 Alok Agrawal
Alok Agrawal Donald N. Ngwa1
Donald N. Ngwa1 Sanjay K. Singh
Sanjay K. Singh- 1Department of Biomedical Sciences, College of Medicine, East Tennessee State University, Johnson City, TN, United States
- 2Centre for Amyloidosis and Acute Phase Proteins, Division of Medicine, University College London, London, United Kingdom
C-reactive protein (CRP) protects mice during the initial stages of Streptococcus pneumoniae infection. In order to be protective against all stages of infection, we hypothesize that CRP binds to two different ligands on pneumococci. In its native form, CRP binds to phosphocholine residues of C-polysaccharide to activate complement. In its altered form, CRP binds to amyloid-like structures (amyloids) formed on complement inhibitors recruited by pneumococci. We employed CRP knockout mice to test this hypothesis. In one approach, both wild-type CRP and E42Q/F66A/T76Y/E81A mutant CRP (E-CRP-1) were administered together. E-CRP-1 does not bind to phosphocholine but binds to amyloids. In another approach, Y40F/E42Q mutant CRP (E-CRP-2) was administered. E-CRP-2 binds to both phosphocholine and amyloids. When CRP was administered to mice 12 h after inoculation, then unlike wild-type CRP by itself, the combination of wild-type CRP and E-CRP-1 was protective and E-CRP-2 alone was protective. We also detected amyloids on pneumococci. The serum levels of the amyloid-binding protein, serum amyloid P component (SAP), were higher in CRP knockout mice than in wild-type mice. Also, the basal SAP levels were higher in female than in male mice and, conversely, male mice were more susceptible than female mice to severe infection. We conclude that the protection against prolonged pneumococcal infection requires structural changes in CRP and binding to both phosphocholine and amyloids on pneumococci. The sources of amyloids can be virulence factors or recruited complement inhibitors or both. Combined data also raise the possibility that SAP cooperates with CRP in reducing bacteremia and bacterial load.
Introduction
C-reactive protein (CRP) is an evolutionarily conserved protein, which suggests that CRP performs host-defense functions in all organisms from arthropods to humans (1–4). In humans, CRP is a component of the acute phase response; the serum level of CRP increases thousand-fold or more in acute inflammatory states (5). CRP is composed of five identical subunits arranged in a cyclic pentameric symmetry (6). CRP binds to phosphocholine (PCh)-containing substances, such as C-polysaccharide of the cell wall of Streptococcus pneumoniae, in a Ca2+-dependent manner (7). All five subunits of CRP have a PCh-binding site consisting of amino acid residues Phe66, Thr76 and Glu81 (8–10). PCh-complexed CRP activates the classical pathway of the complement system, leading to the destruction of the ligand (11). Human CRP activates mouse complement system also and therefore mice are widely used to investigate the in vivo functions of CRP (12–14).
The native pentameric conformation of CRP is altered under experimental inflammatory conditions such as in the presence of acidic pH or reactive oxygen species (15–18). At acidic pH, CRP has been shown to bind to amyloid-β peptide 1-42 (Aβ) (16, 19, 20). Some proteins, when immobilized, express Aβ-like structures (amyloids), and acidic pH-treated CRP binds to such immobilized proteins through the exposed amyloids (21, 22). The existence of non-native CRP in vivo and their deposition at sites of inflammation have been demonstrated by employing antibodies specific for non-native CRP (23, 24); however, it is not evident whether CRP seen at the sites of inflammation was monomeric CRP or non-native pentameric CRP. Like the PCh-binding function of CRP, the amyloid-binding function of CRP has also been conserved throughout evolution (25).
Human CRP protects against lethal pneumococcal infection by decreasing bacteremia in mouse models of the disease (26–30). Complement activation by ligand-complexed CRP is necessary for CRP-mediated protection against infection (31–33). In mouse models in which human CRP was passively administered to mice, CRP was protective only when administered 6 h before to 2 h after inoculation with pneumococci (34). It has been shown that pneumococci recruit complement-inhibitory proteins on their surface to become complement attack-resistant (35–42). The presence of complement-inhibitory proteins on pneumococci could be the reason for the inability of CRP to protect when administered 2 h after inoculation (Figures 1A, B).
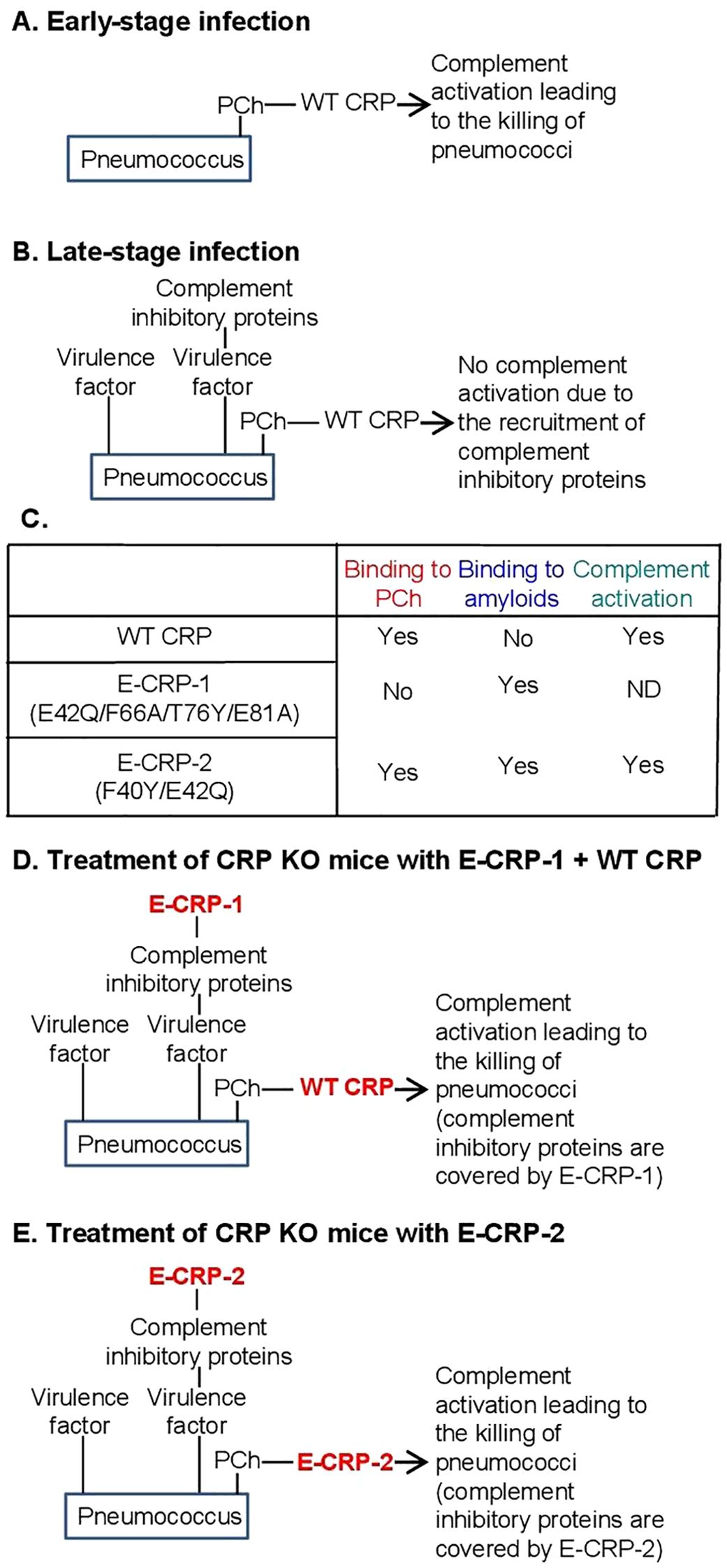
Figure 1. (A, B) Suggested mechanism of action of CRP in pneumococcal infection. (C) Ligand-recognition and effector functions of CRP molecules employed in this study. (D, E) Hypothesis for the mechanism of action of exogenously administered CRP in protecting CRP KO mice against prolonged pneumococcal infection.
It remains to be established that the ability of structurally altered pentameric CRP to bind to amyloids contributes to protection against pneumococcal infection (43). Since the acidic pH-induced changes in CRP are reversible at physiological pH, acidic pH-treated CRP cannot be administered to mice for in vivo studies (16). Therefore, recombinant CRP mutants have been created that mimic the amyloid-binding property of acidic pH-treated CRP (20, 21, 44). Previously, two such CRP mutants have been used as tools to investigate the host-defense functions of structurally altered CRP in vivo: E-CRP-1 and E-CRP-2 (22, 43). The ligand-recognition and effector functions of wild-type (WT) CRP, E-CRP-1 and E-CRP-2 are summarized in Figure 1C. E-CRP-1 (E42Q/F66A/T76Y/E81A CRP mutant) does not bind to PCh due to the mutations in the PCh-binding site (43, 45). E-CRP-1, however, binds to amyloids due to the presence of the E42Q mutation (20, 21). In contrast to E-CRP-1, E-CRP-2 (Y40F/E42Q CRP mutant) retains the ability to bind to PCh and, in addition, also binds to amyloids (20, 21, 43, 46). Biochemical analyses of acidic pH-treated WT CRP and of CRP mutants suggested that CRP gains the amyloid-binding property due to the loss of one Ca2+ from CRP and that the cholesterol-binding region of CRP which contains the Ca2+-binding site may be involved (16, 20, 21, 47).
Our earlier findings that immobilized proteins express amyloids (21) prompted us to hypothesize that complement inhibitory proteins recruited by pneumococci may express amyloids. It is not known whether the virulence factors present on pneumococci also express amyloids. In this study, we tested the overall hypothesis that if the amyloid-expressing proteins on the pneumococcal surface are blocked by E-CRP-1/-2, then WT CRP should be able to protect mice against all stages of pneumococcal infection; for example, WT CRP should be able to protect mice against infection even when administered to mice 12 h post-inoculation. Two approaches were employed in this study. In the first approach, both WT CRP (for PCh-binding) and E-CRP-1 (for amyloid-binding) were administered to mice. In this approach, E-CRP-1 can bind to amyloid-expressing proteins and PCh-bound WT CRP can then activate complement and kill the bacteria (Figure 1D). In the second approach, E-CRP-2 (for both PCh-binding and amyloid-binding) was administered to mice, assuming that some E-CRP-2 molecules would bind to amyloids and some to PCh (Figure 1E). PCh-bound E-CRP-2 can then activate complement and kill the bacteria. CRP knockout (KO) mice were used in this study since our hypothesis could only be tested in CRP-deficient mice.
Materials and methods
Preparation of CRP
The cDNAs for E42Q/F66A/T76Y/E81A mutant CRP (E-CRP-1) and Y40F/E42Q mutant CRP (E-CRP-2) were constructed, expressed in CHO cells using the ExpiCHO Expression System (Thermo Fisher Scientific) and purified from the cell culture supernatants, as described previously (43, 46, 48). E-CRP-1 was purified by employing Ca2+-dependent affinity chromatography on a phosphoethanolamine-conjugated Sepharose column, followed by ion-exchange chromatography on a MonoQ column and gel filtration on a Superose12 column. E-CRP-2 from cell culture supernatants and native WT CRP from discarded human pleural fluid were purified by employing Ca2+-dependent affinity chromatography on a PCh-conjugated Sepharose column, followed by ion-exchange chromatography on a MonoQ column and gel filtration on a Superose12 column. The purity of CRP preparations was confirmed by denaturing 4-20% SDS-PAGE under reducing conditions. Purified CRP was dialyzed against 10 mM Tris-HCl, pH 7.2, containing 150 mM NaCl and 2 mM CaCl2, and was subsequently treated with Detoxi-Gel Endotoxin Removing Gel (Thermo Fisher Scientific). The concentration of endotoxin in CRP preparations was determined by using the Limulus Amebocyte Lysate kit QCL-1000 (Lonza). Purified CRP was stored at 4°C and used within a week.
Pneumococci
S. pneumoniae type 3, strain WU2, was obtained from Dr. David Briles (University of Alabama at Birmingham, Birmingham, AL, USA) and used as described previously (43, 45). In brief, pneumococci were made virulent by sequential i.v. passages in mice and were stored in aliquots at -80°C. For each experiment, a separate aliquot of pneumococci was thawed and cultured. Cultured pneumococci were resuspended in normal saline and the concentration of pneumococci (cfu/ml) was adjusted based on the absorbance of the suspension at 600 nm (A600 = 1.00 = 1.2 x 109 cfu/ml). Within 2 h, 100 µl of pneumococci suspension containing the required number (cfu) of pneumococci, as mentioned in the figures, was injected into mice. The concentration of pneumococci was confirmed next day by plating.
Mice
The method for the generation of pure-line C57BL/6 CRP KO mice has been previously described (49). The breeding colony of CRP KO mice was maintained in the Division of Laboratory Animal Research of our university. Male and female WT C57BL/6 mice were purchased from Jackson Laboratories. Mice were brought up and maintained according to protocols approved by the University Committee on Animal Care. Mice were 8–10 weeks old when used in experiments.
Mouse protection experiments
Mouse protection experiments were performed exactly as described previously (29, 43). In brief, mice were inoculated with pneumococci; the numbers of pneumococci (cfu) are mentioned in the figures. CRP (25 μg) was administered at different time points as mentioned in the figures. The amount of endotoxin in 25 μg of all CRP preparations was <1.0 endotoxin units. Survival of mice was recorded three times per day for 7 days. Survival curves were generated using the GraphPad Prism 9 software. To determine p-values for the differences in the survival curves among various groups, the survival curves were compared using the software’s Logrank (Mantel-Cox) test.
To determine bacteremia in the surviving mice, blood was collected daily for 5 days from the tip of the tail vein, diluted in normal saline, and plated on sheep blood agar for colony counting. The bacteremia value for dead mice was recorded as 109 cfu/ml because mice died when the bacteremia exceeded 108 cfu/ml. The scatter plots of the bacteremia data and the median bacteremia value for each group were generated using the GraphPad Prism 9 software. The software’s Mann-Whitney test was used to determine p-values for the differences in bacteremia among various groups at each time point. The median values shown in the scatter plots for each group of mice were also plotted separately for easier comparison of all groups of mice in a single figure.
Assay for the detection of amyloids on pneumococci
Microtiter wells (Corning, 9018) were coated with increasing numbers of pneumococci in 100 µl TBS, pH 7.2, in duplicate, and incubated overnight at 4°C. The unreacted sites in the wells were blocked with TBS containing 0.5% gelatin for 45 min at room temperature. Both, polyclonal anti-Aβ antibodies (Novus, NBP2-25093) and monoclonal anti-Aβ antibodies (Novus, NBP2-13075) were used to detect the amyloids on the coated pneumococci. Normal rabbit IgG and normal mouse IgG were used as controls for the antibodies. The anti-Aβ antibodies (10 μg/ml) diluted in TBS containing 0.1% gelatin and 0.02% Tween 20 were added to the wells and incubated at 37°C for 1 h. After washing the wells, bound polyclonal anti-Aβ antibodies were detected by using HRP-conjugated donkey anti-rabbit IgG (GE Healthcare) and bound monoclonal anti-Aβ antibodies were detected by using HRP-conjugated goat anti-mouse IgG (Thermo Fisher Scientific). Color was developed, and the OD was read at 405 nm.
Assays for the measurement of mouse CRP and serum amyloid P component
The levels of mouse CRP (moCRP) in the sera obtained from the blood of WT mice were measured by using Mouse CRP Quantikine ELISA Kit (R&D, catalog number MCRP00). The levels of mouse serum amyloid P component (SAP) in the sera from WT and KO mice were measured by using Mouse SAP Quantikine ELISA Kit (R&D, catalog number MPTX20).
Results
Male mice are more susceptible than female mice to infection
To establish a CRP KO mouse model of infection with S. pneumoniae type 3, strain WU2, we titrated the dose of bacteria needed for inoculation. In order to determine whether there was any difference between WT and KO mice for their susceptibility to infection and whether there was any difference between male and female mice, both WT and KO mice and both male and female mice were employed. The median survival time (MST, the time taken for the death of 50% of mice) for each dose of bacteria was then determined.
As shown in the survival curves for all four types of mice (Figures 2A–D), and as expected, the MST decreased as the inoculation dose of bacteria increased. To determine the relative susceptibility of each type of mice to infection in terms of the dose of bacteria and the corresponding survival times, the data (Figures 2A–D) were compiled together and presented as MST curves (Figure 2E). The survival time of 50 h and the dose of 3 x 107 cfu of bacteria were chosen for comparing the four MST curves. As shown, for each type of mice to survive for 50 h after inoculation, the doses of bacteria were 13 x 107, 8 x 107, 5 x 107 and 2 x 107 cfu for WT female, KO female, WT male and KO male mice, respectively. Similarly, for mice inoculated with 3 x 107 cfu of bacteria, the survival times were 112 h, 86 h, 63 h and 36 h for WT female, KO female, WT male and KO male mice, respectively. These results and the statistical analyses of the MST curves indicated that male mice were significantly more susceptible than female mice to infection. Also, CRP KO mice were more susceptible than WT mice to infection, although the difference between the MST curves for female WT and female KO mice was not statistically significant.
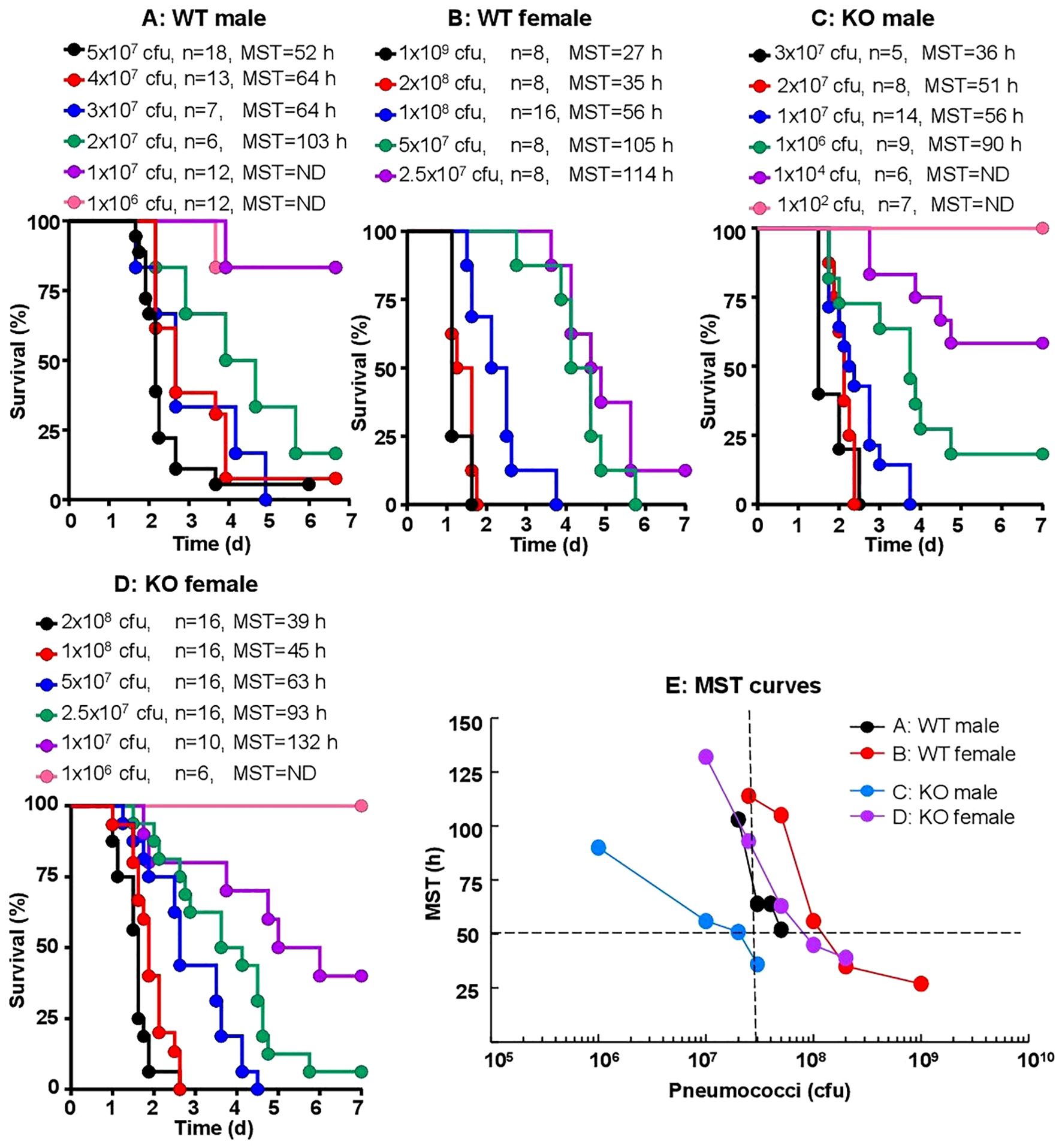
Figure 2. Sex-specificity and susceptibility of mice to infection. Mice were injected with 102–109 cfu of pneumococci, as shown for each group of mice (n, number of mice). The MST values are shown for each dose of pneumococci for each group of mice (ND, MST not determined since >50% mice survived). The data are combined from two separate experiments with six to nine mice for each dose of pneumococci in each group of mice. (A) Survival of male WT mice. (B) Survival of female WT mice. (C) Survival of male CRP KO mice. (D) Survival of female CRP KO mice. (E) The MST values for each group of mice shown in A-D are plotted together for comparing the susceptibility of all four types of mice. To determine p-values for the differences in the MST curves, the curves were subjected to two-way ANOVA followed by Tukey’s multiple comparison test. The p-values for the differences in the MST curves between groups A B, C D, A C and B D were 0.07, 0.01, 0.03 and 0.31, respectively.
CRP KO male mice inoculated with 107 cfu of pneumococci were employed as the mouse model for all CRP-mediated protection experiments, unless otherwise mentioned.
KO mice are most protected when WT CRP is administered prior to or within an hour of inoculation
The protective effects of WT CRP administered 30 min prior to and 15 min to 4 h after inoculation into KO male mice (Figure 3A) were determined first, by analyzing the survival curves of mice. Statistical analysis of the data showed that, when compared to group A mice (bacteria alone), CRP was clearly protective for groups B-F of mice, that is, if injected 30 min before to 2 h after inoculation. The p-value between group A and group G (CRP injected 4 h after inoculation), however, was 0.05, suggesting that CRP may still be protective. When compared to group B mice in which CRP was administered 30 min prior to inoculation, CRP was found to be protective only when injected 45 min after inoculation, but not later. Overall, as shown, as the interval between inoculation and CRP administration increased, the MST decreased. Mice survived longest and the MST could not be determined when CRP was given to mice 30 min before inoculation. Mice survived shortest when CRP was given to mice 4 h after inoculation. These results were similar to previously published data on the effects of WT CRP in WT mice: CRP was protective only when administered either prior to inoculation or at most 2 h post-inoculation. To confirm that WT CRP was protective against infection in KO female mice also, the 30 min time point for CRP injection prior to inoculation was chosen. As shown in Figure 3B, CRP protected female mice also. In all subsequent protection experiments, only CRP KO male mice were employed.
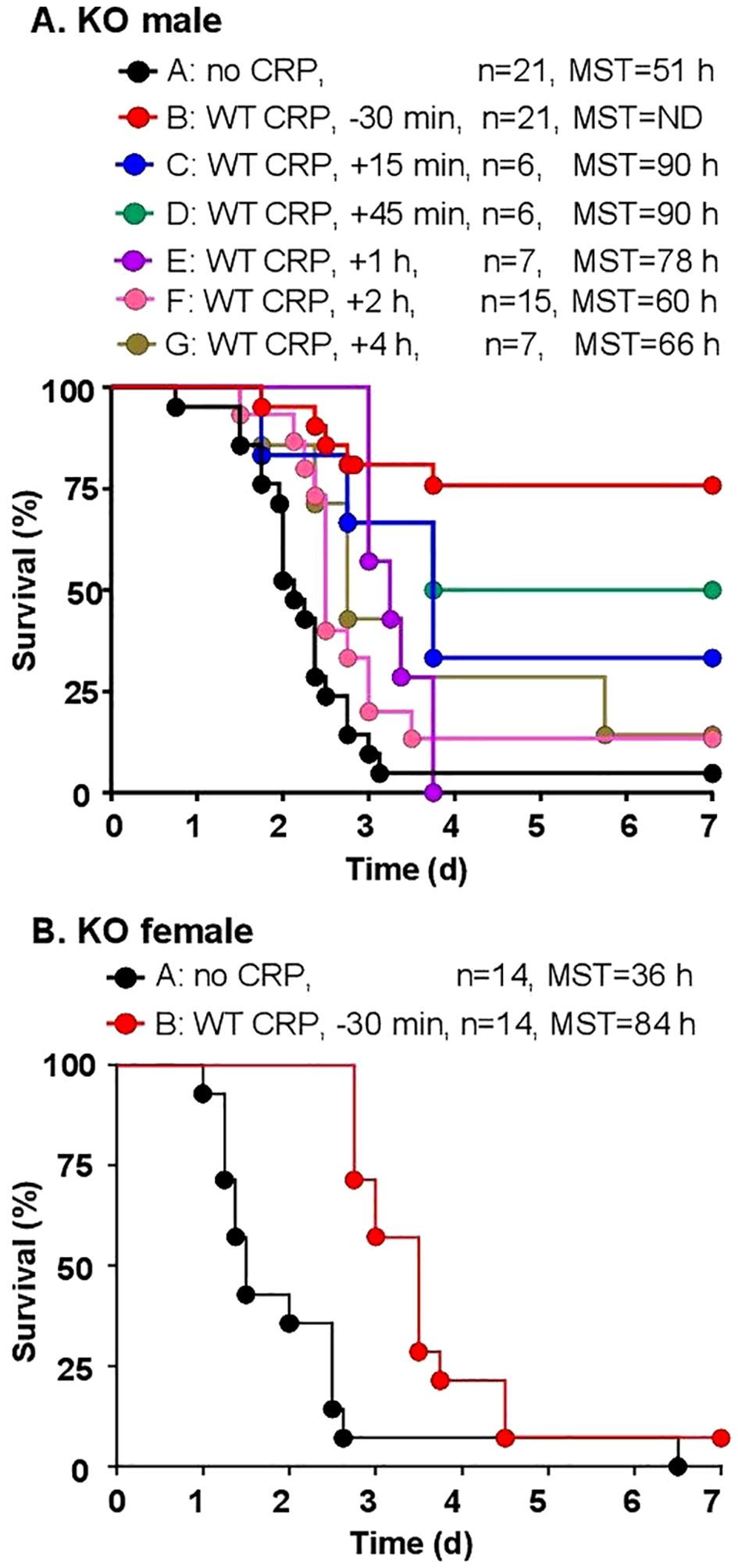
Figure 3. Survival of CRP KO mice infected with pneumococci and treated with WT CRP. The data are combined from two to three separate experiments with six to eight mice in each group in each experiment (n, number of mice). The MST values are shown for each group of mice (ND, MST not determined since >50% mice survived). (A) Male CRP KO mice. CRP was injected at various time points in different groups of mice, from 30 min before to 4 h after inoculation with pneumococci (107 cfu). The p-value for the difference in the survival curves between groups A and B was <0.001. The p-values for the differences between A C, A D, A E and A F were 0.01. The p-value for the difference between A and G was 0.05. The p-values for the differences between B C and B D were >0.05. The p-values for the differences between B E, B F and B G were <0.005. (B) Female CRP KO mice. CRP was injected 30 min prior to inoculation with pneumococci (108 cfu). The p-value for the difference between groups A and B was <0.05.
WT CRP and E-CRP-1 together protect KO mice even when administered 12 h after inoculation
We determined the protective effects of E-CRP-1 which does not bind to PCh and instead binds to amyloids. WT CRP administered 30 min prior to inoculation was included as a control for the animal model. As shown in Figure 4, and as has been reported earlier (43), WT CRP protected mice when administered 30 min prior to inoculation (group B) but did not protect when administered 12 h after inoculation (group D). The survival curve of mice treated with E-CRP-1, 30 min prior to inoculation (group C), was found to be significantly different from both group A (no CRP) and group B. However, like WT CRP, E-CRP-1 was not protective when administered 12 h after inoculation (group E). Although WT CRP and E-CRP-1 were not protective when either one was administered 12 h after inoculation, the survival curve of mice treated with the combination of WT CRP and E-CRP-1 was significantly different from both groups D and E and also different from group A. The survival times of mice treated with both WT CRP and E-CRP-1 together were longer than the survival times of mice treated with either WT CRP or E-CRP-1 alone and 10% of mice survived at the end of the experiment.
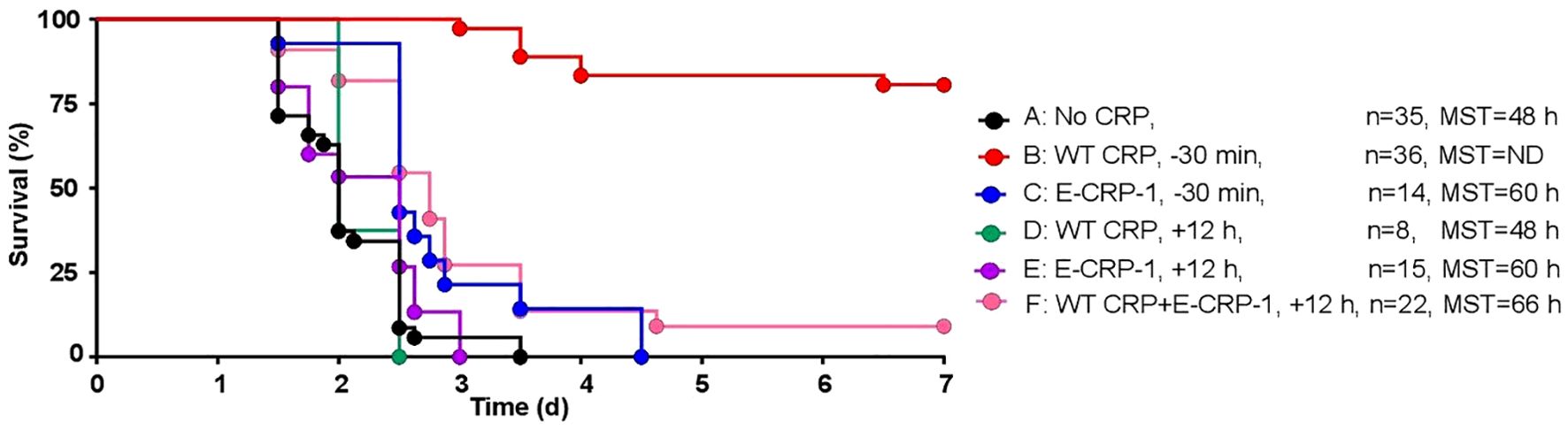
Figure 4. Survival of CRP KO mice infected with pneumococci and treated with E-CRP-1. The data are combined from two to four separate experiments with six to nine mice in each group in each experiment (n, number of mice). The MST values are shown for each group of mice (ND, MST not determined since >50% mice survived). CRP was injected 30 min prior to inoculation with pneumococci (107 cfu) in groups B and C and 12 h after inoculation in groups D-F. The p-values for the differences in the survival curves between groups A B, A C and A F were <0.001. The p-values for the differences between A D and A E were >0.05. The p-values for the differences between B C, B D, B E and B F were <0.001. The p-values for the differences between C D, C E and C F were 0.004, 0.04 and 0.46, respectively. The p-value for the difference between E and F was 0.01.
To determine whether the increased survival of mice was due to reduced bacteremia, we measured bacteremia in each surviving mouse in all six groups of mice (Figure 5). After 60 h of inoculation, bacteremia reached >108 cfu/ml in all groups of mice except in mice treated with WT CRP 30 min prior to inoculation. For statistical analysis and to compare bacteremia between any two groups of mice, the median values of bacteremia (Figure 5) for each time point up to 60 h for all groups of mice were plotted together (Figure 6). The bacteremia in mice treated with either WT CRP (group D) or E-CRP-1 (group E) was not significantly different from bacteremia in untreated mice (group A). However, the bacteremia in mice treated with both WT CRP and E-CRP-1 (group F) was found to be significantly lower than bacteremia in untreated mice (group A) at time points 36 h and 44 h. Consistent with the survival curves of mice treated with E-CRP-1 alone 30 min prior to inoculation (Figure 4), these mice had significantly lower bacteremia than in untreated mice and at the same time, significantly higher than mice treated with WT CRP alone 30 min prior to inoculation.
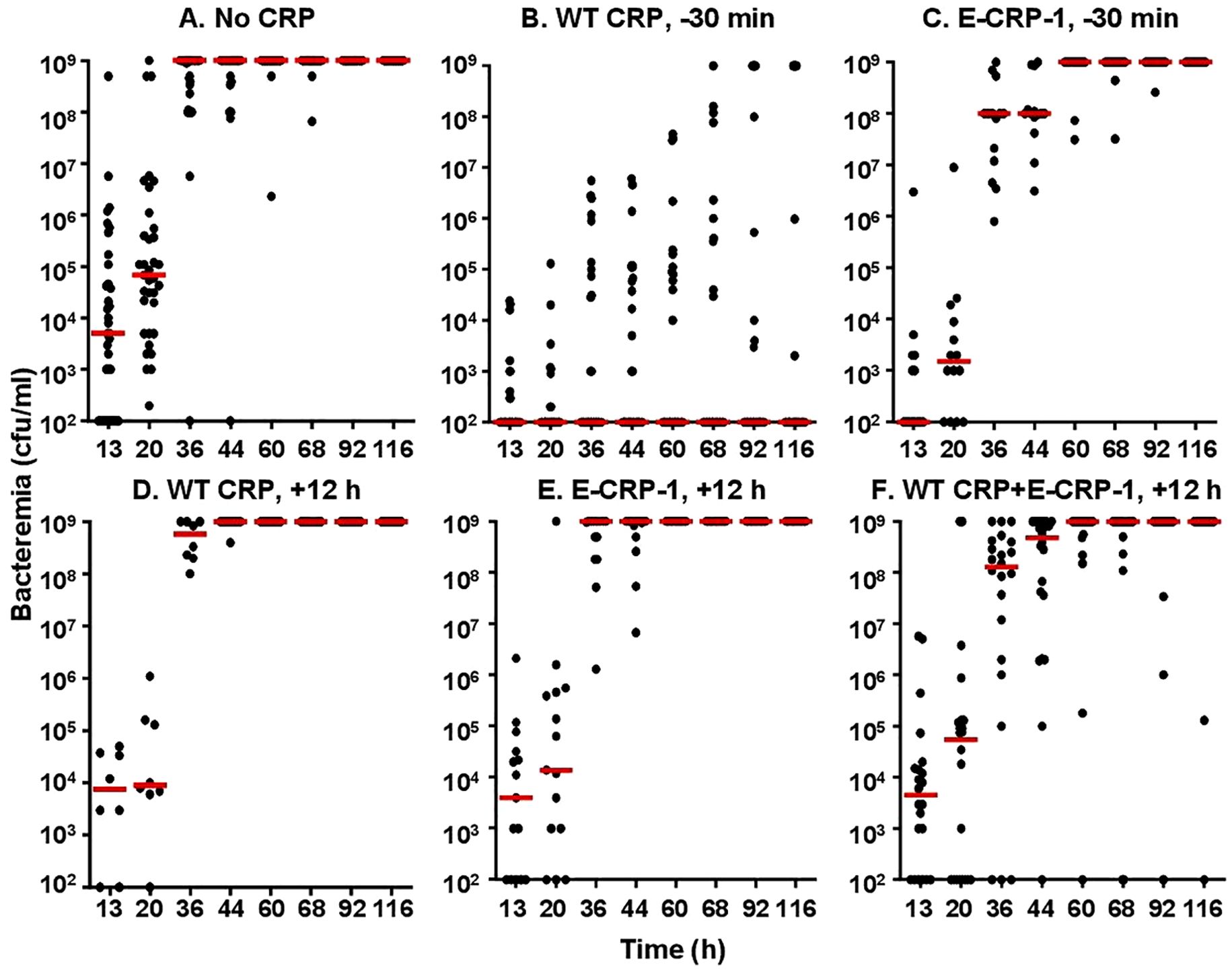
Figure 5. Bacteremia in CRP KO mice inoculated with pneumococci and treated with E-CRP-1 (A–F). Blood was collected from each surviving mouse shown in Figure 4. Bacteremia was determined by plating. The bacteremia values for dead mice were recorded as 109 cfu/ml. Bacteremia values of 0–100 were plotted as 100 and bacteremia values of >108 cfu/ml were plotted as 109 cfu/ml. The red horizontal line in each group of mice represents median bacteremia.
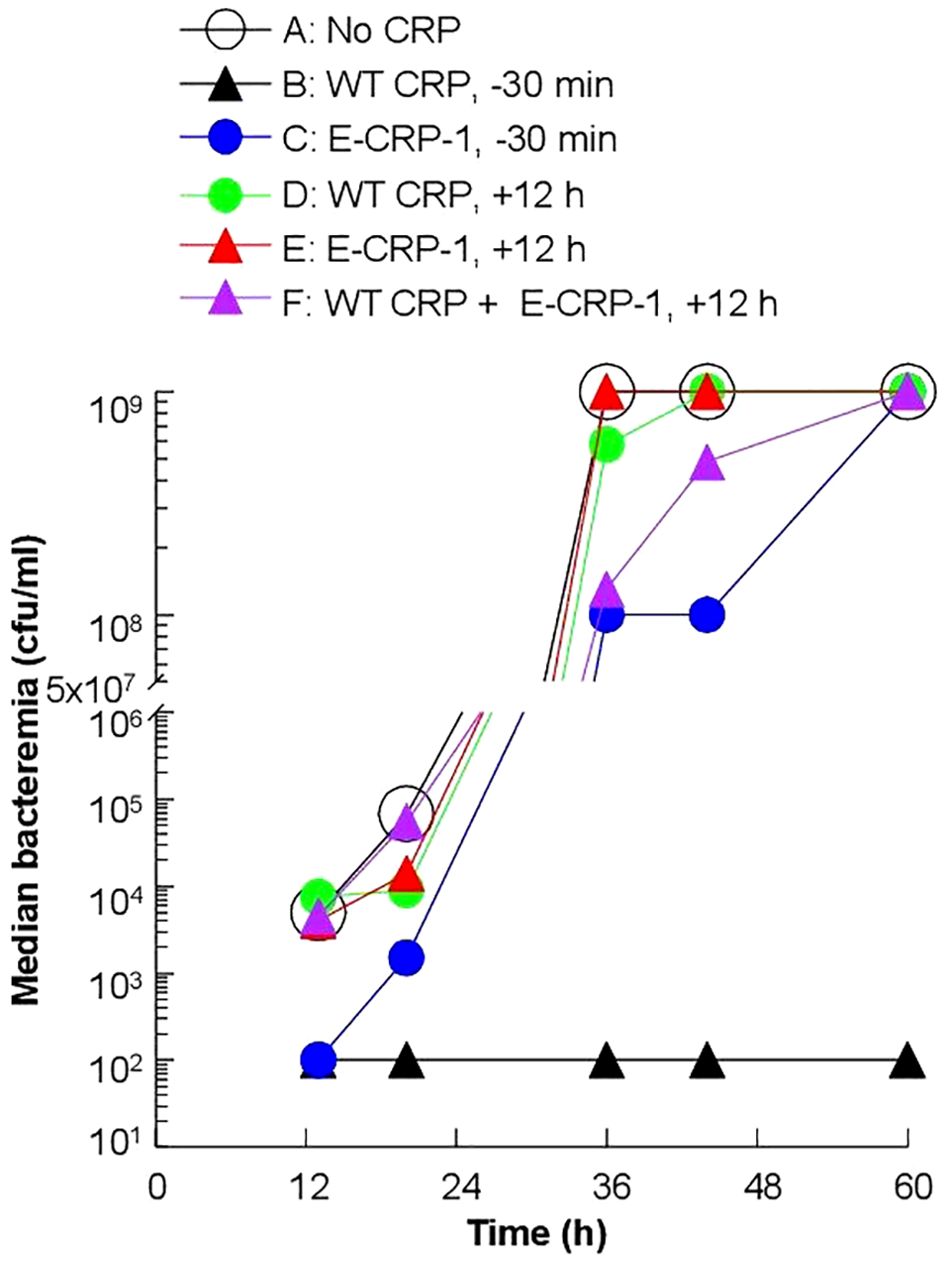
Figure 6. Median values of bacteremia in mice inoculated with pneumococci with and without E-CRP-1. The median bacteremia values for all six groups of mice shown in panels (A-F) in Figure 5 are plotted together for comparison. For all time points, the p-values for the differences between groups A, C were <0.01. For all time points, the p-values for the differences between groups A, D and between groups A, E were >0.05. For 36 h and 44 h, the p-values for the difference between groups A, F were <0.01.
E-CRP-2 by itself protects KO mice when administered 12 h after inoculation
The protective effects of E-CRP-2 which binds to both PCh and amyloids was determined next. As shown in Figure 7, WT CRP was protective when administered 30 min prior to inoculation (group B) but was not protective when administered 12 h after inoculation (group D). In contrast to WT CRP, E-CRP-2 was protective irrespective of whether E-CRP-2 was administered 30 min prior to inoculation (group C) or 12 h after inoculation (group E). However, the survival curve of mice in which E-CRP-2 was administered 12 h after inoculation was found to be significantly different from both group A (no CRP) and groups B and C. When CRP was administered 12 h after inoculation, the survival times of mice treated with E-CRP-2 were found to be longer than the survival times of mice treated with WT CRP.
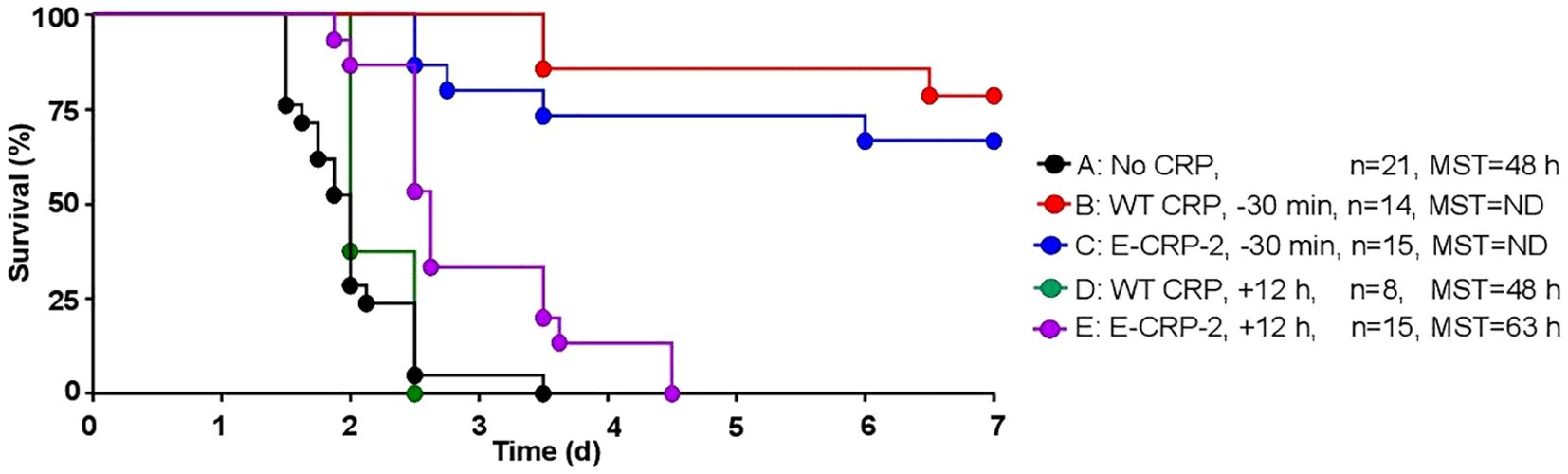
Figure 7. Survival of CRP KO mice inoculated with pneumococci and treated with E-CRP-2. The data are combined from two to three separate experiments with six to eight mice in each group in each experiment (n, number of mice). The MST values are shown for each group of mice (ND, MST not determined since >50% mice survived). CRP was injected 30 min prior to inoculation with pneumococci (107 cfu) in groups B and C and 12 h after inoculation in groups D, E. The p-values for the differences in the survival curves between groups A B, A C and A E were 0.001. The p-value for the differences between A, D was 0.22. The p-value for the difference between B, C was 0.40. The p-values for the differences between B D and B E were <0.001. The p-values for the differences between C D, C E and D E were <0.005.
Next, we measured bacteremia in each surviving mouse from all five groups of mice (Figure 8). After 66 h of inoculation, bacteremia reached >108 cfu/ml in all groups of mice except in mice treated with either WT CRP or E-CRP-2, 30 min prior to inoculation. For statistical analysis and to compare bacteremia between any two groups of mice, the median values of bacteremia (Figure 8) for each time point up to 66 h for all groups of mice were plotted together (Figure 9). Bacteremia was significantly reduced in mice treated with either CRP species 30 min prior to inoculation for the entire duration of the experiment. Bacteremia was not reduced in mice treated with WT CRP 12 h after inoculation, except at 36 h time point. In contrast, when E-CRP-2 was administered 12 h after inoculation, bacteremia was significantly reduced sometime after 20 h post-inoculation and remained low till at least 44 h. The bacteremia data were largely consistent with the survival data shown in Figure 7.
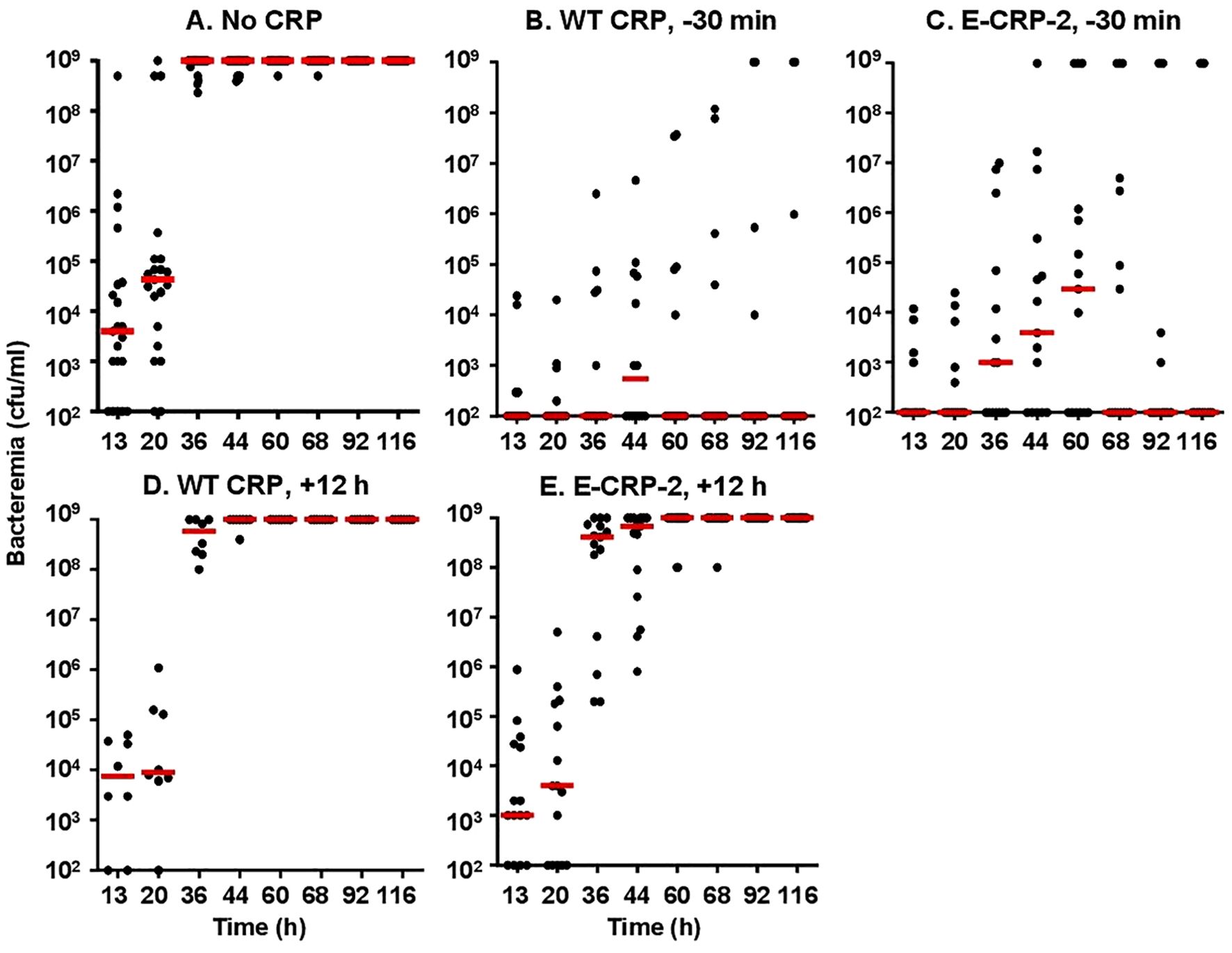
Figure 8. Bacteremia in CRP KO mice inoculated with pneumococci and treated with E-CRP-2 (A–E). Blood was collected from each surviving mouse shown in Figure 7. Bacteremia was determined by plating. The bacteremia values for dead mice were recorded as 109 cfu/ml. Bacteremia values of 0–100 were plotted as 100 and bacteremia values of >108 cfu/ml were plotted as 109 cfu/ml. The red horizontal line in each group of mice represents median bacteremia.
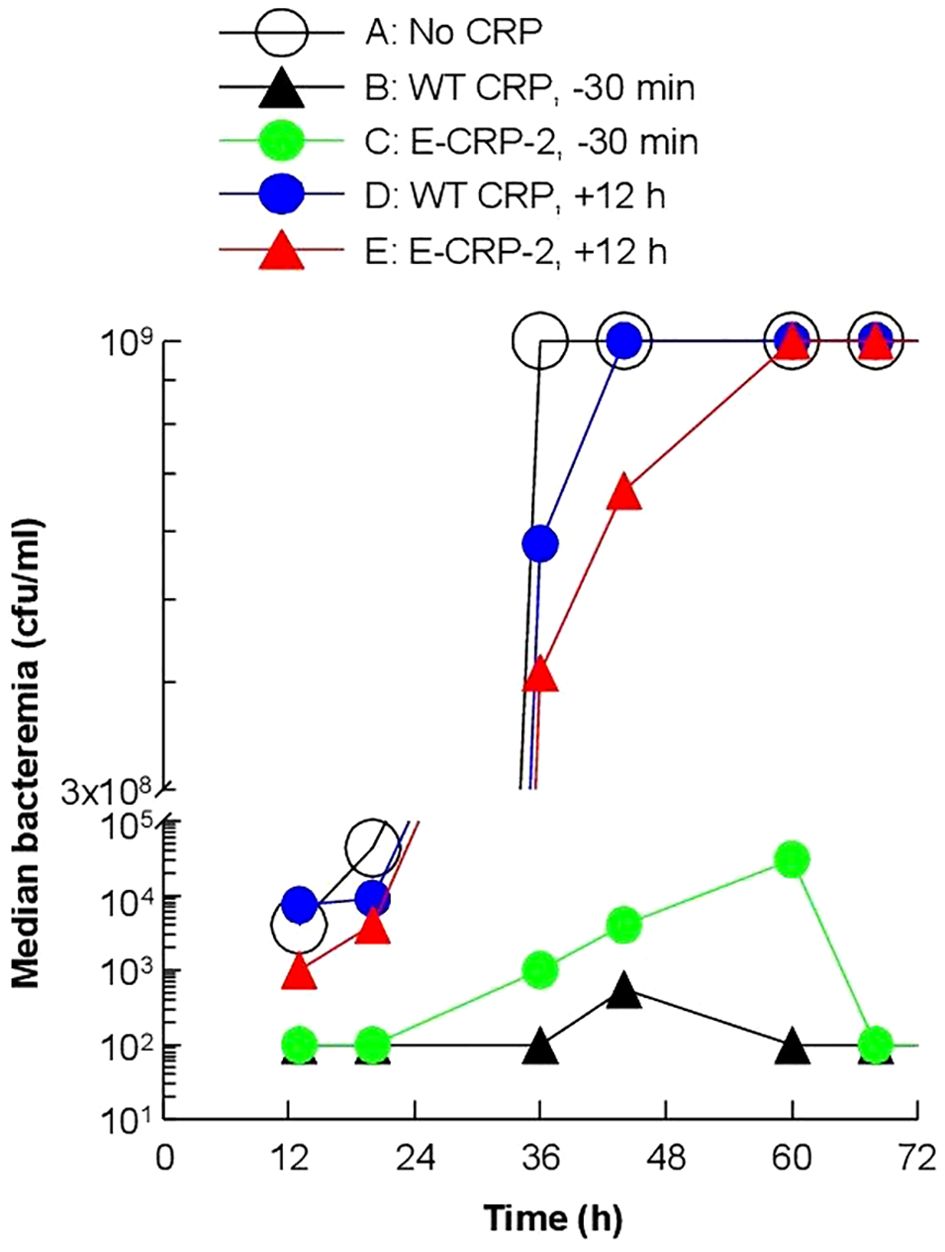
Figure 9. Median values of bacteremia in mice inoculated with pneumococci with and without E-CRP-2. The median bacteremia values for all five groups of mice shown in panels (A-F) in Figure 7 are plotted together for comparison. For all time points, the p-values for the differences between groups A and C were <0.01. For all time points, the p-values for the differences between groups A and D were >0.05, except at 36 h where the p-value was <0.05. The p values for the difference between groups A and E for time points 13 h and 20 h were >0.05 and for 36 h and 44 h were <0.005.
Amyloids are present on the pneumococcal surface
First, we determined whether amyloids were present on pneumococci which were cultured in vitro in broth and used to inoculate mice. As shown in Figure 10, pneumococci were reactive with both polyclonal and monoclonal anti-Aβ antibodies. These data suggested that amyloids were already present on the surface of pneumococci even if the bacteria were not grown in the serum in the presence of complement inhibitor proteins. For this reason, we did not investigate the presence of amyloids on pneumococci which were grown in vivo in mice and then isolated from the blood.
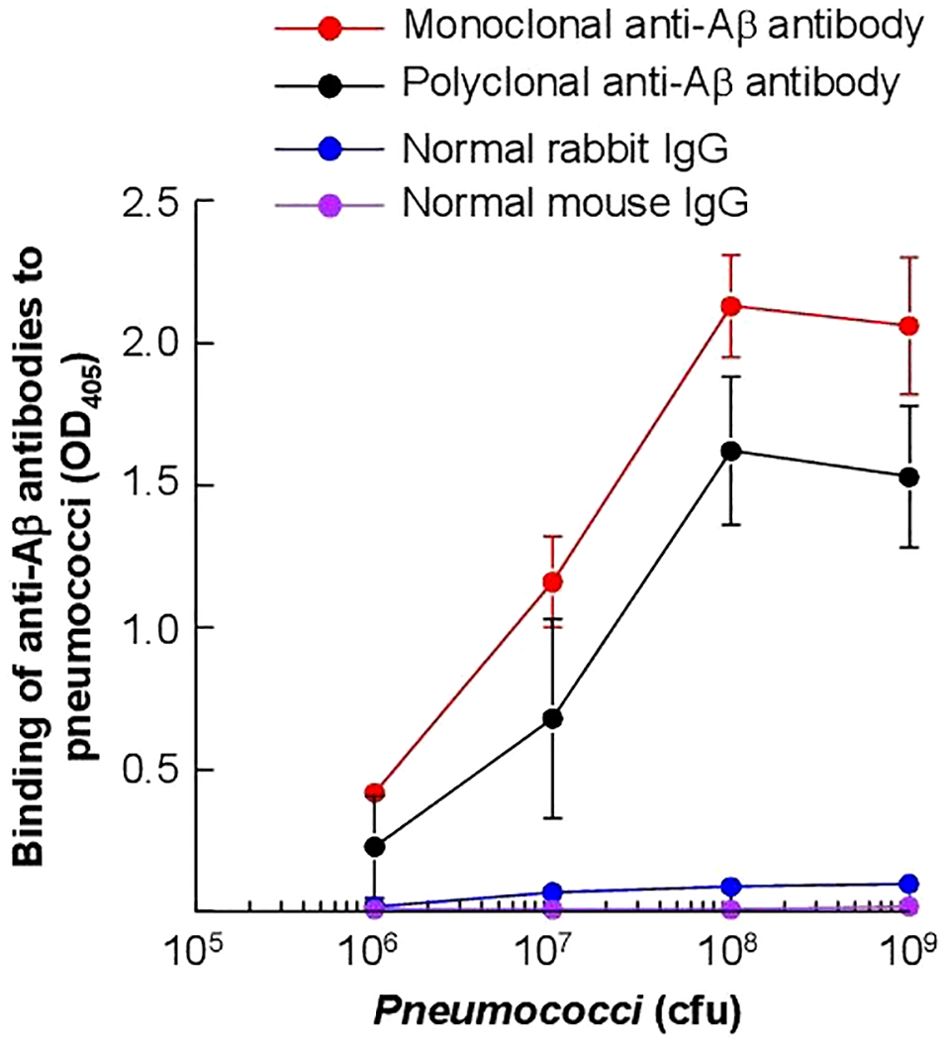
Figure 10. Detection of Aβ epitopes on the surface of S. pneumoniae. Microtiter wells were coated with pneumococci. Anti-Aβ antibodies, both monoclonal and polyclonal, were then added to the wells. Normal mouse IgG and rabbit IgG were included as controls. Bound monoclonal anti-Aβ antibodies and normal mouse IgG were detected by using HRP-conjugated goat anti-mouse IgG. Bound polyclonal anti-Aβ antibodies and normal rabbit IgG were detected by using HRP-conjugated donkey anti-rabbit IgG. The OD of the developed color was read at 405 nm. Data shown are mean ± SEM of three experiments.
SAP levels are higher in KO mice than in WT mice
In mice, CRP is a minor acute phase protein (50). SAP, an amyloid-binding protein, is the major acute phase protein in mice (51–54). Since the four types of mice (WT female and male, KO female and male) responded differently to the severity of infection in terms of their MST, we measured the serum levels of moCRP in WT mice and of SAP in WT and KO mice. As shown in Figure 11A, the basal levels of SAP in the sera (0 h) were approximately five-fold higher in KO mice than in WT mice, irrespective of the sex of mice (p = <0.005). The induced levels of SAP in mice after 36 h of inoculation was also higher in KO mice than in WT mice, irrespective of the sex of mice (p = <0.005). These data suggest that the absence of endogenous CRP can trigger acute phase response and that SAP can substitute CRP.
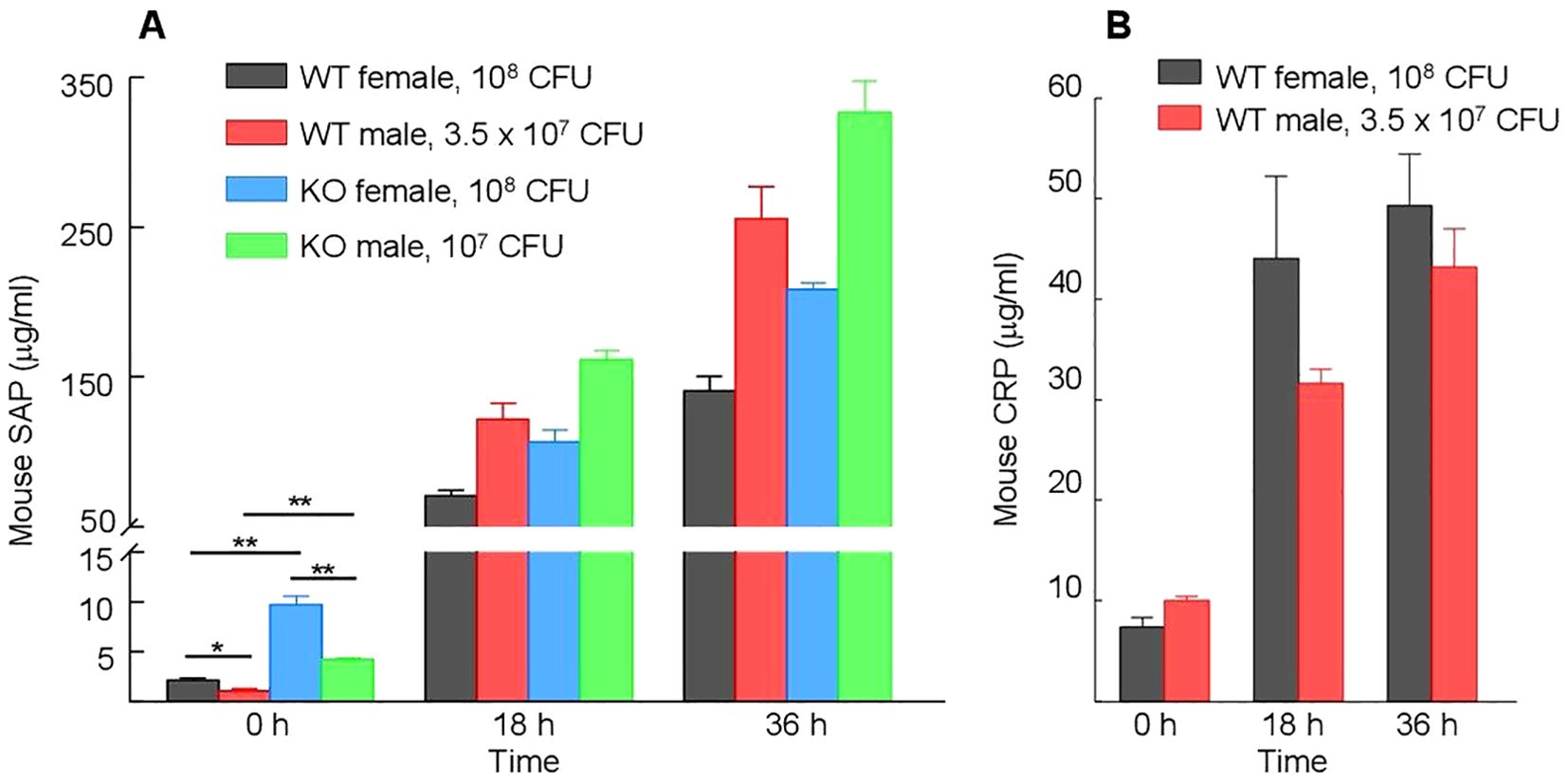
Figure 11. Serum levels of endogenous moCRP and SAP. Different types of mice were inoculated with different doses of pneumococci; the doses of pneumococci were chosen based on the relative susceptibility of each type of mouse to infection. Blood was collected just prior to inoculation (time zero) and at 18 h and 36 h post-inoculation. The data shown are mean ± SEM of three assays. At time zero, there were 8 mice in each group. At 18 h and 36 h time points, there were 5 mice in both groups of female mice and 8 mice in both groups of male mice, since 3 WT and 3 KO female mice died after infection. The p-values are shown as *p < 0.05 and **p < 0.005. (A) Levels of CRP in WT mice before and after inoculation. (B) Levels of SAP in WT and KO mice before and after inoculation.
Basal SAP levels are higher in female mice than in male mice
In both WT and KO mice, the basal levels of SAP were significantly higher (p = <0.005) in female mice than in male mice (Figure 11A). At 36 h post-inoculation, however, the levels of SAP were significantly higher in males than in females (p < 0.005), probably due to greater induction of SAP expression in male mice in response to inoculation. In WT mice, the increase in the levels of SAP was ~250-fold in male mice and ~50-fold in female mice. Similarly, in KO mice, the increase in the levels of SAP was ~80-fold in male mice and ~25-fold in female mice. In contrast to SAP, both the basal and induced serum levels of moCRP were not significantly different (p = >0.05) in male and female mice (Figure 11B).
Discussion
WT CRP protects mice against pneumococcal infection if administered to mice prior to inoculation with bacteria and does not protect if administered a few hours after inoculation (30, 34). Thus, WT CRP is not protective against prolonged infection. We tested the hypothesis that the amyloid-binding function of structurally altered CRP is required for CRP-mediated protection against prolonged infection. In this study, we employed CRP KO mice and investigated the effects of the amyloid-binding CRP molecules E-CRP-1 and E-CRP-2 on the survival of and bacteremia in mice when administered 12 h after inoculation. There were three major findings: 1. Amyloids were detected on the surface of broth-cultured pneumococci. 2. Unlike WT CRP alone, the combination of WT CRP and E-CRP-1 was protective when administered to mice 12 h after inoculation. E-CRP-2 by itself protected mice when administered 12 h after inoculation. 3. Serum SAP levels were higher in KO mice than in WT mice. Also, the basal SAP levels were higher in female mice than in male mice and, conversely, male mice were more susceptible than female mice to infection. Thus, there was an inverse relationship between the susceptibility of mice to infection and basal SAP levels in their sera.
In a previous study, WT mice were employed to investigate the effects of E-CRP-1 on pneumococcal infection (43). It was reported that E-CRP-1 was protective in WT mice against infection when administered 12 h after inoculation. The explanation was that E-CRP-1 blocked the complement inhibitor proteins recruited by pneumococci and then endogenous moCRP could activate complement by complexing with the PCh groups on pneumococci. Subsequently, it was reported that many proteins, when immobilized on microtiter plates, expressed amyloids (21), which raised the possibility that complement inhibitors expressed amyloids once immobilized on the pneumococcal surface and that the amyloids were the ligands for E-CRP-1. In this study, we found that pneumococci cultured in broth, with no exposure to serum complement inhibitors, had amyloids on their surface. The presence of functional amyloids on bacterial surfaces has been demonstrated previously (55). The sources of the amyloids on broth-grown pneumococci are not known; however, it is possible that the surface virulence factors are amyloidogenic proteins. The possibility that E-CRP-1 directly binds to virulence factors has been raised previously based on the following findings: the binding of E-CRP-1 to pneumococci was 99% less than the binding of WT CRP to pneumococci, and that the residual binding of E-CRP-1 to pneumococci occurred in the absence of Ca2+ (43). Since broth-cultured pneumococci were already amyloid-positive, we did not test pneumococci isolated from infected mice for the presence of surface amyloids. However, the possibility that both the virulence factors and the complement inhibitor proteins recruited by virulence factors express amyloids still exists.
In the current study, instead of WT mice, KO mice were employed to investigate the effects of E-CRP-1 on pneumococcal infection. In this animal model, E-CRP-1 by itself was not protective but the combination of exogenous WT human CRP and E-CRP-1 was protective when administered to mice 12 h after inoculation. Like the combination of WT CRP and E-CRP-1, E-CRP-2 by itself was protective. These findings provide a proof of concept that CRP can protect against prolonged infection provided that both PCh and amyloids on the bacterial surface are occupied by either structurally altered CRP molecules mimicking E-CRP-2 or by WT CRP and structurally altered CRP molecules mimicking E-CRP-1, respectively. Although both E-CRP-1 and E-CRP-2 were protective when administered to mice 12 h after inoculation, the protection was not as good as the protection seen when CRP was administered to mice 30 min prior to inoculation. It is possible that multiple injections of WT CRP and E-CRP-1 or E-CRP-2 or higher doses of each CRP species would result in better protection than seen with the single injection of structurally altered CRP reported here.
It is not known whether structurally altered CRP mimicking the ligand-binding properties of E-CRP-1 or E-CRP-2 is formed at sites of inflammation in vivo during pneumococcal infection. Since CRP mutants E-CRP-1 and E-CRP-2 mimic the ligand-binding properties of acidic pH-treated CRP and H2O2-treated CRP, these mutants provide us with a tool to identify the actual CRP species present at sites of inflammation. E-CRP-1 and E-CRP-2 can be used to generate a library of monoclonal antibodies which can then be screened to identify the antibodies which neither react with WT CRP nor with monomeric CRP. The antibodies specific for E-CRP-1 and E-CRP-2 can then be employed to detect structurally altered pentameric CRP in vivo and to locate the sites where WT CRP is converted to amyloid-binding forms of CRP.
Our data suggest that the recognition of bacterial amyloids by structurally altered CRP is critical for protection against prolonged infection. In theory then, any amyloid-binding protein administered to mice should give the results similar to that of E-CRP-1 and E-CRP-2. Since SAP is an amyloid-binding protein, SAP should be able to bind to bacterial amyloids and contribute to CRP-mediated protection. It is also possible that the binding of SAP to bacterial amyloids reduces the virulence of pneumococci. It has been shown previously that SAP binds to pneumococci and activates complement (56); however, it has been suggested that the ligands of SAP on pneumococci were surface carbohydrates since SAP also binds to carbohydrates.
While developing the KO mouse model for this study, we noted sex-specificity in the expression of the SAP gene, in the susceptibility of both WT and KO mice to infection and in the SAP levels in the sera from both WT and KO mice. There was no sex-specificity in the expression of the moCRP gene, although it has been reported previously that the expression of the human CRP transgene in mice was sex-specific (28, 57). We found that female mice were less susceptible than male mice to infection; these results are consistent with a previously published report that, in both animals and humans, males are generally more susceptible than females to bacterial infections and that women have stronger immune responses to foreign antigens than men (58). We propose that female mice were less susceptible than male mice to infection because female mice had higher basal SAP levels in their sera than in the sera of male mice. The inverse relationship between the susceptibility of mice to infection and the basal levels of SAP in the sera further suggests that SAP plays a role in protecting against pneumococcal infection.
As described above, the capability of SAP to bind amyloids and the relationship between basal SAP levels and susceptibility to infection both suggested the involvement of SAP in protection against pneumococcal infection. Another evidence to support the role of SAP in pneumococcal infection came from the data on SAP levels seen in KO mice which was higher than in WT mice. The higher expression of the SAP gene in the absence of the CRP gene supports our interpretation that SAP plays a role in protecting against pneumococcal infection. Indeed, employing SAP KO mice, it has been shown previously that mouse SAP participates in protection against pneumococcal infection (56).
We conclude that the protection against prolonged pneumococcal infection involves conformational changes in CRP, binding of CRP to both PCh and amyloids on the pneumococcal surface, and complement activation by PCh-complexed CRP. The two recognition functions of CRP, PCh-binding and amyloid-binding, can be exhibited by two different CRP molecules as exemplified here by the combination of WT CRP and E-CRP-1 or by a single structurally altered CRP molecule if the PCh-binding function is retained in the structurally altered form as exemplified here by E-CRP-2. The combined data reported here and published previously (56) suggest that SAP cooperates with CRP in protection against pneumococcal infection. We propose that, in circulation, CRP cooperates with SAP to reduce bacteremia. At sites of inflammation in the organs, where the conformation of CRP can be altered, WT CRP cooperates with structurally altered CRP to reduce bacterial load in the organs. Future investigations employing double SAP KO and CRP KO mice would provide definitive proof for the cooperation between SAP and CRP in controlling pneumococcal infection.
Data availability statement
All data generated for this study is presented in this article. Further inquiries can be directed to corresponding authors.
Ethics statement
The animal study was approved by University Committee of Animal Care, East Tennessee State University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
AA: Conceptualization, Formal analysis, Funding acquisition, Supervision, Writing – original draft. DN: Investigation, Writing – review & editing. JS: Methodology, Resources, Writing – review & editing. SS: Investigation, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by National Institutes of Health Grant AI151561.
Acknowledgments
We thank Nicole Lewis, Ph.D., for statistical analysis of the data shown in Figure 2E.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
Aβ, Amyloid-β peptide; Amyloids, amyloid-like structures; cfu, colony-forming units; CRP, C-reactive protein; E-CRP, engineered CRP; KO, knockout; moCRP, mouse CRP; MST, median survival time; PCh, phosphocholine; SAP, serum amyloid P component; S. pneumoniae, Streptococcus pneumoniae; WT, wild-type.
References
1. Pathak A and Agrawal A. Evolution of C-reactive protein. Front Immunol. (2019) 10:943. doi: 10.3389/fimmu.2019.00943
2. Torzewski M. C-reactive protein: Friend or foe? Phylogeny from heavy metals to modified lipoproteins and SARS-CoV-2. Front Cardiovasc Med. (2022) 9:797116. doi: 10.3389/fcvm.2022.797116
3. Ji S-R, Zhang S-H, Chang Y, Li H-Y, Wang M-Y, Lv J-M, et al. C-reactive protein: The most familiar stranger. J Immunol. (2023) 210:699–707. doi: 10.4049/jimmunol.2200831
4. Saralahti AK, Harjula SE, Rantapero T, Uusi-Mäkelä MIE, Kaasinen M, Junno M, et al. Characterization of the innate immune response to Streptococcus pneumoniae infection in zebrafish. PloS Genet. (2023) 19:1010586. doi: 10.1371/journal.pgen.1010586
5. Kushner I. The phenomenon of the acute phase response. Ann N Y Acad Sci. (1982) 389:39–48. doi: 10.1111/j.1749-6632.1982.tb22124.x
6. Osmand AP, Friedenson B, Gewurz H, Painter RH, Hofmann T, and Shelton E. Characterization of C-reactive protein and the complement subcomponent C1t as homologous proteins displaying cyclic pentameric symmetry (pentraxins). Proc Natl Acad Sci USA. (1977) 74:739–43. doi: 10.1073/pnas.74.2.739
7. Volanakis JE and Kaplan MH. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc Soc Exp Biol Med. (1971) 136:612–4. doi: 10.3181/00379727-136-35323
8. Shrive AK, Cheetham GMT, Holden D, Myles DAA, Turnell WG, Volanakis JE, et al. Three-dimensional structure of human C-reactive protein. Nat Struct Biol. (1996) 3:346–54. doi: 10.1038/nsb0496-346
9. Agrawal A, Lee S, Carson M, Narayana SVL, Greenhough TJ, and Volanakis JE. Site-directed mutagenesis of the phosphocholine-binding site of human C-reactive protein: Role of Thr76 and Trp67. J Immunol. (1997) 158:345–50. doi: 10.4049/jimmunol.158.1.345
10. Agrawal A, Simpson MJ, Black S, Carey M-P, and Samols D. A C-reactive protein mutant that does not bind to phosphocholine and pneumococcal C-polysaccharide. J Immunol. (2002) 169:3217–22. doi: 10.4049/jimmunol.169.6.3217
11. Kaplan MH and Volanakis JE. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin, and sphingomyelin. J Immunol. (1974) 112:2135–47. doi: 10.4049/jimmunol.112.6.2135
12. Suresh MV, Singh SK, Ferguson DA Jr, and Agrawal A. Role of the property of C-reactive protein to activate the classical pathway of complement in protecting mice from pneumococcal infection. J Immunol. (2006) 176:4369–74. doi: 10.4049/jimmunol.176.7.4369
13. Li H-Y, Tang Z-M, Wang Z, Lv J-M, Liu X-L, Liang Y-L, et al. C-reactive protein protects against acetaminophen-induced liver injury by preventing complement overactivation. Cell Mol Gastroenterol Hepatol. (2022) 13:289–307. doi: 10.1016/j.jcmgh.2021.09.003
14. Ma YJ, Parente R, Zhong H, Sun Y, Garlanda C, and Doni A. Complement-pentraxins synergy: Navigating the immune battlefield and beyond. BioMed Pharmacother. (2023) 169:115878. doi: 10.1016/j.biopha.2023.115878
15. Suresh MV, Singh SK, and Agrawal A. Interaction of calcium-bound C-reactive protein with fibronectin is controlled by pH: In vivo implications. J Biol Chem. (2004) 279:52552–7. doi: 10.1074/jbc.M409054200
16. Hammond DJ Jr, Singh SK, Thompson JA, Beeler BW, Rusiñol AE, Pangburn MK, et al. Identification of acidic pH-dependent ligands of pentameric C-reactive protein. J Biol Chem. (2010) 285:36235–44. doi: 10.1074/jbc.M110.142026
17. Singh SK, Thirumalai A, Pathak A, Ngwa DN, and Agrawal A. Functional transformation of C-reactive protein by hydrogen peroxide. J Biol Chem. (2017) 292:3129–36. doi: 10.1074/jbc.M116.773176
18. Li S-L, Feng J-R, Zhou H-H, Zhang C-M, Lv G-B, Tan Y-B, et al. Acidic pH promotes oxidation-induced dissociation of C-reactive protein. Mol Immunol. (2018) 104:47–53. doi: 10.1016/j.molimm.2018.09.021
19. Singh SK, Hammond DJ Jr, Beeler BW, and Agrawal A. The binding of C-reactive protein, in the presence of phosphoethanolamine, to low-density lipoproteins is due to phosphoethanolamine-generated acidic pH. Clin Chim Acta. (2009) 409:143–4. doi: 10.1016/j.cca.2009.08.013
20. Singh SK, Thirumalai A, Hammond DJ Jr, Pangburn MK, Mishra VK, Johnson DA, et al. Exposing a hidden functional site of C-reactive protein by site-directed mutagenesis. J Biol Chem. (2012) 287:3550–8. doi: 10.1074/jbc.M111.310011
21. Ngwa DN and Agrawal A. Structurally altered, not wild-type, pentameric C-reactive protein inhibits formation of amyloid-β fibrils. J Immunol. (2022) 209:1180–8. doi: 10.4049/jimmunol.2200148
22. Singh SK, Prislovsky A, Ngwa DN, Munkhsaikhan U, Abidi AH, Brand DD, et al. C-reactive protein lowers the serum level of IL-17, but not TNF-α, and decreases the incidence of collagen-induced arthritis in mice. Front Immunol. (2024) 15:1385085. doi: 10.3389/fimmu.2024.1385085
23. Olson ME, Hornick MG, Stefanski A, Albanna HR, Gjoni A, Hall GD, et al. A biofunctional review of C-reactive protein (CRP) as a mediator of inflammatory and immune responses: Differentiating pentameric and modified CRP isoform effects. Front Immunol. (2023) 14:1264383. doi: 10.3389/fimmu.2023.1264383
24. Zeller J, Cheung Tung Shing KS, Nero TL, McFadyen JD, Krippner G, Bogner B, et al. A novel phosphocholine-mimetic inhibits a pro-inflammatory conformational change in C-reactive protein. EMBO Mol Med. (2023) 15:16236. doi: 10.15252/emmm.202216236
25. Agrawal A, Pathak A, Ngwa DN, Thirumalai A, Armstrong PB, and Singh SK. An evolutionarily conserved function of C-reactive protein is to prevent the formation of amyloid fibrils. Front Immunol. (2024) 15:1466865. doi: 10.3389/fimmu.2024.1466865
26. Mold C, Nakayama S, Holzer TJ, Gewurz H, and Du Clos TW. C-reactive protein is protective against Streptococcus pneumoniae infection in mice. J Exp Med. (1981) 154:1703–8. doi: 10.1084/jem.154.5.1703
27. Yother J, Volanakis JE, and Briles DE. Human C-reactive protein is protective against fatal Streptococcus pneumoniae infection in mice. J Immunol. (1982) 128:2374–6. doi: 10.4049/jimmunol.128.5.2374
28. Szalai AJ, Briles DE, and Volanakis JE. Human C-reactive protein is protective against fatal Streptococcus pneumoniae infection in transgenic mice. J Immunol. (1995) 155:2557–63. doi: 10.4049/jimmunol.155.5.2557
29. Gang TB, Hanley GA, and Agrawal A. C-reactive protein protects mice against pneumococcal infection via both phosphocholine-dependent and phosphocholine-independent mechanisms. Infect Immun. (2015) 83:1845–52. doi: 10.1128/IAI.03058-14
30. Ngwa DN and Agrawal A. Structure-function relationships of C-reactive protein in bacterial infection. Front Immunol. (2019) 10:166. doi: 10.3389/fimmu.2019.00166
31. Szalai AJ, Briles DE, and Volanakis JE. Role of complement in C-reactive protein-mediated protection of mice from Streptococcus pneumoniae. Infect Immun. (1996) 64:4850–3. doi: 10.1128/iai.64.11.4850-4853.1996
32. Mold C, Rodic-Polic B, and Du Clos TW. Protection from Streptococcus pneumoniae infection by C-reactive protein and natural antibody requires complement but not Fcγ receptors. J Immunol. (2002) 168:6375–81. doi: 10.4049/jimmunol.168.12.6375
33. Singh SK, Ngwa DN, and Agrawal A. Complement activation by C-reactive protein is critical for protection of mice against pneumococcal infection. Front Immunol. (2020) 11:1812. doi: 10.3389/fimmu.2020.01812
34. Nakayama S, Gewurz H, Holzer T, Du Clos TW, and Mold C. The role of the spleen in the protective effect of C-reactive protein in Streptococcus pneumoniae infection. Clin Exp Immunol. (1983) 54:319–26.
35. Janulczyk R, Iannelli F, Sjoholm AG, Pozzi G, and Bjorck L. Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J Biol Chem. (2000) 275:37257–63. doi: 10.1074/jbc.M004572200
36. Dave S, Brooks-Walter A, Pangburn MK, and McDaniel LS. PspC, a pneumococcal surface protein, binds human factor H. Infect Immun. (2001) 69:3435–7. doi: 10.1128/IAI.69.5.3435-3437.2001
37. Jarva H, Janullczyk R, Hellwage J, Zipfel PF, Björck L, and Meri S. Streptococcus pneumoniae evades complement attack and opsonophagocytosis by expressing the pspC locus encoded Hic protein that binds to short consensus repeats 8–11 of factor H. J Immunol. (2002) 168:1886–94. doi: 10.4049/jimmunol.168.4.1886
38. Dieudonné-Vatran A, Krentz S, Blom AM, Meri S, Henriques-Normark B, Riesbeck K, et al. Clinical isolates of Streptococcus pneumoniae bind the complement inhibitor C4b-binding protein in a PspC allele-dependent fashion. J Immunol. (2009) 182:7865–77. doi: 10.4049/jimmunol.0802376
39. Agarwal V, Hammerschmidt S, Malm S, Bergmann S, Riesbeck K, and Blom AM. Enolase of Streptococcus pneumoniae binds human complement inhibitor C4b-binding protein and contributes to complement evasion. J Immunol. (2012) 189:3575–84. doi: 10.4049/jimmunol.1102934
40. Andre GO, Converso TR, Politano WR, Ferraz LFC, Ribeiro ML, Leite LCC, et al. Role of Streptococcus pneumoniae proteins in evasion of complement-mediated immunity. Front Microbiol. (2017) 8:224. doi: 10.3389/fmicb.2017.00224
41. Du S, Vilhena C, King S, Sahagún-Ruiz A, Hammerschmidt S, Skerka C, et al. Molecular analyses identify new domains and structural differences among Streptococcus pneumoniae immune evasion proteins PspC and Hic. Sci Rep. (2021) 11:1701. doi: 10.1038/s41598-020-79362-3
42. Gil E, Noursadeghi M, and Brown JS. Streptococcus pneumoniae interactions with the complement system. Front Cell Infect Microbiol. (2022) 12:929483. doi: 10.3389/fcimb.2022.929483
43. Ngwa DN, Singh SK, Gang TB, and Agrawal A. Treatment of pneumococcal infection by using engineered human C-reactive protein in a mouse model. Front Immunol. (2020) 11:586669. doi: 10.3389/fimmu.2020.586669
44. Pathak A, Singh SK, Thewke DP, and Agrawal A. Conformationally altered C-reactive protein capable of binding to atherogenic lipoproteins reduces atherosclerosis. Front Immunol. (2020) 11:1780. doi: 10.3389/fimmu.2020.01780
45. Gang TB, Hammond DJ Jr, Singh SK, Ferguson DA Jr, Mishra VK, and Agrawal A. The phosphocholine-binding pocket on C-reactive protein is necessary for initial protection of mice against pneumococcal infection. J Biol Chem. (2012) 287:43116–25. doi: 10.1074/jbc.M112.427310
46. Agrawal A, Xu Y, Ansardi D, Macon KJ, and Volanakis JE. Probing the phosphocholine-binding site of human C-reactive protein by site-directed mutagenesis. J Biol Chem. (1992) 267:25352–8. doi: 10.1016/S0021-9258(19)74047-2
47. Lv J-M, Huang X-P, Chen J-Y, Cheng B, Chen W-Z, Yuan P, et al. Cholesterol-binding sequence is a key regulatory motif of cellular folding and conformational activation for C-reactive protein. Mol Immunol. (2022) 152:123–8. doi: 10.1016/j.molimm.2022.10.010
48. Thirumalai A, Singh SK, Hammond DJ Jr, Gang TB, Ngwa DN, Pathak A, et al. Purification of recombinant C-reactive protein mutants. J Immunol Methods. (2017) 443:26–32. doi: 10.1016/j.jim.2017.01.011
49. Simons JP, Loeffler JM, Al-Shawi R, Ellmerich S, Hutchinson WL, Tennent GA, et al. C-reactive protein is essential for innate resistance to pneumococcal infection. Immunology. (2014) 142:414–20. doi: 10.1111/imm.12266
50. Whitehead AS, Zahedi K, Rits M, Mortensen RF, and Lelias JM. Mouse C-reactive protein: Generation of cDNA clones, structural analysis, and induction of mRNA during inflammation. Biochem J. (1990) 266:283290. doi: 10.1042/bj2660283
51. Pepys MB, Baltz M, Gomer K, Davies AJ, and Doenhoff M. Serum amyloid P-component is an acute-phase reactant in the mouse. Nature. (1979) 278:259–61. doi: 10.1038/278259a0
52. Pepys MB, Dyck RF, de Beer FC, Skinner M, and Cohen AS. Binding of serum amyloid P-component (SAP) by amyloid fibrils. Clin Exp Immunol. (1979) 38:284–93.
53. Janciauskiene S, de Frutos PG, Carlemalm E, Dahlbäck B, and Eriksson S. Inhibition of Alzheimer β-peptide fibril formation by serum amyloid P component. J Biol Chem. (1995) 270:26041–4. doi: 10.1074/jbc.270.44.26041
54. Hamazaki H. Ca2+-dependent binding of human serum amyloid P component to Alzheimer’s β-amyloid peptide. J Biol Chem. (1995) 270:10392–4. doi: 10.1074/jbc.270.18.10392
55. Van Gerven N, van der Verren SE, Reiter DM, and Remaut H. The role of functional amyloids in bacterial virulence. J Mol Biol. (2018) 430:3657–84. doi: 10.1016/j.jmb.2018.07.010
56. Yuste J, Botto M, Bottoms SE, and Brown JS. Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PloS Pathog. (2007) 3:1208–19. doi: 10.1371/journal.ppat.0030120
57. Szalai AJ, van Ginkel FW, Dalrymple SA, Murray R, McGhee JR, and Volanakis JE. Testosterone and IL-6 requirements for human C-reactive protein gene expression in transgenic mice. J Immunol. (1998) 160:5294–9. doi: 10.4049/jimmunol.160.11.5294
Keywords: C-reactive protein, Streptococcus pneumoniae, bacterial amyloids, complement evasion, serum amyloid P component
Citation: Agrawal A, Ngwa DN, Simons JP and Singh SK (2025) Protection against prolonged pneumococcal infection involves structural changes in C-reactive protein and subsequent binding to both phosphocholine and amyloids on the bacterial surface. Front. Immunol. 16:1631409. doi: 10.3389/fimmu.2025.1631409
Received: 19 May 2025; Accepted: 30 June 2025;
Published: 16 July 2025.
Edited by:
Bin Cheng, Lanzhou University of Technology, ChinaReviewed by:
Tayyaba Zainab, Pir Mehr Ali Shah Arid Agriculture University, PakistanChayan Munshi, Ethophilia Research Foundation, India
Hao An, Lanzhou University, China
Copyright © 2025 Agrawal, Ngwa, Simons and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alok Agrawal, YXZkc185OUB5YWhvby5jb20=; Sanjay K. Singh, c2luZ2hzQGV0c3UuZWR1
†Present address: Alok Agrawal, Retired, Johnson City, TN, United States