 Kohei Shiroshita1†
Kohei Shiroshita1† Yoshitaka Zaimoku2,3†
Yoshitaka Zaimoku2,3† Himari Kudo1
Himari Kudo1 Naoshi Obara4
Naoshi Obara4 Kazuyoshi Hosomichi5Yui Kano6Miku Kobayashi7
Kazuyoshi Hosomichi5Yui Kano6Miku Kobayashi7 Eriko Morishita7
Eriko Morishita7 Hiroyuki Takamatsu2,8Takaaki Toyama1
Hiroyuki Takamatsu2,8Takaaki Toyama1 Shinji Nakao2*
Shinji Nakao2*- 1Department of Hematology, Federation of National Public Service Personnel Mutual Aid Associations Tachikawa Hospital, Tokyo, Japan
- 2Department of Hematology, Kanazawa University, Kanazawa, Japan
- 3Innovative Clinical Research Center, Kanazawa University Hospital, Kanazawa, Japan
- 4Department of Medical Science, Faculty of Medicine, University of Tsukuba, Tsukuba, Ibaraki, Japan
- 5Laboratory of Computational Genomics, School of Life Science, Tokyo University of Pharmacy and Life Sciences, Tokyo, Japan
- 6Department of Clinical Laboratory Science, School of Health Sciences, College of Medical, Pharmaceutical and Health Sciences, Kanazawa University, Kanazawa, Japan
- 7Department of Clinical Laboratory Science, Division of Health Sciences, Graduate School of Medical Science, Kanazawa University, Kanazawa, Japan
- 8Faculty of Transdisciplinary Sciences for Innovation, Institute of Transdisciplinary Sciences for Innovation, Kanazawa University, Kanazawa, Japan
T-cell-mediated severe aplastic anemia (SAA) is typically fatal without prompt hematopoietic stem cell transplantation or intensive immunosuppressive therapy. Although rare cases of spontaneous remission have been reported, the underlying mechanisms remain poorly understood. A 24-year-old woman was incidentally found to have mild pancytopenia during a routine workplace health checkup. Over the subsequent 12 months, her pancytopenia gradually worsened, resulting in exertional dyspnea, purpura, and a diagnosis of SAA. Remarkably, her blood counts began to improve spontaneously 11 days after the diagnosis without any treatment or transfusions. She no longer met the criteria for SAA by day 27 and achieved complete hematologic normalization within three months. At 22 months, flow cytometry and targeted sequencing revealed that 69% of her granulocytes lacked the HLA-A*02:01-C*03:04-B*40:02-DRB1*14:54 haplotype due to acquired loss of heterozygosity, while 23% were glycosylphosphatidylinositol-deficient owing to PIGA mutations. Retrospective digital polymerase chain reaction of diagnostic bone marrow demonstrated that nearly all non-lymphoid cells had already been replaced by HLA allele-lacking clones, whereas glycosylphosphatidylinositol-deficient erythrocytes constituted only 0.25%. These findings suggest that hematologic recovery occurred through the selective expansion of mutant hematopoietic stem cells capable of evading persistent T-cell-mediated destruction. Early identification of HLA allele-lacking leukocytes may help predict spontaneous remission and avoid unnecessary intensive therapy in patients with SAA.
Introduction
Aplastic anemia (AA) is a life-threatening bone marrow failure disorder characterized by pancytopenia resulting from a marked reduction in hematopoietic stem cells (HSCs) (1). Idiopathic severe AA (SAA) is most commonly caused by T cell-mediated destruction, for which prompt treatment with intensive immunosuppressive therapy (IST) or hematopoietic stem cell transplantation is essential for survival (2–4).
Although rare, spontaneous remission of SAA has been reported (5–7). Lee et al. described 18 cases of AA, including eight with SAA, in which spontaneous remission occurred at a median of 14 days (range, 4–332) (5). However, they concluded that most cases likely represented recovery from transient bone marrow suppression triggered by external factors, such as medications or infections, rather than true idiopathic SAA with an immune pathogenesis. Similarly, spontaneous remission of pregnancy-associated AA has been observed following delivery (6).
The peripheral blood of patients with immune-mediated SAA often contains progeny of mutant HSCs that evade T-cell attacks. These include blood cells deficient in glycosylphosphatidylinositol (GPI) due to PIGA mutations (8, 9), as well as cells lacking expression of HLA class I alleles due to copy-neutral loss of heterozygosity on chromosome 6p (6pLOH) or loss-of-function mutations in HLA genes (10–17). These immune escape HSC clones may contribute to spontaneous remission, although definitive evidence supporting this hypothesis is lacking.
We herein report the first documented case of spontaneous remission in idiopathic SAA, attributed to the concurrent expansion of HLA allele-lacking and GPI-deficient HSCs.
Case description
A 24-year-old woman underwent a routine physical examination at her workplace every six months. Eighteen months prior to presentation, her blood count was within normal range. Twelve months prior, mild pancytopenia with macrocytic anemia was first detected: white blood cell count (WBC), 3.54 × 109/L; hemoglobin, 92 g/L; mean corpuscular volume, 110 fL; and platelet count, 112 × 109/L. Over the subsequent six months, her pancytopenia worsened: WBC, 2.71 × 109/L; hemoglobin, 72 g/L; and platelet count, 59 × 109/L. Despite developing exertional dyspnea and petechiae in her lower extremities, she did not seek medical attention, as her symptoms remained mild. Two weeks before her admission to our hospital, she visited a local clinic because of a fever that had lasted for a week. A blood test revealed worsening leukopenia (WBC, 2.10 × 109/L) and thrombocytopenia (platelet count, 36 × 109/L), prompting referral to our hospital for further evaluation.
By the time of admission, the fever had resolved. The patient had no history of medication use, chemical exposure, or menstrual irregularities. Physical examination was unremarkable, with no signs of bleeding tendency, hepatosplenomegaly, or congenital anomalies suggestive of inherited bone marrow failure syndrome. Laboratory tests revealed progression of pancytopenia with an inadequate reticulocyte response: neutrophil count, 0.53 × 109/L; hemoglobin, 76 g/L; reticulocyte count, 36 × 109/L; and platelet count, 15 × 109/L (Table 1).
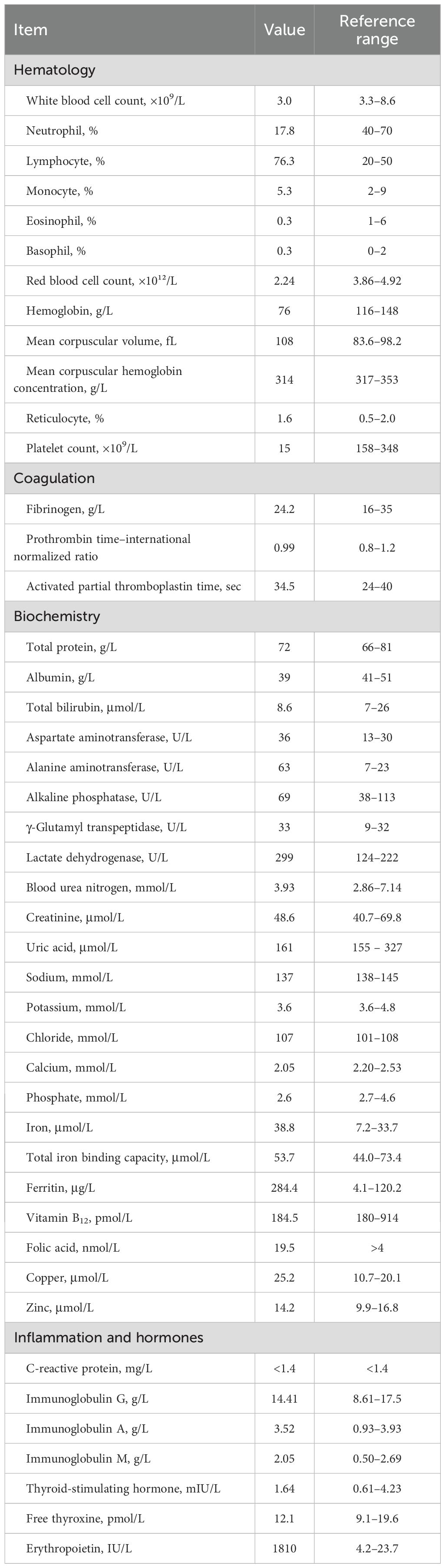
Table 1. Laboratory test results at the diagnosis.
Bone marrow examination revealed severe hypocellularity (Figure 1A), with no evidence of dysplasia or increased blasts, and cytogenetic analysis showed normal karyotype. Magnetic resonance imaging of the thoracolumbar spine revealed that most of the marrow space was replaced by fatty tissue, with patchy areas of residual hematopoietic activity (Figure 1B). Flow cytometry using anti-CD55 and anti-CD59 monoclonal antibodies identified 0.25% of erythrocytes and 0.2% of granulocytes as GPI-deficient (Figure 1C). These findings were all consistent with a diagnosis of idiopathic SAA.
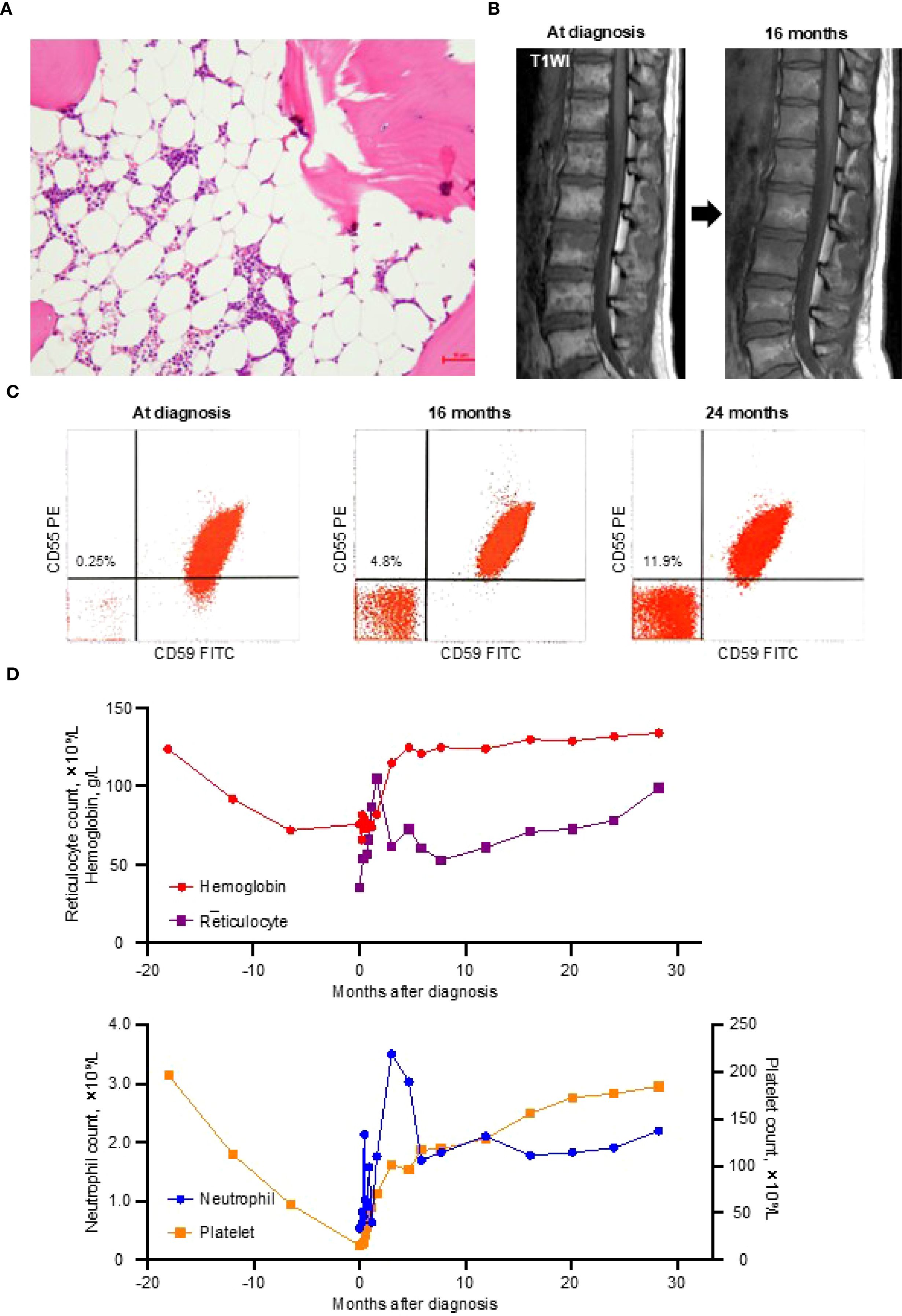
Figure 1. Diagnostic findings and the clinical course. (A) A bone marrow trephine biopsy at diagnosis showing severe hypocellularity with fatty replacement. (B) T1-weighted magnetic resonance imaging (T1WI) of the thoracolumbar spine demonstrating fatty marrow conversion (high signal intensity) at the diagnosis and improvement at 16 months. (C) Glycosylphosphatidylinositol-deficient (CD55–CD59–) erythrocytes detected by flow cytometry at the diagnosis and at 16 and 24 months thereafter. (D) Trends in blood cell counts from 18 months before the diagnosis to 28 months after the diagnosis. FITC, fluorescein isothiocyanate; PE, phycoerythrin.
During the 11-day diagnostic period, her reticulocyte and platelet counts increased to 54 × 109/L and 18 × 109/L, respectively, without any medication or blood transfusion. This unexpected improvement led the attending physician to withhold the planned IST. By day 27 after presentation, her blood counts no longer met the criteria for SAA (neutrophil count, 1.58 × 109/L; hemoglobin, 76 g/L; reticulocyte count, 66 × 109/L; platelet count, 46 × 109/L), and all parameters normalized within three months (Figure 1D).
This hematologic recovery was accompanied by a gradual increase in GPI-deficient erythrocytes, reaching 4.8% at 16 months and 11.9% at 24 months (Figure 1C), without signs of intravascular hemolysis (lactate dehydrogenase at 24 months, 199 U/L). Follow-up magnetic resonance imaging at 16 months showed partial resolution of the fatty marrow changes (Figure 1B). As of May 2025, the patient has remained in complete remission without any treatment for 30 months.
Diagnostic assessment
To investigate the mechanism underlying her spontaneous remission, we assessed the presence of HLA allele-lacking and GPI-deficient cells 22 months after the diagnosis. Flow cytometry using anti-HLA-A2 monoclonal antibodies and fluorescently labeled inactivated aerolysin, as previously described (15), revealed that 69% of granulocytes and 75% of monocytes lacked HLA-A0201 expression. In addition, GPI-deficient cells retaining HLA expression accounted for 24% of granulocytes and 17% of monocytes (Figure 2A). In contrast, 89% of lymphocytes retained normal expression of both HLA and GPI, with only 10% lacking HLA-A0201 and 0.8% being GPI-deficient.
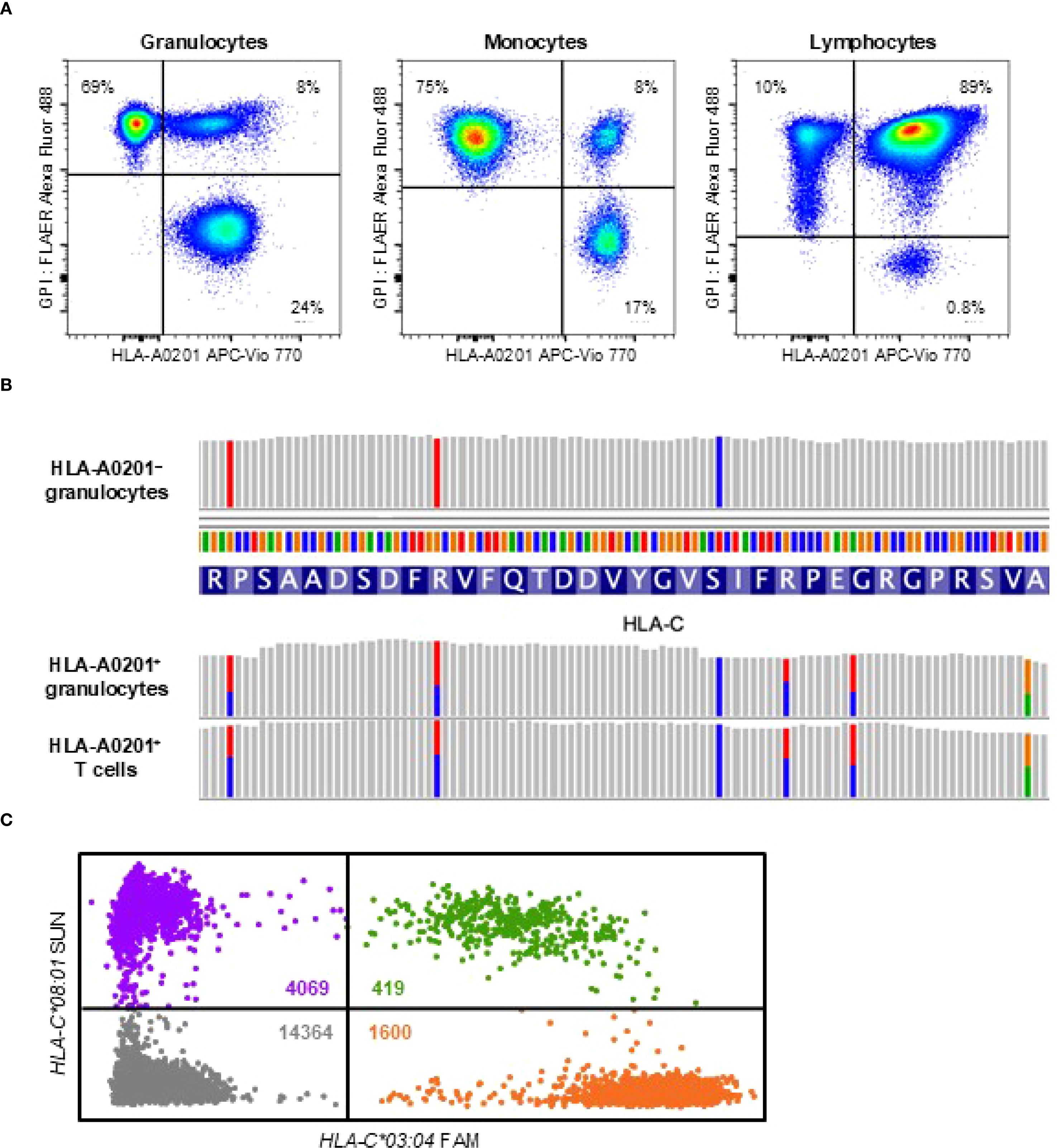
Figure 2. HLA loss analysis. (A) Flow cytometry at 22 months after the diagnosis showing HLA-A0201-lacking cells (left upper quadrant), GPI-deficient cells (right lower quadrant), and wild-type cells expressing both HLA and GPI (right upper quadrant) across three leukocyte subsets: high side-scatter (SSChi) CD45dimCD33dim granulocytes, high forward-scatter (FSChi) CD45hiCD33hi monocytes, and CD45hiCD33– lymphocytes. (B) Targeted sequencing of HLA genes revealing loss of heterozygosity at the HLA-C locus in sorted HLA-A0201-lacking granulocytes, while heterozygosity was preserved in HLA-A0201-expressing granulocytes and T cells. (C) Digital polymerase chain reaction analysis of a bone marrow smear obtained at the diagnosis showing significantly fewer microwells containing HLA-C*03:04 (1600 orange dots) compared to those containing HLA-C*08:01 (4069 purple dots), indicating loss of HLA-C*03:04 due to 6pLOH. Green dots represent microwells containing both alleles and grey dots represent those with neither allele. The estimated concentrations of HLA-C*03:04 and HLA-C*08:01 in the reaction mixture, calculated using the Poisson distribution, were 240.6 copies/µL and 573.5 copies/µL, respectively. The clonal burden of the 6pLOH cells among total bone marrow nucleated cells was calculated to be 40.9% using the following formula: Clonal burden (%) = (573.5 − 240.6)/(573.5 + 240.6) × 100. APC, allophycocyanin; FAM, 6-carboxyfluorescein; FLAER, fluorescently labeled inactivated aerolysin; GPI, glycosylphosphatidylinositol.
Targeted sequencing and HLA genotyping of sorted cell populations, using T cells expressing both GPI and HLA-A0201 as the germline control, revealed that all HLA-A0201-lacking granulocytes lost the HLA haplotype A*02:01-C*03:04-B*40:02-DRB1*14:54 due to 6pLOH (Table 2; Figure 2B). GPI-deficient granulocytes harbored two distinct PIGA frameshift mutations: c.577_581delGTACT (variant allele frequency, 43%) and c.845delA (variant allele frequency, 4%). No mutations in any of the 51 genes associated with myeloid malignancies were detected in the wild-type, GPI-deficient, or HLA-lacking granulocytes. At 28 months after the diagnosis—six months following the initial HLA loss analysis —the percentages of HLA-A0201-lacking and GPI-deficient cells remained unchanged.
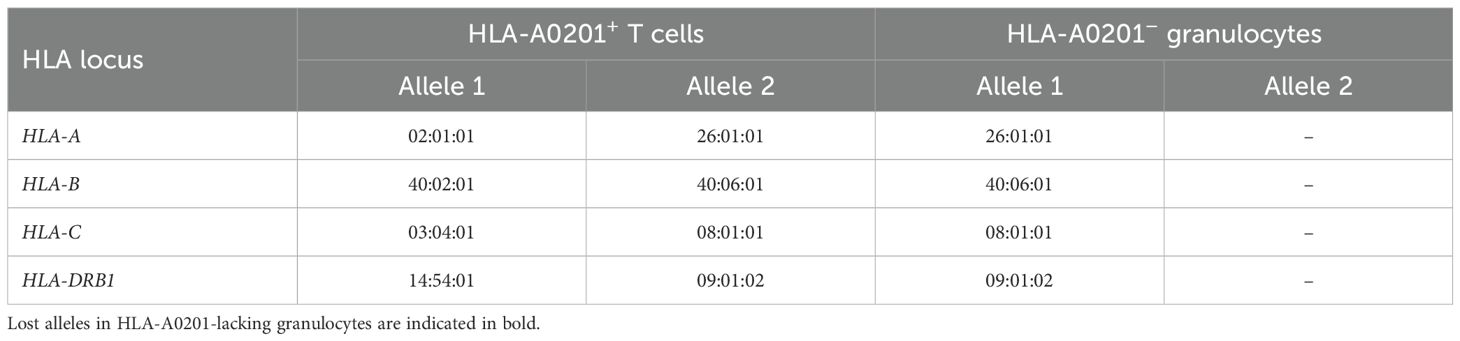
Table 2. HLA genotyping results of sorted cell populations.
To determine whether HLA allele-lacking cells due to 6pLOH were present at the time of diagnosis, we extracted genomic DNA from initial bone marrow smears and performed digital polymerase chain reaction to quantify allele-specific copy numbers of HLA-C, as previously described (12). This analysis revealed that 41% of bone marrow nucleated cells harbored 6pLOH (Figure 2C). Given that lymphocytes accounted for 62% of the nucleated bone marrow cells, it was estimated that nearly all non-lymphoid hematopoietic cells had already been replaced by HLA allele-lacking cells at the time of diagnosis. However, it is possible that bone marrow aspirates were obtained from a hematopoietic niche dominated by HLA-A0201-lacking stem cells, while wild-type cells continued to be produced at other sites, as cells expressing both HLA and GPI accounted for 8% of granulocytes and monocytes at 22 months after the diagnosis.
Discussion
HSCs deficient in HLA class I or GPI can evade T-cell-mediated destruction and sustain clonal or oligoclonal hematopoiesis for an extended period following IST (12, 15, 18–21). However, all previously reported cases of remission with escape hematopoiesis had been treated with immunosuppressive therapy or anabolic steroids, spontaneous complete remission of SAA mediated by such immune-escaping HSC clones has not been previously reported. This phenomenon may be underrecognized, as most patients receive standard therapy shortly after diagnosis in accordance with current guidelines, and HLA loss is not routinely assessed in clinical practice.
HLA loss due to copy-neutral 6pLOH in AA was first identified in 2011 through copy number analysis using single nucleotide polymorphism array (10, 11). Although 6pLOH provides evidence of immune-mediated HSC depletion and can aid in diagnosing bone marrow failure of autoimmune origin (22–24), the relatively low prevalence of this abnormality (~13%) among AA patients limits its clinical utility (11, 25). More recent studies have identified loss-of-function mutations in HLA class I genes as an additional and more frequent mechanism of HLA loss (12–17). HLA allele-lacking leukocytes, arising from either 6pLOH or loss-of-function mutations, can be detected in 25-43% of patients with SAA using sensitive flow cytometry with HLA allele-specific monoclonal antibodies (15, 26). The impact of HLA loss on response to immunosuppressive therapy and prognosis of AA differs depending on the HLA class I alleles that is lost (13, 15).
In our case, T-cell-mediated HSC destruction appears to have begun 12–18 months prior to the diagnosis, potentially triggered by the loss of tolerance to self-antigens presented by HLA-A0201 or HLA-B4002 (11, 12). The gradual progression of the disease may have allowed sufficient time for the selective expansion of immune-evading HSC clones. The steep recovery observed during the first four weeks, which did not align with the subsequent hematologic recovery, may have been influenced by the fever the patient experienced prior to hospitalization. By the time of the diagnosis, most residual HSCs had been replaced by 6pLOH clones, along with a small population of GPI-deficient cells. These immune-escaping clones jointly restored hematopoiesis in the absence of any treatment, leading to complete recovery within three months. The absence of driver gene mutations in HLA-lacking or GPI-deficient granulocytes supports the notion that immune pressure alone was sufficient to drive their expansion.
In AA patients who harbor both HLA allele-lacking and GPI-deficient cell populations, one population can occasionally outcompete the other; however, it remains unpredictable which will preferentially expand (27). Nevertheless, we initially expected that GPI-deficient cells in the present case would eventually disappear during the expansion of HLA allele-lacking HSCs, based on a previously reported case of SAA treated with cyclosporine and methenolone (21). In that case, although the initial treatment response was poor, durable remission was ultimately achieved through the gradual expansion of HLA-A*02:06-deficient cells, accompanied by a decline and eventual disappearance of GPI-deficient cells. This pattern is thought to reflect the complete immune evasion of HLA-lacking HSCs from antigen-specific CD8+ T-cell-mediated cytotoxicity (22), whereas GPI-deficient HSCs, which retain HLA expression, remain partially susceptible to CD8+ T-cell attack.
In contrast, in our case, GPI-deficient cells gradually expanded during spontaneous recovery. This observation may indicate the coexistence of a distinct, yet incompletely understood, immune mechanism—such as CD4+ T cell-mediated marrow suppression—that selectively spares GPI-deficient HSCs while targeting HLA class I allele-lacking HSCs (28–30). This raises concerns about the potential for future relapse and progression to paroxysmal nocturnal hemoglobinuria and highlights the need to explore therapeutic strategies specifically targeting this mechanism.
This case report provides valuable clinical insight. A high percentage of HLA allele-lacking leukocytes at the time of the AA diagnosis may predict spontaneous remission. Even when the percentage is low, treatment with thrombopoietin receptor agonist, with or without cyclosporine, might facilitate the proliferation of immune-escaping HSCs and reduce the need for intensive IST or hematopoietic stem cell transplantation. Therefore, detection of HLA-lacking leukocytes may be considered as part of the diagnostic evaluation of AA to help assess the potential for spontaneous remission. Further studies are needed to clarify the incidence and long-term outcomes of spontaneous remission mediated by the immune-evading mutant HSCs. A nationwide prospective study is currently underway to validate the prognostic value of detecting the immune-escaping clones in treatment-naïve patients with AA.
Data availability statement
All data supporting the findings of this study are included in the article and its supplementary information files. Additional details are available from the corresponding author upon reasonable request.
Ethics statement
The studies involving humans were approved by The Institutional Review Board and Ethics Committee of Kanazawa University, the University of Tsukuba, and Tachikawa Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
KS: Visualization, Data curation, Writing – review & editing, Formal analysis, Writing – original draft. YZ: Investigation, Writing – original draft, Funding acquisition, Formal analysis, Data curation, Writing – review & editing, Methodology, Conceptualization. HK: Visualization, Writing – review & editing, Data curation, Formal analysis. NO: Supervision, Project administration, Writing – review & editing. KH: Methodology, Data curation, Investigation, Writing – review & editing, Formal analysis. YK: Data curation, Writing – review & editing, Investigation, Methodology, Formal analysis. MK: Investigation, Writing – review & editing. EM: Writing – review & editing, Investigation. HT: Investigation, Writing – review & editing. TT: Data curation, Writing – review & editing, Formal analysis. SN: Supervision, Methodology, Conceptualization, Funding acquisition, Writing – review & editing, Formal analysis, Project administration, Writing – original draft, Investigation, Validation, Data curation.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Numbers JP22K08473 and JP24K19196) and Mochida Memorial Foundation for Medical and Pharmaceutical Research.
Acknowledgments
The authors thank the medical staff at the Tachikawa Hospital for their excellent clinical care and assistance. This work was the result of using research equipment shared in MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0440300024.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as potential conflicts of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
2. Champlin R, Ho W, and Gale RP. Antithymocyte globulin treatment in patients with aplastic anemia: a prospective randomized trial. N Engl J Med. (1983) 308:113–8. doi: 10.1056/NEJM198301203080301
3. Bacigalupo A. How I treat acquired aplastic anemia. Blood. (2017) 129:1428–36. doi: 10.1182/blood-2016-08-693481
4. Scheinberg P and Young NS. How I treat acquired aplastic anemia. Blood. (2012) 120:1185–96. doi: 10.1182/blood-2011-12-274019
5. Lee JH, Lee JH, Shin YR, Lee JS, Kim WK, Chi HS, et al. Spontaneous remission of aplastic anemia: a retrospective analysis. Haematologica. (2001) 86:928–33.
6. Choudhry VP, Gupta S, Gupta M, Kashyap R, and Saxena R. Pregnancy associated aplastic anemia–a series of 10 cases with review of literature. Hematology. (2002) 7:233–8. doi: 10.1080/1024533021000024067
7. Saitoh T, Saiki M, Kumagai T, Kura Y, Sawada U, and Horie T. Spontaneous clinical and cytogenetic remission of aplastic anemia in a patient with del(13q). Cancer Genet Cytogenet. (2002) 136:126–8. doi: 10.1016/s0165-4608(02)00519-8
8. Sugimori C, Chuhjo T, Feng X, Yamazaki H, Takami A, Teramura M, et al. Minor population of CD55-CD59- blood cells predicts response to immunosuppressive therapy and prognosis in patients with aplastic anemia. Blood. (2006) 107:1308–14. doi: 10.1182/blood-2005-06-2485
9. Luzzatto L and Risitano AM. Advances in understanding the pathogenesis of acquired aplastic anaemia. Br J Haematol. (2018) 182:758–76. doi: 10.1111/bjh.15443
10. Afable MG 2nd, Wlodarski M, Makishima H, Shaik M, Sekeres MA, Tiu RV, et al. SNP array-based karyotyping: differences and similarities between aplastic anemia and hypocellular myelodysplastic syndromes. Blood. (2011) 117:6876–84. doi: 10.1182/blood-2010-11-314393
11. Katagiri T, Sato-Otsubo A, Kashiwase K, Morishima S, Sato Y, Mori Y, et al. Frequent loss of HLA alleles associated with copy number-neutral 6pLOH in acquired aplastic anemia. Blood. (2011) 118:6601–9. doi: 10.1182/blood-2011-07-365189
12. Zaimoku Y, Takamatsu H, Hosomichi K, Ozawa T, Nakagawa N, Imi T, et al. Identification of an HLA class I allele closely involved in the autoantigen presentation in acquired aplastic anemia. Blood. (2017) 129:2908–16. doi: 10.1182/blood-2016-11-752378
13. Babushok DV, Duke JL, Xie HM, Stanley N, Atienza J, Perdigones N, et al. Somatic HLA mutations expose the role of class I-mediated autoimmunity in aplastic anemia and its clonal complications. Blood Adv. (2017) 1:1900–10. doi: 10.1182/bloodadvances.2017010918
14. Mizumaki H, Hosomichi K, Hosokawa K, Yoroidaka T, Imi T, Zaimoku Y, et al. A frequent nonsense mutation in exon 1 across certain HLA-A and -B alleles in leukocytes of patients with acquired aplastic anemia. Haematologica. (2021) 106:1581–90. doi: 10.3324/haematol.2020.247809
15. Zaimoku Y, Patel BA, Adams SD, Shalhoub R, Groarke EM, Lee AAC, et al. HLA associations, somatic loss of HLA expression, and clinical outcomes in immune aplastic anemia. Blood. (2021) 138:2799–809. doi: 10.1182/blood.2021012895
16. Olson TS, Frost BF, Duke JL, Dribus M, Xie HM, Prudowsky ZD, et al. Pathogenicity and impact of HLA class I alleles in aplastic anemia patients of different ethnicities. JCI Insight. (2022) 7:e163040. doi: 10.1172/jci.insight.163040
17. Pagliuca S, Gurnari C, Hercus C, Hergalant S, Nadarajah N, Wahida A, et al. Molecular landscape of immune pressure and escape in aplastic anemia. Leukemia. (2023) 37:202–11. doi: 10.1038/s41375-022-01723-w
18. Hillmen P, Lewis SM, Bessler M, Luzzatto L, and Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med. (1995) 333:1253–8. doi: 10.1056/NEJM199511093331904
19. Imi T, Katagiri T, Hosomichi K, Zaimoku Y, Hoang Nguyen V, Nakagawa N, et al. Sustained clonal hematopoiesis by HLA-lacking hematopoietic stem cells without driver mutations in aplastic anemia. Blood Adv. (2018) 2:1000–12. doi: 10.1182/bloodadvances.2017013953
20. Hosokawa K, Mizumaki H, Yoroidaka T, Maruyama H, Imi T, Tsuji N, et al. HLA class I allele-lacking leukocytes predict rare clonal evolution to MDS/AML in patients with acquired aplastic anemia. Blood. (2021) 137:3576–80. doi: 10.1182/blood.2020010586
21. Zaimoku Y, Sakai K, Tsuji N, Hosomichi K, Yamada S, Tran DC, et al. Haematopoietic regeneration by HLA-A*0206-deficient clones in severe aplastic anaemia without definitive immunosuppressive treatment. Br J Haematol. (2024) 205:1995–9. doi: 10.1111/bjh.19712
22. Espinoza JL, Elbadry MI, Chonabayashi K, Yoshida Y, Katagiri T, Harada K, et al. Hematopoiesis by iPSC-derived hematopoietic stem cells of aplastic anemia that escape cytotoxic T-cell attack. Blood Adv. (2018) 2:390–400. doi: 10.1182/bloodadvances.2017013342
23. Shah YB, Priore SF, Li Y, Tang CN, Nicholas P, Kurre P, et al. The predictive value of PNH clones, 6p CN-LOH, and clonal TCR gene rearrangement for aplastic anemia diagnosis. Blood Adv. (2021) 5:3216–26. doi: 10.1182/bloodadvances.2021004201
24. Zaimoku Y, Katagiri T, Nakagawa N, Imi T, Maruyama H, Takamatsu H, et al. HLA class I allele loss and bone marrow transplantation outcomes in immune aplastic anemia. Transplant Cell Ther. (2024) 30:281.e1–281.e13. doi: 10.1016/j.jtct.2023.11.013
25. Yoshizato T, Dumitriu B, Hosokawa K, Makishima H, Yoshida K, Townsley D, et al. Somatic mutations and clonal hematopoiesis in aplastic anemia. N Engl J Med. (2015) 373:35–47. doi: 10.1056/NEJMoa1414799
26. Ishiyama K, Suzuki R, Mitani K, Maruyama H, Onishi Y, Usuki K, et al. Efficacy and safety of eltrombopag added to rabbit anti-human thymocyte globulin+cyclosporine in aplastic anemia: results from a prospective multicenter phase II study [abstract. Blood. (2024) 144:2689. doi: 10.1182/blood-2024-193818
27. Yoroidaka T, Hosokawa K, Imi T, Mizumaki H, Katagiri T, Ishiyama K, et al. Hematopoietic stem progenitor cells lacking HLA differ from those lacking GPI-anchored proteins in the hierarchical stage and sensitivity to immune attack in patients with acquired aplastic anemia. Leukemia. (2021) 35:3257–67. doi: 10.1038/s41375-021-01202-8
28. Murakami Y, Kosaka H, Maeda Y, Nishimura J, Inoue N, Ohishi K, et al. Inefficient response of T lymphocytes to glycosylphosphatidylinositol anchor-negative cells: implications for paroxysmal nocturnal hemoglobinuria. Blood. (2002) 100:4116–22. doi: 10.1182/blood-2002-06-1669
29. Tsuji N, Hosokawa K, Urushihara R, Tanabe M, Zaimoku Y, Katagiri T, et al. Frequent HLA-DR loss on hematopoietic stem progenitor cells in patients with cyclosporine-dependent aplastic anemia carrying HLA-DR15. Leukemia. (2022) 36:1666–75. doi: 10.1038/s41375-022-01549-6
Keywords: severe aplastic anemia, spontaneous remission, immune escape, HLA loss, paroxysmal nocturnal hemoglobinuria, somatic mutation, chromosome 6p loss of heterozygosity
Citation: Shiroshita K, Zaimoku Y, Kudo H, Obara N, Hosomichi K, Kano Y, Kobayashi M, Morishita E, Takamatsu H, Toyama T and Nakao S (2025) Case report: Spontaneous remission of severe aplastic anemia mediated by mutant hematopoietic stem cells evading T-cell attack. Front. Immunol. 16:1635943. doi: 10.3389/fimmu.2025.1635943
Received: 27 May 2025; Accepted: 15 September 2025;
Published: 01 October 2025.
Edited by:
Rachel Koldej, Royal Melbourne Hospital, AustraliaReviewed by:
Daria Babushok, University of Pennsylvania, United StatesToru Kawakami, Matsumoto Medical Center, Japan
Copyright © 2025 Shiroshita, Zaimoku, Kudo, Obara, Hosomichi, Kano, Kobayashi, Morishita, Takamatsu, Toyama and Nakao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shinji Nakao, c25ha2FvODIwNUBzdGFmZi5rYW5hemF3YS11LmFjLmpw
†These authors have contributed equally to this work and share first authorship