 Mengnan Liu1†
Mengnan Liu1† Mengting Yang2,3†
Mengting Yang2,3† Yue Qi2,3†Yuting Ma2,3†Qulian Guo2,3
Yue Qi2,3†Yuting Ma2,3†Qulian Guo2,3 Ling Guo2,3Chunyan Liu2,3
Ling Guo2,3Chunyan Liu2,3 Wenjun Liu2,3*Lan Xiao2,3*
Wenjun Liu2,3*Lan Xiao2,3* You Yang2,3*
You Yang2,3*- 1Department of Cardiovascular Medicine, Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, China
- 2Department of Pediatrics (Children Hematological Oncology), Birth Defects and Childhood Hematological Oncology Laboratory, The Affiliated Hospital of Southwest Medical University, Sichuan Clinical Research Center for Birth Defects, Luzhou, Sichuan, China
- 3Department of Pediatrics, Southwest Medical University, Luzhou, Sichuan, China
Immunotherapy has emerged as a cornerstone strategy for augmenting therapeutic efficacy in acute myeloid leukemia (AML). The immunosuppressive AML microenvironment, characterized by profound immune dysfunction, critically impairs anti-leukemic immune surveillance. This immunologically hostile niche is principally governed by specialized immunosuppressive cell populations—notably regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), leukemia-associated macrophages (LAMs), and regulatory B cells (Bregs)—which collectively establish an immune-privileged sanctuary for leukemic cells. This review critically examines three fundamental aspects of these immunosuppressive regulators in AML pathogenesis: (1) their recruitment dynamics within the leukemic niche, (2) the molecular mechanisms underlying their immunosuppressive functions, and (3) current and emerging therapeutic approaches designed to neutralize their inhibitory effects. Through this comprehensive analysis, we aim to provide a mechanistic framework for developing more effective immunotherapeutic interventions against AML.
1 Introduction
Acute myeloid leukemia (AML) is a highly aggressive hematologic malignancy characterized by uncontrolled clonal proliferation of immature myeloid cells, resulting in the accumulation of abnormal blast cells in the bone marrow (BM) and impairment of normal hematopoietic function (1). AML is the most prevalent form of leukemia in adults, with an annual incidence rate of approximately 3 to 5 cases per 100,000 individuals (2–4). AML patients typically have a poor prognosis, marked by a short survival time and unsatisfactory clinical outcomes. AML is a profoundly heterogeneous hematologic malignancy with multifaceted pathophysiology involving: genomic instability and mutational accumulation, oncogenic fusion events, epigenetic reprogramming, immune dysregulation and inflammatory cascades, apoptosis resistance mechanisms, metabolic pathway derangements, cellular senescence evasion, growth suppression circumvention, and sustained proliferative signaling (5–12).
Current AML treatment strategies include conventional chemotherapy, targeted therapies (FLT3/IDH/BCL-2 inhibitors), hematopoietic stem cell transplantation, and emerging immunotherapies (CAR-T, checkpoint inhibitors) with microenvironment-modulating approaches (13). Although advancements in treatment have led to improvements in AML prognosis, challenges such as chemoresistance, relapse, and refractory disease persist as significant barriers (14).
Emerging evidence underscores the pivotal role of bone marrow niche dysregulation in AML pathogenesis (9, 10, 15). During disease progression, the microenvironment undergoes profound cellular and functional remodeling, creating a permissive ecosystem that sustains leukemic cell survival (16). Notably, the AML microenvironment exhibits prominent immunosuppressive characteristics (17). Key immunosuppressive cell populations—including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), leukemia-associated macrophages (LAMs), regulatory B cells (Bregs) and leukemia-associated neutrophils (LANs)—employ diverse mechanisms to facilitate immune evasion by leukemic cells. Therapeutic targeting of these immunosuppressive populations represents a promising strategic approach for AML immunotherapy. A comprehensive understanding of the regulatory networks of these immunosuppressive cells is crucial for developing novel immunotherapeutic strategies. This review provides a comprehensive analysis of the role and mechanisms of crucial immunosuppressive cells within the AML microenvironment, including Tregs, MDSCs, LAMs, Bregs and LANs, to serve as a reference for future research in this field.
2 The famous immunosuppressive cell: regulatory T cell
2.1 The phenotype of Treg
Tregs represent a heterogeneous population of T cells, exhibiting diverse origins, phenotypes, and effects. The traditional classification of Tregs comprises two primary subsets: thymic Tregs (tTregs), also referred to as natural Tregs (nTregs), and peripheral Tregs (pTregs), alternatively known as induced Tregs (iTregs) or adaptive Tregs (aTregs), depending on their distinct sources (18). In the thymus, a subset of CD4 single-positive autoreactive cells successfully undergo negative selection by expressing FOXP3, leading to their differentiation into thymic Tregs (tTregs). These tTregs make up approximately 5% to 10% of CD4+ T cells present in the peripheral blood (PB) (19, 20). pTregs are generated from naive CD4+ T cells in the peripheral tissues in response to various stimuli, including antigens, as well as factors like TGF-β and IL-2 (21, 22). Interestingly, Treg cells display a relatively anergic state and are unable to produce IL-2 due to the transcriptional repressive effects of FOXP3 (23), despite the fact that IL-2 is essential for the generation, survival, and activation of Tregs (24). Aside from the conventional CD4+ Treg cells mentioned previously, several other T cell subsets have been identified to possess immunosuppressive capabilities. These include CD8+ T cells (25), IL-17+ Treg cells (26), ICOS+ Treg cells (27), Type II NKT cells (28, 29), and γδT cells (30). A comprehensive summary detailing the phenotypes of T cells exhibiting regulatory properties can be found in Table 1.
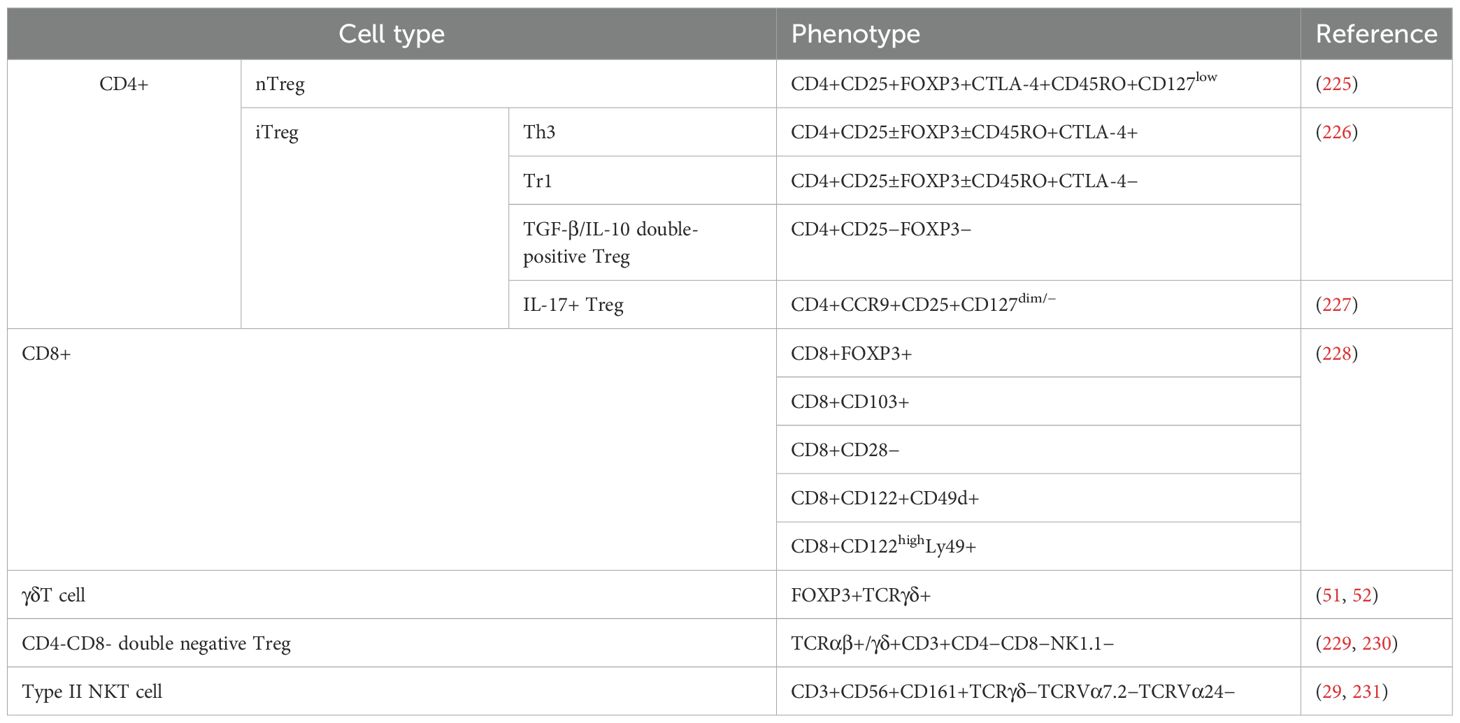
Table 1. Phenotypes of T cells with regulatory properties.
Currently, the primary markers employed for the identification of conventional Tregs are CD25high, CD127low/−, and FOXP3+ (31). Furthermore, several supplementary molecules, including CD45RA (32), CD39/CD73 (33), CD26 (34), CD6 (35), NRP-1 (36), TIM-3 (37), and others (38), can serve as surface markers for Tregs.
2.2 Treg accumulation and its mechanisms in AML
Numerous studies have demonstrated an elevated frequency of Tregs in the BM and PB of AML patients. The heightened accumulation of Tregs within the AML microenvironment not only facilitates the development and advancement of AML but also amplifies treatment resistance and the likelihood of relapse.
2.2.1 Elevated Tregs observed in AML occurrence, drug resistance, and relapse
Elevated percentages of Tregs contribute to the establishment of an immunosuppressive microenvironment in AML, providing favorable conditions for the survival and proliferation of malignant AML cells. Consequently, this immunosuppressive milieu plays a facilitating role in the progression and pathogenesis of the disease. Wang et al. discovered that individuals newly diagnosed with AML exhibited an increased proportion of CD4+CD25high Tregs in both PB and BM. Notably, these Tregs displayed a more robust state of renewal, characterized by heightened rates of proliferation and apoptosis, when compared to healthy donors (39). The elevated presence of Tregs in newly diagnosed AML patients results in a reduced ratio of Th17/Treg cells. This finding confirms the immunosuppressive polarization of the bone marrow microenvironment in AML (40). In the PB of AML patients, circulating T follicular regulatory cells (cTfr), defined as CD4+CXCR5+PD-1+FOXP3+, were elevated, indicating increased suppression of B cell responses (41). Additional studies have consistently identified greater proportions of Tregs in the BM and PB of patients diagnosed with AML compared to healthy control subjects (42, 43). These findings underscore the abundant presence of Tregs in AML and their role in establishing an immunosuppressive microenvironment. Contrary to previous beliefs, a recent report suggests that the proportion of Tregs in the BM is similar between individuals with AML and healthy donors. However, it was observed that AML patients exhibit higher proportions of effector Tregs (CD45RA− Tregs). Furthermore, the study found a significant increase in PD1+/TIGIT+ Tregs in the BM of AML patients with a high leukemia burden (44). This suggests that the AML microenvironment may intensify the regulatory function of Tregs, and the number of Tregs present is influenced by the extent of leukemia burden.
In addition to the involvement in pathogenesis, Tregs have also been demonstrated a connection to chemotherapy resistance and disease relapse. Szczepanski et al. conducted a study that reaffirmed the observation of elevated percentages of Tregs and their suppressive activity in the PB of AML patients. Remarkably, the study found that patients with a lower frequency of Tregs at the time of diagnosis exhibited a more positive response to induction chemotherapy (45). Ersvaer et al. observed persistent high frequency of Tregs in AML patients both prior to chemotherapy and throughout the period of cytopenia induced by intensive chemotherapy. Additionally, these proportions remained elevated during the regeneration phase following treatment (46). Moreover, several other research groups have reported an increase in Treg expansion in the PB during the recovery of lymphocytes after intensive chemotherapy and during cytotoxic maintenance chemotherapy (47, 48). Several studies have indicated that patients with AML who achieved complete remission (CR) experienced a notable decrease in Treg frequency compared to those at the time of diagnosis (42, 49), and Zhang et al. further observed a sudden increase in Tregs during relapse, suggesting that monitoring Treg frequency after achieving CR could serve as a valuable predictor of relapse (49). Additionally, findings from a phase IV clinical trial (NCT01347996) revealed that the accumulation of Tregs in the PB as a result of immunotherapy with HDC/IL-2 is associated with the risk of relapse in AML. In cycle 3 of the treatment, a decrease in Treg accumulation was indicative of a lower risk of relapse, supporting the notion that the prolonged presence of Tregs may adversely affect the prognosis of AML (50). Strikingly, in Szczepanski’s study, patients who achieved CR still maintained an increased frequency of Tregs, which was counterintuitive and inconsistent with the observations of other researchers. They proposed an interesting conclusion that Tregs are resistant to conventional chemotherapy (45). In addition to the conventional Tregs, studies have also shown that γδ Treg cells are increased in AML patients and correlated with unfavorable clinical outcomes (51, 52). Therefore, the assessment of Treg frequency holds considerable importance in understanding the progression of leukemia, treatment response, and prognosis in AML patients. A compilation of studies focusing on Treg accumulation in AML can be found in Supplementary Table S1.
2.2.2 Accumulation mechanisms of Tregs in AML microenvironment
Numerous studies have elucidated the mechanisms underlying the accumulation of Tregs within the microenvironment of AML. These well-established mechanisms encompass the secretion of specific factors, interactions between receptors and ligands, chemotactic effects, and metabolic advantages (Figure 1). Subsequently, we will delve into each of these mechanisms in detail.
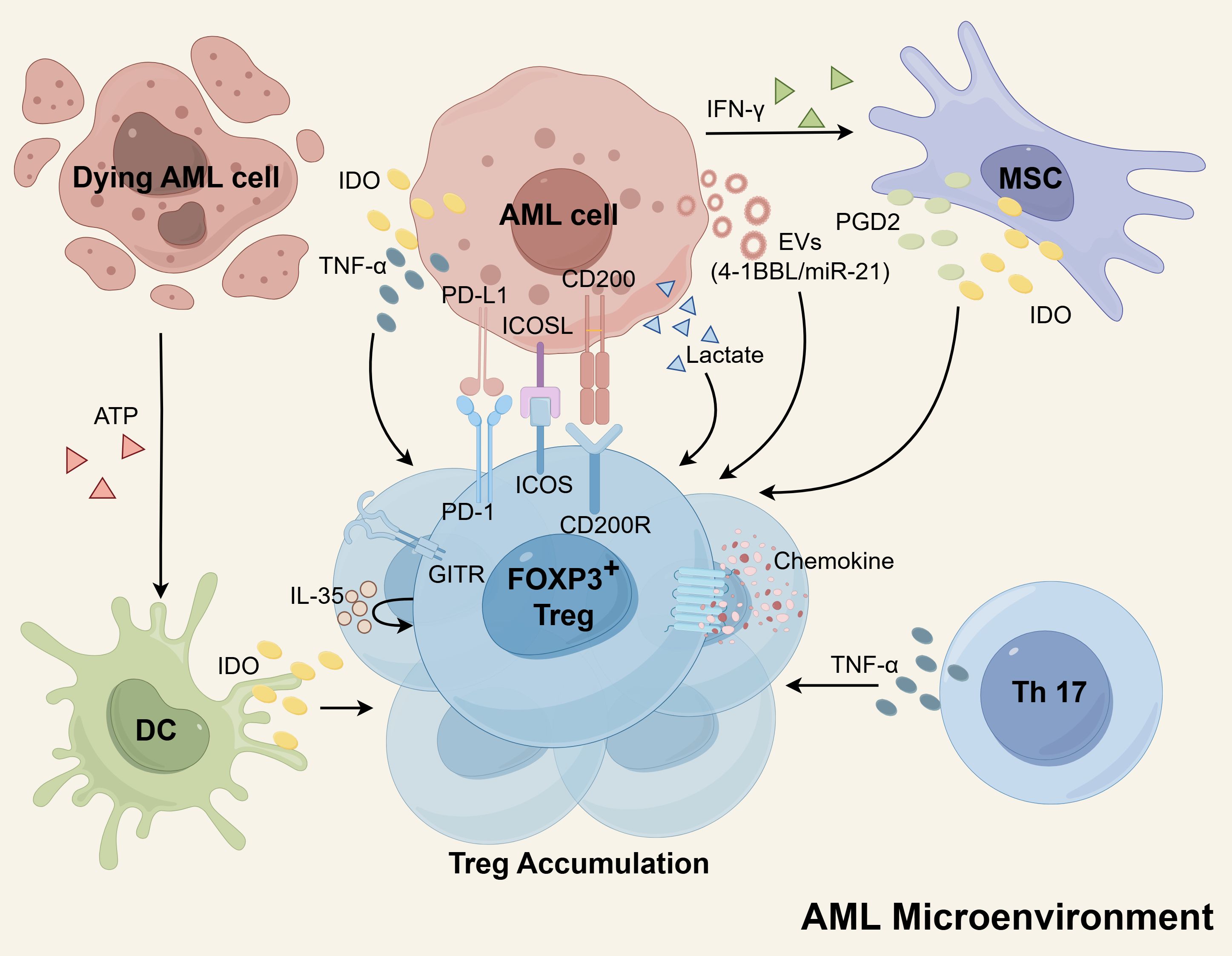
Figure 1. The mechanisms of Treg cells accumulation in the AML microenvironment. The secretion of EVs by AML cells plays a role in increasing Tregs, as these EVs contain molecules such as miR-21 and 4-1BBL that promote Treg expansion. Additionally, AML cells, DCs, and MSCs can produce IDO, which induces proliferation of Tregs. MSCs also release PGD2 to enhance Treg numbers. Both Th17 cells and AML cells express TNF-α, which supports the expansion of Tregs. Furthermore, Tregs themselves express high levels of IL-35, which can further amplify Treg proliferation. The interaction between AML cells and Tregs through receptor-ligand interactions, including PD-L1/PD-1, ICOSL/ICOS, and CD200/CD200R, also promotes Treg expansion. Tregs possess enhanced chemokine receptors, facilitating robust migration and contributing to their aggregation. Moreover, Tregs have a metabolic advantage as they can utilize lactate for metabolism, indirectly contributing to their accumulation. Schematic figure was drawn by Figdraw (www.figdraw.com).
Recent findings have revealed that extracellular vesicles (EVs) derived from AML cells and containing 4-1BBL play a pivotal role in augmenting the expression of FOXP3 and the effector phenotype in Tregs, thereby bolstering their activity. Treg cells actively internalize EVs carrying the costimulatory ligand 4-1BBL, resulting in the upregulation of STAT5 and the suppression of mTOR-S6 signaling. Consequently, this process promotes the immunosuppressive effector Treg cells (53). In addition, miR-21 originating from AML-derived EVs has been demonstrated to promote the expression of genes recognized as markers for Tregs and immunosuppression. These genes include IL-10, FOXP3, CTLA-4, and others. Intriguingly, the transfer of miR-21 into leukemia-infiltrating T lymphocyte cells yielded the acquisition of a Treg cell phenotype, accompanied by a notable increase in FOXP3 levels in AML (54).
Indoleamine 2,3-dioxygenase (IDO) is an enzyme with immunomodulatory properties that facilitates the conversion of tryptophan (Trp) into kynurenines (Kyn). These Kyn metabolites have the ability to promote the generation of Treg (55). The generation of this inducible Treg can be significantly hindered by the IDO inhibitor, 1-methyl tryptophan (1-MT) (56, 57). Arandi et al. revealed that elevated expression of IDO in patients with AML may contribute to an increase in the number of Treg (58). Furthermore, in vitro studies have demonstrated the presence of functionally active IDO proteins within AML cells, which have the capability to stimulate the proliferation of Treg (56, 59). In a study by Curti et al., it was reported that a notable proportion of primary blast cells derived from adult patients with AML constitutively express the active form of IDO protein (60). Conversely, a multicenter study involving pediatric AML patients indicated that blast cells do not exhibit constitutive expression of IDO protein. However, functional IDO protein was found to be upregulated in approximately half of the AML samples in response to IFN-γ stimulation (61). IDO is an IFN-γ-inducible enzyme, whose expression is transcriptionally activated through the JAK-STAT1 signaling pathway in coordination with the transcription factor IRF1 (62). These studies suggest that regardless of whether IDO protein is constitutively expressed or induced, it is evident that AML cells have the capability to produce and release IDO protein. This leads to an elevation of IDO concentration within the microenvironment, consequently promoting the expansion of Treg. Additionally, dendritic cells (DCs) are known to express functional IDO protein, which can hinder the T-cell response by facilitating the expansion of Tregs (62). DCs derived from AML cells have been suggested as potential leukemia vaccines due to their increased immunogenicity. However, one challenge is that these DCs show upregulation of IDO, which can negatively impact immune responses by activating powerful Tregs (63). Clinical sample analysis has demonstrated that adenosine triphosphate (ATP) released by dying AML cells, specifically those targeted by chemotherapy, plays a role in the induction of Tregs. The release of ATP from AML cells treated with chemotherapy leads to the upregulation of IDO1 in DCs. These DCs, in turn, are fully capable of inducing Tregs through the IDO1 pathway in vitro (64). Moreover, bone marrow mesenchymal stem cells (MSCs) derived from AML patients exhibited considerable upregulation of IDO and released heightened levels of PGD2. These factors collectively contributed to the expansion of Tregs (65, 66). PGD2 derived from MSCs engages the receptor CRTH2 on type 2 innate lymphoid cells (ILC2s) to promote the overproduction of IL-5, which specifically expands CD4+CD25+IL5Rα+ Tregs (66). Furthermore, experimental evidence has shown that the release of IFN-γ by AML cells in vitro triggers the upregulation of IDO expression in MSCs. Consequently, this upregulation contributes to the proliferation of Tregs (67, 68).
In AML patients, abnormally high levels of TNF-α secreted by Th17 cells promote Treg proliferation through the TNF-α receptor 2 (TNFR2) pathway expressed by Tregs (69). Additionally, AML blast cells also generate significant quantities of TNF-α, which have the potential to induce the proliferation of Tregs by upregulating the expression of TNFR2 and FOXP3 on T cells (70, 71). Further research has shown that TNF-α binding to TNFR2 activates the p38 MAPK signaling pathway, which upregulates the surface expression of TNFR2 and Foxp3 on Tregs, thereby driving their proliferation and expansion (72, 73). Azacitidine combined with lenalidomide or panobinostat therapy can reduce TNFR2+ Tregs in vivo, which may contribute to the maintenance of clinical remission (70, 74). Previous reports indicate that azacitidine promotes Treg expansion by hypomethylation of the CpG island associated with the promoter of the FOXP3 gene (75, 76). This potentially contradictory finding can be explained by several reasons. First, the combined drugs, lenalidomide or panobinostat, might reverse this effect of azacitidine. In vitro studies have provided evidence that lenalidomide can decrease the expression of FOXP3 and inhibit the expansion of Tregs mediated by IL-2 (77). Similarly, studies have shown that administering low doses of panobinostat can lead to a reduction in FOXP3 expression and Treg frequency (78). Additionally, azacitidine treatment indirectly decreases TNFR2+ Tregs by reducing the population of residual blast cells, as blast cells secrete TNF to stimulate Treg expansion (70, 79). Furthermore, within the AML microenvironment, Tregs express elevated levels of IL-35, which can further contribute to the expansion of Tregs themselves (80).
The expansion of Tregs is facilitated by the interaction between AML cells and Treg cells through receptor-ligand interactions. This includes the interaction of PD-L1 (B7-H1) on the surface of AML cells with PD-1 on Tregs, as well as the ICOSL/ICOS and CD200/CD200R interactions. The expression of PD-L1 on AML cells increases the population of PD1+ Tregs and suppresses anti-leukemia immunity (81, 82). The PD-L1/PD-1 pathway has been found to have a role in driving the conversion of naive T cells into FOXP3+ Tregs by antagonizing the Akt-mTOR signaling pathway (82). Blocking the PD-L1/PD-1 signaling pathway using anti-PD-L1 antibodies has been shown to reduce Treg production and delay the progression of AML in mouse models (83, 84). Han et al. revealed that AML cells possess the ability to express ICOSL, which interacts with ICOS on the surface of Tregs and fosters their proliferation. Through the utilization of an antibody targeting ICOSL, they successfully impeded the generation of ICOS-positive Tregs and effectively retarded the advancement of AML in a murine model (85). Studies have reported that elevated levels of CD200 expression in AML blasts promote the induction of Tregs (86, 87). Inhibition of the interaction between CD200 and its receptor CD200R has been shown to decrease the intensity of FOXP3 (87). Research has demonstrated that the GITR plays a role in promoting the differentiation and expansion of Tregs (88). Furthermore, studies have indicated that surface expression of GITR is increased in Treg of AML patients (45). However, further studies are needed to determine if and how GITR can promote Treg accumulation in AML. Zhou et al. found that Gal-9 defective mice were more resistant to AML cells than wild-type mice, which was associated with less Treg accumulation, hinting that Gal-9 on AML cells may be engaged in expansion of Treg (89). The Gal-9/TIM-3 signaling pathway has been found to contribute to excessive proliferation and activation of Treg cells in chronic lymphocytic leukemia (CLL) (90). Additional evidence is required to determine if a similar role exists in AML.
The expression of chemokine receptors has been demonstrated to play a role in the excessive accumulation of Tregs (91, 92). Specifically in AML, there is an increased presence of TNFR2+ Tregs, which exhibit a heightened capacity for migration towards the BM (74). Additionally, study has reported that the frequencies of Tregs in the BM are significantly higher compared to PB in the same patients with AML (49). In vitro research has also demonstrated that AML-induced DCs exert a significant chemotactic effect on Tregs, which may contribute to the accumulation of Tregs at the site of leukemia (93). Tregs in AML have been shown to display strong migration towards the BM due to their increased expression of the chemokine receptor CXCR4 (43). It has been found that blocking the CCL3-CCR1/CCR5 and CXCL12-CXCR4 axes can slow down AML progression by inhibiting the migration of Tregs into the leukemic hematopoietic microenvironment (94).
Additionally, the metabolic profile of Tregs provides them with a competitive advantage, indirectly promoting aggregation. The hypermetabolic state of tumor cells creates a low-glucose and lactate-rich microenvironment, which is unfavorable for immune effector cells. Tregs possess the ability to reprogram their metabolic profile by regulation of FOXP3, thereby conferring upon them a metabolic edge and enhanced adaptive capacity within this environment (95). In the B16-F10 melanoma mouse model, tumor-infiltrating Treg cells have the capability to utilize lactate as a source of energy to sustain their proliferation and functional activity in a glucose-deficient environment (96). Consistent with this, higher lactate concentrations were observed in BM of AML (97). Zhang et al. reaffirmed the contribution of AML cells to the lactate-rich TME, and then they employed the lactate transporter inhibitor Syrosingopine to reduce lactate production, which resulted in a reduction of Treg. Based on these findings, the researchers concluded that lactate produced by AML cells actively promotes the aggregation of Treg cells (44). Additionally, Tregs in AML displayed an enrichment of pathways linked to fatty acid metabolism, providing further evidence that Tregs have the capacity to enhance energy production through the utilization of fatty acids present in their surrounding environment (98).
2.3 The immunosuppressive mechanisms of Treg in the AML microenvironment
Tregs play a pivotal role in the inhibition of immune effector cells, ultimately leading to the impairment of anti-leukemia immune responses in AML. Tregs achieve this immunosuppressive effect through two ways: cell-to-cell contact and contact-independent pathways (Figure 2). The contact-dependent mechanism primarily involves intricate receptor-ligand interactions between cells, while the contact-independent mechanism predominantly relies on cytokine secretion and other non-secretory means. Subsequently, this section will provide an elaborate elucidation of how Treg cells effectively suppress immune effector cells in AML by employing these two mechanisms.
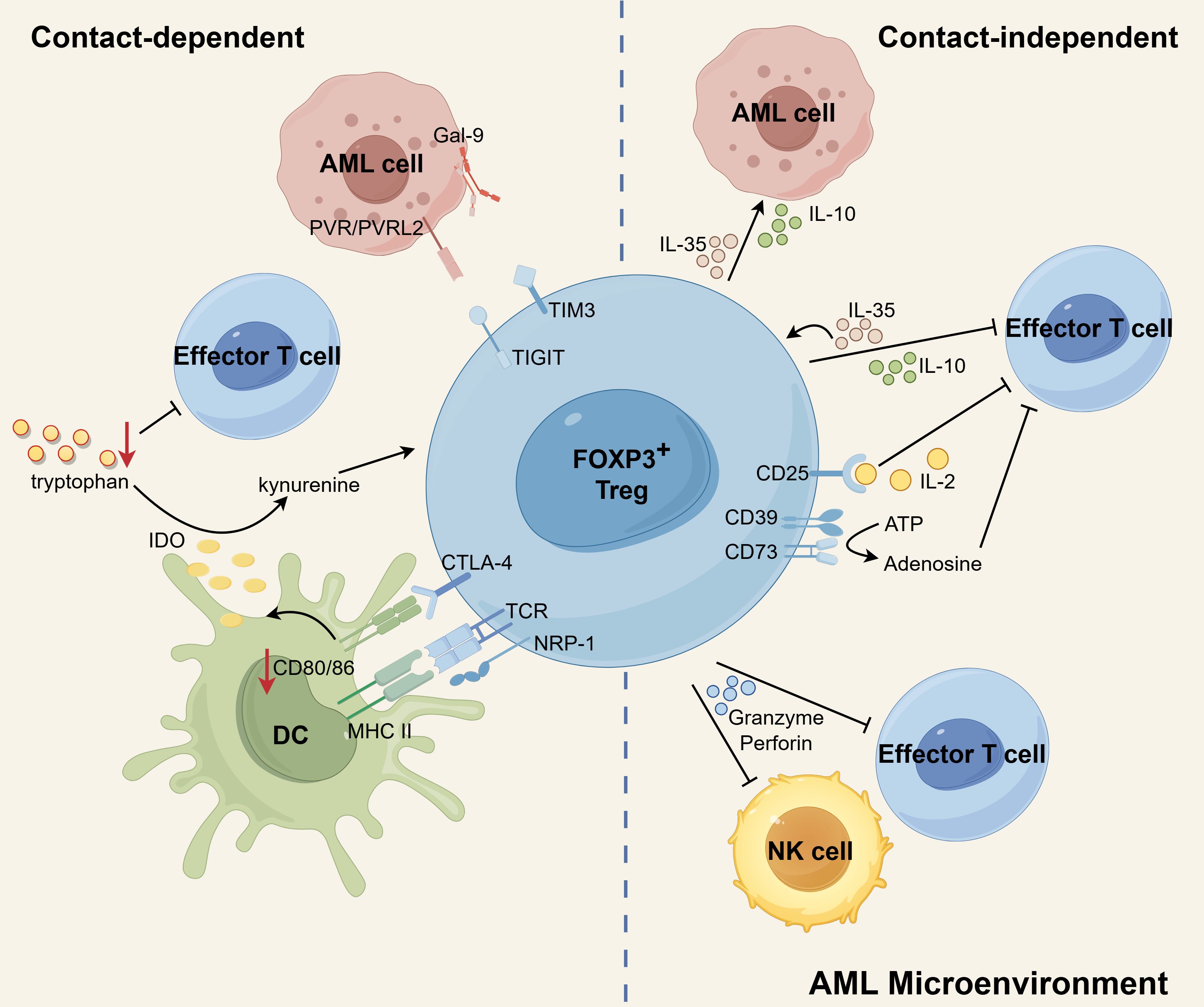
Figure 2. The immunosuppressive mechanisms of Tregs in AML microenvironment. CTLA-4 expressed by Tregs binds to CD80/86 on DCs, leading to inhibition of co-stimulation of Teffs, downregulation of CD80/86 on DCs, and elevated expression of IDO in DCs. By degrading tryptophan to kynurenines, IDO contributes to the induction of Tregs and the suppression of T-cell responses. Additionally, NRP1 prolongs the MHC-II molecule-dependent interactions between Tregs and DCs, which effectively restricts the recruitment of MHC-II peptides to immune synapses, ultimately inhibiting immune responses. Treg-derived IL-10 diminishes anti-leukemia immunity by suppressing the activity of Teffs. IL-35 released by Treg can suppress Teff functions and proliferation while also expanding a population of inducible Tregs. IL-10 and IL-35 also stimulate the proliferation of AML blasts. Additionally, Tregs can induce cell death in NK and Teff cells by utilizing granzyme and perforin. CD25, expressed on Tregs, allows for continuous uptake of IL-2, leading to the cytokine deprivation-induced apoptosis of Teff cells. Tregs express membrane surface enzymes CD39 and CD73, which can hydrolyze ATP to generate adenosine. Adenosine, in turn, inhibits cytokine production and proliferation of Teff cells, further contributing to the suppressive function of Tregs. In addition, the possible existence of TIGIT-PVR/PVRL2 and TIM3-Gal9 signaling pathways between Tregs and AML cells may contribute to a propensity for leukemia progression. Schematic figure was drawn by Figdraw (www.figdraw.com).
2.3.1 Contact-dependent mechanism
Contact-dependent immunosuppression heavily relies on the interaction between surface molecules expressed by Tregs and other cells. Notably, investigations have revealed that Tregs in AML enhance the expression of specific suppressive surface molecules. In particular, Tregs derived from individuals with AML have demonstrated elevated levels of CTLA-4 expression (45, 50). The expression of CTLA-4 by Tregs hinders the co-stimulation of effector T cells (Teffs) by outcompeting CD28 for binding to CD80/86 on antigen-presenting cells (APCs) (99). Additionally, CTLA-4 on Tregs downregulates the expression of CD80/86 on DCs, thereby impeding the activation of Teffs (99, 100). Furthermore, the interaction between CTLA-4 and CD80/86 triggers an upregulation of IDO in DCs (62, 101). IDO, in turn, degrades tryptophan within the microenvironment, leading to the suppression of T-cell responses (102) and the generation of Tregs (55). In acute leukemia patients, there is an observed increase in the expression of NRP-1 on Tregs. Interestingly, the introduction of exogenous Sema3A, which serves as a ligand for NRP-1, can effectively downregulate NRP-1 expression on Tregs and facilitate the apoptosis of leukemia cells (103). Notably, NRP-1 is highly expressed on intratumoral Tregs (104), and enables prolonged interactions between Tregs and DCs that are dependent on MHC-II molecules. This, in turn, restricts the recruitment of MHC-II peptide complexes to immune synapses, ultimately impeding immune responses (105).
There are some studies implicating that Tregs may interact with AML cells through TIGIT and TIM-3 to help them escape immune surveillance. TIGIT, as a co-inhibitory receptor, was found to be ubiquitously expressed on the Tregs in AML (44, 53). The activation of TIGIT signaling leads to the upregulation of suppressive genes (such as Pdcd1, IL10, Prf1, and Havcr2) in TIGIT-positive Tregs, resulting in the manifestation of a highly activated suppressive phenotype (106). Stamm et al. conducted a study demonstrating that AML cell lines and patient samples exhibit high expression levels of the TIGIT ligands, PVR and PVRL2, which correlates with a poor prognosis. They further revealed that blocking PVR/PVRL2 on AML cells or inhibiting TIGIT on immune cells enhances the anti-leukemic effects in vitro (107). Moreover, TIGIT+ Tregs were found to upregulate the expression of the co-inhibitory receptor TIM-3, suggesting a collaborative suppression of antitumor responses by TIM-3 and TIGIT (106). Indeed, it was observed that TIM-3+ Treg cells significantly increased in de novo AML patients (108). High levels of Gal-9 (the ligand of TIM-3) were also observed on leukemia blasts in AML samples (109, 110). Interestingly, TIM-3 is also expressed on leukemic stem cells in AML (111, 112), and even Gal-9 has been shown to be expressed on activated Treg (113). These studies illustrate that Gal-9 and TIM-3 may engage in complex interactions within the AML microenvironment.
2.3.2 Contact-independent mechanism
Cytokines, granzyme and perforin are involved in a contact-independent mechanism (40, 80, 114). Newly diagnosed AML patients have been found to exhibit heightened levels of Treg-associated cytokines, specifically IL-10 and IL-35 (115). The immunosuppressive factor IL-10, derived from Tregs, plays a crucial role in diminishing anti-tumor immune responses by suppressing the activity of Teffs and APCs (116). IL-35 has the ability to suppress the functions and proliferation of Teffs, while simultaneously promoting the expansion of inducible Tregs (117, 118). In the AML microenvironment, both IL-10 and IL-35 not only exert inhibitory effects on immune cells but also contribute to the stimulation of AML blast proliferation. The highly expressed cytokine IL-10 by Tregs has been shown to enhance the stemness of AML cells by activating the PI3K/AKT signaling pathway. In AML/ETO c-kitmut (A/Ec) leukemia mice, blocking the IL10/IL10R/PI3K/AKT signaling pathway extended their survival and significantly reduced the stemness of A/Ec leukemia cells. Furthermore, a positive correlation was found between the proportion of Tregs and leukemia stem cells (LSCs) in patient samples. AML patients with high Treg infiltration also exhibited stronger activation of the PI3K/AKT pathway in CD34+ primary AML cells (119). Additionally, IL-35 has been shown to directly promote the proliferation of AML blasts and inhibit their apoptosis (80). The expression of perforin and granzyme B is upregulated in Tregs of patients with AML compared to healthy individuals. Additionally, Tregs in AML patients have been shown to exert immunosuppressive effects by utilizing perforin and granzyme B (45). Tregs have the ability to induce apoptosis in natural killer (NK) cells and CD8+ T cells by utilizing granzyme B and perforin. Research indicates that mice lacking granzyme B show improved efficacy in clearing AML cells in comparison to mice with intact granzyme B functionality. Moreover, when wild-type Treg cells are introduced into granzyme B-deficient mice, there is a discernible suppression of AML clearance (114).
In addition to the secretion, the uptake and enzymatic hydrolysis of factors from the microenvironment also occur independently of contact. The constitutive expression of CD25, which represents high affinity IL-2 receptors, allows Treg cells to continually absorb IL-2. This uptake of IL-2 leads to cytokine deprivation-induced apoptosis of Teff cells (120). Tregs constitutively express the membrane surface enzymes CD39 and CD73. These enzymes have the ability to hydrolyze ATP or ADP, resulting in the production of adenosine. Consequently, the levels of adenosine in the microenvironment are elevated. Adenosine, in turn, interacts with the adenosine receptor A2A on the surface of Teff cells, leading to the inhibition of cytokine production and proliferation (33). Indeed, study has shown that CD39 and CD73 are expressed on CD4+CD25high Tregs isolated from patients with AML. Interestingly, Tregs obtained from AML patients have been shown to have a higher ability to hydrolyze ATP into adenosine compared to Tregs from healthy individuals (45).
2.4 Potential immunotherapy strategies targeting Treg in AML
Currently, immunotherapy for AML targeting Tregs represents an extremely promising treatment, with a main focus on reducing the number of Tregs (Table 2). Evidence suggests that the downregulation of Tregs coincides with an increase in antileukemic reactivity (121). The combination therapy of Ara-C, a CXCR4 inhibitor, and PD-L1 mAb has been shown to enhance the eradication of leukemic myeloid blast cells by effectively suppressing Tregs (122). In mouse models, it has been shown that the depletion of Tregs using anti-CD25 antibodies prior to DC vaccination against AML significantly enhances the immune response against leukemia. This approach facilitates the development of robust and long-lasting immune responses (123). The depletion of Tregs using anti-CD25 antibody (124) or interleukin-2 diphtheria toxin (IL-2DT) (NCT01106950) (125) prior to IL-2 administration has demonstrated enhanced antileukemic effects mediated by NK cells. Similarly, IL-2DT can eliminate Tregs, increasing the quantity of transferred cytotoxic T lymphocytes (CTL) at AML disease sites and reducing tumor burden (126). Clinical trials (NCT00675831, NCT00987987) have shown that a donor lymphocyte infusion depleted of CD25+ Tregs can lead to enhanced anti-tumor efficacy in patients with hematologic malignancies who have experienced relapse after undergoing allo-HSCT (127, 128). The safety and efficacy of the combined treatment strategy of infusion of Treg-depleted T lymphocytes and WT1 antigen-specific cancer immunotherapeutic in patients with WT1-positive AML are under evaluation (NCT01513109). Various targets highly expressed on Treg cells, including LAG3, TIM3, VISTA, TIGIT, OX40, ICOS, and chemokine receptors such as CCR4, CCR5, and CCR8, have been suggested as potential targets for eliminating Treg cells (116). These studies suggest that reducing the population of Treg cells may hold therapeutic benefits in the treatment of AML.
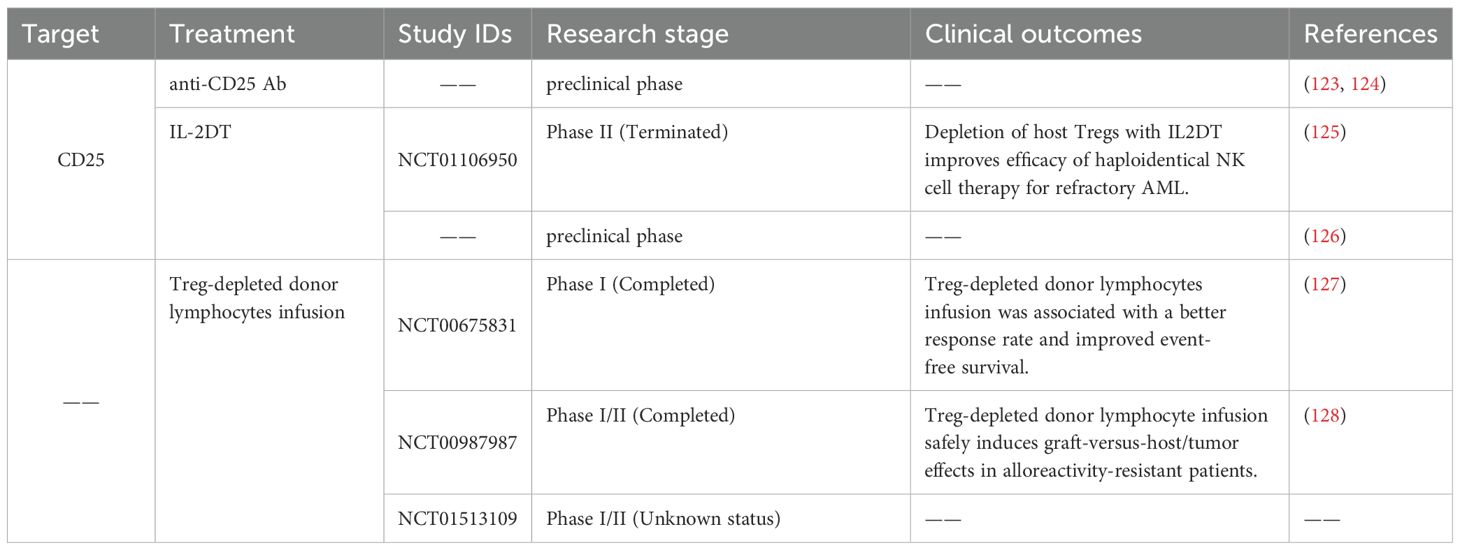
Table 2. AML treatment through reducing Treg numbers.
3 The other usual one: myeloid-derived suppressor cell
3.1 The phenotype of MDSC
The TME impedes the normal differentiation of hematopoietic stem cells, resulting in the emergence of a subset of immature and heterogeneous myeloid cells called MDSCs (129). MDSCs can be broadly classified into two main categories: monocytic MDSCs (M-MDSCs) and polymorphonuclear MDSCs (PMN-MDSCs). M-MDSCs are characterized as Lin−(CD3, CD19, CD56)CD11b+CD15−CD14+HLA-DRlow/−, while PMN-MDSCs are defined as Lin−CD11b+CD15+CD14−CD66b+HLA-DRlow/− (130, 131). M-MDSCs exhibit phenotypic and morphological similarities to monocytes, while PMN-MDSCs share closer resemblance to neutrophils (129). In humans, M-MDSCs can be distinguished from monocytes by the absence of MHC class II molecules, and the population of PMN-MDSCs can be identified using LOX-1 as a marker to differentiate them from neutrophils (132, 133). PMN-MDSCs comprise the majority of MDSCs, accounting for more than 75% of the population, whereas M-MDSCs make up only 10-20% (133). However, it is important to highlight that M-MDSCs possess a higher immunosuppressive potential compared to PMN-MDSCs (133, 134). In recent years, researchers have identified a small population of human bone marrow progenitor and precursor cells that exhibit colony-forming activity. These cells, known as early myeloid-derived suppressor cells (eMDSCs), are characterized by their labeling as Lin−HLA-DRlow/−CD11b+CD14−CD15−CD33+ (131).
3.2 MDSC accumulation and its mechanisms in AML
Substantial evidence suggests that MDSCs are expanded in AML and significantly contributes to poor prognosis. Specifically, in C57BL/6 mice engrafted with TIB-49 AML, an expansion of CD11b+Gr11+ MDSCs was observed in both the BM and spleen (135). Clinical studies have demonstrated that adult patients with AML exhibit significantly elevated frequency of MDSCs in their BM. These MDSCs are identified by CD33highCD11b+HLA-DRlow/neg. Importantly, it has been observed that the proportion of MDSCs decreased after patients achieve CR. Additionally, the frequency of MDSCs is positively correlated with minimal residual disease (MRD) levels, suggesting that these cells may impact the clinical course and prognosis of AML (136). Studies have provided evidence that circulating M-MDSCs are increased in individuals with AML. Moreover, the presence of elevated M-MDSC percentage has been associated with a low CR rate, a high relapse/refractory rate, and poor long-term survival in AML patients (137–139). In a monocentric prospective study on AML, two independent negative prognostic indicators for overall survival were identified: an initial peripheral percentage of M-MDSCs exceeding 0.55% of leukocytes at the time of diagnosis, and a subsequent decrease in the percentage of M-MDSCs following induction therapy (140). Research conducted by Hyun et al. demonstrated that AML patients with a heightened frequency of MDSC-like blasts, characterized by elevated levels of ARG-1 and iNOS, exhibited the ability to suppress T cell proliferation, thereby contributing to an unfavorable prognosis (141).
Extensive research has been conducted to investigate the mechanisms of MDSC accumulation. AML-derived EVs are an important factor contributing to the accumulation of MDSCs in AML. Specifically, palmitoylated proteins present on the surface of AML-EVs activate Toll-like receptor 2 (TLR2) of monocytes and trigger MDSC induction controlled by Akt/mTOR signaling pathway (142). Therefore, targeting protein palmitoylation could serve as a potential approach to disrupt the differentiation of MDSCs. Additionally, AML cells employ a MUC1-dependent mechanism to secrete EVs containing c-myc, when co-cultured with MDSCs. The presence of these EVs subsequently prompts the upregulation of cyclin D2 and cyclin E1 in MDSCs, suggesting that the c-myc-containing EVs potentially enhance MDSC proliferation (135). Cytarabine (Ara-C) treatment prompted AML cells to express and secret TNF-α, which subsequently facilitated the expansion of MDSCs and enhanced their function and survival through activating IL-6/STAT3 and NFκB pathways (143). Additionally, Gao et al. proposed the hypothesis that TIM-3 on AML stem cells interacts with Gal-9 on MDSCs, thereby promoting the expansion of MDSCs and their differentiation into tumor-associated macrophages (TAMs) (144). However, further research is necessary to validate this hypothesis. Theoretically, if the increase in MDSCs could be inhibited based on these mechanisms, it may offer a potential rescue strategy for AML patients. The mechanisms underlying MDSC accumulation within the AML microenvironment are illustrated in Figure 3.
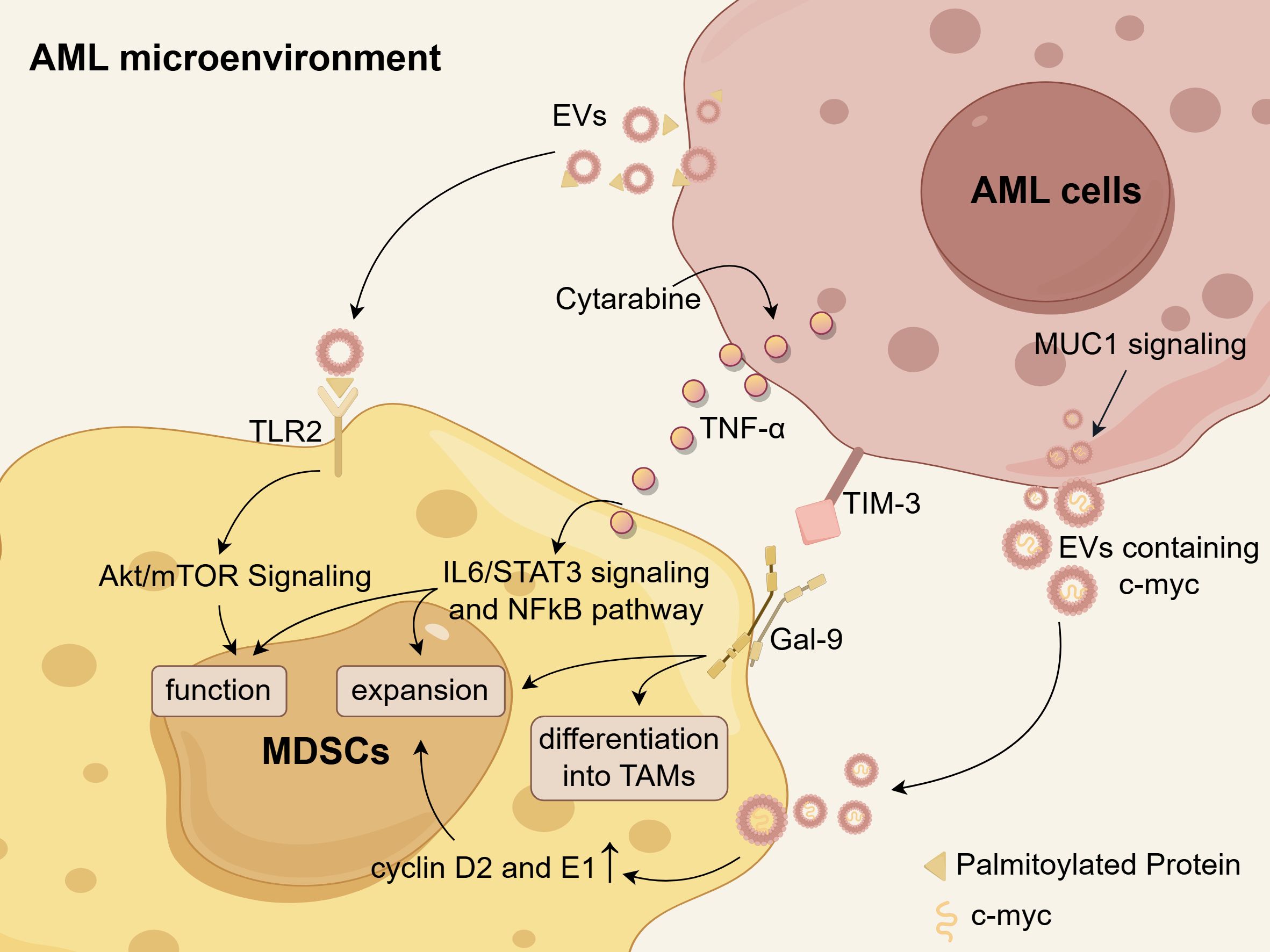
Figure 3. The mechanisms of MDSC accumulation in the AML microenvironment. Palmitoylated proteins present on the surface of AML-derived EVs activate TLR2, triggering the Akt/mTOR-dependent induction of MDSCs. Cytarabine-induced TNF-α secretion from AML cells leads to an expansion of MDSCs and enhances their functions and survival by activating IL-6/STAT3 signaling and NFκB pathways. AML cells secrete EVs containing c-myc in a MUC1-dependent manner, which facilitates MDSC proliferation through upregulation of cyclin D2 and E1. There is a hypothesis that the Tim-3/Gal-9 pathway may promote the expansion of MDSCs and their differentiation into TAMs in AML. Schematic figure was drawn by Figdraw (www.figdraw.com).
3.3 The immunosuppressive mechanisms of MDSC in the AML microenvironment
MDSCs exhibit immunosuppressive activities that hinder effective anti-leukemic immune responses. MDSCs accumulated in the PB of AML patients exhibit high expression of VISTA, which is thought to be associated with the suppression of the T-cell response. Evidence suggests that VISTA exerts an inhibitory effect on the anti-leukemia T-cell response, as demonstrated by the effective reduction of MDSC-mediated CD8+ T-cell inhibition in AML following VISTA knockdown using specific siRNA (145). However, the precise mechanisms by which MDSCs operate within the AML microenvironment remain unclear at present, underscoring the urgent need for a more detailed investigation of their functional roles.
3.4 Potential immunotherapy strategies targeting MDSC in AML
Targeted intervention of MDSCs has the potential to attenuate their immunosuppressive capabilities and strengthen the immune response against leukemia. In an AML mouse model, Hwang et al. demonstrated that a triple combination therapy consisting of Ara-C, a CXCR4 inhibitor, and a PD-L1 mAb resulted in a significant reduction of MDSCs and a potent eradication of leukemic myeloid blast cells (122). In addition, a clinical trial (NCT01347996) demonstrated a notable decrease in peripheral M-MDSCs among AML patients treated with histamine dihydrochloride (HDC) and low-dose IL-2 for relapse prevention, heralding a promising clinical outcome (146). Given the prevalent expression of CD33 on MDSCs, CD33 is frequently employed as a target of MDSCs (147). The CD33/CD3-bispecific T-cell engaging (BiTE®) antibody (AMG 330) exhibited notable efficacy in combating leukemia by specifically targeting CD33+ MDSCs in AML (148). A multicenter clinical trial (NCT03214666) is currently underway to investigate the potential of CD16/IL-15/CD33 tri-specific killer cell engager (GTB-3550 TriKE®) in targeting CD33+ MDSCs. The 123NL CAR-T therapy, which has been designed to target CD123 and NKG2DL, has demonstrated the ability to effectively eliminate M-MDSCs in AML (149). In a murine AML model, treatment with the hypomethylating agent guadecitabine (SGI-110) has been shown to reduce the MDSC burden, subsequently resulting in an increase proportion of functionally active leukemia-specific T cells (150). These studies suggest that targeted decrease of MDSCs is advantageous for the AML treatment. Immunotherapy strategies targeting MDSC in AML are summarized in Table 3.
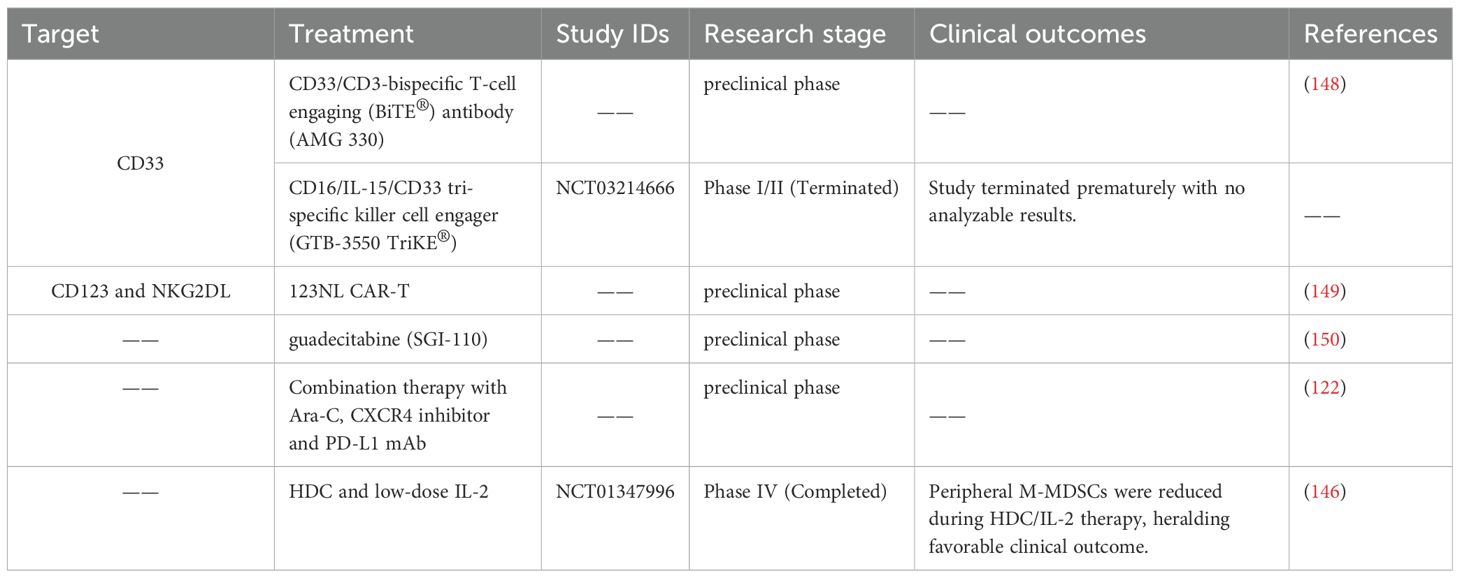
Table 3. AML treatment through targeting MDSC.
4 The other developing one: leukemia-associated macrophage
4.1 The phenotype of LAM
Tumor-associated macrophages within the leukemia microenvironment, specifically referred to as LAMs, have been documented to play a significant role in the progression of leukemia. Macrophages can undergo polarization from the M0 state into classically activated (M1) macrophages, which demonstrate anti-leukemic and immunostimulatory capabilities, or alternatively activated (M2) macrophages, which exhibit pro-leukemic and immunosuppressive characteristics (151, 152). LAMs share functional characteristics with both M1- and M2-like macrophages. However, they predominantly align with the pro-leukemic properties of M2 macrophages (151, 153). M2 macrophages are characterized by the expression of surface markers such as CD163, CD206, and the M-CSF receptor CD115. Additionally, they secrete arginase II (Arg2), chitinase-3-like protein 1 (CHI3L1/YKL-40), and the anti-inflammatory cytokines IL-10 and TGF-β, which contribute to their immunosuppressive and tumor-promoting roles (154).
4.2 LAM accumulation and its mechanisms in AML
The expansion of M2-like LAMs in AML is a contributor to a negative prognosis. Al-Matary et al. demonstrated that M2-like macrophages were elevated in the BM of AML patients and mice (155). It has been observed that more M2-like LAMs are associated with a worse prognosis in AML patients (156, 157). Tian et al. found that the proportion and number of LAMs were higher in patients with refractory AML than in those who achieved CR (156). Consistent with this finding, a study by Brauneck et al. demonstrated an increased frequency of BM-infiltrating immunosuppressive M2 macrophages expressing TIGIT, TIM-3, and LAG-3 in patients with newly diagnosed and relapsed AML (157). Xu et al. reaffirmed that M2-like LAMs, characterized by CD206 positivity, are predominantly enriched within the AML microenvironment, and a high infiltration of M2 macrophages is correlated with adverse clinical outcomes (158). Patients with AML exhibiting elevated levels of CD163 transcripts demonstrated a diminished likelihood of survival (159). This finding aligns with the results reported by Guo et al. through single-cell RNA sequencing, which identified a specific monocyte/macrophage cluster characterized by high CD163 expression that correlates with a reduced probability of survival in AML patients (160).
The mechanisms underlying the increase of M2-like LAMs has been comprehensively investigated. There is increasing evidence that the factors influencing M1 and M2 characteristics are imbalanced within the AML microenvironment, resulting in a greater accumulation of M2-like LAMs (Figure 4). Using in vitro and in vivo models, Mussai et al. provided the first reports demonstrating that the secretion of arginase II by AML blasts induces the polarization of monocytes into an immunosuppressive M2-like phenotype, marked by the increased expression of CD206 (161). The transcription factor Gfi1 expression was about two-fold upregulated in LAMs of AML compared to non-leukemic macrophages, and it promote the polarization of macrophages to a leukemia-supporting state (155). Recently, Tian et al. identified let-7b as a potential aberrant gene implicated in conferring M2-like characteristics and demonstrated its significant upregulation in LAMs from refractory AML mice. Knockdown of let-7b in LAMs was shown to suppress AML progression by reprogramming LAMs toward an M1-like phenotype, mediated through the activation of the Toll-like receptor and NF-κB signaling pathways (156). Jiang et al. discovered that low levels of MOZ correlate with poor prognosis in AML. They observed that the loss of MOZ led to reduced M1 activation in macrophages and heightened resistance to chemotherapeutic agents (162). Similarly, IRF7, a key contributor to M1 polarization, was found to be underexpressed in the more immunosuppressive phenotype of spleen-derived LAMs. IRF7 promotes M1 characteristics by activating the SAPK/JNK pathway in macrophages, and stimulation of this pathway was shown to significantly extend the survival duration of AML mice (159).
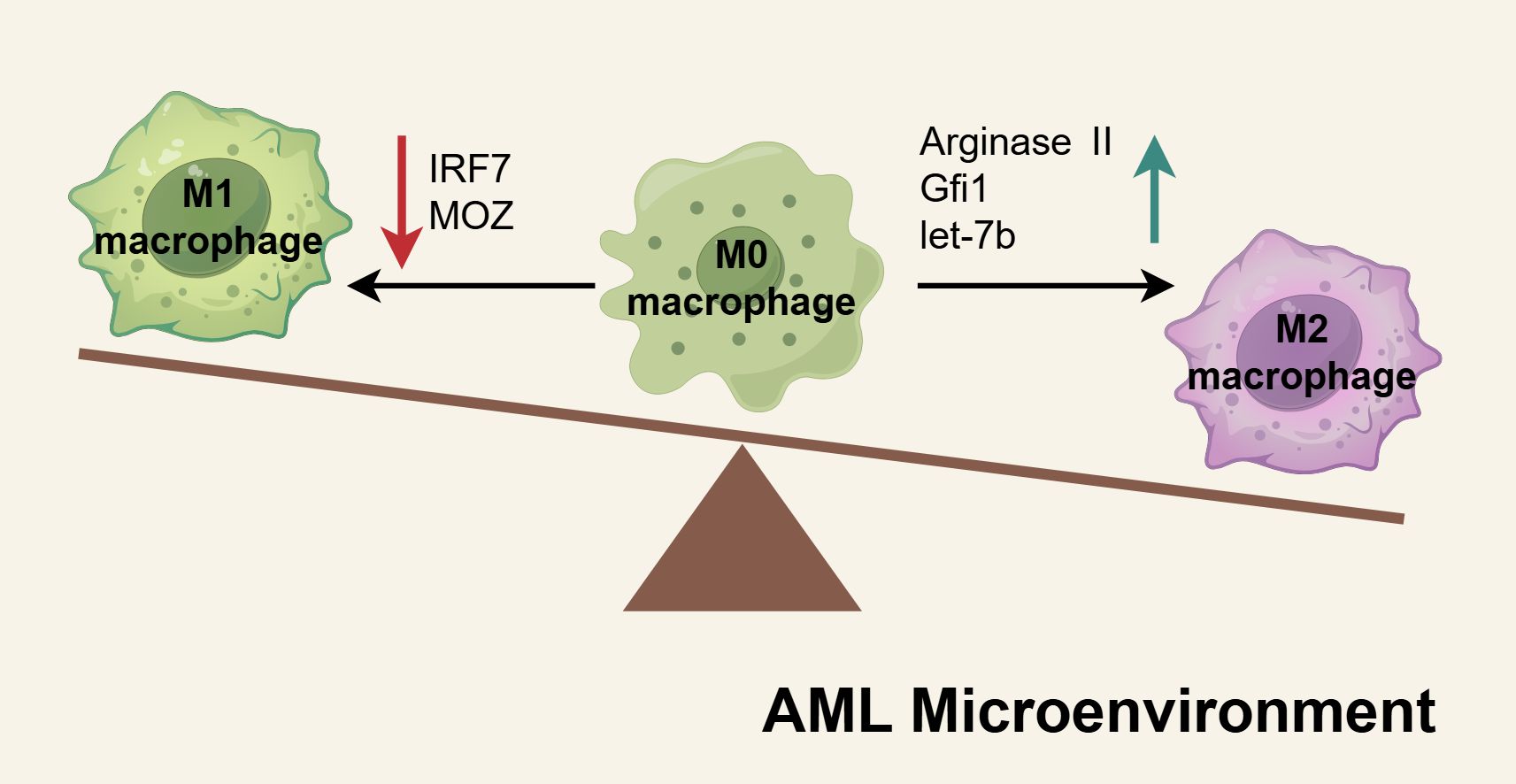
Figure 4. The mechanisms of M2-like LAMs accumulated in AML microenvironment. In the AML microenvironment, the factors regulating M1 or M2 macrophage polarization are dysregulated, creating an imbalance in macrophage differentiation. Pro-M1 factors such as IRF7 and MOZ are downregulated in AML macrophages, resulting in diminished M1 activation. Conversely, elevated levels of pro-M2 factors, including arginase II, Gfi1, and let-7b, drive increased polarization toward the M2 phenotype. These shifts culminate in the accumulation of M2-like macrophages within the AML microenvironment, fostering an immunosuppressive milieu that supports leukemia progression. Schematic figure was drawn by Figdraw (www.figdraw.com).
4.3 The immunosuppressive mechanisms of LAM in the AML microenvironment
The interplay between LAMs and AML blasts enhances AML cell survival. M2-like macrophages secrete soluble factors such as CCL2 and CXCL8, which activate pro-survival pathways and suppress apoptosis in leukemic blasts (151). Williams et al. show that M2-like macrophages protect the U937 and THP-1 AML cell lines against daunorubicin-induced apoptosis (163). While the role of TAMs in solid tumors has been extensively studied (164), the significance of LAMs in leukemia has only recently gained attention due to the unique and heterogeneous nature of leukemic microenvironments. Overall, the precise mechanisms by which LAMs influence AML remain poorly understood.
4.4 Potential immunotherapy strategies targeting LAM in AML
To counteract the immunosuppressive and leukemia-promoting effects mediated by M2-like LAMs in AML, current effective strategies primarily focus on depletion and reprogramming. In a mouse model of MLL-AF9-driven AML, Keech et al. demonstrated that targeted depletion of CD169+/SIGLEC1+ macrophages via diphtheria toxin injection significantly extended median survival in mice treated with cytarabine and doxorubicin (165). Furthermore, the 123NL CAR-T therapy designed to target CD123 and NKG2DL, has proven effective in eliminating M2 macrophages in AML (149). Experimental evidence indicates that knockdown of let-7b in LAMs causes M1-like polarization, thereby significantly inhibiting the progression of AML in a mouse model driven by MLL-AF9 (156). Additionally, Liu et al. revealed that chenodeoxycholic acid (CDCA) inhibited the polarization of M2-like LAMs and curtailed their proliferation-promoting effects on AML cells (166). Moreover, in vitro blockade of TIGIT reprograms M2 LAMs toward an M1 phenotype and enhances anti-CD47-mediated phagocytosis of AML cells (157). Immunotherapy strategies targeting LAMs in AML are comprehensively summarized in Table 4.
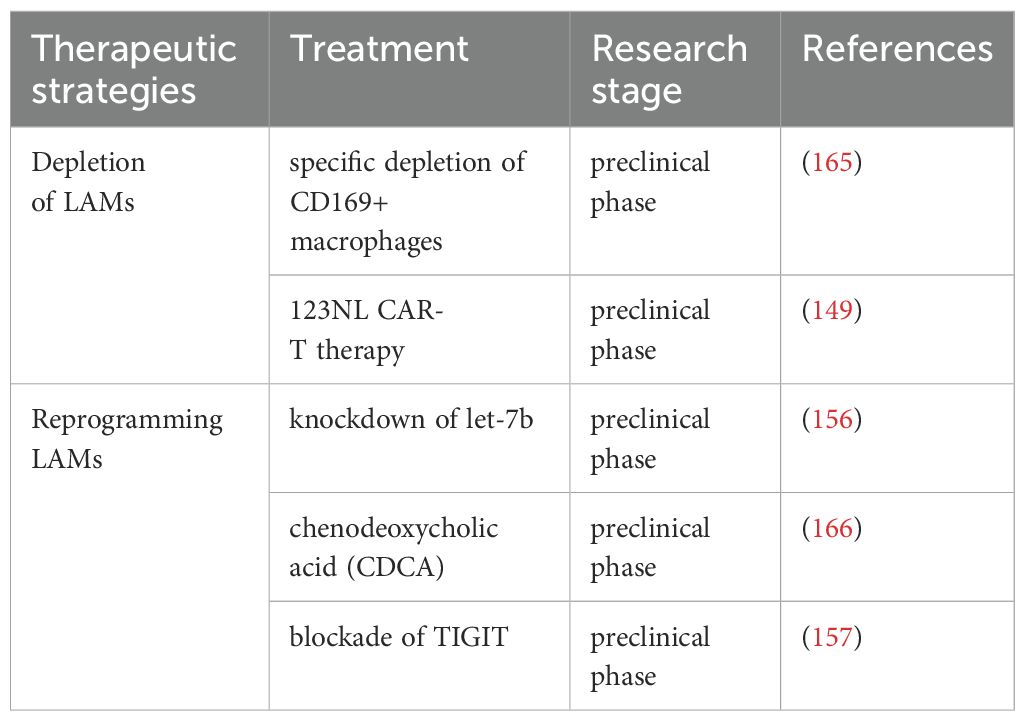
Table 4. AML treatment through targeting LAM.
5 The other emerging one: regulatory B cell
As early as the 1970s, researchers proposed that certain B cells could exert immunosuppressive function by secreting inhibitory cytokines (167). In 2002, Mizoguchi identified a subset of B cells characterized by up-regulation of CD1d in the mesenteric lymph nodes of intestinal inflammation murine models, which inhibited the progression of enteritis by producing IL-10, and defined this group of B cells with immunomodulatory functions as regulatory B cells (Breg) (168). Currently, the origin and development of Breg cells are poorly understood. It is widely accepted that immature and mature B cells, as well as plasmablasts, can differentiate into Breg cells under appropriate stimulation and timing, resulting in a heterogeneous Breg population (169). Several different subtypes of Breg cells have been identified in humans and mice, though specific biomarkers for Breg cell activation have yet to be established (169, 170). The phenotypes of major Breg subsets are summarized in Table 5. The most well-characterized human Breg phenotypes include CD19+CD24highCD38high (171) and CD19+CD24highCD27+ (172).
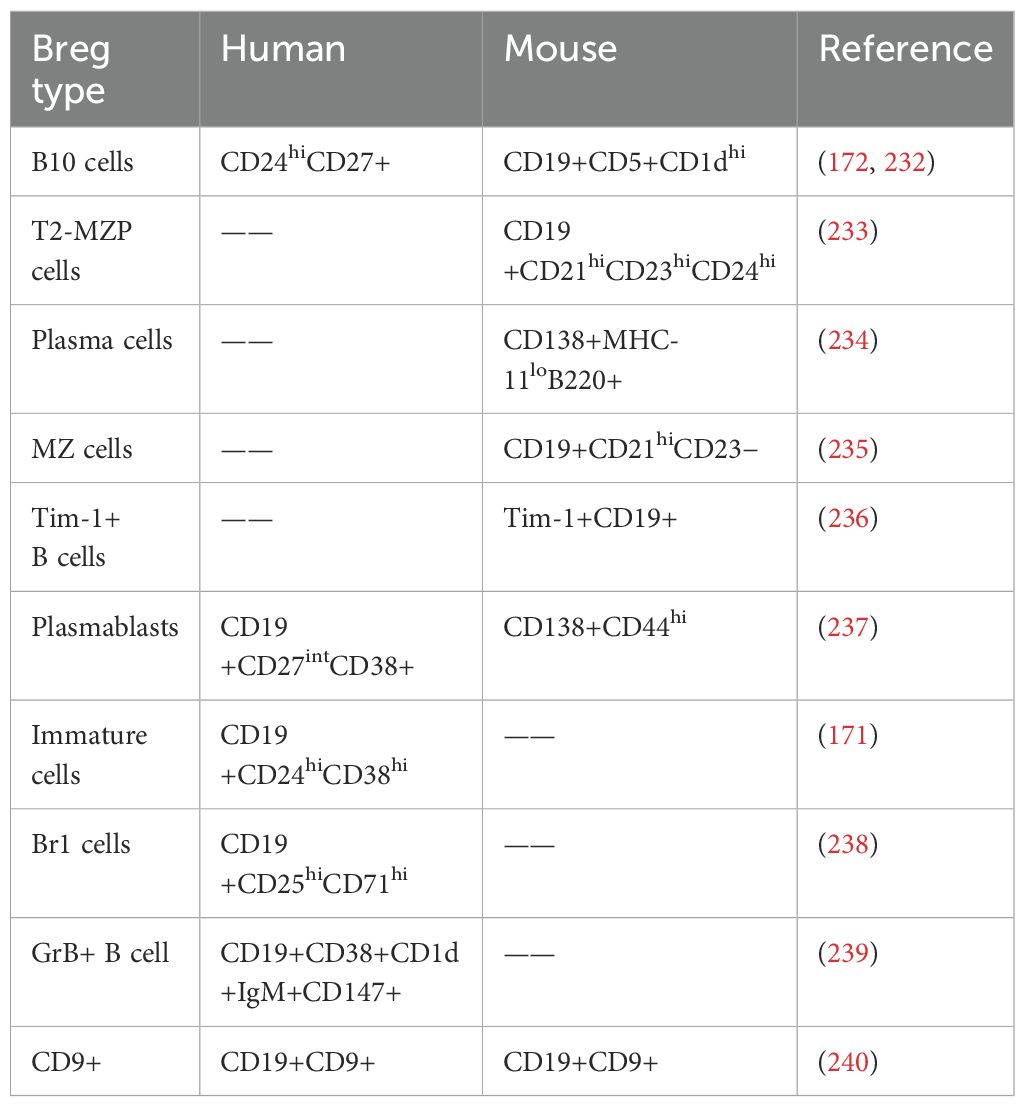
Table 5. Phenotypes of Breg subsets in humans and mice.
The absence of definitive biomarkers for Breg cells considerably impedes research advancements, particularly in the context of AML, where investigations remain markedly constrained. Wan et al. demonstrated a significant elevation in the proportion of CD19+CD24highCD38high Breg cells within the BM of AML patients (43). This aligns with the findings of Lv et al., who observed an elevated frequency of Breg cells in both PB and BM of AML patients compared to healthy controls, and this increased frequency was associated with a shorter overall survival (173). However, a subsequent study by Dong et al. found that patients with newly diagnosed AML exhibited a significantly lower Breg frequency in PB than healthy controls (174). Interestingly, all three studies utilized CD19, CD24, and CD38 as markers to define Breg cells, yet their results exhibited notable inconsistencies. Wan’s study enrolled 45 patients, Lv’s included 46, and Dong’s involved 40. This divergence may be attributed to their relatively limited sample sizes. Furthermore, the inclusion of samples from both PB and BM sources could have introduced variability, potentially compromising the accuracy of the findings. To resolve this controversy, more extensive, well-replicated studies are imperative. Shi et al. demonstrated that PD-L1 expression was elevated on Breg cells from AML patients, with higher PD-L1 levels correlating with poorer prognosis (175). Research on Breg in AML is indeed quite scarce, underscoring both the significance and urgency of this investigative focus.
6 The other newly identified one: leukemia-associated neutrophils
Within the microenvironment of AML, leukemia/tumor-associated neutrophils (LANs/TANs) have emerged as a critical cellular component with increasingly recognized pathophysiological significance.
TANs are neutrophils recruited to tumor sites via chemokines (including CXCL1, CXCL2, and IL-8) secreted by tumor cells and stromal cells. Functionally, TANs can be polarized into anti-tumor N1 and pro-tumor N2 phenotypes. N1-type TANs are characterized by high expression of ICAM-1 and CD95, exerting anti-tumor effects through the release of ROS and cytokines such as IFN-γ. In contrast, N2-type TANs exhibit elevated expression of CCL2, IL-8, and ARG1, promoting tumor progression via angiogenesis induction, extracellular matrix remodeling, and immunosuppressive microenvironment formation (176).
In AML, LANs’ functional role is unclear, but an FGFR1-driven murine model revealed leukemogenesis polarizes neutrophils into six subsets (notably Ly6g+ and Camk1d+), which upregulate MMP8/9 to migrate from bone marrow to blood and differentiate into PMN-MDSCs; MMP inhibition with Ilomastat blocked migration and improved survival, while clinical data linked high MMP8 to poor AML outcomes, highlighting MMP8 as a potential therapeutic target to disrupt immune evasion (177).
7 Discussions and future prospects
The immunosuppressive role in AML orchestrated by immunosuppressive cells persists as a critical impediment to eliciting a robust anti-leukemic immune response. Despite significant advancements in understanding these cells, the development of viable therapeutic strategies remains an ongoing challenge, requiring further innovation and exploration.
7.1 Tregs in AML: current understanding and future directions
While research on Tregs in AML remains challenging, the function mechanisms of Treg in the solid tumor have been more clearly elucidated. Tumor-infiltrating Tregs heightened activation and potent immunosuppressive capabilities, characterized by elevated expression of LAG-3, LFA-1, TGF-β, EVs, and others (116). LAG-3 expressed on the surface of Treg could bind with a high affinity to MHC class II molecules on the surface of DCs, effectively inhibiting the maturation and immunostimulatory capacity of DCs (178). Treg-expressed LFA-1 has been shown to be involved in downregulating CD80/86 on DCs (179). TGF-β produced by intratumoral Tregs directly inhibited proliferation and differentiation of immunocompetent cells (180). Contrary to observations in solid tumors, AML demonstrates distinct TGF-β dynamics, with studies reporting either unchanged or reduced TGF-β levels in AML patients (115, 174). The underlying mechanisms for this differential expression remain unclear and warrant further investigation. Through gap junctions, Tregs deliver substantial quantities of cAMP to Teff cells, inducing metabolic interference that culminates in Teff suppression and apoptosis (181, 182). Additionally, recent studies have identified a novel suppression mechanism involving Treg-derived EVs. These EVs serve as bioactive carriers of proteins, lipids, and nucleic acids, orchestrating intercellular communication networks and modulating anti-tumor immunity (183). It was demonstrated that EVs derived from natural CD8+CD25+ Treg cells, containing LAMP-1 and CD9, were observed to significantly inhibit CTL responses and anti-tumor immunity in a B16 melanoma model (184). While established mechanisms of Treg-mediated immunosuppression in solid tumors provide a valuable framework for investigating their role in AML, critical distinctions must be acknowledged. The TME exhibits remarkable complexity, with Treg populations demonstrating substantial functional and phenotypic heterogeneity that varies significantly across different tumor subtypes (185). Additionally, emerging evidence suggests that Tregs may develop distinct functional properties within the unique leukemic microenvironment. TIGIT was ubiquitously expressed on the Tregs in AML (44, 53), and its ligands PVR and PVRL2 have been reported to be highly expressed on AML cell lines and patient samples (107). Moreover, antibody blockade of PVR or PVRL2 on AML cell lines or primary AML cells or TIGIT blockade on immune cells could enhance the anti-leukemic effects (107). It is possible that TIGIT on Treg cells may engage with PVR/PVRL2 on AML cells, thereby protecting leukemic cells from immune attack. However, there is no direct evidence so far. Further research is needed to determine the function of these molecules. A marked increase in TIM-3+ Treg cell populations was observed among de novo AML cases (108). Previous studies have reported that TIM-3+ Tregs in CLL drive immunosuppression via its ligand soluble Gal-9 (90). High levels of Gal-9 expression were also observed on blasts in primary AML samples (109, 110). Whether a similar situation exists in AML requires further study. Interestingly, TIM-3 is also expressed on AML stem cells (111, 112), and even Gal-9 has been shown to be expressed on activated Treg (113). These studies illustrate that the interaction between Gal-9 and TIM-3 in the AML immune microenvironment is complex and needs further exploration.
In AML treatment strategies, therapeutic depletion of Tregs can potentiate antileukemic immunity and improve clinical outcomes. However, any pharmacological approaches to reduce Treg frequency should be carefully optimized to mitigate potential adverse effects, including autoimmune reactions or uncontrolled inflammatory responses resulting from Treg dysregulation. Given the pivotal role of Treg homeostasis, targeting the molecular mechanisms underlying their accumulation represents a promising therapeutic avenue. Disrupting these pathways—such as with the IDO inhibitor 1-MT, which has demonstrated efficacy in suppressing Treg expansion—could offer a novel and clinically viable strategy for AML immunotherapy (56, 57). To facilitate clinical translation, rigorous evaluation of therapeutic feasibility remains essential, alongside the development of novel agents with optimized efficacy and safety profiles. Alternatively, attenuating Treg functionality represents a viable strategy to counteract the immunosuppressive AML microenvironment. OX40 activation has been shown to diminish Treg-mediated immunosuppression (186, 187), and targeting other immune checkpoint proteins and kinase signaling pathways in Tregs similarly disrupts their suppressive capacity (188). However, most investigations remain confined to preclinical studies or solid tumor trials, with AML-specific research notably limited. To realize effective Treg-targeted therapies in AML and maximize clinical benefits, comprehensive mechanistic elucidation and dedicated clinical validation are urgently required.
7.2 MDSCs in AML: current understanding and future directions
Similarly, insights into MDSC biology in AML may benefit greatly from an understanding of its mode of function in solid tumors and pan-cancer models. In TME, MDSCs highly express arginase-1 (ARG-1) (189) and inducible nitric oxide synthase (iNOS) (190), and transfer the metabolite methylglyoxal to CD8+ T cells (191), all of which degrade L-arginine and thus prevent T cell proliferation (192). In addition, MDSCs suppress T-cell activation by depleting cystine and cysteine (193). Within the TME, M-MDSCs exhibit heightened glucose uptake and consumption, thereby disrupting the metabolic activity of neighboring immune cells (194). Notably, in breast cancer models, MDSC-mediated tryptophan catabolism via IDO has been shown to drive Treg expansion while concurrently inducing T-cell autophagy, cell cycle arrest, and cell death (195). Adenosine production by CD39/CD73-expressing MDSCs further potentiates their expansion and enhances immunosuppressive activity in lung cancer models (196, 197). The immunosuppressive capacity of MDSCs is mediated through excessive generation of reactive oxygen species (ROS) (198, 199), nitric oxide (NO), and peroxynitrite (PNT) (200), which collectively impair T-cell function. Additionally, tumor-infiltrating MDSCs engage with T cells through multiple immune checkpoint interactions—including PD-L1/PD-1, Gal-9/TIM-3, CD80\CD86/CTLA-4, CD155/TIGIT, VISTA/VISTAL, and FasL/Fas—inducing T-cell anergy and apoptosis (201). In murine tumor models, tumor-expanded MDSCs can suppress NK cell function via membrane-bound TGF-β1 (202). However, the existence and relative contribution of these MDSC-mediated immunosuppressive mechanisms in AML remain unclear and warrant further investigation.
Therapeutic targeting of MDSCs represents a promising strategy to augment anti-leukemic immunity through multiple approaches: inhibiting their generation, promoting differentiation into immunocompetent mature cells, suppressing their immunosuppressive activity, or selectively depleting MDSC populations (203, 204). AML-derived EVs, characterized by surface palmitoylated proteins or c-Myc cargo, potently drive MDSC expansion (135, 142). EV inhibition represents a theoretically viable approach to curtail MDSC generation, and experimental validation remains essential. In addition, reprogramming existing MDSCs into immunocompetent mature cells serves as an alternative strategy. Preclinical studies demonstrate that all-trans retinoic acid (ATRA) effectively reprogram MDSCs into mature APCs, thereby restoring T-cell functionality in both renal carcinoma and pulmonary malignancy models (203, 205). Thus, pharmacological induction of MDSC differentiation into non-immunosuppressive myeloid lineages represents a viable therapeutic strategy for AML. The suppression of MDSC activity may be based on its immunosuppressive mechanisms, such as the reduction of ROS and NO production. Targeting depletion of MDSCs through agents like gemtuzumab ozogamicin (GO) has demonstrated significant clinical potential. As a CD33-directed antibody-drug conjugate (ADC) approved for CD33+ AML treatment, GO has shown both efficacy and a manageable safety profile in multiple clinical trials (206). The constitutive expression of CD33 across MDSC subtypes makes it an attractive therapeutic target, with a study by Fultang et al. demonstrating GO’s ability to increase MDSC death, consequently restoring T-cell response and enhancing tumor cell clearance (147). This study encompassed multiple tumor subtypes; however, AML samples were not included, warranting further investigation in the AML context. These findings provide a strong rationale for developing novel MDSC-targeted therapies in AML, potentially leading to significant advances in treatment outcomes.
7.3 LAMs in AML: current understanding and future directions
The AML microenvironment is characterized by significant infiltration of M2-like LAMs, which actively support leukemic cell survival and disease progression. These cells represent the leukemic counterpart of TAMs observed in solid malignancies. TAMs exhibit pro-tumorigenic properties through multiple mechanisms: (1) direct promotion of malignant cell proliferation and metastasis, (2) suppression of T cell-mediated anti-tumor immunity, and (3) facilitation of angiogenic processes. TAMs can facilitate the proliferation of tumor cells by producing growth factors, cytokines, and chemokines, including FGF-2, TGF-β, PDGF, IL-10, CXCL, and so on (207). Evidence demonstrates that TAMs significantly enhance osteosarcoma metastasis and invasion through activating the COX-2/STAT3 axis and epithelial-mesenchymal transition (208). TAMs suppress antitumor immunity by inhibiting T cells, B cells, NK cells, and DCs, while promoting Tregs, Th17, γδT cells, MDSCs, angiogenesis, and metastasis (207). TAMs can induce tumor angiogenesis through the secretion of cytokines, including VEGF, COX-2, and PDGF (209). Building on the well-characterized role of TAMs in solid tumors, investigating LAMs in AML represents a promising research direction.
Given the established pro-tumor functions of TAMs, targeting LAMs may offer novel therapeutic strategies to disrupt AML progression and improve treatment outcomes. Therapeutic reprogramming of LAMs from a pro-tumorigenic to an anti-tumor M1-like phenotype emerges as a promising strategy for AML treatment. Experimental evidence demonstrates that let-7b knockdown in LAMs induces M1-like polarization, resulting in significant suppression of AML progression and extended survival in MLL-AF9-driven murine leukemia models (156). RNA-seq profiling of AML patient-derived LAMs identified let-7b as a potential target, though its downstream mechanisms remain undefined. Future work should characterize let-7b effector pathways and assess whether targeting either the microRNA itself or its products offer therapeutic benefit in AML.
7.4 Bregs in AML: current understanding and future directions
Breg cells have been found to be increased in AML and are thought to be involved in the negative immunoregulation of the hematopoietic microenvironment of AML. However, so far, no specific marker has been identified for Breg cells to define their phenotype. These findings suggest that Breg cells may not represent a distinct lineage, but rather reflect a functional state adopted by B cells at various developmental stages in response to microenvironmental stimuli (169). Nevertheless, the possibility remains that specific Breg markers exist but were not identified in the current study. Further investigation is required to fully elucidate the origin, developmental pathways, and phenotypic characteristics of Breg cells. While their phenotype remains incompletely defined, their functional significance in immune regulation has become increasingly evident. Breg research in solid malignancies has revealed their critical immunosuppressive role, with IL-10 emerging as the prototypical functional marker of Breg (168, 210). Recent advances have revealed that Breg cells employ a broader immunomodulatory factor to mediate immune suppression, including TGF-β, IL-35, CD1d and PD-L1 (211). Breg cells suppress immune responses by inhibiting CD4+ T cell proliferation and cytokine secretion (212), while also blocking TNF-α production in monocyte-macrophages (172). Given the nascent state of Breg research in AML, systematic efforts are needed to map their ontogeny, functional heterogeneity, and clinical relevance. Such studies could unlock Breg-targeted therapies to complement existing AML immunotherapies.
7.5 LANs in AML: current understanding and future directions
While research on LANs in AML remains limited, their mechanistic roles in CLL have been well characterized (213). In CLL, LANs promote leukemic cell proliferation and survival via IL-17/IL-6 secretion while fostering immunosuppression through T-cell inhibition. Notably, LANs enhance bone marrow homing and maintain leukemic stemness via the CXCR4/CXCL12 axis (213, 214). These findings offer valuable insights for AML research, particularly regarding LANs-leukemic stem cell crosstalk and the therapeutic potential of modulating LANs polarization. Key unresolved questions include (1): spatiotemporal dynamics of LANs subsets in AML progression, and (2) mechanistic interactions between LANs and the leukemic stem cell niche. Addressing these gaps could advance precision immunotherapy strategies for AML.
7.6 The likely coordinated network of immunosuppressive cells in AML
The development of an immunosuppressive microenvironment in AML involves a coordinated interplay of multiple regulatory cell populations. While studies have individually characterized the leukemia-promoting effects of Tregs, MDSCs, LAMs, and Bregs, accumulating evidence suggests these cells function synergistically to establish a potent immunosuppressive network that facilitates immune evasion and disease progression (Figure 5). As demonstrated by Flores-Borja et al., CD19+CD24highCD38high Bregs in healthy individuals can induce regulatory properties in CD4+CD25− T cells through IL-10-dependent mechanisms (212). However, in the study by Wan et al., the researchers observed that Bregs from healthy controls failed to promote the conversion of CD4+CD25− T cells into CD4+CD25+FOXP3+ Tregs, irrespective of whether the T cells originated from healthy individuals or AML patients. In contrast, BM-derived CD19+CD24highCD38high Bregs of AML patients possessed this conversion capability. Furthermore, this conversion appeared to be primarily mediated through direct cell-to-cell contact, as cytokine profiling revealed no significant alterations in the expression levels of soluble factors (43). More investigations are required to elucidate the precise mechanisms underlying Treg and Breg interactions within the AML microenvironment. In the TME, it has been demonstrated that Bregs promote Treg tumorigenicity through secretion of IL-21, IL-35, and TGF-β (215). Emerging evidence demonstrates functional reciprocity between Tregs and MDSCs across diverse tumor models. This bidirectional crosstalk establishes self-reinforcing immunosuppressive circuits, wherein factors (such as TGF-β, IL-10) produced by each population reciprocally stimulate expansion and activation, thereby amplifying immune suppression within the TME (216). M-MDSCs in CLL exhibit elevated IDO expression, which drives enhanced Treg differentiation (217). In breast cancer, MDSCs promote the development of PD-L1+ Bregs through PD-1/PD-L1-mediated activation of the PI3K/AKT/NF-κB signaling axis in B lymphocytes (218). MDSCs can drive macrophage polarization toward an immunosuppressive M2-like phenotype via IL-10 secretion, thereby facilitating solid tumor progression (219). Additionally, M2 cells secrete CCL2 into the TME to recruit MDSCs and Tregs (220). A reciprocal regulatory axis further connects M2-polarized macrophages and Tregs within the TME (215). Tregs promote monocyte differentiation into M2 macrophages through the release of IL-10, VEGF and STAT3 signaling (215, 221). In turn, M2 cells secrete IL-6 (222) and IL-10 (223) to activate Tregs. M2 cells release CCL22 and recruit more CCR4-expressing Tregs to infiltrate the tumor microenvironment (224). Evidences suggests a coordinated network of immunosuppressive cells collectively fosters tumor progression in AML and other malignancies. While these cooperative mechanisms remain incompletely characterized, their systematic investigation represents a crucial frontier in tumor. A comprehensive elucidation of these cellular interactions potentially informing novel immunomodulatory approaches for AML.
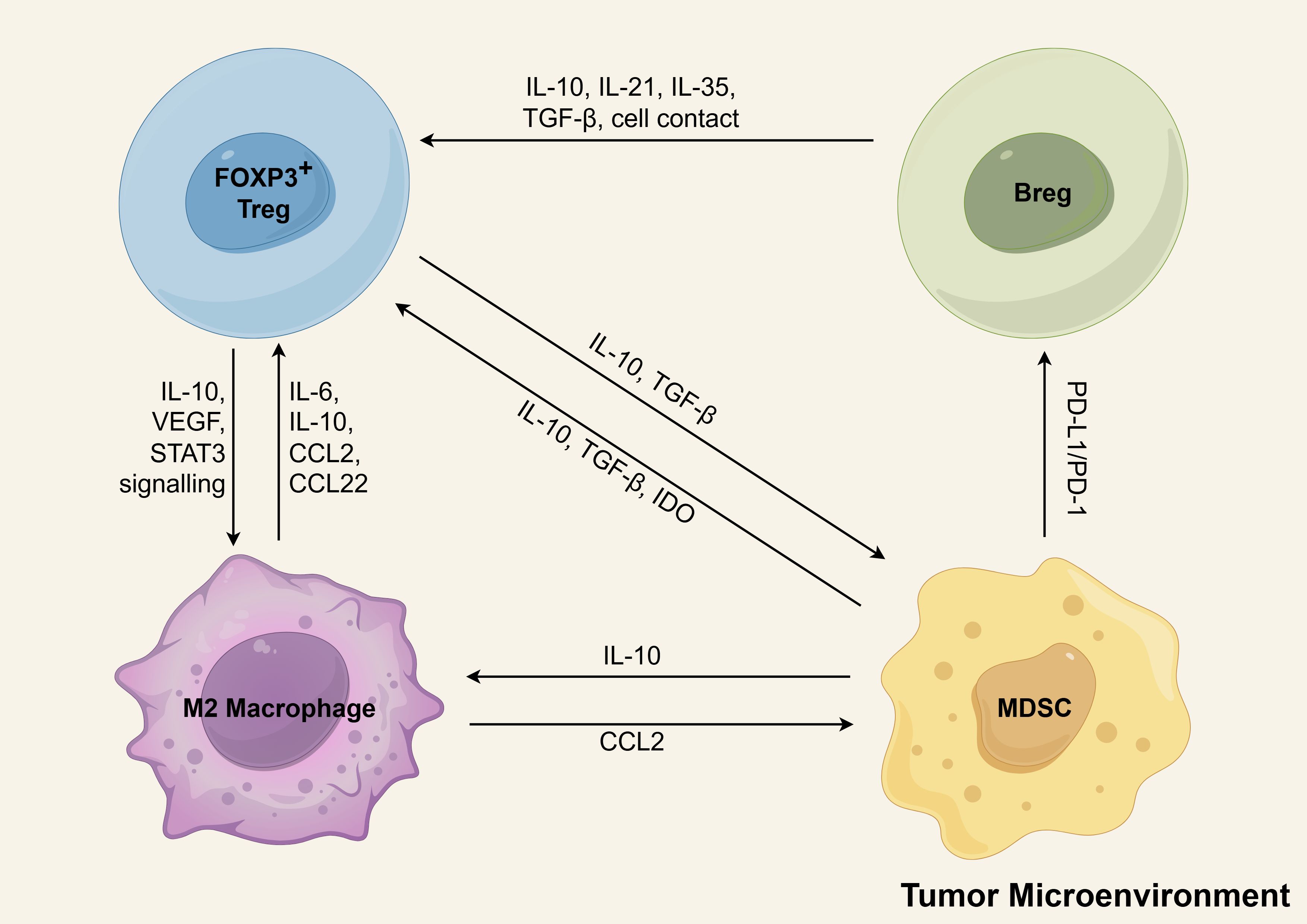
Figure 5. The positive feedback loops of immunosuppressive cells in tumor microenvironment. Tregs, MDSCs, M2 macrophages, and Bregs form interlinked positive feedback loops that reinforce immune suppression and drive immune evasion and AML progression. Key interactions include: (1) Breg-mediated enhancement of Treg function via IL-10, IL-21, IL-35, TGF-β, and direct cell contact; (2) Treg-induced monocyte-to-M2 differentiation through IL-10, VEGF, and STAT3 signaling; (3) M2 macrophage secretion of IL-6/IL-10 for Treg activation and CCL2/CCL22 for Treg recruitment; (4) Reciprocal TGF-β/IL-10-mediated activation between Tregs and MDSCs; (5) MDSC-driven Treg differentiation via IDO upregulation; (6) PD-1/PD-L1-dependent MDSC induction of PD-L1+ Bregs; and (7) IL-10-mediated MDSC promotion of M2 polarization. M2-derived CCL2 further recruits MDSCs to the TME. Schematic figure was drawn by Figdraw (www.figdraw.com).
7.7 Advantages and challenges of targeting immunosuppressive cells
7.7.1 Advantages of targeting Tregs in AML immunotherapy
Targeting Tregs in AML immunotherapy offers multiple benefits. Depleting Tregs via anti-CD25 antibodies, IL-2DT, or CXCR4 inhibitors significantly enhances NK/CTL-mediated antileukemic activity, with preclinical studies demonstrating durable immune responses (124–126). Combination therapies (e.g., Treg depletion with DC vaccines) synergistically improve leukemic cell clearance (123), while clinical trials show that Treg-depleted donor lymphocyte infusions boost graft-versus-leukemia effects post-allo-HSCT (127, 128). Multiple targetable markers (LAG3/TIM3/CCR family) enable precise interventions, and existing regimens (e.g., IL-2DT) exhibit acceptable safety profiles (116).
7.7.2 Challenges of targeting Tregs in AML immunotherapy
This approach faces critical limitations. Systemic Treg depletion risks triggering GVHD or autoimmune toxicity, and non-specific agents like CXCR4 inhibitors may compromise effector T cells. Tumor microenvironment complexity leads to compensatory immunosuppression (e.g., MDSC expansion) and drug delivery barriers, while Treg populations often rebound post-treatment. Clinical translation remains challenging, with current efficacy largely confined to murine models or post-transplant settings, limited responses in advanced AML, and a lack of predictive biomarkers for personalized therapy. These hurdles underscore the need for more precise Treg-targeting strategies and optimized combination regimens.
7.7.3 Advantages of targeting MDSCs in AML immunotherapy
Targeting MDSCs in AML presents multiple therapeutic benefits, including the ability to reverse immunosuppression and restore anti-leukemic immune responses through various approaches such as CXCR4 inhibition (122), CD33-targeting agents (e.g., BiTE® antibodies AMG 330 and TriKE® engagers GTB-3550) (148, 149), and hypomethylating agents (150). These strategies have demonstrated efficacy in reducing MDSC populations and enhancing T-cell function in both preclinical models and early clinical trials. Additionally, combination therapies integrating MDSC-targeted interventions with chemotherapy or immune checkpoint blockade show synergistic effects, improving leukemic cell clearance and potentially overcoming treatment resistance (122).
7.7.4 Challenges of targeting MDSCs in AML immunotherapy
However, MDSC-targeted therapies face significant hurdles, including the heterogeneity of MDSC subsets (e.g., M-MDSCs vs. PMN-MDSCs) with distinct immunosuppressive mechanisms, complicating broad-spectrum targeting. CD33-directed therapies may also deplete normal myeloid cells, leading to myelosuppression and infection risks. Furthermore, while preclinical studies are promising, clinical translation remains inconsistent, with variable patient responses and a lack of standardized biomarkers for patient selection. The tumor microenvironment’s adaptability, including compensatory recruitment of alternative immunosuppressive cells, further limits sustained efficacy, underscoring the need for more precise and combination-based strategies.
7.7.5 Advantages of targeting LAMs in AML immunotherapy
Targeting LAMs in AML offers several therapeutic advantages. First, strategies such as CD169+/SIGLEC1+ macrophage depletion (165) and CD123/NKG2DL-targeted CAR-T therapy (149) have demonstrated significant efficacy in disrupting the immunosuppressive tumor microenvironment and directly eliminating pro-leukemic M2-like LAMs, leading to improved survival in preclinical models. Second, innovative approaches like TIGIT blockade (157) and let-7b knockdown (156) not only reduce M2 polarization but also actively reprogram LAMs toward anti-tumor M1 phenotypes, enhancing phagocytic activity and synergizing with therapies like anti-CD47. These dual-action mechanisms provide a multifaceted attack against AML progression while potentially restoring immune surveillance.
7.7.6 Challenges of targeting LAMs in AML immunotherapy
Despite these advantages, LAM-targeted therapies face notable limitations. A major concern is the risk of off-target effects, as broad macrophage depletion may damage beneficial tissue-resident macrophages, potentially leading to unintended toxicity. Additionally, the plasticity of LAM phenotypes poses a challenge, as reprogrammed M1-like macrophages can revert to immunosuppressive M2 states under persistent tumor microenvironment pressures, undermining long-term therapeutic efficacy. Finally, while preclinical models (e.g., MLL-AF9-driven AML) show promise, translating these findings to human patients remains difficult due to the heterogeneity of LAM populations in AML and the lack of validated biomarkers for patient stratification. These hurdles highlight the need for more selective targeting strategies and robust combination approaches to maximize clinical benefit.
8 Conclusion
The therapeutic landscape of AML has been reshaped by immunotherapy advances, yet clinical outcomes remain suboptimal for most patients, with limited agents specifically targeting immunosuppressive cells. Critical challenges endure in characterizing these inhibitory immune populations, as key molecular signatures for distinct subsets remain undefined. While preclinical studies constitute most current research, few therapeutic strategies have advanced to clinical testing, highlighting crucial unmet needs in bridging the laboratory-to-clinic translation gap for immunotherapeutic development.
Author contributions
ML: Conceptualization, Visualization, Writing – original draft, Writing – review & editing. MY: Visualization, Writing – original draft, Writing – review & editing. YQ: Writing – review & editing. YM: Data curation, Writing – original draft, Writing – review & editing. QG: Conceptualization, Funding acquisition, Methodology, Supervision, Writing – review & editing. LG: Formal Analysis, Investigation, Writing – review & editing. CL: Conceptualization, Methodology, Writing – review & editing. WL: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. LX: Conceptualization, Methodology, Supervision, Writing – review & editing. YY: Funding acquisition, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Doctoral Research Initiation Fund of the Affiliated Hospital of Southwest Medical University; The Science and Technology Strategic Cooperation Programs of Luzhou Municipal People’s Government and Southwest Medical University (2024LZXNYDJ102); Southwest Medical University Program (2022ZD007); China Postdoctoral Science Foundation (2023M732926); Sichuan Science and Technology Program (2022YFS0622, 2024YFFK0271); Sichuan Science and Technology Plan Joint Innovation Program (2022YFS0622-A2; 2022YFS0622-A3; 2022YFS0622-A4; 2022YFS0622-A5; 2022YFS0622-B5), and Southwest Medical University Innovation and Entrepreneurship Training Program (S202310632168).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1627161/full#supplementary-material
References
1. Döhner H, Weisdorf DJ, and Bloomfield CD. Acute myeloid leukemia. N Engl J Med. (2015) 373:1136–52. doi: 10.1056/NEJMra1406184
2. Shallis RM, Wang R, Davidoff A, Ma X, and Zeidan AM. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. (2019) 36:70–87. doi: 10.1016/j.blre.2019.04.005
3. Yi M, Li A, Zhou L, Chu Q, Song Y, and Wu K. The global burden and attributable risk factor analysis of acute myeloid leukemia in 195 countries and territories from 1990 to 2017: estimates based on the global burden of disease study 2017. J Hematol Oncol. (2020) 13:72. doi: 10.1186/s13045-020-00908-z
4. Dong Y, Shi O, Zeng Q, Lu X, Wang W, Li Y, et al. Leukemia incidence trends at the global, regional, and national level between 1990 and 2017. Exp Hematol Oncol. (2020) 9:14. doi: 10.1186/s40164-020-00170-6
5. Kelly LM and Gilliland DG. Genetics of myeloid leukemias. Annu Rev Genomics Hum Genet. (2002) 3:179–98. doi: 10.1146/annurev.genom.3.032802.115046
6. Moore CG, Stein A, Fathi AT, and Pullarkat V. Treatment of relapsed/refractory aml-novel treatment options including immunotherapy. Am J Hematol. (2025) 100 Suppl 2:23–37. doi: 10.1002/ajh.27584
7. Abelson S, Collord G, Ng SWK, Weissbrod O, Mendelson Cohen N, Niemeyer E, et al. Prediction of acute myeloid leukaemia risk in healthy individuals. Nature. (2018) 559:400–4. doi: 10.1038/s41586-018-0317-6
8. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
9. Kusumbe AP, Ramasamy SK, Itkin T, Mäe MA, Langen UH, Betsholtz C, et al. Age-dependent modulation of vascular niches for haematopoietic stem cells. Nature. (2016) 532:380–4. doi: 10.1038/nature17638
10. Battula VL, Le PM, Sun JC, Nguyen K, Yuan B, Zhou X, et al. Aml-induced osteogenic differentiation in mesenchymal stromal cells supports leukemia growth. JCI Insight. (2017) 2(13):e90036. doi: 10.1172/jci.insight.90036
11. Ellegast JM, Alexe G, Hamze A, Lin S, Uckelmann HJ, Rauch PJ, et al. Unleashing cell-intrinsic inflammation as a strategy to kill aml blasts. Cancer Discov. (2022) 12:1760–81. doi: 10.1158/2159-8290.Cd-21-0956
12. Wachter F and Pikman Y. Pathophysiology of acute myeloid leukemia. Acta Haematol. (2024) 147:229–46. doi: 10.1159/000536152
13. Liu H. Emerging agents and regimens for aml. J Hematol Oncol. (2021) 14:49. doi: 10.1186/s13045-021-01062-w
14. Zarnegar-Lumley S, Caldwell KJ, and Rubnitz JE. Relapsed acute myeloid leukemia in children and adolescents: current treatment options and future strategies. Leukemia. (2022) 36:1951–60. doi: 10.1038/s41375-022-01619-9
15. Winkler IG, Barbier V, Nowlan B, Jacobsen RN, Forristal CE, Patton JT, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. (2012) 18:1651–7. doi: 10.1038/nm.2969
16. Tettamanti S, Pievani A, Biondi A, Dotti G, and Serafini M. Catch me if you can: how aml and its niche escape immunotherapy. Leukemia. (2022) 36:13–22. doi: 10.1038/s41375-021-01350-x
17. Zha C, Yang X, Yang J, Zhang Y, and Huang R. Immunosuppressive microenvironment in acute myeloid leukemia: overview, therapeutic targets and corresponding strategies. Ann Hematol. (2024) 103:4883–99. doi: 10.1007/s00277-024-06117-9
18. Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, et al. Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol. (2013) 14:307–8. doi: 10.1038/ni.2554
19. Sakaguchi S. Naturally arising foxp3-expressing cd25+Cd4+ Regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. (2005) 6:345–52. doi: 10.1038/ni1178
20. Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, et al. Thymic selection of cd4+Cd25+ Regulatory T cells induced by an agonist self-peptide. Nat Immunol. (2001) 2:301–6. doi: 10.1038/86302
21. Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, et al. Conversion of peripheral cd4+Cd25- naive T cells to cd4+Cd25+ Regulatory T cells by tgf-beta induction of transcription factor foxp3. J Exp Med. (2003) 198:1875–86. doi: 10.1084/jem.20030152
22. Coombes JL, Siddiqui KR, Arancibia-Cárcamo CV, Hall J, Sun CM, Belkaid Y, et al. A functionally specialized population of mucosal cd103+ Dcs induces foxp3+ Regulatory T cells via a tgf-beta and retinoic acid-dependent mechanism. J Exp Med. (2007) 204:1757–64. doi: 10.1084/jem.20070590
23. Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, et al. Foxp3 controls regulatory T-cell function by interacting with aml1/runx1. Nature. (2007) 446:685–9. doi: 10.1038/nature05673
24. Fontenot JD, Rasmussen JP, Gavin MA, and Rudensky AY. A function for interleukin 2 in foxp3-expressing regulatory T cells. Nat Immunol. (2005) 6:1142–51. doi: 10.1038/ni1263
25. Li S, Xie Q, Zeng Y, Zou C, Liu X, Wu S, et al. A naturally occurring cd8(+)Cd122(+) T-cell subset as a memory-like treg family. Cell Mol Immunol. (2014) 11:326–31. doi: 10.1038/cmi.2014.25
26. Mercer F, Khaitan A, Kozhaya L, Aberg JA, and Unutmaz D. Differentiation of il-17-producing effector and regulatory human T cells from lineage-committed naive precursors. J Immunol. (2014) 193:1047–54. doi: 10.4049/jimmunol.1302936
27. Vocanson M, Rozieres A, Hennino A, Poyet G, Gaillard V, Renaudineau S, et al. Inducible costimulator (Icos) is a marker for highly suppressive antigen-specific T cells sharing features of th17/th1 and regulatory T cells. J Allergy Clin Immunol. (2010) 126:280–9. doi: 10.1016/j.jaci.2010.05.022
28. Dasgupta S and Kumar V. Type ii nkt cells: A distinct cd1d-restricted immune regulatory nkt cell subset. Immunogenetics. (2016) 68:665–76. doi: 10.1007/s00251-016-0930-1
29. Yang Zhou J, Werner JM, Glehr G, Geissler EK, Hutchinson JA, and Kronenberg K. Identification and isolation of type ii nkt cell subsets in human blood and liver. Front Immunol. (2022) 13:898473. doi: 10.3389/fimmu.2022.898473
30. Hu G, Wu P, Cheng P, Zhang Z, Wang Z, Yu X, et al. Tumor-infiltrating cd39(+)Γδtregs are novel immunosuppressive T cells in human colorectal cancer. Oncoimmunology. (2017) 6:e1277305. doi: 10.1080/2162402x.2016.1277305
31. Savage PA, Klawon DEJ, and Miller CH. Regulatory T cell development. Annu Rev Immunol. (2020) 38:421–53. doi: 10.1146/annurev-immunol-100219-020937
32. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two foxp3(+)Cd4(+) T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med. (2016) 22:679–84. doi: 10.1038/nm.4086
33. Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by cd39 and cd73 expressed on regulatory T cells mediates immune suppression. J Exp Med. (2007) 204:1257–65. doi: 10.1084/jem.20062512
34. Salgado FJ, Pérez-Díaz A, Villanueva NM, Lamas O, Arias P, and Nogueira M. Cd26: A negative selection marker for human treg cells. Cytometry A. (2012) 81:843–55. doi: 10.1002/cyto.a.22117
35. Garcia Santana CA, Tung JW, and Gulnik S. Human treg cells are characterized by low/negative cd6 expression. Cytometry A. (2014) 85:901–8. doi: 10.1002/cyto.a.22513
36. Bruder D, Probst-Kepper M, Westendorf AM, Geffers R, Beissert S, Loser K, et al. Neuropilin-1: A surface marker of regulatory T cells. Eur J Immunol. (2004) 34:623–30. doi: 10.1002/eji.200324799
37. Yan J, Zhang Y, Zhang JP, Liang J, Li L, and Zheng L. Tim-3 expression defines regulatory T cells in human tumors. PloS One. (2013) 8:e58006. doi: 10.1371/journal.pone.0058006
38. Wegrzyn AS, Kedzierska AE, and Obojski A. Identification and classification of distinct surface markers of T regulatory cells. Front Immunol. (2022) 13:1055805. doi: 10.3389/fimmu.2022.1055805
39. Wang X, Zheng J, Liu J, Yao J, He Y, Li X, et al. Increased population of cd4(+)Cd25(High), regulatory T cells with their higher apoptotic and proliferating status in peripheral blood of acute myeloid leukemia patients. Eur J Haematol. (2005) 75:468–76. doi: 10.1111/j.1600-0609.2005.00537.x
40. Tian T, Yu S, Liu L, Xue F, Yuan C, Wang M, et al. The profile of T helper subsets in bone marrow microenvironment is distinct for different stages of acute myeloid leukemia patients and chemotherapy partly ameliorates these variations. PloS One. (2015) 10:e0131761. doi: 10.1371/journal.pone.0131761
41. Guo Z, Chen Z, Xu Y, Zhang Y, Hu L, Yu F, et al. The association of circulating T follicular helper cells and regulatory cells with acute myeloid leukemia patients. Acta Haematol. (2020) 143:19–25. doi: 10.1159/000500588
42. Yang W and Xu Y. Clinical significance of treg cell frequency in acute myeloid leukemia. Int J Hematol. (2013) 98:558–62. doi: 10.1007/s12185-013-1436-3
43. Wan Y, Zhang C, Xu Y, Wang M, Rao Q, Xing H, et al. Hyperfunction of cd4 cd25 regulatory T cells in de novo acute myeloid leukemia. BMC Cancer. (2020) 20:472. doi: 10.1186/s12885-020-06961-8
44. Zhang Y, Huang Y, Hong Y, Lin Z, Zha J, Zhu Y, et al. Lactate acid promotes pd-1(+) tregs accumulation in the bone marrow with high tumor burden of acute myeloid leukemia. Int Immunopharmacol. (2024) 130:111765. doi: 10.1016/j.intimp.2024.111765
45. Szczepanski MJ, Szajnik M, Czystowska M, Mandapathil M, Strauss L, Welsh A, et al. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin Cancer Res. (2009) 15:3325–32. doi: 10.1158/1078-0432.Ccr-08-3010
46. Ersvaer E, Liseth K, Skavland J, Gjertsen BT, and Bruserud Ø. Intensive chemotherapy for acute myeloid leukemia differentially affects circulating tc1, th1, th17 and treg cells. BMC Immunol. (2010) 11:38. doi: 10.1186/1471-2172-11-38
47. Kanakry CG, Hess AD, Gocke CD, Thoburn C, Kos F, Meyer C, et al. Early lymphocyte recovery after intensive timed sequential chemotherapy for acute myelogenous leukemia: peripheral oligoclonal expansion of regulatory T cells. Blood. (2011) 117:608–17. doi: 10.1182/blood-2010-04-277939
48. Lichtenegger FS, Lorenz R, Gellhaus K, Hiddemann W, Beck B, and Subklewe M. Impaired nk cells and increased T regulatory cell numbers during cytotoxic maintenance therapy in aml. Leuk Res. (2014) 38:964–9. doi: 10.1016/j.leukres.2014.05.014
49. Shenghui Z, Yixiang H, Jianbo W, Kang Y, Laixi B, Yan Z, et al. Elevated frequencies of cd4+ Cd25+ Cd127lo regulatory T cells is associated to poor prognosis in patients with acute myeloid leukemia. Int J Cancer. (2011) 129:1373–81. doi: 10.1002/ijc.25791
50. Sander FE, Nilsson M, Rydström A, Aurelius J, Riise RE, Movitz C, et al. Role of regulatory T cells in acute myeloid leukemia patients undergoing relapse-preventive immunotherapy. Cancer Immunol Immunother. (2017) 66:1473–84. doi: 10.1007/s00262-017-2040-9
51. Zheng J, Qiu D, Jiang X, Zhao Y, Zhao H, Wu X, et al. Increased pd-1(+)Foxp3(+) Γδ T cells associate with poor overall survival for patients with acute myeloid leukemia. Front Oncol. (2022) 12:1007565. doi: 10.3389/fonc.2022.1007565
52. Jin Z, Ye W, Lan T, Zhao Y, Liu X, Chen J, et al. Characteristic of tigit and dnam-1 expression on foxp3+ Γδ T cells in aml patients. BioMed Res Int. (2020) 2020:4612952. doi: 10.1155/2020/4612952
53. Swatler J, Turos-Korgul L, Brewinska-Olchowik M, De Biasi S, Dudka W, Le BV, et al. 4-1bbl-containing leukemic extracellular vesicles promote immunosuppressive effector regulatory T cells. Blood Adv. (2022) 6:1879–94. doi: 10.1182/bloodadvances.2021006195
54. Moussa Agha D, Rouas R, Najar M, Bouhtit F, Fayyad-Kazan H, Lagneaux L, et al. Impact of bone marrow mir-21 expression on acute myeloid leukemia T lymphocyte fragility and dysfunction. Cells. (2020) 9(9):2053. doi: 10.3390/cells9092053
55. Wells G, Kennedy PT, and Dahal LN. Investigating the role of indoleamine 2,3-dioxygenase in acute myeloid leukemia: A systematic review. Front Immunol. (2021) 12:651687. doi: 10.3389/fimmu.2021.651687
56. Curti A, Pandolfi S, Valzasina B, Aluigi M, Isidori A, Ferri E, et al. Modulation of tryptophan catabolism by human leukemic cells results in the conversion of cd25- into cd25+ T regulatory cells. Blood. (2007) 109:2871–7. doi: 10.1182/blood-2006-07-036863
57. Trabanelli S, Lecciso M, Salvestrini V, Cavo M, Očadlíková D, Lemoli RM, et al. Pge2-induced ido1 inhibits the capacity of fully mature dcs to elicit an in vitro antileukemic immune response. J Immunol Res. (2015) 2015:253191. doi: 10.1155/2015/253191
58. Arandi N, Ramzi M, Safaei F, and Monabati A. Overexpression of indoleamine 2,3-dioxygenase correlates with regulatory T cell phenotype in acute myeloid leukemia patients with normal karyotype. Blood Res. (2018) 53:294–8. doi: 10.5045/br.2018.53.4.294
59. Curti A, Trabanelli S, Salvestrini V, Baccarani M, and Lemoli RM. The role of indoleamine 2,3-dioxygenase in the induction of immune tolerance: focus on hematology. Blood. (2009) 113:2394–401. doi: 10.1182/blood-2008-07-144485
60. Curti A, Aluigi M, Pandolfi S, Ferri E, Isidori A, Salvestrini V, et al. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia. (2007) 21:353–5. doi: 10.1038/sj.leu.2404485
61. Folgiero V, Goffredo BM, Filippini P, Masetti R, Bonanno G, Caruso R, et al. Indoleamine 2,3-dioxygenase 1 (Ido1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget. (2014) 5:2052–64. doi: 10.18632/oncotarget.1504
62. Mellor AL and Munn DH. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat Rev Immunol. (2004) 4:762–74. doi: 10.1038/nri1457
63. Curti A, Trabanelli S, Onofri C, Aluigi M, Salvestrini V, Ocadlikova D, et al. Indoleamine 2,3-dioxygenase-expressing leukemic dendritic cells impair a leukemia-specific immune response by inducing potent T regulatory cells. Haematologica. (2010) 95:2022–30. doi: 10.3324/haematol.2010.025924
64. Lecciso M, Ocadlikova D, Sangaletti S, Trabanelli S, De Marchi E, Orioli E, et al. Atp release from chemotherapy-treated dying leukemia cells elicits an immune suppressive effect by increasing regulatory T cells and tolerogenic dendritic cells. Front Immunol. (2017) 8:1918. doi: 10.3389/fimmu.2017.01918
65. Mansour I, Zayed RA, Said F, and Latif LA. Indoleamine 2,3-dioxygenase and regulatory T cells in acute myeloid leukemia. Hematology. (2016) 21:447–53. doi: 10.1080/10245332.2015.1106814
66. Wu L, Lin Q, Ma Z, Chowdhury FA, Mazumder MHH, and Du W. Mesenchymal pgd(2) activates an ilc2-treg axis to promote proliferation of normal and Malignant hspcs. Leukemia. (2020) 34:3028–41. doi: 10.1038/s41375-020-0843-8
67. Ferrell PB and Kordasti S. Hostile takeover: tregs expand in ifnγ-rich aml microenvironment. Clin Cancer Res. (2022) 28:2986–8. doi: 10.1158/1078-0432.Ccr-22-1030
68. Corradi G, Bassani B, Simonetti G, Sangaletti S, Vadakekolathu J, Fontana MC, et al. Release of ifnγ by acute myeloid leukemia cells remodels bone marrow immune microenvironment by inducing regulatory T cells. Clin Cancer Res. (2022) 28:3141–55. doi: 10.1158/1078-0432.Ccr-21-3594
69. Wang M, Zhang C, Tian T, Zhang T, Wang R, Han F, et al. Increased regulatory T cells in peripheral blood of acute myeloid leukemia patients rely on tumor necrosis factor (Tnf)-α-tnf receptor-2 pathway. Front Immunol. (2018) 9:1274. doi: 10.3389/fimmu.2018.01274
70. Govindaraj C, Madondo M, Kong YY, Tan P, Wei A, and Plebanski M. Lenalidomide-based maintenance therapy reduces tnf receptor 2 on cd4 T cells and enhances immune effector function in acute myeloid leukemia patients. Am J Hematol. (2014) 89:795–802. doi: 10.1002/ajh.23746
71. Hamano R, Huang J, Yoshimura T, Oppenheim JJ, and Chen X. Tnf optimally activatives regulatory T cells by inducing tnf receptor superfamily members tnfr2, 4-1bb and ox40. Eur J Immunol. (2011) 41:2010–20. doi: 10.1002/eji.201041205
72. He T, Zhao Y, Zhao P, Zhao L, Zakaria J, and Wang K. Signaling pathway(S) of tnfr2 required for the immunoregulatory effect of cd4(+)Foxp3(+) regulatory T cells. Int Immunopharmacol. (2022) 108:108823. doi: 10.1016/j.intimp.2022.108823
73. He T, Liu S, Chen S, Ye J, Wu X, Bian Z, et al. The P38 mapk inhibitor sb203580 abrogates tumor necrosis factor-induced proliferative expansion of mouse cd4(+)Foxp3(+) regulatory T cells. Front Immunol. (2018) 9:1556. doi: 10.3389/fimmu.2018.01556
74. Govindaraj C, Tan P, Walker P, Wei A, Spencer A, and Plebanski M. Reducing tnf receptor 2+ Regulatory T cells via the combined action of azacitidine and the hdac inhibitor, panobinostat for clinical benefit in acute myeloid leukemia patients. Clin Cancer Res. (2014) 20:724–35. doi: 10.1158/1078-0432.Ccr-13-1576
75. Goodyear OC, Dennis M, Jilani NY, Loke J, Siddique S, Ryan G, et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (Aml). Blood. (2012) 119:3361–9. doi: 10.1182/blood-2011-09-377044
76. Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of foxp3 expression in regulatory T cells by DNA methylation. J Immunol. (2009) 182:259–73. doi: 10.4049/jimmunol.182.1.259
77. Galustian C, Meyer B, Labarthe MC, Dredge K, Klaschka D, Henry J, et al. The anti-cancer agents lenalidomide and pomalidomide inhibit the proliferation and function of T regulatory cells. Cancer Immunol Immunother. (2009) 58:1033–45. doi: 10.1007/s00262-008-0620-4
78. Shen L and Pili R. Class I histone deacetylase inhibition is a novel mechanism to target regulatory T cells in immunotherapy. Oncoimmunology. (2012) 1:948–50. doi: 10.4161/onci.20306
79. Schuh AC, Döhner H, Pleyer L, Seymour JF, Fenaux P, and Dombret H. Azacitidine in adult patients with acute myeloid leukemia. Crit Rev Oncol Hematol. (2017) 116:159–77. doi: 10.1016/j.critrevonc.2017.05.010
80. Tao Q, Pan Y, Wang Y, Wang H, Xiong S, Li Q, et al. Regulatory T cells-derived il-35 promotes the growth of adult acute myeloid leukemia blasts. Int J Cancer. (2015) 137:2384–93. doi: 10.1002/ijc.29563
81. Ge W, Ma X, Li X, Wang Y, Li C, Meng H, et al. B7-H1 up-Regulation on Dendritic-Like Leukemia Cells Suppresses T Cell Immune Function through Modulation of Il-10/Il-12 Production and Generation of Treg Cells. Leuk Res. (2009) 33:948–57. doi: 10.1016/j.leukres.2009.01.007
82. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. Pd-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
83. Dong Y, Han Y, Huang Y, Jiang S, Huang Z, Chen R, et al. Pd-L1 is expressed and promotes the expansion of regulatory T cells in acute myeloid leukemia. Front Immunol. (2020) 11:1710. doi: 10.3389/fimmu.2020.01710
84. Zhou Q, Munger ME, Highfill SL, Tolar J, Weigel BJ, Riddle M, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. (2010) 116:2484–93. doi: 10.1182/blood-2010-03-275446
85. Han Y, Dong Y, Yang Q, Xu W, Jiang S, Yu Z, et al. Acute myeloid leukemia cells express icos ligand to promote the expansion of regulatory T cells. Front Immunol. (2018) 9:2227. doi: 10.3389/fimmu.2018.02227
86. Coles SJ, Hills RK, Wang EC, Burnett AK, Man S, Darley RL, et al. Increased cd200 expression in acute myeloid leukemia is linked with an increased frequency of foxp3+ Regulatory T cells. Leukemia. (2012) 26:2146–8. doi: 10.1038/leu.2012.75
87. Memarian A, Nourizadeh M, Masoumi F, Tabrizi M, Emami AH, Alimoghaddam K, et al. Upregulation of cd200 is associated with foxp3+ Regulatory T cell expansion and disease progression in acute myeloid leukemia. Tumour Biol. (2013) 34:531–42. doi: 10.1007/s13277-012-0578-x
88. Ronchetti S, Ricci E, Petrillo MG, Cari L, Migliorati G, Nocentini G, et al. Glucocorticoid-induced tumour necrosis factor receptor-related protein: A key marker of functional regulatory T cells. J Immunol Res. (2015) 2015:171520. doi: 10.1155/2015/171520
89. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of tim-3 and pd-1 identifies a cd8+ T-cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. (2011) 117:4501–10. doi: 10.1182/blood-2010-10-310425
90. Pang N, Alimu X, Chen R, Muhashi M, Ma J, Chen G, et al. Activated galectin-9/tim3 promotes treg and suppresses th1 effector function in chronic lymphocytic leukemia. FASEB J. (2021) 35:e21556. doi: 10.1096/fj.202100013R
91. Zou L, Barnett B, Safah H, Larussa VF, Evdemon-Hogan M, Mottram P, et al. Bone marrow is a reservoir for cd4+Cd25+ Regulatory T cells that traffic through cxcl12/cxcr4 signals. Cancer Res. (2004) 64:8451–5. doi: 10.1158/0008-5472.Can-04-1987
92. Iellem A, Mariani M, Lang R, Recalde H, Panina-Bordignon P, Sinigaglia F, et al. Unique chemotactic response profile and specific expression of chemokine receptors ccr4 and ccr8 by cd4(+)Cd25(+) regulatory T cells. J Exp Med. (2001) 194:847–53. doi: 10.1084/jem.194.6.847
93. Olsnes AM, Ryningen A, Ersvaer E, and Bruserud Ø. In vitro induction of a dendritic cell phenotype in primary human acute myelogenous leukemia (Aml) blasts alters the chemokine release profile and increases the levels of T cell chemotactic ccl17 and ccl22. J Interferon Cytokine Res. (2008) 28:297–310. doi: 10.1089/jir.2007.0052
94. Wang R, Feng W, Wang H, Wang L, Yang X, Yang F, et al. Blocking migration of regulatory T cells to leukemic hematopoietic microenvironment delays disease progression in mouse leukemia model. Cancer Lett. (2020) 469:151–61. doi: 10.1016/j.canlet.2019.10.032
95. Angelin A, Gil-de-Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low-glucose, high-lactate environments. Cell Metab. (2017) 25:1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
96. Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, et al. Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature. (2021) 591:645–51. doi: 10.1038/s41586-020-03045-2
97. Chen Y, Feng Z, Kuang X, Zhao P, Chen B, Fang Q, et al. Increased lactate in aml blasts upregulates tox expression, leading to exhaustion of cd8(+) cytolytic T cells. Am J Cancer Res. (2021) 11:5726–42.
98. Abolhalaj M, Sincic V, Lilljebjörn H, Sandén C, Aab A, Hägerbrand K, et al. Transcriptional profiling demonstrates altered characteristics of cd8(+) cytotoxic T-cells and regulatory T-cells in tp53-mutated acute myeloid leukemia. Cancer Med. (2022) 11:3023–32. doi: 10.1002/cam4.4661
99. Krummel MF and Allison JP. Cd28 and ctla-4 have opposing effects on the response of T cells to stimulation. J Exp Med. (1995) 182:459–65. doi: 10.1084/jem.182.2.459
100. Yamaguchi T, Kishi A, Osaki M, Morikawa H, Prieto-Martin P, Wing K, et al. Construction of self-recognizing regulatory T cells from conventional T cells by controlling ctla-4 and il-2 expression. Proc Natl Acad Sci U.S.A. (2013) 110:E2116–25. doi: 10.1073/pnas.1307185110
101. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. (2003) 4:1206–12. doi: 10.1038/ni1003
102. Li R, Wei F, Yu J, Li H, Ren X, and Hao X. Ido inhibits T-cell function through suppressing vav1 expression and activation. Cancer Biol Ther. (2009) 8:1402–8. doi: 10.4161/cbt.8.14.8882
103. Yang ZG, Wen RT, Qi K, Li J, Zheng GX, Wang YF, et al. The neuropilin-1 ligand, sema3a, acts as a tumor suppressor in the pathogenesis of acute leukemia. Anat Rec (Hoboken). (2019) 302:1127–35. doi: 10.1002/ar.24016
104. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-Γ Drives T(Reg) fragility to promote anti-tumor immunity. Cell. (2017) 169:1130–41.e11. doi: 10.1016/j.cell.2017.05.005
105. Sarris M, Andersen KG, Randow F, Mayr L, and Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity. (2008) 28:402–13. doi: 10.1016/j.immuni.2008.01.012
106. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. Tigit predominantly regulates the immune response via regulatory T cells. J Clin Invest. (2015) 125:4053–62. doi: 10.1172/jci81187
107. Stamm H, Klingler F, Grossjohann EM, Muschhammer J, Vettorazzi E, Heuser M, et al. Immune checkpoints pvr and pvrl2 are prognostic markers in aml and their blockade represents a new therapeutic option. Oncogene. (2018) 37:5269–80. doi: 10.1038/s41388-018-0288-y
108. Huang S, Liang C, Zhao Y, Deng T, Tan J, Zha X, et al. Increased tox expression concurrent with pd-1, tim-3, and cd244 expression in T cells from patients with acute myeloid leukemia. Cytometry B Clin Cytom. (2022) 102:143–52. doi: 10.1002/cyto.b.22049
109. Gleason MK, Lenvik TR, McCullar V, Felices M, O’Brien MS, Cooley SA, et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood. (2012) 119:3064–72. doi: 10.1182/blood-2011-06-360321
110. Folgiero V, Cifaldi L, Li Pira G, Goffredo BM, Vinti L, and Locatelli F. Tim-3/gal-9 interaction induces ifnγ-dependent ido1 expression in acute myeloid leukemia blast cells. J Hematol Oncol. (2015) 8:36. doi: 10.1186/s13045-015-0134-4
111. Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, et al. Prospective separation of normal and leukemic stem cells based on differential expression of tim3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U.S.A. (2011) 108:5009–14. doi: 10.1073/pnas.1100551108
112. Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, et al. Tim-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. (2010) 7:708–17. doi: 10.1016/j.stem.2010.11.014
113. Wang F, Wan L, Zhang C, Zheng X, Li J, and Chen ZK. Tim-3-galectin-9 pathway involves the suppression induced by cd4+Cd25+ Regulatory T cells. Immunobiology. (2009) 214:342–9. doi: 10.1016/j.imbio.2008.10.007
114. Cao X, Cai SF, Fehniger TA, Song J, Collins LI, Piwnica-Worms DR, et al. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity. (2007) 27:635–46. doi: 10.1016/j.immuni.2007.08.014
115. Wu H, Li P, Shao N, Ma J, Ji M, Sun X, et al. Aberrant expression of treg-associated cytokine il-35 along with il-10 and tgf-β in acute myeloid leukemia. Oncol Lett. (2012) 3:1119–23. doi: 10.3892/ol.2012.614
116. Qin D, Zhang Y, Shu P, Lei Y, Li X, and Wang Y. Targeting tumor-infiltrating tregs for improved antitumor responses. Front Immunol. (2024) 15:1325946. doi: 10.3389/fimmu.2024.1325946
117. Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine il-35 contributes to regulatory T-cell function. Nature. (2007) 450:566–9. doi: 10.1038/nature06306
118. Castellani ML, Anogeianaki A, Felaco P, Toniato E, De Lutiis MA, Shaik B, et al. Il-35, an anti-inflammatory cytokine which expands cd4+Cd25+ Treg cells. J Biol Regul Homeost Agents. (2010) 24:131–5.
119. Xu Y, Mou J, Wang Y, Zhou W, Rao Q, Xing H, et al. Regulatory T cells promote the stemness of leukemia stem cells through il10 cytokine-related signaling pathway. Leukemia. (2022) 36:403–15. doi: 10.1038/s41375-021-01375-2
120. Pandiyan P, Zheng L, Ishihara S, Reed J, and Lenardo MJ. Cd4+Cd25+Foxp3+ Regulatory T cells induce cytokine deprivation-mediated apoptosis of effector cd4+ T cells. Nat Immunol. (2007) 8:1353–62. doi: 10.1038/ni1536
121. Pepeldjiyska E, Li L, Gao J, Seidel CL, Blasi C, Özkaya E, et al. Leukemia derived dendritic cell (Dc(Leu)) mediated immune response goes along with reduced (Leukemia-specific) regulatory T-cells. Immunobiology. (2022) 227:152237. doi: 10.1016/j.imbio.2022.152237
122. Hwang HS, Han AR, Lee JY, Park GS, Min WS, and Kim HJ. Enhanced anti-leukemic effects through induction of immunomodulating microenvironment by blocking cxcr4 and pd-L1 in an aml mouse model. Immunol Invest. (2019) 48:96–105. doi: 10.1080/08820139.2018.1497057
123. Delluc S, Hachem P, Rusakiewicz S, Gaston A, Marchiol-Fournigault C, Tourneur L, et al. Dramatic efficacy improvement of a dc-based vaccine against aml by cd25 T cell depletion allowing the induction of a long-lasting T cell response. Cancer Immunol Immunother. (2009) 58:1669–77. doi: 10.1007/s00262-009-0678-7
124. Hallett WHD, Ames E, Álvarez M, Barao I, Taylor PA, Blazar BR, et al. Combination therapy using il-2 and anti-cd25 results in augmented natural killer cell-mediated antitumor responses. Biol Blood Marrow Transplant. (2008) 14:1088–99. doi: 10.1016/j.bbmt.2008.08.001
125. Bachanova V, Cooley S, Defor TE, Verneris MR, Zhang B, McKenna DH, et al. Clearance of acute myeloid leukemia by haploidentical natural killer cells is improved using il-2 diphtheria toxin fusion protein. Blood. (2014) 123:3855–63. doi: 10.1182/blood-2013-10-532531
126. Zhou Q, Bucher C, Munger ME, Highfill SL, Tolar J, Munn DH, et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood. (2009) 114:3793–802. doi: 10.1182/blood-2009-03-208181
127. Nikiforow S, Kim HT, Daley H, Reynolds C, Jones KT, Armand P, et al. A phase I study of cd25/regulatory T-cell-depleted donor lymphocyte infusion for relapse after allogeneic stem cell transplantation. Haematologica. (2016) 101:1251–9. doi: 10.3324/haematol.2015.141176
128. Maury S, Lemoine FM, Hicheri Y, Rosenzwajg M, Badoual C, Cheraï M, et al. Cd4+Cd25+ Regulatory T cell depletion improves the graft-versus-tumor effect of donor lymphocytes after allogeneic hematopoietic stem cell transplantation. Sci Transl Med. (2010) 2:41ra52. doi: 10.1126/scitranslmed.3001302
129. Gabrilovich DI, Ostrand-Rosenberg S, and Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. (2012) 12:253–68. doi: 10.1038/nri3175
130. Kapor S and Santibanez JF. Myeloid-derived suppressor cells and mesenchymal stem/stromal cells in myeloid Malignancies. J Clin Med. (2021) 10(13):2788. doi: 10.3390/jcm10132788
131. Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. (2016) 7:12150. doi: 10.1038/ncomms12150
132. Condamine T, Dominguez GA, Youn JI, Kossenkov AV, Mony S, Alicea-Torres K, et al. Lectin-type oxidized ldl receptor-1 distinguishes population of human polymorphonuclear myeloid-derived suppressor cells in cancer patients. Sci Immunol. (2016) 1(2):aaf8943. doi: 10.1126/sciimmunol.aaf8943
133. Wu Y, Yi M, Niu M, Mei Q, and Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. (2022) 21:184. doi: 10.1186/s12943-022-01657-y
134. Youn JI, Nagaraj S, Collazo M, and Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol. (2008) 181:5791–802. doi: 10.4049/jimmunol.181.8.5791
135. Pyzer AR, Stroopinsky D, Rajabi H, Washington A, Tagde A, Coll M, et al. Muc1-mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood. (2017) 129:1791–801. doi: 10.1182/blood-2016-07-730614
136. Sun H, Li Y, Zhang ZF, Ju Y, Li L, Zhang BC, et al. Increase in myeloid-derived suppressor cells (Mdscs) associated with minimal residual disease (Mrd) detection in adult acute myeloid leukemia. Int J Hematol. (2015) 102:579–86. doi: 10.1007/s12185-015-1865-2
137. Lv J, Zhao Y, Zong H, Ma G, Wei X, and Zhao Y. Increased levels of circulating monocytic- and early-stage myeloid-derived suppressor cells (Mdsc) in acute myeloid leukemia. Clin Lab. (2021) 67. doi: 10.7754/Clin.Lab.2020.200719
138. Wang H, Tao Q, Wang Z, Zhang Q, Xiao H, Zhou M, et al. Circulating monocytic myeloid-derived suppressor cells are elevated and associated with poor prognosis in acute myeloid leukemia. J Immunol Res. (2020) 2020:7363084. doi: 10.1155/2020/7363084
139. Ren X, Tao Q, Wang H, Zhang Q, Zhou M, Liu L, et al. Monocytic myeloid-derived suppressor cells but not monocytes predict poor prognosis of acute myeloid leukemia. Turk J Haematol. (2022) 39:230–6. doi: 10.4274/tjh.galenos.2022.2022.0137
140. Peterlin P, Debord C, Eveillard M, Garnier A, Le Bourgeois A, Guillaume T, et al. Peripheral Levels of Monocytic Myeloid-Derived Suppressive Cells before and after First Induction Predict Relapse and Survivals in Aml Patients. J Cell Mol Med. (2022) 26:5486–92. doi: 10.1111/jcmm.17576
141. Hyun SY, Na EJ, Jang JE, Chung H, Kim SJ, Kim JS, et al. Immunosuppressive role of cd11b(+) cd33(+) hla-dr(-) myeloid-derived suppressor cells-like blast subpopulation in acute myeloid leukemia. Cancer Med. (2020) 9:7007–17. doi: 10.1002/cam4.3360
142. Tohumeken S, Baur R, Böttcher M, Stoll A, Loschinski R, Panagiotidis K, et al. Palmitoylated proteins on aml-derived extracellular vesicles promote myeloid-derived suppressor cell differentiation via tlr2/akt/mtor signaling. Cancer Res. (2020) 80:3663–76. doi: 10.1158/0008-5472.Can-20-0024
143. Bai H, Peng Y, Li Y, Duan J, Fu W, Liang X, et al. Cytarabine-induced tnfα Promotes the expansion and suppressive functions of myeloid-derived suppressor cells in acute myeloid leukaemia. Scand J Immunol. (2022) 95:e13158. doi: 10.1111/sji.13158
144. Gao L, Yu S, and Zhang X. Hypothesis: tim-3/galectin-9, a new pathway for leukemia stem cells survival by promoting expansion of myeloid-derived suppressor cells and differentiating into tumor-associated macrophages. Cell Biochem Biophys. (2014) 70:273–7. doi: 10.1007/s12013-014-9900-0
145. Wang L, Jia B, Claxton DF, Ehmann WC, Rybka WB, Mineishi S, et al. Vista is highly expressed on mdscs and mediates an inhibition of T cell response in patients with aml. Oncoimmunology. (2018) 7:e1469594. doi: 10.1080/2162402x.2018.1469594
146. Grauers Wiktorin H, Nilsson MS, Kiffin R, Sander FE, Lenox B, Rydström A, et al. Histamine targets myeloid-derived suppressor cells and improves the anti-tumor efficacy of pd-1/pd-L1 checkpoint blockade. Cancer Immunol Immunother. (2019) 68:163–74. doi: 10.1007/s00262-018-2253-6
147. Fultang L, Panetti S, Ng M, Collins P, Graef S, Rizkalla N, et al. Mdsc targeting with gemtuzumab ozogamicin restores T cell immunity and immunotherapy against cancers. EBioMedicine. (2019) 47:235–46. doi: 10.1016/j.ebiom.2019.08.025
148. Jitschin R, Saul D, Braun M, Tohumeken S, Völkl S, Kischel R, et al. Cd33/cd3-bispecific T-cell engaging (Bite®) antibody construct targets monocytic aml myeloid-derived suppressor cells. J Immunother Cancer. (2018) 6:116. doi: 10.1186/s40425-018-0432-9
149. Jin X, Xie D, Sun R, Lu W, Xiao X, Yu Y, et al. Car-T cells dual-target cd123 and nkg2dls to eradicate aml cells and selectively target immunosuppressive cells. Oncoimmunology. (2023) 12:2248826. doi: 10.1080/2162402x.2023.2248826
150. Nahas MR, Stroopinsky D, Rosenblatt J, Cole L, Pyzer AR, Anastasiadou E, et al. Hypomethylating agent alters the immune microenvironment in acute myeloid leukaemia (Aml) and enhances the immunogenicity of a dendritic cell/aml vaccine. Br J Haematol. (2019) 185:679–90. doi: 10.1111/bjh.15818
151. Miari KE, Guzman ML, Wheadon H, and Williams MTS. Macrophages in acute myeloid leukaemia: significant players in therapy resistance and patient outcomes. Front Cell Dev Biol. (2021) 9:692800. doi: 10.3389/fcell.2021.692800
152. Zhang D, Cui X, Li Y, Wang R, Wang H, Dai Y, et al. Sox13 and M2-like leukemia-associated macrophages contribute to endogenous il-34 caused accelerated progression of acute myeloid leukemia. Cell Death Dis. (2023) 14:308. doi: 10.1038/s41419-023-05822-z
153. Wang L and Zheng G. Macrophages in leukemia microenvironment. Blood Sci. (2019) 1:29–33. doi: 10.1097/bs9.0000000000000014
154. Mantovani A and Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. (2015) 212:435–45. doi: 10.1084/jem.20150295
155. Al-Matary YS, Botezatu L, Opalka B, Hönes JM, Lams RF, Thivakaran A, et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a growth factor independence 1 dependent manner. Haematologica. (2016) 101:1216–27. doi: 10.3324/haematol.2016.143180
156. Tian C, Li Y, Si J, Kang J, Chen Z, Nuermaimaiti R, et al. Knockdown of let-7b in leukemia associated macrophages inhibit acute myeloid leukemia progression. Hematol Oncol. (2022) 41(3):510–9. doi: 10.1002/hon.3120
157. Brauneck F, Fischer B, Witt M, Muschhammer J, Oelrich J, da Costa Avelar PH, et al. Tigit blockade repolarizes aml-associated tigit(+) M2 macrophages to an M1 phenotype and increases cd47-mediated phagocytosis. J Immunother Cancer. (2022) 10(12):e004794. doi: 10.1136/jitc-2022-004794
158. Xu ZJ, Gu Y, Wang CZ, Jin Y, Wen XM, Ma JC, et al. The M2 macrophage marker cd206: A novel prognostic indicator for acute myeloid leukemia. Oncoimmunology. (2020) 9:1683347. doi: 10.1080/2162402x.2019.1683347
159. Yang X, Feng W, Wang R, Yang F, Wang L, Chen S, et al. Repolarizing heterogeneous leukemia-associated macrophages with more M1 characteristics eliminates their pro-leukemic effects. Oncoimmunology. (2018) 7:e1412910. doi: 10.1080/2162402x.2017.1412910
160. Guo R, Lü M, Cao F, Wu G, Gao F, Pang H, et al. Single-cell map of diverse immune phenotypes in the acute myeloid leukemia microenvironment. biomark Res. (2021) 9:15. doi: 10.1186/s40364-021-00265-0
161. Mussai F, De Santo C, Abu-Dayyeh I, Booth S, Quek L, McEwen-Smith RM, et al. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood. (2013) 122:749–58. doi: 10.1182/blood-2013-01-480129
162. Jiang M, Zhang J, Qian L, Miao Y, Song W, Liu H, et al. Moz forms an autoregulatory feedback loop with mir-223 in aml and monocyte/macrophage development. iScience. (2019) 11:189–204. doi: 10.1016/j.isci.2018.12.016
163. Williams M. (2020). Investigating the role of the anti-cell death protein myeloid cell leukaemia 1 in macrophage mediated chemoresistance in acute myeloid leukaemia, in: British Pharmacological Society (BPS), Pharmacology 2019, .
164. Xiang X, Wang J, Lu D, and Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. (2021) 6:75. doi: 10.1038/s41392-021-00484-9
165. Keech T, McGirr C, Winkler IG, and Levesque J-P. Macrophage involvement in the response of acute myeloid leukaemia to chemotherapy. Blood. (2017) 130:5069. doi: 10.1182/blood.V130.Suppl_1.5069.5069
166. Liu J, Wei Y, Jia W, Can C, Wang R, Yang X, et al. Chenodeoxycholic acid suppresses aml progression through promoting lipid peroxidation via ros/P38 mapk/dgat1 pathway and inhibiting M2 macrophage polarization. Redox Biol. (2022) 56:102452. doi: 10.1016/j.redox.2022.102452
167. Turk JL, Parker D, and Poulter LW. Functional aspects of the selective depletion of lymphoid tissue by cyclophosphamide. Immunology. (1972) 23:493–501.
168. Mizoguchi A, Mizoguchi E, Takedatsu H, Blumberg RS, and Bhan AK. Chronic intestinal inflammatory condition generates il-10-producing regulatory B cell subset characterized by cd1d upregulation. Immunity. (2002) 16:219–30. doi: 10.1016/s1074-7613(02)00274-1
169. Jansen K, Cevhertas L, Ma S, Satitsuksanoa P, Akdis M, and van de Veen W. Regulatory B cells, a to Z. Allergy. (2021) 76:2699–715. doi: 10.1111/all.14763
170. Rosser EC and Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
171. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. Cd19(+)Cd24(Hi)Cd38(Hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. (2010) 32:129–40. doi: 10.1016/j.immuni.2009.11.009
172. Iwata Y, Matsushita T, Horikawa M, Dilillo DJ, Yanaba K, Venturi GM, et al. Characterization of a rare il-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. (2011) 117:530–41. doi: 10.1182/blood-2010-07-294249
173. Lv Y, Wang H, and Liu Z. The role of regulatory B cells in patients with acute myeloid leukemia. Med Sci Monit. (2019) 25:3026–31. doi: 10.12659/msm.915556
174. Dong Q, Li G, Fozza C, Wang S, Yang S, Sang Y, et al. Levels and clinical significance of regulatory B cells and T cells in acute myeloid leukemia. BioMed Res Int. (2020) 2020:7023168. doi: 10.1155/2020/7023168
175. Shi Y, Liu Z, and Wang H. Expression of pd-L1 on regulatory B cells in patients with acute myeloid leukaemia and its effect on prognosis. J Cell Mol Med. (2022) 26:3506–12. doi: 10.1111/jcmm.17390
176. Que H, Fu Q, Lan T, Tian X, and Wei X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim Biophys Acta Rev Cancer. (2022) 1877:188762. doi: 10.1016/j.bbcan.2022.188762
177. Hu T, Cheng B, Matsunaga A, Zhang T, Lu X, Fang H, et al. Single-cell analysis defines highly specific leukemia-induced neutrophils and links mmp8 expression to recruitment of tumor associated neutrophils during fgfr1 driven leukemogenesis. Exp Hematol Oncol. (2024) 13:49. doi: 10.1186/s40164-024-00514-6
178. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of mhc class ii. J Immunol. (2008) 180:5916–26. doi: 10.4049/jimmunol.180.9.5916
179. Onishi Y, Fehervari Z, Yamaguchi T, and Sakaguchi S. Foxp3+ Natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U.S.A. (2008) 105:10113–8. doi: 10.1073/pnas.0711106105
180. Wang J, Zhao X, and Wan YY. Intricacies of tgf-β Signaling in treg and th17 cell biology. Cell Mol Immunol. (2023) 20:1002–22. doi: 10.1038/s41423-023-01036-7
181. Bopp T, Becker C, Klein M, Klein-Hessling S, Palmetshofer A, Serfling E, et al. Cyclic adenosine monophosphate is a key component of regulatory T cell-mediated suppression. J Exp Med. (2007) 204:1303–10. doi: 10.1084/jem.20062129
182. Li C, Jiang P, Wei S, Xu X, and Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. (2020) 19:116. doi: 10.1186/s12943-020-01234-1
183. Lin C, Guo J, and Jia R. Roles of regulatory T cell-derived extracellular vesicles in human diseases. Int J Mol Sci. (2022) 23(19):11206. doi: 10.3390/ijms231911206
184. Xie Y, Zhang X, Zhao T, Li W, and Xiang J. Natural cd8+25+ Regulatory T cell-secreted exosomes capable of suppressing cytotoxic T lymphocyte-mediated immunity against B16 melanoma. Biochem Biophys Res Commun. (2013) 438:152–5. doi: 10.1016/j.bbrc.2013.07.044
185. Wing JB, Tanaka A, and Sakaguchi S. Human foxp3(+) regulatory T cell heterogeneity and function in autoimmunity and cancer. Immunity. (2019) 50:302–16. doi: 10.1016/j.immuni.2019.01.020
186. Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, and Colombo MP. Triggering of ox40 (Cd134) on cd4(+)Cd25+ T cells blocks their inhibitory activity: A novel regulatory role for ox40 and its comparison with gitr. Blood. (2005) 105:2845–51. doi: 10.1182/blood-2004-07-2959
187. Piconese S, Valzasina B, and Colombo MP. Ox40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med. (2008) 205:825–39. doi: 10.1084/jem.20071341
188. Togashi Y, Shitara K, and Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
189. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res. (2004) 64:5839–49. doi: 10.1158/0008-5472.Can-04-0465
190. García-Ortiz A and Serrador JM. Nitric oxide signaling in T cell-mediated immunity. Trends Mol Med. (2018) 24:412–27. doi: 10.1016/j.molmed.2018.02.002
191. Baumann T, Dunkel A, Schmid C, Schmitt S, Hiltensperger M, Lohr K, et al. Regulatory myeloid cells paralyze T cells through cell-cell transfer of the metabolite methylglyoxal. Nat Immunol. (2020) 21:555–66. doi: 10.1038/s41590-020-0666-9
192. Yu S, Ren X, and Li L. Myeloid-derived suppressor cells in hematologic Malignancies: two sides of the same coin. Exp Hematol Oncol. (2022) 11:43. doi: 10.1186/s40164-022-00296-9
193. Srivastava MK, Sinha P, Clements VK, Rodriguez P, and Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. (2010) 70:68–77. doi: 10.1158/0008-5472.Can-09-2587
194. Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. (2021) 593:282–8. doi: 10.1038/s41586-021-03442-1
195. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through ido expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. (2013) 190:3783–97. doi: 10.4049/jimmunol.1201449
196. Ryzhov S, Novitskiy SV, Goldstein AE, Biktasova A, Blackburn MR, Biaggioni I, et al. Adenosinergic regulation of the expansion and immunosuppressive activity of cd11b+Gr1+ Cells. J Immunol. (2011) 187:6120–9. doi: 10.4049/jimmunol.1101225
197. Li J, Wang L, Chen X, Li L, Li Y, Ping Y, et al. Cd39/cd73 upregulation on myeloid-derived suppressor cells via tgf-β-mtor-hif-1 signaling in patients with non-small cell lung cancer. Oncoimmunology. (2017) 6:e1320011. doi: 10.1080/2162402x.2017.1320011
198. Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, et al. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol. (2009) 182:5693–701. doi: 10.4049/jimmunol.0900092
199. Kusmartsev S and Gabrilovich DI. Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J Leukoc Biol. (2003) 74:186–96. doi: 10.1189/jlb.0103010
200. Raber PL, Thevenot P, Sierra R, Wyczechowska D, Halle D, Ramirez ME, et al. Subpopulations of myeloid-derived suppressor cells impair T cell responses through independent nitric oxide-related pathways. Int J Cancer. (2014) 134:2853–64. doi: 10.1002/ijc.28622
201. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. (2021) 6:362. doi: 10.1038/s41392-021-00670-9
202. Li H, Han Y, Guo Q, Zhang M, and Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of nk cells through membrane-bound tgf-beta 1. J Immunol. (2009) 182:240–9. doi: 10.4049/jimmunol.182.1.240
203. Law AMK, Valdes-Mora F, and Gallego-Ortega D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells. (2020) 9(3):561. doi: 10.3390/cells9030561
204. Bewersdorf JP and Zeidan AM. Myeloid-derived suppressor cells: A grey eminence in the aml tumor microenvironment? Expert Rev Anticancer Ther. (2022) 22:239–41. doi: 10.1080/14737140.2022.2030227
205. Kusmartsev S, Su Z, Heiser A, Dannull J, Eruslanov E, Kübler H, et al. Reversal of myeloid cell-mediated immunosuppression in patients with metastatic renal cell carcinoma. Clin Cancer Res. (2008) 14:8270–8. doi: 10.1158/1078-0432.Ccr-08-0165
206. Cortes JE, de Lima M, Dombret H, Estey EH, Giralt SA, Montesinos P, et al. Prevention, recognition, and management of adverse events associated with gemtuzumab ozogamicin use in acute myeloid leukemia. J Hematol Oncol. (2020) 13:137. doi: 10.1186/s13045-020-00975-2
207. Li C, Xu X, Wei S, Jiang P, Xue L, and Wang J. Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J Immunother Cancer. (2021) 9(1):e001341. doi: 10.1136/jitc-2020-001341
208. Han Y, Guo W, Ren T, Huang Y, Wang S, Liu K, et al. Tumor-associated macrophages promote lung metastasis and induce epithelial-mesenchymal transition in osteosarcoma by activating the cox-2/stat3 axis. Cancer Lett. (2019) 440-441:116–25. doi: 10.1016/j.canlet.2018.10.011
209. Pan Y, Yu Y, Wang X, and Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
210. Michaud D, Steward CR, Mirlekar B, and Pylayeva-Gupta Y. Regulatory B cells in cancer. Immunol Rev. (2021) 299:74–92. doi: 10.1111/imr.12939
211. Catalán D, Mansilla MA, Ferrier A, Soto L, Oleinika K, Aguillón JC, et al. Immunosuppressive mechanisms of regulatory B cells. Front Immunol. (2021) 12:611795. doi: 10.3389/fimmu.2021.611795
212. Flores-Borja F, Bosma A, Ng D, Reddy V, Ehrenstein MR, Isenberg DA, et al. Cd19+Cd24hicd38hi B cells maintain regulatory T cells while limiting th1 and th17 differentiation. Sci Transl Med. (2013) 5:173ra23. doi: 10.1126/scitranslmed.3005407
213. Wachowska M, Wojciechowska A, and Muchowicz A. The role of neutrophils in the pathogenesis of chronic lymphocytic leukemia. Int J Mol Sci. (2021) 23(1):365. doi: 10.3390/ijms23010365
214. Podaza E, Risnik D, Colado A, Elías E, Almejún MB, Fernandez Grecco H, et al. Chronic lymphocytic leukemia cells increase neutrophils survival and promote their differentiation into cd16(High) cd62l(Dim) immunosuppressive subset. Int J Cancer. (2019) 144:1128–34. doi: 10.1002/ijc.31762
215. Najafi M, Farhood B, and Mortezaee K. Contribution of regulatory T cells to cancer: A review. J Cell Physiol. (2019) 234:7983–93. doi: 10.1002/jcp.27553
216. Haist M, Stege H, Grabbe S, and Bros M. The functional crosstalk between myeloid-derived suppressor cells and regulatory T cells within the immunosuppressive tumor microenvironment. Cancers (Basel). (2021) 13(2):210. doi: 10.3390/cancers13020210
217. Jitschin R, Braun M, Büttner M, Dettmer-Wilde K, Bricks J, Berger J, et al. Cll-cells induce idohi cd14+Hla-drlo myeloid-derived suppressor cells that inhibit T-cell responses and promote tregs. Blood. (2014) 124:750–60. doi: 10.1182/blood-2013-12-546416
218. Liu M, Wei F, Wang J, Yu W, Shen M, Liu T, et al. Myeloid-derived suppressor cells regulate the immunosuppressive functions of pd-1(-)Pd-L1(+) bregs through pd-L1/pi3k/akt/nf-κb axis in breast cancer. Cell Death Dis. (2021) 12:465. doi: 10.1038/s41419-021-03745-1
219. Beury DW, Parker KH, Nyandjo M, Sinha P, Carter KA, and Ostrand-Rosenberg S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J Leukoc Biol. (2014) 96:1109–18. doi: 10.1189/jlb.3A0414-210R
220. Chang AL, Miska J, Wainwright DA, Dey M, Rivetta CV, Yu D, et al. Ccl2 produced by the glioma microenvironment is essential for the recruitment of regulatory T cells and myeloid-derived suppressor cells. Cancer Res. (2016) 76:5671–82. doi: 10.1158/0008-5472.Can-16-0144
221. Ou W, Thapa RK, Jiang L, Soe ZC, Gautam M, Chang JH, et al. Regulatory T cell-targeted hybrid nanoparticles combined with immuno-checkpoint blockage for cancer immunotherapy. J Control Release. (2018) 281:84–96. doi: 10.1016/j.jconrel.2018.05.018
222. Chen L, Wang S, Wang Y, Zhang W, Ma K, Hu C, et al. Il-6 influences the polarization of macrophages and the formation and growth of colorectal tumor. Oncotarget. (2018) 9:17443–54. doi: 10.18632/oncotarget.24734
223. Vera-Lozada G, Minnicelli C, Segges P, Stefanoff G, Kristcevic F, Ezpeleta J, et al. Interleukin 10 (Il10) proximal promoter polymorphisms beyond clinical response in classical hodgkin lymphoma: exploring the basis for the genetic control of the tumor microenvironment. Oncoimmunology. (2018) 7:e1389821. doi: 10.1080/2162402x.2017.1389821
224. Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP, et al. Marginal zone cd169+ Macrophages coordinate apoptotic cell-driven cellular recruitment and tolerance. Proc Natl Acad Sci U.S.A. (2014) 111:4215–20. doi: 10.1073/pnas.1320924111
225. Yi H, Zhen Y, Jiang L, Zheng J, and Zhao Y. The phenotypic characterization of naturally occurring regulatory cd4+Cd25+ T cells. Cell Mol Immunol. (2006) 3:189–95.
226. Ustun C, Miller JS, Munn DH, Weisdorf DJ, and Blazar BR. Regulatory T cells in acute myelogenous leukemia: is it time for immunomodulation? Blood. (2011) 118:5084–95. doi: 10.1182/blood-2011-07-365817
227. Ma F, Li S, Gao X, Zhou J, Zhu X, Wang D, et al. Interleukin-6-mediated ccr9(+) interleukin-17-producing regulatory T cells polarization increases the severity of necrotizing enterocolitis. EBioMedicine. (2019) 44:71–85. doi: 10.1016/j.ebiom.2019.05.042
228. Mishra S, Srinivasan S, Ma C, and Zhang N. Cd8(+) regulatory T cell - a mystery to be revealed. Front Immunol. (2021) 12:708874. doi: 10.3389/fimmu.2021.708874
229. Bafor EE, Valencia JC, and Young HA. Double negative T regulatory cells: an emerging paradigm shift in reproductive immune tolerance? Front Immunol. (2022) 13:886645. doi: 10.3389/fimmu.2022.886645
230. Fischer K, Voelkl S, Heymann J, Przybylski GK, Mondal K, Laumer M, et al. Isolation and characterization of human antigen-specific tcr alpha beta+ Cd4(-)Cd8- double-negative regulatory T cells. Blood. (2005) 105:2828–35. doi: 10.1182/blood-2004-07-2583
231. Singh AK, Tripathi P, and Cardell SL. Type ii nkt cells: an elusive population with immunoregulatory properties. Front Immunol. (2018) 9:1969. doi: 10.3389/fimmu.2018.01969
232. Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, and Tedder TF. A regulatory B cell subset with a unique cd1dhicd5+ Phenotype controls T cell-dependent inflammatory responses. Immunity. (2008) 28:639–50. doi: 10.1016/j.immuni.2008.03.017
233. Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. (2007) 178:7868–78. doi: 10.4049/jimmunol.178.12.7868
234. Shen P, Roch T, Lampropoulou V, O’Connor RA, Stervbo U, Hilgenberg E, et al. Il-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. (2014) 507:366–70. doi: 10.1038/nature12979
235. Bankoti R, Gupta K, Levchenko A, and Stäger S. Marginal zone B cells regulate antigen-specific T cell responses during infection. J Immunol. (2012) 188:3961–71. doi: 10.4049/jimmunol.1102880
236. Xiao S, Brooks CR, Zhu C, Wu C, Sweere JM, Petecka S, et al. Defect in regulatory B-cell function and development of systemic autoimmunity in T-cell ig mucin 1 (Tim-1) mucin domain-mutant mice. Proc Natl Acad Sci U.S.A. (2012) 109:12105–10. doi: 10.1073/pnas.1120914109
237. Matsumoto M, Baba A, Yokota T, Nishikawa H, Ohkawa Y, Kayama H, et al. Interleukin-10-producing plasmablasts exert regulatory function in autoimmune inflammation. Immunity. (2014) 41:1040–51. doi: 10.1016/j.immuni.2014.10.016
238. van de Veen W, Stanic B, Yaman G, Wawrzyniak M, Söllner S, Akdis DG, et al. Igg4 production is confined to human il-10-producing regulatory B cells that suppress antigen-specific immune responses. J Allergy Clin Immunol. (2013) 131:1204–12. doi: 10.1016/j.jaci.2013.01.014
239. Lindner S, Dahlke K, Sontheimer K, Hagn M, Kaltenmeier C, Barth TF, et al. Interleukin 21-induced granzyme B-expressing B cells infiltrate tumors and regulate T cells. Cancer Res. (2013) 73:2468–79. doi: 10.1158/0008-5472.Can-12-3450
Keywords: acute myeloid leukemia, regulatory T cells, regulatory B cells, myeloid-derived suppressor cells, leukemia-associated macrophages, leukemia-associated neutrophils
Citation: Liu M, Yang M, Qi Y, Ma Y, Guo Q, Guo L, Liu C, Liu W, Xiao L and Yang Y (2025) Immunosuppressive cells in acute myeloid leukemia: mechanisms and therapeutic target. Front. Immunol. 16:1627161. doi: 10.3389/fimmu.2025.1627161
Received: 12 May 2025; Accepted: 08 July 2025;
Published: 23 July 2025.
Edited by:
Akhileshwar Namani, Sri Shankara Cancer Hospital and Research Centre, IndiaReviewed by:
Nikoleta Bizymi, University of Crete, GreeceFábio Magalhães-Gama, Oswaldo Cruz Foundation (Fiocruz), Brazil
Jiapei Yuan, Peking Union Medical College Hospital (CAMS), China
Copyright © 2025 Liu, Yang, Qi, Ma, Guo, Guo, Liu, Liu, Xiao and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenjun Liu, d2VuanVuX2xpdUBzd211LmVkdS5jbg==; Lan Xiao, ODE0NTEwODUxQHFxLmNvbQ==; You Yang, eW91eWFuZzA5MUBzd211LmVkdS5jbg==
†These authors have contributed equally to this work