 Magdalena Niedzielska1
Magdalena Niedzielska1 Amy Chalmers1
Amy Chalmers1 Martyna C. Popis1
Martyna C. Popis1 Efrat Altman-Sharoni1
Efrat Altman-Sharoni1 Stephen Addis1
Stephen Addis1 Rebekka Beulen1Nils-Petter Rudqvist1
Rebekka Beulen1Nils-Petter Rudqvist1 Eleni Chantzoura1
Eleni Chantzoura1 Marco A. Purbhoo1
Marco A. Purbhoo1 Dhan Chand2
Dhan Chand2 Mark A. Exley1,3,4*
Mark A. Exley1,3,4*- 1Research & Development, MiNK Therapeutics Inc., Lexington, MA, United States
- 2Research & Development, Agenus Inc., Lexington, MA, United States
- 3Gastroenterology, Brigham & Women’s Hospital, Boston, MA, United States
- 4Life Sciences, Imperial College, London, United Kingdom
Despite significant advances in cancer therapies, many malignancies remain resistant to current treatments due to complex immunosuppressive mechanisms, limited neoantigen expression, and dynamic tumor adaptations, underscoring the need for innovative therapeutic strategies. Adoptive cell therapy (ACT), particularly with chimeric antigen receptors (CARs and recombinant TCRs) targeting cancer-associated antigens, has emerged as a transformative strategy. However, conventional CAR-T cell therapies face substantial limitations such as manufacturing challenges, severe toxicities, and limited efficacy against solid tumors. Invariant natural killer T (iNKT) cells, a unique lymphocyte subset bridging innate and adaptive immunity, have emerged as a compelling alternative platform for CAR-based therapies, due to their distinctive ability to persist, penetrate in and remodel the tumor microenvironment (TME). Unlike conventional T cells, iNKT cells exhibit rapid activation without priming, potent cytotoxicity, and extensive immunomodulatory functions. Furthermore, the inherent immunomodulatory properties of iNKT cells through interactions with the monomorphic antigen-presenting molecule CD1d or stress ligands augment endogenous anti-tumor immunity by activating NK cells and cytotoxic T lymphocytes, promoting dendritic cell maturation, and reducing immunosuppressive myeloid cells, unlike other Innate T cells. CAR-engineered iNKT (CAR-iNKT) cells therefore leverage multiple targeting mechanisms through their native semi-invariant T-cell receptor (TCR), NK receptors (NKRs) and engineered CARs, enabling broader and more effective tumor recognition while actively reshaping immunosuppressive TME. Notably, iNKT cells lack alloreactivity, circumventing the risk of graft-versus-host disease (GvHD), positioning CAR-iNKT cells as ideal candidates for “off-the-shelf” allogeneic therapies that can overcome the limitations of existing immunotherapies.
Introduction
Cancer remains a major global health and socioeconomic challenge, with steadily rising incidence rates, particularly among adults under 50, and an expected 77% increase in new cases by 2050 (1, 2), underscoring the urgent need for innovative therapeutic strategies. A revolution in understanding cancer and immunotherapy over the past decade has fundamentally transformed treatment paradigms by exploiting the immune system’s intrinsic ability to recognize and eliminate malignant cells (3).
Adoptive cell therapy (ACT) using chimeric antigen receptor (CAR) technologies, antibody-like membrane bound antigen receptors which redirect immune cells toward specific tumour antigens with high precision, represents one of the most significant breakthroughs in immunotherapy. Notably, autologous CAR-T cells have demonstrated exceptional efficacy in hematological malignancies, resulting in multiple FDA approvals for products targeting CD19 and B-cell maturation antigen (BCMA) (4, 5). In some patients, these “living drugs” persist for many years, functioning as long-term sentinels that sustain remission and further underscore the curative potential of engineered cellular immunotherapy (6).
Despite these impressive achievements, CAR-T cell efficacy against solid tumors remains limited to date, primarily restricted by a highly immunosuppressive TME that impairs T cell trafficking and infiltration into tumor sites, hostile metabolic challenges, tumor antigen heterogeneity and escape (7–9). Moreover, life-threatening toxicities, including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) (10, 11), coupled with prohibitive manufacturing and treatment costs that limit accessibility (12, 13), remain significant clinical issues and limit eligibility. To overcome these barriers, researchers are reimagining CAR therapy by engineering autologous and allogeneic (healthy donor) non-conventional immune effector cells with improved tumor-homing capacity, persistence, intrinsic TME-modulating activity, cytotoxicity and in the case of allogeneic cells, reduced alloreactivity for universal use.
Invariant Natural Killer T (iNKT) cells, a subset of T lymphocytes with innate-like activity have emerged as promising candidates for next-generation CAR-based therapies. These unique lymphocytes are defined by their semi-invariant T cell receptor (TCR), which recognizes lipid antigens presented by CD1d, a conserved non-polymorphic MHC class I-like molecule (14, 15). This distinct recognition mechanism enables iNKT cells to bypass traditional HLA restrictions, a critical feature for developing truly “off-the-shelf” allogeneic therapies with broader patient reach than traditional CAR-T cells. Functionally, iNKT cells offer several advantages over conventional T cells for cancer immunotherapy. Coupling TCR specificity with innate-like speed, iNKT cells are rapidly activated without requiring priming and demonstrate potent cytotoxicity (via perforin, granzyme B and FasL/CD95) against various tumor types. Moreover, iNKT cells secrete high levels of multiple cytokines and actively remodel the TME towards a proinflammatory anti-tumor phenotype by removing key barriers such as tumor associated macrophages (TAMs) and myeloid-derived suppressor cells (MDSC). iNKT cells also enhance broader immune surveillance via activation/maturation of dendritic cells and lymphocytes, including NK cells and cytotoxic T lymphocytes (16–19). Given these unique attributes, engineered iNKT cells (e.g., CAR-iNKT, TCR-iNKT) are being developed to further optimize tumor killing and extend the reach of cellular immunotherapy to immunologically ‘cold’ and difficult-to-treat solid tumors (20–22).
In this review, we examine the unique biological properties of iNKT cells, recent advances in CAR-iNKT and TCR-iNKT engineering, emerging clinical data, and strategies to overcome current challenges, providing a comprehensive assessment of how this innovative platform is redefining cellular cancer immunotherapy.
iNKT cell identity and functional heterogeneity
CD1d-restricted Natural Killer T (NKT) cells represent a heterogenous T cell subset characterized by their ability to recognize lipid antigens presented by the MHC class I-like molecule, CD1d. Among NKT cells, iNKT cells, also known as type I NKT cells, are the best-characterized subpopulation with relative homogeneity but functional heterogeneity. This subset of unconventional αβ T lymphocytes are defined by their semi-invariant TCR, comprising a near-germline TCRα chain (specifically Vα24-Jα18 in humans and Vα14-Jα18 in mice) paired with a limited TCRβ chains (predominantly Vβ11 in humans and Vβ8, Vβ7, or Vβ2 in mice). Additionally, iNKT cells express several NK cell-associated surface molecules, including CD161 or NKG2D (18, 23) reflecting their hybrid phenotype between innate and adaptive lymphocyte.
The recognition of lipid antigens by iNKT cells occurs via binding to CD1d, a monomorphic antigen-presenting molecule predominantly expressed on professional antigen-presenting cells (APCs), such as dendritic cells and macrophages, as well as B cells. Alpha-galactosylceramide (α-GalCer), originally isolated from marine sponges, is considered the prototypical and potent lipid antigen recognized by iNKT cells (24). However, α-GalCer and structurally related lipids are also produced by bacteria including gut microbiota (25), and have recently been identified as endogenous mammalian antigens for iNKT cells (26). Beyond α-GalCer, additional endogenous iNKT cell ligands include phospholipids and glycosphingolipids, such as glucosylceramide (27). A distinctive feature of iNKT is their unique developmental programming. Unlike conventional T cells that require priming, iNKT exit the thymus as fully functional effectors, capable of rapid cytokine production and cytotoxic responses (18, 28). As such, iNKT can play pivotal roles in modulating immune responses during infections and in tumor immune surveillance, partly by promoting DC maturation and activating NK cells as well as CD8+ T lymphocytes (18).
Despite their common TCR architecture and therefore specificity, iNKT cells exhibit substantial phenotypic and functional diversity. iNKT cells are rare cells and need precise detection reagents, including a monoclonal antibody (6B11), that recognizes primate iTCR rearrangements (29) and lipid antigen-loaded CD1d-multimers (30). Human iNKT can be classified based on CD4 and CD8 expression into four main subsets: CD4+CD8- (CD4+), CD4-CD8+ (CD8+), CD4-CD8- (double negative; DN), and CD4+CD8+ (double positive; DP, mainly thymic early iNKT) (29, 30). These subsets exhibit distinct tissue homing properties through differential chemokine receptor expression. One of 2 major peripheral subsets, CD4+ iNKT cells predominantly express CCR4, a chemokine receptor associated with homing to (e.g.) the lung (18, 31–33). In contrast, the rarer CD8+ and other major subset, DN iNKT preferentially express CCR1, CCR6, and CXCR6, with the latter being associated with iNKT homing, retention and survival within hepatic and pulmonary tissues (23, 30).
Functionally, iNKT cell subsets parallel the classical T helper (Th) cell lineages. Th1-like iNKT, predominantly found within the DN and CD8 subsets, express the transcription factor T-bet, secrete IFN-γ and TNF-α upon activation, express high levels of the activating receptor NKG2D, and exhibit potent cytotoxicity (23, 30). Mixed Th1-Th2-like iNKT, primarily CD4+, express GATA-3, produce IL-4, IL-13, and IFN-γ, and support humoral immunity (23, 34). Th17-like iNKT express RORγt and secrete IL-17, IL-21, and IL-22 and promote mucosal immunity and inflammation. Additional functional subsets include Treg-like iNKT that express FOXP3 and secrete IL-10, and T follicular helper (Tfh)-like iNKT, which produce IL-21 and support B cell function (23, 35).
A defining characteristic of iNKT cells is their remarkable functional plasticity. Their cytokine production profiles adapt in response to specific microenvironmental cues and immunological contexts. This functional flexibility allows iNKT to respond dynamically to diverse pathological conditions, highlighting their pivotal role in bridging innate and adaptive immunity (23). Such adaptability is potentially valuable in cancer, where iNKT can recalibrate their effector functions in response to the complex and evolving tumor microenvironment, as well as other immunotherapies.
From entry to eradication: how iNKT cells infiltrate tumors and drive anti-tumor immunity
Effective tumor infiltration by immune cells is critical for shaping the tumor microenvironment and can significantly influence disease progression and response to immunotherapy. Emerging evidence demonstrates that iNKT cells possess remarkable capabilities to infiltrate tumor tissues and orchestrate multi-faceted anti-tumor responses through both direct and indirect mechanisms (19).
For example, in MC38 colorectal tumor models using Vα14 Tg Cxcr6^Gfp mice, iNKT preferentially migrate to sites of disease, rather than normal colon tissue (36). These tumor-infiltrating iNKT cells exhibit an activated phenotype, characterized by CD69 and FasL expression, and production of effector molecules such as IFN-γ and Granzyme B (36, 37). Similarly, in the TRAMP prostate cancer model, iNKT are recruited to the TME via the CCL2-CCR5 chemokine axis (38). Notably, human prostate tumor and epithelial cells can express CD1d, enabling direct interaction with iNKT cells (38, 39). However, like conventional T cells, intratumoral iNKT often accumulate at the tumor periphery, suggesting that physical and immunological barriers restrict deeper infiltration into tumor cores. These barriers are largely mediated by interactions with CD1d-expressing TAM. Strategies aimed at enhancing iNKT mobility and modulating these interactions represent promising approaches to overcome these limitations (37).
Within the tumor, iNKT cells can mount a multi-pronged attack to recondition the TME and enhance anti-tumor immunity (Figure 1). They can directly lyse CD1d-positive malignancies, including hematologic and select solid tumors, and stress ligand-expressing tumor cells via Fas/FasL interactions, TRAIL-mediated apoptosis, or release of cytotoxic granules containing perforin and granzymes (23, 39, 40). Beyond their direct cytotoxic functions, iNKT serve as powerful orchestrators of broader anti-tumor immune responses. Activation through the TCR leads to upregulation of IL-12 receptors and CD40L, which in turn promotes DC maturation. This bidirectional interaction creates a positive feedback loop that amplifies IFNγ secretion, upregulates MHC class I and II antigen presentation machinery, and promotes Th1 polarized immune responses (18, 39, 41, 42). Furthermore, the activation cascade initiated by iNKT extends to multiple immune compartments, leading to enhanced NK cell activation, and potentiation of B cell and cytotoxic T cell responses, effectively bridging innate and adaptive immunity to generate a coordinated, multi-faceted anti-tumor response.
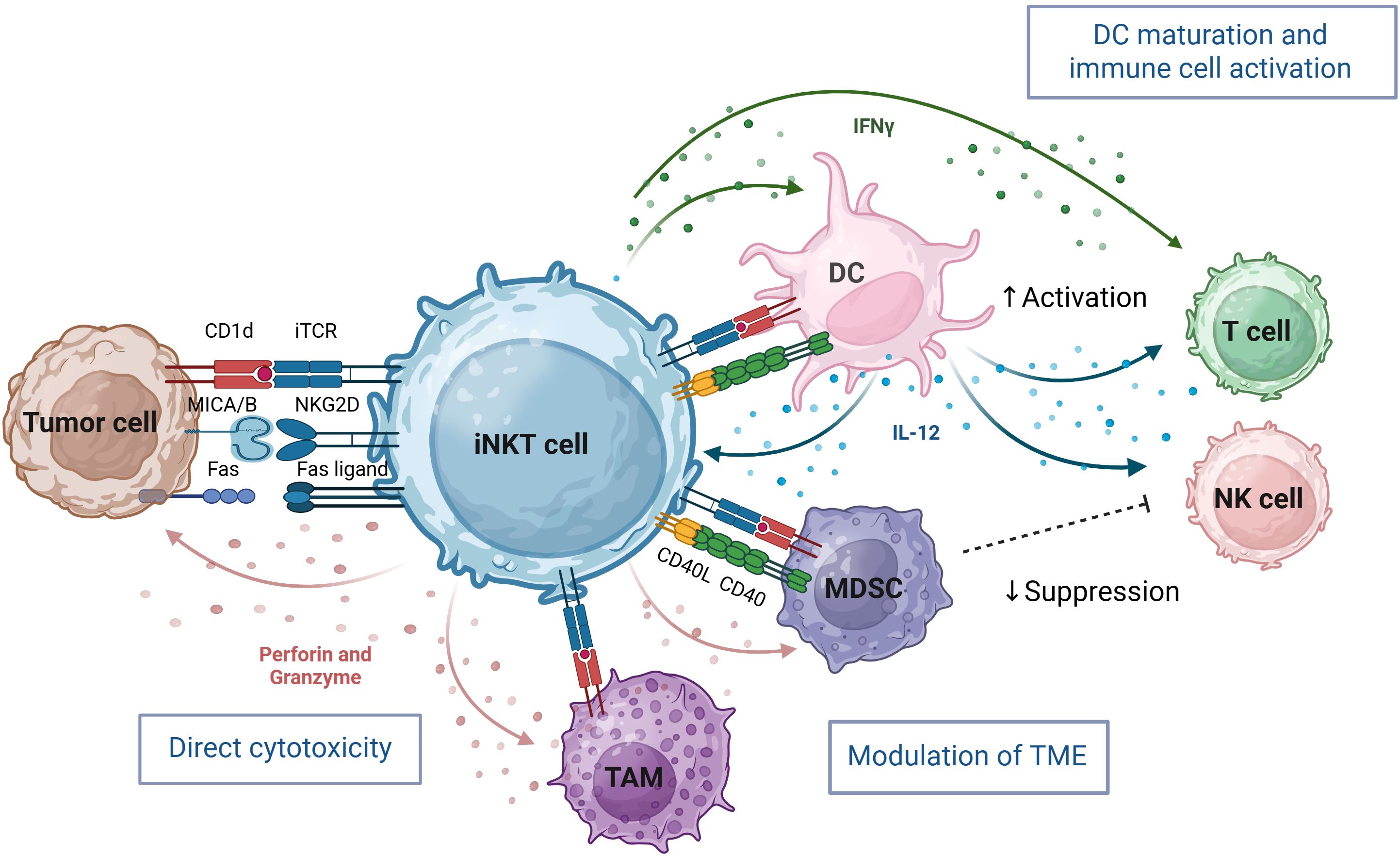
Figure 1. iNKT cells bridge innate and adaptive immunity through cytotoxic activity and immune-enhancing adjuvant effects. iNKT cells orchestrate potent anti-tumor responses through direct cytotoxicity, immune cell cross-talk, and tumor microenvironment (TME) remodeling. They directly kill tumor cells via invariant iTCR recognition of CD1d–lipid complexes and through NK cell receptors such as NKG2D which recognize stress ligands on tumor cells. iNKT cells also target immunosuppressive tumor-associated macrophages (TAM) and Myeloid-derived suppressor cells (MDSC), creating a more permissive environment for NK and T cell infiltration and function. Indirectly, iNKT-derived IFN-γ activates NK cells, promotes dendritic cell (DC) maturation, and enhances T cell responses. Bidirectional activation with DCs—mediated by CD1d, CD40–CD40L, and IL-12 further amplifies iNKT effector functions. iNKT cells enlarged for clarity, not to scale relative to others. Image created in BioRender.
Notably, iNKT cells also directly target immunosuppressive components within the TME. In preclinical neuroblastoma models, iNKT eliminate CD1d-expressing TAM and inhibit the function of myeloid-derived suppressor cells (MDSC) in a CD1d- and CD40-dependent manner, thereby creating a more favorable microenvironment for effector immune responses (23, 43, 44). This combination of direct cytotoxicity, immunomodulation and TME reprogramming positions iNKT as promising candidates for next-generation cellular engineered immunotherapies.
Clinical snapshot: harnessing unmodified iNKT cells for immunotherapy
Insights from preclinical murine models to early clinical studies have provided compelling evidence for the intrinsic anti-tumor capacity of iNKT. Retrospective analyses across a broad spectrum of malignancies, including colorectal and hepatocellular carcinoma, acute myeloid leukemia, head-and-neck squamous cell carcinoma, neuroblastoma, upper-gastrointestinal cancers, prostate and lung cancers, show that elevated intratumoral or peripheral iNKT frequencies correlate with improved overall survival (45). These observations provided a strong rationale for therapeutic approaches designed to enhance iNKT cell frequency and function in cancer patients.
Initial clinical trials focused on adoptively transferred, ex vivo expanded autologous and later allogeneic iNKT cells in patients with non-small cell lung cancer, head and neck squamous cell carcinoma, hepatocellular carcinoma, pancreatic cancer and advanced melanoma (Table 1). These early-phase studies demonstrated encouraging clinical benefits and a favorable safety profile, with only five treatment-related adverse events of grade ≥2 reported among many patients treated (46–53), underscoring the exceptional tolerability of this approach compared to other cellular immunotherapies.
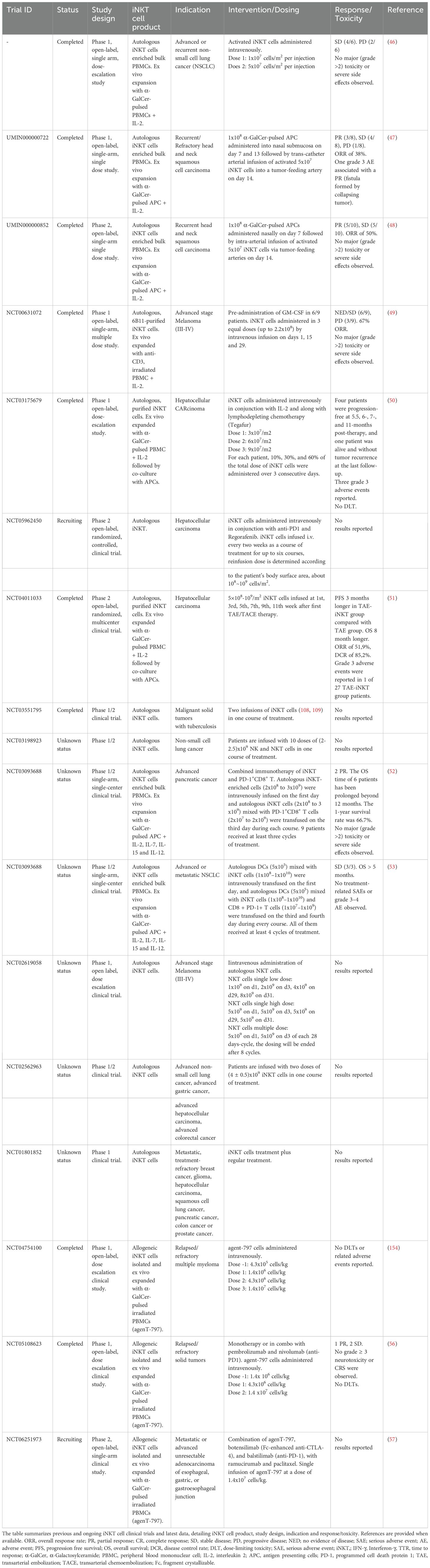
Table 1. I Summary of iNKT cell-based clinical trials for adoptive cellular therapy of cancer.
Unlike conventional T cells, which pose significant risks when used across HLA barriers, iNKT cells can be safely transferred between unmatched donors and recipients without causing GvHD. Their restricted TCR recognizes the non-polymorphic CD1d molecule rather than variable HLA complexes, enabling their use as universal donor cells. Recently, an allogeneic iNKT therapy, agenT-797 (MiNK Therapeutics), has entered clinical development as an off-the-shelf immunotherapy, comprising of over 99.5% unmodified human iNKT cells isolated from healthy donors mononuclear cell apheresis and vastly expanded ex vivo to treat many recipients per donor. To accumulate as much as practical of this allogeneic product, where quality and quantity are more important than speed, expansions take about 6 weeks with simply low dose IL-2. AgenT-797 functions as a potent immune modulator with clinical proof-of-concept demonstrated across multiple disease indications, including acute respiratory distress syndrome (ARDS), hematologic and solid tumors (54–56). Notably, among 82 treated patients, there were no occurrences of severe CRS, GvHD, or ICANS events (54, 56, 57), reflecting the non−alloreactive nature of iNKT cells. Interestingly, there was also no evidence of depletion of CD1d+ monocytes, B cells or other cells that iNKT interact with, presumably since no high avidity exogenous antigen was simultaneously administered (54, 56, 57).
In a heavily pre−treated solid−tumor cohort (N=34), agenT-797 demonstrated meaningful clinical activity, including partial responses in third-line gastric cancer and testicular cancer, and prolonged disease stabilization in nine additional patients (55, 56). In the gastric cancer cohort, paired tumor biopsies revealed a transformation from ‘cold’ immune deserts to inflamed microenvironments following agenT-797 therapy, characterized by enhanced infiltration of cytotoxic T cells and dendritic cells, and activation of both central and effector memory T cells (55, 58). Additionally, peripheral blood analyses demonstrated elevated levels of pro-inflammatory cytokines, including IFN-γ post-treatment (55, 58). While direct detection of infused iNKTs within tumor tissue remains limited, post-treatment biopsies showed robust immune remodeling, consistent with their functional activity in the tumor and active engagement of innate immune cascades (55, 58). Moreover, mechanistic studies using clinically relevant human models demonstrated that agenT-797 rescues exhausted T cells, activates dendritic cells, and selectively targets immunosuppressive M2 macrophages while sparing M1 macrophages, providing a mechanistic basis for agenT-797’s ability to reshape the TME toward the more immunostimulatory state observed in post-treatment clinical specimens (54, 55, 58).
Collectively, these clinical findings underscore the transformative potential of iNKT cell-based therapies in cancer treatment. Their unique ability to directly kill tumor cells, reprogram the TME and activate multiple immune effector mechanisms with favorable safety profiles positions them as a promising approach and platform to overcome critical barriers associated with current cellular immunotherapies and expanded therapeutic potential across a broader spectrum of malignancies.
CAR design and optimization for iNKT cell-based immunotherapy
Adding a CAR to iNKT cells enhances their tumor-targeting specificity and cytotoxicity, combining the innate-like, tissue-homing and immunoregulatory properties of iNKT cells with the precision of CAR-mediated antigen recognition for improved anti-tumor efficacy. CARs were originally designed for conventional T cells, and the majority of work has been done with this cell type. With the emergence of novel cellular platforms, such as iNKT cells, CARs are increasingly being adapted and integrated into diverse immune cell types due to their standardized design and proven efficacy.
CARs are made up of 4 main components: the antigen binding domain displayed extracellularly, a spacer/hinge region, a transmembrane domain (TMD) and one or more intracellular domains (ICD) which mediate signal transduction (Figure 2) (59, 60). The antigen binding domain recognizes the given antigen and is most commonly a single-chain variable fragment (scFv) due to their high specificity and affinity. scFv’s are derived from the variable heavy and light chains of monoclonal antibodies connected by a flexible peptide linker. When selecting a scFv for use in CAR T-cell therapies, it’s crucial to carefully consider its affinity. The scFv must bind strongly enough to trigger a significant activation response, but not so strongly that it causes excessive stimulation. Unlike monoclonal antibodies, CARs with too high an affinity can lead to detrimental effects, such as activation-induced cell death, T cell exhaustion, or toxicity due to CRS. Balancing the affinity is key to achieving effective immune responses while minimizing these risks (61–63). The optimal scFv for CARs success must also consider scFv epitope location, density of the target antigen and ligand-independent tonic signaling (64, 65). The flexibility and length of the linker between the variable heavy and light chains can also significantly influence CAR function by affecting the stability and proper folding of the scFv (66).
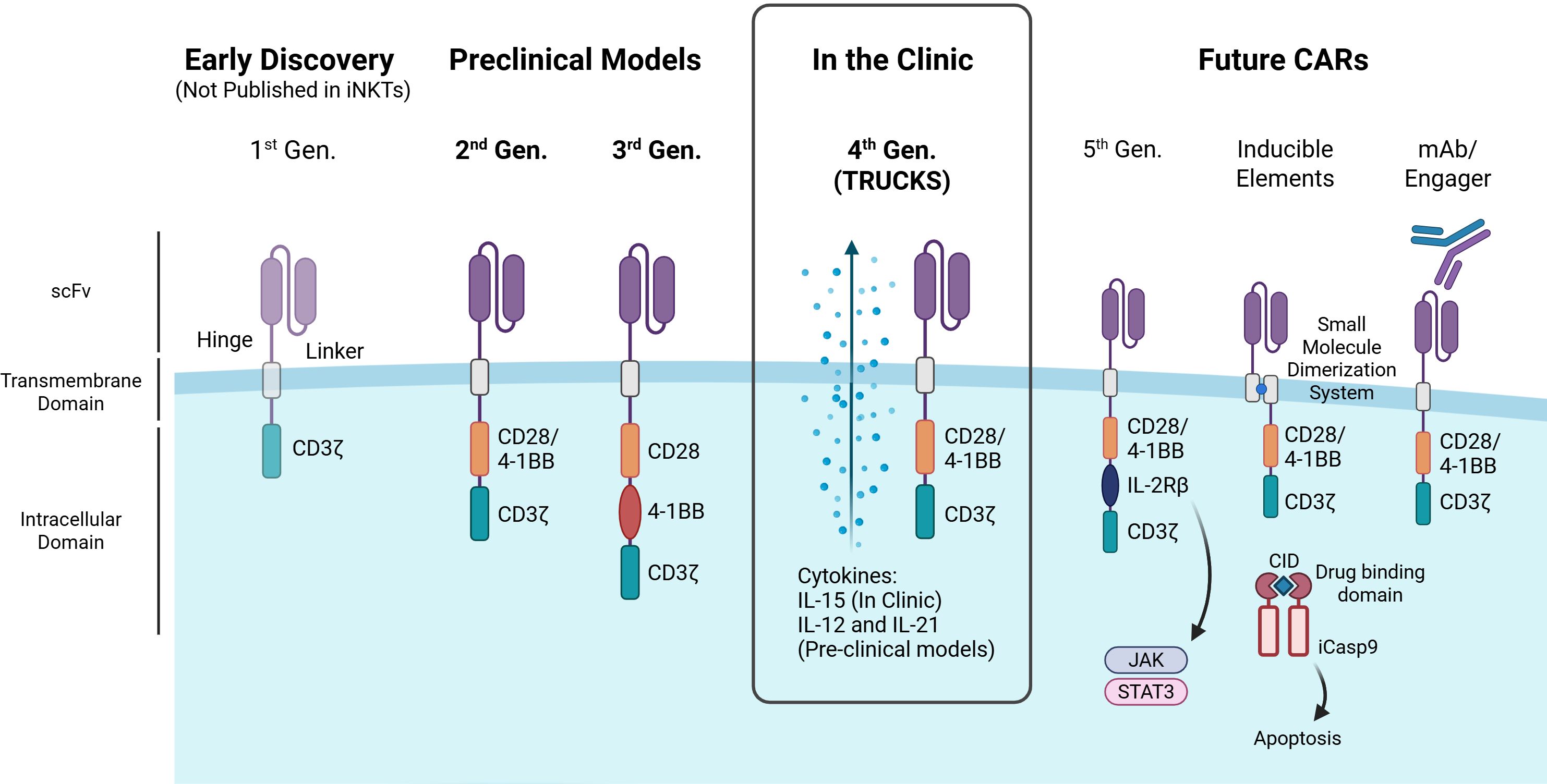
Figure 2. Advances in CAR design and their application in iNKT cells. The figure illustrates the evolution of chimeric antigen receptor (CAR) generations, highlighting key structural components and advancements. First-generation CARs contain a single CD3ζ signaling domain, but have not been reported in iNKTs. Second-generation CARs enhance T cell activation with a costimulatory domain (e.g., CD28, 4-1BB), while third-generation CARs integrate multiple domains for improved function. Both second- and third-generation CARs have been tested in iNKT preclinical models. Fourth-generation CARs (aka ‘TRUCKs’) have been used in iNKT clinical trials and introduce cytokine expression for better iNKT persistence, cytotoxicity and/or expansion. Promising future CAR designs for iNKT cells include: fifth-generation CARs with cytokine receptor domains (e.g., IL-2Rβ) linked to JAK-STAT signaling to boost proliferation, persistence, and anti-tumor activity; CARs with inducible safety features such as small molecule dimerization systems or kill switches (e.g., iCasp9); and CARs that target tumor cells via monoclonal antibodies or engager molecules. CID – Chemical Inducer of Dimerization. Image created in BioRender.
The scFv is attached to the transmembrane domain via the spacer (hinge) component which is commonly derived from amino acid sequences found in CD8, CD28, IgG1 or IgG4 (67). Although the hinge region has been less researched than the antigen binding domain or ICDs, studies indicate altering the length or flexibility of the hinge region cause significant effects on CAR-T cytotoxicity (68–70). Zhang et al., indicated by removing 2 consecutive glycine residues in a CD8 hinge, lowering the flexibility, can cause lower cytokine production of IFN-γ, IL-2, TNF-α and IL-6 in T cells (71). IgG hinges have also been associated with decreased CAR-T persistence in vivo due to their interaction with Fcγ domains therefore more origins for a variety of flexibilities and length hinges are being explored (72, 73).
The TMD anchors the CAR to the surface of the effector cell and facilitates the transfer of antigen binding signals inside the cell. Generally, CAR TMD’s are derived from type-I single spanning proteins such as CD3ζ, CD4, CD8α or CD28 (74). The TMD is the least researched component of CARs but has been shown to have effects on CAR expression level and stability. For example, CARs with TMD originating from CD8α and CD28 have been related to higher surface expression and increased CAR expression stability enhancing cytotoxic capacity (75). Though most CARs are designed as homodimers, CAR heterodimerization can also be mediated by certain TMDs such as CD3ζ TMD dimerization with endogenous TCR complexes and CD28 TMD dimerization with CD28 receptors, which can increase CAR signaling/sensitivity but potentially raise on-target off-tumor toxicity risks (76, 77).
Effective lymphocyte activation by a CAR relies on its ICDs to determine the highest cytotoxicity while maintaining safety. The first 3 generations of CAR were determined by the number of ICDs, with the original first-generation CAR intracellular domain consisting solely of a CD3ζ primary activation domain, second including one costimulatory domain and third including two costimulatory domains. The CD3ζ, derived from the TCR complex, is the crucial signaling component of the CAR providing signal 1 of activation into the cell. It contains immunoreceptor tyrosine-based activation motifs (ITAMs) that initiate downstream signaling cascades and cell activation, as a result of antigen binding to CAR. Costimulatory domains, such as CD28 and 4-1BB, provide signal 2 of activation, enhancing internal activation and improving persistence and determine the function of the CAR (78). Due to the more recent advent of iNKT cell-based therapies, 1st generation CARs have not been studied in iNKT cells, only second, third and fourth generation CARs are currently in preclinical models or clinical trials (Figure 2). Currently, CAR-iNKT cell engineering relies on conventional CAR signaling domains, due to the limited research on optimal domains specifically for iNKTs, with 4-1BB emerging as a key enhancer of cytotoxic activity (79, 80). Heczey et al., found that incorporating 4-1BB as a costimulatory domain shifted the cytokine secretion profile of iNKT cells toward a Th1-dominant response, unlike CD28 or CD3ζ (80). To support this, Poels et al. demonstrated that iNKT cells expressing 4-1BB-containing CARs specific for BCMA and CD38 exhibited a similar Th1-skewed polarization (79). Third generation CARs incorporated two costimulatory domains in series with the CD3ζ to enhance persistence, cytokine secretion, and cytotoxicity compared to second generation CARs (81). Though the results of 3rd generation CARs in iNKTs are limited, the combination of CD28 and 4-1BB has been promising in vivo with GD2 and CD19 CARs demonstrating prolonged survival and increased CAR-iNKT cell persistence and anti-tumor activity (80, 82). However, the use of other intracellular domains in NK cells and emerging in αβ T cells, such as DAP12, Dectin-1 and ICOS, may hold promise for enhancing innate-like killing, reducing exhaustion, and improving persistence (83–85).
Fourth-generation CARs also known as TRUCKs (T cells Redirected for Universal Cytokine Killing) incorporated additional DNA sequences encoding cytokines to be secreted upon activation of the cell, also known as cytokine armor. Armored CAR-iNKT cells can enhance persistence and expansion of iNKTs, boost anti-tumor activity and reprogram the TME to enhance tumor infiltration and reduce suppressive signals. IL-15 is a key enhancement for CAR-iNKTs, as evidenced by its inclusion in all CAR-iNKTs currently in clinical use, due to its well-established role in iNKT cell persistence and anti-tumor activity (86, 87). IL-21 armored B7-H3 CAR-iNKT cells demonstrate improved persistence and therapeutic efficacy in lung metastatic in vivo mouse models along with an increased percentage of CD62L-positive memory-like iNKT cells (88). A recent study also incorporated IL-12 armor into CAR-iNKT cells resulting in an increased polarization of iNKT cells to Th1 phenotype with memory features, prolonged persistence and antitumoral capability (89).
Recently, fifth-generation CARs have been developed by modifying second-generation CARs with a truncated IL-2 receptor β-chain domain featuring a binding site for the transcription factor STAT3. In this way, fifth-generation CARs, upon antigen recognition, can activate the JAK-STAT pathway, enhancing T cell expansion, survival and resistance to exhaustion, however, fifth-generation CARs are yet to be established in iNKT cells (90).
The unpredictable expansion and associated toxicities of CAR-T cells such as CRS or ICANS have driven the search for inducible elements, engineered mechanisms that allow precise control over CAR cell activation, function, or depletion in response to specific stimuli. While similar toxicities have not yet been observed in CAR-iNKT cells, their potential for extensive expansion and persistence and the utmost importance of patient safety necessitates the consideration of safety measures. Kill switches take advantage of intrinsic or extrinsic apoptotic pathways or antibody-dependent cell-mediated cytotoxicity (ADCC) and phagocytosis of remaining cell components, allowing for the rapid elimination of a therapeutic agent on demand in the instance of excessive expansion or toxicity (91). The most effective, clinically proven and widely used kill switch utilizes a modified Caspase-9 (iC9), which triggers apoptosis upon dimerization with FK506-binding proteins (FKBPs) (92). The dimerization of these proteins can be modulated with small molecule chemical inducer of dimerization (CID) agents such as AP1903 (Rimiducid), eliminating up to 90% of the peripheral blood CAR T cells within hours (93, 94). Drug-inducible activation switches, such as rapamycin-based dimerization systems, can also assemble CAR signaling domains only in the presence of a small molecule allowing regulation of activation (95). Altogether, CAR design continues to evolve to enhance safety and efficacy, with iNKT-specific optimization offering exciting potential for next-generation cell therapies.
Engineered iNKT cells: combining innate sensing with CAR-targeted precision for superior anti-tumor immunity
CAR-engineered iNKT cells offer distinct advantages over conventional CAR-T therapies by integrating CAR-mediated precision targeting with innate immunomodulatory capabilities, tumor microenvironment reprogramming, and coordinated anti-tumor immunity. Preclinical and clinical evidence increasingly highlights their distinct advantages over conventional cellular therapies, particularly in their ability to infiltrate tumors, modulate myeloid populations, and establish durable immune responses.
A fundamental limitation of conventional CAR-T therapies is their inefficient trafficking to, and infiltration of, solid tumors. Heczey et al. demonstrated that iNKT cells engineered with CARs targeting GD2 ganglioside (CAR.GD2), a disialoganglioside highly expressed on neuroblastoma and other solid tumors (96), exhibited significantly enhanced tumor infiltration compared to similarly engineered conventional T cells in neuroblastoma xenograft models (80). This enhanced tumor-homing capacity, likely attributable to the distinct chemokine receptor profiles of iNKT cells, represents a critical advantage for targeting solid malignancies where limited infiltration often undermines therapeutic efficacy.
The most distinctive feature of engineered iNKT cells is their capacity to remodel immunosuppressive tumor microenvironments into immunostimulatory ones through coordinated immunomodulatory mechanisms. In preclinical mouse studies, autologous CAR-iNKT exhibit superior anti-tumor activity compared to conventional CAR-T cells across multiple syngeneic tumor models, including subcutaneous and lung metastatic B16-OVA-hCD19 tumors and D8-mB7-H3 ovarian cancer (97). Detailed mechanistic analyses using single-cell RNA sequencing, flow cytometry, and immunohistochemistry revealed that CAR-iNKT cells selectively depleted immunosuppressive myeloid populations through CD1d-dependent elimination of M2-like macrophages, while preserving CD1d-low M1-like macrophages with pro-inflammatory functions. This selective CD1d-dependent remodeling of the myeloid compartment was also observed with TCR-engineered iNKT cells, which controlled multiple tumors expressing cognate antigen more effectively than either non-transduced iNKT cells or CD8+ T cells engineered with the same TCR (98), further validating the translational potential of engineered iNKT platforms.
Beyond TME remodeling, CAR-iNKT promoted robust epitope spreading, eliciting broad immune responses against both engineered targets and endogenous tumor neoantigens, and enhanced the expansion of T cell memory, thereby establishing durable anti-tumor immunity that persists even after clearance of the transferred cells themselves (97, 99). While CAR-iNKT directly reduced tumor burden through CAR-mediated cytotoxicity in a syngeneic murine model of B cell CD19+ lymphoma, complete anti-tumor efficacy required dendritic cell-mediated cross-priming of host CD8+ T cells (99). Similarly, next-generation IL-15-armored, FAP-targeting CAR-iNKT therapy MiNK-215, designed to target the tumor stroma, further enhanced endogenous anti-tumor activity in human organoid models of treatment-resistant colorectal cancer liver metastases (22, 100, 101). MiNK-215, depleted FAP+ hepatic stellate cells and reduced CXCL12 expression to alleviate immune suppression, enhanced secretion of chemokines (CX3CL1, CXCL9, CCL3) to promote T cell trafficking, and boosted T cell activation and cytotoxicity through increased levels of effector molecules (sCD137, CD40L, IFN-γ, GZMB, sFASL) (100). This capacity to induce epitope spreading and activate broader host immunity represents a powerful strategy to overcome tumor heterogeneity and antigen escape mechanisms, which frequently limit the efficacy of conventional CAR approaches.
iNKT cells as a universal platform for engineering safe, scalable, and effective off-the-shelf immunotherapy
Promising safety profile makes CAR-iNKT cells ideal candidates for allogeneic ‘off-the-shelf’ development (Figure 3). In xenogeneic neuroblastoma models, GD2-targeting CAR-iNKT exhibited superior safety compared to conventional GD2-CAR-T cells, with no evidence of GvHD or multiorgan toxicity despite repeated administration, underscoring their preserved non-alloreactive properties post-engineering (80). More recently, PSCA CAR_sIL-15 iNKT and T cells showed similar antitumor efficacy in humanized mice, significantly suppressing tumor growth of metastatic pancreatic ductal adenocarcinoma (PDAC). However, only CAR-T cell–treated mice developed severe GvHD, splenomegaly, and elevated CRS-related cytokines. In contrast, CAR-iNKT treatment did not cause notable GvHD or CRS, indicating a safer therapeutic profile with comparable efficacy (102). This intrinsic non-alloreactivity represents a valuable advantage for developing allogeneic therapies, as conventional allogeneic T cell approaches can require genetic modification to overcome HLA barriers, including elimination of endogenous TCRs for GvHD and HLA molecules for host-versus-graft (103, 104). Such modifications introduce substantial manufacturing challenges. TCR removal impairs ex vivo expansion by preventing expression of the CD3 complex for T cell activation, signaling, and proliferation (105), while HLA deletion can render cells susceptible to NK cell-mediated elimination (106). Furthermore, the extensive gene editing required raises regulatory concerns regarding potential off-target effects and chromosomal abnormalities (107–109). iNKT cells circumvent these fundamental limitations, requiring no genetic manipulation to prevent GvHD alloreactivity and persistence has so far been encouraging without HLA manipulation (54–58).
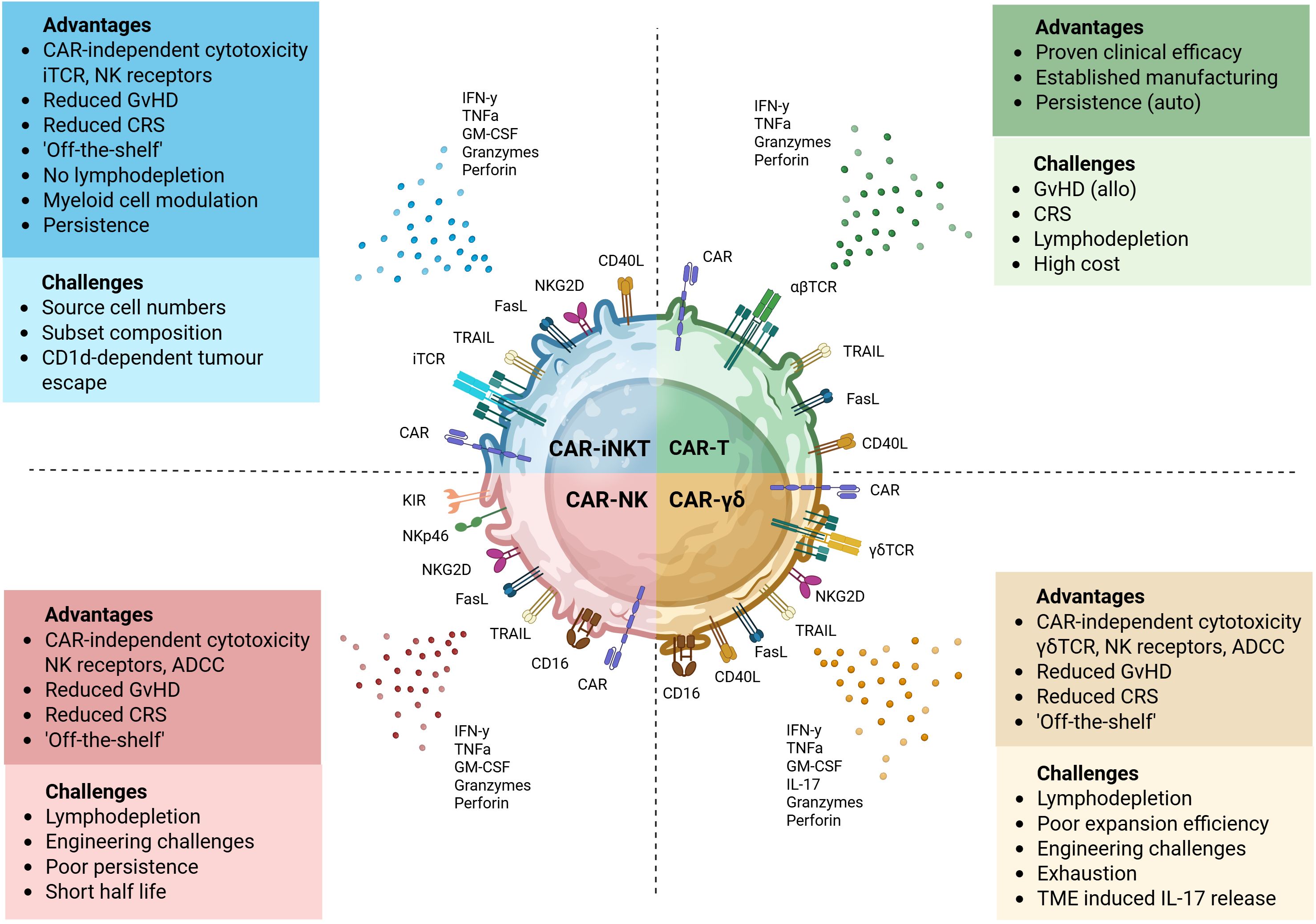
Figure 3. Key attributes of CAR-engineered lymphocyte platforms. Each CAR-engineered platform offers unique therapeutic benefits and presents specific limitations. Autologous CAR-T cells can provide potent and sustained tumor control, particularly in hematological malignancies. However, they are associated with significant safety concerns, including CRS, neurotoxicity and GvHD in allogeneic settings. CAR-iNKT, -NK and -γδ T cells offer simpler “off-the-shelf” manufacturing potential. Innate cytotoxicity of CAR-NK through activating receptors and cytokine secretion supports antigen-independent killing, but they may have reduced persistence and are difficult to engineer. CAR-iNKT cells combine adaptive and innate-like features, recognizing glycolipid antigens via their invariant TCR and mediating cytotoxicity through FasL, TRAIL, and NK receptors. They can also modulate the tumor microenvironment by promoting dendritic cell maturation and influencing macrophage phenotype. Their dual targeting capacity and lower alloreactivity make them promising. Ongoing studies are investigating how tumor-driven modulation of CD1d expression and lipid antigen presentation impairs iNKT recognition and function. In parallel, efforts aim to understand the functional differences between iNKT subsets and optimize their composition to improve the efficacy of CAR-iNKT therapies. CAR-γδT cells recognize a broad range of stress-induced or metabolic antigens independently of MHC and can mediate ADCC via Fc receptors like CD16. They can exhibit both pro- and anti-tumor activity and expansion protocols remain areas of active development. Image created in BioRender.
Complementing their inherent allogeneic-related properties, the clinical advantages of iNKT extend further to patient preparation protocols. While conventional CAR-T therapies typically require intensive lymphodepletion regimens, which limit eligibility and could impact efficacy (110, 111) and is often associated with toxicities, including leukopenia, reactive myelopoiesis, and prolonged hospitalization (112, 113), iNKT cells demonstrate remarkable persistence and function in HLA-mismatched recipients without such intensive preconditioning (114). Clinical evaluation of agenT-797 confirmed this advantage, with cells persisting up to 6 months post-infusion without lymphodepletion or HLA matching (56).
Compared to alternative “off-the-shelf” approaches utilizing γδ T cells or NK cells, iNKT offer several practical advantages (Figure 3). NK cells, while potent effectors, are challenging to transduce due to innate immune sensors that trigger apoptosis following viral transduction, and they demonstrate poor persistence in vivo (115–117). Similarly, γδ T cells face limitations including poor expansion efficiency, difficulties in genetic engineering, and susceptibility to activation-induced cell death (118–121). Collectively, these intrinsic and engineered advantages position engineered iNKT cells as uniquely suited candidates for scalable, safe, and effective next-generation cellular immunotherapies, offering compelling alternatives to conventional CAR-T, NK, and γδ T-cell strategies.
CAR-iNKT cells in action: a look at the clinical landscape
While extensive preclinical data supports the therapeutic potential of CAR-iNKT, their translation to clinical applications remains in early stages (21, 122–124). To date, only seven clinical trials evaluating CAR-iNKT have been initiated, with emerging data providing preliminary insights into their safety profile and therapeutic efficacy across both hematologic and solid malignancies (Table 2). Most use low dose IL-2 to expand CAR-iNKT cells with additional “armoring” with mainly IL-15 gene to date.
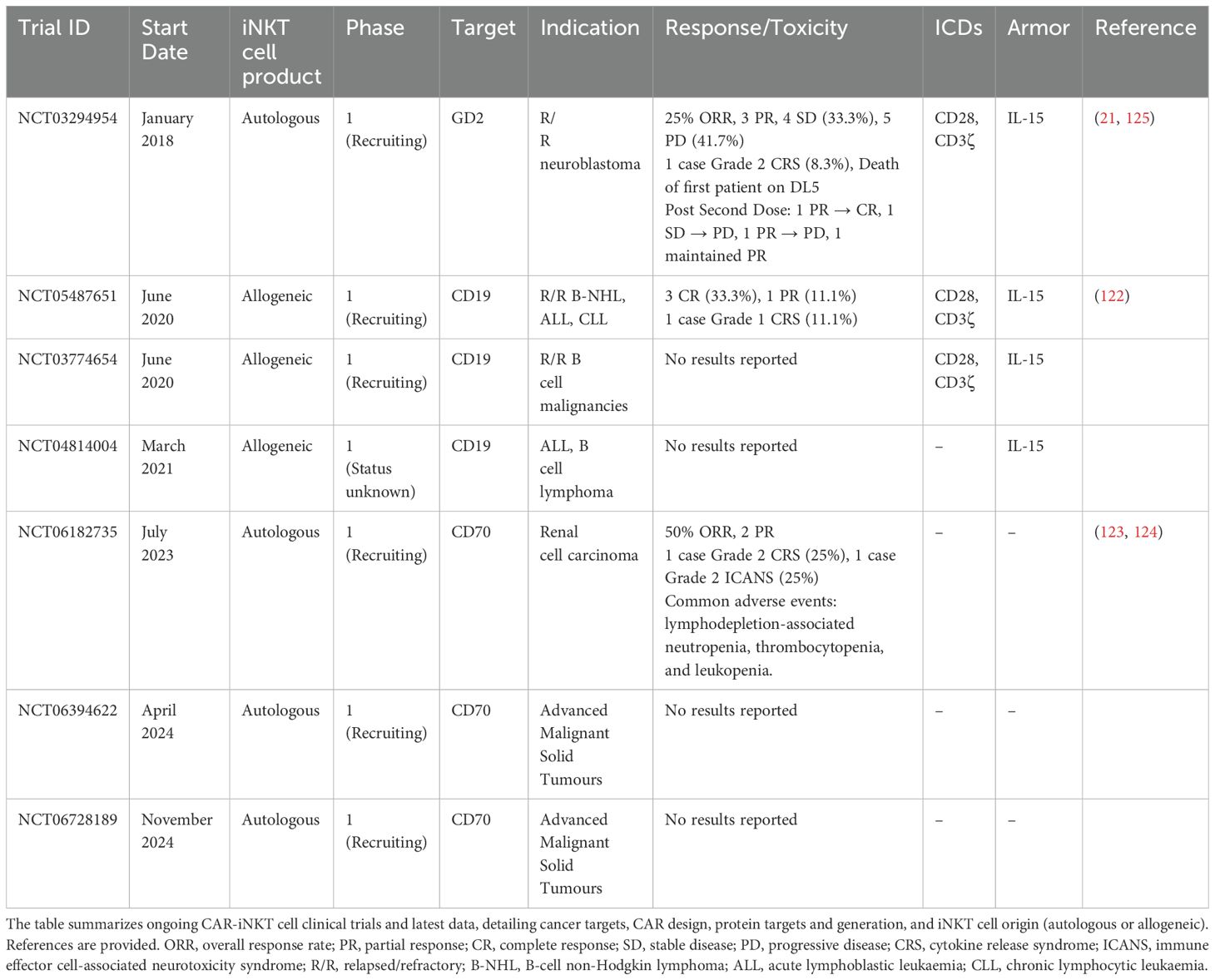
Table 2. Clinical trials of CAR-iNKT cells.
Three of the seven clinical trials target CD19-expressing hematologic malignancies. The ANCHOR trial (NCT03774654) and its follow-up ANCHOR2 (NCT05487651) are evaluating allogeneic CD19-targeting CAR-iNKTs in patients with relapsed or refractory B-cell malignancies, including B-cell non-Hodgkin lymphoma (NHL) and acute/chronic lymphocytic leukemia (ALL/CLL). These CAR-iNKTs incorporate CD28/CD3ζ intracellular signaling domains, IL-15 armoring for enhanced persistence, and express shRNAs targeting β2M and CD74 to downregulate HLA class I and II expression, respectively (122). In the phase 1 dose-escalation ANCHOR trial, nine patients (seven with NHL and two with ALL) received treatment across three dose levels. Encouraging efficacy signals were observed, particularly in the NHL cohort where three of seven patients achieved initial partial responses (PR), with two subsequently converting to complete responses (CR). Additionally, one of the two ALL patients achieved a CR with incomplete hematologic recovery. Importantly, CAR-iNKT demonstrated dose-dependent in vivo expansion, with cells detectable in peripheral blood one week post-infusion at higher dose levels. Although circulating CAR-iNKT were not detected beyond three hours in the lowest dose cohort, cells were identified in post-treatment tumor biopsies, confirming their ability to infiltrate target tissues even at low doses. The safety profile of this allogeneic product appears favorable, with only one patient experiencing mild CRS (122). A parallel approach is being evaluated in a separate trial (NCT04814004) using CD19-CAR-iNKTs armored with IL-15 in patients with acute lymphoblastic leukemia, B-cell lymphoma, or chronic lymphocytic leukemia, though results from this study have not yet been reported.
The application of CAR-iNKT to solid tumors has so far been explored in four clinical trials targeting distinct malignancies. The GINAKIT2 trial (NCT03294954) evaluated autologous GD2-CAR-iNKT incorporating CD28/CD3ζ intracellular domains and IL-15 secretion in pediatric patients with relapsed or refractory neuroblastoma. Initial results from twelve treated patients demonstrated a 25% overall response rate, including three partial responses, four cases of stable disease, and five instances of progressive disease (21). Among four patients who received a second infusion, one converted from PR to CR, one maintained PR, and two experienced disease progression. Importantly, this trial provided valuable insights into factors affecting therapeutic efficacy. Higher levels of CD62L+ CAR-iNKTs in the infused product correlated with improved patient outcomes, consistent with preclinical findings highlighting the importance of this memory-like subset for enhanced persistence and anti-tumor activity (21). However, the study faced a significant setback following a patient death, prompting temporary suspension by the FDA. Subsequent investigation attributed this adverse event to hyperleukocytosis resulting from manufacturing changes implemented to achieve higher cell doses. Analysis of the pre-infusion product revealed cell-autonomous expansion and outgrowth of NK cells (125).
More recently, autologous CD70-CAR-iNKTs (CGC729, NCT06182735) have shown promising activity in patients with relapsed and refractory metastatic renal cell carcinoma (mRCC). Preliminary results from four patients who received a single dose following lymphodepletion demonstrated an ORR of 50% (2/4) and a DCR of 75% (3/4) (123, 124). Notably, both patients with CD70-expressing RCC responded to therapy despite low antigen expression levels, suggesting that CAR-iNKT cells may effectively target tumors with limited antigen density; a significant advantage for solid tumor applications where heterogeneous and low antigen expression often limit conventional CAR-T efficacy. CGC729 exhibited favorable pharmacokinetics, with peak expansion between days 14 and 28 and persistence in circulation up to week 20. The durability of its biological activity was further evidenced by sustained reduction in peripheral CD70+ T cells through week 20. The treatment demonstrated a manageable safety profile with no dose-limiting toxicities. Most adverse events were attributable to lymphodepletion rather than the cellular product itself, primarily consisting of transient cytopenias. Only one patient experienced grade 2 CRS and grade 2 ICANS, both of which resolved rapidly with standard management (123, 124). Based on these encouraging results, CGC729 is now being evaluated in additional solid tumor indications (NCT06394622, NCT06728189), with data anticipated to further elucidate its potential across diverse malignancies.
As these clinical trials mature and additional studies commence, a more comprehensive understanding of the therapeutic potential CAR-iNKT therapy in humans will emerge. Nevertheless, These early findings already demonstrate the versatility, safety, and promising efficacy of CAR-iNKT therapies across diverse cancer indications, with significant potential to transform treatment paradigms where conventional cellular immunotherapies face significant limitations.
Challenges in the development of CAR-iNKT cell therapy
Despite the significant therapeutic potential of CAR-iNKT cells, several critical challenges must be addressed to fully realize their clinical promise. These challenges span manufacturing hurdles, functional heterogeneity considerations, and tumor microenvironment-mediated resistance mechanisms.
A fundamental challenge in developing CAR-iNKT therapies arises from the inherent scarcity of iNKT cells, which constitute only 0.01–1% of human peripheral blood lymphocytes (29, 30), necessitating extensive ex vivo expansion to generate clinically relevant doses. This challenge is particularly pronounced in autologous settings, where cancer impacts endogenous iNKT (18, 19) and patients may have undergone multiple rounds of immunosuppressive treatments that further deplete iNKT populations and compromise their functionality. Despite these limitations, several successful expansion protocols have been developed, typically involving stimulating naïve iNKT cells with α-GalCer-pulsed antigen-presenting cells or anti-CD3 antibodies, combined with supportive cytokines such as IL-2, IL-7, IL-15, or IL-21 (126). The clinical feasibility of this approach was demonstrated in the GINAKIT2 trial conducted by Heczey and colleagues, where autologous GD2 CAR-iNKT expressing IL-15 were manufactured for children with neuroblastoma. Despite the low blood volume available and low starting frequency of iNKT cells (¾0.1% of peripheral blood lymphocytes) and patients’ histories of myeloablative and lymphodepleting chemotherapy, the expansion process achieved a median of more than 60% CAR+ cells and 108 CAR+ iNKT per product, enabling repeat dosing for most patients (21, 127).
Alternative manufacturing approaches utilizing induced pluripotent stem cells (iPSCs) or hematopoietic stem or progenitor cells (HS/PC) as starting materials are being actively explored to circumvent the need for patient-derived iNKT cells (128–130). Yamada et al. successfully generated functional iNKT from iPSC, which demonstrated robust antitumor activity and enhanced NK cell responses in preclinical models (131). Additionally, universal CAR-engineered iNKT (UCAR-NKT) have been developed through ex vivo HSC differentiation, iNKT TCR engineering, HLA gene editing, producing large-scale, potent CAR-NKT populations with promising preclinical antitumor efficacy and safety profiles (130). Notably a single cord blood donor containing approximately 5 × 106 CD34+ HSPC could potentially yield an estimated 10¹² mature allogeneic CAR+ iNKT, sufficient for ~1,000+ clinical doses (132). These advances establish a robust foundation for large-scale manufacturing of engineered iNKT as an “off-the-shelf” immunotherapy platform.
The functional heterogeneity of iNKT cells represents both an opportunity and a challenge for therapeutic development. As mentioned above, iNKT can be broadly classified into distinct subsets based on CD4 expression, with CD4+ cells demonstrating polyfunctional cytokine production (GM-CSF, TNF-α, IFN-γ, IL-4, IL-2 etc.) and CD4- iNKT specializing in cytotoxicity and relatively more Th1 cytokines (133). The therapeutic relevance of these subsets was demonstrated in a xenograft model of Epstein-Barr virus-driven B-lymphoma, where CD4+ iNKT cells functioned in an adjuvant-like manner to enhance anti-tumor responses by antigen-specific T cells (134), while CD62L expression further distinguished memory-like iNKT, correlating with enhanced persistence, proliferation, and antitumor activity (135, 136). This preclinical observation was subsequently validated clinically, where the frequency of CD62L+ GD2-specific iNKT cells in pre-infusion products correlated with CAR-iNKT expansion in patients and was significantly higher in responders compared to non-responders (21). Interestingly, HS/PC-derived iNKT cells lack the CD4+ subpopulation commonly found in endogenous human iNKT, suggesting that peripheral blood-derived iNKT may serve as a more suitable starting material for generating functionally complete CAR-iNKT cell products (130, 132). Additional functionally distinct iNKT subsets include CD244+CXCR6+ iNKT with NK cell-like features, high IFN-γ and granzyme production, and enhanced anti-tumor activity (137). These cells belong exclusively to the CD4- subset, further defining functional specialization among iNKT populations (137). Optimizing the composition and functional properties of these distinct iNKT subsets represents a key opportunity to enhance both therapeutic outcomes and predictability of CAR-iNKT therapies for specific clinical applications from cancer to immune/inflammatory, infectious and other diseases.
Lastly, while CAR-iNKT cells demonstrate remarkable capabilities to reshape the TME, they remain susceptible to various immunosuppressive mechanisms that could limit CAR-iNKT therapeutic efficacy. The lipid composition within tumors can significantly alter iNKT function, with many tumors accumulating immunoregulatory lipids that skew iNKT cells towards a tolerogenic phenotype (138, 139). In hepatocellular carcinoma, accumulated long-chain acylcarnitine induced premature iNKT cell senescence and impaired responsiveness to activating stimuli (140). CD1d-mediated interactions between iNKT cells and other immune populations also shape anti-tumor responses, sometimes in unexpected ways. In colorectal cancer (CRC), the iNKT-neutrophil axis paradoxically suppresses anti-tumor immunity, with tumor-infiltrating iNKT exhibiting a pro-tumorigenic phenotype characterized by GM-CSF and IL-17 secretion and expression of exhaustion markers (PD-1, TIGIT, TIM-3) (141). In contrast, iNKT in adjacent non-tumor tissue predominantly produced IFNγ (141), highlighting the critical impact of the local microenvironment on iNKT functional polarization. Metabolic challenges further compound these issues, as tumor-derived lactic acid suppresses PPARγ expression in iNKT, impairing IFNγ production and anti-tumor function (142, 143). Will allogeneic healthy donor iNKT restore the anti-tumor balance or be eventually impacted by the TME, perhaps therefore requiring further dosing. Elucidating these mechanisms presents both challenges and opportunities for the rational design of next-generation CAR-iNKT with broad anti-tumor efficacy and persistence. Promising strategies include engineering resistance to metabolic stress, incorporation of checkpoint blockade elements, and cytokine armoring to maintain functionality within a hostile TME (144, 145).
The road ahead: opportunities and takeaways
While pre-clinical and early clinical data for CAR-iNKT demonstrate promising safety and efficacy, multiple opportunities exist to enhance their therapeutic potential and expand applications to difficult-to-treat cancers. Emerging strategies include combination therapies targeting complementary immune pathways, engineering approaches to enhance persistence and functionality, and innovative targeting strategies that address the broader TME beyond malignant cells themselves.
Strategic combinations of CAR-iNKT with checkpoint blockade and T-cell engagers (TCEs) represent a compelling opportunity to deepen clinical efficacy and overcome resistance mechanisms. Activation-induced programmed cell death protein 1 (PD-1) expression on iNKT has been documented in both mouse models (146, 147) and non-small cell lung cancer patients (148), suggesting that combining CAR-iNKT with checkpoint inhibitors could prevent exhaustion and enhance therapeutic outcomes. Pre-clinical data show that PD-1 blockade (nivolumab) enhanced CAR-iNKT proliferation, cytokine production (IL-2, TNF-α, IFN-γ, and granzyme B) and effectively reversed CAR-iNKT exhaustion upon tumor rechallenge, translating into deeper tumor control (149). Translating these findings to the clinic, a first-in-human study (NCT06251793) combining unmodified iNKT (agenT-797) with Fc-enhanced anti-CTLA-4 (botensilimab) and anti-PD-1 (balstilimab) antibodies demonstrated potent intratumoral immune activation and enhanced effector cell responses in PD-1–refractory gastrointestinal cancers (57), suggesting that combination with dual checkpoint inhibition may overcome resistance in PD-1-refractory solid tumors. Likewise, co-infusion of agenT-797 with CD3-based T cell Engagers (TCE, e.g., DLL3, HER2, Muc16 and CLDN18.2 bispecifics) broadens the therapeutic window in multiple tumor models by providing a pre-primed, co-stimulation-independent effector pool, enabling lower, and therefore safer TCE dosing (150). Optimization of sequencing and dosing will be essential to unlock the full clinical benefit of these combinations.
Long-term persistence of CAR effector cells correlates with durable clinical responses (151, 152), however tumor recurrence in preclinical models has been associated with limited CAR-iNKT cell persistence (153). While IL-15 armoring has been incorporated in current clinical trials (Table 2), emerging evidence suggests that alternative cytokine strategies may further enhance CAR-iNKT anti-tumor activity. In leukemia and neuroblastoma models, IL-12-expressing CAR-iNKT cells demonstrate superior anti-tumor efficacy compared to IL-15-expressing counterparts by promoting Th1 polarization and molecular reprogramming that enhances activation and proliferation while limiting exhaustion and generating long-lived memory iNKT cells (89). Similarly, IL-21 armoring enhances CAR-iNKT efficacy by increasing CD62L+ memory-like cell frequency and activating STAT3 signaling pathways critical for survival and effector function (88). In renal cancer xenograft models, these IL-21-armed cells achieved durable, recurrence-free tumor control without evidence of cytokine-related toxicity (88). These findings position IL-12 and IL-21 as promising armoring candidates to overcome exhaustion and enhance durability, while future approaches targeting metabolic fitness and resistance to tumor-derived immunosuppressive factors could further optimize CAR-iNKT efficacy across diverse malignancies.
Lastly, expanding targets beyond tumor cells could represent a transformative opportunity for CAR-iNKT cells to simultaneously address both malignant cells and the immunosuppressive components of the tumor microenvironment that enable immune evasion. This dual-targeting approach is exemplified by MiNK-215, an allogeneic IL-15-armored FAP-targeting CAR-iNKT therapy that demonstrated remarkable efficacy in models of treatment-resistant microsatellite-stable colorectal cancer liver metastases by eliminating FAP+ cancer-associated fibroblasts and promoting T cell infiltration and activation in tumors refractory to checkpoint blockade (22, 100, 101).
Conclusion
iNKT therapy in general and CAR-iNKT cell therapy in particular represent transformative approaches in cellular immunotherapy. The latter combines the precision targeting of chimeric antigen receptors with the unique immunomodulatory properties of iNKT. Inherent advantages of iNKT cells include their remarkable proliferative capacity without exhaustion, capability to be produced ultra-pure with mono- iTCR specificity, iTCR-specific control in vivo, non-alloreactivity, enhanced tumor infiltration capacity, cytotoxic potential, and ability to reshape the tumor microenvironment. These properties address many limitations of conventional CAR-T and other CAR+ cell approaches. Early clinical trials have demonstrated encouraging safety and efficacy across both hematologic and solid malignancies, while preclinical studies continue to reveal innovative strategies to enhance persistence, functionality, and therapeutic range. As research progresses, CAR-iNKT platforms are likely to expand beyond oncology into inflammatory, autoimmune, and age-related conditions, leveraging their versatile immunomodulatory properties. By capitalizing on the unique advantages of iNKT cells, CAR-iNKT therapy is poised to emerge as a next-generation approach in cellular immunotherapy with broad applications across human disease.
Author contributions
MN: Writing – original draft, Writing – review & editing. AC: Writing – original draft. MCP: Writing – original draft. EA-S: Writing – original draft. SA: Writing – original draft. RB: Writing – original draft. N-PR: Writing – review & editing. EC: Writing – review & editing. MAP: Writing – review & editing, Writing – original draft. DC: Writing – review & editing. ME: Writing – review & editing, Conceptualization, Data curation, Funding acquisition, Investigation, Supervision, Writing – original draft.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We would like to thank colleagues for discussions and apologize for references omitted for space reasons. The content of this manuscript has been presented in part at the Society for Immunotherapy of Cancer 37th Annual Meeting (22, 154), Society for Immunotherapy of Cancer 38th Annual Meeting (56), Society for Immunotherapy of Cancer 39th Annual Meeting (20, 150), International Cancer Immunotherapy Conference 2023 (101), American Association for Cancer Research Annual Meeting 2024 (100), American Association for Cancer Research Immuno-Oncology 2025 (58), American Society of Clinical Oncology Gastrointestinal Cancers Symposium 2025 (57).
Conflict of interest
MN, AC, MCP, EA-S, SA, RB, N-PR, EC and MAP are employees of, and current holders of stock options with MiNK Therapeutics Inc. DC is an employee of, and current holders of stock options with Agenus Inc. MAE is a founder, scientific advisor for and holds stock and stock options in MiNK Therapeutics Inc.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Laversanne M, Sung H, Ferlay J, Siegel RL, Soerjomataram I, et al. Global cancer statistics 2022: globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2024) 74:229–63. doi: 10.3322/caac.21834
2. Ugai T, Sasamoto N, Lee HY, Ando M, Song M, Tamimi RM, et al. Is early-onset cancer an emerging global epidemic? Current evidence and future implications. Nat Rev Clin Oncol. (2022) 19:656–73. doi: 10.1038/s41571-022-00672-8
3. Cha JH, Chan LC, Song MS, and Hung MC. New approaches on cancer immunotherapy. Cold Spring Harb Perspect Med. (2020) 10. doi: 10.1101/cshperspect.a036863
4. Lee A. Obecabtagene autoleucel: first approval. Mol Diagn Ther. (2025) 29:419–423. doi: 10.1007/s40291-025-00771-z
5. Zhang X, Zhu L, Zhang H, Chen S, and Xiao Y. Car-T cell therapy in hematological Malignancies: current opportunities and challenges. Front Immunol. (2022) 13:927153. doi: 10.3389/fimmu.2022.927153
6. Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of cd4(+) car T cells. Nature. (2022) 602:503–9. doi: 10.1038/s41586-021-04390-6
7. Michaelides S, Obeck H, Kechur D, Endres S, and Kobold S. Migratory engineering of T cells for cancer therapy. Vaccines (Basel). (2022) 10. doi: 10.3390/vaccines10111845
8. Liu G, Rui W, Zhao X, and Lin X. Enhancing car-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. (2021) 18:1085–95. doi: 10.1038/s41423-021-00655-2
9. Albelda SM. Car T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat Rev Clin Oncol. (2024) 21:47–66. doi: 10.1038/s41571-023-00832-4
10. Morris EC, Neelapu SS, Giavridis T, and Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
11. Brudno JN and Kochenderfer JN. Current understanding and management of car T cell-associated toxicities. Nat Rev Clin Oncol. (2024) 21:501–21. doi: 10.1038/s41571-024-00903-0
12. Elsallab M and Maus MV. Expanding access to car T cell therapies through local manufacturing. Nat Biotechnol. (2023) 41:1698–708. doi: 10.1038/s41587-023-01981-8
13. Cliff ERS, Kelkar AH, Russler-Germain DA, Tessema FA, Raymakers AJN, Feldman WB, et al. High cost of chimeric antigen receptor T-cells: challenges and solutions. Am Soc Clin Oncol Educ Book. (2023) 43:e397912. doi: 10.1200/EDBK_397912
14. Kawano T, Cui J, Koezuka Y, Toura I, Kaneko Y, Motoki K, et al. Cd1d-restricted and tcr-mediated activation of valpha14 nkt cells by glycosylceramides. Science. (1997) 278:1626–9. doi: 10.1126/science.278.5343.1626
15. Borg NA, Wun KS, Kjer-Nielsen L, Wilce MC, Pellicci DG, Koh R, et al. Cd1d-lipid-antigen recognition by the semi-invariant nkt T-cell receptor. Nature. (2007) 448:44–9. doi: 10.1038/nature05907
16. Li YR, Wilson M, and Yang L. Target tumor microenvironment by innate T cells. Front Immunol. (2022) 13:999549. doi: 10.3389/fimmu.2022.999549
17. Delfanti G, Dellabona P, Casorati G, and Fedeli M. Adoptive immunotherapy with engineered inkt cells to target cancer cells and the suppressive microenvironment. Front Med (Lausanne). (2022) 9:897750. doi: 10.3389/fmed.2022.897750
18. Wolf BJ, Choi JE, and Exley MA. Novel approaches to exploiting invariant nkt cells in cancer immunotherapy. Front Immunol. (2018) 9:384. doi: 10.3389/fimmu.2018.00384
19. Exley MA, Dellabona P, and Casorati G. Exploiting cd1-restricted T cells for clinical benefit. Mol Immunol. (2021) 132:126–31. doi: 10.1016/j.molimm.2020.12.015
20. Ibbett P, Dijk MV, Chantzoura E, Antonopoulos G, Sans GR, Niedzielska M, et al. 374 prame-tcr inkt cell therapy: opportunity for best-in-class off-the-shelf solid tumor therapy targeting prame. J ImmunoTher Cancer. (2024) 12:A430. doi: 10.1136/jitc-2024-SITC2024.0374
21. Heczey A, Xu X, Courtney AN, Tian G, Barragan GA, Guo L, et al. Anti-gd2 car-nkt cells in relapsed or refractory neuroblastoma: updated phase 1 trial interim results. Nat Med. (2023) 29:1379–88. doi: 10.1038/s41591-023-02363-y
22. Michelet X, Popis M, Boi S, Niedzielska M, Masakayan R, Sharoni EA, et al. 358 development of an allogenic fap car inkt product to target tumor stroma and modulate the tumor microenvironment. J ImmunoTher Cancer. (2022) 10:A377. doi: 10.1136/jitc-2022-SITC2022.0358
23. Look A, Burns D, Tews I, Roghanian A, and Mansour S. Towards a better understanding of human inkt cell subpopulations for improved clinical outcomes. Front Immunol. (2023) 14:1176724. doi: 10.3389/fimmu.2023.1176724
24. Rossjohn J, Pellicci DG, Patel O, Gapin L, and Godfrey DI. Recognition of cd1d-restricted antigens by natural killer T cells. Nat Rev Immunol. (2012) 12:845–57. doi: 10.1038/nri3328
25. Wieland Brown LC, Penaranda C, Kashyap PC, Williams BB, Clardy J, Kronenberg M, et al. Production of alpha-galactosylceramide by a prominent member of the human gut microbiota. PloS Biol. (2013) 11:e1001610. doi: 10.1371/journal.pbio.1001610
26. Hosono Y, Tomiyasu N, Kasai H, Ishikawa E, Takahashi M, Imamura A, et al. Identification of alpha-galactosylceramide as an endogenous mammalian antigen for inkt cells. J Exp Med. (2025) 222. doi: 10.1084/jem.20240728
27. Venkataswamy MM and Porcelli SA. Lipid and glycolipid antigens of cd1d-restricted natural killer T cells. Semin Immunol. (2010) 22:68–78. doi: 10.1016/j.smim.2009.10.003
28. Kronenberg M. When less is more: T lymphocyte populations with restricted antigen receptor diversity. J Immunol. (2014) 193:975–6. doi: 10.4049/jimmunol.1401491
29. Montoya CJ, Pollard D, Martinson J, Kumari K, Wasserfall C, Mulder CB, et al. Characterization of human invariant natural killer T subsets in health and disease using a novel invariant natural killer T cell-clonotypic monoclonal antibody, 6b11. Immunology. (2007) 122:1–14. doi: 10.1111/j.1365-2567.2007.02647.x
30. Gumperz JE, Miyake S, Yamamura T, and Brenner MB. Functionally distinct subsets of cd1d-restricted natural killer T cells revealed by cd1d tetramer staining. J Exp Med. (2002) 195:625–36. doi: 10.1084/jem.20011786
31. Yang D and Chertov O. Oppenheim JJ. Participation of mammalian defensins and cathelicidins in anti-microbial immunity: receptors and activities of human defensins and cathelicidin (Ll-37). J Leukoc Biol. (2001) 69:691–7. doi: 10.1189/jlb.69.5.691
32. Kim CH, Johnston B, and Butcher EC. Trafficking machinery of nkt cells: shared and differential chemokine receptor expression among V alpha 24(+)V beta 11(+) nkt cell subsets with distinct cytokine-producing capacity. Blood. (2002) 100:11–6. doi: 10.1182/blood-2001-12-0196
33. Godiska R, Chantry D, Raport CJ, Sozzani S, Allavena P, Leviten D, et al. Human macrophage-derived chemokine (Mdc), a novel chemoattractant for monocytes, monocyte-derived dendritic cells, and natural killer cells. J Exp Med. (1997) 185:1595–604. doi: 10.1084/jem.185.9.1595
34. Lee PT, Benlagha K, Teyton L, and Bendelac A. Distinct functional lineages of human V(Alpha)24 natural killer T cells. J Exp Med. (2002) 195:637–41. doi: 10.1084/jem.20011908
35. Snyder-Cappione JE, Tincati C, Eccles-James IG, Cappione AJ, Ndhlovu LC, Koth LL, et al. A comprehensive ex vivo functional analysis of human nkt cells reveals production of mip1-alpha and mip1-beta, a lack of il-17, and a th1-bias in males. PloS One. (2010) 5:e15412. doi: 10.1371/journal.pone.0015412
36. Tachibana T, Onodera H, Tsuruyama T, Mori A, Nagayama S, Hiai H, et al. Increased intratumor valpha24-positive natural killer T cells: A prognostic factor for primary colorectal carcinomas. Clin Cancer Res. (2005) 11:7322–7. doi: 10.1158/1078-0432.CCR-05-0877
37. Tian C, Wang Y, Su M, Huang Y, Zhang Y, Dou J, et al. Motility and tumor infiltration are key aspects of invariant natural killer T cell anti-tumor function. Nat Commun. (2024) 15:1213. doi: 10.1038/s41467-024-45208-z
38. Nowak M, Arredouani MS, Tun-Kyi A, Schmidt-Wolf I, Sanda MG, Balk SP, et al. Defective nkt cell activation by cd1d+ Tramp prostate tumor cells is corrected by interleukin-12 with alpha-galactosylceramide. PloS One. (2010) 5:e11311. doi: 10.1371/journal.pone.0011311
39. Stolk D, van der Vliet HJ, de Gruijl TD, van Kooyk Y, and Exley MA. Positive & Negative roles of innate effector cells in controlling cancer progression. Front Immunol. (2018) 9:1990. doi: 10.3389/fimmu.2018.01990
40. Waldowska M, Bojarska-Junak A, and Rolinski J. A brief review of clinical trials involving manipulation of invariant nkt cells as a promising approach in future cancer therapies. Cent Eur J Immunol. (2017) 42:181–95. doi: 10.5114/ceji.2017.69361
41. Nowak M and Schmidt-Wolf IG. Natural killer T cells subsets in cancer, functional defects in prostate cancer and implications for immunotherapy. Cancers (Basel). (2011) 3:3661–75. doi: 10.3390/cancers3033661
42. Nair S and Dhodapkar MV. Natural killer T cells in cancer immunotherapy. Front Immunol. (2017) 8:1178. doi: 10.3389/fimmu.2017.01178
43. Song L, Asgharzadeh S, Salo J, Engell K, Wu HW, Sposto R, et al. Valpha24-invariant nkt cells mediate antitumor activity via killing of tumor-associated macrophages. J Clin Invest. (2009) 119:1524–36. doi: 10.1172/JCI37869
44. Metelitsa LS. Anti-tumor potential of type-I nkt cells against cd1d-positive and cd1d-negative tumors in humans. Clin Immunol. (2011) 140:119–29. doi: 10.1016/j.clim.2010.10.005
45. Alhamawi RM, Aloufi N, Alamri AF, Altubayli FA, Alsairi RT, Alhamad RA, et al. Prognostic impact of invariant natural killer T cells in solid and hematological tumors; systematic review and meta-analysis. Cancer biomark. (2024) 41:155–64. doi: 10.3233/CBM-240069
46. Motohashi S, Ishikawa A, Ishikawa E, Otsuji M, Iizasa T, Hanaoka H, et al. A phase I study of in vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin Cancer Res. (2006) 12:6079–86. doi: 10.1158/1078-0432.CCR-06-0114
47. Kunii N, Horiguchi S, Motohashi S, Yamamoto H, Ueno N, Yamamoto S, et al. Combination therapy of in vitro-expanded natural killer T cells and alpha-galactosylceramide-pulsed antigen-presenting cells in patients with recurrent head and neck carcinoma. Cancer Sci. (2009) 100:1092–8. doi: 10.1111/j.1349-7006.2009.01135.x
48. Yamasaki K, Horiguchi S, Kurosaki M, Kunii N, Nagato K, Hanaoka H, et al. Induction of nkt cell-specific immune responses in cancer tissues after nkt cell-targeted adoptive immunotherapy. Clin Immunol. (2011) 138:255–65. doi: 10.1016/j.clim.2010.11.014
49. Exley MA, Friedlander P, Alatrakchi N, Vriend L, Yue S, Sasada T, et al. Adoptive transfer of invariant nkt cells as immunotherapy for advanced melanoma: A phase I clinical trial. Clin Cancer Res. (2017) 23:3510–9. doi: 10.1158/1078-0432.CCR-16-0600
50. Gao Y, Guo J, Bao X, Xiong F, Ma Y, Tan B, et al. Adoptive transfer of autologous invariant natural killer T cells as immunotherapy for advanced hepatocellular carcinoma: A phase I clinical trial. Oncologist. (2021) 26:e1919–e30. doi: 10.1002/onco.13899
51. Guo J, Bao X, Liu F, Guo J, Wu Y, Xiong F, et al. Efficacy of invariant natural killer T cell infusion plus transarterial embolization vs transarterial embolization alone for hepatocellular carcinoma patients: A phase 2 randomized clinical trial. J Hepatocell Carcinoma. (2023) 10:1379–88. doi: 10.2147/JHC.S416933
52. Wang J, Cheng X, Jin Y, Xia B, Qin R, Zhang W, et al. Safety and clinical response to combined immunotherapy with autologous inkt cells and pd-1(+)Cd8(+) T cells in patients failing first-line chemotherapy in stage iv pancreatic cancer. Cancer Res Commun. (2023) 3:991–1003. doi: 10.1158/2767-9764.CRC-23-0137
53. Cheng X, Wang J, Qiu C, Jin Y, Xia B, Qin R, et al. Feasibility of inkt cell and pd-1+Cd8+ T cell-based immunotherapy in patients with lung adenocarcinoma: preliminary results of a phase I/ii clinical trial. Clin Immunol. (2022) 238:108992. doi: 10.1016/j.clim.2022.108992
54. Hammond TC, Purbhoo MA, Kadel S, Ritz J, Nikiforow S, Daley H, et al. A phase 1/2 clinical trial of invariant natural killer T cell therapy in moderate-severe acute respiratory distress syndrome. Nat Commun. (2024) 15:974. doi: 10.1038/s41467-024-44905-z
55. Hadfield MJ, Safran H, Purbhoo MA, Grossman JE, Buell JS, and Carneiro BA. Overcoming resistance to programmed cell death protein 1 (Pd-1) blockade with allogeneic invariant natural killer T-cells (Inkt). Oncogene. (2024) 43:758–62. doi: 10.1038/s41388-024-02948-y
56. Moon J, Ryu S, Ramos IR, Garmezy B, Hamm J, Carneiro B, et al. 735 peripheral and tissue persistence of agent-797, an allogeneic inkt cell-based cell therapy for the treatment of cancer. J ImmunoTher Cancer. (2023) 11:A828. doi: 10.1136/jitc-2023-SITC2023.0735
57. Cytryn SL, Maron SB, and Janjigian YY. A phase ii study of agent-797 (Invariant natural killer T-cells), botensilimab (Fc-enhanced ctla-4 inhibitor) and balstilimab (Anti-pd-1) in patients with advanced, refractory gastroesophageal adenocarcinoma. J Clin Oncol. (2025) 43:TPS515–TPS. doi: 10.1200/JCO.2025.43.4_suppl.TPS515
58. Cytryn SL, Evans P, Song J, Hieronymi S, Segal M, Tsai C, et al. Biomarker Analysis from Phase 2 Study of Agent-797 (Invariant Natural Killer T-Cells), Botensilimab (a Fc-Enhanced Ctla-4 Inhibitor) with Balstilimab (Anti-Pd-1) in Pd-1 Refractory Gastroesophageal Cancer (Gec). Los Angeles, CA: American Association for Cancer Research Immuno-Oncology (2025).
59. Mitra A, Barua A, Huang L, Ganguly S, Feng Q, and He B. From bench to bedside: the history and progress of car T cell therapy. Front Immunol. (2023) 14:1188049. doi: 10.3389/fimmu.2023.1188049
60. Hossian A, Hackett CS, Brentjens RJ, and Rafiq S. Multipurposing cars: same engine, different vehicles. Mol Ther. (2022) 30:1381–95. doi: 10.1016/j.ymthe.2022.02.012
61. Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity gd2-specific car T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. (2018) 6:36–46. doi: 10.1158/2326-6066.CIR-17-0211
62. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing erbb2. Mol Ther. (2010) 18:843–51. doi: 10.1038/mt.2010.24
63. Han X, Wang Y, Wei J, and Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. (2019) 12:128. doi: 10.1186/s13045-019-0813-7
64. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. (2015) 3:356–67. doi: 10.1158/2326-6066.CIR-14-0186
65. Arcangeli S, Rotiroti MC, Bardelli M, Simonelli L, Magnani CF, Biondi A, et al. Balance of anti-cd123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol Ther. (2017) 25:1933–45. doi: 10.1016/j.ymthe.2017.04.017
66. Shen Z, Yan H, Zhang Y, Mernaugh RL, and Zeng X. Engineering peptide linkers for scfv immunosensors. Anal Chem. (2008) 80:1910–7. doi: 10.1021/ac7018624
67. Zhang X, Zhang H, Lan H, Wu J, and Xiao Y. Car-T cell therapy in multiple myeloma: current limitations and potential strategies. Front Immunol. (2023) 14:1101495. doi: 10.3389/fimmu.2023.1101495
68. Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated muc1: the evolution of a chimeric antigen receptor. J Immunol. (2008) 180:4901–9. doi: 10.4049/jimmunol.180.7.4901
69. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. (2015) 3:125–35. doi: 10.1158/2326-6066.CIR-14-0127
70. Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ror1-specific chimeric antigen receptor T cells. Clin Cancer Res. (2013) 19:3153–64. doi: 10.1158/1078-0432.CCR-13-0330
71. Zhang A, Sun Y, Du J, Dong Y, Pang H, Ma L, et al. Reducing hinge flexibility of car-T cells prolongs survival in vivo with low cytokines release. Front Immunol. (2021) 12:724211. doi: 10.3389/fimmu.2021.724211
72. Hombach A, Hombach AA, and Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the igg1 fc ‘Spacer’ Domain in the extracellular moiety of chimeric antigen receptors avoids ‘Off-target’ Activation and unintended initiation of an innate immune response. Gene Ther. (2010) 17:1206–13. doi: 10.1038/gt.2010.91
73. Almasbak H, Walseng E, Kristian A, Myhre MR, Suso EM, Munthe LA, et al. Inclusion of an igg1-fc spacer abrogates efficacy of cd19 car T cells in a xenograft mouse model. Gene Ther. (2015) 22:391–403. doi: 10.1038/gt.2015.4
74. Mazinani M and Rahbarizadeh F. Car-T cell potency: from structural elements to vector backbone components. biomark Res. (2022) 10:70. doi: 10.1186/s40364-022-00417-w
75. Zhang T, Wu MR, and Sentman CL. An nkp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. (2012) 189:2290–9. doi: 10.4049/jimmunol.1103495
76. Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, et al. The cd28-transmembrane domain mediates chimeric antigen receptor heterodimerization with cd28. Front Immunol. (2021) 12:639818. doi: 10.3389/fimmu.2021.639818
77. Degirmencay A, Thomas S, Holler A, Burgess S, Morris EC, and Stauss HJ. Exploitation of cd3zeta to enhance tcr expression levels and antigen-specific T cell function. Front Immunol. (2024) 15:1386132. doi: 10.3389/fimmu.2024.1386132
78. van der Stegen SJ, Hamieh M, and Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. (2015) 14:499–509. doi: 10.1038/nrd4597
79. Poels R, Drent E, Lameris R, Katsarou A, Themeli M, van der Vliet HJ, et al. Preclinical evaluation of invariant natural killer T cells modified with cd38 or bcma chimeric antigen receptors for multiple myeloma. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22031096
80. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant nkt cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood. (2014) 124:2824–33. doi: 10.1182/blood-2013-11-541235
81. Zheng Z, Li S, Liu M, Chen C, Zhang L, and Zhou D. Fine-tuning through generations: advances in structure and production of car-T therapy. Cancers (Basel). (2023) 15. doi: 10.3390/cancers15133476
82. Rotolo A, Caputo V, and Karadimitris A. The prospects and promise of chimeric antigen receptor immunotherapy in multiple myeloma. Br J Haematol. (2016) 173:350–64. doi: 10.1111/bjh.13976
83. Topfer K, Cartellieri M, Michen S, Wiedemuth R, Muller N, Lindemann D, et al. Dap12-based activating chimeric antigen receptor for nk cell tumor immunotherapy. J Immunol. (2015) 194:3201–12. doi: 10.4049/jimmunol.1400330
84. Liang X, Huang Y, Li D, Yang X, Jiang L, Zhou W, et al. Distinct functions of car-T cells possessing a dectin-1 intracellular signaling domain. Gene Ther. (2023) 30:411–20. doi: 10.1038/s41434-021-00257-7
85. Guedan S, Posey AD Jr., Shaw C, Wing A, Da T, Patel PR, et al. Enhancing car T cell persistence through icos and 4-1bb costimulation. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.96976
86. Matsuda JL, Gapin L, Sidobre S, Kieper WC, Tan JT, Ceredig R, et al. Homeostasis of V alpha 14i nkt cells. Nat Immunol. (2002) 3:966–74. doi: 10.1038/ni837
87. Liu D, Song L, Wei J, Courtney AN, Gao X, Marinova E, et al. Il-15 protects nkt cells from inhibition by tumor-associated macrophages and enhances antimetastatic activity. J Clin Invest. (2012) 122:2221–33. doi: 10.1172/JCI59535
88. Liu Y, Dang Y, Zhang C, Liu L, Cai W, Li L, et al. Il-21-armored B7h3 car-inkt cells exert potent antitumor effects. iScience. (2024) 27:108597. doi: 10.1016/j.isci.2023.108597
89. Landoni E, Woodcock MG, Barragan G, Casirati G, Cinella V, Stucchi S, et al. Il-12 reprograms car-expressing natural killer T cells to long-lived th1-polarized cells with potent antitumor activity. Nat Commun. (2024) 15:89. doi: 10.1038/s41467-023-44310-y
90. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, and Kobold S. Teaching an old dog new tricks: next-generation car T cells. Br J Cancer. (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
91. Glowacki P and Rieske P. Application and design of switches used in car. Cells. (2022) 11. doi: 10.3390/cells11121910
92. Straathof KC, Pule MA, Yotnda P, Dotti G, Vanin EF, Brenner MK, et al. An inducible caspase 9 safety switch for T-cell therapy. Blood. (2005) 105:4247–54. doi: 10.1182/blood-2004-11-4564
93. Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering cd19-specific T lymphocytes with interleukin-15 and a suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. (2010) 24:1160–70. doi: 10.1038/leu.2010.75
94. Budde LE, Berger C, Lin Y, Wang J, Lin X, Frayo SE, et al. Combining a cd20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PloS One. (2013) 8:e82742. doi: 10.1371/journal.pone.0082742
95. Leung WH, Gay J, Martin U, Garrett TE, Horton HM, Certo MT, et al. Sensitive and adaptable pharmacological control of car T cells through extracellular receptor dimerization. JCI Insight. (2019) 5. doi: 10.1172/jci.insight.124430
96. Nazha B, Inal C, and Owonikoko TK. Disialoganglioside gd2 expression in solid tumors and role as a target for cancer therapy. Front Oncol. (2020) 10:1000. doi: 10.3389/fonc.2020.01000
97. Zhou X, Wang Y, Dou Z, Delfanti G, Tsahouridis O, Pellegry CM, et al. Car-redirected natural killer T cells demonstrate superior antitumor activity to car-T cells through multimodal cd1d-dependent mechanisms. Nat Cancer. (2024) 5:1607–21. doi: 10.1038/s43018-024-00830-0
98. Delfanti G, Cortesi F, Perini A, Antonini G, Azzimonti L, de Lalla C, et al. Tcr-engineered inkt cells induce robust antitumor response by dual targeting cancer and suppressive myeloid cells. Sci Immunol. (2022) 7:eabn6563. doi: 10.1126/sciimmunol.abn6563
99. Simonetta F, Lohmeyer JK, Hirai T, Maas-Bauer K, Alvarez M, Wenokur AS, et al. Allogeneic car invariant natural killer T cells exert potent antitumor effects through host cd8 T-cell cross-priming. Clin Cancer Res. (2021) 27:6054–64. doi: 10.1158/1078-0432.CCR-21-1329
100. Krishnan S, Joshi B, Keith J, Frazier R, Kalinowska B, Choi JS, et al. Abstract 1331: mink-215, an il-15 armored fap-targeting car inkt cell therapy, effectively treats human organoid models of treatment-refractory mss colorectal cancer (Crc) liver metastases. Cancer Res. (2024) 84:1331. doi: 10.1158/1538-7445.AM2024-1331
101. Boi S, Popis M, Niedzielska M, Masakayan R, Altman-Sharoni E, Venkatraman V, et al. (2023). Counteracting immune suppression and enhancing cytotoxic T cell activity with allogeneic invariant natural killer T (Inkt) cells expressing an armored car targeting fibroblast activation protein in an advanced solid tumor model system, in: International Cancer Immunotherapy Conference, Milan, Italy. CRI-ENCI-AACR 7th International Cancer Immunotherapy Conference (Milano, 2023). Book of abstracts: Poster P537, page 703
102. Dai Z, Zhu Z, Li Z, Tian L, Teng KY, Chen H, et al. Off-the-shelf invariant nkt cells expressing anti-psca car and il-15 promote pancreatic cancer regression in mice. J Clin Invest. (2025) 135. doi: 10.1172/JCI179014
103. Lv Z, Luo F, and Chu Y. Strategies for overcoming bottlenecks in allogeneic car-T cell therapy. Front Immunol. (2023) 14:1199145. doi: 10.3389/fimmu.2023.1199145
104. Depil S, Duchateau P, Grupp SA, Mufti G, and Poirot L. ‘Off-the-shelf’ Allogeneic car T cells: development and challenges. Nat Rev Drug Discov. (2020) 19:185–99. doi: 10.1038/s41573-019-0051-2
105. Galetto R, Lebuhotel C, Poirot L, Gouble A, Toribio ML, Smith J, et al. Pre-tcralpha supports cd3-dependent reactivation and expansion of tcralpha-deficient primary human T-cells. Mol Ther Methods Clin Dev. (2014) 1:14021. doi: 10.1038/mtm.2014.21
106. Wang B, Iriguchi S, Waseda M, Ueda N, Ueda T, Xu H, et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat BioMed Eng. (2021) 5:429–40. doi: 10.1038/s41551-021-00730-z
107. Guo C, Ma X, Gao F, and Guo Y. Off-target effects in crispr/cas9 gene editing. Front Bioeng Biotechnol. (2023) 11:1143157. doi: 10.3389/fbioe.2023.1143157
108. Aparicio C, Acebal C, and Gonzalez-Vallinas M. Current approaches to develop “Off-the-shelf” Chimeric antigen receptor (Car)-T cells for cancer treatment: A systematic review. Exp Hematol Oncol. (2023) 12:73. doi: 10.1186/s40164-023-00435-w
109. Sasu BJ, Opiteck GJ, Gopalakrishnan S, Kaimal V, Furmanak T, Huang D, et al. Detection of chromosomal alteration after infusion of gene-edited allogeneic car T cells. Mol Ther. (2023) 31:676–85. doi: 10.1016/j.ymthe.2022.12.004
110. Jain MD, Zhao H, Wang X, Atkins R, Menges M, Reid K, et al. Tumor interferon signaling and suppressive myeloid cells are associated with car T-cell failure in large B-cell lymphoma. Blood. (2021) 137:2621–33. doi: 10.1182/blood.2020007445
111. Innamarato P, Kodumudi K, Asby S, Schachner B, Hall M, Mackay A, et al. Reactive myelopoiesis triggered by lymphodepleting chemotherapy limits the efficacy of adoptive T cell therapy. Mol Ther. (2020) 28:2252–70. doi: 10.1016/j.ymthe.2020.06.025
112. Bechman N and Maher J. Lymphodepletion strategies to potentiate adoptive T-cell immunotherapy - what are we doing; where are we going? Expert Opin Biol Ther. (2021) 21:627–37. doi: 10.1080/14712598.2021.1857361
113. Lickefett B, Chu L, Ortiz-Maldonado V, Warmuth L, Barba P, Doglio M, et al. Lymphodepletion - an essential but undervalued part of the chimeric antigen receptor T-cell therapy cycle. Front Immunol. (2023) 14:1303935. doi: 10.3389/fimmu.2023.1303935
114. Rotolo A, Whelan EC, Atherton MJ, Kulikovskaya I, Jarocha D, Fraietta JA, et al. Unedited allogeneic inkt cells show extended persistence in mhc-mismatched canine recipients. Cell Rep Med. (2023) 4:101241. doi: 10.1016/j.xcrm.2023.101241
115. Peng L, Sferruzza G, Yang L, Zhou L, and Chen S. Car-T and car-nk as cellular cancer immunotherapy for solid tumors. Cell Mol Immunol. (2024) 21:1089–108. doi: 10.1038/s41423-024-01207-0
116. Ruppel KE, Fricke S, Kohl U, and Schmiedel D. Taking lessons from car-T cells and going beyond: tailoring design and signaling for car-nk cells in cancer therapy. Front Immunol. (2022) 13:822298. doi: 10.3389/fimmu.2022.822298
117. McErlean EM and McCarthy HO. Non-viral approaches in car-nk cell engineering: connecting natural killer cell biology and gene delivery. J Nanobiotechnol. (2024) 22:552. doi: 10.1186/s12951-024-02746-4
118. Tsiverioti CA, Gottschlich A, Trefny M, Theurich S, Anders HJ, Kroiss M, et al. Beyond car T cells: exploring alternative cell sources for car-like cellular therapies. Biol Chem. (2024) 405:485–515. doi: 10.1515/hsz-2023-0317
119. Yan W, Dunmall LSC, Lemoine NR, Wang Y, Wang Y, and Wang P. The capability of heterogeneous gammadelta T cells in cancer treatment. Front Immunol. (2023) 14:1285801. doi: 10.3389/fimmu.2023.1285801
120. Yuan M, Wang W, Hawes I, Han J, Yao Z, and Bertaina A. Advancements in gammadeltat cell engineering: paving the way for enhanced cancer immunotherapy. Front Immunol. (2024) 15:1360237. doi: 10.3389/fimmu.2024.1360237
121. Ganapathy T, Radhakrishnan R, Sakshi S, and Martin S. Car gammadelta T cells for cancer immunotherapy. Is the field more yellow than green? Cancer Immunol Immunother. (2023) 72:277–86. doi: 10.1007/s00262-022-03260-y
122. Ramos CA, Courtney AN, Lulla PD, Hill LC, Kamble RT, Carrum G, et al. Off-the-shelf cd19-specific car-nkt cells in patients with relapsed or refractory B-cell Malignancies. Transplant Cell Ther. (2024) 30:S41–S2. doi: 10.1016/j.jtct.2023.12.072
123. Lin Y, Zhang Y, He S, and Ye D. Interim safety and efficacy of anti-cd70 car-nkt (Cgc729) for patients with refractory metastatic renal cell carcinoma. Mol Ther. (2024) 32:1–17. doi: 10.1016/j.ymthe.2024.04.021
124. Wang Y, Wang S, and Li N. Accelerating the clinical translation of cd70-targeted chimeric antigen receptor-based cell therapies in oncology: A comprehensive clinical investigation panorama analysis based on the trialtrove database. Cancer Lett. (2025) 614:217510. doi: 10.1016/j.canlet.2025.217510
125. Tian G, Courtney AN, Yu H, Bhar S, Xu X, Barragan GA, et al. Hyperleukocytosis in a neuroblastoma patient after treatment with natural killer T cells expressing a gd2-specific chimeric antigen receptor and il-15. J Immunother Cancer. (2025) 13. doi: 10.1136/jitc-2024-010156
126. Takami M and Motohashi S. Comparative assessment of autologous and allogeneic inkt cell transfer in inkt cell-based immunotherapy. Front Immunol. (2024) 15:1457771. doi: 10.3389/fimmu.2024.1457771
127. Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-gd2 car-nkt cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med. (2020) 26:1686–90. doi: 10.1038/s41591-020-1074-2
128. Fang M, Allen A, Luo C, and Finn JD. Unlocking the potential of ipsc-derived immune cells: engineering ink and it cells for cutting-edge immunotherapy. Front Immunol. (2024) 15:1457629. doi: 10.3389/fimmu.2024.1457629
129. Li YR, Zhou Y, Yu J, Kim YJ, Li M, Lee D, et al. Generation of allogeneic car-nkt cells from hematopoietic stem and progenitor cells using a clinically guided culture method. Nat Biotechnol. (2025) 43:329–44. doi: 10.1038/s41587-024-02226-y
130. Li YR, Zhou Y, Yu J, Zhu Y, Lee D, Zhu E, et al. Engineering allorejection-resistant car-nkt cells from hematopoietic stem cells for off-the-shelf cancer immunotherapy. Mol Ther. (2024) 32:1849–74. doi: 10.1016/j.ymthe.2024.04.005
131. Yamada D, Iyoda T, Vizcardo R, Shimizu K, Sato Y, Endo TA, et al. Efficient regeneration of human valpha24(+) invariant natural killer T cells and their anti-tumor activity in vivo. Stem Cells. (2016) 34:2852–60. doi: 10.1002/stem.2465
132. Li YR, Fang Y, Niu S, Zhu Y, Chen Y, Lyu Z, et al. Allogeneic cd33-directed car-nkt cells for the treatment of bone marrow-resident myeloid Malignancies. Nat Commun. (2025) 16:1248. doi: 10.1038/s41467-025-56270-6
133. Bharadwaj NS and Gumperz JE. Harnessing invariant natural killer T cells to control pathological inflammation. Front Immunol. (2022) 13:998378. doi: 10.3389/fimmu.2022.998378
134. Baiu DC, Sharma A, Schehr JL, Basu J, Smith KA, Ohashi M, et al. Human cd4(+) inkt cell adoptive immunotherapy induces anti-tumour responses against cd1d-negative ebv-driven B lymphoma. Immunology. (2024) 172:627–40. doi: 10.1111/imm.13799
135. Kasuya H, Zhang H, Ito Y, Yoshikawa T, Nakashima T, Li Y, et al. High cd62l expression predicts the generation of chimeric antigen receptor T cells with potent effector functions. Int Immunol. (2024) 36:353–64. doi: 10.1093/intimm/dxae015
136. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. Cd62l+ Nkt cells have prolonged persistence and antitumor activity in vivo. J Clin Invest. (2016) 126:2341–55. doi: 10.1172/JCI83476
137. Cui G, Shimba A, Jin J, Ogawa T, Muramoto Y, Miyachi H, et al. A circulating subset of inkt cells mediates antitumor and antiviral immunity. Sci Immunol. (2022) 7:eabj8760. doi: 10.1126/sciimmunol.abj8760
138. Tiwary S, Berzofsky JA, and Terabe M. Altered lipid tumor environment and its potential effects on nkt cell function in tumor immunity. Front Immunol. (2019) 10:2187. doi: 10.3389/fimmu.2019.02187
139. Lee MS and Webb TJ. Novel lipid antigens for nkt cells in cancer. Front Immunol. (2023) 14:1173375. doi: 10.3389/fimmu.2023.1173375
140. Cheng X, Tan X, Wang W, Zhang Z, Zhu R, Wu M, et al. Long-chain acylcarnitines induce senescence of invariant natural killer T cells in hepatocellular carcinoma. Cancer Res. (2023) 83:582–94. doi: 10.1158/0008-5472.CAN-22-2273
141. Lattanzi G, Strati F, Diaz-Basabe A, Perillo F, Amoroso C, Protti G, et al. Inkt cell-neutrophil crosstalk promotes colorectal cancer pathogenesis. Mucosal Immunol. (2023) 16:326–40. doi: 10.1016/j.mucimm.2023.03.006
142. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. (2021) 20:28. doi: 10.1186/s12943-021-01316-8
143. Fu S, He K, Tian C, Sun H, Zhu C, Bai S, et al. Impaired lipid biosynthesis hinders anti-tumor efficacy of intratumoral inkt cells. Nat Commun. (2020) 11:438. doi: 10.1038/s41467-020-14332-x
144. Tang L, Pan S, Wei X, Xu X, and Wei Q. Arming car-T cells with cytokines and more: innovations in the fourth-generation car-T development. Mol Ther. (2023) 31:3146–62. doi: 10.1016/j.ymthe.2023.09.021
145. Li W, Pan X, Chen L, Cui H, Mo S, Pan Y, et al. Cell metabolism-based optimization strategy of car-T cell function in cancer therapy. Front Immunol. (2023) 14:1186383. doi: 10.3389/fimmu.2023.1186383
146. Chang WS, Kim JY, Kim YJ, Kim YS, Lee JM, Azuma M, et al. Cutting edge: programmed death-1/programmed death ligand 1 interaction regulates the induction and maintenance of invariant nkt cell anergy. J Immunol. (2008) 181:6707–10. doi: 10.4049/jimmunol.181.10.6707
147. Parekh VV, Lalani S, Kim S, Halder R, Azuma M, Yagita H, et al. Pd-1/pd-L blockade prevents anergy induction and enhances the anti-tumor activities of glycolipid-activated invariant nkt cells. J Immunol. (2009) 182:2816–26. doi: 10.4049/jimmunol.0803648
148. Kamata T, Suzuki A, Mise N, Ihara F, Takami M, Makita Y, et al. Blockade of programmed death-1/programmed death ligand pathway enhances the antitumor immunity of human invariant natural killer T cells. Cancer Immunol Immunother. (2016) 65:1477–89. doi: 10.1007/s00262-016-1901-y
149. Moraes Ribeiro E, Secker KA, Nitulescu AM, Schairer R, Keppeler H, Wesle A, et al. Pd-1 checkpoint inhibition enhances the antilymphoma activity of cd19-car-inkt cells that retain their ability to prevent alloreactivity. J Immunother Cancer. (2024) 12. doi: 10.1136/jitc-2023-007829
150. Popis M, Sharoni EA, Krishnan S, Wright D, Raheja R, Chand D, et al. 753 agent-797 cell therapy can be combined with next-generation immune checkpoint inhibitors (Ici) and/or bi-specific engagers to improve anti-cancer response. J ImmunoTher Cancer. (2024) 12:A857. doi: 10.1136/jitc-2024-SITC2024.0753
151. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
152. Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. Cd19 car-T cells of defined cd4+:Cd8+ Composition in adult B cell all patients. J Clin Invest. (2016) 126:2123–38. doi: 10.1172/JCI85309
153. Xu X, Huang W, Heczey A, Liu D, Guo L, Wood M, et al. Nkt cells coexpressing a gd2-specific chimeric antigen receptor and il15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin Cancer Res. (2019) 25:7126–38. doi: 10.1158/1078-0432.CCR-19-0421
154. Stevens D, Mo C, Garmezy B, Hamm J, Carneiro B, Wilky B, et al. 647 phase I studies of agent-797, a novel allogeneic invariant natural killer T (Inkt) cell therapy, for the treatment of patients with solid tumors or multiple myeloma. J ImmunoTher Cancer. (2022) 10:A678. doi: 10.1136/jitc-2022-SITC2022.0647
Keywords: iNKT cells, CD1d, cancer immunotherapy, CAR, adoptive cell therapy (ACT)
Citation: Niedzielska M, Chalmers A, Popis MC, Altman-Sharoni E, Addis S, Beulen R, Rudqvist N-P, Chantzoura E, Purbhoo MA, Chand D and Exley MA (2025) CAR-iNKT cells: redefining the frontiers of cellular immunotherapy. Front. Immunol. 16:1625426. doi: 10.3389/fimmu.2025.1625426
Received: 08 May 2025; Accepted: 09 June 2025;
Published: 11 July 2025.
Edited by:
Youwei Wang, Tianjin University, ChinaReviewed by:
Joseph Clara, University of Virginia, United StatesJianhong Chu, Soochow University, China
Changxian Shen, The Ohio State University, United States
Copyright © 2025 Niedzielska, Chalmers, Popis, Altman-Sharoni, Addis, Beulen, Rudqvist, Chantzoura, Purbhoo, Chand and Exley. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark A. Exley, bWFyay5leGxleUBtaU5LVGhlcmFwZXV0aWNzLmNvbQ==