, NF-κB signaling is consistently implicated in proinflammatory gene expression in LN podocytes, driving production of cytokines and apoptotic markers (55). Besides, the NF-κB pathway also orchestrates cell survival and tissue repair mechanisms. For instance, selective inhibition of canonical NF-κB mitigates glomerular inflammation, while blockade of noncanonical branches could disrupt podocyte adaptation and regeneration (29, 56). Future studies need to clarify which subunits and downstream effectors of NF-κB signaling represent optimal therapeutic windows in LN (57). The extent of podocyte injury closely parallels proteinuria severity, as research shows that foot process effacement (FPE) in both proliferative and non-proliferative LN correlates with significant protein loss, alongside diminished expression of mature podocyte markers like synaptopodin, nephrin, and GLEPP1 in proliferative forms (34). Additionally, these injured podocytes exacerbate LN-related inflammation by releasing cytokines such as IL-1β, TNF-α, IFN-α, and IFN-γ, which sustain renal damage (58). Osteopontin (OPN), produced by T cells, enhances macrophage recruitment into glomeruli (59) and may also modulate podocyte signaling and movement, thereby contributing to proteinuria onset and progression (60).
4 Noninvasive urinary biomarkers in lupus nephritis
Urinary biomarkers serve as crucial tools for the noninvasive assessment and longitudinal tracking of LN, offering insights into renal immune dysfunction and inflammatory damage (61, 62). Among individual biomarkers, neutrophil gelatinase-associated lipocalin (NGAL) demonstrates strong associations with disease severity and therapeutic efficacy (63). Similarly, tumor necrosis factor-related weak inducer of apoptosis (TWEAK) is implicated in NF-κB-mediated inflammatory pathways and LN-specific renal pathology (64). Monocyte chemoattractant protein-1 (MCP-1), a key chemokine, reflects macrophage accumulation and aids in histopathological classification, while vascular cell adhesion molecule-1 (VCAM-1) facilitates leukocyte migration and correlates with disease exacerbations (65–67). Multiparametric strategies, such as the Renal Activity Index for Lupus (RAIL), improve diagnostic precision by combining several biomarkers, such as NGAL, KIM-1, and MCP-1, to forecast treatment responses. Urinary proteomic profiling further enhances detection by identifying renal-specific pathological patterns, though its clinical implementation faces hurdles due to preanalytical inconsistencies and methodological disparities (68–71). Although standalone biomarkers exhibit limited discriminatory capacity, composite panels like RAIL show enhanced predictive value, highlighting their utility in real-time LN monitoring. Nevertheless, validation across diverse populations and standardization of analytical protocols are essential for widespread clinical adoption (Figure 2).
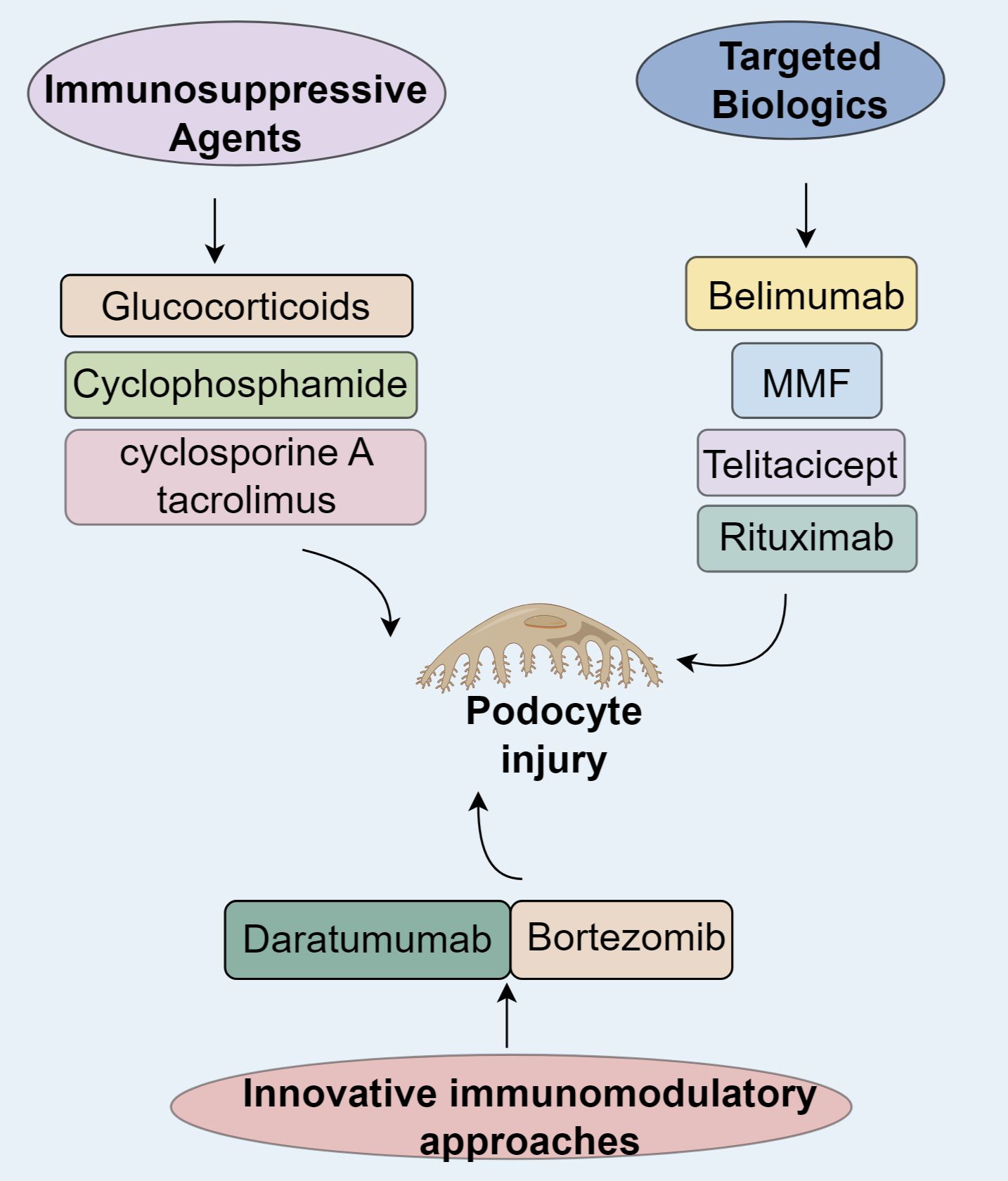
Figure 2. Immunotherapy in lupus nephritis.
5 Immunotherapy in lupus nephritis
5.1 Traditional immunosuppressive agents
Immunosuppressive drugs exhibit podocyte-preserving properties in LN by modulating slit diaphragm components. Glucocorticoids reinforce the actin cytoskeleton, stimulate RhoA/ROCK pathways, and enhance nephrin synthesis, recovering podocin immunofluorescence to 82% of baseline in NZB/W mice (72). Cyclophosphamide diminishes podocyte apoptosis by inhibiting IL-6/JAK2/STAT3 cascades, with combination therapies increasing nephrin-podocin colocalization by 2.3-fold (72). Calcineurin inhibitors (cyclosporine A, tacrolimus) attenuate foot process effacement by obstructing NFAT signaling and facilitating podocin reintegration into slit diaphragms, thus restoring glomerular charge selectivity (73).
5.2 Targeted biologics of immune inhibitors in lupus nephritis
Belimumab, a humanized IgG1λ monoclonal antibody, neutralizes soluble BAFF to prevent BAFFR/BCMA/TACI receptors and curtailing pathogenic B-cell expansion and autoantibody generation (74–78). The landmark BLISS-LN phase III trial revealed that belimumab adjunctive therapy significantly enhanced primary and complete renal responses at 104 weeks compared to standard care alone, with a safety profile matching placebo (79, 80). However, the trial’s findings must be interpreted with caution, as its relatively short follow-up duration and underrepresentation of patients with severe proteinuria or class V LN limit generalizability. Efficacy was more pronounced in patients on mycophenolate mofetil (MMF) induction, possibly due to baseline disease heterogeneity (79, 81). While proliferative LN with mild proteinuria showed 9–10% improvements in PERR/CRR, membranous LN or high-proteinuria subgroups derived no significant benefit (82). East Asian populations confirmed its robust efficacy, demonstrating a 63% reduction in renal risk (83), culminating in its 2019 FDA approval as the first SLE-specific biologic.
Telitacicept simultaneously inhibits BAFF and APRIL, disrupting their binding to BCMA/TACI and impairing long-lived plasma cell survival and B-cell hyperactivity (84). A retrospective analysis of 72 active SLE patients (34 with LN) reported median reductions of 77% in 24-hour proteinuria and 75% in anti-dsDNA titers at 24 weeks, with PRR and CRR rates of 76.24% and 70.58%, respectively. By 52 weeks, CRR reached 66.67% (6/9), with infections being the most frequent adverse event (23.6%) (85). Following global phase III trial approvals (NMPA/EMA), large-scale efficacy validation is underway. In refractory LN, sequential telitacicept-rituximab therapy achieved 100% CRR at 19 months, surpassing belimumab monotherapy without increased infection risk (15), suggesting superior efficacy in treatment-resistant cases pending further verification. Rituximab is a chimeric anti-CD20 IgG1κ antibody that depletes CD20+ B cells through ADCC and CDC mechanisms while dampening T-cell-driven autoimmunity (86, 87). The LUNAR phase III trial did not meet its primary endpoint, with no significant renal response improvement at 52 weeks in class III/IV LN, potentially due to CDC impairment in hypocomplementemic states and BAFF rebound (88–90). Nevertheless, refractory LN cohorts demonstrate clinical utility: a meta-analysis of 300 patients revealed a 74% overall response rate, with superior outcomes in class III LN (91, 92). Prospective studies corroborate its glucocorticoid-sparing effects and acceptable safety (93), leading to its classification as a second-line refractory LN therapy in the 2024 KDIGO guidelines (94).
5.3 Innovative immunomodulatory approaches
The proteasome inhibitor bortezomib induces plasma cell apoptosis via endoplasmic reticulum stress and NF-κB pathway inhibition. In NZB/W F1 mice, bortezomib significantly depleted splenic and bone marrow-resident long-lived plasma cells (95–97). A Japanese multicenter randomized controlled trial in refractory SLE reported a 75% SLE Responder Index at 12 weeks, compared to 40% in placebo, despite similar anti-dsDNA levels at 24 weeks (98). In China, five patients with high-activity LN showed partial renal response after four cycles, with 60% achieving complete renal response (CRR) and 20% progressing to end-stage renal disease over three years (99). Another Spanish retrospective study found that bortezomib reduced median SLEDAI from 27 to 0, with CRR and PRR rates of 8.3% and 83.3%, respectively, though hypogammaglobulinemia (IgG <500 mg/dL) occurred in 50% of patients (100). However, the immunosuppressive side effects of bortezomib require serious attention, with infection being the most frequently observed adverse event (101). These safety concerns, combined with small sample sizes and limited follow-up constrain conclusions regarding long-term efficacy and safety (98–100). Alternatively, daratumumab, a CD38-targeting IgGκ monoclonal antibody, depletes >90% of CD38+plasma cells via antibody-dependent cellular cytotoxicity (ADCC) in bone marrow and inflamed tissues (97, 102, 103). In Germany, two refractory LN cases treated with daratumumab-belimumab showed a 50% reduction in anti-dsDNA antibodies and a 62 ± 8% decline in interferon signature gene expression by week 12, with no severe adverse events (102). Similarly, an Italian cohort of six refractory LN patients demonstrated a 66% reduction in SLEDAI, 86% decrease in proteinuria, 35% improvement in serum creatinine, and 1.8-fold rise in C4 after 12 months (104). Daratumumab demonstrates comparable autoantibody clearance efficacy to bortezomib with a trend toward lower infectious adverse events (98, 104, 105), supporting its therapeutic promise in refractory LN.
5.4 Natural compounds reprogram TME in lupus nephritis
Macrophages, as a major subset of innate immune cells, represent the predominant infiltrating cell population in the kidneys of LN patients. Compounds targeting macrophage polarization include total glucosides of paeony, which upregulate PD-L2 expression via STAT6 phosphorylation, inducing M2-like macrophage polarization and exerting immunosuppressive effects to attenuate glomerular damage in LN mice (106). Dihydroartemisinin (DHA), a metabolite of artemisinin with potent antimalarial properties, also demonstrates remarkable anti-inflammatory and immunomodulatory activities. By inhibiting ITK signaling to suppress T follicular helper (Tfh) cells and reducing serum levels of IgG, IgM, IgA and anti-dsDNA antibodies, DHA significantly alleviates LN symptoms (107). Genetically engineered macrophage membranes overexpressing CCR2 can enhance the targeted delivery of DHA-loaded nanoparticles to inflammatory sites in LN, thereby reducing monocyte/macrophage infiltration and reprogramming the M1/M2 macrophage balance to modulate the renal immune microenvironment and ameliorate kidney injury (108). Artesunate, a semi-synthetic derivative of artemisinin, improves lupus nephritis symptoms by regulating the T follicular regulatory to T follicular helper cell ratio and activating the JAK2-STAT3 signaling pathway, consequently decreasing renal anti-dsDNA antibody deposition and reducing pathogenic cytokine levels including IL-6, IFN-γ, and IL-21 (109). Furthermore, LLDT-8, a novel triptolide analog, has been demonstrated by ZHANG et al. (28) to exhibit therapeutic effects against LN through suppressing chemokine and IL-6 expression, decreasing renal macrophage and neutrophil infiltration, and reducing glomerular IgG deposition in LN mice (110).
6 Conclusion
LN is driven by a multifaceted network of autoimmunity in which podocytes are not merely passive victims but active contributors to renal inflammation and immune dysregulation. As the field progresses, a paradigm shift is emerging that positions podocytes as both biomarkers and therapeutic targets. Advances in our understanding of podocyte-specific signaling, such as VEGF Notch, iNOS autophagy, and cytokine-mediated injury, have catalyzed the development of more refined, histotype-specific interventions. The rise of targeted biologics, including belimumab and telitacicept, marks a turning point in LN management with promising efficacy in modulating B cell and plasma cell function. Novel immunomodulators like bortezomib and daratumumab further expand treatment options, particularly in refractory LN characterized by persistent autoantibody production. These agents also hold the potential to indirectly preserve or restore podocyte function by reducing the burden of circulating immune complexes and complement activation.
Moving forward, integrating urinary podocyte-associated biomarkers into clinical practice may enable real-time disease stratification and therapeutic monitoring. Moreover, therapies aimed directly at stabilizing podocyte architecture, modulating cell-to-cell signaling with glomerular endothelial cells, or enhancing autophagic resilience represent exciting frontiers. Longitudinal studies addressing the durability of remission, infection risks, and population-specific responses to therapy are urgently needed. Ultimately, a podocyte-centered therapeutic framework guided by immunological profiling and biomarker-informed personalization may offer a path toward durable renal protection and reduced reliance on nonspecific immunosuppression in LN.
Author contributions
CD: Writing – original draft. MW: Writing – original draft. HL: Writing – original draft. KY: Writing – original draft. SM: Writing – original draft. YG: Writing – original draft. YL: Writing – original draft, Writing – review & editing. XL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by Science and Technology Development Plan Project of Jilin Province (YDZJ202301ZYTS200), the Outstanding Program of the Jilin Provincial Youth Fund (20240101007JJ) and 2024 Provincial Budgeted Basic Construction Funds (2024C012-13).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. De Vriese AS, Sethi S, and Fervenza FC. Lupus nephritis: redefining the treatment goals. Kidney Int. (2025) 107:198–211. doi: 10.1016/j.kint.2024.10.018
2. Halfon M, Tankeu AT, and Ribi C. Mitochondrial dysfunction in systemic lupus erythematosus with a focus on lupus nephritis. Int J Mol Sci. (2024) 25:6162. doi: 10.3390/ijms25116162
3. Perl A. Systems biology of lupus: mapping the impact of genomic and environmental factors on gene expression signatures, cellular signaling, metabolic pathways, hormonal and cytokine imbalance, and selecting targets for treatment. Autoimmunity. (2010) 43:32–47. doi: 10.3109/08916930903374774
4. Wang H, Liang Y, Boor PJ, Khanipov K, Zhang Y, Yu X, et al. Protective role of dietary short-chain fatty acid propionate against autoimmune responses and pathology of systemic lupus erythematosus in MRL-lpr mice. J Nutr. (2025) 155:3116–27. doi: 10.1016/j.tjnut.2025.06.031
5. Said D, Rashad NM, Abdelrahmanc NS, and Dawaa GA. Antineutrophil cytoplasmic antibody in lupus nephritis: correlation with clinicopathological characteristics and disease activity. Curr Rheumatol Rev. (2021) 17:213–21. doi: 10.2174/1573397116999201208213422
6. Sakhi H, Moktefi A, Bouachi K, Audard V, Hénique C, Remy P, et al. Podocyte injury in lupus nephritis. J Clin Med. (2019) 8:1340. doi: 10.3390/jcm8091340
7. Davidson A. What is damaging the kidney in lupus nephritis? Nat Rev Rheumatol. (2016) 12:143–53. doi: 10.1038/nrrheum.2015.159
8. Goldwich A, Burkard M, Olke M, Daniel C, Amann K, Hugo C, et al. Podocytes are nonhematopoietic professional antigen-presenting cells. J Am Soc Nephrol. (2013) 24:906–16. doi: 10.1681/ASN.2012020133
9. Liu T, Lin Z, Zhang X, Yang Y, Huang G, Yu Y, et al. Targeting nanotherapeutics for highly efficient diagnosis and treatment of systemic lupus erythematosus through regulation of immune response. Small Sci. (2025) 5:2400521. doi: 10.1002/smsc.202400521
10. Liu T, Yang YL, Zhou Y, and Jiang YM. Noninvasive biomarkers for lupus nephritis. Lab Med. (2024) 55:535–42. doi: 10.1093/labmed/lmae015
11. Appel GB, Contreras G, Dooley MA, Ginzler EM, Isenberg D, Jayne D, et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J Am Soc Nephrol. (2009) 20:1103–12. doi: 10.1681/ASN.2008101028
12. Tektonidou MG, Dasgupta A, and Ward MM. Risk of end-stage renal disease in patients with lupus nephritis, 1971-2015: A systematic review and bayesian meta-analysis. Arthritis Rheumatol. (2016) 68:1432–41. doi: 10.1002/art.39594
13. Atisha-Fregoso Y, Malkiel S, Harris KM, Byron M, Ding L, Kanaparthi S, et al. Phase II randomized trial of rituximab plus cyclophosphamide followed by belimumab for the treatment of lupus nephritis. Arthritis Rheumatol. (2021) 73:121–31. doi: 10.1002/art.41466
14. Furie R, Rovin BH, Houssiau F, Contreras G, Teng YKO, Curtis P, et al. Safety and efficacy of belimumab in patients with lupus nephritis: open-label extension of BLISS-LN study. Clin J Am Soc Nephrol. (2022) 17:1620–30. doi: 10.2215/CJN.02520322
15. Chen Y, Shi N, Lei X, Ren P, Lan L, Chen L, et al. The efficacy of rituximab plus belimumab or telitacicept in refractory lupus nephritis. Rheumatol (Oxford). (2025) 64:221–7. doi: 10.1093/rheumatology/kead674
16. Siegel CH and Sammaritano LR. Systemic lupus erythematosus: A review. Jama. (2024) 331:1480–91. doi: 10.1001/jama.2024.2315
17. Roveta A, Parodi EL, Brezzi B, Tunesi F, Zanetti V, Merlotti G, et al. Lupus nephritis from pathogenesis to new therapies: an update. Int J Mol Sci. (2024) 25:8981. doi: 10.3390/ijms25168981
18. Lee YS, Woo JS, Jhun J, Choi JW, Lee AR, Lee KH, et al. SARS-CoV-2 spike aggravates lupus nephritis and lung fibrosis in systemic lupus erythematosus. Lupus Sci Med. (2024) 11:e001104. doi: 10.1136/lupus-2023-001104
19. He G, Wang Y, Cheng C, Guo J, Lin Z, Liang Z, et al. PM(2.5) constituents associated with mortality and kidney failure in childhood-onset lupus nephritis: A 19-year cohort study. Sci Tot Environ. (2024) 949:175333. doi: 10.1016/j.scitotenv.2024.175333
20. Bhatnagar M, Agaronov A, Sarkisyan E, Sotoudeh Deilamy I, Pepito D, and Akhondi H. Overlapping drug-induced vasculitis, ANCA-associated vasculitis, and lupus nephritis caused by low-dose hydralazine. Int J Rheum Dis. (2023) 26:2272–7. doi: 10.1111/1756-185X.14809
21. Villanueva V, Li X, Jimenez V, Faridi HM, and Gupta V. CD11b agonists offer a novel approach for treating lupus nephritis. Transl Res. (2022) 245:41–54. doi: 10.1016/j.trsl.2022.03.001
22. Waterman HR, Dufort MJ, Posso SE, Ni M, Li LZ, Zhu C, et al. Lupus IgA1 autoantibodies synergize with IgG to enhance plasmacytoid dendritic cell responses to RNA-containing immune complexes. Sci Transl Med. (2024) 16::eadl3848. doi: 10.1126/scitranslmed.adl3848
23. Bruschi M, Angeletti A, Kajana X, Moroni G, Sinico RA, Fredi M, et al. Evidence for charge-based mimicry in anti dsDNA antibody generation. J Autoimmun. (2022) 132:102900. doi: 10.1016/j.jaut.2022.102900
24. Hanrotel-Saliou C, Segalen I, Le Meur Y, Youinou P, and Renaudineau Y. Glomerular antibodies in lupus nephritis. Clin Rev Allergy Immunol. (2011) 40:151–8. doi: 10.1007/s12016-010-8204-4
25. Onishi S, Adnan E, Ishizaki J, Miyazaki T, Tanaka Y, Matsumoto T, et al. Novel autoantigens associated with lupus nephritis. PLoS One. (2015) 10:e0126564. doi: 10.1371/journal.pone.0126564
26. Wirestam L, Arve S, Linge P, and Bengtsson AA. Neutrophils-important communicators in systemic lupus erythematosus and antiphospholipid syndrome. Front Immunol. (2019) 10:2734. doi: 10.3389/fimmu.2019.02734
27. Gilkeson GS, Mashmoushi AK, Ruiz P, Caza TN, Perl A, and Oates JC. Endothelial nitric oxide synthase reduces crescentic and necrotic glomerular lesions, reactive oxygen production, and MCP1 production in murine lupus nephritis. PLoS One. (2013) 8:e64650. doi: 10.1371/journal.pone.0064650
28. Olaru F, Döbel T, Lonsdorf AS, Oehrl S, Maas M, Enk AH, et al. Intracapillary immune complexes recruit and activate slan-expressing CD16+ monocytes in human lupus nephritis. JCI Insight. (2018) 3:e96492. doi: 10.1172/jci.insight.96492
29. Liu Y, Deng W, Meng Q, Qiu X, Sun D, and Dai C. CD8+iTregs attenuate glomerular endothelial cell injury in lupus-prone mice through blocking the activation of p38 MAPK and NF-κB. Mol Immunol. (2018) 103:133–43. doi: 10.1016/j.molimm.2018.09.006
30. Bajema IM, Wilhelmus S, Alpers CE, Bruijn JA, Colvin RB, Cook HT, et al. Revision of the International Society of Nephrology/Renal Pathology Society classification for lupus nephritis: clarification of definitions, and modified National Institutes of Health activity and chronicity indices. Kidney Int. (2018) 93:789–96. doi: 10.1016/j.kint.2017.11.023
31. Zago C, Oliveira BF, Uehara G, da Silva ALC, Rocha LP, Custódio FB, et al. Influence of podocyte injury on the development of class IV lupus nephritis. Int J Nephrol Renovasc Dis. (2024) 17:215–25. doi: 10.2147/IJNRD.S473616
32. Yuan M, Tan Y, Wang Y, Wang SX, Yu F, and Zhao MH. The associations of endothelial and podocyte injury in proliferative lupus nephritis: from observational analysis to in vitro study. Lupus. (2019) 28:347–58. doi: 10.1177/0961203319828509
33. Oliva-Damaso N, Payan J, Oliva-Damaso E, Pereda T, and Bomback AS. Lupus podocytopathy: an overview. Adv Chronic Kidney Dis. (2019) 26:369–75. doi: 10.1053/j.ackd.2019.08.011
34. Rezende GM, Viana VS, Malheiros DM, Borba EF, Silva NA, Silva C, et al. Podocyte injury in pure membranous and proliferative lupus nephritis: distinct underlying mechanisms of proteinuria? Lupus. (2014) 23:255–62. doi: 10.1177/0961203313517152
35. Li M, Armelloni S, Mattinzoli D, Ikehata M, Chatziantoniou C, Alfieri C, et al. Crosstalk mechanisms between glomerular endothelial cells and podocytes in renal diseases and kidney transplantation. Kidney Res Clin Pract. (2024) 43:47–62. doi: 10.23876/j.krcp.23.071
36. Avihingsanon Y, Benjachat T, Tassanarong A, Sodsai P, Kittikovit V, and Hirankarn N. Decreased renal expression of vascular endothelial growth factor in lupus nephritis is associated with worse prognosis. Kidney Int. (2009) 75:1340–8. doi: 10.1038/ki.2009.75
37. Ghazali WSW, Iberahim R, and Ashari NSM. Serum vascular endothelial growth factor (VEGF) as a biomarker for disease activity in lupus nephritis. Malays J Med Sci. (2017) 24:62–72. doi: 10.21315/mjms2017.24.5.7
38. Muniz MPR, Brito L, Vale PHC, Guedes FL, Oliveira TKM, de Araújo Brito DJ, et al. Renal involvement in systemic lupus erythematosus: additional histopathological lesions. Arch Med Sci. (2023) 19:1398–409. doi: 10.5114/aoms.2020.96617
39. Almaani S and Parikh SV. Membranous lupus nephritis: A clinical review. Adv Chronic Kidney Dis. (2019) 26:393–403. doi: 10.1053/j.ackd.2019.08.009
40. Ponticelli C, Moroni G, and Fornoni A. Lupus membranous nephropathy. Glomerular Dis. (2021) 1:10–20. doi: 10.1159/000512278
41. Wang Y, Yu F, Song D, Wang SX, and Zhao MH. Podocyte involvement in lupus nephritis based on the 2003 ISN/RPS system: a large cohort study from a single centre. Rheumatol (Oxford). (2014) 53:1235–44. doi: 10.1093/rheumatology/ket491
42. Nawata A, Hisano S, Shimajiri S, Wang KY, Tanaka Y, and Nakayama T. Podocyte and endothelial cell injury lead to nephrotic syndrome in proliferative lupus nephritis. Histopathology. (2018) 72:1084–92. doi: 10.1111/his.13454
43. Iwamoto T, Dorschner JM, Selvaraj S, Mezzano V, Jensen MA, Vsetecka D, et al. High systemic type I interferon activity is associated with active class III/IV lupus nephritis. J Rheumatol. (2022) 49:388–97. doi: 10.3899/jrheum.210391
44. Noris M, Mele C, and Remuzzi G. Podocyte dysfunction in atypical haemolytic uraemic syndrome. Nat Rev Nephrol. (2015) 11:245–52. doi: 10.1038/nrneph.2014.250
45. Kim HS, Lee HK, Kim K, Ahn GB, Kim MS, Lee TY, et al. Mesenchymal stem cells enhance CCL8 expression by podocytes in lupus-prone MRL.Fas(lpr) mice. Sci Rep. (2023) 13:13074. doi: 10.1038/s41598-023-40346-8
46. Mashmoushi AK and Oates JC. Lipopolysaccharide induces inducible nitric oxide synthase-dependent podocyte dysfunction via a hypoxia-inducible factor 1α and cell division control protein 42 and Ras-related C3 botulinum toxin substrate 1 pathway. Free Radic Biol Med. (2015) 84:185–95. doi: 10.1016/j.freeradbiomed.2015.02.031
47. Jin J, Zhao L, Zou W, Shen W, Zhang H, and He Q. Activation of cyclooxygenase-2 by ATF4 during endoplasmic reticulum stress regulates kidney podocyte autophagy induced by lupus nephritis. Cell Physiol Biochem. (2018) 48:753–64. doi: 10.1159/000491904
48. Oates JC, Ruiz P, Alexander A, Pippen AM, and Gilkeson GS. Effect of late modulation of nitric oxide production on murine lupus. Clin Immunol Immunopathol. (1997) 83:86–92. doi: 10.1006/clin.1997.4332
49. Njoku C, Self SE, Ruiz P, Hofbauer AF, Gilkeson GS, and Oates JC. Inducible nitric oxide synthase inhibitor SD-3651 reduces proteinuria in MRL/lpr mice deficient in the NOS2 gene. J Investig Med. (2008) 56:911–9. doi: 10.2310/JIM.0b013e3181889e13
50. Perysinaki GS, Moysiadis DK, Bertsias G, Giannopoulou I, Kyriacou K, Nakopoulou L, et al. Podocyte main slit diaphragm proteins, nephrin and podocin, are affected at early stages of lupus nephritis and correlate with disease histology. Lupus. (2011) 20:781–91. doi: 10.1177/0961203310397412
51. Perez-Hernandez J, Olivares MD, Forner MJ, Chaves FJ, Cortes R, and Redon J. Urinary dedifferentiated podocytes as a non-invasive biomarker of lupus nephritis. Nephrol Dial Transplant. (2016) 31:780–9. doi: 10.1093/ndt/gfw002
52. Yung S, Cheung KF, Zhang Q, and Chan TM. Anti-dsDNA antibodies bind to mesangial annexin II in lupus nephritis. J Am Soc Nephrol. (2010) 21:1912–27. doi: 10.1681/ASN.2009080805
53. Bruschi M, Sinico RA, Moroni G, Pratesi F, Migliorini P, Galetti M, et al. Glomerular autoimmune multicomponents of human lupus nephritis in vivo: α-enolase and annexin AI. J Am Soc Nephrol. (2014) 25:2483–98. doi: 10.1681/ASN.2013090987
54. Lu J, Hu ZB, Chen PP, Lu CC, Zhang JX, Li XQ, et al. Urinary podocyte microparticles are associated with disease activity and renal injury in systemic lupus erythematosus. BMC Nephrol. (2019) 20:303. doi: 10.1186/s12882-019-1482-z
55. Shoctor NA, Brady MP, Lightman RR, Overton KN, Tandon S, Mathis SP, et al. Serum NF-κB-regulated biomarkers of proliferative lupus nephritis. Glomerular Dis. (2025) 5:328–43. doi: 10.1159/000547044
56. Chen J, Liu L, Liu M, Qin M, Yan S, Wu C, et al. Knockdown of TRIM22 regulates the expression of NF-κB/NLRP3 and alleviates inflammation and renal injury in mice with lupus nephritis. Allergol Immunopathol (Madr). (2025) 53:98–105. doi: 10.15586/aei.v53i3.1313
57. Cui JH and Xie X. UCH-L1 expressed by podocytes: a potentially therapeutic target for lupus nephritis? Inflammation. (2017) 40:657–65. doi: 10.1007/s10753-017-0512-x
58. Wright RD and Beresford MW. Podocytes contribute, and respond, to the inflammatory environment in lupus nephritis. Am J Physiol Renal Physiol. (2018) 315:F1683–f1694. doi: 10.1152/ajprenal.00512.2017
59. Ma R, Jiang W, Li Z, Sun Y, and Wei Z. Intrarenal macrophage infiltration induced by T cells is associated with podocyte injury in lupus nephritis patients. Lupus. (2016) 25:1577–86. doi: 10.1177/0961203316646861
60. Schordan S, Schordan E, Endlich K, and Endlich N. AlphaV-integrins mediate the mechanoprotective action of osteopontin in podocytes. Am J Physiol Renal Physiol. (2011) 300:F119–132. doi: 10.1152/ajprenal.00143.2010
61. Mok CC and Mohan C. Urinary biomarkers in lupus nephritis: are we there yet? Arthritis Rheumatol. (2021) 73:194–6. doi: 10.1002/art.41508
62. Vodehnal S and Mohan C. Urinary biomarkers for active Lupus Nephritis that have survived independent validation across cohorts. Kidney Int. (2024) 106:1135–45. doi: 10.1016/j.kint.2024.09.007
63. Barguil Macedo M, Wang T, Jönsen A, Bengtsson AA, Gunnarsson I, Svenungsson E, et al. Neutrophil gelatinase-associated lipocalin (NGAL) in lupus nephritis and beyond. Lupus Sci Med. (2025) 12:e001418. doi: 10.1136/lupus-2024-001418
64. Sun F, Teng J, Yu P, Li W, Chang J, and Xu H. Involvement of TWEAK and the NF-κB signaling pathway in lupus nephritis. Exp Ther Med. (2018) 15:2611–9. doi: 10.3892/etm.2018.5711
65. Gasparin AA, de Andrade NPB, Hax V, Palominos PE, Siebert M, Marx R, et al. Urinary soluble VCAM-1 is a useful biomarker of disease activity and treatment response in lupus nephritis. BMC Rheumatol. (2020) 4:67. doi: 10.1186/s41927-020-00162-3
66. Mok CC, Soliman S, Ho LY, Mohamed FA, Mohamed FI, and Mohan C. Urinary angiostatin, CXCL4 and VCAM-1 as biomarkers of lupus nephritis. Arthritis Res Ther. (2018) 20:6. doi: 10.1186/s13075-017-1498-3
67. Xia YR, Mao YM, Wang JP, Li QR, Fan YG, Pan HF, et al. Elevated urinary and blood vascular cell adhesion molecule-1 as potential biomarkers for active systemic lupus erythematosus: A meta-analysis. Curr Pharm Des. (2020) 26:5998–6006. doi: 10.2174/1381612826666200826135929
68. Brunner HI, Bennett MR, Abulaban K, Klein-Gitelman MS, O’Neil KM, Tucker L, et al. Development of a novel renal activity index of lupus nephritis in children and young adults. Arthritis Care Res (Hoboken). (2016) 68:1003–11. doi: 10.1002/acr.22762
69. Liu X, Zhao X, Duan X, Wang X, Wang T, Feng S, et al. Knockout of NGAL aggravates tubulointerstitial injury in a mouse model of diabetic nephropathy by enhancing oxidative stress and fibrosis. Exp Ther Med. (2021) 21:321. doi: 10.3892/etm.2021.9752
70. Buonafine M, Martinez-Martinez E, and Jaisser F. More than a simple biomarker: the role of NGAL in cardiovascular and renal diseases. Clin Sci (Lond). (2018) 132:909–23. doi: 10.1042/CS20171592
71. Yan C, Yuanjie T, Zhengqun X, Jiayan C, and Kongdan L. Neutrophil gelatinase-associated lipocalin attenuates ischemia/reperfusion injury in an in vitro model via autophagy activation. Med Sci Monit. (2018) 24:479–85. doi: 10.12659/MSM.908158
72. Moysiadis DK, Perysinaki GS, Bertsias G, Stratakis S, Kyriacou K, Nakopoulou L, et al. Early treatment with glucocorticoids or cyclophosphamide retains the slit diaphragm proteins nephrin and podocin in experimental lupus nephritis. Lupus. (2012) 21:1196–207. doi: 10.1177/0961203312451784
73. Shen X, Jiang H, Ying M, Xie Z, Li X, Wang H, et al. Calcineurin inhibitors cyclosporin A and tacrolimus protect against podocyte injury induced by puromycin aminonucleoside in rodent models. Sci Rep. (2016) 6:32087. doi: 10.1038/srep32087
74. Bossen C, Tardivel A, Willen L, Fletcher CA, Perroud M, Beermann F, et al. Mutation of the BAFF furin cleavage site impairs B-cell homeostasis and antibody responses. Eur J Immunol. (2011) 41:787–97. doi: 10.1002/eji.201040591
75. Müller-Winkler J, Mitter R, Rappe JCF, Vanes L, Schweighoffer E, Mohammadi H, et al. Critical requirement for BCR, BAFF, and BAFFR in memory B cell survival. J Exp Med. (2021) 218:e20191393. doi: 10.1084/jem.20191393
76. Derudder E, Herzog S, Labi V, Yasuda T, Köchert K, Janz M, et al. Canonical NF-κB signaling is uniquely required for the long-term persistence of functional mature B cells. Proc Natl Acad Sci U.S.A. (2016) 113:5065–70. doi: 10.1073/pnas.1604529113
77. Smulski CR and Eibel H. BAFF and BAFF-receptor in B cell selection and survival. Front Immunol. (2018) 9:2285. doi: 10.3389/fimmu.2018.02285
78. Baker KP, Edwards BM, Main SH, Choi GH, Wager RE, Halpern WG, et al. Generation and characterization of LymphoStat-B, a human monoclonal antibody that antagonizes the bioactivities of B lymphocyte stimulator. Arthritis Rheum. (2003) 48:3253–65. doi: 10.1002/art.11299
79. Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, et al. Two-Year, randomized, controlled trial of belimumab in lupus nephritis. N Engl J Med. (2020) 383:1117–28. doi: 10.1056/NEJMoa2001180
80. Furie RA, Aroca G, Cascino MD, Garg JP, Rovin BH, Alvarez A, et al. B-cell depletion with obinutuzumab for the treatment of proliferative lupus nephritis: a randomised, double-blind, placebo-controlled trial. Ann Rheum Dis. (2022) 81:100–7. doi: 10.1136/annrheumdis-2021-220920
81. Ward M and Tektonidou MG. Belimumab as add-on therapy in lupus nephritis. N Engl J Med. (2020) 383:1184–5. doi: 10.1056/NEJMe2027516
82. Rovin BH, Furie R, Teng YKO, Contreras G, Malvar A, Yu X, et al. A secondary analysis of the Belimumab International Study in Lupus Nephritis trial examined effects of belimumab on kidney outcomes and preservation of kidney function in patients with lupus nephritis. Kidney Int. (2022) 101:403–13. doi: 10.1016/j.kint.2021.08.027
83. Yu X, Chen N, Xue J, Mok CC, Bae SC, Peng X, et al. Efficacy and safety of belimumab in patients with lupus nephritis: subgroup analyses of a phase 3 randomized trial in the east asian population. Am J Kidney Dis. (2023) 81:294–306.e291. doi: 10.1053/j.ajkd.2022.06.013
84. Dhillon S:. Telitacicept: first approval. Drugs. (2021) 81:1671–5. doi: 10.1007/s40265-021-01591-1
85. Jin HZ, Li YJ, Wang X, Li Z, Ma B, Niu L, et al. Efficacy and safety of telitacicept in patients with systemic lupus erythematosus: a multicentre, retrospective, real-world study. Lupus Sci Med. (2023) 10:e001074. doi: 10.1136/lupus-2023-001074
86. Rougé L, Chiang N, Steffek M, Kugel C, Croll TI, Tam C, et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science. (2020) 367:1224–30. doi: 10.1126/science.aaz9356
87. Casan JML, Wong J, Northcott MJ, and Opat S. Anti-CD20 monoclonal antibodies: reviewing a revolution. Hum Vaccin Immunother. (2018) 14:2820–41. doi: 10.1080/21645515.2018.1508624
88. Rovin BH, Furie R, Latinis K, Looney RJ, Fervenza FC, Sanchez-Guerrero J, et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: the Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. (2012) 64:1215–26. doi: 10.1002/art.34359
89. Hartinger JM, Kratky V, Hruskova Z, Slanar O, and Tesar V. Implications of rituximab pharmacokinetic and pharmacodynamic alterations in various immune-mediated glomerulopathies and potential anti-CD20 therapy alternatives. Front Immunol. (2022) 13:1024068. doi: 10.3389/fimmu.2022.1024068
90. Ehrenstein MR and Wing C. The BAFFling effects of rituximab in lupus: danger ahead? Nat Rev Rheumatol. (2016) 12:367–72. doi: 10.1038/nrrheum.2016.18
91. Weidenbusch M, Römmele C, Schröttle A, and Anders HJ. Beyond the LUNAR trial. Efficacy of rituximab in refractory lupus nephritis. Nephrol Dial Transplant. (2013) 28:106–11. doi: 10.1093/ndt/gfs285
92. Teng S, Tian Y, Luo N, Zheng Q, Shao M, and Li L. Efficacy and safety of an anti-CD20 monoclonal antibody, rituximab, for lupus nephritis: A meta-analysis. Int J Rheum Dis. (2022) 25:101–9. doi: 10.1111/1756-185X.14240
93. Condon MB, Ashby D, Pepper RJ, Cook HT, Levy JB, Griffith M, et al. Prospective observational single-centre cohort study to evaluate the effectiveness of treating lupus nephritis with rituximab and mycophenolate mofetil but no oral steroids. Ann Rheum Dis. (2013) 72:1280–6. doi: 10.1136/annrheumdis-2012-202844
94. KDIGO. Clinical practice guideline for the management of LUPUS NEPHRITIS. Kidney Int. (2024) 105:S1–s69. doi: 10.1016/j.kint.2023.09.002
95. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, and Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. (2006) 107:4907–16. doi: 10.1182/blood-2005-08-3531
96. Neubert K, Meister S, Moser K, Weisel F, Maseda D, Amann K, et al. The proteasome inhibitor bortezomib depletes plasma cells and protects mice with lupus-like disease from nephritis. Nat Med. (2008) 14:748–55. doi: 10.1038/nm1763
97. Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dörner T, et al. Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol. (2006) 6:741–50. doi: 10.1038/nri1886
98. Ishii T, Tanaka Y, Kawakami A, Saito K, Ichinose K, Fujii H, et al. Multicenter double-blind randomized controlled trial to evaluate the effectiveness and safety of bortezomib as a treatment for refractory systemic lupus erythematosus. Mod Rheumatol. (2018) 28:986–92. doi: 10.1080/14397595.2018.1432331
99. Zhang H, Liu Z, Huang L, Hou J, Zhou M, Huang X, et al. The short-term efficacy of bortezomib combined with glucocorticoids for the treatment of refractory lupus nephritis. Lupus. (2017) 26:952–8. doi: 10.1177/0961203316686703
100. Segarra A, Arredondo KV, Jaramillo J, Jatem E, Salcedo MT, Agraz I, et al. Efficacy and safety of bortezomib in refractory lupus nephritis: a single-center experience. Lupus. (2020) 29:118–25. doi: 10.1177/0961203319896018
101. Walhelm T, Gunnarsson I, Heijke R, Leonard D, Trysberg E, Eriksson P, et al. Clinical experience of proteasome inhibitor bortezomib regarding efficacy and safety in severe systemic lupus erythematosus: A nationwide study. Front Immunol. (2021) 12:756941. doi: 10.3389/fimmu.2021.756941
102. Ostendorf L, Burns M, Durek P, Heinz GA, Heinrich F, Garantziotis P, et al. Targeting CD38 with daratumumab in refractory systemic lupus erythematosus. N Engl J Med. (2020) 383:1149–55. doi: 10.1056/NEJMoa2023325
103. Lokhorst HM, Plesner T, Laubach JP, Nahi H, Gimsing P, Hansson M, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med. (2015) 373:1207–19. doi: 10.1056/NEJMoa1506348
104. Roccatello D, Fenoglio R, Caniggia I, Kamgaing J, Naretto C, Cecchi I, et al. Daratumumab monotherapy for refractory lupus nephritis. Nat Med. (2023) 29:2041–7. doi: 10.1038/s41591-023-02479-1
105. Alexander T, Sarfert R, Klotsche J, Kühl AA, Rubbert-Roth A, Lorenz HM, et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. (2015) 74:1474–8. doi: 10.1136/annrheumdis-2014-206016
106. Liang CL, Jiang H, Feng W, Liu H, Han L, Chen Y, et al. Total Glucosides of Paeony Ameliorate Pristane-Induced Lupus Nephritis by Inducing PD-1 ligands(+) Macrophages via Activating IL-4/STAT6/PD-L2 Signaling. Front Immunol. (2021) 12:683249. doi: 10.3389/fimmu.2021.683249
107. Zhang Z, Guo M, Zhao S, Shao J, and Zheng S. ROS-JNK1/2-dependent activation of autophagy is required for the induction of anti-inflammatory effect of dihydroartemisinin in liver fibrosis. Free Radic Biol Med. (2016) 101:272–83. doi: 10.1016/j.freeradbiomed.2016.10.498
108. Zhao L, Zhang Y, Wang R, Yang J, Wu R, Wu Z, et al. Engineered macrophage membrane-coated dihydroartemisinin nanoparticles with enhanced CCR2 expression improved symptoms in MRL/LPR mice by metabolic reprogramming of proinflammatory macrophages. J Transl Med. (2025) 23:608. doi: 10.1186/s12967-025-06574-4
109. Dang WZ, Li H, Jiang B, Nandakumar KS, Liu KF, Liu LX, et al. Therapeutic effects of artesunate on lupus-prone MRL/lpr mice are dependent on T follicular helper cell differentiation and activation of JAK2-STAT3 signaling pathway. Phytomedicine. (2019) 62:152965. doi: 10.1016/j.phymed.2019.152965
Keywords: lupus nephritis, biomarkers, immunotherapy, podocyte injury, targeted therapy, systemic lupus erythematosus
Citation: Dong C, Wu M, Liu H, Yang K, Ma S, Guo Y, Lan Y and Lu X (2025) Breaking the cycle: immune complexes, complement activation, and novel immunotherapies in lupus nephritis. Front. Immunol. 16:1624850. doi: 10.3389/fimmu.2025.1624850
Received: 08 May 2025; Accepted: 23 September 2025;
Published: 09 October 2025.
Edited by:
Andrea Angioi, G. Brotzu Hospital, ItalyReviewed by:
Melissa Anne Cunningham, Medical University of South Carolina, United StatesZhuolun Sun, Ludwig Maximilian University of Munich, Germany
Mattia Congia, University of Cagliari, Italy
Copyright © 2025 Dong, Wu, Liu, Yang, Ma, Guo, Lan and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuejiao Lan, MTM4NDMyMjU2ODVAMTYzLmNvbQ==; Xiaodan Lu, bHV4aWFvZGFuQGNjc2Z1LmVkdS5jbg==
†These authors have contributed equally to this work