 Xiaoying Zhao1†
Xiaoying Zhao1† Xinyue Yang1†
Xinyue Yang1† Yumeng Lin2†
Yumeng Lin2† Ruolan Lei3Wei Ding4Xinying He1Ying Cao1
Ruolan Lei3Wei Ding4Xinying He1Ying Cao1 Dechou Zhang1
Dechou Zhang1 Ping Liu1Meirou Liang5
Ping Liu1Meirou Liang5 Zhongyu Han1,6*Yu Jiang1,7*
Zhongyu Han1,6*Yu Jiang1,7*- 1Department of Gerontology, The Affiliated Traditional Chinese Medicine Hospital, Southwest Medical University, Luzhou, China
- 2Health Management Center, Nanjing Tongren Hospital, School of Medicine, Southeast University, Nanjing, China
- 3Department of Nutrition, The First People’s Hospital of Zigong, Zigong, China
- 4The First Affiliated Hospital of Jinzhou Medical University, Jinzhou, China
- 5The First Clinical Medical College of Shaanxi University of Chinese Medicine, Xi’an, China
- 6Zhongda Hospital, School of Medicine, Southeast University, Nanjing, China
- 7College of Traditional Chinese Medicine, Chongqing College of Traditional Chinese Medicine, Chongqing, China
Cardiovascular diseases (CVDs) pose a significant threat to the health of the elderly population. As the global population ages and medical management remains imperfect, reducing the medical burden of CVDs is of great importance. Aging is a complex process that contributes to the development and progression of CVDs through various mechanisms. The manuscript reviews the mechanisms of aging and their impact on the cardiovascular system. We explore the role of aging in the cardiac microenvironment, highlighting the changes that occur in the heart’s cellular and molecular landscape as a result of the aging process.
1 Introduction
Enhanced living conditions and medical advancements have significantly prolong human lifespan (1). The proportion of the population aged 65 years and over is predicted to increase substantially worldwide by 2030, accounting for approximately 19% of the total population (2). Old age is generally regarded as a major and nonmodifiable risk factor for chronic, life-threatening conditions (3), including CVDs, cancer (4, 5), and neurodegenerative diseases (6) (Figure 1). Among these, CVDs represent the leading cause of mortality among the elderly (7). During body ageing, the accumulation of senescent cells may adversely affect tissue homeostasis (8, 9). Therefore, reducing the accumulation of senescent cells is important for slowing the onset and progression of ageing-related CVDs.
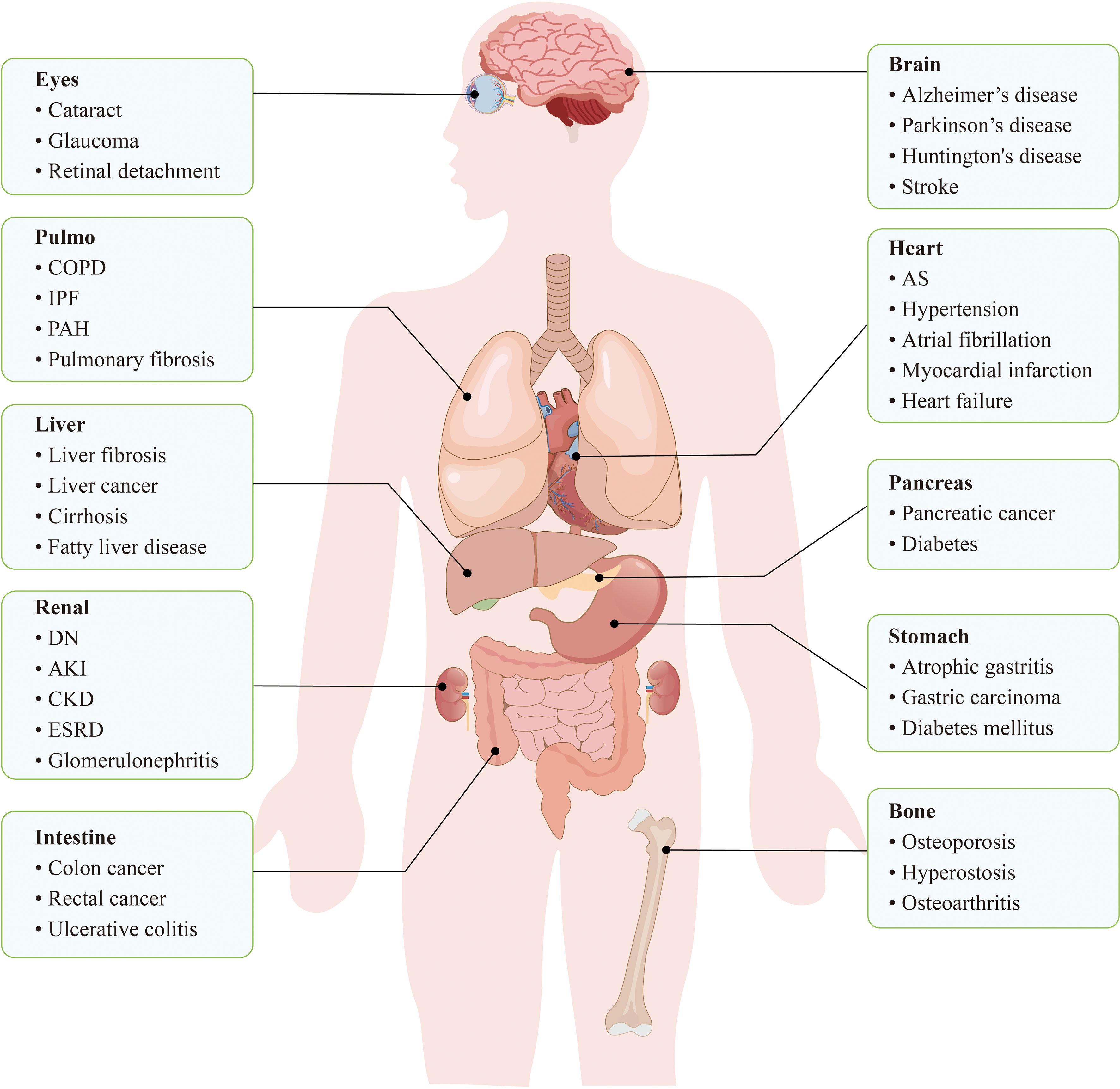
Figure 1. Cellular senescence is closely related to a variety of diseases throughout the body. In the cardiovascular system, cellular senescence leads to dysfunction of ECs, VSMCs, etc., which in turn increases the risk of diseases such as hypertension, AS, and cardiac infarction. In the nervous system, neuronal senescence may trigger neurodegenerative diseases, such as Alzheimer’s disease and Parkinson ‘s disease. In conclusion, cellular senescence accumulates in systemic tissues and becomes a potential trigger for a variety of diseases.
In the cardiac environment, aging emerges as a stress response triggered by numerous stimuli, such as telomere attrition, virus infection, hypoxia, oxidative stress, mitochondrial dysfunction, protein imbalance, and impaired autophagy (10). Increasing evidence illustrates the complex associations between cardiovascular cellular senescence and the pathogenesis as well as progression of CVDs, including atherosclerosis (AS), arterial stiffening, aortic aneurysms, myocardial fibrosis and heart failure (11).
This manuscript discusses the phenotypic expressions and underlying molecular pathways correlated with the ageing process and their contribution to the development of CVDs. Additionally, we assessed the advantages and challenges of targeting senescent cells in preventing and managing ageing-related CVDs. The entire framework of the article is depicted in Figure 2.
Figure 2. Overview of this review.
2 Molecular mechanisms of ageing
The ageing process in the heart is marked by several key molecular mechanisms (Figure 3). Aging hinders tissue regeneration, whereas the accumulation of progerin disrupts nuclear function. Impaired autophagy and mTOR pathway dysfunction lead to the accumulation of damaged cellular components. Mitochondrial issues impact energy metabolism and contribute to oxidative stress. When dysregulated, the cGAS-STING signaling pathway can trigger inflammation. Telomeres shorten with age, triggering a DNA damage response (DDR) that can lead to senescence. The senescence-associated secretory phenotype results in the release of inflammatory factors that degrade the surrounding tissue. Epigenetic changes also influence the expression of genes related to cardiac ageing. Together, these factors contribute to the ageing characteristics of the heart.
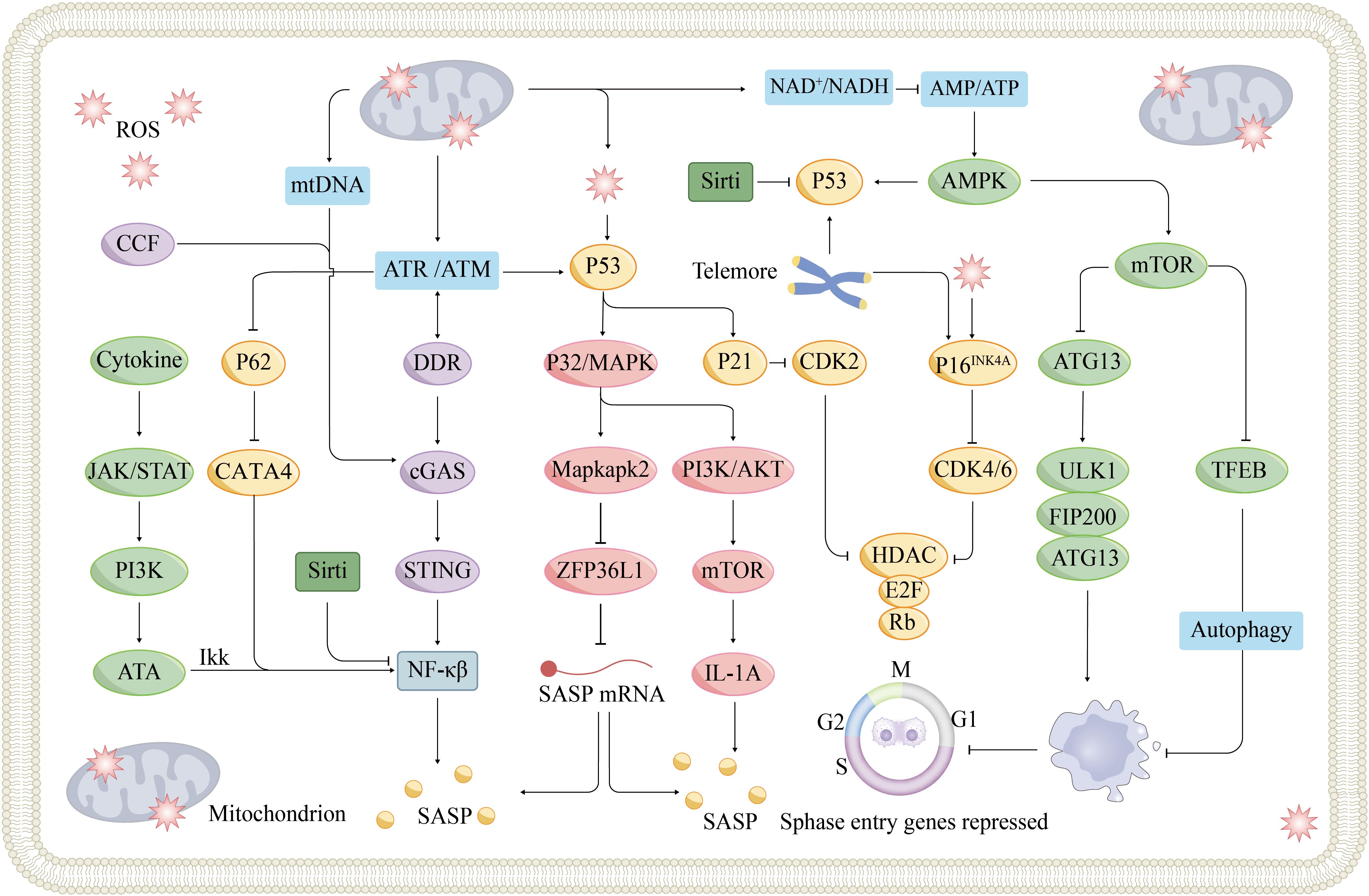
Figure 3. Signaling pathways and mechanisms of cellular senescence. Cellular senescence is a cellular state triggered by stress injury and some physiological processes, which is mainly divided into replicative senescence and non-replicative senescence. Replicative senescence is caused by telomere shortening due to continuous division of normal cells, and when telomeric DNA shortens to a certain extent, cells automatically turn on the ageing program and prevent cell cycle progression through the p53/p21 and p16INK4A/Rb signaling pathways. Non-replicative ageing can be triggered by various stress factors, and nuclear DNA damage is an important role. DNA damage-activated DDR can lead to p53 activation via ATM or ATR kinase activation, which in turn triggers cell cycle arrest. DDR can also induce SASP secretion via the cGAS-STING pathway. SASP components secreted by nearby senescent cells, such as IL-6, trigger the JAK-STAT signaling pathway, the so-called paracrine-induced senescence. In addition, mitochondrial dysfunction has been implicated as a driver of cellular senescence, mainly through three different mechanisms, which have been described in detail previously. ATM, Ataxia-Telangiectasia mutated; ATR, Ataxia-Telangiectasia and Rad3-related protein; CCF, cytoplasmic chromatin fragments; MAPK, mitogen-activated protein kinases; mtDNA, Mitochondrial DNA; ULK1, UNC-51-like kinases 1.
2.1 Cellular senescence and ageing
Cellular senescence, characterized by irreversible exit from the cell cycle and entry into a state of growth arrest, was initially proposed by Hayflick and Moorhead in the 1960s (12, 13). This phenomenon, initially described as limited in the ability of human diploid cells to proliferate in vitro, is now recognized as a response to various stressors, including telomere shortening, oxidative stress, and chromatin structure abnormalities (14). During cellular senescence, the retinoblastoma protein (Rb) is typically dephosphorylated or hypophosphorylated via the p53/p21WAF1/CIP1 or p16INK4a/Rb signaling pathway. This process halts cell cycle progression, ultimately leading to senescence (10).
Senescent cells are characterized by the following traits (15, 16) (1): increased expression and activity of senescence-associated β-galactosidase (SA-β-gal) (2); increased levels of p21 and p16 (3); the presence of nuclear senescence-associated heterochromatin foci (SAHFs) (4); a senescence-associated secretory phenotype (SASP); and (5) an abnormally enlarged cell size and flattened morphology. Among these, the SASP is a distinctive secretory profile specific to senescent cells and is a critical marker of cellular ageing (17). Below, we extensively discuss the molecular mechanisms of ageing that may occur in different cell types.
2.2 Progerin accumulation and ageing
Nuclear structural abnormalities emerging during cellular senescence are dominated by progerin, a truncated form of Lamin A generated by mutations in the LMNA gene (18, 19). Progerin accumulation disrupts nuclear integrity and accelerates the aging process, especially in the cardiovascular system.
Hutchinson – Gilford progeria syndrome (HGPS) is associated with LMNA gene mutations leading to abnormal lamin levels. Patients with HGPS exhibit calcification and abrasion of vascular smooth muscle cells (VSMCs), along with significant adventitial fibrosis, leading to severe premature arteriosclerosis (20). However, progerin overexpression in different cardiac cells leads to different cardiac diseases in the HGPS mouse model. Selective overexpression of VSMC-derived progerin induces endoplasmic reticulum (ER) stress and atherogenesis (AS) (21, 22). Progerin accumulation in endothelial cells(ECs) leads to cardiac fibrosis and cardiac hypertrophy (23). Simultaneously, At the same time, progerin is also elevated in individuals with dilated cardiomyopathy, which is strongly associated with left ventricular remodeling and myocardial ageing (24).
In HGPS mouse model, the massive accumulation of prelamin A (Lamin A precursor) resulting from knockdown of Zmpste24 similarly causes nuclear lamina defects and accelerates VSMC premature senescence (25). In human arteries, prelamin A is prevalent in the media of VSMCs or atherosclerotic lesions in older individuals, whereas it rarely accumulates in young and healthy vessels. Consequently, prelamin A may emerge as a novel biomarker for cardiovascular ageing and may participate in the development of CVDs. Reducing prelamin A/progerin by injecting CRISPR/Cas9 improves HGPS symptoms in mice, which highlights a new therapeutic approach for improving age-induced CVDs (26, 27).
2.3 Impaired autophagy and ageing
Autophagy is able to eliminate misfolded proteins and dysfunctional organelles. Maintaining efficient autophagy is also necessary for many cellular processes associated with lifespan extension (28). Age-related decreases in autophagic activity, attributed to diminished lysosomal function as well as decreased expression of genes associated with autophagy, such as ATG7, contribute substantially to cardiovascular ageing (29–31). In VSMCs, defective autophagy accelerates aging and promotes atherosclerotic plaque formation; whereas in ECs, it exacerbates vascular inflammation and impairs NO bioavailability, thereby aggravating arterial stiffness and hypertension. In cardiomyocytes, impaired autophagy leads to accumulation of dysfunctional mitochondria and damaged proteins, which trigger myocardial fibrosis and contractile dysfunction.
Mice with impaired autophagy exhibit worsened cardiac dysfunction, whereas enhancing autophagy can enhance cardiac function and alleviate age-related heart problems by eliminating proteins with damage, dysfunctional organelles, and altered DNA (32).
Key mechanisms of autophagy include the inhibition of the target of rapamycin (mTOR) or the activation of 5-AMP-activated protein kinase (AMPK) (33). mTOR inhibits autophagy in two ways. First, mTOR directly inhibits unc-51-like kinase 1 (ULK1), which is a critical initiator of the autophagic process. Second, mTOR exerts an inhibitory effect on autophagy by hindering lysosome development, which is facilitated by impeding the nuclear translocation of TFEB (34). mTORC1, a protein complex formed by mTOR, is pivotal in the regulation of translational processes. Inhibition of mTORC1 decelerates the rate of protein translation, increasing the accuracy of mRNA translation into proteins and improving protein folding precision. This process contributes to slowing the ageing process by reducing proteotoxicity and the accumulation of oxidative stress (35, 36). The inhibition of mTOR expression to activate autophagy has been shown to suppress VSMC replicative senescence and stabilize progressive atherosclerotic plaques (37, 38).
2.4 Mitochondrial dysfunction and ageing
Mitochondrial dysfunction is a prominent feature of cellular senescence, primarily driven by dysregulated mitochondrial dynamics, mitochondrial DNA (mtDNA) damage, and oxidative stress (39, 40). Imbalanced mitochondrial dynamics—including hyperfusion (mediated by MFN1/2 and OPA1) and impaired fission (due to reduced DRP1/FIS1 levels)—compromise cardiomyocyte function and promote ageing (41–43). Mitochondria are the factory of cell energy, and their dynamic imbalance will damage the efficiency of ATP synthesis and produce ROS. Excessive ROS generation directly damages ECs and vascular smooth muscle cells, leading to arterial stiffening and plaque vulnerability. In cardiomyocytes, persistent mitochondrial dysfunction impairs ATP generation and activates pro-fibrotic pathways, contributing to maladaptive remodeling and heart failure. Inflammatory signaling triggered by mtDNA release through the cGAS–STING pathway further links mitochondrial senescence to chronic vascular inflammation and AS. Mitochondrial division contributes to the removal of dysfunctional mitochondria by mitophagy. Consequently, disruptions in fission-fusion balance (as evidenced by hyperfusion) accelerates the accumulation of abnormal mitochondria and oxidative proteins and triggers downstream inflammatory signaling pathways. Currently, mitochondria can promote cardiomyocyte senescence through three different mechanisms. Anderson et al. reported that excessive reactive oxygen species (ROS) production directly induces DNA and telomere damage (44, 45). Chung et al. suggested that mtDNA activates the cGAS-STING pathway, thereby stimulating SASP release (45, 46). A third view suggests that mitochondria can act on the AMPK-p53 signaling pathway, thereby accelerating cellular senescence.
2.5 Inflammation and ageing
The cGAS-STING pathway and SASP constitute two interrelated core elements of inflammation, a prominent feature of cardiovascular ageing. Cyclic GMP-AMP synthase (cGAS) can recognize exogenous DNA (bacterial viruses, dead cells, tumor cells, etc.) and endogenous DNA (damaged chromosomes, mitochondria, etc.) and bind to the obtained double-stranded DNA (dsDNA) to form cyclic GMP-AMP (cGAMP) (47, 48). cGAMP binds and initiates the STING protein located in the ER, initiating the activation of its downstream signaling (49, 50). CDNs, DNA damage, ER stress, and inherited gain-of-function mutations in the gene encoding STING can directly activate STING, bypassing the need for cGAMP (51, 52). Activated STING can initiate the phosphorylation and nuclear translocation of IFN regulatory factor 3(IRF3) and nuclear factor-kappa B(NF-κB), which further promotes the synthesis of IFN-I, tumor necrosis factor (TNF), and IL-6 by cells (53). These inflammatory factors are also prominent components of the SASP. Therefore, the cGAS-STING pathway contributes to inflammatory ageing by facilitating the secretion of SASP components from cells.
The mechanism of the SASP involves the activation of transcription factors such as NF-κB, C/EBPβ, and GATA4, which are closely related to the chronic DDR, and the mTOR and p38 MAPK pathways (54). Many SASP-associated genes contain binding sites for NF-kappaB and C/EBPβ b in their cis-regulatory regions, and the upregulation of expression at these sites promotes a positive feedback cycle. This cycle further consolidates the ageing state of cells by communicating with the microenvironment through NOTCH signaling, ROS, the cytoplasmic bridge, and the secretion of small extracellular vesicles (sEVs) (10).
Persistent SASP and cGAS–STING activation fuel chronic vascular inflammation, enhance endothelial dysfunction, destabilize atherosclerotic plaques, and promote myocardial fibrosis, thereby linking cellular senescence to CVDs progression.
2.6 Telomeres and ageing
Telomeres are protective caps at chromosome ends consisted of repetitive TTAGGG sequences and associated proteins that prevent chromosome degradation and fusion (55). These proteins help avoid the recognition of telomeres as DNA damage, initiating the DDR. Telomeres shorten as cell division repeats, and the shielding proteins no longer protect DNA after a critical telomere length is reached, thereby activating the DDR mechanism (56). This process inhibits cell cycle progression by inducing the expression of p21 and p16. The activation of the telomeric DDR (tDDR) also leads to the generation of telomere-associated DDR sites (TAFs) or telomere-induced DNA damage sites (TIFs), which are regarded as markers of tissue ageing and cellular senescence in vitro (57).
Activation of the tDDR and the accumulation of TAFs are also often causally linked to various age-related phenomena, including mitochondrial dysfunction, altered nutrient perception, impaired autophagy, a loss of proteostasis, and epigenetic dysregulation (57). These findings suggest that many ageing hallmarks revolve around a unified “telomere-centric” mechanistic principle (58).
2.7 Epigenetic regulation and ageing
Epigenetics pertains to biological mechanisms that influence gene activity without modifying DNA sequences, impacting gene expression through DNA methylation, histone modifications, and noncoding RNAs (59). DNA methylation changes gene expression through DNA methyltransferases (DNMTs) without altering the DNA sequence, affecting the cell cycle, DNA repair capacity, and cellular processes associated with cellular senescence. Thus, DNA methylation is a marker of ageing and a critical regulator of cellular ageing (60). Research indicates a link between the onset and progression of CVDs such as coronary heart disease (CHD), heart failure, and hypertension with DNA methylation (61, 62).
Modifications to histones have the ability to modify the binding strength between histones and DNA double helices while recruiting various adaptor proteins or effector proteins to remodel chromatin. Sirtuins (SIRTs) represent a group of histone deacetylases that effectively counteract ageing characteristics across various cell types (63). In cardiomyocytes, SIRT3 prevents TGFβ-induced fibrosis by activating GSK3β (64). In ECs, SIRT1 regulates endothelial nitric oxide synthase (eNOS) to mitigate oxidative damage (65) and deacetylates p65 to disrupt the interaction between acetyltransferase P300 and NF-κB, thereby inhibiting NF-kappa B activity (66).
Noncoding RNAs, including microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), are vital in regulating ageing processes and CVDs. For example, when miRNA-22 is actively expressed, it accelerates the ageing and migration of cardiac fibroblasts (CFs) (67). LncRNAs bidirectionally regulate cardiac regeneration and development. Linc1405 and the lncRNAs PANCR and Hdn were found to induce the transformation of CFs into cardiomyocytes, promoting cell differentiation and heart development (68, 69). The repression of cardiac regeneration and differentiation is observed in the presence of the lncRNA CAREL (70). In summary, the regulation of cellular senescence involves various stimulatory factors and pathways (Figure 3).
3 Cardiac ageing microenvironment
Cardiovascular resident cells and immune cells together constitute the cardiac ageing microenvironment. Cardiomyocytes, ECs, VSMCs, and fibroblasts in this microenvironment undergo senescence in this microenvironment, accelerating cardiac structural abnormalities and functional deterioration. The ageing immune microenvironment includes monocytes/macrophages, dendritic cells (DCs), and T cells, which has an impact on tissue homeostasis by modulating the inflammatory response. All these cells and their interactions shape the cardiac ageing microenvironment and influence the resilience and ageing process of the heart.
3.1 Ageing in cardiomyocyte
Multiple cells collaborate to sustain the normal physiological function of the heart. Therefore, the senescence of certain cell types increases the risk of CVDs (Figure 4). Cardiac cells constitute approximately 30%-40% of cardiomyocytes, which are essential for generating the force required for the heart’s pumping function (71). Cardiomyocytes that have reached senescence exhibit DNA damage, ER stress, impaired mitochondrial function, and compromised contractile performance and regulate the microenvironment through the paracrine secretion of the SASP to induce local noncardiomyocyte ageing (72). Many mechanisms that induce cardiomyocyte ageing have been identified, including telomere shortening (73), epigenetic changes (74), and the SASP (44), but metabolic dysfunction is a key factor contributing to cardiomyocyte ageing and decreased cardiac function (75).
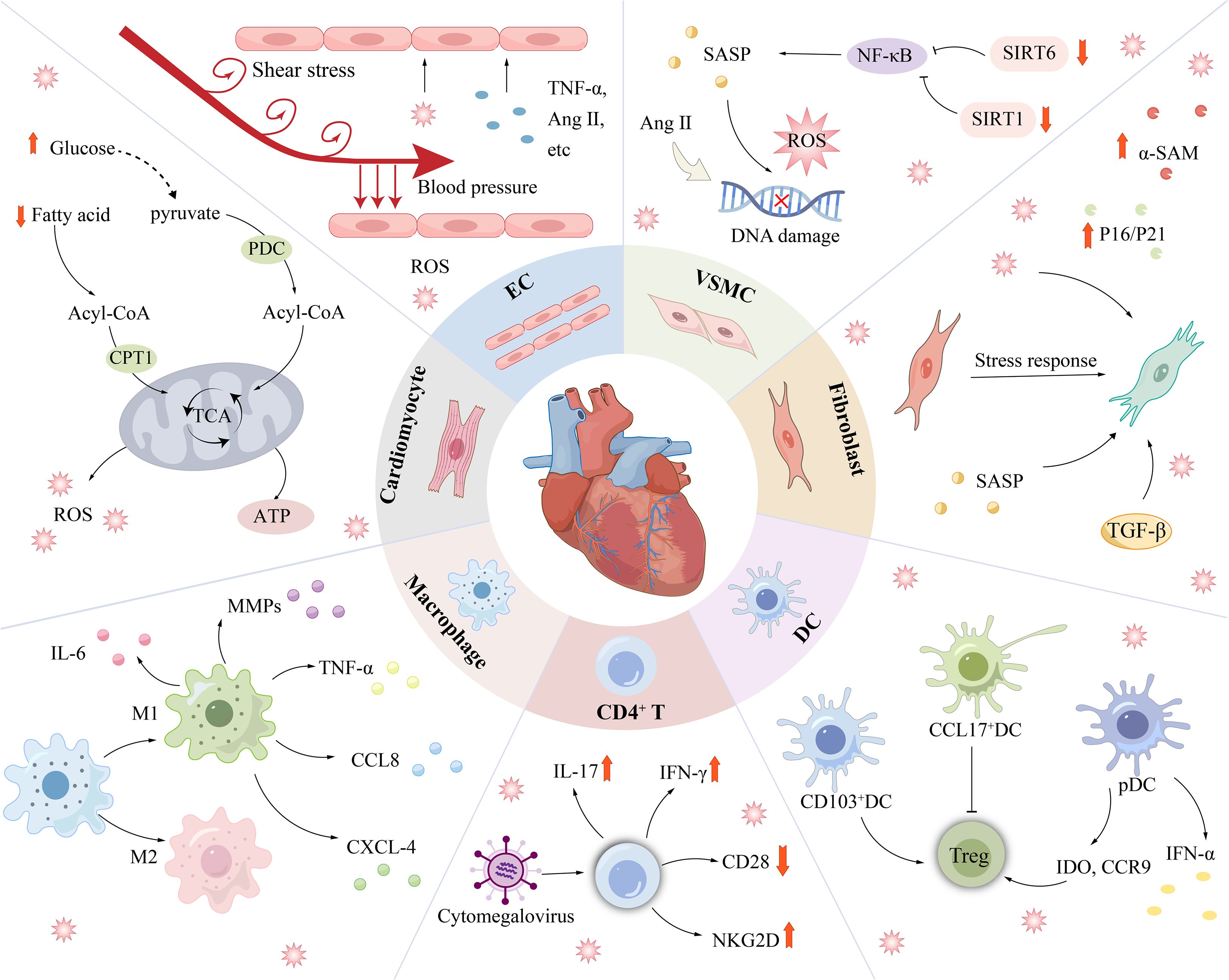
Figure 4. Cardiovascular disease is associated with senescence of a variety of cells. Senescent cardiomyocytes mainly showed decreased fatty acid oxidation ability and enhanced glucose oxidation pathway. ECs are continuously exposed to unique injury-stimulating environments (blood flow pressure, blood flow shear stress, circulating factors, pathogenic stimuli, etc.) and are therefore highly susceptible to injury. In addition to being affected by DNA damage, oxidative stress, etc., SIRT6 deficiency and SIRT1 inactivation can lead to senescence in VSMCs. Under stress conditions, the phenotype of CFs is irreversibly altered, as shown by an increase in ageing markers such as α-SMA. Immune-related cells such as DCs, Macrophages, and T cells regulate the progression of cardiovascular disease mainly through changes in inflammatory factors. α-SMA, myofibroblast marker; Ang II, Angiotensin II; CPT1, carnitine palmitoyl transterase-1; SIRT1/6, Sirtuin 1/6.
Compared with other cells, cardiomyocytes exhibit a distinct metabolic profile, relying primarily on fatty acids and glucose for energy provision. The ratio of fatty acids and glucose in the energy supply is dynamically regulated by developmental, physiological, and pathological responses (76, 77). Fatty acyl-CoA (CoA) and pyruvate serve as the primary substrates for ATP generation in the mitochondria of cardiomyocytes and are produced through the oxidation of fatty acids and glucose, respectively. CoA and pyruvate are regulated mainly by carnitine-palmitoyltransferase-1 (CPT1) and pyruvate dehydrogenase (PDH) (78), which are rate-limiting enzymes in mitochondria. CPT1 levels are significantly reduced during ageing (79) and may lead to cardiac complications (80). A lack of CPT1 exacerbates the ageing process in cardiomyocytes and contributes to lipotoxic cardiac hypertrophy (81).
In addition, the expression levels of peroxisome proliferator-activated receptor α (PPARα) and PGC-lα, which are essential regulators of fatty acid metabolism, decrease with ageing (82). In mice prone to accelerated ageing, decreases in PPARα mRNA and protein levels lead to increases in ceramide levels, which correlate with the development of cardiac hypertrophy (83). Aged hearts demonstrate a reduced capacity for fatty acid oxidation and rely more on augmented glucose oxidation pathways to meet their metabolic needs (84).
The insulin signaling pathway is essential for glucose metabolism in cardiomyocytes and can be activated by insulin growth factor receptor (IGFR) to induce the SASP and promote cardiomyocyte senescence (85). More importantly, metabolic dysfunction impairs mitochondrial function, impacting all substrates, including increasing ROS generation (86). Defective mitochondria persist in the heart, leading to exacerbated oxidative stress and injury, alongside the activation of oxidative signaling pathways.
Senescent cardiomyocytes display diminished contractile function and disrupted conduction patterns, resulting in cardiomyopathy and arrhythmias (87). Alterations in the mitochondrial membrane potential and telomere shortening were observed in cardiomyocytes from mice with Duchenne muscular dystrophy (DMD), suggesting cellular senescence (88). Anthracyclines cause a dilated cardiomyopathy phenotype linked to cardiomyocyte senescence, as shown by increased mtDNA levels (89). In addition, ageing rat cardiomyocytes display a decreased mitochondrial membrane potential, increased ROS levels, and an attenuated ability to undergo electrical pacing, indicating an increased risk of arrhythmia (90).
3.2 Ageing in endothelial cell
ECs are highly active monolayers that line the inner layers of blood vessels and cover the inner surface of the entire circulatory system (71). ECs not only form the vascular barrier, which helps maintain blood flow, but also regulate vascular tone and blood pressure by synthesizing vasoactive substances and growth factors (91). However, ECs are highly susceptible to injury because they are located between circulating blood and semisolid tissues and are continuously exposed to unique injury–irritating environments (hemodynamically generated pressures, circulating factors, pathogenic stimuli, etc.). One of the consequences of EC injury is cellular senescence, which causes impaired vasodilation and vascular dysfunction. Senescent ECs can be observed in the hearts of patients with diseases such as AS, heart failure, and aneurysms (92). EC senescence is mainly caused by oxidative stress and vascular inflammation. The senescence of ECs is expedited by metabolic factors such as hyperuricemia or dysregulation of the RAAS (93). Many molecules and pathways, such as SIRT, Klotho, RAAS, IGFBP, NRF2, and mTOR, are associated with promoting EC senescence (94).
Aging and impaired function of ECs play critical roles in the development of CVDs. SIRT6 deficiency, miR-217 overexpression or NOX activity accelerate EC senescence, leading to AS (95, 96). In addition, EC senescence can mediate thrombosis by increasing plasminogen activator inhibitor-1 (PAI-1) (97). Heart failure with a preserved ejection fraction (HFpEF) represents a category of age-related CVDs closely linked to EC senescence and myocardial fibrosis (98). More importantly, mouse models of accelerated ageing have shown that EC senescence contributes to HFpEF, as evidenced by diastolic dysfunction, interstitial fibrosis, left atrial dilation, and left ventricular hypertrophy (92).
The incidence of atrial fibrillation (AF) is higher in older individuals. The onset of AF correlates with the senescence of ECs and fibroblasts (99, 100). The downregulation of eNOS and abnormalities in miRNAs are associated with EC dysfunction (101). Angiotensin II (Ang II) can induce EC senescence through an AT1R-mediated pathway, increasing ROS generation, inflammation, extracellular matrix remodeling, and vascular tone (102, 103).
3.3 Ageing in vascular smooth muscle cell
VSMCs are vital for regulating vascular wall tension and maintaining blood pressure (104). VSMC senescence promotes arterial stiffness and arterial calcification, leading to reduced arterial compliance and elastic reservoir dysfunction, which are the pathological foundations of diseases such as hypertension and independent risk factors for heart failure (105). Senescent VSMCs considerably influence AS development (106) and are closely associated with aortic aneurysm (107), pulmonary hypertension (108), and fibrotic neointima formation (109).
Telomere shortening, DNA damage, oxidative stress, and autophagic dysfunction can all cause VSMC senescence (110). The activation of SIRT family proteins plays a multifaceted antiaging role (111), and SIRT6 deficiency and SIRT1 inactivation can lead to senescence in VSMCs (112). Abnormal processing of Prelamin A to lamin A results in defects in the nuclear layer, increasing the vulnerability of DNA to damage and accelerating cellular senescence (25). Moreover, sustained DNA damage signals promote the transformation of VSMCs into osteoblastic vascular smooth muscle cells, leading to subsequent vascular calcification and AS (113, 114).
Interestingly, the replicative senescence of VSMCs mediates their phenotypic transformation through runt-related transcription factor-2 (RUNX-2) and induces age-related medial arterial calcification (115). In addition, senescent VSMCs exhibit elevated levels of inflammatory cytokines and reduced expression of anti-inflammatory factors (116). IL-1a activates the SASP in local cells and increases IL-6 secretion, inducing local inflammation in the cardiac microenvironment (117).
Like other heart cells, VSMC senescence also leads to CVDs, most commonly AS. Matthews et al. detected a large amount of senescent VSMCs in atherosclerotic fibrous caps (118). Compared with normal VSMCs, plaque VSMCs are distinguished by shorter telomeres, higher p16 and p21 expression, stronger SAβ-gal activity, and a flatter cell morphology. In addition, telomere shortening in intimal VSMCs is positively correlated with the severity of AS. VSMC senescence also leads to plaque instability, resulting in myocardial infarction (MI) and stroke. This instability may be related to the secretion of MCP1, MIP1a/b, and CCL3/4, which promote the accumulation of monocytes, macrophages, and lymphocytes (119, 120). Ang II also induces premature VSMC senescence, thereby accelerating the development of AS (103). The overexpression of TRF2 decreases DNA damage and inhibits senescence in VSMCs, thereby attenuating plaque vulnerability (119).
Additionally, VSMC senescence may also participate in the pathophysiological processes of pulmonary arterial hypertension through the SASP (121). The existing literature suggests that VSMC senescence is associated with the development of aortic aneurysms. Liao et al. were the first researchers to document that medial VSMCs from patients with AAA display enhanced replicative senescence. Compared with VSMCs from the same patient’s inferior mesenteric artery (IMA), AAA-derived VSMCs are more extensive and rounder, and their proliferative capacity is significantly diminished (122). Angiotensin converting enzyme, Ang II, and RAS accelerate VSMC ageing and lead to the formation of AAAs by stimulating the production of proinflammatory cytokines, ROS, and the ageing phenotype in VSMCs (123).
3.4 Ageing in cardiac fibroblasts
CFs are important components of cardiac noncardiomyocytes. CFs maintain the extracellular matrix (ECM) structure and adhesion integrity by expressing integrins and matrix metalloproteinases (MMPs) (124). In addition, CFs can also participate in paracrine secretion to regulate the hypertrophy, proliferation, growth, and ageing of surrounding cells (125).
Under stress conditions, CFs change their phenotype and transform into myofibroblasts. CFs undergo irreversible senescence upon sustained stimulation by stressors. Notably, the expression of ageing biomarkers such as p16 and p21 is significantly increased in the hearts of mice following MI (126). Costaining of α-SMA (a marker for myofibroblasts) with p53 or p16 revealed an increased presence of senescent fibroblasts within the border zone of the infarct (127). Similarly, senescent fibroblasts have been detected in mouse models of cardiac hypertrophy and remodeling (128). In conclusion, senescent fibroblasts are ubiquitous in fibrotic areas and are involved in the pathological processes associated with myocardial fibrosis.
CF senescence has a dual impact on cardiac health. On the one hand, as cardiac fibroblasts enter a senescent state, their ability to secrete collagen decreases, which may delay the initial stage of the wound healing process. On the other hand, fibrosis can be reduced and cardiac function can be improved by inducing CF senescence. Conversely, if the natural ageing process of fibroblasts is delayed, it may exacerbate the degree of myocardial fibrosis and ultimately lead to cardiac dysfunction. Therefore, balancing the ageing of fibroblasts is essential for maintaining heart health.
Following acute MI, the activated cardiac fibroblast phenotype undergoes dynamic changes from an inflammatory to a noninflammatory state, driving extracellular matrix regulation and ultimately supporting scar formation (129). Premature ageing of CFs reduces the production of ECM components, such as collagen, and may lead to the inhibition of reparative fibrosis in wounds during healing. However, under chronic pressure loading, the premature ageing of CFs may play a protective role by reducing ECM deposition and preventing excessive fibrosis, thereby preventing further decreases in cardiac stiffness and function (127, 128). Furthermore, the overexpression of matricellular protein (CCN1) may induce CF senescence, thereby reducing myocardial fibrosis and enhancing cardiac function post-MI, thus playing a beneficial role in acute ischemia (84). These findings suggest potential positive effects of fibroblast senescence in some cases.
However, some studies indicate that the beneficial effects of CF senescence require a balance with the potentially deleterious effects of ageing. Gavin D. Richardson et al. found that following myocardial ischemia/reperfusion injury (IRI), biological processes associated with fibrosis and inflammation are attenuated upon the administration of the antiaging agent navitoclax, thereby improving cardiac function and reducing the scar size (130).
Interestingly, NEIL3 is an enzyme involved in DNA repair processes that minimizes oxidative damage to DNA by recognizing and removing oxidized bases. CFs proliferate excessively in the hearts of Neil3 −/− mice, but the risk of cardiac rupture remains (131). DNA damage caused by Neil3 deletion may initiate the ageing phenotype in the cardiac microenvironment via SASP-mediated paracrine signaling. This process increases MMP2 expression, leading to ECM degradation and, ultimately, cardiac rupture (131, 132). Cardiac fibrosis tends to increase with age and is correlated with HFpEF (133). Some molecules, such as miR-1468-3p and SIRT6, promote the ageing of CFs by regulating TGF-β1 signaling, which in turn increases the occurrence of myocardial fibrosis (134). These studies suggest that regulating the ageing balance of fibroblasts is crucial for treating CVD.
3.5 Ageing in monocyte/macrophage
Stoneman et al. showed that the quantity of monocytes/macrophages significantly promote the development of atherosclerotic plaques, including increasing the collagen content in plaques and the formation of necrotic cores (135). Monocytes undergo a metabolic shift toward glycolysis and enhance pro-inflammatory signaling upon stimulation with oxidized low-density lipoprotein (ox-LDL) (136). Within the intima, these monocytes differentiate into macrophages under macrophage colony-stimulating factor (M-CSF) regulation (137). The resulting M1 macrophages promote inflammatory responses through abundant secretion of growth factors and cytokines, particularly TNF-α and IL-1β - two central mediators of atherosclerosis-related inflammatory pathways (138). These activated M1 macrophages further stimulate CFs via the Smad3 signaling pathway by releasing profibrotic factors (particularly TGF-β1), thereby upregulating collagen and MMPs production, which ultimately leads to abnormal extracellular matrix deposition and remodeling (139). In contrast, M2 macrophages exhibit anti-inflammatory properties through IL-4, IL-13, and IL-10 secretion.
Ageing macrophages have a greater effect on plaque formation. Senescent macrophages can undergo polarization towards the M1 phenotype and release SASP factors, including TNF-α, IL-6, IL-1β, CCL2, and MMP 9, the collagenase enzyme. In addition, senescent macrophages have impaired efferocytosis capacity, increasing the expansion and vulnerable plaque shape of necrotic cores (140). Their collective actions contribute to the accelerated advancement of atherosclerotic plaques (141). Senescent macrophages accumulate in the subendothelial area during the early stage of AS and drive the pathological development of AS by increasing the expression of inflammatory cytokines and chemokines. In the late stages of AS, macrophages increase plaque instability, which is characteristic of elastic fiber fragmentation and fibrous cap thinning, by increasing metalloproteinases (142).
3.6 Ageing in Dendritic cell
In CVDs, DCs act as antigen-presenting cells (APCs) to influence the progression of AS by regulating Tregs (143). Senescent DCs exhibit downregulated expression of MHC-I/II molecules, leading to impaired T cell activation and compromised immune surveillance functions. However, different DC subsets present within the vessel wall each have unique functions, which reflect their diversity and complexity in CVDs. For example, CD103+ DCs are present in the normal arterial wall and exert anti-AS effects mainly by inducing Tregs, whereas CCL17+ DCs exert pro-AS effects mainly by limiting Treg production. In Ldlr−/− mice, impaired autophagy in CD11b+ DCs due to Atg16l1 deficiency promotes aortic CD4+ Treg cells expansion and reduced AS (144). The role of pDCs in regulating AS is also complex. On the one hand, pDCs promote Treg differentiation by releasing indoleamine 2,3-dioxygenase (IDO) and chemokine (C-C motif) receptor 9 (CCR9), thereby producing IL-10 and mitigating AS progression (145). On the other hand, pDCs also accelerate AS formation by producing IFN-α.
Notably, senescence is associated with increased DC activation and lipid contents in DCs compared with the characteristics of DCs in young adult and aged mice. The regulation of lipid accumulation and activation of DC subsets may be attributed to the decrease in the response to infection with ageing (146). Although increased accumulation of DCs and Tregs has been reported in the murine atherosclerotic intima, the role of senescent DCs in CVD development remains unclear (147).
3.7 Ageing in T cell
During the development of AS, antigen-presenting cells (APCs) present antigens produced from components such as LDL to naïve CD4+T cells. This process results in the stimulation of antigen-specific CD4+T cells and the secretion of the proinflammatory cytokines IFN-γ and TNF or the anti-inflammatory cytokine IL-10 to regulate macrophage polarization (148). Therefore, T cells are bifaceted in the regulation of the establishment and stability of atherosclerotic plaques, which can not only exert beneficial inhibitory effects but also contribute to facilitating the formation of plaques.
Ageing T cells are associated with CVD pathological progression. In older individuals, an increase in CD4+ T-cell populations with high expression levels of IL-17 and IFN-γ has been observed. These cells also display characteristics commonly associated with ageing, such as decreased CD28 expression and elevated NKG2D levels. Interestingly, these changes are strongly linked to metabolic risk factors for CVDs (149). Recent findings have shown that cytomegalovirus (CMV) seropositivity, a widely recognized driver of T-cell senescence, is closely linked to the incidence of CHD. Additionally, there is a positive correlation between CMV seropositivity and the risk of stroke, MI, and mortality from CVDs (150, 151).
Furthermore, the presence of aged T cells in the bloodstream is linked to disease relapse and the emergence of additional CVDs among individuals diagnosed with acute coronary syndrome (152). Indeed, the detrimental impact of ageing-related T cells on CVDs has been documented in mice. Specifically, in a mouse model of hypertension induced by Ang II, the introduction of T cells from aged mice into young recipients expedited cardiac and renal damage through an increase in IFN-γ secretion, thereby fostering inflammation and fibrosis. A recent study revealed that ageing-related cardiovascular changes, such as aortic dilatation, partial rupture, and myocardial dysfunction, developed in a mouse model of premature T-cell failure due to mitochondrial dysfunction (153). The results of this study suggest that the presence of aged T cells may directly impact the progression of CVDs.
4 Endocrine ageing and sex-specific differences in cardiovascular ageing
Endocrine ageing, a core aspect of the biology of aging, has garnered increasing attention. Research indicates that the decline in estrogen, testosterone, growth hormone (GH), and thyroid hormone (TH) levels is closely associated with cardiovascular dysfunction, increased vascular stiffness, elevated inflammation, and myocardial remodeling. Furthermore, sex differences are evident throughout the spectrum of cardiovascular disease. Women experience relatively stronger cardiovascular protection before menopause, but this risk rises rapidly post-menopause. In contrast, men exhibit a higher vascular risk profile due to age-related declines in androgen from midlife onward. These findings suggest a significant interaction between endocrine aging and biological sex differences in the process of cardiovascular ageing.
4.1 Estrogen and cardiovascular protection
Sufficient literature demonstrates that estrogen exerts multiple protective effects on the cardiovascular system, including promoting vasodilation, protecting endothelial function, improving lipid metabolism, reducing inflammation and mitigating oxidative stress (154). Estrogen primarily exerts its pleiotropic protective effects through nuclear receptors (ERα/ERβ) and membrane-associated receptors (GPER).
In the regulation of vascular tone, ERα rapidly activates eNOS through the PI3K/Akt signaling pathway, mediating the rapid release of NO from ECs (155).NO serves as a crucial vasodilator that effectively dilates blood vessels, improves endothelial function, and exerts anti-atherosclerotic effects. Conversely, reduced NO levels diminish vascular antioxidant capacity and exacerbate inflammatory responses.
Estrogen exerts a positive regulatory effect on lipid metabolism. It enhances the production of high-density lipoprotein (HDL) by inhibiting hepatic lipase activity and accelerates the clearance of low-density lipoprotein (LDL) through upregulation of LDL receptor expression. During the menopausal transition, decreased estrogen levels accompanied by a relative increase in androgen levels may lead to disordered lipid metabolism, thereby increasing the risk of AS (156). Estrogen regulates lipid metabolism mainly through genomic and non-genomic effects mediated by estrogen receptors (ERs). Among them, ERα mainly promotes the transport of cholesterol from peripheral tissues (such as arterial wall macrophages) to the liver by regulating apolipoprotein E (APOE) and cholesterol reverse transporter ABCA1/ABCG1, thereby enhancing HDL biosynthesis (157).
In addition, estrogen plays a role in vascular protection through various mechanisms such as anti-oxidation, promoting NO production and inhibiting inflammatory signaling pathways. Activation of ERα can inhibit NF-κB and NLRP3 inflammasome signaling pathways, reduce the release of inflammatory factors such as IL-6 and TNF-α, thereby reducing vascular endothelial inflammation (158). ERβ inhibits LDL oxidative modification by enhancing the activity of superoxide dismutase (SOD) and glutathione peroxidase (GPx), reducing the accumulation of reactive ROS (159).
Although a large amount of evidence supports that estrogen has a protective effect on the cardiovascular system, the clinical application of its alternative therapy (ERT) is still controversial. A number of large-scale clinical trials have suggested that ERT may increase the risk of stroke and thromboembolic events, so its benefits and safety should be carefully evaluated in translational applications (160).
4.2 Androgens and cardiovascular homeostasis
The effect of androgen on CVDs is a complex and controversial topic. However, most studies suggest that elevated TES levels have a protective effect on the cardiovascular system.
Early clinical studies have found that the incidence of hypertension and coronary artery disease in men is higher than that in premenopausal women, thus forming the view that TES and other androgens may be detrimental to cardiovascular health (161). However, the latest clinical and animal research evidence overturns the traditional understanding that androgens have significant benefits for male blood pressure and metabolism - both of which are key risk factors for CVDs (162). Systematic follow-up evaluations of early epidemiological investigations, clinical studies, and animal experiments revealed that these initial studies had many methodological flaws in experimental design, model selection, and data analysis (163, 164). Epidemiological studies have shown that low androgen levels are an independent risk factor for CVDs (165, 166). Low TES is often accompanied by lipid metabolism disorders, insulin resistance and central obesity.
The protective effect of TES on the heart is mainly manifested in its diastolic vascular function and endothelial protection. The core mechanism of TES relaxing blood vessels is to activate cGMP-PKG signaling pathway by promoting NO synthesis, and then open BKca channel (167, 168).In rat aortic tissue, TES significantly enhances NO synthesis through the androgen receptor and calcium influx, whereas the calcium channel blocker verapamil attenuates TES-induced NO production (169). Cardiovascular ageing is closely associated with reduced NO synthesis in ECs. Androgens help counteract this process by promoting eNOS activity and NO production, thereby enhancing the antioxidant capacity of ECs—a mechanism aligned with cardiovascular anti-ageing pathways.
At physiological levels, androgens can improve endothelial function and enhance antioxidant capacity. However, supraphysiological doses may lead to adverse effects, such as hypertensive heart disease, increased risk of venous thrombosis, and recurrence in patients with prostate cancer (170, 171). However, meta-analyses have also indicated that TES replacement therapy is safe in the short to medium term, with no higher risk of cardiovascular events compared to men not receiving TES treatment (172). In summary, current clinical evidence is insufficient to support the beneficial effect of androgen replacement therapy on CVDs, and further large-scale clinical trials are needed to evaluate its efficacy and safety.
4.3 Growth hormone and the cardiovascular system
With the increase of age, the secretion of growth hormone (GH) decreases gradually. Some elderly people have age-related GH deficiency. Since the GH/IGF-1 axis plays a critical role in the development and functional regulation of the cardiovascular system, reduced GH secretion is considered to be closely associated with metabolic disorders and an increased risk of CVDs.
The GH/IGF-1 axis maintains cardiac structure and metabolic homeostasis by promoting myocardial gene expression, enhancing amino acid uptake and protein synthesis, and regulating cardiomyocyte size. It upregulates muscle protein mRNA, augments type I calcium channel activity, improves calcium sensitivity, and increases Ca ² -ATPase levels, thereby optimizing calcium handling and contractility (173). Physiological GH/IGF-1 signaling is crucial for normal heart mass and function.
In the vascular system, GH/IGF-1 receptors are widely expressed. Experimental studies indicate that GH/IGF-1 exerts angiogenic factor-like effects by inducing the proliferation and migration of vascular endothelial cells and promoting the formation of new capillaries (174). Furthermore, it enhances vascular endothelial function and regulates vasomotion through stimulating NO synthesis, thereby playing a key role in maintaining vascular homeostasis.
In addition, GH exerts metabolic effects including promoting protein synthesis, stimulating lipolysis, and suppressing glucose utilization. It also modulates vascular tone, thereby influencing peripheral resistance and blood pressure. Consequently, abnormal GH secretion not only contributes to metabolic disorders but may also disrupt blood pressure homeostasis, elevating the risk of atherosclerosis and other cardiovascular diseases.
Clinical evidence indicates that GH replacement therapy improves the lipid profile (reducing LDL-C and increasing HDL-C), restores vascular endothelial function, and lowers inflammatory markers—such as high-sensitivity C-reactive protein, IL-6, and TNF-α—in patients with growth hormone deficiency, while also reducing carotid intima-media thickness (175). Some studies further suggest that GH treatment can enhance cardiac function, exemplified by reduced left ventricular end-systolic volume and improved ejection fraction (176). However, large-scale prospective clinical trials using cardiovascular events as primary endpoints are still lacking, and the long-term cardiovascular benefits of such therapy require further validation (177).
4.4 Thyroid hormone and cardiovascular system
Thyroid hormones (TH) play a critical role in maintaining cardiovascular homeostasis by regulating heart rate, myocardial contractility, and systemic vascular resistance. Thyroid dysfunction is frequently observed in patients with CVDs, with subclinical hypothyroidism (SCH) being the most common form (178). Epidemiological evidence consistently indicates that the prevalence of overt hypothyroidism and SCH increases with advancing age and is strongly associated with dyslipidemia, hypertension, diabetes, and other cardiovascular risk factors (179–181).
Thyroid dysfunction affects cardiovascular function by altering the levels of T3, T4 and TSH. T3 binds to nuclear thyroid hormone receptors (TRs) in cardiomyocytes, promoting the synthesis of contractile proteins such as myosin heavy chain V3, and enhances myocardial contractility by upregulating β1-adrenergic receptor expression (182). In addition, T3 increases intracellular cAMP levels, which upregulates Ca2+-ATPase activity and thereby improves diastolic relaxation. Moreover, thyroid hormones can activate the PI3K/AKT signaling pathway to stimulate NO production in vascular endothelial cells, ultimately reducing systemic vascular resistance (183).
Thyroid hormone also regulates lipid metabolism by acting on genes such as the LDL receptor (184). TSH is positively associated with elevated lipids, insulin resistance, and hyperglycemia. TSH not only affects lipid metabolism indirectly by regulating TH levels, but also acts directly on hepatic TSH receptors to activate cAMP/PKA/CREB signaling pathways and promote cholesterol synthesis. This explains the phenomenon that SCH patients have elevated lipids despite normal TH levels (185).
Levothyroxine is a commonly used drug for the treatment of hypothyroidism. Available studies have shown that levothyroxine replacement appears to improve left ventricular function, endothelial function, and lipid metabolism and partially reverse the pathological effects of hypothyroidism on the cardiovascular system (186). However, there remains a lack of consistent evidence for its cardiovascular benefit in SCH patients, which needs to be verified by further large-scale prospective studies.
In general, these changes in hormone levels directly or indirectly contribute to the development and progression of CVDs mainly through the regulation of lipid metabolism, the impact of inflammatory factors, and the SASP. From the therapeutic perspective, although hormone replacement therapy (HRT) and selective estrogen receptor modulators (SERMs) can improve endothelial function and lipid metabolism disorders, their long-term safety remains controversial. Although TES replacement therapy is increasingly active, the results of studies on cardiovascular outcomes are variable and require strict weighing of risks versus benefits. Compared with systemic sex hormone intervention, targeting the clearance of senescent cells and SASP may more accurately and safely intervene in the endocrine ageing process.
5 Targeting aging to alleviate CVDs
A large body of data suggests that ageing cardiovascular cells add to and accelerate the development and progression of CVDs. Hence, the targeted clearance of senescent cells represents a promising therapy for averting or managing age-related ailments such as CVDs (187). While senescent cells can originate from various tissues, diseases, and cell types, they exhibit common ageing mechanisms and biochemical characteristics, which opens the possibility of treating or delaying ageing-related diseases by removing senescent cells. As early as 2004, a report noted that the burden of senescent cells in mammals is inversely proportional to their healthy lifespan. This insight has prompted researchers to explore the development of targeted therapies to eradicate these ageing cells (188). Since then, the therapeutic elimination of senescent cells has emerged as a groundbreaking strategy to decelerate ageing and potentially inhibit disease progression.
5.1 Drug therapy
5.1.1 Senolytics: selective removal of senescent cells
Senolytics, compounds designed to target and eliminate senescent cells selectively, facilitate this process primarily by inhibiting antiapoptotic factors. In 2015, the Kirkland trial at the Mayo Clinic in the United States first reported the first group of senolytics, dasatinib and quercetin (Table 1) (206). Dasatinib, a commonly employed medication for leukemia treatment in clinical settings, effectively inhibits both Bcr-Abl fusion gene I and Src tyrosine kinase (207). Quercetin, a flavonol compound, can suppress PI3K activity, increase SIRT1–213 expression (208) and impede mTOR signaling (209). The combination of dasatinib and quercetin (D+Q) enhances the clearance of senescent cells and promotes improvements in cardiac function and carotid vascular reactivity in older mice.
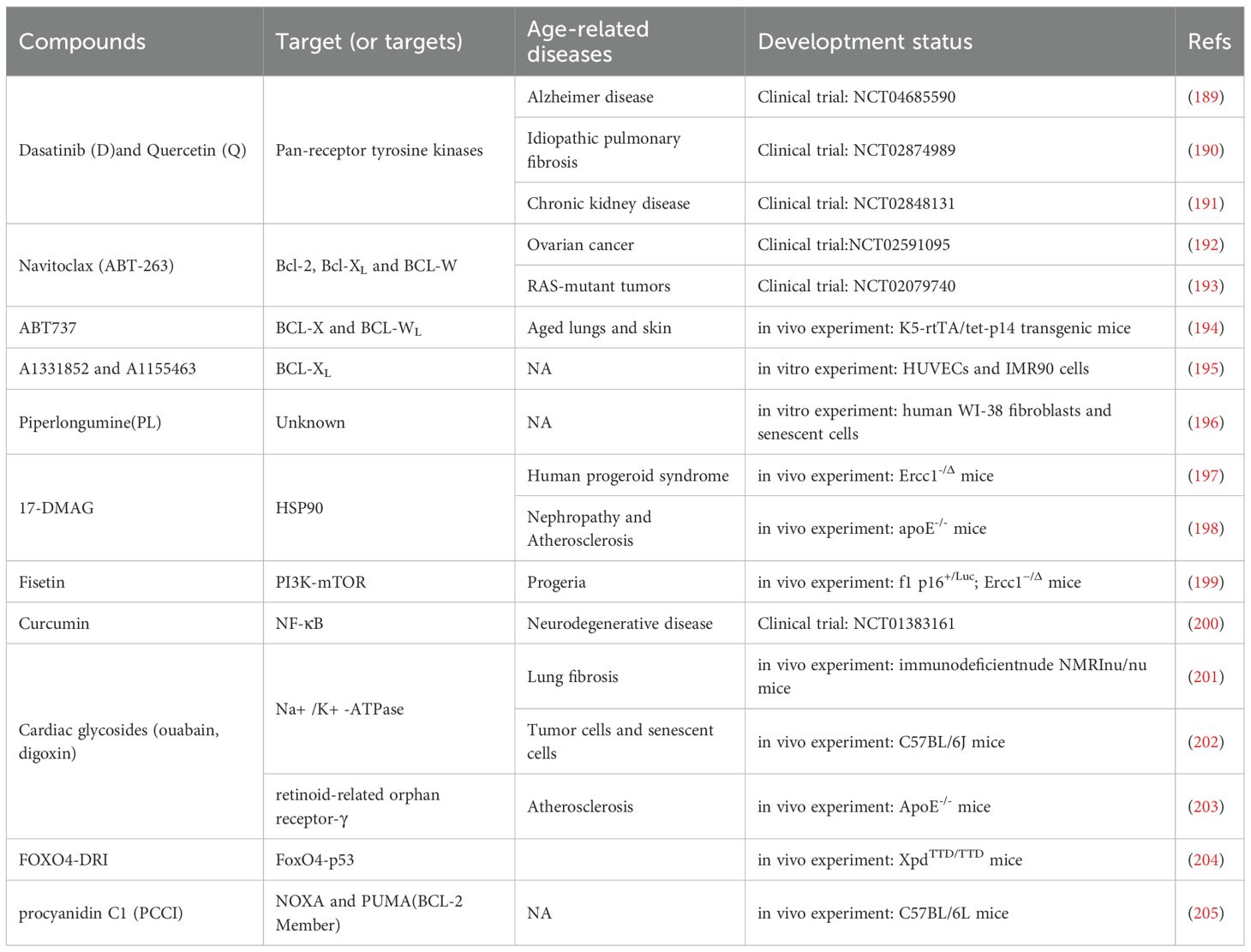
Table 1. Senolytics treatment in age-related diseases.
Interestingly, at the time, the Kirkland team noticed an essential phenomenon: the activity of proapoptotic pathways increased significantly in senescent cells. Based on this result, they proposed a bold hypothesis: senescent cells rely on senescent cell antiapoptotic pathways (SCAPs) to antagonize apoptosis, thus allowing them to eventually survive (206). The theoretical hypothesis of SCAPs proposed by the Kirkland team at the time was confirmed by a series of subsequent studies in multiple laboratories; at the same time, many novel senolytics emerged based on this feature of senescent cells (Table 1).
Subsequently, navitoclax (ABT-263), an inhibitor of the synthetic BCL-2 protein family (which includes Bcl-2, Bcl-XL, and Bcl-w), was identified as a third-generation senolytic drug (210, 211). Experiments performed by Childs et al. demonstrated that the depletion of senescent cells by ABT-263 (navitoclax) significantly inhibited AS in the aortic arch of Ldlr−/−mice (142). ABT-263 also promotes the clearance of senescent cardiomyocytes, thereby reducing myocardial fibrosis and cardiomyocyte hypertrophy (212). ABT-263 administration in mice with simulated MI alleviates myocardial remodeling, enhances diastolic function, and increases the overall survival of aged mice (213).
Piperlongumine (PL) is also a senolytic that promotes apoptosis in senescent cells. PL kills WI-38 fibroblasts, but does not induce ROS generation, by inducing apoptosis (196). The combined use of PL with ABT-263 resulted in enhanced antiaging activity. These findings suggest that we can reduce the dose of ABT-263 when administered in combination with the other two drugs, significantly reducing the adverse effects of ABT-263. However, the antiaging mechanism of PL needs to be clarified. Notably, senescent cells share survival traits with cancer cells. Thus, PL has shown promise in inducing apoptosis in these cells by suppressing the PI3K/Akt/mTOR signaling pathway (206).
In 2017, scientists such as Kirkland discovered that drugs such as fisetin and the BCL-XL inhibitors A1331852 and A1155463 also have basic antiaging effects (195). In recent years, a growing array of senolytics with antiaging potential has been identified, including sexual small molecules, natural products and their key components, as well as peptide inhibitors designed to target known SCAPs (e.g., FOXO4-DRI) (204). FOXO4-DRI can interfere with the interplay between FoxO4 and p53 in senescent cells and trigger apoptosis in senescent but unhealthy cells by releasing and activating p53.
Interestingly, most reported senolytics appear to clear only one or several specific types of senescent cells. For example, fisetin explicitly triggers programmed cell death in aged human umbilical vein endothelial cells (HUVECs). However, it does not have any senescence-inducing effects on aged IMR90 cells, human lung fibroblast lines, or primary human preadipocytes (195). Navitoclax, A1331852, and A1155463 exhibit the ability to trigger programmed cell death in aged HUVECs and IMR90 cells but show limited efficacy in inducing apoptosis in senescent preadipocytes (214). In contrast, dasatinib selectively induces apoptosis in senescent human preadipocytes more efficiently than in HUVECs (206). Individual senolytic drugs have different effects even when they act on a specific type of cell. For example, navitoclax has apoptosis-inducing effects on senescent embryonic fibroblasts such as IMR-90 cells. However, its efficacy is relatively low for senescent primary lung fibroblasts (211). Hence, accurately defining or drawing conclusions about the generalizability and effectiveness of particular senolytics without thorough empirical examinations is difficult.
A recent study revealed that procyanidin C1 (PCC1) can safely and efficiently clear various cell types and senescent cells generated by different senescence triggers (205). In addition, PCC1 significantly improved the physiological function and lifespan of ageing mice, and the creatinine, body weight, urea and immunity of the mice were not affected throughout the process. Phytochemical senolytics of natural origin, similar to PCC1, deserve in-depth exploration as potential antiaging agents.
5.1.2 Senomorphics: SASP inhibition
The SASP contributes to both the generation of senescent cells and the enhancement of senescence within the microenvironment through paracrine and autocrine signaling mechanisms. Senomorphics, which inhibit the SASP without killing senescent cells, are another approach to alleviate tissue disturbances, organ regression, and body ageing caused by cellular ageing.
Senomorphics can lower SASP expression levels in senescent cells either directly or indirectly. This process is achieved by inhibiting various transcription factors, such as NF-κB, the JAK2/STAT3 signal transduction pathway, the TRAF6/TAK1 inflammatory signal transduction pathway, the mTOR protein kinase, and other signaling pathways involved in inducing and sustaining the SASP (215).
Prior research has demonstrated that NF-κB inhibitors can reduce the expression of proinflammatory components of the SASP, especially cytokines and chemokines (216). Resveratrol and epigallocatechin gallate (EGCG) are both NF-κB inhibitors. The former downregulates the levels of SASP-related proinflammatory cytokines such as IL-8 and TNF-α by inhibiting the SIRT1/NF-κB signaling pathway (217). The latter can directly downregulate the production of TNF-α and IL-6 in 3T3-L1 preadipocytes (218).
Similar natural compounds include naringenin, apigenin, pterostilbene, kaempferol, and catechin, which are relatively safer than synthetic compounds and have better application prospects (219, 220). Rapamycin and its analogues (rapalogs), on the other hand, reduce SASP expression levels by inhibiting mTOR activity and can prolong the healthy lifespan and overall lifespan of mice (221, 222). Metformin, a drug that effectively treats the symptoms of individuals with type 2 diabetes mellitus (T2DM), can inhibit SASP expression and alleviate age-related chronic diseases (223). It impedes tumor development by reducing SASP production through the inhibition of IKK/NF-κB activity (224). Ruxolitinib, a tyrosine kinase inhibitor, is a JAK1/JAK2-STAT3 pathway-targeting agent that inhibits the development and progression of the SASP in vitro and in vivo (225). In a population of older individuals diagnosed with myelodysplastic syndrome and a median age of 65 years, the administration of ruxolitinib alleviated the intensity of asthenia symptoms, encompassing factors such as weight, strength, and excessive appetite (226).
However, the issue that arises from the inhibition of intracellular pro-SASP signaling is the potential increase in cancer risk due to the disruption of SASP factor expression. For example, in mouse lymphoma models, downregulation of the SASP by the inhibition of NF-κB attenuates immune surveillance following therapeutic ageing and synergizes with p53 insufficiency to lead to ageing escape, resulting in treatment resistance and relapse (216). Future in-depth clinical studies specifically addressing these issues are still needed.
Although so many senolytics and senomorphics have been found, most of them are still in the stage of in vitro and animal experiments (Table 1). Animal models offer the advantage of rapidly validating theories and mechanisms, but their limitation lies in the gap from the pathological ageing environment in humans. More importantly, in the same aging research, different laboratories may use natural aging mice, transgenic models, drug-induced models, etc., resulting in significant differences in the effect of drugs on scavenging senescent cells. Moreover, the disease course in animal models progresses much faster than human natural ageing, which may exaggerate drug efficacy or obscure long-term adverse effects. In addition, many senolytics or senomorphics demonstrate promising results in animal studies, but their effectiveness is highly dependent on cell type. For instance, navitoclax effectively eliminates senescent VSMCs in mice, yet shows limited efficacy in other cell types.
At present, clinical trials of anti-ageing drugs targeting CVDs have not yet been initiated and remain largely confined to metabolic diseases or osteoarthritis. Although theoretical foundations and animal experiments provide important mechanistic insights, the results should be interpreted with caution when translated into clinical practice. The development of anti-aging drugs specifically targeting CVDs will still require a long process of exploration.
5.2 Increasing immune surveillance of senescent cells
Under physiological conditions, senescent cell clearance primarily relies on apoptosis and immune-mediated mechanisms. However, most senescent cells acquire anti-apoptotic properties, rendering the immune system crucial for their elimination. Currently, two principal immunotherapeutic strategies exist for cardiac injury repair: molecular-level interventions targeting IL-1β to mitigate inflammatory responses, and cellular-level approaches utilizing senescence-specific ligands to direct immune cell-mediated recognition and clearance.
The IL-1β-IL6-CRP axis constitutes a central inflammatory pathway in atherosclerosis and cardiovascular disease, where IL-1β serves as the upstream regulator that activates the NLRP3 inflammasome to induce ECs expression of adhesion molecules, thereby promoting inflammatory cell recruitment and macrophage activation. Furthermore, IL-1β enhances IL-6 production, which stimulates hepatic synthesis of CRP, fibrinogen, and plasminogen activator inhibitors (227). The CANTOS clinical trial demonstrated that IL-1β inhibition with canakinumab significantly reduced cardiovascular risk in patients with elevated inflammation (hs-CRP >2 mg/dL) independent of LDL modulation, but there were limitations of increased risk of infection and no improvement in mortality (228). Conversely, the broad-spectrum anti-inflammatory drug methotrexate showed no cardiovascular benefit, confirming the need for pathway-specific interventions (229). These findings highlight the therapeutic potential of developing novel interventions against specific pathogenic pathways in AS, including chemokine-receptor interactions, immune checkpoint, immunemetabolic modulation, and hormonal/lipid mediator networks, which may collectively overcome the limitations of conventional anti-inflammatory strategies while providing more precise therapeutic effects (230).
Emerging findings indicate that various immune cells, such as macrophages, NK cells, neutrophils, and cytotoxic T cells, are involved in the immunosurveillance of senescent cells (231). Senescent cells recruit corresponding immune cells for recognition and clearance by expressing different ligands on their surface. For example, senescent IMR-90 fibroblasts increase the expression of MICA and ULBP2, the corresponding ligands of the NK cell-activating receptor NKG2D, which triggers their targeted clearance by NK cells (232). Furthermore, specific markers such as major histocompatibility complex class II (MHCII) molecules may be expressed by senescent cells, enabling their precise identification and subsequent elimination by CD4+ T cells within the immune system (233). However, how these immune cells clear apoptotic or senescent cardiovascular cells remains unknown. Notably, chimeric antigen receptor (CAR)-T cells may be a potential immune surveillance tool for ageing.
The efficacy and specificity of cytotoxic T cells decline with age, despite their crucial roles in identifying and eradicating foreign entities within the human body. CAR-T cells represent a form of live-cell therapy that allows T cells to more precisely identify cancer cell surface markers by introducing chimeric antigen receptors (CARs) onto the surface of T cells using genetic engineering techniques. This technology has already shown considerable efficacy in the treatment of a range of cancers (234) and is FDAI1-approved for treating certain leukemias and lymphomas (235). In recent years, CAR-T-cell therapy has been considered to target the elimination of noncancerous cells, such as senescent cells.
Fortunately, CAR-T cells have progressed successively as antiaging drugs. High expression of fibroblast-activating protein (FAP) in CFs leads to myocardial fibrosis and myocardial disease. A reversal of cardiac fibrosis and restoration of function were observed in mice exposed to Ang II and phenylephrine following the adoptive transfer of FAP-targeting CD8+ T cells generated using CAR-T-cell technology (236). Corina Amor et al. reported that urokinase-type plasminogen activator receptor (uPAR), a cell membrane protein, is significantly upregulated with age. They have also successfully documented the efficacy of CAR-T cells targeting uPAR in clearing senescent cells both in vivo and in vitro (237). UPAR-CAR-T cells improve exercise capacity, reverse liver fibrosis, and ameliorate metabolic dysfunction in aged mice and mice fed a high-fat diet (237).
Unlike senolytics, which are not system-specific and require long-term repeated administration, uPAR-CAR-T cells demonstrate enhanced targeted clearance and can achieve long-term therapeutic and preventive effects with a single low dose administration (238). These findings confirm the strong therapeutic activity of antiaging CAR-T cells in addressing ageing-related disorders. Notably, XuDong Zhao et al. recently reported that NKG2D ligand (NKG2DL) was upregulated in senescent cells (239). Accordingly, the team developed NKG2D-CAR-T-cell therapy that selectively targets the consumption of NKG2DL-expressing senescent cells in mice and juvenile nonhuman animals while improving the function of multiple organs. Nevertheless, pertinent evidence indicating that ageing cardiovascular cells can produce NKG2DL is insufficient. In summary, the utilization of specialized CAR-T cells for the targeted elimination of aged cardiovascular cells holds great potential as a viable approach.
5.3 Cell replacement
Specific induction conditions can facilitate the differentiation of stem cells into contractile cardiomyocytes, ECs, and smooth muscle cells so that myocardial contractile function, vascular regeneration, and myocardial regeneration are enhanced, thereby improving cardiac function. Consequently, stem cell therapy represents a promising method for replenishing regenerative cells. Currently, induced pluripotent stem cells (iPSCs), mesenchymal stem cells (MSCs), cardiac stem cells (CSCs) and embryonic stem cells (ESCs) are among the primary types of stem cells employed for treating CVDs. Among these cells, the clinical application of ESCs is constrained by ethical considerations and the potential for immune rejection.
IPSCs are cells with self-renewal and pluripotent differentiation abilities obtained from autologous mature somatic cells after reprogramming. Since Takahashi et al. discovered iPSCs in 2006, their potential therapeutic effects on diseases, especially CVDs, have been explored (240). Animal experiments indicated that iPSCs could successfully differentiate into vascular ECs, cardiomyocytes and VSMCs. Furthermore, injection of iPSCs into ischemic myocardial tissue of rats has been shown to increase cardiac ejection fraction and reduce fibrosis (241, 242). Another study revealed that the integration of iPSC-derived cardiomyocytes, ECs, and VSMCs into the ischemic myocardium of pigs via intramyocardial microsphere transplantation enhanced the left ventricular ejection fraction, improved myocardial metabolism, and reduced the infarct size (243). In addition to the above animal experiments, Osaka University officially performed a phase I clinical trial of hiPSC-CM myocardial patches in January 2020 to assess their safety and potential efficacy in the hearts of patients with ischemic cardiomyopathy (237). The above evidence suggests that iPSC-CMs can be used as a new method for cardiac regenerative therapy.
MSCs are a subset of adult stem cells with the capacity to differentiate into mesodermal derivatives (chondrocytes, osteocytes, and adipocytes), have powerful multilineage differentiation potential and self-renewal ability, and have been widely used to alleviate ageing-related diseases. MSCs primarily treat ischemic CVDs through the following mechanisms (1): MSCs stimulate the proliferation and differentiation of cardiac cells, as well as angiogenesis (240) (2). MSCs promote cardiac repair and reduce myocardial apoptosis by secreting growth factors and exerting paracrine effects (244). Clinical trials of MSCs in CVD treatment are also more mature, multiple trials have been completed, and the expected results have been obtained.
In 2005, Hare et al. first used MSC transplantation to treat MI, and this study yielded crucial findings regarding the safety and effectiveness of allogeneic bone marrow stem cell applications (245). A clinical trial conducted in 2015 involved a controlled, multicenter randomized study of patients with chronic ischemic cardiomyopathy. The aim of this study was to evaluate the safety and efficacy of intramyocardial transplantation of allogeneic human MSCs derived from the umbilical cords of different individuals (246). In addition, Bartolucci et al. assessed the safety and effectiveness of administering intravenous infusions of MSCs derived from human umbilical cords to individuals diagnosed with chronic heart failure (247).
The above experiments demonstrated the safety of MSC transplantation as well as the efficacy of improving myocardial perfusion after MI. However, current clinical trials in CVD patients are still at a very early stage, and some potential risks associated with the systemic application of MSCs, such as embolism and inflammation, still exist. In addition, the potential differences in efficacy between MSCs from different sources must be circumvented and elucidated in future studies.
Whether CSCs can be used for the treatment of CVDs is controversial. In 2003, Beltrami et al. concluded that endogenous stem cells exist in the heart and that c-Kit+ cardiomyocytes cultured in vitro, enriched, and injected into necrotic cardiomyocyte areas were able to repair most necrotic areas and improve cardiac systolic function (248). The study had significant repercussions in academia, followed by the successive discovery of CSCs with different surface markers, and the locations and proportions of various CSC distributions have varied (249, 250).
However, the same approach was used by Jesty et al. (251) but did not replicate the findings of Beltrami et al. (248) that c-Kit+ CSCs can be converted into cardiomyocytes in the infarcted myocardium of adults. Van Berlo et al. (252) also questioned the role of CSCs in treating MI reported by Beltrami et al. In 2018, after an investigation by Harvard University and other relevant authorities, Beltrami et al. were suspected of fabricating data and paper fraud, which basically halted clinical trials of CSCs for the treatment of CVDs.
However, in some clinical trials, such as the SCIPIO trial (253) and the CADUCEUS trial (250), an intracoronary injection of endogenous CSCs has been observed to enhance the left ventricular ejection fraction, reduce the size of the myocardial infarct and the amount of scar tissue, and enhanced regional systolic function in patients with myocardial ischemia. Therefore, although CSCs with different surface markers cannot differentiate into cardiomyocytes, the paracrine effects of these cells can potentially enhance the movement, growth, specialization, and angiogenesis by cardiac stem cells within the body. Moreover, they can enhance the recruitment of endogenous CSCs, hinder cell apoptosis in the infarct region, inhibit myocardial remodeling, and thus improve cardiac function (254, 255). Given that adult cardiomyocytes still possess a relatively sluggish capacity for cell division, enhancing their ability to proliferate into cardiomyocytes and replace necrotic cardiomyocytes following myocardial ischemia could emerge as a prominent area of focus in future research.
5.4 Other factors
In addition to pharmacological interventions, increasing evidence suggests that modifiable lifestyle and environmental factors play an important role in modulating cardiac ageing.
Exercise has emerged as an effective strategy for the prevention and rehabilitation of CVDs. Exercise promotes mitochondrial biogenesis through AMPK regulation, increases cellular energy metabolism, and enhances functional reserve in the cardiovascular system. In addition, regular aerobic exercise can reduce oxidative stress in ECs and suppress aging-related inflammatory processes (256).
Dietary patterns significantly influence the ageing process. Both caloric restriction (CR) and intermittent fasting (IF) have been shown to delay aging through telomere lengthening and modulation of key signaling pathways including AMPK, PKB/AKT, and mTOR (257). Adherence to the EAT-Lancet dietary pattern - characterized by increased consumption of vegetables, fruits, whole grains, and nuts, along with reduced intake of animal-derived foods, red meat, added sugars, and saturated fats - has been associated with decelerated biological aging and extended life expectancy (258). This dietary approach provides abundant bioactive compounds such as omega-3 fatty acids, antioxidants (vitamin C, carotenoids, and polyphenols), zinc, and vitamin D, which exert multi-faceted anti-ageing effects. Its mechanisms of action mainly include enhancing innate and adaptive immune function, reducing oxidative stress damage, and improving cellular metabolic homeostasis, thereby effectively delaying aging-related inflammatory processes (259). The potential synergy between dietary interventions and senolytic therapies represents an emerging research frontier. Interestingly, β-hydroxybutyrate (β-HB) may serve as a crucial mediator connecting ketogenic diet, intermittent fasting (IF), and exercise with extended health span. The underlying mechanisms involve its anti-inflammatory properties, attenuation of vascular aging processes, and maintenance of immune homeostasis through CD8+T cell regulation (260).
Conversely, environmental toxins significantly accelerate aging processes through sustained genotoxic stress. Chronic exposure to airborne particulate matter (PM2.5) promotes DNA damage, micronuclei formation, and cGAS activation (261). Notably, smoking cessation represents a key lifestyle intervention that reduces inflammation and improves immune function (262).
6 Conclusions
With scientific and technological advancements and the evolution of society, the ageing population trend has become an inevitable social and medical problem in various countries around the world. Ageing research has experienced unprecedented momentum and potential in recent years. Previous findings indicate that the accumulation of senescent cells potentially plays a role in the progression of pathological states in different regions of the cardiovascular system. The rapid development of antiaging drugs and ageing intervention technologies will significantly benefit various aspects, such as human health, medical progress, and socioeconomic status. Despite these advancements, the comprehension of the specific molecular mechanisms underlying cardiovascular cell senescence remains limited, for example, how the ageing of a single cardiac cell type leads to a specific disease phenotype and how to screen highly selective markers for ageing cardiovascular cells (10). These conditions have hindered the development of effective methods to prevent or treat CVDs.
Investigational treatments are currently being explored with the goal of achieving overall suppression of senescence and/or clearance of senescent cells. Compared with senomorphics, which transiently reduce SASP levels, senolytics have rapidly become a reasonably effective and advantageous therapeutic strategy to prevent, delay, or reduce various age-related diseases and organ dysfunctions in recent years (263). Although multiple clinical trials on senolytic interventions are currently being conducted, none have targeted CVDs. In the future, more extensive randomized controlled trials must be conducted to accurately assess and ensure medication safety and treatment benefits and validate the preliminary results of early clinical trials. In addition, gaining a more comprehensive comprehension of the molecular mechanisms underlying immune response is imperative. Furthermore, the specific identification and targeting of ageing cardiovascular cells are crucial. Additionally, advancements in genetic, epigenetic, or metabolomic mechanisms related to cardiac cell senescence may provide enhanced personalized therapeutic options for individuals suffering from CVDs.
Author contributions
XZ: Writing – original draft. XY: Writing – original draft. YL: Writing – original draft. RL: Writing – original draft. WD: Writing – original draft. XH: Writing – original draft. YC: Writing – original draft. DZ: Writing – original draft. PL: Writing – original draft. ML: Writing – original draft. ZH: Writing – original draft, Writing – review & editing. YJ: Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The work was supported by Sichuan Provincial Department of Science and Technology Scientific Research Project (2022YFS0613); Sichuan Provincial Administration of Traditional Chinese Medicine (2024MS208); Sichuan Provincial Department of Science and Technology Scientific Research Project (2022YFS0618); Sichuan Provincial Department of Science and Technology Scientific Research Project (2022ZDZX0022); The Science and Technology Strategic Cooperation Programs of Luzhou Municipal People’s Government and Southwest Medical University(2024LZXNYDJ042); High-Level Breakthrough Project - Innovation Research on Traditional Chinese Medicine and the Practical Platform Construction Project for Characteristic Traditional Chinese Medicine Talents (2023120015;2023CZJ002).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Foreman KJ, Marquez N, Dolgert A, Fukutaki K, Fullman N, McGaughey M, et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: reference and alternative scenarios for 2016–40 for 195 countries and territories. Lancet. (2018) 392:2052–90. doi: 10.1016/s0140-6736(18)31694-5
2. Kontis V, Bennett JE, Mathers CD, Li G, Foreman K, and Ezzati M. Future life expectancy in 35 industrialised countries: projections with a bayesian model ensemble. Lancet. (2017) 389:1323–35. doi: 10.1016/s0140-6736(16)32381-9
3. Niccoli T and Partridge L. Ageing as a risk factor for disease. Curr Biol. (2012) 22:R741–52. doi: 10.1016/j.cub.2012.07.024
4. Hosseini L, Shahabi P, Fakhari A, Zangbar HS, Seyedaghamiri F, Sadeghzadeh J, et al. Aging and age-related diseases with a focus on therapeutic potentials of young blood/plasma. Naunyn Schmiedebergs Arch Pharmacol. (2023) 397:1–13. doi: 10.1007/s00210-023-02657-5
5. Shakeri H, Lemmens K, Gevaert AB, De Meyer GRY, and Segers VFM. Cellular senescence links aging and diabetes in cardiovascular disease. Am J Physiol Heart Circ Physiol. (2018) 315:H448–h62. doi: 10.1152/ajpheart.00287.2018
6. Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch SG, Croteau DL, et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. (2019) 15:565–81. doi: 10.1038/s41582-019-0244-7
7. Roth GA, Johnson C, Abajobir A, Abd-Allah F, Abera SF, Abyu G, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. (2017) 70:1–25. doi: 10.1016/j.jacc.2017.04.052
8. Sapieha P and Mallette FA. Cellular senescence in postmitotic cells: beyond growth arrest. Trends Cell Biol. (2018) 28:595–607. doi: 10.1016/j.tcb.2018.03.003
9. von Zglinicki T, Wan T, and Miwa S. Senescence in post-mitotic cells: A driver of aging? Antioxid Redox Signal. (2021) 34:308–23. doi: 10.1089/ars.2020.8048
10. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
11. Gude NA, Broughton KM, Firouzi F, and Sussman MA. Cardiac ageing: extrinsic and intrinsic factors in cellular renewal and senescence. Nat Rev Cardiol. (2018) 15:523–42. doi: 10.1038/s41569-018-0061-5
12. Hayflick L and Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. (1961) 25:585–621. doi: 10.1016/0014-4827(61)90192-6
13. Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. (1965) 37:614–36. doi: 10.1016/0014-4827(65)90211-9
14. Mao Z, Ke Z, Gorbunova V, and Seluanov A. Replicatively senescent cells are arrested in G1 and G2 phases. Aging (Albany NY). (2012) 4:431–5. doi: 10.18632/aging.100467
15. Hernandez-Segura A, Nehme J, and Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. (2018) 28:436–53. doi: 10.1016/j.tcb.2018.02.001
16. Matjusaitis M, Chin G, Sarnoski EA, and Stolzing A. Biomarkers to identify and isolate senescent cells. Ageing Res Rev. (2016) 29:1–12. doi: 10.1016/j.arr.2016.05.003
17. Zhang L, Pitcher LE, Yousefzadeh MJ, Niedernhofer LJ, Robbins PD, and Zhu Y. Cellular senescence: A key therapeutic target in aging and diseases. J Clin Invest. (2022) 132:e158450. doi: 10.1172/jci158450
18. Cisneros B, García-Aguirre I, De Ita M, Arrieta-Cruz I, and Rosas-Vargas H. Hutchinson-gilford progeria syndrome: cellular mechanisms and therapeutic perspectives. Arch Med Res. (2023) 54:102837. doi: 10.1016/j.arcmed.2023.06.002
19. Kim PH, Luu J, Heizer P, Tu Y, Weston TA, Chen N, et al. Disrupting the linc complex in smooth muscle cells reduces aortic disease in a mouse model of hutchinson-gilford progeria syndrome. Sci Transl Med. (2018) 10:eaat7163. doi: 10.1126/scitranslmed.aat7163
20. Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, et al. Cardiovascular pathology in hutchinson-gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. (2010) 30:2301–9. doi: 10.1161/atvbaha.110.209460
21. Hamczyk MR, Villa-Bellosta R, Gonzalo P, Andrés-Manzano MJ, Nogales P, Bentzon JF, et al. Vascular smooth muscle-specific progerin expression accelerates atherosclerosis and death in a mouse model of hutchinson-gilford progeria syndrome. Circulation. (2018) 138:266–82. doi: 10.1161/circulationaha.117.030856
22. Hamczyk MR, Villa-Bellosta R, Quesada V, Gonzalo P, Vidak S, Nevado RM, et al. Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol Med. (2019) 11:e9736. doi: 10.15252/emmm.201809736
23. Osmanagic-Myers S, Kiss A, Manakanatas C, Hamza O, Sedlmayer F, Szabo PL, et al. Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J Clin Invest. (2019) 129:531–45. doi: 10.1172/jci121297
24. Messner M, Ghadge SK, Goetsch V, Wimmer A, Dörler J, Pölzl G, et al. Upregulation of the aging related lmna splice variant progerin in dilated cardiomyopathy. PloS One. (2018) 13:e0196739. doi: 10.1371/journal.pone.0196739
25. Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, et al. Prelamin a acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. (2010) 121:2200–10. doi: 10.1161/circulationaha.109.902056
26. Beyret E, Liao HK, Yamamoto M, Hernandez-Benitez R, Fu Y, Erikson G, et al. Single-dose crispr-cas9 therapy extends lifespan of mice with hutchinson-gilford progeria syndrome. Nat Med. (2019) 25:419–22. doi: 10.1038/s41591-019-0343-4
27. Santiago-Fernández O, Osorio FG, Quesada V, Rodríguez F, Basso S, Maeso D, et al. Development of a crispr/cas9-based therapy for hutchinson-gilford progeria syndrome. Nat Med. (2019) 25:423–6. doi: 10.1038/s41591-018-0338-6
28. Anding AL and Baehrecke EH. Cleaning house: selective autophagy of organelles. Dev Cell. (2017) 41:10–22. doi: 10.1016/j.devcel.2017.02.016
29. Leidal AM, Levine B, and Debnath J. Autophagy and the cell biology of age-related disease. Nat Cell Biol. (2018) 20:1338–48. doi: 10.1038/s41556-018-0235-8
30. Sasaki Y, Ikeda Y, Iwabayashi M, Akasaki Y, and Ohishi M. The impact of autophagy on cardiovascular senescence and diseases. Int Heart J. (2017) 58:666–73. doi: 10.1536/ihj.17-246
31. Grootaert MO, da Costa Martins PA, Bitsch N, Pintelon I, De Meyer GR, Martinet W, et al. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. (2015) 11:2014–32. doi: 10.1080/15548627.2015.1096485
32. Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, and Sadoshima J. Aging and autophagy in the heart. Circ Res. (2016) 118:1563–76. doi: 10.1161/circresaha.116.307474
33. Hansen M, Rubinsztein DC, and Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. (2018) 19:579–93. doi: 10.1038/s41580-018-0033-y
34. Yang M, Lu Y, Piao W, and Jin H. The translational regulation in mtor pathway. Biomolecules. (2022) 12:802. doi: 10.3390/biom12060802
35. Syntichaki P, Troulinaki K, and Tavernarakis N. Eif4e function in somatic cells modulates ageing in caenorhabditis elegans. Nature. (2007) 445:922–6. doi: 10.1038/nature05603
36. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, and Benzer S. Regulation of lifespan in drosophila by modulation of genes in the tor signaling pathway. Curr Biol. (2004) 14:885–90. doi: 10.1016/j.cub.2004.03.059
37. Luo Z, Xu W, Ma S, Qiao H, Gao L, Zhang R, et al. Moderate autophagy inhibits vascular smooth muscle cell senescence to stabilize progressed atherosclerotic plaque via the mtorc1/ulk1/atg13 signal pathway. Oxid Med Cell Longev. (2017) 2017:3018190. doi: 10.1155/2017/3018190
38. Tan P, Wang YJ, Li S, Wang Y, He JY, Chen YY, et al. The pi3k/akt/mtor pathway regulates the replicative senescence of human vsmcs. Mol Cell Biochem. (2016) 422:1–10. doi: 10.1007/s11010-016-2796-9
39. Chapman J, Fielder E, and Passos JF. Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett. (2019) 593:1566–79. doi: 10.1002/1873-3468.13498
40. Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. (2016) 23:303–14. doi: 10.1016/j.cmet.2015.11.011
41. Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, et al. Mitochondrial fission and fusion mediators, hfis1 and opa1, modulate cellular senescence. J Biol Chem. (2007) 282:22977–83. doi: 10.1074/jbc.M700679200
42. Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, and Cho H. Loss of march5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. (2010) 123:619–26. doi: 10.1242/jcs.061481
43. Song M, Franco A, Fleischer JA, Zhang L, and Dorn GW 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab. (2017) 26:872–83.e5. doi: 10.1016/j.cmet.2017.09.023
44. Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. (2019) 38:e100492. doi: 10.15252/embj.2018100492
45. Chung KW, Dhillon P, Huang S, Sheng X, Shrestha R, Qiu C, et al. Mitochondrial damage and activation of the sting pathway lead to renal inflammation and fibrosis. Cell Metab. (2019) 30:784–99.e5. doi: 10.1016/j.cmet.2019.08.003
46. Maekawa H, Inoue T, Ouchi H, Jao TM, Inoue R, Nishi H, et al. Mitochondrial damage causes inflammation via cgas-sting signaling in acute kidney injury. Cell Rep. (2019) 29:1261–73.e6. doi: 10.1016/j.celrep.2019.09.050
47. Barber GN. Sting-dependent cytosolic DNA sensing pathways. Trends Immunol. (2014) 35:88–93. doi: 10.1016/j.it.2013.10.010
48. West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. (2015) 520:553–7. doi: 10.1038/nature14156
49. Sun L, Wu J, Du F, Chen X, and Chen ZJ. Cyclic gmp-amp synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
50. Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic gmp-amp is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. (2013) 339:826–30. doi: 10.1126/science.1229963
51. Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, et al. Sting senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell. (2017) 171:809–23.e13. doi: 10.1016/j.cell.2017.09.034
52. Yin Q, Tian Y, Kabaleeswaran V, Jiang X, Tu D, Eck MJ, et al. Cyclic di-gmp sensing via the innate immune signaling protein sting. Mol Cell. (2012) 46:735–45. doi: 10.1016/j.molcel.2012.05.029
53. Oduro PK, Zheng X, Wei J, Yang Y, Wang Y, Zhang H, et al. The cgas-sting signaling in cardiovascular and metabolic diseases: future novel target option for pharmacotherapy. Acta Pharm Sin B. (2022) 12:50–75. doi: 10.1016/j.apsb.2021.05.011
54. Herranz N, Gallage S, Mellone M, Wuestefeld T, Klotz S, Hanley CJ, et al. Mtor regulates mapkapk2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. (2015) 17:1205–17. doi: 10.1038/ncb3225
55. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. (2005) 19:2100–10. doi: 10.1101/gad.1346005
56. d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. (2003) 426:194–8. doi: 10.1038/nature02118
57. Rossiello F, Jurk D, Passos JF, and d’Adda di Fagagna F. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. (2022) 24:135–47. doi: 10.1038/s41556-022-00842-x
58. Chakravarti D, LaBella KA, and DePinho RA. Telomeres: history, health, and hallmarks of aging. Cell. (2021) 184:306–22. doi: 10.1016/j.cell.2020.12.028
59. Berger S, Kouzarides T, Shiekhattar R, and Shilatifard A. An operational definition of epigenetics. Genes Dev. (2009) 23:781–3. doi: 10.1101/gad.1787609
60. Noroozi R, Ghafouri-Fard S, Pisarek A, Rudnicka J, Spólnicka M, Branicki W, et al. DNA methylation-based age clocks: from age prediction to age reversion. Ageing Res Rev. (2021) 68:101314. doi: 10.1016/j.arr.2021.101314
61. Navas-Acien A, Domingo-Relloso A, Subedi P, Riffo-Campos AL, Xia R, Gomez L, et al. Blood DNA methylation and incident coronary heart disease: evidence from the strong heart study. JAMA Cardiol. (2021) 6:1237–46. doi: 10.1001/jamacardio.2021.2704
62. Luo X, Hu Y, Shen J, Liu X, Wang T, Li L, et al. Integrative analysis of DNA methylation and gene expression reveals key molecular signatures in acute myocardial infarction. Clin Epigenet. (2022) 14:46. doi: 10.1186/s13148-022-01267-x
63. Grootaert MOJ, Finigan A, Figg NL, Uryga AK, and Bennett MR. Sirt6 protects smooth muscle cells from senescence and reduces atherosclerosis. Circ Res. (2021) 128:474–91. doi: 10.1161/circresaha.120.318353
64. Luo YX, Tang X, An XZ, Xie XM, Chen XF, Zhao X, et al. Sirt4 accelerates ang ii-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J. (2017) 38:1389–98. doi: 10.1093/eurheartj/ehw138
65. Man AWC, Li H, and Xia N. The role of sirtuin1 in regulating endothelial function, arterial remodeling and vascular aging. Front Physiol. (2019) 10:1173. doi: 10.3389/fphys.2019.01173
66. Shinozaki S, Chang K, Sakai M, Shimizu N, Yamada M, Tanaka T, et al. Inflammatory stimuli induce inhibitory S-nitrosylation of the deacetylase sirt1 to increase acetylation and activation of P53 and P65. Sci Signal. (2014) 7:ra106. doi: 10.1126/scisignal.2005375
67. Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A, et al. Microrna-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr). (2013) 35:747–62. doi: 10.1007/s11357-012-9407-9
68. Guo X, Xu Y, Wang Z, Wu Y, Chen J, Wang G, et al. A linc1405/eomes complex promotes cardiac mesoderm specification and cardiogenesis. Cell Stem Cell. (2018) 22:893–908.e6. doi: 10.1016/j.stem.2018.04.013
69. Ritter N, Ali T, Kopitchinski N, Schuster P, Beisaw A, Hendrix DA, et al. The lncrna locus handsdown regulates cardiac gene programs and is essential for early mouse development. Dev Cell. (2019) 50:644–57.e8. doi: 10.1016/j.devcel.2019.07.013
70. Cai B, Ma W, Ding F, Zhang L, Huang Q, Wang X, et al. The long noncoding rna carel controls cardiac regeneration. J Am Coll Cardiol. (2018) 72:534–50. doi: 10.1016/j.jacc.2018.04.085
71. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. (2016) 118:400–9. doi: 10.1161/circresaha.115.307778
72. Tang X, Li PH, and Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front Endocrinol (Lausanne). (2020) 11:280. doi: 10.3389/fendo.2020.00280
73. Alam P, Haile B, Arif M, Pandey R, Rokvic M, Nieman M, et al. Inhibition of senescence-associated genes rb1 and meis2 in adult cardiomyocytes results in cell cycle reentry and cardiac repair post-myocardial infarction. J Am Heart Assoc. (2019) 8:e012089. doi: 10.1161/jaha.119.012089
74. El-Nachef D, Oyama K, Wu YY, Freeman M, Zhang Y, and MacLellan WR. Repressive histone methylation regulates cardiac myocyte cell cycle exit. J Mol Cell Cardiol. (2018) 121:1–12. doi: 10.1016/j.yjmcc.2018.05.013
75. Li H, Hastings MH, Rhee J, Trager LE, Roh JD, and Rosenzweig A. Targeting age-related pathways in heart failure. Circ Res. (2020) 126:533–51. doi: 10.1161/circresaha.119.315889
76. Tang X, Luo YX, Chen HZ, and Liu DP. Mitochondria, endothelial cell function, and vascular diseases. Front Physiol. (2014) 5:175. doi: 10.3389/fphys.2014.00175
77. Lopaschuk G, Ussher J, Folmes C, Jaswal J, and Stanley W. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
78. Kolwicz SC Jr., Purohit S, and Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. (2013) 113:603–16. doi: 10.1161/circresaha.113.302095
79. Zhang X, Liu C, Liu C, Wang Y, Zhang W, and Xing Y. Trimetazidine and L−Carnitine prevent heart aging and cardiac metabolic impairment in rats via regulating cardiac metabolic substrates. Exp Gerontol. (2019) 119:120–7. doi: 10.1016/j.exger.2018.12.019
80. Bogazzi F, Raggi F, Ultimieri F, Russo D, D’Alessio A, Manariti A, et al. Regulation of cardiac fatty acids metabolism in transgenic mice overexpressing bovine gh. J Endocrinol. (2009) 201:419–27. doi: 10.1677/joe-08-0194
81. He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, et al. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation. (2012) 126:1705–16. doi: 10.1161/circulationaha.111.075978
82. Dillon LM, Rebelo AP, and Moraes CT. The role of pgc-1 coactivators in aging skeletal muscle and heart. IUBMB Life. (2012) 64:231–41. doi: 10.1002/iub.608
83. Rodríguez-Calvo R, Serrano L, Barroso E, Coll T, Palomer X, Camins A, et al. Peroxisome proliferator-activated receptor alpha down-regulation is associated with enhanced ceramide levels in age-associated cardiac hypertrophy. J Gerontol A Biol Sci Med Sci. (2007) 62:1326–36. doi: 10.1093/gerona/62.12.1326
84. Cui S, Xue L, Yang F, Dai S, Han Z, Liu K, et al. Postinfarction hearts are protected by premature senescent cardiomyocytes via gata 4-dependent ccn 1 secretion. J Am Heart Assoc. (2018) 7:e009111. doi: 10.1161/jaha.118.009111
85. Ock S, Lee WS, Ahn J, Kim HM, Kang H, Kim HS, et al. Deletion of igf-1 receptors in cardiomyocytes attenuates cardiac aging in male mice. Endocrinology. (2016) 157:336–45. doi: 10.1210/en.2015-1709
86. Lesnefsky EJ, Chen Q, and Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. (2016) 118:1593–611. doi: 10.1161/circresaha.116.307505
87. Chadda KR, Ajijola OA, Vaseghi M, Shivkumar K, Huang CL, and Jeevaratnam K. Ageing, the autonomic nervous system and arrhythmia: from brain to heart. Ageing Res Rev. (2018) 48:40–50. doi: 10.1016/j.arr.2018.09.005
88. Chang AC, Ong SG, LaGory EL, Kraft PE, Giaccia AJ, Wu JC, et al. Telomere shortening and metabolic compromise underlie dystrophic cardiomyopathy. Proc Natl Acad Sci U.S.A. (2016) 113:13120–5. doi: 10.1073/pnas.1615340113
89. Mitry MA, Laurent D, Keith BL, Sira E, Eisenberg CA, Eisenberg LM, et al. Accelerated cardiomyocyte senescence contributes to late-onset doxorubicin-induced cardiotoxicity. Am J Physiol Cell Physiol. (2020) 318:C380–c91. doi: 10.1152/ajpcell.00073.2019
90. Masoud S, McDonald F, Bister D, Kotecki C, Bootman MD, and Rietdorf K. Examining cardiomyocyte dysfunction using acute chemical induction of an ageing phenotype. Int J Mol Sci. (2019) 21:197. doi: 10.3390/ijms21010197
91. Colliva A, Braga L, Giacca M, and Zacchigna S. Endothelial cell-cardiomyocyte crosstalk in heart development and disease. J Physiol. (2020) 598:2923–39. doi: 10.1113/jp276758
92. Gevaert AB, Shakeri H, Leloup AJ, Van Hove CE, De Meyer GRY, Vrints CJ, et al. Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ Heart Fail. (2017) 10:e003806. doi: 10.1161/circheartfailure.116.003806
93. Jia G, Aroor AR, Jia C, and Sowers JR. Endothelial cell senescence in aging-related vascular dysfunction. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:1802–9. doi: 10.1016/j.bbadis.2018.08.008
94. Hwang HJ, Kim N, Herman AB, Gorospe M, and Lee JS. Factors and pathways modulating endothelial cell senescence in vascular aging. Int J Mol Sci. (2022) 23:10135. doi: 10.3390/ijms231710135
95. de Yébenes VG, Briones AM, Martos-Folgado I, Mur SM, Oller J, Bilal F, et al. Aging-associated mir-217 aggravates atherosclerosis and promotes cardiovascular dysfunction. Arterioscler Thromb Vasc Biol. (2020) 40:2408–24. doi: 10.1161/atvbaha.120.314333
96. Lee OH, Woo YM, Moon S, Lee J, Park H, Jang H, et al. Sirtuin 6 deficiency induces endothelial cell senescence via downregulation of forkhead box M1 expression. Aging (Albany NY). (2020) 12:20946–67. doi: 10.18632/aging.202176
97. McDonald AP, Meier TR, Hawley AE, Thibert JN, Farris DM, Wrobleski SK, et al. Aging is associated with impaired thrombus resolution in a mouse model of stasis induced thrombosis. Thromb Res. (2010) 125:72–8. doi: 10.1016/j.thromres.2009.06.005
98. Ovchinnikov AG, Arefieva TI, Potekhina AV, Filatova AY, Ageev FT, and Boytsov SA. The molecular and cellular mechanisms associated with a microvascular inflammation in the pathogenesis of heart failure with preserved ejection fraction. Acta Naturae. (2020) 12:40–51. doi: 10.32607/actanaturae.10990
99. Jesel L, Abbas M, Park SH, Matsushita K, Kindo M, Hasan H, et al. Atrial fibrillation progression is associated with cell senescence burden as determined by P53 and P16 expression. J Clin Med. (2019) 9:36. doi: 10.3390/jcm9010036
100. Xie J, Chen Y, Hu C, Pan Q, Wang B, Li X, et al. Premature senescence of cardiac fibroblasts and atrial fibrosis in patients with atrial fibrillation. Oncotarget. (2017) 8:57981–90. doi: 10.18632/oncotarget.19853
101. Desantis V, Potenza MA, Sgarra L, Nacci C, Scaringella A, Cicco S, et al. Micrornas as biomarkers of endothelial dysfunction and therapeutic target in the pathogenesis of atrial fibrillation. Int J Mol Sci. (2023) 24:5307. doi: 10.3390/ijms24065307
102. Herbert KE, Mistry Y, Hastings R, Poolman T, Niklason L, and Williams B. Angiotensin ii-mediated oxidative DNA damage accelerates cellular senescence in cultured human vascular smooth muscle cells via telomere-dependent and independent pathways. Circ Res. (2008) 102:201–8. doi: 10.1161/circresaha.107.158626
103. Kunieda T, Minamino T, Nishi J, Tateno K, Oyama T, Katsuno T, et al. Angiotensin ii induces premature senescence of vascular smooth muscle cells and accelerates the development of atherosclerosis via a P21-dependent pathway. Circulation. (2006) 114:953–60. doi: 10.1161/circulationaha.106.626606
104. Owens GK, Kumar MS, and Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
105. Demer L and Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. (2008) 117:2938–48. doi: 10.1161/circulationaha.107.743161
106. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, and Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/s41569-019-0227-9
107. Cafueri G, Parodi F, Pistorio A, Bertolotto M, Ventura F, Gambini C, et al. Endothelial and smooth muscle cells from abdominal aortic aneurysm have increased oxidative stress and telomere attrition. PloS One. (2012) 7:e35312. doi: 10.1371/journal.pone.0035312
108. van der Feen DE, Bossers GPL, Hagdorn QAJ, Moonen JR, Kurakula K, Szulcek R, et al. Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med. (2020) 12:eaaw4974. doi: 10.1126/scitranslmed.aaw4974
109. Komaravolu RK, Waltmann MD, Konaniah E, Jaeschke A, and Hui DY. Apoer2 (Apolipoprotein E receptor-2) deficiency accelerates smooth muscle cell senescence via cytokinesis impairment and promotes fibrotic neointima after vascular injury. Arterioscler Thromb Vasc Biol. (2019) 39:2132–44. doi: 10.1161/atvbaha.119.313194
110. Brozovich FV, Nicholson CJ, Degen CV, Gao YZ, Aggarwal M, and Morgan KG. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev. (2016) 68:476–532. doi: 10.1124/pr.115.010652
111. Csiszar A, Labinskyy N, Jimenez R, Pinto JT, Ballabh P, Losonczy G, et al. Anti-oxidative and anti-inflammatory vasoprotective effects of caloric restriction in aging: role of circulating factors and sirt1. Mech Ageing Dev. (2009) 130:518–27. doi: 10.1016/j.mad.2009.06.004
112. Gorenne I, Kumar S, Gray K, Figg N, Yu H, Mercer J, et al. Vascular smooth muscle cell sirtuin 1 protects against DNA damage and inhibits atherosclerosis. Circulation. (2013) 127:386–96. doi: 10.1161/circulationaha.112.124404
113. Liu Y, Drozdov I, Shroff R, Beltran LE, and Shanahan CM. Prelamin a accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. (2013) 112:e99–109. doi: 10.1161/circresaha.111.300543
114. Durham A, Speer M, Scatena M, Giachelli C, and Shanahan C. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
115. Nakano-Kurimoto R, Ikeda K, Uraoka M, Nakagawa Y, Yutaka K, Koide M, et al. Replicative senescence of vascular smooth muscle cells enhances the calcification through initiating the osteoblastic transition. Am J Physiol Heart Circ Physiol. (2009) 297:H1673–84. doi: 10.1152/ajpheart.00455.2009
116. Fang C, Du L, Gao S, Chen Y, Chen Z, Wu Z, et al. Association between premature vascular smooth muscle cells senescence and vascular inflammation in takayasu’s arteritis. Ann Rheum Dis. (2024) 83:1522–35. doi: 10.1136/ard-2024-225630
117. Gardner SE, Humphry M, Bennett MR, and Clarke MC. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α-dependent senescence-associated secretory phenotype. Arterioscler Thromb Vasc Biol. (2015) 35:1963–74. doi: 10.1161/atvbaha.115.305896
118. Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, et al. Vascular smooth muscle cells undergo telomere-based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. (2006) 99:156–64. doi: 10.1161/01.RES.0000233315.38086.bc
119. Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, et al. Vascular smooth muscle cell senescence promotes atherosclerosis and features of plaque vulnerability. Circulation. (2015) 132:1909–19. doi: 10.1161/circulationaha.115.016457
120. Lin J, Kakkar V, and Lu X. Impact of mcp-1 in atherosclerosis. Curr Pharm Des. (2014) 20:4580–8. doi: 10.2174/1381612820666140522115801
121. Saker M, Lipskaia L, Marcos E, Abid S, Parpaleix A, Houssaini A, et al. Osteopontin, a key mediator expressed by senescent pulmonary vascular cells in pulmonary hypertension. Arterioscler Thromb Vasc Biol. (2016) 36:1879–90. doi: 10.1161/atvbaha.116.307839
122. Liao S, Curci JA, Kelley BJ, Sicard GA, and Thompson RW. Accelerated replicative senescence of medial smooth muscle cells derived from abdominal aortic aneurysms compared to the adjacent inferior mesenteric artery. J Surg Res. (2000) 92:85–95. doi: 10.1006/jsre.2000.5878
123. Hiromi T, Yokoyama U, Kurotaki D, Mamun A, Ishiwata R, Ichikawa Y, et al. Excessive ep4 signaling in smooth muscle cells induces abdominal aortic aneurysm by amplifying inflammation. Arterioscler Thromb Vasc Biol. (2020) 40:1559–73. doi: 10.1161/atvbaha.120.314297
124. Civitarese RA, Kapus A, McCulloch CA, and Connelly KA. Role of integrins in mediating cardiac fibroblast-cardiomyocyte cross talk: A dynamic relationship in cardiac biology and pathophysiology. Basic Res Cardiol. (2017) 112:6. doi: 10.1007/s00395-016-0598-6
125. Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, and Omens JH. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat Rev Cardiol. (2019) 16:361–78. doi: 10.1038/s41569-019-0155-8
126. Jia L, Zhang W, Ma Y, Chen B, Liu Y, Piao C, et al. Haplodeficiency of ataxia telangiectasia mutated accelerates heart failure after myocardial infarction. J Am Heart Assoc. (2017) 6:e006349. doi: 10.1161/jaha.117.006349
127. Zhu F, Li Y, Zhang J, Piao C, Liu T, Li HH, et al. Senescent cardiac fibroblast is critical for cardiac fibrosis after myocardial infarction. PloS One. (2013) 8:e74535. doi: 10.1371/journal.pone.0074535
128. Meyer K, Hodwin B, Ramanujam D, Engelhardt S, and Sarikas A. Essential role for premature senescence of myofibroblasts in myocardial fibrosis. J Am Coll Cardiol. (2016) 67:2018–28. doi: 10.1016/j.jacc.2016.02.047
129. Daseke MJ 2nd, Tenkorang MAA, Chalise U, Konfrst SR, and Lindsey ML. Cardiac fibroblast activation during myocardial infarction wound healing: fibroblast polarization after mi. Matrix Biol. (2020) 91-92:109–16. doi: 10.1016/j.matbio.2020.03.010
130. Dookun E, Walaszczyk A, Redgrave R, Palmowski P, Tual-Chalot S, Suwana A, et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell. (2020) 19:e13249. doi: 10.1111/acel.13249
131. Olsen MB, Hildrestrand GA, Scheffler K, Vinge LE, Alfsnes K, Palibrk V, et al. Neil3-dependent regulation of cardiac fibroblast proliferation prevents myocardial rupture. Cell Rep. (2017) 18:82–92. doi: 10.1016/j.celrep.2016.12.009
132. Özcan S, Alessio N, Acar MB, Mert E, Omerli F, Peluso G, et al. Unbiased analysis of senescence associated secretory phenotype (Sasp) to identify common components following different genotoxic stresses. Aging (Albany NY). (2016) 8:1316–29. doi: 10.18632/aging.100971
133. Schelbert EB, Fridman Y, Wong TC, Abu Daya H, Piehler KM, Kadakkal A, et al. Temporal relation between myocardial fibrosis and heart failure with preserved ejection fraction: association with baseline disease severity and subsequent outcome. JAMA Cardiol. (2017) 2:995–1006. doi: 10.1001/jamacardio.2017.2511
134. Lin R, Rahtu-Korpela L, Magga J, Ulvila J, Swan J, Kemppi A, et al. Mir-1468-3p promotes aging-related cardiac fibrosis. Mol Ther Nucleic Acids. (2020) 20:589–605. doi: 10.1016/j.omtn.2020.04.001
135. Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, et al. Monocyte/macrophage suppression in cd11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ Res. (2007) 100:884–93. doi: 10.1161/01.Res.0000260802.75766.00
136. Groh L, Keating ST, Joosten LAB, Netea MG, and Riksen NP. Monocyte and macrophage immunometabolism in atherosclerosis. Semin Immunopathol. (2018) 40:203–14. doi: 10.1007/s00281-017-0656-7
137. Park I, Goddard ME, Cole JE, Zanin N, Lyytikäinen LP, Lehtimäki T, et al. C-type lectin receptor clec4a2 promotes tissue adaptation of macrophages and protects against atherosclerosis. Nat Commun. (2022) 13:215. doi: 10.1038/s41467-021-27862-9
138. Roy P, Orecchioni M, and Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. (2022) 22:251–65. doi: 10.1038/s41577-021-00584-1
139. Sun K, Li YY, and Jin J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct Target Ther. (2021) 6:79. doi: 10.1038/s41392-020-00455-6
140. Seneviratne AN, Edsfeldt A, Cole JE, Kassiteridi C, Swart M, Park I, et al. Interferon regulatory factor 5 controls necrotic core formation in atherosclerotic lesions by impairing efferocytosis. Circulation. (2017) 136:1140–54. doi: 10.1161/circulationaha.117.027844
141. Vellasamy DM, Lee SJ, Goh KW, Goh BH, Tang YQ, Ming LC, et al. Targeting immune senescence in atherosclerosis. Int J Mol Sci. (2022) 23:13059. doi: 10.3390/ijms232113059
142. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, and van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
143. Dietel B, Cicha I, Voskens CJ, Verhoeven E, Achenbach S, and Garlichs CD. Decreased numbers of regulatory T cells are associated with human atherosclerotic lesion vulnerability and inversely correlate with infiltrated mature dendritic cells. Atherosclerosis. (2013) 230:92–9. doi: 10.1016/j.atherosclerosis.2013.06.014
144. Clement M, Raffort J, Lareyre F, Tsiantoulas D, Newland S, Lu Y, et al. Impaired autophagy in cd11b(+) dendritic cells expands cd4(+) regulatory T cells and limits atherosclerosis in mice. Circ Res. (2019) 125:1019–34. doi: 10.1161/circresaha.119.315248
145. Yun TJ, Lee JS, Machmach K, Shim D, Choi J, Wi YJ, et al. Indoleamine 2,3-dioxygenase-expressing aortic plasmacytoid dendritic cells protect against atherosclerosis by induction of regulatory T cells. Cell Metab. (2016) 23:852–66. doi: 10.1016/j.cmet.2016.04.010
146. Gardner JK, Mamotte CD, McGonigle T, Dye DE, Jackaman C, and Nelson DJ. Lipid-laden partially-activated plasmacytoid and cd4(-)Cd8α(+) dendritic cells accumulate in tissues in elderly mice. Immun Ageing. (2014) 11:11. doi: 10.1186/1742-4933-11-11
147. Zernecke A. Dendritic cells in atherosclerosis: evidence in mice and humans. Arterioscler Thromb Vasc Biol. (2015) 35:763–70. doi: 10.1161/atvbaha.114.303566
148. Carrasco E, Gómez de Las Heras MM, Gabandé-Rodríguez E, Desdín-Micó G, Aranda JF, and Mittelbrunn M. The role of T cells in age-related diseases. Nat Rev Immunol. (2022) 22:97–111. doi: 10.1038/s41577-021-00557-4
149. Phoksawat W, Jumnainsong A, Sornkayasit K, Srisak K, Komanasin N, and Leelayuwat C. Il-17 and ifn-Γ Productions by cd4+ T cells and T cell subsets expressing nkg2d associated with the number of risk factors for cardiovascular diseases. Mol Immunol. (2020) 122:193–9. doi: 10.1016/j.molimm.2020.04.003
150. Spyridopoulos I, Martin-Ruiz C, Hilkens C, Yadegarfar ME, Isaacs J, Jagger C, et al. Cmv seropositivity and T-cell senescence predict increased cardiovascular mortality in octogenarians: results from the newcastle 85+ Study. Aging Cell. (2016) 15:389–92. doi: 10.1111/acel.12430
151. Wang H, Peng G, Bai J, He B, Huang K, Hu X, et al. Cytomegalovirus infection and relative risk of cardiovascular disease (Ischemic heart disease, stroke, and cardiovascular death): A meta-analysis of prospective studies up to 2016. J Am Heart Assoc. (2017) 6:e005025. doi: 10.1161/jaha.116.005025
152. Bergström I, Backteman K, Lundberg A, Ernerudh J, and Jonasson L. Persistent accumulation of interferon-Γ-producing cd8+Cd56+ T cells in blood from patients with coronary artery disease. Atherosclerosis. (2012) 224:515–20. doi: 10.1016/j.atherosclerosis.2012.07.033
153. Desdín-Micó G, Soto-Heredero G, Aranda JF, Oller J, Carrasco E, Gabandé-Rodríguez E, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science. (2020) 368:1371–6. doi: 10.1126/science.aax0860
154. Xin Y, Yuan Z, Wang J, and Li S. Complex interplay between estrogen and aging via lipid metabolism and inflammation forms the novel treatment strategies for atherosclerosis. FASEB J. (2025) 39:e70877. doi: 10.1096/fj.202500244RRR
155. Kalaitzidis D and Gilmore TD. Transcription factor cross-talk: the estrogen receptor and nf-kappab. Trends Endocrinol Metab. (2005) 16:46–52. doi: 10.1016/j.tem.2005.01.004
156. Kim H, Jung JH, Han K, Lee DY, Fava M, Mischoulon D, et al. Ages at menarche and menopause, hormone therapy, and the risk of depression. Gen Hosp Psychiatry. (2023) 83:35–42. doi: 10.1016/j.genhosppsych.2023.04.001
157. Wang H, Liu Y, Zhu L, Wang W, Wan Z, Chen F, et al. 17β-estradiol promotes cholesterol efflux from vascular smooth muscle cells through a liver X receptor A-dependent pathway. Int J Mol Med. (2014) 33:550–8. doi: 10.3892/ijmm.2014.1619
158. Nasser SA, Afify EA, Kobeissy F, Hamam B, Eid AH, and El-Mas MM. Inflammatory basis of atherosclerosis: modulation by sex hormones. Curr Pharm Des. (2021) 27:2099–111. doi: 10.2174/1381612827666210122142811
159. Sarchielli E, Guarnieri G, Idrizaj E, Squecco R, Mello T, Comeglio P, et al. The G protein-coupled oestrogen receptor, gper1, mediates direct anti-inflammatory effects of oestrogens in human cholinergic neurones from the nucleus basalis of meynert. J Neuroendocrinol. (2020) 32:e12837. doi: 10.1111/jne.12837
160. El Khoudary SR, Aggarwal B, Beckie TM, Hodis HN, Johnson AE, Langer RD, et al. Menopause transition and cardiovascular disease risk: implications for timing of early prevention: A scientific statement from the american heart association. Circulation. (2020) 142:e506–e32. doi: 10.1161/cir.0000000000000912
161. Kalin MF and Zumoff B. Sex hormones and coronary disease: A review of the clinical studies. Steroids. (1990) 55:330–52. doi: 10.1016/0039-128x(90)90058-j
162. Perusquía M, Contreras D, and Herrera N. Hypotestosteronemia is an important factor for the development of hypertension: elevated blood pressure in orchidectomized conscious rats is reversed by different androgens. Endocrine. (2019) 65:416–25. doi: 10.1007/s12020-019-01978-x
163. Morgentaler A, Miner MM, Caliber M, Guay AT, Khera M, and Traish AM. Testosterone therapy and cardiovascular risk: advances and controversies. Mayo Clin Proc. (2015) 90:224–51. doi: 10.1016/j.mayocp.2014.10.011
164. Stallone JN and Oloyo AK. Cardiovascular and metabolic actions of the androgens: is testosterone a janus-faced molecule? Biochem Pharmacol. (2023) 208:115347. doi: 10.1016/j.bcp.2022.115347
165. Francomano D, Bruzziches R, Natali M, Aversa A, and Spera G. Cardiovascular effect of testosterone replacement therapy in aging male. Acta BioMed. (2010) 81 Suppl 1:101–6.
166. Lin YH, Lin KJ, Chen JY, Juang HH, and Wu CT. Associations of testosterone and related hormones with all-cause and cardiovascular mortality and incident cardiovascular disease in men. Ann Intern Med. (2025) 178:905–6. doi: 10.7326/annals-25-01146
167. Deenadayalu V, Puttabyatappa Y, Liu AT, Stallone JN, and White RE. Testosterone-induced relaxation of coronary arteries: activation of bkca channels via the cgmp-dependent protein kinase. Am J Physiol Heart Circ Physiol. (2012) 302:H115–23. doi: 10.1152/ajpheart.00046.2011
168. Kelly DM and Jones TH. Testosterone: A vascular hormone in health and disease. J Endocrinol. (2013) 217:R47–71. doi: 10.1530/joe-12-0582
169. Campelo AE, Cutini PH, and Massheimer VL. Testosterone modulates platelet aggregation and endothelial cell growth through nitric oxide pathway. J Endocrinol. (2012) 213:77–87. doi: 10.1530/joe-11-0441
170. Weikert C, Pischon T, and Weikert S. Adverse events associated with testosterone administration. N Engl J Med. (2010) 363:1865. doi: 10.1056/NEJMc1009326
171. Fontaine C, Gosset A, Davezac M, Buscato M, Grouthier V, Renault MA, et al. From sex hormone decrease to hormonal treatment: impacts on cardiovascular risk with ageing. Cardiovasc Res. (2025) 121:1551–65. doi: 10.1093/cvr/cvaf086
172. Hudson J, Cruickshank M, Quinton R, Aucott L, Aceves-Martins M, Gillies K, et al. Adverse cardiovascular events and mortality in men during testosterone treatment: an individual patient and aggregate data meta-analysis. Lancet Healthy Longev. (2022) 3:e381–e93. doi: 10.1016/s2666-7568(22)00096-4
173. Lombardi G, Di Somma C, Grasso LF, Savanelli MC, Colao A, and Pivonello R. The cardiovascular system in growth hormone excess and growth hormone deficiency. J Endocrinol Invest. (2012) 35:1021–9. doi: 10.3275/8717
174. Messias de Lima CF, Dos Santos Reis MD, da Silva Ramos FW, Ayres-Martins S, and Smaniotto S. Growth hormone modulates in vitro endothelial cell migration and formation of capillary-like structures. Cell Biol Int. (2017) 41:577–84. doi: 10.1002/cbin.10747
175. Liu H, Bravata DM, Olkin I, Nayak S, Roberts B, Garber AM, et al. Systematic review: the safety and efficacy of growth hormone in the healthy elderly. Ann Intern Med. (2007) 146:104–15. doi: 10.7326/0003-4819-146-2-200701160-00005
176. Huang X, Blackman MR, Herreman K, Pabst KM, Harman SM, and Caballero B. Effects of growth hormone and/or sex steroid administration on whole-body protein turnover in healthy aged women and men. Metabolism. (2005) 54:1162–7. doi: 10.1016/j.metabol.2005.03.023
177. Chanson P. The heart in growth hormone (Gh) deficiency and the cardiovascular effects of gh. Ann Endocrinol (Paris). (2021) 82:210–3. doi: 10.1016/j.ando.2020.03.005
178. Jabbar A, Ingoe L, Thomas H, Carey P, Junejo S, Addison C, et al. Prevalence, predictors and outcomes of thyroid dysfunction in patients with acute myocardial infarction: the thyrami-1 study. J Endocrinol Invest. (2021) 44:1209–18. doi: 10.1007/s40618-020-01408-0
179. Li Y, Teng D, Ba J, Chen B, Du J, He L, et al. Efficacy and safety of long-term universal salt iodization on thyroid disorders: epidemiological evidence from 31 provinces of mainland China. Thyroid. (2020) 30:568–79. doi: 10.1089/thy.2019.0067
180. Bensenor IM, Olmos RD, and Lotufo PA. Hypothyroidism in the elderly: diagnosis and management. Clin Interv Aging. (2012) 7:97–111. doi: 10.2147/cia.S23966
181. Ding X, Zhao Y, Zhu CY, Wu LP, Wang Y, Peng ZY, et al. The association between subclinical hypothyroidism and metabolic syndrome: an update meta-analysis of observational studies. Endocr J. (2021) 68:1043–56. doi: 10.1507/endocrj.EJ20-0796
182. Nadal-Ginard B and Mahdavi V. Molecular basis of cardiac performance. Plasticity of the myocardium generated through protein isoform switches. J Clin Invest. (1989) 84:1693–700. doi: 10.1172/jci114351
183. Razvi S, Jabbar A, Pingitore A, Danzi S, Biondi B, Klein I, et al. Thyroid hormones and cardiovascular function and diseases. J Am Coll Cardiol. (2018) 71:1781–96. doi: 10.1016/j.jacc.2018.02.045
184. Tian L, Song Y, Xing M, Zhang W, Ning G, Li X, et al. A Novel Role for Thyroid-Stimulating Hormone: Up-Regulation of Hepatic 3-Hydroxy-3-Methyl-Glutaryl-Coenzyme a Reductase Expression through the Cyclic Adenosine Monophosphate/Protein Kinase a/Cyclic Adenosine Monophosphate-Responsive Element Binding Protein Pathway. Hepatology. (2010) 52:1401–9. doi: 10.1002/hep.23800
185. Zhong F, Guan Q, Zhang H, Zhang X, Zhao M, Yuan Z, et al. Association of longitudinal changes in serum lipids with the natural history of subclinical hypothyroidism: A retrospective cohort study using data from the reaction study. EClinicalMedicine. (2022) 53:101629. doi: 10.1016/j.eclinm.2022.101629
186. Ilic S, Tadic M, Ivanovic B, Caparevic Z, Trbojevic B, and Celic V. Left and right ventricular structure and function in subclinical hypothyroidism: the effects of one-year levothyroxine treatment. Med Sci Monit. (2013) 19:960–8. doi: 10.12659/msm.889621
187. Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. (2017) 16:718–35. doi: 10.1038/nrd.2017.116
188. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, et al. Ink4a/arf expression is a biomarker of aging. J Clin Invest. (2004) 114:1299–307. doi: 10.1172/jci22475
189. Gonzales MM, Garbarino VR, Kautz T, Palavicini JP, Lopez-Cruzan M, Dehkordi SK, et al. Senolytic therapy to modulate the progression of Alzheimer's Disease (SToMP-AD) - Outcomes from the first clinical trial of senolytic therapy for Alzheimer's disease. Res Sq. (2023) 71:1781–96. doi: 10.21203/rs.3.rs-2809973/v1
190. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
191. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
192. Joly F, Fabbro M, Follana P, Lequesne J, Medioni J, Lesoin A, et al. A phase II study of Navitoclax (ABT-263) as single agent in women heavily pretreated for recurrent epithelial ovarian cancer: The MONAVI - GINECO study. Gynecol Oncol. (2022) 165:30–9. doi: 10.1016/j.ygyno.2022.01.021
193. Corcoran RB, Do KT, Kim JE, Cleary JM, Parikh AR, Yeku OO, et al. Phase I/II Study of Combined BCL-xL and MEK Inhibition with Navitoclax and Trametinib in KRAS or NRAS Mutant Advanced Solid Tumors. Clin Cancer Res. (2024) 30:1739–49. doi: 10.1158/1078-0432.Ccr-23-3135
194. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. (2016) 7:11190. doi: 10.1038/ncomms11190
195. Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, et al. New agents that target senescent cells: the flavone, fisetin, and the bcl-X(L) inhibitors, A1331852 and A1155463. Aging (Albany NY). (2017) 9:955–63. doi: 10.18632/aging.101202
196. Wang Y, Chang J, Liu X, Zhang X, Zhang S, Zhang X, et al. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging (Albany NY). (2016) 8:2915–26. doi: 10.18632/aging.101100
197. Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, Brooks RW, et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun. (2017) 8:422. doi: 10.1038/s41467-017-00314-z
198. Lazaro I, Oguiza A, Recio C, Mallavia B, Madrigal-Matute J, Blanco J, et al. Targeting HSP90 Ameliorates Nephropathy and Atherosclerosis Through Suppression of NF-κB and STAT Signaling Pathways in Diabetic Mice. Diabetes. (2015) 64:3600–13. doi: 10.2337/db14-1926
199. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
200. Small GW, Siddarth P, Li Z, Miller KJ, Ercoli L, Emerson ND, et al. Memory and Brain Amyloid and Tau Effects of a Bioavailable Form of Curcumin in Non-Demented Adults: A Double-Blind, Placebo-Controlled 18-Month Trial. Am J Geriatr Psychiatry. (2018) 26:266–77. doi: 10.1016/j.jagp.2017.10.010
201. Triana-Martínez F, Picallos-Rabina P, Da Silva-Álvarez S, Pietrocola F, Llanos S, Rodilla V, et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat Commun. (2019) 10:4731. doi: 10.1038/s41467-019-12888-x
202. Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, et al. Cardiac glycosides are broad-spectrum senolytics. Nat Metab. (2019) 1:1074–88. doi: 10.1038/s42255-019-0122-z
203. Shi H, Mao X, Zhong Y, Liu Y, Zhao X, Yu K, et al. Digoxin reduces atherosclerosis in apolipoprotein E-deficient mice. Br J Pharmacol. (2016) 173:1517–28. doi: 10.1111/bph.13453
204. Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeois BRM, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. (2017) 169:132–47.e16. doi: 10.1016/j.cell.2017.02.031
205. Xu Q, Fu Q, Li Z, Liu H, Wang Y, Lin X, et al. The flavonoid procyanidin C1 has senotherapeutic activity and increases lifespan in mice. Nat Metab. (2021) 3:1706–26. doi: 10.1038/s42255-021-00491-8
206. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, et al. The achilles’ Heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. (2015) 14:644–58. doi: 10.1111/acel.12344
207. Montero JC, Seoane S, Ocaña A, and Pandiella A. Inhibition of src family kinases and receptor tyrosine kinases by dasatinib: possible combinations in solid tumors. Clin Cancer Res. (2011) 17:5546–52. doi: 10.1158/1078-0432.Ccr-10-2616
208. Tang J, Lu L, Liu Y, Ma J, Yang L, Li L, et al. Quercetin improve ischemia/reperfusion-induced cardiomyocyte apoptosis in vitro and in vivo study via sirt1/pgc-1α Signaling. J Cell Biochem. (2019) 120:9747–57. doi: 10.1002/jcb.28255
209. Olson ER, Melton T, Dickinson SE, Dong Z, Alberts DS, and Bowden GT. Quercetin potentiates uvb-induced C-fos expression: implications for its use as a chemopreventive agent. Cancer Prev Res (Phila). (2010) 3:876–84. doi: 10.1158/1940-6207.Capr-09-0220
210. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, et al. Identification of a novel senolytic agent, navitoclax, targeting the bcl-2 family of anti-apoptotic factors. Aging Cell. (2016) 15:428–35. doi: 10.1111/acel.12445
211. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, et al. Clearance of senescent cells by abt263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. (2016) 22:78–83. doi: 10.1038/nm.4010
212. Jia K, Dai Y, Liu A, Li X, Wu L, Lu L, et al. Senolytic agent navitoclax inhibits angiotensin ii-induced heart failure in mice. J Cardiovasc Pharmacol. (2020) 76:452–60. doi: 10.1097/fjc.0000000000000878
213. Zhu M, Meng P, Ling X, and Zhou L. Advancements in therapeutic drugs targeting of senescence. Ther Adv Chronic Dis. (2020) 11:2040622320964125. doi: 10.1177/2040622320964125
214. Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun. (2017) 8:14532. doi: 10.1038/ncomms14532
215. Chaib S, Tchkonia T, and Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. (2022) 28:1556–68. doi: 10.1038/s41591-022-01923-y
216. Chien Y, Scuoppo C, Wang X, Fang X, Balgley B, Bolden JE, et al. Control of the senescence-associated secretory phenotype by nf-Kb promotes senescence and enhances chemosensitivity. Genes Dev. (2011) 25:2125–36. doi: 10.1101/gad.17276711
217. Liu S, Zheng Z, Ji S, Liu T, Hou Y, Li S, et al. Resveratrol reduces senescence-associated secretory phenotype by sirt1/nf-Kb pathway in gut of the annual fish nothobranchius guentheri. Fish Shellfish Immunol. (2018) 80:473–9. doi: 10.1016/j.fsi.2018.06.027
218. Kumar R, Sharma A, Kumari A, Gulati A, Padwad Y, and Sharma R. Epigallocatechin gallate suppresses premature senescence of preadipocytes by inhibition of pi3k/akt/mtor pathway and induces senescent cell death by regulation of bax/bcl-2 pathway. Biogerontology. (2019) 20:171–89. doi: 10.1007/s10522-018-9785-1
219. Gurău F, Baldoni S, Prattichizzo F, Espinosa E, Amenta F, Procopio AD, et al. Anti-senescence compounds: A potential nutraceutical approach to healthy aging. Ageing Res Rev. (2018) 46:14–31. doi: 10.1016/j.arr.2018.05.001
220. Aliper A, Jellen L, Cortese F, Artemov A, Karpinsky-Semper D, Moskalev A, et al. Towards natural mimetics of metformin and rapamycin. Aging (Albany NY). (2017) 9:2245–68. doi: 10.18632/aging.101319
221. Lamming DW, Ye L, Sabatini DM, and Baur JA. Rapalogs and mtor inhibitors as anti-aging therapeutics. J Clin Invest. (2013) 123:980–9. doi: 10.1172/jci64099
222. Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schröder S, Adler T, et al. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest. (2013) 123:3272–91. doi: 10.1172/jci67674
223. Kulkarni AS, Gubbi S, and Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. (2020) 32:15–30. doi: 10.1016/j.cmet.2020.04.001
224. Moiseeva O, Deschênes-Simard X, St-Germain E, Igelmann S, Huot G, Cadar AE, et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with ikk/nf-Kb activation. Aging Cell. (2013) 12:489–98. doi: 10.1111/acel.12075
225. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. Jak inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U.S.A. (2015) 112:E6301–10. doi: 10.1073/pnas.1515386112
226. Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of incb018424, a jak1 and jak2 inhibitor, in myelofibrosis. N Engl J Med. (2010) 363:1117–27. doi: 10.1056/NEJMoa1002028
227. Libby P. Interleukin-1 Beta as a Target for Atherosclerosis therapy: Biological Basis of Cantos and beyond. J Am Coll Cardiol. (2017) 70:2278–89. doi: 10.1016/j.jacc.2017.09.028
228. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
229. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. (2019) 380:752–62. doi: 10.1056/NEJMoa1809798
230. Lutgens E, Atzler D, Döring Y, Duchene J, Steffens S, and Weber C. Immunotherapy for cardiovascular disease. Eur Heart J. (2019) 40:3937–46. doi: 10.1093/eurheartj/ehz283
231. Burton DG and Krizhanovsky V. Physiological and pathological consequences of cellular senescence. Cell Mol Life Sci. (2014) 71:4373–86. doi: 10.1007/s00018-014-1691-3
232. Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. (2008) 134:657–67. doi: 10.1016/j.cell.2008.06.049
233. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. (2011) 479:547–51. doi: 10.1038/nature10599
234. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC. Car T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
235. Ghobadi A. Chimeric antigen receptor T cell therapy for non-hodgkin lymphoma. Curr Res Transl Med. (2018) 66:43–9. doi: 10.1016/j.retram.2018.03.005
236. Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting cardiac fibrosis with engineered T cells. Nature. (2019) 573:430–3. doi: 10.1038/s41586-019-1546-z
237. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic car T cells reverse senescence-associated pathologies. Nature. (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
238. Amor C, Fernández-Maestre I, Chowdhury S, Ho YJ, Nadella S, Graham C, et al. Prophylactic and long-lasting efficacy of senolytic car T cells against age-related metabolic dysfunction. Res Sq. (2023) 4:336–49. doi: 10.21203/rs.3.rs-3385749/v1
239. Yang D, Sun B, Li S, Wei W, Liu X, Cui X, et al. Nkg2d-car T cells eliminate senescent cells in aged mice and nonhuman primates. Sci Transl Med. (2023) 15:eadd1951. doi: 10.1126/scitranslmed.add1951
240. Takahashi K and Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
241. Carpenter L, Carr C, Yang CT, Stuckey DJ, Clarke K, and Watt SM. Efficient differentiation of human induced pluripotent stem cells generates cardiac cells that provide protection following myocardial infarction in the rat. Stem Cells Dev. (2012) 21:977–86. doi: 10.1089/scd.2011.0075
242. Citro L, Naidu S, Hassan F, Kuppusamy ML, Kuppusamy P, Angelos MG, et al. Comparison of human induced pluripotent stem-cell derived cardiomyocytes with human mesenchymal stem cells following acute myocardial infarction. PloS One. (2014) 9:e116281. doi: 10.1371/journal.pone.0116281
243. Ye L, Chang YH, Xiong Q, Zhang P, Zhang L, Somasundaram P, et al. Cardiac repair in a porcine model of acute myocardial infarction with human induced pluripotent stem cell-derived cardiovascular cells. Cell Stem Cell. (2014) 15:750–61. doi: 10.1016/j.stem.2014.11.009
244. Zhang Z, Yang J, Yan W, Li Y, Shen Z, and Asahara T. Pretreatment of cardiac stem cells with exosomes derived from mesenchymal stem cells enhances myocardial repair. J Am Heart Assoc. (2016) 5:e002856. doi: 10.1161/jaha.115.002856
245. Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP, et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (Prochymal) after acute myocardial infarction. J Am Coll Cardiol. (2009) 54:2277–86. doi: 10.1016/j.jacc.2009.06.055
246. Can A, Ulus AT, Cinar O, Topal Celikkan F, Simsek E, Akyol M, et al. Human umbilical cord mesenchymal stromal cell transplantation in myocardial ischemia (Huc-heart trial). A study protocol of a phase 1/2, controlled and randomized trial in combination with coronary artery bypass grafting. Stem Cell Rev Rep. (2015) 11:752–60. doi: 10.1007/s12015-015-9601-0
247. Bartolucci J, Verdugo FJ, González PL, Larrea RE, Abarzua E, Goset C, et al. Safety and efficacy of the intravenous infusion of umbilical cord mesenchymal stem cells in patients with heart failure: A phase 1/2 randomized controlled trial (Rimecard trial [Randomized clinical trial of intravenous infusion umbilical cord mesenchymal stem cells on cardiopathy]). Circ Res. (2017) 121:1192–204. doi: 10.1161/circresaha.117.310712
248. Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. (2003) 114:763–76. doi: 10.1016/s0092-8674(03)00687-1
249. Bollini S, Smart N, and Riley PR. Resident cardiac progenitor cells: at the heart of regeneration. J Mol Cell Cardiol. (2011) 50:296–303. doi: 10.1016/j.yjmcc.2010.07.006
250. Itzhaki-Alfia A, Leor J, Raanani E, Sternik L, Spiegelstein D, Netser S, et al. Patient characteristics and cell source determine the number of isolated human cardiac progenitor cells. Circulation. (2009) 120:2559–66. doi: 10.1161/circulationaha.109.849588
251. Jesty SA, Steffey MA, Lee FK, Breitbach M, Hesse M, Reining S, et al. C-kit+ Precursors support postinfarction myogenesis in the neonatal, but not adult, heart. Proc Natl Acad Sci U.S.A. (2012) 109:13380–5. doi: 10.1073/pnas.1208114109
252. van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, et al. C-kit+ Cells minimally contribute cardiomyocytes to the heart. Nature. (2014) 509:337–41. doi: 10.1038/nature13309
253. Limana F, Capogrossi MC, and Germani A. The epicardium in cardiac repair: from the stem cell view. Pharmacol Ther. (2011) 129:82–96. doi: 10.1016/j.pharmthera.2010.09.002
254. Urbanek K, Rota M, Cascapera S, Bearzi C, Nascimbene A, De Angelis A, et al. Cardiac stem cells possess growth factor-receptor systems that after activation regenerate the infarcted myocardium, improving ventricular function and long-term survival. Circ Res. (2005) 97:663–73. doi: 10.1161/01.Res.0000183733.53101.11
255. Ellison GM, Torella D, Dellegrottaglie S, Perez-Martinez C, Perez de Prado A, Vicinanza C, et al. Endogenous cardiac stem cell activation by insulin-like growth factor-1/hepatocyte growth factor intracoronary injection fosters survival and regeneration of the infarcted pig heart. J Am Coll Cardiol. (2011) 58:977–86. doi: 10.1016/j.jacc.2011.05.013
256. Carapeto PV and Aguayo-Mazzucato C. Effects of exercise on cellular and tissue aging. Aging (Albany NY). (2021) 13:14522–43. doi: 10.18632/aging.203051
257. Li Q, Zhang H, Xiao N, Liang G, Lin Y, Yang X, et al. Aging and lifestyle modifications for preventing aging-related diseases. FASEB J. (2025) 39:e70575. doi: 10.1096/fj.202402797RR
258. Cai YW, Gao JW, Wu MX, Xie YX, You S, Liao GH, et al. Adherence to eat-lancet diet, biological aging, and life expectancy in the uk biobank: A cohort study. Am J Clin Nutr. (2025) 122:29–38. doi: 10.1016/j.ajcnut.2025.04.030
259. Pangrazzi L and Meryk A. Molecular and cellular mechanisms of immunosenescence: modulation through interventions and lifestyle changes. Biol (Basel). (2024) 14:17. doi: 10.3390/biology14010017
260. Song P, An J, and Zou MH. Immune clearance of senescent cells to combat ageing and chronic diseases. Cells. (2020) 9:671. doi: 10.3390/cells9030671
261. Wu T, Xu S, Chen B, Bao L, Ma J, Han W, et al. Ambient pm2.5 exposure causes cellular senescence via DNA damage, micronuclei formation, and cgas activation. Nanotoxicology. (2022) 16:757–75. doi: 10.1080/17435390.2022.2147460
262. Qiu F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, et al. Impacts of cigarette smoking on immune responsiveness: up and down or upside down? Oncotarget. (2017) 8:268–84. doi: 10.18632/oncotarget.13613
Keywords: cardiac microenvironment, aging, cardiac aging, therapy, inflammation
Citation: Zhao X, Yang X, Lin Y, Lei R, Ding W, He X, Cao Y, Zhang D, Liu P, Liang M, Han Z and Jiang Y (2025) Mechanisms of aging in the cardiovascular system: challenges and opportunities. Front. Immunol. 16:1635736. doi: 10.3389/fimmu.2025.1635736
Received: 27 May 2025; Accepted: 24 October 2025;
Published: 05 November 2025.
Edited by:
Nisha Jain Garg, University of Texas Medical Branch at Galveston, United StatesReviewed by:
Carolina Dalmasso, University of Kentucky, United StatesSuman Asalla, The Ohio State University, United States
Copyright © 2025 Zhao, Yang, Lin, Lei, Ding, He, Cao, Zhang, Liu, Liang, Han and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongyu Han, aHp5Y3p5MTk5N0AxNjMuY29t; Yu Jiang, amlhbmd5dS0yMkAxMjYuY29t
†These authors have contributed equally to this work