 Xiaoqian Zhang
Xiaoqian Zhang Zhenya Fang1
Zhenya Fang1 Xietong Wang
Xietong Wang- 1Key Laboratory of Maternal & Fetal Medicine of National Health Commission of China, Shandong Provincial Maternal and Child Health Care Hospital Affiliated to Qingdao University, Jinan, China
- 2Department of Obstetrics and Gynaecology, Shandong Provincial Hospital Affiliated to Shandong First Medical University, Jinan, China
- 3School of Clinical and Basic Medicine (Institute of Basic Medicine), Shandong First Medical University (Shandong Academy of Medical Sciences), Jinan, China
Chronic Histiocytic Intervillositis (CHI) is a severe placental inflammatory response caused by various atypical antigens, attracting attention due to its high recurrence rate, which results in adverse pregnancy outcomes such as miscarriage and fetal growth restriction. The pathogenesis of CHI is still poorly understood. Immune factors such as autoimmune diseases or viral infections, maternal-fetal genetic compatibility, and other factors cause immune imbalance at the maternal-fetal interface. Disorders of immune tolerance in CHI includes abnormal activity of Cytotrophoblasts, mononuclear macrophages, and CD8+/CD4+ T lymphocytes. Additionally, pro-inflammatory factors such as IL-1β, TNF-α, and anti-inflammatory molecules like IL-10, TGF-β, and fibrin are crucial in regulating the pathological formation of CHI. Histopathological sections and staining, serological screening, and medical imaging techniques are the primary methods for diagnosing CHI. Patients with CHI may benefit from treatments including immunosuppressants, anticoagulants, and monoclonal antibodies.
1 Introduction
Chronic histiocytic villous interstitial inflammation (CHI), also known as chronic villous interstitial inflammation of unknown etiology (CIUE), is a severe inflammatory disease that significantly affects the villous spaces (1–3). CHI distinguishes from chronic villitis, chronic deciduitis, or chronic chorionic amnionitis, although CHI may coexist with chronic villitis in the same inflamed placenta (4–9) (Table 1). CHI patients often exhibit severe adverse pregnancy outcomes and a high recurrence rate, associated with complications such as miscarriage and fetal growth restriction (10). However, the understanding of the pathogenesis, diagnosis, and treatment strategies for CHI remains in its infancy.
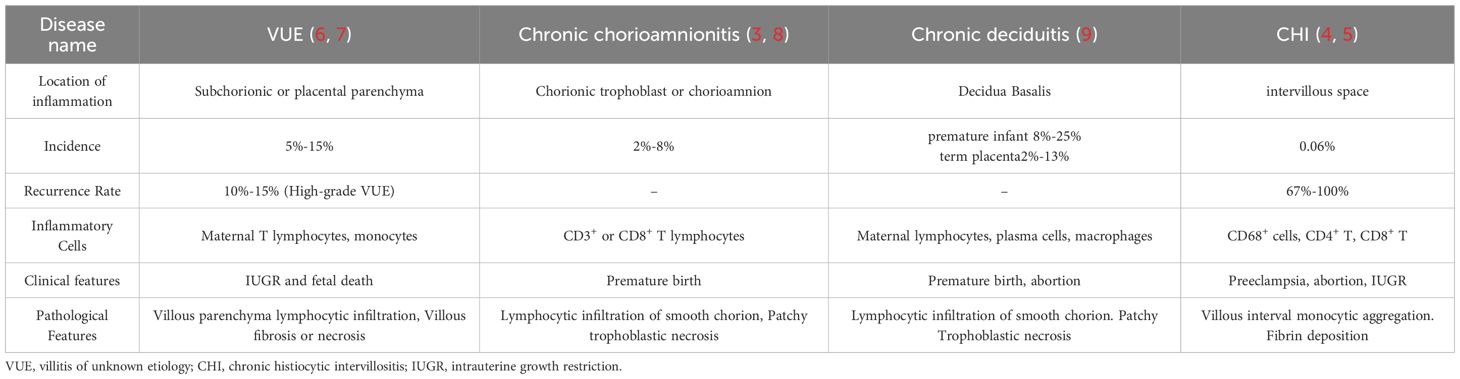
Table 1. Comparison of chronic inflammation of the placenta.
Although maternal-fetal interface immune tolerance imbalance plays a crucial role in the pathogenesis of CHI, the mechanisms by which maternal-fetal genetic compatibility or immune factors regulate the etiology of CHI, as well as the underlying pathophysiological processes, remain poorly understood. This paper reviews the epidemiological characteristics of CHI, summarizes the biological mechanisms underlying its etiology and pathogenesis, and outlines current diagnostic and clinical treatment approaches, aiming to provide new insights into the regulation of maternal-fetal interface immunity and explore novel strategies for the treatment of placental inflammation.
2 CHI is characterized by adverse pregnancy outcome and high recurrence
The prevalence of recurrence for CHI can range from 25% to 100%, displaying significant heterogeneity (10). A study involving 24 patients found that the recurrence rate of adverse outcomes in CHI patients reached 67% (11). Another multicenter prospective study indicated that the recurrence rate of adverse pregnancy outcomes in CHI patients could be as high as 30%. Additionally, patients with a history of severe placental lesions face an even higher risk of CHI recurrence (12).
The following example uses severe adverse pregnancy outcomes such as fetal growth restriction and miscarriage to illustrate the relationship between CHI and severe adverse pregnancy outcomes, as well as their epidemiological characteristics.
2.1 CHI and fetal growth restriction
Patients with CHI exhibit significant pathological changes in placental function, and multiple studies have confirmed its association with fetal growth restriction (FGR). Research indicates that the incidence of fetal growth restriction among CHI patients ranges from 51.6% to 73%. Clinical observations of 111 CHI patients revealed that the rate of FGR was significantly higher than that of the control group (70.4% vs. 0.9%, p < 0.001) (13). An analysis of 69 CHI cases from 1977 to 2009 revealed an FGR incidence of 66.7%, with a live birth rate of less than 54%. Similarly, a Dutch cohort study found that among 38 CHI patients, the incidence of FGR was 51.6%. These cases were also associated with microstructural abnormalities, including placental vascular lesions and fibrosis in the chorionic mesenchyme.
2.2 CHI and miscarriage
CHI is correlated with fetal miscarriage. Statistics indicate that the rate of spontaneous miscarriage among CHI patients ranges from 9.5% to 33%, with early miscarriages occurring more frequently than late ones. A pathological analysis of 178 placentas in France revealed that 73% of CHI cases were associated with fetal growth restriction, with 9.5% resulting in miscarriage (5). A study involving 69 pregnancies with CHI found that the rate of early spontaneous miscarriage was 30.4%, while the rate for late miscarriage was 13.0% (14). An analysis of 151 CHI cases in France from 2000 to 2020 indicated that early miscarriages constituted 20.0%, while late miscarriages accounted for 4.8% (5). Additionally, the occurrence of CHI is often associated with recurrent miscarriages. In a study conducted in Leiden, Netherlands, the miscarriage rate among 38 women with CHI reached as high as 42% (15).
3 The pathophysiological mechanism of CHI
The fetus, as a semi-allogeneic transplant, inducing the mother to form specific tolerance due to multiple synergistic mechanisms. First, trophoblast cells selectively express non-classical HLA molecules (such as HLA-G, HLA-C) to avoid recognition and attack by the mother’s T cells and NK cells, and together with the decidua form a physical barrier (16, 17). Second, local immune cells are reprogrammed. Induced by the high expression of inhibitory HLA-G/E/F receptors on trophoblast cells, regulatory T cells (Treg) and M2 macrophages proliferate, while uterine NK cells and tolerogenic dendritic cells secrete IL-10 and TGF-β, suppressing Th1/Th17 inflammatory responses and maintaining Th2 dominance (18, 19). Concurrently, cytokines promote and maintain an immune tolerance environment. Indoleamine 2,3-dioxygenase depletes tryptophan, while immune checkpoint molecules such as PD-L1/CTLA-4 block T cell activation (20). Trophoblast exosomes carrying miRNAs train maternal immune cells; additionally, the embryo actively “educates” maternal immunity by secreting TSLP and IL-35 (21).
Maternal immune dysfunction is the primary factor breakdown of maternal–fetal immune tolerance and trigger CHI. Studies have shown that patients with autoimmune diseases have an increased risk of CHI. Patients with autoimmune diseases exhibit significant maternal-fetal immune dysfunction and disruption of immune tolerance. Among CHI patients, 58.3% (7/12 cases) had concomitant autoimmune diseases such as systemic lupus erythematosus or antiphospholipid antibodies. A German research group reported in 2023 that 29% (7/24) of 24 CHI patients had autoimmune diseases, and those with positive autoantibodies had significantly increased placental inflammation (p<0.05) (12). Pathogen infections during pregnancy can also trigger similar CHI symptoms (22). Pathogen infections can cause maternal immune dysfunction and maternal-fetal interface homeostasis imbalance during pregnancy. SARS-CoV-2 placental infection during pregnancy can lead to pathological changes in the villous spaces, manifested as chronic histiocytic villous interstitial inflammation combined with trophoblast necrosis (23). Pathological analysis of infection-related CHI revealed that viral particles were detectable in 18.2% of Hofbauer cells and 9.1% of villous capillary endothelial cells, accompanied by acute placental dysfunction and fetal hypoxia (24). Cytomegalovirus can also cause villous inflammatory pathological changes similar to CHI (25).
However, in transplant rejection reactions, MHC molecule recognition plays a central role, so poor maternal-fetal MHC compatibility is a potential triggering factor for CHI. Studies indicate that 30–40% of pregnant women can be detected to express antibodies against paternal HLA during pregnancy, with the proportion increasing with the number of deliveries (26). CHI has been confirmed to meet the Banff antibody-mediated rejection criteria (defined by diffuse infiltration of monocytes) (27). Additionally, twin pregnancy studies have confirmed that genetic factors play a significant role in the development of CHI. In monozygotic twins, CHI occurred bilaterally in the placenta (3/5 cases, with identical genetic backgrounds in both placentas (8), while in dizygotic twins, CHI incidence was inconsistent between the two placentas (3/3 cases, with non-identical genetic backgrounds in both placentas) (28).
However, unlike rejection reactions caused by organ transplantation, the semi-allogeneic transplantation rejection reaction at the maternal-fetal interface is centered on monocytes, with T lymphocytes playing a primary role.
4 CHI cytopathic alterations
The cellular pathological mechanism of CHI is primarily characterized by an imbalance proliferation and migration of immune cells at maternal fetal interface (29). Under normal circumstances, the mother maintains immunological tolerance for the fetus through special immune system, such as Treg-mediated immunosuppression and decidual natural killer cells (dNK cells). However, in CHI, this maternal-fetal interface becomes unbalanced (30). Changes in the maternal-fetal interface and immune disorders and alterations in CHI include large number of CD68+ monocyte-macrophages are abnormally aggregated in the intervillous space, while T cells subsets predominantly composed of CD4+/CD8+ T lymphocytes significantly infiltrate this space, Treg cells reduction, secretion of immune factors such as IL-1β and TGFβR1, and release of immune effector molecules such as MMPs.
4.1 Trophoblast
The syncytiotrophoblast (STB) develops from the fusion of trophoblast cells in the blastocyst’s outer layer, serving as the core functional unit of the maternal-fetal interface. It is central to nutrient and material exchange in the placenta, secreting hormones like human chorionic gonadotropin (hCG) and progesterone to maintain placental development and function. Importantly, the STB also plays a crucial role in immune tolerance and regulation at the maternal-fetal interface, In the placentas of CHI patients, STB was observed in the inflammatory lesion region (31). Previous studies found that normal syncytiotrophoblasts do not express MHC class II molecules. However, recent research has shown that MHC class II molecules could be detected on syncytiotrophoblasts layer in a pathological condition, which may underlie immune recognition and inflammatory responses in CHI (32).
CD200 is a type I membrane glycoprotein widely expressed in fetal-derived placental trophoblast cells (33). At the maternal-fetal interface, the high expression of CD200 on fetal membrane glycoproteins binds to CD200R on the surface of maternal myeloid immune cells, inhibiting the NF-κB/MAPK pathway through phosphatase cascade. Thereby, the signals break the maternal immune system (34). It inhibits dendritic cell maturation and antigen presentation, drives macrophages toward anti-inflammatory M2 polarization (35). simultaneously downregulates Th1-type pro-inflammatory factors (IFN-γ, TNF-α), and upregulates Th2-type anti-inflammatory factors IL-10, thereby inducing Treg expansion and Th2 shift (33). The CD200/CD200R axis also synergizes with molecules such as indoleamine 2,3-dioxygenase (IDO) and FasL to form a fetal “invisibility cloak,” preventing maternal rejection (36). The expression level of the anti-inflammatory molecule CD200R in CHI placental villous trophoblast cells is significantly reduced, which may be associated with abnormal Treg proliferation and immune homeostasis imbalance at the maternal-fetal interface (37).
Besides, the exonuclease CD39 is an immune signaling molecule highly expressed on the surface of trophoblast cells. CD39 hydrolyzes extracellular pro-inflammatory ATP/ADP to generate AMP, which is further converted by CD73 into the immunosuppressive adenosine, thereby establishing a “high adenosine-low ATP” microenvironment at the maternal-fetal interface (38). Adenosine is the core molecule in the formation of an inhibitory immune homeostasis. Adenosine binds to the A2A receptor on maternal immune cells, inhibiting NK cell cytotoxicity, T cell activation, and DC maturation, while promoting Treg expansion and M2 macrophage polarization (39, 40). It also synergizes with tolerance molecules such as PD-L1 and HLA-G to prevent maternal rejection of the semi-allogeneic fetus (41). The reduced expression of the immunosuppressive enzyme CD39 in the trophoblast cells of the placenta disrupts the immune tolerance at the maternal-fetal interface (42).
Additionally, the elevated expression of the adhesion molecule ICAM-1 in the trophoblast cells enhances the adhesion capacity of macrophages, facilitating their accumulation in the intervillous space (43). Furthermore, the increased expression of the inflammatory receptor TLR1 in the trophoblast layer promotes the formation of inflammation in the intervillous space (44).
In CHI placental lesions, trophoblast cells induce large numbers of CD68+ monocytes in the villous spaces, which is also the primary pathological feature of CHI.
4.2 Macrophages
Macrophages are one of the primary immune cells at the maternal-fetal interface. Research indicates that in the CHI placenta, macrophages primarily originate from maternal circulation rather than from fetal Hofbauer cells. The infiltration of macrophages predominates in this lesion, accounting for 80% of inflammatory cells (45). Macrophages play crucial roles in antigen presentation, immune modulation, and the repair of inflammatory damage. In the CHI lesion, macrophages display a mixed M1/M2 polarization phenotype. The M1 subset typically expresses high levels of pro-inflammatory factors such as Tumor Necrosis Factor-alpha (TNF-α) and Interleukin-6 (IL-6), while the M2 subset suppresses local T cell activation by secreting Interleukin-10 (IL-10) and Transforming Growth Factor-beta (TGF-β) (46). Furthermore, studies on CHI have found that macrophages synthesize and release Interleukin-1 beta (IL-1β) through the NLR family pyrin domain containing 3 (NLRP3) inflammasome pathway, leading to its maturation and release, which in turn induces placental inflammation (47). It is important to note that macrophages mediate the remodeling of the extracellular matrix and tissue repair during placental inflammation in preeclampsia (48), and they are also a significant source of proteins for collagen deposition and fibrosis fetal interface in parturition and preterm birth (49).
Macrophages are the primary cells involved in immune regulation and antigen presentation. In classical immune responses, macrophages present antigens bound to MHC class II molecules to CD4+ T lymphocytes, which constitutes a critical step in the immune response.
4.3 T lymphocytes
As the most numerous immune cells in the body, T lymphocytes play a central role in immune recognition and response at the maternal-fetal interface (50). CD4+ T cells and CD8+ T cells are the effector cells responsible for self-limiting immune recognition of antigens, which trigger immune rejection responses. In CHI, there is a significant presence of CD4+ T cells and CD8+ T cells in the placental villous space, with their ratio being nearly 1:1.
A plethora of research has indicated that the expression of cytotoxic T lymphocytes (CTL), a population of immune cells differentiated from CD8+ T cells, is elevated in patients diagnosed with Chronic Hypertension in Pregnancy in comparison to levels observed during a normal pregnancy (51). During the immune induction phase of inflammation in CHI, CD8+ T cells may recognize paternal antigen-MHC I complexes presented by macrophages, which is similar to the classical immune recognition pathway. This recognition leads to their activation, proliferation, and differentiation into CTL cells, exerting cytotoxic effects. CTL cells could induce target cell apoptosis by expressing perforin and granzyme B through the Fas/FasL pathway.
Besides, CD4+ T cells play the core role in immune transplant rejection. CD4+ T cells are activated by recognizing MHC-II molecule-antigen complex and differentiates into Th1 lymphocytes, which assist CD8+ T cells in exerting cytotoxic functions. The expression of MHC II-antigen complex by trophoblasts in CHI is a potential activation binding site for CD4+ T cells. CD4+ T cells may play a role in CHI similar to that in liver transplant rejection, where IFN-γ and TNF-α secreted by Th1 cells exacerbate the inflammatory response, disrupt immune tolerance, and trigger a cascade amplification of the inflammatory response (52).
Additionally, Treg cells are vital for the development and maintenance of placental function. The appropriate number and functionality of Tregs are critical for achieving maternal-fetal immune tolerance. Insufficient Treg numbers and activity can lead to complications such as recurrent miscarriage and preeclampsia (53). Tregs bind to CD80 and CD86 on antigen-presenting cells (APCs) through cytotoxic T lymphocyte-associated protein 4 (CTLA-4), competitively inhibiting costimulatory signals for CD4+T cells (54). A significant infiltration of Tregs is observed in the CHI chorionic space, accompanied by reduced Foxp3 expression. This situation reflects a compensatory mechanism for the imbalance at the maternal-fetal interface and immune tolerance in CHI (55). Additionally, Treg cells secrete immunomodulatory factors like TGF-β to suppress the activity of CD4+ T cells, preventing maternal immune rejection of the fetus (56).
CHI placental pathology is multi-layered and multi-faceted, involving not only cellular changes but also significant molecular alterations.
5 Cytokines in CHI placental inflammation
The core pathological mechanism of CHI involves a dynamic imbalance in the cytokine network. These cytokines regulate the inflammatory process through a complex interactive network. They can be categorized into pro-inflammatory and anti-inflammatory cytokines based on their functional characteristics. Pro-inflammatory cytokines (such as IL-1, IL-6, and TNF-α) serve as the primary drivers of the inflammatory cascade, facilitating the recruitment and activation of immune cells. Anti-inflammatory cytokines (such as IL-10 and TGF-β) help suppress excessive immune responses and maintain immune tolerance and homeostasis.
5.1 IL-1β
IL-1β plays a crucial role in placental inflammation and is implicated in various pregnancy complications, particularly in placental immune regulation. It is likely that IL-1β is secreted by infiltrating macrophages and trophoblast cells via the NLRP3 inflammasome pathway. IL-1β promotes trophoblast invasion and angiogenesis by activating the PI3K/Akt-VEGF pathway. It also enhances maternal-fetal immune tolerance by inducing NK cells in the decidua to secrete IL-8 and GM-CSF (57). In addition, low concentrations of IL-1β upregulate HLA-G expression in trophoblast cells while simultaneously suppressing the excessive activation of maternal T cells (58).
In the placenta of CHI, the expression level of the IL-1β gene increases by 3.9 times, according to reference (59). Under pathological conditions (such as preeclampsia and chronic villitis), macrophages trigger the secretion and synthesis of sFLT-1 through a pro-inflammatory cascade involving NLRP3 inflammasomes and Gasdermin D (GSDMD) (60). These proteins antagonize the binding of VEGF-A and PlGF to the vascular endothelial cell receptor VEGFR-1/2, leading to increased NO synthesis, elevated ROS production, and heightened vascular permeability, thereby inducing vascular spasm (61). Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. Additionally, IL-1β recruits CD8+ T cells and neutrophils, causing damage to the villi (57). This recruitment also stimulates prostaglandin synthesis, which can lead to preterm labor (62).
Once IL-1β presses the “start button” for inflammation, TNF-α immediately takes the stage, acting as an “accelerator” that amplifies and sustains this inflammatory storm.
5.2 Tumor necrosis factor-alpha
Tumor necrosis factor-alpha (TNF-α) is a multifunctional pro-inflammatory cytokine that plays a role in immune regulation, apoptosis, and vascular function during both physiological and pathological processes in the placenta (63). In patients with chronic hypoxia-induced (including CHI) conditions, the TNF-α levels in placental tissue show a significant positive correlation with the risk of fetal growth restriction and preterm birth (64). TNF-α is primarily secreted by infiltrating macrophages and CD8+ T cells, with a smaller secreted by trophoblasts under stress conditions (65). In the placenta of cases with idiopathic FGR, TNF-α expression is significantly increased in trophoblastic giant cells and vascular endothelial cells. This increase may lead to fetal developmental restriction by inhibiting placental angiogenesis or directly damaging trophoblast function (66). In preeclampsia and gestational diabetes, elevated placental TNF-α levels is associated with insulin resistance, abnormal expression of advanced glycation end products and their receptor for advanced glycation end products. This pro-inflammatory microenvironment can induce localized oxidative stress and endothelial dysfunction in the placenta (67), which may lead to maternal hypertension and proteinuria (68).
TNF-α expression is essential for the formation of immune tolerance in early pregnancy. In the normal placenta development, TNF-α induces trophoblasts to express HLA-G, which inhibits the cytotoxicity of NK cells, thereby maintaining maternal-fetal immune tolerance (69). The soluble tumor necrosis factor receptor 1 (sTNFR1) secreted by placental tissue specifically neutralizes the pro-inflammatory effects of TNF-α, and this local immune regulation mechanism effectively suppresses autoimmune responses (70).
TNF-α-driven placental inflammatory signals can activate and amplify the complement system cascade reaction, which is a key marker of immune-mediated tissue damage.
5.3 The complement molecule C4d
C4d is a breakdown product activated by the classical complement pathway and is commonly associated with antibody-mediated immune responses (71). The deposition of the complement breakdown product C4d in placental inflammation is a significant pathological marker for pregnancy complications related to antiphospholipid antibodies (aPL) (72). Recent studies have shown that C4d plays a crucial role in organ transplant rejection and maternal-fetal immune tolerance (73). In the placenta of CHI, the deposition of C4d is distributed either diffusely or focally, and the amount of deposition is positively correlated with the severity of the disease (74). The abnormal expression of HLA class II molecules (such as HLA-DR and HLA-DQ) in placental trophoblast cells is significantly positively correlated with C4d deposition. This finding supports the idea that maternal anti-HLA antibodies may drive inflammatory responses and macrophage infiltration through the classical complement pathway (32).
The deposition of complement fragment C4d in CHI may be involved in the formation of pathological deposits in intercellular spaces due to the activation of the complement cascade (75). C4d is produced during the complement cascade and induces the formation of the membrane attack complex (MAC), which directly damages vascular endothelial and stromal cells while releasing anaphylatoxins C3a and C5a (76). Chemokines recruit monocytes, exacerbating local inflammatory responses. This leads to chronic inflammatory infiltration of the villous stroma and disruption of the epithelial barrier (77). Furthermore, C4d may enhance the pro-fibrotic microenvironment in conjunction with TGF-β. The deposition of C4d can directly promote the conversion of local fibrinogen to fibrin, exacerbating the formation of fibrotic networks in the villous space and the deposition of extracellular matrix, ultimately hindering maternal-fetal blood exchange (78).
In addition, although some studies have used C4d expression levels as an auxiliary diagnostic criterion, given lack of studies specifically investigating C4d as a diagnostic marker (27). the discriminatory value of C4d in CHI remains controversial (75). Along with C4d-mediated complement attack and inflammatory damage, changes in TGF-β signaling are also commonly observed in placental tissue. Changes in TGFβR1 expression and activity may reflect the body’s attempt to curb excessive inflammation and promote tissue repair.
5.4 Transforming growth factor beta receptor 1
In the placenta, transforming growth factor-β receptor 1 (TGF-βR1) acts as a receptor for transforming growth factor-β (TGF-β), playing a multifaceted role in regulating trophoblast differentiation, maintaining immune balance, and supporting angiogenesis to uphold pregnancy stability (79). TGF-βR1 and type II receptor (TβRII) are primarily expressed in the syncytiotrophoblast of the placenta, as well as in extravillous trophoblasts and the chorionic plate (80).
TGFβR1 is significantly upregulated in the placenta of CHI, and it may influence the pathological process of CHI by inhibiting trophoblast invasion, regulating maternal-fetal immune tolerance, and modulating inflammatory repair. It is a key cytokine that regulates the pathological changes in chronic placental inflammation (81). In preeclampsia, TGFβR1 is found to function in the remodeling of uterine spiral arteries by inhibiting trophoblast invasion (82). Besides, TGFβR1 plays an important role in immune tolerance and inflammatory repair at the maternal-fetal interface (83). In patients with recurrent miscarriage, the expression and activity of TGF-β are reduced, which can inhibit Treg proliferation and differentiation, leading to immune dysfunction at the maternal-fetal interface (84). The decreased ability of monocytes/macrophages to synthesize TGF-β1 fails to effectively suppress excessive inflammatory responses, leading to chronic inflammation (85). Furthermore, in the repair of inflammatory damage associated with chronic liver disease and pulmonary fibrosis, pro-inflammatory factors in the tissue microenvironment, such as TNF-α and IL-6, collaborate with TGF-β to induce fibroblast differentiation and extracellular matrix deposition (86).
5.5 Matrix metalloproteinases
Matrix metalloproteinases (MMPs) are a superfamily of proteases that depend on metal ions, such as zinc and calcium, as cofactors. They are capable of degrading critical components of the extracellular matrix (ECM), including collagen and elastin, leading to the disruption and remodeling of tissue structure, playing roles in placental development, immune regulation, and tissue remodeling (87). Tissue inhibitor of metalloproteinases (TIMPs) maintains tissue microenvironment homeostasis by inhibiting MMP-driven matrix degradation and excessive inflammatory responses, thereby preventing inflammation spread and tissue damage.
During tissue repair, MMP-2 participates in the clearance of damaged ECM, while TIMP-1 helps control the extent of degradation and promotes the deposition of new matrix (87, 88). Increased TIMP-1 activity is considered a key factor in promoting ECM accumulation and fibrosis formation (89). Abnormal MMP expression is the mechanism underlying the formation of massive perivillous fibrin deposition in CHI pathology (90–92). Additionally, TIMP-1 activation of the CD63/β1 integrin receptor complex in oligodendrocytes promotes the conversion of macrophages to an anti-inflammatory phenotype (M2 type), increasing IL-10 and TGF-β secretion while inhibiting the release of pro-inflammatory factors such as TNF-α and IL-6 (93).
In addition to MMPs, fibronectin is another important active molecule in the process of massive deposition around villi.
5.6 Fibronectin
Fibronectin deposition is a biomarker for diagnosing placental diseases and predicting recurrences (94). Fibronectin is a core molecule of the coagulation system, formed from fibrinogen after activation by thrombin. Its dynamic balance is crucial for hemostasis, inflammation, and tissue repair (95). In the placenta, fibronectin moderately deposits in the intervillous space, covering approximately 5%-10% of the area. This forms a temporary scaffold that supports the branching of the chorionic tree and the structural integrity of the maternal-fetal interface (96). Fibronectin promotes the fusion of trophoblast cells into a multinucleated syncytiotrophoblast through integrin (αvβ3) signaling, enhancing hormone secretion (such as hCG) and barrier function (97).
A significant feature of CHI pathology is the massive perivillous fibrin deposition (MPFD) around the chorionic villi. In this context, fibrin serves both as a product of inflammation-coagulation cross-reaction and as a key mediator driving placental damage (98). The maternal interface immune rejection activates mononuclear-macrophages, which release pro-inflammatory factors such as IL-6 and IL-1β. This process leads to endothelial damage in the intervillous space and thrombin generation, subsequently promoting the conversion and accumulation of fibrinogen into fibrin (99). Additionally, the necrosis of placental trophoblasts releases cellular debris and mitochondrial DNA. This activates Toll-like receptor 9 and the complement system, such as C5a, enhancing the local coagulation cascade and exacerbating fibrin deposition (100). Furthermore, under chronic hypoxic conditions, HIF-1α promotes the synthesis of plasminogen activator inhibitor-1 and inhibits plasmin activity. This ultimately hinders the clearance of fibrin (101). Together, these mechanisms contribute to increased fibrin deposition around the chorionic villi, negatively affecting placental function.
The impact of fibrin deposition covering the placental intervillous space on villous function manifests in two ways: physical obstruction and disruption of cellular signaling. The physical barrier created by fibrin mechanically hinders the exchange of oxygen and nutrients between mother and fetus. It also obstructs the migration of trophoblasts and immune cells, thereby suppressing placental development and villous vascularization (102). In cases of chronic hypoxia-induced (CHI), the area of fibrin deposition is negatively correlated with the birth weight percentile of newborns, indicating that fibrin accumulation affects placental exchange efficiency (103). Pathological observations reveal that in severe cases of CHI, the area affected by fibrin can reach 40%-60% of the placental volume, leading to extensive placental infarction and functional impairment (104). Furthermore, fibrin recruit macrophages and stimulates the activation of complement components C3d and C4d in the intervillous space, exacerbating immune rejection and inflammatory responses at the maternal-fetal interface (1).
6 Clinical assessment and research updates on CHI
6.1 Pathological diagnosis of CHI
CHI pathological diagnosis is based on placental histological examination, with routine examination content including observation of placental tissue morphology, CD68+ histiocyte positivity screening, and examination of fibrin deposition around the villi. Quantitative immunohistochemical analysis of placental pathology in CHI showed that the density of CD68+ macrophages in the CHI group was 88 ± 23 per unit area (HPF 40×), while in the control group it was 8 ± 5 per unit area (P < 0.001) (45). CHI pathology is classified into three grades—Grade 1 (5%-10%), Grade 2 (10%-50%), and Grade 3 (>50%)—which exhibit a significant dose-response relationship with perinatal outcomes (P<0.0001). The neonatal survival rate for Grade 3 patients was only 16.1%, significantly lower than that for Grade 2 (59%) and Grade 1 (86.5%) (P = 0.0002) (45). The risk of disease recurrence for Grade 2 and Grade 3 patients was 3.8 times higher than that for Grade 1 patients. Pathological grading is of great significance for predicting pregnancy outcomes and recurrence (105).
6.2 Biochemical diagnostics placental alkaline phosphatase
Placental alkaline phosphatase (PLAP) is an enzyme specifically expressed in placental cells. It regulates active transport across cell membranes and calcium-phosphate metabolism, providing nutrition to the fetus. There is academic debate regarding the clinical significance of alkaline phosphatase (ALP) levels in CHI patients from different regions. A retrospective cohort study from Canada involving 33 patients found that 31.6% (10/33) of CHI cases exhibited elevated serum ALP levels (>125 U/L), suggesting that ALP could serve as a reference indicator for inflammatory activity (106). However, a prospective cohort study from Japan in 2017 (n=58) conducted a multivariable regression analysis and found no significant correlation between fluctuations in ALP levels related to CHI (elevated group vs. normal group) and adverse pregnancy outcomes such as fetal growth restriction (OR = 1.32, 95% CI 0.75-2.34) or preterm birth (OR = 1.15, 95% CI 0.82-1.61) (p>0.05). This study also emphasized that the clinical value of PLAP as a specific diagnostic marker for CHI needs to be validated through large-scale multicenter studies (107).
6.3 HLA antibodies
The role of HLA antibodies as biochemical markers for CHI remains contentious within the international academic community. Some studies report that anti-paternal HLA-I/II antibodies may be detected in the placental tissue of CHI patients, with positivity rates reaching up to 75% in certain case reports (40). HLA antibodies may be associated with fluctuations in the expression levels of HLA-B and HLA-DRB1 alleles in peripheral blood. Additionally, some cases exhibit abnormal deposition of complement C4d in placental tissue (107). However, a recent cohort study from the UK found no statistically significant difference in the overall positivity rates of anti-HLA antibodies between the CHI group and healthy controls, indicating that the diagnostic specificity of a single HLA antibody marker has not yet met clinical requirements.
7 Progress in the treatment of CHI
Currently, there is no standardized treatment protocol for chronic histiocytic villous interstitial inflammation (CHI). However, several international clinical studies have shown that anticoagulation, anti-inflammatory, and biological therapies may have positive implications for improving pregnancy outcomes. Nevertheless, due to the limited sample sizes of existing studies and the lack of high-level evidence-based medical evidence, there remains significant controversy regarding the efficacy, safety, and applicability of various treatment methods.
Regarding anticoagulant therapy, research findings remain inconsistent (Table 2). A French clinical study involving 21 CHI patients showed that while the use of aspirin and low-molecular-weight heparin (LMWH) alone did not reduce the risk of preterm birth (still at 30%), the live birth rate significantly increased from 32% to 67% (12). In 2017, Japanese scholars proposed a triple therapy combining low-dose aspirin, corticosteroids, and LMWH. This strategy further improved the live birth rate and demonstrated superior clinical benefits compared to single anticoagulant regimens (107). A UK study involving 28 patients with refractory CHI demonstrated that adding hydroxychloroquine (200 mg/day) and prednisolone (20 mg/day) to aspirin (75–150 mg/day) and LMWH therapy significantly improved the live birth rate (from 61.5% to 86.2%, p < 0.05) (11). However, due to the small sample sizes of all studies and the lack of randomized controlled trials, the exact efficacy and applicability of combined anticoagulation and immunosuppression therapy in a broader population remain controversial.
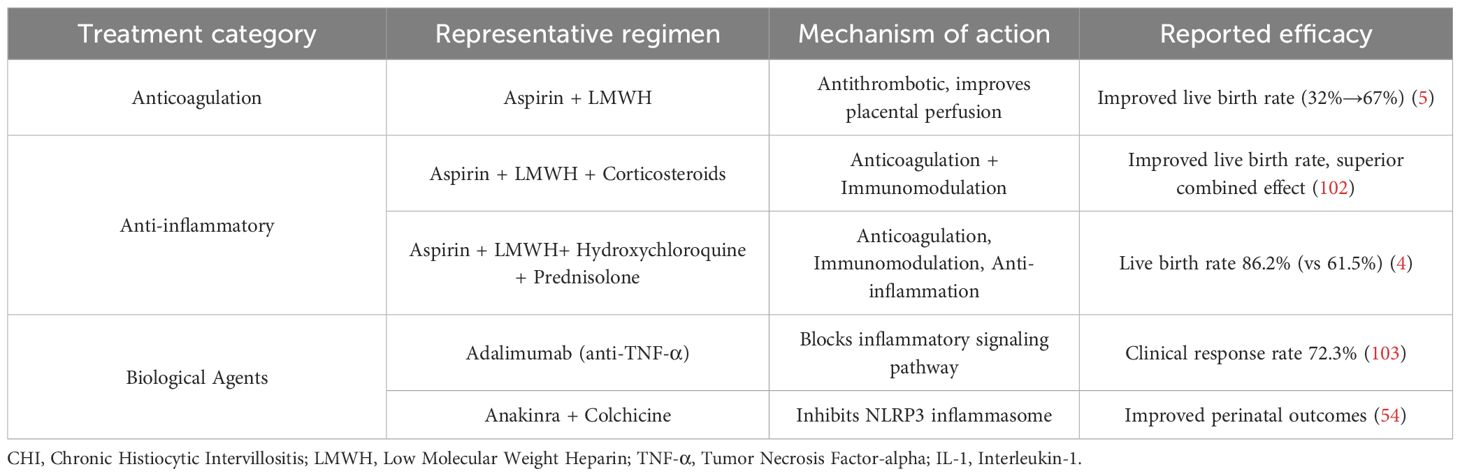
Table 2. Treatment strategies for CHI.
In terms of anti-inflammatory therapy, immune modulation strategies are primarily used to suppress excessive inflammatory responses. Glucocorticoids are commonly used drugs and are often combined with anticoagulants. Studies in Japan and the UK have suggested that adding glucocorticoids can further improve live birth rates and expand clinical benefits. Hydroxychloroquine is also commonly used to inhibit immune inflammatory pathways, and its combination with low-molecular-weight heparin and glucocorticoids has shown synergistic anti-inflammatory and immunomodulatory effects (11). However, this class of treatment still faces challenges such as significant individual response variability, unclear long-term safety, and lack of consensus on optimal treatment regimens.
In terms of biological therapy, agents targeting specific inflammatory factors or signaling pathways offer new directions for refractory CHI. Anti-TNF-α monoclonal antibodies such as adalimumab (40 mg every two weeks) achieve a clinical remission rate of 72.3% in refractory cases, with the mechanism involving blocking the TNF-α signaling pathway, inhibiting abnormal macrophage activation, and reducing inflammatory infiltration in the villous spaces (108). The IL-1 receptor antagonist anakinra combined with colchicine can inhibit NLRP3 inflammasome activation, and reports indicate it can improve perinatal outcomes in patients with recurrent CHI (59). However, such biologics are currently limited to case reports or small case series, and their safety, timing of administration, and long-term maternal and infant outcomes require further research validation.
Additionally, various natural immune modulators exhibit good anti-inflammatory effects and have the potential to become drugs for CHI treatment. The flavonoid quercetin effectively improves endothelial dysfunction in preeclampsia (109, 110).the flavonoid hesperidin exhibits excellent primary villus antioxidant activity (111). Research indicates that liposoluble vitamin D3 is a key regulatory factor in placental and fetal development (112, 113). Additionally, plant estrogens such as soy isoflavones can inhibit Th17 expression in the placenta, promote Treg expansion, reduce CD68+ macrophages in the placenta, and mitigate inflammatory responses (114).
8 Discussion
Chronic histiocytic intervillositis (CHI) is a rare, placenta-specific immune-inflammatory disorder characterized by disrupted maternal–fetal immune tolerance. The maintenance and breakdown of immune tolerance at the maternal–fetal interface are regulated by multiple factors, including genetic compatibility, maternal autoimmune status, and infections (Figure 1). During the induction of immune tolerance in normal pregnancy, HLA molecules play an important role. Trophoblasts do express classical HLA-A/B molecules but highly express non-classical HLA-G, which acts in coordination with locally enriched regulatory T cells (Tregs) to effectively suppress maternal immune activation and maintain immune tolerance. Under conditions such as poor histocompatibility between mother and fetus, autoimmune diseases, or pathogen infection, the tolerant balance at the maternal–fetal interface is disrupted. Trophoblasts aberrantly upregulate HLA-A/B expression, exhibit reduced HLA-G levels, and are accompanied by a decrease in Treg numbers, collectively leading to a breakdown in maternal–fetal immune tolerance and a shift toward pathological responses as CHI.
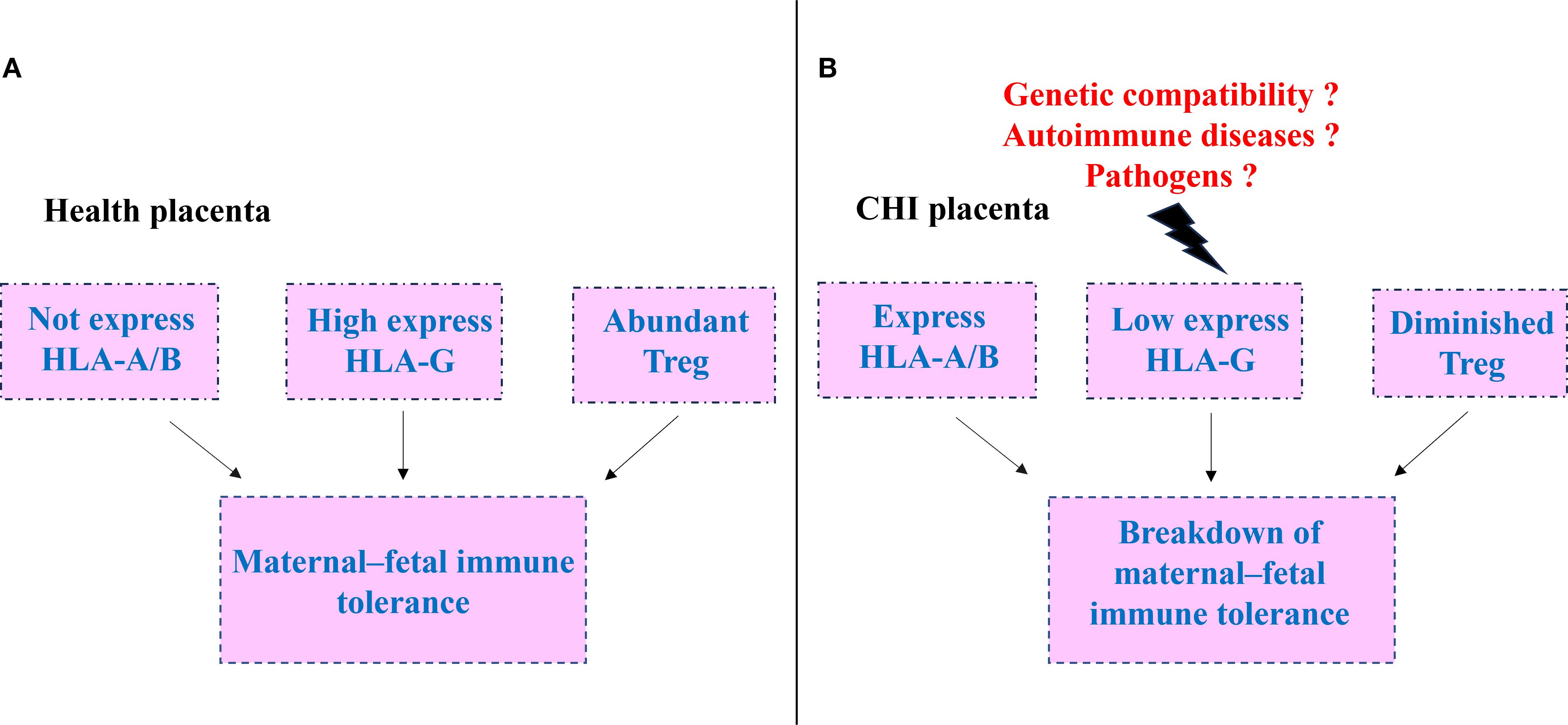
Figure 1. Schematic of tolerance in normal pregnancy and its breakdown in chronic histiocytic intervillositis (CHI). (A) Normal maternal -fetal immune. extravillous trophoblasts express high levels of HLA-G, lack HLA-A/B expression, and are accompanied by abundant regulatory T (Treg) cells, sustaining immune tolerance at the maternal -fetal interface. (B) Tolerance collapse in CHI.HLA-G expression is markedly diminished, HLA-A/B is up-regulated, and Treg cells are reduced, precipitating the breakdown of maternal -fetal immune tolerance.
CHI represents an aberrant maternal immune response to semi-allogeneic fetal antigens. This review summarizes the cellular and molecular players implicated in the pathogenesis of CHI and proposes a two-phase model (Figure 2): (A)inflammation initiation and (B)tissue repair. During the initiation phase, down-regulation of CD200 and CD39 on syncytiotrophoblasts compromises maternal–fetal tolerance. Concomitantly, interleukin-1β (IL-1β) is up-regulated and secreted into the interstitial space, triggering an inflammatory cascade. Macrophages infiltrate the placental bed and polarize, while T cells migrate and become activated. In the subsequent repair phase, monocytes differentiate into M2 macrophages. Chronic inflammation promotes the release of cytokines such as gasdermin D, inducing tissue cell apoptosis. Extensive deposition of complement split product C4d and fibrin results in abundant perivillous fibrinoid material.
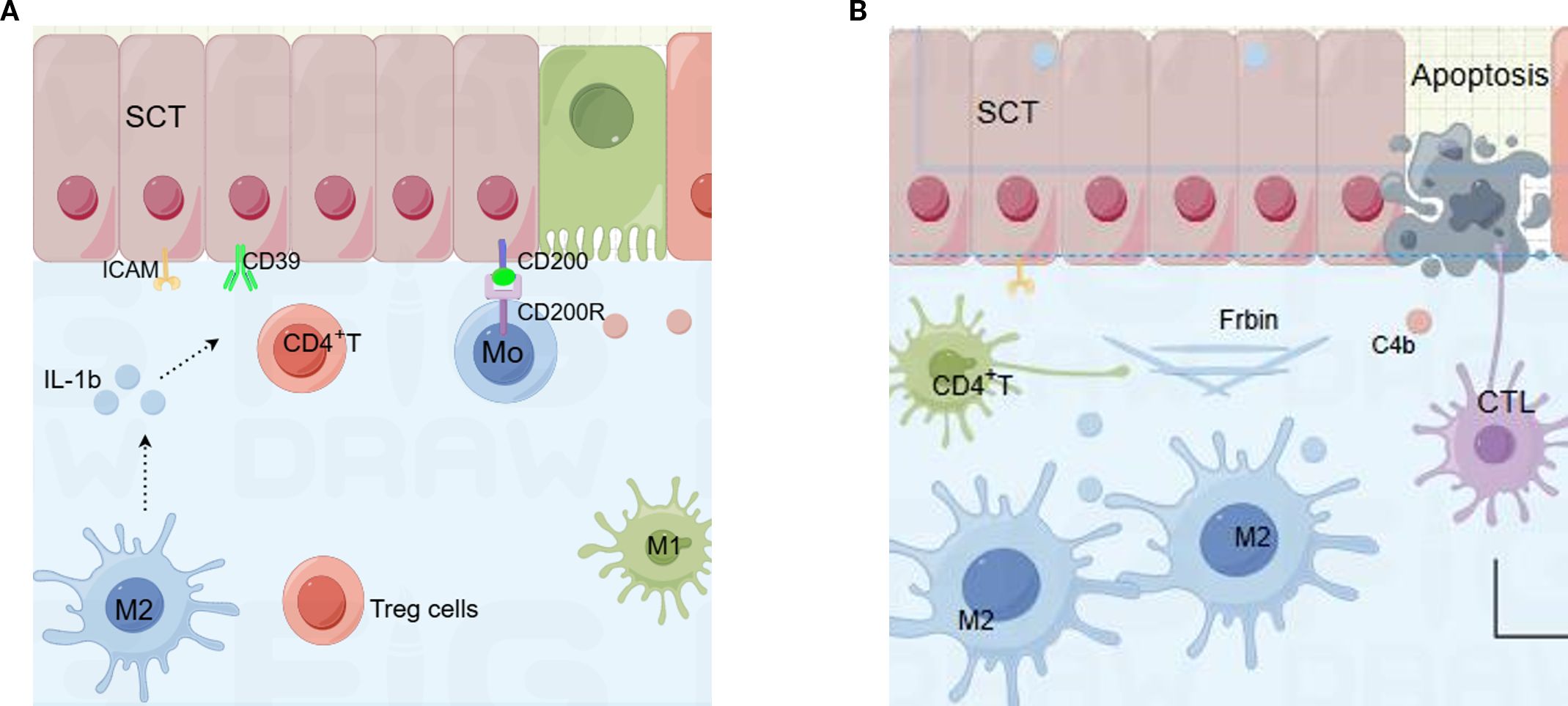
Figure 2. Cellular and molecular components in two processes of chronic histiocytic intervillositis (CHI). (A) Initiators of inflammation in CHI. Cells includes the syncytiotrophoblast (SCT), macrophages, CD4⁺ T cells, and soluble meditheators including interleukin-1β (IL-1β), CD200 -CD200R axis, ectonucleoside triphosphate diphosphohydrolase-1 (CD39), and intercellular adhesion molecule-1 (ICAM-1). (B) Mediators of injury and repair in CHI: fibrin deposition and complement component 4b (C4b). SCT, syncytiotrophoblast; IL-1β, interleukin-1β; ICAM-1, intercellular adhesion molecule-1; C4b, complement component 4b.
9 Outlook
Looking ahead, research on the mechanisms, diagnosis, and treatment of CHI is gaining momentum, with several key areas for future investigation outlined below:
Some immunosuppressants have shown promising therapeutic effects. There is a good application potential for developing inhibitors targeting key proteins involved in CHI regulation, such as chemokines and NLRP3 inflammasome pathways.
The serological diagnosis of CHI aims to develop non-invasive biomarkers based on cell-free fetal DNA or exosomes from maternal blood, including HLA antibodies and specific microRNAs.
Some immunosuppressants have shown promising therapeutic effects. There is a good potential for developing inhibitors targeting key proteins involved in CHI regulation, such as chemokines and the NLRP3 inflammasome. Additionally, we will explore the use of placenta-derived mesenchymal stem cells or gene-edited CAR-Treg cells for localized delivery to the chorionic space to promote immune tolerance.
Author contributions
XZ: Formal Analysis, Writing – original draft, Data curation. ZF: Methodology, Writing – original draft. XW: Data curation, Investigation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by Shandong medical and health science and technology development plan (202405020585).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cornish EF, McDonnell T, and Williams DJ. Chronic inflammatory placental disorders associated with recurrent adverse pregnancy outcome. Front Immunol. (2022) 13:825075. doi: 10.3389/fimmu.2022.825075
2. Crawford A, Moore L, Bennett G, Savarirayan R, Manton N, Khong Y, et al. Recurrent chronic histiocytic intervillositis with intrauterine growth restriction, osteopenia, and fractures. Am J Med Genet A. (2016) 170:2960–4. doi: 10.1002/ajmg.a.37856
3. Feist H, Blocker T, and Hussein K. Massive perivillous fibrin deposition, chronic histiocytic intervillositis and villitis of unknown etiology: lesions of the placenta at the fetomaternal interface with risk of recurrence. Pathologe. (2015) 36:355–61. doi: 10.1007/s00292-014-2051-7
4. Traeder J, Jonigk D, Feist H, Brocker V, Langer F, Kreipe H, et al. Pathological characteristics of a series of rare chronic histiocytic intervillositis of the placenta. Placenta. (2010) 31:1116–9. doi: 10.1016/j.placenta.2010.09.012
5. Mawa A, Bizet G, Stichelbout M, Devisme L, Pauchet M, Gobert S, et al. Caracteristics of women presenting with chronic histiocytic intervillositis during pregnancy: A case-control study. J gynecology obstetrics Hum Reprod. (2025) 54:102882. doi: 10.1016/j.jogoh.2024.102882
6. Sebastian T, Ravikumar G, and Crasta J. Villitis of unknown etiology (Vue): effect on placental size and association with clinical parameters. J Matern Fetal Neonatal Med. (2022) 35:1695–702. doi: 10.1080/14767058.2020.1767577
7. Aldahmash W, Aljerian K, and Alwasel S. Prevalence of fetal inflammatory response syndrome and villitis of unknown etiology in the placenta of saudi women and their association with baby sex. Life. (2024) 14:79. doi: 10.3390/life14010079
8. Feist H, Lehmann U, Bajwa S, Bruschke C, and Schaumann N. Villitis of unknown etiology, chronic deciduitis, chronic chorioamnionitis and chronic histiocytic intervillositis in monozygotic and dizygotic twin pregnancies. A retrospective analysis of 16 cases. Placenta. (2023) 133:32–9. doi: 10.1016/j.placenta.2023.02.002
9. Khong TY, Bendon RW, Qureshi F, Redline RW, Gould S, Stallmach T, et al. Chronic deciduitis in the placental basal plate: definition and interobserver reliability. Hum Pathol. (2000) 31:292–5. doi: 10.1016/s0046-8177(00)80241-5
10. Brady CA, Williams C, Sharps MC, Shelleh A, Batra G, Heazell AEP, et al. Chronic histiocytic intervillositis: A breakdown in immune tolerance comparable to allograft rejection? Am J Reprod Immunol. (2021) 85:e13373. doi: 10.1111/aji.13373
11. Brady CA, Williams C, Batra G, Church E, Tower CL, Crocker IP, et al. Immunomodulatory therapy reduces the severity of placental lesions in chronic histiocytic intervillositis. Front Med. (2021) 8:753220. doi: 10.3389/fmed.2021.753220
12. Mekinian A, Costedoat-Chalumeau N, Masseau A, Botta A, Chudzinski A, Theulin A, et al. Chronic histiocytic intervillositis: outcome, associated diseases and treatment in a multicenter prospective study. Autoimmunity. (2015) 48:40–5. doi: 10.3109/08916934.2014.939267
13. Homatter C, Stichelbout M, Devisme L, Chudzinski A, Debarge V, Garabedian C, et al. Is chronic histiocytic intervillositis a severe placental disease? A case-control study. Placenta. (2020) 91:31–6. doi: 10.1016/j.placenta.2019.12.020
14. Marchaudon V, Devisme L, Petit S, Ansart-Franquet H, Vaast P, and Subtil D. Chronic histiocytic intervillositis of unknown etiology: clinical features in a consecutive series of 69 cases. Placenta. (2011) 32:140–5. doi: 10.1016/j.placenta.2010.11.021
15. Bos M, Harris-Mostert E, van der Meeren LE, Baelde JJ, Williams DJ, Nikkels PGJ, et al. Clinical outcomes in chronic intervillositis of unknown etiology. Placenta. (2020) 91:19–23. doi: 10.1016/j.placenta.2020.01.001
16. de Boer EN, Corsten-Janssen N, Wierenga E, Bijma T, Knapper JT, Meerman GJT, et al. Limitations of semi-automated immunomagnetic separation of hla-G-positive trophoblasts from papanicolaou smears for prenatal genetic diagnostics. Diagnostics. (2025) 15:386. doi: 10.3390/diagnostics15030386
17. Alexandrova M, Manchorova D, Vangelov I, Terzieva A, Dimitrova V, Mor G, et al. First trimester extravillous trophoblast secretes hla class I molecules via small extracellular vesicles. Placenta. (2025) 167:11–21. doi: 10.1016/j.placenta.2025.04.023
18. Peng X, Chinwe Oluchi-Amaka I, Kwak-Kim J, and Yang X. A comprehensive review of the roles of T-cell immunity in preeclampsia. Front Immunol. (2025) 16:1476123. doi: 10.3389/fimmu.2025.1476123
19. Nakamura K. Immunotoxicological disruption of pregnancy as a new research area in immunotoxicology. J Immunotoxicol. (2025) 22:2475772. doi: 10.1080/1547691X.2025.2475772
20. Tripathi S, Varma K, and Misra V. Role of pd-L1 in the pathogenesis of pre-eclampsia and its association with adverse fetal outcomes. Turk Patoloji Derg. (2025) 41:I-VIII. doi: 10.5146/tjpath.2025.13658
21. Lin Y, Zhong Y, Shen W, Chen Y, Shi J, Di J, et al. Tslp-induced placental dc activation and il-10(+) nk cell expansion: comparative study based on balb/C X C57bl/6 and nod/scid X C57bl/6 pregnant models. Clin Immunol. (2008) 126:104–17. doi: 10.1016/j.clim.2007.09.006
22. Kirisri S, Vongsakulyanon A, Kantachuvesiri S, Razonable RR, and Bruminhent J. Predictors of cmv infection in cmv-seropositive kidney transplant recipients: impact of pretransplant cmv-specific humoral immunity. Open Forum Infect Dis. (2021) 8:ofab199. doi: 10.1093/ofid/ofab199
23. Arora M and Lakshmi R. Vaccines - safety in pregnancy. Best Pract Res Clin obstetrics gynaecology. (2021) 76:23–40. doi: 10.1016/j.bpobgyn.2021.02.002
24. Ivert A, Lindblad Wollmann C, and Pettersson K. A case series on pregnant patients with mild covid-19 infection and signs of severe placental insufficiency. Case Rep Obstet Gynecol. (2023) 2023:2018551. doi: 10.1155/2023/2018551
25. Taweevisit M, Sukpan K, Siriaunkgul S, and Thorner PS. Chronic histiocytic intervillositis with cytomegalovirus placentitis in a case of hydrops fetalis. Fetal Pediatr Pathol. (2012) 31:394–400. doi: 10.3109/15513815.2012.659405
26. Joo JS, Lee D, and Hong JY. Multi-layered mechanisms of immunological tolerance at the maternal-fetal interface. Immune Netw. (2024) 24:e30. doi: 10.4110/in.2024.24.e30
27. Albersammer L, Leon J, Martinovic J, Dagobert J, Lebraud E, Bessieres B, et al. Histologic and molecular features shared between antibody-mediated rejection of kidney allografts and chronic histiocytic intervillositis support common pathogenesis. J Pathol. (2025) 266:177–91. doi: 10.1002/path.6413
28. van der Meeren LE, Krop J, Dijkstra KL, Bloemenkamp KWM, Cornish EF, Nikkels PGJ, et al. One-sided chronic intervillositis of unknown etiology in dizygotic twins: A description of 3 cases. Int J Mol Sci. (2021) 22:4786. doi: 10.3390/ijms22094786
29. Collin-Bund V, Poindron V, Van Quyen PL, Boudier E, Minella C, Langer B, et al. Controversies in chronic histiocytic intervillositis. J gynecology obstetrics Hum Reprod. (2025) 54:102931. doi: 10.1016/j.jogoh.2025.102931
30. Wienke J, Brouwers L, van der Burg LM, Mokry M, Scholman RC, Nikkels PG, et al. Human tregs at the materno-fetal interface show site-specific adaptation reminiscent of tumor tregs. JCI Insight. (2020) 5:e137926. doi: 10.1172/jci.insight.137926
31. Angelova DM, Tsolova A, Prater M, Ballasy N, Bacon W, Hamilton RS, et al. Single-cell rna sequencing identifies cxadr as a fate determinant of the placental exchange surface. Nat Commun. (2025) 16:142. doi: 10.1038/s41467-024-55597-w
32. Brady CA, Ford LB, Moss C, Zou Z, Crocker IP, and Heazell AEP. Virtual crossmatching reveals upregulation of placental hla-class ii in chronic histiocytic intervillositis. Sci Rep. (2024) 14:18714. doi: 10.1038/s41598-024-69315-5
33. Clark DA, Dmetrichuk JM, McCready E, Dhesy-Thind S, and Arredondo JL. Changes in expression of the cd200 tolerance-signaling molecule and its receptor (Cd200r) by villus trophoblasts during first trimester missed abortion and in chronic histiocytic intervillositis. Am J Reprod Immunol. (2017) 78:e12665. doi: 10.1111/aji.12665
34. Zhu B, Yu Y, Liu X, Han Q, Kang Y, and Shi L. Cd200 modulates S. Aureus-induced innate immune responses through suppressing P38 signaling. Int J Mol Sci. (2019) 20:659. doi: 10.3390/ijms20030659
35. Lin M, Chen D, Shao Z, Liu Q, Hao Z, Xin Z, et al. Inflammatory dendritic cells restrain cd11b(+)Cd4(+) ctls via cd200r in human nsclc. Cell Rep. (2024) 43:113767. doi: 10.1016/j.celrep.2024.113767
36. Clark DA. Tolerance signaling molecules. Chem Immunol Allergy. (2005) 89:36–48. doi: 10.1159/000087911
37. Wang LQ, Yan CF, Zhao Y, Chu J, and Yu XW. Reduced cd200 and cd200r1 expression in human chorionic villi contributes to early spontaneous abortion. Acta obstetricia gynecologica Scandinavica. (2014) 93:1248–54. doi: 10.1111/aogs.12476
38. Gao H, Zhang T, Li K, and Li X. Cd73: A new immune checkpoint for leukemia treatment. Front Immunol. (2025) 16:1486868. doi: 10.3389/fimmu.2025.1486868
39. Riccio LGC, Andres MP, Deho IZ, Fontanari GO, and Abrao MS. Foxp3(+)Cd39(+)Cd73(+) regulatory T-cells are decreased in the peripheral blood of women with deep infiltrating endometriosis. Clinics (Sao Paulo). (2024) 79:100390. doi: 10.1016/j.clinsp.2024.100390
40. Gao X, Hao Z, Du J, Zhang X, Yang S, Hu T, et al. Compromised adenosine-A2ar axis contributes to recurrent spontaneous abortion by promoting proinflammatory macrophage polarization. Int Immunopharmacol. (2025) 158:114838. doi: 10.1016/j.intimp.2025.114838
41. Forstner D, Guettler J, Brugger BA, Lyssy F, Neuper L, Daxboeck C, et al. Cd39 abrogates platelet-derived factors induced il-1beta expression in the human placenta. Front Cell Dev Biol. (2023) 11:1183793. doi: 10.3389/fcell.2023.1183793
42. Krop J, van der Meeren LE, van der Hoorn MP, Ijsselsteijn ME, Dijkstra KL, Kapsenberg H, et al. Identification of a unique intervillous cellular signature in chronic histiocytic intervillositis. Placenta. (2023) 139:34–42. doi: 10.1016/j.placenta.2023.05.007
43. Labarrere CA, Bammerlin E, Hardin JW, and Dicarlo HL. Intercellular adhesion molecule-1 expression in massive chronic intervillositis: implications for the invasion of maternal cells into fetal tissues. Placenta. (2014) 35:311–7. doi: 10.1016/j.placenta.2014.02.006
44. Freitag L, von Kaisenberg C, Kreipe H, and Hussein K. Expression analysis of leukocytes attracting cytokines in chronic histiocytic intervillositis of the placenta. Int J Clin Exp Pathol. (2013) 6:1103–11. doi: 10.62347/IJCEP061103
45. Heller DS. Cd68 immunostaining in the evaluation of chronic histiocytic intervillositis. Arch Pathol Lab Med. (2012) 136:657–9. doi: 10.5858/arpa.2011-0328-OA
46. Brady CA, Riley T, Batra G, Crocker I, and Heazell AEP. Characterizing histopathologic features in pregnancies with chronic histiocytic intervillositis using computerized image analysis. Arch Pathol Lab Med. (2024) 148:430–42. doi: 10.5858/arpa.2022-0494-OA
47. Hussein K, Stucki-Koch A, Muller AM, Arnold R, Kreipe H, and Feist H. Complement receptor-associated cd163(+)/cd18(+)/cd11c(+)/cd206(-)/cd209(-) expression profile in chronic histiocytic intervillositis of the placenta. Placenta. (2019) 78:23–8. doi: 10.1016/j.placenta.2019.02.007
48. Camesi A, Wettstein R, Valido E, Nyfeler N, Stojic S, Glisic M, et al. Advancements in cell-based therapies for the treatment of pressure injuries: A systematic review of interventional studies. J Tissue Eng. (2023) 14:20417314231201071. doi: 10.1177/20417314231201071
49. Lv M, Jia Y, Dong J, Wu S, and Ying H. The landscape of decidual immune cells at the maternal-fetal interface in parturition and preterm birth. Inflammation Res. (2025) 74:44. doi: 10.1007/s00011-025-02015-6
50. Dogra P and Farber DL. Stealth killing by uterine nk cells for tolerance and tissue homeostasis. Cell. (2020) 182:1074–6. doi: 10.1016/j.cell.2020.08.018
51. Reus AD, van Besouw NM, Molenaar NM, Steegers EA, Visser W, de Kuiper RP, et al. An immunological basis for chronic histiocytic intervillositis in recurrent fetal loss. Am J Reprod Immunol. (2013) 70:230–7. doi: 10.1111/aji.12125
52. Lee HM, Ghill BK, Park E, Park CY, Choi WK, and Lee J. Changes in the ratio of T helper 1 to T helper 2 signature cytokines in patients undergoing living donor liver transplantation surgery: A prospective controlled study. Transplant Proc. (2018) 50:3621–5. doi: 10.1016/j.transproceed.2018.08.055
53. Robertson SA, Care AS, and Moldenhauer LM. Regulatory T cells in embryo implantation and the immune response to pregnancy. J Clin Invest. (2018) 128:4224–35. doi: 10.1172/JCI122182
54. Tekguc M, Wing JB, Osaki M, Long J, and Sakaguchi S. Treg-expressed ctla-4 depletes cd80/cd86 by trogocytosis, releasing free pd-L1 on antigen-presenting cells. Proc Natl Acad Sci U.S.A. (2021) 118:e2023739118. doi: 10.1073/pnas.2023739118
55. Capuani C, Meggetto F, Duga I, Danjoux M, March M, Parant O, et al. Specific infiltration pattern of foxp3+ Regulatory T cells in chronic histiocytic intervillositis of unknown etiology. Placenta. (2013) 34:149–54. doi: 10.1016/j.placenta.2012.12.004
56. Ishigame H, Zenewicz LA, Sanjabi S, Licona-Limon P, Nakayama M, Leonard WJ, et al. Excessive th1 responses due to the absence of tgf-beta signaling cause autoimmune diabetes and dysregulated treg cell homeostasis. Proc Natl Acad Sci U.S.A. (2013) 110:6961–6. doi: 10.1073/pnas.1304498110
57. Hirata Y, Shimazaki S, Suzuki S, Henmi Y, Komiyama H, Kuwayama T, et al. Beta-hydroxybutyrate suppresses nlrp3 inflammasome-mediated placental inflammation and lipopolysaccharide-induced fetal absorption. J Reprod Immunol. (2021) 148:103433. doi: 10.1016/j.jri.2021.103433
58. Scott LM, Bryant AH, Rees A, Down B, Jones RH, and Thornton CA. Production and regulation of interleukin-1 family cytokines at the materno-fetal interface. Cytokine. (2017) 99:194–202. doi: 10.1016/j.cyto.2017.07.005
59. Mattuizzi A, Sauvestre F, Fargeix T, White E, Leibler C, Cargou M, et al. Inflammasome-targeted therapy might prevent adverse perinatal outcomes of recurrent chronic intervillositis of unknown etiology. Nat Commun. (2024) 15:9396. doi: 10.1038/s41467-024-53591-w
60. Tanaka H, Ozawa R, Henmi Y, Hosoda M, Karasawa T, Takahashi M, et al. Gasdermin D regulates soluble fms-like tyrosine kinase 1 release in macrophages. Reprod Biol. (2024) 24:100857. doi: 10.1016/j.repbio.2024.100857
61. Mulla MJ, Salmon JE, Chamley LW, Brosens JJ, Boeras CM, Kavathas PB, et al. A role for uric acid and the nalp3 inflammasome in antiphospholipid antibody-induced il-1beta production by human first trimester trophoblast. PLoS One. (2013) 8:e65237. doi: 10.1371/journal.pone.0065237
62. Reis AS, Barboza R, Murillo O, Barateiro A, Peixoto EPM, Lima FA, et al. Inflammasome activation and il-1 signaling during placental malaria induce poor pregnancy outcomes. Sci Adv. (2020) 6:eaax6346. doi: 10.1126/sciadv.aax6346
63. Trampont P, Roudier M, Andrea AM, Nomal N, Mignot TM, Leborgne-Samuel Y, et al. The placental-umbilical unit in sickle cell disease pregnancy: A model for studying in vivo functional adjustments to hypoxia in humans. Hum Pathol. (2004) 35:1353–9. doi: 10.1016/j.humpath.2004.07.003
64. Lodge-Tulloch NA, Pare JF, Couture C, Bernier E, Cotechini T, Girard S, et al. Maternal innate immune reprogramming after complicated pregnancy. Am J Reprod Immunol. (2024) 92:e13908. doi: 10.1111/aji.13908
65. You Y, Stelzl P, Joseph DN, Aldo PB, Maxwell AJ, Dekel N, et al. Tnf-alpha regulated endometrial stroma secretome promotes trophoblast invasion. Front Immunol. (2021) 12:737401. doi: 10.3389/fimmu.2021.737401
66. Almasry SM, Eldomiaty MA, Elfayomy AK, and Habib FA. Expression pattern of tumor necrosis factor alpha in placentae of idiopathic fetal growth restriction. J Mol Histol. (2012) 43:253–61. doi: 10.1007/s10735-012-9410-6
67. Fukushima K, Miyamoto S, Tsukimori K, Kobayashi H, Seki H, Takeda S, et al. Tumor necrosis factor and vascular endothelial growth factor induce endothelial integrin repertories, regulating endovascular differentiation and apoptosis in a human extravillous trophoblast cell line. Biol Reprod. (2005) 73:172–9. doi: 10.1095/biolreprod.104.039479
68. Xian N, Chen W, Zhang Y, Li J, Zhang N, and Ye Y. Correlation of the expressions of advanced glycation end products and its receptor in serum and placenta with the pathogenesis of preeclampsia. Zhonghua fu chan ke za zhi. (2015) 50:493–9. doi: 10.3760/cma.j.issn.0529-567x.2015.07.005
69. Sasaki H, Xu XC, Smith DM, Howard T, and Mohanakumar T. Hla-G expression protects porcine endothelial cells against natural killer cell-mediated xenogeneic cytotoxicity. Transplantation. (1999) 67:31–7. doi: 10.1097/00007890-199901150-00005
70. Landek-Salgado MA, Rose NR, and Caturegli P. Placenta suppresses experimental autoimmune hypophysitis through soluble tnf receptor 1. J Autoimmun. (2012) 38:J88–96. doi: 10.1016/j.jaut.2011.07.001
71. Ricklin D, Reis ES, and Lambris JD. Complement in disease: A defence system turning offensive. Nat Rev Nephrol. (2016) 12:383–401. doi: 10.1038/nrneph.2016.70
72. Viall CA and Chamley LW. Histopathology in the placentae of women with antiphospholipid antibodies: A systematic review of the literature. Autoimmun Rev. (2015) 14:446–71. doi: 10.1016/j.autrev.2015.01.008
73. Mehra NK and Baranwal AK. Clinical and immunological relevance of antibodies in solid organ transplantation. Int J Immunogenet. (2016) 43:351–68. doi: 10.1111/iji.12294
74. Bendon RW, Coventry S, Thompson M, Rudzinski ER, Williams EM, and Oron AP. Significance of C4d immunostaining in placental chronic intervillositis. Pediatr Dev Pathol. (2015) 18:362–8. doi: 10.2350/14-12-1582-OA.1
75. Chan ES, de Koning L, Yu W, and Chadha R. C4d staining is present in normal placentas from pregnancies prior to pregnancy loss associated with chronic histiocytic intervillositis and is reduced by immunomodulatory therapy in subsequent pregnancies. Pediatr Dev Pathol. (2023) 26:374–87. doi: 10.1177/10935266231176682
76. Valenzuela NM, Thomas KA, Mulder A, Parry GC, Panicker S, and Reed EF. Complement-mediated enhancement of monocyte adhesion to endothelial cells by hla antibodies, and blockade by a specific inhibitor of the classical complement cascade, tnt003. Transplantation. (2017) 101:1559–72. doi: 10.1097/TP.0000000000001486
77. He Y, Xu B, Song D, Yu F, Chen Q, and Zhao M. Expression of the complement system’s activation factors in plasma of patients with early/late-onset severe pre-eclampsia. Am J Reprod Immunol. (2016) 76:205–11. doi: 10.1111/aji.12541
78. Marton T, Hargitai B, Hunter K, Pugh M, and Murray P. Massive perivillous fibrin deposition and chronic histiocytic intervillositis a complication of sars-cov-2 infection. Pediatr Dev Pathol. (2021) 24:450–4. doi: 10.1177/10935266211020723
79. Xuan YH, Choi YL, Shin YK, Ahn GH, Kim KH, Kim WJ, et al. Expression of tgf-beta signaling proteins in normal placenta and gestational trophoblastic disease. Histol histopathology. (2007) 22:227–34. doi: 10.14670/HH-22.227
80. Horvat Mercnik M, Schliefsteiner C, Sanchez-Duffhues G, and Wadsack C. Tgfbeta signalling: A nexus between inflammation, placental health and preeclampsia throughout pregnancy. Hum Reprod Update. (2024) 30:442–71. doi: 10.1093/humupd/dmae007
81. Wen B, Liao H, Lin W, Li Z, Ma X, Xu Q, et al. The role of tgf-beta during pregnancy and pregnancy complications. Int J Mol Sci. (2023) 24:16882. doi: 10.3390/ijms242316882
82. Fang L, Yan Y, Gao Y, Wu Z, Wang Z, Yang S, et al. Tgf-Beta1 Inhibits Human Trophoblast Cell Invasion by Upregulating Kisspeptin Expression through Erk1/2 but Not Smad Signaling Pathway. Reprod Biol endocrinology: RB&E. (2022) 20:22. doi: 10.1186/s12958-022-00902-9
83. Jia Y, Xie H, Zhang J, and Ying H. Induction of tgf-beta receptor I expression in a DNA methylation-independent manner mediated by dnmt3a downregulation is involved in early-onset severe preeclampsia. FASEB J. (2020) 34:13224–38. doi: 10.1096/fj.202000253RR
84. Keller CC, Eikmans M, van der Hoorn MP, and Lashley L. Recurrent miscarriages and the association with regulatory T cells; a systematic review. J Reprod Immunol. (2020) 139:103105. doi: 10.1016/j.jri.2020.103105
85. Grygorczuk S, Chmielewski T, Zajkowska J, Swierzbinska R, Pancewicz S, Kondrusik M, et al. Concentration of tgf-beta1 in the supernatant of peripheral blood mononuclear cells cultures from patients with early disseminated and chronic lyme borreliosis. Adv Med Sci. (2007) 52:174–8. doi: 10.1016/s0001-5180(07)80039-8
86. Matsuzaki K. Smad phosphoisoform signals in acute and chronic liver injury: similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. (2012) 347:225–43. doi: 10.1007/s00441-011-1178-6
87. Araki-Taguchi M, Nomura S, Ino K, Sumigama S, Yamamoto E, Kotani-Ito T, et al. Angiotensin ii mimics the hypoxic effect on regulating trophoblast proliferation and differentiation in human placental explant cultures. Life Sci. (2008) 82:59–67. doi: 10.1016/j.lfs.2007.10.017
88. Bai SX, Wang YL, Qin L, Xiao ZJ, Herva R, and Piao YS. Dynamic expression of matrix metalloproteinases (Mmp-2, -9 and -14) and the tissue inhibitors of mmps (Timp-1, -2 and -3) at the implantation site during tubal pregnancy. Reproduction. (2005) 129:103–13. doi: 10.1530/rep.1.00283
89. Han H, Dai D, Du R, Hu J, Zhu Z, Lu L, et al. Oncostatin M promotes infarct repair and improves cardiac function after myocardial infarction. Am J Transl Res. (2021) 13:11329–40. doi: 10.62347/AJTR0129180
90. Li S, Mohamedi AH, Senkowsky J, Nair A, and Tang L. Imaging in chronic wound diagnostics. Adv Wound Care (New Rochelle). (2020) 9:245–63. doi: 10.1089/wound.2019.0967
91. Uxa R, Baczyk D, Kingdom JC, Viero S, Casper R, and Keating S. Genetic polymorphisms in the fibrinolytic system of placentas with massive perivillous fibrin deposition. Placenta. (2010) 31:499–505. doi: 10.1016/j.placenta.2010.03.013
92. Mao Q, Chu S, Shapiro S, Young L, Russo M, and De Paepe ME. Placental sars-cov-2 distribution correlates with level of tissue oxygenation in covid-19-associated necrotizing histiocytic intervillositis/perivillous fibrin deposition. Placenta. (2022) 117:187–93. doi: 10.1016/j.placenta.2021.12.002
93. Nicaise AM, Johnson KM, Willis CM, Guzzo RM, and Crocker SJ. Timp-1 promotes oligodendrocyte differentiation through receptor-mediated signaling. Mol Neurobiol. (2019) 56:3380–92. doi: 10.1007/s12035-018-1310-7
94. Snir A, Brenner B, Paz B, Ohel G, and Lanir N. The role of fibrin matrices and tissue factor in early-term trophoblast proliferation and spreading. Thromb Res. (2013) 132:477–83. doi: 10.1016/j.thromres.2013.08.023
95. Castillo MM, Yang Q, Sigala AS, McKinney DT, Zhan M, Chen KL, et al. The endothelial protein C receptor plays an essential role in the maintenance of pregnancy. Sci Adv. (2020) 6:eabb6196. doi: 10.1126/sciadv.abb6196
96. Lanir N, Aharon A, and Brenner B. Haemostatic mechanisms in human placenta. Best Pract Res Clin Haematol. (2003) 16:183–95. doi: 10.1016/s1521-6926(02)00098-1
97. Kaufmann P, Huppertz B, and Frank HG. The fibrinoids of the human placenta: origin, composition and functional relevance. Ann Anat. (1996) 178:485–501. doi: 10.1016/S0940-9602(96)80102-6
98. Farran SH, Rabah R, and Simon C. Sars-cov-2 placentitis: A review of pathologic findings and discussion of differential diagnosis. Arch Pathol Lab Med. (2025) 149:e291–97. doi: 10.5858/arpa.2024-0247-RA
99. Schwartz DA. Stillbirth after covid-19 in unvaccinated mothers can result from Sars-Cov-2 placentitis, placental insufficiency, and hypoxic ischemic fetal demise, not direct fetal infection: potential role of maternal vaccination in pregnancy. Viruses. (2022) 14:458. doi: 10.3390/v14030458
100. Shao W, Huang Y, Wang L, Li P, Jia Y, and Zhang J. Expression of fibrinogen-like protein 2 (Fgl2) on toll-like receptor 9 (Tlr9) expression in autoimmune myelitis. Int Immunopharmacol. (2023) 114:109539. doi: 10.1016/j.intimp.2022.109539
101. Hannibal RL, Cardoso-Moreira M, Chetty SP, Lau J, Qi Z, Gonzalez-Maldonado E, et al. Investigating human placentation and pregnancy using first trimester chorionic villi. Placenta. (2018) 65:65–75. doi: 10.1016/j.placenta.2018.03.005
102. Pierleoni C, Castellucci M, Kaufmann P, Lund LR, and Schnack Nielsen B. Urokinase receptor is up-regulated in endothelial cells and macrophages associated with fibrinoid deposits in the human placenta. Placenta. (2003) 24:677–85. doi: 10.1016/s0143-4004(03)00082-1
103. El-Dessouki AM, Alzokaky AA, Raslan NA, Ibrahim S, Selim H, and Al-Karmalawy AA. Dabigatran attenuates methotrexate-induced hepatotoxicity by regulating coagulation, endothelial dysfunction, and the nf-kb/il-1beta/mcp-1 and tlr4/nlrp3 signaling pathways. Naunyn Schmiedebergs Arch Pharmacol. (2024) 398:5129–45. doi: 10.1007/s00210-024-03567-w
104. Baergen RN, Heller DS, and Goldstein JA. Placental pathology in covid-19. Am J Clin Pathol. (2020) 154:279. doi: 10.1093/ajcp/aqaa101
105. Sauvestre F, Mattuizzi A, Sentilhes L, Poingt M, Blanco P, Houssin C, et al. Chronic intervillositis of unknown etiology: development of a grading and scoring system that is strongly associated with poor perinatal outcomes. Am J Surg Pathol. (2020) 44:1367–73. doi: 10.1097/PAS.0000000000001549
106. Koby L, Keating S, Malinowski AK, and D’Souza R. Chronic histiocytic intervillositis - clinical, biochemical and radiological findings: an observational study. Placenta. (2018) 64:1–6. doi: 10.1016/j.placenta.2018.02.002
107. Ozawa N, Yamaguchi K, Shibata M, Sugibayashi R, Yagi H, Sago H, et al. Chronic histiocytic intervillositis in three consecutive pregnancies in a single patient: differing clinical results and pathology according to treatment used. J obstetrics gynaecology Res. (2017) 43:1504–8. doi: 10.1111/jog.13404
108. Moar L, Simela C, Nanda S, Marnerides A, Al-Adnani M, Nelson-Piercy C, et al. Chronic histiocytic intervillositis (Chi): current treatments and perinatal outcomes, a systematic review and a meta-analysis. Front Endocrinol (Lausanne). (2022) 13:945543. doi: 10.3389/fendo.2022.945543
109. Chirumbolo S, Marzotto M, Conforti A, Vella A, Ortolani R, and Bellavite P. Bimodal action of the flavonoid quercetin on basophil function: an investigation of the putative biochemical targets. Clin Mol Allergy. (2010) 8:13. doi: 10.1186/1476-7961-8-13
110. Machha A, Achike FI, Mustafa AM, and Mustafa MR. Quercetin, a flavonoid antioxidant, modulates endothelium-derived nitric oxide bioavailability in diabetic rat aortas. Nitric Oxide. (2007) 16:442–7. doi: 10.1016/j.niox.2007.4.001
111. Ebegboni VJ, Dickenson JM, and Sivasubramaniam SD. Antioxidative effects of flavonoids and their metabolites against hypoxia/reoxygenation-induced oxidative stress in a human first trimester trophoblast cell line. Food Chem. (2019) 272:117–25. doi: 10.1016/j.foodchem.2018.08.036
112. Xie G, Zhang Q, Dong J, Fang Z, Che L, Lin Y, et al. Maternal vitamin D3 supplementation in an oxidized-oil diet protects fetus from developmental impairment and ameliorates oxidative stress in mouse placenta and fetus. J Nutr. (2024) 154:2920–31. doi: 10.1016/j.tjnut.2024.07.025
113. Stenhouse C, Suva LJ, Gaddy D, Wu G, and Bazer FW. Phosphate, calcium, and vitamin D: key regulators of fetal and placental development in mammals. Adv Exp Med Biol. (2022) 1354:77–107. doi: 10.1007/978-3-030-85686-1_5
Keywords: CHI, placental inflammation, macrophage, maternal-fetal interface, alloantigen
Citation: Zhang X, Fang Z and Wang X (2025) Gaps in maternal-fetal interface rejection response: chronic histiocytic intervillositis. Front. Immunol. 16:1625701. doi: 10.3389/fimmu.2025.1625701
Received: 12 May 2025; Accepted: 05 September 2025;
Published: 19 September 2025.
Edited by:
Uday Kishore, United Arab Emirates University, United Arab EmiratesReviewed by:
Panicos Shangaris, King’s College London, United KingdomVladimir S. Rogovskii, Pirogov Russian National Research Medical University, Russia
Copyright © 2025 Zhang, Fang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xietong Wang, enhxdGcyMDI0QDE2My5jb20=