 Yuzhang Yuan1†
Yuzhang Yuan1† Haozhe Zhang1†
Haozhe Zhang1† Zehua Wang1
Zehua Wang1 Luying Huang2
Luying Huang2 Derya Kabacaoglu3
Derya Kabacaoglu3 Boxing Zhang4
Boxing Zhang4 Liang Song4
Liang Song4 Jiaoyu Ai2*
Jiaoyu Ai2*- 1Department of Oncology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 2Department of Gastroenterology, The First Affiliated Hospital of Nanchang University, Nanchang, China
- 3Comprehensive Cancer Center Munich CCCM, Technical University of Munich, Munich, Germany
- 4Medical Experiment Center, Shaanxi University of Chinese Medicine, Xianyang, China
Pancreatic cancer (PC) is an aggressive malignancy with a poor prognosis, and the tumor microenvironment (TME) plays a pivotal role in its initiation, progression, and response to treatment. Recent studies have highlighted the critical involvement of inflammatory factors in shaping and sustaining the PC microenvironment. Chronic inflammation is a hallmark of this cancer, with inflammatory molecules such as cytokines, chemokines, proteases, and other immune-modulatory factors driving tumor cell proliferation, metastasis, and resistance to therapy. These inflammatory factors exert their effects by modulating immune cell infiltration, extracellular matrix (ECM) remodeling, and angiogenesis. This review provides an overview of the diverse roles of inflammatory factors in the PC TME and explores their potential as therapeutic targets. It offers new perspectives for developing novel immunotherapies and inflammation-modulating strategies to improve the treatment of PC.
1 Introduction
PC represents a growing public health concern, currently ranking as the third-leading cause of cancer-related mortality in the United States, with an annual increase in mortality of 0.3% since 2000 (1). Projections suggest it will become the second-leading cause of cancer-related deaths by 2030 (2). The prognosis for PC remains dismal, primarily due to late-stage diagnosis, as the majority of patients are diagnosed after the disease has already metastasized (3–6). Furthermore, the limited effectiveness of current treatment options, coupled with inherent resistance to standard therapies, contributes to a 5-year survival rate of 13.3% (7, 8). Given these challenges, it is imperative to further investigate the pathogenesis of PC and develop novel therapeutic strategies.
The profound influence of the TME on tumor progression has been well-documented in numerous studies. PC, however, exhibits distinct TME characteristics that differentiate it from other malignancies. In the PC TME, T cell dysfunction, driven by a variety of mechanisms, plays a pivotal role in tumor progression and is a central contributor to the immunosuppressive milieu that characterizes this cancer (9). Moreover, the presence of extensive fibrosis and the resulting low perfusion further complicate the development of effective therapies targeting the microenvironment, presenting an additional challenge in the treatment of PC (10).
Inflammatory factors play a pivotal role in the aforementioned alterations of the TME. Inflammation is a complex physiological process initiated by the immune system, wherein immune cells are mobilized to respond to signals such as wounds, infections, or other irritants. Upon activation, these immune cells release a variety of chemical mediators that recruit additional specialized immune cells to the site of injury, thereby driving the inflammatory response. This accumulation of immune cells and the release of pro-inflammatory molecules characterize the inflammatory process. Previous studies have established a strong correlation between pancreatic inflammation and the onset and progression of PC (11), with inflammatory factors playing a central role in this association (12). In one respect, inflammation promotes the survival and proliferation of cancer cells through the secretion of inflammatory mediators such as cytokines. In another respect, inflammatory cytokines serve as key modulators of TME changes, thus contributing to tumor progression. Given the significant role of inflammation in the development and progression of PC, targeting inflammatory cytokines has emerged as a promising therapeutic strategy. This review aims to provide an overview of the mechanisms by which inflammatory factors influence the TME and, consequently, tumor progression in PC, while also discussing the feasibility and potential of existing therapeutic approaches that target these inflammatory pathways.
In light of the aforementioned background, we have endeavored to summarize the current clinical trials targeting inflammation-mediated alterations in the TME, as well as the potential therapeutic targets for PC. The ultimate aim of this effort is to enhance the prognosis of patients with PC.
2 The key features of the TME in PC
The tumorigenesis of PC is a complex, multistep process (13). The most prevalent form of PC is pancreatic ductal adenocarcinoma (PDAC), which accounts for approximately 85% of all pancreatic malignancies (14). The onset of cancer involves two primary types of changes: genetic mutations in the pancreatic ductal cells and alterations in the composition and function of the pancreatic stroma. Pancreatic intraepithelial neoplasia (PanIN) is considered the most likely precursor lesion of PDAC, characterized by preneoplastic mucinous lesions with ductal morphology (15). KRAS mutations play a pivotal role in the initiation of this process (16), while mutations in other genes such as TP53, CDKN2A, and SMAD4 also contribute significantly, working in concert with KRAS mutations to drive the progression of PDAC (17, 18). In addition to the mutations within the pancreatic ductal cells, the influence of stromal changes is equally critical, as various alterations in the stromal microenvironment play an indispensable role in the development of PC.
2.1 Immunosuppressive TME
Compared to other cancers with high mutational burdens, PC exhibits a relatively low mutational burden, resulting in a limited number of potential targets for immune recognition (19). Consequently, immunotherapy through checkpoint inhibition has yielded only modest success in patients with PDAC, in contrast to its more substantial benefits in other malignancies. Research has shown that the TME of many PDACs is characterized by an increased prevalence of immunosuppressive cell types, including T-regulatory cells (Tregs), tumor-associated macrophages (TAMs) with M2 polarization, and myeloid-derived suppressor cells (MDSCs), all of which contribute significantly to the immunosuppressive milieu (20–22). The accumulation of these immunosuppressive cells correlates with enhanced malignancy and aggressiveness of PC (23). These cells primarily inhibit antitumor immunity by reducing both the number and the functional activity of effector T cells (Figure 1). Oncogenic KRAS-induced upregulation of granulocyte-macrophage colony-stimulating factor (GM-CSF) fosters the recruitment of GR1(+)CD11b(+) MDSCs, a process that is mediated by CD8(+) T cells. Consequently, the presence of GR1(+)CD11b(+) myeloid cells suppresses the proliferation of CD8+ T cells (24). Additionally, Treg cells have been shown to inhibit the production of costimulatory ligands necessary for the activation of CD8+ T cells by restricting the immunogenic functions of tumor-associated CD11c+ dendritic cells (DCs), thereby limiting CD8+ T cell responses (25). Moreover, TAMs play a crucial role in PC tumorigenesis. An inflammatory feedback loop has been identified between TAMs expressing interleukin-1β (IL-1β) and tumor cells, which serves as a precursor to pancreatic carcinogenesis (26). Further studies have demonstrated that TAMs contribute to pancreatic acinar-ductal metaplasia through the secretion of tumor necrosis factor (TNF), RANTES, and the induction of matrix metalloproteinase 9 (MMP9) (27). These findings indicate that the immunosuppressive effects of these cell populations are largely mediated by inflammatory cytokines and their receptors, thus offering valuable insights for the identification of novel therapeutic targets in the treatment of PC.
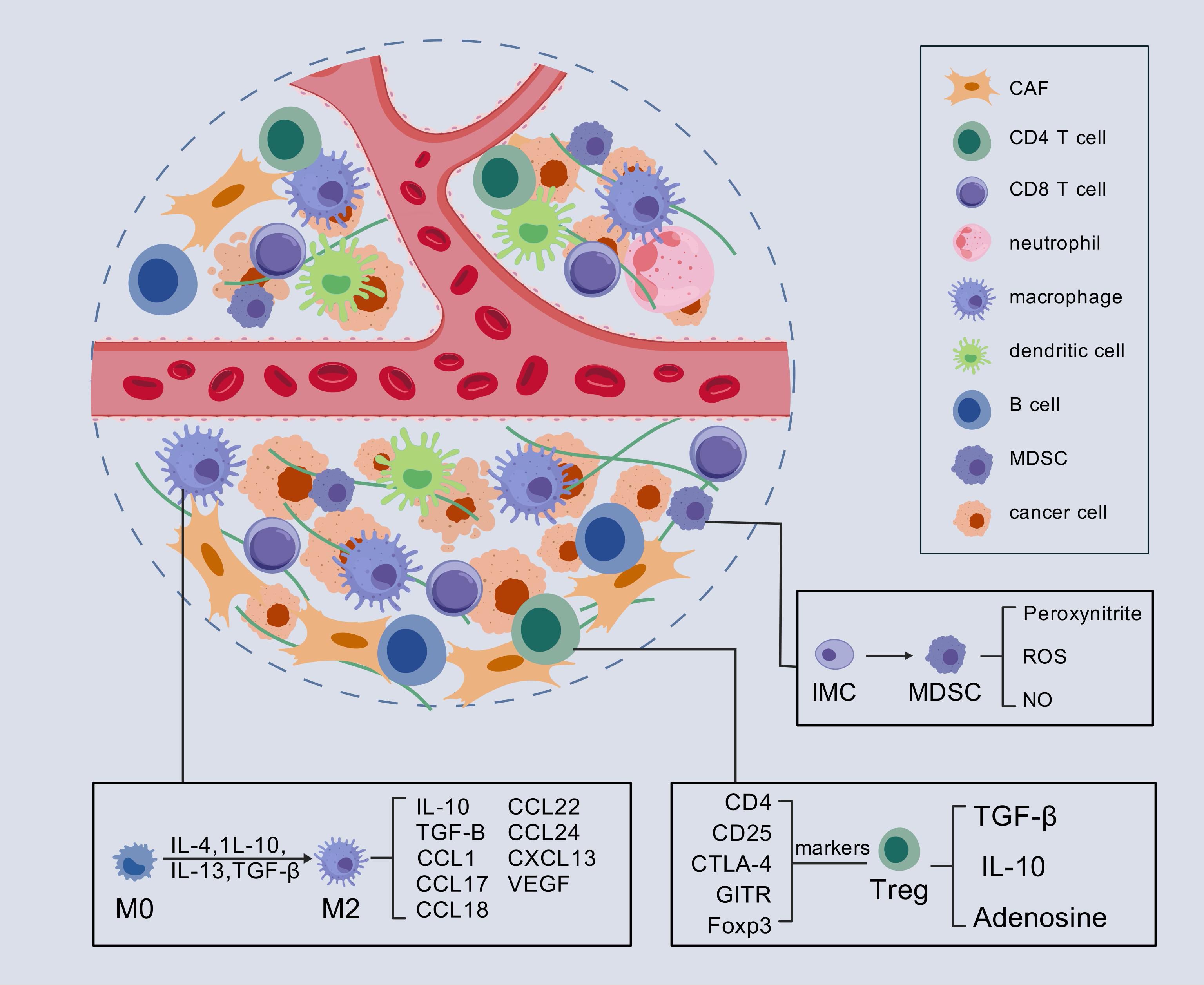
Figure 1. Immunosuppressive TME in PC. In the immunosuppressive TME of PC, Tregs, TAMs with M2 polarization, and MDSCs play key roles. They alter signaling pathways within the TME by secreting various inflammatory factors, which in turn lead to the suppression of T cell function. CAF, cancer-associated fibroblasts; IMC, immature myeloid cell; MDSC, myeloid-derived suppressor cell; Treg, regulatory T cells. ROS, reactive oxygen species.
2.2 Dense desmoplastic stroma and cancer-associated fibroblasts
The TME of PC is characterized by extensive fibrosis, with stromal components constituting the majority of its volume (28). Key ECM components, such as collagen and hyaluronan (HA), are prominently involved, and elevated levels of HA in PDAC are strongly associated with poor prognosis (29, 30). CAFs play a significant role in the fibrosis process. The sources of CAFs in PDAC are extensive, such as resident fibroblasts, mesenchymal stem cells (MSCs), tissue-resident fibroblasts, and pancreatic stellate cells (PSCs) (31–33). These CAFs are functionally classified into three major subtypes: myofibroblastic CAFs (myCAFs) that promote extracellular matrix stiffening and create mechanical tumor barriers, inflammatory CAFs (iCAFs) which secrete cytokines to establish an immunosuppressive microenvironment, and antigen-presenting CAFs (apCAFs). Importantly, these functionally distinct CAF subtypes demonstrate plasticity and can interconvert under specific microenvironmental conditions, as evidenced by recent single-cell and spatial transcriptomic studies (34). CAFs play a crucial role in promoting fibrosis within the TME. For instance, the knockout of NID2 has been shown to suppress CAF activation, thereby inhibiting both tumor fibrosis and metastasis, while concurrently modulating tumor vasculature and enhancing therapeutic efficacy (35). Traditionally, fibrosis in tumors has been understood through the perspective that the ECM acts as a physical barrier that obstructs drug delivery, thereby reducing the efficacy of therapeutic interventions. However, recent insights suggest a more complex interaction wherein CAFs, in conjunction with their native ECM, function as an integrated entity (36). This CAF-ECM complex not only serves as a physical barrier but also acts as a reservoir for secretory factors, which play a pivotal role in tumor progression (37, 38).
Moreover, CAFs can influence tumor growth by modulating key metabolic pathways, including lipid, amino acid, and polyamine metabolism (39–41). Among these, lipid metabolism has been particularly well-explored. A recent study demonstrated that the loss of Setd2 results in an aberrant increase of H3K27Ac at the gene body of Bmp2, which subsequently drives the differentiation of adjacent CAFs into a lipid-enriched phenotype. These lipid-laden CAFs, in turn, supply lipids to Setd2-deficient tumor cells, thereby supporting mitochondrial oxidative phosphorylation (OXPHOS) and promoting tumor growth (42).
Several therapeutic strategies aimed at targeting CAFs or disrupting their interactions with other immune cells have demonstrated varying degrees of success in both preclinical studies and clinical trials (NCT01130142 and NCT03563248). The ECM acts as both a physical and metabolic barrier to drug delivery, contributing to the resistance of PDAC to chemotherapy, targeted therapies, and even immunotherapy (43). Recent research has highlighted that targeting CAFs’ autophagic processes can enhance the efficacy of immunochemotherapy in PC by alleviating adaptive immune resistance. This approach holds promise as a potential novel therapeutic target for the treatment of PC in the future (44).
2.3 Hypoperfusion
To ensure an adequate supply of nutrients and support tumor metastasis, most tumors secrete a variety of pro-angiogenic factors to promote angiogenesis (45). In PC, the interstitial fluid pressure (IFP) is significantly elevated compared to other cancers, primarily due to a pronounced fibroinflammatory response. This elevated IFP leads to hypoperfusion within the tumor, resulting in poor delivery of small molecule drugs and the creation of a hypoxic TME (46). The former contributes to drug resistance, while the latter induces the overexpression of hypoxia-inducible factor (HIF)-1α in PC cells. HIF-1 is a key regulator of the cellular response to hypoxia, and the expression of HIF-1α is closely associated with tumor progression and metastasis (47). HIF-1α functions by directly binding to hypoxia-response elements (HREs) in the promoters of target genes, activating downstream pathways that enable cells to adapt to low oxygen conditions. However, recent perspectives suggest that the regulation of HIF-1α in tumors is more complex than merely promoting tumor development. As such, rather than targeting HIF-1α directly, addressing the hypoxic TME itself may offer more effective therapeutic outcomes in PC (48, 49).
Notably, these characteristics may be interrelated and potentially causative, rather than entirely independent from one another (Figure 2). For instance, stromal fibrosis can physically obstruct the infiltration of immune cells and encapsulate tumor tissue, thereby contributing to the formation of a hypoxic microenvironment. Hypoxia, in turn, activates Lactate dehydrogenase A (LDHA), which subsequently promotes the production and secretion of L-2HG. This cascade of events ultimately results in the inhibition of T cell proliferation and migration (50).
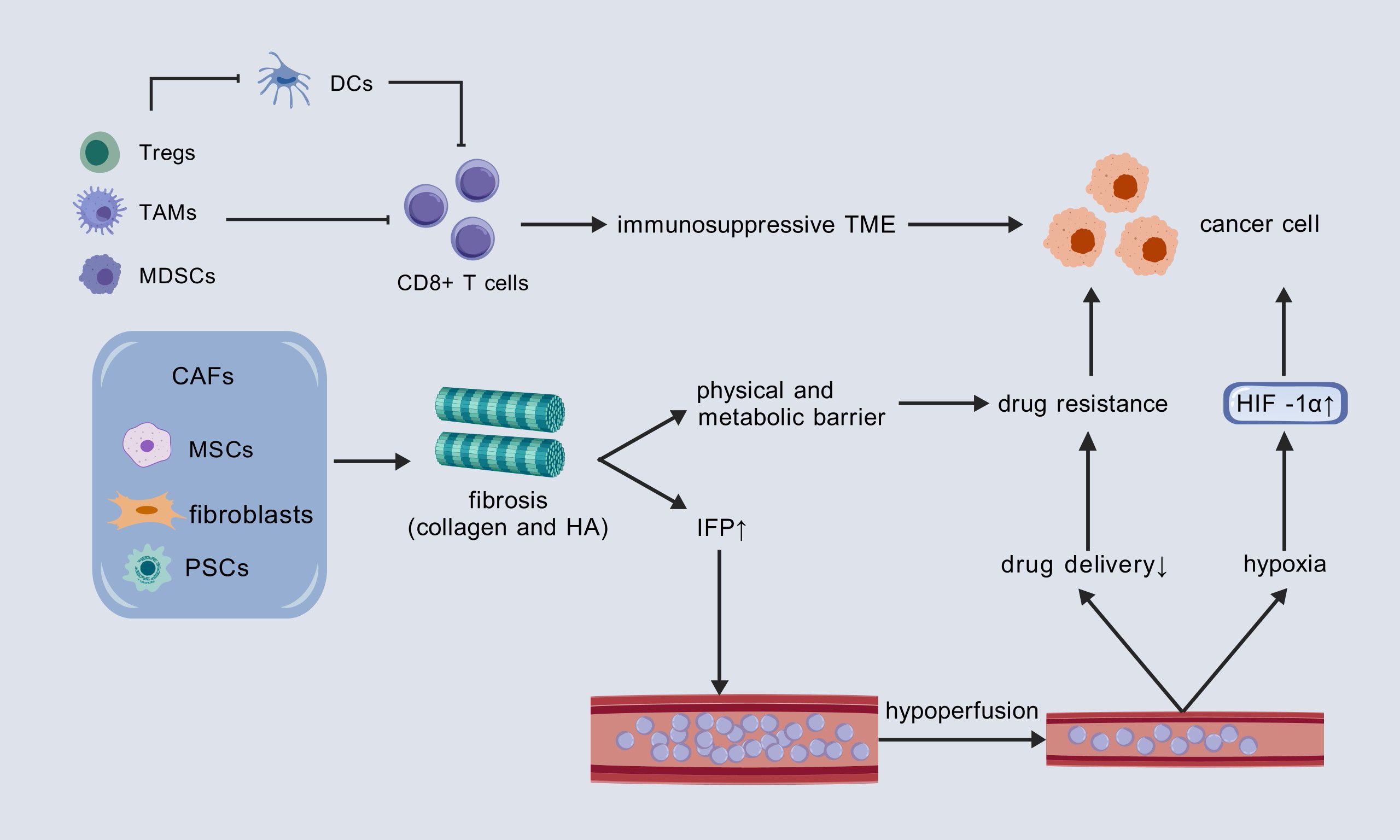
Figure 2. Three core mechanisms promoting tumor progression in the PC TME and their interconnections. DCs, dendritic cells; TAM, tumor-associated macrophage; MDSC, myeloid-derived suppressor cell; CAF, cancer-associated fibroblast; MSC, mesenchymal stem cell; PSCs, pancreatic stellate cells; HA, hyaluronic acid; IFP, interstitial fluid pressure; HIF-1α, hypoxia-inducible factor 1-alpha.
3 Inflammatory factors’ impact on PC TME
Inflammatory factors serve as a crucial link between the TME and tumor cells through complex regulatory mechanisms. Acute inflammation typically promotes the immune response, whereas chronic inflammation may lead to immunosuppression. In the context of chronic inflammation, several inflammatory mediators, including IL-6, IL-1, IL-17A, IL-22, and transforming growth factor beta (TGF-β), have been shown to exert pro-tumorigenic effects. These factors, which influence cell fate, can be hijacked by mutated cells, thereby activating key signaling pathways such as mitogen-activated protein kinases (MAPK), phosphatidylinositol-3-kinase (PI3K)-Akt, Janus kinase (JAK)-STAT, and NF-κB, thereby increasing the risk of tumorigenesis (51, 52). Inflammatory mediators play a significant role in the early malignant progression of various cancers, including esophageal squamous cell carcinoma, hepatocellular carcinoma, and intrahepatic cholangiocarcinoma (53). Building upon existing research, we review the inflammatory factors that contribute to the initiation and progression of PC through their influence on the TME (Figure 3).
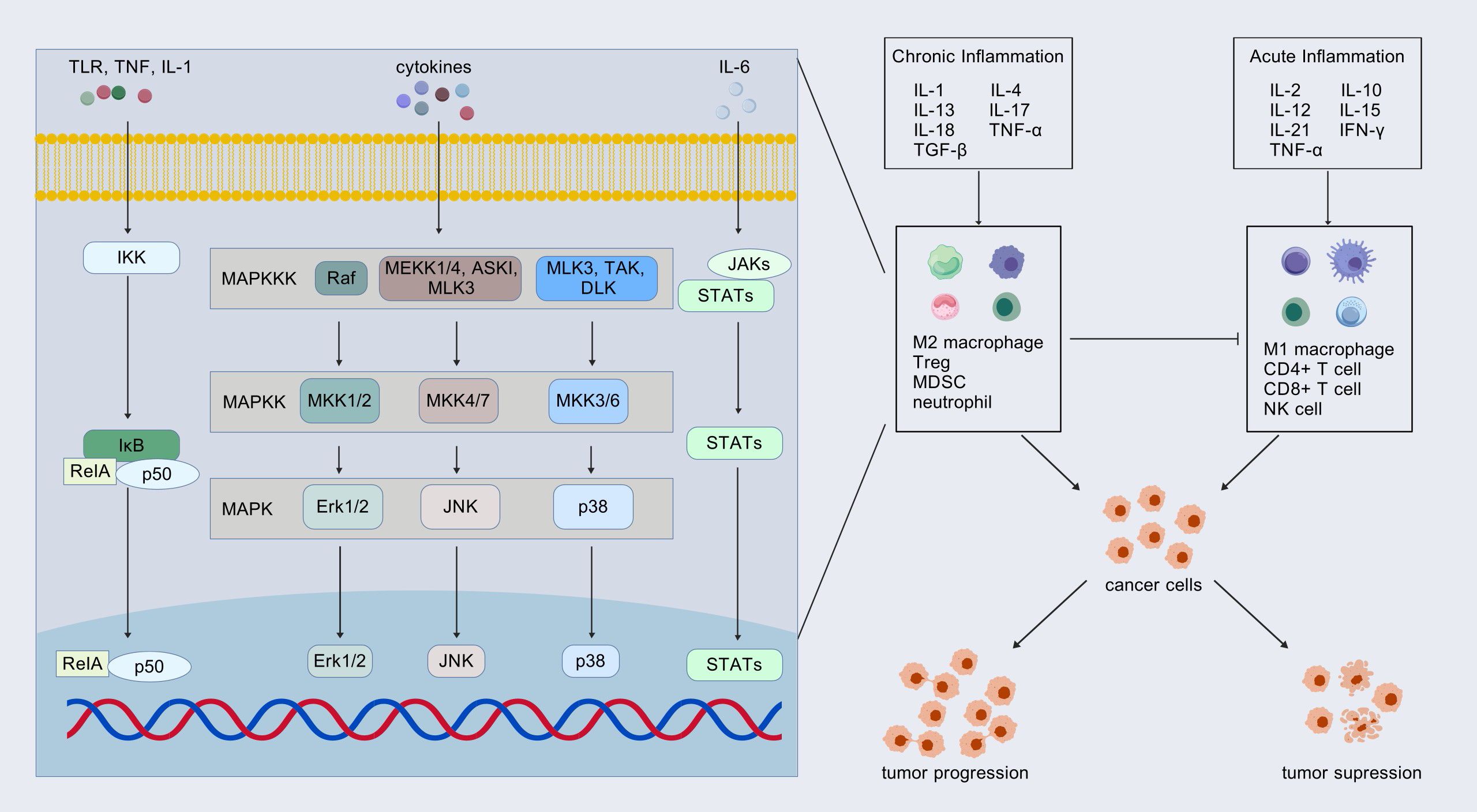
Figure 3. Inflammatory factors: friends or foes? Inflammatory factors in acute and chronic inflammation play opposite roles in tumor progression by affecting the TME. These inflammatory factors transfer information into the cell through signaling pathways. The most common pathways involved include mitogen-activated protein kinases (MAPK), Janus kinase (JAK)-STAT, and NF-κB pathways. Treg, regulatory T cell; MDSC, myeloid-derived suppressor cell.
3.1 Interleukin-1β
Among these cytokines, IL-1β stands out as a key player in PC pathogenesis. IL-1β, a factor secreted by various cells, has pleiotropic effects on immune cells, angiogenesis, cancer cell proliferation, migration and metastasis. In the context of PC, tumor cell expression of IL-1β in vivo was driven by microbial-dependent activation of Toll-like receptor 4 (TLR4) signaling and subsequent engagement of the NLRP3 inflammasome. As a cancer-promoting factor, IL-1β promotes tumor immune escape by interacting with stromal cells (54). As a bridge, it connects various stromal cells such as fibroblasts, macrophages, stellate cells and PC cells, and plays an important role in the occurrence and development of PC. As a cancer-promoting factor, IL-1β promotes tumor immune escape by interacting with stromal cells. As a bridge, it connects various stromal cells such as fibroblasts, macrophages, stellate cells and PC cells, and plays an important role in the occurrence and development of PC. Recent studies have demonstrated that tumor-infiltrating monocytes differentiate into IL-1β-producing TAMs following exposure to prostaglandin E2 (PGE2) and TNF. These IL-1β+ TAMs subsequently interact with IL-1β-reactive PC cells, thereby promoting tumor progression. This process highlights the PGE2–IL-1β axis as a critical driver of spatial and transcriptional heterogeneity within both immune and tumor cells in PDAC. Furthermore, inflamed PDAC tissue and TAMs engage in a positive feedback loop that exacerbates cancer progression (26). In addition to macrophages, a similar positive feedback pathway is observed in PSCs. ESE3, a transcription factor, induces the transcription of α-SMA, collagen I, and IL-1β by binding to specific ESE3 response elements on their promoters. As a result, IL-1β upregulates ESE3 expression in PSCs through NF-κB activation, and ESE3 is essential for PSC activation by tumor-derived IL-1β (55). Furthermore, technological advances in single-cell and spatial transcriptomics have enabled precise characterization of the role and mechanisms of IL-1β within tumor tissues. IL-1β exhibits non-uniform spatial distribution in tumors. These studies revealed that IL-1β+ TAMs preferentially localize to hypoxic stromal regions and directly neighbor tumor cells expressing the IL-1β Response Signature (TIRS). Critically, IL-1β+ TAMs activate adjacent TIRS+ PDAC cells through IL-1β secretion, triggering the release of PGE2 and TNF. These factors then synergistically drive monocyte differentiation into IL-1β+ TAMs, establishing a self-amplifying circuit that perpetuates tumor progression (26).
3.2 Interleukin-4
Building on the role of IL-1β, IL-4 also exerts a profound influence on PC. Significantly elevated levels of IL-4 have been observed in cancer cases compared to control participants (56). Interleukin-4 (IL-4) exerts a dual effect in human PC, acting both directly on the tumor cells themselves and indirectly within the TME. PC cells serve as both a source and target of IL-4, creating an autocrine loop that directly promotes tumor cell proliferation. This pro-proliferative effect can be counteracted by neutralizing antibodies against IL-4 (57). Beyond its autocrine action, IL-4 acts paracrinely to establish an immunosuppressive TME. A key mechanism involves IL-4 inducing the polarization of macrophages towards an immunosuppressive M2 phenotype (58). In addition to shaping macrophage function, IL-4 directly impairs anti-tumor immunity by inhibiting T cell effector functions. IL-4 secreted within the TME binds to the IL-4 receptor on T cell surfaces, suppressing their activity and facilitating tumor immune escape. Consequently, strategies aimed at blocking IL-4 signaling within this immunosuppressive TME, such as engineering CAR-T cells to co-express IL-4/IL-15-based inverted cytokine receptors, represent promising therapeutic approaches. This strategy could enhance immunotherapy efficacy, particularly in tumors like PC characterized by elevated IL-4 levels (59).
3.3 Interleukin-13
IL-13 follows a similar pattern of dysregulation in PC. IL-13 is expressed at significantly higher levels in PC compared to normal tissues (60), and its elevated expression is considered an unfavorable prognostic factor for PC (61). The primary receptor subunits for IL-13 are IL-13Rα1 and IL-13Rα2, both of which are involved in promoting the progression of PC by acting on cancer cells (61, 62). Notably, while IL-13Rα1 is ubiquitously expressed in healthy tissues, IL-13Rα2 exhibits tumor-specific overexpression with minimal distribution in normal organs, positioning it as a high-value target for chimeric antigen receptor (CAR) T-cell therapy in PDAC. Engineered IL-13 mutein-based CARs have thus been developed to exploit this selectivity for PDAC treatment. Compared to conventional single-chain variable fragment (scFv)-based CARs, these ligand-directed designs not only mitigate immunogenicity risks (63) but also offer potential multi-tumor targeting capabilities with reduced engineering complexity (64). Within the TME, IL-13 serves as a key mediator bridging PSCs and macrophages. PSCs, which are a major source of IL-4 and IL-13 (65), contribute to the tumorigenic process. In lesions such as acinar-to-ductal metaplasia (ADM) and panIN, IL-13 induces the polarization of inflammatory macrophages into Ym1+ alternatively activated macrophages. These Ym1+ macrophages, in turn, secrete factors such as CCL2 and IL-1 receptor antagonist (IL-1Ra), which further support tumorigenesis and pancreatic fibrogenesis (66).
3.4 Interleukin-15
Shifting focus to another cytokine, IL-15 predominantly stimulates the proliferation and cytotoxic activity of CD8+ T cells and NK cells, enhancing antitumor immune responses. Although research on the role of IL-15 in PC remains limited, recent studies have highlighted its potential as a mediator in exercise-induced tumor immunity. Kurz et al. demonstrated that aerobic exercise inhibited PC growth in mice by activating the immune system, particularly through the activation of CD8+ T cells. Moreover, Niz985, an IL-15 super-agonist, was shown to replicate the beneficial effects of exercise on tumor immunity. Notably, both Niz985 and exercise significantly enhanced the therapeutic sensitivity of otherwise intractable pancreatic tumors in mice. This pioneering study revealed that exercise promotes antitumor immunity in PC via activation of the IL-15/IL-15Rα signaling axis, suggesting that this pathway may represent a promising therapeutic strategy (67). As a central regulator of NK cell survival, proliferation, and cytotoxicity, IL-15 expression in CAFs is suppressed by nociceptor neuron-derived CGRP. Consequently, NK cell infiltration and activation are inhibited. Multiplex immunofluorescence of PDAC tissues showed reduced CAF-derived IL-15 and diminished NK cell presence in regions with high CGRP+ nerve density (e.g., tumor periphery or perineural invasion sites). Spatial analysis confirmed a significant inverse correlation between CGRP+ nerve density and IL-15/NK cell levels, indicating concentrated IL-15 suppression in nerve-rich regions (68).
3.5 Interleukin-17
IL-17 also plays a crucial role in PC initiation and progression by regulating CAFs, T cells, and neutrophils. Primarily secreted by CD4+ and γδ T cells, IL-17 promotes the formation of panIN lesions (69, 70). In the context of PC, IL-17 contributes to the maintenance of immunosuppression by decreasing the recruitment of CD8+ T cells while simultaneously increasing the infiltration of neutrophils into the TME. Furthermore, IL-17 triggers the formation of neutrophil NETs, which leads to the remodeling of the ECM and surrounding tissue. This process facilitates the development, spread, and metastasis of the cancer (71). IL-17 also exerts significant regulatory effects on CAFs. Specifically, Tc17 cells, a subset of CD8+ T cells that produce IL-17A, promote the transformation of IL-17RA+ CAFs into an inflammatory phenotype through the secretion of IL-17A and TNF. This, in turn, fosters the progression of PDAC by altering the transcriptome of PC cells, thereby enhancing tumor cell proliferation (72). In addition, a recent study has revealed that IL-17 may promote tumor progression by inhibiting fibrosis in CAFs, which challenges the conventional view that fibrosis within the TME enhances PC progression. This finding underscores the need for a more nuanced classification of the various components within the TME, which could aid in refining therapeutic strategies and improving treatment precision (73). Moreover, IL-17 interacts with a variety of other cytokines, further complicating its role in tumor biology. For instance, the IL-17B/IL-17RB axis activates the expression of chemokines such as CCL20, CXCL1, IL-8, and TFF1 via the ERK1/2 signaling pathway, which in turn influences tumor metastasis and the recruitment of macrophages and endothelial cells. This highlights the critical involvement of IL-17 in the TME and its potential as a therapeutic target to enhance treatment efficacy (74). Beyond its role within the TME, circulating IL-17 has also been implicated in the carcinogenesis and metastasis of PC, further emphasizing the complexity of IL-17’s functions in cancer biology (75).
3.6 Interleukin-18
IL-18, recognized as a pro-cancer factor, is strongly associated with poor prognosis in patients (76). Similar to other cytokines previously studied, IL-18 plays a significant regulatory role in macrophages and CD8+ T cells within the PC microenvironment, thereby contributing to tumor progression. Recent studies have identified IL-18 as a key downstream mediator of GFPT2, wherein GFPT2-mediated O-GlcNAcylation of YBX1 enhances its nuclear translocation and promotes the transcription of IL-18. This process is critical for M2 macrophage polarization in PC (77). Furthermore, research into T cell exhaustion has highlighted the crucial role of IL-18 receptor (IL-18R) signaling in this phenomenon. It has been demonstrated that IL-18, acting as a downstream molecule of NLRP3, activates both the IL-2/STAT5 and AKT/mTOR pathways upon binding to IL-18R. This signaling cascade promotes CD8+ T cell exhaustion, contributing to immune evasion in tumors (76). In addition to these immune-suppressive mechanisms, other studies have shown that IL-18 can drive eosinophil accumulation, leading to eosinophilic chronic inflammation. This inflammation promotes pancreatic tissue remodeling and fibrosis, which is closely linked to the initiation and progression of pancreatic ADM and panIN. These processes may represent the critical early steps in the transition from chronic pancreatitis (CP) to PC (78).
3.7 Tumor necrosis factor
Finally, TNF-α has been extensively studied for its pivotal role in PC initiation and progression (79, 80). TNF-α is predominantly secreted by macrophages and exerts its biological effects through binding to two distinct receptors: the death-domain-containing TNF receptor 1 (TNFR1) and the tissue-restricted TNF receptor 2 (TNFR2) on the cell surface (81–83). TNFR1 is broadly expressed across various cell types, including tumor cells and CAFs, and it mediates pro-apoptotic pathways through the activation of its death domain. In contrast, TNFR2 is primarily expressed on immune cells and endothelial cells, and its signaling promotes the activation of NF-κB through a non-classical pathway, without inducing cell death. Both of these pathways are implicated in the pathogenesis of PC. Some studies have indicated that TNF-α, through its interaction with TNFR1, can inhibit the infiltration and activation of antigen-presenting DCs. Blockade of TNFR1 has been shown to attenuate this inhibitory effect, thereby restoring T cell-mediated anti-tumor immunity and effectively impeding tumor progression (84). On the other hand, with respect to the major microenvironmental characteristics associated with resistance in PC—specifically, the inflammatory polarization of CAFs and T cell dysfunction—recent research has highlighted the significant role of neutrophil-derived transmembrane TNF-TNFR2 interactions in these processes (85). Furthermore, a separate study has suggested that the inhibition of TNF-α expression can lead to an upregulation of IL-33 in tumor cells, which in turn enhances the activity of DCs and cytotoxic T cells, thereby promoting anti-tumor immunity. This may represent one of the downstream mechanisms through which TNF-α exerts its immunosuppressive effects (81). Taken together, these findings indicate that targeting TNF-α or specifically the pro-tumor immunosuppressive TAMs that produce TNF-α may represent a promising therapeutic strategy to counteract the immunosuppressive microenvironment and improve treatment outcomes in PC. Spatial multi-omics (IMC/transcriptomics) reveal TNF operates via direct cell-contact: KRAS-TP53 mutant tumor islands autonomously secrete CXCL1, recruiting CXCR2+ neutrophils (PMN-MDSCs) into close proximity (<10 μm). This interaction excludes CD8+ T cells (>95 μm), creating immunosuppressive niches. Mechanistically, tmTNF+ neutrophils engage TNFR2 on tumor cells, inducing reciprocal CXCL1/IL-6 upregulation (feedforward loop) and driving iCAF differentiation. Resultant iCAFs promote fibrosis and activate therapy-resistant IL-6/STAT3 signaling (86).
3.8 Prostaglandins
PGs are a class of lipid mediators derived from arachidonic acid via cyclooxygenase (COX) catalysis, with major subtypes including PGE2, PGD2, and thromboxane. In PC, upregulation of the cyclooxygenase-2 (COX-2) pathway is prevalent, and its carcinogenic effects are largely attributed to the overproduction of PGE2 (87–89). Prospective studies provide further evidence supporting the involvement of the COX-2 pathway in pancreatic carcinogenesis and suggest that urinary prostaglandin E metabolite (PGE-M) may serve as a biomarker for predicting PC risk (90). It is noteworthy that PC cells undergoing endoplasmic reticulum (ER) stress release PGE2, which can transfer this stress signal to DCs, impairing their immune function and contributing to immunosuppression (91). Simultaneously, the role of PGE2 on CAFs within the TME is crucial. On one hand, PGE2 acts on cancer cells to induce their secretion of fibroblast growth factor 1 (FGF1), which subsequently stimulates CAF proliferation and enhances their fibrotic activity. On the other hand, activated CAFs increase the expression of vascular endothelial growth factor A (VEGFA), promoting angiogenesis. These processes collectively drive the formation of a fibrotic TME, not only promoting the initiation and progression of pancreatic tumor tissue but also creating a physical barrier through increased collagen secretion that impedes chemotherapeutic drug delivery (92, 93). Expanding the focus to therapeutic applications, the interaction between PGE2 signaling and CAR-T therapy effectiveness is a subject of ongoing research, despite the well-characterized role of PGE2 in PC pathogenesis. Existing evidence demonstrates that PGE2 significantly suppresses T cell (including CAR-T cell) proliferation and impairs their antitumor function through EP2/EP4 receptor signaling. This underscores the potential value of targeting the PGE2 signaling pathway as a strategy to enhance CAR-T cell efficacy and improve therapeutic responses in PC (94).
4 The clinical value of inflammatory factors in PC
In recent years, research on inflammatory factors in PC has accumulated certain findings in terms of prognosis evaluation, clinical treatment, and new drug development (Table 1).
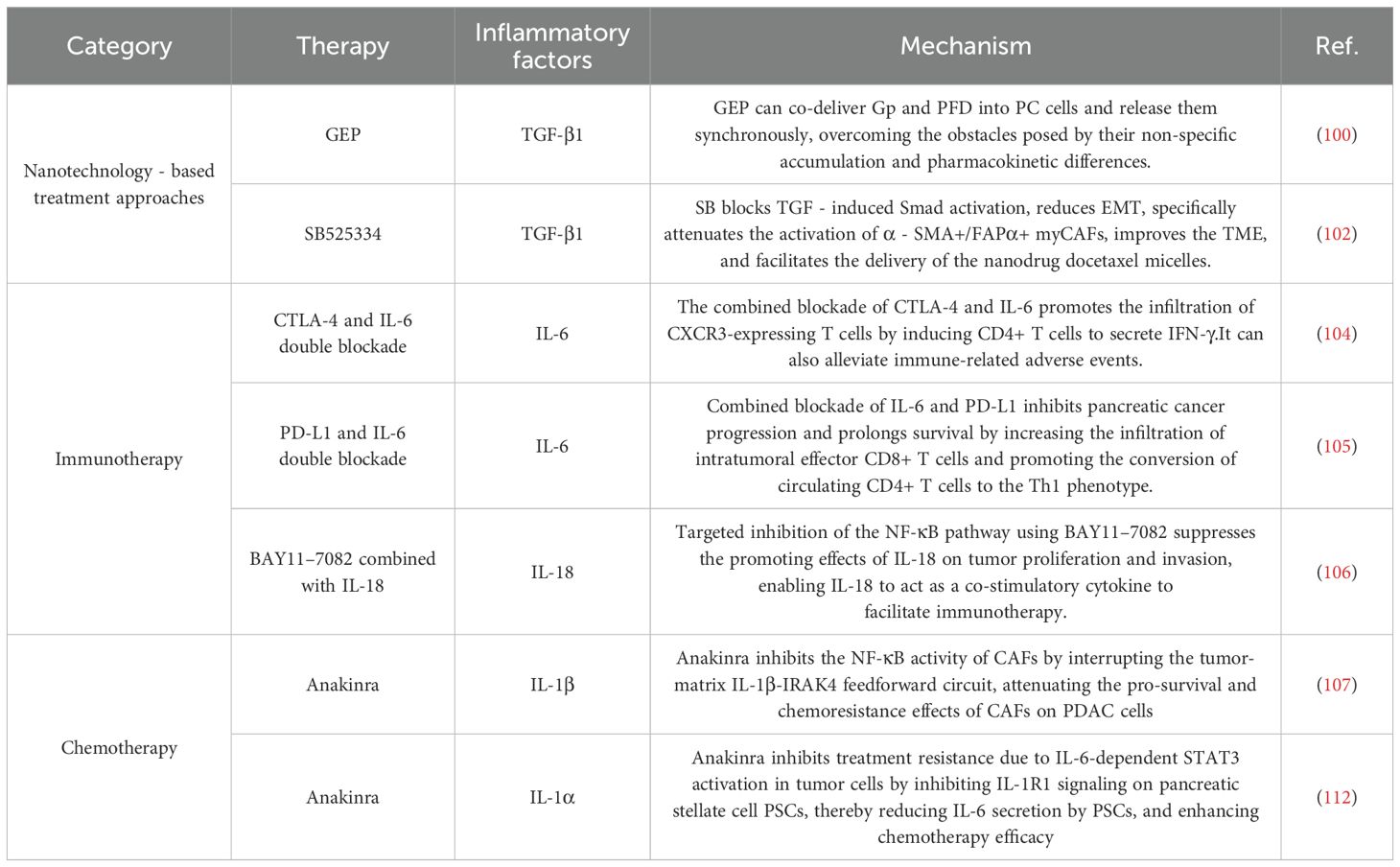
Table 1. The role of inflammatory factors in PC therapy.
4.1 The evaluation of the prognosis of PC
In the diagnosis of PC, existing biomarkers have certain limitations. Take CA19-9, which is universally used in clinical practice, for example. Its sensitivity and specificity for early detection of PC are limited. Therefore, the search for more reliable diagnostic markers is extremely urgent. Notably, although a single biomarker performs poorly in diagnosis, the combined detection of CA19-9, CEA, CA125, and CA242 is significantly more accurate than a single serum biomarker. Their sensitivity and specificity are as high as 90.4% and 93.8%, respectively (95). Currently, the role of inflammatory factors in the diagnosis of PC remains unclear. However, emerging studies have indicated that they are gradually demonstrating value in predicting disease prognosis.
S Mitsunaga et al. discovered that the serum levels of IL-6 and IL-1β can predict the therapeutic efficacy of Gemcitabine (GEM) and the prognosis of patients with advanced PC. They measured the levels of pro-inflammatory cytokines in the serum of advanced PC patients receiving single-agent GEM treatment. Multivariate analysis showed that high levels of IL-6 and IL-1β are poor prognostic factors for overall survival. Compared with patients with low levels of both IL-6 and IL-1β, patients with high levels of both IL-6 and IL-1β have significantly shorter overall survival and progression-free survival, a lower tumor control rate, and a reduced high-dose intensity of GEM (96).
Moreover, multiple studies have collectively demonstrated that the inflammatory factor IL-13 is of great significance in evaluating patient prognosis. Rachel F Gabitass et al. conducted a comprehensive analysis of circulating MDSC and Tregs in patients with pancreatic, esophageal, and gastric cancers. They found that the MDSC value is an independent prognostic factor for patients with pancreatic and esophagogastric cancers. For every one - unit increase in MDSC, the risk of death in patients increases by 22%. Meanwhile, when evaluating a series of plasma cytokines, they found that the cytokine IL-13 in the plasma of patients was significantly elevated, and this elevation was positively correlated with the MDSC level (60). This suggests that IL-13 may play an important role in the process of MDSC exerting its functions. The study by Mandruzzato S et al. also confirmed this view: among tumor-induced CD11b(+) splenocytes, IL-4Rα(+) cells produce large amounts of inhibitory IL-13 and IFN-γ, while IL-4Rα (-) cells do not constitutively secrete these cytokines and are non-inhibitory. The full inhibitory function of MDSC in tumor-conditioned mice requires the coordinated action of IL-13 and IFNα released by cells in an autocrine manner (97). Additionally, Formentin et al. detected high levels of IL-13 in pancreatic ductal carcinoma cells, while normal pancreatic cells did not contain IL-13. They also found that in some PC cell lines, IL-13 can induce dose - dependent cell growth, and this phenomenon can be inhibited in a dose-dependent manner by an IL-13 neutralizing antibody, indicating that IL-13 is an autocrine growth factor in PC (98).
4.2 Therapeutic strategies targeting inflammatory factors to remodel TME
The unique TME of PC is a primary contributor to treatment failure with conventional monotherapies. As previously described, its key features include: An immunosuppressive microenvironment, Dense desmoplastic stroma, Hypoperfusion. The complex network of stromal cells and signaling pathways during TME formation poses significant therapeutic challenges. However, inflammatory factors serve as pivotal mediators connecting tumor cells, stromal components, and signaling cascades. Thus, targeting core inflammatory factors represents a viable strategy to remodel or reverse the pro-fibrotic/immunosuppressive TME, thereby enhancing existing therapies.
4.2.1 Targeting pro-fibrotic factors: reversing the stromal barrier
TGF-β is a central pro-fibrotic driver of stromal remodeling in PC. Inhibiting its signaling pathway effectively suppresses cancer-associated fibroblast (CAF) activation and remodels the dense extracellular matrix (ECM), improving drug delivery and alleviating immune suppression.
Early studies reported gabapentin’s ability to inhibit ketogenic acid production in CAFs (99). Paradoxically, gabapentin dose-dependently elevates TGF-β1 levels in cancer cells. Jin Zhang et al. demonstrated that combining pirfenidone with gabapentin synergistically inhibits PDAC growth and overcomes apoptosis resistance. They subsequently developed GEP—a coordination nanomedicine incorporating gabapentin, pirfenidone, and epigallocatechin gallate (EGCG). This agent remodels TME by altering CAF phenotypes, overcoming “gabapentin resistance” caused by TGF-β1 upregulation while significantly increasing intratumoral functional CD8+ T cell infiltration (100).
Notably, myofibroblast-like CAFs (myCAFs; α-SMA+FAPα+) have recently gained attention as TGF-β-driven architects of pro-tumorigenic TME (101). Ning Pang et al. introduced SB525334—a selective TGF-β receptor I inhibitor—as a pioneering agent preceding nanochemotherapy. It ablates TGF-β signaling without compensatory autocrine secretion, disrupting the CAF barrier while normalizing microvasculature and improving TME perfusion. This paves the way for enhanced tumor accumulation and delivery of docetaxel-loaded micelles, ultimately boosting antitumor efficacy (102).
4.2.2 Blocking immunosuppressive factors: restoring anti-tumor immunity
IL-6 and IL-18 are key immunosuppressive factors maintaining PC’s immunosuppressive microenvironment by suppressing T cell function.
PC’s poor immunogenicity, scarce neoantigens, and profoundly immunosuppressive TME render single-agent immune checkpoint inhibitors (ICIs) clinically ineffective. Although IL-6 typically mediates immune defense, accumulating evidence reveals its dual role in PC: directly promoting tumor proliferation/survival while driving immune escape via T cell exclusion and functional impairment (103). Preclinical studies confirm that targeting IL-6/IL-6R reverses TME immunosuppression: IL-6 blockade combined with immunotherapy significantly enhances intratumoral T cell infiltration, dismantles immunosuppressive barriers, and restores anti-tumor immunity, thereby potentiating ICI efficacy and suppressing tumor progression in murine PC models (104, 105).
Beyond cytokine blockade, a “function-selective modulation” strategy has been proposed to address the context-dependent duality of cytokines such as IL-18. This principle recognizes that cytokines may exert divergent biological effects in distinct microenvironments—a critical constraint for cytokine-based therapies. As demonstrated by Xingjun Guo et al., IL-18 exemplifies this duality in PC: systemic elevation correlates with anti-tumor immunity and prolonged patient survival, whereas high intratumoral levels activate NF-κB to promote invasion, metastasis, and reduced survival (Figure 4). Consequently, IL-18 monotherapy exhibits marginal efficacy. To resolve this, Guo et al. pioneered a novel co-targeting strategy combining IL-18 with NF-κB inhibitors. This approach selectively neutralizes IL-18’s pro-tumor effects within the TME while preserving its systemic immunostimulatory potential, significantly improving survival in murine PC models. This establishes IL-18/NF-κB co-targeting as a paradigm for overcoming context-dependent functional limitations of ambivalent cytokines (106).
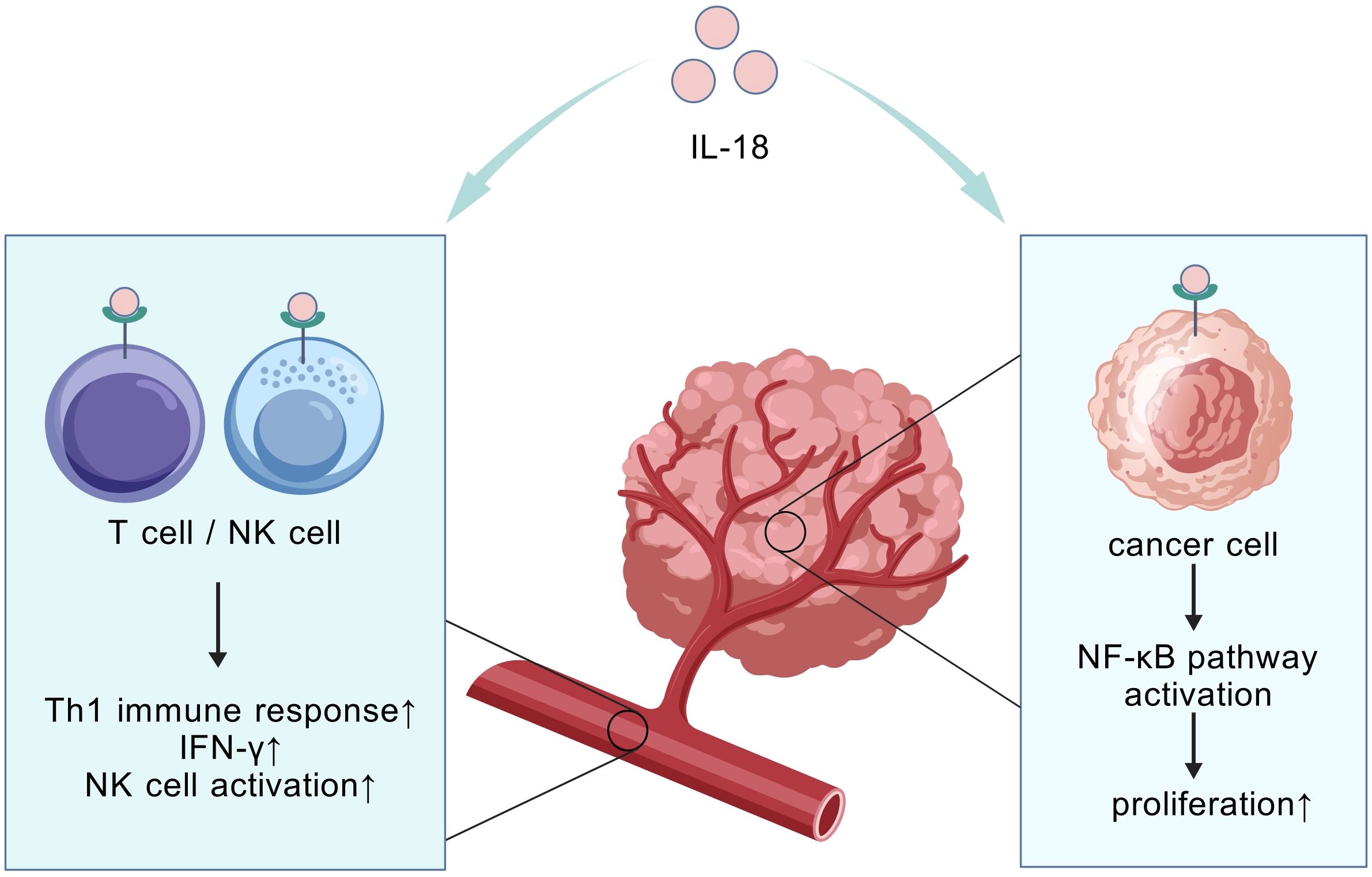
Figure 4. Compartment-specific IL-18 effects. The compartment-specific effect refers to the spatially dependent dual role of IL-18. It activates systemic anti-tumor immunity in circulation (correlating with improved survival), while locally within tumor tissue it promotes immunosuppression and tumor progression via NF-κB pathway activation.
4.2.3 Modulating inflammatory signaling to sensitize chemotherapy
IL-1-mediated chemoresistance represents a major limitation for conventional therapies (e.g., gemcitabine). Targeting this inflammatory signaling hub disrupts resistance pathways and reverses TME-driven chemotherapy refractoriness.
Zhang et al. discovered that IRAK4 activation in CAFs promotes NF-κB-dependent IL-1β secretion, establishing a feedforward loop. Within this circuit, IL-1β further activates the IRAK4-NF-κB signaling pathway in both CAFs and PDAC cells. This pathway activation drives CAFs to secrete excessive profibrotic factors (e.g., collagen type I), leading to the formation of a dense, fibrotic stroma. Critically, this fibrotic stroma exerts dual barrier effects: it physically impedes the penetration of chemotherapeutic agents (e.g., gemcitabine) while simultaneously creating a protective, pro-survival niche for PDAC cells. In preclinical studies, combining the IRAK4 inhibitor AS2444697 or IL-1β-neutralizing antibodies with gemcitabine significantly suppressed tumor growth and reduced fibrosis, highlighting the therapeutic potential of disrupting this signaling loop (107).
Separately, constitutive activation of the pro-inflammatory STAT3 pathway is a key biomarker of PDAC chemoresistance. PSCs release IL-6 to cross-talk with tumor cells, activating STAT3 signaling and promoting invasive phenotypes. STAT3 inhibition suppresses PDAC growth/invasion and profoundly remodels stroma to improve drug delivery and therapeutic response (108–111). Austin R. Dosch et al. further identified tumor-derived IL-1α as an upstream mediator of PSC-driven IL-6 release and STAT3 activation in TME. Consequently, IL-1R1 inhibitor anakinra partially overcomes STAT3-mediated chemoresistance in PDAC (112).
Collectively, these findings position anakinra combined with chemotherapy as a promising strategy to counteract IL-1α/β-driven resistance mechanisms.
5 Conclusion
With the deepening of research in recent years, the understanding of tumors has expanded from focusing solely on tumor cells to also considering the complex regulatory roles of the TME. In the TME, inflammatory factors play a key role in intercellular communication, directly or indirectly interacting with tumor cells and exerting significant regulatory effects on tumor progression. This review highlights several important factors in PC TME research in recent years, such as IL-1β, IL-4, IL-13, IL-15, IL-17, IL-18, and TNF-α. These factors influence the phenotypes of cells in the microenvironment through pathways such as MAPK, PI3K-Akt, JAK-STAT, and NF-κB. leading to changes such as an immunosuppressive TME, dense desmoplastic stroma, and hypoperfusion. These changes are critical nodes in the development and progression of PC and represent three distinct characteristics of the PC microenvironment compared to other tumors. They are closely associated with poor prognosis and treatment resistance in PC (Table 2).
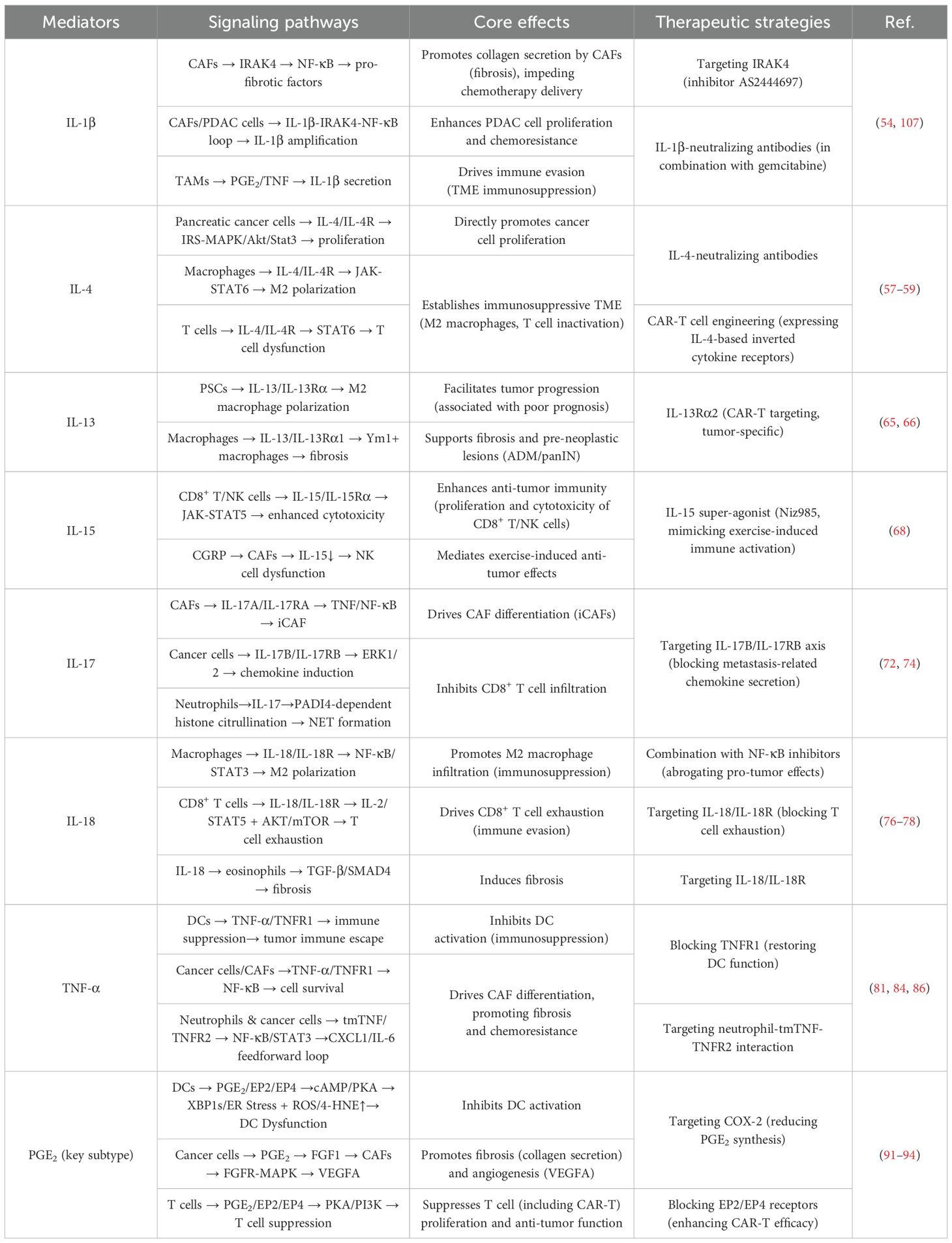
Table 2. Summary table: key inflammatory mediators in pancreatic cancer.
Clinically, inflammatory factors serve as potential biomarkers and therapeutic targets. Treatments such as nanotechnology, immunotherapy (e.g., IL-6 blockade with anti-CTLA-4), and chemotherapy (using agents like IL-1β antagonists) are being explored. However, challenges persist. The mechanisms of inflammatory factors are not fully understood, and their complex effects hinder therapy development. Future research should focus on deepening our understanding through molecular, cellular, and in vivo studies to enable personalized treatments.
Author contributions
YY: Writing – original draft. HZ: Writing – original draft. ZW: Writing – review & editing. LH: Writing – review & editing. DK: Writing – review & editing. BZ: Writing – review & editing. LS: Writing – review & editing. JA: Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82160451).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Giaquinto AN, and Jemal A. Cancer statistics, 2024. CA: A Cancer J Clin. (2024) 74:12–49. doi: 10.3322/caac.21820
2. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, and Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. (2014) 74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155
3. Walter FM, Mills K, Mendonça SC, Abel GA, Basu B, Carroll N, et al. Symptoms and patient factors associated with diagnostic intervals for pancreatic cancer (Symptom pancreatic study): A prospective cohort study. Lancet Gastroenterol Hepatol. (2016) 1:298–306. doi: 10.1016/S2468-1253(16)30079-6
4. Luo G, Jin K, Deng S, Cheng H, Fan Z, Gong Y, et al. Roles of Ca19–9 in pancreatic cancer: biomarker, predictor and promoter. Biochim Et Biophys Acta Rev Cancer. (2021) 1875:188409. doi: 10.1016/j.bbcan.2020.188409
5. Kartal E, Schmidt TSB, Molina-Montes E, Rodríguez-Perales S, Wirbel J, Maistrenko OM, et al. A faecal microbiota signature with high specificity for pancreatic cancer. Gut. (2022) 71:1359–72. doi: 10.1136/gutjnl-2021-324755
6. Siegel RL, Miller KD, and Jemal A. Cancer statistics, 2015. CA: Cancer J Clin. (2015) 65:5–29. doi: 10.3322/caac.21254
7. Chen X, Zeh HJ, Kang R, Kroemer G, and Tang D. Cell death in pancreatic cancer: from pathogenesis to therapy. Nat Rev Gastroenterol Hepatol. (2021) 18:804–23. doi: 10.1038/s41575-021-00486-6
8. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, and Jemal A. Cancer statistics, 2025. CA Cancer J Clin. (2025) 75:10–45. doi: 10.3322/caac.21871
9. Maru SY and Jaffee EM. Pancreatic cancer is feeling the heat. J Immunother Cancer. (2024) 12:e010124. doi: 10.1136/jitc-2024-010124
10. Shah A, Ganguly K, Rauth S, Sheree SS, Khan I, Ganti AK, et al. Unveiling the resistance to therapies in pancreatic ductal adenocarcinoma. Drug Resist Updates: Rev Commentaries Antimicrob Anticancer Chemother. (2024) 77:101146. doi: 10.1016/j.drup.2024.101146
11. Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. (2007) 11:291–302. doi: 10.1016/j.ccr.2007.01.012
12. Roshani R, McCarthy F, and Hagemann T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. (2014) 345:157–63. doi: 10.1016/j.canlet.2013.07.014
13. Strobel O, Dor Y, Alsina J, Stirman A, Lauwers G, Trainor A, et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology. (2007) 133:1999–2009. doi: 10.1053/j.gastro.2007.09.009
14. Klimstra DS. Nonductal neoplasms of the pancreas. Modern Pathol: Off J United States Can Acad Pathol Inc. (2007) 20 Suppl 1:S94–112. doi: 10.1038/modpathol.3800686
15. Hruban RH, Wilentz RE, and Maitra A. Identification and analysis of precursors to invasive pancreatic cancer. Methods Mol Med. (2005) 103:1–13. doi: 10.1385/1-59259-780-7:001
16. Waters AM and Der CJ. Kras: the critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor Perspect Med. (2018) 8:a031435. doi: 10.1101/cshperspect.a031435
17. Knudsen ES, O’Reilly EM, Brody JR, and Witkiewicz AK. Genetic diversity of pancreatic ductal adenocarcinoma and opportunities for precision medicine. Gastroenterology. (2016) 150:48–63. doi: 10.1053/j.gastro.2015.08.056
18. Bardeesy N, Cheng K-H, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. (2006) 20:3130. doi: 10.1101/gad.1478706
19. Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. (2015) 518:495–501. doi: 10.1038/nature14169
20. Carstens JL, Correa de Sampaio P, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun. (2017) 8:15095. doi: 10.1038/ncomms15095
21. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
22. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T cells from pancreatic carcinomas in mice is regulated by Ly6c(Low) F4/80(+) extratumoral macrophages. Gastroenterology. (2015) 149:201–10. doi: 10.1053/j.gastro.2015.04.010
23. Hiraoka N, Onozato K, Kosuge T, and Hirohashi S. Prevalence of Foxp3+ Regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res: Off J Am Assoc Cancer Res. (2006) 12:5423–34. doi: 10.1158/1078-0432.CCR-06-0369
24. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, and Bar-Sagi D. Oncogenic Kras-induced Gm-Csf production promotes the development of pancreatic neoplasia. Cancer Cell. (2012) 21:836–47. doi: 10.1016/j.ccr.2012.04.024
25. Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, and Bar-Sagi D. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep. (2017) 20:558–71. doi: 10.1016/j.celrep.2017.06.062
26. Caronni N, La Terza F, Vittoria FM, Barbiera G, Mezzanzanica L, Cuzzola V, et al. Il-1β+ Macrophages fuel pathogenic inflammation in pancreatic cancer. Nature. (2023) 623:415–22. doi: 10.1038/s41586-023-06685-2
27. Liou G-Y, Döppler H, Necela B, Krishna M, Crawford HC, Raimondo M, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through Nf-Kb and Mmps. J Cell Biol. (2013) 202:563–77. doi: 10.1083/jcb.201301001
28. Haqq J, Howells LM, Garcea G, Metcalfe MS, Steward WP, and Dennison AR. Pancreatic stellate cells and pancreas cancer: current perspectives and future strategies. Eur J Cancer (Oxford England: 1990). (2014) 50:2570–82. doi: 10.1016/j.ejca.2014.06.021
29. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C, et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res: Off J Am Assoc Cancer Res. (2015) 21:3561–8. doi: 10.1158/1078-0432.CCR-14-1051
30. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. (2013) 62:112–20. doi: 10.1136/gutjnl-2012-302529
31. Miyazaki Y, Oda T, Mori N, and Kida YS. Adipose-derived mesenchymal stem cells differentiate into pancreatic cancer-associated fibroblasts in vitro. FEBS Open Bio. (2020) 10:2268–81. doi: 10.1002/2211-5463.12976
32. Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. (2008) 68:2085–93. doi: 10.1158/0008-5472.CAN-07-2477
33. Nair N, Calle AS, Zahra MH, Prieto-Vila M, Oo AKK, Hurley L, et al. A cancer stem cell model as the point of origin of cancer-associated fibroblasts in tumor microenvironment. Sci Rep. (2017) 7:6838. doi: 10.1038/s41598-017-07144-5
34. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov. (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
35. Pereira BA, Ritchie S, Chambers CR, Gordon KA, Magenau A, Murphy KJ, et al. Temporally resolved proteomics identifies nidogen-2 as a cotarget in pancreatic cancer that modulates fibrosis and therapy response. Sci Adv. (2024) 10:eadl1197. doi: 10.1126/sciadv.adl1197
36. Gardiner JC and Cukierman E. Meaningful connections: interrogating the role of physical fibroblast cell-cell communication in cancer. Adv Cancer Res. (2022) 154:141–68. doi: 10.1016/bs.acr.2022.01.004
37. Tian C, Öhlund D, Rickelt S, Lidström T, Huang Y, Hao L, et al. Cancer cell-derived matrisome proteins promote metastasis in pancreatic ductal adenocarcinoma. Cancer Res. (2020) 80:1461–74. doi: 10.1158/0008-5472.CAN-19-2578
38. Tian C, Clauser KR, Öhlund D, Rickelt S, Huang Y, Gupta M, et al. Proteomic analyses of ecm during pancreatic ductal adenocarcinoma progression reveal different contributions by tumor and stromal cells. Proc Natl Acad Sci United States America. (2019) 116:19609–18. doi: 10.1073/pnas.1908626116
39. Murthy D, Attri KS, Shukla SK, Thakur R, Chaika NV, He C, et al. Cancer-associated fibroblast-derived acetate promotes pancreatic cancer development by altering polyamine metabolism via the Acss2-Sp1-Sat1 axis. Nat Cell Biol. (2024) 26:613–27. doi: 10.1038/s41556-024-01372-4
40. Zhu Y, Fang S, Fan B, Xu K, Xu L, Wang L, et al. Cancer-associated fibroblasts reprogram cysteine metabolism to increase tumor resistance to ferroptosis in pancreatic cancer. Theranostics. (2024) 14:1683–700. doi: 10.7150/thno.89805
41. Han X, Burrows M, Kim LC, Xu JP, Vostrejs W, Van Le TN, et al. Cancer-associated fibroblasts maintain critical pancreatic cancer cell lipid homeostasis in the tumor microenvironment. Cell Rep. (2024) 43:114972. doi: 10.1016/j.celrep.2024.114972
42. Niu N, Shen X, Wang Z, Chen Y, Weng Y, Yu F, et al. Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell. (2024) 42:869–84.e9. doi: 10.1016/j.ccell.2024.03.005
43. Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, et al. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res: Off J Am Assoc Cancer Res. (2014) 20:3422–33. doi: 10.1158/1078-0432.CCR-13-2972
44. Zhang X, Lao M, Yang H, Sun K, Dong Y, He L, et al. Targeting cancer-associated fibroblast autophagy renders pancreatic cancer eradicable with immunochemotherapy by inhibiting adaptive immune resistance. Autophagy. (2024) 20:1314–34. doi: 10.1080/15548627.2023.2300913
45. Folkman J. Tumor angiogenesis: therapeutic implications. New Engl J Med. (1971) 285:1182–6. doi: 10.1056/NEJM197111182852108
46. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, and Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. (2012) 21:418–29. doi: 10.1016/j.ccr.2012.01.007
47. Matsuo Y, Ding Q, Desaki R, Maemura K, Mataki Y, Shinchi H, et al. Hypoxia inducible factor-1 alpha plays a pivotal role in hepatic metastasis of pancreatic cancer: an immunohistochemical study. J Hepatobil Pancreat Sci. (2014) 21:105–12. doi: 10.1002/jhbp.6
48. Koong AC, Mehta VK, Le QT, Fisher GA, Terris DJ, Brown JM, et al. Pancreatic tumors show high levels of hypoxia. Int J Radiat Oncol Biol Phys. (2000) 48:919–22. doi: 10.1016/s0360-3016(00)00803-8
49. Chen Y, Cairns R, Papandreou I, Koong A, and Denko NC. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PloS One. (2009) 4:e7033. doi: 10.1371/journal.pone.0007033
50. Gupta VK, Sharma NS, Durden B, Garrido VT, Kesh K, Edwards D, et al. Hypoxia-driven oncometabolite L-2hg maintains stemness-differentiation balance and facilitates immune evasion in pancreatic cancer. Cancer Res. (2021) 81:4001. doi: 10.1158/0008-5472.CAN-20-2562
51. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. (2021) 6:263. doi: 10.1038/s41392-021-00658-5
52. Grivennikov SI, Greten FR, and Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
53. Zhang S, Xiao X, Yi Y, Wang X, Zhu L, Shen Y, et al. Tumor initiation and early tumorigenesis: molecular mechanisms and interventional targets. Signal Transduct Target Ther. (2024) 9:1–36. doi: 10.1038/s41392-024-01848-7
54. Das S, Shapiro B, Vucic EA, Vogt S, and Bar-Sagi D. Tumor cell-derived Il1β Promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res. (2020) 80:1088–101. doi: 10.1158/0008-5472.CAN-19-2080
55. Zhao T, Xiao D, Jin F, Sun X, Yu J, Wang H, et al. Ese3-positive Pscs drive pancreatic cancer fibrosis, chemoresistance and poor prognosis via tumour-stromal Il-1β/Nf-Kb/Ese3 signalling axis. Br J Cancer. (2022) 127:1461–72. doi: 10.1038/s41416-022-01927-y
56. Yako YY, Brand M, Smith M, and Kruger D. Inflammatory cytokines and angiogenic factors as potential biomarkers in South African pancreatic ductal adenocarcinoma patients: A preliminary report. Pancreatol: Off J Int Assoc Pancreatol (IAP) [et al]. (2017) 17:438–44. doi: 10.1016/j.pan.2017.03.003
57. Prokopchuk O, Liu Y, Henne-Bruns D, and Kornmann M. Interleukin-4 enhances proliferation of human pancreatic cancer cells: evidence for autocrine and paracrine actions. Br J Cancer. (2005) 92:921–8. doi: 10.1038/sj.bjc.6602416
58. Liu C-Y, Xu J-Y, Shi X-Y, Huang W, Ruan T-Y, Xie P, et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through Tlr4/Il-10 signaling pathway. Lab Invest; J Tech Methods Pathol. (2013) 93:844–54. doi: 10.1038/labinvest.2013.69
59. Zhou Y, Farooq MA, Ajmal I, He C, Gao Y, Guo D, et al. Co-expression of Il-4/Il-15-based inverted cytokine receptor in car-T cells overcomes Il-4 signaling in immunosuppressive pancreatic tumor microenvironment. Biomed Pharmacother = Biomed Pharmacother. (2023) 168:115740. doi: 10.1016/j.biopha.2023.115740
60. Gabitass RF, Annels NE, Stocken DD, Pandha HA, and Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother: CII. (2011) 60:1419–30. doi: 10.1007/s00262-011-1028-0
61. Fujisawa T, Shimamura T, Goto K, Nakagawa R, Muroyama R, Ino Y, et al. A novel role of interleukin 13 receptor alpha2 in perineural invasion and its association with poor prognosis of patients with pancreatic ductal adenocarcinoma. Cancers. (2020) 12:1294. doi: 10.3390/cancers12051294
62. Shi J, Shen X, Kang Q, Yang X, Denzinger M, Kornmann M, et al. Loss of interleukin-13-receptor-alpha-1 induces apoptosis and promotes Emt in pancreatic cancer. Int J Mol Sci. (2022) 23:3659. doi: 10.3390/ijms23073659
63. Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of car T cells in cancer therapy. Nat Rev Clin Oncol. (2021) 18:379–93. doi: 10.1038/s41571-021-00476-2
64. Stern LA, Gholamin S, Moraga I, Yang X, Saravanakumar S, Cohen JR, et al. Engineered Il13 variants direct specificity of Il13rα2-targeted car T cell therapy. Proc Natl Acad Sci United States America. (2022) 119:e2112006119. doi: 10.1073/pnas.2112006119
65. Xue J, Sharma V, Hsieh MH, Chawla A, Murali R, Pandol SJ, et al. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. (2015) 6:7158. doi: 10.1038/ncomms8158
66. Liou G-Y, Bastea L, Fleming A, Döppler H, Edenfield BH, Dawson DW, et al. The presence of interleukin-13 at pancreatic Adm/Panin lesions alters macrophage populations and mediates pancreatic tumorigenesis. Cell Rep. (2017) 19:1322–33. doi: 10.1016/j.celrep.2017.04.052
67. Kurz E, Hirsch CA, Dalton T, Shadaloey SA, Khodadadi-Jamayran A, Miller G, et al. Exercise-induced engagement of the Il-15/Il-15rα Axis promotes anti-tumor immunity in pancreatic cancer. Cancer Cell. (2022) 40:720–37.e5. doi: 10.1016/j.ccell.2022.05.006
68. Wang K, Ni B, Xie Y, Li Z, Yuan L, Meng C, et al. Nociceptor neurons promote pdac progression and cancer pain by interaction with cancer-associated fibroblasts and suppression of natural killer cells. Cell Res. (2025) 35:362–80. doi: 10.1038/s41422-025-01098-4
69. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic kras activates a hematopoietic-to-epithelial Il-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell. (2014) 25:621–37. doi: 10.1016/j.ccr.2014.03.014
70. Zhang Y, Zoltan M, Riquelme E, Xu H, Sahin I, Castro-Pando S, et al. Immune cell production of interleukin 17 induces stem cell features of pancreatic intraepithelial neoplasia cells. Gastroenterology. (2018) 155:210–23.e3. doi: 10.1053/j.gastro.2018.03.041
71. Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J Exp Med. (2020) 217:e20190354. doi: 10.1084/jem.20190354
72. Picard FSR, Lutz V, Brichkina A, Neuhaus F, Ruckenbrod T, Hupfer A, et al. Il-17a-producing Cd8+ T cells promote Pdac via induction of inflammatory cancer-associated fibroblasts. Gut. (2023) 72:1510–22. doi: 10.1136/gutjnl-2022-327855
73. Mucciolo G, Curcio C, Roux C, Li WY, Capello M, Curto R, et al. Il17a critically shapes the transcriptional program of fibroblasts in pancreatic cancer and switches on their protumorigenic functions. Proc Natl Acad Sci United States America. (2021) 118:e2020395118. doi: 10.1073/pnas.2020395118
74. Wu H-H, Hwang-Verslues WW, Lee W-H, Huang C-K, Wei P-C, Chen C-L, et al. Targeting Il-17b-Il-17rb signaling with an anti-Il-17rb antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. J Exp Med. (2015) 212:333–49. doi: 10.1084/jem.20141702
75. Khan IA, Singh N, Gunjan D, Gopi S, Dash NR, Gupta S, et al. Increased circulating Th17 cell populations in patients with pancreatic ductal adenocarcinoma. Immunogenetics. (2023) 75:433–43. doi: 10.1007/s00251-023-01318-4
76. Lutz V, Hellmund VM, Picard FSR, Raifer H, Ruckenbrod T, Klein M, et al. Il18 receptor signaling regulates tumor-reactive Cd8+ T-cell exhaustion via activation of the Il2/Stat5/Mtor pathway in a pancreatic cancer model. Cancer Immunol Res. (2023) 11:421–34. doi: 10.1158/2326-6066.CIR-22-0398
77. Zhang H-R, Li T-J, Yu X-J, Liu C, Wu W-D, Ye L-Y, et al. The Gfpt2-O-glcnacylation-Ybx1 axis promotes Il-18 secretion to regulate the tumor immune microenvironment in pancreatic cancer. Cell Death Dis. (2024) 15:244. doi: 10.1038/s41419-024-06589-7
78. Kandikattu HK, Manohar M, Verma AK, Kumar S, Yadavalli CS, Upparahalli Venkateshaiah S, et al. Macrophages-induced Il-18-mediated eosinophilia promotes characteristics of pancreatic Malignancy. Life Sci Alliance. (2021) 4:e202000979. doi: 10.26508/lsa.202000979
79. Alam MS, Gaida MM, Bergmann F, Lasitschka F, Giese T, Giese NA, et al. Selective inhibition of the P38 alternative activation pathway in infiltrating T cells inhibits pancreatic cancer progression. Nat Med. (2015) 21:1337–43. doi: 10.1038/nm.3957
80. Adjuto-Saccone M, Soubeyran P, Garcia J, Audebert S, Camoin L, Rubis M, et al. Tnf-a Induces endothelial-mesenchymal transition promoting stromal development of pancreatic adenocarcinoma. Cell Death Dis. (2021) 12:649. doi: 10.1038/s41419-021-03920-4
81. Dixit A, Sarver A, Zettervall J, Huang H, Zheng K, Brekken RA, et al. Targeting Tnf-a-producing macrophages activates antitumor immunity in pancreatic cancer via Il-33 signaling. JCI Insight. (2022) 7:e153242. doi: 10.1172/jci.insight.153242
82. Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. (1997) 385:729–33. doi: 10.1038/385729a0
83. Moss ML, Jin SL, Milla ME, Bickett DM, Burkhart W, Carter HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-alpha. Nature. (1997) 385:733–6. doi: 10.1038/385733a0
84. Alam MS, Gaida MM, Witzel HR, Otsuka S, Abbasi A, Guerin T, et al. Tnfr1 signaling promotes pancreatic tumor growth by limiting dendritic cell number and function. Cell Rep Med. (2024) 5:101696. doi: 10.1016/j.xcrm.2024.101696
85. Dickey EM, Bianchi A, Amirian H, Hosein PJ, Faustman D, Brambilla R, et al. Transmembrane Tnf-Tnfr2 signaling as a critical immunoregulatory node in pancreatic cancer. Oncoimmunology. (2024) 13:2326694. doi: 10.1080/2162402X.2024.2326694
86. Bianchi A, De Castro Silva I, Deshpande NU, Singh S, Mehra S, Garrido VT, et al. Cell-autonomous cxcl1 sustains tolerogenic circuitries and stromal inflammation via neutrophil-derived Tnf in pancreatic cancer. Cancer Discov. (2023) 13:1428–53. doi: 10.1158/2159-8290.CD-22-1046
87. Markosyan N, Li J, Sun YH, Richman LP, Lin JH, Yan F, et al. Tumor cell-intrinsic Epha2 suppresses anti-tumor immunity by regulating Ptgs2 (Cox-2). J Clin Invest. (2019) 129:3594–609. doi: 10.1172/JCI127755
88. Murthy D and Attri KS. Ptges expression is associated with metabolic and immune reprogramming in pancreatic ductal adenocarcinoma. Int J Mol Sci. (2023) 24:7304. doi: 10.3390/ijms24087304
89. Markosyan N, Kim I-K, Arora C, Quinones-Ware L, Joshi N, Cheng N, et al. Pivotal roles for cancer cell-intrinsic Mpges-1 and autocrine Ep4 signaling in suppressing antitumor immunity. JCI Insight. (2024) 9:e178644. doi: 10.1172/jci.insight.178644
90. Cui Y, Shu X-O, Li H-L, Yang G, Wen W, Gao Y-T, et al. Prospective study of urinary prostaglandin E2 metabolite and pancreatic cancer risk. Int J Cancer. (2017) 141:2423–9. doi: 10.1002/ijc.31007
91. Gilardini Montani MS, Benedetti R, Piconese S, Pulcinelli FM, Timperio AM, Romeo MA, et al. Pge2 released by pancreatic cancer cells undergoing Er stress transfers the stress to Dcs impairing their immune function. Mol Cancer Ther. (2021) 20:934–45. doi: 10.1158/1535-7163.MCT-20-0699
92. Xie C, Xu X, Wang X, Wei S, Shao L, Chen J, et al. Cyclooxygenase-2 induces angiogenesis in pancreatic cancer mediated by prostaglandin E2. Oncol Lett. (2018) 16:940–8. doi: 10.3892/ol.2018.8786
93. Bu L, Yonemura A, Yasuda-Yoshihara N, Uchihara T, Ismagulov G, Takasugi S, et al. Tumor microenvironmental 15-Pgdh depletion promotes fibrotic tumor formation and angiogenesis in pancreatic cancer. Cancer Sci. (2022) 113:3579–92. doi: 10.1111/cas.15495
94. Akbari B, Soltantoyeh T, Shahosseini Z, Jadidi-Niaragh F, Hadjati J, Brown CE, et al. Pge2-Ep2/Ep4 signaling elicits mesocar T cell immunosuppression in pancreatic cancer. Front Immunol. (2023) 14:1209572. doi: 10.3389/fimmu.2023.1209572
95. Gu Y-L, Lan C, Pei H, Yang S-N, Liu Y-F, and Xiao L-L. Applicative value of serum Ca19-9, Cea, Ca125 and Ca242 in diagnosis and prognosis for patients with pancreatic cancer treated by concurrent chemoradiotherapy. Asian PacifI J Cancer Prevent: APJCP. (2015) 16:6569–73. doi: 10.7314/apjcp.2015.16.15.6569
96. Mitsunaga S, Ikeda M, Shimizu S, Ohno I, Furuse J, Inagaki M, et al. Serum levels of Il-6 and Il-1β can predict the efficacy of gemcitabine in patients with advanced pancreatic cancer. Br J Cancer. (2013) 108:2063–9. doi: 10.1038/bjc.2013.174
97. Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, et al. Il4ralpha+ Myeloid-derived suppressor cell expansion in cancer patients. J Immunol (Baltimore Md: 1950). (2009) 182:6562–8. doi: 10.4049/jimmunol.0803831
98. Formentini A, Prokopchuk O, Sträter J, Kleeff J, Grochola LF, Leder G, et al. Interleukin-13 exerts autocrine growth-promoting effects on human pancreatic cancer, and its expression correlates with a propensity for lymph node metastases. Int J Colorectal Dis. (2009) 24:57–67. doi: 10.1007/s00384-008-0550-9
99. Li J-T, Yin M, Wang D, Wang J, Lei M-Z, Zhang Y, et al. Bcat2-mediated Bcaa catabolism is critical for development of pancreatic ductal adenocarcinoma. Nat Cell Biol. (2020) 22:167–74. doi: 10.1038/s41556-019-0455-6
100. Zhang J, Zhang J, Lin R, Hou P, Zheng L, Jiang C, et al. Pirfenidone antagonizes Tgf-B1-mediated gabapentin resistance via reversal of desmoplasia and the ‘Cold’ Microenvironment in pancreatic cancer. Cancer Lett. (2024) 605:217287. doi: 10.1016/j.canlet.2024.217287
101. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
102. Pang N, Yang Z, Zhang W, Du Y, Zhang L, Li X, et al. Cancer-associated fibroblasts barrier breaking via Tgf-B Blockade paved way for docetaxel micelles delivery to treat pancreatic cancer. Int J Pharm. (2024) 665:124706. doi: 10.1016/j.ijpharm.2024.124706
103. Fisher DT, Appenheimer MM, and Evans SS. The two faces of Il-6 in the tumor microenvironment. Semin Immunol. (2014) 26:38–47. doi: 10.1016/j.smim.2014.01.008
104. Ware MB, Phillips M, McQuinn C, Zaidi MY, Knochelmann HM, Greene E, et al. Dual Il-6 and Ctla-4 blockade regresses pancreatic tumors in a T cell- and Cxcr3-dependent manner. JCI Insight. (2023) 8:e155006. doi: 10.1172/jci.insight.155006
105. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. Il-6 and Pd-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. (2018) 67:320–32. doi: 10.1136/gutjnl-2016-311585
106. Guo X, Zheng L, Jiang J, Zhao Y, Wang X, Shen M, et al. Blocking Nf-Kb is essential for the immunotherapeutic effect of recombinant Il18 in pancreatic cancer. Clin Cancer Res: Off J Am Assoc Cancer Res. (2016) 22:5939–50. doi: 10.1158/1078-0432.CCR-15-1144
107. Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J, et al. Tumor-stroma Il1β-Irak4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res. (2018) 78:1700–12. doi: 10.1158/0008-5472.CAN-17-1366
108. Nagaraj NS, Washington MK, and Merchant NB. Combined blockade of Src kinase and epidermal growth factor receptor with gemcitabine overcomes Stat3-mediated resistance of inhibition of pancreatic tumor growth. Clin Cancer Res: Off J Am Assoc Cancer Res. (2011) 17:483–93. doi: 10.1158/1078-0432.CCR-10-1670
109. Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R, et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology. (2015) 149:1932–43.e9. doi: 10.1053/j.gastro.2015.07.058
110. Dosch AR, Dai X, Reyzer ML, Mehra S, Srinivasan S, Willobee BA, et al. Combined Src/Egfr inhibition targets Stat3 signaling and induces stromal remodeling to improve survival in pancreatic cancer. Mol Cancer Res: MCR. (2020) 18:623–31. doi: 10.1158/1541-7786.MCR-19-0741
111. Nagathihalli NS, Castellanos JA, VanSaun MN, Dai X, Ambrose M, Guo Q, et al. Pancreatic stellate cell secreted Il-6 stimulates Stat3 dependent invasiveness of pancreatic intraepithelial neoplasia and cancer cells. Oncotarget. (2016) 7:65982–92. doi: 10.18632/oncotarget.11786
Keywords: pancreatic cancer, inflammatory factors, tumor microenvironment, immunology, targeted therapy
Citation: Yuan Y, Zhang H, Wang Z, Huang L, Kabacaoglu D, Zhang B, Song L and Ai J (2025) The role of inflammatory factors in the tumor microenvironment of pancreatic cancer. Front. Immunol. 16:1625114. doi: 10.3389/fimmu.2025.1625114
Received: 08 May 2025; Accepted: 04 August 2025;
Published: 28 August 2025.
Edited by:
Ravi Kumar Sharma, Chandigarh University, IndiaReviewed by:
Nune Markosyan, University of Pennsylvania, United StatesTariq Ahmad Najar, New York University, United States
Copyright © 2025 Yuan, Zhang, Wang, Huang, Kabacaoglu, Zhang, Song and Ai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jiaoyu Ai, bmR5ZnkwNTcxOEBuY3UuZWR1LmNu
†These authors have contributed equally to this work