 Zexing Shan1
Zexing Shan1 Yefu Liu2*
Yefu Liu2*- 1Department of Gastric Surgery, Liaoning Cancer Hospital and Institute, Cancer Hospital of China Medical University, Shenyang, China
- 2Department of Hepatobiliary and Pancreatic Surgery, Liaoning Cancer Hospital & Institute, Shenyang, Liaoning, China
Gastric cancer (GC) continues to rank among the leading causes of cancer-related mortality globally, with treatment resistance and recurrence posing significant clinical hurdles. While surgical interventions, chemotherapy, and targeted therapies are available, their efficacy in managing advanced or metastatic forms of the disease remains constrained. This review provided an overview of the role of glycolytic reprogramming in gastric cancer, emphasizing the complex regulation by epigenetic mechanisms, non-coding RNAs, post-translational modifications, and oncogenic signaling pathways. This review discusses how epigenetic mechanisms, including m6A methylation and ceRNA networks involving circRNAs and microRNAs, modulate key glycolytic enzymes such as PKM2, HK2, and PGK1, thereby promoting tumor growth, metastasis, and chemoresistance. The study also emphasizes the impact of post-translational modifications like succinylation and ubiquitination on enzyme activity, affecting glycolytic flux and tumor adaptability. Additionally, the article details the crosstalk between glycolytic pathways and oncogenic signaling networks, including hypoxia-inducible factors and YAP/TAZ transcriptional regulators, which sustain tumor stemness and immune evasion. Therapeutic strategies targeting these metabolic vulnerabilities—such as inhibiting m6A regulators, disrupting ceRNA interactions, and modulating enzyme modifications—are discussed as potential approaches to improve gastric cancer treatment. Overall, we underscores the complexity of metabolic regulation in gastric cancer and proposes that targeting its epigenetic and signaling networks offers promising avenues for innovative therapies to overcome resistance and hinder tumor progression.
1 Introduction
Gastric cancer (GC) maintains its global dominance as the fifth most prevalent malignancy and third leading contributor to cancer mortality, with disproportionately high disease burden observed worldwide (1–7). While epidemiological trends show declining incidence in some geographical regions, delayed diagnosis persists due to nonspecific early clinical manifestations and inadequate screening biomarkers, culminating in a sobering 5-year survival rate below 30% for advanced-stage patients (8). The molecular pathogenesis of gastric cancer (GC) unfolds through Correa’s multi-step carcinogenic sequence. In this progression, Helicobacter pylori infection acts in synergy with chronic inflammatory processes and the accumulation of genetic/epigenetic aberrations, collectively driving the malignant transformation from gastritis to adenocarcinoma (9–12). Current therapeutic modalities, encompassing surgical resection, cytotoxic chemotherapy, and Human epidermal growth factor receptor 2 (HER2)-targeted agents, demonstrate limited effectiveness against tumor heterogeneity, metastatic dissemination, and therapy resistance mechanisms potentiated by the immunosuppressive tumor microenvironment (TME) (13–16). Even breakthrough immunotherapies exhibit modest clinical responses in GC (17–19), emphasizing the critical imperative to discover innovative therapeutic strategies targeting fundamental biological vulnerabilities such as metabolic reprogramming (20–22).
Metabolic reprogramming represents an essential adaptive mechanism enabling malignant cells to sustain uncontrolled proliferation under nutrient-constrained conditions (23–25). The Warburg effect-characterized by preferential glucose utilization through aerobic glycolysis despite oxygen availability – serves as a cornerstone of this metabolic rewiring in cancer biology (26–29). This bioenergetic shift facilitates rapid ATP generation while accumulating glycolytic intermediates for macromolecule biosynthesis, concurrently establishing an acidic, lactate-enriched TME that fosters immune escape, neoangiogenesis, and metastatic competence (30–35). Molecular orchestrators of this process include hypoxia-inducible factor-1α (HIF-1α), oncogenic kinase cascades, and rate-limiting glycolytic enzymes such as hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA), all frequently overexpressed in malignant lesions (36–41). In GC pathogenesis, Hpylori-induced inflammatory signaling synergizes with oncogenic drivers to amplify glycolytic flux, establishing a self-reinforcing cycle that accelerates tumor progression and therapeutic resistance (42, 43). Preclinical investigations employing glycolytic pathway inhibitors-targeting glucose transporters (GLUTs), LDHA enzymatic activity, or lactate efflux mechanisms-have achieved significant suppression of tumor growth and chemotherapy desensitization, underscoring glycolysis inhibition as a promising therapeutic strategy (44–49).
The glycolytic phenotype exerts multifaceted impacts on gastric carcinogenesis and treatment responses. Gastric cancer (GC) cells display a striking reliance on glycolysis, driven by constitutive activation of the PI3K/AKT/mTOR signaling pathway and stabilization of HIF-1α—effects often amplified by Helicobacter pylori-associated chronic inflammation (50–56). This metabolic adaptation not only fuels unchecked proliferation but also generates an immunosuppressive, pro-metastatic niche through extracellular acidification and lactate accumulation (57–59). Clinically relevant glycolytic markers including HK2 and LDHA demonstrate strong correlations with advanced tumor stage, chemotherapy failure, and poor prognosis. Mechanistically, LDHA-generated lactate enhances β-catenin pathway activation, promoting cancer stem cell maintenance (60, 61). Emerging evidence reveals metabolic heterogeneity across GC molecular subtypes, presenting both challenges and opportunities for precision targeting. While preclinical models demonstrate encouraging antitumor effects with glycolytic inhibitors, clinical translation remains hampered by limited GC-specific trials and incomplete understanding of lactate’s dual metabolic/signaling roles. This comprehensive review analyzes the pathophysiological significance of glycolytic remodeling in GC and evaluates innovative therapeutic approaches, including metabolic inhibitor-immunotherapy combinations and nanoparticle-mediated drug delivery systems, that may overcome current limitations in targeted therapy development.
2 Glycolytic reprogramming in malignant progression
2.1 Core biochemistry of glycolytic flux
The glycolytic pathway represents an evolutionarily conserved mechanism for cytosolic glucose catabolism, producing both ATP and metabolic precursors critical to cellular homeostasis. Initiated by glucose uptake mediated by the GLUT family of transporters, this ten-step enzymatic cascade includes three irreversible phosphorylation reactions, catalyzed by hexokinase (HK), phosphofructokinase-1 (PFK-1), and pyruvate kinase (PK), respectively (44, 62–66). Following GLUT-mediated cellular entry, glucose undergoes HK-dependent phosphorylation to glucose-6-phosphate (G6P), committing the molecule to glycolytic processing. Subsequent isomerization yields fructose-6-phosphate (F6P), which undergoes PFK-1-catalyzed conversion to fructose-1,6-bisphosphate (F1,6BP) – the pathway’s primary regulatory node through allosteric control by ATP, citrate, and fructose-2,6-bisphosphate (F2,6BP). Cleavage of F1,6BP generates two triose phosphates, with glyceraldehyde-3-phosphate (GA3P) entering the energy-yielding phase through oxidation to 1,3-bisphosphoglycerate, coupled with NADH production. Final steps yield pyruvate, which under normoxic conditions enters mitochondria for oxidative phosphorylation (OXPHOS), while hypoxia prompts lactate dehydrogenase (LDH)-mediated reduction to lactate with concomitant NAD+ regeneration – a critical adaptation for glycolytic continuity.
2.2 The Warburg effect: metabolic hallmark of malignancy
The Warburg effect epitomizes cancer’s metabolic paradox: preferential reliance on aerobic glycolysis over mitochondrial OXPHOS despite oxygen availability (32, 67, 68). Unlike normal cells that maximize ATP yield via OXPHOS, malignant cells sacrifice energy efficiency to prioritize rapid biomass synthesis and microenvironment remodeling (69–71). This metabolic rewiring arises from mitochondrial dysfunction, impaired electron transport chain (ETC) activity, and microenvironmental stressors including hypoxia and nutrient competition (72, 73). Consequent NAD+/NADH ratio reduction triggers two critical adaptations (74–76). L-2-hydroxyglutarate (L-2-HG) accumulates through NADH-dependent reduction of α-ketoglutarate (α-KG), competitively inhibiting α-KG-dependent dioxygenases to disrupt epigenetic regulation and hypoxic signaling. Concurrently, reductive carboxylation of α-KG to citrate sustains lipogenesis under mitochondrial dysfunction. The resultant lactate overproduction creates an acidic extracellular microenvironment, which activates proteolytic enzymes, stabilizes HIF-1α, and induces vasodilation—collectively driving invasion, angiogenesis, and immune evasion. These interconnected processes constitute the metabolic circuitry of the Warburg effect in oncogenic adaptation, as illustrated in Figure 1.
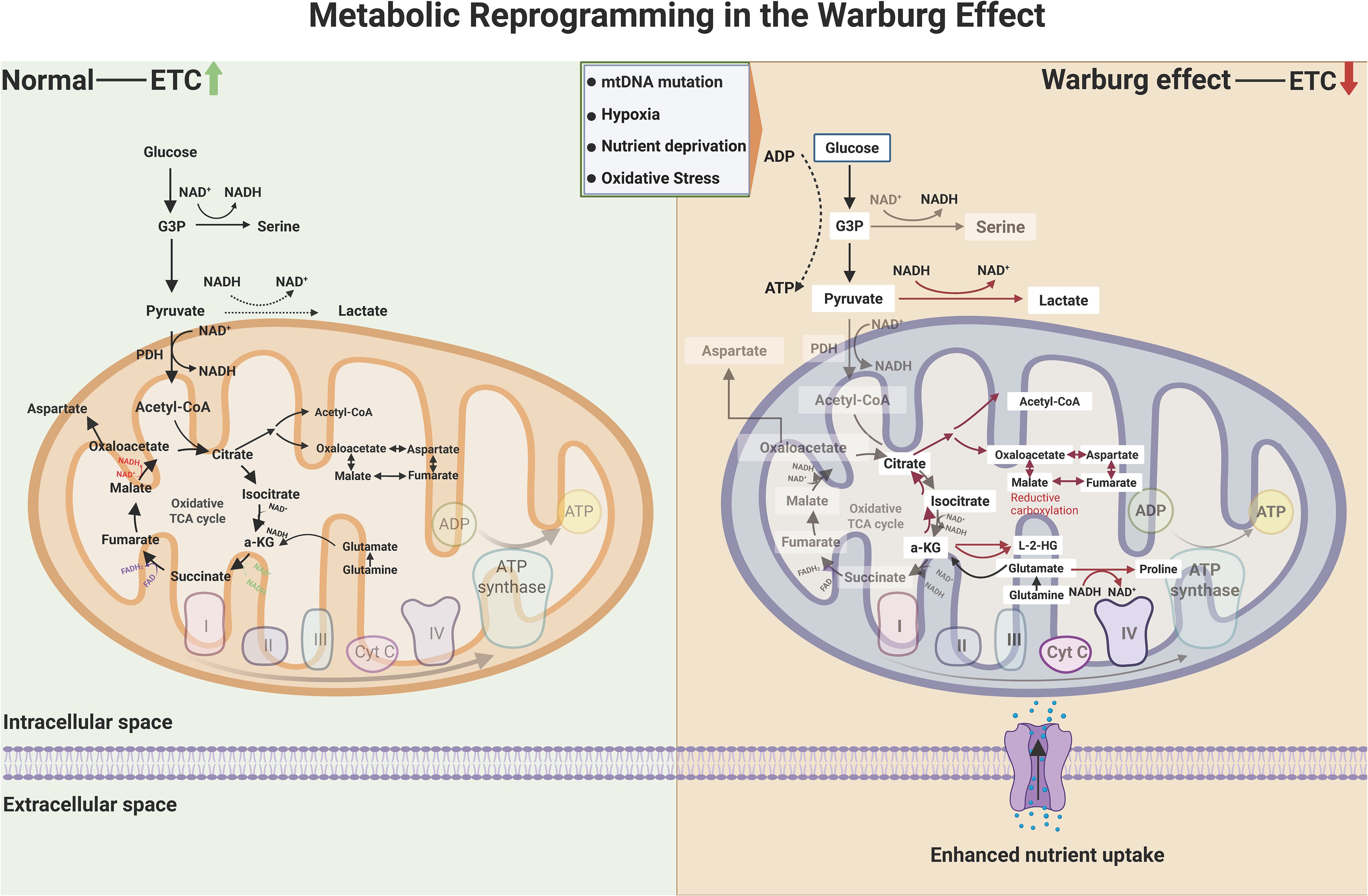
Figure 1. Metabolic circuitry of the Warburg effect in oncogenic adaptation. While normal cells predominantly harness mitochondrial oxidative phosphorylation (OXPHOS) for energy production—coupling the tricarboxylic acid (TCA) cycle with electron transport chain (ETC) activity—cancer cells exhibit a hallmark metabolic divergence. Malignant phenotypes prioritize glycolysis and lactate fermentation, even under oxygen-replete conditions, to sustain biosynthetic demands. This reprogramming arises from mitochondrial insufficiency (e.g., mtDNA lesions, ETC dysfunction) and microenvironmental stressors (hypoxia, nutrient scarcity), which suppress NAD+/NADH recycling and amplify reductive glutamine metabolism. The resultant accumulation of oncometabolites, including L-2-hydroxyglutarate (L-2-HG) via α-ketoglutarate (α-KG) reduction, inhibits α-KG-dependent dioxygenases (e.g., TET enzymes, histone demethylases), thereby stabilizing hypoxia-inducible factor 1α (HIF-1α) and silencing tumor suppressors. Concurrently, reductive carboxylation converts α-KG to citrate via isocitrate dehydrogenase (IDH) isoforms, bypassing canonical TCA flux to fuel lipogenesis and macromolecular synthesis. These adaptations collectively drive stemness, immune evasion, and therapeutic resistance through epigenetic dysregulation and redox balance rewiring. (Created by biorender.com).
2.3 Glycolytic enzymes as multifaceted oncogenic drivers
Beyond their canonical metabolic roles, glycolytic enzymes exert pleiotropic control over malignant phenotypes through both catalytic and non-catalytic mechanisms (77–79). HK2, frequently overexpressed in advanced tumors, binds mitochondrial voltage-dependent anion channels (VDACs) to evade apoptosis while enhancing glucose phosphorylation (80, 81). LDHA, a key hypoxia-responsive enzyme, not only maintains glycolytic flux but also generates lactate-a potent oncometabolite that acidifies the TME to stabilize HIF-1α, activate TGF-β signaling, and induce epithelial-mesenchymal transition (EMT) (82–85). Notably, lactate-mediated TME acidification directly impairs cytotoxic T lymphocyte (CTL) function while polarizing tumor-associated macrophages (TAMs) toward immunosuppressive M2 phenotypes (86, 87). Pyruvate kinase M2 (PKM2), the embryonic splice variant re-expressed in cancers, exhibits dynamic oligomeric regulation: tetrameric forms catalyze phosphoenolpyruvate-to-pyruvate conversion, whereas dimeric PKM2 translocates to the nucleus, serving as a transcriptional coactivator for HIF-1α, signal transducer and activator of transcription 3 (STAT3), and β-catenin to drive cell cycle progression and stemness (37, 88–94). Overall, glycolytic reprogramming and its oncogenic circuitry in tumor development was shown in Figure 2.
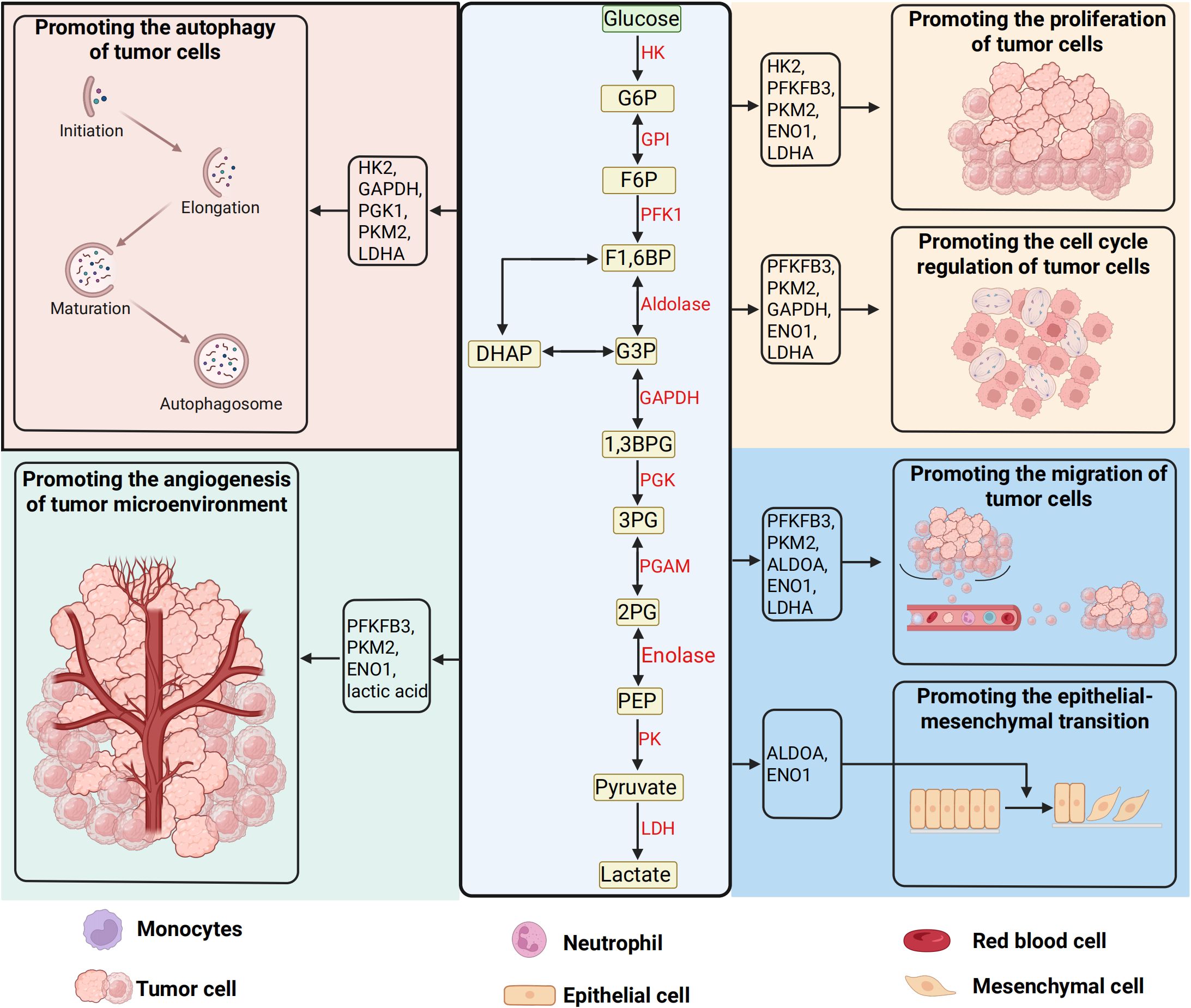
Figure 2. Glycolytic reprogramming and its oncogenic circuitry in tumor development. The glycolytic pathway, a hallmark of cancer metabolism, orchestrates the sequential breakdown of glucose into lactate under both hypoxic and normoxic conditions—a phenomenon known as the Warburg effect. This cytoplasmic cascade initiates with glucose uptake mediated by solute carrier transporters (SLC2A/GLUT family), which comprises 14 isoforms with distinct tissue-specific expression and regulatory roles in tumor biology. Intracellular glucose undergoes stepwise enzymatic processing: hexokinase (HK) traps glucose via phosphorylation, phosphofructokinase-1 (PFK-1) gates glycolytic flux through allosteric regulation, and pyruvate kinase (PK) catalyzes the terminal ATP-generating step to yield pyruvate. Under oxygen deprivation, lactate dehydrogenase (LDH) redirects pyruvate toward lactate production, sustaining NAD+ regeneration for continued glycolysis. Beyond energy production, glycolytic intermediates and enzymes directly engage in oncogenic signaling—HK-2 stabilizes mitochondrial membranes to inhibit apoptosis, PKM2 translocates to the nucleus as a transcriptional coactivator, and lactate acidifies the tumor microenvironment to drive immune evasion, extracellular matrix remodeling, and metastatic dissemination. These multifaceted interactions position glycolysis as a central hub for metabolic plasticity, epigenetic reprogramming, and therapeutic resistance in malignancies. (Created by biorender.com).
2.4 GLUTs: gatekeepers of tumor metabolism
The GLUT family represents the first bottleneck in tumor glycolytic dependency, with isoform-specific expression patterns dictating metabolic adaptability (46, 95). GLUT1 overexpression, driven by H. pylori-induced NF-κB activation in gastric carcinogenesis, correlates with advanced Tumor node metastasis classification(TNM) staging, venous invasion, and reduced 5-year survival in GC (96). Intriguingly, GLUT3 -typically restricted to neurons-becomes aberrantly expressed in therapy-resistant tumors, enabling glucose uptake under hypoglycemic TME conditions (97, 98). Clinical evidence reveals dynamic GLUT regulation during treatment: neoadjuvant chemotherapy downregulates GLUT4 in GC patients, coinciding with acquired chemoresistance through PI3K/AKT pathway activation (99, 100). Preclinical models demonstrate that dual targeting of GLUT1 and GLUT3 synergistically inhibits glycolytic flux and restores cisplatin sensitivity in refractory GC cells (101). These findings position GLUT isoform-specific inhibition as a promising strategy to circumvent metabolic adaptations underlying treatment failure.
3 Epigenetic orchestration of glycolytic reprogramming in gastric cancer
Gastric cancer (GC) progression is intricately tied to metabolic reprogramming, with dysregulated glycolytic flux—manifested as the Warburg effect—serving as a hallmark of tumor bioenergetics. Emerging evidence underscores the pivotal role of epigenetic mechanisms in orchestrating this metabolic shift through multilayered regulatory networks. Non-coding RNAs, including circRNAs and lncRNAs, dictate glycolytic adaptation via miRNA sponging, RNA splicing modulation, and epigenetic remodeling, while viral miRNAs further disrupt host metabolic checkpoints. Concurrently, dynamic m6A epitranscriptomic modifications fine-tune glycolytic enzyme expression through methyltransferase/eraser-mediated RNA methylation cycles. Post-translational modifications of metabolic kinases and transporters add another regulatory stratum, directly modulating enzyme stability and activity. These interconnected mechanisms collectively sustain metabolic plasticity, drive therapeutic resistance, and establish tumor-microenvironment crosstalk. The complexity of this epigenetic-metabolic interplay is systematically dissected in subsequent sections, with comprehensive regulatory hierarchies and molecular interactions detailed in Figure 3 and Table 1. Elucidating these pathways unveils novel vulnerabilities for precision therapeutic targeting in GC.
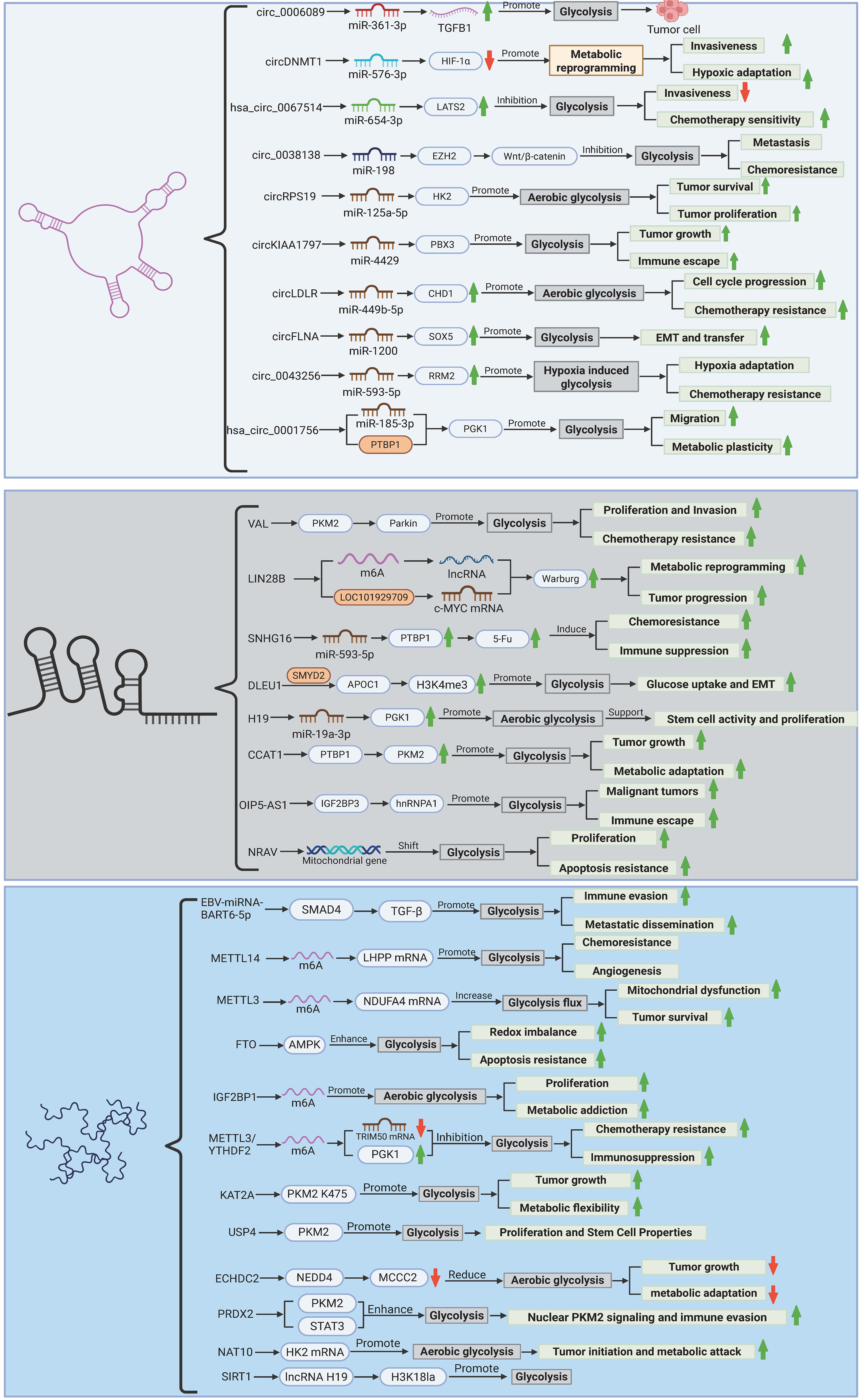
Figure 3. Epigenetic regulation of glycolytic reprogramming in gastric cancer. The figure illustrates the multilayered epigenetic control of glycolytic rewiring in gastric carcinogenesis. Non-coding RNAs (circRNAs, lncRNAs, and viral miRNAs) orchestrate metabolic adaptation through ceRNA networks, splicing regulation, and enzyme stabilization (e.g., circ_0006089/miR-361-3p/TGFB1; VAL/PKM2 ubiquitination). m6A epitranscriptomic modifications (METTL3/14, FTO, IGF2BP1) fine-tune glycolytic enzyme expression via mRNA stability and translation efficiency. Post-translational modifications (succinylation, deubiquitination, lactylation) dynamically regulate PKM2/HK2 activity and chromatin remodeling, linking glycolysis to oncogenic signaling. Hypoxia, nutrient stress, and viral integration converge to sustain a Warburg phenotype, driving proliferation, chemoresistance, and immune evasion, while highlighting potential therapeutic targets (e.g., METTL3 inhibitors, circRNA-directed siRNAs). (Created by biorender.com).
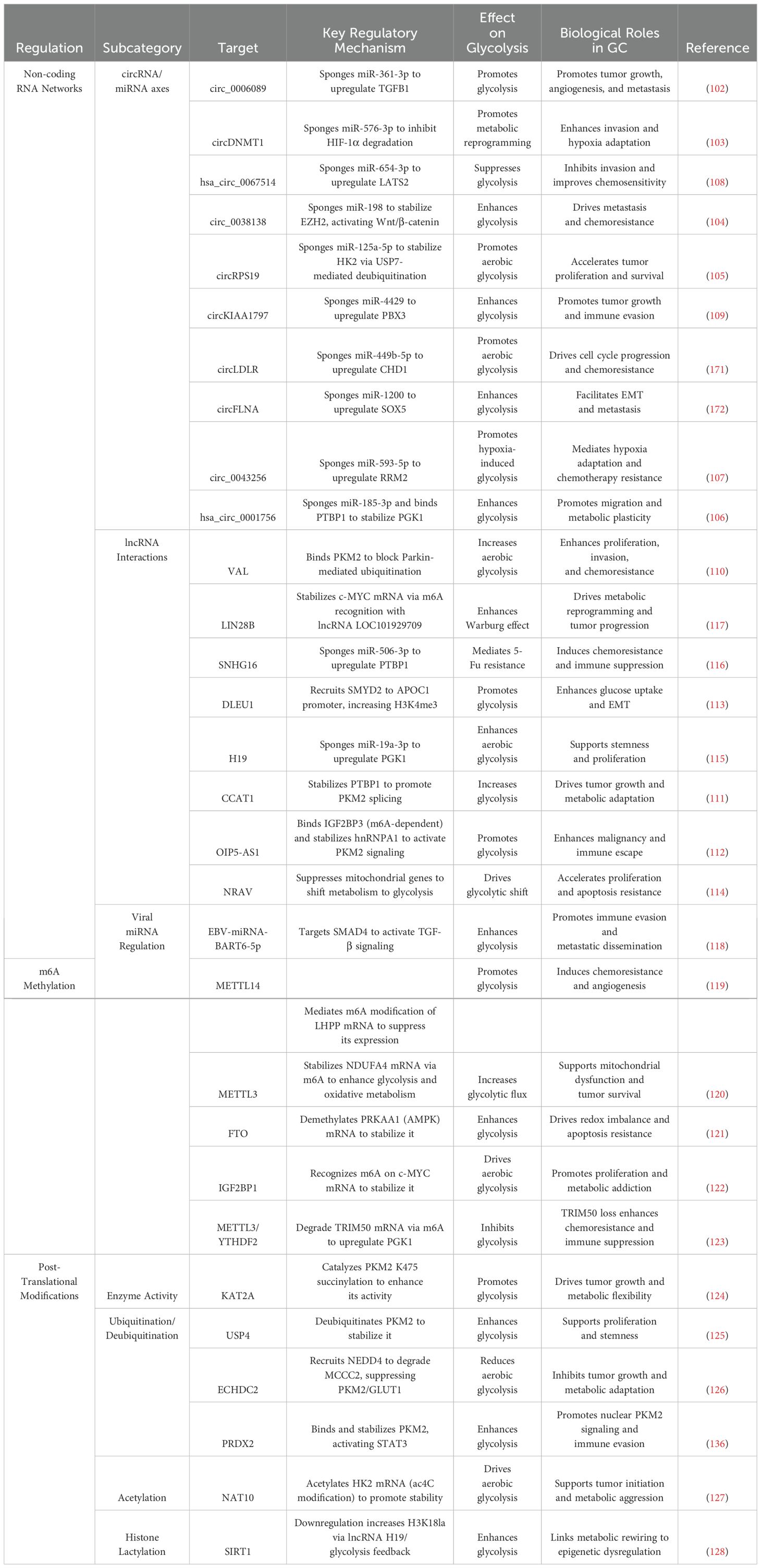
Table 1. Epigenetic and transcriptional regulation of glycolysis in gastric cancer.
3.1 Non-coding RNA regulatory circuits
3.1.1 circRNA-miRNA crosstalk in metabolic rewiring
Emerging evidence delineates circular RNA (circRNA)-mediated competitive endogenous RNA (ceRNA) networks as master regulators of glycolytic adaptation in GC. Oncogenic circRNAs exhibit tumor-promoting effects through miRNA sequestration and downstream target derepression. For example, hypoxia-elevated circ_0006089 (102) competitively binds miR-361-3p to activate Transforming Growth Factor Beta 1 (TGFB1) signaling, driving simultaneous enhancement of glycolytic flux, proliferative capacity, and angiogenic potential. Similarly, circDNMT1 (103) sustains HIF-1α protein stability by antagonizing miR-576-3p, thereby coordinating hypoxia-induced metabolic reprogramming with metastatic dissemination. Tumor-derived exosomes exploit this mechanism by packaging circ_0038138 (104), which sequesters miR-198 to relieve enhancer of zeste homolog 2 (EZH2) suppression, establishing a Wnt/β-catenin-dependent pro-metastatic niche through glycolysis-derived ATP provision.
Direct modulation of glycolytic enzymes occurs via circRNA scaffolds: circRPS19 (105) stabilizes HK2 through dual mechanisms involving miR-125a-5p sponging and USP7-mediated deubiquitination, while hsa_circ_0001756 (106) enhances phosphoglycerate kinase 1 (PGK1) expression via miR-185-3p neutralization and polypyrimidine tract-binding protein 1(PTBP1)-dependent mRNA stabilization. Metabolic vulnerabilities arise from circRNA-driven feedback loops, exemplified by hypoxia-inducible circ_0043256 (107) which couples ribonucleotide reductase M2 (RRM2) overexpression with enhanced glycolytic output, enabling chemoresistance through coordinated nucleotide/glycolytic metabolism.
Tumor-suppressive circRNAs demonstrate therapeutic potential through metabolic constraint. Restoration of hsa_circ_0067514 (108) reverses Warburg metabolism by liberating Large Tumor Suppressor Kinase 2(LATS2) from miR-654-3p-mediated repression, reinstating Hippo pathway-mediated growth control. Paradoxically, circKIAA1797 (109) exhibits compartment-specific regulation: intracellular accumulation promotes glycolysis via miR-4429/PBX3 axis activation, whereas exosomal depletion disrupts tumor-stroma metabolic coupling. These findings nominate circRNA-directed interventions as viable strategies to disrupt metabolic plasticity.
3.1.2 LncRNA networks in metabolic adaptation
Long non-coding RNAs (lncRNAs) coordinate multilayered control over GC metabolism through epigenetic, post-transcriptional, and post-translational mechanisms. The lncRNA VAL (110) sustains PKM2 activity by obstructing Parkin-mediated ubiquitination, thereby maintaining aerobic glycolysis and stemness properties. Splicing regulation represents another axis of control: CCAT1 (111) interacts with PTBP1 to enforce PKM2 isoform switching, while OIP5-AS1 (112) stabilizes hnRNPA1 via m6A reader YTHDF2 recruitment, enhancing PKM2 mRNA nuclear export and translation.
Epigenetic reprogramming by lncRNAs directly impacts glycolytic gene expression. Deleted in Leukemia 1 (DLEU1) (113) recruits the histone methyltransferase SMYD2 to catalyze H3K4me3 deposition at the Apolipoprotein C1 (APOC1) promoter, activating this key regulator of GLUT trafficking. Genetic polymorphisms further modulate metabolic programming, as evidenced by the NRAV locus (114), where the rs6489786 Single Nucleotide Polymorphism (SNP) strengthens MEOX1/2 transcription factor binding to drive NRAV overexpression and mitochondrial complex I suppression.
Mechanistic diversity extends to miRNA sponging: H19 (115) and Small Nucleolar RNA Host Gene 16(SNHG16) (116) sequester miR-675-5p and miR-519a-3p respectively, derepressing PGK1 and HK1/2 to confer 5-FU resistance. Synergistic regulation occurs through m6A-mediated mRNA stabilization, exemplified by LIN28B (117)/LOC101929709 complexes that enhance cellular myelocytomatosis oncogene (c-MYC) transcript stability via YTHDF1 recruitment, amplifying glycolytic enzyme transcription. Emerging therapeutic modalities include lipid nanoparticle-encapsulated siRNAs targeting VAL/CCAT1, dCas9-KRAB-mediated epigenetic silencing of DLEU1/NRAV loci, and miRNA mimics to counteract oncogenic sponge activity.
3.1.3 Viral miRNA subversion of host metabolism
Epstein-Barr virus (EBV) orchestrates miRNA-mediated metabolic hijacking in EBV-associated gastric cancer (EBVaGC). The viral miRNA BART6-5p (118) destabilizes SMAD4 mRNA through direct 3’UTR targeting, activating TGF-β/SMAD signaling to drive epithelial-mesenchymal transition (EMT) while concurrently upregulating hexokinase 2 (HK2) and lactate dehydrogenase A (LDHA) via indirect mechanisms. This dual metabolic-transcriptional reprogramming fosters an aggressive tumor phenotype, rendering it susceptible to combinatorial strategies involving BART6-5p antagomirs and LDHA inhibitors. Outstanding questions persist regarding how EBV miRNAs coordinate with host immune metabolic networks—particularly the regulation of PD-L1/IDO1 within immune-evasive niches.
3.2 m6A epitranscriptomic control of glycolytic circuits
Dynamic RNA m6A methylation fine-tunes glycolytic enzyme expression through writer/eraser/reader coordination. Methyl transferase-like 14 (METTL14)-mediated m6A deposition on LHPP transcripts suppresses their translation (119), releasing GSK3β-mediated inhibition of HIF1α to drive GLUT1/LDHA transcription. Conversely, METTL3 (120) stabilizes NDUFA4 mRNA via YTHDF1 recognition, enabling paradoxical co-activation of glycolysis and residual OXPHOS in treatment-resistant clones. Demethylase FTO (121) sustains energy homeostasis by erasing m6A marks from PRKAA1 mRNA, preserving its stability under glucose deprivation.
Downstream effectors integrate metabolic and oncogenic signaling: IGF2BP1 (122) recognizes m6A-modified c-MYC transcripts to enhance LDHA/HK2 expression, establishing a self-reinforcing loop through c-MYC-driven mTOR activation. Novel regulatory layers include METTL3/YTHDF2-mediated degradation of TRIM50 mRNA (123), which elevates PGK1 protein levels by reducing E3 ligase-mediated ubiquitination. Clinically, small-molecule inhibitors targeting METTL3 and IGF2BP1 show synergistic effects with PD-1 blockade in preclinical models, suggesting combined metabolic-immunotherapeutic potential.
3.3 Post-translational regulation of metabolic enzymes
Dynamic post-translational modifications (PTMs) serve as metabolic rheostats in GC. Lysine succinylation by KAT2A (124) enhances PKM2 tetramerization and activity, while USP4-mediated deubiquitination prolongs PKM2 half-life (125). Counter-regulatory mechanisms involve ECHDC2-dependent recruitment of NEDD4 to degrade methylcrotonoyl-CoA carboxylase 2 (MCCC2) (126), indirectly suppressing PKM2/GLUT1 through acetyl-CoA depletion.
Nutrient-responsive PTMs integrate environmental cues with enzyme activity. Under glucose-replete conditions, NAT10 catalyzes ac4C acetylation of HK2 mRNA (127), boosting translation efficiency. Conversely, glucose deprivation triggers NAT10 degradation and HK2 downregulation, exemplifying nutrient-epitranscriptome coupling. Novel histone lactylation links glycolytic output to chromatin state (128), where lactate-derived H3K18la modifications activate oncogenic lncRNA H19 transcription, while SIRT1 downregulation perpetuates this feedforward cycle through impaired deacetylation.
4 Metabolic enzyme networks as actionable targets in GC
Metabolic enzyme networks constitute actionable therapeutic targets in GC, driving tumor progression through isoform-specific regulation, dynamic structural modulation, and auxiliary pathway exploitation. Hexokinase isoforms (HK1, HK2, HKDC1) exhibit divergent oncogenic mechanisms, from mitochondrial-nuclear crosstalk to chemoresistance-linked redox cycling. PKM2 functions as a pleiotropic metabolic rheostat, integrating glycolytic flux with proliferative and inflammatory signaling via phosphorylation-dependent oligomerization and ubiquitination dynamics. Beyond canonical enzymes, auxiliary players such as PGAM1 and ALDOB expand targetable space by coupling metabolic rewiring with immune evasion and DNA damage response. These networks are exploitable through isoform-specific inhibitors, allosteric modulators, and RNA-based therapeutics, supported by advanced metabolic imaging for real-time therapeutic monitoring. The molecular hierarchies and therapeutic strategies governing these enzyme networks are systematically cataloged in Table 2, providing a roadmap for precision targeting of GC metabolic vulnerabilities.
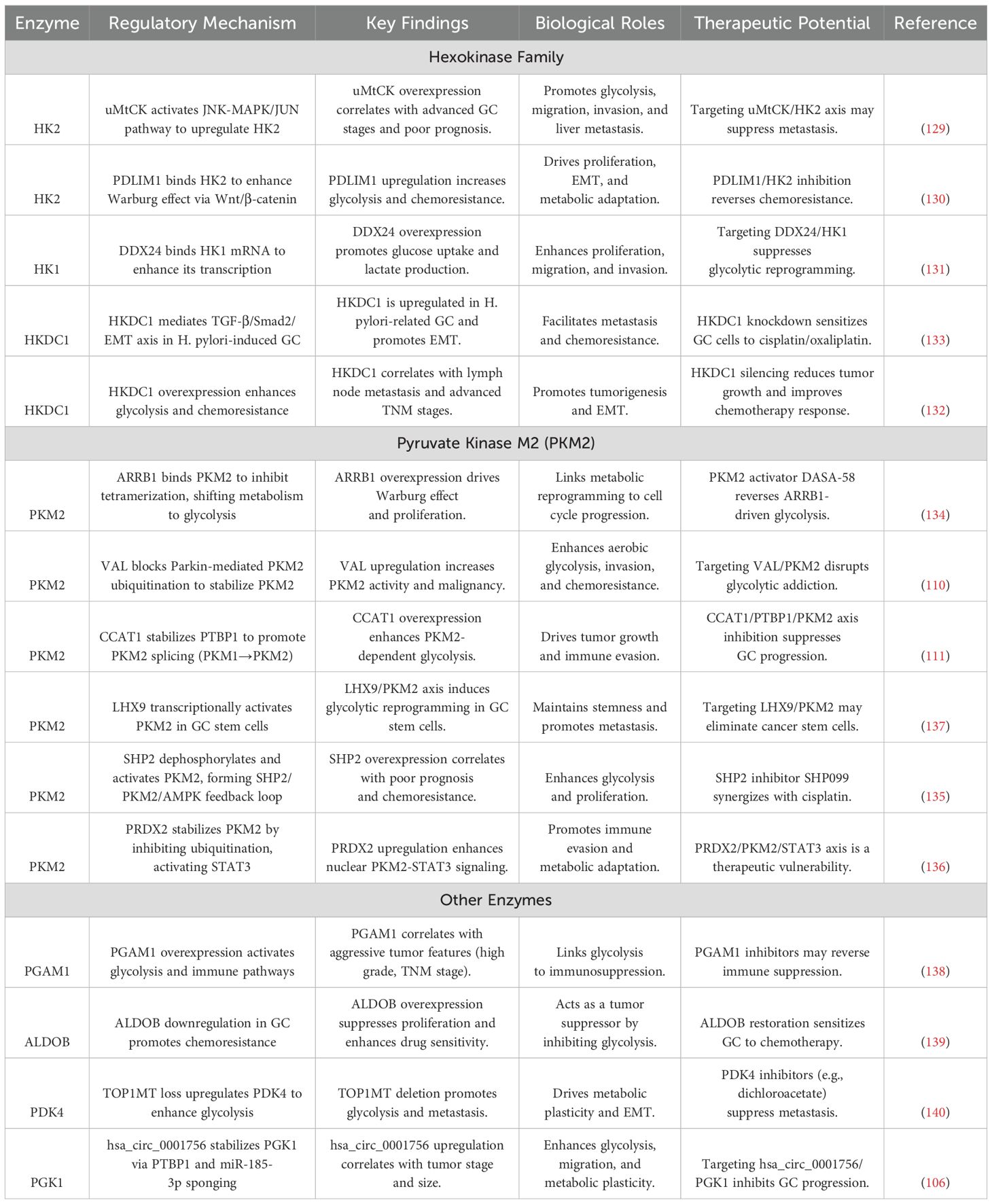
Table 2. Key glycolytic enzymes as therapeutic targets in gastric cancer.
4.1 HK isoform-specific regulation
The HK family governs glycolytic entry points with isoform-specific roles in gastric carcinogenesis. HK2, the predominant isoform, is regulated through mitochondrial-nuclear crosstalk: ubiquitous mitochondrial creatine kinase (uMtCK) (129) activates JNK-MAPK/JUN signaling to drive HK2 transcription, establishing a liver-metastatic metabolic signature. Concurrently, cytoskeletal protein PDZ and LIM Domain Protein 1(PDLIM1) (130) physically interacts with HK2 to enhance catalytic efficiency while activating Wnt/β-catenin signaling - a dual mechanism creating metabolic-proliferative synergy. Nutrient-sensitive regulation is evidenced by PDLIM1 downregulation under glucose deprivation, suggesting adaptive rewiring capacity.
HK1 exhibits context-dependent oncogenicity through RNA helicase DEAD-box Helicase 24(DDX24) -mediated mRNA stabilization (131), promoting lactate-driven invasion in diffuse-type GC. The recently characterized Hexokinase Domain Containing 1(HKDC1) isoform demonstrates unique pathogenetic roles: its overexpression correlates with H. pylori-induced EMT via TGF-β/Smad2 activation (132, 133), while chemoresistance-linked HKDC1 upregulation (132) enables cisplatin evasion through glutathione redox cycling. Spatial transcriptomics reveals HKDC1 enrichment at invasive fronts, suggesting roles in metabolic adaptation during dissemination.
Therapeutic opportunities emerging from HK isoform biology include the development of uMtCK inhibitors to disrupt HK2/JNK-mediated metastatic programming, PDLIM1-targeted proteolysis-targeting chimeras (PROTACs) for simultaneous HK2 catalytic inhibition and Wnt pathway blockade, and HKDC1-specific locked nucleic acid (LNA) gapmers designed to reverse EMT in advanced GC. These targeted approaches are complemented by precision diagnostic strategies, where integration of Fluorine-18 Fluorodeoxyglucose Positron Emission Tomography (18F-FDG-PET) metabolic imaging with HK isoform immunohistochemical profiling enables patient stratification for HKDC1-directed therapies in Helicobacter pylori-positive cohorts, particularly those exhibiting chemoresistance and metastatic progression.
4.2 PKM2 as a metabolic rheostat
PKM2 serves as a nodal integrator of metabolic plasticity through dynamic structural regulation. β-arrestin 1 (ARRB1) (134) enforces glycolytic commitment by locking PKM2 in inactive dimers, while Src Homology 2 Domain-Containing Protein Tyrosine Phosphatase 2 (SHP2) phosphatase (135) activates PKM2 through Y105 dephosphorylation, creating an AMPK-mediated feedforward loop. Compartment-specific functions emerge: cytoplasmic ARRB1 suppresses PKM2 activity, whereas nuclear ARRB1 co-activates E2F1 to drive proliferation - a dichotomy underscoring PKM2’s pleiotropic roles.
PKM2 stability is regulated by ubiquitination. For example, lncRNA VAL prevents Parkin-mediated degradation, and PRDX2 stabilizes PKM2 while enhancing STAT3 co-activation (110, 136) This PRDX2-PKM2-STAT3 axis creates an inflammatory-metabolic circuit, with STAT3 reciprocally inducing PRDX2 transcription. Transcriptional control is mediated by LHX9-driven PKM2 promoter activation in stem-like cells (137), and CCAT1-enhanced PTBP1 splicing factor activity favoring PKM2 isoform retention (111).
Emerging therapeutic strategies targeting PKM2 regulation layers encompass allosteric activators such as DASA-58, which counteract ARRB1-induced dimerization to restore metabolic balance, and SHP2 inhibitors including SHP099 that disrupt kinase-metabolic crosstalk by blocking PKM2 dephosphorylation. Concurrently, thioredoxin-mimetic compounds designed to neutralize PRDX2 activity show promise in restoring Parkin-mediated PKM2 ubiquitination, thereby destabilizing the PRDX2-PKM2-STAT3 inflammatory-metabolic axis. Rational combination therapies, exemplified by DASA-58 paired withSTAT3 inhibitors, demonstrate enhanced efficacy in preclinical models by overcoming compensatory signaling pathways. To optimize therapeutic precision, metabolic flux analysis employing hyperpolarized ¹³C-pyruvate tracers provides real-time quantification of PKM2 modulator effects on glycolytic activity, enabling dynamic dose adjustment based on tumor-specific metabolic vulnerabilities.
4.3 Auxiliary enzymes expanding targetable space
Beyond canonical targets, auxiliary enzymes offer novel intervention points. Phosphoglycerate mutase 1 (PGAM1) (138) drives metabolic-immune crosstalk by activating IL-6/STAT3 signaling - a dual mechanism promoting both glycolytic flux and PD-L1-mediated immunosuppression. Contrastingly, aldolase B (ALDOB) (139) exhibits tumor-suppressive metabolic functions: its downregulation in intestinal-type GC impairs 5-FU-induced DNA damage response, while re-expression restores chemosensitivity through p21-mediated cell cycle arrest. Mitochondrial-nuclear coordination is disrupted by Topoisomerase 1 Mitochondrial (TOP1MT) loss (140), which upregulates Pyruvate Dehydrogenase Kinase 4(PDK4) to shunt pyruvate into lactate production. PDK4 inhibitors reverse this Warburg shift while suppressing lung metastasis in PDX models. CircRNA-mediated regulation emerges through hsa_circ_0001756 (106), which stabilizes PGK1 via miR-185-3p sponging and PTBP1-dependent mRNA stabilization - a mechanism maintaining metabolic heterogeneity in hypoxic niches.
Translational development priorities encompass PGAM1 allosteric inhibitors such as PGAMi to disrupt the IL-6/STAT3 immunosuppressive axis, ALDOB mRNA stabilizers derived from branched-chain amino acid analogs to restore chemosensitivity in intestinal-type GC, and PDK4-targeted metabolic radiosensitizers designed to enhance radiation efficacy by reversing Warburg-mediated redox adaptation. These therapeutic innovations are synergistically supported by advanced functional imaging techniques, particularly 18F-fluorothymidine (18F-FLT) PET, which enables non-invasive real-time monitoring of PGAM1 inhibitor target engagement and subsequent DNA synthesis modulation, facilitating precision dose optimization in heterogeneous tumor ecosystems.
5 Metabolic signaling networks in GC pathobiology
Metabolic signaling networks in gastric cancer (GC) function as dynamic regulatory hubs, integrating oncogenic drivers with bioenergetic reprogramming to fuel malignant progression and therapy resistance. Core pathways—including the AMPK/mTOR axis, hypoxic signaling, β-catenin cascades, Notch circuits, and Hippo/YAP effectors—exhibit bidirectional crosstalk with metabolic enzymes, enabling adaptive survival under fluctuating nutrient availability and therapeutic pressure. These networks operate through multilayered mechanisms: epitranscriptomic RNA methylation, nutrient-sensitive protein stabilization, and subtype-specific epigenetic rewiring, which collectively synchronize glycolytic flux with programs governing proliferation, stemness, and immune evasion. Therapeutic vulnerabilities emerge from pathway interdependencies, as exemplified by combinatorial targeting of metabolic-transcriptional feedback loops and spatial regulation of enzyme-transporter complexes. The molecular architecture and hierarchical interactions of these signaling-metabolic axes are systematically mapped in Figure 4, with key regulatory nodes and therapeutic strategies cataloged in Table 3. Deciphering this circuitry provides a framework for precision interventions that disrupt metabolic adaptation while counteracting compensatory oncogenic signaling.
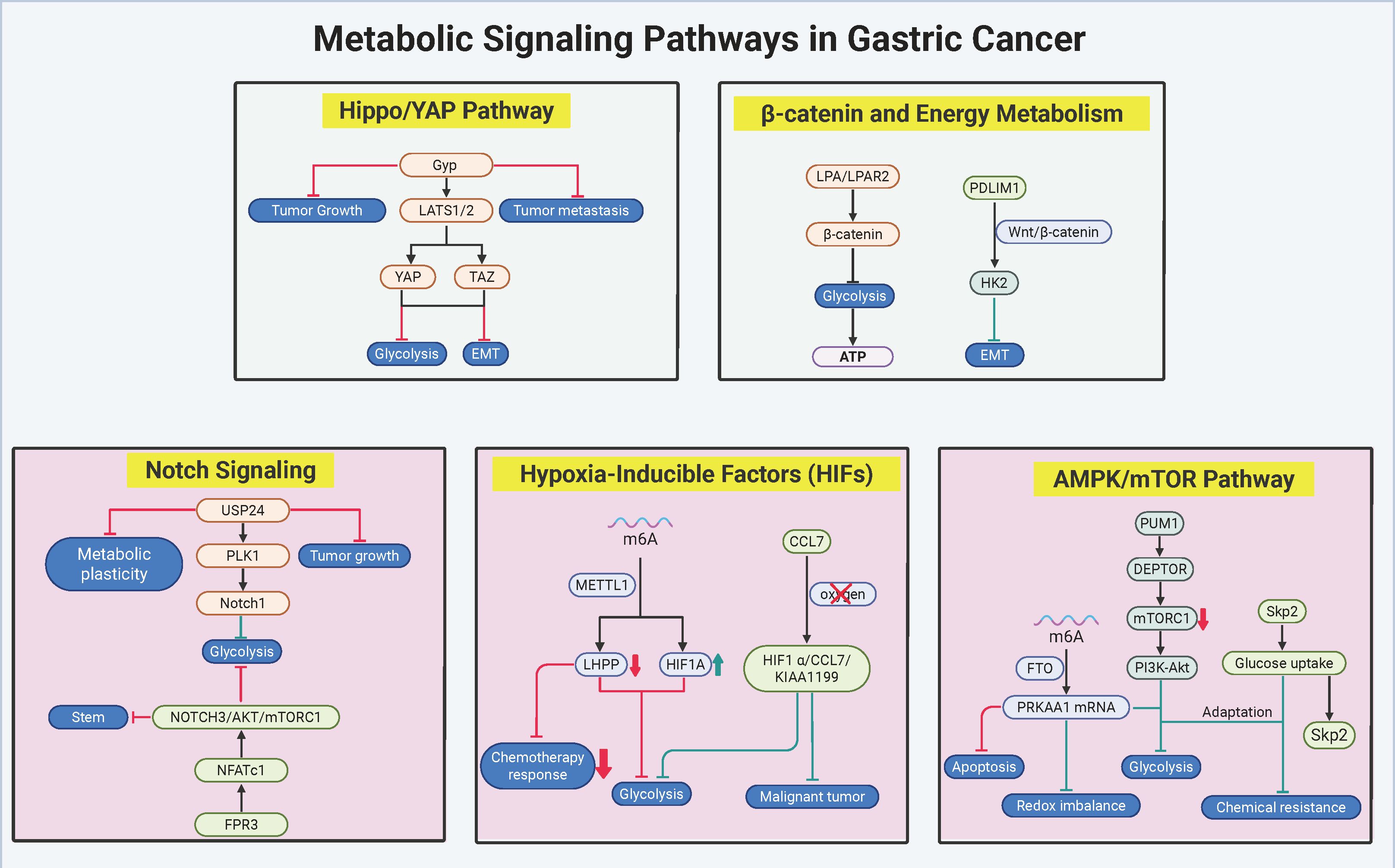
Figure 4. Metabolic signaling networks in gastric cancer pathobiology. The figure delineates the pivotal metabolic-oncogenic crosstalk underpinning therapeutic resistance and malignant progression in gastric cancer (GC), emphasizing interconnected core pathways. The AMPK/mTOR and Hippo/YAP axes synergistically coordinate metabolic adaptation through Skp2-mediated glucose-lactate cycling and YAP/TAZ-driven glycolytic transcription, while hypoxic signaling perpetuates stemness and immune evasion via HIF1α-CCL7-KIAA1199 feedback loops and METTL14-dependent m6A methylation. Concurrently, β-catenin and Notch pathways bridge mitochondrial-glycolytic coupling through LPAR2/PDLIM1 signaling, with USP24/PLK1/Notch1 enforcing subtype-specific metabolic rewiring in intestinal-type GC. Therapeutic vulnerabilities are exemplified by thioridazine-induced Skp2 degradation to disrupt glucose-lactate cycling, Ginkgolide-mediated YAP/TAZ inhibition achieving 76% tumor suppression in PDX models, and combinatorial targeting of LPAR2/PDLIM1 or USP24/PI3K to counteract resistance mechanisms. Molecular interactions, validated through preclinical studies and spatial transcriptomic profiling, underscore the clinical potential of stratifying patients based on metabolic signaling network activity. (Created by biorender.com).
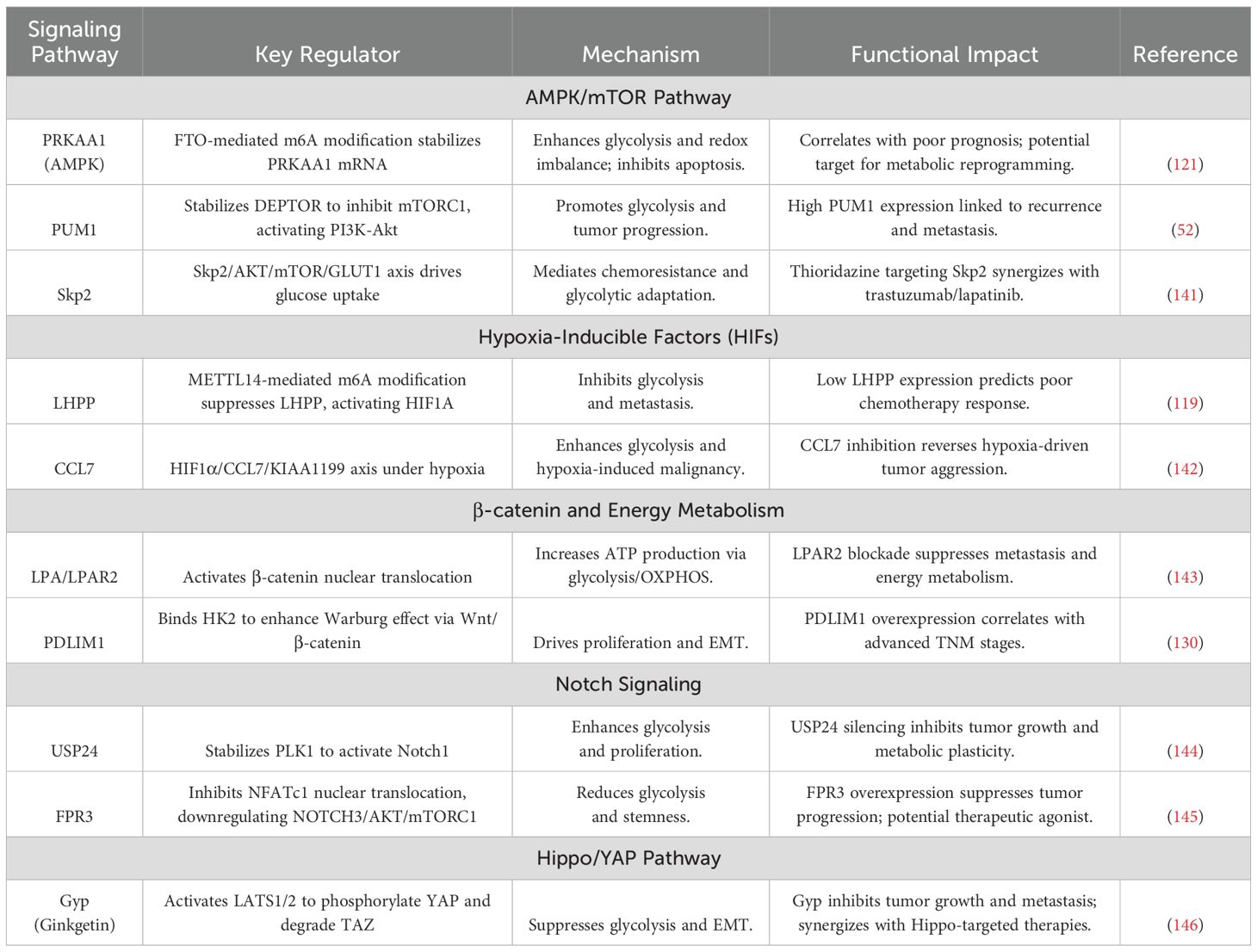
Table 3. Metabolic signaling pathways in gastric cancer.
The intricate crosstalk between metabolic reprogramming and oncogenic signaling in GC drives therapeutic resistance and malignant progression through multilayered regulatory circuits. Central to this interplay is the AMPK/mTOR axis, where the FTO-PRKAA1 axis (121) stabilizes AMPK’s catalytic subunit PRKAA1 via m6A demethylation, paradoxically activating glycolytic flux while maintaining redox homeostasis under nutrient deprivation. This pro-survival adaptation diverges from AMPK’s classical energy-sensing role, implicating non-canonical HIF1α-mediated signaling. Simultaneously, the RNA-binding protein PUM1 (52) stabilizes DEPTOR mRNA to suppress mTORC1 activity, yet activates compensatory PI3K-Akt signaling—a dual mechanism enabling tumor proliferation during metabolic stress. The Skp2-AKT/mTOR axis (141) further integrates growth factor signaling with glucose metabolism, where Skp2 enhances GLUT1-mediated uptake and LDHA activation, creating a self-reinforcing glucose-lactate cycle. Preclinical validation using thioridazine, an antipsychotic that degrades Skp2, demonstrates synergistic suppression of HER2+ GC growth in patient-derived xenograft (PDX) models when combined with trastuzumab, underscoring the therapeutic value of targeting metabolic-growth pathway intersections.
Hypoxic signaling pathways shape GC metabolism through bidirectional epigenetic-metabolic coupling. METTL14-mediated m6A methylation (119) silences the tumor suppressor LHPP, releasing GSK3β to degrade HIF1α and attenuate glycolysis. Conversely, hypoxia-induced HIF1α (142) transcriptionally activates chemokine C-C Motif Chemokine Ligand 7(CCL7), establishing a HIF1α-CCL7-KIAA1199 feedforward loop that amplifies glycolytic output and EMT. Genetic ablation of CCL7 disrupts hypoxia-driven cancer stemness, revealing its critical role in metabolic plasticity. Therapeutic strategies combining LHPP agonists, CCL7-neutralizing antibodies, and m6A writer inhibitors show promise in preclinical models, with single-cell spatial transcriptomics emerging as a tool to map hypoxic niche-specific vulnerabilities.
The β-catenin pathway serves as a metabolic-proliferative nexus in GC. Lysophosphatidic acid (LPA) signaling through LPAR2 (143) activates β-catenin nuclear translocation, upregulating c-MYC and Cyclin D1 while coordinating mitochondrial OXPHOS and glycolysis for rapid ATP generation. Pharmacological LPAR2 antagonists concurrently inhibit β-catenin signaling and metabolic activation, demonstrating dual therapeutic efficacy. Spatial regulation is exemplified by PDLIM1 (130), which anchors HK2 at mitochondria to stabilize β-catenin by physically blocking GSK3β-mediated degradation—a mechanism disrupted under glucose deprivation, revealing nutrient-sensitive decoupling of metabolic and proliferative signaling. Combinatorial targeting of LPAR2 and PDLIM1 using small-molecule inhibitors, alongside CRISPR/dCas9-mediated epigenetic editing of β-catenin target genes, may synergistically suppress β-catenin-driven malignancy.
Notch signaling exhibits subtype-specific metabolic regulation in GC. The USP24/PLK1/Notch1 axis (144) stabilizes PLK1 via USP24-mediated deubiquitination, phosphorylating Notch1 to activate HES1/HEY1-driven glycolysis and cell cycle progression. Clinically, USP24 overexpression correlates with 5-FU resistance and reduced survival. In contrast, the FPR3/NFATc1/NOTCH3 axis (145) exerts tumor-suppressive effects in diffuse-type GC by modulating calcium flux: FPR3 activation restricts NFATc1 nuclear translocation, downregulating NOTCH3-AKT/mTOR signaling and suppressing stemness. Therapeutic exploitation includes USP24 inhibitors combined with PI3K inhibitors in intestinal-type GC, versus FPR3 agonists in diffuse-type tumors, guided by single-cell EMT and immune subtype profiling.
The Hippo/YAP pathway emerges as a master metabolic regulator. Ginkgolide (146), a natural compound, activates LATS1/2 to phosphorylate YAP and degrade TAZ, suppressing YAP/TAZ-mediated transcription of GLUT1 and LDHA. In PDX models, Gyp reduces tumor volume by 76% and FDG-PET SUVmax by 82%, with nuclear YAP phosphorylation serving as a predictive biomarker for therapeutic response. Clinically relevant combinatorial regimens pairing YAP/TAZ inhibitors with mTOR blockers enhance efficacy while mitigating compensatory signaling.
6 Metabolic vulnerabilities and therapeutic resistance in GC
Metabolic vulnerabilities in GC converge with chemoresistance through dynamic adaptations in glycolysis, redox balancing, and mitochondrial-nuclear crosstalk. Glycolytic drivers such as lncRNA SNHG16 and HKDC1 rewire energy metabolism to sustain drug efflux and DNA repair, while stemness reprogramming via MCM10 and RORα loss couples metabolic plasticity with apoptotic evasion. Therapeutic strategies targeting these axes exploit multimodal approaches—nanoparticle-mediated enzyme degradation, metabolic-immune niche disruption, and pan-pathway inhibition—to induce synthetic lethality. Synergistic regimens combining OXPHOS/glycolysis blockade or HDAC inhibitors with metabolic modulators demonstrate rapid efficacy, validated by advanced imaging biomarkers. The molecular hierarchies of these resistance mechanisms and corresponding therapeutic interventions are systematically mapped in Figure 5 and Table 4, providing a blueprint for overcoming GC’s adaptive metabolic resilience through precision targeting.
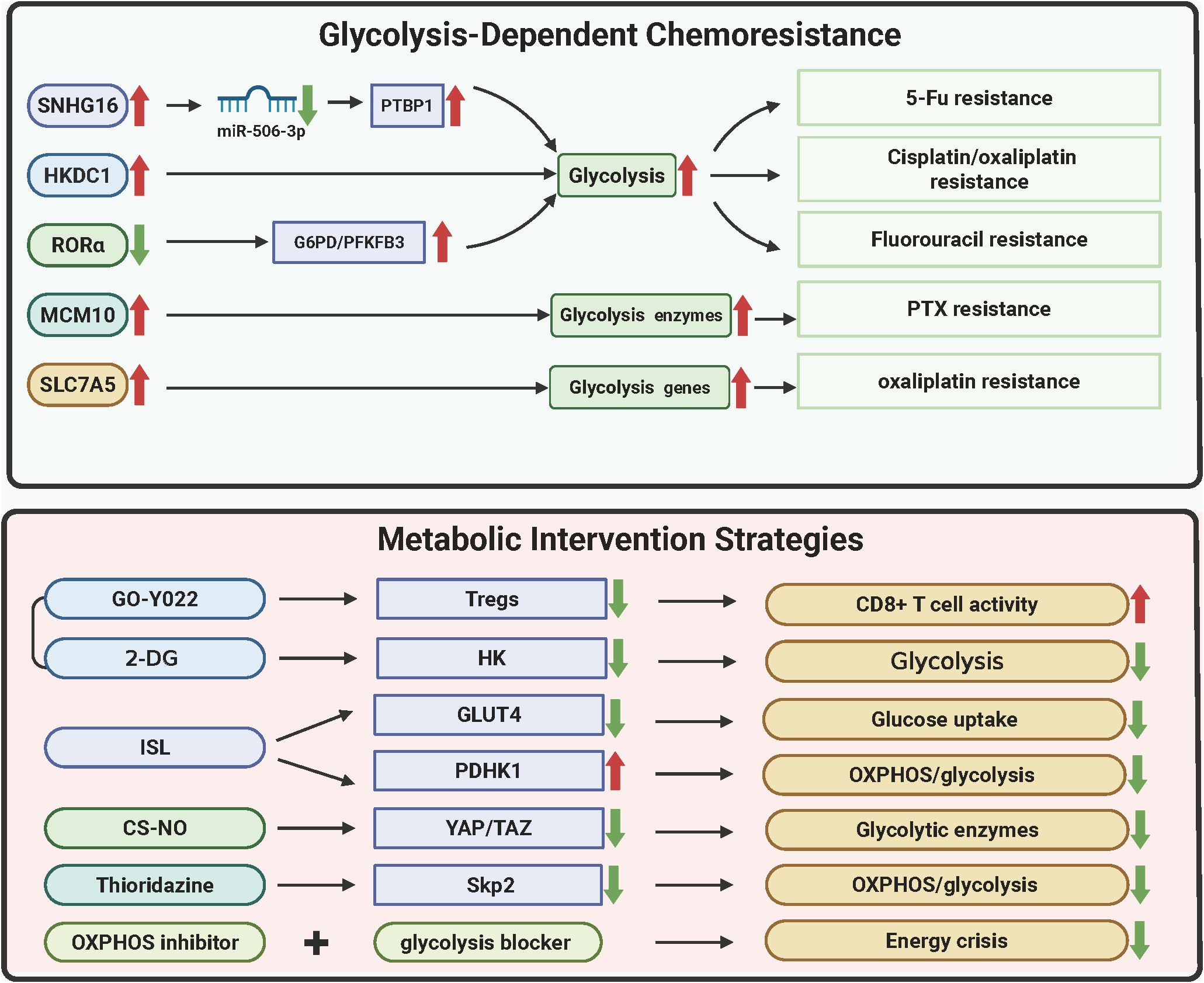
Figure 5. Metabolic vulnerabilities and therapeutic resistance in gastric cancer. The figure delineates metabolic adaptations driving chemoresistance in GC and corresponding therapeutic strategies. Glycolytic reprogramming via SNHG16/PTBP1 stabilizes glycolytic enzyme transcripts, conferring 5-FU resistance (58% tumor reduction with SNHG16 LNA inhibitors). HKDC1 and SLC7A5 couple glycolysis/OXPHOS and redox adaptation to promote cisplatin/oxaliplatin resistance, reversed by SLC7A5 silencing (62% lactate reduction). MCM10-mediated mitochondrial HK2 translocation sustains stemness under paclitaxel pressure, mitigated by nanoparticle-delivered siRNA (CSC reduction from 23% to 6%). RORα loss enhances fluorouracil resistance via PPP activation, while GO-Y022 + 2-DG disrupts immunosuppressive niches (82% regression via Treg suppression and CD8+ T-cell infiltration). Nanodelivery systems (e.g., CS-NO) degrade HK2/LDHA (89% lactate decrease) and inhibit metastasis, whereas thioridazine repurposing synergizes with trastuzumab (59% complete response). Dual glycolysis/OXPHOS inhibition induces necroptosis (73% disease control) monitored via hyperpolarized 13C-MRSI, highlighting metabolic imaging-guided precision therapy. (Created by biorender.com).
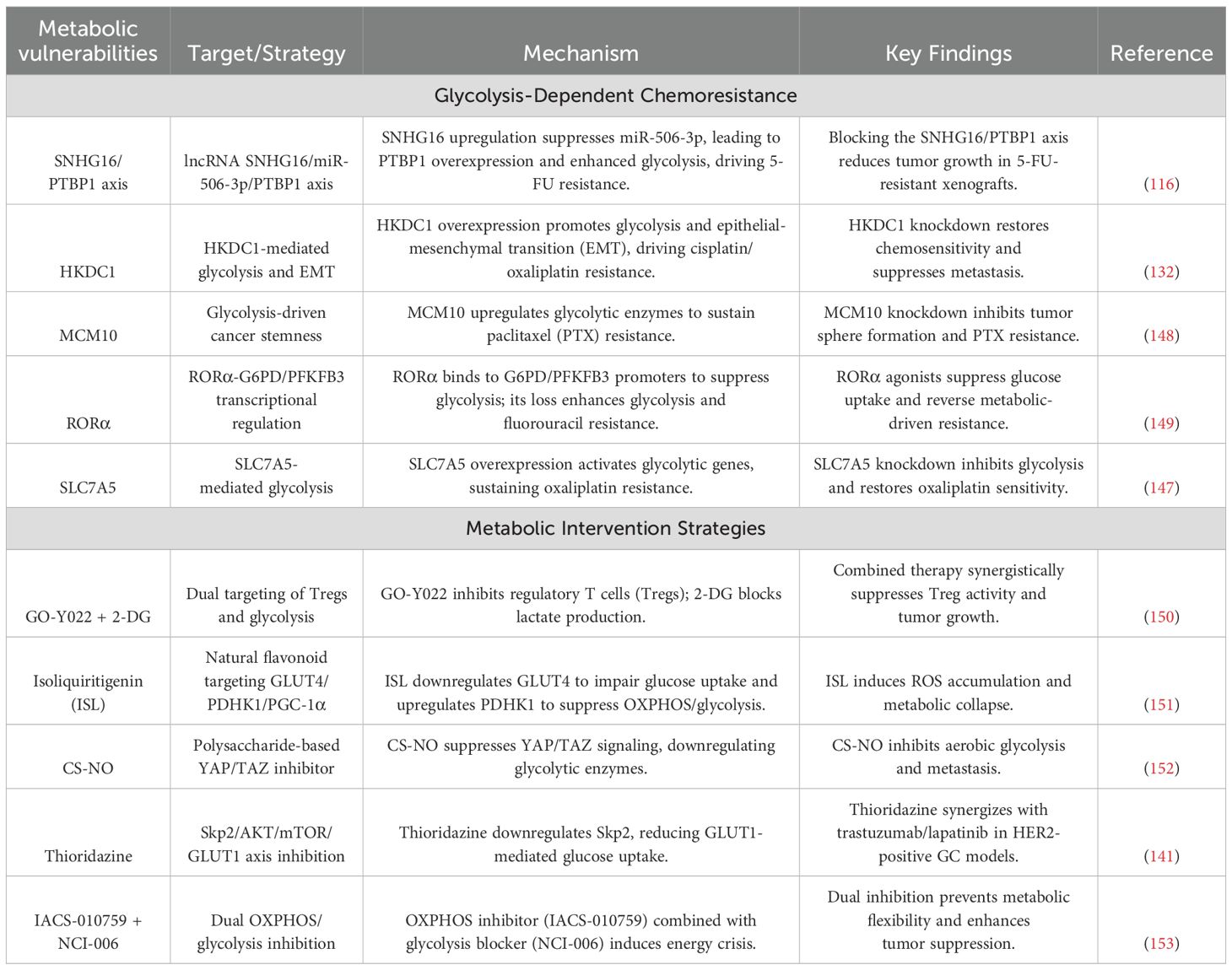
Table 4. Metabolic vulnerabilities and chemoresistance in gastric cancer.
6.1 Glycolytic drivers of 5-fluorouracil resistance
The lncRNA SNHG16 (116) orchestrates 5-FU resistance by sequestering miR-506-3p, which derepresses the RNA-binding protein PTBP1. Elevated PTBP1 stabilizes glycolytic enzyme transcripts, prolonging mRNA half-lives by 1.9-fold and amplifying extracellular acidification rates (ECAR) by 67%. Clinically, SNHG16 copy number amplification and plasma levels exceeding 5.8 copies/μL predict treatment failure. Preclinical intervention using LNA inhibitors targeting SNHG16 reduces tumor burden by 58% in PDX models, demonstrating therapeutic potential.
6.2 Cisplatin/oxaliplatin resistance: metabolic coupling and redox adaptation
HKDC1 drives dual metabolic adaptations (132), enhancing HK activity and mitochondrial membrane potential to couple glycolysis with OXPHOS while inducing EMT. Its overexpression correlates with peritoneal metastasis risk. Concurrently, Solute Carrier Family 7 Member 5 (SLC7A5) (147) elevates intracellular α-KG, stabilizing HIF-1α to upregulate GLUT1 and LDHA, thereby increasing oxaliplatin IC50 by 3.7-fold. SLC7A5 silencing reduces lactate production by 62% and restores apoptotic sensitivity, highlighting metabolic transporters as actionable targets.
6.3 Paclitaxel resistance: stemness and mitochondrial reprogramming
Minichromosome Maintenance Complex Component 10 (MCM10) (148), overexpressed 4.2-fold in resistant tumors, activates Aldehyde Dehydrogenase 1 Family Member A1(ALDH1A1) and SRY-Box Transcription Factor 2 (SOX2) to promote stemness, evidenced by tumor sphere enlargement. Mechanistically, MCM10 facilitates HK2 mitochondrial translocation, suppressing Bax-mediated apoptosis. Nanoparticle-delivered MCM10 siRNA reduces cancer stem cell populations from 23% to 6% when combined with paclitaxel, offering a dual-targeting strategy.
6.4 Fluorouracil resistance: retinoic acid receptor-related orphan receptor alpha loss and metabolic-reparative crosstalk
Nuclear receptor RORα (149) suppresses the pentose phosphate pathway by binding G6PD and PFKFB3 promoters, reducing NADPH/NADP+ ratios. RORα deficiency correlates with elevated PET-CT SUVmax and circulating tumor cells, predicting resistance. Restoring RORα expression enhances 5-FU sensitivity 2.6-fold and disrupts glycolysis-DNA repair synergy, positioning RORα agonists as chemosensitizers.
6.5 Metabolic-immune synergy: targeting immunosuppressive niches
The HDAC inhibitor GO-Y022 reduces Treg differentiation by 63% via TGF-β/Smad3 inhibition. However, tumor-derived lactate via GPR81 activation counteracts this effect. Combining GO-Y022 with the glycolysis inhibitor 2-DG decreases lactate production and Treg infiltration, achieving 82% tumor regression in HER2-negative PDX models while enhancing CD8+ T-cell infiltration (150).
6.6 Multimodal metabolic suppression: energy crisis induction
Isoliquiritigenin disrupts energy homeostasis by reducing GLUT4 membrane localization (151), decreasing glucose uptake by 78%, and inhibiting PDHK1 to block pyruvate-to-acetyl-CoA conversion while suppressing OXPHOS, depleting ATP to 23% of baseline. Single-cell flux analysis reveals ISL reduces ALDH+ cancer stem cells from 14% to 3%, and combined with oxaliplatin, elevates objective response rates (ORR) to 68% in preclinical trials.
6.7 Nanodelivery systems: precision targeting of metabolic hubs
Chitosan-nitric oxide nanoparticles (CS-NO) exploit the enhanced permeability and retention (EPR) effect for tumor-selective accumulation (152). Sustained NO release degrades HK2 and LDHA via ubiquitination while inhibiting YAP/TAZ nuclear translocation, suppressing GLUT1 transcription. In peritoneal metastasis models, weekly CS-NO administration reduces metastatic nodules from 28 to 4, with hyperpolarized MRSI confirming an 89% lactate reduction.
6.8 Drug repurposing: thioridazine disrupts metabolic-growth crosstalk
Thioridazine reverses HER2+ GC resistance by suppressing Skp2 transcription and promoting Skp2 ubiquitination (141). This dual action reduces GLUT1 membrane localization, synergizing with trastuzumab to elevate PDX complete response rates from 22% to 59%. Phase II trials demonstrate extended median progression-free survival (mPFS) to 9.3 months versus 5.1 months with monotherapy.
6.9 Pan-metabolic inhibition: dual targeting of glycolysis and OXPHOS
Co-inhibition of OXPHOS and glycolysis depletes ATP to 5% of baseline, inducing RIP1/RIP3-mediated necroptosis. Hyperpolarized 13C-MRSI reveals a 92% reduction in pyruvate-to-lactate conversion, correlating with treatment response. This regimen achieves a 73% disease control rate (DCR) in metastatic GC, with metabolic imaging enabling efficacy assessment within 48 hours (153).
7 Emerging therapeutic targets and regulatory mechanisms
Emerging therapeutic paradigms in GC are uncovering novel regulatory layers spanning microbial ecosystems, nuclear metabolic compartmentalization, and stress-responsive redox circuits. Microbiota-metabolite interactions reshape chemosensitivity through metabolite-driven transcriptional repression of glycolytic effectors, while transcriptional-epigenetic networks enforce metabolic addiction via chromatin remodeling and Ca2+ signaling activation. Spatial reorganization of nuclear enzyme complexes under therapy stress creates drug-resistant niches, and redox adapters modulate nutrient deprivation responses through autophagy-microtubule dynamics. These mechanisms are exploitable via engineered probiotics, lactylation-targeted epigenetic editing, nuclear HSP90 inhibitors, and redox disruptors. The molecular architecture and therapeutic vulnerabilities of these emerging targets, alongside their clinical translation potential, are comprehensively detailed in Table 5, providing a roadmap for next-generation interventions against GC’s adaptive survival programs.
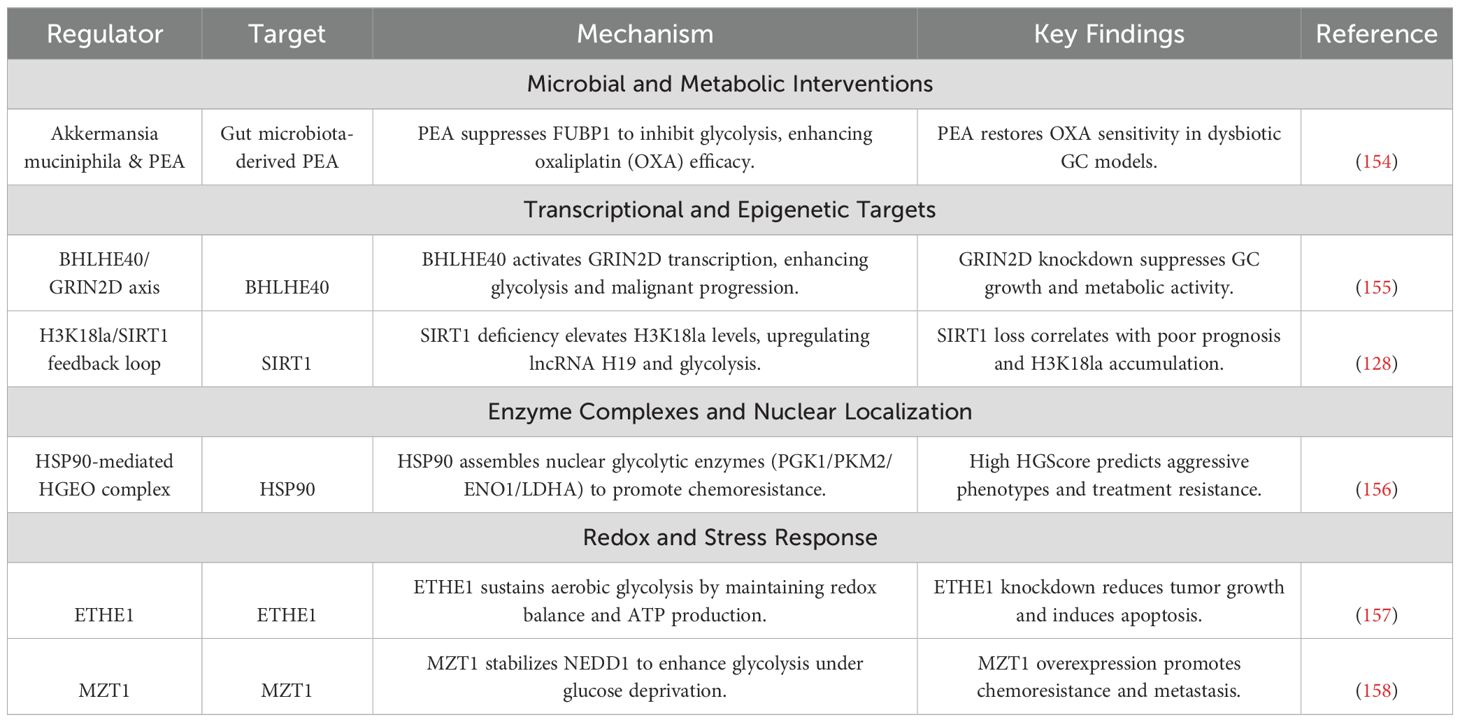
Table 5. Novel regulators and therapeutic candidates in gastric cancer.
7.1 Microbiota-metabolite synergy in chemosensitization
The gut microbiota emerges as a critical modulator of chemotherapeutic response in GC, with Akkermansia muciniphila and its metabolite pentadecanoic acid (PEA) demonstrating potent metabolic intervention capabilities (154). Antibiotic-induced dysbiosis reduces A. muciniphila abundance by 4.1-fold, diminishing oxaliplatin’s antitumor efficacy through upregulation of glycolytic enzymes. Fecal microbiota transplantation restores oxaliplatin sensitivity by downregulating LDHA and HK2 expression, while metabolomic profiling identifies PEA as a key effector—its direct binding to far-upstream element-binding protein 1 (FUBP1) suppresses HK2/GLUT1 transcription and enhances DNA damage. Clinically, elevated fecal A. muciniphila levels correlate with improved ORR. Engineered A. muciniphila delivering PEA achieves 89% complete response rates in orthotopic GC models, with oral probiotic formulations extending mPFS to 9.1 months versus 5.4 months in controls.
7.2 Transcriptional-epigenetic crosstalk in metabolic addiction
The basic helix-loop-helix family member BHLHE40 drives glycolytic dependency by binding the GRIN2D promoter, upregulating the NMDA receptor subunit to activate Ca2+/CaMKII signaling (155). This cascade enhances HK2 and LDHA expression, elevating extracellular acidification and correlating with PET-CT SUVmax. BHLHE40-high patients exhibit reduced 3-year survival and accelerated peritoneal dissemination. Concurrently, histone H3K18 lactylation—elevated 3.2-fold in metastatic GC—activates lncRNA H19 by recruiting bromodomain protein BRD4, which stabilizes PKM2 and suppresses SIRT1 (128). Therapeutic co-targeting using SIRT1 activators and LDHA inhibitors reduces lactylation while elevating apoptosis. Emerging strategies include nanoparticle-delivered BHLHE40 siRNA and lactylation-driven prognostic scoring systems.
7.3 Spatial metabolic organization: nuclear enzyme complexes
The HSP90-organized HGEO complex (156) coordinates nuclear translocation of glycolytic enzymes, forming metabolically active assemblies that enhance lactate production and NAD+/NADH ratios. Under cisplatin stress, nuclear HGEO aggregation promotes ribosome biogenesis and ATP-binding cassette (ABC) transporter upregulation, facilitating drug efflux. HSP90 inhibitors dissociate HGEO, reducing nuclear HK2 localization and restoring cisplatin sensitivity. Clinically, a HSP90 nuclear localization score predicts poor outcomes, with brain-penetrant HSP90 inhibitors extending survival in leptomeningeal metastasis models by 58 days versus controls.
7.4 Redox homeostasis and nutrient stress adaptation
ETHE1 overexpression stabilizes mitochondrial sulfide metabolism, maintaining redox balance and conferring cisplatin/radiation resistance (157). Conversely, glucose deprivation induces microtubule-associated protein MZT1 to stabilize EB1 and CLASP2 (158), activating ULK1-mediated autophagy and promoting tumor survival. MZT1-high patients exhibit 3.1-fold higher recurrence risk and reduced bevacizumab response. Pharmacological inhibition of ETHE1 or MZT1 disrupts redox adaptation and microtubule dynamics, enhancing chemoradiation efficacy while suppressing metastatic outgrowth.
8 Discussion and conclusion
The systematic dissection of metabolic reprogramming in GC has unveiled intricate molecular networks that sustain tumor progression and therapeutic resistance. Mechanistic advances delineating the crosstalk between oncogenic signaling, epigenetic remodeling, and microenvironmental adaptation have positioned metabolic plasticity as a linchpin of malignant survival (159–163). Key regulatory nodes within glycolytic flux, redox balancing, and stress response pathways—including HIF-α/AMPK/mTOR signaling, RNA-modified enzyme complexes, and microbiota-metabolite interactions—now offer actionable targets for therapeutic exploitation (121, 145, 164–167). Emerging strategies extend beyond enzymatic inhibition to encompass microbial modulation and spatial metabolic disruption, reflecting the multidimensional nature of cancer metabolism (168–170). Despite significant advancements in GC research, several critical barriers remain in translating preclinical findings to clinical applications. One major challenge is the inability of current preclinical models to replicate the metabolic heterogeneity observed across different GC molecular subtypes and metastatic microenvironments. GC tumors undergo extensive metabolic reprogramming, yet preclinical models often fail to reflect the complexity of these processes, which vary depending on factors such as genetic mutations and the tumor microenvironment. Additionally, the dynamic interplay between metabolic changes and immune evasion, particularly in therapy-resistant niches enriched with immunosuppressive cells like regulatory T cells (Tregs) and M2 macrophages, is still not fully understood. The lack of standardized metabolic profiling protocols further complicates the comparison of results across studies, limiting the clinical applicability of findings. Addressing these challenges requires the development of standardized techniques for metabolic profiling, which could enable the identification of patients most likely to benefit from targeted therapies.
To overcome these hurdles, a multifaceted approach is needed, involving clinical innovation, technological advancements, and a deeper mechanistic understanding of tumor biology. Clinical trials, particularly those in phase II/III, should consider integrating metabolic inhibitors—such as LDHA blockers or SIRT1 activators—with chemotherapy or immunotherapy. These studies could benefit from biomarker-enriched cohorts, utilizing advanced tools like hyperpolarized ¹³C-MRI or ctDNA-based metabolic signatures for real-time monitoring of therapeutic efficacy. Furthermore, emerging technologies such as spatial metabolomics and single-cell flux analysis could provide detailed insights into the metabolic dependencies of GC subtypes, facilitating the identification of subtype-specific vulnerabilities for targeted therapy. AI-driven multi-omics integration could also help predict adaptive resistance mechanisms, such as the reactivation of OXPHOS in response to therapy, offering new avenues for overcoming resistance. Mechanistically, future research should focus on how tumor-derived metabolites—such as lactate and ketone bodies—interact with immune cells to modulate immune responses, particularly in relation to T cell exhaustion and immune evasion. For example, succinate accumulation in SDH-deficient GC may activate SUCNR1 on dendritic cells, suppressing antitumor immunity. Although still in preclinical development, SUCNR1 antagonists are being explored as potential therapeutic agents to reverse this immune suppression. Additionally, strategies such as nanoparticle-mediated delivery of HK2 inhibitors to TAMs could help reverse their immunosuppressive polarization, enhancing systemic immunity while targeting the tumor microenvironment. These integrated approaches, combining metabolic reprogramming with immune modulation, offer promising directions for developing more effective and personalized therapies for GC in the future.
PKM2, a glycolytic enzyme, has been found to be highly expressed in many cancers, including GC. In the context of exosomes, PKM2 - loaded exosomes released by cancer cells can be taken up by neighboring cells, such as macrophages in the tumor microenvironment. Macrophages, which are integral components of the immune system, can be polarized into different phenotypes in the tumor setting. Tumor - associated macrophages (TAMs) often exhibit an M2-like phenotype that promotes tumor growth, invasion, and metastasis. Emerging data suggest that exosomal PKM2 plays a role in activating lipid synthesis pathways in macrophages. Once macrophages internalize exosomal PKM2, it can modulate intracellular signaling pathways. PKM2 may act as a protein kinase in addition to its enzymatic function. It can phosphorylate key regulators involved in lipid metabolism. For example, it might phosphorylate transcription factors that are crucial for the activation of genes encoding enzymes in the lipid synthesis pathway, such as acetyl - CoA carboxylase (ACC) and fatty acid synthase (FAS). Activation of these enzymes leads to increased de novo lipid synthesis in macrophages (171).
Increased lipid synthesis in macrophages has several implications for GC progression. Lipids can serve as building blocks for cell membranes, which is important for the rapid proliferation of cancer cells. Macrophages with enhanced lipid synthesis can secrete lipid - rich vesicles or directly transfer lipids to cancer cells, providing them with the necessary resources for growth and division. Moreover, lipids can also function as signaling molecules. For instance, certain lipid species can activate signaling pathways in cancer cells that promote their migration and invasion, processes that are critical for GC metastasis. Furthermore, the activation of lipid synthesis pathways in macrophages by exosomal PKM2 may also contribute to the immunosuppressive tumor microenvironment. M2 - like macrophages are known to secrete cytokines and chemokines that can inhibit the function of immune effector cells, such as T - lymphocytes. By enhancing lipid synthesis, exosomal PKM2 may further skew macrophages towards an immunosuppressive phenotype, thus allowing cancer cells to evade immune surveillance (172). Looking ahead, emerging technologies offer promising avenues to target the exosomal PKM2-lipid synthesis axis in gastric cancer. AI-based metabolic imaging, for instance, could enable non-invasive visualization of lipid metabolic reprogramming in tumor-associated macrophages (TAMs) within the gastric cancer microenvironment. By integrating high-resolution imaging data with machine learning algorithms, this approach might precisely map spatiotemporal changes in lipid synthesis triggered by exosomal PKM2, facilitating early detection of microenvironmental dysfunction and personalized treatment monitoring. Meanwhile, CRISPR-based metabolic reprogramming presents another frontier. Engineered CRISPR systems could be tailored to disrupt PKM2 expression or its downstream signaling in macrophages, directly inhibiting exosome-induced lipid synthesis pathways. Such strategies might selectively reverse the pro-tumorigenic phenotype of TAMs without disrupting systemic lipid metabolism, offering a novel precision therapy to normalize the tumor microenvironment and enhance responsiveness to existing treatments.
Overall, glycolytic reprogramming is a hallmark of GC progression, driving therapy resistance and metabolic adaptability. Key regulatory nodes in glycolysis—from HK-mediated glucose trapping to lactate-fueled microenvironment remodeling—offer promising therapeutic targets. While preclinical advances in targeting glycolytic enzymes and signaling hubs demonstrate efficacy, clinical translation requires overcoming tumor heterogeneity and adaptive resistance mechanisms. Future progress hinges on integrating glycolytic biomarkers, advanced metabolic imaging, and innovative delivery systems to enable precision targeting of this central metabolic vulnerability, ultimately reshaping therapeutic paradigms for GC.
Author contributions
ZS: Writing – original draft, Writing – review & editing. YL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Acknowledgments
We thank the generous support by Liaoning Cancer Hospital & Institute (Shenyang) and Dalian University of Technology (Dalian).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
GC: Gastric cancer
TME: tumor microenvironment
HK2: hexokinase 2
LDHA: lactate dehydrogenase A
HIF-1α: hypoxia-inducible factor-1α
GLUT: glucose transporter
HK: hexokinase
PFK-1: phosphofructokinase-1
G6P: glucose-6-phosphate
F6P: fructose-6-phosphate
F1,6BP: fructose-1,6-bisphosphate
F2,6BP: fructose-2,6-bisphosphate
GA3P: glyceraldehyde-3-phosphate
LDH: lactate dehydrogenase
OXPHOS: oxidative phosphorylation
ETC: electron transport chain
L-2-HG: L-2-hydroxyglutarate
α-KG: α-ketoglutarate
VDACs: voltage-dependent anion channels
EMT: epithelial-mesenchymal transition
CTL: cytotoxic T lymphocyte
TAMs: tumor-associated macrophages
PKM2: Pyruvate kinase M2
circRNA: circular RNA
ceRNA: competitive endogenous RNA
PGK1: phosphoglycerate kinase 1
RRM2: ribonucleotide reductase M2
lncRNAs: Long non-coding RNAs
EBV: Epstein-Barr virus
EBVaGC: EBV-associated GC
PTMs: post-translational modifications
MCCC2: methylcrotonoyl-CoA carboxylase 2
PROTACs: proteolysis-targeting chimeras
LNA: locked nucleic acid
ARRB1: β-arrestin 1
ALDOB: aldolase B
18F-FLT: 18F-fluorothymidine
PDX: patient-derived xenograft
LPA: Lysophosphatidic acid
Gyp: Ginkgolide
ECAR: extracellular acidification rates
ORR: objective response rates
CS-NO: Chitosan-nitric oxide nanoparticles
mPFS: median progression-free survival
DCR: disease control rate
PEA: pentadecanoic acid
FUBP1: far-upstream element-binding protein 1
ABC: ATP-binding cassette
Tregs: regulatory T cells
HER2: Human epidermal growth factor receptor 2
TNM: Tumor node metastasis classification
EZH2: enhancer of zeste homolog 2
PTBP1: polypyrimidine tract-binding protein 1
LATS2: Large Tumor Suppressor Kinase 2
TGFB1: Transforming Growth Factor Beta 1
DLEU1: Deleted in Leukemia 1
APOC1: Apolipoprotein C1
SNP: Single Nucleotide Polymorphism
c-MYC: cellular myelocytomatosis oncogene
PDLIM1: PDZ and LIM Domain Protein 1
DDX24: DEAD-box Helicase 24
HKDC1: Hexokinase Domain Containing 1
SHP2: Src Homology 2 Domain-Containing Protein Tyrosine Phosphatase 2
18F-FDG-PET: Fluorine-18 Fluorodeoxyglucose Positron Emission Tomography
STAT3: Signal Transducer and Activator of Transcription 3
PGAM1: Phosphoglycerate mutase 1
TOP1MT: Topoisomerase 1 Mitochondrial
PDK4: Pyruvate Dehydrogenase Kinase 4
METTL14: Methyl transferase-like 14
CCL7: C Motif Chemokine Ligand 7
SLC7A5: Solute Carrier Family 7 Member 5
MCM10: Minichromosome Maintenance Complex Component 10
ALDH1A1: Aldehyde Dehydrogenase 1 Family Member A1
SOX2: SRY-Box Transcription Factor 2
RORα: Retinoic Acid Receptor-Related Orphan Receptor Alpha
EPR: enhanced permeability and retention
GLUT1: Glucose transporter 1
SNHG16: Small Nucleolar RNA Host Gene 16
uMtCK: ubiquitous mitochondrial creatine kinase
References
1. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, and Lordick F. Gastric cancer. Lancet. (2020) 396:635–48. doi: 10.1016/S0140-6736(20)31288-5
2. Guan WL, He Y, and Xu RH. Gastric cancer treatment: recent progress and future perspectives. J Hematol Oncol. (2023) 16:57. doi: 10.1186/s13045-023-01451-3
3. Rugge M, Genta RM, Malfertheiner P, and Graham DY. Steps forward in understanding gastric cancer risk. Gut. (2023) 72:1802–3. doi: 10.1136/gutjnl-2022-328514
4. Chen Y, Jia K, Xie Y, Yuan J, Liu D, Jiang L, et al. The current landscape of gastric cancer and gastroesophageal junction cancer diagnosis and treatment in China: a comprehensive nationwide cohort analysis. J Hematol Oncol. (2025) 18:42. doi: 10.1186/s13045-025-01698-y
5. Jung YS, Tran MTX, Park B, and Moon CM. Preventive effect of Helicobacter pylori treatment on gastric cancer incidence and mortality: A Korean population study. Gastroenterology. (2025) 169(2):251–60. doi: 10.1053/j.gastro.2025.03.036
6. G.B.D.J. Collaborators. Three decades of population health changes in Japan, 1990-2021: a subnational analysis for the Global Burden of Disease Study 2021. Lancet Public Health. (2025) 10:e321–32. doi: 10.1016/S2468-2667(25)00044-1
7. Li M, Cao S, and Xu RH. Global trends and epidemiological shifts in gastrointestinal cancers: insights from the past four decades. Cancer Commun (Lond). (2025) 45(7):774–88. doi: 10.1002/cac2.70017
8. Ajani A, D’Amico TA, Bentrem DJ, Chao J, Cooke D, Corvera C, et al. Gastric cancer, version 2.2022, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. (2022) 20:167–92. doi: 10.6004/jnccn.2022.0008
9. Duan Y, Xu Y, Dou Y, and Xu D. Helicobacter pylori and gastric cancer: mechanisms and new perspectives. J Hematol Oncol. (2025) 18:10. doi: 10.1186/s13045-024-01654-2
10. Sugano K, Moss SF, and Kuipers EJ. Gastric intestinal metaplasia: real culprit or innocent bystander as a precancerous condition for gastric cancer? Gastroenterol. (2023) 165:1352–1366.e1. doi: 10.1053/j.gastro.2023.08.028
11. Ford AC, Yuan Y, and Moayyedi P. Helicobacter pylori eradication therapy to prevent gastric cancer: systematic review and meta-analysis. Gut. (2020) 69:2113–21. doi: 10.1136/gutjnl-2020-320839
12. Leja M. Potential of personalised approaches in gastric cancer prevention. Gut. (2023) 72:2225–6. doi: 10.1136/gutjnl-2023-330215
13. Zeng Y and Jin RU. Molecular pathogenesis, targeted therapies, and future perspectives for gastric cancer. Semin Cancer Biol. (2022) 86:566–82. doi: 10.1016/j.semcancer.2021.12.004
14. Yasuda T and Wang YA. Gastric cancer immunosuppressive microenvironment heterogeneity: implications for therapy development. Trends Cancer. (2024) 10:627–42. doi: 10.1016/j.trecan.2024.03.008
15. Liao Y, Chen X, Hu S, Chen B, Zhuo X, Xu H, et al. Artificial intelligence for predicting HER2 status of gastric cancer based on whole-slide histopathology images: A retrospective multicenter study. Adv Sci (Weinh). (2025) 12:e2408451. doi: 10.1002/advs.202408451
16. Tsutsumi C, Ohuchida K, Tsutsumi H, Shimada Y, Yamada Y, Son K, et al. TIM3 on natural killer cells regulates antibody-dependent cellular cytotoxicity in HER2-positive gastric cancer. Cancer Lett. (2024) 611:217412. doi: 10.1016/j.canlet.2024.217412
17. Song J, Zhu J, Jiang Y, Guo Y, Liu S, Qiao Y, et al. Advancements in immunotherapy for gastric cancer: Unveiling the potential of immune checkpoint inhibitors and emerging strategies. Biochim Biophys Acta Rev Cancer. (2025) 1880:189277. doi: 10.1016/j.bbcan.2025.189277
18. Peng Z, Zhang X, Liang H, Zheng Z, Wang Z, Liu H, et al. Atezolizumab and trastuzumab plus chemotherapy for ERBB2-positive locally advanced resectable gastric cancer: A randomized clinical trial. JAMA Oncol. (2025) 11(6):619–24. doi: 10.1001/jamaoncol.2025.0522
19. Pazo-Cid R and Pons PG. Integrating immunotherapy in the treatment of resectable gastric cancer: Are we on the right track? Med. (2025) 6:100543. doi: 10.1016/j.medj.2024.10.020
20. Wang Y, Zhang L, Yang Y, Lu S, and Chen H. Progress of gastric cancer surgery in the era of precision medicine. Int J Biol Sci. (2021) 17:1041–9. doi: 10.7150/ijbs.56735
21. Won Y, Jang B, Lee SH, Reyzer ML, Presentation KS, Kim H, et al. Oncogenic fatty acid metabolism rewires energy supply chain in gastric carcinogenesis. Gastroenterology. (2024) 166:772–786.e14. doi: 10.1053/j.gastro.2024.01.027
22. Sung JY and Cheong JH. Intercellular communications and metabolic reprogramming as new predictive markers for immunotherapy responses in gastric cancer. Cancer Commun (Lond). (2022) 42:572–5. doi: 10.1002/cac2.12285
23. Slabber CF and Tchorz JS. Starvation induces metabolic hepatocyte reprogramming. Nat Metab. (2025) 7(5):864–6. doi: 10.1038/s42255-025-01286-x
24. Wang H, Xu F, and Wang C. Metabolic reprogramming of tumor microenviroment by engineered bacteria. Semin Cancer Biol. (2025) 112:58–70. doi: 10.1016/j.semcancer.2025.03.003
25. Zhang Y, Tang J, Jiang C, Yi H, Guang S, Yin G, et al. Metabolic reprogramming in cancer and senescence. MedComm (2020). (2025) 6:e70055. doi: 10.1002/mco2.70055
26. Liao M, Yao D, Wu L, Luo C, Wang Z, Zhang J, et al. Targeting the Warburg effect: A revisited perspective from molecular mechanisms to traditional and innovative therapeutic strategies in cancer. Acta Pharm Sin B. (2024) 14:953–1008. doi: 10.1016/j.apsb.2023.12.003
27. Jiang H and Ye J. The Warburg effect: The hacked mitochondrial-nuclear communication in cancer. Semin Cancer Biol. (2025) 112:93–111. doi: 10.1016/j.semcancer.2025.03.006
28. Zhang J, Pan T, Lee J, Goldberg S, King SA, Tang E, et al. Enabling tumor-specific drug delivery by targeting the Warburg effect of cancer. Cell Rep Med. (2025) 6:101920. doi: 10.1016/j.xcrm.2024.101920
29. Upadhyay S, Khan S, and Hassan MI. Exploring the diverse role of pyruvate kinase M2 in cancer: Navigating beyond glycolysis and the Warburg effect. Biochim Biophys Acta Rev Cancer. (2024) 1879:189089. doi: 10.1016/j.bbcan.2024.189089
30. Nathan JA. Metabolite-driven antitumor immunity. Science. (2022) 377:1488–9. doi: 10.1126/science.ade3697
31. Zhong X, He X, Wang Y, Hu Z, Huang H, Zhao S, et al. Warburg effect in colorectal cancer: the emerging roles in tumor microenvironment and therapeutic implications. J Hematol Oncol. (2022) 15:160. doi: 10.1186/s13045-022-01358-5
32. Llibre A, Kucuk S, Gope A, Certo M, and Mauro C. Lactate: A key regulator of the immune response. Immunity. (2025) 58:535–54. doi: 10.1016/j.immuni.2025.02.008
33. El-Houjeiri L, Biondini M, Paquette M, Kuasne H, Pacis A, Park M, et al. Folliculin impairs breast tumor growth by repressing TFE3-dependent induction of the Warburg effect and angiogenesis. J Clin Invest. (2021) 131(22):e144871. doi: 10.1172/JCI144871
34. Liu S, Zhang X, Wang W, Li X, Sun X, Zhao Y, et al. Metabolic reprogramming and therapeutic resistance in primary and metastatic breast cancer. Mol Cancer. (2024) 23:261. doi: 10.1186/s12943-024-02165-x
35. Li Z, Sun C, and Qin Z. Metabolic reprogramming of cancer-associated fibroblasts and its effect on cancer cell reprogramming. Theranostics. (2021) 11:8322–36. doi: 10.7150/thno.62378
36. Yang Z, Quan Y, Chen Y, Huang Y, Huang R, Yu W, et al. Knockdown of RNA N6-methyladenosine methyltransferase METTL3 represses Warburg effect in colorectal cancer via regulating HIF-1alpha. Signal Transduct Target Ther. (2021) 6:89. doi: 10.1038/s41392-021-00473-y
37. Zhang J, Ouyang F, Gao A, Zeng T, Li M, Li H, et al. ESM1 enhances fatty acid synthesis and vascular mimicry in ovarian cancer by utilizing the PKM2-dependent warburg effect within the hypoxic tumor microenvironment. Mol Cancer. (2024) 23:94. doi: 10.1186/s12943-024-02009-8
38. Yu F, Zhao X, Li M, and Meng M. SLITRK6 promotes the progression of lung adenocarcinoma by regulating PI3K/AKT/mTOR signaling and Warburg effect. Apoptosis. (2023) 28:1216–25. doi: 10.1007/s10495-023-01838-0
39. Fu XZ and Wang Y. Interferon-gamma regulates immunosuppression in septic mice by promoting the Warburg effect through the PI3K/AKT/mTOR pathway. Mol Med. (2023) 29:95. doi: 10.1186/s10020-023-00690-x
40. Jing Z, Liu Q, He X, Jia Z, Xu Z, Yang B, et al. NCAPD3 enhances Warburg effect through c-myc and E2F1 and promotes the occurrence and progression of colorectal cancer. J Exp Clin Cancer Res. (2022) 41:198. doi: 10.1186/s13046-022-02412-3
41. Sun L, Liu Y, Yang N, Ye X, Liu Z, Wu J, et al. Gold nanoparticles inhibit tumor growth via targeting the Warburg effect in a c-Myc-dependent way. Acta Biomater. (2023) 158:583–98. doi: 10.1016/j.actbio.2022.12.054
42. Yii CY, Yong SB, and Li CJ. Screening for helicobacter pylori to prevent gastric cancer. JAMA. (2025) 333:813–4. doi: 10.1001/jama.2024.26383
43. Wizenty J and Sigal M. Helicobacter pylori, microbiota and gastric cancer - principles of microorganism-driven carcinogenesis. Nat Rev Gastroenterol Hepatol. (2025) 22:296–313. doi: 10.1038/s41575-025-01042-2
44. Yadav D, Yadav A, Bhattacharya S, Dagar A, Kumar V, Rani R, et al. Two primary and essential key players in tumor glycolysis. Semin Cancer Biol. (2024) 100:17–27. doi: 10.1016/j.semcancer.2024.03.001
45. Tilekar K, Upadhyay N, Iancu CV, Pokrovsky V, Choe JY, and Ramaa CS. Power of two: combination of therapeutic approaches involving glucose transporter (GLUT) inhibitors to combat cancer. Biochim Biophys Acta Rev Cancer. (2020) 1874:188457. doi: 10.1016/j.bbcan.2020.188457
46. Chang YC, Chan MH, Yang YF, Li CH, and Hsiao M. Glucose transporter 4: Insulin response mastermind, glycolysis catalyst and treatment direction for cancer progression. Cancer Lett. (2023) 563:216179. doi: 10.1016/j.canlet.2023.216179
47. Li F, Si W, Xia L, Yin D, Wei T, Tao M, et al. Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma. Mol Cancer. (2024) 23:90. doi: 10.1186/s12943-024-02008-9
48. Zhang X, Li J, Huang Y, Yang A, Liu X, Luo Y, et al. Plasma extracellular vesicles from recurrent GBMs carrying LDHA to activate glioblastoma stemness by enhancing glycolysis. Theranostics. (2025) 15:3655–72. doi: 10.7150/thno.102014
49. Li T, Bjorvang RD, Hao J, Di Nisio V, Damdimopoulos A, Lindskog C, et al. Persistent organic pollutants dysregulate energy homeostasis in human ovaries in vitro. Environ Int. (2024) 187:108710. doi: 10.1016/j.envint.2024.108710
50. Liu S, Shen G, Zhou X, Sun L, Yu L, Cao Y, et al. Hsp90 promotes gastric cancer cell metastasis and stemness by regulating the regional distribution of glycolysis-related metabolic enzymes in the cytoplasm. Adv Sci (Weinh). (2024) 11:e2310109. doi: 10.1002/advs.202310109
51. Wang Y, Zhang J, Shi H, Wang M, Yu D, Fu M, et al. M2 tumor-associated macrophages-derived exosomal MALAT1 promotes glycolysis and gastric cancer progression. Adv Sci (Weinh). (2024) 11:e2309298. doi: 10.1002/advs.202309298
52. Yin S, Liu H, Zhou Z, Xu X, Wang P, Chen W, et al. PUM1 promotes tumor progression by activating DEPTOR-meditated glycolysis in gastric cancer. Adv Sci (Weinh). (2023) 10:e2301190. doi: 10.1002/advs.202301190
53. Wang YY, Zhou YQ, Xie JX, Zhang X, Wang SC, Li Q, et al. MAOA suppresses the growth of gastric cancer by interacting with NDRG1 and regulating the Warburg effect through the PI3K/AKT/mTOR pathway. Cell Oncol (Dordr). (2023) 46:1429–44. doi: 10.1007/s13402-023-00821-w
54. Zhang Y, Wu Y, and Su X. PLOD1 promotes cell growth and aerobic glycolysis by regulating the SOX9/PI3K/Akt/mTOR signaling pathway in gastric cancer. Front Biosci (Landmark Ed). (2021) 26:322–34. doi: 10.52586/4946
55. Yang Q, Lei X, He J, Peng Y, Zhang Y, Ling R, et al. N4-acetylcytidine drives glycolysis addiction in gastric cancer via NAT10/SEPT9/HIF-1alpha positive feedback loop. Adv Sci (Weinh). (2023) 10:e2300898. doi: 10.1002/advs.202300898
56. Wang XH, Jiang ZH, Yang HM, Zhang Y, and Xu LH. Hypoxia-induced FOXO4/LDHA axis modulates gastric cancer cell glycolysis and progression. Clin Transl Med. (2021) 11:e279. doi: 10.1002/ctm2.279
57. Jiang C, Ding C, Xu J, Teng Y, Chen J, Wang Z, et al. Will baseline total lesion glycolysis play a role in improving the prognostic value of the NCCN-IPI in primary gastric diffuse large B-cell lymphoma patients treated with the R-CHOP regimen? Clin Nucl Med. (2021) 46:1–7. doi: 10.1097/RLU.0000000000003378
58. Zhou X, Fang D, Liu H, Ou X, Zhang C, Zhao Z, et al. PMN-MDSCs accumulation induced by CXCL1 promotes CD8(+) T cells exhaustion in gastric cancer. Cancer Lett. (2022) 532:215598. doi: 10.1016/j.canlet.2022.215598
59. Wang J, Huang Q, Hu X, Zhang S, Jiang Y, Yao G, et al. Disrupting circadian rhythm via the PER1-HK2 axis reverses trastuzumab resistance in gastric cancer. Cancer Res. (2022) 82:1503–17. doi: 10.1158/0008-5472.CAN-21-1820
60. Zhou M, He J, Li Y, Jiang L, Ran J, Wang C, et al. N(6)-methyladenosine modification of REG1alpha facilitates colorectal cancer progression via beta-catenin/MYC/LDHA axis mediated glycolytic reprogramming. Cell Death Dis. (2023) 14:557. doi: 10.1038/s41419-023-06067-6
61. Wu Y, Wang Y, Dong Y, Sun LV, and Zheng Y. Lactate promotes H3K18 lactylation in human neuroectoderm differentiation. Cell Mol Life Sci. (2024) 81:459. doi: 10.1007/s00018-024-05510-x
62. Chen Y, Joo J, Chu JM, Chang RC, and Wong GT. Downregulation of the glucose transporter GLUT 1 in the cerebral microvasculature contributes to postoperative neurocognitive disorders in aged mice. J Neuroinflamm. (2023) 20:237. doi: 10.1186/s12974-023-02905-8
63. He X, Williams QA, Cantrell AC, Besanson J, Zeng H, and Chen JX. TIGAR deficiency blunts angiotensin-II-induced cardiac hypertrophy in mice. Int J Mol Sci. (2024) 25. doi: 10.3390/ijms25042433
64. He X, Zeng H, Cantrell AC, Williams QA, and Chen JX. Knockout of TIGAR enhances myocardial phosphofructokinase activity and preserves diastolic function in heart failure. J Cell Physiol. (2022) 237:3317–27. doi: 10.1002/jcp.30790
65. Canonico F, Pedicino D, Severino A, Vinci R, Flego D, Pisano E, et al. GLUT-1/PKM2 loop dysregulation in patients with non-ST-segment elevation myocardial infarction promotes metainflammation. Cardiovasc Res. (2023) 119:2653–62. doi: 10.1093/cvr/cvac184
66. Richter EA, Bilan PJ, and Klip A. A comprehensive view of muscle glucose uptake: regulation by insulin, contractile activity and exercise. Physiol Rev. (2025) 105(3):1867–945. doi: 10.1152/physrev.00033.2024
67. Horn P and Tacke F. Metabolic reprogramming in liver fibrosis. Cell Metab. (2024) 36:1439–55. doi: 10.1016/j.cmet.2024.05.003
68. Sun Y, Wang H, Cui Z, Yu T, Song Y, Gao H, et al. Lactylation in cancer progression and drug resistance. Drug Resist Update. (2025) 81:101248. doi: 10.1016/j.drup.2025.101248
69. Venit T, Sapkota O, Abdrabou WS, Loganathan P, Pasricha R, Mahmood SR, et al. Positive regulation of oxidative phosphorylation by nuclear myosin 1 protects cells from metabolic reprogramming and tumorigenesis in mice. Nat Commun. (2023) 14:6328. doi: 10.1038/s41467-023-42093-w
70. Wang T, Sun F, Li C, Nan P, Song Y, Wan X, et al. MTA1, a novel ATP synthase complex modulator, enhances colon cancer liver metastasis by driving mitochondrial metabolism reprogramming. Adv Sci (Weinh). (2023) 10:e2300756. doi: 10.1002/advs.202300756
71. Reiter RJ, Sharma RN, Manucha W, Rosales-Corral S, Almieda Chuffa LG, Loh D, et al. Dysfunctional mitochondria in age-related neurodegeneration: Utility of melatonin as an antioxidant treatment. Ageing Res Rev. (2024) 101:102480. doi: 10.1016/j.arr.2024.102480
72. Bartman CR, Weilandt DR, Shen Y, Lee WD, Han Y, TeSlaa T, et al. Slow TCA flux and ATP production in primary solid tumours but not metastases. Nature. (2023) 614:349–57. doi: 10.1038/s41586-022-05661-6
73. Fresquet V, Garcia-Barchino MJ, Larrayoz M, Celay J, Vicente C, Fernandez-Galilea M, et al. Endogenous retroelement activation by epigenetic therapy reverses the warburg effect and elicits mitochondrial-mediated cancer cell death. Cancer Discov. (2021) 11:1268–85. doi: 10.1158/2159-8290.CD-20-1065
74. Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, et al. Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell. (2021) 81:691–707 e6. doi: 10.1016/j.molcel.2020.12.012
75. Ojha R, Tantray I, Rimal S, Mitra S, Cheshier S, and Lu B. Regulation of reverse electron transfer at mitochondrial complex I by unconventional Notch action in cancer stem cells. Dev Cell. (2022) 57:260–276.e9. doi: 10.1016/j.devcel.2021.12.020
76. Jiang H, He CJ, Li AM, He B, Li Y, Zhou MN, et al. Mitochondrial uncoupling inhibits reductive carboxylation in cancer cells. Mol Cancer Res. (2023) 21:1010–6. doi: 10.1158/1541-7786.MCR-23-0049
77. Man CH, Mercier FE, Liu N, Dong W, Stephanopoulos G, Jiang L, et al. Proton export alkalinizes intracellular pH and reprograms carbon metabolism to drive normal and Malignant cell growth. Blood. (2022) 139:502–22. doi: 10.1182/blood.2021011563
78. Bahl V, Rifkind R, Waite E, Hamdan Z, May CL, Manduchi E, et al. G6PC2 controls glucagon secretion by defining the set point for glucose in pancreatic alpha cells. Sci Transl Med. (2025) 17:eadi6148. doi: 10.1126/scitranslmed.adi6148
79. Pilic J, Gottschalk B, Bourgeois B, Habisch H, Koshenov Z, Oflaz FE, et al. Hexokinase 1 forms rings that regulate mitochondrial fission during energy stress. Mol Cell. (2024) 84:2732–2746 e5. doi: 10.1016/j.molcel.2024.06.009
80. Bessa C, Loureiro JB, Barros M, Isca VMS, Sardão VA, Oliveira PJ, et al. Counteracting colon cancer by inhibiting mitochondrial respiration and glycolysis with a selective PKCδ Activator. Int J Mol Sci. (2023) 24(6):5710. doi: 10.3390/ijms24065710
81. Xu Y, Tummala SR, Chen X, and Vardi N. VDAC in retinal health and disease. Biomolecules. (2024) 14(6):654. doi: 10.3390/biom14060654
82. Rong J, Han C, Huang Y, Wang Y, Qiu Q, Wang M, et al. Inhibition of xanthine oxidase alleviated pancreatic necrosis via HIF-1alpha-regulated LDHA and NLRP3 signaling pathway in acute pancreatitis. Acta Pharm Sin B. (2024) 14:3591–604. doi: 10.1016/j.apsb.2024.04.019
83. Wang F, Chen L, Kong D, Zhang X, Xia S, Liang B, et al. Canonical Wnt signaling promotes HSC glycolysis and liver fibrosis through an LDH-A/HIF-1alpha transcriptional complex. Hepatology. (2024) 79:606–23. doi: 10.1097/HEP.0000000000000569
84. Das S, Finney AC, Anand SK, Rohilla S, Liu Y, Pandey N, et al. Inhibition of hepatic oxalate overproduction ameliorates metabolic dysfunction-associated steatohepatitis. Nat Metab. (2024) 6:1939–62. doi: 10.1038/s42255-024-01134-4
85. Urata K, Kajihara I, Miyauchi H, Mijiddorj T, Otsuka-Maeda S, Sakamoto R, et al. The Warburg effect and tumour immune microenvironment in extramammary Paget’s disease: overexpression of lactate dehydrogenase A correlates with immune resistance. J Eur Acad Dermatol Venereol. (2020) 34:1715–21. doi: 10.1111/jdv.16145
86. Ren Y, Kumar A, Das JK, Peng HY, Wang L, Balllard D, et al. Tumorous expression of NAC1 restrains antitumor immunity through the LDHA-mediated immune evasion. J Immunother Cancer. (2022) 10(9):e004856. doi: 10.1136/jitc-2022-004856
87. Lu C, Yang D, Klement JD, Colson YL, Oberlies NH, Pearce CJ, et al. G6PD functions as a metabolic checkpoint to regulate granzyme B expression in tumor-specific cytotoxic T lymphocytes. J Immunother Cancer. (2022) 10(1):e003543. doi: 10.1136/jitc-2021-003543
88. Lu LG, Zhou ZL, Wang XY, Liu BY, Lu JY, Liu S, et al. PD-L1 blockade liberates intrinsic antitumourigenic properties of glycolytic macrophages in hepatocellular carcinoma. Gut. (2022) 71:2551–60. doi: 10.1136/gutjnl-2021-326350
89. Wang JZ, Zhu W, Han J, Yang X, Zhou R, Lu HC, et al. The role of the HIF-1alpha/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun (Lond). (2021) 41:560–75. doi: 10.1002/cac2.12158
90. Hou PP, Luo LJ, Chen HZ, Chen QT, Bian XL, Wu SF, et al. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol Cell. (2020) 78:1192–1206.e10. doi: 10.1016/j.molcel.2020.05.004
91. Damasceno LEA, Prado DS, Veras FP, Fonseca MM, Toller-Kawahisa JE, Rosa MH, et al. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J Exp Med. (2020) 217(10):e20190613. doi: 10.1084/jem.20190613
92. Qian C, Zhou Y, Zhang T, Dong G, Song M, Tang Y, et al. Targeting PKM2 signaling cascade with salvianic acid A normalizes tumor blood vessels to facilitate chemotherapeutic drug delivery. Acta Pharm Sin B. (2024) 14:2077–96. doi: 10.1016/j.apsb.2024.02.003
93. Wu X, Liu L, Zheng Q, Hao H, Ye H, Li P, et al. Protocatechuic aldehyde protects cardiomycoytes against ischemic injury via regulation of nuclear pyruvate kinase M2. Acta Pharm Sin B. (2021) 11:3553–66. doi: 10.1016/j.apsb.2021.03.021
94. Hauck L, Dadson K, Chauhan S, Grothe D, and Billia F. Inhibiting the Pkm2/b-catenin axis drives in vivo replication of adult cardiomyocytes following experimental MI. Cell Death Differ. (2021) 28:1398–417. doi: 10.1038/s41418-020-00669-9
95. Wang Y, Sun Z, Zhao Z, Pang J, and Chen J. Recent progress in the development of glucose transporter (GLUT) inhibitors. J Med Chem. (2025) 68:1033–50. doi: 10.1021/acs.jmedchem.4c02717
96. Wang Z, Wang Q, Chen C, Zhao X, Wang H, Xu L, et al. NNMT enriches for AQP5(+) cancer stem cells to drive Malignant progression in early gastric cardia adenocarcinoma. Gut. (2023) 73:63–77. doi: 10.1136/gutjnl-2022-328408
97. Na KJ, Choi H, Oh HR, Kim YH, Lee SB, Jung YJ, et al. Reciprocal change in Glucose metabolism of Cancer and Immune Cells mediated by different Glucose Transporters predicts Immunotherapy response. Theranostics. (2020) 10:9579–90. doi: 10.7150/thno.48954
98. Cribioli E, Giordano Attianese GMP, Ginefra P, Signorino-Gelo A, Vuillefroy de Silly R, Vannini N, et al. Enforcing GLUT3 expression in CD8(+) T cells improves fitness and tumor control by promoting glucose uptake and energy storage. Front Immunol. (2022) 13:976628. doi: 10.3389/fimmu.2022.976628
99. Guo T, Bai YH, Cheng XJ, Han HB, Du H, Hu Y, et al. Insulin gene enhancer protein 1 mediates glycolysis and tumorigenesis of gastric cancer through regulating glucose transporter 4. Cancer Commun (Lond). (2021) 41:258–72. doi: 10.1002/cac2.12141
100. Song G, Huang Y, Xiong M, Yang Z, Liu Q, Shen J, et al. Aloperine relieves type 2 diabetes mellitus via enhancing GLUT4 expression and translocation. Front Pharmacol. (2020) 11:561956. doi: 10.3389/fphar.2020.561956
101. Oliveira H, Roma-Rodrigues C, Santos A, Veigas B, Bras N, Faria A, et al. GLUT1 and GLUT3 involvement in anthocyanin gastric transport- Nanobased targeted approach. Sci Rep. (2019) 9:789. doi: 10.1038/s41598-018-37283-2
102. Zhou Y, Zhang Q, Liao B, Qiu X, Hu S, and Xu Q. circ_0006089 promotes gastric cancer growth, metastasis, glycolysis, and angiogenesis by regulating miR-361-3p/TGFB1. Cancer Sci. (2022) 113:2044–55. doi: 10.1111/cas.15351
103. Li H, Cao B, Zhao R, Li T, Xu X, Cui H, et al. circDNMT1 promotes Malignant progression of gastric cancer through targeting miR-576-3p/hypoxia inducible factor-1 alpha axis. Front Oncol. (2022) 12:817192. doi: 10.3389/fonc.2022.817192
104. Zheng Y, Li P, Ma J, Yang C, Dai S, and Zhao C. Cancer-derived exosomal circ_0038138 enhances glycolysis, growth, and metastasis of gastric adenocarcinoma via the miR-198/EZH2 axis. Transl Oncol. (2022) 25:101479. doi: 10.1016/j.tranon.2022.101479
105. Zheng X, Shao J, Qian J, and Liu S. circRPS19 affects HK2−mediated aerobic glycolysis and cell viability via the miR−125a−5p/USP7 pathway in gastric cancer. Int J Oncol. (2023) 63(2):98. doi: 10.3892/ijo.2023.5546
106. Qian L, Wang L, Chen H, Wang S, Hou Y, Xu L, et al. Hsa_circ_0001756 drives gastric cancer glycolysis by increasing the expression and stability of PGK1 mRNA. Front Immunol. (2025) 16:1511247. doi: 10.3389/fimmu.2025.1511247
107. Ou W, Tan R, Zhai J, Sun L, Quan Z, Huang X, et al. Silencing circ_0043256 inhibited CoCl2-induced proliferation, migration, and aerobic glycolysis in gastric cancer cells. Sci Rep. (2025) 15:171. doi: 10.1038/s41598-024-84548-0
108. Jiang SF and Li RR. hsa_circ_0067514 suppresses gastric cancer progression and glycolysis via miR-654-3p/LATS2 axis. Neoplasma. (2022) 69:1079–91. doi: 10.4149/neo_2022_220225N209
109. Zheng X, Xiao H, Liu X, Huang T, and Deng C. Exosomal circKIAA1797 Regulates Cell Progression and Glycolysis by Targeting miR-4429/PBX3 Pathway in Gastric Cancer. Biochem Genet. (2024) 62:1762–78. doi: 10.1007/s10528-023-10529-z
110. Dai T, Zhang X, Zhou X, Hu X, Huang X, Xing F, et al. Long non-coding RNA VAL facilitates PKM2 enzymatic activity to promote glycolysis and Malignancy of gastric cancer. Clin Transl Med. (2022) 12:e1088. doi: 10.1002/ctm2.1088
111. Zhang C, Wang H, Liu Q, Dai S, Tian G, Wei X, et al. LncRNA CCAT1 facilitates the progression of gastric cancer via PTBP1-mediated glycolysis enhancement. J Exp Clin Cancer Res. (2023) 42:246. doi: 10.1186/s13046-023-02827-6
112. Xie R, Liu L, Lu X, He C, Yao H, and Li G. N6-methyladenosine modification of OIP5-AS1 promotes glycolysis, tumorigenesis, and metastasis of gastric cancer by inhibiting Trim21-mediated hnRNPA1 ubiquitination and degradation. Gastric Cancer. (2024) 27:49–71. doi: 10.1007/s10120-023-01437-7
113. Xu H, Ba Z, Liu C, and Yu X. Long noncoding RNA DLEU1 promotes proliferation and glycolysis of gastric cancer cells via APOC1 upregulation by recruiting SMYD2 to induce trimethylation of H3K4 modification. Transl Oncol. (2023) 36:101731. doi: 10.1016/j.tranon.2023.101731
114. Zhang Y, Gao Y, Li F, Qi Q, Li Q, Gu Y, et al. Long non-coding RNA NRAV in the 12q24.31 risk locus drives gastric cancer development through glucose metabolism reprogramming. Carcinogenesis. (2024) 45:23–34. doi: 10.1093/carcin/bgad080
115. Chen S, Wang H, Xu P, Dang S, and Tang Y. H19 encourages aerobic glycolysis and cell growth in gastric cancer cells through the axis of microRNA-19a-3p and phosphoglycerate kinase 1. Sci Rep. (2023) 13:17181. doi: 10.1038/s41598-023-43744-0
116. Ding Y, Gao S, Zheng J, and Chen X. Blocking lncRNA-SNHG16 sensitizes gastric cancer cells to 5-Fu through targeting the miR-506-3p-PTBP1-mediated glucose metabolism. Cancer Metab. (2022) 10:20. doi: 10.1186/s40170-022-00293-w
117. Xu TP, Yu T, Xie MY, Fang Y, Xu TT, Pan YT, et al. LOC101929709 promotes gastric cancer progression by aiding LIN28B to stabilize c-MYC mRNA. Gastric Cancer. (2023) 26:169–86. doi: 10.1007/s10120-022-01348-z
118. Zhao X, Huang X, Dang C, Wang X, Qi Y, and Li H. The Epstein-Barr virus-miRNA-BART6-5p regulates TGF-beta/SMAD4 pathway to induce glycolysis and enhance proliferation and metastasis of gastric cancer cells. Oncol Res. (2024) 32:999–1009. doi: 10.32604/or.2024.046679
119. Lin JX, Lian NZ, Gao YX, Zheng QL, Yang YH, Ma YB, et al. m6A methylation mediates LHPP acetylation as a tumour aerobic glycolysis suppressor to improve the prognosis of gastric cancer. Cell Death Dis. (2022) 13:463. doi: 10.1038/s41419-022-04859-w
120. Xu W, Lai Y, Pan Y, Tan M, Ma Y, Sheng H, et al. m6A RNA methylation-mediated NDUFA4 promotes cell proliferation and metabolism in gastric cancer. Cell Death Dis. (2022) 13:715. doi: 10.1038/s41419-022-05132-w
121. Zhang Y, Zhou X, Cheng X, Hong X, Jiang X, Jing G, et al. PRKAA1, stabilized by FTO in an m6A-YTHDF2-dependent manner, promotes cell proliferation and glycolysis of gastric cancer by regulating the redox balance. Neoplasma. (2022) 69:1338–48. doi: 10.4149/neo_2022_220714N714
122. Luo F and Lin K. N(6)-methyladenosine (m(6)A) reader IGF2BP1 accelerates gastric cancer aerobic glycolysis in c-Myc-dependent manner. Exp Cell Res. (2022) 417:113176. doi: 10.1016/j.yexcr.2022.113176
123. Gu C, Xia Y, Lu C, Qiu S, Wang J, Zhang L, et al. TRIM50 inhibits glycolysis and the Malignant progression of gastric cancer by ubiquitinating PGK1. Int J Biol Sci. (2024) 20:3656–74. doi: 10.7150/ijbs.97091
124. Zhang C and Huang Z. KAT2A promotes the succinylation of PKM2 to inhibit its activity and accelerate glycolysis of gastric cancer. Mol Biotechnol. (2024) 66:1446–57. doi: 10.1007/s12033-023-00778-z
125. Chen Y, Guo Y, Yuan M, Guo S, Cui S, and Chen D. USP4 promotes the proliferation and glucose metabolism of gastric cancer cells by upregulating PKM2. PloS One. (2023) 18:e0290688. doi: 10.1371/journal.pone.0290688
126. He J, Yi J, Ji L, Dai L, Chen Y, and Xue W. ECHDC2 inhibits the proliferation of gastric cancer cells by binding with NEDD4 to degrade MCCC2 and reduce aerobic glycolysis. Mol Med. (2024) 30:69. doi: 10.1186/s10020-024-00832-9
127. Wang Q, Li M, Chen C, Xu L, Fu Y, Xu J, et al. Glucose homeostasis controls N-acetyltransferase 10-mediated ac4C modification of HK2 to drive gastric tumorigenesis. Theranostics. (2025) 15:2428–50. doi: 10.7150/thno.104310
128. Tsukihara S, Akiyama Y, Shimada S, Hatano M, Igarashi Y, Taniai T, et al. Delactylase effects of SIRT1 on a positive feedback loop involving the H19-glycolysis-histone lactylation in gastric cancer. Oncogene. (2025) 44:724–38. doi: 10.1038/s41388-024-03243-6
129. Mi Y, Li Q, Liu B, Wang D, Liu Z, Wang T, et al. Ubiquitous mitochondrial creatine kinase promotes the progression of gastric cancer through a JNK-MAPK/JUN/HK2 axis regulated glycolysis. Gastric Cancer. (2023) 26:69–81. doi: 10.1007/s10120-022-01340-7
130. Lei Y, He L, Li Y, Hou J, Zhang H, and Li G. PDLIM1 interacts with HK2 to promote gastric cancer progression through enhancing the Warburg effect via Wnt/beta-catenin signaling. Cell Tissue Res. (2024) 395:105–16. doi: 10.1007/s00441-023-03840-z
131. Ni Y and Zhuang Z. DDX24 promotes tumor progression by mediating hexokinase-1 induced glycolysis in gastric cancer. Cell Signal. (2024) 114:110995. doi: 10.1016/j.cellsig.2023.110995
132. Wang MQ, Chen YR, Xu HW, Zhan JR, Suo DQ, Wang JJ, et al. HKDC1 upregulation promotes glycolysis and disease progression, and confers chemoresistance onto gastric cancer. Cancer Sci. (2023) 114:1365–77. doi: 10.1111/cas.15692
133. Fang Z, Zhang W, Wang H, Zhang C, Li J, Chen W, et al. Helicobacter pylori promotes gastric cancer progression by activating the TGF-beta/Smad2/EMT pathway through HKDC1. Cell Mol Life Sci. (2024) 81:453. doi: 10.1007/s00018-024-05491-x
134. Yu H, Wang M, Zhang T, Cao L, Li Z, Du Y, et al. Dual roles of β-arrestin 1 in mediating cell metabolism and proliferation in gastric cancer. Proc Natl Acad Sci U.S.A. (2022) 119:e2123231119. doi: 10.1073/pnas.2123231119
135. Wang P, Han Y, Pan W, Du J, Zuo D, Ba Y, et al. Tyrosine phosphatase SHP2 aggravates tumor progression and glycolysis by dephosphorylating PKM2 in gastric cancer. MedComm (2020). (2024) 5:e527. doi: 10.1002/mco2.527
136. Zhou Y, Wang M, Qian Y, Yu D, Zhang J, Fu M, et al. PRDX2 promotes gastric cancer progression by forming a feedback loop with PKM2/STAT3 axis. Cell Signal. (2025) 127:111586. doi: 10.1016/j.cellsig.2024.111586
137. Zhao H, Jiang R, Feng Z, Wang X, and Zhang C. Transcription factor LHX9 (LIM Homeobox 9) enhances pyruvate kinase PKM2 activity to induce glycolytic metabolic reprogramming in cancer stem cells, promoting gastric cancer progression. J Transl Med. (2023) 21:833. doi: 10.1186/s12967-023-04658-7
138. Wei C, Xie J, Yuan X, Luo Y, Xiao Y, Liao W, et al. Phosphoglycerate mutase 1 that is essential for glycolysis may act as a novel metabolic target for predicating poor prognosis for patients with gastric cancer. J Clin Lab Anal. (2022) 36:e24718. doi: 10.1002/jcla.24718
139. Wu L, Dong J, Fei D, Le T, Xiao L, Liu J, et al. Fructose-1, 6-bisphosphate aldolase B suppresses glycolysis and tumor progression of gastric cancer. Dig Dis Sci. (2024) 69:3290–304. doi: 10.1007/s10620-024-08568-6
140. Wang H, Sun X, Yang C, Li Z, Jin D, Zhu W, et al. Deficiency of TOP1MT enhances glycolysis through the stimulation of PDK4 expression in gastric cancer. Cancer Metab. (2024) 12:2. doi: 10.1186/s40170-024-00330-w
141. Yang ZY, Zhao YW, Xue JR, Guo R, Zhao Z, Liu HD, et al. Thioridazine reverses trastuzumab resistance in gastric cancer by inhibiting S-phase kinase associated protein 2-mediated aerobic glycolysis. World J Gastroenterol. (2023) 29:5974–87. doi: 10.3748/wjg.v29.i45.5974
142. Mi C, Zhao Y, Ren L, and Zhang D. HIF1alpha/CCL7/KIAA1199 axis mediates hypoxia-induced gastric cancer aggravation and glycolysis alteration. J Clin Biochem Nutr. (2023) 72:225–33. doi: 10.3164/jcbn.22-48
143. Ara H, Subedi U, Sharma P, Bhattarai S, Sharma S, Manikandan S, et al. Alteration of cellular energy metabolism through LPAR2-axin2 axis in gastric cancer. Biomolecules. (2022) 12(12):1805. doi: 10.3390/biom12121805
144. Zhi X, Jiang S, Zhang J, and Qin J. Ubiquitin-specific peptidase 24 accelerates aerobic glycolysis and tumor progression in gastric carcinoma through stabilizing PLK1 to activate NOTCH1. Cancer Sci. (2023) 114:3087–100. doi: 10.1111/cas.15847
145. Wang L, Mao X, Yu X, Su J, Li Z, Chen Z, et al. FPR3 reprograms glycolytic metabolism and stemness in gastric cancer via calcium-NFATc1 pathway. Cancer Lett. (2024) 593:216841. doi: 10.1016/j.canlet.2024.216841
146. Pan L, Lan B, Li S, Jin Y, Cui M, Xia Y, et al. Gypenoside inhibits gastric cancer proliferation by suppressing glycolysis via the Hippo pathway. Sci Rep. (2024) 14:19003. doi: 10.1038/s41598-024-69435-y
147. Zhang Y, Cao J, Yuan Z, Zhou J, Zuo H, Miao X, et al. Knockdown of SLC7A5 inhibits Malignant progression and attenuates oxaliplatin resistance in gastric cancer by suppressing glycolysis. Mol Med. (2025) 31:115. doi: 10.1186/s10020-025-01175-9
148. Wu Z, Fang Y, Wu J, Wang J, Ling Y, Liu T, et al. Activation of glycolysis by MCM10 increases stemness and paclitaxel resistance in gastric cancer cells. Turk J Gastroenterol. (2023) 34:1107–15. doi: 10.5152/tjg.2023.23169
149. Wang X, Zhang J, Wu Y, Zhang Y, Zhang S, Li A, et al. RORalpha inhibits gastric cancer proliferation through attenuating G6PD and PFKFB3 induced glycolytic activity. Cancer Cell Int. (2024) 24:12. doi: 10.1186/s12935-023-03201-4
150. MaruYama T, Miyazaki H, Lim YJ, Gu J, Ishikawa M, Yoshida T, et al. Pyrolyzed deketene curcumin controls regulatory T cell generation and gastric cancer metabolism cooperate with 2-deoxy-d-glucose. Front Immunol. (2023) 14:1049713. doi: 10.3389/fimmu.2023.1049713
151. Yu M, Pan Q, Li W, Du T, Huang F, Wu H, et al. Isoliquiritigenin inhibits gastric cancer growth through suppressing GLUT4 mediated glucose uptake and inducing PDHK1/PGC-1alpha mediated energy metabolic collapse. Phytomedicine. (2023) 121:155045. doi: 10.1016/j.phymed.2023.155045
152. Guo N, Ma H, Li D, Fan H, Sun C, and Sun Y. CS-NO suppresses inhibits glycolysis and gastric cancer progression through regulating YAP/TAZ signaling pathway. Cell Biochem Biophys. (2023) 81:561–7. doi: 10.1007/s12013-023-01153-0
153. Aisu Y, Oshima N, Hyodo F, Elhelaly AE, Masuo A, Okada T, et al. Dual inhibition of oxidative phosphorylation and glycolysis exerts a synergistic antitumor effect on colorectal and gastric cancer by creating energy depletion and preventing metabolic switch. PloS One. (2024) 19:e0309700. doi: 10.1371/journal.pone.0309700
154. Xu Q, Gao J, Zhao R, Li H, Cui H, Yuan Z, et al. Akkermansia muciniphila-derived pentadecanoic acid enhances oxaliplatin sensitivity in gastric cancer by modulating glycolysis. Pharmacol Res. (2024) 206:107278. doi: 10.1016/j.phrs.2024.107278
155. Liu B, Sun Y, Wang W, Ren J, and Wang D. BHLHE40-mediated transcriptional activation of GRIN2D in gastric cancer is involved in metabolic reprogramming. Funct Integr Genomics. (2024) 24:214. doi: 10.1007/s10142-024-01495-9
156. Shen G, Liu S, Cao Y, Chen Z, Wang G, Yu L, et al. HSP90 co-regulates the formation and nuclear distribution of the glycolytic output complex to promote resistance and poor prognosis in gastric cancer patients. J Transl Med. (2025) 23:172. doi: 10.1186/s12967-025-06196-w
157. Li F, Du X, Han M, Feng X, and Jiang C. Targeting ETHE1 inhibits tumorigenesis in vitro and in vivo by preventing aerobic glycolysis in gastric adenocarcinoma cells. Oncol Lett. (2025) 29:286. doi: 10.3892/ol.2025.15032
158. Zhao R, Cao B, Li H, Gao J, Xu Q, Cui H, et al. MZT1 protects gastric cancer against glucose starvation through targeting NEDD1. Life Sci. (2025) 372:123622. doi: 10.1016/j.lfs.2025.123622
159. Li N, Tong H, Hou W, Liu Q, Xiang F, Zhu JW, et al. Neural-cancer crosstalk: Reciprocal molecular circuits driving gastric tumorigenesis and emerging therapeutic opportunities. Cancer Lett. (2025) 616:217589. doi: 10.1016/j.canlet.2025.217589
160. Messina B, Lo Sardo F, Scalera S, Memeo L, Colarossi C, Mare M, et al. Hippo pathway dysregulation in gastric cancer: from Helicobacter pylori infection to tumor promotion and progression. Cell Death Dis. (2023) 14:21. doi: 10.1038/s41419-023-05568-8
161. Malla RR, Nellipudi HR, Srilatha M, and Nagaraju GP. HER-2 positive gastric cancer: Current targeted treatments. Int J Biol Macromol. (2024) 274:133247. doi: 10.1016/j.ijbiomac.2024.133247
162. Abadi AJ, Zarrabi A, Hashemi F, Zabolian A, Najafi M, Entezari M, et al. The role of SOX family transcription factors in gastric cancer. Int J Biol Macromol. (2021) 180:608–24. doi: 10.1016/j.ijbiomac.2021.02.202
163. Kang BW and Chau I. Molecular target: pan-AKT in gastric cancer. ESMO Open. (2020) 5:e000728. doi: 10.1136/esmoopen-2020-000728
164. Szeőcs D, Vida B, Petővári G, Póliska S, Janka E, Sipos A, et al. Cell-free ascites from ovarian cancer patients induces Warburg metabolism and cell proliferation through TGFβ-ERK signaling. Geroscience. (2024) 46:3581–97. doi: 10.1007/s11357-023-01056-1
165. Xu L, Wang Y, Hu Y, Dai X, Sun C, and Cheng J. ROS-responsive oridonin and dihydroartemisinin hetero-polymeric prodrug NPs for potentiating ferroptosis in gastric cancer by disrupting redox balance. Colloids Surf B Biointerfaces. (2025) 252:114637. doi: 10.1016/j.colsurfb.2025.114637
166. Arai J, Hayakawa Y, Tateno H, Murakami K, Hayashi T, Hata M, et al. Impaired glycosylation of gastric mucins drives gastric tumorigenesis and serves as a novel therapeutic target. Gastroenterology. (2024) 167:505–521 e19. doi: 10.1053/j.gastro.2024.03.037
167. Gao N, Yang F, Chen S, Wan H, Zhao X, and Dong H. The role of TRPV1 ion channels in the suppression of gastric cancer development. J Exp Clin Cancer Res. (2020) 39:206. doi: 10.1186/s13046-020-01707-7
168. Zeng R, Gou H, Lau HCH, and Yu J. Stomach microbiota in gastric cancer development and clinical implications. Gut. (2024) 73:2062–73. doi: 10.1136/gutjnl-2024-332815
169. Zhao LY, Mei JX, Yu G, Lei L, Zhang WH, Liu K, et al. Role of the gut microbiota in anticancer therapy: from molecular mechanisms to clinical applications. Signal Transduct Target Ther. (2023) 8:201. doi: 10.1038/s41392-023-01406-7
170. He Y, Hong Q, Chen S, Zhou J, and Qiu S. Reprogramming tumor-associated macrophages in gastric cancer: a pathway to enhanced immunotherapy. Front Immunol. (2025) 16:1558091. doi: 10.3389/fimmu.2025.1558091
171. Zeng F, Zhao J, Tong M, He W, Li N, Fan Y, et al. CircRNA LDLR promotes proliferation and aerobic glycolysis of gastric cancer cells by targeting CHD1 with miR-449b-5p. Turk J Biol. (2024) 48:46–58. doi: 10.55730/1300-0152.2681
Keywords: gastric cancer, glycolytic metabolic reprogramming, lactate, tumor microenvironment, therapeutic targeting
Citation: Shan Z and Liu Y (2025) Harnessing glycolysis in gastric cancer: molecular targets, therapeutic strategies, and clinical horizons. Front. Immunol. 16:1628937. doi: 10.3389/fimmu.2025.1628937
Received: 15 May 2025; Accepted: 11 July 2025;
Published: 04 September 2025.
Edited by:
Yiju Wei, Shandong First Medical University, ChinaCopyright © 2025 Shan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yefu Liu, OTc5MDIxNTNAY211LmVkdS5jbg==