 Xuege Guo
Xuege Guo Hanlu Zhang1
Hanlu Zhang1- 1Department of Hematology, The Second Hospital and Clinical Medical School, Lanzhou University, Lanzhou, China
- 2Gansu Provincial Hematology Clinical Medical Research Center (National Branch), The Second Hospital of Lanzhou University, Lanzhou, China
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy driven by diverse genetic mutations that shape tumor progression, immune evasion, and clinical outcomes. While molecular profiling has improved AML classification, the precise impact of specific mutations on immune cell infiltration and dysregulation remains insufficiently understood. This review examines the immunologic consequences of common AML mutations—including FLT3-ITD, NPM1, DNMT3A, TP53, IDH1/2, and NRAS—and their role in remodeling the immune microenvironment. We further explore the dynamic shifts in immune responses across different AML risk stratifications, emphasizing the balance between immune activation and suppression, which is influenced by specific genetic alterations. Additionally, we highlight the emerging potential of immunotherapies targeting neoepitopes derived from driver mutations, offering promising avenues to overcome immune escape and enhance anti-tumor immune responses. By integrating genetic mutations and immunologic insights, this review outlines a framework for developing more precise and effective immunotherapies for AML.
1 Introduction
Acute myeloid leukemia (AML) is a highly aggressive hematologic malignancy that accounts for approximately 80% of all acute leukemias in adults (1). The pathogenesis of AML is complex, involving a combination of genetic mutations, chromosomal abnormalities, and environmental factors, all of which lead to abnormal hematopoietic stem cell development. These aberrant cells proliferate uncontrollably, progressively replacing normal hematopoietic tissue and impairing the production of healthy blood cells. This disruption manifests clinically as anemia, bleeding, and increased susceptibility to infections (2). In 1976, the French-American-British (FAB) Cooperative Group, consisting of hematology experts from France, the United States, and the United Kingdom, first proposed diagnostic and classification criteria for AML (FAB classification), dividing AML into eight subtypes (M0 to M7). Accurate classification of leukemia is essential for selecting appropriate treatment strategies. However, the FAB classification, which is based primarily on the morphology, differentiation status, and chemical staining of bone marrow (BM) leukemia cells, has become increasingly insufficient to meet the demands of modern clinical diagnosis and treatments. In response, the World Health Organization incorporated molecular genetic, molecular biology, and immunological characteristics into the classification of AML for the first time in its 2001 “Classification of Hematopoietic and Lymphoid Tumors”. This revision aimed to provide a more biologically relevant classification system and was updated in 2008 and 2016 based on new research findings. Unlike the morphology-based FAB classification, the World Health Organization classification places greater emphasis on the role of genetic mutations and chromosomal abnormalities in AML classification, particularly in the context of prognosis and risk stratification (3). Today, the cytogenetic and molecular characteristics of AML are not only fundamental to accurate disease classification but also crucial for guiding clinical decision-making. Through the detection of specific genetic mutations and chromosomal abnormalities, clinicians can more precisely predict outcomes such as the complete remission (CR) rate, disease-free survival, relapse risk, and overall survival (OS), enabling the development of personalized treatment regimens tailored to individual patients (2).
In the initiation and progression of AML, the tumor microenvironment (TME) is no longer a passive “bystander”. It functions both as an “inhibitor”, slowing leukemogenesis by impeding the proliferation of malignant cells, and as a potent “catalyst”, playing a critical role in sustaining and promoting leukemia development. The TME is a complex network composed of immune cells, cytokines, extracellular matrix components, and other immune-regulatory molecules, with its dynamic alterations directly influencing tumor progression and therapeutic responses (4). Recent studies have highlighted the crucial role of genetic mutations in AML in shaping and regulating the immune microenvironment. These mutations impact immune cell infiltration patterns and immune function via various pathways, exhibiting significant heterogeneity. For instance, mutations associated with favorable prognosis, such as NPM1 mutations, are typically linked to immune activation and enhanced anti-tumor responses (5), while mutations associated with poor prognosis, such as TP53 mutations, often lead to immune suppression or escape, impairing immune surveillance (6). Importantly, the AML immune microenvironment shows both immune activation and suppression at the same time. Immune cells may become dysfunctional in suppressive conditions, but can also be reactivated by signals from the tumor. This balance between immune response and escape reflects the complexity of AML and varies with different genetic mutations.
Although significant progress has been made in genetic research on AML, which has gradually been incorporated into risk stratification systems, a comprehensive understanding of how gene mutations influence immune cell infiltration, the expression of immune regulatory molecules, and immune evasion mechanisms remains lacking. This review aims to explore how common genetic mutations in AML shape the immune microenvironment through various mechanisms and how these alterations impact patient prognosis.
2 AML tumor cell remodeling of the immune microenvironment
The immune system in AML patients exhibits significant heterogeneity and dysfunction, with both the innate and adaptive immune systems being suppressed and dysregulated. In the context of the innate immune system, AML patients show a marked reduction in the number of natural killer (NK) cells, particularly in the CD56dimCD16+ functional subset (7). The imbalance between immature and overmature NK cell subpopulations varies significantly among individuals (8, 9), which may be linked to specific genetic mutations. Furthermore, tumor cells further impair NK cell function by downregulating the activating ligand HLA-E, secreting soluble ligands such as MICA/B (10), and upregulating inhibitory receptors like TIM-3, KIR, and CD159a (8–11). As the number of regulatory T cells (Tregs) increases, NK cell dysfunction becomes more pronounced. Despite the widespread expression of ligands for the NK cell-activating receptor NKG2D on AML cells (12), NK cell responses to cytokine stimulation remain diminished, as evidenced by significantly reduced expression of granzyme B and IFN-γ (13). Additionally, macrophages in AML patients undergo a phenotypic shift from the anti-tumor M1 type to the immunosuppressive M2 type. M2 macrophages further promote the immunosuppressive environment through the high expression of inhibitory receptors (14). Regarding dendritic cells (DCs), although the overall number of DCs is increased, the conventional dendritic cell type 1 (cDC1) subset is significantly reduced (15, 16), indicating dysfunction within this population.
There are significant individual differences in lymphocyte counts among AML patients. In some cases, the lymphocyte count is approximately five times higher than normal, while in others, it remains within the normal range (1). Furthermore, lymphocyte distribution shows heterogeneity, with a slightly lower proportion in the BM and a slight increase in the peripheral blood (PB), though there are no significant changes in relative proportions (15, 17). T cell function is significantly impaired in AML patients. In general, T cells exhibit reduced proliferative capacity (18–21), increased apoptosis (19, 22), diminished expression of costimulatory molecules (19), and upregulated expression of inhibitory receptors (15, 17, 19, 23–25) often with increased co-expression of these receptors (19, 26, 27). These changes lead to a decrease in the secretion of pro-inflammatory cytokines, such as IFN-γ, TNF-α, and IL-2 (21), thus weakening the anti-tumor immune response. Although studies have shown that the number of CD8+ T cells in the BM is elevated (19, 24), and that these cells are predominantly effector memory T cells (Tem) (17), their functionality remains compromised. In the PB, there is an increase in the proportion of terminally differentiated effector cells, while the proportion of naive T cells (Tn) decreases (19, 21, 28). However, in a study by Oscar Brück, it was found that compared to healthy individuals, T cells in the BM of AML patients exhibit high expression of PD-1 and low expression of LAG-3 and TIM-3 (20). This suggests that the immunological characteristics of T cells in AML may be influenced by multiple factors. Additionally, T cells in the BM of AML patients show impaired immune synapse formation, with reduced F-actin polymerization and insufficient recruitment of signaling molecules (19). This may be related to the dysfunction of AML cells as antigen-presenting cells (18). Moreover, AML patients have reduced T helper 1 (Th1) cells and decreased IFN-γ secretion (21), while T helper 17 (Th17) cells are increased and secrete IL-17, promoting AML cell proliferation and inhibiting Th1 differentiation (16, 21). There is also an increase in CD4+ T cells in the BM expressing PD-1+/OX40+, ICOS+ (26), and TIM-3+ (29). Although these CD4+ T cells are partially activated, their function remains relatively weak. In certain patients, there is a notably higher frequency of double-positive T cell subsets in the BM (26). Additionally, the increased number of Tregs suppresses the anti-leukemic function of effector T cells (Teffs) (1, 24), and removing Tregs can partially restore T cell functionality (21). Unlike typical NKT cells, CD3+CD56+ T cells in AML patients exhibit significantly reduced cytotoxic potential (18). The BM of AML patients contains atypical B cells (30), although their exact role remains unclear. Overall, the immune microenvironment in AML patients is characterized by immune suppression, which hinders anti-tumor immune responses and promotes tumor immune escape and disease progression.
3 Common gene mutations in AML patients and the immune microenvironment
In different patient populations, the immunogenicity of AML cells and the quality of the immune response are shaped by specific oncogenic driver mutations. Even in the presence of the same mutations, variations in co-mutations or other genetic background differences can lead to distinct pathways of AML progression, resulting in differing prognoses. Although AML is typically characterized by a low mutation burden, certain high-frequency driver mutations, such as FLT3-ITD and NPM1 mutations, can generate immunogenic peptides that act as tumor-specific antigens, triggering targeted immune responses (31, 32). Therefore, understanding the impact of these genetic mutations on the immune microenvironment is essential for the development of effective immunotherapy strategies for AML.
3.1 FLT3-ITD mutation
FLT3 is a receptor tyrosine kinase predominantly expressed in DCs (33). The FLT3 signaling pathway regulates the differentiation and mobilization of precursor DCs, as well as the homeostatic division of cDCs in peripheral lymph nodes (29). In patients with AML, approximately 20-25% harbor FLT3-ITD mutations, while 5-7% have mutations in the FLT3-TKD (34). These mutations lead to constitutive activation of the FLT3 receptor, resulting in enhanced cell proliferation and inhibition of apoptosis (10).
3.1.1 T cell dysfunction and immune escape in FLT3-ITD mutant AML
T cells can specifically recognize FLT3-ITD-mutated AML cells and induce cell lysis by secreting IFN-γ, granzyme B, and perforin (16). In patients with FLT3-ITD mutations, the proportion of CD3+ T cells (1), including both CD4+ and CD8+ subsets, is significantly increased (35), which contrasts with the significantly reduced percentages of B cells, plasmablasts (1, 36), and NKT cells (37). However, despite the increase in T cells, the anti-leukemia immune response is impaired due to multiple immune escape mechanisms, such as the predominant expansion of Tregs among CD4+ T cells. Studies have shown that Tregs are significantly enriched in the BM and spleen of FLT3-ITD-mutant AML mice (16), suggesting that the increase in CD4+ T lymphocytes is primarily driven by the expansion of Tregs.
In addition, immune evasion is also achieved through the following mechanisms: upregulation of co-expressed immunosuppressive molecules on CD8+ T cells (38), and the elevated expression of immune checkpoint receptors like TIM-3 and LAG-3 (29). Although the proportion of CD8+ T cells is increased in these patients, they typically exhibit a TIGIT+PD-1+DNAM-1− phenotype (38). The co-expression of these immunosuppressive molecules is associated with poorer prognosis (39). However, although no significant differences in the expression of TIGIT and PD-1 were observed between the FLT3-ITD mutant and wild-type groups when analyzed in the overall T cell population in some samples (15), it is important to note that the immunosuppressive effects are primarily determined by the expression of immune checkpoint receptors on T cells that specifically recognize leukemia antigens (40). Additionally, following mutation, the FLT3 receptor remains aberrantly activated, and in combination with the effect of FLT3 ligand, this leads to elevated expression of TIM-3 in T cells (29). The autocrine or paracrine signaling pathways of TIM-3 promote leukemia cell proliferation and anti-apoptotic activity, while also suppressing the function of distant immune cells (29). TIM-3 expression is accompanied by galectin-9 secretion, which inhibits T cell activity. TIM-3 transcript levels correlate with CLIP levels (29), suggesting that immune evasion mechanisms are often co-activated. Moreover, In the context of FLT3-ITD mutation, the expression of the immune checkpoint LAG-3 is significantly increased in T cell subsets (27). LAG-3 impairs T cell receptor (TCR)-mediated signaling, thereby affecting the proliferation and function of Teffs cells. High LAG-3 expression is associated with shorter OS and disease-free survival in AML patients (27, 39) (Figure 1).
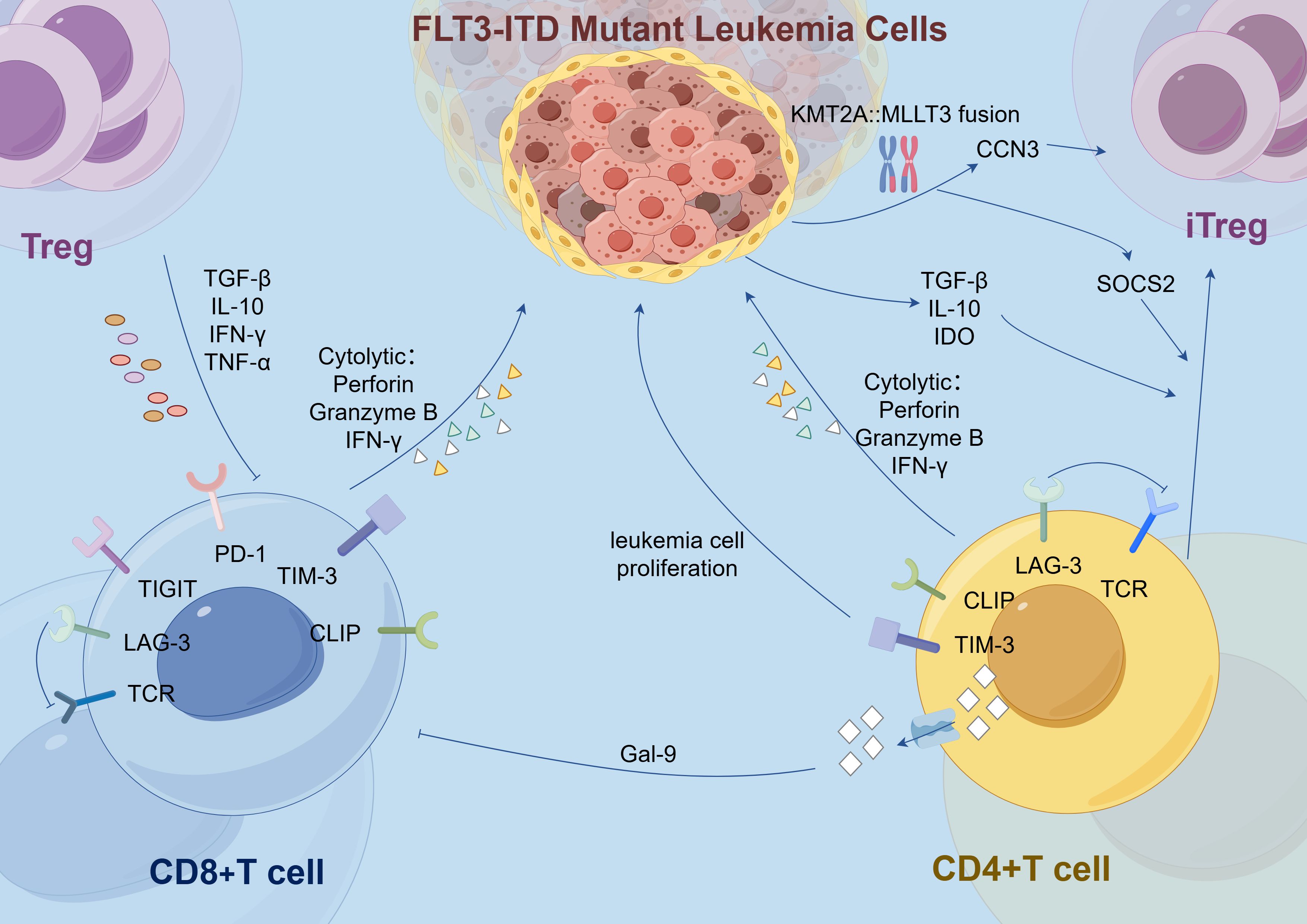
Figure 1. In the FLT3-ITD-mutated AML microenvironment, elevated T cell subsets, including CD8+ and CD4+ T cells, are suppressed by overexpression of immune checkpoints (e.g., PD-1, TIGIT, LAG-3, TIM-3) and their ligands (e.g., Gal-9). Additionally, in cells harboring both FLT3-ITD mutations and the KMT2A::MLLT3 fusion, activated CCN3 enhances Treg functionality, while SOCS2 promotes the polarization of iTregs from CD4+ T cells. FLT3-ITD, Fms-like tyrosine kinase 3 internal tandem duplication; Treg, Regulatory T cell; iTreg, Inducible Regulatory T cells; CD8+ T Cell, Cluster of Differentiation 8 Positive T Cell; CD4+ T Cell, Cluster of Differentiation 4 Positive T Cell; TGF: Transforming Growth Factor; IL: Interleukin; IFN, Interferon; TNF: Tumor Necrosis Factor; TCR, T Cell Receptor; LAG, Lymphocyte activation gene; TIGIT, T cell immunoreceptor with Ig and ITIM domains; PD-1, Programmed cell death protein 1; TIM, T-cell immunoglobulin and mucin-domain containing; CLIP, Class II-associated invariant chain peptide; Gal-9, Galectin-9; IDO, Indoleamine 2,3-dioxygenase; CCN3, Cysteine-rich angiogenic protein 3; SOCS2, Suppressor of Cytokine Signaling 2.
3.1.2 DC expansion and dysfunction in FLT3-ITD mutant AML
Compared to wild-type AML patients, those with FLT3-ITD mutations exhibit a significant expansion of DCs, particularly common DC progenitors and precursor DCs. In mouse models, the effect of FLT3-ITD on DCs is allele dose-dependent; the more copies of the mutation present, the greater the expansion of DCs. This expansion promotes the proliferation of Tregs, a phenomenon that becomes especially pronounced in the BM as the mutation burden increases (36, 41). Concurrently, DCs undergo abnormal phenotypic changes. The frequency of XCR1/cDC1 double-negative cDCs is markedly elevated, and these cells display impaired antigen presentation capabilities (16). In FLT3-ITD mutant patients, CLIP on the surface of cDCs remains bound to and exposed on HLA molecules. CLIP is an invariant chain polypeptide essential for HLA class II antigen presentation and can also be cross-presented on HLA class I molecules. Persistent exposure to CLIP disrupts T cell activation and is associated with poorer prognosis (29). Additionally, compared to healthy mice, the cDC phenotype in FLT3-ITD mutant mice is skewed toward T-bet-expressing cDC2. Under the influence of specific cytokines, these cDC2 cells effectively polarize naive CD4+ T cells into Th17 cells, leading to increased production of IL-17A. This Th17 subpopulation has been linked to unfavorable prognosis in AML (16) (Figure 2). Meanwhile, the immune evasion mechanisms are further complicated by alterations in TNF secretion by macrophages. The secretion of TNF by macrophages is decreased in patients with FLT3-ITD mutations (42), but TNF exerts a dual effect on tumor cells. Under normal conditions, TNF is involved in regulating T lymphocyte-mediated homeostasis and anti-tumor responses, thereby improving CR rates and extending event-free survival. However, studies have shown that lower levels of TNF may promote the death of tumor-infiltrating T cells, enhance tumor cell differentiation, and facilitate the migration of myeloid cells, thereby accelerating leukemia progression (1). Therefore, the role of TNF in AML is complex, and changes in its levels may have varying effects on disease progression.
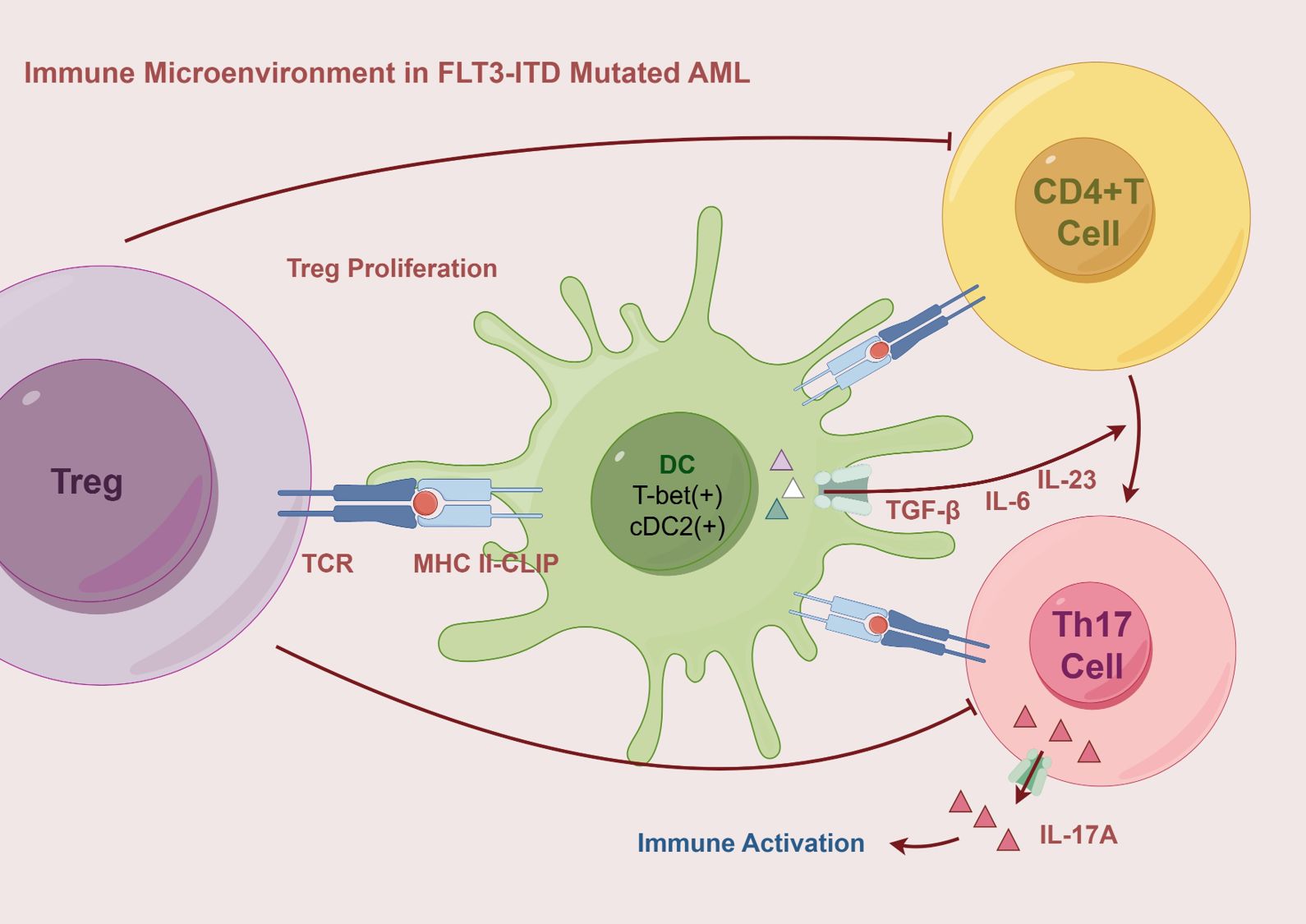
Figure 2. The immune microenvironment in FLT3-ITD mutated AML is characterized by the expansion of DCs, particularly the cDC2 subset, which exhibits a T-bet-positive phenotype and persistent CLIP exposure, leading to impaired antigen presentation. Through HLA II-CLIP, DCs promote Treg proliferation and secrete TGF-β, IL-6, and IL-23, driving the polarization of CD4+ T cells into Th17 cells. These Th17 cells produce IL-17A, which contributes to immune activation and inflammatory responses. AML, Acute Myeloid Leukemia; MHC, Major Histocompatibility Complex Class; T-bet, T-box transcription factor expressed in T cells; DC, Dendritic cell; cDC2, Conventional Dendritic Cell Subtype 2; Th17, T-helper 17 Cell.
3.1.3 Macrophage polarization and immune modulation in FLT3-ITD mutant AML
The frequency of TIGIT+ M2 macrophages is elevated in AML patients with the FLT3-ITD mutation. The high infiltration and expression of TIGIT in M2 macrophages are significantly associated with poor prognosis in AML (14). In AML cells harboring both FLT3-ITD mutations and the KMT2A::MLLT3 fusion, the genes CCN3 and SOCS2 become activated (43). Activation of CCN3 recruits macrophages and promotes their differentiation into the M2 phenotype, downregulates the expression of CD36 and SRA1, and consequently reduces phagocytic function (44). Additionally, as a target gene of FoxO1, CCN3 activation enhances the functionality of Tregs (45). SOCS2 plays a multifaceted role by not only inhibiting the expression of pro-inflammatory cytokines and the development of T helper 2 (Th2) cells in DCs but also promoting the polarization of CD4+ T cells into inducible regulatory T cells (iTregs) (46) (Figure 1). It maintains the stable expression of Foxp3 in iTregs by inhibiting the IL-4 signaling pathway, thereby sustaining the anti-inflammatory phenotype and cellular stability of iTregs (47). Moreover, excessive activation of SOCS2 leads to the inhibition of IL-8 secretion and upregulation of RANTES expression, both of which are associated with poor prognosis (48). These findings align with observations in AML, where elevated levels of SOCS2 correlate with reduced OS (43).
3.1.4 Altered NK cell proportions and function in FLT3-ITD mutant AML
In addition, the proportion of NK cells in AML patients with FLT3-ITD mutations was significantly elevated, and the copy number, ITD length, and mutant allele frequency of FLT3-ITD mutations were positively correlated with the proportion of NK cells. However, patients with a high proportion of NK cells at the initial stage of AML tend to have a poor prognosis. This may be due to a reduced number of mature NK cells and their limited cytotoxic function (37). Furthermore, the expression of inhibitory receptors is generally increased across all NK cell subsets, although it exhibits heterogeneity (8). In a study by Cianga’s team analyzing BM samples from eight AML patients, the proportion of overmature NK cells in patients with FLT3 mutations was significantly increased (9). Conversely, in another study by the same team examining PB from 20 newly diagnosed AML patients, those with FLT3 mutations exhibited an extremely low proportion of NK cells and markedly abnormal expression levels of the inhibitory receptor CD159a (8). These discrepancies may be attributed to differences in patient cohorts, variations between the BM and PB environments, and the regulatory influence of the TME on NK cell development.
3.1.5 Dual role of MAIT cells in tumor surveillance and immune evasion in FLT3-ITD mutant AML
AML patients with FLT3-ITD mutations have increased numbers of mucosal-associated invariant T (MAIT) cells, which predominantly exhibit effector memory or terminally differentiated phenotypes (35), indicating high activation but also signs of aging and exhaustion, characterized by upregulated PD-1 and downregulated CD161 expression (49). Moreover, MAIT cell function is compromised, with reduced Th1-type cytokine production (IFN-γ and TNF-α) and increased secretion of Th17-type cytokines (IL-17A and IL-8), granzyme B, and perforin (49). We speculate that in AML patients, despite reduced Th1-type cytokine production, MAIT cells may exert anti-tumor effects primarily through granzyme B and perforin-mediated degranulation and cytokine activation, as indicated by the increased secretion of Th17-type cytokines and cytotoxic molecules. These findings suggest that MAIT cells have a dual role in AML, functioning both as anti-tumor agents and potentially as tumor promoters. However, the changes in PD-1 and CD161 expression and the secretion of immune effector molecules in the context of FLT3-ITD mutations require further investigation.
3.2 NPM1 mutation
NPM1 mutations occur in approximately 20% to 30% of adult AML patients. Over 80% of these mutations are type A, characterized by a frameshift insertion at the fourth nucleotide position. This mutation alters the last 11 amino acids at the C-terminus of the NPM1 protein, resulting in its abnormal retention in the cytoplasm, referred to as NPM1c (50–52).
3.2.1 NPM1 mutation-specific T cell responses in AML
The abnormal cytoplasmic localization of NPM1c leads to the generation of novel neoepitopes, including AIQDLCLAV (AIQ) (5) and CLAVEEVSL (CLA) (53), which can be recognized by specific TCRs. The AIQ epitope, presented by HLA-A2, can bind to specific TCRs, and T cells engineered to express these TCRs effectively kill NPM1c+HLA-A2+ AML cells. AML patients with NPM1c+ who exhibit AIQ-specific CD8+ T cell responses have significantly longer survival (5). In contrast, the CLA epitope does not elicit a significant T cell response (53), suggesting that CLA may be suppressed by the TME in vivo. NPM1 mutation-specific CD8+ T cells can directly lyse leukemia cells harboring NPM1 mutations, whereas CD4+ T cells support CD8+ T cell function and induce HLA class II-mediated anti-tumor cytotoxic responses. These specific T cells predominantly express CD107a (54), indicating their activated state and involvement in cytotoxic activity (Figure 3). Notably, specific T cell responses can still be detected after morphological CR in AML patients. This suggests that even when leukemic cells are substantially reduced, these T cells continue to eliminate minimal residual disease (MRD), thereby maintaining long-term CR and reducing relapse risk. A decrease in specific T cells is associated with disease relapse, and patients exhibiting these T cell responses have longer OS (54), further indicating a strong correlation with better prognosis. However, it remains to be investigated whether the frequency and intensity of NPM1 mutation-specific T cell responses vary based on patients’ molecular characteristics.
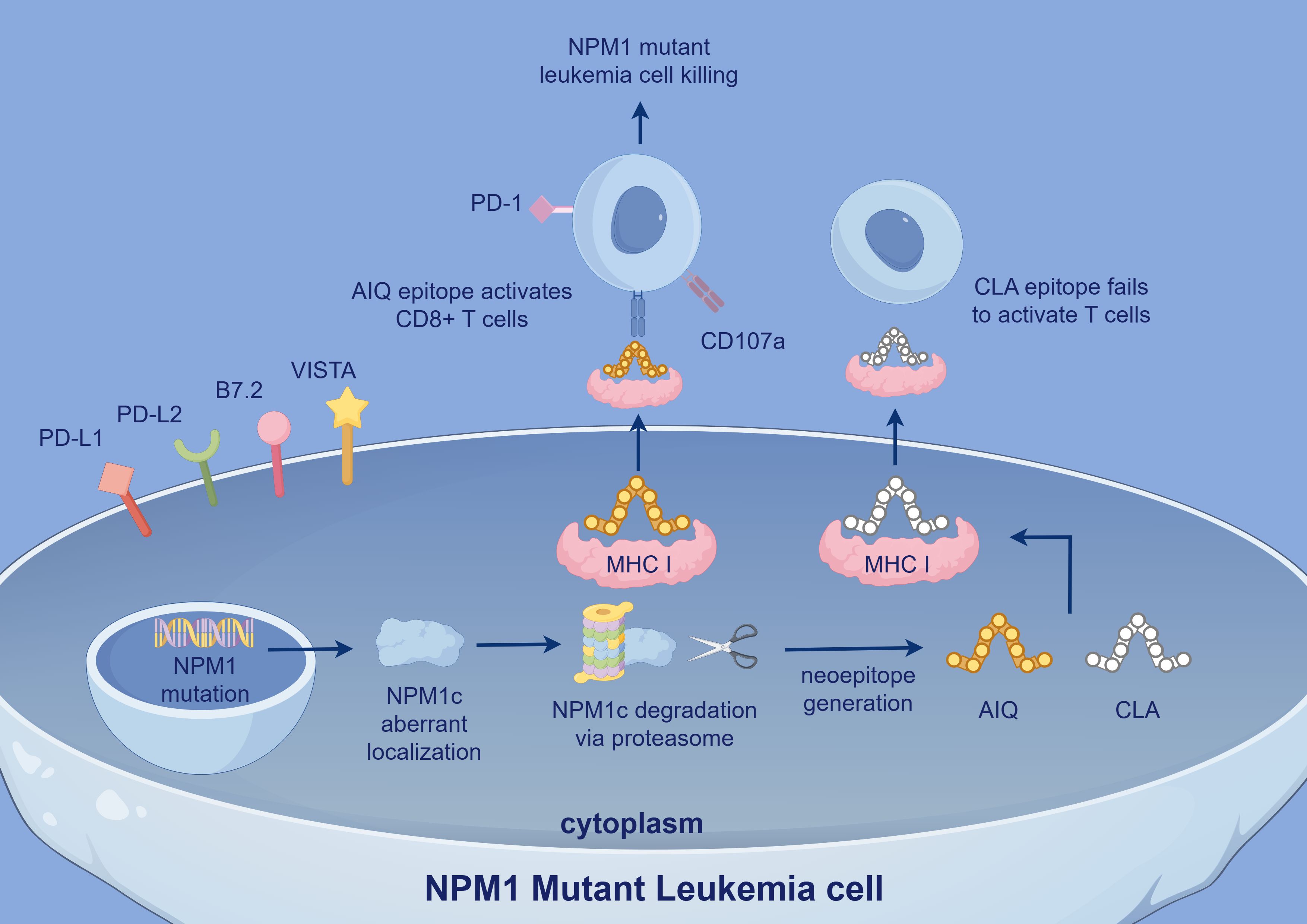
Figure 3. In NPM1 mutant leukemia cells, aberrant NPM1c localization leads to the generation of neoepitopes, including AIQ and CLA. The AIQ epitope activates CD8+ T cells via MHC I presentation and induces leukemia cell killing, with CD107a expression indicating T cell activation. In contrast, the CLA epitope fails to activate T cells. Immune checkpoint molecules such as PD-1, VISTA, and PD-L1 also contribute to immune evasion. NPM1, Nucleophosmin 1; NPM1c, Cytoplasmic Nucleophosmin 1 (mutated form of NPM1 abnormally localized to cytoplasm); PD-L2: Programmed Death-Ligand 2; VISTA, V-domain Ig Suppressor of T cell Activation; AIQ, AIQDLCLAV; CLA, CLAVEEVSL.
3.2.2 Immune checkpoint regulation and inhibitory ligands in NPM1-mutated AML
AML cells can still evade immune surveillance through multiple mechanisms. In particular, under the influence of neoantigenic epitopes produced by NPM1 mutations, leukemic cells predominantly express inhibitory B7 family ligands (such as PD-L1, PD-L2, B7.2, and VISTA) (51, 55). Elevated PD-L1 expression significantly enhances the immunosuppression of NPM1 mutation-specific CD8+ T cells (56). This high level of PD-L1 is strongly associated with poorer patient outcomes, in part due to the expansion of Tregs (57), especially in patients with concurrent FLT3-ITD mutations (51, 54). Although some studies suggest that NPM1 is essential for PD-L1 expression and that NPM1 mutations may slightly reduce PD-L1 expression, these differences are not statistically significant (58), and the underlying mechanisms remain to be elucidated. Furthermore, In patients with NPM1-mutated AML, TIM-3 transcript levels are also significantly reduced (58). TIM-3 plays a complex role in immune function, correlating with T cell exhaustion while also enhancing NK cell cytotoxicity. Some studies indicate that low TIM-3 expression may be associated with favorable prognosis in NPM1-mutated AML, whereas higher TIM-3 expression is linked to significantly lower CR and survival rates at one-year follow-up (58). These findings suggest that TIM-3 may serve as a potential indicator of poor prognosis. Notably, patients with a greater number of TIM-3+ NK cells exhibit better prognoses (59), emphasizing the dual nature of TIM-3 in AML. Overexpression of the pro-inflammatory mediator LTB4R is positively correlated with the expression of inhibitory immune checkpoint molecules such as PD-1 and TIM-3, while showing a negative correlation with immune effector cell populations (60). Although the mechanistic link remains unclear, this suggests that LTB4R may contribute to the immunosuppressive landscape in NPM1-mutated AML. In addition, NPM1 mutations have been shown to enhance the expression of CD47, a key ‘don’t eat me’ signal, which interacts with SIRPα on macrophages to inhibit phagocytosis. This mechanism further protects leukemic cells from immune clearance by the innate immune system (58). Although activating signals such as ULBP1 can stimulate NK and T cell responses through engagement with the NKG2D receptor, their immunostimulatory effects may be attenuated by the concurrent upregulation of immune checkpoint molecules, further enabling immune evasion in NPM1-mutated AML (51).
3.2.3 NPM1 mutation-mediated modulation of HLA expression and antigen presentation in AML
The antigen presentation process is also inhibited. In the absence of DNMT3A mutations, NPM1 mutations lead to the downregulation of the CIITA gene (51), thereby inhibiting the expression of CLIP protein and HLA molecules, which helps leukemia cells evade recognition by CD8+ T cells. However, AML cells partially retain HLA expression, and cells with low HLA expression do not exhibit higher NK cell lysis rates (58), suggesting that leukemia cells may balance NK and T cell attacks in immunoediting by regulating HLA levels. Furthermore, studies have found that the frequency of specific HLA-I alleles in patients with NPM1 mutations is significantly lower than that in healthy controls and NPM1 wild-type AML patients (54). This suggests that HLA alleles capable of effectively presenting NPM1 peptides may reduce the risk of developing NPM1-mutated AML. Even among patients carrying such alleles who develop the disease, specific immune responses may contribute to disease remission. DNMT3A mutations can weaken the effect of NPM1 mutations on HLA expression. Interestingly, in samples with high HLA-DR expression, NPM1 mutations are associated with higher CLIP levels, indicating a complex regulation of antigen presentation and immune responses (58).
3.2.4 Metabolic and costimulatory dysregulation in NPM1-mutated AML
In addition to modulating classical immune checkpoint pathways, NPM1 mutations suppress immune function through a range of noncanonical mechanisms. One such mechanism involves the regulation of small extracellular vesicle-mediated signaling. The NPM1c/CTCF/PABPC1 signaling axis controls the secretion of miR-19a-3p via small extracellular vesicles, which are subsequently internalized by CD8+ T cells. This process inhibits the expression of creatine transporters, prevents creatine uptake, reduces ATP production, and consequently impairs the immune function of CD8+ T cells (53). Moreover, NPM1 mutations significantly upregulate SPINK2 expression and downregulate ALCAM, both of which contribute to impaired T cell activation (61).
3.3 DNMT3A mutation
Approximately 25% of AML patients harbor mutations in the DNMT3A gene, with the R882H variant being the most prevalent. This mutation reduces DNMT3A’s methyltransferase activity and is associated with genome-wide hypomethylation. Some studies suggest that this hypomethylation represents an early event initiated by the mutation, whereas DNMT3A-dependent CpG island hypermethylation may emerge during AML progression (62). In addition, DNMT3A mutations are frequently associated with increased chemotherapy resistance (63–65). Although study results have varied (66), such discrepancies may be attributed to differences in patient population characteristics.
3.3.1 T cell subset imbalance and functional consequences in DNMT3A-mutated AML
DNMT3A plays a pivotal role in shaping immune cell fate, particularly by maintaining the differentiation stability of CD4+ T cells and restricting both the formation of long-term memory CD8+ T cells and the pool of memory precursor effector cells (28, 67). In the context of AML, DNMT3A mutations have been associated with notable alterations in T cell infiltration and subset composition. In AML patients, DNMT3A mutations are associated with increased T cell infiltration; however, the distribution of T cell subsets is aberrant. Specifically, in patients with wild-type DNMT3A, T cells tend to undergo terminal differentiation, resulting in a reduced proportion of memory T cells (28). In contrast, patients with DNMT3A mutations exhibit a reduction in CD8+ Tn and CD4+ Tem in the BM, accompanied by an increase in CD4+ central memory T cells. Clinical studies have demonstrated that using donors with a higher proportion of CD8+ Tn for lymphocyte infusion can contribute to long-term remission in AML patients (28). Notably, similar to other mutations affecting DNA methylation regulators such as NPM1, IDH2, and CEBPA, DNMT3A mutations have also been shown to upregulate tumor-specific antigens, which in turn can activate antigen-specific clonal T cell responses (68). Conversely, lower ratios of CD8+ Tn and CD4+ Tem are associated with adverse genetic risks and poorer relapse-free survival and event-free survival (28). The specific impact of Tem cells on prognosis remains controversial. Some studies, such as those by Ling Xu and Adam J. Lamble, have found that an increased proportion of Tem cells is associated with enhanced T cell proliferation and higher CR rates. However, Maddalena Noviello’s team reported that the proportion of Tem cells also increases in relapsed AML patients (28). Moreover, DNMT3A mutations are linked to a higher risk of acute graft-versus-host disease following allogeneic hematopoietic stem cell transplantation, primarily by promoting CD4+ T cell polarization toward a Th1 phenotype and enhancing IFN-γ production (67). These findings collectively highlight the profound impact of DNMT3A mutations on T cell differentiation, function, and clinical outcomes in AML.
3.3.2 Immunosuppressive mechanisms and innate immune impairment driven by DNMT3A mutations
Beyond modulating adaptive immunity, DNMT3A mutations also disrupt innate immune signaling and promote an immunosuppressive TME. One such mechanism involves the hypomethylation-induced upregulation of miR-196b, which directly inhibits the Toll-like receptors (TLR) 7/8 signaling pathway, thereby weakening the immune response. In normal immune cells, TLR7 activates type I IFN and cytokines through the MyD88 pathway, indirectly activating Stat1 signaling to enhance the activity of Th1 cells and monocytes while promoting DC differentiation (69). Consequently, inhibition of TLR7/8 may lead to a diminished overall immune response. DNMT3A mutations are also frequently accompanied by increased infiltration of Tregs (70). Previous studies have shown that Tregs are highly adaptable to different tissue environments, and DNMT3A-dependent de novo DNA methylation facilitates this adaptability by establishing tissue-specific epigenetic memory, thereby refining and modulating their functions. However, in AML, DNMT3A mutations may impair methyltransferase activity, making it difficult for Tregs to adapt to various environments and thereby affecting their immune regulatory functions (71). Additionally, DNMT3A-dependent de novo DNA methylation is essential for silencing Foxp3 transcription. Mutations in DNMT3A may impair the effective silencing of Foxp3 transcription in Tregs, allowing them to continuously maintain their immunosuppressive functions, which may further promote immune escape in AML (72). In addition to Treg-mediated suppression and impaired TLR signaling, DNMT3A-mutated AML cells also exhibit elevated levels of immunosuppressive cytokines such as IL-10 and TGF-β (62). Furthermore, the concurrent upregulation of immune checkpoint molecules including PD-L1, CLIP, and TIM-3 indicates a coordinated activation of multiple immune evasion pathways, collectively contributing to a highly suppressive TME and poor prognosis (58).
3.3.3 Myeloid reprogramming and TAM polarization in DNMT3A-mutated AML
In addition to its effects on lymphoid immunity, DNMT3A mutation significantly alters the myeloid compartment. AML cells harboring DNMT3A mutations have been shown to possess an enhanced capacity to chemoattract monocytes, thereby modifying the TME to favor immune suppression. These AML cells inhibit the activity of the AP-1 binding site, leading to the downregulation of pro-inflammatory cytokines such as MIP-1α, MIP-1β, and IL-1β (62). The suppression of these key mediators impairs M1 macrophage polarization and diminishes their cytotoxic functions against tumor cells. In vivo studies using murine models have demonstrated a marked increase in the proportion of M2-polarized tumor-associated macrophages (TAMs) in the presence of DNMT3A mutations. These M2 macrophages express high levels of CD163 and CD206 and secrete chemokines such as CCL17, CCL22, and CCL24, which recruit Th2 cells to the leukemia microenvironment (62). The accumulation of M2 TAMs and Th2 cells creates an anti-inflammatory milieu that correlates strongly with reduced patient survival. Furthermore, DNMT3A-mutated AML cells exhibit resistance to macrophage-mediated killing and can differentiate into monocyte-like cells with immunosuppressive properties, further contributing to the inhibition of effective anti-leukemic T cell responses (62). These findings underscore a critical role for DNMT3A mutations in reprogramming the myeloid landscape, skewing macrophage polarization toward an immunosuppressive phenotype, and establishing a tumor-permissive microenvironment.
3.4 TP53 mutation
In AML, the detection rate of TP53 gene mutations is approximately 5-10%. However, this rate is significantly higher in treatment-related AML and in elderly patients with complex karyotypes, reaching as high as 70-80%. TP53 mutations are recognized as an independent prognostic factor for poor outcomes in AML patient (73).
3.4.1 T cell dysfunction, exhaustion, and Tregs expansion in TP53-mutated AML
TP53 mutations in AML lead to substantial alterations in the function and composition of T cells. Despite the increased infiltration of T cells, these cells exhibit signs of exhaustion and impaired functionality (71). Specifically, the expression of activation markers such as CD25, HLA-DR, and CD127 is low (74), while the immune checkpoint receptor CTLA-4 is upregulated (75). Moreover, the expression of cytotoxic molecules such as perforin and granzyme B is diminished (76), and the secretion of Th1 cytokines is significantly reduced (77). The T cell subsets are also significantly altered. Unsupervised clustering analysis revealed that CD8+ T cells predominantly exhibit an ICOS+/4-1BB+/PD-1+ phenotype (78), suggesting a dysfunctional, exhausted state. In contrast, Th cells predominantly express ICOS (6), which, despite being a costimulatory molecule, contributes to immune evasion in this context. The increase in PD-1+ cytotoxic T lymphocytes and PD-L1+ BM blasts further supports this hypothesis, as PD-1 signaling has been shown to inhibit T cell function and induce exhaustion (79). The upregulation of PD-L1 is closely associated with the downregulation of miR-34a and the overexpression of the MYC gene. Under normal conditions, wild-type p53 induces the transcription of miR-34a, which targets MYC mRNA and promotes its degradation, thereby negatively regulating MYC expression. However, in TP53-mutated AML, miR-34a expression is significantly reduced, leading to the upregulation of MYC and the induction of PD-L1 expression. The downregulation of miR-34a weakens its binding to the 3′ untranslated region of PD-L1 mRNA, reducing the inhibition of PD-L1 expression and thereby promoting T cell exhaustion (6, 78) (Figure 4). Transcriptional analysis revealed that, compared to healthy controls, cytotoxic T lymphocytes from TP53-mutated AML patients exhibited upregulation of inhibitory molecules (such as CD244, CD160, LILRB1, CD300A, and PVRIG) and downregulation of stimulatory molecules (such as CD40LG, CD28, TNFSF8, TMIGD2, and TNFRSF25). These alterations were not significant in other AML molecular subtypes (79).
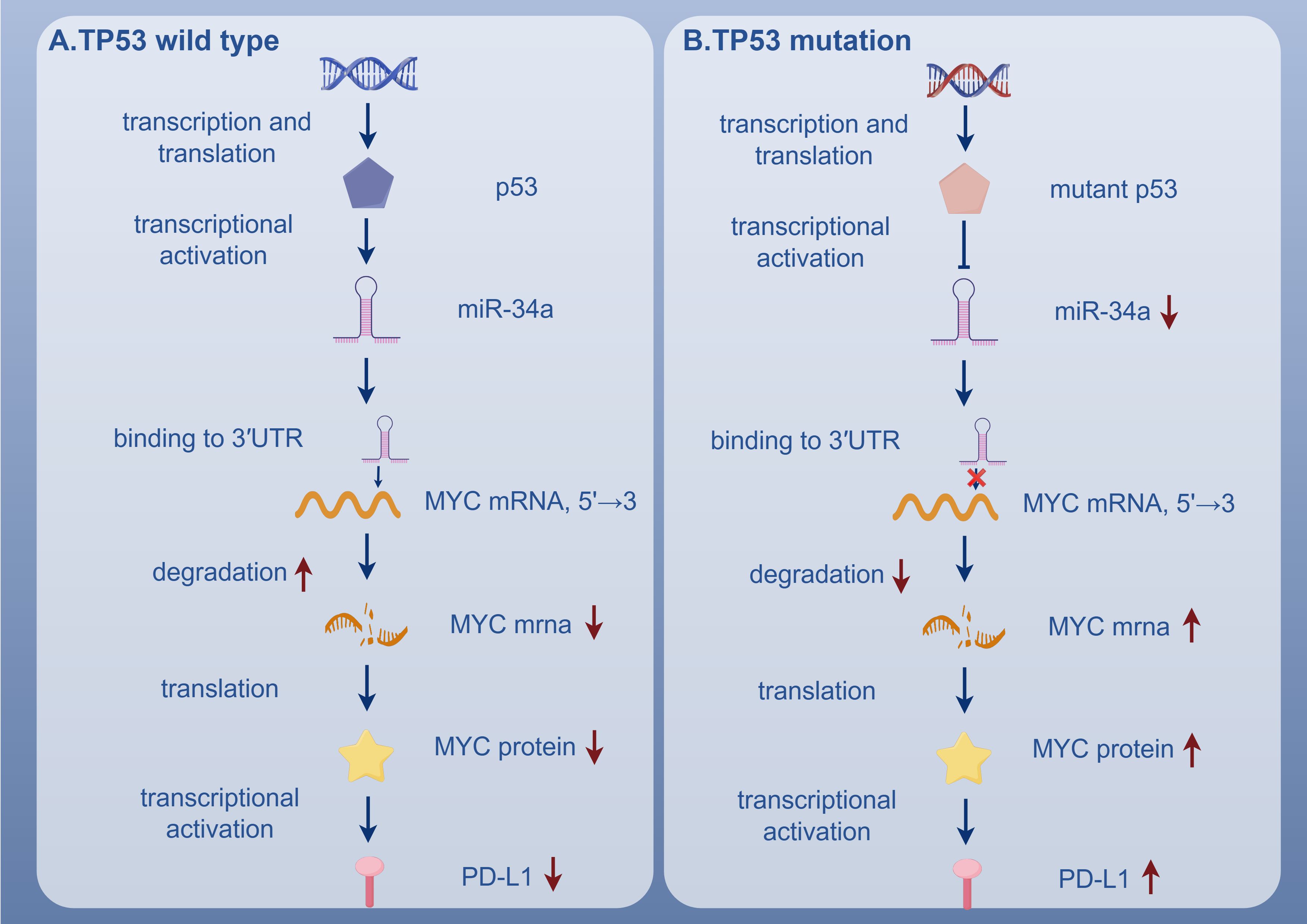
Figure 4. In TP53 wild-type AML, p53 promotes miR-34a expression, leading to MYC mRNA degradation and suppression of PD-L1 expression. In contrast, TP53-mutated AML shows reduced miR-34a, resulting in MYC upregulation and PD-L1 overexpression. TP53, Tumor Suppressor P53 Gene; p53, Tumor Protein 53; miR-34a, MicroRNA-34a; 3'UTR, 3' Untranslated Region; MYC Mrna, Myelocytomatosis Oncogene Messenger RNA; PD-L1, Programmed Death-Ligand 1.
In contrast, Tregs demonstrated metabolic adaptations and proliferative advantages. Although OX40+ Tregs are markedly reduced in the BM, the highly immunosuppressive ICOShigh/PD-1neg Tregs are significantly expanded (6). These Tregs exhibit enhanced proliferative capacity and are implicated in the suppression of anti-tumor immunity, which has been identified as an independent predictor of poor OS (78). The IL-2/STAT5 signaling axis further promotes Treg differentiation and stabilization (74, 80), with elevated FOXP3 expression (74) and increased secretion of immunosuppressive cytokines such as TGF-β and IL-10 (76). Together, these findings illustrate a significant alteration in T cell functionality, with exhaustion of Teffs and expansion of suppressive Tregs, contributing to the immune escape observed in TP53-mutated AML.
3.4.2 Dysregulation of innate immune pathways and pro-inflammatory microenvironment in TP53-mutated AML
TP53 mutations in AML significantly affect innate immune and leads to the formation of a pro-inflammatory microenvironment. One important mechanism is the downregulation of HLA molecules, which impairs antigen presentation, thus preventing the immune system from effectively recognizing and eliminating leukemia cells (74, 76). In addition, despite promoting significant infiltration of TAMs (80), the overexpression of CD47 on leukemia stem cells interacts with the SIRPα receptor on TAMs, inhibiting their phagocytic activity (81). Additionally, TP53 mutations lead to the upregulation of JAK/STAT, PI3K-Akt, and NF-κB signaling pathways, which are associated with increased production of pro-inflammatory cytokines such as CXCL1, CXCL2, CXCL8/IL-8, and IFN-induced products like CCL2, IL33, and IL6 (75). This results in the formation of a pro-inflammatory microenvironment, with IFN-γ playing a dominant role, which has been linked to poor responses to induction chemotherapy. Moreover, TP53 mutations inhibit IRF3 transcriptional activity and affect IFN expression through two mechanisms (1): by binding to TBK1, preventing the formation of the STING-TBK1-IRF3 complex (82), and (2) by inducing the overexpression of PLK4, which further inhibits the activation of the cGAS-STING-TBK1-IRF3 pathway (83).
3.5 IDH1/2 mutation
Mutations in IDH1/2 are present in approximately 15-20% of AML patients. Gain-of-function mutations in these enzymes can lead to a blockade in hematopoietic cell differentiation and promote leukemic transformation (84). In AML, mutant IDH1/2 enzymes convert α-ketoglutarate into the oncometabolite 2-hydroxyglutarate (2-HG), which accumulates in tumor tissues and patient serum, thereby suppressing immune function. 2-HG limits the secretion of CXCL10 by tumor cells, reducing T cell recruitment to tumor sites (85). Additionally, it inhibits the differentiation of monocytes into DCs, decreases the expression of HLA-DQ and HLA-DR on DCs, induces a tolerant phenotype, and suppresses the upregulation of DC markers such as CD1a and DC-SIGN. This results in reduced IL-12 and increased IL-10 secretion, thereby weakening the ability of DCs to stimulate T cells. AML cells harboring IDH mutations also exhibit decreased HLA-DP expression and demonstrate increased resistance to lysis by HLA-DP-specific T cells (86). However, Sunthankar KI reported that AML cells with the IDH2 R140Q mutation show increased HLA-DR expression and are capable of inducing T cell immune responses (84). Furthermore, once absorbed by immune cells, 2-HG inhibits histone and DNA demethylation in mouse CD8+ T cells, activates HIF-1α, and impairs T cell proliferation and effector functions. In human T cells, 2-HG destabilizes HIF-1α, promotes oxidative phosphorylation, enhances differentiation into CD4+CD25+FOXP3+ Tregs, and inhibits Th17 cell differentiation. Additionally, 2-HG is transported into T cells via SLC13A3, where it interferes with NFATC1 signaling, limits T cell proliferation and function, and induces ATP depletion by inhibiting oxidative phosphorylation (85), thereby further enhancing immunosuppression. In stromal cells, 2-HG upregulates NF-κB and enhances the NF-κB phosphorylation response of IDH2 mutant cells under IL-1β stimulation, leading to abnormal cytokine secretion (84). Most of these mechanisms facilitate AML progression and tumor immune evasion. However, the immune microenvironment also contains anti-tumor effector cells. For instance, IDH1/2 mutations can induce a significant increase in MAIT cells (33) and CD4+ Teffs (26).
3.6 NRAS mutation
NRAS mutations are found in approximately 15-20% of AML patient (87). Multiple studies have shown that NRAS mutations alone have no significant impact on prognosis, but are associated with higher survival rates after adjusting for age and other factors (88). This suggests that NRAS mutations may predict a better prognosis under certain conditions, but further verification is needed. NRAS mutations show strong antigen presentation potential. Specifically, in the NRAS^G12D mutant AML mouse model, hematopoietic stem/progenitor cells upregulated the expression of MHC class molecules, driving a potent anti-leukemia response. When the RUNX1-RUNX1T1 fusion gene is present, the expression levels of H2-Db and H2-Kb of MHC class I molecules are also significantly increased (89). Compared with the normal control group, the proportion of CD4+ T cells in the mutant group of mice was significantly reduced and the proportion of CD8+ T cells was significantly increased, indicating that the adaptive immune response was activated. However, expression levels of the PD-1 gene were increased in T cells, suggesting that NRAS^G12D AML cells evade immune surveillance by activating and depleting T cells. When T cells express inhibitory receptors and enter a state of exhaustion, disease is more likely to develop. NRAS^G12D AML, which is highly immunogenic, exhibits immunoediting in mice and upregulates PD-L1 expression. In mutation models, anti-PD-1 treatment has limited effect on relieving T cell suppression and the recovery of anti-leukemia immune responses is also limited, suggesting that NRAS mutant AML evades immune system surveillance through multiple mechanisms (89). When NRAS and ASXL1 are double mutated, AML cells also hyperactivate the MEK/ERK/AP-1 signaling pathway, leading to the upregulation of AP-1-related genes and inhibitory immune checkpoint ligands PD-L2, CD80, CD86, and CD155. This further inhibits the anti-leukemia activity of CD8+ T cells, NK cells and γδ T cells (25).
In summary, various driver gene mutations in AML regulate the immune microenvironment through multiple mechanisms (Supplementary Table 1). These mutations can either promote specific immune responses or lead to immune escape and immunosuppression, thereby influencing disease progression and prognosis. A thorough investigation of the relationship between these gene mutations and immune responses will enhance our understanding of the dynamic changes within the AML TME. This understanding provides a theoretical foundation for elucidating the mechanisms of immune escape and developing precise treatment strategies for the disease.
4 Immune microenvironment and genetic risk stratification
The TME comprises a diverse array of cellular components (90) and plays a well-established role in supporting tumor survival and progression across both solid and hematological malignancies (91). In AML, the TME exhibits a dual role: while it fosters leukemogenesis, it may also limit the expansion of malignant clones and contribute to their immune-mediated clearance (92). Within this context, the immune microenvironment of AML is highly complex and heterogeneous. Interactions between leukemic cells and immune components are central to disease initiation, progression, immune escape, and therapeutic resistance.
Recent transcriptomic analyses, such as those using TCGA-LAML data, have revealed distinct immune signatures across cytogenetic risk categories (93). To further investigate this, we examined immune microenvironmental profiles based on ELN 2022 (31) and 2024 (32) genetic risk stratifications. According to these guidelines, mutations in NPM1 and IDH2 R140 are categorized as low risk; FLT3-ITD, NRAS, and DNMT3A as intermediate risk; and TP53 and IDH2 R172 as high risk. Our synthesis suggests that AML patients exhibit markedly different immune landscapes depending on their genetic risk category. Patients with low-risk mutations tend to have immunologically active environments, characterized by elevated effector T cell infiltration, increased pro-inflammatory cytokines, robust antigen presentation, and reduced expression of inhibitory checkpoint molecules (5, 53–55, 94–96). In contrast, high-risk mutation profiles are associated with suppressed T cell activity, impaired antigen presentation, an abundance of Tregs, and increased levels of immunosuppressive cytokines (6, 71, 74–81). Intermediate-risk groups appear to exhibit a transitional immune state with both pro-inflammatory and immunosuppressive features, reflecting a dynamic equilibrium. These observations are well illustrated in Figure 5.
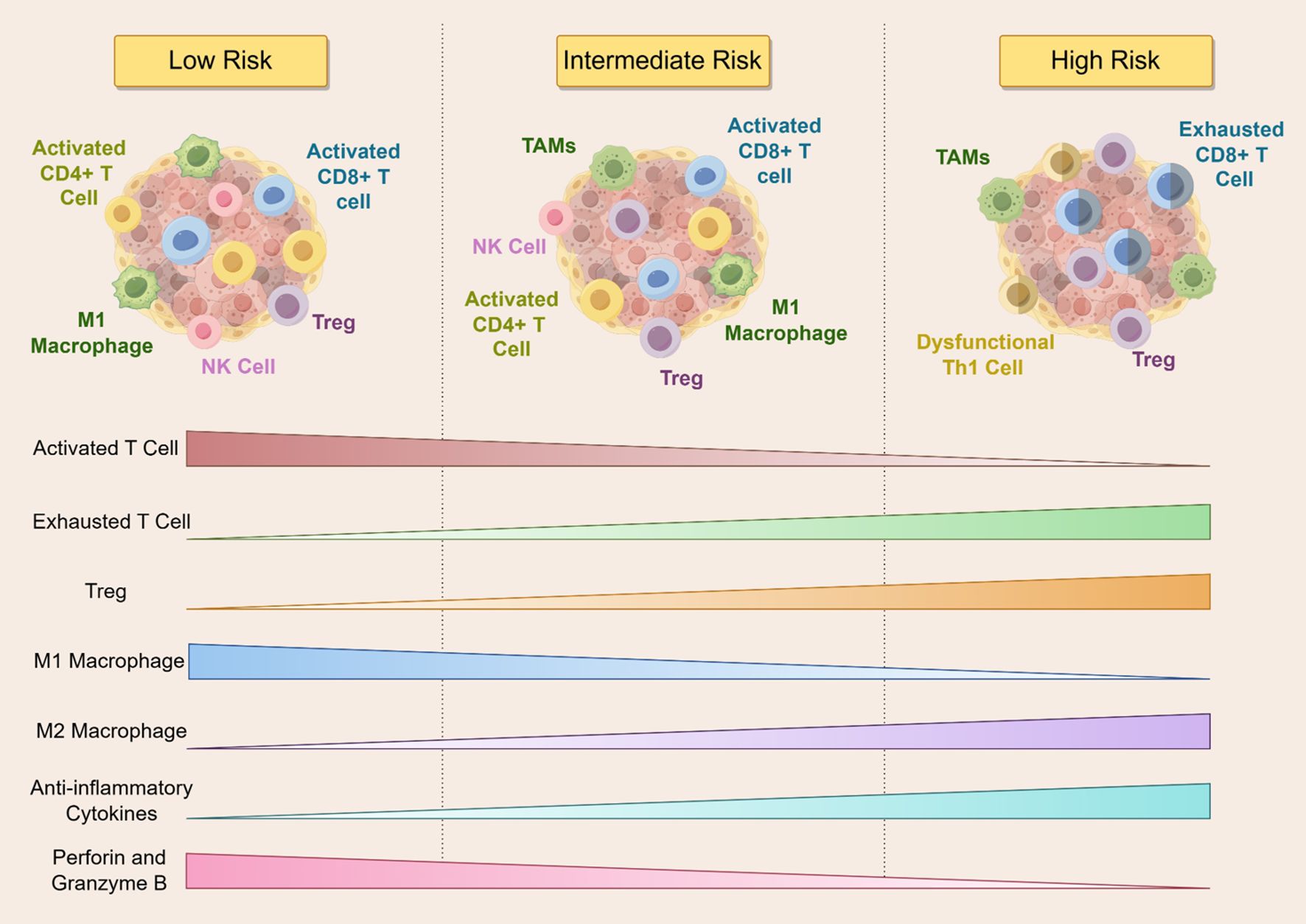
Figure 5. The figure illustrates the dynamics of immune cell infiltration across different risk stratifications in AML patients. As the risk level increases, activated T cells, M1 macrophages, and cytotoxic molecules such as perforin and granzyme B gradually decline, whereas exhausted T cells, Tregs, M2 macrophages, and anti-inflammatory cytokines progressively increase. Low-risk patients exhibit robust anti-tumor immune activity, while high-risk patients are characterized by a markedly immunosuppressive microenvironment. NK Cell, Natural Killer Cell; TAMs, tumor-associated macrophages; Th1, T-helper 1 Cell.
While these trends are compelling, several limitations warrant consideration. Many referenced studies rely on bulk transcriptomic data or immune deconvolution algorithms, which may not accurately capture the spatial and functional heterogeneity of immune cells in the AML microenvironment. Moreover, although immune activation is generally associated with favorable prognosis, the clinical significance of certain immune signatures—particularly in high-risk subtypes—remains controversial. Some studies have reported paradoxical findings, such as activated T cell phenotypes coexisting with immune dysfunction in high-risk groups (97). Additionally, immune infiltration is modulated by variables such as clonal hematopoiesis (98), treatment history (99), and the bone marrow niche (100), complicating its prognostic interpretation. Future studies incorporating single-cell and spatial transcriptomics, functional assays, and longitudinal immune profiling will be essential to disentangle the complex relationships between genetic mutations, immune remodeling, and patient outcomes in AML. While our review attempts to summarize and synthesize the current understanding, we acknowledge the rapidly evolving nature of this field and the need for continued critical evaluation of emerging evidence.
5 Future developments: immunotherapy strategies targeting neoepitopes
In recent years, the therapeutic landscape of AML has evolved beyond conventional intensive chemotherapy, driven by the urgent need to address high relapse rates and poor long-term survival (101), particularly in older patients and those with comorbidities (102, 103). Despite initial responses, even MRD-negative patients remain at substantial risk of relapse within three years, with rates approaching 70–80% (104). While targeted therapies such as FLT3 inhibitors, such as midostaurin and gilteritinib (105–107), and IDH inhibitors have improved survival in molecularly defined subgroups (108), their durability is limited. Resistance mechanisms—including secondary kinase domain mutations and bypass signaling activation—commonly emerge, often without significantly altering the immunosuppressive TME (108). Immunotherapy has revolutionized the treatment of several hematologic malignancies, yet its impact in AML has remained modest (109). One of the key challenges is the lack of leukemia-specific antigens that distinguish malignant from normal hematopoietic cells, thereby increasing the risk of off-tumor toxicity (101). Furthermore, AML is characterized by profound immune evasion strategies. As such, most current immunotherapeutic strategies benefit only a subset of patients, and their effectiveness is constrained by the highly suppressive immune milieu.
With advances in genomic profiling, driver mutations in AML have emerged not only as prognostic markers but also as potential sources of neoepitopes. These mutant-derived peptides, absent in healthy cells, can be presented via MHC molecules and recognized by T cells (52, 110), rendering them attractive targets for precision immunotherapy (111). Notably, AML harbors a high frequency of insertion/deletion (indel) mutations, which generate disproportionately more high-affinity neoepitopes compared to single nucleotide variants or gene fusions (101). Among the most extensively studied neoepitopes are those derived from NPM1 mutations. AIQ-specific CD8+ T cells have shown cytotoxic activity and correlate with improved survival, suggesting potential for adoptive T cell therapy (112, 113). While NPM1-derived peptides can stimulate immune responses in preclinical models and relapse settings, tumor-driven HLA loss and immune editing may limit long-term effectiveness. Furthermore, studies remain inconsistent regarding the frequency and robustness of these responses across patient subgroups, highlighting the need for standardized immunomonitoring. Similarly, FLT3-ITD mutations, which are present in both leukemic blasts and stem cells, generate neoepitopes such as the YVD/A1 peptide (114). TCR-engineered T cells targeting FLT3D835Y have demonstrated specificity and efficacy in preclinical settings (115). However, the heterogeneity in ITD insertion sites and lengths raises concerns regarding peptide variability and inconsistent T cell responses across patients (116). Moreover, while the immunogenicity of some FLT3-derived peptides is promising (115), their therapeutic potential has yet to be validated in clinical trials. Neoepitopes from DNMT3A R882H and IDH2 R140Q mutations have also been identified, capable of binding to HLA-A01:01 and HLA-B07:02 respectively (111). These peptides have been shown to elicit memory T cell responses, although supporting evidence in clinical or in vivo contexts remains limited. For example, 2-HG, a metabolite produced by mutant IDH enzymes, suppresses T cell activation, complicating efforts to harness these neoepitopes for therapy (117). Likewise, while TP53 mutations are associated with immunosuppressive TME (118, 119), few studies have successfully identified immunogenic peptides from TP53 variants with therapeutic applicability.
However, a higher neoepitope burden may not translate into improved immunogenicity or therapeutic responsiveness. In contrast to observations in solid tumors, where high neoantigen load often correlates with better immune activation, AML appears to exhibit the opposite trend: chronic exposure to neoantigens may promote T cell exhaustion and immune tolerance, thereby impairing effective antitumor immunity (101). Additionally, neoantigen heterogeneity and clonal evolution further complicate therapeutic targeting, as subclonal neoepitopes may be poorly presented or lack broad applicability (101). These findings underscore the need for caution when interpreting neoepitope quantity as a surrogate for immunotherapeutic potential. Neoepitope-targeted strategies in AML must be carefully integrated into comprehensive therapeutic approaches that also address the profoundly immunosuppressive tumor microenvironment and the dynamic nature of leukemic clonal architecture.
For patients in remission or with low disease burden, neoepitope-based vaccines or adoptive T cell therapies may offer an opportunity to eliminate MRD and prolong survival. Nonetheless, monotherapy approaches targeting neoantigens are unlikely to suffice. Combination strategies—pairing neoepitope-based interventions with checkpoint blockade, metabolic modulators, or cytokine support—will likely be necessary to overcome the barriers imposed by the AML TME. In conclusion, while neoepitope-targeted immunotherapy represents a conceptually appealing avenue for AML treatment, its translation into clinical practice is fraught with challenges. A balanced assessment must recognize both its potential and its limitations. Future studies should prioritize rigorous validation of immunogenic peptides, explore inter-patient variability, and incorporate strategies to overcome T cell exhaustion and antigenic heterogeneity. Only through such integrative approaches can the promise of personalized immunotherapy in AML be fully realized.
6 Conclusions
This review underscores the multifaceted complexity of the immune microenvironment in AML, highlighting its dynamic and heterogeneous nature across genetic and clinical contexts. Genetic mutations in AML not only alter the intrinsic behavior of leukemic cells but also remodel the surrounding immune milieu—affecting immune cell infiltration, polarization, and effector function. These alterations underlie diverse immune evasion mechanisms that contribute to immunosuppression, disease progression, and treatment resistance. Importantly, these immunologic changes are not uniform but vary significantly across ELN-defined genetic risk categories, with low-risk mutations often associated with more immunologically active profiles, and high-risk mutations linked to profound immune dysfunction. This suggests that effective therapeutic strategies must account for both genetic and immune stratification. While traditional chemotherapy remains the cornerstone of AML treatment, its efficacy is limited, particularly in patients harboring high-risk molecular lesions. Recent progress in targeted therapies and immunotherapies—especially those directed against neoepitopes derived from AML driver mutations—has provided new hope for achieving disease control. However, multiple barriers remain, including immune exhaustion, neoepitope heterogeneity, and the deeply immunosuppressive bone marrow microenvironment. Future research should therefore prioritize the development of integrative treatment strategies that not only target specific genetic lesions but also modulate the immune contexture. Combining neoepitope-based interventions with immune checkpoint blockade, T cell engineering, or microenvironment-modulating agents may help overcome resistance and enhance long-term therapeutic responses. Ultimately, the advancement of personalized immunotherapy—guided by both molecular and immunologic profiling—holds the greatest promise for improving outcomes in AML.
Author contributions
XG: Writing – original draft. HZ: Writing – review & editing. XW: Writing – review & editing. LL: Writing – review & editing. LZ: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by the National Natural Science Foundation of China, grant number 82360029; Gansu Province Graduate Student Innovation Star Program, grant numbers 2025CXZX-211 and 2025CXZX-213; and Gansu Province Key Research and Development Program, grant number 24YFFA046.
Acknowledgments
Thank you to Figdraw 2.0 for providing the drawing materials (ID: Figure 1: PRURA75ea8; Figure 2: RUUTUbad15; Figure 3: TTYOY6cee3; Figure 4: IWYPI4da4d; Figure 5: TWAOS92330).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1635111/full#supplementary-material
References
1. Reis R, Müller GS, Santos MM, Santos AS, Santos H, Santos LS, et al. Description of lymphocyte and cytokine profiles in individuals with acute myeloid leukemia associated with FLT3-ITD and NPM1 mutation status. Eur J Cancer Prev. (2024) 34(2):115–23. doi: 10.1097/CEJ.0000000000000905
2. Prada-Arismendy J, Arroyave JC, and Röthlisberger S. Molecular biomarkers in acute myeloid leukemia. Blood Rev. (2017) 31:63–76. doi: 10.1016/j.blre.2016.08.005
3. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
4. Mumme H, Thomas BE, Bhasin SS, Krishnan U, Dwivedi B, Perumalla P, et al. Single-cell analysis reveals altered tumor microenvironments of relapse- and remission-associated pediatric acute myeloid leukemia. Nat Commun. (2023) 14:6209. doi: 10.1038/s41467-023-41994-0
5. Xie G, Ivica NA, Jia B, Li Y, Dong H, Liang Y, et al. CAR-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat Biomed engineering. (2021) 5:399–413. doi: 10.1038/s41551-020-00625-5
6. Zhao Y, Chen W, Yu J, Pei S, Zhang Q, Shi J, et al. TP53 in MDS and AML: Biological and clinical advances. Cancer letters. (2024) 588:216767. doi: 10.1016/j.canlet.2024.216767
7. Park SH, Bae MH, Park CJ, Cho YU, Jang S, Lee JH, et al. Effect of changes in lymphocyte subsets at diagnosis in acute myeloid leukemia on prognosis: association with complete remission rates and relapse free survivals. J hematopathology. (2023) 16:73–84. doi: 10.1007/s12308-023-00536-9
8. Cianga VA, Rusu C, Pavel-Tanasa M, Dascalescu A, Danaila C, Harnau S, et al. Combined flow cytometry natural killer immunophenotyping and KIR/HLA-C genotyping reveal remarkable differences in acute myeloid leukemia patients, but suggest an overall impairment of the natural killer response. Front Med. (2023) 10:1148748. doi: 10.3389/fmed.2023.1148748
9. Cianga VA, Campos Catafal L, Cianga P, Pavel Tanasa M, Cherry M, Collet P, et al. Natural killer cell subpopulations and inhibitory receptor dynamics in myelodysplastic syndromes and acute myeloid leukemia. Front Immunol. (2021) 12:665541. doi: 10.3389/fimmu.2021.665541
10. Leifheit ME, Johnson G, Kuzel TM, Schneider JR, Barker E, Yun HD, et al. Enhancing therapeutic efficacy of FLT3 inhibitors with combination therapy for treatment of acute myeloid leukemia. Int J Mol Sci. (2024) 25(17):9448. doi: 10.3390/ijms25179448
11. Straube J, Janardhanan Y, Haldar R, and Bywater MJ. Immune control in acute myeloid leukemia. Exp hematology. (2024) 138:104256. doi: 10.1016/j.exphem.2024.104256
12. Wu Z, Zhang H, Wu M, Peng G, He Y, Wan N, et al. Targeting the NKG2D/NKG2D-L axis in acute myeloid leukemia. Biomedicine pharmacotherapy = Biomedecine pharmacotherapie. (2021) 137:111299. doi: 10.1016/j.biopha.2021.111299
13. Chretien AS, Devillier R, Granjeaud S, Cordier C, Demerle C, Salem N, et al. High-dimensional mass cytometry analysis of NK cell alterations in AML identifies a subgroup with adverse clinical outcome. Proc Natl Acad Sci United States America. (2021) 118(22):e2020459118. doi: 10.1073/pnas.2020459118
14. Brauneck F, Fischer B, Witt M, Muschhammer J, Oelrich J, da Costa Avelar PH, et al. TIGIT blockade repolarizes AML-associated TIGIT(+) M2 macrophages to an M1 phenotype and increases CD47-mediated phagocytosis. J immunotherapy Cancer. (2022) 10(12):e004794. doi: 10.1136/jitc-2022-004794
15. Xu L, Liu L, Yao D, Zeng X, Zhang Y, Lai J, et al. PD-1 and TIGIT are highly co-expressed on CD8(+) T cells in AML patient bone marrow. Front Oncol. (2021) 11:686156. doi: 10.3389/fonc.2021.686156
16. Flynn PA, Long MD, Kosaka Y, Long N, Mulkey JS, Coy JL, et al. Leukemic mutation FLT3-ITD is retained in dendritic cells and disrupts their homeostasis leading to expanded Th17 frequency. Front Immunol. (2024) 15:1297338. doi: 10.3389/fimmu.2024.1297338
17. Jia B, Wang L, Claxton DF, Ehmann WC, Rybka WB, Mineishi S, et al. Bone marrow CD8 T cells express high frequency of PD-1 and exhibit reduced anti-leukemia response in newly diagnosed AML patients. Blood Cancer J. (2018) 8:34. doi: 10.1038/s41408-018-0069-4
18. Le Dieu R, Taussig DC, Ramsay AG, Mitter R, Miraki-Moud F, Fatah R, et al. Peripheral blood T cells in acute myeloid leukemia (AML) patients at diagnosis have abnormal phenotype and genotype and form defective immune synapses with AML blasts. Blood. (2009) 114:3909–16. doi: 10.1182/blood-2009-02-206946
19. Knaus HA, Berglund S, Hackl H, Blackford AL, Zeidner JF, Montiel-Esparza R, et al. Signatures of CD8+ T cell dysfunction in AML patients and their reversibility with response to chemotherapy. JCI Insight. (2018) 3(21):e120974. doi: 10.1172/jci.insight.120974
20. Brück O, Dufva O, Hohtari H, Blom S, Turkki R, Ilander M, et al. Immune profiles in acute myeloid leukemia bone marrow associate with patient age, T-cell receptor clonality, and survival. Blood advances. (2020) 4:274–86. doi: 10.1182/bloodadvances.2019000792
21. Hao F, Sholy C, Wang C, Cao M, and Kang X. The role of T cell immunotherapy in acute myeloid leukemia. Cells. (2021) 10(12):3376. doi: 10.3390/cells10123376
22. Luciano M, Krenn PW, and Horejs-Hoeck J. The cytokine network in acute myeloid leukemia. Front Immunol. (2022) 13:1000996. doi: 10.3389/fimmu.2022.1000996
23. Forsberg M and Konopleva M. SOHO state of the art updates and next questions: understanding and overcoming venetoclax resistance in hematologic Malignancies. Clin lymphoma myeloma leukemia. (2024) 24:1–14. doi: 10.1016/j.clml.2023.10.006
24. Zhong F, Yao F, Jiang J, Yu X, Liu J, Huang B, et al. CD8 + T cell-based molecular subtypes with heterogeneous immune landscapes and clinical significance in acute myeloid leukemia. Inflammation Res. (2024) 73:329–44. doi: 10.1007/s00011-023-01839-4
25. You X, Liu F, Binder M, Vedder A, Lasho T, Wen Z, et al. Asxl1 loss cooperates with oncogenic Nras in mice to reprogram the immune microenvironment and drive leukemic transformation. Blood. (2022) 139:1066–79. doi: 10.1182/blood.2021012519
26. Williams P, Basu S, Garcia-Manero G, Hourigan CS, Oetjen KA, Cortes JE, et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer. (2019) 125:1470–81. doi: 10.1002/cncr.31896
27. El Dosoky W, Aref S, El Menshawy N, Ramez A, Abou Zaid T, Aref M, et al. Prognostic effect of CTLA4/LAG3 expression by T-cells subsets on acute myeloid leukemia patients. Asian Pacific J Cancer prevention: APJCP. (2024) 25:1777–85. doi: 10.31557/APJCP.2024.25.5.1777
28. Sun K, Shi ZY, Wang YZ, Xie DH, Liu YR, Jiang Q, et al. The profile and prognostic significance of bone marrow T-cell differentiation subsets in adult AML at diagnosis. Front Immunol. (2024) 15:1418792. doi: 10.3389/fimmu.2024.1418792
29. Shapoorian H, Zalpoor H, and Ganjalikhani-Hakemi M. The correlation between Flt3-ITD mutation in dendritic cells with TIM-3 expression in acute myeloid leukemia. Blood Sci (Baltimore Md). (2021) 3:132–5. doi: 10.1097/BS9.0000000000000092
30. Lasry A, Nadorp B, Fornerod M, Nicolet D, Wu H, Walker CJ, et al. An inflammatory state remodels the immune microenvironment and improves risk stratification in acute myeloid leukemia. Nat cancer. (2023) 4:27–42. doi: 10.1038/s43018-022-00480-0
31. Rausch C, Rothenberg-Thurley M, Dufour A, Schneider S, Gittinger H, Sauerland C, et al. Validation and refinement of the 2022 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia. (2023) 37:1234–44. doi: 10.1038/s41375-023-01884-2
32. Döhner H, DiNardo CD, Wei AH, Löwenberg B, Appelbaum F, Craddock C, et al. Genetic risk classification for adults with AML receiving less-intensive therapies: the 2024 ELN recommendations. Blood. (2024) 144(21):2169–73. doi: 10.1182/blood.2024025409
33. Chen H, Wu M, Xia H, Du S, Zhou G, Long G, et al. FLT3LG and IFITM3P6 consolidate T cell activity in the bone marrow microenvironment and are prognostic factors in acute myelocytic leukemia. Front Immunol. (2022) 13:980911. doi: 10.3389/fimmu.2022.980911
34. Tecik M and Adan A. Emerging DNA methylome targets in FLT3-ITD-positive acute myeloid leukemia: combination therapy with clinically approved FLT3 inhibitors. Curr Treat options Oncol. (2024) 25:719–51. doi: 10.1007/s11864-024-01202-7
35. Comont T, Nicolau-Travers ML, Bertoli S, Recher C, Vergez F, and Treiner E. MAIT cells numbers and frequencies in patients with acute myeloid leukemia at diagnosis: association with cytogenetic profile and gene mutations. Cancer immunology immunotherapy: CII. (2022) 71:875–87. doi: 10.1007/s00262-021-03037-9
36. Lau CM, Nish SA, Yogev N, Waisman A, Reiner SL, and Reizis B. Leukemia-associated activating mutation of Flt3 expands dendritic cells and alters T cell responses. J Exp Med. (2016) 213:415–31. doi: 10.1084/jem.20150642
37. Hu Z, Yang Y, Li J, and Hu Z. Genetic mutations and immune microenvironment: unveiling the connection to AML prognosis. Hematol (Amsterdam Netherlands). (2024) 29:2346965. doi: 10.1080/16078454.2024.2346965
38. Kaito Y, Hirano M, Futami M, Nojima M, Tamura H, Tojo A, et al. CD155 and CD112 as possible therapeutic targets of FLT3 inhibitors for acute myeloid leukemia. Oncol letters. (2022) 23:51. doi: 10.3892/ol.2021.13169
39. Chen C, Liang C, Wang S, Chio CL, Zhang Y, Zeng C, et al. Expression patterns of immune checkpoints in acute myeloid leukemia. J Hematol Oncol. (2020) 13:28. doi: 10.1186/s13045-020-00853-x
40. Brodská B, Otevřelová P, Šálek C, Fuchs O, Gašová Z, and Kuželová K. High PD-L1 expression predicts for worse outcome of leukemia patients with concomitant NPM1 and FLT3 mutations. Int J Mol Sci. (2019) 20(11):2823. doi: 10.3390/ijms20112823
41. Mendez LM, Posey RR, and Pandolfi PP. The interplay between the genetic and immune landscapes of AML: mechanisms and implications for risk stratification and therapy. Front Oncol. (2019) 9:1162. doi: 10.3389/fonc.2019.01162
42. Kupsa T, Vanek J, Zak P, Jebavy L, and Horacek JM. Serum levels of selected cytokines and soluble adhesion molecules in acute myeloid leukemia: Soluble receptor for interleukin-2 predicts overall survival. Cytokine. (2020) 128:155005. doi: 10.1016/j.cyto.2020.155005
43. Zhang Q, Falqués-Costa T, Pilheden M, Sturesson H, Ovlund T, Rissler V, et al. Activating mutations remodel the chromatin accessibility landscape to drive distinct regulatory networks in KMT2A-rearranged acute leukemia. HemaSphere. (2024) 8:e70006. doi: 10.1002/hem3.70006
44. Peng L, Wei Y, Shao Y, Li Y, Liu N, and Duan L. The emerging roles of CCN3 protein in immune-related diseases. Mediators inflammation. (2021) 2021:5576059. doi: 10.1155/2021/5576059
45. Naughton M, Moffat J, Eleftheriadis G, de la Vega Gallardo N, Young A, Falconer J, et al. CCN3 is dynamically regulated by treatment and disease state in multiple sclerosis. J neuroinflammation. (2020) 17:349. doi: 10.1186/s12974-020-02025-7
46. Guzylack-Piriou L, Gausseres B, Tasca C, Hassel C, Tabouret G, and Foucras G. A loss of function mutation in SOCS2 results in increased inflammatory response of macrophages to TLR ligands and Staphylococcus aureus. Front Immunol. (2024) 15:1397330. doi: 10.3389/fimmu.2024.1397330
47. Knosp CA, Schiering C, Spence S, Carroll HP, Nel HJ, Osbourn M, et al. Regulation of Foxp3+ inducible regulatory T cell stability by SOCS2. J Immunol (Baltimore Md: 1950). (2013) 190:3235–45. doi: 10.4049/jimmunol.1201396
48. Sarajlic M, Neuper T, Föhrenbach Quiroz KT, Michelini S, Vetter J, Schaller S, et al. IL-1β Induces SOCS2 expression in human dendritic cells. Int J Mol Sci. (2019) 20(23):5931. doi: 10.3390/ijms20235931
49. Peng Q, Huang R, Wang H, Xiao H, Wang Y, Zhai Z, et al. Immune characteristics and prognostic implications of mucosal-associated invariant T cells in acute myeloid leukemia. Cancer immunology immunotherapy: CII. (2023) 72:4399–414. doi: 10.1007/s00262-023-03574-5
50. Issa GC, Bidikian A, Venugopal S, Konopleva M, DiNardo CD, Kadia TM, et al. Clinical outcomes associated with NPM1 mutations in patients with relapsed or refractory AML. Blood advances. (2023) 7:933–42. doi: 10.1182/bloodadvances.2022008316
51. Ranieri R, Pianigiani G, Sciabolacci S, Perriello VM, Marra A, Cardinali V, et al. Current status and future perspectives in targeted therapy of NPM1-mutated AML. Leukemia. (2022) 36:2351–67. doi: 10.1038/s41375-022-01666-2
52. van der Lee DI, Reijmers RM, Honders MW, Hagedoorn RS, de Jong RC, Kester MG, et al. Mutated nucleophosmin 1 as immunotherapy target in acute myeloid leukemia. J Clin Invest. (2019) 129:774–85. doi: 10.1172/JCI97482
53. Peng M, Ren J, Jing Y, Jiang X, Xiao Q, Huang J, et al. Tumour-derived small extracellular vesicles suppress CD8+ T cell immune function by inhibiting SLC6A8-mediated creatine import in NPM1-mutated acute myeloid leukaemia. J extracellular vesicles. (2021) 10:e12168. doi: 10.1002/jev2.12168
54. Forghieri F, Riva G, Lagreca I, Barozzi P, Bettelli F, Paolini A, et al. Neoantigen-Specific T-cell immune responses: the paradigm of NPM1-mutated acute myeloid leukemia. Int J Mol Sci. (2021) 22(17):9159. doi: 10.3390/ijms22179159
55. Antohe I, Dǎscǎlescu A, Dǎnǎilǎ C, Titieanu A, Zlei M, Ivanov I, et al. B7-positive and B7-negative acute myeloid leukemias display distinct T cell maturation profiles, immune checkpoint receptor expression, and european leukemia net risk profiles. Front Oncol. (2020) 10:264. doi: 10.3389/fonc.2020.00264
56. Greiner J, Goetz M, Schuler PJ, Bulach C, Hofmann S, Schrezenmeier H, et al. Enhanced stimulation of antigen-specific immune responses against nucleophosmin 1 mutated acute myeloid leukaemia by an anti-programmed death 1 antibody. Br J haematology. (2022) 198:866–74. doi: 10.1111/bjh.18326
57. Dong Y, Han Y, Huang Y, Jiang S, Huang Z, Chen R, et al. PD-L1 is expressed and promotes the expansion of regulatory T cells in acute myeloid leukemia. Front Immunol. (2020) 11:1710. doi: 10.3389/fimmu.2020.01710
58. Kuželová K, Brodská B, Marková J, Petráčková M, Schetelig J, Ransdorfová Š, et al. NPM1 and DNMT3A mutations are associated with distinct blast immunophenotype in acute myeloid leukemia. Oncoimmunology. (2022) 11:2073050. doi: 10.1080/2162402X.2022.2073050
59. Kikushige Y and Miyamoto T. TIM-3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. Int J hematology. (2013) 98:627–33. doi: 10.1007/s12185-013-1433-6
60. Zhang X, Zhang X, Liu P, Liu K, Li W, Chen Q, et al. Prognostic implications and functional enrichment analysis of LTB4R in patients with acute myeloid leukemia. Nan fang yi ke da xue xue bao = J South Med University. (2022) 42:309–20. doi: 10.12122/j.issn.1673-4254.2022.03.01
61. Pitts HA, Cheng CK, Cheung JS, Sun MK, Yung YL, Chan HY, et al. SPINK2 protein expression is an independent adverse prognostic marker in AML and is potentially implicated in the regulation of ferroptosis and immune response. Int J Mol Sci. (2023) 24(11):9696. doi: 10.3390/ijms24119696
62. Que Y, Li H, Lin L, Zhu X, Xiao M, Wang Y, et al. Study on the immune escape mechanism of acute myeloid leukemia with DNMT3A mutation. Front Immunol. (2021) 12:653030. doi: 10.3389/fimmu.2021.653030
63. Jafari PA, Bagheri R, Lavasani S, and Goudarzi S. DNMT3A-R882: a mutation with many paradoxes. Ann Hematol. (2024) 103(12):4981–8. doi: 10.1007/s00277-024-05874-x
64. Guryanova OA, Shank K, Spitzer B, Luciani L, Koche RP, Garrett-Bakelman FE, et al. DNMT3A mutations promote anthracycline resistance in acute myeloid leukemia via impaired nucleosome remodeling. Nat Med. (2016) 22:1488–95. doi: 10.1038/nm.4210
65. Chu X, Zhong L, Dan W, Wang X, Zhang Z, Liu Z, et al. DNMT3A R882H mutation drives daunorubicin resistance in acute myeloid leukemia via regulating NRF2/NQO1 pathway. Cell communication signaling: CCS. (2022) 20:168. doi: 10.1186/s12964-022-00978-1
66. Elsayed GM, Abd Elgawad AF, Shafik NF, Elshimy RA, Abd Elhakeem HK, and Attea SA. Study of DNA methyl transferase 3A mutation in acute myeloid leukemic patients. Egyptian J Med Human Genet. (2018) 19:315–9. doi: 10.1016/j.ejmhg.2018.05.005
67. Wei X, Huang S, Gu Z, Liu J, Li M, Jin X, et al. Clonal hematopoiesis-associated gene mutations affect acute graft-versus-host disease after hematopoietic stem cell transplantation in AML patients. Ann transplantation. (2024) 29:e943688. doi: 10.12659/AOT.943688
68. Ehx G, Larouche JD, Durette C, Laverdure JP, Hesnard L, Vincent K, et al. Atypical acute myeloid leukemia-specific transcripts generate shared and immunogenic MHC class-I-associated epitopes. Immunity. (2021) 54:737–52.e10. doi: 10.1016/j.immuni.2021.03.001
69. Gamlen HA, Romer-Seibert JS, Lawler ME, Versace AM, Goetz ML, Feng Y, et al. miR-196b-TLR7/8 signaling axis regulates innate immune signaling and myeloid maturation in DNMT3A-mutant AML. Clin Cancer Res. (2022) 28:4574–86. doi: 10.1158/1078-0432.CCR-22-1598
70. Xu Q and Guo T. Somatic mutation-associated risk index based on lncRNA expression for predicting prognosis in acute myeloid leukemia. Hematol (Amsterdam Netherlands). (2022) 27:659–71. doi: 10.1080/16078454.2022.2056677
71. Zeidan AM, Bewersdorf JP, Hasle V, Shallis RM, Thompson E, de Menezes DL, et al. Integrated genetic, epigenetic, and immune landscape of TP53 mutant AML and higher risk MDS treated with azacitidine. Ther Adv hematology. (2024) 15:20406207241257904. doi: 10.1177/20406207241257904
72. Bai L, Hao X, Keith J, and Feng Y. DNA methylation in regulatory T cell differentiation and function: challenges and opportunities. Biomolecules. (2022) 12(9):1282. doi: 10.3390/biom12091282
73. Zhu G, Cai J, Fu W, Sun Y, Wang T, and Zhong H. Elucidating the immune landscape and potential prognostic model in acute myeloid leukemia with TP53 mutation. Hematol (Amsterdam Netherlands). (2024) 29:2400620. doi: 10.1080/16078454.2024.2400620
74. Vadakekolathu J, Lai C, Reeder S, Church SE, Hood T, Lourdusamy A, et al. TP53 abnormalities correlate with immune infiltration and associate with response to flotetuzumab immunotherapy in AML. Blood advances. (2020) 4:5011–24. doi: 10.1182/bloodadvances.2020002512
75. Urabe A, Chi S, and Minami Y. The immuno-oncology and genomic aspects of DNA-hypomethylating therapeutics in acute myeloid leukemia. Int J Mol Sci. (2023) 24(4):3727. doi: 10.3390/ijms24043727
76. Chomczyk M, Gazzola L, Dash S, Firmanty P, George BS, Mohanty V, et al. Impact of p53-associated acute myeloid leukemia hallmarks on metabolism and the immune environment. Front Pharmacol. (2024) 15:1409210. doi: 10.3389/fphar.2024.1409210
77. Daver NG, Maiti A, Kadia TM, Vyas P, Majeti R, Wei AH, et al. TP53-mutated myelodysplastic syndrome and acute myeloid leukemia: biology, current therapy, and future directions. Cancer discovery. (2022) 12:2516–29. doi: 10.1158/2159-8290.CD-22-0332
78. Sallman DA, McLemore AF, Aldrich AL, Komrokji RS, McGraw KL, Dhawan A, et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood. (2020) 136:2812–23. doi: 10.1182/blood.2020006158
79. Abolhalaj M, Sincic V, Lilljebjörn H, Sandén C, Aab A, Hägerbrand K, et al. Transcriptional profiling demonstrates altered characteristics of CD8(+) cytotoxic T-cells and regulatory T-cells in TP53-mutated acute myeloid leukemia. Cancer Med. (2022) 11:3023–32. doi: 10.1002/cam4.4661
80. Wen XM, Xu ZJ, Jin Y, Xia PH, Ma JC, Qian W, et al. Association analyses of TP53 mutation with prognosis, tumor mutational burden, and immunological features in acute myeloid leukemia. Front Immunol. (2021) 12:717527. doi: 10.3389/fimmu.2021.717527
81. Shallis RM, Bewersdorf JP, Stahl MF, Halene S, and Zeidan AM. Are we moving the needle for patients with TP53-mutated acute myeloid leukemia? Cancers. (2022) 14(10):2434. doi: 10.3390/cancers14102434
82. Brummer T and Zeiser R. The role of the MDM2/p53 axis in antitumor immune responses. Blood. (2024) 143:2701–9. doi: 10.1182/blood.2023020731
83. Palm-Apergi C. PLK4, a potential target against AML. Blood. (2023) 142:1941–2. doi: 10.1182/blood.2023021950
84. Sunthankar KI, Jenkins MT, Cote CH, Patel SB, Welner RS, and Ferrell PB. Isocitrate dehydrogenase mutations are associated with altered IL-1β responses in acute myeloid leukemia. Leukemia. (2022) 36:923–34. doi: 10.1038/s41375-021-01487-9
85. Galluzzi L and Kroemer G. Potent immunosuppressive effects of the oncometabolite R-2-hydroxyglutarate. Oncoimmunology. (2018) 7:e1528815. doi: 10.1080/2162402X.2018.1528815
86. Hammon K, Renner K, Althammer M, Voll F, Babl N, Decking SM, et al. D-2-hydroxyglutarate supports a tolerogenic phenotype with lowered major histocompatibility class II expression in non-malignant dendritic cells and acute myeloid leukemia cells. Haematologica. (2024) 109:2500–14. doi: 10.3324/haematol.2023.283597
87. Testa U, Castelli G, and Pelosi E. TP53-mutated myelodysplasia and acute myeloid leukemia. Mediterr J Hematol Infect diseases. (2023) 15:e2023038. doi: 10.4084/MJHID.2023.038
88. Mustafa Ali MK, Williams MT, Corley EM, AlKaabba F, and Niyongere S. Impact of KRAS and NRAS mutations on outcomes in acute myeloid leukemia. Leukemia lymphoma. (2023) 64:962–71. doi: 10.1080/10428194.2023.2190432
89. Austin RJ, Straube J, Halder R, Janardhanan Y, Bruedigam C, Witkowski M, et al. Oncogenic drivers dictate immune control of acute myeloid leukemia. Nat Commun. (2023) 14:2155. doi: 10.1038/s41467-023-37592-9
90. Hinshaw DC and Shevde LA. The tumor microenvironment innately modulates cancer progression. Cancer Res. (2019) 79:4557–66. doi: 10.1158/0008-5472.CAN-18-3962
91. Lyu A, Nam SH, Humphrey RS, Horton TM, and Ehrlich LIR. Cells and signals of the leukemic microenvironment that support progression of T-cell acute lymphoblastic leukemia (T-ALL). Exp Mol Med. (2024) 56(11):2337–47. doi: 10.1038/s12276-024-01335-7
92. Menter T and Tzankov A. Tumor microenvironment in acute myeloid leukemia: adjusting niches. Front Immunol. (2022) 13:811144. doi: 10.3389/fimmu.2022.811144
93. Chen T, Zhang Y, Zhang D, and Zhou H. Immune-based subgroups uncover diverse tumor immunogenicity and implications for prognosis and precision therapy in acute myeloid leukemia. Front Immunol. (2024) 15:1451486. doi: 10.3389/fimmu.2024.1451486
94. Nelde A, Schuster H, Heitmann JS, Bauer J, Maringer Y, Zwick M, et al. Immune surveillance of acute myeloid leukemia is mediated by HLA-presented antigens on leukemia progenitor cells. Blood Cancer discovery. (2023) 4:468–89. doi: 10.1158/2643-3230.BCD-23-0020
95. Forghieri F, Riva G, Lagreca I, Barozzi P, Vallerini D, Morselli M, et al. Characterization and dynamics of specific T cells against nucleophosmin-1 (NPM1)-mutated peptides in patients with NPM1-mutated acute myeloid leukemia. Oncotarget. (2019) 10:869–82. doi: 10.18632/oncotarget.26617
96. Wei X, Li Y, Zhang G, Wang N, Mi M, Xin Y, et al. IL-37 was involved in progress of acute myeloid leukemia through regulating IL-6 expression. Cancer Manage Res. (2021) 13:3393–402. doi: 10.2147/CMAR.S303017
97. Radpour R, Riether C, Simillion C, Höpner S, Bruggmann R, and Ochsenbein AF. CD8(+) T cells expand stem and progenitor cells in favorable but not adverse risk acute myeloid leukemia. Leukemia. (2019) 33:2379–92. doi: 10.1038/s41375-019-0441-9
98. Kang TG, Lan X, Mi T, Chen H, Alli S, Lim SE, et al. Epigenetic regulators of clonal hematopoiesis control CD8 T cell stemness during immunotherapy. Sci (New York NY). (2024) 386:eadl4492. doi: 10.1126/science.adl4492
99. Lee JB, Khan DH, Hurren R, Xu M, Na Y, Kang H, et al. Venetoclax enhances T cell-mediated antileukemic activity by increasing ROS production. Blood. (2021) 138:234–45. doi: 10.1182/blood.2020009081
100. Saadh MJ, Torabi Fard N, Hussein A, Mirzazadeh A, Siavashi M, SeyedMoharami F, et al. Mesenchymal stem cells in the bone marrow microenvironment: a double-edged sword for AML. J Cancer Res Clin Oncol. (2025) 151:193. doi: 10.1007/s00432-025-06244-4
101. Jin P, Shen J, Zhao M, Yu J, Jin W, Jiang G, et al. Driver mutation landscape of acute myeloid leukemia provides insights for neoantigen-based immunotherapy. Cancer letters. (2024) 611:217427. doi: 10.1016/j.canlet.2024.217427
102. Sahib N, Mohamed JS, Rashid M, Jayalakshmi LYC, Lin YC, Chee YL, et al. A combinatorial functional precision medicine platform for rapid therapeutic response prediction in AML. Cancer Med. (2024) 13:e70401. doi: 10.1002/cam4.70401
103. Tettamanti S, Pievani A, Biondi A, Dotti G, and Serafini M. Catch me if you can: how AML and its niche escape immunotherapy. Leukemia. (2022) 36:13–22. doi: 10.1038/s41375-021-01350-x
104. Goulart H, Wei AH, and Kadia TM. Maintenance therapy in AML: what is the future potential? Am J Hematol. (2025) 100 Suppl 2:38–49. doi: 10.1002/ajh.27583
105. Kantarjian HM, DiNardo CD, Kadia TM, Daver NG, Altman JK, Stein EM, et al. Acute myeloid leukemia management and research in 2025. CA Cancer J Clin. (2025) 75:46–67. doi: 10.3322/caac.21873
106. Saadh MJ, KA W, Ashurova D, Sanghvi G, Ballal S, Sharma R, et al. FLT3-mutated AML: immune evasion through exosome-mediated mechanisms and innovative combination therapies targeting immune escape. Expert Rev Anticancer Ther. (2025) 25:143–50. doi: 10.1080/14737140.2025.2461632
107. Bruzzese A, Martino EA, Labanca C, Mendicino F, Lucia E, Olivito V, et al. Advances and challenges in quizartinib-based FLT3 inhibition for acute myeloid leukemia: mechanisms of resistance and prospective combination therapies. Eur J Haematol. (2025) 114(4):584–95. doi: 10.1111/ejh.14383
108. Yamaguchi H. Advances in pathogenesis research and challenges in treatment development for acute myeloid leukemia. Int J hematology. (2024) 120:414–6. doi: 10.1007/s12185-024-03837-6
109. Zhang Y and Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. (2020) 17:807–21. doi: 10.1038/s41423-020-0488-6
110. Greiner J, Ono Y, Hofmann S, Schmitt A, Mehring E, Götz M, et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood. (2012) 120:1282–9. doi: 10.1182/blood-2011-11-394395
111. Struckman NE, de Jong RCM, Honders MW, Smith SI, van der Lee DI, Koutsoumpli G, et al. Hotspot DNA methyltransferase 3A (DNMT3A) and isocitrate dehydrogenase 1 and 2 (IDH1/2) mutations in acute myeloid leukemia and their relevance as targets for immunotherapy. Biomedicines. (2024) 12(5):1086. doi: 10.3390/biomedicines12051086
112. Greiner J, Schneider V, Schmitt M, Gotz M, Dohner K, Wiesneth M, et al. Immune responses against the mutated region of cytoplasmatic NPM1 might contribute to the favorable clinical outcome of AML patients with NPM1 mutations (NPM1mut). Blood. (2013) 122:1087–8. doi: 10.1182/blood-2013-04-496844
113. Greiner J, Mohamed E, Fletcher DM, Schuler PJ, Schrezenmeier H, Gotz M, et al. Immunotherapeutic potential of mutated NPM1 for the treatment of acute myeloid leukemia. Cancers. (2024) 16(20):3443. doi: 10.3390/cancers16203443
114. Graf C, Heidel F, Tenzer S, Radsak MP, Solem FK, Britten CM, et al. A neoepitope generated by an FLT3 internal tandem duplication (FLT3-ITD) is recognized by leukemia-reactive autologous CD8+ T cells. Blood. (2007) 109:2985–8. doi: 10.1182/blood-2006-07-032839
115. Giannakopoulou E, Lehander M, Virding Culleton S, Yang W, Li Y, Karpanen T, et al. A T cell receptor targeting a recurrent driver mutation in FLT3 mediates elimination of primary human acute myeloid leukemia in vivo. Nat Cancer. (2023) 4:1474–90. doi: 10.1038/s43018-023-00642-8
116. Rombola G, Crocchiolo R, Falco M, Iozzi S, Marseglia G, Amoriello R, et al. Selective HLA haplotype loss in npm1-positive acute myeloid leukaemia: A model of immunological escape. Hla. (2025) 105:e70058. doi: 10.1111/tan.70058
117. Jung S, Nelde A, Maringer Y, Denk M, Zieschang L, Kammer C, et al. AML-VAC-XS15-01: protocol of a first-in-human clinical trial to evaluate the safety, tolerability and preliminary efficacy of a multi-peptide vaccine based on leukemia stem cell antigens in acute myeloid leukemia patients. Front Oncol. (2024) 14:1458449. doi: 10.3389/fonc.2024.1458449
118. Vadakekolathu J, Minden MD, Hood T, Church SE, Reeder S, Altmann H, et al. Immune landscapes predict chemotherapy resistance and immunotherapy response in acute myeloid leukemia. Sci Trans Med. (2020) 12(546):eaaz0463. doi: 10.1126/scitranslmed.aaz0463
Keywords: acute myeloid leukemia, gene mutation, immune microenvironment, risk stratification, immunotherapy, neoepitopes
Citation: Guo X, Zhang H, Wang X, Li L and Zhang L (2025) The interaction between common genetic mutations in AML and the immune landscape: mechanisms and implications for immune response. Front. Immunol. 16:1635111. doi: 10.3389/fimmu.2025.1635111
Received: 26 May 2025; Accepted: 25 July 2025;
Published: 11 August 2025.
Edited by:
Avishek Bhuniya, Wistar Institute, United StatesReviewed by:
Matteo Caforio, Bambino Gesù Children’s Hospital (IRCCS), ItalyJasmin Straube, QIMR Berghofer Medical Research Institute, Australia
Copyright © 2025 Guo, Zhang, Wang, Li and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lijuan Li, ZG9jdG9yanVhbkBzaW5hLmNvbQ==; Liansheng Zhang, ZG9jdG9yemhhbmdsc2hAc2luYS5jb20=