 Giovanni Almanzar1*†
Giovanni Almanzar1*† Juan Carlos Alarcon2,3†
Juan Carlos Alarcon2,3† Ruth Garzon4Ana Maria Navarro5
Ruth Garzon4Ana Maria Navarro5 Alejandro Ondo-Méndez3
Alejandro Ondo-Méndez3 Martina Prelog1
Martina Prelog1- 1Department of Pediatrics, Pediatric Rheumatology/Special Immunology, University Hospital Würzburg, Würzburg, Germany
- 2Center for Proteomics and Metabolomics, Leiden University Medical Center (LUMC), Leiden, Netherlands
- 3Clinical Research Group, School of Medicine and Health Sciences, Universidad del Rosario, Bogotá, Colombia
- 4Facultad de Ciencias, Departamento de Química, Universidad Nacional de Colombia, Bogotá, Colombia
- 5Fundación Universitaria Ciencias de la Salud (FUCS), Servicio de Genética Médica, Bogotá, Colombia
As a part of the tumor microenvironment, hypoxia is an important hallmark in the tumor progression. Hypoxia is a condition in which the oxygen supply is not sufficient to sustain the cell demand. In addition to its known impact in tumor progression, hypoxia seems to play a principal role in the generation and evolution of several autoimmune diseases. Both tumor and autoimmune diseases can be modulated by the hypoxia inducible factor alpha (HIF-1α) sharing similar molecular mechanisms. Here, we outline the links between cancer and autoimmunity regarding hypoxia-induced factors, such as HIF-1α, and describe the role of hypoxia in the modulation of the autoimmune response.
Introduction
Clinically, hypoxia causes cellular stress resulting in inflammatory reactions leading to tissue and organ damage. From the cellular perspective, hypoxia triggers the activation of cell signaling pathways related to metabolism and inflammation trying to adapt to the new oxygen conditions (1). Autoimmune diseases are known as a broad spectrum of conditions in which humoral and cellular components of the adaptive immune system react to cells, matrix structures, and nuclear molecules. Autoimmune conditions are accompanied by inflammatory cytokines and induction of molecular signaling pathways, which lead to abundance of inflammatory helper, cytotoxic T cell types, and to dysfunctional regulatory mechanisms, e.g. mediated by regulatory T cells (Treg) or anti-inflammatory mediators (2). Additionally, B cells contribute also to tissue- and cell-destruction by production of autoantibodies (3). The etiopathogenesis of autoimmune inflammation is multifactorial and mostly based on genetic susceptibility which may be triggered by pathogen- and damage-associated molecular patterns (PAMPs and DAMPs, respectively) driven by microbial antigens and lead to the destruction of tissue or cell structures (4). Further, abundance of cellular and nuclear materials may fire the reactogenicity of the innate and adaptive humoral and cellular immune system. Consequently, hypoxia, which causes cell damage, activation of inflammatory mechanisms, and revealing cell and matrix surfaces may constitute a significant factor in the breakthrough of peripheral immune tolerance mechanisms, and, thus, set an individual at risk to develop autoimmunity and autoinflammation (5). The present narrative review aims to unveil links between hypoxia and autoimmune reactivity with particular emphasis on T cell-specific factors that highlights parallels to inflammatory mechanisms driven by tumor-induced hypoxia.
Hypoxia pathway and hypoxia-inducible factor
Suitable oxygenation is essential for ensuring cell survival and appropriate cell function. Normal oxygenation of tissues lays in a narrow range, from 5 to 21 percent of oxygen, depending on the tissue and its metabolic requirements. For example, while there could be 21% of oxygen in lung tissue, tissues like the bone marrow are under lower oxygen tension (2 – 8% pO2) (6–9). The variability of oxygenation among tissues makes it difficult to establish a universal definition of hypoxia, so it has become more useful to define it in a functional way, in which hypoxia arises because of the imbalance of oxygen supply and consumption. In this situation, oxygen delivery does not meet the demands of the tissue resulting in anaerobic metabolism (10–12).
Tissue hypoxia can result from physiological factors such as physical activity and high altitude as well as from pathological factors, like pulmonary diseases, anemia, ischemia, chronic vascular disease, and cancer (13–16). Although hypoxia could be dangerous for every type of cell, metabolic and enzymatic plasticity provides the cells with the adequate mechanisms to adapt and survive under such harsh conditions. The metabolic shift is driven mainly by the activation of the hypoxia inducible factor (HIF) by increasing glucose uptake and the accumulation of metabolites such as lactate, citrate, and pyruvate, among others (17, 18). After the modulation of glycolytic enzymes such as hexokinase (HK) and phosphofructokinase (PKF) (19). This metabolic rewiring enables cells to generate ATP anaerobically under low levels of oxygen (20) and allows them to adapt and survive under hypoxic conditions. Another cell protecting mechanism activated by HIF are the activation of the superoxide dismutase (SOD) to neutralize the effect of reactive oxygen species (ROS) (21) and the increment in mitophagy to reduce mitochondrial damage under hypoxic conditions (22).
To maintain oxygen homeostasis, the human body senses oxygen concentration and responds to acute or chronic hypoxia. In sharp changes of oxygen, lasting seconds to minutes, changes in phosphorylation of cell signaling molecules and redox state in the cell are the principal response. Cells can switch their metabolism from aerobic to anaerobic energy generation through the increased expression of glucose transporters and glycolytic enzymes (18, 20, 21). Under chronic hypoxia, changes in gene expression in the cell are typical. The upregulation of HIF-1α and HIF-2α leads to transcriptional changes reprogramming metabolic processes switching to an inflammatory profile affecting macrophages and T cells (23, 24). Moreover, hypoxia affects macrophages polarization driving them to a proinflammatory phenotype M1-like (25). In the case of T cells, cytotoxic capacity can be impaired by a reduction in the IFNγ production (26), increasing regulatory T cell populations (27), and/or facilitating Th17 differentiation, all of them relevant for immune modulation. Tissue under this condition needs to reduce the total number of cells by the blocking cell cycle and cell proliferation, or by inducing apoptosis and necrosis as prominent cell death associated mechanisms (28, 29). Autophagy appears as a substitute mechanism when this adaptive response is not sufficient to overcome low oxygen conditions (30).
The completely adaptive response is under control at the molecular level by the rapid accumulation and activation of HIF-1 (Figure 1). HIF-1 is a heterodimeric transcription factor constituted by two subunits, the regulatory hypoxia response factor HIF-1α, and the constitutive receptor nuclear translocator HIF-1β (31, 32). These subunits are members of a large family of transcription factors which share a basic helix-loop-helix region and a Per-Arnt-Sim (PAS) domain; these regions are necessary for the formation of the dimer, and to the recognition of specific DNA sequences to transcription factor binding in the nucleus (5´- XCGTG – 3´, X represents adenine or guanine) (33).
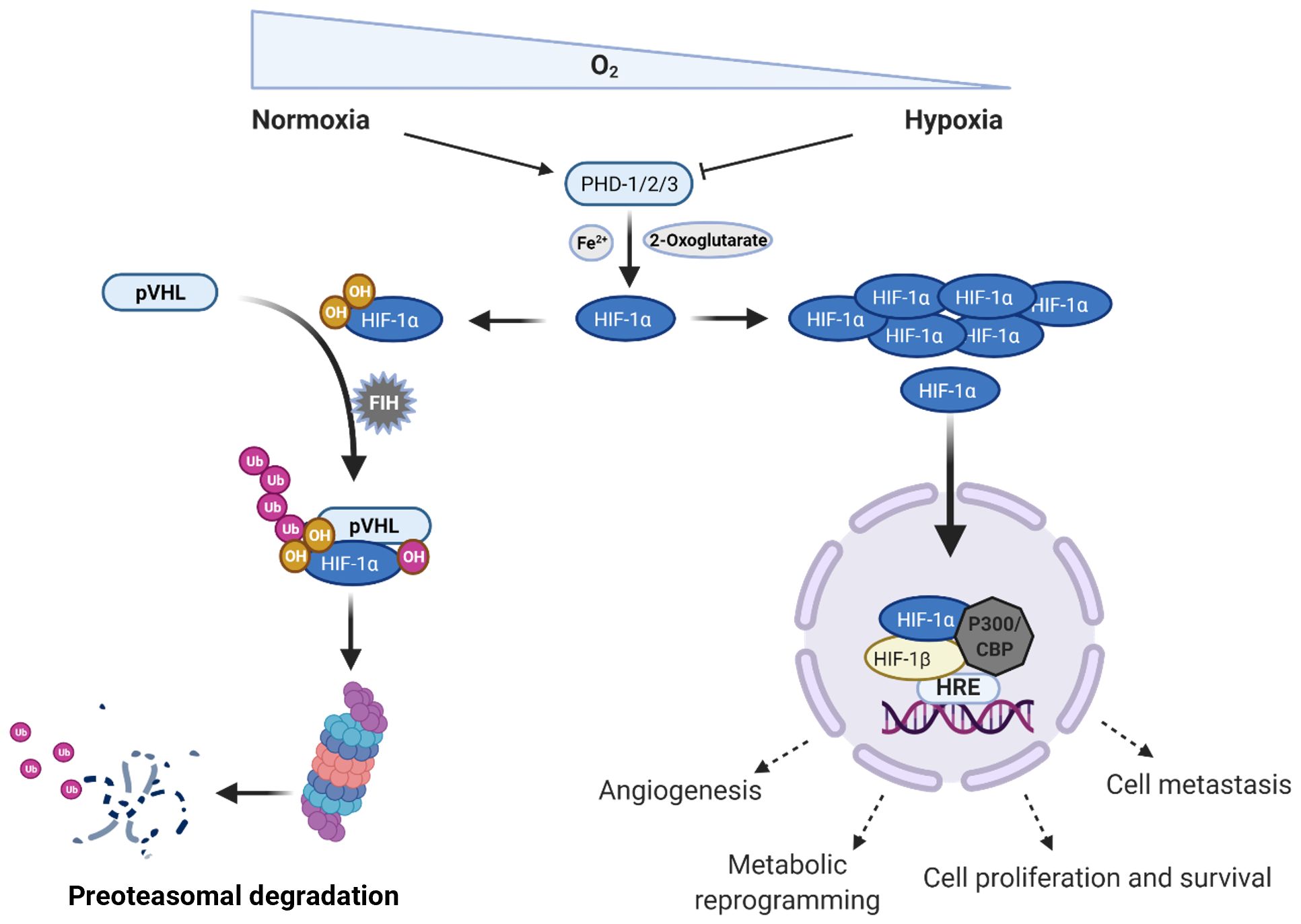
Figure 1. Regulation of the hypoxia inducible factor 1 alpha (HIF-1α) in normoxic and hypoxic conditions. Under normoxia, intracellular concentration of HIF-1α is regulated by different types of enzymes inducing its rapid proteasomal degradation; however, when the oxygen levels drop, HIF-1α can translocate to the nucleus and together with HIF-1β starts the transcription of genes related to different cell processes, such as angiogenesis, metabolic reprogramming, cell proliferation, and survival as well as metastasis. PHD, prolyl hydroxylases; pVHL, phosphorylated Von Hippel – Lindau tumor suppressor protein; FIH, factor inhibiting HIF-1 protein; HRE; hypoxia response elements; CBP, CREB-binding protein; Ub, Ubiquitin; OH, hydroxylation. Created in BioRender. Almanzar, G. (2025) https://BioRender.com/b1lf2y0.
Low oxygen levels lead to accumulation of HIF-1α, which can translocate to the nucleus and form a dimer with the β subunit. This dimer interacts with the CH1 pocket of the transcriptional activator CBP/P300 and binds to hypoxia response elements (HREs), thereby activating the transcription of multiple target genes, such as phosphoglycerate kinase (PGK1), vascular endothelial growth factor A (VEGFA), glucose transporter (GLUT1), among others. Cellular processes that are regulated by HIF pathway include angiogenesis, glycolysis, growth-factor signaling, immortalization, genetic instability, tissue invasion and metastasis, apoptosis and pH regulation (34, 35).
There are two other isoforms for the regulatory subunit, HIF-2α and HIF-3α. The last one is less studied, but it has been described as an inhibitor of HIF-1α through a negative feedback that involves the inhibitory Per/Arnt/Sim domain protein (IPAS) promoter (36, 37). On the other side, HIF-2α has a similar structure to HIF-1α but it is only expressed in certain tissues (38). In addition to its tissue specific expression, right after dimerization with HIF-1β, HIF-2α promotes the activation of different genes principally related to tumor growth, cell proliferation and cell pluripotency (39).
Nevertheless, under non-hypoxic conditions, HIF-1α is degraded upon hydroxylation, and ubiquitylation. HIF-1α is hydroxylated on proline residues 402 and/or 564 by prolyl hydroxylases (PHD – 1/2/3), which are tetrameric enzymes with two hydroxylases subunits and two di-sulfide isomerase subunits (31, 35). These enzymes show high oxygen affinity and use oxygen as a co-substrate to introduce the hydroxylation in proline residues generating a 4 – hydroxyproline (40, 41). The hydroxyproline is recognized by the Von Hippel – Lindau (VHL) tumor suppressor protein that is the recognition component of an E3 ubiquitin-protein ligase complex. Thus, finally leads to the polyubiquitylation of HIF-1α and its proteasomal degradation by the 26S subunit. The prolyl hydroxylases also require a ferrous ion (Fe2+), a Krebs cycle intermediate (2 – oxoglutarate), and ascorbate as co-factor to reduce the Fe3+ (31, 35, 42). Transcriptional activity of HIF-1α can also be hampered by asparaginyl hydroxylase enzyme factor inhibiting HIF-1 (FIH), which disturbs its interaction with the CBP/P300 transcriptional activator (Figure 1) (35).
In conclusion, hypoxia response through HIF-1α is meant to increase the cell adaptive response to lower oxygen conditions by regulating energy and redox status, thereby promoting cell survival and proliferation. This mechanism is highly regulated in cells under normal oxygen levels; hence, its dysregulation conveys the appearance of many metabolic and cell deviations.
Hypoxia and tumor microenvironment
Tumor microenvironment (TME) is the result of the paracrine and autocrine crosstalk of different cell types, controlling a variety of molecular processes with the aim to maintain and promote the tumor growth. It is considered as a region distinguished in space and function, which is comprised by different cell populations, namely, cancer cells, epithelial cells, extracellular matrix, stromal fibroblasts, immune cells (lymphocytes, macrophages, and mast cells), and vascular cells (endothelial, pericytes, and smooth muscle cells) (43, 44). One important feature of the TME is the presence of intermittent hypoxia which induces a higher glycolytic metabolism, resulting in extracellular acidosis; low nutrient availability, and probably failure of pH regulation (45). These specific conditions produce cellular stress, genetic variability, and up-regulation of genes such as the Eukaryotic translation initiation factor 4A, isoform 2 (EIF4A2), and Ribosomal protein L37 (RPL37) involved in supporting translation process, cell survival and invasion (46). In tumors, hypoxia is triggered by the up-regulation of cell proliferation that increases the oxygen requirements, which subsequently cause neovascular formation and cellular adaptation through metabolic and enzymatic adjustment (19, 47).
An interesting example comes from the 26S proteasome non-ATPase subunit 4 (PSMD4) gene, which encodes a ubiquitin-binding protein and constitutes one of the major ubiquitin receptor of 26S proteasome. A recent study showed that hypoxic conditions led to the upregulation of this gene through the direct binding of HF1 to the HRE in the PSMD4 promoter (48). PSMD4 overexpression correlated with poor survival rates in colon carcinoma and in breast cancer (49). Additionally, in hepatocellular carcinoma (HCC), the expression of HF1A and PMSD4 showed to be highly correlated and both being strong indicators of the disease progression (50). Under hypoxia a metabolic shift from oxidative phosphorylation to glycolysis (the Warburg effect) takes place. HIF-1α induces upregulation of GLUT1 (SLC2A) facilitating glucose uptake across the plasma membrane, the hexokinase 2 (HK2) which phosphorylates glucose to glucose-6-phosphate, and conversion of pyruvate into lactate, regenerating NAD+ for glycolytic flux (51). However, to prevent intracellular acidification, cancer cells upregulate monocarboxylate transporter 4 (MCT4) to export lactate (52). Accumulation of lactate in the TME promotes angiogenesis, immune evasion, and matrix remodeling (53).
Hypoxic cancer cell survival is mediated by the overexpression of the anti-apoptotic Bcl-2 family member Mcl-1 in solid tumors, which is regulated by HIF-1α and correlates to poor survival (54). Additionally, HIF-1α activates BNIP3 inducing selective autophagy and reducing ROS (55). The carbonic anhydrase IX (CA9) plays an important role in regulating intracellular pH acting as buffer catalyzing the hydration of carbon dioxide (CO2) to bicarbonate (HCO3-) and a proton (H+) reducing the acidic environment created by lactate accumulation (56). The overexpression of CA9, specially in hypoxic tumor regions, is associated with metastasis and poor prognosis (57). The phosphoinositide 3-kinase (PI3K)/Akt signaling pathway is involved in hypoxia-ischemia and it is a major signaling pathway in various types of cancer controlling cell survival, metastasis, and metabolism increasing the production of insulin-like growth factor 2 (IGF2) (58). This activation drives phosphorylation of the PIK3 to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3) through phosphorylation of phosphatidylinositol-4,5-bisphosphate (PIP2), recruiting Akt to the cell membrane, where it is activated by PDK1 (59). One of the downstream effectors of Akt, the mechanistic target of rapamicyn (mTOR) associates several other proteins involved in cancer progression (60). Thus, several components of the PI3K/Akt/mTOR pathway frequently show mutations and are activated in cancer (61, 62).
Hypoxic TME favors endothelial cell (EC) proliferation and migration. Deletion of p53 in cancer cells increases HIF-1α levels and enhances transcriptional activation of HIF-1α-dependent VEGF and erythropoietin (EPO) in response to hypoxia, thus promoting EC proliferation, migration, and angiogenesis (63, 64). Angiogenesis initiated from Tumor activated-endothelial cells (TECs), resulting in the production of vascular endothelial growth factor A (VEGF-A), angiopoietin-like 4 (ANGPTL4), placental growth factor (PIGF), and platelet-derived growth factor B (PDGF-B), all of them, contribute to the endothelial cell proliferation, migration, and capillary formation. However, the hypoxia-angiogenesis cycle occurs because the newly generated vasculature is usually immature, leaky, and poorly perfused, which reduces the oxygen supply and reinforces hypoxia (65–67).
TECs maintain close contact with circulating immune cells, both innate and adaptive, and mediates their interactions with tumor stroma, thus, TECs play a pivotal role as sentinels and immune regulators performing tissue- and vessel-type-specific immunomodulatory functions, including recruitment, activation and antigen presentation of immune cells. Activated ECs recruit effector immune cells guiding their infiltration into the tumor microenvironment through a multi-staged adhesion process which includes binding of the integrins lymphocyte function-associated antigen-1 (LFA-1) and very late antigen-4 (VLA-4) on T cells to the respective ligands intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion protein-1 (VCAM-1) on TECs (68). The tumor-derived cytokines such as vascular endothelial growth factor (VEGF), endothelin-1 (ET1), EGF-like domain-containing protein 7 (EGFL7), and fibroblast growth factor 2 (FGF2) downregulate gene expression and protein expression of adhesion molecules and chemoattractants (e.g., CCL2, CXCL10, and CXCL7) resulting into inhibition of the immune cell infiltration (69).
Cancer associated fibroblasts (CAFs), an important component of the tumor stroma in many types of cancer such as colorectal and breast cancer, illustrate another example of the role of hypoxia in the tumor progression. Although CAF cells can be derived from multiple cells types (resident fibroblast, endothelial, epithelia, mesenchymal stem cells, pericytes, adipocytes) they lack the protein expression for endothelial, epithelial on hematopoietic cells, but they do express mesenchymal biomarkers such as vimentin or platelet derived growth factor receptor alpha (PDGFR-α) (70). These cells can be regulated by both HIF-dependent and HIF-independent mechanisms (71). Under hypoxic conditions, activated CAFs cells can mediate cancer progression by activating pathways that regulate extracellular matrix remodeling, immune response, metabolic reprogramming, angiogenesis and metastasis (72).
Interleukin-6 (IL-6) is the predominantly cytokine released by CAFs in response to the activation of the signal transducer and activator of transcription 3 (STAT3) via HIF-1α. This downstream cascade target PKM2 and facilitates the expansion of CAFs (70). Furthermore, hypoxia increases the mRNA and protein expression of anti-tumor immunity factors including TGF-β, IL-10, VEGF, CXCL12, PD-L1, FasL, and CD39 on CAFs. Therefore, hypoxia improves the immunosuppressive function of CAFs in the TME (71).
In addition to promoting the hypoxia dependent CAF expansion, IL-6 is also produced in the early stage of the TME development, it is strongly associated with the proinflammatory response during infection and immune cell activation. IL-6 is important for the differentiation of naïve CD4+ T cells into IL-17-producing T helper (Th17) cells in association with TGF-β (72). In contrast, IL-6 inhibits TGF-β-induced Treg differentiation (73) contributing to the dysbalance between Th17/Treg described in several T cell mediated autoimmune diseases (74–76) such as psoriasis, rheumatoid arthritis (RA), and inflammatory bowel diseases (IBD) (75–77). IL-6 exacerbates inflammation facilitating differentiation of CD4+ T cells into Th17, inducing tissue damage and supporting fibrosis by increasing the levels of extracellular matrix components (78, 79). High levels of IL-6 and the soluble IL-6 receptor alpha chain (IL-6Rα) play an important role in suppression of the immune response in tumor progression of non-small cell lung cancer (NSCLC) (80). IL-6 has been reported to induce and maintain the cancer stem cell state (81, 82). IL-6 can be secreted by resident macrophages (83), fibroblasts upon activation (84), or by the presence of T cells during cellular response (85). Thus, data regarding the key cytokine IL-6 suggests a link between the role of hypoxia in tumor progression and the mechanisms used by hypoxia in the progression of inflammation and autoimmunity.
Immune evasion in the hypoxic tumor microenvironment
Hypoxia profoundly shapes the immunological landscape of the tumor microenvironment by promoting immune evasion through multiple mechanisms. A central feature of this process is the HIF-1α–mediated upregulation of immune checkpoint ligands, particularly programmed death-ligand 1 (PD-L1), on tumor cells and associated stromal cells (86). PD-L1 binding to its receptor PD-1 on cytotoxic CD8+ T cells leading to T cell exhaustion and diminished antitumor immunity (87, 88). HIF-1α binds directly to HRE in the promoter region of the PD-L1 gene, enhancing the transcriptional upregulation of the ligand in several tumor types such as lung, breast, renal, head and neck cancers (86). PD-L1 expressed on cancer cells binds to PD-1 on activated CD8+ T cells inhibiting T cell proliferation, reducing IFNγ and IL-2 production, and cytolytic activity (89, 90). As consequence, features of T cell exhaustion are identified and enable tumor cells to resist immune-mediated elimination. PD-L1 expression under hypoxia is induced on tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), CAFs, and endothelial cells creating an immune-privileged niche. PD-L1+ TAMs bind to PD-1 on NK cells impairing their cytotoxic function (91). MDSCs are recruited to hypoxic tumor areas via CCL2 and CXCL12, express PD-L1 and suppress antigen presenting cells function affecting DC maturation and impairing T cell priming (92). CAFs remodel the extracellular matrix and express PD-L1 inhibiting T cell proliferation (93), while PD-L1+ endothelial cells suppress trans-endothelial migration of T cells blocking T cell activation at tumor entry. In fact, PD-L1 expression induced by hypoxia acts as a gatekeeper mechanism allowing Treg or MDSCs to enter the tumor while excluding CD8+ cytotoxic T cells (Figure 2).
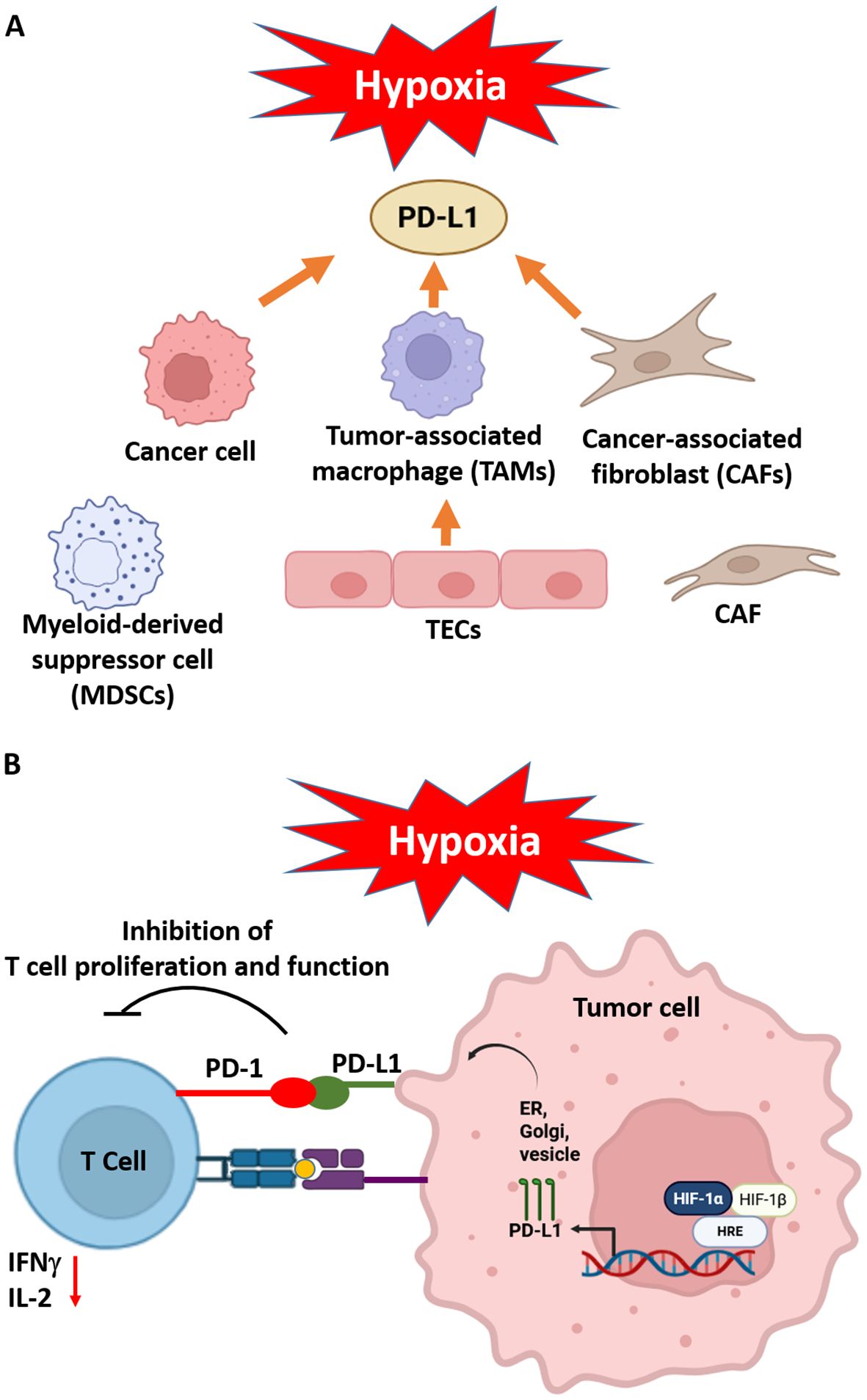
Figure 2. Hypoxia generates an immunosuppressive environment inducing PD-L1 expression in tumor microenvironment components such as cancer cells, tumor-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs) and tumor endothelial cells (TEC) (A) Hypoxia increased levels of HIF-1α binds to HRE in the PD-L1 promoter stimulates the PD-L1 production, which is transported to the surface of the tumor cell (B) The presence of PD-L1 on cells in the tumor microenvironment (TME) results in the binding of PD-1, which in turn inhibits the activity of tumor T cells reducing IFNγ and IL-2 levels and consequently leads to tumor progression. Adapted from (94). Created in BioRender. Almanzar, G. (2025) https://BioRender.com/b1lf2y0.
In parallel, hypoxic tumor cells secrete a repertoire of immunosuppressive factors, including transforming growth factor-beta (TGFβ), interleukin-10 (IL-10), and vascular endothelial growth factor (VEGF) (95). These soluble mediators not only suppress the activity of effector T cells and NK cells but also assist the recruitment and expansion of immunosuppressive populations such as Treg and MDSCs. Collectively, these hypoxia-driven alterations establish a profoundly immunosuppressive microenvironment that supports tumor-associated immune escape and limits the efficacy of immunotherapeutic interventions.
Regulatory T cells in the hypoxic microenvironment
Treg are characterized by the high expression of the IL-2 Rα (CD25) and the transcription factor forkhead-box-protein 3 (FoxP3) in CD4+ T cells, which are responsible for immune tolerance and homeostasis. However, the TME actively promotes Treg recruitment, stability, and suppressive function increasing resistance to anti-tumor immunity (Figure 3).
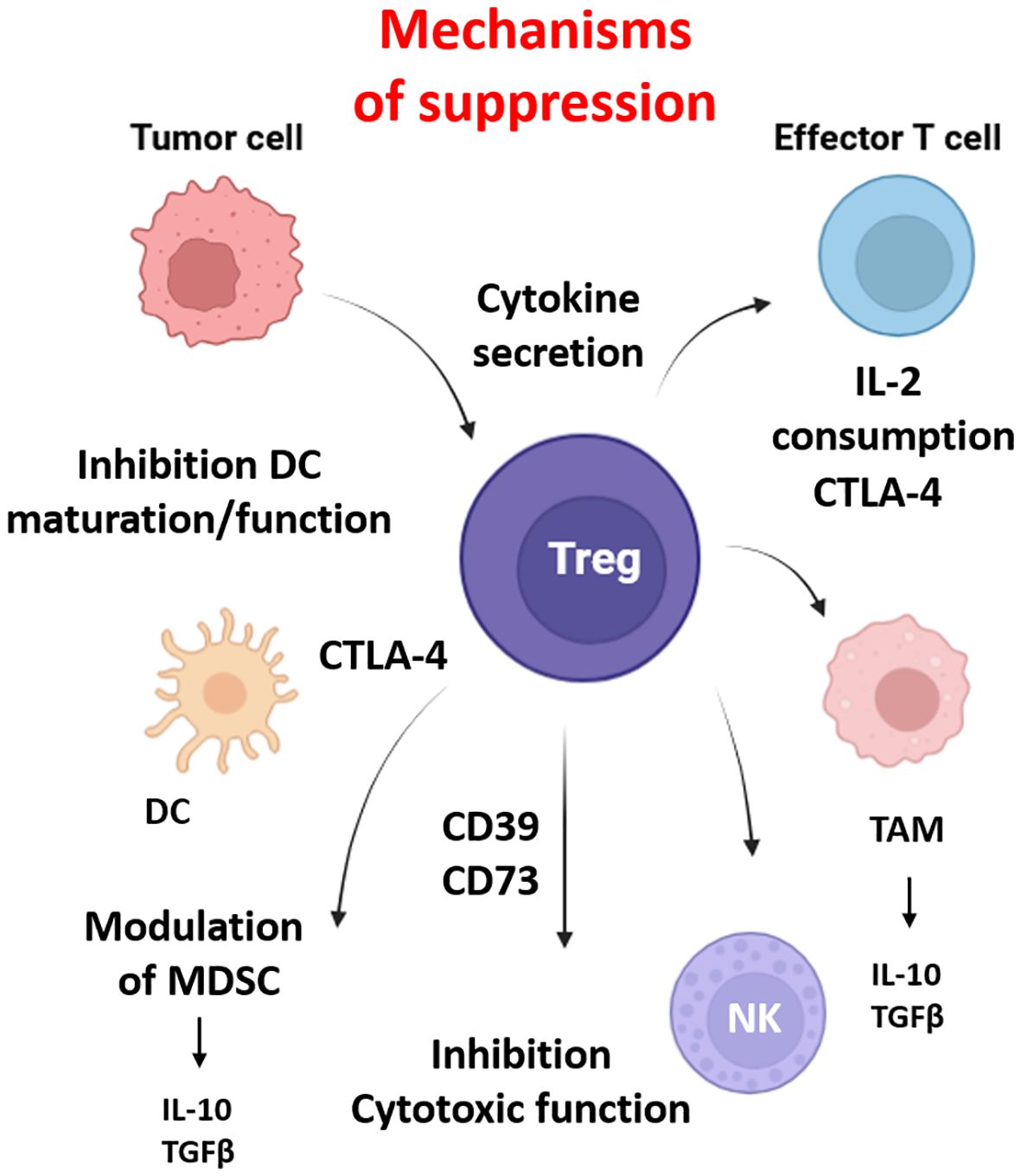
Figure 3. Regulatory T cell (Treg) mediated immunosuppression and cellular interactions in the tumor microenvironment. Regulatory T cells suppress anti-tumor immunity within the tumor microenvironment (TME). Tregs inhibit CD8+ cytotoxic T lymphocytes via CTLA-4–mediated competition for co-stimulation, IL-2 consumption, and secretion of suppressive cytokines (IL-10, TGF-β), which are produced by myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophage (TAMs). They impair dendritic cell (DC) maturation and antigen presentation, convert extracellular ATP to immunosuppressive adenosine through CD39/CD73, and can directly kill effector cells via granzyme B/perforin. Tregs also interact with tumor-associated macrophages (TAMs), promoting M2-like polarization and reinforcing the immunosuppressive niche. Together, these actions contribute to immune evasion, therapy resistance, and tumor progression in hypoxic and immunosuppressive regions of solid tumors. Created in BioRender. Almanzar, G. (2025) https://BioRender.com/b1lf2y0.
In hypoxic tumors, HIF-1α enhances the expression of chemokines such as CCL20, CCL22, CCL28, and CXCL12 establishing a chemotactic gradient that selectively recruit CCR6-, CCR10- and CCR4-expressing Treg (96). Treg in hypoxia also upregulate CTLA-4, IL-10, and TGFβ, which increase suppressive activity on effector T cells, NK, and DCs. CTLA-4 binds to CD80/CD86 on DCs downregulating co-stimulatory molecules and inducing indoleamine 2,3-dioxygenase (IDO) depleting tryptophan and stopping T cell proliferation. IL-10 and TGFβ are produced by TAMs and MDSCs contributing to the Treg differentiation (97, 98). Additionally, the expression of CD39 and CD73 on Treg generate adenosine from extracellular ATP. Adenosine interacts through the G-coupled purinergic type 1 receptor such as A2A receptors on effector cells and NK cells to inhibit cytotoxic function and cytokine production such as IFNγ and IL-2 (99, 100). IL-2 consumption via CD25 also contributes to reduce the effect of conventional T cells (101).
HIF and inflammation
Inflammation describes a clinical phenomenon related to increased flow in local blood vessels, increased cellular infiltration, and changes in tissue metabolism and oxygen supply. The inflammatory microenvironment is characterized not only by low molecular weights mediators, such as lipid mediators, cytokines, and chemokines secreted by different cell types (leucocytes and monocytes) but also by low levels in oxygen and nutrients (102–104). In acute and extensive inflammatory conditions, oxygen supply is decreased by poor tissue perfusion due to the microvasculature disruption e.g. by edema, vasculitis or vasoconstriction as found in many autoimmune conditions (105–107).
During an inflammatory condition, hypoxia and many other mechanisms from the immune response can induce a stable expression of HIF-1α. For example, during inflammation cytokines like IL-1β, IL-6 and TNFα increase the translation of the mRNA of HIF-1α in a NF-κB dependent way, through the activation of the PI3K/AKT/mTOR pathway (102, 108). Another mechanism involves the bacterial lipopolysaccharides (LPS), which can bind to the cell surface receptors CD14 and TLR-4 stimulating the p44/MAPK/ERK1/2 pathway leading to the activation of the transcription factor NF-κB, and the up-regulation of HIF-1α mRNA (102, 109). The inflammatory condition is also associated with perturbations in oxidative stress and redox cell state. For example, it was reported that IL-1β, IL-6, IL-8, and TNFα promote the activation, stabilization, and translocation of HIF-1α in a ROS depending way (110). One more example is the nitric oxide (NO) produced by macrophages and granulocytes, which are important players of the innate immune system during inflammation. NO has been shown to stabilize HIF-1α by two mechanisms, the first one, by attenuating HIF-1α ubiquitylation and reducing its degradation; the second one is related with the inhibition of PHD by sequestering of Fe(II) ion co-factor (102, 111).
HIF-1α has been associated with the regulation of metabolism, vascularization, and matrix remodeling. During inflammation for instance, HIF-1α promotes the transcription of genes like VEGF, SLC2A1 (encodes GLUT1) and those related to metalloproteinases (112–114). In macrophages and neutrophils, HIF-1α has been defined as the main regulator of glycolytic energy production. Impaired aggregation and motility have been observed in mice with deletion of myeloid HIF-1α (115). The HIF-1α – NFκB pathway is also involved in stimulating macrophages and reducing apoptosis in neutrophils through proinflammatory cytokine release (114, 116). Protein expression of FIH, prolyl hydroxylase domain enzymes 1 (PHD1) and 2 (PHD2) are not affected by hypoxia, whereas up-regulation of PHD3 has been observed in neutrophils under hypoxic conditions or isolated from rheumatoid patients (117, 118).
Macrophages and neutrophilic granulocytes also express HIF-2α in response to hypoxia. While HIF-1α is essential for survival, HIF-2α stimulates prevalence in the course of sterile inflammation in neutrophils (117).
Th17 and Treg cell regulation and HIF
Treg are essential for maintaining immune tolerance and preventing autoimmunity, but in the context of cancer, their immunosuppressive function becomes detrimental by enabling tumor immune evasion. A balance that is intricately regulated alongside Th17 cell differentiation by HIFs within the tumor microenvironment. HIF-1α regulates inflammation responses by modulating the activation and differentiation of Th17 and Treg cells in several manners (Figure 4).
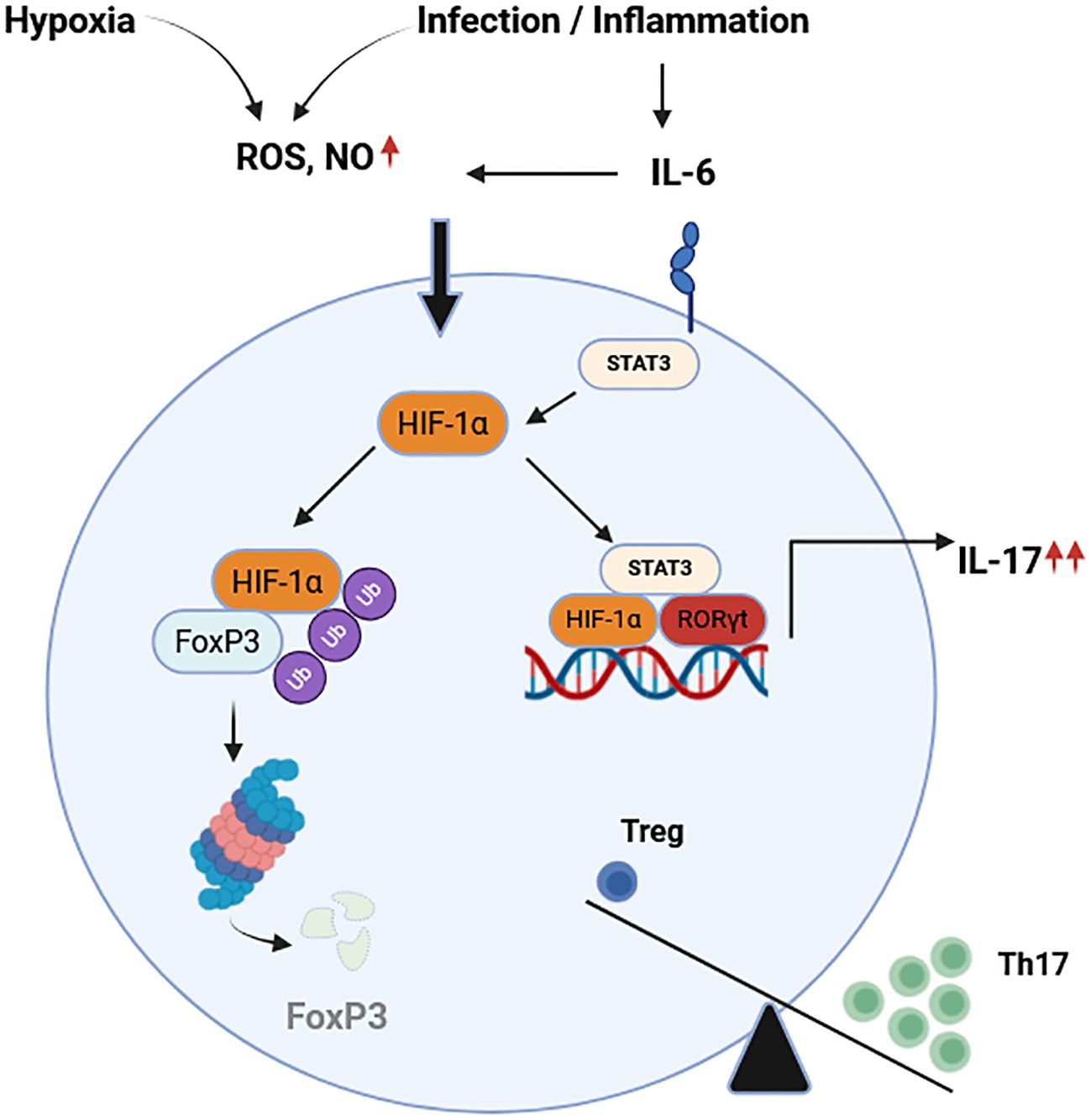
Figure 4. Hypoxia and infection or inflammation control the balance of Th17/Treg. Hypoxia and inflammatory conditions increase the levels of ROS, NO, and pro-inflammatory cytokines such as IL-6. On one hand, HIF-1α promotes the degradation of FoxP3 in proteasome regulating the differentiation of Treg. On the other hand, HIF-1α and STAT3 enhance expression of RORγt driving transcription of the Th17 genes and protein expression. These factors contribute to the imbalance between Th17 and Treg described in several T cell mediated autoimmune diseases. HIF-1α, hypoxia inducible factor 1 alpha; FoxP3, Forkhead-Box-Protein 3; STAT3, signal transducer and activator of transcription 3; ROS, reactive oxygen species; NO, nitric oxide; RORγt, retinoid-related orphan receptor gamma t; IL-6. Interleukin 6; IL-17, interleukin 17; Th17, T-helper 17 cells; Treg, regulatory T cells. Adapted from (119). Created in BioRender. Almanzar, G. (2025) https://BioRender.com/b1lf2y0.
This regulation involves a metabolic shift stemmed from the cell fate rewiring and the boost in cell expansion (120). For example, compared with other cell populations (Th1, Th2 or Treg), Th17 differentiation requires up-regulation in the glycolytic pathway in a HIF-1α dependent manner (121). During this process, both HIF-1α translation and transcription increased under independent pathways (122). To date, two pathways have been described, the first involves T cell receptor (TCR) and phosphoinositol-3-phosphate/mammalian target of rapamycin PI3K/mTOR pathway, in which the TCR can also induce the expression of splicing isoforms of HIF-1α mRNA whose function remains still unclear (123, 124). The second mechanism includes IL-6 and STAT3 pathway, both of which are associated in the differentiation of naïve T cells into Th17 cells. Although the molecular regulation and machinery involved are still unknown, many studies report a STAT3 mediated Th17 differentiation which also involves the HIF-1α induction in a hypoxia independent manner (24, 122).
Once HIF-1α is up-regulated in T cells, it readily associates with the promoter of the retinoid-orphan receptor gamma t (RORγt) which is necessary to stabilize the Th17 polarization. The combination of gene reporters and co-immunoprecipitation has shown that HIF-1α not only promotes RORγt gene expression, but also promotes the differentiation into Th17 cells (122). Moreover, HIF-1α regulates the expression of FoxP3 under inflammatory conditions (119). Studies including stimulation of HIF-1α knockout T cells showed no changes in the levels of FoxP3 mRNA in wild type (WT) and HIF-1 α-/- T cells stimulated under Th17 conditions. However, FoxP3 protein expression was diminished in this condition. Additionally, induction of Treg in WT and HIF-1 α-/- T cells upon in vitro treatment with IL-2 and TGFβ showed a reduction in the expression of the FoxP3 on the knockout cells compared to WT. Thus, HIF-1α down-regulates FoxP3 protein expression not affect the FoxP3 mRNA levels, suggest a mechanism that promotes the association of HIF-1α and FoxP3 and their further degradation via ubiquitination in the proteasome (119). This observation has been supported by other studies in which the absence of HIF-1α in T cells resulted in an up-regulation of FoxP3 and hence modulating cell metabolism in favor of lipid oxidation and down-regulating glycolytic metabolism (120).
The Th17 cell population can be derived from naïve cells by blocking the Aryl Hydrocarbon Receptor (AhR) (87). The AhR is regulated via STAT3, through the JAK/STAT pathway and the upregulation of IL-27. AhR and STAT3 are important transcription factors up-stream of RORγt for polarization of naïve T cells into the Th17 lineage, contribute to the membrane expression of CD39 enzyme. Through this enzyme the Treg population reduces the extracellular ATP and hence to the regulation of inflammation. Extracellular ATP is important for the differentiation of naïve T cells into Th17 cells (125, 126). Under hypoxic conditions, HIF-1α induces ubiquitination and degradation of AhR in proteasomes resulting in differentiation into regulatory type 1 T cells (Tr1). Tr1 produce IL-10 and play an important role in suppress autoimmune reactions (127). In Rheumatoid arthritis (RA) patients have shown negative correlations between expression of HIF-1α in Treg and disease activity score 28 (DAS28) (128). In contrast, the upregulation of HIF-1α promotes Th17 cell differentiation and hence to progression of the disease. Upregulation of HIF-1α has been associated with many other autoimmune and autoinflammatory conditions. For instance, HIF-1α over-expression in colon biopsies of patients with active IBD has been associated to increased expression of the macrophage inflammatory protein 3alpha (MIP-3α), and the presence of high serum levels of VEGF (129). Accumulation of HIF-1α has been identified in persistent pathofibrogenesis and may play a role in progression of systemic sclerosis (SSc), an inflammatory autoimmune disease resulting in abnormal fibroblast activation and fibrosis leading to skin thickening and organ insufficiency. HIF-1α promotes the deposition of extracellular matrix (ECM) and, thus, increases fibrosis (130). High levels of circulating Th17 cells and IL-17-producing “Th17-like” Treg have been identified previously in SSc patients. Functional assays demonstrated an impaired suppressive capacity of Treg cells that may be associated with the progression of the disease (76). Therefore, hypoxia may play a multifactor role in the progression of the SSc disease contributing to tissue remodeling, expansion of Th17 cells, and plasticity of Treg towards inflammatory helper T cell types.
Tumor-induced HIF-1α, Th17, and pathogenesis of autoimmunity
Table 1 summarized some features of the effect of hypoxia on tumor development and autoimmunity. Reduced levels of oxygen may induce a high rate of cancer cells proliferation, generating areas where hypoxia can spread heterogeneously within the tumor mass, especially in solid tumors (132). It has been estimated that 50-60% of tumors contain hypoxic regions, which have been associated with cancer metastasis and resistance to chemo and radiotherapy, suggesting hypoxia as indicator of poor prognosis in many types of cancer such as hepatocellular carcinoma, breast, pancreas, gastric and colorectal (133). In particular, it has been observed that adaptation in metabolite-driven gene regulation can be a powerful hallmark for measuring tumorigenesis (134).
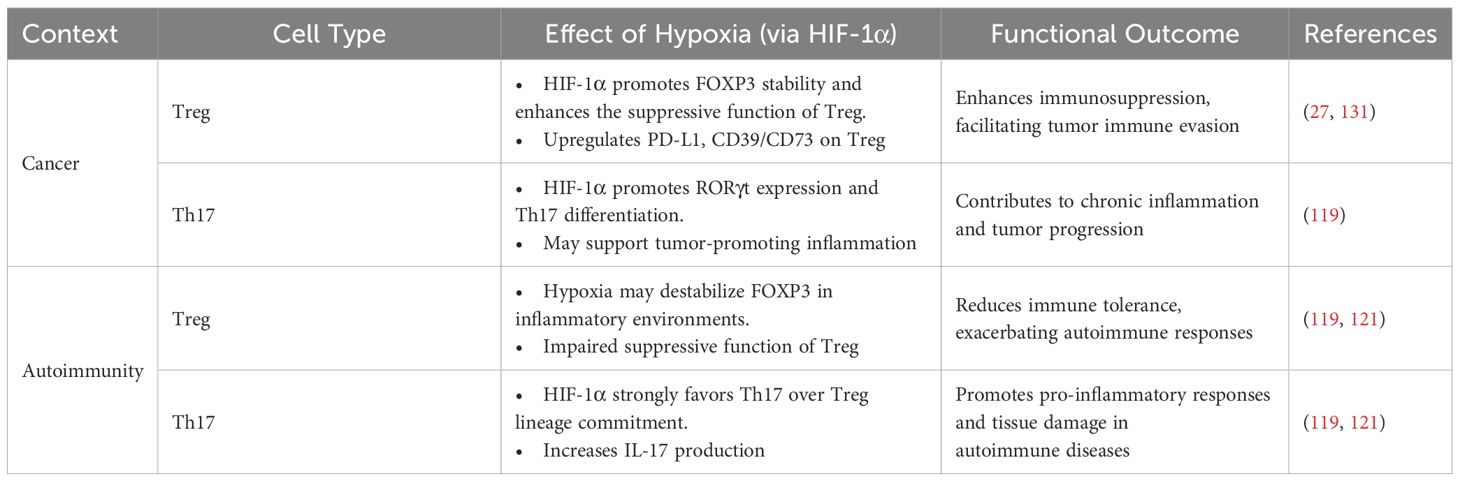
Table 1. Regulation of Treg and Th17 cells by hypoxia in cancer and autoimmunity.
Under cancer conditions, hypoxic levels in the cellular microenvironment inhibited the normal physiological processes, leading to stable accumulation of HIF-1α and HIF-2α (134). As a result, the HIF-1 complex can bind to HRE and recruit their coactivators p300 and the cAMP-response element binding protein (CREB)-binding protein to activate the transcription of multiple genes that respond to hypoxia (133). Namely, VEGFA, endocrine-gland-derived VEGF (EGVEGF), insulin-like growth factor-2 (IGF2), P53 and P21, transforming growth factor-β (TGF-β), SLC2A1, lactate dehydrogenase-A (LDHA), matrix metalloproteinases (MMPs), and nitric oxide synthase (NOS2) (132).
HIF-1α and HIF-1β are primary oxygen-limiting sensors and their induction support cancer cell proliferation during hypoxia and diverse metabolic alterations. HIF-2 α is sensitive to the availability of oxygen in tumors and promotes tumor progression through the lactate axis of macrophages (134).
Consequently, the TME particularly under hypoxic conditions significantly reprograms glucose metabolism to favor the pentose phosphate pathway (PPP) over oxidative phosphorylation, a phenomenon often associated with the Warburg effect (135, 136). This metabolic shift is primarily driven by hypoxia-inducible factor-1 alpha (HIF-1α) (137, 138),. HIF-1α upregulates the expression of glycolytic enzymes and glucose transporters, increasing glucose uptake and its diversion into the PPP (139). Therefore, part of the glucose-6-phospahte is diverted to generate nicotinamide adenine dinucleotide phosphate (NADPH), which neutralized ROS by glutathione reductase and thioredoxin systems maintaining redox balance (140, 141). NADPH is vital for maintaining cellular redox homeostasis by reducing oxidative stress, a common challenge in rapidly proliferating cancer cells, thus protecting them from ROS-mediated damage and apoptosis (135, 136). Additionally, the PPP supplies ribose-5-phosphate which server as a precursor for nucleotide biosynthesis, supporting the high proliferative demand of cancer cells and contributing to genomic instability (135, 142). This metabolic adaptation provides cancer cells with a significant advantage, enabling sustained proliferation, evasion of immune surveillance, and the production of growth factors that stimulate angiogenesis and ultimately facilitate metastasis (143–146). Therefore, the deviation of glucose to the PPP, orchestrated by factors like HIF-1α in the TME, is a critical mechanism by which cancer cells acquire hallmarks of malignancy, making HIF-1α and PPP enzymes promising targets for therapeutic intervention (137, 138, 147–149).
In activated immune cells, such as macrophages, dendritic cells, and T cells, NADPH promotes anabolic growth and the generation of ROS via NADPH oxidase for process such as microbial killing and immune signaling (150–152). However, dysregulated PPP flux in macrophages and T cells alters redox balance and promotes inflammatory pathways, skewing toward Th17 responses and impairing clearance of apoptotic debris (153) and on the other hand, G6PD-deficient immune cells exhibit poor oxidative burst and impaired bacterial killing, highlighting NADPH’s dual role in both ROS production and antioxidant defense (136).
Dysregulation of PPP activity has been implicated in the pathogenesis of autoimmune diseases, particularly systemic lupus erythematosus (SLE) (154). In SLE, elevated PPP flux has been observed in both T cells and monocytes. This contributes to increased NADPH production and ROS generation, exacerbating oxidative stress and promoting aberrant immune activation (155, 156). For example, the hyperactivation of the PPP in CD4+ T cells from patients with SLE correlates with increased mTOR signaling, T cell survival, and pro-inflammatory cytokine production (157–160). Furthermore, excessive NADPH supports lipid synthesis and nucleotide biosynthesis, thereby facilitating the proliferation and survival of autoreactive lymphocytes (161). These metabolic alterations amplify inflammatory responses and impair regulatory mechanisms, thereby sustaining autoimmunity (162–164). Preclinical studies have shown that targeting key enzymes in the PPP, such as glucose-6-phosphate dehydrogenase (G6PD), can reduce oxidative stress and restore immune balance (165). In rheumatoid arthritis (RA), high levels of serum toll-like receptor 2 (TLR2) and increased TLR2 expression on CD4+ T cells have been observed. This is associated with TNFα secretion and increased production of ROS, as well as reprogramming of glucose metabolism via HK2, which promotes the PPP (166). This suggests that modulating the PPP may offer novel therapeutic strategies for SLE and related autoimmune conditions.
The PPP plays an important role in regulating Treg function. However, deletion of the enzyme transketolase (TKT) in mice induces impaired Treg suppressive function, despite normal FoxP3 expression levels and leads to unhampered oxidative phosphorylation causing fatal autoimmune disease (167).
Hypoxia may also contribute to tumor escape from the immune system affecting the number and functionality of the natural killer cells (NK), T cells, and dendritic cells (DCs) by regulating the expression of HIF-1α (168). Additionally, in some autoimmune diseases such as rheumatoid arthritis, diabetes mellitus type 1, multiple sclerosis and intestinal inflammatory disease, among others, high concentrations of HIF-1α have been reported, which correlates with a higher activity of the disease (169). Thus, HIF-1α is not only involved in tumor progression and escape from immune surveillance but may also directly affect T cell functionality.
Increased levels of IFNγ have been shown in activated T cells under hypoxia (170). Reduced levels of oxygen and presence of an inflammation cytokine milieu contribute to the tissue damage (171, 172), and stimulate IFNγ production in effector cells under Th17-driving conditions, suggesting a switch towards Th1 responses (1).
Low oxygen levels stabilize HIF-1α disrupting mitochondrial function and increasing ROS impairing metabolic fitness in activated T cells (173). These metabolic stresses trigger intrinsic apoptotic pathways, characterized by mitochondrial outer membrane permeabilization, cytochrome C release, and activation of caspase-9 and caspase-3 (174–176). Hypoxia also enhances the expression of pro-apoptotic molecules such as Bcl-2-interacting mediator of cell death (BIM) and the p53 upregulated modulator of apoptosis (PUMA), while downregulating survival factors like Bcl-2 (177). Moreover, tumor and stromal cells in hypoxic regions upregulate PD-L1, Fas ligand (FasL), and galectin-9, which engage death receptors on T cells (e.g., PD-1, Fas, TIM-3) to initiate extrinsic apoptosis (89, 178, 179).
Previously, Sun et al. demonstrated that T cells undergoing apoptosis under hypoxic conditions showed an inhibition in the expression of chemokine C receptor 7 (CCR7) (180). T cells lose their homing migration capacity to regional secondary lymphatic organs reducing tumor-specific antigen-presentation. Therefore, increased T cell apoptosis probably impairs the antitumor T cell response resulting in tumor progression (181).
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterized by lymphocyte tissue infiltration, cytokine release, and joint damage (182). High levels of HIF-1α in serum and increased number of HIF-1α expressing cells have been detected in synovial tissue from RA patients (183). Under hypoxic conditions and high levels of IL-17, HIF-1α promotes synovial angiogenesis by increasing VEGF expression and exacerbating migration of fibroblast-like synovial cells through enzymatic activity of the metalloproteases 2 and 9 (MMP2 and MMP9) escalating bone destruction (184). Moreover, high levels of HIF-1α in fibroblast-like synovial cells stimulate expansion of Th1 and Th17 cells and increase concentrations of IFNγ and IL-17 (185).
Psoriasis arthritis (PsA) is characterized by inflammation of synovial tissues with hyperplasia of the joint surface, enhanced vascularity, and presence of inflammatory cells (186). Serum levels of VEGF and HIF-1α are significantly elevated in PsA patients compared to healthy controls or PsA patients in remission (187) suggesting a direct effect of the HIF-1α factor in the increased expression of VEGF which induces proliferation, migration, and differentiation of endothelial cells (188, 189).
Inflammatory bowel diseases (IBD) are chronical intestinal inflammatory diseases characterized by dysfunction of the intestinal barrier and tissue hypoxia (190). HIF-1α stimulates production of IL-1β inducing conversion of macrophages into the M1 phenotype (191). The M1 phenotype is characterized by high levels of inducible nitric oxide synthase (iNOS) (192), expression of HLA-DR and CCR7 (193). M1 macrophages secrete proinflammatory cytokines such as TNFα, IL-6 and iNOS promoting colitis (194, 195). HIF-1α is abnormally secreted in inflamed tissue from Crohn’s disease activating VEGF signaling promoting angiogenesis. Supporting this observation, HIF-1α-/- mice showed weight loss and displayed severe intestinal inflammation suggesting a protective role of HIF-1α in inflammatory mechanisms involved in the immune regulation of IBD (196). Taken together, HIF1-α may display a protective and anti-pathogenic role in autoimmune colitis.
Epigenetic changes by hypoxia in inflammation
Tumor infiltrating lymphocytes have a predominantly Th17 phenotype due to the presence of high levels of Th17-polarizing cytokine in the tumor microenvironment described in several cancer types (197, 198). IL-6 and the signal transducer and activator transcription 3 (STAT3) upregulate the histone motif H3K4me3 on the IL-17 locus. Epigenetic regulators of the tripartite motif-containing 28 (TRIM28) are activated through TCR signaling and together with H3K4me3 permissive histone mark allow binding of the RORγt leading to the production of IL-17 (199). Th17 cells can be recruited to the tumor sites by the expression of CCR6 and binding to its natural ligand CCL20 highly expressed on tumor tissues (197, 200), which is coordinated by long non-coding RNA (lncRNA-u50535) (197). Interestingly, CCR6 and CXCR3-Th17/Treg cells secreting IL-17 have been described in colorectal and esophageal cancer, while the presence of IL-17-producing-FoxP3+ cells is related with poor prognosis because they have been shown to promote tumor progression (201, 202). Th17 plasticity is associated with epigenetic modifications. Mark of H3K27me3 to the IL17 and IL21 loci in Treg correlated to permissive marks of H3K4me3 in Th17 cells (203). Taking together, hypoxia may compromise epigenetic modifications that accelerate and explain conversion of Th0 into Th17 cells promoting tumor proliferation and impairing T cell response to tumor antigens.
The critical role of hypoxia in cancer therapy
Understanding the molecular and biochemical mechanisms of tumor hypoxia is key to improving cancer treatments. Recognition of hypoxia as a major factor in treatment resistance dates back to the 1950s. Gray et al. (1953) (204) showed that hypoxic tissues require much higher radiation doses than oxygenated ones, identifying hypoxia as a cause of radio resistance. This finding highlighted the challenge of oxygen deficiency in solid tumors. Later research confirmed that hypoxia promotes tumor growth, aggressiveness, and resistance to therapies like chemotherapy and radiotherapy (205, 206).
Hypoxia drives immune evasion, abnormal angiogenesis, and metabolic shifts, leading to poor treatment response. In the context of radiotherapy, treatment efficacy depends on the generation of reactive oxygen species (ROS) to induce DNA damage in cancer cells. Hypoxic conditions, characterized by low oxygen levels, directly compromise this mechanism, as oxygen is essential for fixing radiation-induced DNA damage. This results in radio resistance, requiring higher radiation doses to achieve tumor control (207). The hypoxic microenvironment also promotes autophagy contributing to radio resistance by enabling cancer cells to survive under stress conditions (208). Studies using nasopharyngeal carcinoma xenografts have shown that hypoxia reduces radiation sensitivity, and overcoming this requires strategies that suppress the repair of DNA double-strand breaks under hypoxic conditions (209). Imaging techniques such as the 18F-fluoromisonidazole (18F- FMISO PET) have been developed to localize hypoxic regions within tumors, offering the potential to guide adaptive radiotherapy and escalate radiation doses to resistant areas (210, 211). However, clinical implementation of these approaches faces challenges related to cost, time, and accessibility (211).
Chemotherapeutic agents—particularly those whose mechanisms of action depend on oxygen—are significantly impacted by tumor hypoxia. Hypoxia can reduce drug uptake, alter metabolism, and induce cellular adaptations that confer resistance. For example, mitomycin C (MMC) treatment in ovarian cancer cells under hypoxic conditions exhibits extracellular matrix (ECM) modifications rather than ribosome-related pathways currently observed under normoxia (212). The efficacy of cisplatin (CisPt) in ovarian cancer cells is diminished under hypoxia, and even natural compounds like resveratrol, often used as adjuvants, lose their cytotoxic effects in low-oxygen environments (213).
Immunotherapy using checkpoint inhibitors (ICIs) (e.g., anti-PD-1, anti-PD-L1, anti-CTLA-4) is limited by hypoxia, which upregulates immunosuppressive molecules and attracts regulatory T cells and suppressor cells (214, 215). Therefore, immunotherapy is more effective in tumors with high immune cell infiltration. Thus, use of anti-angiogenic drugs such as bevacizumab or anlotinib may reduce hypoxia through vascular normalization to enhance infiltration of immune cells in tumors. Therapeutic approaches including use of ICIs and anti-hypoxia may represent a promising strategy to reduce tumor progression (215). The tumor microenvironment, particularly hypoxic regions, plays a major role in modulating immune responses, frequently leading to immunosuppression and resistance to immunotherapies. Hypoxia reinforces immunosuppresion inhibiting cytotoxic T cell and NK cell function (206). However, the use of hypoxia-activated prodrug evofosfamide has shown promise in enhancing breast cancer cell sensitivity to apoptosis and NK cell-mediated cytotoxicity under hypoxic conditions, in part by preserving type I interferon signaling, which is otherwise suppressed in hypoxia (216). Modulation of TME by targeting hypoxia may enhance effectiveness of immunotherapy (217).
Nanoparticle-based delivery systems offer an alternative therapeutic approach to specifically targeting hypoxic tumor cells. These nanoparticles can deliver anti-hypoxic agents directly to the TME improving oxygenation and boosting the effectiveness of conventional therapies (218). In hepatocellular carcinoma (HCC), the combination of HIF-1α inhibitors with metabolic modulators such as palmitic acid and L-carnitine has shown potential in inducing apoptosis in hypoxic HCC cells by preventing lipid metabolism reprogramming (138). Interestingly, is the possibility to combine nanoparticles carrying metabolic inhibitors with ICIs to increase T cell function diminishing T cell exhaustion (219).
In the era of genetic and molecular medicine, gene-editing technologies such as CRISPR-Cas9 aim to permanently eliminate the HIF-1α gene (220), thereby reducing tumor growth and enhancing T cell-mediated immunity. RNA interference therapies using siRNA or shrank can silence HIF-1α mRNA, preventing its translation and reducing hypoxia-driven angiogenesis and immunosuppression (221).
Conclusions
Inflammatory and tumor microenvironments are similar regarding the composition of cells and features involved in both pathogenetic processes (222). Increased and stable expression of HIF-1α due to a reduction of the oxygen concentrations is a common feature observed in both pathologic conditions, tumor progression and (auto)inflammation. HIF-1α translocates into the nucleus and binds to HRE to initiate transcription of target genes involved in angiogenesis, invasion/metastasis, and chemoresistance (223). On the other hand, HIF-1α is associated with plasticity of Th1, Th17 and Treg cells toward Th17-like phenotype, suggesting that HIF-1α plays an important role in tumor progression and T cell immune response modulation by induction of inflammatory T cell phenotypes. The presence of Th17 polarizing cytokines such as IL-6 in inflamed microenvironment and the upregulation of HIF-1α contribute to the functional imbalance between pro-inflammatory T cells and Treg cells. In addition, hypoxia may support plasticity of Treg cells in detriment of their regulatory function on one side accelerating the degradation of the FoxP3 in the proteasome and enhancing proportions of Treg with a Th17 –like phenotype described also in several autoimmune diseases (75, 224). Thus, the evidence suggests a common mechanism in tumor progression and autoimmune disease development involving the regulation of HIF-1α, which contributes to the differentiation of T cells into pro-inflammatory cells characteristic of many autoimmune diseases (Figure 5).
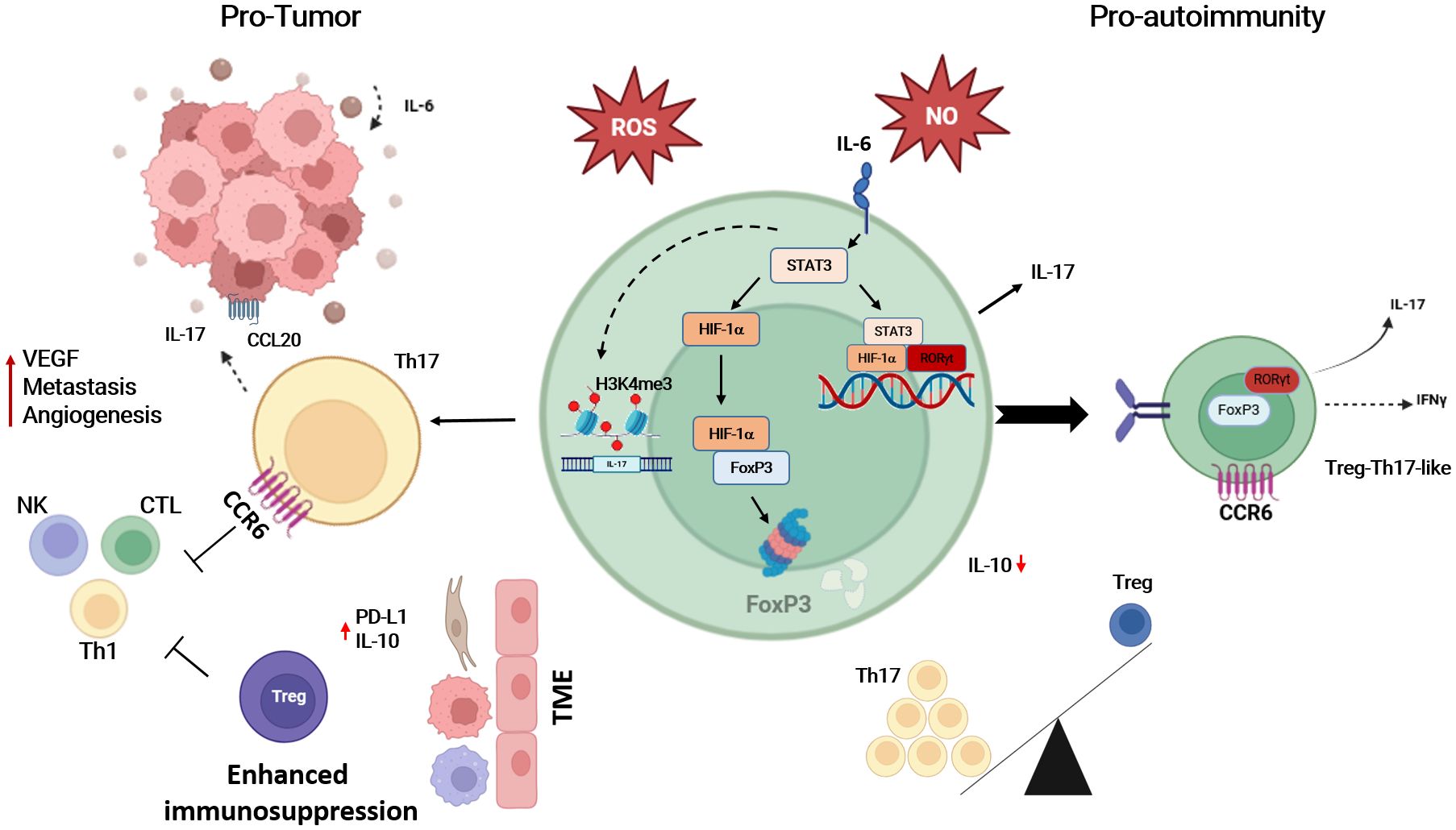
Figure 5. The up-regulation of HIF-1α under hypoxic conditions is likely to be the key to T cell differentiation into Th17 cells, which contributes to tumor progression and/or the triggering of autoimmune diseases. The abundance of IL-17 is not only a product of STAT3 induction, but hypoxia also drives the recruitment of Th17 cells to the tumor microenvironment (TME). This blocks the anti-tumor effect of NK, Th1 cells, and CTLs, and is also responsible for the impaired function of Treg, either by degrading FoxP3 or by enhancing Treg plasticity towards Treg-Th17-like T cells. On the other hand, TME increased expression of PD-L1 and IL-10 recruiting Treg and enhancing immunosuppression. Th17, T-helper-17 cells; Treg, regulatory T cells; NK, natural killer cell; Th1, T-helper-1 cells; CTL, cytotoxic T cells; IL-17, interleukin-17; IL-6, interleukin-6; IL-10, interleukin-10; IFNγ, interferon-gamma; CCR6, chemokine receptor 6; CCL20, chemokine ligand 20; HIF-1α, hypoxia inducible factor 1 alpha; FoxP3, Forkhead-Box-Protein 3; STAT3, signal transducer and activator of transcription 3; RORγt, retinoid-related orphan receptor gamma t; ROS, reactive oxygen species; NO, nitric oxide. Created in BioRender. Almanzar, G. (2025) https://BioRender.com/b1lf2y0.
Due to immunological dysregulation, and in part also fired by immunosuppressive therapy in autoimmune disorders, cancer rates are higher in those patients with autoimmune conditions compared to the general population (225–227). HIF-1α and other hypoxia-induced mechanisms may be the missing link to understand the increased risk of tumorigenesis in those patients suffering from autoimmune diseases. Unraveling the molecular and epigenetic pathways that govern HIF-1α expression in T cell regulation under inflammatory and tumor-promoting conditions is essential to develop targeted therapies to interrupt the signaling pathways towards hypoxia-induced inflammation promoting tumor progression or manifestation of autoimmunity. Therefore, it is mandatory to gain better insights to define common hypoxia-associated hallmarks for (auto)inflammation and cancer that may further be used for the development of new therapeutic treatments.
Author contributions
GA: Funding acquisition, Writing – original draft, Formal Analysis, Conceptualization, Writing – review & editing, Methodology, Investigation. JA: Investigation, Writing – review & editing, Formal Analysis, Writing – original draft, Methodology, Conceptualization. RG: Validation, Writing – review & editing, Supervision, Writing – original draft, Visualization. AN: Writing – review & editing, Writing – original draft. AO-M: Supervision, Writing – review & editing, Writing – original draft. MP: Writing – review & editing, Writing – original draft, Supervision.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by Bayerisches Hochschulzentrum für Lateinamerika (BAYLAT) JA is supported by a grant of the Administrative Department of Science, Technology and Innovation, COLCIENCIAS, in Colombia (Call 756/2016).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. During the preparation of this work the authors used DeepL (DeepL SE, Cologne, Germany) to improve language only. After using this tool, the authors reviewed and edited the content as need and take full responsibility for the content of the publication.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Luo Z, Tian M, Yang G, Tan Q, Chen Y, Li G, et al. Hypoxia signaling in human health and diseases: implications and prospects for therapeutics. Signal Transduct Target Ther. (2022) 7:218. doi: 10.1038/s41392-022-01080-1
2. Xiang Y, Zhang M, Jiang D, Su Q, and Shi J. The role of inflammation in autoimmune disease: a therapeutic target. Front Immunol. (2023) 14:1267091. doi: 10.3389/fimmu.2023.1267091
3. Martin F and Chan AC. Pathogenic roles of B cells in human autoimmunity; insights from the clinic. Immunity. (2004) 20:517–27. doi: 10.1016/s1074-7613(04)00112-8
4. Olson LB, Hunter NI, Rempel RE, and Sullenger BA. Targeting DAMPs with nucleic acid scavengers to treat lupus. Transl Res. (2022) 245:30–40. doi: 10.1016/j.trsl.2022.02.007
5. Ghorani E, Swanton C, and Quezada SA. Cancer cell-intrinsic mechanisms driving acquired immune tolerance. Immunity. (2023) 56:2270–95. doi: 10.1016/j.immuni.2023.09.004
6. Chow DC, Wenning LA, Miller WM, and Papoutsakis ET. Modeling pO(2) distributions in the bone marrow hematopoietic compartment. I. Krogh’s model. Biophys J. (2001) 81:675–84. doi: 10.1016/s0006-3495(01)75732-3
7. Nathan AT and Singer M. The oxygen trail: tissue oxygenation. Br Med Bull. (1999) 55:96–108. doi: 10.1258/0007142991902312
8. Papandreou I, Powell A, Lim AL, and Denko N. Cellular reaction to hypoxia: sensing and responding to an adverse environment. Mutat Research/Fundamental Mol Mech Mutagenesis. (2005) 569:87–100. doi: 10.1016/j.mrfmmm.2004.06.054
9. Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. (2012) 148:399–408. doi: 10.1016/j.cell.2012.01.021
10. Chicco AJ, Le CH, Gnaiger E, Dreyer HC, Muyskens JB, D’Alessandro A, et al. Adaptive remodeling of skeletal muscle energy metabolism in high-altitude hypoxia: Lessons from AltitudeOmics. J Biol Chem. (2018) 293:6659–71. doi: 10.1074/jbc.RA117.000470
11. Horscroft JA and Murray AJ. Skeletal muscle energy metabolism in environmental hypoxia: climbing towards consensus. Extrem Physiol Med. (2014) 3:19. doi: 10.1186/2046-7648-3-19
12. Pescador N, Villar D, Cifuentes D, Garcia-Rocha M, Ortiz-Barahona A, Vazquez S, et al. Hypoxia promotes glycogen accumulation through hypoxia inducible factor (HIF)-mediated induction of glycogen synthase 1. PloS One. (2010) 5:e9644. doi: 10.1371/journal.pone.0009644
13. Burtscher M, Haider T, Domej W, Linser T, Gatterer H, Faulhaber M, et al. Intermittent hypoxia increases exercise tolerance in patients at risk for or with mild COPD. Respir Physiol Neurobiol. (2009) 165:97–103. doi: 10.1016/j.resp.2008.10.012
14. Harris AL. Hypoxia — a key regulatory factor in tumour growth. Nat Rev Cancer. (2002) 2:38–47. doi: 10.1038/nrc704
15. Höckel M and Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. (2001) 93:266–76. doi: 10.1093/jnci/93.4.266
16. Tuder RM, Yun JH, Bhunia A, and Fijalkowska I. Hypoxia and chronic lung disease. J Mol Med (Berl). (2007) 85:1317–24. doi: 10.1007/s00109-007-0280-4
17. Liang B, Deng L, and Zhou X. Targeting hypoxia-related metabolism molecules: How to improve tumour immune and clinical treatment? BioMed Pharmacother. (2022) 156:113917. doi: 10.1016/j.biopha.2022.113917
18. Hayashi M, Sakata M, Takeda T, Yamamoto T, Okamoto Y, Sawada K, et al. Induction of glucose transporter 1 expression through hypoxia-inducible factor 1alpha under hypoxic conditions in trophoblast-derived cells. J Endocrinol. (2004) 183:145–54. doi: 10.1677/joe.1.05599
19. Vaupel P and Mayer A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. (2007) 26:225–39. doi: 10.1007/s10555-007-9055-1
20. Lubec B, Chiappe-Gutierrez M, Hoeger H, Kitzmueller E, and Lubec G. Glucose transporters, hexokinase, and phosphofructokinase in brain of rats with perinatal asphyxia. Pediatr Res. (2000) 47:84–8. doi: 10.1203/00006450-200001000-00016
21. Wang Y, Branicky R, Noe A, and Hekimi S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. (2018) 217:1915–28. doi: 10.1083/jcb.201708007
22. Wen L, Shao M, Li Y, Zhang Y, Peng C, Yu H, et al. Unveiling the hypoxia-induced mitophagy process through two-channel real-time imaging of NTR and viscosity under the same excitation. Talanta. (2024) 266:125028. doi: 10.1016/j.talanta.2023.125028
23. Staples KJ, Sotoodehnejadnematalahi F, Pearson H, Frankenberger M, Francescut L, Ziegler-Heitbrock L, et al. Monocyte-derived macrophages matured under prolonged hypoxia transcriptionally up-regulate HIF-1alpha mRNA. Immunobiology. (2011) 216:832–9. doi: 10.1016/j.imbio.2010.12.005
24. Tao JH, Barbi J, and Pan F. Hypoxia-inducible factors in T lymphocyte differentiation and function. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol. (2015) 309:C580–9. doi: 10.1152/ajpcell.00204.2015
25. Henze AT and Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. (2016) 126:3672–9. doi: 10.1172/JCI84427
26. Ma S, Zhao Y, Lee WC, Ong LT, Lee PL, Jiang Z, et al. Hypoxia induces HIF1alpha-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat Commun. (2022) 13:4118. doi: 10.1038/s41467-022-31764-9
27. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U.S.A. (2012) 109:E2784–93. doi: 10.1073/pnas.1202366109
28. Shimizu S, Eguchi Y, Kamiike W, Itoh Y, Hasegawa J-i, Yamabe K, et al. Induction of apoptosis as well as necrosis by hypoxia and predominant prevention of apoptosis by Bcl-2 and Bcl-XL. Cancer Res. (1996) 56:2161–6.
29. Weinmann M, Jendrossek V, Handrick R, Güner D, Goecke B, and Belka C. Molecular ordering of hypoxia-induced apoptosis: critical involvement of the mitochondrial death pathway in a FADD/caspase-8 independent manner. Oncogene. (2004) 23:3757–69. doi: 10.1038/sj.onc.1207481
30. Mazure NM and Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. (2010) 22:177–80. doi: 10.1016/j.ceb.2009.11.015
31. Lahiri S, Roy A, Baby S, Hoshi T, Semenza G, and Prabhakar N. Oxygen sensing in the body. Prog biophysics Mol Biol. (2006) 91:249–86. doi: 10.1016/j.pbiomolbio.2005.07.001
32. Semenza GL. Hypoxia, clonal selection, and the role of HIF-1 in tumor progression. Crit Rev Biochem Mol Biol. (2000) 35:71–103. doi: 10.1080/10409230091169186
33. Semenza GL, Jiang B-H, Leung SW, Passantino R, Concordet J-P, Maire P, et al. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. (1996) 271:32529–37. doi: 10.1074/jbc.271.51.32529
34. Fong G-H. Mechanisms of adaptive angiogenesis to tissue hypoxia. Angiogenesis. (2008) 11:121–40. doi: 10.1007/s10456-008-9107-3
35. Maxwell PH. Hypoxia-inducible factor as a physiological regulator. Exp Physiol. (2005) 90:791–7. doi: 10.1113/expphysiol.2005.030924
36. Makino Y, Uenishi R, Okamoto K, Isoe T, Hosono O, Tanaka H, et al. Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1) A NEGATIVE FEEDBACK REGULATORY CIRCUIT IN HIF-1-MEDIATED SIGNALING IN HYPOXIC CELLS. J Biol Chem. (2007) 282:14073–82. doi: 10.1074/jbc.M700732200
37. Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. (2010) 29:625–34. doi: 10.1038/onc.2009.441
38. Wiesener MS, Jurgensen JS, Rosenberger C, Scholze CK, Horstrup JH, Warnecke C, et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. (2003) 17:271–3. doi: 10.1096/fj.02-0445fje
39. Loboda A, Jozkowicz A, and Dulak J. HIF-1 versus HIF-2—is one more important than the other? Vasc Pharmacol. (2012) 56:245–51. doi: 10.1016/j.vph.2012.02.006
40. Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. (2001) 107:43–54. doi: 10.1016/S0092-8674(01)00507-4
41. Ivan M, Haberberger T, Gervasi DC, Michelson KS, Günzler V, Kondo K, et al. Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci. (2002) 99:13459–64. doi: 10.1073/pnas.192342099
42. Kuiper C and Vissers MC. Ascorbate as a co-factor for fe- and 2-oxoglutarate dependent dioxygenases: physiological activity in tumor growth and progression. Front Oncol. (2014) 4:359. doi: 10.3389/fonc.2014.00359
43. Ariztia EV, Lee CJ, Gogoi R, and Fishman DA. The tumor microenvironment: key to early detection. Crit Rev Clin Lab Sci. (2006) 43:393–425. doi: 10.1080/10408360600778836
44. Mbeunkui F and Johann DJ Jr. Cancer and the tumor microenvironment: a review of an essential relationship. Cancer chemother Pharmacol. (2009) 63:571–82. doi: 10.1007/s00280-008-0881-9
45. Laconi E. The evolving concept of tumor microenvironments. Bioessays. (2007) 29:738–44. doi: 10.1002/bies.20606
46. Sorensen BS, Busk M, Overgaard J, Horsman MR, and Alsner J. Simultaneous hypoxia and low extracellular pH suppress overall metabolic rate and protein synthesis in vitro. PloS One. (2015) 10:e0134955. doi: 10.1371/journal.pone.0134955
47. Höckel M and Vaupel P. Biological consequences of tumor hypoxia. Semin Oncol. (2001) 28(2 Suppl 8):36–41. doi: 10.1016/S0093-7754(01)90211-8
48. AydoGan TS, Dayi G, and K. OF. Upregulation of PSMD4 gene by hypoxia in prostate cancer cells. Turk J Biol. (2020) 44:275–83. doi: 10.3906/biy-2002-71
49. Fejzo MS, Anderson L, Chen HW, Guandique E, Kalous O, Conklin D, et al. Proteasome ubiquitin receptor PSMD4 is an amplification target in breast cancer and may predict sensitivity to PARPi. Genes Chromosomes Cancer. (2017) 56:589–97. doi: 10.1002/gcc.22459
50. Wang GL, Jiang BH, Rue EA, and Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U.S.A. (1995) 92:5510–4. doi: 10.1073/pnas.92.12.5510
51. DeWaal D, Nogueira V, Terry AR, Patra KC, Jeon SM, Guzman G, et al. Hexokinase-2 depletion inhibits glycolysis and induces oxidative phosphorylation in hepatocellular carcinoma and sensitizes to metformin. Nat Commun. (2018) 9:446. doi: 10.1038/s41467-017-02733-4
52. Yamaguchi A, Mukai Y, Sakuma T, Narumi K, Furugen A, Yamada Y, et al. Monocarboxylate transporter 4 involves in energy metabolism and drug sensitivity in hypoxia. Sci Rep. (2023) 13:1501. doi: 10.1038/s41598-023-28558-4
53. Chen J HZ, Chen Y, Tian H, Chai P, Shen Y, Yao Y, et al. Lactate and lactylation in cancer. Signal Transduction Targeted Ther. (2025) 10:1–26. doi: 10.1038/s41392-024-02082-x
54. Ding Q, He X, Xia W, Hsu JM, Chen CT, Li LY, et al. Myeloid cell leukemia-1 inversely correlates with glycogen synthase kinase-3beta activity and associates with poor prognosis in human breast cancer. Cancer Res. (2007) 67:4564–71. doi: 10.1158/0008-5472.CAN-06-1788
55. Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, and Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. (2001) 61:6669–73.
56. Benej M, Svastova E, Banova R, Kopacek J, Gibadulinova A, Kery M, et al. CA IX stabilizes intracellular pH to maintain metabolic reprogramming and proliferation in hypoxia. Front Oncol. (2020) 10:1462. doi: 10.3389/fonc.2020.01462
57. McDonald PC, Winum JY, Supuran CT, and Dedhar S. Recent developments in targeting carbonic anhydrase IX for cancer therapeutics. Oncotarget. (2012) 3:84–97. doi: 10.18632/oncotarget.422
58. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. (2014) 505:495–501. doi: 10.1038/nature12912
59. Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, and Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. (2010) 11:329–41. doi: 10.1038/nrm2882
60. Hennessy BT, Smith DL, Ram PT, Lu Y, and Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. (2005) 4:988–1004. doi: 10.1038/nrd1902
61. Hanker AB, Kaklamani V, and Arteaga CL. Challenges for the clinical development of PI3K inhibitors: strategies to improve their impact in solid tumors. Cancer Discov. (2019) 9:482–91. doi: 10.1158/2159-8290.CD-18-1175
62. Thorpe LM, Yuzugullu H, and Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. (2015) 15:7–24. doi: 10.1038/nrc3860
63. Nagy JA, Chang SH, Shih SC, Dvorak AM, and Dvorak HF. Heterogeneity of the tumor vasculature. Semin Thromb Hemost. (2010) 36:321–31. doi: 10.1055/s-0030-1253454
64. Zhang C, Liu J, Wang J, Zhang T, Xu D, Hu W, et al. The interplay between tumor suppressor p53 and hypoxia signaling pathways in cancer. Front Cell Dev Biol. (2021) 9:648808. doi: 10.3389/fcell.2021.648808
65. Laakkonen JP, Lahteenvuo J, Jauhiainen S, Heikura T, and Yla-Herttuala S. Beyond endothelial cells: Vascular endothelial growth factors in heart, vascular anomalies and placenta. Vascul Pharmacol. (2019) 112:91–101. doi: 10.1016/j.vph.2018.10.005
66. Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. (2013) 153:13–9. doi: 10.1093/jb/mvs136
67. Tan MJ, Teo Z, Sng MK, Zhu P, and Tan NS. Emerging roles of angiopoietin-like 4 in human cancer. . Mol Cancer Res. (2012) 10:677–88. doi: 10.1158/1541-7786.MCR-11-0519
68. Peschen M, Lahaye T, Hennig B, Weyl A, Simon JC, and Vanscheidt W. Expression of the adhesion molecules ICAM-1, VCAM-1, LFA-1 and VLA-4 in the skin is modulated in progressing stages of chronic venous insufficiency. Acta Derm Venereol. (1999) 79:27–32. doi: 10.1080/000155599750011651
69. Leone P, Malerba E, Susca N, Favoino E, Perosa F, Brunori G, et al. Endothelial cells in tumor microenvironment: insights and perspectives. Front Immunol. (2024) 15:1367875. doi: 10.3389/fimmu.2024.1367875
70. Xu Y, Kuai R, Chu YM, Zhou L, Zhang HQ, and Li J. Hypoxia facilitates the proliferation of colorectal cancer cells by inducing cancer-associated fibroblast-derived IL6. Neoplasma. (2021) 68(5):1015–22. doi: 10.4149/neo_2021_210308N296
71. Ziani L, Buart S, Chouaib S, and Thiery J. Hypoxia increases melanoma-associated fibroblasts immunosuppressive potential and inhibitory effect on T cell-mediated cytotoxicity. Oncoimmunology. (2021) 10:1950953. doi: 10.1080/2162402X.2021.1950953
72. Korn T, Bettelli E, Oukka M, and Kuchroo VK. IL-17 and th17 cells. Annu Rev Immunol. (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
73. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. (2006) 441:235–8. doi: 10.1038/nature04753
74. Kimura A and Kishimoto. T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. (2010) 40:1830–5. doi: 10.1002/eji.201040391
75. Holzer MT, Almanzar G, Woidich R, Hugle B, Haas JP, and Prelog M. Mitigated suppressive function of regulatory T cells (Treg) upon Th17-inducing cytokines in oligo- and polyarticular Juvenile Idiopathic Arthritis (JIA) patients. Pediatr Rheumatol Online J. (2022) 20:26. doi: 10.1186/s12969-022-00680-z
76. Almanzar G, Klein M, Schmalzing M, Hilligardt D, El Hajj N, Kneitz H, et al. Disease manifestation and inflammatory activity as modulators of th17/treg balance and RORC/foxP3 methylation in systemic sclerosis. Int Arch Allergy Immunol. (2016) 171:141–54. doi: 10.1159/000450949
77. Chen YT, Chang YM, Chen YL, Su YH, Liao CC, Chiang TH, et al. N-ethyl-N-nitrosourea (ENU)-induced C-terminal truncation of Runx3 results in autoimmune colitis associated with Th17/Treg imbalance. Immunol Lett. (2024) 268:106869. doi: 10.1016/j.imlet.2024.106869
78. Karpathiou G, Peoc’h M, Sundaralingam A, Rahman N, and Froudarakis ME. Inflammation of the pleural cavity: A review on pathogenesis, diagnosis and implications in tumor pathophysiology. Cancers (Basel). (2022) 14:1415. doi: 10.3390/cancers14061415
79. Wolfram D, Rabensteiner E, Grundtman C, Bock G, Mayerl C, Parson W, et al. T regulatory cells and TH17 cells in peri-silicone implant capsular fibrosis. Plast Reconstr Surg. (2012) 129:327e–37e. doi: 10.1097/PRS.0b013e31823aeacf
80. Donnenberg AD, Luketich JD, and Donnenberg VS. Secretome of pleural effusions associated with non-small cell lung cancer (NSCLC) and Malignant mesothelioma: therapeutic implications. Oncotarget. (2019) 10:6456–65. doi: 10.18632/oncotarget.27290
81. Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. (2013) 73:3470–80. doi: 10.1158/0008-5472.CAN-12-4524-T
82. Lu H, Clauser KR, Tam WL, Frose J, Ye X, Eaton EN, et al. A breast cancer stem cell niche supported by juxtacrine signalling from monocytes and macrophages. Nat Cell Biol. (2014) 16:1105–17. doi: 10.1038/ncb3041
83. Martin WJ, Walton M, and Harper J. Resident macrophages initiating and driving inflammation in a monosodium urate monohydrate crystal-induced murine peritoneal model of acute gout. Arthritis Rheum. (2009) 60:281–9. doi: 10.1002/art.24185
84. Bergqvist A, Bruse C, Carlberg M, and Carlstrom K. Interleukin 1beta, interleukin-6, and tumor necrosis factor-alpha in endometriotic tissue and in endometrium. Fertil Steril. (2001) 75:489–95. doi: 10.1016/s0015-0282(00)01752-0
85. Tiegs G and Lohse AW. Immune tolerance: what is unique about the liver. J Autoimmun. (2010) 34:1–6. doi: 10.1016/j.jaut.2009.08.008
86. Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. (2014) 211:781–90. doi: 10.1084/jem.20131916
87. Blank C and Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: an update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother. (2007) 56:739–45. doi: 10.1007/s00262-006-0272-1
88. Wu SP, Liao RQ, Tu HY, Wang WJ, Dong ZY, Huang SM, et al. Stromal PD-L1-Positive Regulatory T cells and PD-1-Positive CD8-Positive T cells Define the Response of Different Subsets of Non-Small Cell Lung Cancer to PD-1/PD-L1 Blockade Immunotherapy. J Thorac Oncol. (2018) 13:521–32. doi: 10.1016/j.jtho.2017.11.132
89. Han Y, Liu D, and Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. (2020) 10:727–42.
90. Jiang Y, Li Y, and Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
91. Chen BJ, Chapuy B, Ouyang J, Sun HH, Roemer MG, Xu ML, et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated Malignancies. Clin Cancer Res. (2013) 19:3462–73. doi: 10.1158/1078-0432.CCR-13-0855
92. Groth C, Hu X, Weber R, Fleming V, Altevogt P, Utikal J, et al. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br J Cancer. (2019) 120:16–25. doi: 10.1038/s41416-018-0333-1
93. Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y, et al. Roles of cancer-associated fibroblasts (CAFs) in anti- PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. (2023) 22:29. doi: 10.1186/s12943-023-01731-z
94. Viry E, Noman MZ, Arakelian T, Lequeux A, Chouaib S, Berchem G, et al. Hijacker of the antitumor immune response: autophagy is showing its worst facet. Front Oncol. (2016) 6:246. doi: 10.3389/fonc.2016.00246
95. Zhao H, Wu L, Yan G, Chen Y, Zhou M, Wu Y, et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct Target Ther. (2021) 6:263. doi: 10.1038/s41392-021-00658-5
96. Sarkar T, Dhar S, and Sa G. Tumor-infiltrating T-regulatory cells adapt to altered metabolism to promote tumor-immune escape. Curr Res Immunol. (2021) 2:132–41. doi: 10.1016/j.crimmu.2021.08.002
97. Dong S, Guo X, Han F, He Z, and Wang Y. Emerging role of natural products in cancer immunotherapy. Acta Pharm Sin B. (2022) 12:1163–85. doi: 10.1016/j.apsb.2021.08.020
98. Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest. (2016) 126:4157–73. doi: 10.1172/JCI87252
99. Elsaghir A, El-Sabaa EMW, Ahmed AK, Abdelwahab SF, Sayed IM, and El-Mokhtar MA. The role of cluster of differentiation 39 (CD39) and purinergic signaling pathway in viral infections. Pathogens. (2023) 12:279. doi: 10.3390/pathogens12020279
100. Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, and Ernst PB. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. (2006) 177:2765–9. doi: 10.4049/jimmunol.177.5.2765
101. Nickle RA, DeOca KB, Garcia BL, and Mannie MD. Soluble CD25 imposes a low-zone IL-2 signaling environment that favors competitive outgrowth of antigen-experienced CD25(high) regulatory and memory T cells. Cell Immunol. (2023) 384:104664. doi: 10.1016/j.cellimm.2023.104664
102. Frede S, Berchner-Pfannschmidt U, and Fandrey J. Regulation of hypoxia-inducible factors during inflammation. Methods enzymol. (2007) 435:403–19. doi: 10.1016/S0076-6879(07)35021-0
103. Larsen GL and Henson PM. Mediators of inflammation. Annu Rev Immunol. (1983) 1:335–59. doi: 10.1146/annurev.iy.01.040183.002003
104. Sacca R, Cuff CA, and Ruddle NH. Mediators of inflammation. Curr Opin Immunol. (1997) 9:851–7. doi: 10.1016/S0952-7915(97)80189-6
105. Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, and Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. (2004) 114:1098–106. doi: 10.1172/JCI200421086
106. Taylor CT. Interdependent roles for hypoxia inducible factor and nuclear factor-κB in hypoxic inflammation. J Physiol. (2008) 586:4055–9. doi: 10.1113/jphysiol.2008.157669
107. Wakefield A, Dhillon A, Rowles P, Sawyerr A, Pittilo R, Lewis A, et al. Pathogenesis of Crohn’s disease: multifocal gastrointestinal infarction. Lancet. (1989) 334:1057–62. doi: 10.1016/S0140-6736(89)91078-7
108. Haddad JJ and Harb HL. Cytokines and the regulation of hypoxia-inducible factor (HIF)-1α. Int Immunopharmacol. (2005) 5:461–83. doi: 10.1016/j.intimp.2004.11.009
109. Scholz CC and Taylor CT. Targeting the HIF pathway in inflammation and immunity. Curr Opin Pharmacol. (2013) 13:646–53. doi: 10.1016/j.coph.2013.04.009
110. Malkov MI, Lee CT, and Taylor CT. Regulation of the hypoxia-inducible factor (HIF) by pro-inflammatory cytokines. Cells. (2021) 10:2340. doi: 10.3390/cells10092340
111. Dehne N and Brüne B. HIF-1 in the inflammatory microenvironment. Exp Cell Res. (2009) 315:1791–7. doi: 10.1016/j.yexcr.2009.03.019
112. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/S0092-8674(03)00154-5
113. Elson DA, Ryan HE, Snow JW, Johnson R, and Arbeit JM. Coordinate up-regulation of hypoxia inducible factor (HIF)-1α and HIF-1 target genes during multi-stage epidermal carcinogenesis and wound healing. Cancer Res. (2000) 60:6189–95.
114. Walmsley SR, Cadwallader KA, and Chilvers ER. The role of HIF-1α in myeloid cell inflammation. Trends Immunol. (2005) 26:434–9. doi: 10.1016/j.it.2005.06.007
115. Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. (2003) 112:645–57. doi: 10.1016/s0092-8674(03)00154-5
116. Walmsley SR, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, Sobolewski A, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1α–dependent NF-κB activity. J Exp Med. (2005) 201:105–15. doi: 10.1084/jem.20040624
117. Walmsley SR, Print C, Farahi N, Peyssonnaux C, Johnson RS, Cramer T, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. (2005) 201:105–15. doi: 10.1084/jem.20040624
118. Walmsley SR, Chilvers ER, Thompson AA, Vaughan K, Marriott HM, Parker LC, et al. Prolyl hydroxylase 3 (PHD3) is essential for hypoxic regulation of neutrophilic inflammation in humans and mice. J Clin Invest. (2011) 121:1053–63. doi: 10.1172/JCI43273
119. Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. (2011) 146:772–84. doi: 10.1016/j.cell.2011.07.033
120. Palazon A, Goldrath AW, Nizet V, and Johnson RS. HIF transcription factors, inflammation, and immunity. Immunity. (2014) 41:518–28. doi: 10.1016/j.immuni.2014.09.008
121. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. (2011) 208:1367–76. doi: 10.1084/jem.20110278
122. Barbi J, Pardoll D, and Pan F. Metabolic control of the Treg/Th17 axis. Immunol Rev. (2013) 252:52–77. doi: 10.1111/imr.12029
123. Lukashev D, Ohta A, and Sitkovsky M. Hypoxia-dependent anti-inflammatory pathways in protection of cancerous tissues. Cancer Metastasis Rev. (2007) 26:273–9. doi: 10.1007/s10555-007-9054-2
124. Sitkovsky MV, Kjaergaard J, Lukashev D, and Ohta A. Hypoxia-adenosinergic immunosuppression: tumor protection by T regulatory cells and cancerous tissue hypoxia. . Clin Cancer Res. (2008) 14:5947–52. doi: 10.1158/1078-0432.CCR-08-0229
125. Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med. (2015) 21:638–46. doi: 10.1038/nm.3868
126. Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. (2008) 455:808–12. doi: 10.1038/nature07240
127. Jia X, Zhai T, Wang B, Yao Q, Li Q, Mu K, et al. Decreased number and impaired function of type 1 regulatory T cells in autoimmune diseases. J Cell Physiol. (2019) 234:12442–50. doi: 10.1002/jcp.28092
128. Paradowska-Gorycka A, Wajda A, Romanowska-Prochnicka K, Walczuk E, Kuca-Warnawin E, Kmiolek T, et al. Th17/treg-related transcriptional factor expression and cytokine profile in patients with rheumatoid arthritis. Front Immunol. (2020) 11:572858. doi: 10.3389/fimmu.2020.572858
129. deZoeten EF, Battista KD, Colson SB, Lovell MA, Kessler BE, Isfort RW, et al. Markers of hypoxia correlate with histologic and endoscopic severity of colitis in inflammatory bowel disease. Hypoxia (Auckl). (2020) 8:1–12. doi: 10.2147/HP.S219049
130. Xiong A and Liu Y. Targeting hypoxia inducible factors-1alpha as a novel therapy in fibrosis. Front Pharmacol. (2017) 8:326. doi: 10.3389/fphar.2017.00326
131. Izawa M, Tanaka N, Murakami T, Anno T, Teranishi Y, Takamatsu K, et al. Single-cell phenotyping of CD73 expression reveals the diversity of the tumor immune microenvironment and reflects the prognosis of bladder cancer. Lab Invest. (2023) 103:100040. doi: 10.1016/j.labinv.2022.100040
132. Rashid M, Zadeh LR, Baradaran B, Molavi O, Ghesmati Z, Sabzichi M, et al. Up-down regulation of HIF-1alpha in cancer progression. Gene. (2021) 798:145796. doi: 10.1016/j.gene.2021.145796
133. Wang X, Zhao D, Xie H, and Hu Y. Interplay of long non-coding RNAs and HIF-1alpha: A new dimension to understanding hypoxia-regulated tumor growth and metastasis. Cancer Lett. (2021) 499:49–59. doi: 10.1016/j.canlet.2020.11.007
134. Li T, Mao C, Wang X, Shi Y, and Tao Y. Epigenetic crosstalk between hypoxia and tumor driven by HIF regulation. J Exp Clin Cancer Res. (2020) 39:224. doi: 10.1186/s13046-020-01733-5
135. Qiao J, Yu Z, Zhou H, Wang W, Wu H, and Ye J. The pentose phosphate pathway: from mechanisms to implications for gastrointestinal cancers. Int J Mol Sci. (2025) 26:610. doi: 10.3390/ijms26020610
136. TeSlaa T, Ralser M, Fan J, and Rabinowitz JD. The pentose phosphate pathway in health and disease. Nat Metab. (2023) 5:1275–89. doi: 10.1038/s42255-023-00863-2
137. Han D, Li Z, Luo L, and Jiang H. Targeting hypoxia and HIF1alpha in triple-negative breast cancer: new insights from gene expression profiling and implications for therapy. Biol (Basel). (2024) 13:577. doi: 10.3390/biology13080577
138. Huynh KN, Rao S, Roth B, Bryan T, Fernando DM, Dayyani F, et al. Targeting hypoxia-inducible factor-1alpha for the management of hepatocellular carcinoma. Cancers (Basel). (2023) 15:2738. doi: 10.3390/cancers15102738
139. Zhao Y, Zhong B, Cao JY, Wang Q, Liang J, Lu K, et al. Tumor hypoxia: Classification, detection, and its critical role in cancer progression. Biomol BioMed. (2025) 25(8):1672–90. doi: 10.17305/bb.2025.12318
140. He L, He T, Farrar S, Ji L, Liu T, and Ma X. Antioxidants maintain cellular redox homeostasis by elimination of reactive oxygen species. Cell Physiol Biochem. (2017) 44:532–53. doi: 10.1159/000485089
141. Patra KC and Hay N. The pentose phosphate pathway and cancer. Trends Biochem Sci. (2014) 39:347–54. doi: 10.1016/j.tibs.2014.06.005
142. Ravi D, Beheshti A, Abermil N, Lansigan F, Kinlaw W, Matthan NR, et al. Oncogenic integration of nucleotide metabolism via fatty acid synthase in non-hodgkin lymphoma. Front Oncol. (2021) 11:725137. doi: 10.3389/fonc.2021.725137
143. DeBerardinis RJ, Lum JJ, Hatzivassiliou G, and Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. (2008) 7:11–20. doi: 10.1016/j.cmet.2007.10.002
144. Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, et al. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. (2011) 13:310–6. doi: 10.1038/ncb2172
145. Marini C, Ravera S, Buschiazzo A, Bianchi G, Orengo AM, Bruno S, et al. Discovery of a novel glucose metabolism in cancer: The role of endoplasmic reticulum beyond glycolysis and pentose phosphate shunt. Sci Rep. (2016) 6:25092. doi: 10.1038/srep25092
146. Tsachaki M, Mladenovic N, Stambergova H, Birk J, and Odermatt A. Hexose-6-phosphate dehydrogenase controls cancer cell proliferation and migration through pleiotropic effects on the unfolded-protein response, calcium homeostasis, and redox balance. FASEB J. (2018) 32:2690–705. doi: 10.1096/fj.201700870RR
147. Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, and Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L287–300. doi: 10.1152/ajplung.00229.2014
148. Xu K, He Z, Chen M, Wang N, Zhang D, Yang L, et al. HIF-1alpha regulates cellular metabolism, and Imatinib resistance by targeting phosphogluconate dehydrogenase in gastrointestinal stromal tumors. Cell Death Dis. (2020) 11:586. doi: 10.1038/s41419-020-02768-4
149. Zhao F, Mancuso A, Bui TV, Tong X, Gruber JJ, Swider CR, et al. Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1alpha-induced metabolic reprograming. Oncogene. (2010) 29:2962–72. doi: 10.1038/onc.2010.67
150. Yurkanova MD, Kosheleva NV, Teplova AA, Timashev PS, and Vlasova II. Pro-inflammatory properties of M1 phenotypes of human macrophages: prolongation of myeloperoxidase-mediated oxidative stress. Free Radic Res. (2025) 59:452–61. doi: 10.1080/10715762.2025.2519528
151. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, and Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
152. Grund EM, Clarkson BD, Pucci S, Westphal MS, Partida CM, Muhammad SA, et al. Pentose phosphate pathway inhibition metabolically reprograms CD8+ T cells and disrupts CNS autoimmunity. JCI Insight. (2025) 10:e184240. doi: 10.1172/jci.insight.184240
153. Banki K and Perl A. Cell type-specific regulation of the pentose phosphate pathway during development and metabolic stress-driven autoimmune diseases: Relevance for inflammatory liver, renal, endocrine, cardiovascular and neurobehavioral comorbidities, carcinogenesis, and aging. Autoimmun Rev. (2025) 24:103781. doi: 10.1016/j.autrev.2025.103781
154. Li W, Kolios AGA, Pan W, Burbano C, Karino K, Vichos T, et al. Gluconolactone restores immune regulation and alleviates skin inflammation in lupus-prone mice and in patients with cutaneous lupus. Sci Transl Med. (2025) 17:eadp4447. doi: 10.1126/scitranslmed.adp4447
155. Yin Y, Choi SC, Xu Z, Zeumer L, Kanda N, Croker BP, et al. Glucose oxidation is critical for CD4+ T cell activation in a mouse model of systemic lupus erythematosus. J Immunol. (2016) 196:80–90. doi: 10.4049/jimmunol.1501537
156. Kishore M, Cheung KCP, Fu H, Bonacina F, Wang G, Coe D, et al. Regulatory T cell migration is dependent on glucokinase-mediated glycolysis. Immunity. (2017) 47:875–89 e10. doi: 10.1016/j.immuni.2017.10.017
157. Lee HT, Wu TH, Lin CS, Lee CS, Wei YH, Tsai CY, et al. The pathogenesis of systemic lupus erythematosus - From the viewpoint of oxidative stress and mitochondrial dysfunction. Mitochondrion. (2016) 30:1–7. doi: 10.1016/j.mito.2016.05.007
158. Katsuyama T, Tsokos GC, and Moulton VR. Aberrant T cell signaling and subsets in systemic lupus erythematosus. Front Immunol. (2018) 9:1088. doi: 10.3389/fimmu.2018.01088
159. Guleria A, Pratap A, Dubey D, Rawat A, Chaurasia S, Sukesh E, et al. NMR based serum metabolomics reveals a distinctive signature in patients with Lupus Nephritis. Sci Rep. (2016) 6:35309. doi: 10.1038/srep35309
160. Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. (2009) 206:1983–94. doi: 10.1084/jem.20090480
161. Chen J, Liu C, Chernatynskaya AV, Newby B, Brusko TM, Xu Y, et al. NADPH oxidase 2-derived reactive oxygen species promote CD8+ T cell effector function. J Immunol. (2024) 212:258–70. doi: 10.4049/jimmunol.2200691
162. Glennon-Alty L, Hackett AP, Chapman EA, and Wright HL. Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free Radic Biol Med. (2018) 125:25–35. doi: 10.1016/j.freeradbiomed.2018.03.049
163. Perl A. Activation of mTOR (mechanistic target of rapamycin) in rheumatic diseases. Nat Rev Rheumatol. (2016) 12:169–82. doi: 10.1038/nrrheum.2015.172
164. Zeng MY, Miralda I, Armstrong CL, Uriarte SM, and Bagaitkar J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol Oral Microbiol. (2019) 34:27–38. doi: 10.1111/omi.12252
165. Diaz-Flores M, Ibanez-Hernandez MA, Galvan RE, Gutierrez M, Duran-Reyes G, Medina-Navarro R, et al. Glucose-6-phosphate dehydrogenase activity and NADPH/NADP+ ratio in liver and pancreas are dependent on the severity of hyperglycemia in rat. Life Sci. (2006) 78:2601–7. doi: 10.1016/j.lfs.2005.10.022
166. Lin Q, Zhang C, Huang H, Bai Z, Liu J, Zhang Y, et al. TLR2 reprograms glucose metabolism in CD4(+) T cells of rheumatoid arthritis patients to mediate cell hyperactivation and TNF-alpha secretion. Clin Rheumatol. (2024) 43:3537–49. doi: 10.1007/s10067-024-07125-w
167. Liu Q, Zhu F, Liu X, Lu Y, Yao K, Tian N, et al. Non-oxidative pentose phosphate pathway controls regulatory T cell function by integrating metabolism and epigenetics. Nat Metab. (2022) 4:559–74. doi: 10.1038/s42255-022-00575-z
168. Long J, Hu Z, Xue H, Wang Y, Chen J, Tang F, et al. Vascular endothelial growth factor (VEGF) impairs the motility and immune function of human mature dendritic cells through the VEGF receptor 2-RhoA-cofilin1 pathway. Cancer Sci. (2019) 110:2357–67. doi: 10.1111/cas.14091
169. Garchow B, Maque Acosta Y, and Kiriakidou M. HIF-1alpha and miR-210 differential and lineage-specific expression in systemic lupus erythematosus. Mol Immunol. (2021) 133:128–34. doi: 10.1016/j.molimm.2021.02.019
170. Roman J, Rangasamy T, Guo J, Sugunan S, Meednu N, Packirisamy G, et al. T-cell activation under hypoxic conditions enhances IFN-gamma secretion. Am J Respir Cell Mol Biol. (2010) 42:123–8. doi: 10.1165/rcmb.2008-0139OC
171. Cavailles A, Brinchault-Rabin G, Dixmier A, Goupil F, Gut-Gobert C, Marchand-Adam S, et al. Comorbidities of COPD. Eur Respir Rev. (2013) 22:454–75. doi: 10.1183/09059180.00008612
172. Mirchandani AS, Jenkins SJ, Bain CC, Sanchez-Garcia MA, Lawson H, Coelho P, et al. Hypoxia shapes the immune landscape in lung injury and promotes the persistence of inflammation. Nat Immunol. (2022) 23:927–39. doi: 10.1038/s41590-022-01216-z
173. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. (2005) 1:401–8. doi: 10.1016/j.cmet.2005.05.001
174. Boland K, Flanagan L, and Prehn JH. Paracrine control of tissue regeneration and cell proliferation by Caspase-3. Cell Death Dis. (2013) 4:e725. doi: 10.1038/cddis.2013.250
175. Bratton SB and Salvesen GS. Regulation of the apaf-1-caspase-9 apoptosome. J Cell Sci. (2010) 123:3209–14. doi: 10.1242/jcs.073643
176. Martinou JC, Desagher S, and Antonsson B. Cytochrome c release from mitochondria: all or nothing. Nat Cell Biol. (2000) 2:E41–3. doi: 10.1038/35004069
177. Nelson DA, Tan TT, Rabson AB, Anderson D, Degenhardt K, and White E. Hypoxia and defective apoptosis drive genomic instability and tumorigenesis. Genes Dev. (2004) 18:2095–107. doi: 10.1101/gad.1204904
178. Kam NW, Lau CY, Lau JYH, Dai X, Liang Y, Lai SPH, et al. Cell-associated galectin 9 interacts with cytotoxic T cells confers resistance to tumor killing in nasopharyngeal carcinoma through autophagy activation. Cell Mol Immunol. (2025) 22:260–81. doi: 10.1038/s41423-024-01253-8
179. Saff RR, Spanjaard ES, Hohlbaum AM, and Marshak-Rothstein A. Activation-induced cell death limits effector function of CD4 tumor-specific T cells. J Immunol. (2004) 172:6598–606. doi: 10.4049/jimmunol.172.11.6598
180. Sun J, Zhang Y, Yang M, Zhang Y, Xie Q, Li Z, et al. Hypoxia induces T-cell apoptosis by inhibiting chemokine C receptor 7 expression: the role of adenosine receptor A(2). Cell Mol Immunol. (2010) 7:77–82. doi: 10.1038/cmi.2009.105
181. Hopken UE, Droese J, Li JP, Joergensen J, Breitfeld D, Zerwes HG, et al. The chemokine receptor CCR7 controls lymph node-dependent cytotoxic T cell priming in alloimmune responses. Eur J Immunol. (2004) 34:461–70. doi: 10.1002/eji.200324690
182. McInnes IB and Schett G. Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol. (2007) 7:429–42. doi: 10.1038/nri2094
183. Brouwer E, Gouw AS, Posthumus MD, van Leeuwen MA, Boerboom AL, Bijzet J, et al. Hypoxia inducible factor-1-alpha (HIF-1alpha) is related to both angiogenesis and inflammation in rheumatoid arthritis. Clin Exp Rheumatol. (2009) 27:945–51.
184. Li G, Zhang Y, Qian Y, Zhang H, Guo S, Sunagawa M, et al. Interleukin-17A promotes rheumatoid arthritis synoviocytes migration and invasion under hypoxia by increasing MMP2 and MMP9 expression through NF-kappaB/HIF-1alpha pathway. Mol Immunol. (2013) 53:227–36. doi: 10.1016/j.molimm.2012.08.018
185. Hu F, Liu H, Xu L, Li Y, Liu X, Shi L, et al. Hypoxia-inducible factor-1alpha perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. Eur J Immunol. (2016) 46:742–51. doi: 10.1002/eji.201545784
186. Veale DJ, Ritchlin C, and FitzGerald O. Immunopathology of psoriasis and psoriatic arthritis. Ann Rheum Dis. (2005) 64 Suppl 2:ii26–9. doi: 10.1136/ard.2004.031740
187. Rahat MA, Safieh M, Simanovich E, Pasand E, Gazitt T, Haddad A, et al. The role of EMMPRIN/CD147 in regulating angiogenesis in patients with psoriatic arthritis. Arthritis Res Ther. (2020) 22:240. doi: 10.1186/s13075-020-02333-6
188. Watanabe A, Kamata M, Shimizu T, Uchida H, Sakurai E, Suzuki S, et al. Serum levels of angiogenesis-related factors in patients with psoriasis. J Dermatol. (2023) 50:222–8. doi: 10.1111/1346-8138.16588
189. Kilic Y, Guzel Erdogan D, Baykul M, and Nas K. Examining the functions of the vascular endothelial growth factor/hypoxia-inducible factor signaling pathway in psoriatic arthritis. Arch Rheumatol. (2023) 38:579–89. doi: 10.46497/ArchRheumatol.2023.9898
190. Wu MM, Wang QM, Huang BY, Mai CT, Wang CL, Wang TT, et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol Res. (2021) 172:105796. doi: 10.1016/j.phrs.2021.105796
191. Galvan-Pena S and O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. (2014) 5:420. doi: 10.3389/fimmu.2014.00420
192. Palmieri EM, Gonzalez-Cotto M, Baseler WA, Davies LC, Ghesquiere B, Maio N, et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun. (2020) 11:698. doi: 10.1038/s41467-020-14433-7
193. Gao L, Wang FQ, Li HM, Yang JG, Ren JG, He KF, et al. CCL2/EGF positive feedback loop between cancer cells and macrophages promotes cell migration and invasion in head and neck squamous cell carcinoma. Oncotarget. (2016) 7:87037–51. doi: 10.18632/oncotarget.13523
194. Arranz A, Doxaki C, Vergadi E, Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, et al. Akt1 and Akt2 protein kinases differentially contribute to macrophage polarization. Proc Natl Acad Sci U.S.A. (2012) 109:9517–22. doi: 10.1073/pnas.1119038109
195. Bloemendaal FM, Levin AD, Wildenberg ME, Koelink PJ, McRae BL, Salfeld J, et al. Anti-tumor necrosis factor with a glyco-engineered fc-region has increased efficacy in mice with colitis. Gastroenterology. (2017) 153:1351–62 e4. doi: 10.1053/j.gastro.2017.07.021
196. Fluck K, Breves G, Fandrey J, and Winning S. Hypoxia-inducible factor 1 in dendritic cells is crucial for the activation of protective regulatory T cells in murine colitis. Mucosal Immunol. (2016) 9:379–90. doi: 10.1038/mi.2015.67
197. Yu X, Yuan Z, Yang Z, Chen D, Kim T, Cui Y, et al. The novel long noncoding RNA u50535 promotes colorectal cancer growth and metastasis by regulating CCL20. Cell Death Dis. (2018) 9:751. doi: 10.1038/s41419-018-0771-y
198. Rezalotfi A, Ahmadian E, Aazami H, Solgi G, and Ebrahimi M. Gastric cancer stem cells effect on th17/treg balance; A bench to beside perspective. Front Oncol. (2019) 9:226. doi: 10.3389/fonc.2019.00226
199. Jiang Y, Liu Y, Lu H, Sun SC, Jin W, Wang X, et al. Epigenetic activation during T helper 17 cell differentiation is mediated by Tripartite motif containing 28. Nat Commun. (2018) 9:1424. doi: 10.1038/s41467-018-03852-2
200. Yu Q, Lou XM, and He Y. Preferential recruitment of Th17 cells to cervical cancer via CCR6-CCL20 pathway. PloS One. (2015) 10:e0120855. doi: 10.1371/journal.pone.0120855
201. Yang S, Wang B, Guan C, Wu B, Cai C, Wang M, et al. Foxp3+IL-17+ T cells promote development of cancer-initiating cells in colorectal cancer. J Leukoc Biol. (2011) 89:85–91. doi: 10.1189/jlb.0910506
202. Huang C and Fu ZX. Localization of IL-17+Foxp3+ T cells in esophageal cancer. Immunol Invest. (2011) 40:400–12. doi: 10.3109/08820139.2011.555489
203. Renaude E, Kroemer M, Loyon R, Binda D, Borg C, Guittaut M, et al. The fate of th17 cells is shaped by epigenetic modifications and remodeled by the tumor microenvironment. Int J Mol Sci. (2020) 21:1673. doi: 10.3390/ijms21051673
204. Gray LH, Conger AD, Ebert M, Hornsey S, and Scott OC. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br J Radiol. (1953) 26:638–48. doi: 10.1259/0007-1285-26-312-638
205. Ciepla J and Smolarczyk R. Tumor hypoxia unveiled: insights into microenvironment, detection tools and emerging therapies. Clin Exp Med. (2024) 24:235. doi: 10.1007/s10238-024-01501-1
206. Obeagu EI. Oxygen deprivation in breast cancer: mechanisms, pathways, and implications. Ann Med Surg (Lond). (2025) 87:3635–59. doi: 10.1097/MS9.0000000000003334
207. Baskar R, Dai J, Wenlong N, Yeo R, and Yeoh KW. Biological response of cancer cells to radiation treatment. Front Mol Biosci. (2014) 1:24. doi: 10.3389/fmolb.2014.00024
208. Hill RM, Fok M, Grundy G, Parsons JL, and Rocha S. The role of autophagy in hypoxia-induced radioresistance. Radiother Oncol. (2023) 189:109951. doi: 10.1016/j.radonc.2023.109951
209. Dong J, Wang C, Zhang T, Yu X, Peng H, Xiao Z, et al. Establishment and application of novel hypoxia-driven dual-reporter model to investigate hypoxic impact on radiation sensitivity in human nasopharyngeal carcinoma xenografts. J Cancer. (2024) 15:4345–59. doi: 10.7150/jca.96378
210. Marks C and Leech M. Optimising hypoxia PET imaging and its applications in guiding targeted radiation therapy for non-small cell lung cancer: a scoping review. J Med Radiat Sci. (2025) 72:106–18. doi: 10.1002/jmrs.831
211. Sambasivan K, Barrington SF, Connor SE, Witney TH, Blower PJ, and Urbano TG. Is there a role for [(18)F]-FMISO PET to guide dose adaptive radiotherapy in head and neck cancer? A review of the literature. Clin Transl Imaging. (2024) 12:137–55. doi: 10.1007/s40336-023-00607-y
212. Gawrylak A, Brodaczewska K, Iwanicka-Nowicka R, Koblowska M, Synowiec A, Bodnar L, et al. Hypoxia alters the response of ovarian cancer cells to the mitomycin C drug. Front Cell Dev Biol. (2025) 13:1575134. doi: 10.3389/fcell.2025.1575134
213. Synowiec A, Brodaczewska K, Wcislo G, Majewska A, Borkowska A, Filipiak-Duliban A, et al. Hypoxia, but not normoxia, reduces effects of resveratrol on cisplatin treatment in A2780 ovarian cancer cells: A challenge for resveratrol use in anticancer adjuvant cisplatin therapy. Int J Mol Sci. (2023) 24:5715. doi: 10.3390/ijms24065715
214. Chen G, Wu K, Li H, Xia D, and He T. Role of hypoxia in the tumor microenvironment and targeted therapy. Front Oncol. (2022) 12:961637. doi: 10.3389/fonc.2022.961637
215. Fan P, Zhang N, Candi E, Agostini M, Piacentini M, Centre TOR, et al. Alleviating hypoxia to improve cancer immunotherapy. Oncogene. (2023) 42:3591–604. doi: 10.1038/s41388-023-02869-2
216. Das S, Venkatesh GH, Elsayed WSM, Abou Khouzam R, Mahmood AS, Nawafleh HH, et al. Evofosfamide enhances sensitivity of breast cancer cells to apoptosis and natural-killer-cell-mediated cytotoxicity under hypoxic conditions. Cancers (Basel). (2025) 17:1988. doi: 10.3390/cancers17121988
217. McNeal KC, Reeves KM, Song PN, and Lapi SE. A. G. Sorace,B. M. Larimer. [(18)F]FMISO-PET imaging reveals the role of hypoxia severity in checkpoint blockade response. Nucl Med Biol. (2024) 134-135:108918. doi: 10.1016/j.nucmedbio.2024.108918
218. Debnath SK, Debnath M, Ghosh A, Srivastava R, and Omri A. Targeting tumor hypoxia with nanoparticle-based therapies: challenges, opportunities, and clinical implications. Pharm (Basel). (2024) 17:1389. doi: 10.3390/ph17101389
219. Wei J, Hu M, and Du H. Improving cancer immunotherapy: exploring and targeting metabolism in hypoxia microenvironment. Front Immunol. (2022) 13:845923. doi: 10.3389/fimmu.2022.845923
220. Uddin F, Rudin CM, and Sen T. CRISPR gene therapy: applications, limitations, and implications for the future. Front Oncol. (2020) 10:1387. doi: 10.3389/fonc.2020.01387
221. Shi S, Ou X, Liu C, Wen H, and Ke J. Research progress of HIF-1a on immunotherapy outcomes in immune vascular microenvironment. Front Immunol. (2025) 16:1549276. doi: 10.3389/fimmu.2025.1549276
222. Gajewski TF, Schreiber H, and Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. (2013) 14:1014–22. doi: 10.1038/ni.2703
223. Semenza GL. Hypoxia-inducible factors: mediators of cancer progression and targets for cancer therapy. Trends Pharmacol Sci. (2012) 33:207–14. doi: 10.1016/j.tips.2012.01.005
224. Kleinewietfeld M and Hafler DA. The plasticity of human Treg and Th17 cells and its role in autoimmunity. Semin Immunol. (2013) 25:305–12. doi: 10.1016/j.smim.2013.10.009
225. Mark-Christensen A, Erichsen R, Veres K, Laurberg S, and Sorensen HT. Extracolonic cancer risk after total colectomy for inflammatory bowel disease: A population-based cohort study. J Crohns Colitis. (2020) 14:630–5. doi: 10.1093/ecco-jcc/jjz199
226. Olen O, Erichsen R, Sachs MC, Pedersen L, Halfvarson J, Askling J, et al. Colorectal cancer in ulcerative colitis: a Scandinavian population-based cohort study. Lancet. (2020) 395:123–31. doi: 10.1016/S0140-6736(19)32545-0
Keywords: hypoxia, HIF-1α, helper T cells, cancer, autoimmunity
Citation: Almanzar G, Alarcon JC, Garzon R, Navarro A M, Ondo-Méndez A and Prelog M (2025) Hypoxia and activation of hypoxia inducible factor alpha as influencers of inflammatory helper T cells in autoimmune disease – a link between cancer and autoimmunity. Front. Immunol. 16:1633845. doi: 10.3389/fimmu.2025.1633845
Received: 23 May 2025; Accepted: 13 August 2025;
Published: 02 September 2025.
Edited by:
Andras Perl, Upstate Medical University, United StatesReviewed by:
James Joseph Driscoll, University Hospitals of Cleveland, United StatesOlalekan Chris Akinsulie, Washington State University, United States
Copyright © 2025 Almanzar, Alarcon, Garzon, Navarro, Ondo-Méndez and Prelog. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giovanni Almanzar, QWxtYW56YXJfR0B1a3cuZGU=
†These authors have contributed equally to this work