Abstract
Introduction:
In severe COVID-19, direct-acting antiviral drugs were not effective in hyperinflammatory stages and steroid treatment may weaken host immunity. The MEK inhibitor zapnometinib, as a host-targeting drug, has demonstrated promising efficacy against severe acute viral infections. Proof-of-concept for the innovative approach was presented in a clinical Phase 2 trial with hospitalized COVID-19 patients.
Methods:
The antiviral and immunomodulatory potential of zapnometinib was investigated in samples obtained from COVID-19 patients enrolled in a Phase 2 clinical trial (RESPIRE), as well as in a SARS-CoV-2 Syrian hamster model, an acute lung injury mouse model, and in cell culture. The antiviral activity of zapnometinib was assessed using viral load reduction assays and RT-qPCR. Cytokines and chemokines were analyzed via ELISA and RT-qPCR. Alterations in T and B cells from COVID-19 patients were analyzed using flow cytometry. Biomarker analysis in hamster serum was conducted to monitor potential toxic effects.
Results:
Zapnometinib reduced SARS-CoV-2 viral load in hospitalized COVID-19 patients, in the hamster model and in various highly pathogenic coronaviruses in vitro. Pro-inflammatory cytokines and chemokines decreased in COVID-19 patients, in a lung injury mouse model, and in vitro in primary human blood cells treated with zapnometinib. In the hamster model, zapnometinib alleviated SARS-CoV-2-mediated lung pathology. In patients with COVID-19, zapnometinib increased T and plasma B cells.
Conclusion:
Unlike direct-acting antivirals, zapnometinib’s dual effect highlights its therapeutic potential in the treatment of severe acute viral infections, with favorable antiviral and immunomodulatory properties.
1 Introduction
In late 2019, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which causes coronavirus disease 2019 (COVID-19), emerged in humans. The World Health Organization (WHO) declared COVID-19 a pandemic due to the rapid person-to-person transmission of SARS-CoV-2 and the significant morbidity and mortality it caused worldwide (1–3). The severity of COVID-19 varies widely from asymptomatic infection to severe disease, with the latter being associated with lower respiratory tract symptoms and pathological hyperinflammation (4–6) which may lead to multiorgan dysfunction, including acute respiratory distress syndrome (ARDS), neurological symptoms, cardiovascular events, and coagulopathies (7–9). While the viral load is high at onset and in the early stages of COVID-19, the hyperinflammatory response plays a more important role in determining disease severity in later stages (10). Hyperinflammation is evidenced by significantly elevated levels of certain proinflammatory cytokines and chemokines observed in the majority of COVID-19 patients, particularly in those with severe disease requiring intensive care (11).
Currently available treatments can be categorized into therapeutic antibodies, repurposed direct-acting antivirals (DAA) like nirmatrelvir, remdesivir, or molnupiravir, and immunomodulating drugs, such as dexamethasone or anti-IL-6 antibodies (12–16). They all have their limitations. Therapeutic antibodies have become less effective due to the emergence of new SARS-CoV-2 variants, underscoring the issue of viral escape leading to resistance. Direct-acting antivirals must be administered very early in the onset of infection and have not proven to be highly effective in treating severely ill patients. Various immunomodulatory approaches, some with promising results, were employed for treating lung inflammation in COVID‐19 patients. Examples of these include the intake of probiotics, prebiotics, vitamins, or zinc. However, the appropriate dosage and timing of intake still need to be evaluated, and any potential benefits should clearly outweigh the risks (17–19). Approved immunomodulating drugs such as dexamethasone have demonstrated significant efficacy in advanced stages of infection. Nevertheless, administering them too early could potentially impede the initiation of an antiviral immune response. Thus, a drug that is effective throughout the full course of the disease is still missing. Targeting cellular factors presents a promising avenue for addressing this therapeutic gap. The cellular Raf/MEK/ERK signaling pathway is mainly involved in cell proliferation and regulation of cytokine and chemokine expression (20). Furthermore, the inhibition of this signaling pathway induces a shift in the adaptive immune response towards an effector immune reaction (21–23). Severe cases of COVID-19 are characterized by aberrant adaptive immunity, specifically evidenced by lymphopenia and especially impaired CD8+ T cell functionality (24, 25). Consequently, targeting this signaling pathway may ameliorate hyperactive innate and adaptive immune responses.
MEK (mitogen-activated protein kinase kinase) inhibitors prevent the phosphorylation of ERK (extracellular signal-regulated kinase) in cells where the signaling pathway is activated, such as in cancer cells or cells attacked by certain viruses. In most of our somatic cells, the signaling pathway is not highly activated. Several MEK inhibitors are already in development or approved for the treatment of various cancers (26). We previously demonstrated that targeting the cellular Raf/MEK/ERK signaling pathway leads to a reduction in viral load and protects mice from lethal infection with another health-relevant respiratory virus, the influenza A virus (IAV) (27–29). Additionally, apart from IAV, various RNA-viruses, like Respiratory Syncytial virus (RSV), Zika virus (ZIKV), Dengue virus (DENV), Yellow fever virus (YFV), Borna disease virus (BDV) or Mouse hepatitis virus (MHV), require activation of the Raf/MEK/ERK signaling pathway to ensure their propagation. Inhibition of this pathway disrupts various stages of their viral life cycle (27, 30–33). There are also certain viruses that are not sensitive to MEK inhibition (34, 35). This prompted us to analyze whether inhibition of the pathway would also result in inhibition of SARS-CoV-2 replication. Indeed, we were able to demonstrate that zapnometinib (ATR-002, PD 0184264), an oral MEK inhibitor, not only exhibits antiviral activity against the influenza virus in both in vitro and in vivo settings (28, 29), but also against SARS-CoV-2 in vitro in cell lines as well as in primary air-liquid-interphase epithelial cell cultures, with a safe and selective treatment window (36). Furthermore, the in vitro synergistic effect of zapnometinib was demonstrated in combination with baloxavir-marboxil against IAV (29), and also with various direct-acting SARS-CoV-2 drugs (37). In the life cycle of IAV, the transport of viral ribonucleoprotein complexes relies on active MEK (27). Despite SARS-CoV-2 lacking a nuclear phase in its replication cycle, MEK activation assumes a crucial function in the initial phases of infection (36). It is noteworthy from the aforementioned study that the immunomodulatory impact of zapnometinib operates autonomously from its antiviral properties. It could be shown that MEK inhibition leads to reduced expression of pro-inflammatory cytokines and chemokines without interfering with the interferon-induced antiviral response (38, 39). In the subsequent stages of drug development, it was possible to demonstrate that zapnometinib showed no toxicity and was well tolerated in humans in two phase 1 studies in healthy volunteers (NCT04385420; EudraCT 2021-005225-25). The correlation between pharmacokinetics, pharmacodynamics and antiviral efficacy could be demonstrated in various animal models and in humans, confirming both safety and efficacy and highlighting the drug’s significant therapeutic potential (40). Moreover, an absorption, distribution, metabolism, and excretion (ADME) study in rats showed a favorable distribution and metabolism of the drug (41). In the phase 2 clinical trial of hospitalized COVID-19 patients (RESPIRE; NCT04776044), zapnometinib treatment was found to be safe and well-tolerated. Consistent trends toward better outcomes were observed with zapnometinib, especially among patients with severe disease (42).
Here, our aim was to evaluate the therapeutic potential of zapnometinib against various coronaviruses, demonstrating its dual effect in a phase 2 clinical trial with hospitalized COVID-19 patients as well as through different pre-clinical approaches. Zapnometinib treatment leads to a reduction in viral load and favorable immunomodulatory effects on top of standard of care. These results are supported by preclinical data from a Syrian hamster SARS-CoV-2 infection model, wherein zapnometinib significantly reduces viral load and lung pathology without causing toxic effects. Furthermore, our findings demonstrate that zapnometinib can significantly alleviate hyperinflammation induced by cytokines and chemokines associated with severe COVID-19 in an acute lung injury (ALI) mouse model and in primary human blood cells.
These findings support the potential of zapnometinib as a promising broad-spectrum antiviral therapeutic with a dual effect, particularly in severe respiratory viral infections leading to hyperinflammation such as COVID-19 or influenza.
2 Material and methods
2.1 Ethics
The hamster experiments were carried out in the Central Animal Facilities of Viroclinics-DDL, Cerba Research Company, Schaijk, the Netherlands, in accordance with the standards of the Dutch law for animal experimentation (2010/63/EU). Ethical approval for the present study was obtained from the institutional Animal Welfare Body under the number AVD277002015283-WP24.
The mouse experiments for the ALI mouse model were conducted following review and approval by the Regional Council Tuebingen (35/9185.81-2/IM 1/17) and in compliance with the institutional Principles of Animal Welfare and Animal Research.
Human sputum and nasopharyngeal swabs were taken from samples collected for the RESPIRE trial (NCT04776044) in 18 hospitals from 5 countries that was approved by respective regulatory authorities and by the Ethics Committees concerned (for details, please see the clinical trial protocol provided in (42).
2.2 Animals
Seven- to nine-week-old male Syrian hamsters (Janvier, France) with a body weight ranging from 102 to 134 g at the time of drug administration were utilized for the pharmacokinetic study. The same animals, aged 11 to 13 weeks and a body weight ranging from 113 to 148 g, were included for the efficacy study. The animals were housed in elongated type 2 IVC (individually ventilated cages) group cages with two animals per cage under BSL-II conditions during acclimatization and in elongated type 2 group cages under BSL-III conditions (isolators) during challenge, with sawdust as bedding. Daily checks were conducted for overt signs of disease, standard food was provided, and drinking water was available ad libitum. All biotechnical procedures were performed under 3% isoflurane anesthesia.
Six- to eight-week-old female C57BL/6J mice weighing 20–27 g from the Mouse Facility of the University of Tuebingen (Interfaculty Institute for Cell Biology) were utilized to establish an inflammatory acute lung injury (ALI) model. The animals were maintained in elongated type 2 IVC cages housing four mice each under BSL-II conditions and provided with standard food and drinking water ad libitum.
2.3 Virus and cells
SARS-CoV-2 (BetaCoV/Munich/BavPat1/2020, containing the D614G substitution in the S1 fragment, kindly provided by Dr. C. Drosten, Berlin, Germany) was passaged once in Vero-TMPRSS2 cells and three times in Vero E6 cells and titered in a TCID50 assay as described in the section “Determination of replication-competent virus (TCID50 assay)”. SARS-CoV-2 “Omicron” (B.1.1.592) (kindly provided by Prof. Dr. M. Schindler, Institute of Medical Virology and Epidemiology, University Hospital Tübingen) was passaged on Calu-3 cells and titrated by plaque assay, as previously described in Koch-Heier et al., 2021 (43). Briefly, Vero E6 cells were infected with a 6-fold serial dilution of the virus stock, overlayed with Avicel medium and incubated for 72h at 37°C, 5% CO2. After fixation and staining of the cell layer with crystal violet solution, the virus titer was determined based on the number of plaques counted per well and the respective dilution factor. Vero E6 (Cat# CRL-1586), Vero cells (Cat# CCL-81), MRC-5 (Cat# CCL-171) and Calu-3 cells (Cat# HTB-55) used for in vitro assays were obtained from ATCC (Manassas, Virginia, USA). The virus strains used for in vitro studies at a MOI of 0.001 were the following: SARS-CoV-1: SARS-CoV-HKU-39849; SARS-CoV-2 BavPat1: Human 2019-nCoV Isolate, EVAg Ref-SKU:026V-03883; SARS-CoV-2 “Omicron” B.1.1.592 (described in (44):); HCoV-229E (Cat# VR-740; ATCC). HCoV-OC43 (Cat# VR-1558; ATCC) was used at MOI 0.1.
Human peripheral blood mononuclear cells (PBMCs) were isolated from healthy volunteers registered with the Biobank of the Department of Immunology at the University of Tuebingen, after informed consent documented in writing. Ethical approval was obtained upon review by the Ethics Board of the Medical Faculty of the Eberhard Karls University Tuebingen and the University Hospital Tuebingen (887/2020BO2). Briefly, 50 ml whole blood (roughly 5x107 PBMCs) of two donors were taken in 50 ml syringes (BD Biosciences, Franklin Lakes, New Jersey, USA) supplied with heparin-natrium (100.000 I.E./ml; Braun, Melsungen, Germany). The whole blood was mixed with PBS (Gibco, Carlsbad, California, USA) in a 1:1 ratio in 50 ml tubes (Greiner Bio-One, Frickenhausen, Germany), and PBMCs were subsequently isolated from the whole blood by density gradient centrifugation using Histopaque®-1077 (Sigma-Aldrich, St. Louis, Missouri, USA) as density gradient medium. Five millilitres of plasma per donor were preserved for cell culture medium preparation. The PBMCs were then purified by washing them twice with PBS.
2.4 Antiviral activity assays
The virus yield reduction assay was conducted at Viroclinics Biosciences BV (Rotterdam, the Netherlands) following the company’s standard operating procedures (SOPs). In brief, Vero E6 cells (for SARS-CoV-2) or Vero cells (for SARS-CoV-1) were seeded at a density of 1.5x104 cells in 96-well plates the day before infection and inoculated with 0.001 MOI of the appropriate virus in the absence (virus control wells) and presence of 100, 75, 50, 25, 12.5, 6.25, 3.125, 1.5625, 0.78125, 0.390625 or 0 µM zapnometinib. The 96-well plates were incubated for 24 and 48 h at 37°C. The supernatants were harvested after the incubation, and the viral titer was determined in four replicates by TrueBlue Immunostaining and spot counting using a CTL Immunospot Image Analyzer. The viral titer measured in the presence of each concentration of zapnometinib was utilized to determine the 50% effective concentration (EC50) using a previously described method (45). A cytotoxicity run was conducted concurrently using a lactate dehydrogenase (LDH) release assay as described in supplementary.
For the virus yield reduction assay of SARS-CoV-2 Omicron (B.1.1.592), Calu-3 cells were seeded at a density of 3x105 cells in 24-well plates two days before infection and inoculated with 0.001 MOI of SARS-CoV-2 Omicron (B.1.1.592) for 1 h. Subsequently, the inoculum was completely removed, and the cells were treated with 100, 75, 62.5, 50, 25, 12.5, or 0 µM zapnometinib. Plates were incubated for 24 h at 37°C. The supernatants were harvested after the incubation, and the viral titer was determined using a real time one-step multiplex RT-qPCR method. Briefly, viral nucleic acid extraction was performed using a QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions. A TaqMan-based RT-qPCR assay was carried out using QuantiNova Pathogen kit (QIAGEN, Hilden, Germany) with specific primers targeting the SARS-CoV-2 N gene (Supplementary Table 1). The viral genome copies were determined based on a standard curve prepared with tenfold serial dilutions of the target gene. For QC, nuclease-free water was included in each set of extractions as a negative control to monitor any possible cross-contamination. Moreover, an artificial exogenous QuantiNova RNA internal control (QIAGEN, Hilden, Germany) was included during the nucleic acid purification to monitor the RNA purification efficiency and the RT-PCR amplification. EC50 values were calculated based on the measured viral titers normalized to the solvent control (1% DMSO).
The virus yield reduction assay of HCoV-229E and HCoV-OC43 was performed on MRC-5 cells seeded at 1.5x105 cells per well in 24-well plates 24 h prior infection with HCoV-229E (MOI 0.001) or HCoV-OC43 (MOI 0.1) for 1 h. The inoculum was completely removed, and the cells were treated with different concentrations of zapnometinib (50 µM to 0.006 µM) and a solvent control in Iscove’s Modified Dulbecco’s Medium (IMDM) containing 2% FBS, 1% Penicillin/Streptomycin at 34°C, 5% CO2 for 24 h (HCoV-OC43) or 48 h (HCoV-229E). Supernatants were harvested and the viral titer was determined by a real-time one-step multiplex RT-qPCR using specific primers targeting the N and NS2 genes for HCoV-229E and HCoV-OC43 respectively (Supplementary Table 1) EC50 values were calculated based on the measured viral titers normalized to the solvent control (1% DMSO) and using the “log(inhibitor) vs. response - Variable slope (four parameters)” analysis in GraphPad Prism v9.
2.5 Drug administration in hamsters
Zapnometinib was synthesized by Chemcon GmbH (Freiburg, Germany). Before administration to Syrian hamsters, it was freshly dissolved in dimethyl sulfoxide (DMSO) (Carl Roth GmbH, Karlsruhe, Germany) and then further dissolved in 15% Kolliphor EL (Sigma-Aldrich, Germany) and 80% phosphate-buffered saline (PBS) (Gibco, Life Technology Europe B.V., Bleiswijk, the Netherlands). Kolliphor EL is used to emulsify and solubilize oils and other water in-soluble substances, it has been used as a pharmaceutical solvent for numerous drugs and has proven in previous in vivo studies with zapnometinib. Zapnometinib was administered orally at the indicated doses in a volume of 500 μL/150 g body weight using a gavage needle.
2.6 Pharmacokinetics
Drug administration and blood sampling in Syrian hamsters were performed by Viroclinics Xplore (Schaijk, the Netherlands) and are described in the sections “Drug administration” and “Sampling after inoculation”. Specifically, at different time points (0, 0.5, 1, 2, 4, 8, 12, and 24 h) after the administration of a single dose of zapnometinib (15, 30, or 60 mg/kg per oral (p.o.)), ~200 µL blood was collected retro-orbitally from three animals per time point per dose and alternating animals between time points (except at 24 h, n = 6 per group) under isoflurane anesthesia using a heparinized capillary tube containing a clot activator (Microvette 500 Z-Gel, Sarstedt). Serum was collected by centrifuging the tubes at 4000 x g for 10 min, aliquoted, and stored at < -70°C until pharmacokinetic analysis. Daily clinical observations included monitoring of signs such as ruffled fur, hunched back posture, accelerated breathing and lethargy, and the body weight was monitored before and 24 h post administration.
Bioanalysis and pharmacokinetic evaluation were performed at Prolytic GmbH (Frankfurt, Germany). High-affinity liquid chromatography tandem mass spectrometry (HPLC-MS/MS) was employed to determine the concentration of zapnometinib in hamster serum in accordance with the company’s SOPs. During the analytical phase, two batches were evaluated. The acceptance criteria for the calibration standards and quality control (QC) samples were fulfilled for both batches as per the company’s SOPs. The calibration curve was in the range of 10–10000 ng/mL for zapnometinib and showed acceptable linearity over the calibration range. In total, 81 study test samples were analyzed. The validity of the method during the analysis of the test samples was ensured by assaying QC samples with known concentrations of zapnometinib; zapnometinib 13C6 was used as the internal standard. The lower limit of quantification (LLOQ) of the method was determined to be 10 ng/mL for zapnometinib in undiluted serum. All serum concentrations falling below LLOQ were assigned a value of 0. ANALYST software was used to calculate the concentrations of the test and QC samples based on the corresponding calibration curves. The obtained mean serum concentrations were used for noncompartmental pharmacokinetic evaluation with Phoenix® WinNonLin software (Pharsight Corporation, Mountain View, CA, USA; version 8.1).
2.7 Efficacy study in the Syrian hamster infection model
On day 0, all animals (n=6 per group) were infected intranasally with SARS-CoV-2 (1x103 median tissue culture infectious dose (TCID50) BetaCoV/Munich/BavPat1/2020 (grown and titered at Viroclinics) in a total volume of 0.1 mL diluted in PBS (Gibco, Paisley, UK) and divided between the nostrils. Zapnometinib (100 mg/kg) was administered p.o. (per os; oral administration) at either +4 h or +24 h post-infection (p.i.). Subsequently, all animals in both groups received a daily dose of 75 mg/kg zapnometinib. The animals in the control group received vehicle +4 h p.i. and once daily thereafter. All animals were euthanized by exsanguination under 3% isoflurane anesthesia on day 4 post-infection (dpi). All individuals conducting clinical observations and laboratory analyses requiring data interpretation were blinded before the conclusion of the study.
2.8 Sampling after inoculation
Respiratory tract samples were collected daily during the study from Syrian hamsters infected and treated as described in the in the section “Efficacy study in the Syrian hamster infection model”. Briefly, throat swabs were collected in virus transport medium (Eagles minimal essential medium containing HEPES buffer, Na bicarbonate solution, L-Glutamine, Penicillin, Streptomycin, BSA fraction V and Amphotericin B), and the samples were aliquoted and stored at < -70°C. Serum was also collected as described for pharmacokinetics at 0 dpi and 4 dpi and frozen at < -70°C until further analysis.
Additionally, upon necropsy, lung, nasal turbinate, and trachea samples were collected and stored in 10% formalin for histopathology or frozen at < -70°C for virological analysis. For virological analysis, the tissue samples were weighed, homogenized in infection medium and centrifuged at 3000 xg for 10 min at room temperature before titration.
2.9 Determination of viral load
Detection of replication-competent virus (TCID50 assay): Quadruplicate 10-fold serial dilutions of throat swabs and tissue homogenate samples were used to determine the viral titers in confluent layers of Vero E6 cells. Briefly, serial dilutions of samples were prepared and incubated on Vero E6 monolayers for 1 h at 37°C. Then, the Vero E6 monolayers were washed once with infection medium (DMEM containing L-Glutamine, Penicillin, Streptomycin, Amphotericin B and Fetal Bovine serum) and incubated for 4–6 days at 37°C, after which the plates were scored using WST8 (colorimetric assessment). The optical density (OD) of the wells was read at 450 nm (OD450) using a microplate reader and compared to the OD of the positive control wells (non-treated, infected cells), which showed a cytopathic effect (CPE). The viral titer (TCID50) per mL or g was calculated using the Spearman-Karber method.
Detection of viral RNA: The viral titer in throat swabs and homogenized tissue samples was determined using an RT-qPCR method. Briefly, RNA was isolated with a Roche MagNA Pure 96 instrument using a MagNA Pure 96 DNA and Viral NA Small Volume Kit, and a TaqMan-based RT-qPCR assay was performed with specific primers (Supplementary Table 1), as described by Corman et al. (46). The number of viral copies (CP per mL or g) per sample was calculated by using the resulting Ct value of the sample and the slope, intercept, and upper and lower limits of detection of a log10 dilution series of a viral stock for which the number of copies per dilution was known.
2.10 Gross pathology
At the time of necropsy, gross pathology was performed for each animal. All lung lobes were inspected, and the percentage of affected lung tissue was estimated from the dorsal view. In addition, any other abnormalities observed in other organs during full-body gross pathology were documented. Then, the left lung lobes, trachea, and nasal turbinates were preserved in 10% neutral buffered formalin for histopathology, while the right lung lobes and nasal turbinates were subsequently homogenized for TaqMan-based RT-qPCR and virus titration.
2.11 Histopathology
Histopathological analysis of the left lung, trachea and left nasal turbinate was performed for all animals. After fixation with 10% formalin for at least 2 weeks, tissue sections were embedded in paraffin, micro-sectioned to 3 μm on glass slides and stained with hematoxylin and eosin (H&E) for histological examination. The H&E-stained tissue sections were examined by light microscopy, using an Olympus BX45 light microscope with magnification steps of 40x, 100x, 200x, and 1000x, for histopathology scoring, as well as for the presence of any other lesions. SARS-CoV-2-associated lesions in the lungs were semi-quantitatively assessed as follows: presence of alveolar oedema, alveolar haemorrhage, and type II pneumocyte hyperplasia: 0 = no, and 1 = yes as described previously (47).
2.12 Candidate biomarker analysis
Serum samples from hamsters collected at pre- infection (0 dpi) and post-infection (4 dpi) and treatment time points were analyzed to evaluate the levels of various candidate safety biomarkers. Serum proteins were enzymatically fragmented through proteolysis, and protein-specific peptides were targeted for quantification of the corresponding protein biomarker levels by immunoaffinity liquid chromatography tandem mass spectrometry (IA-LC-MS/MS) performed as described previously (48, 49). For further details, see Supplementary Methods.
2.13 Inflammatory ALI mouse model
Mice were anesthetized by intraperitoneal (i.p.) injection of ketamine solution (10 mg/kg). Both the treatment and control groups (n = 4 each) received 5 mg/kg lipopolysaccharide (LPS) (O55:B5, Sigma-Aldrich) in PBS (100 µL per mouse) via i.p. injection. At 1 h post-stimulation, the treatment group received 25 mg/kg zapnometinib via oral gavage, while the control group received vehicle (15% Kolliphor, 5% DMSO in PBS). The mice were euthanized at 6 h post-zapnometinib treatment, and the lungs were preserved in RNAlater (Qiagen, Hilden, Germany).
Lungs were homogenized by a bead beating system (FastPrep™, MP Biomedicals) and the RNA was isolated using RNeasy Plus Universal Midi Kit (Qiagen) according to the manufacturer’s instructions. RNA quality (A260/A280 ratio ≥1.8 and A260/A230 in the range of 2.0-2.2) was determined using a NanoDrop spectrophotometer (Thermo Fisher Scientific), and the isolated RNA was reverse transcribed using an RT2 First Strand Kit (Qiagen). Afterward, the generated cDNA was subjected to a real-time RT² Profiler PCR Array using RT² SYBR® Green qPCR Mastermix. For optimal performance, the - RT² Profiler™ Mouse Cytokines & Chemokines arrays (PAMM-150ZR, Qiagen) were customized to include 84 genes and 5 housekeeping genes for data normalization (see Supplementary Table 4). For QC, a mouse genomic DNA contamination test, 3 reverse transcription efficiency tests, and 3 PCR array reproducibility tests were included. The data were analyzed using the GeneGlobe portal (Qiagen) according to the ΔΔCt method. The fold change/regulation was calculated, and the threshold value was 2.
2.14 Analysis of cytokine and chemokine levels in human PBMCs
2x106 human PBMCs from two donors each were suspended in 1 mL RPMI 1640 medium (Gibco) containing 5% autologous plasma, 1% penicillin/streptomycin (Sigma-Aldrich), and 1% L-glutamine (Sigma-Aldrich) and were treated with 1 μg/mL LPS from E. coli O55:B5 (prepared in PBS) and 10 μg/mL zapnometinib (prepared in DMSO and diluted in cell culture medium to a final concentration of 0.1% DMSO) for 6 h at 37°C and 5% CO2. The supernatants (cell culture medium) were collected and stored at -20°C until analysis by ELISA. Ready-to-use ELISA kits (Invitrogen, Waltham, Massachusetts, USA) were used according to the manufacturer’s instructions to determine the levels of the following cytokines: IL-1β, IL-6, IL-8, IP-10, MCP-1, MIP-1α, and TNF-α. Briefly, the samples and standards were applied to ELISA plates pre-coated with specific antibodies targeting the cytokines of interest. Biotinylated detection antibodies binding to the cytokines of interest were added to the wells followed by a horseradish peroxidase (HRP)-streptavidin conjugate, which binds to the biotinylated detection antibodies. Unbound cytokines, detection antibodies, and HRP-streptavidin conjugate were removed by washing. Tetramethylbenzidine (TMB) substrate was added, and the color change in the wells served as the readout for the ELISA. Optical density (OD) at 450 nm was measured using a SpectraMAX Plus 384 microplate reader. For IL-1β, IL-8, MCP-1, and TNF-α the OD was also read at a reference wavelength of 620 nm, and for IL-6 at a reference wavelength of 550 nm. For IP-10 and MIP-1α no reference wavelength was used. Cytokine and chemokine concentrations were calculated following the manufacturer’s instructions for interpolating unknown concentrations from a standard curve.
2.15 Virology and immunology from the RESPIRE trial
RESPIRE was a global randomized, double-blind, placebo-controlled proof-of-concept Phase 2 trial in adults with moderate-to-severe COVID-19 requiring hospitalization (clinical severity status (CSS) 3 or 4, measured on a 7-point ordinal scale). The study details are described in Rohde et al., 2023 (42). Briefly, patients were randomized 1:1 to oral zapnometinib (900 mg on Day 1 (D1); 600 mg daily on Day 2-6) or matching placebo, on top of standard of care according to local guidelines. Patients, investigators, the study team, and the Sponsor remained blinded to treatment allocation throughout the study.
Viral load: Nasopharyngeal swabs and sputum were collected at time points D1, D3, D5, D8, D11, D15 and D30 after treatment start and the viral load was quantified via RT-qPCR at CheckImmune GmbH, Berlin.
Cytokine/chemokine: Parameters from 24 (12 zapnometinib, 12 placebo) study participants at time points D1, D3, D5, D8, D11, D15 and D30 were analyzed using MesoScale V-Plex Proinflammatory Panel I and Chemokine Panel and whole Blood IL-8 (MSD, Rockville, USA). For standard of care treatment see Supplementary Table 5.
Adaptive immune cell parameters from 12 (6 zapnometinib, 6 placebo) study participants at time points D1, D3, D5, D8, D11, D15 and D30 were analyzed using the following panels: activated/exhausted T cells (Duraclone; Beckman Coulter, Krefeld Germany), activated T cells/Tregs (Duraclone; Beckman Coulter, Krefeld Germany), B cell panel (Duraclone; Beckman Coulter, Krefeld Germany). Patients receiving glucocorticoids as standard of care treatment were excluded from the analyses. Experiments were performed by CheckImmune GmbH according to the companies’ SOPs. In short, presence of lymphocytes, T cells and B cells were determined using Flow cytometry analysis. Fresh whole blood from hospitalized SARS-CoV-2 infected patients was stained with the aforementioned DuraClone antibody panels for 15 min at room-temperature and lysed for 15 min at room temperature following the standard staining protocol. Sample acquisition was carried out using the Navios Flow Cytometer (Beckman Coulter). Data analysis was conducted using Kaluza software before being sent to the LIMS system. This procedure is fit for purpose validated under ISO and GCP guidelines.
2.16 Statistical analysis
Unless otherwise noted, data were collected in MS Excel, and statistical analyses were performed using GraphPad Prism software v8 or v9 (La Jolla, CA, USA). Statistical details for each experiment are described in the corresponding figure legends. Differences in viral titer, lung injury severity or body weight were analyzed by one-way ANOVA. Differences in the mRNA expression of cytokines were assessed by analyzing replicate data using one-way ANOVA with the Kruskal-Wallis test or using Student’s t-test (unpaired t-test with Welch’s correction). Differences in cytokine expression at the protein level were analyzed using an unpaired t-test with Welch’s correction for normally distributed data; otherwise, the Mann-Whitney test was applied. Differences in biomarkers were evaluated using Brown-Forsythe ANOVA with the Dunnett T3 correction. P values < 0.05 were considered statistically significant.
3 Results
3.1 Zapnometinib inhibits propagation of coronaviruses in vitro
We started out with an in vitro analysis of the antiviral efficacy of zapnometinib against the SARS-CoV-2 variant “omicron” and other pathogenic human coronaviruses (HCoV OC43, 229E and SARS-CoV-1) using a Virospot or virus yield reduction assay. The viral titer of all tested coronaviruses could be reduced (Figures 1A–D). The half-maximal effective concentration (EC50) values were in a comparable range, with minor variations attributed to virus-specific assay adjustments, from 16.1 µM (HCoV-OC43) to 37.9 µM (SARS-CoV-2 omicron) for all the viruses tested (Table 1). No cytotoxicity was observed in the in vitro antiviral assays, as measured by LDH release (Supplementary Table 2) and calculated by assessing the 50% cytotoxic concentration (CC50) (Supplementary Figure 1; Table 1). These results confirm and extend the data from our recent findings, demonstrating the antiviral efficacy of zapnometinib against the alpha, beta and delta variants of SARS-CoV-2 in cell culture without inducing cytotoxic effects (36). In summary, the in vitro antiviral efficacy of zapnometinib against various coronaviruses has been demonstrated, affirming its broad antiviral therapeutic potential. Moreover, as a host-cell targeting drug, zapnometinib is likely to remain effective against upcoming novel variants or newly emerging zoonotic strains, which is a critical characteristic particularly considering new variants of SARS-CoV-2.
Figure 1
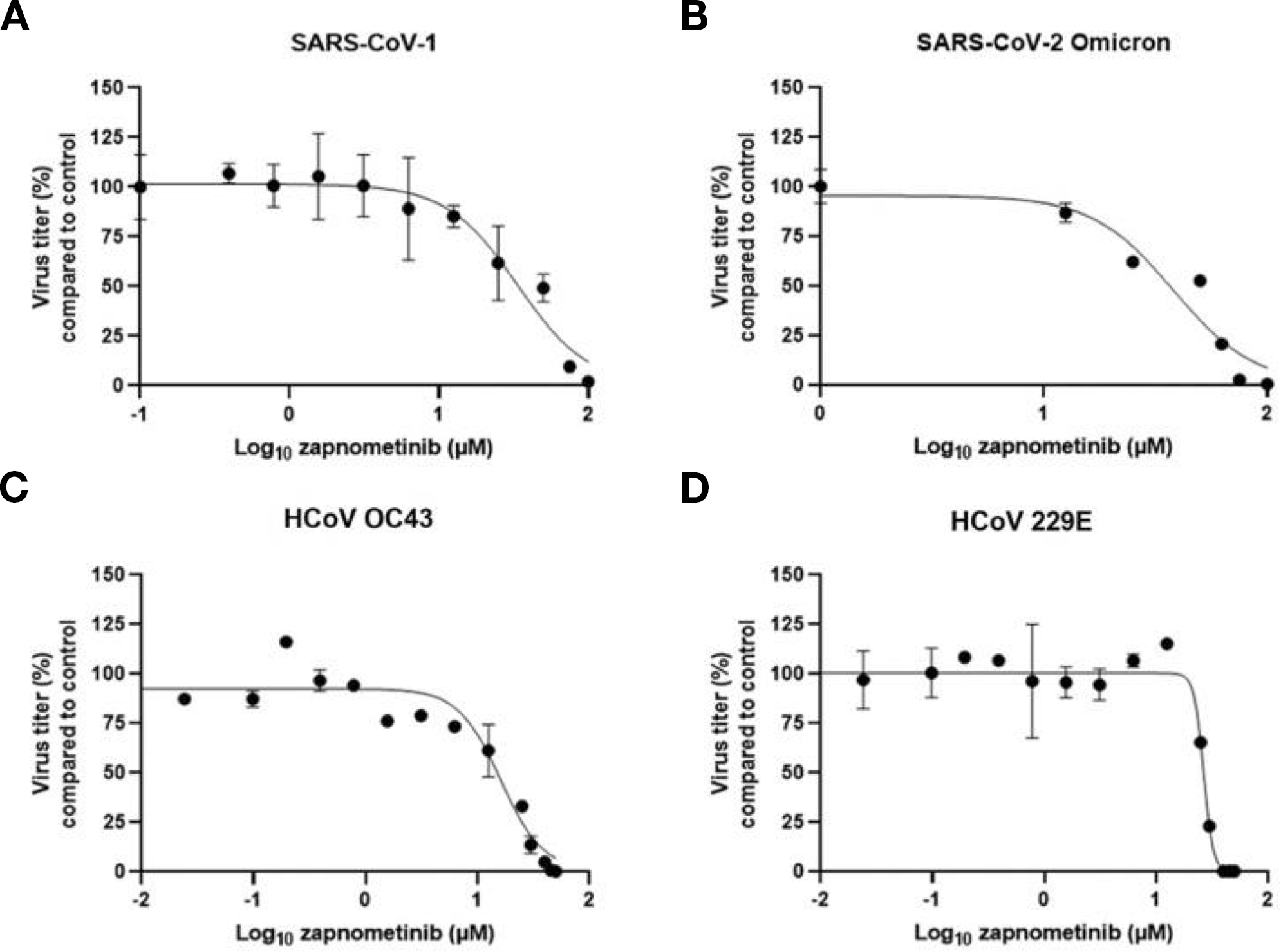
Zapnometinib inhibits various coronaviruses. The antiviral activity of zapnometinib against different Coronaviruses was assessed using a virus yield reduction assay (B) SARS-CoV-2 Omicron, (C) HCoV OC43 and (D) 229E, or virospot reduction assay (A) SARS-CoV-1 and the half-maximal effective concentration (EC50) was calculated by plotting the viral titer (compared to control) vs. the zapnometinib concentration. The data is shown as mean with standard deviation. n(SARS-CoV-1) = 4; n(SARS-CoV-2 Omicron) = 3; n(HCoV OC43) ≥ 3 (n(control) = 9; n(12.5, 25, 50 µM) = 6; n(0.006, 0.024, 0.098, 0.195, 0.391, 0.781, 1.563, 3.125, 6.25, 30, 40, 45 µM) = 3); n(HCoV 229E) = 3 or 9 (n = 9 for the control). The EC50 values are shown in Table 1.
Table 1
| virus strain | EC50 (µM)/(µg/mL) | 95% CI (µM) |
|---|---|---|
| SARS-CoV-1 | 33.6/13.8 | 25.5 - 43.1 |
| SARS-CoV-2 Omicron B1.1.592 | 37.9/15.5 | 27.9 - 57.9 |
| HCoV OC43 | 16.1/6.6 | 13.6 - 18.6 |
| HCoV 229E | 26.6/10.9 | 25.5 - 27.7 |
| hrs after infection | CC50 (µM)/(µg/mL) | 95% CI (µM) |
| 24 h | 532.1/217.9 | 422.7 - n/a |
| 48 h | 471.0/192.9 | 439.5 - 496.6 |
In vitro data. EC50 values of zapnometinib for various viruses calculated with a Virospot or virus yield reduction assay; CC50 values determined on Calu-3 cells with an LDH cytotoxicity assay after 24 or 48 h zapnometinib treatment.
EC50, half maximal effective concentration; CI, confidence interval; SARS-CoV, severe acute respiratory syndrome coronavirus; HCoV, human coronavirus; CC50, 50% toxic concentration
3.2 Pharmacokinetics of zapnometinib in Syrian hamsters
Next to the in vitro efficacy of a drug, we aimed to confirm the therapeutic potential and efficacy in in vivo studies. Prior to the in vivo efficacy study, we investigated the pharmacokinetics of zapnometinib in Syrian hamsters, a favorable animal model for SARS-CoV-2 infections, to better understand the metabolism of zapnometinib in this animal and to determine an appropriate dose for the subsequent efficacy study. In addition, the tolerability of zapnometinib was assessed by monitoring clinical parameters such as ruffled fur, hunched back posture or lethargy. To investigate dose proportionality, clearance, and safety of zapnometinib in hamsters, single doses of 15, 30 or 60 mg/kg zapnometinib were administered p.o. to groups of 6 animals. Blood samples were collected for analysis after 30 min and after 1, 2, 4, 8, 12, and 24 h from 3 alternating animals per group. The serum concentration-time curves for all doses showed a monophasic progression following oral administration. A proportional increase was observed until the maximum observed plasma concentration (Cmax) which was reached after 2 h for the 15 and 30 mg/kg doses and after 4 h for the 60 mg/kg dose (Figure 2). Moreover, the area under the concentration-time curve (AUC) and the Cmax values increased proportionally with the dose. The apparent terminal half-life was comparable for all dose groups, ranging from of 2 to 3 h (Table 2). Notably, at the highest dose (60 mg/kg), the concentration of zapnometinib after 24 h was 5.32 µg/mL (13 µM) (Table 2; Figure 2). Based on previous work in our laboratory, 10 µg/mL (24.4 µM) is required for 50% MEK inhibition (40). Thus, higher doses are required to attain a serum concentration exceeding 10 µg/mL at 24 h, ensuring effective and consistent MEK inhibition. Additionally, all doses were well tolerated, and no adverse events were reported following zapnometinib treatment. Therefore, based on the current data, a loading dose of 100 mg/kg followed by a daily dose of 75 mg/kg was employed for the efficacy study in the Syrian hamster infection model. This regimen was equivalent to the human dosing used in the phase 2 clinical trial, which involved a higher initial loading dose of 900 mg on day 1, followed by daily dosing with 600 mg zapnometinib. This dosing regimen ensures that the drug concentration remains above 10 µg/mL (24.4 µM) throughout the entire treatment period (40, 42).
Figure 2
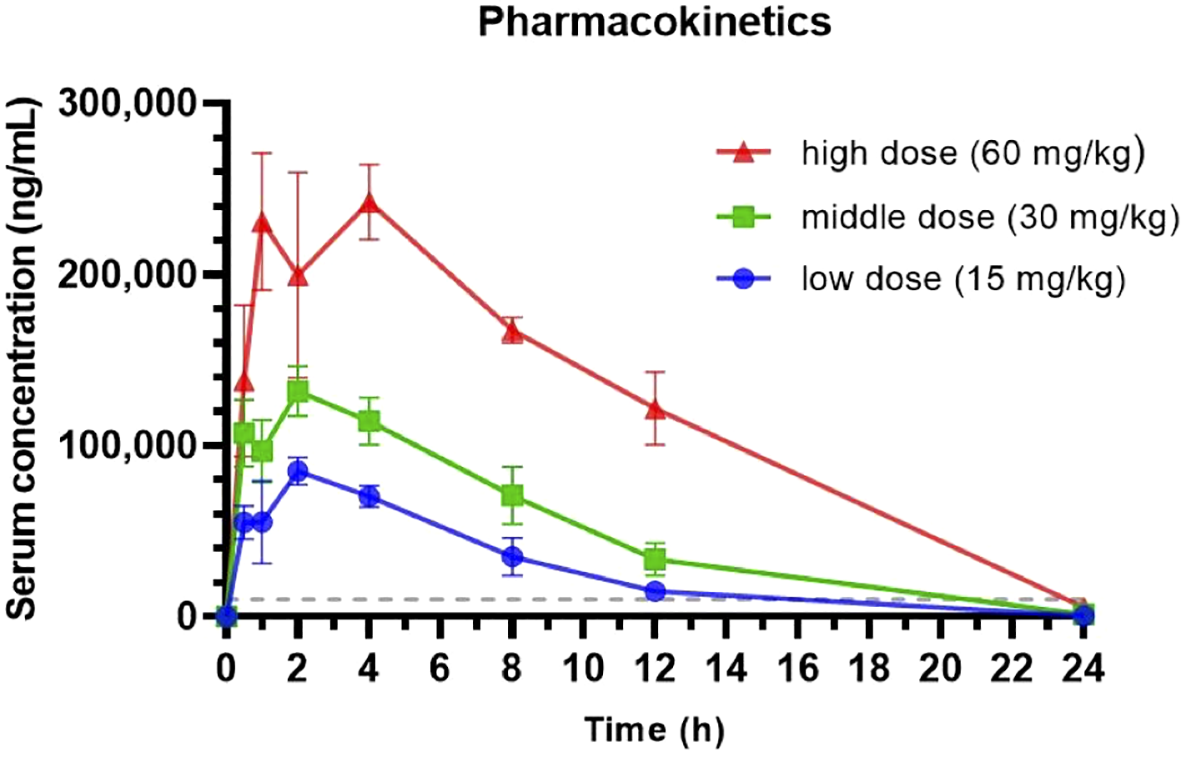
Pharmacokinetic analysis of zapnometinib in Syrian hamsters. Three different doses (15, 30 and 60 mg/kg) of zapnometinib were administered orally to Syrian hamsters (n = 6 per group), and serum was collected 0.5, 1, 2, 4, 8, 12 and 24 h later (alternately from 3 animals per group). The zapnometinib concentration in the serum was determined by HPLC-MS/MS. The data are presented as mean serum concentration ± SD vs. time profiles. The dashed line shows the critical plasma level for MEK inhibition (approx. 10 µg/mL = 24.4 µM).
Table 2
| Group | Dose (mg/kg) | Cmax (ng/mL)/(µM) | Tmax (h) | AUC (h*ng/mL) | T1/2 (h) | C(24 h) (ng/µL)/(µM) |
|---|---|---|---|---|---|---|
| 1 (n = 6) | 60 | 242434/99.2 | 4 | 2970884 | 3.07 | 5319.1/2.18 |
| 2 (n = 6) | 30 | 131720/53.9 | 2 | 1231900 | 2.75 | 1347.23/0.55 |
| 3 (n = 6) | 15 | 85109/34.8 | 2 | 672365 | 2.74 | 636.91/0.26 |
Summary of pharmacokinetic parameters per dose group for zapnometinib in Syrian hamsters.
Cmax, maximum observed plasma concentration; Tmax, time to maximum observed plasma concentration; AUC, area under the concentration-time curve; T1/2, terminal half-life; C(24 h), mean concentration at 24 h
3.2 Efficacy of zapnometinib in the Syrian hamster infection model
An in vivo study was designed to investigate the efficacy of zapnometinib administered p.o. in SARS-CoV-2-infected hamsters. In this efficacy study, all animals were challenged with 1x103 TCID50 SARS-CoV-2 via intranasal administration, which induces both high levels of viral replication in the respiratory tract and significant histopathological changes in the lungs (50). Control animals were treated with vehicle (5% DMSO, 15% Kolliphor in PBS) beginning at +4 h p.i., while the experimental animals were treated with the following regimen: one loading dose of 100 mg/kg zapnometinib (+4 or +24 h p.i.) followed by 75 mg/kg zapnometinib once per day until the day of sacrifice (4 dpi). During the challenge phase, the animals were weighed, clinical observations were monitored following treatment and infection, and throat swabs were taken daily (Figure 3A). At 4 dpi, all animals were euthanized for virological and pathological analyses.
Figure 3
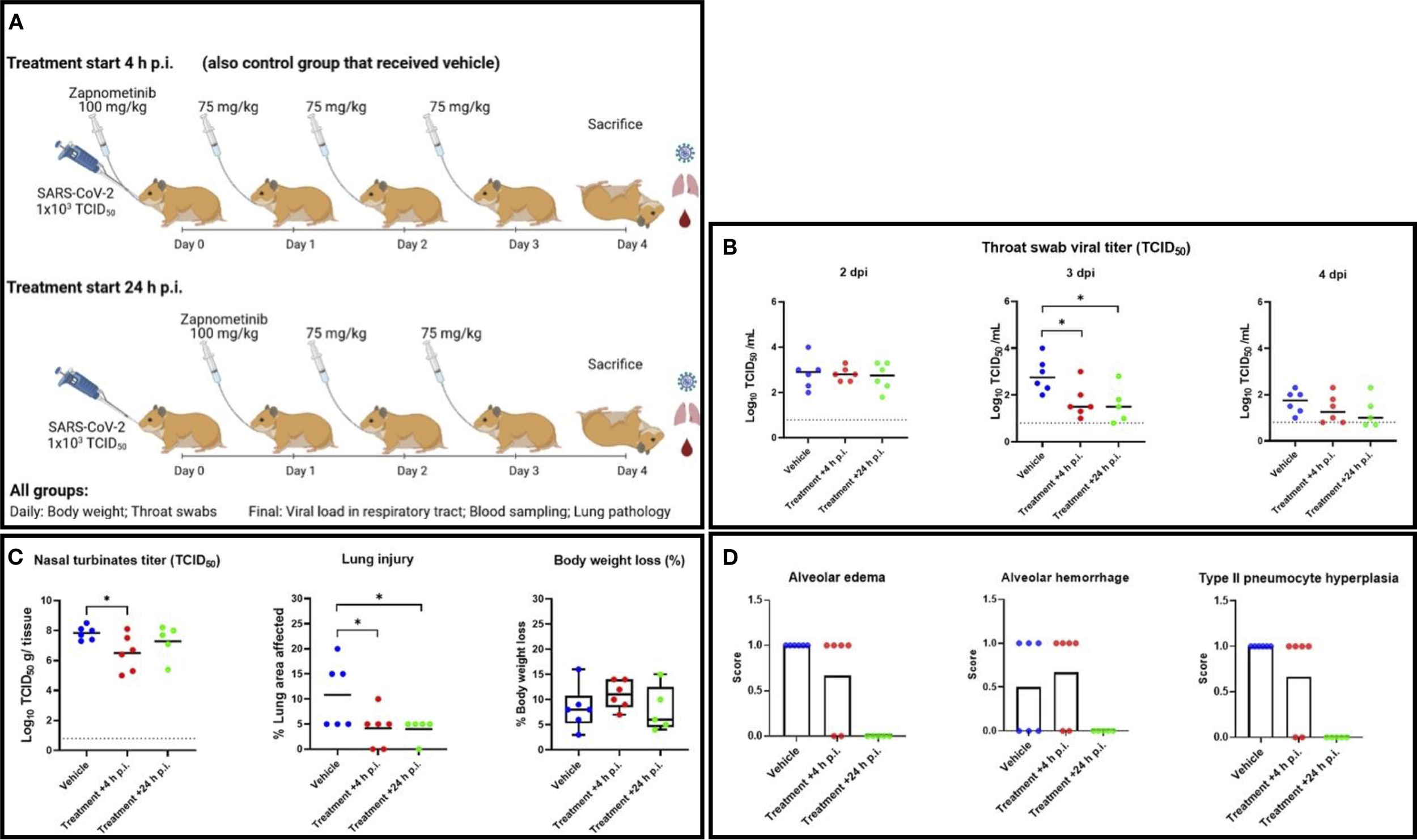
Study of the efficacy of zapnometinib against SARS-CoV-2 in a Syrian hamster infection model. (A) Study schedule. On day 0, all animals were infected intranasally with 1x103 TCID50 SARS-CoV-2. Zapnometinib (100 mg/kg) was given beginning +4 h p.i. (+ control group that received vehicle) or +24 h p.i. Thereafter, both groups were treated with 75 mg/kg zapnometinib once daily and body weight was measured, and throat swabs were taken daily until sacrifice at 4 dpi. Nasal turbinate tissues were subsequently homogenized and subjected to virus titration. Blood was collected for further analysis, the lungs were inspected and collected for histopathological examination. The study schedule was created on biorender.com. (B) Infectious viral titers in throat swabs collected at 2 (left), 3 (middle) and 4 (right) dpi expressed as log10 TCID50 of control (vehicle-treated) and zapnometinib-treated SARS-CoV-2-infected hamsters. (C) left: Infectious viral loads in nasal turbinates collected from control (vehicle-treated) and zapnometinib-treated SARS-CoV-2-infected hamsters at 4 dpi expressed as log10 TCID50 per g nasal turbinate tissue analysed via TCID50 assay. The dashed line represents the lower limit of detection (0.8 log10 TCID50/mL). (C) middle: Percentage of lung area with pathological signs in control (vehicle-treated) and zapnometinib-treated SARS-CoV-2-infected hamsters at 4 dpi. (C) right: Weight loss at 4 dpi as a percentage of the body weight measured at the time of infection. Individual data (dots) and median values (horizontal lines) are presented. The whiskers represent the min. and max. values. (D) Lung slides were examined and scored for the presence of alveolar edema (left), alveolar haemorrhage (middle) and type II pneumocyte hyperplasia (right). Mean values are presented as percent distribution. All data are from a single experiment. The number of animals per group was as follows: n(Vehicle) = 6; n(Treatment +4 h p.i.) = 6; n(Treatment +24 h p.i.) = 5. The data were analysed by one-way ANOVA. *p < 0.05.
For the analysis of viral titers, throat swabs were titered to detect replication-competent virus. Additionally, they were analyzed using TaqMan-based RT-qPCR to detect viral RNA and determine the viral copy number. At 2 dpi, the viral load in the throat swabs was comparable among all three treatment groups (Figure 3B). At 3 dpi, the TCID50 assay indicated that zapnometinib treatment in both treatment groups resulted in a significant reduction in the amount of replication-competent virus in the throat swabs, with reductions of 1.1 log10 (p = 0.0134) and 1.3 log10 (p = 0.0191) noted in the groups treated at +4 h p.i. and +24 h p.i., respectively. These results were supported by the RT-qPCR data, which demonstrated viral titer reductions (number of viral copies) of 1.1 log10 (p = 0.0089) and 1.2-log10 (p = 0.0090) in animals treated at +4 h p.i. and +24 h p.i., respectively, compared to control animals (Supplementary Figure 2A). At 4 dpi, the mean viral titer was still lower in the throat swabs of drug-treated animals, although no statistically significant differences were discernible (Figure 3B). Nevertheless, in the drug-treated animals, a clear decrease in titer over time (2 dpi, 3 dpi, 4 dpi) was observed, more pronounced than in the control animals.
A similar pattern was observed for the infectious viral load in the nasal turbinates at 4 dpi. Treatment starting at +4 h p.i. significantly reduced the infectious viral titer by 1.3 log10 (p = 0.0335) (Figure 3C). These data were further confirmed by RT-qPCR, which showed a reduction of 1.1 log10 (p = 0.0145) when treatment was started at +4 h p.i. (Supplementary Figure 2B). Similar results were obtained when treatment was started at +24 h p.i. showing a trend toward lower mean viral titers in the nasal turbinates at 4 dpi, although the differences did not reach statistical significance (Figure 3C; Supplementary Figure 2B).
To gain further insight into how treatment affects disease progression beside its influence on viral load, the lung lesions of the infected animals were evaluated by gross pathology at 4 dpi, and the percentage of affected lung area was estimated. The percentage of lung injury in lung tissue was significantly reduced in both treated groups (p = 0.0282 for treatment beginning at +4 h p.i., p = 0.0314 for treatment beginning at +24 h p.i.) compared to the vehicle-treated group, indicating a beneficial effect of zapnometinib treatment (Figure 3C). Histopathological evaluation of the lungs revealed that pulmonary changes were mostly mild in all treatment groups. However, inflammation, as indicated by the number of inflammatory cells infiltrating the tissue, consolidation of the tissue, and decreased air containing space in the tissue, was less severe in the zapnometinib-treated groups than in the vehicle-treated control group (Figure 4). Hamsters treated with the vehicle showed alveolitis characterized by numerous inflammatory cells, including macrophages and neutrophils in the alveolar septa and lumina, haemorrhage and oedema in the alveolar lumina and type II pneumocyte hyperplasia. In contrast, hamsters treated with zapnometinib exhibited reduced alveolitis with less cellular inflammatory infiltrate containing fewer macrophages and neutrophils and no haemorrhage, oedema, or type II pneumocyte hyperplasia (Figure 3C). Comparing the lungs of all treatment groups revealed that alveolar edema, seen as intraluminal proteinaceous fluid, was less frequent in animals treated beginning at +4 h p.i. than in control animals and absent in animals treated beginning at +24 h p.i. Additionally, intra-alveolar haemorrhage, suggestive of epithelial damage and observed as extravasated erythrocytes, was comparable between animals treated beginning at +4 h p.i. and vehicle-treated animals, whereas it was absent in animals treated beginning at +24 h p.i. (Figure 3C). The alveolar epithelium showed variable scoring of hyperplasia among the different treatment groups (type II hyperplasia), which is related to regeneration and is seen as plump cuboidal epithelium in the place of the usual flatter alveolar epithelium. Animals treated beginning at +4 h p.i. were less affected, and no hyperplasia was observed in animals treated beginning at +24 h p.i. (Figure 3C).
Figure 4
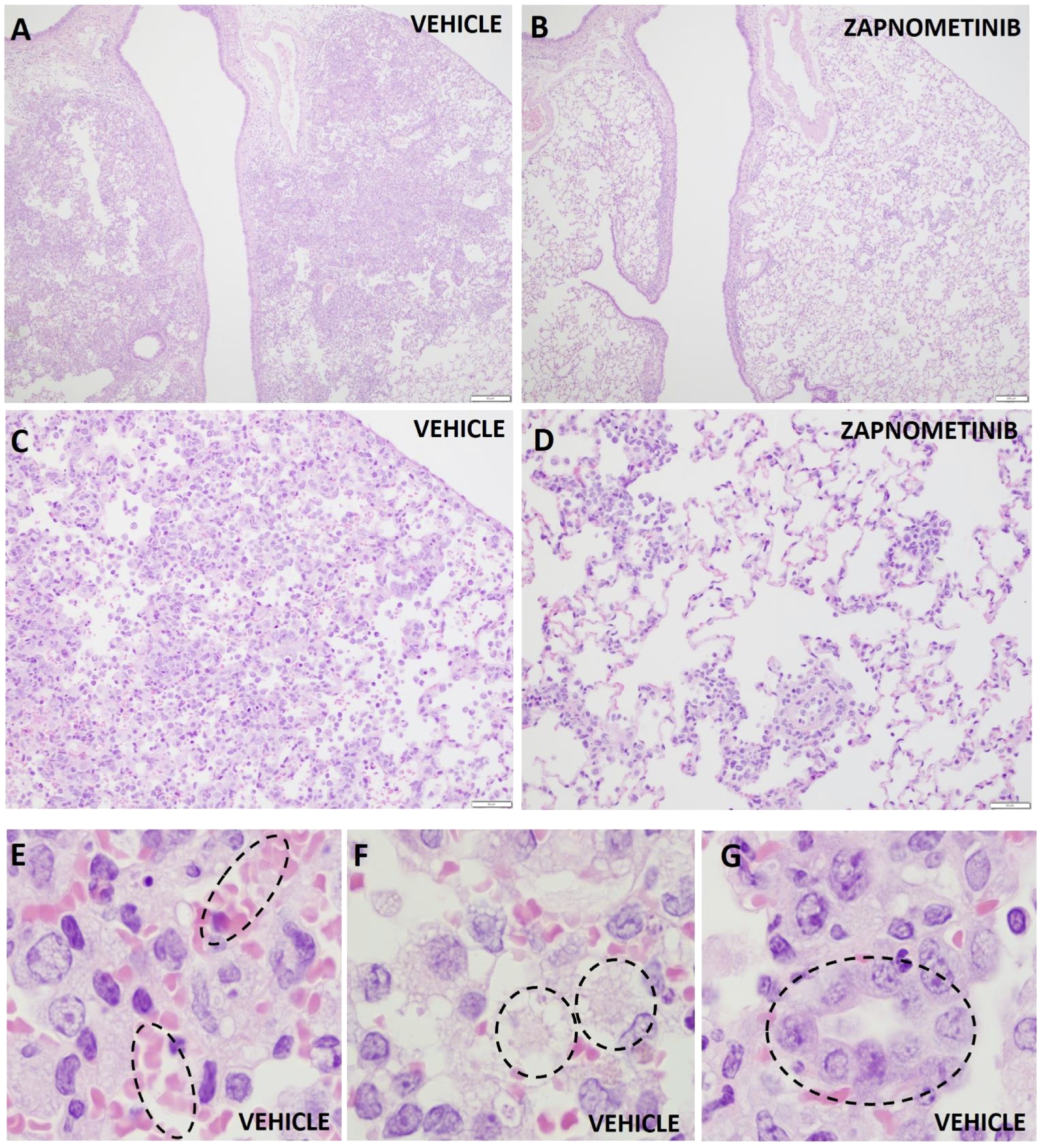
Histopathologic changes in the lungs of hamsters infected with SARS-CoV-2. Representative histopathologic changes at 4 dpi in the lungs of hamsters infected with SARS-CoV-2 and treated with zapnometinib beginning at +24 h p.i. (B, D) or treated with the vehicle only (A, C, E, F, G). Haemorrhage (E), edema (F) and type II pneumocyte hyperplasia (G) were observed in the vehicle only animals (shown in dashed circles). Hematoxylin and eosin staining; 40x magnification (A, B), 200x magnification (C, D) and 1000x (E–G).
Significant disparities in outcomes between the two treatment regimens were not observed; however, initiating treatment earlier (+4 h p.i.) seemed to enhance the antiviral efficacy of zapnometinib in the nasal turbinates (Figure 3C). Conversely, commencing treatment later (+24 h p.i.) exhibited slightly superior effects on certain markers of inflammation in the lungs.
In general, treatment was well tolerated in both groups, as no significant differences in weight were observed among all groups; mean body weight loss was higher than 5% and comparable among all groups (ranging from 8-11%) (Figure 3C). In addition, no treatment-related adverse effects were observed in the zapnometinib-treated groups compared to the vehicle-treated group. Taken together, these results demonstrate that zapnometinib treatment is well tolerated in the Syrian hamster model and can exert beneficial effects depending on the timing of administration—significantly reducing viral load in the respiratory tract when initiated early (+4 h p.i.) or alleviating lung pathology when initiated later (+24 h p.i.).
3.3 Levels of drug safety biomarkers
To further monitor possible drug-related adverse effects in the hamster study, serum samples collected at baseline (0 dpi) and at 4 dpi following infection and treatment with zapnometinib or vehicle. These samples were analyzed for candidate protein biomarkers indicative of organ injury. Various candidate biomarkers were analyzed to evaluate drug-induced vascular injury (DIVI) (51) and drug-induced liver injury (DILI) (51, 52). The levels of high mobility group box 1 (HMGB1; an indicator of necrosis) (53, 54) and glutamate dehydrogenase (GLDH; an indicator of liver damage) (55) were measured to assess liver injury (DILI), and p-selectin (SELP) was used as an early indicator of endothelial damage to blood vessels (DIVI). There were no prominent changes in the levels of the examined biomarkers before and after infection and treatment of the animals, and the levels always remained comparable among all groups (Figure 5). When comparing the values from day 0 to day 4, it can also be observed that there was no significant increase in the biomarkers due to the infection. Collectively, these results provide further evidence that the selected treatment regimen (100 mg/kg zapnometinib followed by 75 mg/kg beginning at +4 or +24 h p.i.) was not toxic to hamsters.
Figure 5
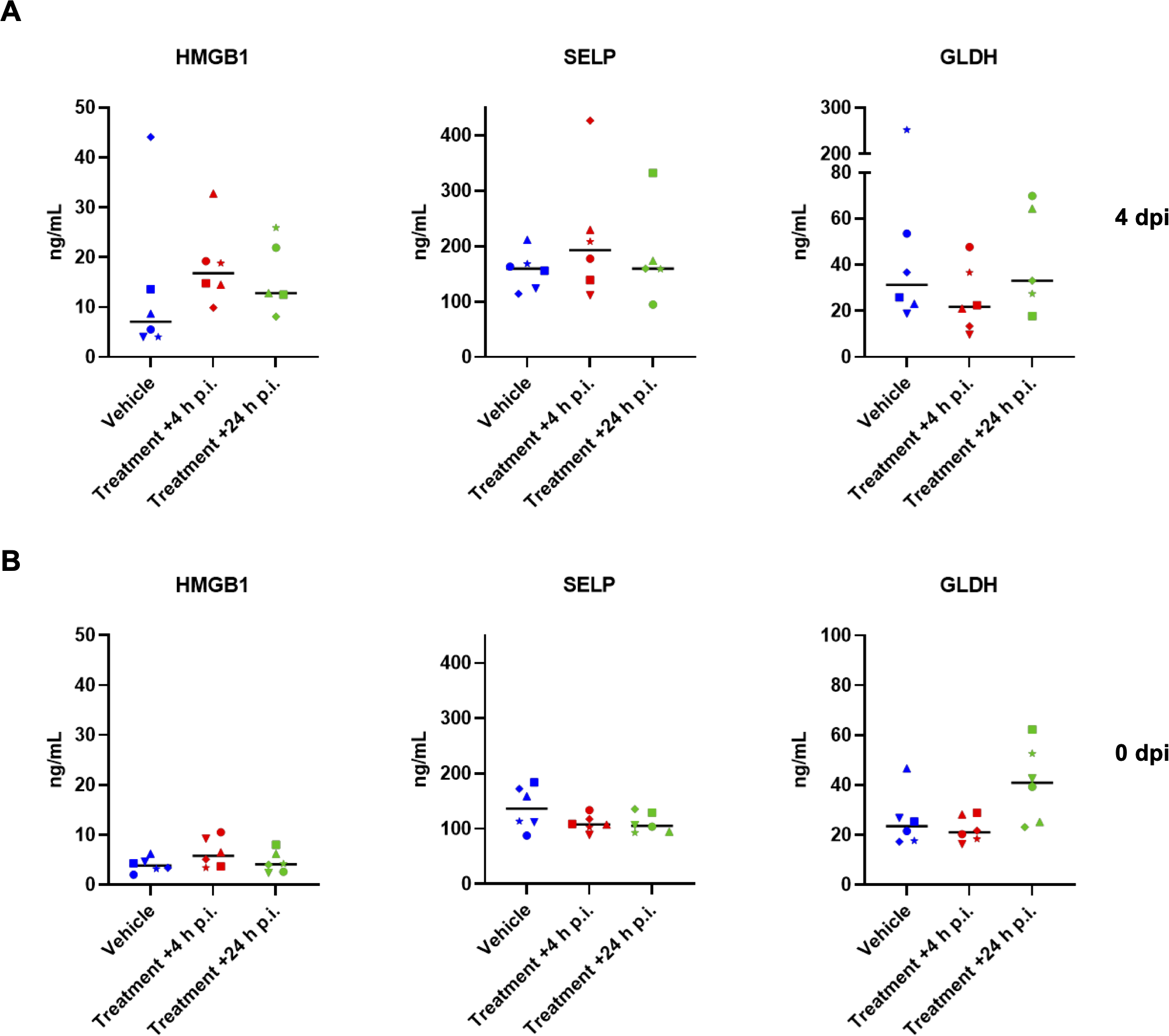
Candidate safety biomarker analysis. The levels of candidate biomarkers in serum samples from hamsters collected (A), at 4 dpi after infection and zapnometinib treatment and (B), before infection with SARS-CoV-2 (0 dpi, prior to infection and zapnometinib treatment) and were analysed by IA-LC-MS/MS. High mobility group box 1 protein (HMGB1, an indicator of necrosis), glutamate dehydrogenase (GLDH, an indicator of liver damage), and p-selectin (SELP, an indicator of endothelial damage) levels in the zapnometinib-treated and vehicle-treated groups are shown. The treatment conditions described below the graphs refer to the treatment that the animals received after infection. Individual data (different symbols) and median values are presented. The number of animals per group was as follows: n(Vehicle) = 6; n(Treatment +4 h p.i.) = 6; n(Treatment +24 h p.i.) = 5. The data were analysed by one-way ANOVA. P values < 0.05 were considered statistically significant.
3.4 Effect on cytokines and chemokines in preclinical models
In addition to demonstrating antiviral efficacy, we also investigated the potential immunomodulatory properties of zapnometinib, focusing on cytokine and chemokine expression. However, in the SARS-CoV-2-challenged Syrian hamster model, the levels of cytokines and chemokines increase only marginally. Therefore, the model is inappropriate for assessing alterations in cytokine and chemokine levels (56). We confirmed this observation in serum samples from the hamsters obtained from the efficacy study using ELISA, with no major differences between the groups after infection and treatment at 4 dpi (Supplementary Figure 3A) and in lung tissue samples analyzed by RT-qPCR from the same animals at 4 dpi (Supplementary Figure 3B). As the effects investigated in the hamster model do not clearly show the immunomodulatory component of zapnometinib, we aimed to demonstrate a direct anti-inflammatory effect in the absence of a dynamic stimulus such as virus infection. We used an LPS-induced acute lung injury (ALI) mouse model to simulate a pathogen-induced hyperinflammatory disease with high severity. Using this virus-independent model, we could demonstrate the direct effect of zapnometinib on cytokine and chemokine expression. Mice were challenged by i.p. injection of 5 mg/kg LPS and then treated with 25 mg/kg zapnometinib 1 h post-stimulation. Alterations in cytokine and chemokine gene expression patterns were evaluated by comparing them with untreated control mice using an RT2 profiler PCR array. As depicted in the volcano plot, the expression of most of the 84 genes encoding key secreted proteins central to inflammation, such as IL-1β (Il1b), IL-6, MCP-1 (ccl2), IL-8 (cxcl1), IP-10 (cxcl10) and MIP-1α (ccl3), was downregulated after zapnometinib treatment compared to control treatment (Figure 6A). Importantly, the levels of the Ifna2 gene, encoding for IFN-α, a key regulator of the innate antiviral immune response, were not significantly downregulated (p = 0.118). The expression of various important genes known to be elevated in COVID-19, such as Il6, Cxcl1 and Tnf (57) was significantly reduced in the lung tissues of zapnometinib-treated mice (Figure 6B). These data represent the first in vivo confirmation of previous in vitro findings that zapnometinib affects the proinflammatory gene expression response while genes of the antiviral type I IFN response remain largely unaffected (36).
Figure 6
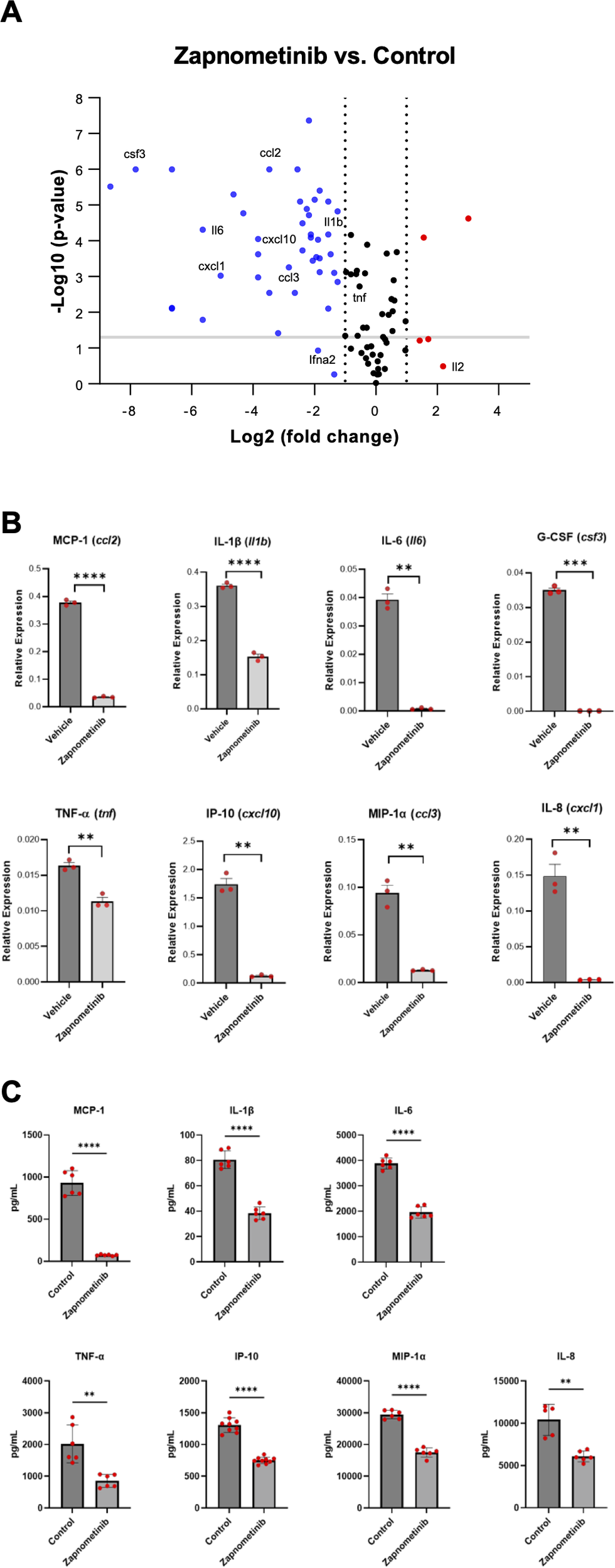
Levels of cytokines and chemokines associated with COVID-19 after zapnometinib treatment. mRNA expression of different cytokines and chemokines in acute lung injury (ALI) model mice after LPS stimulation and zapnometinib treatment 1h post stimulation (control group received vehicle =1% DMSO in PBS). The mRNA levels were determined at 6 h post treatment by an RT2 profiler PCR array. (A) Volcano plot shows gene expression changes. Representative genes mainly associated with COVID-19 are highlighted. Red: upregulation, blue: downregulation, black: no change in gene expression (regardless of statistical significance). The log2 of the fold change in gene expression is plotted on the x-axis, and statistical significance is plotted on the y-axis. The two dashed vertical lines indicate the fold change threshold, while the horizontal line indicates the p value threshold. Genes represented by data points in the far upper left (downregulated) and far upper right (upregulated) sections met the fold change and p value thresholds, while the genes represented by the data points between the two vertical lines showed no change in gene expression. (B) Representative expression. Mean + SD and individual values of n = 3 technical replicates are shown, gene names in brackets. The data were analysed using the ΔΔCt method and normalized to the mean expression levels of 5 housekeeping genes. The P values for the difference in replicate (-DeltaCt) expression data for each gene between the control and treatment groups were calculated by unpaired t-test with Welch’s correction. **p < 0.01, ***p < 0.001, ****p < 0.0001. (C) Reduction in cytokine expression after zapnometinib treatment in LPS-stimulated human PBMCs from two healthy donors. Cells were simultaneously stimulated with 1 µg/mL LPS and treated with 10 µg/mL zapnometinib for 6 h. The expression of cytokines in the supernatants was analysed by ELISA. The data are presented as the mean ± SD of technical replicates. n(IL-1β; IL-6; MCP-1; MIP-1α; TNF-α) = 6; n(IP-10) = 9; n(IL-8 control) = 5; n(IL-8 zapnometinib-treated) = 6. The differences in the levels of all cytokines of interest between the control and zapnometinib-treated groups were analysed with unpaired t-test with Welch’s correction. **p < 0.01; ****p < 0.0001. Results from the second donor are shown in Supplementary Figure 4.
In addition to the animal model, we aimed to demonstrate the immunomodulatory properties of zapnometinib in a primary human cell type, specifically freshly isolated human PBMCs. PBMCs obtained from two healthy donors were challenged with 1µg/mL LPS from E. coli O55:B5 (prepared in PBS) and treated with 10 μg/mL zapnometinib (prepared in 0.1% DMSO) for 6 h. Cytokine and chemokine expression analysis by ELISA confirmed the anti-inflammatory properties of zapnometinib in human PBMCs. Expression levels of IL-1β, IL-6, IL-8, IP-10, MCP-1, MIP-1α, and TNF-α were significantly reduced upon treatment with zapnometinib for 6 h compared to the untreated control group (Figure 6C, Supplementary Figure 4). Taken together, these experiments collectively validate the anti-inflammatory properties of zapnometinib both in vivo and in primary human blood cells, demonstrating a significant reduction of cytokines and chemokines known to be elevated in COVID-19.
3.5 Effect on cytokines and chemokines in the clinical trial
To determine whether the reduction in cytokine and chemokine levels following zapnometinib treatment was also observed in hospitalized COVID-19 patients, serum samples from six patients in each group of a randomized, double-blind, placebo-controlled proof-of-concept Phase 2 trial in adults with moderate-to-severe COVID-19 symptoms (clinical severity status CSS 3 or 4; RESPIRE; NCT04776044) were analyzed. Samples were collected at various time points from day 1 to day 30 (D1, D3, D5, D8, D11, D15 and D30) after treatment initiation using a multiplex ELISA system. Despite the absence of a pronounced inflammatory response suggestive of a cytokine storm in the patients, as reflected by their clinical severity scores (CSS3/4), a significant reduction was observed in MCP-1 (p = <0.0001), MIP-1α (p = <0.0001), and IFNγ (p = <0.0001) levels among individuals treated with zapnometinib compared to those in the placebo cohort throughout the duration of the study period (up to day 30). IL-6 levels were significantly reduced (p = 0.0017), while TNF-α showed no changes and IL-8/CXCL-8 levels displayed a slight but not statistically significant increase (Figure 7A). These results further support the findings from the pre-clinical investigations that zapnometinib treatment leads to a reduction of various pro-inflammatory cytokines and chemokines.
Figure 7
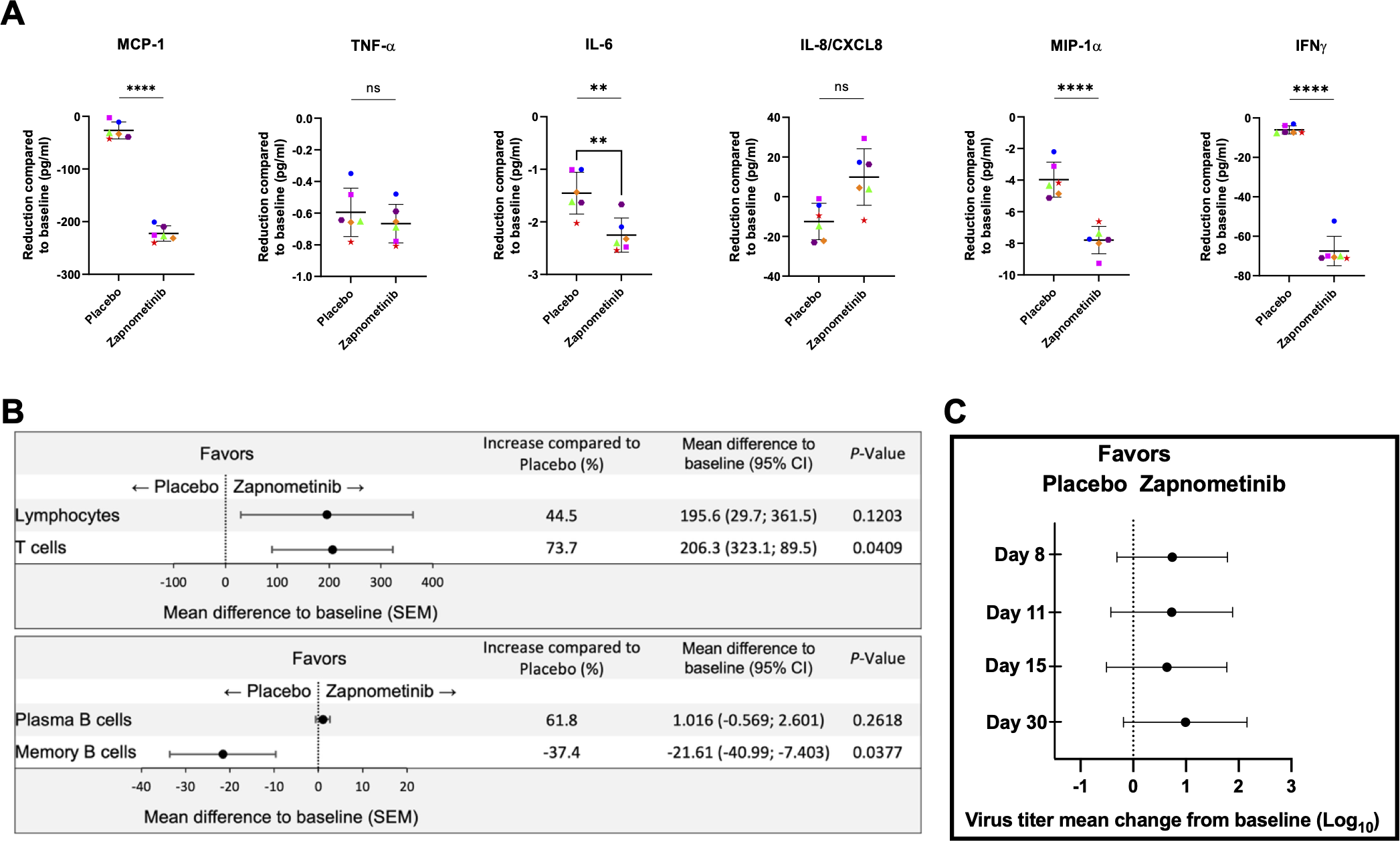
Immunomodulating effects and viral load analysis of patients hospitalized with COVID-19 and treated with either zapnometinib or placebo for 6 days. (A) Reduction of cytokine and chemokine expression compared to baseline values. Analysis was performed with blood from 6 patients from each group using Meso Scale V-Plex. All of them received either no standard of care treatment or antiviral treatment. Patients that received glucocorticoids as standard of care were excluded. Analysis was performed with values from the complete observation period (until day 30), horizontal line showing the mean value ± SEM of all days. Colour and symbols represent the mean values of n = 6 patients on different days: • blue circle = day 3, pink square = day 5; green triangle = day 8; orange rhombus = day 11, aubergine-coloured hexagon = day 15, red star = day 30 ▲. Analysis was performed using a one-tailed t-test. **p < 0.01; ****p < 0.0001.(B) Mean difference ± SEM of the number of lymphocytes, T cells, Plasma B cells and Memory B cells compared to baseline in zapnometinib (n = 34) and placebo treated (n = 34) patients. The graphics represent values from the entire study period (D1, D3, D5, D8, D11, D15 and D30). The data were analysed by one-tailed t-test. (C) RNA virus titer mean change from baseline over time in SARS-CoV-2 sputum samples from hospitalized COVID-19 patients on D8, D11, D15 and D30. Analysis only includes patients with baseline sputum SARS-CoV-2 RNA titer ≥ 500 copies/ml. Difference of mean change from zapnometinib vs. placebo treatment was given as Log10 copies/ml ± SEM, analysed with unpaired t-test with Welch’s correction. Further details of analysis see Supplementary Table 3.
3.6 Modulation of adaptive immune response in the clinical trial
In addition to the cytokine storm observed in the hyperinflammatory stage in moderate to severe cases of COVID-19, there is also a decrease in the adaptive immune response. Therefore, we also investigated the impact of zapnometinib on the overall number of lymphocytes, as well as T and B cells, which were quantified in the blood using FACS analysis at the same time points as the cytokine analysis. Elevated levels of lymphocytes, plasma B cells, and T cells were observed in individuals undergoing treatment with zapnometinib (Figure 7B), indicating a beneficial immune enhancement that augments the antiviral response following zapnometinib administration. Notably, a substantial 44.5% increase in lymphocyte count was documented during the 30-day observation period post-zapnometinib treatment. For T cells, the increase was even more pronounced (73.7%; p = 0.0409). As for Plasma B cells, there was also an increase of 61.8% in favor of zapnometinib, while a reduction was found for memory B cells (-37.4%; p = 0.0377) (Figure 6B). The difference in favor of zapnometinib was consistent across all observation days. Thus, we demonstrate here a positive immunomodulatory effect of zapnometinib on both the innate and adaptive immune responses.
3.7 Antiviral effect of zapnometinib in the clinical trial
Besides the immunomodulatory effect, the antiviral effect of zapnometinib was also investigated in patients participating the above-mentioned clinical trial. Therefore, sputum and nasopharyngeal swabs were collected on days D1, D3, D5, D8, D11, D15, and D30 after treatment initiation, and the viral load was determined by RT-qPCR. In the modified intention-to-treat population, 62 out of 103 patients (60.2%) had quantifiable RNA above 500 copies/ml confirmed in nasopharyngeal samples at baseline, and 74 out of 103 patients (71.8%) had RNA above 500 copies/ml confirmed in sputum samples (Table 3). Patients in the study received either antiviral, glucocorticoid or no standard of care treatment, with the presence of alpha, beta, gamma, delta and omicron strains. Zapnometinib treatment was associated with greater reductions from baseline in mean viral load than placebo in sputum samples at days 8, 11, 15 and 30 (Figure 7C, Table 3). However, at days 3 and 5, there was only a slight reduction in favor of zapnometinib (Table 3). In participants infected with non-Omicron variants, Zapnometinib demonstrated a consistent and clinically meaningful reduction in SARS-CoV-2 RNA titers compared to placebo, with a statistically significant difference of +1.57 log10 on Day 8 (p=0.0101). Although statistical significance was narrowly missed on Days 11, 15, and 30 (p=0.0551–0.0672), the differences remained above 1 log10 at all time points. In nasopharyngeal samples, greater reductions from baseline in mean viral load in favor of zapnometinib were also found, albeit to a lesser extent (Supplementary Table 3).
Table 3
| Sputum | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Visit | Zapnometinib | Placebo | Zapnometinib - Placebo | ||||||||
| n | Baseline Mean | Mean Changea (SD) | n | Baseline Mean | Mean Changea (SD) | Difference | (95% CI) | p-value | |||
| SARS-CoV-2 RNA titer (log10 copies/ml) in participants overall | |||||||||||
| Baseline | 37 | 6.85 | 34 | 6.46 | |||||||
| Day 3 | 36 | 5.79 | 1.08 | (2.08) | 34 | 5.51 | 0.94 | (1.98) | 0.14 | -0.827 to 1.112 | 0.3854 |
| Day 5 | 34 | 5.40 | 1.48 | (1.98) | 31 | 5.01 | 1.38 | (1.56) | 0.10 | -0.785 to 0.977 | 0.4145 |
| Day 8 | 31 | 4.04 | 2.81 | (1.98) | 27 | 4.35 | 2.07 | (1.99) | 0.74 | -0.308 to 1.785 | 0.0814 |
| Day 11 | 28 | 3.39 | 3.68 | (2.18) | 25 | 3.39 | 2.95 | (2.00) | 0.73 | -0.426 to 1.883 | 0.1054 |
| Day 15 | 30 | 2.98 | 3.93 | (2.19) | 4 | 3.74 | 3.36 | (2.11) | 0.56 | -0.570 to 1,697 | 0.1619 |
| Day 30 | 27 | 2.26 | 4.74 | (1.81) | 24 | 2.47 | 3.75 | (2.23) | 0.97 | -0.175 to 2.102 | 0.0476b |
| SARS-CoV-2 RNA titer non omicron (log10 copies/ml) in participants overall | |||||||||||
| Baseline | 24 | 6.75 | 21 | 6.42 | |||||||
| Day 3 | 23 | 5.80 | 1.16 | (2.25) | 21 | 5.47 | 0.96 | (1.90) | 0.21 | -1.057 to 1.472 | 0.3709 |
| Day 5 | 23 | 5.60 | 1.28 | (2.09) | 18 | 5.24 | 1.07 | (1.28) | 0.21 | -0.867 to 1.278 | 0.3501 |
| Day 8 | 20 | 4.24 | 2.63 | (2.26) | 15 | 5.26 | 1.06 | (1.55) | 1.57 | 0.261 to 2.883 | 0.0101b |
| Day 11 | 17 | 3.61 | 3.62 | (2.62) | 14 | 4.05 | 2.20 | (2.17) | 1,42 | -0.341 to 3.174 | 0.0551 |
| Day 15 | 19 | 3.43 | 3.59 | (2.47) | 16 | 3.74 | 2.40 | (2.08) | 1.18 | -0.385 to 2.744 | 0.0672 |
| Day 30 | 16 | 2.52 | 4.61 | (1.92) | 14 | 2.61 | 3.37 | (2.16) | 1.24 | -0.311 to 2.797 | 0.0561 |
| SARS-CoV-2 RNA titer omicron (log10 copies/ml) in participants overall | |||||||||||
| Baseline | 13 | 7.02 | 13 | 6.51 | |||||||
| Day 3 | 13 | 5.78 | 0.94 | (1.82) | 13 | 5.59 | 0.92 | (2.19) | 0.02 | -1.608 to 1.656 | 0.4881 |
| Day 5 | 11 | 5.04 | 1.91 | (1.75) | 13 | 4.69 | 1.82 | (1.86) | 0.09 | -1.442 to 1.618 | 0.4532 |
| Day 8 | 11 | 3.73 | 3.12 | (1.36) | 12 | 3.21 | 3.32 | (3.32) | -0.20 | -1.579 to 1.171 | 0.3801 |
| Day 11 | 11 | 3.10 | 3.77 | (1.38) | 11 | 2.57 | 3.90 | (1.31) | -0.13 | -1.326 to 1.066 | 0.4115 |
| Day 15 | 11 | 2.31 | 4.52 | (1.56) | 12 | 1.90 | 4.64 | (1.39) | -0.13 | -1.410 to 1.162 | 0.4216 |
| Day 30 | 11 | 1.95 | 4.92 | (1.71) | 10 | 2.28 | 4.34 | (2.15) | 0.58 | -1.222 to 2.380 | 0.2534 |
Mean viral titer and mean change from baseline (Day 0) over time (Day 3 – Day 30) in SARS-CoV-2 RNA titer from sputum samples (all randomized MITT population) from patients of the RESPIRE clinical trial treated either with zapnometinib or placebo D1 - D6.
Analysis only includes patients with baseline sputum SARS-CoV-2 RNA titer ≥ 500 copies/ml. Baseline results below this limit were excluded from the analysis.
Mean change from baseline values are based on the measurements from patients with values at both baseline and at the time point assessed. b p < 0.050.
In summary, the positive immunomodulatory properties, and the antiviral effect of zapnometinib demonstrated in various preclinical studies were confirmed in hospitalized patients with a severe viral infection.
4 Discussion
Recent cell culture studies have provided initial evidence that the MEK inhibitor zapnometinib exhibits anti-SARS-CoV-2 activity in vitro and downregulates the expression of candidate cytokines, suggesting a potential dual benefit in COVID-19 treatment. The present study aimed to substantiate this dual effect through extended preclinical work and further confirm it with clinical data. We evaluated whether these in vitro activities translate into efficacy in an infected organism, thereby assessing whether zapnometinib displays a dual therapeutic effect in vivo. Therefore, we analyzed the antiviral efficacy and immunomodulatory impact of zapnometinib using a well-established Syrian hamster SARS-CoV-2 infection model and in other models of hyperinflammatory infectious diseases. This study was a first attempt to demonstrate the efficacy of zapnometinib against SARS-CoV-2 in vivo. Further details, such as an optimized treatment regimen, need to be clarified in further studies. Given that in vitro activities of antiviral compounds are not always reflected in vivo, it is very important that a dual effect of zapnometinib could also be demonstrated in vivo. This is evidenced by reduced levels of both viral RNA and infectious particles in hamster throat swabs and nasal turbinates, an ameliorative effect on lung pathology as well as the results of a clinical phase 2 trial with hospitalized COVID-19 patients. The results emphasize the therapeutic potential of zapnometinib against coronavirus infections correlated with hyperinflammatory symptoms.
Zapnometinib, a host-targeted antiviral (HTA) inhibits the cellular Raf/MEK/ERK signaling pathway that is pivotal for replication of various viruses (58). It is important to note that the EC50 values observed for zapnometinib in vitro (16.1–37.9 μM) are higher than those typically reported for direct-acting antivirals (DAAs). However, zapnometinib as HTA inhibits a cellular signaling pathway essential for viral replication rather than acting directly on viral proteins. This fundamental difference in mode of action means that higher compound concentrations are required in vitro to achieve an antiviral effect, because the drug must sufficiently inhibit the host cell factor to disrupt viral replication indirectly. Such, in vitro EC50 values are therefore not directly comparable to those of DAAs. In vivo, the pharmacokinetic–pharmacodynamic relationship is also governed by tissue distribution, target engagement, and the modulation of host immune responses, which cannot be fully captured by cell culture assays. The clinical feasibility of the dosing has been demonstrated in two human phase 1 studies and in the present phase 2 study, where zapnometinib was well tolerated at doses designed to achieve sufficient systemic exposure for MEK inhibition without notable toxicity. This distinction is especially important when comparing zapnometinib to currently approved DAAs. As of February 2024, the DAAs nirmatrelvir, molnupiravir (emergency use in the U.S.) and remdesivir were considered and approved for treatment against COVID-19 by FDA and/or EMA (16, 59–61). In the Syrian hamster model of SARS-CoV-2 infection, zapnometinib showed a significant effect on viral load reduction at 3 dpi (Figure 3B) comparable to that of the direct-acting nucleoside analogue molnupiravir (59). Furthermore, zapnometinib treatment resulted in a reduction in lung pathology. Based on the data, there is no clear indication which start of treatment (+4 h p.i. or +24 h p.i.) is ultimately more beneficial. The assessment of the precise contribution of the immunomodulatory component of zapnometinib versus the reduction in viral load to the observed positive impact on lung pathology in the hamster model remains indeterminate. Notably, this model did not include an in-depth analysis of the effects on the adaptive immune response. However, our latest publication involving IAV-infected mice demonstrated that zapnometinib positively influences disease pathogenesis by modulating regulatory T cells, indicating that zapnometinib affects not only the expression of cytokines and chemokines (62). Future investigations are warranted to systematically explore and elucidate the specific mode of action of zapnometinib in this context. Nevertheless, the data from the hamster study could also substantiate the therapeutic success observed in the RESPIRE study (42). We do not anticipate zapnometinib, being a host-targeted antiviral agent, to induce mutagenic effects on the virus. In contrast, concerns have been raised regarding the mutagenic potential of nucleoside analogue antivirals like molnupiravir and, less frequently, remdesivir, which could potentially enhance viral evolution in treating viral diseases (63–65). This concern is not applicable to host-targeted antivirals, as they do not interfere with viral RNA replication. Another major advantage of host-targeted antivirals is their high barrier against the emergence of antiviral resistance. This is because the virus faces difficulty in compensating for the absence of cellular functions targeted by the drug. This was evidenced in a resistance study with zapnometinib compared to various direct-acting SARS-CoV-2 drugs (66). Drug resistance to current DAAs has already been documented, presenting a challenging problem that could potentially be mitigated by HTAs (67, 68). Given the stable nature of viral dependence on cellular factors, zapnometinib’s antiviral activity is likely to remain effective against new and emerging variants. Notably, zapnometinib demonstrated efficacy against a spectrum of coronaviruses, including SARS-CoV-1, SARS-CoV-2 Omicron variant, HCoV-OC43, and HCoV-229E (Figure 1), underscoring its broad-spectrum potential. In the phase 2 RESPIRE study, various SARS-CoV-2 variants were identified in the patients (alpha, beta, gamma, delta and omicron). The reduction in propagation observed in both omicron and non-omicron strains after zapnometinib treatment underscores the strong correlation between pre-clinical and clinical data. The time course of viral RNA reduction in the RESPIRE trial also deserves further discussion. The data (Table 3) show that reductions in viral load were modest at days 3 and 5 but became more pronounced and consistent from day 8 onwards, persisting through day 30. Since treatment was discontinued after day 7, the later differences cannot be directly attributed to ongoing drug exposure. One possible explanation is that zapnometinib not only reduces viral replication during treatment but also enhances cellular immune responses that may support clearance of infected cells at later stages. At the same time, the contrasting patterns observed between sputum and nasopharyngeal swabs cannot yet be fully explained. While this discrepancy lies beyond the scope of the present manuscript, the findings nevertheless suggest a potential benefit of zapnometinib even when administered beyond the very early phase of infection.
Interestingly, no viral load reduction was observed after molnupiravir treatment in hospitalized COVID-19 patients with a similar CSS as in RESPIRE (69). In a clinical trial involving nirmatrelvir/ritonavir (Paxlovid™) administered to hospitalized patients, no statistically significant variance was observed in the duration of SARS-CoV-2 RNA clearance or mortality rates (70). For high-risk patients, a favorable impact on the severity of COVID-19 is delineated (71). However, even if the two approved medications effectively reduce the incidence of hospitalizations and fatalities in non-hospitalized adults with mild-to-moderate COVID-19 in clinical trials, there remains an absence of a viable treatment option for severely ill patients, the specific patient population for which zapnometinib is intended (72–74). The efficacy of molnupiravir and Paxlovid™ in hospitalized patients appears to be constrained. This represents a critical gap in available treatments that could potentially be addressed by the introduction of zapnometinib.
Understanding cytokine pathways is crucial for developing effective treatments, with ongoing trials exploring anti-inflammatory and antiviral drugs’ potential in combating cytokine storms. Additionally, dietary interventions, such as zinc or vitamins, are being studied to regulate cytokine levels and alleviate COVID-19 symptoms. Zinc’s anti-inflammatory properties are being evaluated in clinical trials to assess its impact on the immune response and disease prognosis. However, careful risk assessment is necessary for immunomodulatory approaches due to the risk of secondary infections or impairment of viral clearance (17–19).
A major emphasis of the study was to explore the immunomodulatory effects of zapnometinib in vivo, as a second beneficial activity alongside its antiviral action. As anticipated, no significant difference in cytokine and chemokine expression was observed at 4 dpi in the SARS-CoV-2 infected Syrian hamster, indicating its unsuitability for infection-related cytokine studies (56). Therefore, we opted for an in vivo acute lung injury (ALI) model which represents a fulminant hyperinflammatory condition independent of pathogen infection dynamics. While the LPS-induced ALI mouse model does not fully recapitulate the immunopathology of SARS-CoV-2 infection, we deliberately chose this non-viral model to isolate zapnometinib’s direct immunomodulatory effects from its antiviral activity. In a viral infection model, reduced cytokine and chemokine expression could arise indirectly from decreased viral load, making it challenging to distinguish primary immunomodulatory effects from secondary consequences of antiviral action. The ALI model, with its pathogen-independent cytokine induction, allowed us to demonstrate zapnometinib’s direct anti-inflammatory potential without this confounding factor. Nevertheless, we acknowledge that this approach has limitations in faithfully modelling the complexity of viral immunopathology. Future studies using SARS-CoV-2 infection models will be important to confirm and extend these findings in a more clinically relevant setting. In addition to pathogen infection, ALI is an inflammatory condition that can be triggered by infections, sepsis, trauma, ischemia, or reperfusion. The most severe manifestation is ARDS (acquired respiratory distress syndrome), a hallmark of severe COVID-19 (75). In the ALI model, excessive inflammatory response was induced by bacterial LPS stimulation. Zapnometinib treatment led to a massive reduction in hyperinflammatory cytokine responses, highlighting its immunomodulatory potency in vivo. In addition, we assessed zapnometinib’s effectiveness in a primary human cell model, namely freshly isolated PBMCs, where a similar cytokine reduction was observed, confirming its immunomodulatory effect. A limitation of the ex vivo PBMC experiments is the small number of donors (n=2), which may limit generalizability. However, the highly consistent cytokine and chemokine response profiles across both donors support the mechanistic validity of the observed immunomodulatory effects of zapnometinib, in line with previously published MEK inhibitor data.
These pre-clinical findings were confirmed in the RESPIRE study, showing significant reductions in MCP-1, MIP-1a and IFN-γ levels. However, the implications for IFN-γ remain uncertain as it has dual roles as both an inducer and a regulator of inflammation (76). Notably, IL-8/CXCL8 levels showed a slight though not statistically significant increase with zapnometinib treatment. CXCL8, together with its receptors CXCR1/2, triggers neutrophil activity to combat inflammation during bacterial infections (77, 78). Although the overserved increase did not reach statistical significance, this trend may suggest a potentially beneficial role for CXCL8 in modulating immune responses during viral-induced inflammation. Overall, these findings highlight zapnometinib’s significant anti-inflammatory potential.
In recent years, many studies have highlighted the great potential of MEK inhibitors as immunomodulators (23, 79, 80). These inhibitors not only reduce excessive cytokine responses but also modulate the adaptive immune response for improved antiviral efficacy. Severe and moderate stages of COVID-19 are associated with lymphocyte depletion (81). In the RESPIRE study, we demonstrated that treatment with zapnometinib resulted in an increase in the number of lymphocytes, including T cells and antibody-producing plasma B cells, while the quantity of memory B cells was reduced. This shift may suggest an enhanced availability of plasma cells at the expense of memory B cells, potentially supporting a more active antibody response in treated patients (82). Given the scope of this manuscript as an integrated summary of preclinical models and first clinical trial results, we focus here on these initial findings. A more detailed analysis of T cell subpopulations from the RESPIRE trial will be presented in a dedicated follow-up publication. In summary, the positive effect on the adaptive immune response also shows the great potential of treating respiratory viral infections by a MEK inhibitor.
In some patients in the RESPIRE study, glucocorticoids were administered as part of the standard of care. Dexamethasone, a glucocorticoid commonly used to treat severe COVID-19, suppresses cytokine production by mainly targeting NF-κB signaling, as shown in animal models of LPS-induced ALI and in human PBMCs (83, 84). Additionally, dexamethasone down-regulates type I interferons (IFNs), which are crucial in the initial defense against severe viral respiratory infections (75, 85, 86). A drawback of glucocorticoids is their immunosuppressive nature, which can compromise host immunity and prolong viral clearance. In addition, tocilizumab and baricitinib, two immunomodulatory agents recommended for severe COVID-19 treatment, have shown effectiveness in attenuating multiple cytokines and chemokines in animal models of ALI and in human PBMCs (87). The study did not conclusively show a direct impact on interference with type I interferon (IFN). However, as baricitinib inhibits NF-κB, it is presumed that it may influence type I IFN expression since NF-κB plays a crucial role in their regulation (88, 89). In contrast to the NF-κB cascade, inhibiting the Raf/MEK/ERK signaling pathway, as demonstrated in this and previous studies, does not attenuate type I IFNs and downstream IFN-regulated genes (36). This suggests that zapnometinib treatment has a notable advantage over dexamethasone, tocilizumab, and baricitinib, as it does not diminish the type I IFN response. The potential to modulate an excessive proinflammatory cytokine and chemokine response remains comparable.
Overall, the study underscores the noteworthy therapeutic potential of zapnometinib as a novel and broadly effective approach. In recent years, there have been emerging strategies employing HTAs as broad-spectrum antiviral agents, with particular emphasis on repurposing kinase inhibitors. However, most substances have only been tested in proof-of-concept studies so far (90, 91). Our dual effect represents a distinct advantage over current therapies, which typically target either the virus itself or the hyperinflammatory response. In contrast, by inhibiting the Raf/MEK/ERK signaling pathway, zapnometinib exhibits both a reduction in viral load and a modulation rather than complete suppression of the immune response (39, 92). This sets the drug apart from most, if not all, COVID-19 treatments currently licensed or in development.
The findings presented here align with our earlier studies (36) which emphasize the potential value of zapnometinib as a standalone treatment or in combination with other drugs targeting various stages of viral infection and disease progression. Such combinations may represent a promising strategy for COVID-19 control. Furthermore, zapnometinib’s implications extend beyond COVID-19, as evidenced by its recent orphan drug designation by the FDA for hantavirus infections and the initiation of a phase 2 clinical trial for influenza. The robust correlation between preclinical and clinical data for SARS-CoV-2 and COVID-19 suggests a potential treatment benefit for influenza (28, 29, 93). Collectively, these findings underscore the broad-spectrum anti-infective potential of zapnometinib against acute viral infections. Importantly, the clinical impact of zapnometinib has been reported in detail in our companion publication (42), where treatment was associated with improved clinical scores and accelerated symptom resolution in hospitalized COVID-19 patients, in addition to the virological and immunological effects described here.
Statements
Data availability statement
The data that support the findings of this study are available upon reasonable request from the corresponding author.
Ethics statement
Human sputum and nasopharyngeal swabs were taken from samples collected for the RESPIRE trial (NCT04776044) in 18 hospitals from 5 countries that was approved by respective regulatory authorities and by the Ethics Committees concerned (for details, please see the clinical trial protocol provided in (42). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by The hamster experiments were carried out in the Central Animal Facilities of Viroclinics-DDL, Cerba Research Company, Schaijk, the Netherlands, in accordance with the standards of the Dutch law for animal experimentation (2010/63/EU). Ethical approval for the present study was obtained from the institutional Animal Welfare Body under the number AVD277002015283-WP24. The mouse experiments for the ALI mouse model were conducted following review and approval by the Regional Council Tuebingen (35/9185.81-2/IM 1/17) and in compliance with the institutional Principles of Animal Welfare and Animal Research. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
YF: Funding acquisition, Project administration, Resources, Writing – original draft, Writing – review & editing, Data curation, Formal Analysis, Methodology, Software, Validation, Visualization. LS: Writing – review & editing, Data curation, Formal Analysis, Methodology. HaH: Data curation, Formal Analysis, Methodology, Software, Writing – original draft, Writing – review & editing. HeH:Data curation, Formal Analysis, Methodology, Software, Validation, Writing – review & editing. MB: Formal Analysis, Methodology, Project administration, Validation, Writing – review & editing. SS: Data curation, Formal Analysis, Investigation, Methodology, Writing – review & editing. OPo: Investigation, Writing – review & editing, Data curation, Formal Analysis, Methodology, Software, Validation. AS: Data curation, Formal Analysis, Investigation, Methodology, Software, Writing – review & editing. VA: Formal Analysis, Investigation, Methodology, Writing – review & editing. MD: Writing – review & editing, Data curation, Formal Analysis, Methodology, Software. JV:Data curation, Software, Writing – review & editing, Investigation, Supervision, Validation. GV: Data curation, Investigation, Software, Validation, Writing – review & editing, Formal Analysis, Methodology, Project administration. LD: Project administration, Supervision, Writing – review & editing, Data curation, Methodology. SP: Investigation, Supervision, Writing – review & editing, Software, Validation. SL:Conceptualization, Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing, Validation. OPl:Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The Federal Ministry of Education and Research (BMBF 16LW0001), Germany, DFG grants SFB1009B13 and CRU342-P6.
Acknowledgments
The authors thank Annika Schönsiegel for coordinating the shipment of the compound and Eugenia Steinhilber for help with the manuscript. We acknowledge support from the Open Access Publication Fund of the University of Tübingen.
Conflict of interest
Authors YF, LS, HaH, HeH, MB, SS was/were employed by Atriva Therapeutics GmbH.
Authors OPo, AS, VA was/were employed by SIGNATOPE GmbH.
Authors GV, LD was/were employed by Viroclinics-DDL, Cerba Research Company.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declare that this study received funding from Atriva Therapeutics GmbH. Atriva Therapeutics GmbH acted as sponsor of the Phase 1 study, and was responsible for the study design. The sponsor had no additional role in data collection, data analysis, interpretation of data, writing of the manuscript, or the decision to submit it for publication.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1631721/full#supplementary-material
References
1
Khan M Adil SF Alkhathlan HZ Tahir MN Saif S Khan M et al . COVID-19: A global challenge with old history, epidemiology and progress so far. Molecules. (2021) 26:39. doi: 10.3390/molecules26010039
2
O’Driscoll M Ribeiro Dos Santos G Wang L Cummings DAT Azman AS Paireau J et al . Age-specific mortality and immunity patterns of SARS-CoV-2. Nature. (2021) 590:140–5. doi: 10.1038/s41586-020-2918-0
3
Alwani M Yassin A Al-Zoubi RM Aboumarzouk OM Nettleship J Kelly D et al . Sex-based differences in severity and mortality in COVID-19. Rev Med Virol. (2021) 31:e2223. doi: 10.1002/rmv.2223
4
Burkert FR Lanser L Bellmann-Weiler R Weiss G . Coronavirus disease 2019: clinics, treatment, and prevention. Front Microbiol. (2021) 12:761887. doi: 10.3389/fmicb.2021.761887
5
Wang LL Yang JW Xu JF . Coronavirus (SARS-CoV-2) causes lung inflammation and injury. Clin Microbiol Infect. (2022) 28:513–20. doi: 10.1016/j.cmi.2021.11.022
6
Leisman DE Ronner L Pinotti R Taylor MD Sinha P Calfee CS et al . Cytokine elevation in severe and critical COVID-19: a rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir Med. (2020) 8:1233–44. doi: 10.1016/S2213-2600(20)30404-5
7
Fan S Xiao M Han F Xia P Bai X Chen H et al . Neurological manifestations in critically ill patients with COVID-19: A retrospective study. Front Neurol. (2020) 11:806. doi: 10.3389/fneur.2020.00806
8
Aggarwal G Cheruiyot I Aggarwal S Wong J Lippi G Lavie CJ et al . Association of cardiovascular disease with coronavirus disease 2019 (COVID-19) severity: A meta-analysis. Curr Probl Cardiol. (2020) 45:100617. doi: 10.1016/j.cpcardiol.2020.100617
9
Shi W Lv J Lin L . Coagulopathy in COVID-19: Focus on vascular thrombotic events. J Mol Cell Cardiol. (2020) 146:32–40. doi: 10.1016/j.yjmcc.2020.07.003
10
Gustine JN Jones D . Immunopathology of hyperinflammation in COVID-19. Am J Pathol. (2021) 191:4–17. doi: 10.1016/j.ajpath.2020.08.009
11
Costela-Ruiz VJ Illescas-Montes R Puerta-Puerta JM Ruiz C Melguizo-Rodriguez L . SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. (2020) 54:62–75. doi: 10.1016/j.cytogfr.2020.06.001
12
Group RC Horby P Lim WS Emberson JR Mafham M Bell JL et al . Dexamethasone in hospitalized patients with covid-19. N Engl J Med. (2021) 384:693–704. doi: 10.1056/NEJMoa2021436
13
Goletti D Cantini F . Baricitinib therapy in covid-19 pneumonia - an unmet need fulfilled. N Engl J Med. (2021) 384:867–9. doi: 10.1056/NEJMe2034982
14
Stone JH Frigault MJ Serling-Boyd NJ Fernandes AD Harvey L Foulkes AS et al . Efficacy of tocilizumab in patients hospitalized with covid-19. N Engl J Med. (2020) 383:2333–44. doi: 10.1056/NEJMoa2028836
15
Yoon JJ Toots M Lee S Lee ME Ludeke B Luczo JM et al . Orally efficacious broad-spectrum ribonucleoside analog inhibitor of influenza and respiratory syncytial viruses. Antimicrob Agents Chemother. (2018) 62:10.1128/aac.00766-18. doi: 10.1128/AAC.00766-18
16
Owen DR Allerton CMN Anderson AS Aschenbrenner L Avery M Berritt S et al . An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science. (2021) 374:eabl4784. doi: 10.1126/science.abl4784
17
Verma G Dhawan M Saied AA Kaur G Kumar R Emran TB . Immunomodulatory approaches in managing lung inflammation in COVID-19: A double-edge sword. Immun Inflammation Dis. (2023) 11:e1020. doi: 10.1002/iid3.1020
18
Rabaan AA Al-Ahmed SH Muhammad J Khan A Sule AA Tirupathi R et al . Role of inflammatory cytokines in COVID-19 patients: A review on molecular mechanisms, immune functions, immunopathology and immunomodulatory drugs to counter cytokine storm. Vaccines. (2021) 9:436. doi: 10.3390/vaccines9050436
19
Dhawan M Emran TB Priyanaka COP . Immunomodulatory effects of zinc and its impact on COVID-19 severity. Ann Med Surg (Lond). (2022) 77:103638. doi: 10.1016/j.amsu.2022.103638
20
Chang F Steelman LS Lee JT Shelton JG Navolanic PM Blalock WL et al . Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia. (2003) 17:1263–93. doi: 10.1038/sj.leu.2402945
21
Lieske NV Tonby K Kvale D Dyrhol-Riise AM Tasken K . Targeting tuberculosis and HIV infection-specific regulatory T cells with MEK/ERK signaling pathway inhibitors. PloS One. (2015) 10:e0141903. doi: 10.1371/journal.pone.0141903
22
Nakayama T Yamashita M . The TCR-mediated signaling pathways that control the direction of helper T cell differentiation. Semin Immunol. (2010) 22:303–9. doi: 10.1016/j.smim.2010.04.010
23
Verma V Jafarzadeh N Boi S Kundu S Jiang Z Fan Y et al . MEK inhibition reprograms CD8(+) T lymphocytes into memory stem cells with potent antitumor effects. Nat Immunol. (2021) 22:53–66. doi: 10.1038/s41590-020-00818-9
24
Moss P . The T cell immune response against SARS-CoV-2. Nat Immunol. (2022) 23:186–93. doi: 10.1038/s41590-021-01122-w
25
Zhu Q Xu Y Wang T Xie F . Innate and adaptive immune response in SARS-CoV-2 infection-Current perspectives. Front Immunol. (2022) 13:1053437. doi: 10.3389/fimmu.2022.1053437
26
Ram T Singh AK Kumar A Singh H Pathak P Grishina M et al . MEK inhibitors in cancer treatment: structural insights, regulation, recent advances and future perspectives. RSC Medicinal Chem. (2023) 14:1837–57. doi: 10.1039/D3MD00145H
27
Pleschka S Wolff T Ehrhardt C Hobom G Planz O Rapp UR et al . Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. (2001) 3:301–5. doi: 10.1038/35060098
28
Laure M Hamza H Koch-Heier J Quernheim M Muller C Schreiber A et al . Antiviral efficacy against influenza virus and pharmacokinetic analysis of a novel MEK-inhibitor, ATR-002, in cell culture and in the mouse model. Antiviral Res. (2020) 178:104806. doi: 10.1016/j.antiviral.2020.104806
29
Hamza H Shehata MM Mostafa A Pleschka S Planz O . Improved in vitro efficacy of baloxavir marboxil against influenza A virus infection by combination treatment with the MEK inhibitor ATR-002. Front Microbiol. (2021) 12:611958. doi: 10.3389/fmicb.2021.611958
30
Planz O Pleschka S Ludwig S . MEK-specific inhibitor U0126 blocks spread of Borna disease virus in cultured cells. J Virol. (2001) 75:4871–7. doi: 10.1128/JVI.75.10.4871-4877.2001
31
Preugschas HF Hrincius ER Mewis C Tran GVQ Ludwig S Ehrhardt C . Late activation of the Raf/MEK/ERK pathway is required for translocation of the respiratory syncytial virus F protein to the plasma membrane and efficient viral replication. Cell Microbiol. (2019) 21:e12955. doi: 10.1111/cmi.12955
32
Cai Y Liu Y Zhang X . Suppression of coronavirus replication by inhibition of the MEK signaling pathway. J Virol. (2007) 81:446–56. doi: 10.1128/JVI.01705-06
33
Valencia HJ de Aguiar M Costa MA Mendonca DC Reis EV Arias NEC et al . Evaluation of kinase inhibitors as potential therapeutics for flavivirus infections. Arch Virol. (2021) 166:1433–8. doi: 10.1007/s00705-021-05021-1
34
Lee S Yang W Kim DK Kim H Shin M Choi KU et al . Inhibition of MEK-ERK pathway enhances oncolytic vaccinia virus replication in doxorubicin-resistant ovarian cancer. Mol Ther Oncolytics. (2022) 25:211–24. doi: 10.1016/j.omto.2022.04.006
35
Ndjomou J Park IW Liu Y Mayo LD He JJ . Up-regulation of hepatitis C virus replication and production by inhibition of MEK/ERK signaling. PloS One. (2009) 4:e7498. doi: 10.1371/journal.pone.0007498
36
Schreiber A Viemann D Schoning J Schloer S Mecate Zambrano A Brunotte L et al . The MEK1/2-inhibitor ATR-002 efficiently blocks SARS-CoV-2 propagation and alleviates pro-inflammatory cytokine/chemokine responses. Cell Mol Life Sci. (2022) 79:65. doi: 10.1007/s00018-021-04085-1
37
Schreiber A Ambrosy B Planz O Schloer S Rescher U Ludwig S . The MEK1/2 inhibitor ATR-002 (Zapnometinib) synergistically potentiates the antiviral effect of direct-acting anti-SARS-coV-2 drugs. Pharmaceutics. (2022) 14:1776. doi: 10.3390/pharmaceutics14091776
38
Schrader T Dudek SE Schreiber A Ehrhardt C Planz O Ludwig S . The clinically approved MEK inhibitor Trametinib efficiently blocks influenza A virus propagation and cytokine expression. Antiviral Res. (2018) 157:80–92. doi: 10.1016/j.antiviral.2018.07.006
39
Pinto R Herold S Cakarova L Hoegner K Lohmeyer J Planz O et al . Inhibition of influenza virus-induced NF-kappaB and Raf/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antiviral Res. (2011) 92:45–56. doi: 10.1016/j.antiviral.2011.05.009
40
Koch-Heier J Schonsiegel A Waidele LM Volk J Full Y Wallasch C et al . Pharmacokinetics, pharmacodynamics and antiviral efficacy of the MEK inhibitor zapnometinib in animal models and in humans. Front Pharmacol. (2022) 13:893635. doi: 10.3389/fphar.2022.893635
41
Full Y Wallasch C Hilton A Planz O . Pharmacokinetics, absorption, distribution, metabolism and excretion of the MEK inhibitor zapnometinib in rats. Front Pharmacol. (2022) 13:1050193. doi: 10.3389/fphar.2022.1050193
42
Rohde G Stenglein S Prozesky H Manudhane G Sandulescu O Bauer M et al . Efficacy and safety of zapnometinib in hospitalised adult patients with COVID-19 (RESPIRE): a randomised, double-blind, placebo-controlled, multicentre, proof-of-concept, phase 2 trial. eClinicalMedicine. (2023) 102237. doi: 10.1016/j.eclinm.2023.102237
43
Koch-Heier J Hoffmann H Schindler M Lussi A Planz O . Inactivation of SARS-coV-2 through treatment with the mouth rinsing solutions viruProX((R)) and bacterX((R)) pro. Microorganisms. (2021) 9:521. doi: 10.3390/microorganisms9030521
44
Zekri L Ruetalo N Christie M Walker C Manz T Rammensee HG et al . Novel ACE2 fusion protein with adapting activity against SARS-CoV-2 variants in vitro. Front Immunol. (2023) 14:1112505. doi: 10.3389/fimmu.2023.1112505
45
Zielinska E Liu D Wu HY Quiroz J Rappaport R Yang DP . Development of an improved microneutralization assay for respiratory syncytial virus by automated plaque counting using imaging analysis. Virol J. (2005) 2:84. doi: 10.1186/1743-422X-2-84
46
Corman VM Landt O Kaiser M Molenkamp R Meijer A Chu DK et al . Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill. (2020) 25:pii=2000045. doi: 10.2807/1560-7917.ES.2020.25.3.2000045
47
Watterson D Wijesundara DK Modhiran N Mordant FL Li Z Avumegah MS et al . Preclinical development of a molecular clamp-stabilised subunit vaccine for severe acute respiratory syndrome coronavirus 2. Clin Transl Immunol. (2021) 10:e1269. doi: 10.1002/cti2.1269
48
Steinhilber AE Schmidt FF Naboulsi W Planatscher H Niedzwiecka A Zagon J et al . Mass spectrometry-based immunoassay for the quantification of banned ruminant processed animal proteins in vegetal feeds. Anal Chem. (2018) 90:4135–43. doi: 10.1021/acs.analchem.8b00120
49
Anselm V Sommersdorf C Carrasco-Triguero M Katavolos P Planatscher H Steinhilber A et al . Matrix and sampling effects on quantification of protein biomarkers of drug-induced liver injury. J Proteome Res. (2021) 20:4985–94. doi: 10.1021/acs.jproteome.1c00478
50
Munoz-Fontela C Dowling WE Funnell SGP Gsell PS Riveros-Balta AX Albrecht RA et al . Animal models for COVID-19. Nature. (2020) 586:509–15. doi: 10.1038/s41586-020-2787-6
51
Bendjama K Guionaud S Aras G Arber N Badimon L Bamberger U et al . Translation strategy for the qualification of drug-induced vascular injury biomarkers. Toxicol Pathol. (2014) 42:658–71. doi: 10.1177/0192623314527644
52
Church RJ Kullak-Ublick GA Aubrecht J Bonkovsky HL Chalasani N Fontana RJ et al . Candidate biomarkers for the diagnosis and prognosis of drug-induced liver injury: An international collaborative effort. Hepatology. (2019) 69:760–73. doi: 10.1002/hep.29802
53
Scaffidi P Misteli T Bianchi ME . Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. (2002) 418:191–5. doi: 10.1038/nature00858
54
Zemskova M McClain N Niihori M Varghese MV James J Rafikov R et al . Necrosis-released HMGB1 (High mobility group box 1) in the progressive pulmonary arterial hypertension associated with male sex. Hypertension. (2020) 76:1787–99. doi: 10.1161/HYPERTENSIONAHA.120.16118
55
Schomaker S Potter D Warner R Larkindale J King N Porter AC et al . Serum glutamate dehydrogenase activity enables early detection of liver injury in subjects with underlying muscle impairments. PloS One. (2020) 15:e0229753. doi: 10.1371/journal.pone.0229753
56
Nouailles G Wyler E Pennitz P Postmus D Vladimirova D Kazmierski J et al . Temporal omics analysis in Syrian hamsters unravel cellular effector responses to moderate COVID-19. Nat Commun. (2021) 12:4869. doi: 10.1038/s41467-021-25030-7
57
Del Valle DM Kim-Schulze S Huang HH Beckmann ND Nirenberg S Wang B et al . An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
58
Pleschka S . RNA viruses and the mitogenic Raf/MEK/ERK signal transduction cascade. Biol Chem. (2008) 389:1273–82. doi: 10.1515/BC.2008.145
59
Rosenke K Hansen F Schwarz B Feldmann F Haddock E Rosenke R et al . Orally delivered MK-4482 inhibits SARS-CoV-2 replication in the Syrian hamster model. Nat Commun. (2021) 12:2295. doi: 10.1038/s41467-021-22580-8
60
Abdelnabi R Boudewijns R Foo CS Seldeslachts L Sanchez-Felipe L Zhang X et al . Comparing infectivity and virulence of emerging SARS-CoV-2 variants in Syrian hamsters. Ebiomedicine. (2021) 68:103403. doi: 10.1016/j.ebiom.2021.103403
61
Ye ZW Yuan S Chan JF Zhang AJ Yu CY Ong CP et al . Beneficial effect of combinational methylprednisolone and remdesivir in hamster model of SARS-CoV-2 infection. Emerg Microbes Infect. (2021) 10:291–304. doi: 10.1080/22221751.2021.1885998
62
Koch-Heier J Vogel AB Full Y Ebensperger M Schonsiegel A Zinser RS et al . MEK-inhibitor treatment reduces the induction of regulatory T cells in mice after influenza A virus infection. Front Immunol. (2024) 15:1360698. doi: 10.3389/fimmu.2024.1360698
63
Kosakovsky Pond SL Martin D . Anti-COVID drug accelerates viral evolution. Nature. (2023) 623:486–7. doi: 10.1038/d41586-023-03248-3
64
Gandhi S Klein J Robertson AJ Pena-Hernandez MA Lin MJ Roychoudhury P et al . De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: a case report. Nat Commun. (2022) 13:1547. doi: 10.1038/s41467-022-29104-y
65
Standing JF Buggiotti L Guerra-Assuncao JA Woodall M Ellis S Agyeman AA et al . Randomized controlled trial of molnupiravir SARS-CoV-2 viral and antibody response in at-risk adult outpatients. Nat Commun. (2024) 15:1652. doi: 10.1038/s41467-024-45641-0
66
Schreiber A Rodner F Oberberg N Anhlan D Bletz S Mellmann A et al . The host-targeted antiviral drug Zapnometinib exhibits a high barrier to the development of SARS-CoV-2 resistance. Antiviral Res. (2024) 225:105840. doi: 10.1016/j.antiviral.2024.105840
67
Ip JD Wing-Ho Chu A Chan WM Cheuk-Ying Leung R Umer Abdullah SM Sun Y et al . Global prevalence of SARS-CoV-2 3CL protease mutations associated with nirmatrelvir or ensitrelvir resistance. Ebiomedicine. (2023) 91:104559. doi: 10.1016/j.ebiom.2023.104559
68
Hu Y Lewandowski EM Tan H Zhang X Morgan RT Zhang X et al . Naturally occurring mutations of SARS-coV-2 main protease confer drug resistance to nirmatrelvir. ACS Cent Sci. (2023) 9:1658–69. doi: 10.1021/acscentsci.3c00538
69
Arribas JR Bhagani S Lobo SM Khaertynova I Mateu L Fishchuk R et al . Randomized trial of molnupiravir or placebo in patients hospitalized with covid-19. NEJM Evidence. (2022) 1:EVIDoa2100044. doi: 10.1056/EVIDoa2100044
70
Liu J Pan X Zhang S Li M Ma K Fan C et al . Efficacy and safety of Paxlovid in severe adult patients with SARS-Cov-2 infection: a multicenter randomized controlled study. Lancet Reg Health West Pac. (2023) 33:100694. doi: 10.1016/j.lanwpc.2023.100694
71
Najjar-Debbiny R Gronich N Weber G Khoury J Amar M Stein N et al . Effectiveness of paxlovid in reducing severe coronavirus disease 2019 and mortality in high-risk patients. Clin Infect Dis. (2023) 76:e342–e9. doi: 10.1093/cid/ciac443
72
Wan EYF Yan VKC Wong ZCT Chui CSL Lai FTT Li X et al . Effectiveness of molnupiravir vs nirmatrelvir-ritonavir in non-hospitalised and hospitalised patients with COVID-19: a target trial emulation study. EClinicalMedicine. (2023) 64:102225. doi: 10.1016/j.eclinm.2023.102225
73
Brown AN Lang Y Zhou J Franco EJ Hanrahan KC Bulitta JB et al . Why molnupiravir fails in hospitalized patients. mBio. (2022) 13:e0291622. doi: 10.1128/mbio.02916-22
74
Jayk Bernal A Gomes da Silva MM Musungaie DB Kovalchuk E Gonzalez A Delos Reyes V et al . Molnupiravir for oral treatment of covid-19 in nonhospitalized patients. N Engl J Med. (2022) 386:509–20. doi: 10.1056/NEJMoa2116044
75
Sinha S Rosin NL Arora R Labit E Jaffer A Cao L et al . Dexamethasone modulates immature neutrophils and interferon programming in severe COVID-19. Nat Med. (2022) 28:201–11. doi: 10.1038/s41591-021-01576-3
76
Zhang J . Yin and yang interplay of IFN-gamma in inflammation and autoimmune disease. J Clin Invest. (2007) 117:871–3. doi: 10.1172/JCI31860
77
Hartl D Latzin P Hordijk P Marcos V Rudolph C Woischnik M et al . Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat Med. (2007) 13:1423–30. doi: 10.1038/nm1690
78
Ha H Debnath B Neamati N . Role of the CXCL8-CXCR1/2 axis in cancer and inflammatory diseases. Theranostics. (2017) 7:1543–88. doi: 10.7150/thno.15625
79
Li D March ME Gutierrez-Uzquiza A Kao C Seiler C Pinto E et al . ARAF recurrent mutation causes central conducting lymphatic anomaly treata ble with a MEK inhibitor. Nat Med. (2019) 25:1116–22. doi: 10.1038/s41591-019-0479-2
80
Houde N Beuret L Bonaud A Fortier-Beaulieu SP Truchon-Landry K Aoidi R et al . Fine-tuning of MEK signaling is pivotal for limiting B and T cell activation. Cell Rep. (2022) 38:110223. doi: 10.1016/j.celrep.2021.110223
81
Silva MJA Ribeiro LR Lima KVB Lima L . Adaptive immunity to SARS-CoV-2 infection: A systematic review. Front Immunol. (2022) 13:1001198. doi: 10.3389/fimmu.2022.1001198
82
Khodadadi L Cheng Q Radbruch A Hiepe F . The maintenance of memory plasma cells. Front Immunol. (2019) 10:721. doi: 10.3389/fimmu.2019.00721
83
Mokra D Mikolka P Kosutova P Mokry J . Corticosteroids in acute lung injury: the dilemma continues. Int J Mol Sci. (2019) 20:4765. doi: 10.3390/ijms20194765
84
Held HD Boettcher S Hamann L Uhlig S . Ventilation-induced chemokine and cytokine release is associated with activation of nuclear factor-kappaB and is blocked by steroids. Am J Respir Crit Care Med. (2001) 163:711–6. doi: 10.1164/ajrccm.163.3.2003001
85
Gessani S McCandless S Baglioni C . The glucocorticoid dexamethasone inhibits synthesis of interferon by decreasing the level of its mRNA. J Biol Chem. (1988) 263:7454–7. doi: 10.1016/S0021-9258(18)68518-7
86
McCoy CE Carpenter S Palsson-McDermott EM Gearing LJ O’Neill LA . Glucocorticoids inhibit IRF3 phosphorylation in response to Toll-like receptor-3 and -4 by targeting TBK1 activation. J Biol Chem. (2008) 283:14277–85. doi: 10.1074/jbc.M709731200
87
Ibrahim YF Moussa RA Bayoumi AMA Ahmed AF . Tocilizumab attenuates acute lung and kidney injuries and improves survival in a rat model of sepsis via down-regulation of NF-kappaB/JNK: a possible role of P-glycoprotein. Inflammopharmacology. (2020) 28:215–30. doi: 10.1007/s10787-019-00628-y
88
Marconi VC Ramanan AV de Bono S Kartman CE Krishnan V Liao R et al . Efficacy and safety of baricitinib for the treatment of hospitalised adults with COVID-19 (COV-BARRIER): a randomised, double-blind, parallel-group, placebo-controlled phase 3 trial. Lancet Respir Med. (2021) 9:1407–18. doi: 10.1016/S2213-2600(21)00331-3
89
Zhou Q Zhang L Dong Y Wang Y Zhang B Zhou S et al . The role of SARS-CoV-2-mediated NF-kappaB activation in COVID-19 patients. Hypertens Res. (2024) 47:375–84. doi: 10.1038/s41440-023-01460-2
90
Schor S Einav S . Repurposing of kinase inhibitors as broad-spectrum antiviral drugs. DNA Cell Biol. (2018) 37:63–9. doi: 10.1089/dna.2017.4033
91
Kumar N Sharma S Kumar R Tripathi BN Barua S Ly H et al . Host-Directed Antiviral Therapy. Clin Microbiol Rev. (2020) 33:10.1128/cmr.00168-19. doi: 10.1128/CMR.00168-19
92
Wang ZQ Wu DC Huang FP Yang GY . Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. (2004) 996:55–66. doi: 10.1016/j.brainres.2003.09.074
93
Hoffmann H Ebensperger M Schönsiegel A Hamza H Koch-Heier J Schreiber A et al . Influenza A virus replication has a stronger dependency on Raf/MEK/ERK signaling pathway activity than SARS-CoV-2. Front Cell Infection Microbiol. (2023) 13. doi: 10.3389/fcimb.2023.1264983
Summary
Keywords
zapnometinib, MEK inhibitor, host targeting agent, COVID-19, drug development, immunomodulation, broad-spectrum antiviral
Citation
Füll Y, Schüssele LM, Hamza H, Hoffmann H, Bauer M, Stenglein S, Pötz O, Steinhilber A, Anselm V, Delany MW, Van den Brand JMA, Van Amerongen G, De Waal L, Pleschka S, Ludwig S and Planz O (2025) Antiviral and immunomodulatory effect of zapnometinib in animal models and hospitalized COVID-19 patients. Front. Immunol. 16:1631721. doi: 10.3389/fimmu.2025.1631721
Received
20 May 2025
Accepted
25 August 2025
Published
22 September 2025
Volume
16 - 2025
Edited by
Xulin Chen, Jinan University, China
Reviewed by
Kun Yang, Huazhong University of Science and Technology, China
Sayan Das, University of Maryland, United States
Lucy Gordon, University of California San Diego, United States
Updates
Copyright
© 2025 Füll, Schüssele, Hamza, Hoffmann, Bauer, Stenglein, Pötz, Steinhilber, Anselm, Delany, Van den Brand, Van Amerongen, De Waal, Pleschka, Ludwig and Planz.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oliver Planz, oliver.planz@uni-tuebingen.de
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.