 Dan Wu1†
Dan Wu1† Yujia Zhao
Yujia Zhao Wenna Liu
Wenna Liu Qingqing Liu
Qingqing Liu Ying Liang
Ying Liang- 1Precision Pharmacy and Drug Development Center, Department of Pharmacy, Tangdu Hospital, Fourth Military Medical University, Xi’an, Shaanxi, China
- 2Department of Urology, Xijing Hospital, Fourth Military Medical University, Xi’an, Shaanxi, China
- 3Department of Oncology, Tangdu Hospital, Fourth Military Medical University, Xi’an, Shaanxi, China
Gliomas are the most common primary malignant tumors of the central nervous system (CNS), and despite progress in molecular diagnostics and targeted therapies, their prognosis remains poor. In recent years, immunotherapy has emerged as a promising treatment modality in cancer therapy. However, the inevitable immune evasion by tumor cells is a key barrier affecting therapeutic efficacy. Epigenetic regulation, such as DNA methylation, histone modification, and non-coding RNA expression, plays a crucial role in the occurrence, development, and immune evasion of gliomas. These modifications can dynamically regulate gene expression, leading to the silencing of tumor-associated antigens, dysregulation of pro-inflammatory cytokines, and dynamic modulation of immune checkpoints (such as PD-L1). This review systematically elucidates the key mechanisms by which epigenetic regulation promotes immune evasion in gliomas and details three interconnected mechanisms: 1) epigenetic silencing of tumor-associated antigens and antigen-presenting machinery; 2) dysregulation of pro-inflammatory cytokine secretion; and 3) dynamic modulation of PD-L1 expression through chromatin remodeling. We emphasize the potential of combining epigenetic therapies with immunotherapies to enhance anti-tumor immune responses and overcome treatment resistance in gliomas. Future research should focus on developing biomarker-driven epigenetic immunotherapies and exploring the complex interplay between epigenetic modifications, glioma cells, and the tumor immune microenvironment to improve patient outcomes.
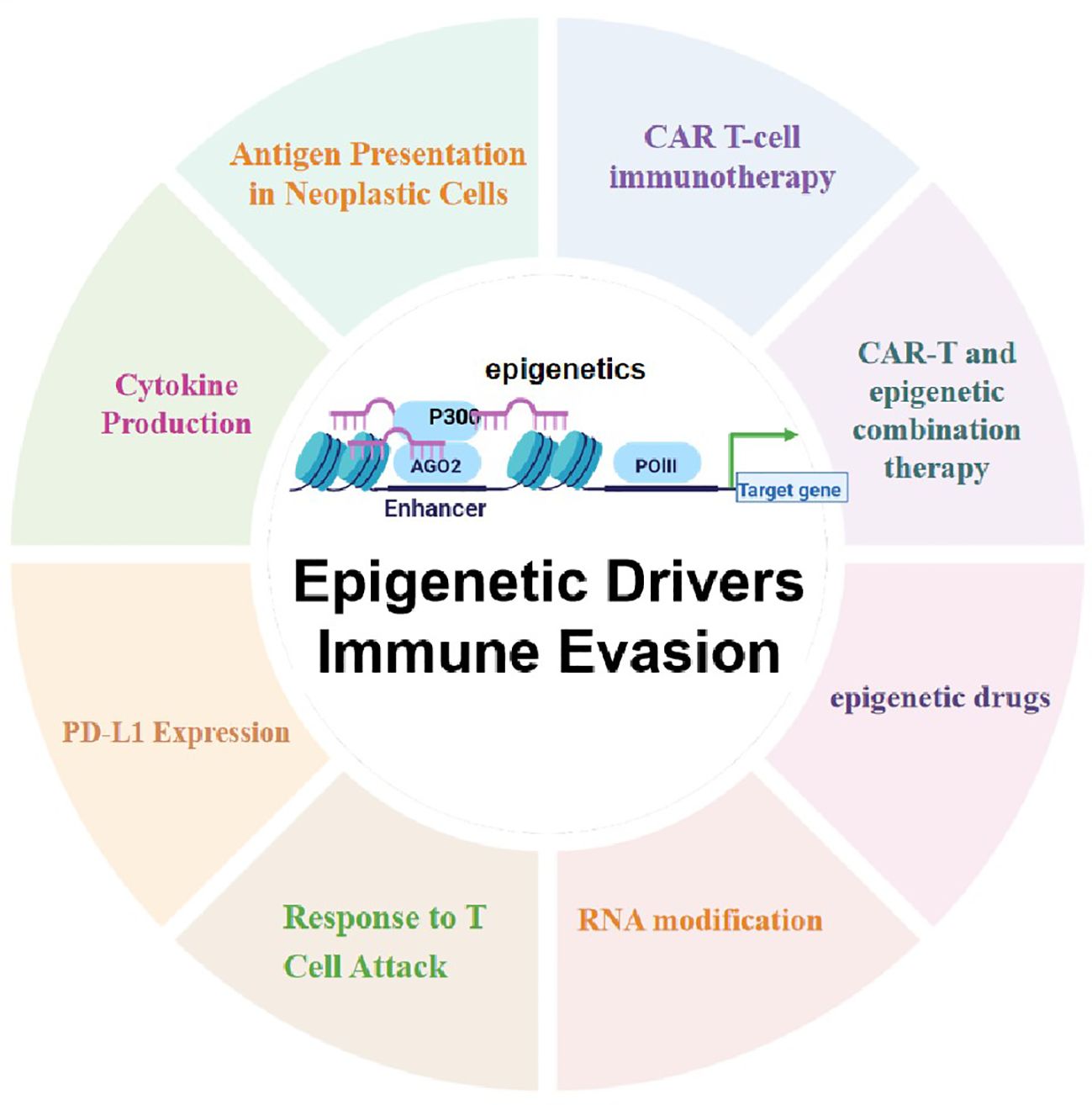
Graphical Abstract.
1 Introduction
Gliomas represent the preeminent primary tumors of the central nervous system, arising from diverse glial cell lineages such as astrocytes, oligodendrocytes, and ependymal cells, these tumors exhibit a high degree of heterogeneity. According to the molecular complexity and the diversity of pathological morphology, the World Health Organization (WHO) categorizes gliomas into four malignancy grades in ascending order, with Grade IV representing the most aggressive form (1). In the adult population, diffuse infiltrating gliomas are mainly classified into three categories: astrocytoma, oligodendroglioma, and glioblastoma. Among them, astrocytoma patients with IDH mutations exhibit substantially improved survival outcomes compared to those with IDH wild-type glioblastoma. Additionally, oligodendroglioma cases with concurrent IDH mutation and 1p/19q codeletion demonstrate the most promising prognostic profile (2, 3). In the pediatric population, pilocytic astrocytoma is relatively common. This tumor is often accompanied by BRAF gene variation and exhibits a relatively well-circumscribed growth pattern, with an overall good prognosis. On the other hand, diffuse midline glioma with H3K27 variation has the highest degree of malignancy and is a key factor leading to death due to glioma in children (4, 5).
Despite significant advances in understanding glioma pathogenesis, molecular diagnosis, and targeted therapy via omics approaches (e.g., gene sequencing, single-cell and spatial transcriptomics) (6–9), prognosis remains poor. Thus, elucidating pathogenic mechanisms and identifying novel therapies are critical to improving patient outcomes and quality of life. Immunotherapy, including immune checkpoint inhibition and CAR-T therapy, has advanced rapidly in oncology, offering therapeutic promise. However, gliomas exhibit strong immunosuppressive properties: glioma cells upregulate immunosuppressive factors (e.g., PD-L1, IDO) to impair antigen presentation, while glioma-associated macrophages and regulatory T cells weaken immunity via inhibitory cytokine secretion and cytotoxic T lymphocyte depletion, respectively (10). This immunosuppressive microenvironment impairs immunotherapy efficacy. For instance, phase III trials showed that the checkpoint inhibitor nivolumab failed to improve survival in newly diagnosed glioblastoma (with standard treatment) or recurrent cases (vs. bevacizumab) (11). Recently, Claudia Galassi et al. inspired by the “three Es” model (Elimination, Equilibrium, Escape) (12), Galassi et al. proposed a “three Cs” framework (camouflage, coercion, cytoprotection) to explain cancer immune evasion (13): evading immune recognition, interfering with effector cell activation, and resisting cytotoxicity. These mechanisms underpin glioma cells’ ability to escape immune attack, critically contributing to immunotherapy failure. Thus, uncovering novel regulators of glioma immune escape is vital for advancing clinical immunotherapy.
Epigenetic modifications, dynamic chemical alterations of chromosomal nucleic acids/proteins, orchestrate gene expression across splicing, transcription, and chromatin architecture without changing DNA sequence. Regulated by DNA methylation, histone modification, chromatin remodeling, and non-coding RNAs (ncRNAs), they play pivotal roles in tumorigenesis and progression (14). Recent studies show histone methylation/acetylation modulates immune checkpoints (e.g., PD-1/PD-L1), affecting immune recognition and elimination of tumor cells (15, 16). Additionally, ncRNAs regulate immune cell activation via immune checkpoints, epithelial-mesenchymal transition (EMT), and the tumor immune microenvironment (TIME), thereby influencing immunotherapy efficacy (17). These findings highlight the critical role of epigenetic mechanisms in immune cell differentiation and function, dictating context-specific gene expression profiles within the tumor immune microenvironment (TIME) that influence immunotherapy outcomes. Given that both tumor neoantigens and autoantigens are immunogenic, epigenetic regulation controls neoantigen generation and reactivates genes restricted to immune-privileged stages (18, 19). Thus, elucidating epigenetic mechanisms underlying tumor immune escape and developing combinatorial strategies with immunosuppressants and epigenetic drugs hold promise for overcoming immune escape in gliomas.
This review systematically outlines key epigenetic mechanisms underlying glioma immune evasion, including silencing of tumor-associated antigens and antigen-presenting machinery, dysregulated pro-inflammatory cytokine secretion, and dynamic PD-L1 modulation via chromatin remodeling. We also evaluate preclinical/clinical strategies targeting such epigenetic-driven immune evasion, offering a framework for developing biomarker-guided epigenetic immunotherapies to overcome glioma treatment resistance.
2 Epigenetics in glioma development and progression
2.1 Epigenetic regulation in glioma subtype classification
Epigenetic alterations are crucial in determining the classification and driving the progression of gliomas. The genetic alteration status of isocitrate dehydrogenase (IDH) is a critical biomarker that distinguishes different glioma subtypes. As a crucial metabolic enzyme, IDH is mainly involved in the cellular energy metabolism process, especially the tricarboxylic acid cycle (TCA cycle). It plays an important role in maintaining normal physiological functions. In glioma cells, IDH mutation is a common molecular alteration, particularly the IDH1-R132H mutation. This mutation leads to changes in enzyme activity, converting α-ketoglutarate (α-KG) into (R)-2-hydroxyglutarate [(R)-2HG]. This metabolite can interfere with normal cellular metabolism and epigenetic regulation, promoting the occurrence and development of tumors (20) (Figure 1).
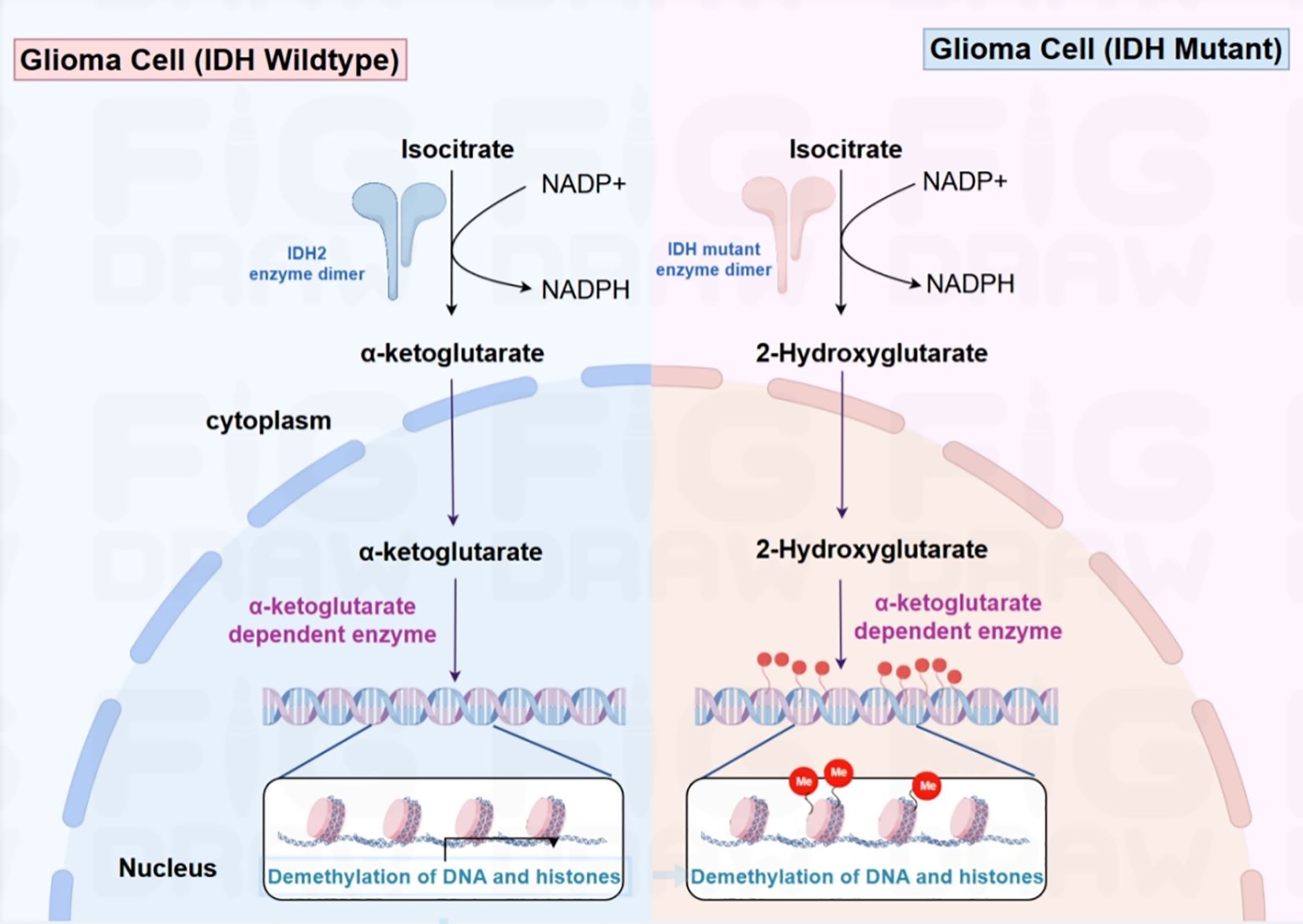
Figure 1. In IDH wild-type glioma cells (left), isocitrate is converted into α-ketoglutarate and NADPH using NADP+ under the action of the IDH2 enzyme dimer. α-ketoglutarate then enters the nucleus and participates in α- ketoglutarate-dependent enzymatic reactions, promoting DNA and histone demethylation. In IDH mutant glioma cells (right), the mutated IDH enzyme catalyzes the conversion of isocitrate to 2-hydroxyglutarate (2-HG), leading to intracellular accumulation of this metabolite. By inhibiting the function of α-ketoglutarate-dependent enzymes, 2-HG obstructs DNA and histone demethylation processes, thereby impacting gene expression modulation.
Interestingly, IDH-mutant gliomas display a CpG island methylator phenotype (G-CIMP), which is characterized by hypermethylation in CpG-rich promoter regions. This epigenetic alteration is associated with distinct clinical outcomes, with the “high” G-CIMP subgroup generally having a better prognosis (21). The dynamic interplay between IDH mutations and epigenetic modifications is further evidenced by the role of G-CIMP in driving the differentiation and aggressiveness of glioma subtypes. For instance, oligodendrogliomas harboring IDH mutation alongside 1p/19q codeletion show a more promising prognosis, whereas glioblastomas with wild-type IDH are associated with a poorer outcome. Compared with untreated patients, patients who experience relapse and exhibit cases with the absence of O(6)-methylguanine-DNA methyltransferase (MGMT) promoter methylation often exhibit a greater load of genetic mutations, indicating that this methylation phenotype may lead to the chemoresistance of tumors (22, 23). These observations underscore the clinical relevance of epigenetic profiling in glioma subtype classification and prognostication.
Even though extensive genomic hypermethylation has been demonstrated to impede the process of cellular differentiation, the role of specific methylation events in tumor progression has been emphasized. This phenomenon involves the transcriptional silencing of tumor suppressor genes (including BRCA1/BRCA2, TP53BP1, SMAD4) and the abnormal activation of oncogenes (such as PDGFRA, PIK3CA, and BRAF), thereby facilitating tumor initiation and progression. These genes are collectively referred to as “methylation-dependent oncogenic drivers” (24). IDH-mutated low-grade gliomas (LGGs) represent a subgroup of brain tumors exhibiting unique molecular features, typically characterized by slow growth and relatively favorable prognosis. However, they carry the potential risk of malignant progression. The progression from low-grade to high-grade malignancy is propelled by the accretion of genetic mutations, epigenetic modifications, and shifts in the tumor microenvironment (TME). Therefore, early identification of the IDH molecular subtypes in glioma patients and targeted treatment have significant clinical implications for patient outcomes. In a recent investigation, Wu and his team constructed a dual-phase framework by probing single-cell RNA sequencing and chromatin accessibility profiles across various glioma grades. During the initial phase, gliomagenesis is driven by oligodendrocyte precursor-like cells in conjunction with epigenetic reprogramming, which deactivate tumor suppressor genes such as CDKN2A and trigger oncogenes like PDGFRA (25). As the disease progresses, tumor expansion is fueled by an increased population of proliferative neural precursor-like cells. Genetic aberrations, including PDGFRA, MYCN, and CDK4 amplifications alongside CDKN2A/B deletions, facilitate tumor advancement. This research underscores the fluctuating impact of IFN signaling during disease progression. In IDH-mutated low-grade gliomas, IFN signaling undergoes epigenetic hypermethylation-mediated suppression, a process reversible by MGMT inhibitors or IDH antagonists, reactivating the IFN pathway. In contrast, high-grade gliomas evade IFN signaling through genetic deletions of IFN genes. These findings indicate a shift from epigenetic to genetic control in glioma progression. Lately, the US Food and Drug Administration (FDA) greenlit vorasidenib, a brain-permeable small molecule targeting mutant IDH1/2 proteins, marking a pivotal change in the therapeutic approach for IDH-mutant gliomas (26).
2.2 Epigenetic alterations shaping the transcriptional landscape of glioma
Epigenetic alterations significantly impact the transcriptional landscape of glioma cells, contributing to their malignant behavior. Research has shown that mIDH1 expression induces transcriptional silencing of the PD-L1-encoding gene CD274 via DNA methylation mechanisms (27, 28). Upon treatment with mIDH1 inhibitors, mIDH1 glioma cells exhibit reduced DNA methylation in the CD274 regulatory region. Concurrently, tumor cell-mediated high PD-L1 expression contributes to T cell exhaustion. Therefore, in gliomas with mIDH1 mutations, the concentration of PD-L1, which restricts T-cell functionality, is notably diminished. A separate study revealed that the absence of ATRX-a key epigenetic regulator linked to adult and pediatric gliomagenesis-epigenetically drives the production of PD-L1 alongside multiple immunosuppressive cytokines. This cascade of events ultimately initiates an immune-tolerance-promoting process in ATRX-mutated gliomas (29–31).
Explorations into the influence of histone modifications on TME modulation are insufficiently documented. Emerging findings highlight that RACK7 (encoded by ZMYND8) displays intensified interaction with the H3.3-G34R histone variant, a mutation linked to pHGG pathogenesis. This augmented binding event results in silencing of specific genomic loci, including CIITA, which plays a pivotal role in governing MHC class II molecule expression (32). The PRC2 complex, a group of histone methyltransferases, plays an essential part in regulating epigenetic silencing, with its activity being modifiable in gliomas. The expression levels of PRC2 proteins are associated with a poor prognosis (33). Recent investigations demonstrate that PRC2-dependent chromatin silencing contributes to immunosuppressive TMEs by inhibiting the transcription of immunostimulatory cytokines in neoplastic cells. Therefore, tumors with PRC2 mutations respond better to ICIs, providing the possibility of enhancing immunotherapy response by epigenetically and pharmacologically restoring PRC2 methyltransferase activity (34). Besides, in mIDH1-mutant gliomas, epigenetic modulation of the Notch signaling cascade is achieved through DNA methylation at CpG dinucleotides within the delta-like ligand 3 (DLL3) genomic locus. As an inhibitory Notch ligand, DLL3 expression exhibits a positive correlation with survival outcomes in mIDH1 gliomas (35), Glioma cases with elevated DLL3 levels demonstrate enhanced immune cell infiltration, suggesting a functional link between Notch pathway activation and immune responsiveness in these neoplasms.
2.3 Non-coding RNA in glioma: epigenetic modulators and beyond
Over the past few decades, it has been revealed that noncoding RNAs, such as microRNAs(miRNAs) and long noncoding RNAs, are crucial in gliomas, where they function as epigenetic regulators among other roles (36), Genomic profiling reveals 27 long non-coding RNAs (lncRNAs) are upregulated and 198 are downregulated in glioblastoma (GBM), underscoring their critical involvement in tumor pathogenesis. Recent findings highlight the role of lncRNAs in modulating GBM cell metabolism. For example, the lncRNA TP53TG1 promotes cell migration and proliferation under glucose-deprived conditions by regulating gene expression of PKM2 and IDH1 in glioma cell cultures (37). Additional research demonstrates that lncRNAs influence glioma biology, progression, and therapeutic responsiveness. For example, HOTAIRM1, an epigenetic modulator, associates with transcriptional start sites to regulate gene expression. LncSNHG6 and lncRNA ZFAT-AS1 facilitate Histone H3 Lysine 27 trimethylation (H3K27me3) deposition, leading to transcriptional silencing. MiRNAs as endogenous regulatory RNAs, function by inhibiting mRNA translation. Depletion of miRNAs that govern key mRNAs- including miR-31 (which targets CDKN2A/B) and miR-34a (involved in EGFR level regulation) (38). T-can drive oncogenic pathways. Variations in miRNA expression across molecular subtypes suggest their role in shaping glioma heterogeneity through transcriptional subtype transitions. MiRNA dysregulates major pathways (TGF-β, PI3K/AKT, EGFR, Notch) in glioma progression likewise. Exosomal miR-148a is related to down-regulated CADM1 expression in GBM samples (39). The antagonistic effect of miR-148a reduces the level of p-STAT3 and subsequently up-regulates the level of the tumor suppressor gene CADM1. In addition, NcRNAs act as intercellular signals; glioma cell-secreted miRNA and lncRNA affect TME behavior. For instance, glioma cell-secreted lncRNA-ATB suppresses miR-204-3p in astrocytes, promoting cell migration. Exosome-secreted lncRNAs have a paracrine effect aiding adaptation and resistance. The ncRNA-mediated communication between glioma cells and TME has therapeutic potential, but its role in TME epigenetic changes, especially in immune cells, requires further study. Furthermore, findings from current research indicate that dysregulated expression of circular RNAs (circRNAs) correlates with glioma initiation and progression (40). Transcriptomic profiling via microarray identified 91 circRNAs with altered expression patterns in glioma tissues compared to matched non-tumor adjacent samples. Notably, elevated expression of circ_0037655 has been shown to promote tumor cell viability and invasive capacity (41, 42). The involvement of noncoding RNAs-including lncRNAs, miRNAs, and circRNAs—in shaping the TME and modulating immune cell functionality broadens therapeutic possibilities. Therapeutic approaches targeting these ncRNAs or their associated regulatory pathways may offer innovative strategies to enhance immunotherapeutic efficacy or circumvent treatment resistance. Nevertheless, the intricate interplay among ncRNAs, TME components, and immune cells requires additional investigation to fully harness their potential as therapeutic targets.
Interestingly, our recent work has revealed a class of miRNAs localized in the nucleus and whose genomic locations highly overlap with enhancers (named NamiRNA, Nuclear Activating miRNA) play a role in enhancing the sensitivity of GBM to temozolomide by activating enhancer activity to positively regulate target genes and induce nucleolar stress (Figure 2). This further suggests the clinical application potential of noncoding RNAs in the treatment of gliomas.
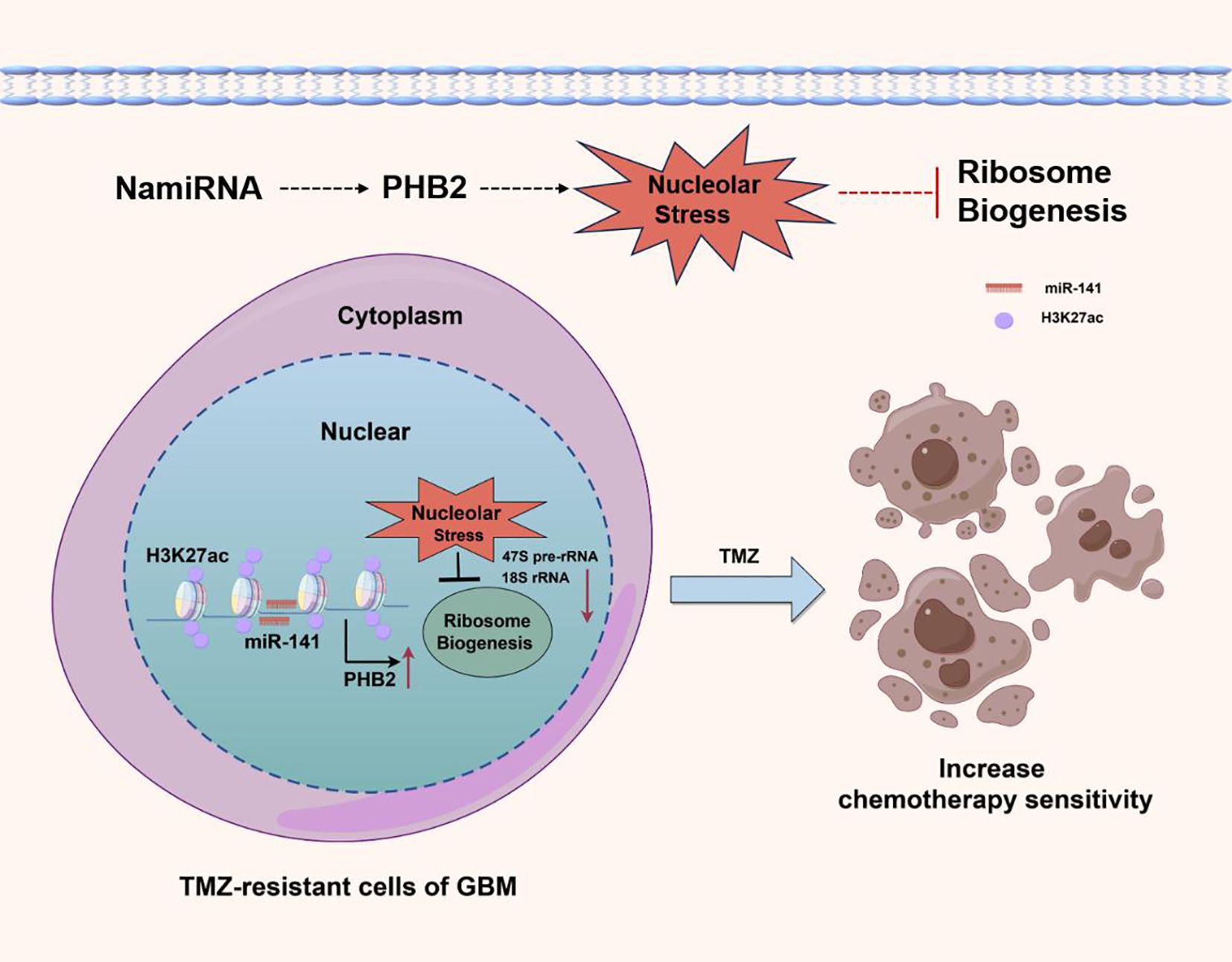
Figure 2. In GBM cells, overexpression of NamiRNA-141 activates enhancer activity, positively regulates PHB2 to induce nucleolar stress, inhibit ribosome biogenesis, and enhance the chemosensitivity of GBM to temozolomide. H3K27ac is an epigenetic marker of active enhancers.
2.4 Epigenetic regulation and glioma stem cells
Glioma stem cells (GSCs) are pivotal in driving glioma initiation, progression, and relapse. Their intrinsic resistance to standard therapies and capacity for tumor re-formation pose substantial challenges in glioma management. Emerging research has underscored the complex interplay between epigenetic mechanisms and the preservation of GSC characteristics (43, 44). Epigenetic alterations, such as DNA methylation, histone alterations, and ncRNA-dependent regulation, are central to sustaining the stem-like properties of GSCs (45). For instance, DNA hypermethylation at specific loci can silence tumor suppressor genes, allowing GSCs to evade cellular senescence and apoptosis. Conversely, hypomethylation can activate oncogenes, promoting proliferation and self-renewal (46). Modifications to histones, with a focus on acetylation and methylation, are vital for the upkeep of GSCs. Histone-modifying enzymes, including histone acetyltransferases (HATs) and histone deacetylases (HDACs) manage how accessible chromatin is to transcription factors, which in turn shapes gene expression. High levels of HDAC activity have been observed in GSCs, promoting a repressive chromatin state that favors the expression of stemness-related genes. Inhibitors of HDACs have shown promise in inducing differentiation and reducing the stemness of GSCs, making them more susceptible to conventional therapies. Besides, enhancers are capable of recruiting a large number of transcription factors and coactivators, Histone H3 lysine 27 acetylation (H3K27ac) functions as an indicator of active enhancers and significantly impacts the regulation of genes associated with cell identity and disease. Gimple etc. analyzed 10 glioblastoma specimens obtained via surgical resection and 15 non-neoplastic brain tissue samples through chromatin immunoprecipitation sequencing (ChIP-seq), and screened out the genes WSCD1, ELOVL2 and KLHDC8A that are regulated by glioma stem cell (GSC)-related enhancers, providing new targets for the treatment of gliomas (47) (Figure 3). In addition, non-coding RNAs, such as miRNAs and lncRNAs, are increasingly recognized as pivotal modulators of GSC phenotypes. MiRNAs may act as oncogenic or tumor suppressive regulators by targeting genetic loci associated with stem cell maintenance, differentiation, and self-renewal pathways. Analogously, lncRNAs have been demonstrated to modulate gene expression through epigenetic mechanisms, influencing chromatin structure and transcription factor activity. A recent study has indicated that LncRNA INHEG regulates the 2’-O-methylation of rRNA and promotes the translation of mRNA to facilitate the renewal and growth of GSCs (48).
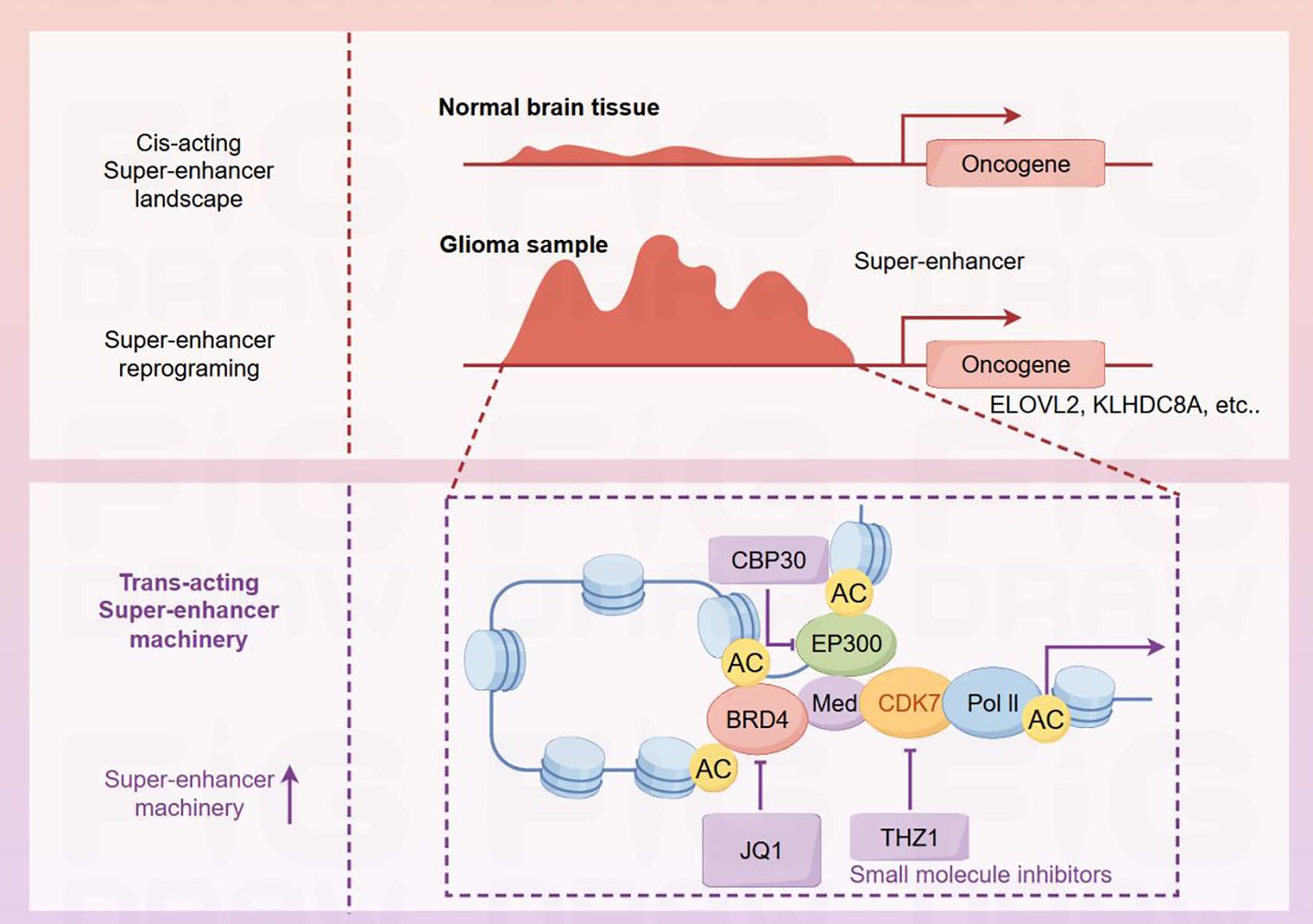
Figure 3. Differences in super-enhancers and related mechanisms of action between normal brain tissues and glioma samples. In normal brain tissues, the cis-acting super-enhancer landscape is relatively stable. In contrast, super-enhancer reprogramming occurs in glioma samples, thereby activating oncogenes such as ELOVL2 and KLHDC8A. The super-enhancer region involves interactions among various proteins, including CBP30, EP300, BRD4, Med, CDK7, and Pol II. The acetylation sites (AC) on these proteins are involved in the regulatory process. The small-molecule inhibitors JQ1 and THZ1 act on BRD4 and CDK7 respectively. By interfering with the super-enhancer machinery, they intervene in the expression of glioma-related genes.
The interplay between epigenetic modifications and GSCs underscores the importance of developing targeted therapies that can disrupt these regulatory networks. By targeting epigenetic modifiers, it may be possible to induce differentiation of GSCs, reducing their tumor-initiating potential and enhancing effectiveness of traditional treatments. Additional investigations into the epigenetic control of GSCs may facilitate the creation of innovative therapeutic approaches to enhance glioma patient outcomes.
3 Epigenetic regulation mediated glioma’s immune evasion
Contrasted with other types of cancers, gliomas possess distinct immune-related traits. The anatomical position of gliomas in the brain results in unique attributes that modify the immunological response against neoplastic cells. Gliomas’ unique immunomodulatory characteristics encompass the neuronal and glial cell populations coexist with the BBB. The maintains CNS stability by controlling the passage of substances in and out (49). Although the BBB usually protects the brain from blood toxins while allowing necessary nutrients through, This semi-permeable barrier is also capable of limiting the entry of peripheral leukocytes and therapeutic agents. Brain inflammation impairs BBB integrity, increasing immune cell infiltration, but gliomas may have already escaped immune surveillance by then (50). Epigenetic mechanisms are central to immune evasion in cancer. Within neoplastic cells, histone and DNA modifications can modulate tumor antigen presentation, suppress anti-tumor cytokine expression, and drive PD-L1 checkpoint induction (46).
3.1 Antigen presentation in neoplastic cells
Epigenetic control influences the dysfunction of antigen-presenting mechanisms in neoplastic cells, allowing tumors to avoid T-cell recognition and fostering immune evasion (51). For the presentation of self- and tumor-specific peptides to CD8 T cells, tumor cell proteins must undergo proteasomal digestion to produce short oligopeptides. The antigen processing-associated transporters 1 and 2 (TAP1 and TAP2) assemble into a heterodimer, facilitating the translocation of these peptides from the cytosol to the endoplasmic reticulum (ER) (52, 53). Within the ER, antigen peptides are incorporated into nascent major histocompatibility complex class I (MHC-I) molecules with the aid of chaperone proteins (54, 55). The MHC-I complex responsible for tumor antigen presentation consists of two polypeptide subunits-human leukocyte antigens (HLA) and β2-microglobulin (B2M)-and is translocated to the cell surface for antigen presentation. Neoplastic cell MHC-I expression can be suppressed by both DNMT and HDAC enzymes. Administration of DNA methyltransferase inhibitors (DNMTi) or histone deacetylase inhibitors (HDACi) to tumor cells can restore MHC-I surface expression (56). In certain instances, MHC-I deficiency stems from the epigenetic repression of auxiliary genes in the antigen-presentation pathway, including B2M, TAP-1, and TAP-2. Intervention with DNMTi in tumor cell models or clinical settings can upregulate gene expression essential for antigen presentation (57).
Currently, in the context of developing therapies against tumor antigens in gliomas, cancer-specific somatic mutation-derived antigens serve as optimal immunotherapeutic targets from multiple perspectives. Their expression is predominantly restricted to neoplastic cells, thereby significantly mitigating the risk of off-tumor toxicity. For example, in children with diffuse midline glioma (DMG), the H3F3A locus, which encodes histone H3.3, harbors a frequently recurring point mutation (58). The K27M substitution at amino acid position 27 (lysine to methionine) reduces H3K27 trimethylation, causing global demethylation and abnormal gene expression by inhibiting PRC2 activity. This mutation is an attractive immunotherapy target. Analogous to the IDH1 R132H alteration in adult gliomas, the H3.3 K27M mutation exhibits uniform distribution across tumor tissues. Recently, Pavlina Chuntova et al. documented that blocking D-2HG results in the increased expression of proinflammatory genes in a new HLA-A2/HLA-DR1 transgenic mouse model expressing IDH1R132H glioma (59). Meanwhile, Gerard L Brien et al. identified the oncogenic activity in human hindbrain neural stem cells, the histone H3.3-K27M modification arises from the simultaneous impairment of both PRC2 activity and enhancer functionality (60).
Furthermore, in addition to affecting transcription, epigenetic disorders exacerbate genetic instability in gliomas by altering chromatin structure: mutations in histones and ATRX lead to genetic instability due to abnormal deposition of histone marks, while mutant IDH epigenetically enhances DNA damage response signaling. This genetic instability is a key driver of neoantigen production-it not only induces cytoplasmic extrachromosomal DNA to activate the cGAS/STING axis, but also directly causes chromosomal aberrations, prompting mutated proteins to express neoantigens; the immune activation or tolerance triggered by neoantigens is determined by the tumor microenvironment (TME). Epigenetic regulation mediated by STING promoter methylation can modulate glioma immune responses, and this process is reversible by MGMT inhibitors. Mutations in the H3-G34 locus in pediatric high-grade gliomas (pHGG) also induce genomic instability, activate the cGAS/STING axis, enhance immune activation, and improve the efficacy of DNA damage-based therapies due to abnormal neoantigen expression (61, 62).
3.2 Cytokine production
Cancer cells can emit signaling molecules that are detected by immune cells at a distance, thereby triggering or inhibiting immune reactions. Glioma cells produce various immunomodulatory factors, such as IL-1β, IL-6, TGF-β, and IL-8. In glioma stem/progenitor-like cells, mIDH1 mediates epigenetic upregulation of G-CSF expression, thereby promoting the remodeling of myeloid cells in the mIDH1-associated TME. Elevated levels of G-CSF stimulate the proliferation of neutrophil precursors and neutrophils, concurrently attenuating the immunosuppressive traits of polymorphonuclear myeloid-derived suppressor cells within the mIDH1-driven tumor microenvironment (22). The epigenetic regulation of tumor cell differentiation impacts the TME. Epigenetic alterations in gliomas, such as H3 mutations in pediatric cases and mIDH1 in adult patients, lead to a halt in cellular differentiation, maintaining cells in a stem-like state. Cells resembling cancer stem cells exhibit reduced immunogenicity and evade immune detection through mechanisms like the downregulation of MHC transcription and the induction of quiescence. A recent investigation revealed that GSCs cultivated in immunocompetent hosts adapt epigenetically and secrete immunosuppressive cytokines (43, 63).
Furthermore, Glioma cells secrete immunomodulatory cytokines like IL-1β, IL-6, TGF-β, and IL-8 to activate or suppress immune response. Studies show GSCs undergo epigenetic adaptation, secreting immunosuppressive cytokines. Additionally, adapted GSCs upregulate interferon regulatory factor 8 (IRF8) secretion. IRF8, a cytokine usually in myeloid cells, may govern myeloid cell lineages and macrophage polarization (64). This implies that epigenetic mechanisms coordinate the functional repatterning of glioma cell populations, which in turn leads to alterations in immune cells and fosters the development of a TME favorable for tumor growth. Independent research has demonstrated that PRC2 constitutes a complex of histone methyltransferases responsible for epigenetic repression. It is hypothesized that PRC2-dependent chromatin repression facilitates immune evasion by suppressing the production of immunostimulatory cytokines in neoplastic cells (65) (Figure 4). Additionally, in gliomas, epigenetic reprogramming of the PI3K/AKT/mTOR signaling axis drives a metabolic shift from oxidative phosphorylation to aerobic glycolysis, and this metabolic reprogramming serves as a core hub for regulating the secretory factor network in the immune microenvironment (66). It exerts its effects through multiple interconnected mechanisms: first, it promotes the massive release of metabolites such as lactate, which not only directly impairs immune cell function and shapes a hypoxia-associated immunosuppressive tumor microenvironment (TME) but also acts as signaling molecules to regulate the expression and secretion of secretory factors; second, it reshapes immune homeostasis via secretory factors through pathways including upregulating TGF-β secretion, inhibiting monocyte differentiation into dendritic cells, and inducing the production of pro-oncogenic cytokines; additionally, metabolic changes in the TME specifically modulate the secretory function of astrocytes, prompting them to release cholesterol to facilitate glioma growth and recruit immunosuppressive macrophages (67). Collectively, these processes reveal that metabolic reprogramming, by systematically regulating the production, release, and function of secretory factors, emerges as a key driver influencing tumor progression and the immune microenvironment.
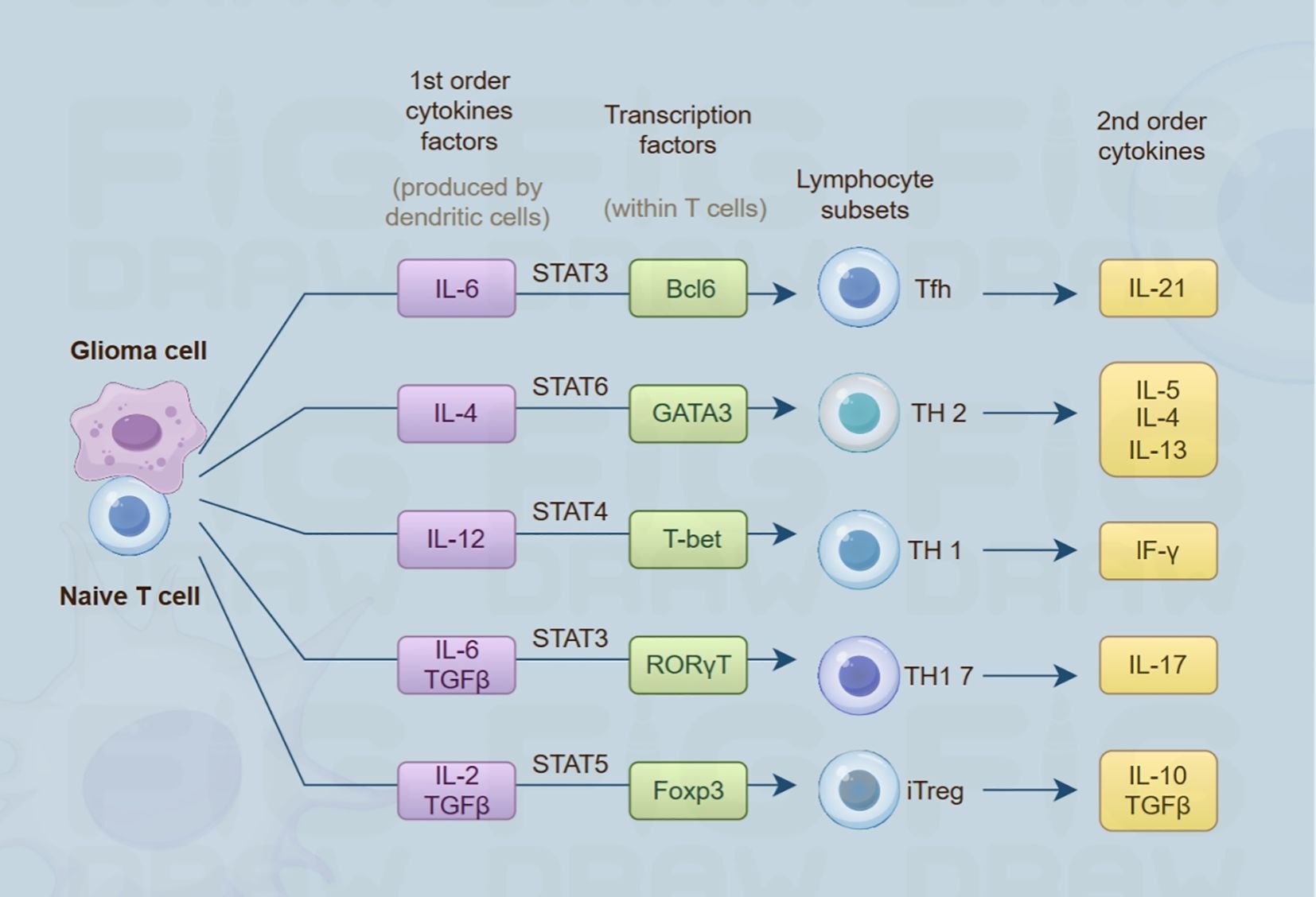
Figure 4. Conceptual illustration of the crosstalk between glioma cell populations and naive T lymphocytes. Glioma-derived cells release primary cytokines (including IL-6, IL-4, IL-12), triggering the activation of cognate transcription factors (such as STAT3, GATA3, T-bet) in naive T cells. These activated transcription factors guide naive T cells to differentiate into different lymphocyte subsets (including Tfh, TH2, TH1, etc.). Subsequently, these subsets produce 2nd-order cytokines (like IL-21, IL-5, IF-γ), which play important roles in the TME and immune regulation.
3.3 PD-L1 expression
The interaction between PD-L1 and the immune checkpoint receptor programmed death-1 (PD-1) on T cells conveys inhibitory signals to T cells. This results in the inhibition of T cell proliferation, cytokine generation, and cytotoxic function (68). This phenotype is referred to as “T cell exhaustion”. Consequently, T cells fail to efficiently identify and eliminate tumor cells. This enables tumor cells to dodge the immune system’s monitoring and elimination processes, thereby attaining immune evasion. Epigenetic modulation incontrovertibly assumes a pivotal role in PD-L1 expression. PD-L1 promoter methylation demonstrates an inverse association with PD-L1 transcriptional activity and clinical outcomes in patients (69). Furthermore, HDAC6, which acts as a histone acetylation “eraser,” lysine methyltransferase 2A (KMT2A), a methylation “writer,” and the bromodomain-containing BET family protein BRD4, an acetylation “reader,” each elevate PD-L1 immunoreactivity across melanomas, pancreatic carcinomas, and ovarian neoplasms. As such, targeting these proteins with small-molecule inhibitors can effectively reduce PD-L1 levels and enhance the body’s anti-tumor immune response (70). Additionally, nivolumab is a fully human immunoglobulin G4 monoclonal antibody targeting the programmed death-1 (PD-1) immune checkpoint receptor. Results from a phase III clinical trial (NCT02017717) comparing the efficacy of Nivolumab monotherapy versus bevacizumab in improving survival rates of patients with recurrent glioblastoma showed that the subgroup of glioblastoma patients with methylated MGMT promoters and no baseline corticosteroid dependence may be most likely to benefit from immune checkpoint inhibition (71, 72).
The immune landscape of gliomas is modulated by IDH1/2 genetic alterations, notably, PD-L1 expression is substantially decreased in mutated tumors, providing a mechanistic basis for immune checkpoint inhibitors (ICIs) in IDH1/2 wild-type (wt) patients. Beyond DNA methylation, additional epigenetic regulators may also modulate immune checkpoint (IC) expression in gliomas (73). MiRNAs directly modulate the expression of immune checkpoint regulators, including CTLA-4, PD-1, and PD-L1, via distinct miRNA species. For instance, miR-155 specifically targets CTLA-4, miR-138 fine-tunes both CTLA-4 and PD-1, miR-424 governs PD-L1 and CD80, miR-28 impacts PD-1, while miR-34a, miR-200, miR-513, and miR-138-5p collectively regulate PD-L1 expression (74, 75). Furthermore, overlapping or distinct miRNAs modulate the production of cytokines such as IFN-γ or transcription factors functioning as either activators or repressors of immune checkpoint pathways thereby establishing a redundant and highly intricate regulatory network.
3.4 Response to T cell attack
Tumor-infiltrating lymphocytes (TILs) exhibit robust antitumor immune capabilities and represent critical components of immunotherapeutic strategies. In the glioma TME, regulatory T cells (Tregs), CD4+ helper T cells (Th cells), and CD8+ cytotoxic T cells undergo infiltration. Tregs potently inhibit the functions of antitumor immune cells while elevating the abundance of other immunosuppressive cell types. Helper T cells-particularly Th1 subsets—and CD8+ T cells mediate antitumor immunity by respectively enhancing pro-inflammatory responses and executing tumor cell cytotoxicity (76). The efficacy of their antitumor actions is impeded due to limited tumor infiltration and the immunosuppressive milieu of glioma-derived TME. The primary driver of immune suppression is the glioma cells themselves, MDSCs, Tregs, and Tumor-Associated Microglia and Macrophages (TAMMs) through PD-L1 expression and TGF-β/IL-10 secretion. Immune-modulatory molecules trigger T cell functional impairment, encompassing anergy and exhaustion phenotypes. Within exhausted Th and CD8+ T cell subsets, proliferative capacity is diminished, and secretion of effector cytokines (e.g., IL-2, TNF-α, IFN-γ) is significantly reduced. Mechanistically, T cell exhaustion is typified by enhanced chromatin openness at immunosuppressive gene loci (PDCD1, CTLA4, LAG3, ENTPD1) and reduced accessibility at cell differentiation gene loci (IL7R, TCF7, LEF1), accompanied by corresponding upregulation and downregulation of transcriptional activity (77). Epigenetic mechanisms have been shown to influence cell-fate determinations in lymphocyte ontogeny (78, 79). Chromatin organization and histone-modifying enzymes represent critical regulators of dendritic cell (DC) functions. For instance, the histone H3K4 demethylase KDM5B exerts negative regulation on the activation of bone marrow-derived dendritic cells, leading to suboptimal T cell responses. Conversely, the methylated DNA “reader” methyl-CpG-binding domain protein 2 (MBD2) is indispensable for the phenotypic activation of dendritic cells and their capacity to prime T cell responses. Moreover, in-depth analysis of enhancer signatures like H3K4me1 and H3K27Ac has demonstrated that enhancers undergo substantial dynamics during T cell activation (80).
3.5 RNA modification
RNA modifications are chemical marks on the bases or riboses of RNA molecules. These modifications are widely present in various types of RNA (such as mRNA, tRNA, rRNA, lncRNA, etc.). To date, more than 150 different modifications have been identified, including methylation (e.g., m6A, m5C, m¹A), pseudouridylation (Ψ), acetylation, and so on. RNA modifications do not alter the sequence of RNA, but can dynamically regulate gene expression by affecting the structure, stability, translation efficiency of RNA and its interaction with other molecules. RNA modifications play a crucial role in the process of tumor immune evasion, reshaping the tumor microenvironment and interfering with immune surveillance mechanisms through multi-dimensional regulation (81, 82). For instance, in GBM, the long non-coding RNA CASC9 is involved in tumor progression by promoting glycolytic metabolism and is significantly associated with poor prognosis in patients. The underlying molecular mechanism involves regulation by N6-methyladenosine (m6A) modification: the m6A “reader” IGF2BP2 can recognize and bind to methylated CASC9, maintaining its function by enhancing the stability of its transcript. The molecular complex formed by these two further acts on the methylated region of hexokinase 2 (HK2) mRNA, directly driving glycolysis in GBM cells by improving the stability of this mRNA. The accumulation of large amounts of lactic acid resulting from enhanced glycolysis acidifies the tumor microenvironment (TME), thereby inducing immunosuppressive effects: the acidic environment not only impairs the proliferation, activation, and cytotoxic functions of immune effector cells such as T lymphocytes and natural killer (NK) cells, but also promotes the differentiation and expansion of regulatory T cells (Tregs). Ultimately, this inhibits systemic immune responses and facilitates tumor immune evasion (83, 84). A separate investigation revealed that targeting the m6A demethylase ALKBH5 disrupts YTHDF2-mediated stability of ZDHHC3 mRNA. This mechanism inhibits PD-L1 expression in gliomas by promoting PD-L1 degradation (85). Another study on immune evasion in gliomas demonstrated that the 28S rRNA methyltransferase NSUN5, via its cysteine 359 (C359)-dependent methyltransferase activity, directly binds to the chromatin-associated RNA (caRNA) of CTNNB1 and deposits 5-methylcytosine (m5C). This modification is oxidized to 5-hydroxymethylcytosine (5hmC) by TET2, which relies on TET2’s binding affinity for Fe²+ and α-KG. Subsequently, 5hmC is recognized by RBFOX2, a 5hmC-specific reader, which promotes the degradation of caRNA. This process downregulates β-catenin, enhances the phagocytic activity of tumor-associated macrophages (TAMs), and impairs the tumor’s ability to evade immunity (86).
4 Clinical implications and therapeutic strategies
The epigenetic panorama of gliomas reveals multiple modifications associated with tumor characteristics. Among gliomas, frequent occurrences include alterations in DNA methylation patterns, histone methylation/acetylation states, and variations in IDH mutation status. Strategies targeting the glioma epigenome are effective in tumor control and a valuable alternative. In glioma cells, epigenetic mechanisms have the ability to regulate immune responses, indicating that therapeutic approaches directed at epigenetic pathways may boost anti-tumor immune capabilities. Currently, several approaches targeting epigenetic alterations are in clinical trials: one targets mutated IDH with small molecule inhibitors and uses it for vaccination in relevant patients; another targets epigenetic modifiers like BET inhibitors (BETi), HDACi, DNMTi, and EZH2 inhibitors (EZH2i). Table 1 summarizes the corresponding clinical trials on clinical-trials.gov.
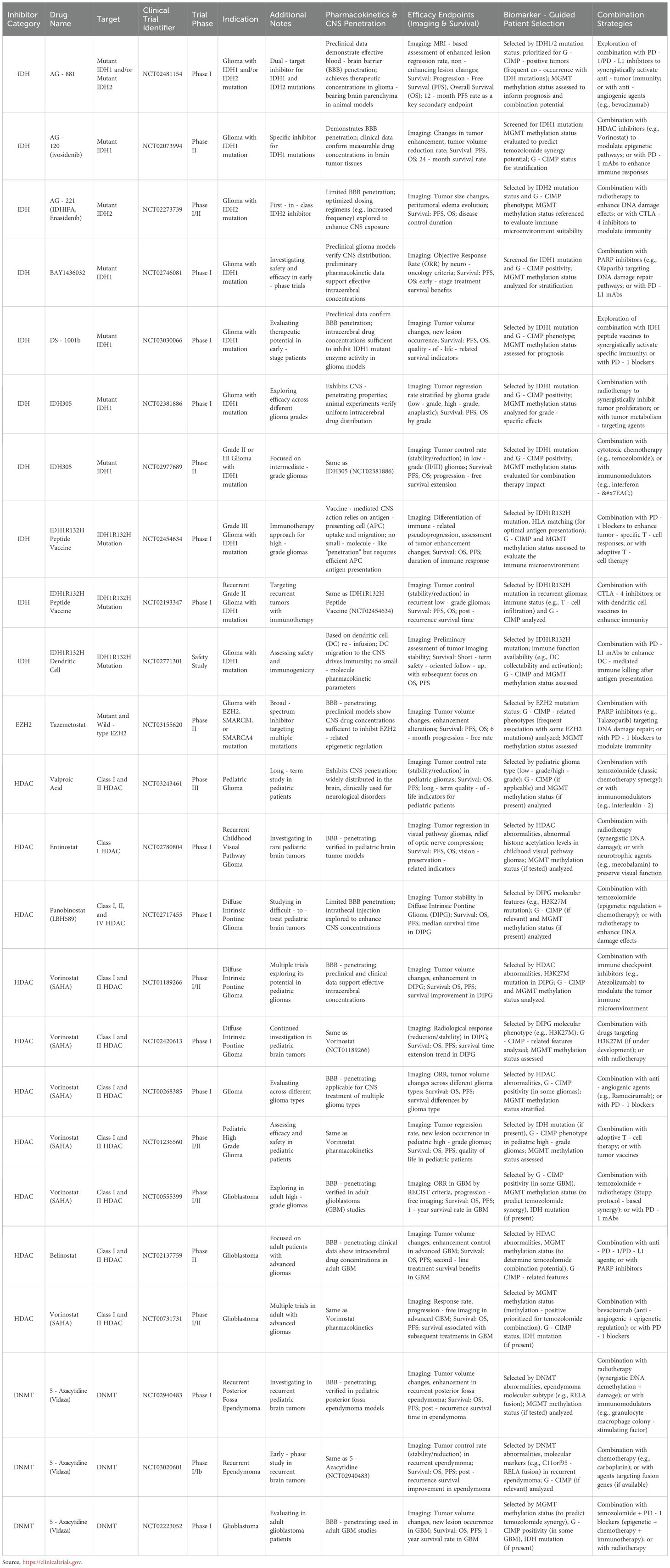
Table 1. Ongoing clinical trials testing epigenetic drugs in glioma.
4.1 Mutant IDH inhibitors
The aim of mutant IDH inhibitors is to block the synthesis of the oncometabolite 2-hydroxyglutarate (2HG). As a rival inhibitor of α-ketoglutarate-dependent dioxygenases, 2HG regulation can be achieved by suppressing its production, thereby reversing DNA hypermethylation and promoting the differentiation of mIDH1 glioma cells. Several mIDH1 inhibitors have shown effectiveness in both laboratory and animal models. Combining current first-line therapies (radiotherapy and temozolomide) with mIDH1 inhibitors and PD-L1-targeted immune checkpoint inhibitors (ICI) has enhanced tumor shrinkage in mIDH1 glioma-bearing mice, alleviated T cell exhaustion, and supported the formation of memory CD8+T cells. Presently, multiple clinical trials are assessing mIDH1 inhibitors for glioma treatment, although these remain in initial stages primarily focused on evaluating safety and the ability to reduce 2HG accumulation (87).
4.2 EZH2 inhibitors
Enhancer of zeste homolog 2 (EZH2), a pivotal histone methyltransferase in the PRC2, undergoes alterations in gliomas that lead to both gain- and loss-of-function mutations. These aberrations in EZH2 expression impact gene regulation by interacting with promoters and modulating methylation patterns, thereby acting as an oncogenic driver. EZH2-dependent silencing of TSGs drives oncogenic proliferation, aggressive invasion, and refractory resistance in cancer. The use of EZH2 inhibitors (EZH2is) has been shown to upregulate p16 expression and effectively curb the progression of gliomas. Research indicates that EZH2 serves as a pivotal regulator in regulating cancer cell immune response and mediates escape by downregulating immune activation genes, upregulating checkpoints, and creating an immunosuppressive TME. Tazemetostat, an EZH2i, is being tested in pediatric gliomas. However, A recent investigation indicates that it might influence the proliferation of primary Histone H3 Lysine 27 to Methionine Mutation (H3K27M)-mutant glioma cells occurs when functional p16INK4A is expressed in GBM cells harboring wild-type H3 and IDH (88). Short-term reduction of EZH2 correlates with decreased cell proliferation. However, recent findings show that extended EZH2 inhibition might induce a cell fate transition, promoting both proliferation and DNA damage repair mechanisms, which ultimately drives tumor progression (89).
4.3 DNA methyltransferase inhibitors
Epigenetic silencing by DNMTs suppresses gene expression via CpG island methylation. DNMTis reverse this, reactivating tumor suppressors and inducing anti-tumor effects. In gliomas, DNMTis upregulate MHC class I presentation, enhancing CD8+ T cell recognition. By preventing DNMT-driven epigenetic reprogramming, DNMTis also counteract T cell exhaustion. Genetic knockout of DNMT3A in CAR T cells mirrors this effect, bolstering anti-tumor immunity (50, 90). Preclinical research has shown DNMTi exhibit efficacy using in vitro and in vivo model systems of IDH-mutant (IDHmt) gliomas. Nevertheless, this preclinical promise has not yet led to successful clinical outcomes for glioma patients treated with DNMTi, potentially due to the S-phase specificity and relatively brief half-life of agents like 5-azacytidine and decitabine. Nevertheless, whether gliomas require general demethylation remains controversial, activation of proto-oncogenes coupled with hypomethylation-mediated reactivation of the DNA repair gene O6- methylguanine-DNA MGMT might confer resistance of glioblastoma to alkylating agents. Notably, low-dose demethylating agents can modulate immunity and potentially trigger an innate immune response through reactivation of retroviruses (91).
4.4 Histone deacetylase inhibitors
Certain frequent epigenetic alterations in neoplastic cells involve dysregulation of histone modifications within oncogene/tumor suppressor gene regulatory domains. Aberrant HDAC expression in cancer cells modifies the cell cycle and initiates tumorigenesis. HDAC expression patterns correlate with glioma malignancy grading; class II and IV HDAC isoforms exhibit reduced expression in GBM compared to low-grade astrocytomas. HDAC1 is excessively expressed in multiple glioma subtypes and associated with diminished overall survival. HDACis modulate antitumor immune responses by facilitating T cell chemokine production, augmenting PD-1-targeted immunotherapy effectiveness, and increasing PD-L1 and HLA-DR surface expression on tumor cells, suggesting a potential cooperative effect between HDACis and ICIs for glioma therapy. Vorinostat, evaluated in a phase II clinical trial for recurrent GBM, showed favorable tolerability as monotherapy and influenced GBM-relevant signaling pathways. Nevertheless, its clinical application is constrained by toxicity and suboptimal efficacy, potentially attributed to inadequate BBB penetration. Integrating HDACis with other therapeutic approaches or enhancing BBB permeability could improve treatment outcomes (92–94).
4.5 BET inhibitors
BRD4 function can be effectively suppressed by inhibitors or degraders. As small-molecule compounds, BRD4 inhibitors have the potential to augment cancer therapy by mimicking acetyl-lysine residues, thereby binding to BET proteins and ultimately interfering with the interaction between BET proteins, acetylated histones, and transcription factors (95). BET proteins play a critical role in driving high-level oncogene expression. BETi can attenuate oncogene transcription by diminishing super-enhancer activity, which consists of large aggregations of transcriptional enhancers that govern gene expression in cell identity and pathological conditions like cancer. Using in vivo RNA interference screening to target chromatin regulators critical for GBM cell viability, BRD4 emerged as a prominent target. Preclinical investigations using orthotopic mouse glioblastoma xenograft models have demonstrated the efficacy of multiple BETi compounds, such as JQ1, I-BET151, and OTX015 (96). Primary glioma cells with IDH mutation (IDHmt) demonstrate significant sensitivity to BET inhibitors JQ1 and GS-626510, exhibiting half-maximal inhibitory concentrations (IC50) 1,000 times lower than that of Temozolomide. A 2015 phase IIa clinical trial evaluating OTX015 for dose optimization in recurrent glioblastoma patients was halted due to insufficient therapeutic response. Alternative BET inhibitors featuring distinct pharmacokinetic properties are currently under preclinical assessment (97, 98).
4.6 Antigen-specific CAR T-cell immunotherapy
Chimeric antigen receptor (CAR)-modified T lymphocyte adoptive transfer emerges as a promising immunotherapeutic approach for central nervous system malignancies. Using genetic modification methods, autologous T cells are reprogrammed to display synthetic antigen receptors on their cellular membranes, enabling targeted recognition of tumor-associated antigens (61, 99). These genetically engineered CAR-T cells exhibit the ability to selectively identify and attach to neoplastic cells, subsequently triggering the immune-effector function of T lymphocytes to efficiently eradicate neoplastic cells (100). Actually, the success of this strategy relies on the recognition of relatively specific cancer targets on the cell surface, effectively overcoming the common immune evasion mechanisms adopted by tumor cells. Clinical investigations into CAR-T cell therapy for glioblastoma have devoted to five distinct antigens: EphA2, EGFRvIII, HER2, IL13Rα2, and GD2. The early results of these targets have been published, revealing some remarkable achievements (101, 102). Although CAR T cell therapy didn’t improve OS significantly, one glioblastoma patient survived 59 months after EGFRvIII-CAR T cell therapy without post-CAR treatment, and some on HER2-CAR T cell therapy had stable disease up to 29 months. A patient treated with anti-IL13Rα2 CAR T cells achieved a full remission within 7.5 months, showing potential.
However, tumor cell immune evasion against CAR T cells and inadequate CAR T cell trafficking to tumor loci represent primary challenges for CAR T cell therapy in solid tumor treatment, with patients frequently experiencing relapse following initial tumor regression. Thus, enhancing CAR T cell infiltration into tumor sites emerges as a critical strategy to circumvent this immune evasion (Figure 5). Studies have shown that upregulated chemokine receptor expression in CAR-T cells can boost their ability to infiltrate tumor microenvironments (103). For example, increased chromatin availability at the CD56 genomic region, together with chemokine upregulation including CXCL9, CXCL10, and CXCL12, enhances tumor infiltration by PD-1/TIM3/LAG3-modified CAR-T cells. Moreover, expression of CASTAT5 (a constantly active STAT5 isoform) fosters CD4+ T cell infiltration and migration to tumor microenvironments through epigenetic reprogramming. In addition, T cell dysfunction and impaired trafficking arise when miR-155 directly inhibits suppressor of cytokine signaling 1, disrupting normal regulatory mechanisms (104); The upregulation of miR-155 in CAR T cells appears to be an interesting strategy to promote the transportation of CAR T cells and an effective anti-tumor response (105); Targeting let-7 miRNA, which suppresses CCR2/CCR5 expression in T cells, might improve CAR-T cell infiltration by restoring chemokine receptor activity (106). Latest studies also show CAR T cell activation, proliferation, and survival are regulated epigenetically (88). For example, DNMT3A (a DNA methyltransferase) regulates epigenetic changes during chronic LCMV infection’s effector-to-exhaustion transition. In vivo, DNMT3A-deficient CAR-T cells outperform wild-type ones, enhanced by IL-10; double KO CAR-T cells confirm this. SUV39H1 (a histone methyltransferase) modulates chromatin and gene expression, and disrupting it in CAR-T cell therapy boosts cell expansion, persistence, and efficacy. TET2 (a methylcytosine dioxygenase) promotes DNA demethylation. BATF3, a key regulator of memory T cell proliferation and differentiation, plays a critical role in T cell exhaustion—particularly in TET2-knockout CAR-T cells—underscoring the therapeutic possibility of epigenetic modulation in CD8+ T lymphocytes (107). Leveraging epigenetic modulation to boosting CAR-T cell effectiveness presents a promising clinical approach to address the current limitations of solid tumor treatment, where conventional CAR-T approaches have shown limited success. Combining epigenetic agents with adoptive cell therapy may overcome barriers by reprogramming the TME and enhancing T cell persistence.
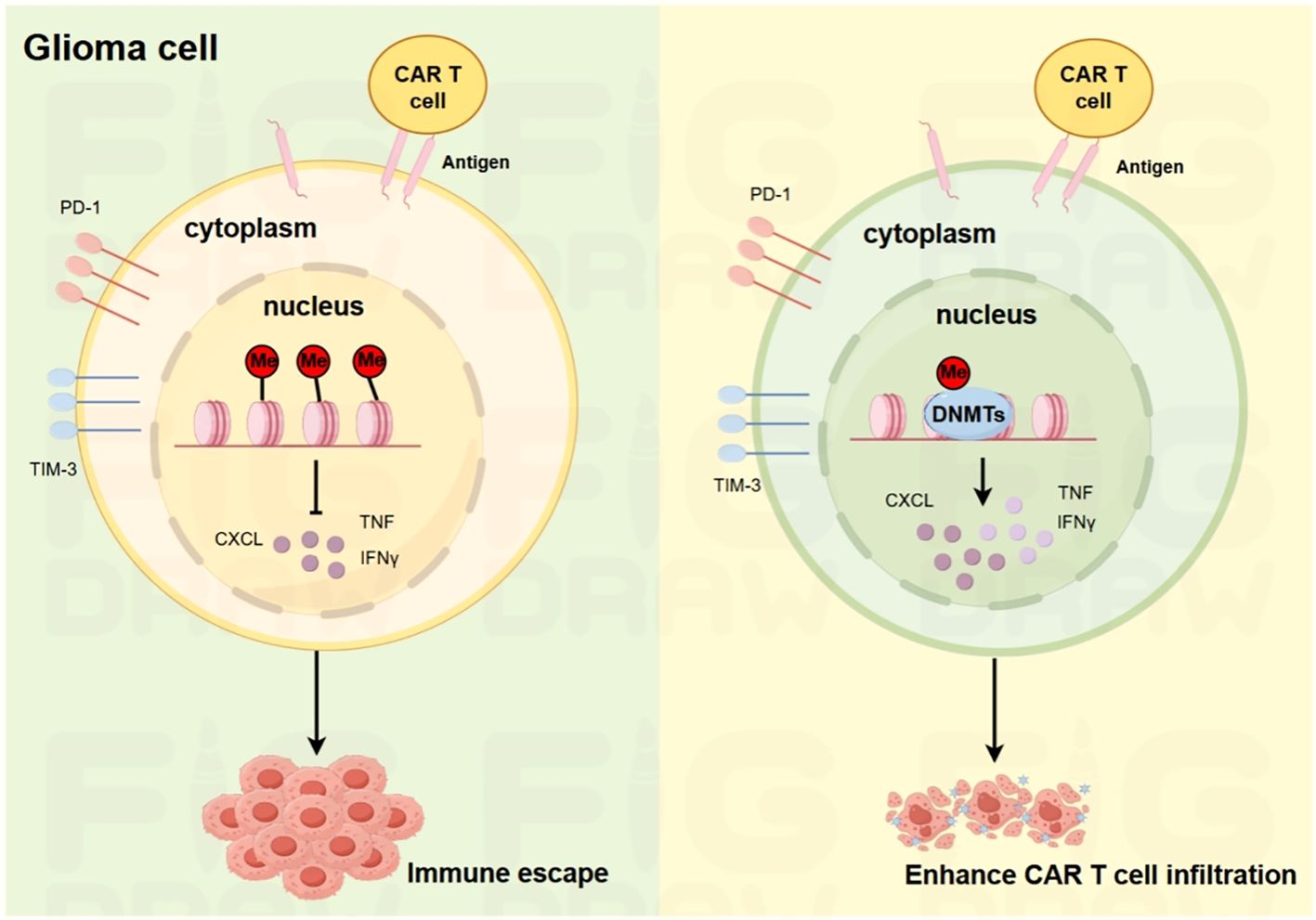
Figure 5. Schematic illustration of the mechanisms underlying glioma immune escape and enhanced CAR-T cell infiltration. On the left, in glioma cells, the expression of PD-1 and TIM-3, along with the release of cytokines such as CXCL, TNF, and IFNγ, contributes to immune escape. On the right, the introduction of DNMT in CAR-T cells modulates the cellular environment, reducing immune-suppressive factors and enhancing the infiltration ability of CAR-T cells into glioma tissues.
5 Toward clinical translation: challenges and opportunities
Immunotherapy has emerged as a rapidly evolving field in glioma treatment, but it is fraught with multiple hurdles. Firstly, the post-treatment local immunosuppressive TME restricts therapeutic efficacy, yielding modest outcomes that only benefit a small subset of patients. Secondly, gliomas are characterized by a scarcity of distinct tumor antigens and significant intratumoral heterogeneity, complicating targeted immunotherapeutic approaches. Thirdly, the chronic immunotoxicity associated with immunotherapies and their long-term sequelae pose substantial concerns. Despite preclinical research and early-stage (I/II) therapeutic trials have produced promising data, and some individual cases have demonstrated success, advancing from phase II/III trials remains a formidable obstacle. Currently, there are no reported Phase III clinical trials that have revealed the efficacy of immunotherapy across large patient populations with glioma (108, 109).
Epigenetic modifications, ubiquitous in tumors, play a key role in establishing and maintaining the heterogeneity of glioblastoma (GBM). Aberrant epigenetic regulation is a major driver of GBM initiation, and dysregulation of epigenetic modulators promotes tumor formation. Epigenetic drugs such as DNMT inhibitors (e.g., 5-azacytidine and decitabine) have shown anti-tumor activity in preclinical GBM models and have been approved by the FDA for the treatment of various tumors (110, 111). However, the clinical efficacy of the EZH2 inhibitor tazemetostat in GBM remains controversial, and its specific benefits require careful evaluation (112, 113); HDAC inhibitors (HDACIs), on the other hand, hold potential as therapeutic agents by regulating oncogene transcription and cell cycle, among other processes (114). Recent studies have identified lactate-derived histone lactylation as a novel modification associated with GBM progression (115). Despite the great potential of epigenetic strategies in glioma treatment and the fact that multiple inhibitors have entered clinical trials, results from large-cohort trials have been suboptimal. Off-target effects, difficulties in crossing the blood-brain barrier, and tumor heterogeneity are the main reasons, making it imperative to optimize drug delivery and targeting strategies.
To overcome the therapeutic bottlenecks of applying epigenetic drugs in GBM, the integration of medicine and engineering has provided new ideas for drug delivery and targeting, such as zinc ionophores, leveraging their tissue specificity, are combined with CpG nanoparticles and AMD-Zn to construct injectable hydrogel systems (imGEL), which enhance drug efficacy and inhibit recurrence through the tissue affinity of zinc particles and the diffusion-retention properties of hydrogels (116); special hydrogel composites combined with GSC-specific CAR-macrophages, when injected into the tumor cavity after GBM resection in mice, exert strong immunotoxicity against tumors to suppress recurrence. In addition, direct intratumoral administration is highly effective: sonodynamic therapy (SDT) eradicates GBM cells by activating photosensitizers with ultrasound to generate reactive oxygen species and cavitation bubbles (117); oncolytic viruses (OVs), after genetic engineering to target tumor cell receptors, can trigger host immune responses to clear cancer cells. Advances in OV delivery technologies have overcome the blood-brain barrier, such as the convection-enhanced delivery of PVSRIPO reported by Desjardins et al., which targets GBM via CD155 (118). Preliminary data show that patients receiving PVSRIPO have higher survival rates than historical controls, and its phase II trial (NCT02986178) alone or in combination with lomustine is underway, with its efficacy in GBM patients highly anticipated (119).
CAR-T therapy succeeds in hematologic malignancies but fails in solid tumors partly due to CAR-T exhaustion, making combination strategies promising for gliomas (120). EZH2, a PRC2 component catalyzing H3K27me3, is key for T-cell functions. A study showed that resting exhausted CAR-T cells with EZH2 inhibitor tazemetostat (D11-15) remodeled exhaustion-related epigenome via EZH2, restoring function (120, 121). Another study identified class I HDACi (e.g., M344, chidamide) as CAR-T enhancers via screening 370 drugs, enhancing memory and anti-exhaustion to induce sustained anti-tumor effects. Mechanistically, HDACi activates Wnt/β-catenin pathway by regulating HDAC1, H3K27ac, and TCF4/LEF1/CTNNB1 (122). Additionally, DNMT3A (a major de novo methyltransferase) upregulates after TCR activation, mediating T-cell polarization via methylation. De novo methylation exacerbates T-cell exhaustion, while DNMT3A deficiency reduces methylation at key loci, enhancing anti-PD-L1 response. Low-dose decitabine pretreatment enhanced T-cell response in mice. In solid tumor (including glioma) CAR-T models, DNMT3A deficiency boosted CAR-T expansion, cytokine secretion, and lysis; after chronic stimulation, these cells showed higher TCF1/LEF1 and stem/naive-like epigenetics, increasing in vivo anti-tumor activity (123, 124).
CAR-T and epigenetic combination therapy offers hope for glioma treatment but faces challenges: glioma heterogeneity limits CAR-T recognition, the complex brain microenvironment inhibits CAR-T function, and patient variability demands personalized plans. However, it provides new insights, with deeper research potentially yielding precise strategies like optimized targets, epigenetic drug combinations, and biomarkers. A GO analysis linked DNMT3A-regulated genes to pathways like TCR signaling, proposing DNMT3A depletion signatures as biomarkers for patient selection and response prediction (123). Another study on pediatric high-grade CNS tumors noted variable B7-H3 expression (a potential marker for CAR-T), recommending tumor tissue testing during B7-H3-targeted trials to assess IHC B7-H3 as a biomarker for tailored treatment (125). Looking forward, single-cell and spatial epigenomic mapping will help address these challenges. They can capture CAR-T cell exhaustion trajectories, functional heterogeneity and epigenetic interactions with tumor cells, and analyze spatial correlations between regional tumor epigenetic landscapes, immune infiltration and CAR-T function. This will advance CAR-T-epigenetic combination therapy from “broad-spectrum exploration” to “precision matching”, supporting personalized treatment of gliomas and other solid tumors.
Author contributions
DW: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Investigation, Validation. DJ: Conceptualization, Writing – original draft. YZ: Writing – original draft, Conceptualization. WL: Conceptualization, Writing – original draft. QL: Supervision, Writing – review & editing. YL: Supervision, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by “Phoenix Introduction Plan” Talent Startover Project of Tangdu Hospital, Fourth Military Medical University (grant number 2022YFJH001) and the National Natural Science Foundation of China (grant number 82203445, 82430047, 82221001).
Acknowledgments
The authors sincerely appreciate Professor Minggao Zhao and Le Yang from Tangdu hospital of Fourth Military Medical University for their critical suggestions. We also would like to thank Editage (www.editage.cn) for the English language improvement.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bi J, Chowdhry S, Wu S, Zhang W, Masui K, and Mischel PS. Altered cellular metabolism in gliomas-an emerging landscape of actionable co-dependency targets. Nat Rev Cancer. (2020) 20:57–70. doi: 10.1038/s41568-019-0226-5
2. Bhatia A, Moreno R, Reiner AS, Nandakumar S, Walch HS, Thomas TM, et al. Tumor volume growth rates and doubling times during active surveillance of IDH-mutant low-grade glioma. Clin Cancer Res. (2024) 30:106–15. doi: 10.1158/1078-0432.CCR-23-1180
3. Dewdney B, Jenkins MR, Best SA, Freytag S, Prasad K, Holst J, et al. From signalling pathways to targeted therapies: unravelling glioblastoma’s secrets and harnessing two decades of progress. Signal Transduct Target Ther. (2023) 8:400. doi: 10.1038/s41392-023-01637-8
4. Rechberger JS, Power BT, Power EA, Nesvick CL, and Daniels DJ. H3K27-altered diffuse midline glioma: a paradigm shifting opportunity in direct delivery of targeted therapeutics. Expert Opin Ther Targets. (2023) 27:9–17. doi: 10.1080/14728222.2023.2177531
5. Ocasio JK, Budd KM, Roach JT, Andrews JM, and Baker SJ. Oncohistones and disrupted development in pediatric-type diffuse high-grade glioma. Cancer Metastasis Rev. (2023) 42:367–88. doi: 10.1007/s10555-023-10105-2
6. Diaz Rosario M, Kaur H, Tasci E, Shankavaram U, Sproull M, Zhuge Y, et al. The next frontier in health disparities-A closer look at exploring sex differences in glioma data and omics analysis, from bench to bedside and back. Biomolecules. (2022) 12:1–19. doi: 10.3390/biom12091203
7. Zhang J, Sun R, Lyu Y, Liu C, Liu Y, Feng Y, et al. Proteomic profiling of gliomas unveils immune and metabolism-driven subtypes with implications for anti-nucleotide metabolism therapy. Nat Commun. (2024) 15:10005. doi: 10.1038/s41467-024-54352-5
8. van den Bent MJ, Geurts M, French PJ, Smits M, Capper D, Bromberg JEC, et al. Primary brain tumours in adults. Lancet. (2023) 402:1564–79. doi: 10.1016/S0140-6736(23)01054-1
9. Yasinjan F, Xing Y, Geng H, Guo R, Yang L, Liu Z, et al. Immunotherapy: a promising approach for glioma treatment. Front Immunol. (2023) 14:1255611. doi: 10.3389/fimmu.2023.1255611
10. Su H, Peng Y, Wu Y, and Zeng X. Overcoming immune evasion with innovative multi-target approaches for glioblastoma. Front Immunol. (2025) 16:1541467. doi: 10.3389/fimmu.2025.1541467
11. Elguindy MM, Young JS, Ho WS, and Lu RO. Co-evolution of glioma and immune microenvironment. J Immunother Cancer. (2024) 12:1–15. doi: 10.1136/jitc-2024-009175
12. Dunn GP, Old LJ, and Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
13. Galassi C, Chan TA, Vitale I, and Galluzzi L. The hallmarks of cancer immune evasion. Cancer Cell. (2024) 42:1825–63. doi: 10.1016/j.ccell.2024.09.010
14. Zhao LY, Song J, Liu Y, Song CX, and Yi C. Mapping the epigenetic modifications of DNA and RNA. Protein Cell. (2020) 11:792–808. doi: 10.1007/s13238-020-00733-7
15. Nanamori H and Sawada Y. Epigenetic modification of PD-1/PD-L1-mediated cancer immunotherapy against melanoma. Int J Mol Sci. (2022) 23:1119. doi: 10.3390/ijms23031119
16. Tao S, Liang S, Zeng T, and Yin D. Epigenetic modification-related mechanisms of hepatocellular carcinoma resistance to immune checkpoint inhibition. Front Immunol. (2022) 13:1043667. doi: 10.3389/fimmu.2022.1043667
17. Qin S, Xie B, Wang Q, Yang R, Sun J, Hu C, et al. New insights into immune cells in cancer immunotherapy: from epigenetic modification, metabolic modulation to cell communication. MedComm (2020). (2024) 5:e551. doi: 10.1002/mco2.551
18. Chow A, Perica K, Klebanoff CA, and Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol. (2022) 19:775–90. doi: 10.1038/s41571-022-00689-z
19. Jones PA, Ohtani H, Chakravarthy A, and De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. (2019) 19:151–61. doi: 10.1038/s41568-019-0109-9
20. Lin MD, Tsai AC, Abdullah KG, McBrayer SK, and Shi DD. Treatment of IDH-mutant glioma in the INDIGO era. NPJ Precis Oncol. (2024) 8:149. doi: 10.1038/s41698-024-00646-2
21. van den Bent MJ, French PJ, Brat D, Tonn JC, Touat M, Ellingson BM, et al. The biological significance of tumor grade, age, enhancement, and extent of resection in IDH-mutant gliomas: How should they inform treatment decisions in the era of IDH inhibitors? Neuro Oncol. (2024) 26:1805–22. doi: 10.1093/neuonc/noae107
22. DeCordova S, Shastri A, Tsolaki AG, Yasmin H, Klein L, Singh SK, et al. Molecular heterogeneity and immunosuppressive microenvironment in glioblastoma. Front Immunol. (2020) 11:1402. doi: 10.3389/fimmu.2020.01402
23. Jegathesan Y, Stephen PP, Sati I, Narayanan P, Monif M, and Kamarudin MNA. MicroRNAs in adult high-grade gliomas: Mechanisms of chemotherapeutic resistance and their clinical relevance. BioMed Pharmacother. (2024) 172:116277. doi: 10.1016/j.biopha.2024.116277
24. Drucker KL, Jenkins RB, and Schramek D. Switching drivers: epigenetic rewiring to genetic progression in glioma. Cancer Res. (2025) 85:836–7. doi: 10.1158/0008-5472.CAN-24-4907
25. Wu J, Gonzalez Castro LN, Battaglia S, El Farran CA, D’Antonio JP, Miller TE, et al. Evolving cell states and oncogenic drivers during the progression of IDH-mutant gliomas. Nat Cancer. (2025) 6:145–57. doi: 10.1038/s43018-024-00865-3
26. de la Fuente MI, Touat M, van den Bent MJ, Preusser M, Peters KB, Young RJ, et al. The role of vorasidenib in the treatment of isocitrate dehydrogenase-mutant glioma. Neuro Oncol. (2024). doi: 10.1093/neuonc/noae259
27. Caccese M, Indraccolo S, Zagonel V, and Lombardi G. PD-1/PD-L1 immune-checkpoint inhibitors in glioblastoma: A concise review. Crit Rev Oncol Hematol. (2019) 135:128–34. doi: 10.1016/j.critrevonc.2018.12.002
28. Sanders S and Debinski W. Challenges to successful implementation of the immune checkpoint inhibitors for treatment of glioblastoma. Int J Mol Sci. (2020) 21:2759. doi: 10.3390/ijms21082759
29. Wu Q, You L, Nepovimova E, Heger Z, Wu W, Kuca K, et al. Hypoxia-inducible factors: master regulators of hypoxic tumor immune escape. J Hematol Oncol. (2022) 15:77. doi: 10.1186/s13045-022-01292-6
30. Karsy M, Guan J, Cohen AL, Jensen RL, and Colman H. New molecular considerations for glioma: IDH, ATRX, BRAF, TERT, H3 K27M. Curr Neurol Neurosci Rep. (2017) 17:19. doi: 10.1007/s11910-017-0722-5
31. Aguilera P and López-Contreras AJ. ATRX, a guardian of chromatin. Trends Genet. (2023) 39:505–19. doi: 10.1016/j.tig.2023.02.009
32. Accolla RS, Ramia E, Tedeschi A, and Forlani G. CIITA-driven MHC class II expressing tumor cells as antigen presenting cell performers: toward the construction of an optimal anti-tumor vaccine. Front Immunol. (2019) 10:1806. doi: 10.3389/fimmu.2019.01806
33. Laugesen A, Højfeldt JW, and Helin K. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol Cell. (2019) 74:8–18. doi: 10.1016/j.molcel.2019.03.011
34. Bieluszewski T, Xiao J, Yang Y, and Wagner D. PRC2 activity, recruitment, and silencing: a comparative perspective. Trends Plant Sci. (2021) 26:1186–98. doi: 10.1016/j.tplants.2021.06.006
35. Yao J, Bergsland E, Aggarwal R, Aparicio A, Beltran H, Crabtree JS, et al. DLL3 as an emerging target for the treatment of neuroendocrine neoplasms. Oncologist. (2022) 27:940–51. doi: 10.1093/oncolo/oyac161
36. Xi J, Sun Q, Ma L, and Kang J. Long non-coding RNAs in glioma progression. Cancer Lett. (2018) 419:203–9. doi: 10.1016/j.canlet.2018.01.041
37. Uddin MS, Mamun AA, Alghamdi BS, Tewari D, Jeandet P, Sarwar MS, et al. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin Cancer Biol. (2022) 83:100–20. doi: 10.1016/j.semcancer.2020.12.015
38. Yu T, Ma P, Wu D, Shu Y, and Gao W. Functions and mechanisms of microRNA-31 in human cancers. BioMed Pharmacother. (2018) 108:1162–9. doi: 10.1016/j.biopha.2018.09.132
39. Li H, Gao J, and Zhang S. Functional and clinical characteristics of cell adhesion molecule CADM1 in cancer. Front Cell Dev Biol. (2021) 9:714298. doi: 10.3389/fcell.2021.714298
40. Wu X, Shi M, Lian Y, and Zhang H. Exosomal circRNAs as promising liquid biopsy biomarkers for glioma. Front Immunol. (2023) 14:1039084. doi: 10.3389/fimmu.2023.1039084
41. Mafi A, Hedayati N, Kahkesh S, Khoshayand S, Alimohammadi M, Farahani N, et al. The landscape of circRNAs in gliomas temozolomide resistance: Insights into molecular pathways. Noncoding RNA Res. (2024) 9:1178–89. doi: 10.1016/j.ncrna.2024.05.010
42. Sun J, Li B, Shu C, Ma Q, and Wang J. Functions and clinical significance of circular RNAs in glioma. Mol Cancer. (2020) 19:34. doi: 10.1186/s12943-019-1121-0
43. Kang QM, Wang J, Chen SM, Song SR, and Yu SC. Glioma-associated mesenchymal stem cells. Brain. (2024) 147:755–65. doi: 10.1093/brain/awad360
44. Agosti E, Antonietti S, Ius T, Fontanella MM, Zeppieri M, and Panciani PP. Glioma stem cells as promoter of glioma progression: A systematic review of molecular pathways and targeted therapies. Int J Mol Sci. (2024) 25:7979. doi: 10.3390/ijms25147979
45. Mulkearns-Hubert EE and Lathia JD. DUB-ling down on the epigenetic regulation of cancer stem cells in glioblastoma. Neuro Oncol. (2023) 25:854–6. doi: 10.1093/neuonc/noad040
46. Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. (2021) 184:2454–70.e26. doi: 10.1016/j.cell.2021.03.023
47. Affronti HC and Wellen KE. Epigenetic control of fatty-acid metabolism sustains glioma stem cells. Cancer Discov. (2019) 9:1161–3. doi: 10.1158/2159-8290.CD-19-0733
48. Liu L, Liu Z, Liu Q, Wu W, Lin P, Liu X, et al. LncRNA INHEG promotes glioma stem cell maintenance and tumorigenicity through regulating rRNA 2’-O-methylation. Nat Commun. (2023) 14:7526. doi: 10.1038/s41467-023-43113-5
49. Martinez PJ, Green AL, and Borden MA. Targeting diffuse midline gliomas: The promise of focused ultrasound-mediated blood-brain barrier opening. J Control Release. (2024) 365:412–21. doi: 10.1016/j.jconrel.2023.11.037
50. McClellan BL, Haase S, Nunez FJ, Alghamri MS, Dabaja AA, Lowenstein PR, et al. Impact of epigenetic reprogramming on antitumor immune responses in glioma. J Clin Invest. (2023) 133:e163450. doi: 10.1172/JCI163450
51. Nejo T, Yamamichi A, Almeida ND, Goretsky YE, and Okada H. Tumor antigens in glioma. Semin Immunol. (2020) 47:101385. doi: 10.1016/j.smim.2020.101385
52. García-López D, Zaragoza-Ojeda M, Eguía-Aguilar P, and Arenas-Huertero F. Endoplasmic reticulum stress in gliomas: exploiting a dual-effect dysfunction through chemical pharmaceutical compounds and natural derivatives for therapeutical uses. Int J Mol Sci. (2024) 25:4078. doi: 10.3390/ijms25074078
53. Obacz J, Avril T, Le Reste PJ, Urra H, Quillien V, Hetz C, et al. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci Signal. (2017) 10:eaal2323. doi: 10.1126/scisignal.aal2323
54. Hirschhorn D, Budhu S, Kraehenbuehl L, Gigoux M, Schroder D, Chow A, et al. T cell immunotherapies engage neutrophils to eliminate tumor antigen escape variants. Cell. (2023) 186:1432–47.e17. doi: 10.1016/j.cell.2023.03.007
55. Sanchez ML, Mangas A, and Covenas R. Glioma and peptidergic systems: oncogenic and anticancer peptides. Int J Mol Sci. (2024) 25:7990. doi: 10.3390/ijms25147990
56. Chen X, Lu Q, Zhou H, Liu J, Nadorp B, Lasry A, et al. A membrane-associated MHC-I inhibitory axis for cancer immune evasion. Cell. (2023) 186:3903–20.e21. doi: 10.1016/j.cell.2023.07.016
57. Sharma P, Goswami S, Raychaudhuri D, Siddiqui BA, Singh P, Nagarajan A, et al. Immune checkpoint therapy-current perspectives and future directions. Cell. (2023) 186:1652–69. doi: 10.1016/j.cell.2023.03.006
58. Aldape K, Zadeh G, Mansouri S, Reifenberger G, and von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol. (2015) 129:829–48. doi: 10.1007/s00401-015-1432-1
59. Chuntova P, Yamamichi A, Chen T, Narayanaswamy R, Ronseaux S, Hudson C, et al. Inhibition of D-2HG leads to upregulation of a proinflammatory gene signature in a novel HLA-A2/HLA-DR1 transgenic mouse model of IDH1R132H-expressing glioma. J Immunother Cancer. (2022) 10:e004644. doi: 10.1136/jitc-2022-004644
60. Brien GL, Bressan RB, Monger C, Gannon D, Lagan E, Doherty AM, et al. Simultaneous disruption of PRC2 and enhancer function underlies histone H3.3-K27M oncogenic activity in human hindbrain neural stem cells. Nat Genet. (2021) 53:1221–32. doi: 10.1038/s41588-021-00897-w
61. Low JT, Brown MC, Reitman ZJ, Bernstock JD, Markert JM, Friedman GK, et al. Understanding and therapeutically exploiting cGAS/STING signaling in glioblastoma. J Clin Invest. (2024) 134:e163452. doi: 10.1172/JCI163452
62. Mackenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. (2017) 548:461–5. doi: 10.1038/nature23449
63. Sabu A, Liu TI, Ng SS, Doong RA, Huang YF, and Chiu HC. Nanomedicines targeting glioma stem cells. ACS Appl Mater Interfaces. (2023) 15:158–81. doi: 10.1021/acsami.2c03538
64. Li L, Zhang T, Xiao M, Lu Y, and Gao L. Brain macrophage senescence in glioma. Semin Cancer Biol. (2024) 104-105:46–60. doi: 10.1016/j.semcancer.2024.07.005
65. Duan R, Du W, and Guo W. EZH2: a novel target for cancer treatment. J Hematol Oncol. (2020) 13:104. doi: 10.1186/s13045-020-00937-8
66. Verdugo E, Puerto I, and Medina M. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun (Lond). (2022) 42:1083–111. doi: 10.1002/cac2.12361
67. Zhang W, Zhang L, Dong H, and Peng H. TGIF2 is a potential biomarker for diagnosis and prognosis of glioma. Front Immunol. (2024) 15:1356833. doi: 10.3389/fimmu.2024.1356833
68. Sun C, Mezzadra R, and Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. (2018) 48:434–52. doi: 10.1016/j.immuni.2018.03.014
69. Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. (2019) 18:10. doi: 10.1186/s12943-018-0928-4
70. Fukumoto T, Fatkhutdinov N, Zundell JA, Tcyganov EN, Nacarelli T, Karakashev S, et al. HDAC6 inhibition synergizes with anti-PD-L1 therapy in ARID1A-inactivated ovarian cancer. Cancer Res. (2019) 79:5482–9. doi: 10.1158/0008-5472.CAN-19-1302
71. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A, et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
72. Baskaran AB, Kozel OA, Venkatesh O, Wainwright DA, Sonabend AM, Heimberger AB, et al. Immune checkpoint inhibitors in glioblastoma IDHwt treatment: A systematic review. Cancers (Basel). (2024) 16:4148. doi: 10.3390/cancers16244148
73. Guo H, Vuille JA, Wittner BS, Lachtara EM, Hou Y, Lin M, et al. DNA hypomethylation silences anti-tumor immune genes in early prostate cancer and CTCs. Cell. (2023) 186:2765–82.e28. doi: 10.1016/j.cell.2023.05.028
74. Davidson CL, Vengoji R, Jain M, Batra SK, and Shonka N. Biological, diagnostic and therapeutic implications of exosomes in glioma. Cancer Lett. (2024) 582:216592. doi: 10.1016/j.canlet.2023.216592
75. Spoerl D, Duroux-Richard I, Louis-Plence P, and Jorgensen C. The role of miR-155 in regulatory T cells and rheumatoid arthritis. Clin Immunol. (2013) 148:56–65. doi: 10.1016/j.clim.2013.03.010
76. Gillard AG, Shin DH, Hampton LA, Lopez-Rivas A, Parthasarathy A, Fueyo J, et al. Targeting innate immunity in glioma therapy. Int J Mol Sci. (2024) 25:947. doi: 10.3390/ijms25020947
77. Cha JH, Chan LC, Li CW, Hsu JL, and Hung MC. Mechanisms controlling PD-L1 expression in cancer. Mol Cell. (2019) 76:359–70. doi: 10.1016/j.molcel.2019.09.030
78. Verykokakis M and Kee BL. Transcriptional and epigenetic regulation of innate-like T lymphocyte development. Curr Opin Immunol. (2018) 51:39–45. doi: 10.1016/j.coi.2018.01.006
79. Sun L, Su Y, Jiao A, Wang X, and Zhang B. T cells in health and disease. Signal Transduct Target Ther. (2023) 8:235. doi: 10.1038/s41392-023-01471-y
80. Koschmann C, Al-Holou WN, Alonso MM, Anastas J, Bandopadhayay P, Barron T, et al. A road map for the treatment of pediatric diffuse midline glioma. Cancer Cell. (2024) 42:1–5. doi: 10.1016/j.ccell.2023.11.002
81. Han D and Xu MM. RNA modification in the immune system. Annu Rev Immunol. (2023) 41:73–98. doi: 10.1146/annurev-immunol-101921-045401
82. Orsolic I, Carrier A, and Esteller M. Genetic and epigenetic defects of the RNA modification machinery in cancer. Trends Genet. (2023) 39:74–88. doi: 10.1016/j.tig.2022.10.004
83. Liu Z, Wang T, She Y, Wu K, Gu S, Li L, et al. N(6)-methyladenosine-modified circIGF2BP3 inhibits CD8(+) T-cell responses to facilitate tumor immune evasion by promoting the deubiquitination of PD-L1 in non-small cell lung cancer. Mol Cancer. (2021) 20:105. doi: 10.1186/s12943-021-01398-4
84. Li C, Li B, Wang H, Qu L, Liu H, Weng C, et al. Role of N6-methyladenosine methylation in glioma: recent insights and future directions. Cell Mol Biol Lett. (2023) 28:103. doi: 10.1186/s11658-023-00514-0
85. Tang W, Xu N, Zhou J, He Z, Lenahan C, Wang C, et al. ALKBH5 promotes PD-L1-mediated immune escape through m6A modification of ZDHHC3 in glioma. Cell Death Discov. (2022) 8:497. doi: 10.1038/s41420-022-01286-w
86. Wu R, Sun C, Chen X, Yang R, Luan Y, Zhao X, et al. NSUN5/TET2-directed chromatin-associated RNA modification of 5-methylcytosine to 5-hydroxymethylcytosine governs glioma immune evasion. Proc Natl Acad Sci U S A. (2024) 121:e2321611121. doi: 10.1073/pnas.2321611121
87. Spitzer A, Gritsch S, Nomura M, Jucht A, Fortin J, Raviram R, et al. Mutant IDH inhibitors induce lineage differentiation in IDH-mutant oligodendroglioma. Cancer Cell. (2024) 42:904–14.e9. doi: 10.1016/j.ccell.2024.03.008
88. Saratsis AM, Knowles T, Petrovic A, and Nazarian J. H3K27M mutant glioma: Disease definition and biological underpinnings. Neuro Oncol. (2024) 26:S92–S100. doi: 10.1093/neuonc/noad164
89. Gusyatiner O and Hegi ME. Glioma epigenetics: From subclassification to novel treatment options. Semin Cancer Biol. (2018) 51:50–8. doi: 10.1016/j.semcancer.2017.11.010
90. Kanwore K, Kanwore K, Adzika GK, Abiola AA, Guo X, Kambey PA, et al. Cancer metabolism: the role of immune cells epigenetic alteration in tumorigenesis, progression, and metastasis of glioma. Front Immunol. (2022) 13:831636. doi: 10.3389/fimmu.2022.831636
91. Butler M, Pongor L, Su YT, Xi L, Raffeld M, Quezado M, et al. MGMT status as a clinical biomarker in glioblastoma. Trends Cancer. (2020) 6:380–91. doi: 10.1016/j.trecan.2020.02.010
92. Chen R, Zhang M, Zhou Y, Guo W, Yi M, Zhang Z, et al. The application of histone deacetylases inhibitors in glioblastoma. J Exp Clin Cancer Res. (2020) 39:138. doi: 10.1186/s13046-020-01643-6
93. Eyüpoglu IY and Savaskan NE. Epigenetics in brain tumors: HDACs take center stage. Curr Neuropharmacol. (2016) 14:48–54. doi: 10.2174/1570159X13666151030162457
94. Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. (2009) 27:2052–8. doi: 10.1200/JCO.2008.19.0694
95. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. (2010) 468:1067–73. doi: 10.1038/nature09504
96. Duan W, Yu M, and Chen J. BRD4: New hope in the battle against glioblastoma. Pharmacol Res. (2023) 191:106767. doi: 10.1016/j.phrs.2023.106767
97. Berenguer-Daizé C, Astorgues-Xerri L, Odore E, Cayol M, Cvitkovic E, Noel K, et al. OTX015 (MK-8628), a novel BET inhibitor, displays in vitro and in vivo antitumor effects alone and in combination with conventional therapies in glioblastoma models. Int J Cancer. (2016) 139:2047–55. doi: 10.1002/ijc.30256
98. Cheng Z, Gong Y, Ma Y, Lu K, Lu X, Pierce LA, et al. Inhibition of BET bromodomain targets genetically diverse glioblastoma. Clin Cancer Res. (2013) 19:1748–59. doi: 10.1158/1078-0432.CCR-12-3066
99. Lin YJ, Mashouf LA, and Lim M. CAR T cell therapy in primary brain tumors: current investigations and the future. Front Immunol. (2022) 13:817296. doi: 10.3389/fimmu.2022.817296
100. Ahn T, Bae EA, and Seo H. Decoding and overcoming T cell exhaustion: Epigenetic and transcriptional dynamics in CAR-T cells against solid tumors. Mol Ther. (2024) 32:1617–27. doi: 10.1016/j.ymthe.2024.04.004
101. Ma K and Hu P. Chimeric antigen receptor T-cell therapy for glioblastoma. Cancers (Basel). (2023) 15:5652. doi: 10.3390/cancers15235652
102. Karschnia P, Teske N, Thon N, Subklewe M, Tonn JC, Dietrich J, et al. Chimeric antigen receptor T cells for glioblastoma: current concepts, challenges, and future perspectives. Neurology. (2021) 97:218–30. doi: 10.1212/WNL.0000000000012193
103. Akbari B, Ghahri-Saremi N, Soltantoyeh T, Hadjati J, Ghassemi S, and Mirzaei HR. Epigenetic strategies to boost CAR T cell therapy. Mol Ther. (2021) 29:2640–59. doi: 10.1016/j.ymthe.2021.08.003
104. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM, et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother. (2010) 33:780–8. doi: 10.1097/CJI.0b013e3181ee6675
105. Dudda JC, Salaun B, Ji Y, Palmer DC, Monnot GC, Merck E, et al. MicroRNA-155 is required for effector CD8+ T cell responses to virus infection and cancer. Immunity. (2013) 38:742–53. doi: 10.1016/j.immuni.2012.12.006
106. Angelou CC, Wells AC, Vijayaraghavan J, Dougan CE, Lawlor R, Iverson E, et al. Differentiation of pathogenic th17 cells is negatively regulated by let-7 microRNAs in a mouse model of multiple sclerosis. Front Immunol. (2019) 10:3125. doi: 10.3389/fimmu.2019.03125
107. Srinivasan S, Armitage J, Nilsson J, and Waithman J. Transcriptional rewiring in CD8(+) T cells: implications for CAR-T cell therapy against solid tumours. Front Immunol. (2024) 15:1412731. doi: 10.3389/fimmu.2024.1412731
108. Xu S, Tang L, Li X, Fan F, and Liu Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. (2020) 476:1–12. doi: 10.1016/j.canlet.2020.02.002
109. Liu J, Peng J, Jiang J, and Liu Y. Clinical immunotherapy in glioma: current concepts, challenges, and future perspectives. Front Immunol. (2024) 15:1476436. doi: 10.3389/fimmu.2024.1476436
110. Liu Y, Ali H, Khan F, Pang L, and Chen P. Epigenetic regulation of tumor-immune symbiosis in glioma. Trends Mol Med. (2024) 30:429–42. doi: 10.1016/j.molmed.2024.02.004
111. Park JW, Sahm F, Steffl B, Arrillaga-Romany I, Cahill D, Monje M, et al. TERT and DNMT1 expression predict sensitivity to decitabine in gliomas. Neuro Oncol. (2021) 23:76–87. doi: 10.1093/neuonc/noaa207
112. Ciccone R, Quintarelli C, Camera A, Pezzella M, Caruso S, Manni S, et al. GD2-targeting CAR T-cell therapy for patients with GD2+ Medulloblastoma. Clin Cancer Res. (2024) 30:2545–57. doi: 10.1158/1078-0432.CCR-23-1880
113. Dhar S, Gadd S, Patel P, Vaynshteyn J, Raju GP, Hashizume R, et al. A tumor suppressor role for EZH2 in diffuse midline glioma pathogenesis. Acta Neuropathol Commun. (2022) 10:47. doi: 10.1186/s40478-022-01336-5
114. Nguyen TTT, Zhang Y, Shang E, Shu C, Torrini C, Zhao J, et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J Clin Invest. (2020) 130:3699–716. doi: 10.1172/JCI129049
115. Liu R, Ren X, Park YE, Feng H, Sheng X, Song X, et al. Nuclear GTPSCS functions as a lactyl-CoA synthetase to promote histone lactylation and gliomagenesis. Cell Metab. (2025) 37:377–94.e9. doi: 10.1016/j.cmet.2024.11.005
116. Zhang Y, You H, Wang Y, Chen Q, Guo Q, Chu Y, et al. A micro-environment regulator for filling the clinical treatment gap after a glioblastoma operation. Adv Healthc Mater. (2022) 11:e2101578. doi: 10.1002/adhm.202101578
117. Liu L, He P, Wang Y, Ma F, Li D, Bai Z, et al. Engineering sonogenetic EchoBack-CAR T cells. Cell. (2025) 188:2621–36.e20. doi: 10.1016/j.cell.2025.02.035
118. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
119. Brown MC, Beasley GM, McKay ZP, Yang Y, Desjardins A, Randazzo DM, et al. Intratumor childhood vaccine-specific CD4(+) T-cell recall coordinates antitumor CD8(+) T cells and eosinophils. J Immunother Cancer. (2023) 11:e006463. doi: 10.1136/jitc-2022-006463
120. Good CR, Aznar MA, Kuramitsu S, Samareh P, Agarwal S, Donahue G, et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell. (2021) 184:6081–100.e26. doi: 10.1016/j.cell.2021.11.016
121. Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, et al. Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science. (2021) 372:eaba1786. doi: 10.1126/science.aba1786
122. Zhu M, Han Y, Gu T, Wang R, Si X, Kong D, et al. Class I HDAC inhibitors enhance antitumor efficacy and persistence of CAR-T cells by activation of the Wnt pathway. Cell Rep. (2024) 43:114065. doi: 10.1016/j.celrep.2024.114065
123. Prinzing B, Zebley CC, Petersen CT, Fan Y, Anido AA, Yi Z, et al. Deleting DNMT3A in CAR T cells prevents exhaustion and enhances antitumor activity. Sci Transl Med. (2021) 13:eabh0272. doi: 10.1126/scitranslmed.abh0272
124. Qin D, Lei Y, Shu P, Zhang Y, Loh YH, Wang Y, et al. Supercharging CAR-T cells through transcriptional and epigenetic armoring. Theranostics. (2025) 15:3345–67. doi: 10.7150/thno.107908
Keywords: gliomas, epigenetic regulations, immune evasion, non-coding RNA, CAR-T cell therapy
Citation: Wu D, Ju D, Zhao Y, Liu W, Liu Q and Liang Y (2025) Unraveling epigenetic drivers of immune evasion in gliomas: mechanisms and therapeutic implications. Front. Immunol. 16:1633338. doi: 10.3389/fimmu.2025.1633338
Received: 22 May 2025; Accepted: 31 July 2025;
Published: 25 August 2025.
Edited by:
Shanzhou Huang, Guangdong Provincial People’s Hospital, ChinaReviewed by:
Amol Tandon, Umeå University, SwedenChristian A. Linares, Barking, Havering and Redbridge University Hospitals NHS Trust, United Kingdom
Copyright © 2025 Wu, Ju, Zhao, Liu, Liu and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Liang, bHlubmUzNjY1QDE2My5jb20=; Qingqing Liu, bGl1cWluZ3FpbmcxMUAxNjMuY29t
†These authors share first authorship