 Renyan Xiao
Renyan Xiao Zhongyu Han
Zhongyu Han Peng Jia1†
Peng Jia1† Meng Gong
Meng Gong Yijin Cai
Yijin Cai Xiangyin Ye
Xiangyin Ye- 1School of Health Preservation and Rehabilitation, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 2Zhongda Hospital, School of Medicine, Southeast University, Nanjing, China
- 3School of Acupuncture and Tuina, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 4School of Medical and Life Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, China
- 5Department of Rehabilitation, Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, China
Ferroptosis is a novel type of programmed cell death that was discovered in recent years and is closely associated with disorders in iron cycling, abnormal lipid metabolism, excessive intracellular reactive oxygen species, and cellular antioxidant-related signaling pathways. Numerous studies have shown that ferroptosis plays a critical role in the development and progression of bone and joint diseases, although the underlying mechanisms remain incompletely understood. This review aims to outline the relevant mechanisms of ferroptosis, its implications in the bone microenvironment, and the mechanisms of action and therapeutic perspectives of ferroptosis in common bone and joint diseases, with the goal of informing future clinical research and treatment strategies targeting ferroptosis under these conditions.
1 Introduction
Degenerative orthopaedic diseases, such as osteoporosis (OP), osteoarthritis (OA), and lumbar disc herniation (LDH), gravely undermine patients’ quality of life. They inflict pain, lead to functional impairment and disability, and generate a massive financial burden for families and society (1). Osteosarcoma (OS) is the most common type of primary malignant bone tumor; it frequently occurs at the metaphysis of long bones in adolescents and strongly affects their growth and development (2).
Ferroptosis was proposed as a novel form of cell death in 2012. It is distinct from traditional forms of programmed cell death, such as apoptosis and pyroptosis, in both biochemical and cellular morphological aspects (3). Ferroptosis is a type of programmed cell death that is driven by iron and is biochemically characterized by the significant accumulation of lipid reactive oxygen species (ROS) within cells. As redox-active trace elements, iron ions are essential regulators of multiple cellular functions. Under pathological conditions, the accumulation of excess iron and ROS can result in ferroptosis. Morphologically, it is primarily observed as the atrophy of mitochondria. The number of cristae in mitochondria may decrease or disappear. While investigations into ferroptosis across different domains are still in the preliminary phase, a growing body of research has recognized its crucial involvement in the pathological mechanisms of orthopaedic ailments. Therefore, comprehensively reviewing and analyzing the connections between ferroptosis and bone and joint diseases is essential.
This review seeks to clarify the molecular mechanisms underlying ferroptosis, its significance within the bone microenvironment, and the modes of action and potential therapeutic targets associated with ferroptosis in conditions such as OP, OA, OS, and LDH. Furthermore, the future prospects of targeting ferroptosis for the treatment of these disorders, as well as the current limitations and challenges within the field, are discussed.
2 Molecular mechanisms of ferroptosis
Ferroptosis, a novel form of cell death distinct from apoptosis, pyroptosis and autophagy, is an iron-dependent programmed form of cell death characterized by an iron metabolism disorder, imbalance of the redox system and the accumulation of lipid peroxides (4). During this form of cell death, a discrepancy exists between intracellular oxidative stress and the antioxidant defense system. This imbalance may lead to cell death by triggering lipid oxidation in the membrane, compromising membrane integrity, inducing lipid cross-linking or causing further oxidative damage to macromolecules. Ferroptosis was discovered a decade ago. However, similar cell death phenomena have been reported in previous scientific research. For example, ‘oxygen toxicity’ describes a form of cell death caused by excess oxidative stress in neuronal cells (5). In the mid-20th century, Harry Eagle et al. made a pioneering discovery that depriving cells of cystine could lead to cell death. They also found that cells with the ability to synthesize cysteine internally could resist ferroptosis (6, 7).
Ferroptosis is an evolutionarily conserved process that occurs in mammals and various organisms (such as plants and microorganisms). It has crucial impacts on the growth and diseases of the abovementioned organisms (8, 9). Ferroptosis involves a complex regulatory network and is regulated by various biological pathways. These pathways include cellular metabolism, such as the intracellular iron and lipid cycles, along with genes that regulate ferroptosis-associated protein expression (10).
2.1 Iron accumulation
Iron is crucial for various life activities of cells (11). An imbalance in the iron redox reaction and the iron ion cycle in cells are important factors leading to ferroptosis in cells (12). Extracellular iron enters the cell by binding to transferrin (TF), which recognizes transferrin receptor 1 (TFR1) on the cell membrane (13). Within the cell, iron is reduced to its divalent form by the six-transmembrane epithelial antigen of the prostate 3 (STEAP3) and is then translocated into the cytoplasm by divalent metal transporter 1 (DMT1) (Figure 1) (14, 15). Intracellular iron is stored either as ferritin or in the labile iron pool (LIP). Iron subsequently exits the cell into the extracellular environment through the iron transport protein ferroportin (FPN) (16, 17).
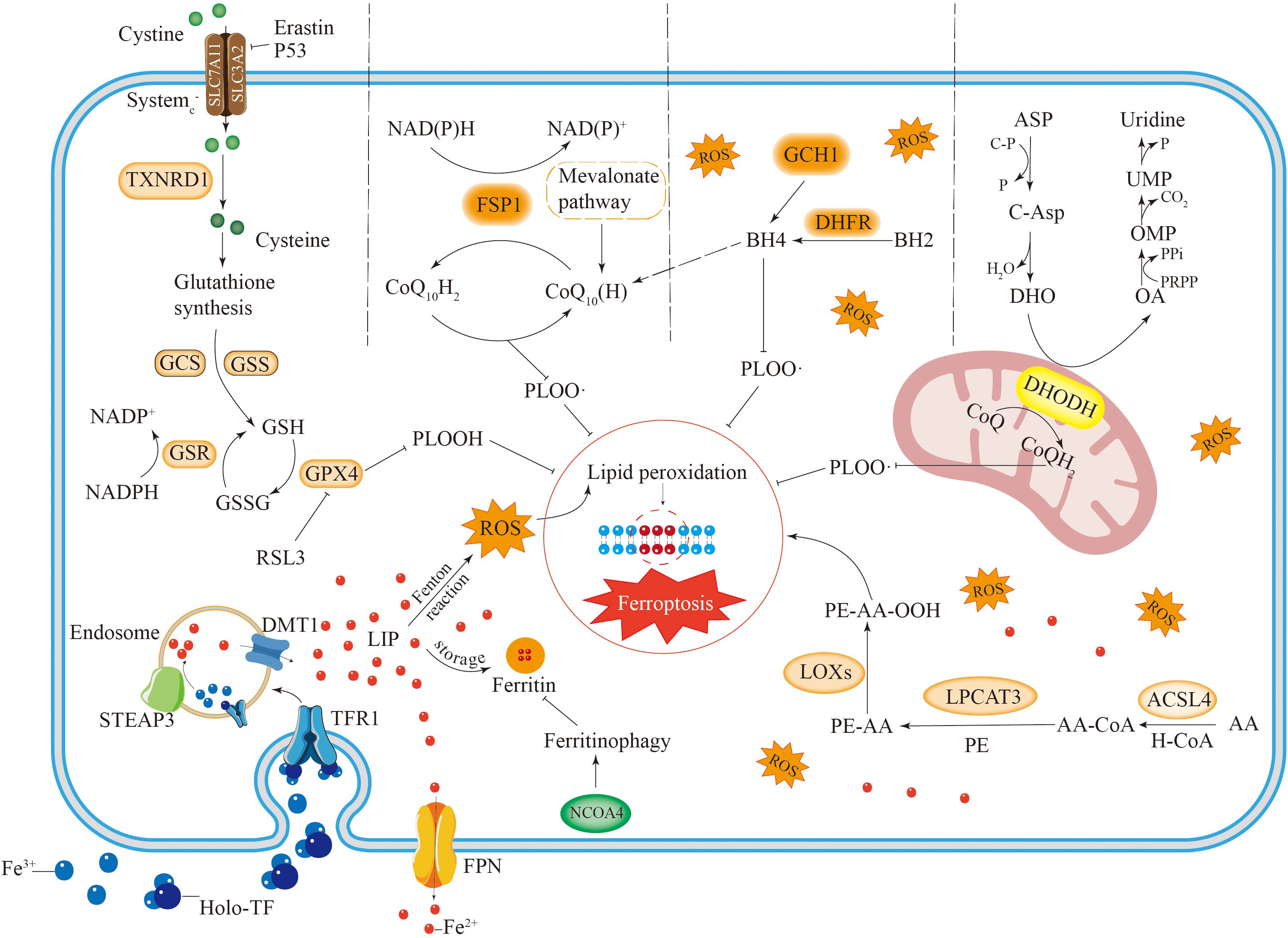
Figure 1. The occurrence and regulatory mechanisms of ferroptosis. Iron metabolism involves iron uptake, storage and export. Iron is mainly stored in cells in the form of free iron (LIP) and ferritin. Reactive iron can trigger the Fenton reaction, leading to the accumulation of ROS and inducing ferroptosis. Autophagy of ferritin mediated by NCOA4 can increase free iron in cells. AA undergoes a series of reactions to cause lipid peroxidation, leading to ferroptosis in cells. There are four antioxidant pathways in ferroptosis, including the System Xc–GSH-GPX4 pathway, NADPH-FSP1-CoQ10 pathway, GCH1-BH4 pathway and DHODH-CoQH2 pathway. They inhibit lipid peroxidation to suppress ferroptosis in cells.
Excessive free iron leads to iron accumulation, which triggers nonenzymatic, iron-dependent Fenton chain reactions that directly produce excessive reactive oxygen species, thereby causing oxidative damage to cells (18). Therefore, excessive iron accumulation promotes ferroptosis in cells. IRP1 and IRP2 are crucial proteins involved in the meticulous regulation of intracellular iron levels. These proteins play significant roles in managing the iron cycle within the cell by engaging in posttranscriptional regulation. This process specifically targets genes that are associated with iron transport inside the cell, ensuring that iron homeostasis is effectively and efficiently maintained (19, 20). Through their regulatory activities, IRP1 and IRP2 help control the availability of iron, which is essential for various cellular functions and overall cellular health. Ferritin, which is composed of a light chain (FTL) and a heavy chain (FTH), has significant antiferroptotic properties. The degradation of ferritin via lysosome-associated ferritin-induced autophagy leads to elevated levels of free reactive iron within cells and an accumulation of iron, which ultimately results in iron-induced cellular death (21, 22). Suppressing ferritin-induced autophagy triggered by nuclear receptor coactivator 4 (NCOA4) promotes the effective use of cellular iron ions, thus reducing the occurrence of cellular ferroptosis (Figure 1).
Poly (RC)-binding proteins (PCBPs), which are iron chaperones that transfer iron to corresponding proteins- can transport Fe2+ to ferritin, thereby increasing ferroptosis resistance in hepatocytes (23). Haem oxygenase 1 (HO-1) facilitates the breakdown of heme, resulting in increased levels of reactive iron ions within the cell, which play a role in inducing cellular iron-related death. Notably, HO-1 has both positive and negative regulatory effects on ferroptosis (24, 25). ATM, a serine–threonine protein kinase, can block metal - regulated transcription factor 1 (MTF1), thereby promoting ferroptosis (26). Furthermore, prominin 2 (PROM2) reduces ferroptosis in breast cancer cells by promoting the export of iron from these cells (27).
Moderate levels of cellular iron are essential for maintaining normal bone health. Bone remodeling and resorption during physiological processes require adequate amounts of iron. Abnormal iron metabolism plays a significant role in various diseases. In the pathology of OA, cellular iron overload can increase localized inflammatory mediators, contributing to OA development (28). Additionally, abnormal iron metabolism can impair osteoclast function and induce oxidative stress, leading to an imbalance in bone homeostasis and bone loss, which is associated with OP formation (29, 30). Iron overload in chondrocytes can elevate markers of chondrocyte catabolism, triggering cartilage degeneration. Furthermore, iron overload in synoviocytes can influence cytokine expression, resulting in synovial inflammation linked to OA formation (28, 31). Therefore, iron metabolism in the bone microenvironment significantly impacts both physiological processes and the pathological development of bone.
2.2 Lipid peroxidation
A disruption of cell membrane integrity, which can lead to cell death, represents one potential mechanism through which ferroptosis occurs. Polyunsaturated fatty acids (PUFAs) play a critical role as key constituents of the cytoplasmic membrane and act as major targets of ROS (32–34). As a result, managing lipid metabolism is essential to the regulatory processes related to ferroptotic cell death. The enzyme adenylate-activated protein kinase (AMPK), which acts as a sensor of cellular energy levels, influences ferroptosis by facilitating the phosphorylation of acetyl coenzyme A carboxylase (ACC) and the production of polyunsaturated fatty acids (35). Additionally, the dissection of its upstream kinase, liver kinase B1 (LKB1), increases the sensitivity of mouse embryonic fibroblasts to ferroptosis (36).
In the process of ferroptosis, PUFAs serve as major targets for lipid peroxidation. Research has shown that oleic acid can effectively prevent erastin-triggered ferroptosis by engaging in competition with PUFAs to be incorporated into PLs (37). PUFAs can be integrated into cellular membranes via acylcarnitine synthase 4 (ACSL4) (38) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) (39), increasing cellular vulnerability to ferroptosis. The oxidation of unsaturated fatty acids may proceed through both enzymatic and nonenzymatic mechanisms. For example, arachidonic acid (AA) is converted by ACSL4 and LPCAT3. Subsequently, the resultant molecule undergoes oxidation through either lipoxygenase (Lox) or a non-enzymatic pathway, leading to the formation of PE-AA-OOH (Figure 1) (40, 41).
Significant lipid peroxidation plays a crucial role in ferroptosis, potentially resulting in cellular death via various mechanisms. These mechanisms involve modifications to the membrane’s lipid bilayer structure, the creation of membrane pores that impair barrier function, a decrease in membrane thickness, and alterations in permeability (42). Furthermore, peroxidated lipids can be decomposed into toxic derivatives, which may induce significant cytotoxicity (43, 44).
Lipid peroxidation serves as a crucial mechanism in ferroptosis, and its occurrence within the bone microenvironment significantly contributes to cellular ferroptosis, thereby impacting bone health. Inhibiting lipid peroxidation in bone microenvironment cells may represent a promising therapeutic approach for treating bone-related diseases. Notably, the inhibition of ACSL4 has been shown to reduce the incidence of neuronal ferroptosis, indicating that ACSL4 could be a viable target for the treatment of bone-related disorders (45). Furthermore, compounds that inhibit the lipid peroxidation process, such as baicalein and zileuton, have demonstrated the ability to enhance bone density by promoting bone formation, suggesting that these agents may serve as potential therapeutic options for OP (46).
2.3 Antioxidant mechanisms in ferroptosis
System XC–GSH–GPX4 pathway is an important antioxidant pathway in cells. Glutathione peroxidase 4 (GPX4) is a key enzyme characterized by the presence of selenocysteine, which plays a vital role in neutralizing phospholipid hydroperoxides (47). Research has shown that when GPX4 is absent, lipid peroxidation leads to non-apoptotic cell death in mouse embryonic fibroblasts (48). Both the expression levels and functionality of GPX4 are influenced by selenium and glutathione (GSH). Selenium participates in the process of GPX4 synthesis and substitutes for the sulfur in cysteine (49). Furthermore, selenium can also induce the upregulation of GPX4 expression through the transcriptional pathway to reduce ferroptosis (50).
GPX4 exerts its physiological functions through GSH. During the process of phospholipid hydroperoxide being reduced to the corresponding phospholipid alcohols, GSH acts as an electron donor, while glutathione disulfide (GSSG) is produced simultaneously (51). Glutathione disulfide reductase (GSR) can recycle oxidized glutathione using electrons sourced from reduced NADPH to restore glutathione (Figure 1) (52).
In the cytoplasm, GSH is synthesized through the catalytic action of glutamate cysteine ligase (GCL) and glutathione synthetase (GSS) (52). In this synthesis reaction, cysteine serves as the critical rate-limiting factor (53). Cysteine enters cells in its oxidized form, known as cystine, through System XC-. Once inside the cell, cystine is reduced to cysteine by the enzyme thioredoxin reductase 1 (TXNRD1) (Figure 1) (54). Furthermore, cysteine may also be synthesized from methionine via the transsulfuration pathway (55). The reverse transporter protein System XC-, which operates as a heterodimeric complex of SLC7A11 and SLC3A2, enables the exchange of cystine and glutamate across the plasma membrane (3, 56). P53 can induce ferroptosis and consequently tumor cell death by inhibiting GSH synthesis through the downregulation of SLC7A11 expression (57). Erastin can directly inhibit cystine uptake, leading to ferroptosis, whereas RSL3 can inhibit GPX4 to induce ferroptosis (Figure 1) (58).
Ferroptosis inhibitory protein 1 (FSP1) serves as an important antioxidant involved in the mechanism of ferroptosis (59). By utilizing NADPH, FSP1 reduces coenzyme Q to ubiquinol, effectively halting lipid autoxidation (Figure 1) (60, 61). Moreover, FSP1 can inhibit cell death through α-tocopherol-mediated antioxidation, which is more potent than the former mechanism (62). The antioxidant effects of the FSP1 pathway are primarily mediated by coenzyme Q10 (CoQ10), an isoprenoid benzoquinone compound (63, 64). Additionally, panthenol (CoQ10H2), serves as an important antioxidant that targets free radicals. It exerts its antiferroptotic effect by directly scavenging lipid peroxidation-inducing free radicals (65).
CoQ10 can be synthesized by the mevalonate (MVA) pathway using acetyl coenzyme A (66). Supplementation with farnesyl pyrophosphate, a product of CoQ10 synthesis, has been shown to inhibit ferroptosis induced by FIN56 (67), highlighting the endogenous inhibitory role of CoQ10 in this process. In addition to its redox enzyme function, FSP1 also has a membrane repair function. It can suppress ferroptosis by triggering the ESCRT-III pathway for membrane repair (55, 68). These findings suggest that FSP1 cannot protect against ferroptosis triggered by the deletion of GPX4. This finding indicates that the NADPH–FSP1–CoQ10 pathway is a separate and parallel system. It functions in conjunction with GPX4 and GSH to inhibit lipid ROS and ferroptosis (60, 61).
Tetrahydrobiopterin (BH4) possesses robust antioxidant properties, enabling it to directly curb lipid peroxidation (69, 70). Additionally, BH4 participates in coenzyme Q10 synthesis, shielding cells from ferroptosis (Figure 1). In addition, BH4 functions as a cofactor for several key enzymes related to dopamine and NO production. Research has demonstrated that both dopamine and NO are associated with ferroptosis (71).
In the synthesis of BH4, GTP cyclohydrolase 1 (GCH1) serves as the key regulatory enzyme, thereby determining the level of BH4 (Figure 1). Moreover, BH4 can be regenerated by dihydrofolate reductase (DHFR). BH4 inhibits ferroptosis by selectively preventing the depletion of phospholipids from two polyunsaturated fatty acid tails (69). A previous study confirmed that dopamine can inhibit ferroptosis induced by erastin (72) and that BH4 is an auxiliary factor for the key enzyme that regulates dopamine synthesis. Additionally, a positive correlation was observed between the expression of GCH1 and the levels of BH4 in cells. Elevated intracellular levels of BH4 inhibit lipid peroxidation and prevent cellular ferroptosis. GCH1, independent of the GPX4 antioxidant pathway, primarily enhances the antioxidant activity of cells by promoting BH4 production and thus protects cells from ferroptosis. The GCH1–BH4–DHFR pathway clearly serves as a key regulatory pathway for ferroptosis. Furthermore, other antioxidant agents, such as vitamin E (73), thioredoxin (74), and aldehyde–ketone reductase family 1, also play certain roles in ferroptosis (75).
Dihydroorotic acid dehydrogenase (DHODH) is a mitochondrial enzyme that contains iron and depends on flavin; it is located in the inner membrane of mitochondria. The primary role of this enzyme is to facilitate the fourth step in pyrimidine synthesis by converting dihydroorotate (DHO) into orotate (Figure 1). At the same time, it reduces ubiquinone to dihydroubiquinone via electron transfer across the mitochondrial inner membrane (76). CoQH2 acts as an antioxidant that traps free radicals, thereby inhibiting lipid peroxidation at the inner mitochondrial membrane and ultimately preventing iron-induced cell death (Figure 1) (77).
An experimental analysis of a group of cancer cell lines with low and high GPX4 expression demonstrated that inhibiting DHODH could induce ferroptosis in tumor cells with low GPX4 expression while enhancing ferroptosis resistance in cancer cells with high GPX4 expression. The DHODH–CoQH2 pathway functions as an antioxidant pathway that works independently of the GPX4 system, providing an antiferroptotic effect by preventing mitochondrial lipid peroxidation (77, 78).
The antioxidant system of cells serves as a critical defense mechanism against ferroptosis. Research has demonstrated that decreased levels of GPX4 and SLC7A11 in the bone tissue of mice with OP correlate with increased bone mineral density and improved bone quality upon elevation of glutathione levels (79). Furthermore, reduced GPX4 levels in chondrocytes heighten their susceptibility to ferroptosis, adversely impacting normal cartilage function (80). These findings suggest that ferroptosis, induced by an imbalance in the antioxidant system, may significantly influence bone health. Consequently, targeting ferroptosis through the activation of the cellular antioxidant system represents a promising avenue for intervention in bone diseases. Furthermore, inducing ferroptosis by inhibiting the cellular antioxidant system presents a potential therapeutic strategy for OS, as various anticancer agents exert their effects by targeting antioxidant system-related proteins such as GPX4 (57).
3 Ferroptosis in bone microenvironment
The bone microenvironment encompasses the local setting where bone tissue resides, comprising key cellular components such as osteoblasts, osteoclasts, and osteocytes within the tissue, along with adipocytes, vascular networks, immune cells, and an abundance of bone marrow and extracellular matrix (81). The bone microenvironment is important for maintaining normal bone physiology and regulating it in pathological settings. Ferroptosis is intricately linked to the bone microenvironment, and the ferroptosis of cells within this microenvironment significantly contributes to various orthopaedic diseases. Ferroptosis of osteoblasts and osteoclasts affects bone growth and repair. Furthermore, a normal bone immune microenvironment is vital for maintaining bone health, and a potential connection exists between immune cell metabolism and the ferroptosis network. Ferroptosis can modulate immune cell function. In particular, the occurrence of this process in immune cells impacts both their numbers and effectiveness, whereas in nonimmune cells, ferroptosis can initiate immune responses via damage-associated molecular patterns.
3.1 Ferroptosis in osteoclasts
Normal iron cycling in cells plays an important regulatory role in maintaining bone health (82). Osteoblasts form bone tissue, while osteoclasts absorb it. The two work in concert in bone formation and remodeling. Increasing evidence suggests that disturbances in iron metabolism can negatively influence the formation and functions of osteoblasts and osteoclasts, thereby disrupting the balance between osteogenesis and bone resorption. Consequently, maintaining iron homeostasis is vital for optimal bone growth and development (83).
During osteoclast differentiation, the demand for iron increases, and the expression of TFR1 is increased through posttranscriptional regulation, thereby increasing iron uptake by the cell. These findings demonstrate the positive regulatory effect of TFR1 on osteoclasts (84). FPN regulates cellular iron efflux (16, 17) and inhibits osteoclast maturation through pathways such as the JNK pathway. During the initial stages of osteoclast maturation, a reduction in the transcription of FPN promotes osteoclast differentiation (85). A study showed that increased plasma iron levels in FPN mutant mice led to decreased osteogenesis, indicating a negative regulatory role of ferroportin in osteoclasts (86, 87).
Osteoclasts, which are specialized cells responsible for bone resorption, arise from the differentiation of myeloid progenitor cells found in the bone marrow (88). The signaling pathway involving receptor activator of nuclear factor-κB ligand (RANKL), its receptor (RANK), and osteoprotegerin (OPG) plays a pivotal role in the regulation of bone resorption. This pathway is essential because it modulates the differentiation and activation of osteoclasts, thereby influencing the overall process of bone resorption (89, 90). Through this intricate signaling mechanism, the balance of bone remodeling is maintained, highlighting the importance of regulating iron levels to ensure optimal bone health.
Osteoclasts require substantial amounts of mitochondrial energy to perform their physiological functions, and iron ions are crucial for mitochondrial synthesis. Iron ion uptake mediated by TFR1 plays a significant regulatory role in osteoclast differentiation (91, 92). It regulates osteoclasts and mitochondria through the activation of the Src–Rac1–WAVE regulatory complex pathway, which in turn modulates mitochondrial respiration (93).
When iron levels are excessively high, the RANKL/OPG ratio increases, leading to the promotion of osteoclast maturation and bone resorption through TFR1-mediated iron uptake (Figure 2). Conversely, iron-chelated lactoferrin inhibits osteoclast-mediated bone resorption by reducing the RANKL/OPG ratio, thereby increasing the bone mineral density (92, 94). TFR1-mediated iron uptake mainly regulates the growth and development of mature osteoclasts, but its impact on osteoclast precursors is relatively limited (93). These findings suggest that iron overload might lead to bone loss by promoting the activity and function of osteoclasts (95, 96).
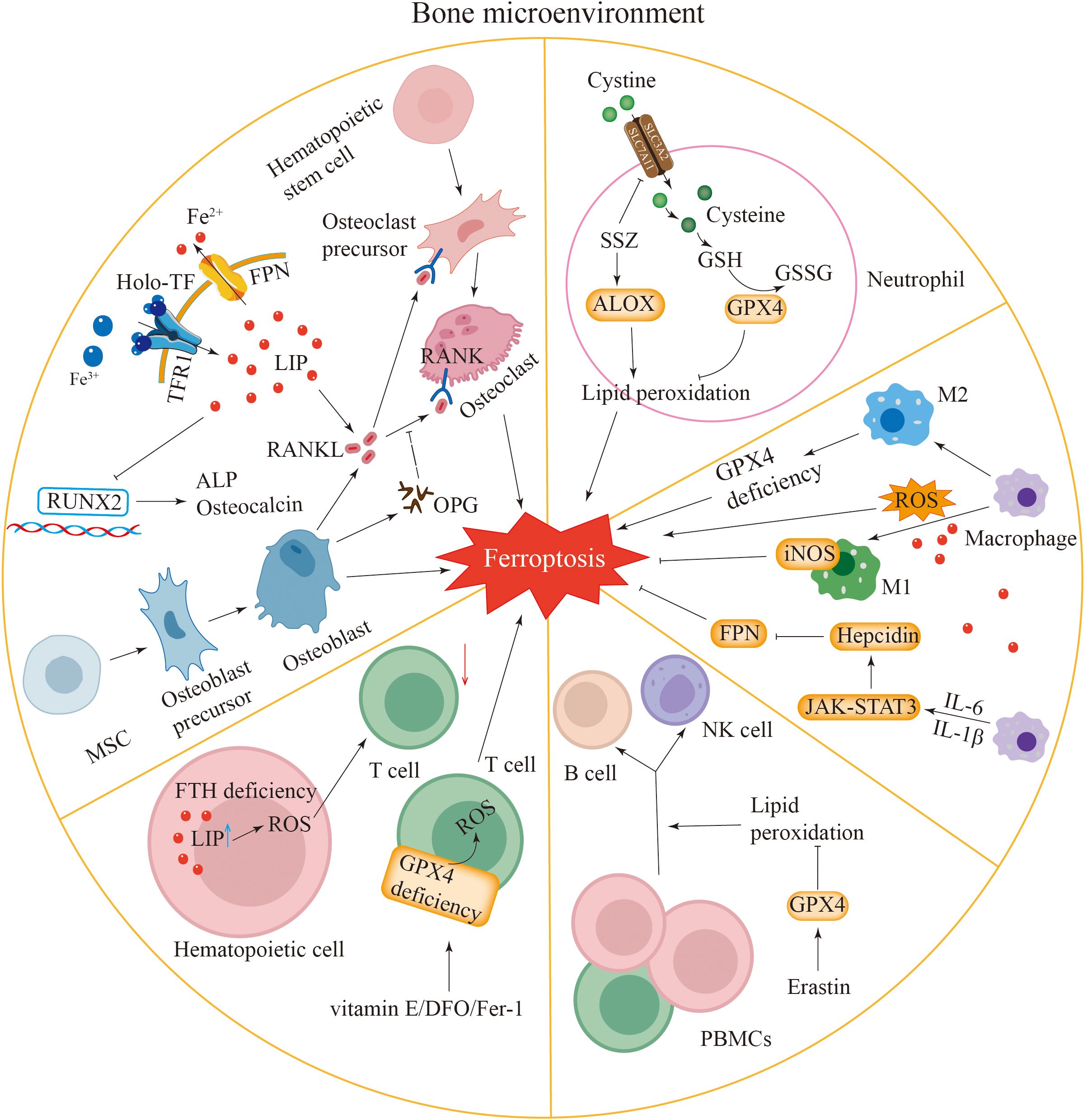
Figure 2. Ferroptosis in the bone microenvironment. Osteoblasts and osteoclasts: iron overload in osteoblasts and osteoclasts induces ferroptosis in these cells. Iron overload inhibits the expression of Runx2, ALP and osteocalcin, thereby affecting osteoblast function. Osteoblasts release RANKL to promote the differentiation of osteoclasts. Enhanced iron uptake mediated by TFR1 leads to an increased RANKL/OPG ratio, promoting osteoclastogenesis. Neutrophils: the expression of GPX4 in neutrophils is inhibited, which can induce ferroptosis. SSZ can promote ferroptosis by inhibiting SLC7A11 and activating ALOX. Macrophages: macrophages have iron overload. Macrophages can promote ferroptosis by generating ROS, and ROS can also promote the transformation of macrophages into M1-type macrophages. Macrophages can activate the JAK-STAT3 pathway by secreting IL-6 and promote the transcription of hepcidin by secreting IL-1β, leading to a decrease in FPN expression and causing ferroptosis. The expression of iNOS in M1-type macrophages inhibits ferroptosis. The loss of GPX4 activity in M2-type macrophages leads to ferroptosis. Lymphocytes: the deletion of H-ferritin gene in hematopoietic cells increases reactive oxygen species and active iron, resulting in a reduction in T cell numbers. Vitamin E, DFO, and Fer-1 can rescue the ferroptosis of GPX4-deficient T cells. Erastin can promote the differentiation of PBMCs into B cells and NKs.
3.2 Ferroptosis in osteoblasts
Osteoblasts, which are essential for bone development, arise from mesenchymal progenitor cells that are vital for managing the processes of bone creation and remodeling (97). Iron primarily exerts its influence by inhibiting the expression of alkaline phosphatase (ALP) and the mineralization of osteoblasts. Notably, trivalent iron has a more pronounced inhibitory effect than does divalent iron (98). Alkaline phosphatase is one of the key factors regulating the mineralization of osteoblasts. The decrease in ALP activity could strengthen the inhibitory effects of Fe3+ and Fe2+ on osteoblasts.
Iron hinders the function of osteoblasts by reducing the expression levels of markers associated with osteogenic differentiation in both C2C12 myoblasts and bone marrow-derived mesenchymal stem cells (BM-MSCs) (99, 100). These findings indicate that iron has a greater inhibitory effect on osteoblast precursors than on mature osteoblasts during osteogenic induction, which is similar to the regulation of osteoclasts by TFR1. Excessive iron accumulation may suppress the function and osteogenic differentiation of osteoblasts. Research has indicated that when iron ions are present at specific concentrations, they can inhibit the activity of Runt-related transcription factor 2 (Runx2), a critical regulator of osteoblast differentiation (Figure 2). This inhibition results in reduced expression of key osteogenic markers, such as alkaline phosphatase (ALP) and osteocalcin, which are essential for the proper differentiation of bone marrow-derived mesenchymal stem cells (BM-MSCs). Moreover, the excessive accumulation of intracellular iron ions further contributes to the downregulation of the osteoblast phenotype, which is essential for maintaining healthy bone formation (101). Thus, iron overload is inferred to cause bone damage by promoting osteoclast activity while simultaneously suppressing the formation of osteoblasts.
3.3 Ferroptosis in neutrophils
Neutrophils are intricate cells with many specialized functions that are vital for managing different pathophysiological processes. Serving as effector cells in the innate immune response, these cells represent the most abundant type of immune cells among human white blood cells. The primary bactericidal role of neutrophils is accomplished via phagocytosis and the creation of neutrophil extracellular traps (NETs), which are composed of degraded chromatin and granular proteins from within the cell, triggered by various stimuli. Even after the death of neutrophils, NETs can still perform their bactericidal function.
Ferroptosis may be involved in the formation of NETs and the regulation of neutrophil recruitment (102). The ferroptosis inhibitor sulfasalazine (SSZ) enhances ferroptosis by blocking SLC7A11 and stimulating the lipoxygenase ALOX (Figure 2) (103). Research indicates that ether-bonded glycerolipids increase cellular sensitivity to ferroptosis, whereas ether lipids are crucial for the formation of the extracellular meshwork induced by sulfasalazine (104). This result suggests a potential association between these two phenomena. Furthermore, ferroptosis is related not only to NETs but also to autoimmune diseases associated with neutrophils. Nevertheless, the impact of neutrophil ferroptosis on the progression of these diseases has yet to be completely verified.
3.4 Ferroptosis in macrophages
Macrophages stem from monocytes and can be divided into two types: M1 and M2. They contribute to both innate and adaptive immunity in vertebrates. Phagocytes primarily function in phagocytosis, which involves the engulfment and digestion of cell debris and pathogens, either as fixed or free-floating cells.
Macrophages are closely associated with ferroptosis. Ferroptotic and M1-type macrophages both exhibit iron accumulation. Excess iron may promote the polarization of macrophages towards the M1 phenotype (Figure 2). In terms of cytokines, macrophages cause inflammation by releasing proinflammatory factors. Similarly, an increase in the levels of proinflammatory factors also occurs within ferroptotic cells. By releasing cytokines, macrophages can regulate the activity of Lox, thereby inducing ferroptosis. Inducible nitric oxide synthase (iNOS) plays a crucial role in M1 macrophages by negatively regulating ferroptosis (Figure 2) (105). An impairment of the GPX4 system in M2-type macrophages can lead to cellular ferroptosis (106). Macrophages are capable of generating ROS, which in turn can drive their differentiation into M1-type macrophages (107, 108). IL-6 secreted by macrophages promotes hepcidin transcription via the activation of the JAK-STAT3 signaling pathway (Figure 2). Furthermore, macrophages secrete IL-1β to upregulate hepcidin expression. These combined actions result in decreased levels of FPN expression, causing an accumulation of intracellular iron and eventually resulting in iron-induced cell death (109, 110).
3.5 Ferroptosis in lymphocytes
Lymphocytes represent the smallest category of white blood cells. They are essential for the body’s immune response. These cells can be divided into three primary types according to their functions: T cells, B cells, and natural killer (NK) cells.
Maintaining proper iron balance within cells is essential for T-cell survival and the performance of their typical physiological roles (111). Elevated levels of ROS and iron in haematopoietic cells lacking the H-ferritin gene lead to a reduction in T-cell populations (Figure 2) (112). T cells deficient in GPX4 undergo ferroptosis, which results in compromised immune function (113). Research has shown that antioxidant interventions, including vitamin E, deferoxamine (DFO), or Fer-1, can alleviate ferroptosis in T cells lacking GPX4 (Figure 2) (114). Additionally, antibodies produced by B cells can influence iron-induced cell death, while ROS also impact the B-cell quantity and normal function (115, 116). Lipid peroxidation induced by the ferroptosis inducer erastin increases the proliferation of human peripheral blood mononuclear cells (PBMCs) and their differentiation into B cells and NKs (116).
3.6 Ferroptosis in other cells
BMSCs that differentiate into osteoblasts or chondrocytes are crucial for the development, reconstruction, and tissue regeneration of bone and cartilage. Iron homeostasis is of vital importance for their proliferation and differentiation.
Studies have confirmed that iron homeostasis imbalance severely damages the bone marrow microenvironment of mice, leading to bone destruction (117). Excessive iron can also trigger oxidative stress, leading to trabecular bone damage and bone loss. Antioxidants can inhibit the formation of osteoclasts and thereby alleviate this process (30). Furthermore, the accumulation of iron in synoviocytes disrupts the balance of various inflammatory factors in the joint, impairing the normal physiological function of cartilage. It increases the capacity of monocytes and synovial fibroblasts within the joint to absorb iron. This process accelerates the connective tissue degradation mediated by histone proteases and consequently induces joint disease (31, 118).
4 Ferroptosis in specific diseases
Ferroptosis is a type of cell death characterized by the accumulation of lipid reactive oxygen species inside cells. Presently, Ferroptosis is linked to a variety of diseases, including stroke, neurodegenerative disorders, cancers, and ischaemia–reperfusion injury (58). Recent studies have revealed that ferroptosis is also a key factor involved in the pathological mechanisms of bone and joint diseases. Ferroptosis exacerbates osteoporosis, triggers osteoarthritis, aggravates lumbar disc herniation, and is a promising target for osteosarcoma therapy.
4.1 Ferroptosis in osteoporosis
Osteoblasts form bone, and osteoclasts absorb bone. The two work in concert to maintain normal bone metabolism. Osteoporosis is a systemic disease characterized by a decreased bone density, destruction of the bone microstructure, and increased bone fragility (119). Osteoporosis can be divided into primary osteoporosis (POP) and secondary osteoporosis (120). Osteoporosis causes an increase in bone fragility, often leading to fractures in elderly and postmenopausal women (121, 122). Statistical data indicate that approximately thirty-three percent of older females and twenty percent of older males are affected (123).
Disrupted calcium metabolism may lead to osteoporosis, and an increasing body of research indicates a potential link between iron-induced cell death and osteoporosis (124). Intracellular iron overload not only inhibits osteoblast function but also enhances osteoclast activity, thereby contributing to osteoporosis. A significant amount of research has indicated that individuals with anaemia frequently experience osteoporosis as well (125, 126), which may be related to iron overload-mediated ferroptosis caused by long-term blood transfusions in anaemia patients (127), and their fracture risk is associated with the frequency of blood transfusions (128). Furthermore, research has revealed a link between haemochromatosis and osteoporosis. In individuals with haemochromatosis, osteoporosis is linked to iron overload (129), and ferroptosis might be the primary factor driving osteoporosis in these populations (130).
Elevated concentrations of reactive iron ions in osteoblasts and osteoclasts result in cellular iron toxicity. With the ongoing accumulation of intracellular iron ions, a notable increase in the levels of iron cycling phase proteins, including DMT1 and TFR1, occur within both osteoblasts and osteoclasts (131, 132). Consequently, iron overload-mediated ferroptosis contributes to the development of osteoporosis through its detrimental effects on osteoblasts, osteoclasts, and BM-MSCs, ultimately disrupting bone homeostasis.
Osteoporosis is a prevalent complication of type II diabetes mellitus, with studies confirming a strong link between iron-mediated cell death and glucose metabolism. Increased serum ferritin levels have been observed in diabetic mice with osteoporosis, accompanied by significantly reduced levels of SLC7A11 and GPX4 in bone tissue (133). High glucose levels are capable of decreasing osteocalcin expression, reducing alkaline phosphatase activity, and suppressing bone mineralization (134, 135). Research has shown that the expression of HO-1 in osteocytes in a high-glucose microenvironment increases. HO-1 facilitates the breakdown of heme, resulting in the release of significant quantities of reactive iron ions, which then promote the Fenton reaction, ultimately producing lipid peroxides and causing ferroptosis in osteoblasts (Figure 3) (124, 136). This process suggests that HO-1 might serve as a potential therapeutic target for treating diabetic osteoporosis.
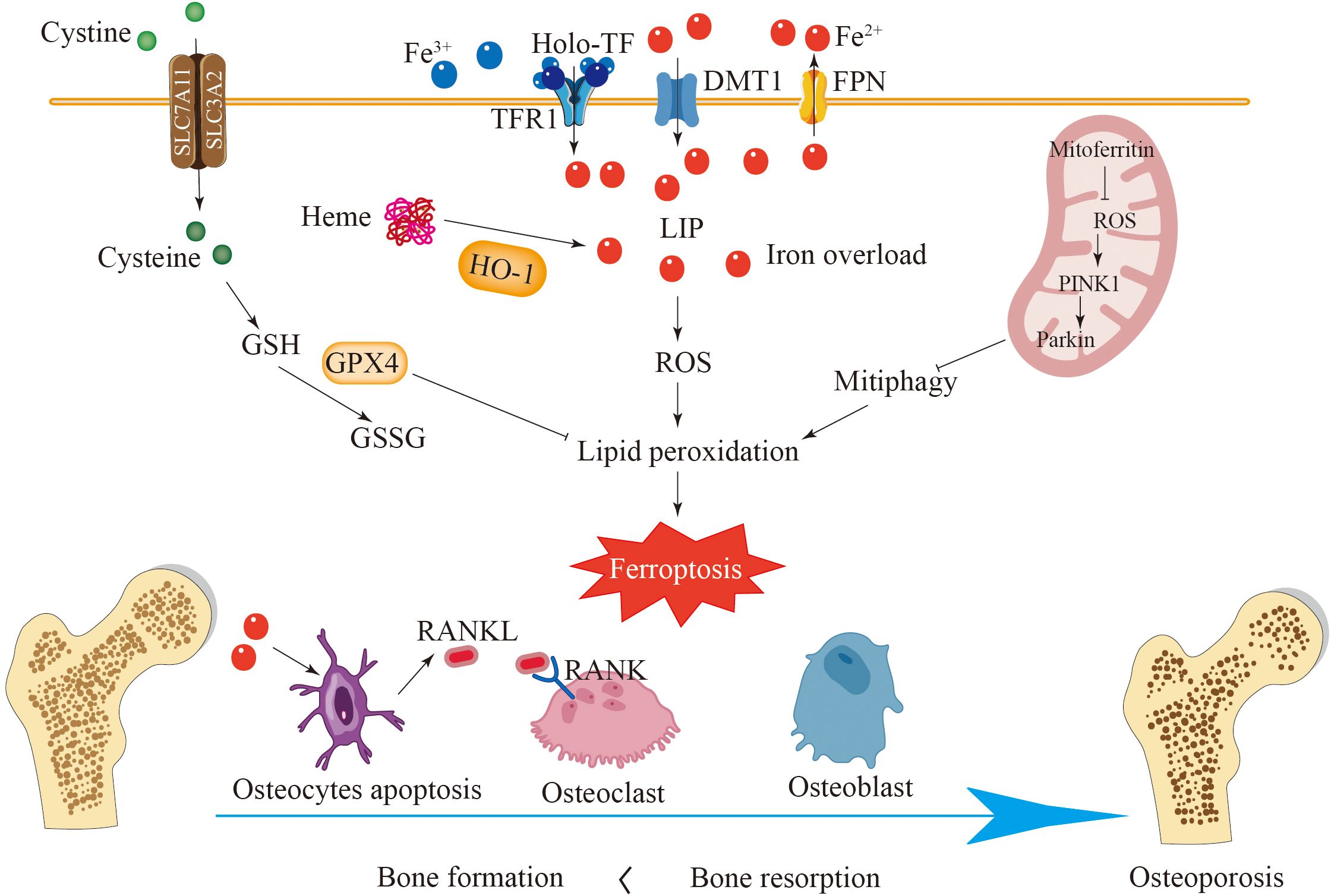
Figure 3. Ferroptosis plays a key role in the development of osteoporosis. In healthy bones, bone formation mediated by osteoblasts and bone resorption mediated by osteoclasts are in balance. Iron overload enhances the expression of DMT1 and TFR1 in osteoblasts and reduces the expression of SLC7A11 and GPX4, leading to ferroptosis of osteoblasts and disruption of bone formation. Iron overload can cause ferroptosis in osteoclasts and osteocytes and promote the secretion of RANKL, promoting the generation of osteoclasts. HO-1 catalyzes the degradation of heme to release a large amount of active iron, accelerating cell ferroptosis. Ferroptosis disrupts the balance between bone formation and bone resorption, leading to osteoporosis.
Ferroptosis is distinct from apoptosis; however, these two modes of cell death are not entirely independent in the pathological progression of OP. The cytokines involved in both processes interact with one another, contributing to the development of OP. Research has demonstrated that among the pathogenic factors associated with osteoporosis, advanced glycosylation end products can induce both ferroptosis and apoptosis in osteoblasts, ultimately leading to OP. Furthermore, DFO has been shown to inhibit this process (137). These findings suggest that targeting the shared key factors of apoptosis and ferroptosis may provide a more effective approach for the treatment of OP.
Mitoferritin located in mitochondria can reduce free iron in mitochondria and thereby decrease the sensitivity of cells to ferroptosis. The overexpression of mitochondrial ferritin has been shown to inhibit oxidative stress, which in turn mitigates ferroptosis (138). In contrast, decreased expression of mitochondrial ferritin can initiate mitochondrial autophagy through the ROS/PINK1/Parkin pathway, leading to the accumulation of ferritin and the enhancement of ferroptosis in osteoblasts (Figure 3) (135). These results suggest that mitochondrial ferritin could represent a promising future avenue for treating diabetic osteoporosis. Based on these findings, iron-induced cell death in osteoblasts is significantly involved in the progression of osteoporosis. Additionally, the pathway associated with iron-mediated death in osteoblasts, which utilizes various mechanisms, may represent a potential approach for the clinical management of osteoporosis.
Research on ferroptosis in OP is on the rise; however, the epigenetic regulatory mechanisms involved remain under investigation. It has been proposed that METTL14-mediated m6A modification plays a crucial role in the expression of GPX4. This indicates that the regulation of ferroptosis in osteoporosis via m6A modification may represent a promising avenue for future treatment strategies for OP (139).
4.2 Ferroptosis in osteoarthritis
Osteoarthritis is a degenerative condition of the joints characterized by the breakdown of cartilage, inflammation of the synovium, and changes in the subchondral bone structure (140, 141). Factors such as age, oestrogen levels, and mechanical stress play roles in increasing the likelihood of developing this disease (142–144). Osteoarthritis is characterized mainly by joint pain, deformity and functional impairment (145, 146). Additionally, it substantially increases the risks of cardiovascular incidents and overall mortality (147, 148). Currently, the number of OA patients worldwide exceeds 300 million (149).
Recent research has demonstrated a significant link between ferroptosis and OA, suggesting that this form of cell death might exacerbate the pathological mechanisms associated with OA (150, 151). Furthermore, the inhibition of ferroptosis represents a novel therapeutic target for OA management (152, 153). Iron overload has been observed in OA patients. Iron overload affects the progression of OA. From an imaging perspective, increased ferritin levels can exacerbate the severity of osteoarthritis (154, 155). Additionally, a positive correlation has been observed between serum iron levels and transferrin saturation and the progression of OA (156). A study conducted on guinea pigs susceptible to osteoporosis, which were placed on a diet low in iron, revealed that lower systemic iron levels delayed the development of cartilage damage. In contrast, when exogenous iron was administered, these animals developed knee osteoarthritis (157). These results highlight the significant impact of iron overload, stemming from iron dysregulation, on the development of OA.
Chondrocytes are involved in maintaining the integrity of the extracellular matrix and controlling the balance of articular cartilage homeostasis, thereby contributing to the deceleration of OA development (158, 159). In the pathological process of OA, cartilage degeneration is a key event. Research has shown that ferroptosis of chondrocytes exacerbates the progression of OA pathology. Both the induction of an inflammatory environment and a redox system imbalance promote ferroptosis in chondrocytes (160). Interleukin-1β (IL-1β), a factor that mimics an inflammatory condition, is capable of triggering alterations in the ROS concentrations within chondrocytes. It also enhances the accumulation of lipid peroxides and alters proteins associated with ferroptosis, such as GPX4, which ultimately result in the ferroptosis of chondrocytes (80, 160). Ferroptosis in chondrocytes increases the levels of MMP13 and IL-1β, simultaneously reducing the expression of type II collagen. This alteration in the balance of the extracellular matrix plays a role in the progression of OA (Figure 4).
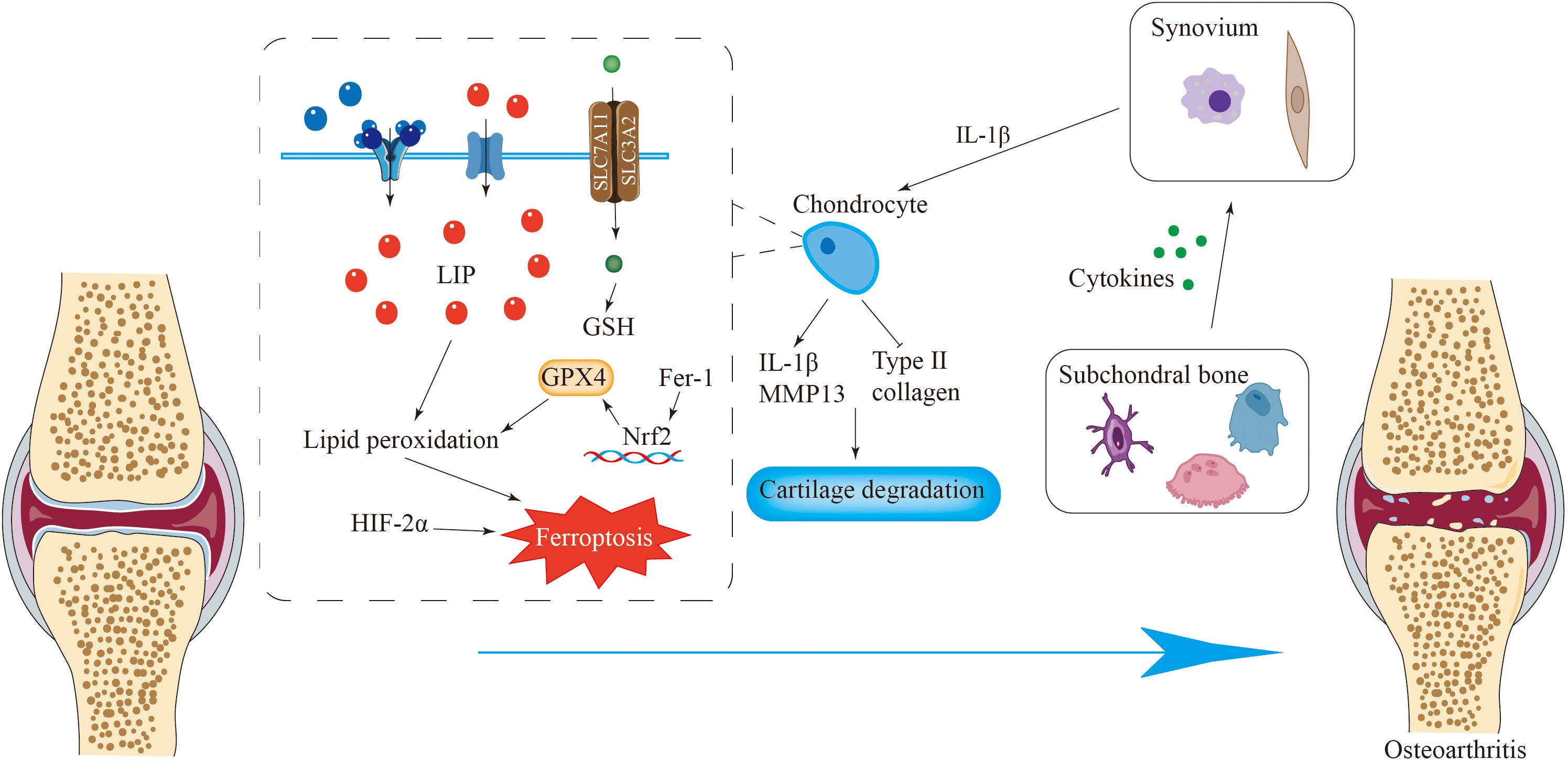
Figure 4. Ferroptosis is closely related to osteoarthritis. Iron overload in chondrocytes causes ferroptosis of chondrocytes, resulting in the destruction of the integrity of the extracellular matrix and the loss of chondrocytes. Fer-1 can activate the Nrf2 antioxidant system to counteract ferroptosis. Various cytokines released by cells such as osteoclasts in subchondral bone promote the release of pro-inflammatory cytokines by synovial cells, inducing ferroptosis of chondrocytes and ultimately leading to osteoarthritis.
Fer-1 activates the Nrf2 antioxidant system, leading to elevated GPX4 levels and increased type II collagen production in chondrocytes. Additionally, Fer-1 mitigates the alterations in ferroptosis-related protein expression induced by IL-1β (160). In an inflammatory setting induced by IL-1β, increased levels of hypoxia-inducible factor-2α (HIF-2α) coupled with diminished GPX4 expression in chondrocytes increase their susceptibility to ferroptosis (Figure 4) (80, 161). Antioxidant and anti-inflammatory substances, such as D-mannose, icaritin (162) and Fer-1, inhibit ferroptosis in chondrocytes by regulating the GPX4 pathway, thereby slowing cartilage degeneration and decelerating the pathological process of OA (80, 163).
Furthermore, subchondral bone is crucial for maintaining normal joint function. Ferroptosis may promote osteoclast bone resorption by inhibiting osteoblast function, thereby affecting the homeostasis and structural integrity of subchondral bone and leading to the occurrence of osteoarthritis (164, 165). Synovial inflammation promotes osteoarthritis. The synovium can secrete proinflammatory cytokines, leading to cartilage damage. Research has shown that excessive iron accumulation occurs in the synovium in haemophilic arthropathy, resulting in synovitis (150, 166). During the development of osteoarthritis, the synovium undergoes interstitial vascularization, fibrosis and hyperplasia (167), and macrophages and fibroblasts in the synovium can release proinflammatory cytokines, etc., which cause synovitis and cartilage degeneration.
RNA LINC00618 promotes apoptosis by increasing the levels of the pro-apoptotic protein, cleaved caspase-3, and also induces ferroptosis by inhibiting the cellular antioxidant system, suggesting a potential interaction between apoptosis and ferroptosis (168). Furthermore, cellular pyroptosis contributes to the progression of bone diseases. In the context of OA, the inflammatory response triggered by cellular pyroptosis exacerbates cartilage degeneration and synovial inflammation, thereby accelerating OA progression (169). These findings suggest that there may be mutual regulation between ferroptosis and other forms of cell death, warranting further investigation into the molecular interaction mechanisms to explore their potential clinical significance.
Epigenetic modifications play a crucial role in regulating ferroptosis in chondrocytes. The histone methyltransferase NSD1 has been identified as having potential therapeutic value by upregulating the H3K36me2 modification, thereby activating the expression of the transcription factor SOX9, which subsequently downregulates the key enzyme for ferroptosis, ACSL4, thus attenuating chondrocyte ferroptosis (170). Furthermore, the lactate metabolizing enzyme LDHB promotes H3K18 histone lactoylation, enhances ACSL4 expression, and induces chondrocyte ferroptosis, representing another epigenetic regulatory target for OA (171).
4.3 Ferroptosis in lumbar disc herniation
One of the most common reasons for chronic low back pain is lumbar disc herniation. After degenerative changes occur in the disc, the annulus fibrosus can break, leading to the protrusion or bulging of the nucleus pulposus, either independently or in conjunction with the annulus fibrosus and the cartilaginous plate. This condition can irritate or compress the spinal nerve roots, resulting in low back pain (172, 173).
Lumbar intervertebral disc protrusion has a high recurrence rate and is more prevalent in middle-aged and elderly individuals, with an increasing incidence among younger individuals. The intervertebral disc is a fibrocartilaginous structure situated between two adjacent lumbar vertebrae that serves to enhance spinal mobility, absorb shock, and safeguard the spinal cord. A tear in the annulus fibrosus, alongside the degeneration of the nucleus pulposus and the breakdown of the cartilage plate, play roles in the degeneration of the intervertebral disc, which can ultimately result in lumbar disc herniation.
Research has recognized ferroptosis as a possible pathogenic mechanism associated with lumbar disc herniation, although detailed mechanistic investigations remain scarce. Research indicates that rats suffering from lumbar degenerative disc disease exhibit reduced levels of the antioxidants GPX4 and FTH within the disc tissue. Conversely, the levels of cyclooxygenase (PTGS2) and ACSL4, both of which are integral to lipid metabolism, are elevated (174). These findings indicate the potential involvement of ferroptosis in the pathological mechanisms related to LDH. Furthermore, iron overload has been shown to exacerbate disc degeneration, ultimately contributing to herniation. In the course of the experiments, tert-butyl hydroperoxide (TBHP) was used to induce oxidative stress in annulus fibrosus cells (AFCs) and nucleus pulposus cells (NPCs). As the concentration of TBHP increased, the expression of FTH and GPX4 decreased, and the lipid ROS levels in the AFCs and NPCs increased. Characteristic features of ferroptosis, such as the crumpling of mitochondria and an increase in the mitochondrial membrane density, were observed at the mitochondrial level. Importantly, these alterations were attenuated by iron-induced death inhibitors (Fer-1 and DFO), further reinforcing the association between ferroptosis and the pathological mechanisms of LDH (Figure 5) (174, 175).
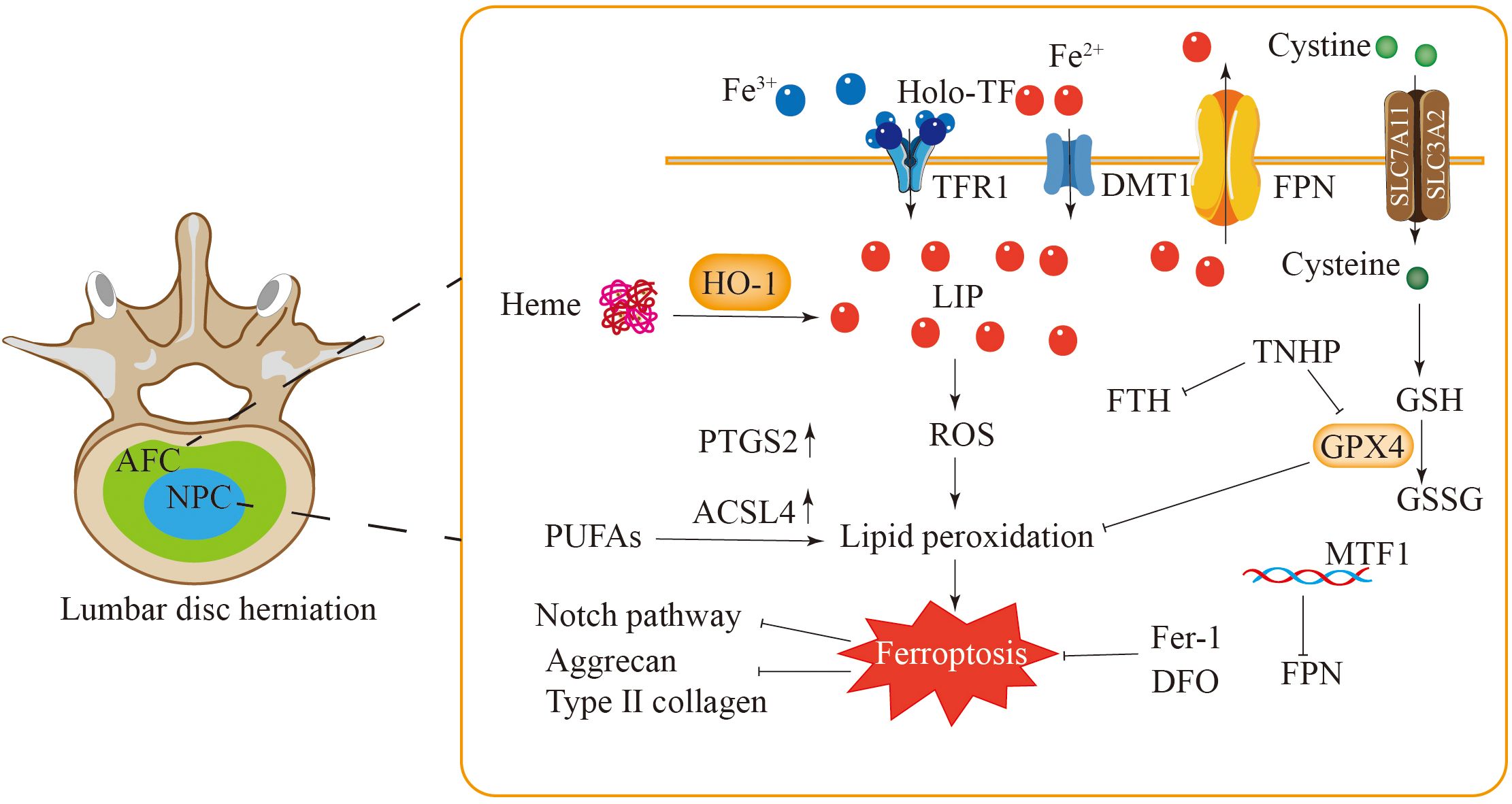
Figure 5. Ferroptosis is closely related to lumbar disc herniation. The levels of GPX4 and FTH in AFC and NPC of patients with lumbar disc herniation are decreased, while the expressions of PTGS2 and ACSL4 are increased, and these changes are inhibited by Fer-1 and DFO. In NPC, the elevated levels of HO-1 and iron intensify lipid peroxidation and induce ferroptosis in NPC. The ferroptosis of NPC may be related to the Notch signaling pathway. Additionally, MTF1 inhibits cell ferroptosis by regulating hepcidin. Ferroptosis in AFC and NPC leads to a decrease in type II collagen and aggrecan levels and extracellular matrix degradation.
Neovascularization is another characteristic of intervertebral disc protrusion. An experiment demonstrated the presence of neoangiogenesis in the herniated nucleus pulposus, which may contribute to tissue damage (176). Compared with patients with normal nucleus pulposus, patients with lumbar disc herniation presented higher haemoglobin concentrations in the disc nucleus pulposus, a higher quantity of dark iron particles following staining, and elevated expression levels of HO-1 (177). Increased concentrations of haemoglobin and haem may trigger ferroptosis, potentially through a mechanism linked to the Notch signaling pathway (Figure 5) (177).
Atypical patterns of NPCs significantly contribute to the pathological progression of lumbar disc degeneration. Although these cells can produce components of the extracellular matrix, reductions in the levels of type II collagen and aggregated proteoglycans are often observed following disc injury (178). Furthermore, the iron overload and lipid peroxidation induced by TBHP in human NPCs can be significantly reversed by Fer-1 or the iron chelator DFO (176).
FPN is an important regulatory factor for cellular iron homeostasis. The overexpression of FPN can alleviate iron ion overload and ferroptosis in intervertebral disc cells. MTF1 eliminates intracellular iron overload by regulating FPN, thereby protecting intervertebral disc cells from ferroptosis (Figure 5) (179). Chondrocytes are essential for maintaining the proper function of intervertebral discs. Research has shown a notable decrease in the chondrocyte population in degenerated intervertebral discs (180), with iron overload being a key contributing factor. In the chondrocytes of the iron overload model, the levels of GPX4 and SLC7A11 within the antioxidant system were lower, whereas lipid peroxidation levels, indicated by 4-HNE, were higher, and their mitochondria exhibited characteristics similar to ferroptosis (175). These findings suggest that the reduction in the number of chondrocytes within degenerated intervertebral discs could be linked to ferroptosis.
Furthermore, a notable rise in the expression levels of genes associated with autophagy, as well as a higher quantity of autophagic vacuoles and lysosomes, has been detected in degenerated intervertebral discs and the annulus fibrosus (181, 182). This finding suggests a potential link between the pathological process of LDH and autophagy. Both autophagy and ferroptosis contribute to the pathological process of disc degeneration. Notably, ferritin autophagy enhances autophagy and ferroptosis in NPC and AFC, whereas Fer-1 inhibits this process (174). Therefore, further investigation into the interaction network between ferroptosis and autophagy in bone diseases may deepen our understanding of their pathogenesis and provide novel insights for the treatment of such conditions.
In LDH, the inhibition of the DNA methyltransferase DNMT3B effectively prevents the onset of ferroptosis in NPCs and enhances cellular activity and mitochondrial function, suggesting a critical role in the regulation of iron homeostasis (183). Furthermore, TBHP can promote the accumulation of iron ions and induce ferroptosis by inhibiting the expression and nuclear export of the FPN. This process is regulated by the JNK/MTF1/FPN pathway, which constitutes a potential therapeutic target (179).
4.4 Ferroptosis in osteosarcoma
Osteosarcoma is a cancerous tumor of the bone characterized by high mortality rates, significant disability, and frequent metastasis and recurrence, primarily affecting the bones and lungs (184). The incidence rate is the highest among children and adolescents, with a higher rate in males than in females. OS often occurs near the growth plates of long bones (185). Clinically, the main treatment approach combines surgery with radiotherapy and chemotherapy. However, the effectiveness of traditional treatment methods for osteosarcoma is still limited, as the 5-year survival rate falls within the range of 60% to 70%. Moreover, significant progress in conventional therapies has been scarce over the past few years.
Ferroptosis has been recognized as a crucial element in the progression of tumors and the immune response, among other biological processes (186). Bavachin exerts its anticancer effects by triggering ferroptosis in osteosarcoma cells through multiple mechanisms. Among them, a reduction in matrix metalloproteinases (MMPs) leads to ferroptosis-like features in mitochondria (187). Furthermore, bavachin upregulated the expression of the transferrin receptor and DMT1 while downregulating the expression of FTL and FTH. These changes resulted in elevated intracellular Fe2+ levels and the downregulation of p-STAT3, SLC7A11, and GPX4 levels (57). Thus, bavachin clearly induces ferroptosis in osteosarcoma cells by modulating genes and proteins associated with cell death. Notably, the upregulation of P53 expression leads to lower expression levels of SLC7A11 and GPX4, promoting the accumulation of ROS and the lipid peroxidation product malondialdehyde (MDA), which in turn induces ferroptosis in osteosarcoma. This process is inhibited by ferroptosis inhibitors (Figure 6) (187).
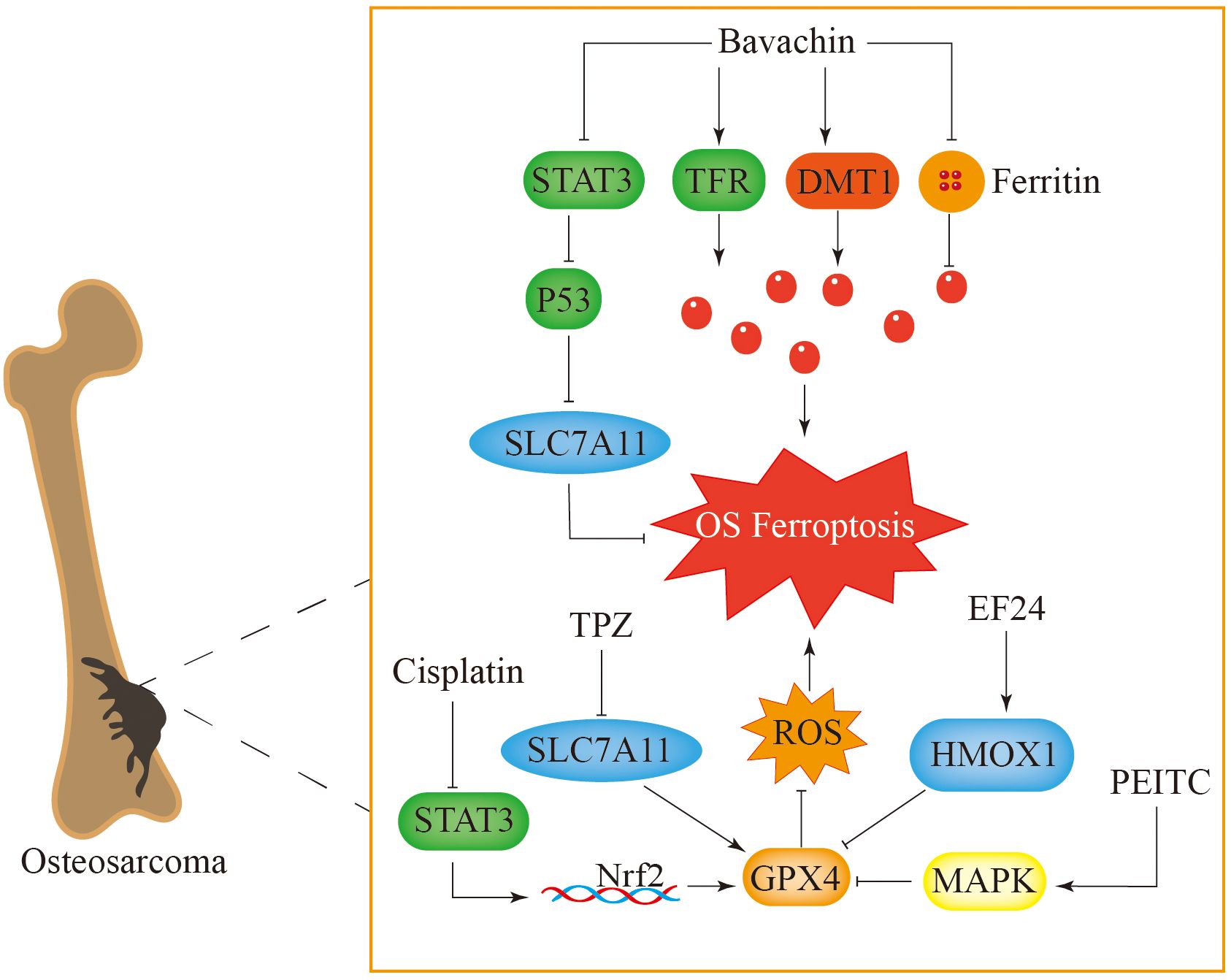
Figure 6. Ferroptosis plays a key role in the treatment of osteosarcoma. Bavachin induces ferroptosis in osteosarcoma cells by increasing intracellular iron levels and inhibiting the STAT3-P53-SLC7A11 pathway. TPZ induces ferroptosis in osteosarcoma cells by inhibiting the expression of SLC7A11 in the classical ferroptosis signaling pathway. Cisplatin causes cell ferroptosis through the STAT3-Nrf2-GPX4 signaling pathway. PEITC and EF24 respectively activate the MAPK and HMOX1 signaling pathways, inhibit GPX4, cause accumulation of reactive oxygen species in cells, and induce ferroptosis.
Tirapazamine (TPZ) is a novel anticancer drug that exerts its antitumour effect under hypoxic conditions (188). Under hypoxic conditions, SLC7A11 expression is reduced, whereas increases in MDA and Fe2+ levels increase ROS accumulation in osteosarcoma cells. This process triggers ferroptosis and suppresses the proliferation and metastasis of osteosarcoma (189). Cisplatin is a commonly used drug for treating osteosarcoma, but osteosarcoma cells can develop resistance to it, which reduces its therapeutic effect (190). Measurements of ferroptosis-associated proteins in osteosarcoma cells indicate that compared with normal cells, drug-resistant cells exhibit elevated GPX4 levels. Moreover, the use of ferroptosis inhibitors increases the sensitivity of these osteosarcoma cells to cisplatin, while diminishing the levels of ferroptosis-related proteins further enhances this sensitivity (191). The signaling pathway involving STAT3/Nrf2/GPX4 is integral to the resistance observed in osteosarcoma cells and may serve as a promising therapeutic target to improve the effectiveness of cisplatin against bone tumors (Figure 6) (191).
Phenethyl isothiocyanate (PEITC) can cause cell cycle arrest and trigger apoptosis in tumor cells. It promotes ferroptosis in bone tumor cells by inducing oxidative stress, depleting GPX4, increasing ROS levels, and activating the MAPK signaling pathway (192, 193). The synthetic derivative of curcumin biphenyl-difluoroketone (EF24) has been reported to trigger cell apoptosis, restrain cell growth, and diminish metastatic capabilities (194, 195). Furthermore, EF24 induces ferroptosis in osteosarcoma cells by increasing the levels of lipid peroxides and intracellular iron ions. It also achieves this effect by increasing the expression of haem oxygenase 1 (HMOX1) and reducing the expression of GPX4 (196). This process can be counteracted with the application of ferroptosis inhibitors.
In recent advances in cancer therapy, it has been found that there may be an interaction between the molecular mechanisms involved in ferroptosis and cellular pyroptosis (197). Anti-tumor immune cells, such as T cells, can induce tumor cells to undergo pyroptosis while simultaneously increasing ROS to trigger ferroptosis, thereby exerting anti-tumor effects (198, 199). However, the molecular interactions between ferroptosis and pyroptosis remain unclear and warrant further investigation.
Induction of cellular ferroptosis significantly enhances the sensitivity of OS cells to both radiotherapy and chemotherapy. Although the specific epigenetic regulatory mechanisms remain incompletely understood, evidence suggests that epigenetic factors, including miRNAs and RNA methylation, may play a role in the therapeutic process by regulating key genes associated with ferroptosis, such as GPX4 and SLC7A11 (200).
5 Therapeutic perspectives and challenges
Ferroptosis is closely related to orthopaedic diseases, and inhibiting ferroptosis in patients with related orthopaedic diseases may constitute a new treatment method. Ferroptosis inducers can induce ferroptosis through different targets. Among these compounds, erastin, sulfasalazine (SAS), RSL3, sorafenib, and FIN56 induce ferroptosis through the inhibition of the GSH/GPX4 pathway (201, 202). t-BuOOH triggers ferroptosis by modulating cellular lipid metabolism (203), whereas erastin and RSL5 induce ferroptosis by altering cellular iron metabolism (204). Temozolomide (TMZ) promotes ferroptosis by upregulating DMT1 (205). FIN56 can also induce ferroptosis in cells by depleting CoQ (52).
In addition, many ferroptosis inhibitors suppress ferroptosis in cells through a series of pathways. The consumption of excessive iron ions in cells is a direct method to inhibit ferroptosis. Compounds that bind iron, such as ciclopirox olamine (CPX), DFO, and deferiprone (DFP), inhibit ferroptosis by chelating iron ions and subsequently reducing ROS production (3, 206, 207). The accumulation of excess lipid ROS in cells is a central feature of ferroptosis. Fer-1, liproxstatin-1, α-tocopherol, VKH2 and other substances inhibit ferroptosis by suppressing lipid peroxidation (208, 209). The GSH/GPX4 pathway serves as a crucial antioxidant mechanism within cells. β-Mercaptoethanol (β-ME) and selenium (Se) are capable of increasing the activity of the GSH/GPX4 pathway via distinct mechanisms, which in turn suppress ferroptosis (50, 210). In addition, prominin 2, miR-522, iNOS, and other molecules inhibit ferroptosis through different pathways (Table 1) (27, 263).
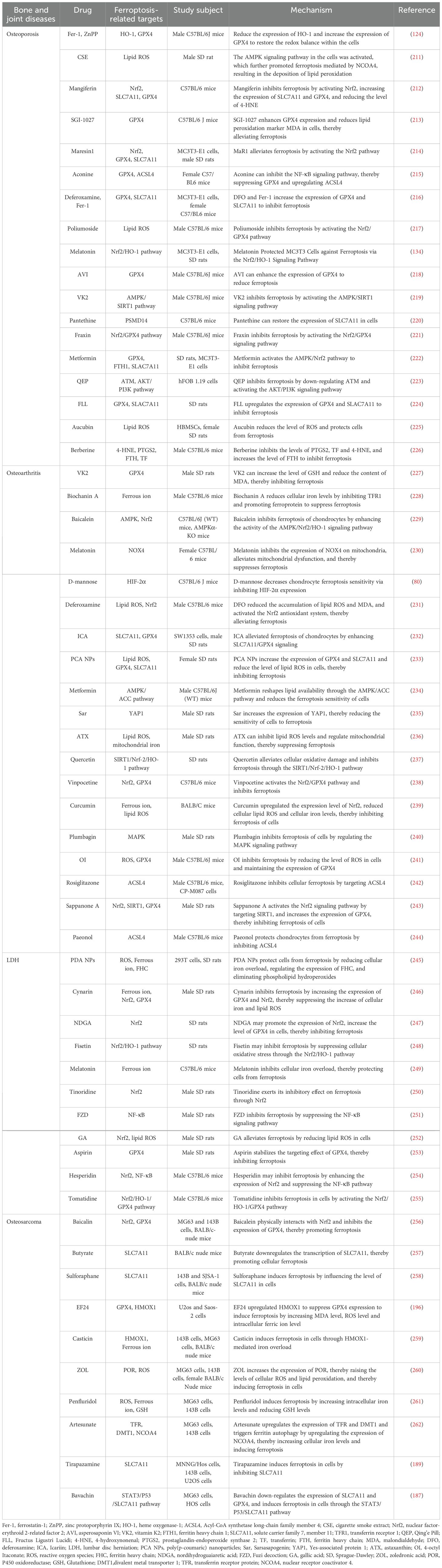
Table 1. The mechanism of ferroptosis-related targets in bone and joint diseases.
Many studies efforts have documented a strong link between cellular ferroptosis and bone health, indicating that manipulating cellular ferroptosis may provide new opportunities for addressing related bone and joint conditions. In osteoporosis, ferroptosis contributes to the pathological process of the disease by altering the functions of osteoblasts, osteoclasts, and mesenchymal stem cells. Notably, HO-1 and mitochondrial ferritin play critical roles in disease progression, and inhibiting their overexpression may represent a novel management strategy for osteoporosis. Furthermore, ferroptosis exacerbates the progression of OA. Inhibitors of ferroptosis, such as Fer-1, D-mannose, and icariin, may mitigate the pathological processes of OA by suppressing ferroptosis in chondrocytes. Ferroptosis may be one of the pathogenic mechanisms of lumbar intervertebral disc protrusion. The ferroptosis inhibitor Fer-1 and the iron chelators DFO and MTF1 can protect intervertebral disc cells from ferroptosis. Ferroptosis plays a crucial role in the pathological progression of osteosarcoma, and agents such as bavachin, TPZ, cisplatin, PEITC, and EF24 induce ferroptosis in osteosarcoma cells, thereby exerting therapeutic effects (Table 1).
Ferroptosis inducers and inhibitors exhibit limitations concerning their specificity and delivery mechanisms. Some inducers promote ferroptosis by inhibiting the GSH/GPX4 pathway (3); however, this pathway is implicated in various physiological and pathological processes, suggesting that the specific induction of ferroptosis may inadvertently disrupt normal cellular functions (264). Regarding delivery, most ferroptosis inducers are small-molecule compounds with diverse chemical properties and stability profiles, complicating their efficient delivery to target cells or tissues. These compounds are prone to rapid metabolism or degradation, leading to inadequate induction in specific tissues or potential toxicity in non-target sites. Similarly, many large-molecule ferroptosis inhibitors struggle to penetrate cell membranes effectively, hindering their ability to exert intracellular effects. Prolonged use of ferroptosis inhibitors may result in toxic accumulation. For instance, extended use of high-dose iron chelators can induce iron deficiency in the body, adversely impacting hematopoietic function, while overdosing on certain antioxidant-based inhibitors may impair coagulation function (58). Some ferroptosis-targeting agents have poor pharmacokinetic properties, such as low solubility, high metabolic clearance, low cellular permeability, and short half-lives (Figure 7).
Figure 7. This figure presents the multiple challenges faced in the clinical transformation of iron-addicted drugs.
The pathogenesis of OP, OA, and OSinvolves multiple factors and signaling pathways. While ferroptosis plays a role in these diseases, it is not the sole mechanism. For example, OP is influenced by factors such as bone metabolism, hormonal regulation, and genetic predisposition. OA involves joint cartilage degeneration, inflammation, and immune responses. OS is characterized by complex genetic mutations and tumor microenvironments. The interplay between ferroptosis and other mechanisms in these diseases requires further research to determine the optimal strategies for targeting ferroptosis in therapy (265).
Identifying reliable biomarkers to monitor ferroptotic activity in vivo and predict treatment efficacy is challenging. Without specific biomarkers, it is difficult to evaluate the therapeutic effects of ferroptosis-targeting agents in clinical trials and adjust treatment plans accordingly. This complicates the precise application of these agents in treating OP, OA, and OS (266). There is significant heterogeneity among patients with OP, OA, and OS. Identifying patient subgroups that are sensitive to ferroptosis-targeting agents and determining the optimal treatment populations remain unresolved issues. This limits the widespread application of ferroptosis-targeting therapies in clinical practice. Similar to other anticancer therapies, ferroptosis-targeting agents may face drug resistance challenges in OS treatment. Tumor cells can develop adaptive mechanisms to resist ferroptosis induction, such as upregulating antioxidant systems or altering iron metabolism pathways. This reduces the efficacy of ferroptosis-targeting agents over time and limits their long-term therapeutic potential (267).
Translating these findings into effective therapies for OP, OA, and OS presents numerous challenges. In OP treatment, there is a notable absence of a single drug that possesses high specificity and efficacy alongside a favorable safety profile. Therefore, exploring combinations of different drugs to achieve synergistic effects while minimizing side effects is essential (268). For OA, the targeted delivery of drugs to affected tissues, such as articular cartilage and synovium, is crucial; however, the current delivery systems struggle to achieve precise targeting, negatively influencing therapeutic outcomes and increasing systemic adverse effects. In OS treatment, the heterogeneity of OS tumors results in variable responses to ferroptosis inducers, with some cells demonstrating resistance. This necessitates extensive research to overcome drug resistance and identify novel strategies.
6 Conclusions
A new form of cell death, termed ferroptosis, was initially characterized in 2012 as distinct from apoptosis and autophagy. Since then, it has become a key area of interest in life science research. Ferroptosis is crucial for regulating bone equilibrium and regeneration. The overaccumulation of iron ions and intracellular oxidative stress are strongly linked to cartilage degeneration and impaired bone metabolism.
The connections between ferroptosis and bone and joint diseases involve mainly iron metabolism, ROS, GPX4, and lipid peroxidation processes. Inhibiting ferroptosis can safeguard osteoblasts from this particular form of cell death, consequently minimizing bone loss and mitigating osteoporosis. Moreover, it can also restrain ferroptosis in chondrocytes, thus delaying the progression of osteoarthritis. An increase in ferroptosis can lead to the death of osteosarcoma cells, suggesting a potential therapeutic approach for osteosarcoma. Various modulators of ferroptosis, including inhibitors and inducers, provide novel perspectives for addressing bone and joint disorders. In addition, our understanding of the iron cycling imbalance and ferroptosis in the pathological context of bone and joint diseases is lacking. The fundamental mechanisms and associated signaling pathways warrant further investigation. Currently, the exploration of ferroptosis in bone and joint diseases has focused mainly on cell and animal models, and future research should pay more attention to clinical studies.
Author contributions
RX: Writing – original draft. ZH: Writing – original draft. PJ: Writing – original draft. PL: Writing – original draft. MG: Writing – original draft. YC: Writing – original draft. LP: Writing – original draft. XY: Writing – original draft. SJ: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cieza A, Causey K, Kamenov K, Hanson SW, Chatterji S, and Vos T. Global estimates of the need for rehabilitation based on the global burden of disease study 2019: A systematic analysis for the global burden of disease study 2019. Lancet. (2021) 396:2006–17. doi: 10.1016/s0140-6736(20)32340-0
2. Ritter J and Bielack SS. Osteosarcoma. Ann Oncol. (2010) 21 Suppl 7:vii320–5. doi: 10.1093/annonc/mdq276
3. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
4. Tang D, Kang R, Berghe TV, Vandenabeele P, and Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
5. Tan S, Schubert D, and Maher P. Oxytosis: A novel form of programmed cell death. Curr Top Med Chem. (2001) 1:497–506. doi: 10.2174/1568026013394741
6. Coltorti M, De Ritis F, and Giusti G. Enzymatic mechanisms of transsulfuration in biology and clinical practice. G Clin Med. (1956) 37:285–323.
7. Eagle H, Piez KA, and Oyama VI. The biosynthesis of cystine in human cell cultures. J Biol Chem. (1961) 236:1425–8. doi: 10.1016/S0021-9258(18)64190-0
8. Distéfano AM, Martin MV, Córdoba JP, Bellido AM, D’Ippólito S, Colman SL, et al. Heat stress induces ferroptosis-like cell death in plants. J Cell Biol. (2017) 216:463–76. doi: 10.1083/jcb.201605110
9. Shen Q, Liang M, Yang F, Deng YZ, and Naqvi NI. Ferroptosis contributes to developmental cell death in rice blast. New Phytol. (2020) 227:1831–46. doi: 10.1111/nph.16636
10. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA, et al. Ferroptosis: mechanisms and links with diseases. Signal Transduction Targeted Ther. (2021) 6:49. doi: 10.1038/s41392-020-00428-9
11. Crielaard BJ, Lammers T, and Rivella S. Targeting iron metabolism in drug discovery and delivery. Nat Rev Drug Discov. (2017) 16:400–23. doi: 10.1038/nrd.2016.248
12. Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of nox4 and inhibition of xct. Cancer Lett. (2018) 428:21–33. doi: 10.1016/j.canlet.2018.04.021
13. Matias C, Belnap DW, Smith MT, Stewart MG, Torres IF, Gross AJ, et al. Citrate and albumin facilitate transferrin iron loading in the presence of phosphate. J Inorg Biochem. (2017) 168:107–13. doi: 10.1016/j.jinorgbio.2016.12.010
14. Dautry-Varsat A, Ciechanover A, and Lodish HF. Ph and the recycling of transferrin during receptor-mediated endocytosis. Proc Natl Acad Sci U.S.A. (1983) 80:2258–62. doi: 10.1073/pnas.80.8.2258
15. Zhang F, Tao Y, Zhang Z, Guo X, An P, Shen Y, et al. Metalloreductase steap3 coordinates the regulation of iron homeostasis and inflammatory responses. Haematologica. (2012) 97:1826–35. doi: 10.3324/haematol.2012.063974
16. Zhang Z, Zhang F, An P, Guo X, Shen Y, Tao Y, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood. (2011) 118:1912–22. doi: 10.1182/blood-2011-01-330324
17. Zhang Z, Zhang F, Guo X, An P, Tao Y, and Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology. (2012) 56:961–71. doi: 10.1002/hep.25746
18. Conrad M and Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. (2019) 15:1137–47. doi: 10.1038/s41589-019-0408-1
19. Andrews NC and Schmidt PJ. Iron homeostasis. Annu Rev Physiol. (2007) 69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337
20. Zhang DL, Ghosh MC, and Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis - an update. Front Pharmacol. (2014) 5:124. doi: 10.3389/fphar.2014.00124
21. Gao M, Monian P, Pan Q, Zhang W, Xiang J, and Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. (2016) 26:1021–32. doi: 10.1038/cr.2016.95
22. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
23. Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, et al. Iron chaperone poly rc binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatology. (2021) 73:1176–93. doi: 10.1002/hep.31328
24. Kwon MY, Park E, Lee SJ, and Chung SW. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget. (2015) 6:24393–403. doi: 10.18632/oncotarget.5162
25. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the P62-keap1-nrf2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatol (Baltimore Md). (2016) 63:173–84. doi: 10.1002/hep.28251
26. Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of atm in regulating iron metabolism. Cell Death Differ. (2020) 27:1008–22. doi: 10.1038/s41418-019-0393-7
27. Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. (2019) 51:575–86.e4. doi: 10.1016/j.devcel.2019.10.007
28. Jing X, Du T, Li T, Yang X, Wang G, Liu X, et al. The detrimental effect of iron on oa chondrocytes: importance of pro-inflammatory cytokines induced iron influx and oxidative stress. J Cell Mol Med. (2021) 25:5671–80. doi: 10.1111/jcmm.16581
29. Zhang H, Wang A, Shen G, Wang X, Liu G, Yang F, et al. Hepcidin-induced reduction in iron content and pgc-1β Expression negatively regulates osteoclast differentiation to play a protective role in postmenopausal osteoporosis. Aging (Albany NY). (2021) 13:11296–314. doi: 10.18632/aging.202817
30. Zhao L, Wang Y, Wang Z, Xu Z, Zhang Q, and Yin M. Effects of dietary resveratrol on excess-iron-induced bone loss via antioxidative character. J Nutr Biochem. (2015) 26:1174–82. doi: 10.1016/j.jnutbio.2015.05.009
31. Telfer JF and Brock JH. Proinflammatory cytokines increase iron uptake into human monocytes and synovial fibroblasts from patients with rheumatoid arthritis. Med Sci Monit. (2004) 10:Br91–5.
32. Gill I and Valivety R. Polyunsaturated fatty acids, part 1: occurrence, biological activities and applications. Trends Biotechnol. (1997) 15:401–9. doi: 10.1016/s0167-7799(97)01076-7
33. Porter NA, Wolf RA, Yarbro EM, and Weenen H. The autoxidation of arachidonic acid: formation of the proposed srs-a intermediate. Biochem Biophys Res Commun. (1979) 89:1058–64. doi: 10.1016/0006-291x(79)92115-6
34. Yin H, Xu L, and Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. (2011) 111:5944–72. doi: 10.1021/cr200084z
35. Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated ampk activation inhibits ferroptosis. Nat Cell Biol. (2020) 22:225–34. doi: 10.1038/s41556-020-0461-8
36. Li C, Dong X, Du W, Shi X, Chen K, Zhang W, et al. Lkb1-ampk axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduction Targeted Ther. (2020) 5:187. doi: 10.1038/s41392-020-00297-2
37. Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. (2019) 26:420–32.e9. doi: 10.1016/j.chembiol.2018.11.016
38. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. Acsl4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
39. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
40. Hishikawa D, Shindou H, Kobayashi S, Nakanishi H, Taguchi R, and Shimizu T. Discovery of a lysophospholipid acyltransferase family essential for membrane asymmetry and diversity. Proc Natl Acad Sci U.S.A. (2008) 105:2830–5. doi: 10.1073/pnas.0712245105
41. Küch EM, Vellaramkalayil R, Zhang I, Lehnen D, Brügger B, Sreemmel W, et al. Differentially localized acyl-coa synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim Biophys Acta. (2014) 1841:227–39. doi: 10.1016/j.bbalip.2013.10.018
42. Wong-Ekkabut J, Xu Z, Triampo W, Tang IM, Tieleman DP, and Monticelli L. Effect of lipid peroxidation on the properties of lipid bilayers: A molecular dynamics study. Biophys J. (2007) 93:4225–36. doi: 10.1529/biophysj.107.112565
43. Feng H and Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PloS Biol. (2018) 16:e2006203. doi: 10.1371/journal.pbio.2006203
44. Ayala A, Muñoz MF, and Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. (2014) 2014:360438. doi: 10.1155/2014/360438
45. Jin ZL, Gao WY, Liao SJ, Yu T, Shi Q, Yu SZ, et al. Paeonol inhibits the progression of intracerebral haemorrhage by mediating the hotair/upf1/acsl4 axis. ASN Neuro. (2021) 13:17590914211010647. doi: 10.1177/17590914211010647
46. Saul D, Gleitz S, Nguyen HH, Kosinsky RL, Sehmisch S, Hoffmann DB, et al. Effect of the lipoxygenase-inhibitors baicalein and zileuton on the vertebra in ovariectomized rats. Bone. (2017) 101:134–44. doi: 10.1016/j.bone.2017.04.011
47. Chen JJ and Galluzzi L. Fighting resilient cancers with iron. Trends Cell Biol. (2018) 28:77–8. doi: 10.1016/j.tcb.2017.11.007
48. Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and aif-mediated cell death. Cell Metab. (2008) 8:237–48. doi: 10.1016/j.cmet.2008.07.005
49. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by gpx4 is required to prevent hydroperoxide-induced ferroptosis. Cell. (2018) 172:409–22.e21. doi: 10.1016/j.cell.2017.11.048
50. Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, et al. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell. (2019) 177:1262–79.e25. doi: 10.1016/j.cell.2019.03.032
51. Forcina GC and Dixon SJ. Gpx4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. (2019) 19:e1800311. doi: 10.1002/pmic.201800311
52. Liang C, Zhang X, Yang M, and Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater. (2019) 31:e1904197. doi: 10.1002/adma.201904197
53. Doll S and Conrad M. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life. (2017) 69:423–34. doi: 10.1002/iub.1616
54. Mandal PK, Seiler A, Perisic T, Kölle P, Banjac Canak A, Förster H, et al. System X(C)- and thioredoxin reductase 1 cooperatively rescue glutathione deficiency. J Biol Chem. (2010) 285:22244–53. doi: 10.1074/jbc.M110.121327
55. Hayano M, Yang WS, Corn CK, Pagano NC, and Stockwell BR. Loss of cysteinyl-trna synthetase (Cars) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. (2016) 23:270–8. doi: 10.1038/cdd.2015.93
56. Bannai S and Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. (1980) 255:2372–6. doi: 10.1016/S0021-9258(19)85901-X
57. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a P53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
58. Jiang X, Stockwell BR, and Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
59. Gong M, Hay S, Marshall KR, Munro AW, and Scrutton NS. DNA binding suppresses human aif-M2 activity and provides a connection between redox chemistry, reactive oxygen species, and apoptosis. J Biol Chem. (2007) 282:30331–40. doi: 10.1074/jbc.M703713200
60. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The coq oxidoreductase fsp1 acts parallel to gpx4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
61. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. Fsp1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
62. Stocker R, Bowry VW, and Frei B. Ubiquinol-10 protects human low density lipoprotein more efficiently against lipid peroxidation than does alpha-tocopherol. Proc Natl Acad Sci U.S.A. (1991) 88:1646–50. doi: 10.1073/pnas.88.5.1646
63. Morré DJ and Morré DM. Non-mitochondrial coenzyme Q. Biofactors. (2011) 37:355–60. doi: 10.1002/biof.156
64. Shukla S and Dubey KK. Coq10 a super-vitamin: review on application and biosynthesis. 3 Biotech. (2018) 8:249. doi: 10.1007/s13205-018-1271-6
65. Ernster L and Forsmark-Andrée P. Ubiquinol: an endogenous antioxidant in aerobic organisms. Clin Investig. (1993) 71:S60–5. doi: 10.1007/bf00226842
66. Friedmann Angeli JP, Krysko DV, and Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. (2019) 19:405–14. doi: 10.1038/s41568-019-0149-1
67. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. (2016) 12:497–503. doi: 10.1038/nchembio.2079
68. Dai E, Zhang W, Cong D, Kang R, Wang J, and Tang D. Aifm2 blocks ferroptosis independent of ubiquinol metabolism. Biochem Biophys Res Commun. (2020) 523:966–71. doi: 10.1016/j.bbrc.2020.01.066
69. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. Gtp cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
70. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol. (2020) 16:1351–60. doi: 10.1038/s41589-020-0613-y
71. Deng G, Li Y, Ma S, Gao Z, Zeng T, Chen L, et al. Caveolin-1 dictates ferroptosis in the execution of acute immune-mediated hepatic damage by attenuating nitrogen stress. Free Radic Biol Med. (2020) 148:151–61. doi: 10.1016/j.freeradbiomed.2019.12.026
72. Wang D, Peng Y, Xie Y, Zhou B, Sun X, Kang R, et al. Antiferroptotic activity of non-oxidative dopamine. Biochem Biophys Res Commun. (2016) 480:602–7. doi: 10.1016/j.bbrc.2016.10.099
73. Carlson BA, Tobe R, Yefremova E, Tsuji PA, Hoffmann VJ, Schweizer U, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. (2016) 9:22–31. doi: 10.1016/j.redox.2016.05.003
74. Llabani E, Hicklin RW, Lee HY, Motika SE, Crawford LA, Weerapana E, et al. Diverse compounds from pleuromutilin lead to a thioredoxin inhibitor and inducer of ferroptosis. Nat Chem. (2019) 11:521–32. doi: 10.1038/s41557-019-0261-6
75. Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. (2014) 3:e02523. doi: 10.7554/eLife.02523
76. Martínez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature. (2020) 585:288–92. doi: 10.1038/s41586-020-2475-6
77. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. Dhodh-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
78. Wang F and Min J. Dhodh tangoing with gpx4 on the ferroptotic stage. Signal Transduct Target Ther. (2021) 6:244. doi: 10.1038/s41392-021-00656-7
79. Oršolić N, Nemrava J, Jeleč Ž, Kukolj M, Odeh D, Jakopović B, et al. Antioxidative and anti-inflammatory activities of chrysin and naringenin in a drug-induced bone loss model in rats. Int J Mol Sci. (2022) 23:2872. doi: 10.3390/ijms23052872
80. Zhou X, Zheng Y, Sun W, Zhang Z, Liu J, Yang W, et al. D-mannose alleviates osteoarthritis progression by inhibiting chondrocyte ferroptosis in a hif-2α-dependent manner. Cell Prolif. (2021) 54:e13134. doi: 10.1111/cpr.13134
81. Hofbauer LC, Bozec A, Rauner M, Jakob F, Perner S, and Pantel K. Novel approaches to target the microenvironment of bone metastasis. Nat Rev Clin Oncol. (2021) 18:488–505. doi: 10.1038/s41571-021-00499-9
82. Balogh E, Paragh G, and Jeney V. Influence of iron on bone homeostasis. Pharm (Basel). (2018) 11:107. doi: 10.3390/ph11040107
83. Jeney V. Clinical impact and cellular mechanisms of iron overload-associated bone loss. Front Pharmacol. (2017) 8:77. doi: 10.3389/fphar.2017.00077
84. Dong D, Yang J, Zhang G, Huyan T, and Shang P. 16 T high static magnetic field inhibits receptor activator of nuclear factor kappa-β Ligand-induced osteoclast differentiation by regulating iron metabolism in raw264.7 cells. J Tissue Eng Regener Med. (2019) 13:2181–90. doi: 10.1002/term.2973
85. Gu Z, Wang H, Xia J, Yang Y, Jin Z, Xu H, et al. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res. (2015) 75:2211–21. doi: 10.1158/0008-5472.Can-14-3804
86. Wang L, Fang B, Fujiwara T, Krager K, Gorantla A, Li C, et al. Deletion of ferroportin in murine myeloid cells increases iron accumulation and stimulates osteoclastogenesis in vitro and in vivo. J Biol Chem. (2018) 293:9248–64. doi: 10.1074/jbc.RA117.000834
87. Ledesma-Colunga MG, Baschant U, Fiedler IAK, Busse B, Hofbauer LC, Muckenthaler MU, et al. Disruption of the hepcidin/ferroportin regulatory circuitry causes low axial bone mass in mice. Bone. (2020) 137:115400. doi: 10.1016/j.bone.2020.115400
88. Soltanoff CS, Yang S, Chen W, and Li YP. Signaling networks that control the lineage commitment and differentiation of bone cells. Crit Rev Eukaryot Gene Expr. (2009) 19:1–46. doi: 10.1615/critreveukargeneexpr.v19.i1.10
89. Teitelbaum SL and Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. (2003) 4:638–49. doi: 10.1038/nrg1122
90. Boyce BF and Xing L. Biology of rank, rankl, and osteoprotegerin. Arthritis Res Ther. (2007) 9 Suppl 1:S1. doi: 10.1186/ar2165
91. Ishii KA, Fumoto T, Iwai K, Takeshita S, Ito M, Shimohata N, et al. Coordination of pgc-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. (2009) 15:259–66. doi: 10.1038/nm.1910
92. Jia P, Xu YJ, Zhang ZL, Li K, Li B, Zhang W, et al. Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. J Orthop Res. (2012) 30:1843–52. doi: 10.1002/jor.22133
93. Das BK, Wang L, Fujiwara T, Zhou J, Aykin-Burns N, Krager KJ, et al. Transferrin receptor 1-mediated iron uptake regulates bone mass in mice via osteoclast mitochondria and cytoskeleton. Elife. (2022) 11:e73539. doi: 10.7554/eLife.73539
94. Hou JM, Xue Y, and Lin QM. Bovine lactoferrin improves bone mass and microstructure in ovariectomized rats via opg/rankl/rank pathway. Acta Pharmacol Sin. (2012) 33:1277–84. doi: 10.1038/aps.2012.83
95. Tsay J, Yang Z, Ross FP, Cunningham-Rundles S, Lin H, Coleman R, et al. Bone loss caused by iron overload in a murine model: importance of oxidative stress. Blood. (2010) 116:2582–9. doi: 10.1182/blood-2009-12-260083
96. Guggenbuhl P, Fergelot P, Doyard M, Libouban H, Roth MP, Gallois Y, et al. Bone status in a mouse model of genetic hemochromatosis. Osteoporos Int. (2011) 22:2313–9. doi: 10.1007/s00198-010-1456-2
97. Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, et al. Tgf-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med. (2009) 15:757–65. doi: 10.1038/nm.1979
98. Lertsuwan K, Nammultriputtar K, Nanthawuttiphan S, Tannop N, Teerapornpuntakit J, Thongbunchoo J, et al. Differential effects of fe2+ and fe3+ on osteoblasts and the effects of 1,25(Oh)2d3, deferiprone and extracellular calcium on osteoblast viability under iron-overloaded conditions. PloS One. (2020) 15:e0234009. doi: 10.1371/journal.pone.0234009
99. Yang Q, Jian J, Abramson SB, and Huang X. Inhibitory effects of iron on bone morphogenetic protein 2-induced osteoblastogenesis. J Bone Miner Res. (2011) 26:1188–96. doi: 10.1002/jbmr.337
100. Balogh E, Tolnai E, Nagy B Jr., Nagy B, Balla G, Balla J, et al. Iron overload inhibits osteogenic commitment and differentiation of mesenchymal stem cells via the induction of ferritin. Biochim Biophys Acta. (2016) 1862:1640–9. doi: 10.1016/j.bbadis.2016.06.003
101. Messer JG, Kilbarger AK, Erikson KM, and Kipp DE. Iron overload alters iron-regulatory genes and proteins, down-regulates osteoblastic phenotype, and is associated with apoptosis in fetal rat calvaria cultures. Bone. (2009) 45:972–9. doi: 10.1016/j.bone.2009.07.073
102. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and tlr4/trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. (2019) 129:2293–304. doi: 10.1172/jci126428
103. Yotsumoto S, Muroi Y, Chiba T, Ohmura R, Yoneyama M, Magarisawa M, et al. Hyperoxidation of ether-linked phospholipids accelerates neutrophil extracellular trap formation. Sci Rep. (2017) 7:16026. doi: 10.1038/s41598-017-15668-z
104. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. (2020) 585:603–8. doi: 10.1038/s41586-020-2732-8
105. Piattini F, Matsushita M, Muri J, Bretscher P, Feng X, Freigang S, et al. Differential sensitivity of inflammatory macrophages and alternatively activated macrophages to ferroptosis. Eur J Immunol. (2021) 51:2417–29. doi: 10.1002/eji.202049114
106. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol. (2020) 16:278–90. doi: 10.1038/s41589-019-0462-8
107. Yuan Y, Chen Y, Peng T, Li L, Zhu W, Liu F, et al. Mitochondrial ros-induced lysosomal dysfunction impairs autophagic flux and contributes to M1 macrophage polarization in a diabetic condition. Clin Sci (Lond). (2019) 133:1759–77. doi: 10.1042/cs20190672
108. Zhang B, Yang Y, Yi J, Zhao Z, and Ye R. Hyperglycemia modulates M1/M2 macrophage polarization via reactive oxygen species overproduction in ligature-induced periodontitis. J Periodontal Res. (2021) 56:991–1005. doi: 10.1111/jre.12912
109. Li B, Gong J, Sheng S, Lu M, Guo S, Zhao X, et al. Increased hepcidin in hemorrhagic plaques correlates with iron-stimulated il-6/stat3 pathway activation in macrophages. Biochem Biophys Res Commun. (2019) 515:394–400. doi: 10.1016/j.bbrc.2019.05.123
110. Kanamori Y, Murakami M, Sugiyama M, Hashimoto O, Matsui T, and Funaba M. Interleukin-1β (Il-1β) transcriptionally activates hepcidin by inducing ccaat enhancer-binding protein Δ (C/ebpδ) expression in hepatocytes. J Biol Chem. (2017) 292:10275–87. doi: 10.1074/jbc.M116.770974
111. Santos M and de Sousa M. In vitro modulation of T-cell surface molecules by iron. Cell Immunol. (1994) 154:498–506. doi: 10.1006/cimm.1994.1094
112. Vanoaica L, Richman L, Jaworski M, Darshan D, Luther SA, and Kühn LC. Conditional deletion of ferritin H in mice reduces B and T lymphocyte populations. PloS One. (2014) 9:e89270. doi: 10.1371/journal.pone.0089270
113. Mak TW, Grusdat M, Duncan GS, Dostert C, Nonnenmacher Y, Cox M, et al. Glutathione primes T cell metabolism for inflammation. Immunity. (2017) 46:1089–90. doi: 10.1016/j.immuni.2017.06.009
114. Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, and Kopf M. T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med. (2015) 212:555–68. doi: 10.1084/jem.20140857
115. Du X, Ma X, Tan Y, Shao F, Li C, Zhao Y, et al. B cell-derived anti-beta 2 glycoprotein I antibody mediates hyperhomocysteinemia-aggravated hypertensive glomerular lesions by triggering ferroptosis. Signal Transduct Target Ther. (2023) 8:103. doi: 10.1038/s41392-023-01313-x
116. Wang D, Xie N, Gao W, Kang R, and Tang D. The ferroptosis inducer erastin promotes proliferation and differentiation in human peripheral blood mononuclear cells. Biochem Biophys Res Commun. (2018) 503:1689–95. doi: 10.1016/j.bbrc.2018.07.100
117. Zhang Y, Zhai W, Zhao M, Li D, Chai X, Cao X, et al. Effects of iron overload on the bone marrow microenvironment in mice. PloS One. (2015) 10:e0120219. doi: 10.1371/journal.pone.0120219
118. Valentino LA. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Haemost. (2010) 8:1895–902. doi: 10.1111/j.1538-7836.2010.03962.x
119. Lane NE. Epidemiology, etiology, and diagnosis of osteoporosis. Am J Obstet Gynecol. (2006) 194:S3–11. doi: 10.1016/j.ajog.2005.08.047
120. Fujita T. Osteoporosis–concept, classification and epidemiology. Nihon Rinsho. (1994) 52:2275–80.
121. Johnston CB and Dagar M. Osteoporosis in older adults. Med Clin North Am. (2020) 104:873–84. doi: 10.1016/j.mcna.2020.06.004
122. Abdalbary M, Sobh M, Elnagar S, Elhadedy MA, Elshabrawy N, Abdelsalam M, et al. Management of osteoporosis in patients with chronic kidney disease. Osteoporos Int. (2022) 33:2259–74. doi: 10.1007/s00198-022-06462-3
123. Sözen T, Özışık L, and Başaran N. An overview and management of osteoporosis. Eur J Rheumatol. (2017) 4:46–56. doi: 10.5152/eurjrheum.2016.048
124. Yang Y, Lin Y, Wang M, Yuan K, Wang Q, Mu P, et al. Targeting ferroptosis suppresses osteocyte glucolipotoxicity and alleviates diabetic osteoporosis. Bone Res. (2022) 10:26. doi: 10.1038/s41413-022-00198-w
125. Dede AD, Trovas G, Chronopoulos E, Triantafyllopoulos IK, Dontas I, Papaioannou N, et al. Thalassemia-associated osteoporosis: A systematic review on treatment and brief overview of the disease. Osteoporos Int. (2016) 27:3409–25. doi: 10.1007/s00198-016-3719-z
126. Wong P, Fuller PJ, Gillespie MT, Kartsogiannis V, Kerr PG, Doery JC, et al. Thalassemia bone disease: A 19-year longitudinal analysis. J Bone Miner Res. (2014) 29:2468–73. doi: 10.1002/jbmr.2266
127. Saki N, Abroun S, Salari F, Rahim F, Shahjahani M, and Javad MA. Molecular aspects of bone resorption in β-thalassemia major. Cell J. (2015) 17:193–200. doi: 10.22074/cellj.2016.3713
128. Handattu K, Aroor S, Kini P, Ramesh Bhat Y, Shivakumar G, Shastry P, et al. Metabolic bone disease in children with transfusion-dependent thalassemia. Indian Pediatr. (2022) 59:920–3. doi: 10.1007/s13312-022-2663-6
129. Valenti L, Varenna M, Fracanzani AL, Rossi V, Fargion S, and Sinigaglia L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int. (2009) 20:549–55. doi: 10.1007/s00198-008-0701-4
130. Simão M and Cancela ML. Musculoskeletal complications associated with pathological iron toxicity and its molecular mechanisms. Biochem Soc Trans. (2021) 49:747–59. doi: 10.1042/bst20200672
131. Xu Z, Sun W, Li Y, Ling S, Zhao C, Zhong G, et al. The regulation of iron metabolism by hepcidin contributes to unloading-induced bone loss. Bone. (2017) 94:152–61. doi: 10.1016/j.bone.2016.09.023
132. Zarjou A, Jeney V, Arosio P, Poli M, Zavaczki E, Balla G, et al. Ferritin ferroxidase activity: A potent inhibitor of osteogenesis. J Bone Miner Res. (2010) 25:164–72. doi: 10.1359/jbmr.091002
133. Lin Y, Shen X, Ke Y, Lan C, Chen X, Liang B, et al. Activation of osteoblast ferroptosis via the mettl3/ask1-P38 signaling pathway in high glucose and high fat (Hghf)-induced diabetic bone loss. FASEB J. (2022) 36:e22147. doi: 10.1096/fj.202101610R
134. Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the nrf2/ho-1 signaling pathway in type 2 diabetic osteoporosis. Oxid Med Cell Longev. (2020) 2020:9067610. doi: 10.1155/2020/9067610
135. Wang X, Ma H, Sun J, Zheng T, Zhao P, Li H, et al. Mitochondrial ferritin deficiency promotes osteoblastic ferroptosis via mitophagy in type 2 diabetic osteoporosis. Biol Trace Elem Res. (2022) 200:298–307. doi: 10.1007/s12011-021-02627-z
136. Tonelli C, Chio IIC, and Tuveson DA. Transcriptional regulation by nrf2. Antioxid Redox Signal. (2018) 29:1727–45. doi: 10.1089/ars.2017.7342
137. He R, Liu B, Xiong R, Geng B, Meng H, Lin W, et al. Itaconate inhibits ferroptosis of macrophage via nrf2 pathways against sepsis-induced acute lung injury. Cell Death Discov. (2022) 8:43. doi: 10.1038/s41420-021-00807-3
138. Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, et al. A human mitochondrial ferritin encoded by an intronless gene. J Biol Chem. (2001) 276:24437–40. doi: 10.1074/jbc.C100141200
139. Deng M, Luo J, Cao H, Li Y, Chen L, and Liu G. Mettl14 represses osteoclast formation to ameliorate osteoporosis via enhancing gpx4 mrna stability. Environ Toxicol. (2023) 38:2057–68. doi: 10.1002/tox.23829
140. Bian Q, Wang YJ, Liu SF, and Li YP. Osteoarthritis: genetic factors, animal models, mechanisms, and therapies. Front Biosci (Elite Ed). (2012) 4:74–100. doi: 10.2741/361
141. Murata K, Yoshitomi H, Tanida S, Ishikawa M, Nishitani K, Ito H, et al. Plasma and synovial fluid micrornas as potential biomarkers of rheumatoid arthritis and osteoarthritis. Arthritis Res Ther. (2010) 12:R86. doi: 10.1186/ar3013
142. Glyn-Jones S, Palmer AJ, Agricola R, Price AJ, Vincent TL, Weinans H, et al. Osteoarthritis. Lancet. (2015) 386:376–87. doi: 10.1016/s0140-6736(14)60802-3
143. Rubio JE, Scanzello C, Felson DT, Au MB, Goggins J, Plunkett A, et al. Correlation between senescence-associated secretory phenotypes factors in synovial fluid and serum and structural changes in osteoarthritis. Eur J Rheumatol. (2020) 7:44–5. doi: 10.5152/eurjrheum.2019.19025
144. Wang S, Li W, Zhang P, Wang Z, Ma X, Liu C, et al. Mechanical overloading induces gpx4-regulated chondrocyte ferroptosis in osteoarthritis via piezo1 channel facilitated calcium influx. J Adv Res. (2022) 41:63–75. doi: 10.1016/j.jare.2022.01.004
145. Bijlsma JW, Berenbaum F, and Lafeber FP. Osteoarthritis: an update with relevance for clinical practice. Lancet. (2011) 377:2115–26. doi: 10.1016/s0140-6736(11)60243-2
146. Lin J, Fransen M, Kang X, Li H, Ke Y, Wang Z, et al. Marked disability and high use of nonsteroidal antiinflammatory drugs associated with knee osteoarthritis in rural China: A cross-sectional population-based survey. Arthritis Res Ther. (2010) 12:R225. doi: 10.1186/ar3212
147. Hawker GA, Croxford R, Bierman AS, Harvey PJ, Ravi B, Stanaitis I, et al. All-cause mortality and serious cardiovascular events in people with hip and knee osteoarthritis: A population based cohort study. PloS One. (2014) 9:e91286. doi: 10.1371/journal.pone.0091286
148. Xing D, Xu Y, Liu Q, Ke Y, Wang B, Li Z, et al. Osteoarthritis and all-cause mortality in worldwide populations: grading the evidence from a meta-analysis. Sci Rep. (2016) 6:24393. doi: 10.1038/srep24393
149. Safiri S, Kolahi AA, Smith E, Hill C, Bettampadi D, Mansournia MA, et al. Global, regional and national burden of osteoarthritis 1990-2017: A systematic analysis of the global burden of disease study 2017. Ann Rheum Dis. (2020) 79:819–28. doi: 10.1136/annrheumdis-2019-216515
150. van Vulpen LFD, Holstein K, and Martinoli C. Joint disease in haemophilia: pathophysiology, pain and imaging. Haemophilia. (2018) 24 Suppl 6:44–9. doi: 10.1111/hae.13449
151. Heiland GR, Aigner E, Dallos T, Sahinbegovic E, Krenn V, Thaler C, et al. Synovial immunopathology in haemochromatosis arthropathy. Ann Rheum Dis. (2010) 69:1214–9. doi: 10.1136/ard.2009.120204
152. Sun K, Guo Z, Hou L, Xu J, Du T, Xu T, et al. Iron homeostasis in arthropathies: from pathogenesis to therapeutic potential. Ageing Res Rev. (2021) 72:101481. doi: 10.1016/j.arr.2021.101481
153. Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X, et al. Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol. (2021) 9:789948. doi: 10.3389/fcell.2021.789948
154. Cai C, Hu W, and Chu T. Interplay between iron overload and osteoarthritis: clinical significance and cellular mechanisms. Front Cell Dev Biol. (2021) 9:817104. doi: 10.3389/fcell.2021.817104
155. Kennish L, Attur M, Oh C, Krasnokutsky S, Samuels J, Greenberg JD, et al. Age-dependent ferritin elevations and hfe C282y mutation as risk factors for symptomatic knee osteoarthritis in males: A longitudinal cohort study. BMC Musculoskelet Disord. (2014) 15:8. doi: 10.1186/1471-2474-15-8
156. Xu J, Zhang S, Tian Y, Si H, Zeng Y, Wu Y, et al. Genetic causal association between iron status and osteoarthritis: A two-sample mendelian randomization. Nutrients. (2022) 14:3683. doi: 10.3390/nu14183683
157. Ru Q, Li Y, Xie W, Ding Y, Chen L, Xu G, et al. Fighting age-related orthopedic diseases: focusing on ferroptosis. Bone Res. (2023) 11:12. doi: 10.1038/s41413-023-00247-y
158. Guilak F, Nims RJ, Dicks A, Wu CL, and Meulenbelt I. Osteoarthritis as a disease of the cartilage pericellular matrix. Matrix Biol. (2018) 71-72:40–50. doi: 10.1016/j.matbio.2018.05.008
159. Zheng L, Zhang Z, Sheng P, and Mobasheri A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res Rev. (2021) 66:101249. doi: 10.1016/j.arr.2020.101249
160. Yao X, Sun K, Yu S, Luo J, Guo J, Lin J, et al. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J Orthop Translat. (2021) 27:33–43. doi: 10.1016/j.jot.2020.09.006
161. Singhal R, Mitta SR, Das NK, Kerk SA, Sajjakulnukit P, Solanki S, et al. Hif-2α Activation potentiates oxidative cell death in colorectal cancers by increasing cellular iron. J Clin Invest. (2021) 131:e143691. doi: 10.1172/jci143691
162. Luo H and Zhang R. Icariin enhances cell survival in lipopolysaccharide-induced synoviocytes by suppressing ferroptosis via the xc-/gpx4 axis. Exp Ther Med. (2021) 21:72. doi: 10.3892/etm.2020.9504
163. Mo Z, Xu P, and Li H. Stigmasterol alleviates interleukin-1beta-induced chondrocyte injury by down-regulatingsterol regulatory element binding transcription factor 2 to regulateferroptosis. Bioengineered. (2021) 12:9332–40. doi: 10.1080/21655979.2021.2000742
164. Zhang J, Hu W, Ding C, Yao G, Zhao H, and Wu S. Deferoxamine inhibits iron-uptake stimulated osteoclast differentiation by suppressing electron transport chain and mapks signaling. Toxicol Lett. (2019) 313:50–9. doi: 10.1016/j.toxlet.2019.06.007
165. Liu F, Zhang WL, Meng HZ, Cai ZY, and Yang MW. Regulation of dmt1 on autophagy and apoptosis in osteoblast. Int J Med Sci. (2017) 14:275–83. doi: 10.7150/ijms.17860
166. Bhat V, Olmer M, Joshi S, Durden DL, Cramer TJ, Barnes RF, et al. Vascular remodeling underlies rebleeding in hemophilic arthropathy. Am J Hematol. (2015) 90:1027–35. doi: 10.1002/ajh.24133
167. Mathiessen A and Conaghan PG. Synovitis in osteoarthritis: current understanding with therapeutic implications. Arthritis Res Ther. (2017) 19:18. doi: 10.1186/s13075-017-1229-9
168. Jing X, Lin J, Du T, Jiang Z, Li T, Wang G, et al. Iron overload is associated with accelerated progression of osteoarthritis: the role of dmt1 mediated iron homeostasis. Front Cell Dev Biol. (2020) 8:594509. doi: 10.3389/fcell.2020.594509
169. An S, Hu H, Li Y, and Hu Y. Pyroptosis plays a role in osteoarthritis. Aging Dis. (2020) 11:1146–57. doi: 10.14336/ad.2019.1127
170. Wang R, Shi D, Pan X, Ren A, and Jiang K. Epigenetic mechanisms of nsd1-mediated histone methylation modifications in chondrocyte ferroptosis in knee osteoarthritis. Biomol BioMed. (2025) 25:894–904. doi: 10.17305/bb.2024.10879
171. Zhang Y, Zhao CY, Zhou Z, Li CC, and Wang Q. The effect of lactate dehydrogenase B and its mediated histone lactylation on chondrocyte ferroptosis during osteoarthritis. J Orthop Surg Res. (2025) 20:493. doi: 10.1186/s13018-025-05894-x
172. Shamji MF, Setton LA, Jarvis W, So S, Chen J, Jing L, et al. Proinflammatory cytokine expression profile in degenerated and herniated human intervertebral disc tissues. Arthritis Rheum. (2010) 62:1974–82. doi: 10.1002/art.27444
173. Zhang AS, Xu A, Ansari K, Hardacker K, Anderson G, Alsoof D, et al. Lumbar disc herniation: diagnosis and management. Am J Med. (2023) 136:645–51. doi: 10.1016/j.amjmed.2023.03.024
174. Yang RZ, Xu WN, Zheng HL, Zheng XF, Li B, Jiang LS, et al. Involvement of oxidative stress-induced annulus fibrosus cell and nucleus pulposus cell ferroptosis in intervertebral disc degeneration pathogenesis. J Cell Physiol. (2021) 236:2725–39. doi: 10.1002/jcp.30039
175. Wang W, Jing X, Du T, Ren J, Liu X, Chen F, et al. Iron overload promotes intervertebral disc degeneration via inducing oxidative stress and ferroptosis in endplate chondrocytes. Free Radic Biol Med. (2022) 190:234–46. doi: 10.1016/j.freeradbiomed.2022.08.018
176. Xiao ZF, Su GY, Hou Y, Chen SD, Zhao BD, He JB, et al. Mechanics and biology interact in intervertebral disc degeneration: A novel composite mouse model. Calcif Tissue Int. (2020) 106:401–14. doi: 10.1007/s00223-019-00644-8
177. Shan L, Xu X, Zhang J, Cai P, Gao H, Lu Y, et al. Increased hemoglobin and heme in maldi-tof ms analysis induce ferroptosis and promote degeneration of herniated human nucleus pulposus. Mol Med. (2021) 27:103. doi: 10.1186/s10020-021-00368-2
178. Zhang Y, Han S, Kong M, Tu Q, Zhang L, and Ma X. Single-cell rna-seq analysis identifies unique chondrocyte subsets and reveals involvement of ferroptosis in human intervertebral disc degeneration. Osteoarthritis Cartilage. (2021) 29:1324–34. doi: 10.1016/j.joca.2021.06.010
179. Lu S, Song Y, Luo R, Li S, Li G, Wang K, et al. Ferroportin-dependent iron homeostasis protects against oxidative stress-induced nucleus pulposus cell ferroptosis and ameliorates intervertebral disc degeneration in vivo. Oxid Med Cell Longev. (2021) 2021:6670497. doi: 10.1155/2021/6670497
180. Bin S, Xin L, Lin Z, Jinhua Z, Rui G, and Xiang Z. Targeting mir-10a-5p/il-6r axis for reducing il-6-induced cartilage cell ferroptosis. Exp Mol Pathol. (2021) 118:104570. doi: 10.1016/j.yexmp.2020.104570
181. Madhu V, Guntur AR, and Risbud MV. Role of autophagy in intervertebral disc and cartilage function: implications in health and disease. Matrix Biol. (2021) 100-101:207–20. doi: 10.1016/j.matbio.2020.12.002
182. Jiang W, Zhang X, Hao J, Shen J, Fang J, Dong W, et al. Sirt1 protects against apoptosis by promoting autophagy in degenerative human disc nucleus pulposus cells. Sci Rep. (2014) 4:7456. doi: 10.1038/srep07456
183. Chen J, Yang X, Li Q, Ma J, Li H, Wang L, et al. Inhibiting DNA methyltransferase dnmt3b confers protection against ferroptosis in nucleus pulposus and ameliorates intervertebral disc degeneration via upregulating slc40a1. Free Radic Biol Med. (2024) 220:139–53. doi: 10.1016/j.freeradbiomed.2024.05.007
184. Panez-Toro I, Muñoz-García J, Vargas-Franco JW, Renodon-Cornière A, Heymann MF, Lézot F, et al. Advances in osteosarcoma. Curr Osteoporos Rep. (2023) 21:330–43. doi: 10.1007/s11914-023-00803-9
185. Hu Z, Wen S, Huo Z, Wang Q, Zhao J, Wang Z, et al. Current status and prospects of targeted therapy for osteosarcoma. Cells. (2022) 11:3507. doi: 10.3390/cells11213507
186. Stockwell BR and Jiang X. The chemistry and biology of ferroptosis. Cell Chem Biol. (2020) 27:365–75. doi: 10.1016/j.chembiol.2020.03.013
187. Luo Y, Gao X, Zou L, Lei M, Feng J, and Hu Z. Bavachin induces ferroptosis through the stat3/P53/slc7a11 axis in osteosarcoma cells. Oxid Med Cell Longev. (2021) 2021:1783485. doi: 10.1155/2021/1783485
188. Wang Y, Xie Y, Li J, Peng ZH, Sheinin Y, Zhou J, et al. Tumor-penetrating nanoparticles for enhanced anticancer activity of combined photodynamic and hypoxia-activated therapy. ACS Nano. (2017) 11:2227–38. doi: 10.1021/acsnano.6b08731
189. Shi Y, Gong M, Deng Z, Liu H, Chang Y, Yang Z, et al. Tirapazamine suppress osteosarcoma cells in part through slc7a11 mediated ferroptosis. Biochem Biophys Res Commun. (2021) 567:118–24. doi: 10.1016/j.bbrc.2021.06.036
190. Rathore R and Van Tine BA. Pathogenesis and current treatment of osteosarcoma: perspectives for future therapies. J Clin Med. (2021) 10:1182. doi: 10.3390/jcm10061182
191. Liu Q and Wang K. The induction of ferroptosis by impairing stat3/nrf2/gpx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol Int. (2019) 43:1245–56. doi: 10.1002/cbin.11121
192. Lv H, Zhen C, Liu J, and Shang P. β-phenethyl isothiocyanate induces cell death in human osteosarcoma through altering iron metabolism, disturbing the redox balance, and activating the mapk signaling pathway. Oxid Med Cell Longev. (2020) 2020:5021983. doi: 10.1155/2020/5021983
193. Soundararajan P and Kim JS. Anti-carcinogenic glucosinolates in cruciferous vegetables and their antagonistic effects on prevention of cancers. Molecules. (2018) 23:2983. doi: 10.3390/molecules23112983
194. He Y, Li W, Hu G, Sun H, and Kong Q. Bioactivities of ef24, a novel curcumin analog: A review. Front Oncol. (2018) 8:614. doi: 10.3389/fonc.2018.00614
195. Wu J, Wu S, Shi L, Zhang S, Ren J, Yao S, et al. Design, synthesis, and evaluation of asymmetric ef24 analogues as potential anti-cancer agents for lung cancer. Eur J Med Chem. (2017) 125:1321–31. doi: 10.1016/j.ejmech.2016.10.027
196. Lin H, Chen X, Zhang C, Yang T, Deng Z, Song Y, et al. Ef24 induces ferroptosis in osteosarcoma cells through hmox1. BioMed Pharmacother. (2021) 136:111202. doi: 10.1016/j.biopha.2020.111202
197. Tang R, Xu J, Zhang B, Liu J, Liang C, Hua J, et al. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J Hematol Oncol. (2020) 13:110. doi: 10.1186/s13045-020-00946-7
198. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme a from cytotoxic lymphocytes cleaves gsdmb to trigger pyroptosis in target cells. Science. (2020) 368:eaaz7548. doi: 10.1126/science.aaz7548
199. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. Cd8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. (2019) 569:270–4. doi: 10.1038/s41586-019-1170-y
200. Han J, Wang C, Yang H, Luo J, Zhang X, and Zhang XA. Novel insights into the links between N6-methyladenosine and regulated cell death in musculoskeletal diseases. Biomolecules. (2024) 14:514. doi: 10.3390/biom14050514
201. Liu T, Liu W, Zhang M, Yu W, Gao F, Li C, et al. Ferrous-supply-regeneration nanoengineering for cancer-cell-specific ferroptosis in combination with imaging-guided photodynamic therapy. ACS Nano. (2018) 12:12181–92. doi: 10.1021/acsnano.8b05860
202. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
203. Wenz C, Faust D, Linz B, Turmann C, Nikolova T, Bertin J, et al. T-buooh induces ferroptosis in human and murine cell lines. Arch Toxicol. (2018) 92:759–75. doi: 10.1007/s00204-017-2066-y
204. Yang WS and Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-ras-harboring cancer cells. Chem Biol. (2008) 15:234–45. doi: 10.1016/j.chembiol.2008.02.010
205. Song Q, Peng S, Sun Z, Heng X, and Zhu X. Temozolomide drives ferroptosis via a dmt1-dependent pathway in glioblastoma cells. Yonsei Med J. (2021) 62:843–9. doi: 10.3349/ymj.2021.62.9.843
206. Radadiya PS, Thornton MM, Puri RV, Yerrathota S, Dinh-Phan J, Magenheimer B, et al. Ciclopirox olamine induces ferritinophagy and reduces cyst burden in polycystic kidney disease. JCI Insight. (2021) 6:e141299. doi: 10.1172/jci.insight.141299
207. Yao X, Zhang Y, Hao J, Duan HQ, Zhao CX, Sun C, et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regener Res. (2019) 14:532–41. doi: 10.4103/1673-5374.245480
208. Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, et al. On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci. (2017) 3:232–43. doi: 10.1021/acscentsci.7b00028
209. Mishima E, Ito J, Wu Z, Nakamura T, Wahida A, Doll S, et al. A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature. (2022) 608:778–83. doi: 10.1038/s41586-022-05022-3
210. Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci U.S.A. (2019) 116:2996–3005. doi: 10.1073/pnas.1819728116
211. Jing Z, Li Y, Zhang H, Chen T, Yu J, Xu X, et al. Tobacco toxins induce osteoporosis through ferroptosis. Redox Biol. (2023) 67:102922. doi: 10.1016/j.redox.2023.102922
212. Deng X, Lin B, Wang F, Xu P, and Wang N. Mangiferin attenuates osteoporosis by inhibiting osteoblastic ferroptosis through keap1/nrf2/slc7a11/gpx4 pathway. Phytomedicine. (2024) 124:155282. doi: 10.1016/j.phymed.2023.155282
213. Ruan B, Dong J, Wei F, Huang Z, Yang B, Zhang L, et al. Dnmt aberration-incurred gpx4 suppression prompts osteoblast ferroptosis and osteoporosis. Bone Res. (2024) 12:68. doi: 10.1038/s41413-024-00365-1
214. Zhang Z, Ji C, Wang YN, Liu S, Wang M, Xu X, et al. Maresin1 suppresses high-glucose-induced ferroptosis in osteoblasts via nrf2 activation in type 2 diabetic osteoporosis. Cells. (2022) 11:2560. doi: 10.3390/cells11162560
215. Xue C, Luo H, Wang L, Deng Q, Kui W, Da W, et al. Aconine attenuates osteoclast-mediated bone resorption and ferroptosis to improve osteoporosis via inhibiting nf-κb signaling. Front Endocrinol (Lausanne). (2023) 14:1234563. doi: 10.3389/fendo.2023.1234563
216. Jiang Z, Wang H, Qi G, Jiang C, Chen K, and Yan Z. Iron overload-induced ferroptosis of osteoblasts inhibits osteogenesis and promotes osteoporosis: an in vitro and in vivo study. IUBMB Life. (2022) 74:1052–69. doi: 10.1002/iub.2656
217. Xu CY, Xu C, Xu YN, Du SQ, Dai ZH, Jin SQ, et al. Poliumoside protects against type 2 diabetes-related osteoporosis by suppressing ferroptosis via activation of the nrf2/gpx4 pathway. Phytomedicine. (2024) 125:155342. doi: 10.1016/j.phymed.2024.155342
218. Wei F, Ruan B, Dong J, Yang B, Zhang G, Kelvin Yeung WK, et al. Asperosaponin vi inhibition of dnmt alleviates gpx4 suppression-mediated osteoblast ferroptosis and diabetic osteoporosis. J Adv Res. (2024) 6:S2090-1232(24)00554-X. doi: 10.1016/j.jare.2024.11.036
219. Jin C, Tan K, Yao Z, Lin BH, Zhang DP, Chen WK, et al. A novel anti-osteoporosis mechanism of vk2: interfering with ferroptosis via ampk/sirt1 pathway in type 2 diabetic osteoporosis. J Agric Food Chem. (2023) 71:2745–61. doi: 10.1021/acs.jafc.2c05632
220. Shi Y, Tang Q, Sheng S, Jiang H, Jin C, Zhou C, et al. Psmd14 stabilizes slc7a11 to ameliorate glucocorticoid-induced osteoporosis by suppressing osteocyte ferroptosis. Adv Sci (Weinh). (2025) 30:e14902. doi: 10.1002/advs.202414902
221. Zheng X, Ye FC, Sun T, Liu FJ, Wu MJ, Zheng WH, et al. Delay the progression of glucocorticoid-induced osteoporosis: fraxin targets ferroptosis via the nrf2/gpx4 pathway. Phytother Res. (2024) 38:5203–24. doi: 10.1002/ptr.8310
222. Liu Y, Fu Z, Wang X, Yang Q, Liu S, and Zhu D. Metformin attenuates diabetic osteoporosis by suppressing ferroptosis via the ampk/nrf2 pathway. Front Pharmacol. (2025) 16:1527316. doi: 10.3389/fphar.2025.1527316
223. Hao J, Bei J, Li Z, Han M, Ma B, Ma P, et al. Qing`E pill inhibits osteoblast ferroptosis via atm serine/threonine kinase (Atm) and the pi3k/akt pathway in primary osteoporosis. Front Pharmacol. (2022) 13:902102. doi: 10.3389/fphar.2022.902102
224. Li P, Wang Y, Yan Q, Yang Y, Zhu R, Ma J, et al. Fructus ligustri lucidi inhibits ferroptosis in ovariectomy−Induced osteoporosis in rats via the nrf2/ho−1 signaling pathway. BioMed Rep. (2024) 20:27. doi: 10.3892/br.2023.1715
225. Zheng Y, Sun R, Yang H, Gu T, Han M, Yu C, et al. Aucubin promotes bmscs proliferation and differentiation of postmenopausal osteoporosis patients by regulating ferroptosis and bmp2 signalling. J Cell Mol Med. (2025) 29:e70288. doi: 10.1111/jcmm.70288
226. Gu S, Wang J, Yu S, Zhang S, Gao T, Yan D, et al. Berberine ameliorates nonalcoholic fatty liver disease-induced bone loss by inhibiting ferroptosis. Bone. (2024) 185:117114. doi: 10.1016/j.bone.2024.117114
227. He Q, Lin Y, Chen B, Chen C, Zeng J, Dou X, et al. Vitamin K2 ameliorates osteoarthritis by suppressing ferroptosis and extracellular matrix degradation through activation gpx4’s dual functions. Biomed. (2024) 175:116697. doi: 10.1016/j.biopha.2024.116697
228. He Q, Yang J, Pan Z, Zhang G, Chen B, Li S, et al. Biochanin a protects against iron overload associated knee osteoarthritis via regulating iron levels and nrf2/system xc-/gpx4 axis. BioMed Pharmacother. (2023) 157:113915. doi: 10.1016/j.biopha.2022.113915
229. Li Y, Zhu X, Wang K, Zhu L, Murray M, and Zhou F. Ginkgo biloba extracts (Gbe) protect human rpe cells from T-bhp-induced oxidative stress and necrosis by activating the nrf2-mediated antioxidant defence. J Pharm Pharmacol. (2023) 75:105–16. doi: 10.1093/jpp/rgac069
230. Wang Q, Qi B, Shi S, Jiang W, Li D, Jiang X, et al. Melatonin alleviates osteoarthritis by regulating nadph oxidase 4-induced ferroptosis and mitigating mitochondrial dysfunction. J Pineal Res. (2024) 76:e12992. doi: 10.1111/jpi.12992
231. Guo Z, Lin J, Sun K, Guo J, Yao X, Wang G, et al. Deferoxamine alleviates osteoarthritis by inhibiting chondrocyte ferroptosis and activating the nrf2 pathway. Front Pharmacol. (2022) 13:791376. doi: 10.3389/fphar.2022.791376
232. Xiao J, Luo C, Li A, Cai F, Wang Y, Pan X, et al. Icariin inhibits chondrocyte ferroptosis and alleviates osteoarthritis by enhancing the slc7a11/gpx4 signaling. Int Immunopharmacol. (2024) 133:112010. doi: 10.1016/j.intimp.2024.112010
233. Guo J, Su K, Wang L, Feng B, You X, Deng M, et al. Poly(P-coumaric acid) nanoparticles alleviate temporomandibular joint osteoarthritis by inhibiting chondrocyte ferroptosis. Bioactive Mater. (2024) 40:212–26. doi: 10.1016/j.bioactmat.2024.06.007
234. Zou Z, Hu W, Kang F, Xu Z, Li Y, Zhang J, et al. Interplay between lipid dysregulation and ferroptosis in chondrocytes and the targeted therapy effect of metformin on osteoarthritis. J Adv Res. (2025) 69:515–29. doi: 10.1016/j.jare.2024.04.012
235. Chen R, Ying C, Zou Y, Lin C, Fu Q, Xiang Z, et al. Sarsasapogenin inhibits yap1-dependent chondrocyte ferroptosis to alleviate osteoarthritis. BioMed Pharmacother. (2023) 168:115772. doi: 10.1016/j.biopha.2023.115772
236. Wang X, Liu Z, Peng P, Gong Z, Huang J, and Peng H. Astaxanthin attenuates osteoarthritis progression via inhibiting ferroptosis and regulating mitochondrial function in chondrocytes. Chem Biol Interact. (2022) 366:110148. doi: 10.1016/j.cbi.2022.110148
237. Ruan H, Zhu T, Wang T, Guo Y, Liu Y, and Zheng J. Quercetin modulates ferroptosis via the sirt1/nrf-2/ho-1 pathway and attenuates cartilage destruction in an osteoarthritis rat model. Int J Mol Sci. (2024) 25:7461. doi: 10.3390/ijms25137461
238. Wang J, Yang J, Fang Y, Lou C, Yu H, Li Y, et al. Vinpocetine protects against osteoarthritis by inhibiting ferroptosis and extracellular matrix degradation via activation of the nrf2/gpx4 pathway. Phytomedicine. (2024) 135:156115. doi: 10.1016/j.phymed.2024.156115
239. Zhou Y, Jia Z, Wang J, Huang S, Yang S, Xiao S, et al. Curcumin reverses erastin-induced chondrocyte ferroptosis by upregulating nrf2. Heliyon. (2023) 9:e20163. doi: 10.1016/j.heliyon.2023.e20163
240. Cui T, Lan Y, Yu F, Lin S, and Qiu J. Plumbagin alleviates temporomandibular joint osteoarthritis progression by inhibiting chondrocyte ferroptosis via the mapk signaling pathways. Aging (Albany NY). (2023) 15:13452–70. doi: 10.18632/aging.205253
241. Pan X, Kong X, Feng Z, Jin Z, Wang M, Lu H, et al. 4-octyl itaconate protects chondrocytes against il-1β-induced oxidative stress and ferroptosis by inhibiting gpx4 methylation in osteoarthritis. Int Immunopharmacol. (2024) 137:112531. doi: 10.1016/j.intimp.2024.112531
242. Cao S, Wei Y, Yue Y, Chen Y, Qian J, Wang D, et al. Rosiglitazone retards the progression of iron overload-induced osteoarthritis by impeding chondrocyte ferroptosis. iScience. (2024) 27:110526. doi: 10.1016/j.isci.2024.110526
243. Zhang G, Yu J, and Wan Y. Usp48 deubiquitination stabilizes slc1a5 to inhibit retinal pigment epithelium cell inflammation, oxidative stress and ferroptosis in the progression of diabetic retinopathy. J Bioenerget Biomembranes. (2024) 56:311–21. doi: 10.1007/s10863-024-10008-z
244. Cao S, Wei Y, Xiong A, Yue Y, Yang J, Wang D, et al. Paeonol inhibits acsl4 to protect chondrocytes from ferroptosis and ameliorates osteoarthritis progression. J Orthop Translat. (2025) 50:1–13. doi: 10.1016/j.jot.2024.10.005
245. Yang X, Chen Y, Guo J, Li J, Zhang P, Yang H, et al. Polydopamine nanoparticles targeting ferroptosis mitigate intervertebral disc degeneration via reactive oxygen species depletion, iron ions chelation, and gpx4 ubiquitination suppression. Adv Sci (Weinh). (2023) 10:e2207216. doi: 10.1002/advs.202207216
246. Zhang P, Rong K, Guo J, Cui L, Kong K, Zhao C, et al. Cynarin alleviates intervertebral disc degeneration via protecting nucleus pulposus cells from ferroptosis. BioMed Pharmacother. (2023) 165:115252. doi: 10.1016/j.biopha.2023.115252
247. Zhang Y, Li H, Chen Y, Li C, Ye H, Qiu J, et al. Nordihydroguaiaretic acid suppresses ferroptosis and mitigates intervertebral disc degeneration through the nrf2/gpx4 axis. Int Immunopharmacol. (2024) 143:113590. doi: 10.1016/j.intimp.2024.113590
248. Xi X, Chen Q, Ma J, Wang X, Zhang J, and Li Y. Sestrin2 ameliorates diabetic retinopathy by regulating autophagy and ferroptosis. J Mol Histol. (2024) 55:169–84. doi: 10.1007/s10735-023-10180-3
249. Dou X, Ma Y, Luo Q, Song C, Liu M, Liu X, et al. Therapeutic potential of melatonin in the intervertebral disc degeneration through inhibiting the ferroptosis of nucleus pulpous cells. J Cell Mol Med. (2023) 27:2340–53. doi: 10.1111/jcmm.17818
250. Yang S, Zhu Y, Shi Y, Su S, Liang H, Li S, et al. Screening of nsaids library identifies tinoridine as a novel ferroptosis inhibitor for potential intervertebral disc degeneration therapy. Free Radic Biol Med. (2024) 221:245–56. doi: 10.1016/j.freeradbiomed.2024.05.040
251. Liu X, Pan F, Sha C, Wang Z, Liu G, Wang H, et al. Fuzi decoction ameliorates intervertebral disc degeneration through ferroptosis modulation by suppressing nf-κb pathway. Int Immunopharmacol. (2025) 148:114155. doi: 10.1016/j.intimp.2025.114155
252. Zhu Z, Huang Z, Zhang C, Xu B, Chen H, Pei S, et al. Gallic acid protects intervertebral disc cells from ferroptosis and alleviates intervertebral disc degeneration by regulating key factors of oxidative stress. Front Pharmacol. (2025) 16:1501725. doi: 10.3389/fphar.2025.1501725
253. Niu H, Qi H, Zhang P, Meng H, Liu N, and Zhang D. Single-cell analysis reveals aspirin restores intervertebral disc integrity via ferroptosis regulation. J Inflammation Res. (2025) 18:6889–905. doi: 10.2147/jir.S519218
254. Zhu J, Sun R, Yan C, Sun K, Gao L, Zheng B, et al. Hesperidin mitigates oxidative stress-induced ferroptosis in nucleus pulposus cells via nrf2/nf-κb axis to protect intervertebral disc from degeneration. Cell Cycle. (2023) 22:1196–214. doi: 10.1080/15384101.2023.2200291
255. Li Z, Cheng P, Xi H, Jiang T, Zheng X, Qiu J, et al. Tomatidine alleviates intervertebral disc degeneration by activating the nrf2/ho-1/gpx4 signaling pathway. Drug Des Devel Ther. (2024) 18:6313–29. doi: 10.2147/dddt.S481714
256. Wen RJ, Dong X, Zhuang HW, Pang FX, Ding SC, Li N, et al. Baicalin induces ferroptosis in osteosarcomas through a novel nrf2/xct/gpx4 regulatory axis. Phytomedicine. (2023) 116:154881. doi: 10.1016/j.phymed.2023.154881
257. Nie J, Ling Y, Jin M, Chen Z, Liu W, Shen W, et al. Butyrate enhances erastin-induced ferroptosis of osteosarcoma cells via regulating atf3/slc7a11 pathway. Eur J Pharmacol. (2023) 957:176009. doi: 10.1016/j.ejphar.2023.176009
258. Zou Q, Zhou X, Lai J, Zhou H, Su J, Zhang Z, et al. Targeting P62 by sulforaphane promotes autolysosomal degradation of slc7a11, inducing ferroptosis for osteosarcoma treatment. Redox Biol. (2025) 79:103460. doi: 10.1016/j.redox.2024.103460
259. Jiwa H, Xie Z, Qu X, Xu J, Huang Y, Huang X, et al. Casticin induces ferroptosis in human osteosarcoma cells through fe(2+) overload and ros production mediated by hmox1 and lc3-ncoa4. Biochem Pharmacol. (2024) 226:116346. doi: 10.1016/j.bcp.2024.116346
260. Jiacong H, Qirui Y, Haonan L, Yichang S, Yan C, and Keng C. Zoledronic acid induces ferroptosis by upregulating por in osteosarcoma. Med Oncol. (2023) 40:141. doi: 10.1007/s12032-023-01988-w
261. Zeng X, Lin GX, Zeng X, Zheng J, Ren C, Luo Z, et al. Penfluridol regulates P62/keap1/nrf2 signaling pathway to induce ferroptosis in osteosarcoma cells. BioMed Pharmacother. (2024) 177:117094. doi: 10.1016/j.biopha.2024.117094
262. Huang R, Xu R, Shi J, Yang Z, Zheng J, and Wei D. Artesunate induces ferroptosis in osteosarcoma through ncoa4-mediated ferritinophagy. FASEB J. (2025) 39:e70488. doi: 10.1096/fj.202403160R
263. Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. Caf secreted mir-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. (2020) 19:43. doi: 10.1186/s12943-020-01168-8
264. Liu P, Wang W, Li Z, Li Y, Yu X, Tu J, et al. Ferroptosis: A new regulatory mechanism in osteoporosis. Oxid Med Cell Longev. (2022) 2022:2634431. doi: 10.1155/2022/2634431
265. Chen H, Han Z, Wang Y, Su J, Lin Y, Cheng X, et al. Targeting ferroptosis in bone-related diseases: facts and perspectives. J Inflammation Res. (2023) 16:4661–77. doi: 10.2147/jir.S432111
266. Hu Y, Wang Y, Liu S, and Wang H. The potential roles of ferroptosis in pathophysiology and treatment of musculoskeletal diseases-opportunities, challenges, and perspectives. J Clin Med. (2023) 12:2125. doi: 10.3390/jcm12062125
267. Wang Y, Yu G, and Chen X. Mechanism of ferroptosis resistance in cancer cells. Cancer Drug Resist. (2024) 7:47. doi: 10.20517/cdr.2024.127
Keywords: ferroptosis, bone microenvironment, bone and joint diseases, mechanisms, therapeutic prospects
Citation: Xiao R, Han Z, Jia P, Li P, Gong M, Cai Y, Pang L, Ye X and Jin S (2025) Ferroptosis and bone health: bridging the gap between mechanisms and therapy. Front. Immunol. 16:1634516. doi: 10.3389/fimmu.2025.1634516
Received: 24 May 2025; Accepted: 30 June 2025;
Published: 16 July 2025.
Edited by:
Qi Feng, First Affiliated Hospital of Zhengzhou University, ChinaReviewed by:
Yufeng Yao, Huazhong University of Science and Technology, ChinaYangYang, First Affiliated Hospital of Zhengzhou University, China
Copyright © 2025 Xiao, Han, Jia, Li, Gong, Cai, Pang, Ye and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Song Jin, MTA0OTE0NzAwMEBxcS5jb20=
†These authors have contributed equally to this work