 Miao Xiang1,2†
Miao Xiang1,2† Panpan Li1,2†
Panpan Li1,2† Xiaofei Yue3†
Xiaofei Yue3† Linlin Liu1,2†Linjing Wang4Nengjin Sun1,2Kaile Wang1,2Yuying Zhang5*
Linlin Liu1,2†Linjing Wang4Nengjin Sun1,2Kaile Wang1,2Yuying Zhang5* Hongyan Wang1,2*
Hongyan Wang1,2*- 1Key Laboratory of Immune Microenvironment and Inflammatory Disease Research in Universities of Shandong Province, School of Basic Medical Sciences, Shandong Second Medical University, Weifang, China
- 2Department of Pathogenic Biology, School of Basic Medical Sciences, Shandong Second Medical University, Weifang, China
- 3Rehabilitation Pharmacy Center, Beijing Rehabilitation Hospital, Capital Medical University, Beijing, China
- 4Department of Oncology, China-Japan Union Hospital, Jilin University, Changchun, China
- 5Department of Gastroenterology, Weifang People’s Hospital, Shandong Second Medical University, Weifang, China
Helicobacter pylori (H. pylori) is a microaerophilic, gram-negative spirochete that primarily colonizes the human gastric mucosa. It is strongly linked to gastritis, ulcers, and the development of malignant tumors. Macrophages, as one of the key components of the innate immune system, play a crucial role in maintaining immune homeostasis through a range of functions, including pathogen phagocytosis, antigen recognition and presentation, inflammation regulation and tumor immune surveillance. Emerging evidence suggests that H. pylori employs diverse molecular mechanisms to evade immune clearance by macrophages. This review provides a comprehensive analysis of how H. pylori infection modulates macrophage functions, including impairing pathogen recognition and phagocytosis, disrupting phagosome maturation and reducing immune clearance capacity. Furthermore, H. pylori infection skews macrophage polarization to promote chronic inflammatory damage, inhibits antigen processing and presentation to evade adaptive immune responses and induces macrophage apoptosis via activation of apoptotic signaling pathways. By unraveling the complex molecular interactions between H. pylori and macrophages, this review highlights strategies for reprogramming macrophage functions, offering innovative approaches to address the limitations of conventional antimicrobial therapies and advancing targeted therapeutic interventions for H. pylori-associated diseases.
1 Introduction
Helicobacter pylori (H. pylori) is a microaerobic, gram-negative spiral bacterium that colonizes the mucous membranes of the human gastrointestinal tract. This globally prevalent pathogen exhibited an infection rate of 58.2% between 1980 and 1990 (1). Although the prevalence has declined in recent years, it remains significantly high at 43.9% between 2015 and 2022 (2). Barry Marshall and Robin Warren first isolated and cultured this bacterium from the gastric mucous membranes of patients with peptic ulcers in 1983 (3). Research has demonstrated that H. pylori infection is a critical pathogenic factor in various gastrointestinal disorders, ranging from superficial gastritis and peptic ulcers to more severe conditions such as gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma (4). Consequently, H. pylori has been classified as a Group 1 carcinogen by the International Agency for Research on Cancer (IARC) of the World Health Organization (WHO), marking it as the first bacterium officially recognized as carcinogenic (5). The pathogenic mechanisms of H. pylori infection are exceedingly complex.
H. pylori infection exhibits pathogenic complexity attributable to its virulence factors, with studies confirming strong correlations between these factors and disease severity (6). Strains are classified as CagA-positive or -negative based on cytotoxin-associated gene A (CagA) expression; CagA-positive variants inject effector proteins into host cells via the type IV secretion system (T4SS), disrupting cytoskeletal dynamics and impairing cellular motility, proliferation, and apoptosis homeostasis. Conversely, ubiquitously expressed vacuolating toxin A (VacA) drives chronic colonization by inducing apoptosis, autophagy, membrane potential collapse, aberrant MAP kinase activation, and T-cell dysfunction (7–9). Urease, the most abundant H. pylori protein, serves dual functions: modulating pH to facilitate colonization while releasing ammonia to neutralize gastric acidity, and subverting host defenses through impaired opsonization, enhanced granulocyte chemotaxis, MHC-II-dependent apoptosis pathway activation, and pro-inflammatory cytokine storm amplification (10, 11). Lipopolysaccharide (LPS) evades pattern recognition receptors via lipid A/O-antigen structures, perpetually activating NF-κB-mediated inflammatory cascades that accelerate the gastritis-ulcer-carcinogenesis sequence (12, 13). The flagellum, essential for motility and chemotaxis, mediates initial mucosal colonization while modifying TLR5 signaling to promote inflammatory polarization and immune evasion (14, 15). Collectively, these virulence factors enable gastric mucosal persistence, subvert immune responses, drive immune escape, and ultimately induce carcinogenesis (16).
Microglia arise directly from yolk sac progenitors during early embryogenesis, whereas most other tissue-resident macrophages are derived from fetal liver monocytes generated through embryonic hematopoiesis (17). In adults, circulating macrophages typically originate from bone marrow–derived monocytes, particularly under inflammatory conditions (17). As a crucial component of the innate immune system, macrophages exhibit potent phagocytic abilities. While both macrophages and neutrophils share phagocytic functions, macrophages have a longer lifespan. In response to pathogen invasion, macrophages act as “scavengers” and, along with neutrophils, are the first responders to infection. As a crucial component of innate immunity, their phagocytic abilities enable direct engulfment and destruction of invading microbes. The pattern recognition receptors (PRRs) on the surface of macrophages recognize pathogen-associated molecular patterns (PAMPs), triggering innate immune signaling pathways to exert antimicrobial effects (18). Beyond their role in pathogen clearance, macrophages also promote inflammation process, present antigens and contribute to tumor immunity, thereby maintaining immune homeostasis (19, 20). Although macrophages serve as the first line of defense against invading pathogens, certain microbes, such as H. pylori, have evolved mechanisms to disrupt the immune regulatory functions of macrophages, thereby facilitating the persistence of infection and contributing to tumorigenesis (21–24). This review explores the mechanisms by which H. pylori infection reprograms macrophage functionality, providing innovative solutions to overcome the limitations of conventional antimicrobial therapy, which may inform macrophage-centric therapeutic development for H. pylori-associated disorders.
2 H. pylori infection and macrophage immune dysfunction: mechanisms and implications
2.1 The impact of H. pylori infection on macrophage recognition and phagocytosis of pathogens
Upon H. pylori infection, innate immune cells such as macrophages recognize bacterial PAMPs through PRRs, including Toll-like receptors. Following recognition, macrophages extend their plasma membranes to engulf H. pylori, forming membrane-bound phagosomes (25). Subsequently, the phagosomes sequentially interact with early and late endosomes, followed by fusion with lysosomes to form phagolysosomes. These phagolysosomes contain fully functional lysosomal hydrolases, potent enzymes capable of degrading pathogens (26, 27). During digestion by hydrolases, small peptides are generated that form complexes with MHC-II. These peptide-MHC-II complexes are then transported via vesicles to the cell surface, where they are presented for antigen recognition (28) (Figure 1A).
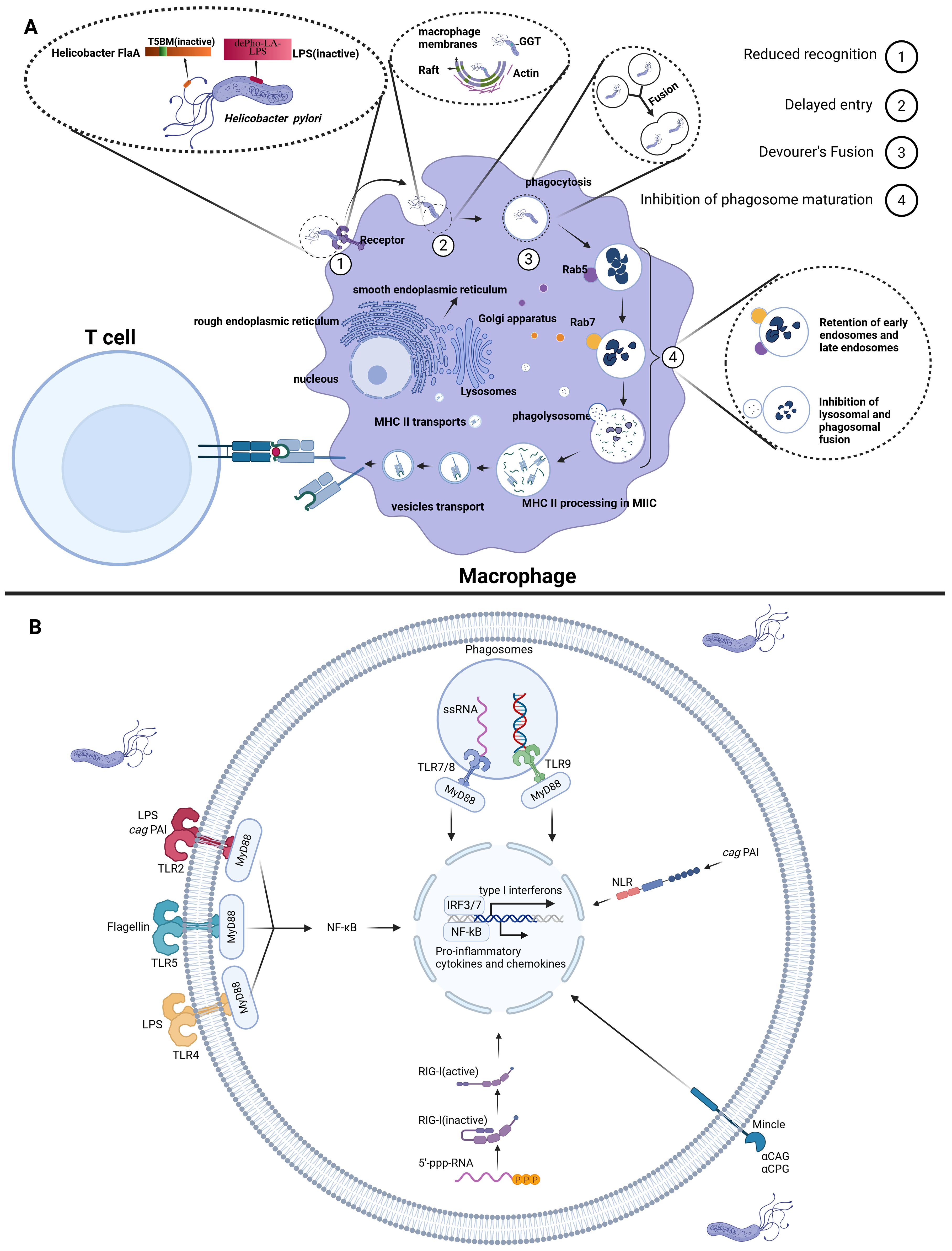
Figure 1. The impact of H. pylori on macrophage recognition and phagocytic function. (A) (1) Reduced Recognition: H. pylori diminishes macrophage PRR-mediated recognition by expressing O-antigens that closely resemble human antigens and through mutations in flagellin (FlaA). (2) Delayed Entry: H. pylori synthesizes CGs, which activates the PI3K pathway, regulating actin dynamics and thereby delaying macrophage phagocytosis. (3) Fusion of Phagocytes: H. pylori promotes the fusion of macrophages that have internalized the bacteria, thereby disrupting normal phagosome maturation and delaying the clearance of the pathogen. (4) Inhibition of Phagosome Maturation: H. pylori enhances resistance in early and late endosomes and lysosomes via GGT; Additionally, it recruits Coronin 1A to delay the formation of phagolysosomes. (B) Recognition of H. pylori by innate immune cells. Created with Biorender.com.
The initial response of macrophages to H. pylori infection is characterized by the specific recognition of PAMPs by PRRs, including Toll-like receptors (TLRs), intracellular RIG-I-like receptors (RLRs), C-type lectin receptors (CLRs) and nucleotide oligomerization domain (NOD)-like receptors (NLRs). TLR4, TLR2 and TLR5 on innate immune cell membranes recognize H. pylori-derived LPS and flagellin, respectively. This recognition triggers activation of the NF-κB signaling pathway and inflammasome complexes, orchestrating the expression of pro-inflammatory cytokines and chemokines (29, 30). The intracellular recognition mechanisms demonstrate greater complexity: TLR9 recognizes unmethylated CpG DNA, while TLR7/8 specifically identifies bacterial single-stranded RNA, with both converging on the MyD88-TRIF signaling axis to activate immune responses (31–33). Concurrently, RIG-I senses 5’-triphosphorylated RNA (5’-ppp-RNA) to initiate type I interferon production (18, 32). Emerging research reveals that the cytotoxin-associated gene pathogenicity island (cag PAI) of H. pylori precisely orchestrates IL-1β synthesis and secretion through synergistic interactions with TLR2, NOD2 and NLRP3 (34). CLRs recognize Cholesteryl acyl α-glucoside (αCAG) and cholesteryl phosphatidyl α-glucoside (αCPG) of H. pylori to exacerbate inflammation (35) (Figure 1B). However, certain PAMPs of H. pylori can evade recognition by macrophage PRRs due to their weak immunogenicity. For instance, H. pylori evades detection by PRRs through the expression of O-antigens that resemble human antigens and the production of low-activity LPS endotoxins (36). Additionally, flagellin, which normally serves as the ligand for TLR5, is recognized as a pro-inflammatory trigger upon detection of the flagella of H. pylori. However, mutations in H. pylori flagellin (FlaA) prevent TLR5 from recognizing the bacterium’s flagellin (37).
Upon successful recognition of H. pylori by macrophages, the cholesterol-α-glucosyltransferase (CGT) of type I H. pylori synthesizes and inserts cholesteryl glucosides (CGs) into the lipid raft regions at the H. pylori-macrophage attachment sites. This action activates class I phosphoinositide 3-kinases (PI3Ks), which regulate actin polymerization and thus delay phagocytosis by macrophages (38–40).
Following the recognition and internalization of H. pylori, the phagosome promptly enters the maturation phase. Experimental evidence demonstrates that phagosomes containing individual H. pylori may undergo homotypic fusion, leading to the formation of megasomes. These megasomes disrupt the normal metabolic processes of both early and late endosomes, thereby impeding maturation of phagosomes (39, 41, 42). Nevertheless, the precise molecular mechanisms underlying the formation of megasomes remain incompletely understood. Early studies suggested that the VacA and/or cag PAI might contribute to this process. However, subsequent research has demonstrated that megasome formation is independent of both VacA and the cag PAI of H. pylori. Instead, the presence of urease has been found to impair the aggregation and fusion of phagosomes (39, 43, 44). In addition to promoting megasome formation, H. pylori GGT has been shown to enhance the resistance of both early and late endosomes, as well as lysosomes, thereby delaying phagolysosome formation (38, 45). Furthermore, H. pylori can directly disrupt the fusion of phagosomes with lysosomes in macrophages by recruiting and retaining Coronin 1A, thus impeding phagosome maturation (46).
These mechanisms demonstrate that H. pylori evades macrophage clearance by comprehensively inhibiting recognition, phagocytosis, digestion and degradation (Figure 1A). Despite substantial research efforts to elucidate H. pylori’s strategies for evading macrophage recognition and phagocytosis, the intricate nature of its PAMPs remains a key research focus.
2.2 The pathogenic role of H. pylori infection on macrophage-mediated inflammatory regulation
Macrophages are inherently heterogeneous and plastic, making them crucial regulators of inflammatory processes and significant contributors to both the initiation and resolution of inflammation (47). Upon exposure to diverse stimuli, macrophages undergo polarization into distinct activation subtypes, each exhibiting unique phenotypic characteristics and cytokine secretion profiles. Classically activated M1 macrophages primarily secrete inflammatory mediators to execute immune defense and combat pathogen invasion. In contrast, alternatively, activated M2 macrophages produce anti-inflammatory factors, promoting tissue repair and wound healing. Recently, beyond the well-characterized M1 and M2 subtypes, regulatory macrophages (Mreg) have been identified. These cells are activated by LPS in combination with immune complexes and secrete anti-inflammatory factors, playing a role in immune regulation (19, 48, 49).
During pathogen infection, macrophages in the gastric mucosa are recruited and activated into M1 macrophages, which produce numerous inflammatory mediators to execute immune defense (48). For example, during Mycobacterium tuberculosis (Mtb) infection, M1 macrophages upregulate innate immune regulatory genes to facilitate the degradation of Mtb (50). However, the Mtb effector PPE36 can inhibit the activation of the ERK signaling pathway, thereby suppressing M1 macrophage polarization and reducing the production of pro-inflammatory cytokines (51). Similarly, Salmonella typhimurium utilizes the type III secretion system (T3SS) encoded by pathogenicity island 1 (SPI-1) and SPI-2 to enhance its survival and replication within macrophages (52). Studies have demonstrated that H. pylori can also inhibit or attenuate the activation of M1-type macrophages through multiple mechanisms, thereby achieving immune evasion and reducing inflammatory responses (Figure 2). The activity of H. pylori’s LPS is significantly lower compared to other Gram-negative bacteria such as Escherichia coli, leading to a diminished potential for M1 macrophage activation (36). Moreover, H. pylori can also inhibit M1-type macrophage polarization through enzymatic activity. Specifically, the bacterium’s urease subunit B (UreB) attenuates LPS-induced M1 polarization in macrophages (53). Through the action of matrix metalloproteinase 7 (MMP7), H. pylori downregulates IL-1β and inducible nitric oxide synthase (iNOS) mRNA expression in macrophages, thereby further impeding M1 polarization (53, 54). Arginase(Arg) plays a crucial role in the host’s anti-inflammatory response (55). H. pylori upregulates Arg2 expression in macrophages, which subsequently restricts M1 macrophage activation (56). This upregulation of Arg2 additionally reduces iNOS protein levels, leading to decreased nitric oxide production and consequently impairing macrophage-mediated bactericidal activity against H. pylori (57).
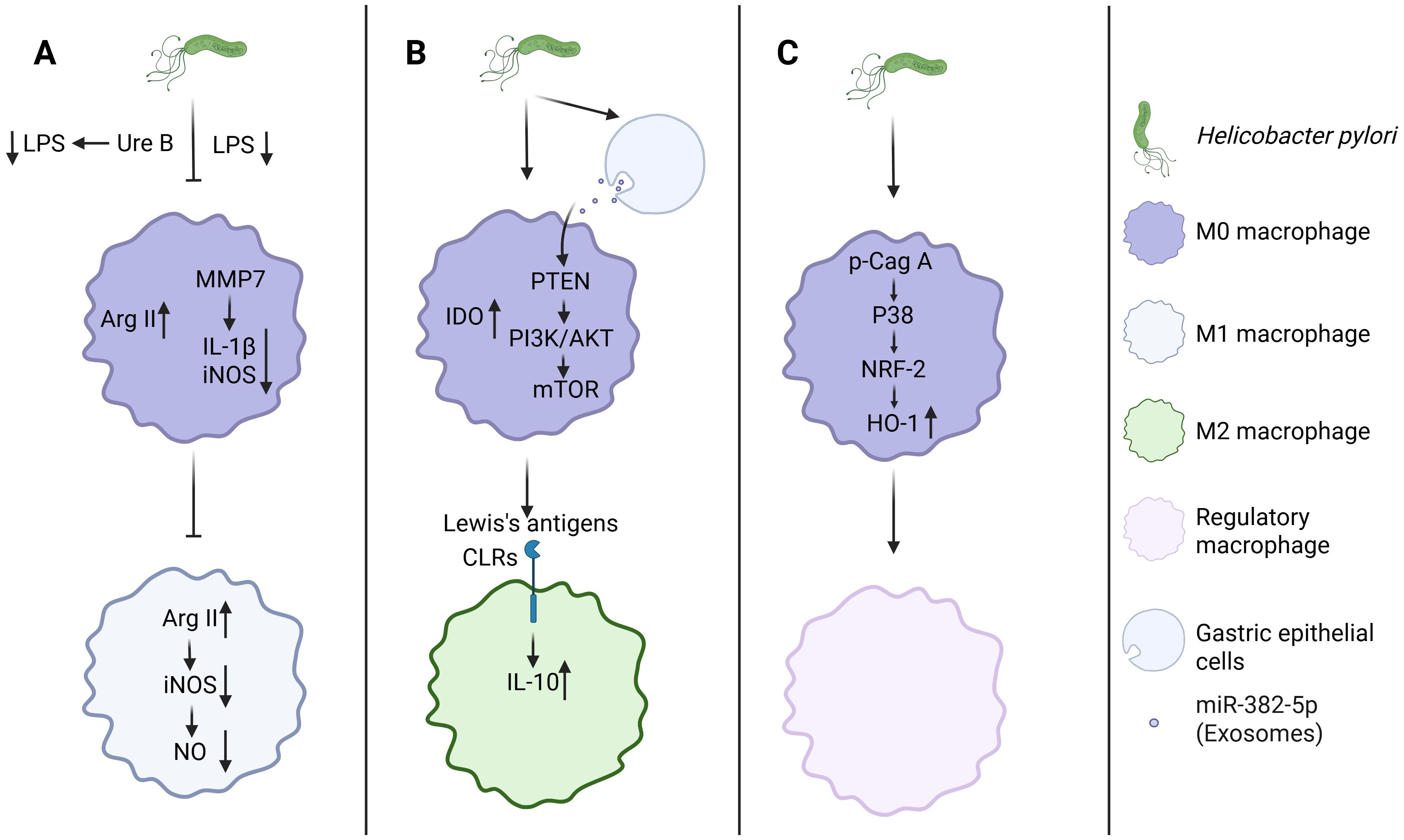
Figure 2. H. pylori orchestrates macrophage polarization through triple-pathway regulation. (A) H. pylori inhibits M1 macrophage polarization. (B, C) H. pylori promotes M2, Mreg macrophage polarization. Created with Biorender.com.
Unlike their pro-inflammatory M1 counterparts, M2 and Mreg actively produce anti-inflammatory mediators that help to suppress inflammatory pathways (58). Stimulating the polarization of macrophages towards the M2 or Mreg phenotype appears to be another strategy employed by H. pylori to evade immune clearance by macrophages (59). Research led by Alain P. Gobert et al. demonstrated that H. pylori can trigger the expression of heme oxygenase 1 (HO-1) in murine macrophages via the p-CagA/p38/NRF-2 signaling axis, thereby promoting the polarization of macrophages towards the Mreg phenotype (60). Additionally, Devi et al. demonstrated that H. pylori LPS Lewis antigens engage CLRs to induce IL-10 production in macrophages (61). Peng et al. indicated that H. pylori upregulates the expression of indoleamine 2,3-dioxygenase (IDO) in macrophages, further driving their polarization towards the M2 phenotype (62). Notably, H. pylori-infected gastric cancer cells secrete exosomal miR-382-5p that targets the PTEN gene in macrophages, thereby activating the PI3K/AKT/mTOR signaling pathway to inhibit autophagy and promote macrophage M2 polarization (63). Despite evidence that H. pylori evades immune surveillance by polarizing macrophages towards M2 or Mreg phenotypes, current research in this area is still in its early stages, with the molecular mechanisms and regulatory networks that need to be fully elucidated.
2.3 The influence of H. pylori infection on antigen processing and presentation
As crucial components of the innate immune system, macrophages function as antigen-presenting cells (APCs) akin to dendritic cells, facilitating the presentation of antigens to T lymphocytes (20, 64). Macrophages internalize antigens through mechanisms such as phagocytosis, trogocytosis, endocytosis and pinocytosis, and subsequently present these antigens to T cells via cross-presentation (65). This antigen-presenting capacity is crucial for macrophages to regulate immune responses. However, H. pylori infection disrupts the antigen recognition and phagocytic functions of macrophages, thereby affecting antigen uptake. Furthermore, H. pylori impairs the antigen-presenting capacity of macrophages by modulating intracellular miRNA expression. Studies have elucidated that H. pylori downregulates miR-4270 in macrophages, which in turn upregulates the immune receptor CD300E, leading to decreased surface expression of MHC-II molecules (66). This hampers the recognition and activation of effector T cells, thereby diminishing the antigen-presenting efficacy of macrophages (66). The application of miRNA microarray profiling in macrophages has identified that MHC-II downregulation post-H. pylori infection is chiefly mediated by the suppression of class II major histocompatibility complex transactivator (CIITA) expression (67). CIITA, a critical regulator of MHC-II gene transcription, is modulated by multiple miRNAs. Studies have demonstrated that H. pylori upregulates miRNAs such as let-7i-5p, miR-146b-5p and miR-185-5p, which inhibit CIITA expression, thereby suppressing the synthesis of HLA-II molecules and further decreasing the antigen-presenting capacity of macrophages (67). Additional studies have demonstrated that H. pylori utilizes ADP-heptose, an intermediate metabolite in the LPS biosynthesis pathway, to upregulate miR-146b expression (68). This leads to CIITA downregulation and subsequent suppression of HLA-II expression, resulting in impaired antigen-presenting function (68). This elucidates H. pylori’s immune evasion strategy through the precise modulation of miRNA expression to impair macrophage antigen-presenting capabilities. Moreover, recent findings indicate that UreB can also inhibit macrophage antigen presentation via its interaction with TLR2 (53). Further studies indicate that H. pylori can also modulate macrophage antigen presentation and immune responses by altering their differentiation states and modulating activation patterns. The differentiation status of macrophages significantly influences their immunological functions, consequently affecting both the efficiency of antigen presentation and the potential for T cell activation (69).
Consequently, H. pylori utilizes a complex immune evasion strategy by modulating macrophage miRNA expression, MHC-II molecule presentation, macrophage subtype differentiation and the action of virulence factors (Figure 3). Nevertheless, a comprehensive elucidation of the mechanisms by which H. pylori infection impacts macrophage antigen-presenting function remains to be fully achieved.
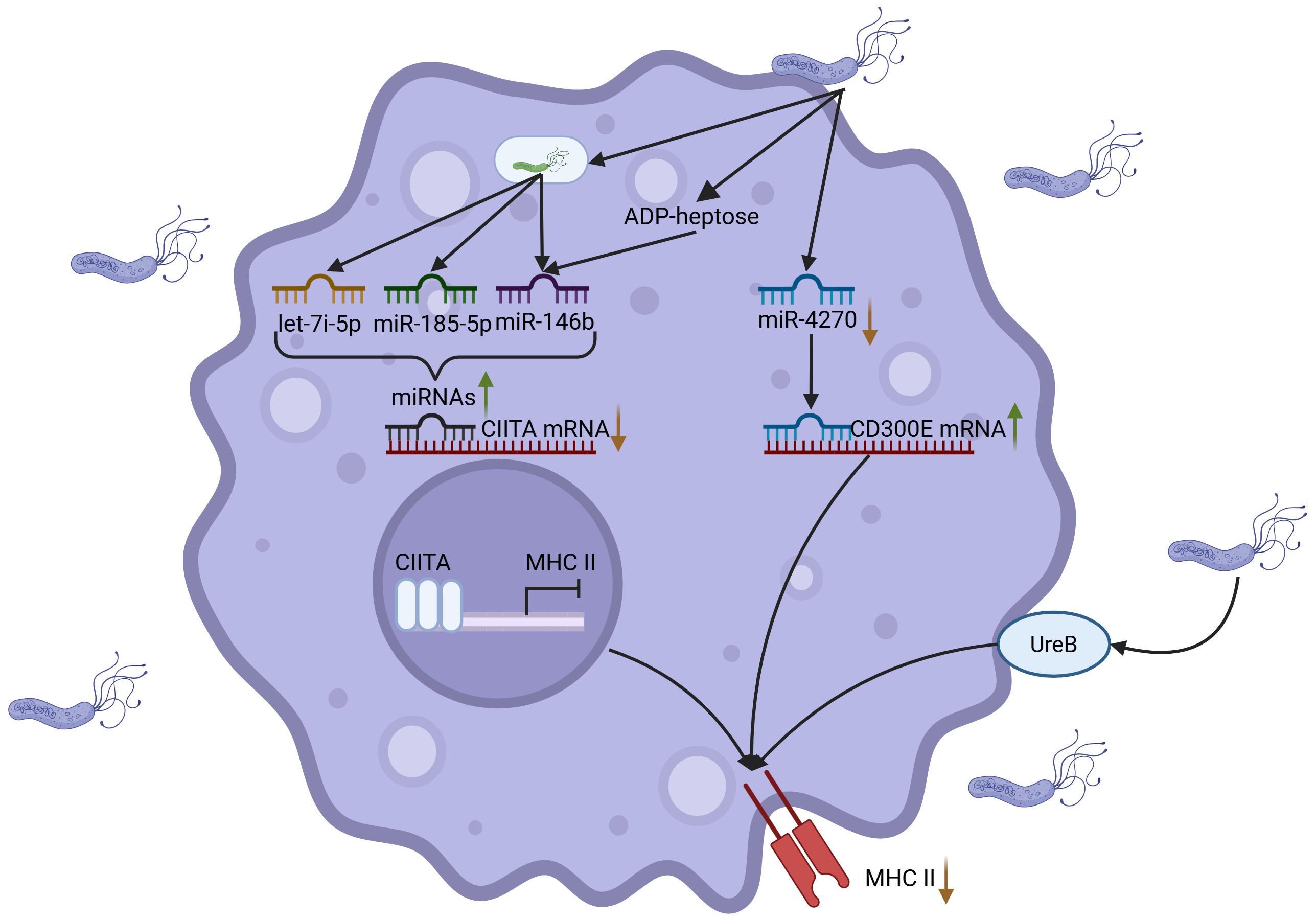
Figure 3. H. pylori inhibits antigen presentation of macrophages. H. pylori suppresses macrophage antigen presentation through coordinated mechanisms: (1) H. pylori downregulates miR-4270 to induce upregulation of CD300E, leading to diminished MHC-II surface expression; (2) H. pylori upregulates miRNAs (including let-7f-5p and miR-146b) to inhibit the transcriptional regulator CIITA, thereby impeding MHC-II biosynthesis; (3) Synergistic action of the metabolite ADP-heptose and surface protein UreB-TLR2 receptor interaction cooperatively impairs antigen-presenting capacity, ultimately facilitating immune evasion. Created with Biorender.com.
2.4 The impact of H. pylori infection on macrophage apoptosis
H. pylori infection modulates the immune microenvironment of gastric cancers through multiple pathways, particularly via induction of macrophage apoptosis, which significantly impairs macrophage function (Figure 4). As essential cells in immune surveillance, macrophages play a pivotal role in anti-tumor immunity. Therefore, any disruption in their apoptotic processes can profoundly affect tumor initiation and progression. Studies have demonstrated that H. pylori can trigger macrophage apoptosis through diverse molecular pathways. Initially, H. pylori triggers macrophage apoptosis by activating Arg2 and c-Myc-induced ornithine decarboxylase (ODC) (70, 71). Subsequent research by Menaker RJ et al. demonstrated that H. pylori-induced macrophage apoptosis is associated with alterations in the mitochondrial pathway (72). Furthermore, studies by Chaturvedi R and Asim M et al. revealed that the activation of polyamine oxidase 1 (PAO1) by H. pylori leads to the production of H2O2, which induces macrophage apoptosis through a mitochondrial-dependent mechanism. Simultaneously, H. pylori activates ERK1/2, leading to the formation of an AP-1 complex that binds to c-Myc, thereby upregulating ODC transcription and generating H2O2, which subsequently triggers apoptosis via mitochondrial membrane depolarization (73, 74). Additionally, H. pylori produces several secretory proteins that regulate macrophage apoptosis. For instance, JHP940 and HP986 from H. pylori trigger apoptosis through Fas and TNFR pathways, while JHP0290 and HP1286 promote c-Myc transcription by forming the AP-1 transcription factor complex via the ERK/MAPK signaling pathway, leading to macrophage apoptosis (75–78). As research advances, more mechanisms involved in H. pylori-induced macrophage apoptosis are being discovered. For example, H. pylori secretes phospholipase A (PldA), which triggers macrophage apoptosis via the p38-MK2 signaling pathway. Additionally, UreB can facilitate macrophage apoptosis by binding to TLR2, and VacA induces macrophage apoptosis through NF-κB activation (53, 79, 80). In addition, H. pylori infection in macrophages induces apoptosis through LPS interactions with TLR2 and TLR4, along with the upregulation of miR-155 via the T4SS. miR-155 promotes macrophage apoptosis by regulating the expression of several apoptosis-related genes, including Tspan14, SOCS1 and TNF-α (81, 82). In conclusion, H. pylori triggers macrophage apoptosis via various mechanisms, which impairs their role as anti-tumor immune surveillance cells, thus promoting the initiation and progression of gastric cancers. This intricate impact is orchestrated via the activation of intracellular signaling pathways and epigenetic modifications, particularly through microRNA regulation, highlighting the bacterium’s crucial role in tumor immune evasion strategies.
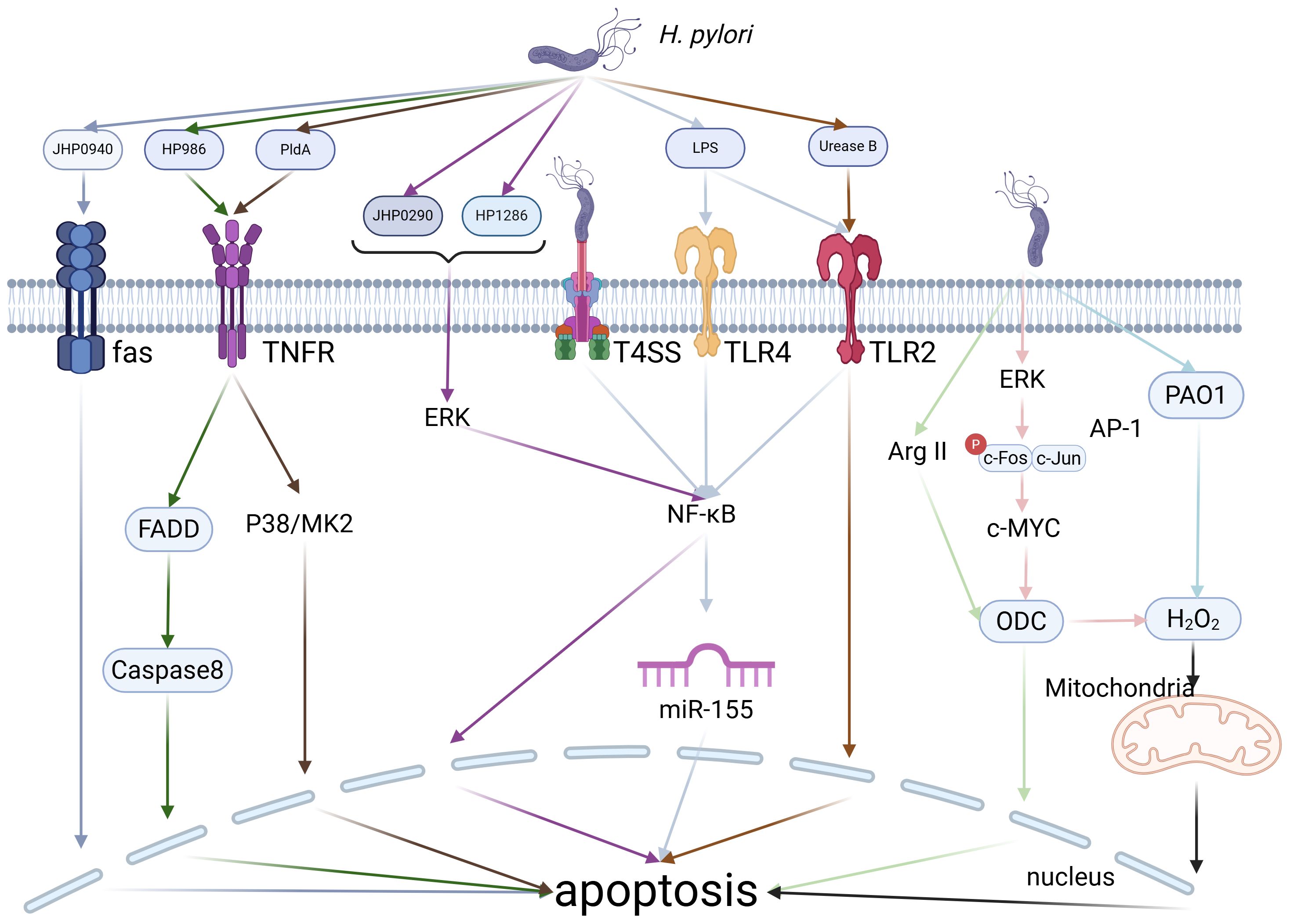
Figure 4. H. pylori-Induced Apoptosis. H. pylori can induce macrophage apoptosis through various pathways, including its own secreted proteins, enzymes, or the modulation of intracellular enzymes and microRNAs within macrophages. Created with biorender.com.
3 The role of macrophages in gastric cancer immunity
H. pylori infection is a major independent risk factor for gastric cancer (GC), with approximately 1-2% of those infected progressing to GC. The development of GC is a complex, multi-step process influenced by various factors, with the tumor microenvironment (TME) playing a crucial role (83). The TME is composed of evolving tumor cells and a complex stromal matrix. This stromal component includes diverse cellular constituents, notably tumor-associated macrophages (TAMs), which represent a significant component of the tumor stroma (84, 85). Circulating monocytes are recruited to the tumor periphery through the action of various chemokines and cytokines secreted by tumor cells or stromal cells. Upon infiltrating tumor tissues or clustering within the solid tumor microenvironment, these macrophages are defined as TAMs (86).
TAMs constitute the principal population of innate immune cells within the tumor immune microenvironment, exhibiting functional versatility with both tumor-promoting and tumor-suppressive potentials mediated via distinct immunological pathways (87–89). H. pylori infection is recognized as a major risk factor for GC development, primarily influencing TAMs by altering macrophage polarization states. Studies have shown that H. pylori infection can disrupt the differentiation of M1 macrophages and promote the formation of M2 macrophages, or even trigger the transdifferentiation of M1 macrophages into M2 macrophages. Subsequently, M2 macrophages further contribute to tumorigenesis and progression by promoting angiogenesis and secreting immunosuppressive factors (90, 91). Research by Bingting Yu et al. demonstrated that H. pylori infection triggers an inflammatory phenotype associated with M1 polarization. However, during chronic infection, sustained autocrine/paracrine IL-6 stimulation promotes the polarization of macrophages towards an M2 phenotype, potentially contributing to the development of gastric cancer (92). Recent studies have also revealed that chronic inflammation induced by M1-like TAM activity may accelerate genomic instability in malignant cells under certain conditions, serving as a driver of tumor progression (93). Furthermore, following H. pylori infection, gastric cancer cells secrete exosomes that subsequently transfer to adjacent immune cells, thereby altering macrophage function (63). This interaction between gastric cancer cells and immune cells highlights the complex interplay between the immune system and tumorigenesis in the context of chronic infection.
Macrophages exposed to these exosomes tend to preferentially secrete the pro-inflammatory cytokine IL-1β and promote tumor cell proliferation, migration and invasion by activating the Akt and MAPK signaling pathways. Since IL-1β plays a critical role in driving the proliferation, migration and invasion of malignant tumors, it is important to further investigate the mechanisms underlying this process (94).
While the tumor-promoting role of TAMs in gastric cancer progression, particularly in relation to H. pylori infection, has garnered considerable attention, further exploration is needed to understand their dual functions within the tumor immune microenvironment. Most studies currently focus on how TAMs promote tumor growth through immunosuppressive mechanisms in the tumor microenvironment. However, TAMs may also exhibit tumor-suppressive potential under certain conditions. Notably, during H. pylori infection, TAMs can be reactivated, leading to the phagocytosis and cytotoxic eradication of malignant cells.
4 Therapeutic approaches for H. pylori infection and macrophage-directed treatment
Currently, common regimens for treating H. pylori infection include a combination of bismuth agents, proton pump inhibitors (PPIs) and antibiotics like metronidazole and clarithromycin (95, 96). Proton pump inhibitors (PPIs), considered the cornerstone drugs in conventional therapy, are widely used in H. pylori treatment for their ability to inhibit H. pylori growth, increase gastric pH, enhance antibiotic concentration in gastric tissues and improve antimicrobial efficacy (97). However, due to the extensive use of antibiotics over the years, the resistance rates of H. pylori to various antibiotics have significantly increased (98). In particular, the rising resistance to clarithromycin, metronidazole and levofloxacin has led to a gradual decline in the efficacy of traditional triple or quadruple therapies (99). The growing problem of antimicrobial resistance makes the development of new treatment strategies essential (100). Macrophages, crucial components of the innate immune system, display significant flexibility and are essential in fighting pathogen infections and aiding tissue repair. Notably, macrophages play a crucial role in the immune clearance of H. pylori, as well as in the development of chronic inflammation, gastric mucosal damage and the progression to gastric cancer caused by H. pylori infection (101, 102). Therefore, macrophages have become a novel therapeutic target for H. pylori treatment (see Table 1). Certain pharmacological agents and natural compounds have been identified to modulate macrophage function, thereby enhancing the therapeutic efficacy against H. pylori infection. Specifically, agents that augment the pathogen-clearing ability of macrophages, such as patchouli alcohol, can significantly reduce the H. pylori ‘s acid resistance, antibiotic resistance and gastric colonization capacity (103). Furthermore, as research progresses, new therapeutic strategies are gradually emerging. For instance, studies reveal that silymarin, naphthopyranone compounds, and specific probiotics (Lactobacillus gasseri Kx110A1, Lacticaseibacillus rhamnosus UCO-25A and Lactobacillus fermentum UCO-979C) effectively suppress macrophage release of pro-inflammatory cytokines such as TNF-α, IL-6, and IL-8. This inhibition consequently prevents inflammatory tissue damage and promotes gastric mucosal repair (104–108). Notably, the probiotic Lacticaseibacillus rhamnosus UCO-25A further modulates the balance between pro-inflammatory and anti-inflammatory factors and promote the production of IL-10, thereby enhancing immunomodulatory effects. Additionally, certain natural compounds like berberine, quercetin and Weizmannia coagulans BCF-01 can inhibit the polarization of macrophages toward the M1 phenotype, thus suppressing inflammation-induced damage (109–111). Moreover, recent studies have found that lactate, a metabolic byproduct, exerts immunomodulatory effects by inhibiting the release of inflammatory factors from macrophages induced by H. pylori. However, its potential as a therapeutic option remains to be further investigated (112).
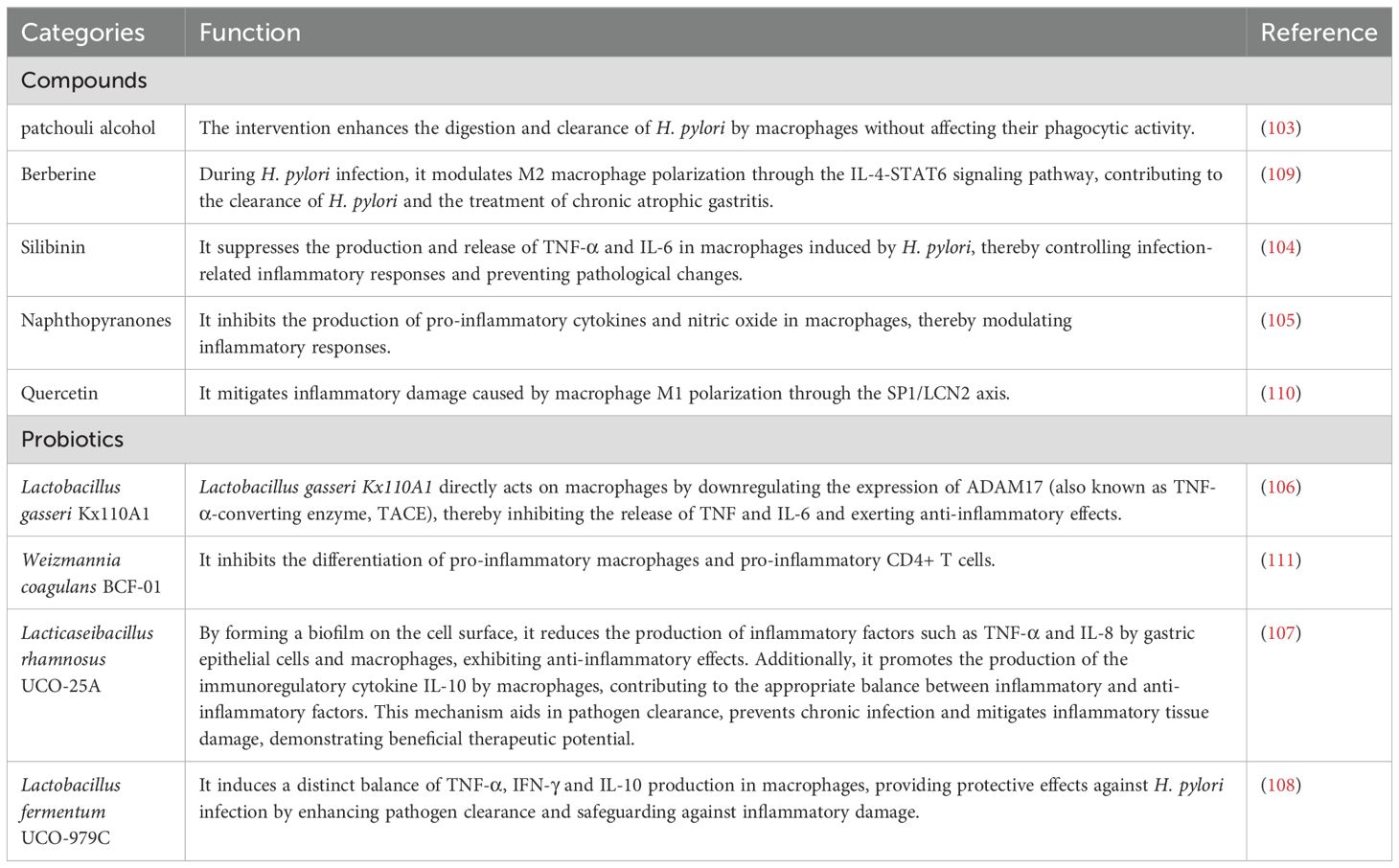
Table 1. Anti-H. pylori drugs targeting macrophage.
Although targeting macrophages for the treatment of H. pylori infection has made some progress, numerous challenges still exist. In drug research, current probiotics are derived from various origins, with limited studies focusing on strains isolated from human gastric mucosa. Utilizing strains from other sources carries significant risks. The long-term application of probiotic interventions may pose a risk of gut microbiota imbalance (113). Moreover, while probiotics act on macrophages to clear H. pylori, H. pylori also produces virulence factors to inhibit the function of probiotics. Furthermore, the specific substances within probiotics that contribute to their anti-H. pylori effects have yet to be fully understood. Additionally, most current compounds are limited to in vitro experiments or small-scale in vivo studies, highlighting a significant gap before practical applications can be achieved. In research on macrophages, their high plasticity offers opportunities for targeted treatments while also presenting significant challenges. The mechanisms underlying the differentiation of macrophages during H. pylori infection remain incompletely elucidated, potentially resulting in instability during therapeutic interventions.
5 Conclusion
In summary, the influence of H. pylori infection on the immunoregulatory functions of macrophages is highly complex. Macrophages, as key components of the immune system, play a crucial role in maintaining immune homeostasis and influencing disease susceptibility. H. pylori establishes persistent infection through sophisticated modulation of macrophage functions, including pattern recognition, phagocytic activity, inflammatory regulation, antigen presentation and tumorigenic processes.
Moreover, treating H. pylori infection continues to face multiple challenges, especially due to increasing antibiotic resistance. Hence, modulating macrophage functions, especially by inhibiting excessive inflammatory responses or promoting their reparative capabilities, may offer novel treatment directions. While significant insights have been obtained, many aspects of the interaction between H. pylori and macrophages remain unclear, highlighting the need for further research to develop new immunotherapy approaches against H. pylori.
Author contributions
MX: Funding acquisition, Writing – review & editing, Writing – original draft. PL: Writing – review & editing, Funding acquisition, Writing – original draft. XY: Writing – original draft, Writing – review & editing. LL: Writing – original draft, Writing – review & editing. LW: Writing – review & editing. NS: Writing – review & editing. KW: Writing – review & editing. YZ: Writing – review & editing, Conceptualization, Supervision. HW: Supervision, Writing – review & editing, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This work was supported by grants from the Natural Science Foundation of Shandong Province (ZR2022MH172, ZR2022QH165), Project of the 2024 Postgraduate Research and Innovation Fund of the Second Medical University of Shandong (2024YJSCX006), Weifang Science and Technology Development Project under Grant (2022ZJ1055, 2023YX042), Affiliated Hospital of Shandong Second Medical University Science and Technology Development Project (2023FYQ013, 2024FYM050).
Acknowledgments
We thank the members of our laboratory for helpful discussion.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Li Y, Choi H, Leung K, Jiang F, Graham DY, and Leung WK. Global prevalence of helicobacter pylori infection between 1980 and 2022: A systematic review and meta-analysis. Lancet Gastroenterol Hepatol. (2023) 8:553–64. doi: 10.1016/S2468-1253(23)00070-5
2. Chen YC, Malfertheiner P, Yu HT, Kuo CL, Chang YY, Meng FT, et al. Global prevalence of helicobacter pylori infection and incidence of gastric cancer between 1980 and 2022. Gastroenterology. (2024) 166:605–19. doi: 10.1053/j.gastro.2023.12.022
3. Marshall BJ and Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. (1984) 1:1311–5. doi: 10.1016/s0140-6736(84)91816-6
4. Fujimori S. Progress in elucidating the relationship between helicobacter pylori infection and intestinal diseases. World J Gastroenterol. (2021) 27:8040–6. doi: 10.3748/wjg.v27.i47.8040
5. Kusters JG, van Vliet AH, and Kuipers EJ. Pathogenesis of helicobacter pylori infection. Clin Microbiol Rev. (2006) 19:449–90. doi: 10.1128/CMR.00054-05
6. Sharndama HC and Mba IE. Helicobacter pylori: an up-to-date overview on the virulence and pathogenesis mechanisms. Braz J Microbiol. (2022) 53:33–50. doi: 10.1007/s42770-021-00675-0
7. Cover TL, Lacy DB, and Ohi MD. The helicobacter pylori cag type iv secretion system. Trends Microbiol. (2020) 28:682–95. doi: 10.1016/j.tim.2020.02.004
8. Tran SC, Bryant KN, and Cover TL. The helicobacter pylori cag pathogenicity island as a determinant of gastric cancer risk. Gut Microbes. (2024) 16:2314201. doi: 10.1080/19490976.2024.2314201
9. Mohammadzadeh R, Menbari S, Pishdadian A, and Farsiani H. Helicobacter pylori virulence factors: subversion of host immune system and development of various clinical outcomes. Expert Rev Mol Med. (2023) 25:e23. doi: 10.1017/erm.2023.17
10. Sidebotham RL, Worku ML, Karim QN, Dhir NK, and Baron JH. How helicobacter pylori urease may affect external ph and influence growth and motility in the mucus environment: evidence from in-vitro studies. Eur J Gastroenterol Hepatol. (2003) 15:395–401. doi: 10.1097/00042737-200304000-00010
11. Baj J, Forma A, Sitarz M, Portincasa P, Garruti G, Krasowska D, et al. Helicobacter pylori virulence factors-mechanisms of bacterial pathogenicity in the gastric microenvironment. Cells. (2020) 10:27. doi: 10.3390/cells10010027
12. Maldonado RF, Sa-Correia I, and Valvano MA. Lipopolysaccharide modification in gram-negative bacteria during chronic infection. FEMS Microbiol Rev. (2016) 40:480–93. doi: 10.1093/femsre/fuw007
13. Silva LM, Correia VG, Moreira ASP, Domingues MRM, Ferreira RM, Figueiredo C, et al. Helicobacter pylori lipopolysaccharide structural domains and their recognition by immune proteins revealed with carbohydrate microarrays. Carbohydr Polym. (2021) 253:117350. doi: 10.1016/j.carbpol.2020.117350
14. Gu H. Role of flagella in the pathogenesis of helicobacter pylori. Curr Microbiol. (2017) 74:863–9. doi: 10.1007/s00284-017-1256-4
15. Clyne M and T OC. Pathogenicity and virulence of helicobacter pylori: A paradigm of chronic infection. Virulence. (2025) 16:2438735. doi: 10.1080/21505594.2024.2438735
16. Lima de Souza Goncalves V, Cordeiro Santos ML, Silva Luz M, Santos Marques H, de Brito BB, Franca da Silva FA, et al. From helicobacter pylori infection to gastric cancer: current evidence on the immune response. World J Clin Oncol. (2022) 13:186–99. doi: 10.5306/wjco.v13.i3.186
17. Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z, et al. Deciphering human macrophage development at single-cell resolution. Nature. (2020) 582:571–6. doi: 10.1038/s41586-020-2316-7
18. Cheok YY, Tan GMY, Lee CYQ, Abdullah S, Looi CY, and Wong WF. Innate immunity crosstalk with helicobacter pylori: pattern recognition receptors and cellular responses. Int J Mol Sci. (2022) 23:7561. doi: 10.3390/ijms23147561
19. Luo M, Zhao F, Cheng H, Su M, and Wang Y. Macrophage polarization: an important role in inflammatory diseases. Front Immunol. (2024) 15:1352946. doi: 10.3389/fimmu.2024.1352946
20. Muntjewerff EM, Meesters LD, and van den Bogaart G. Antigen cross-presentation by macrophages. Front Immunol. (2020) 11:1276. doi: 10.3389/fimmu.2020.01276
21. Santecchia I, Ferrer MF, Vieira ML, Gomez RM, and Werts C. Phagocyte escape of leptospira: the role of tlrs and nlrs. Front Immunol. (2020) 11:571816. doi: 10.3389/fimmu.2020.571816
22. Rosenberger CM and Finlay BB. Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol. (2003) 4:385–96. doi: 10.1038/nrm1104
23. Pham TH and Monack DM. Turning foes into permissive hosts: manipulation of macrophage polarization by intracellular bacteria. Curr Opin Immunol. (2023) 84:102367. doi: 10.1016/j.coi.2023.102367
24. DeLeo FR. Modulation of phagocyte apoptosis by bacterial pathogens. Apoptosis. (2004) 9:399–413. doi: 10.1023/B:APPT.0000031448.64969.fa
25. Vitaliti A, Reggio A, and Palma A. Macrophages and autophagy: partners in crime. FEBS J. (2025) 292:2957–72. doi: 10.1111/febs.17305
26. Uribe-Querol E and Rosales C. Phagocytosis: our current understanding of a universal biological process. Front Immunol. (2020) 11:1066. doi: 10.3389/fimmu.2020.01066
27. Lee HJ, Woo Y, Hahn TW, Jung YM, and Jung YJ. Formation and maturation of the phagosome: A key mechanism in innate immunity against intracellular bacterial infection. Microorganisms. (2020) 8:1298. doi: 10.3390/microorganisms8091298
28. Roche PA and Furuta K. The ins and outs of mhc class ii-mediated antigen processing and presentation. Nat Rev Immunol. (2015) 15:203–16. doi: 10.1038/nri3818
29. Malfertheiner P, Camargo MC, El-Omar E, Liou JM, Peek R, Schulz C, et al. Helicobacter pylori infection. Nat Rev Dis Primers. (2023) 9:19. doi: 10.1038/s41572-023-00431-8
30. Nemati M, Larussa T, Khorramdelazad H, Mahmoodi M, and Jafarzadeh A. Toll-like receptor 2: an important immunomodulatory molecule during helicobacter pylori infection. Life Sci. (2017) 178:17–29. doi: 10.1016/j.lfs.2017.04.006
31. Dooyema SDR, Noto JM, Wroblewski LE, Piazuelo MB, Krishna U, Suarez G, et al. Helicobacter pylori actively suppresses innate immune nucleic acid receptors. Gut Microbes. (2022) 14:2105102. doi: 10.1080/19490976.2022.2105102
32. Rad R, Ballhorn W, Voland P, Eisenacher K, Mages J, Rad L, et al. Extracellular and intracellular pattern recognition receptors cooperate in the recognition of helicobacter pylori. Gastroenterology. (2009) 136:2247–57. doi: 10.1053/j.gastro.2009.02.066
33. Lee CYQ, Chan YT, Cheok YY, Tan GMY, Tang TF, Cheong HC, et al. Helicobacter pylori infection elicits type I interferon response in human monocytes via toll-like receptor 8 signaling. J Immunol Res. (2022) 2022:3861518. doi: 10.1155/2022/3861518
34. Kim DJ, Park JH, Franchi L, Backert S, and Nunez G. The cag pathogenicity island and interaction between tlr2/nod2 and nlrp3 regulate il-1beta production in helicobacter pylori infected dendritic cells. Eur J Immunol. (2013) 43:2650–8. doi: 10.1002/eji.201243281
35. Nagata M, Toyonaga K, Ishikawa E, Haji S, Okahashi N, Takahashi M, et al. Helicobacter pylori metabolites exacerbate gastritis through C-type lectin receptors. J Exp Med. (2021) 218:e20200815. doi: 10.1084/jem.20200815
36. Li H, Liao T, Debowski AW, Tang H, Nilsson HO, Stubbs KA, et al. Lipopolysaccharide structure and biosynthesis in helicobacter pylori. Helicobacter. (2016) 21:445–61. doi: 10.1111/hel.12301
37. Pachathundikandi SK, Tegtmeyer N, and Backert S. Masking of typical tlr4 and tlr5 ligands modulates inflammation and resolution by helicobacter pylori. Trends Microbiol. (2023) 31:903–15. doi: 10.1016/j.tim.2023.03.009
38. Du SY, Wang HJ, Cheng HH, Chen SD, Wang LH, and Wang WC. Cholesterol glucosylation by helicobacter pylori delays internalization and arrests phagosome maturation in macrophages. J Microbiol Immunol Infect. (2016) 49:636–45. doi: 10.1016/j.jmii.2014.05.011
39. Allen LA, Schlesinger LS, and Kang B. Virulent strains of helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. (2000) 191:115–28. doi: 10.1084/jem.191.1.115
40. Allen LA, Allgood JA, Han X, and Wittine LM. Phosphoinositide3-kinase regulates actin polymerization during delayed phagocytosis of helicobacter pylori. J Leukoc Biol. (2005) 78:220–30. doi: 10.1189/jlb.0205091
41. Allen LA. Phagocytosis and persistence of helicobacter pylori. Cell Microbiol. (2007) 9:817–28. doi: 10.1111/j.1462-5822.2007.00906.x
42. Borlace GN, Jones HF, Keep SJ, Butler RN, and Brooks DA. Helicobacter pylori phagosome maturation in primary human macrophages. Gut Pathog. (2011) 3:3. doi: 10.1186/1757-4749-3-3
43. Rittig MG, Shaw B, Letley DP, Thomas RJ, Argent RH, and Atherton JC. Helicobacter pylori-induced homotypic phagosome fusion in human monocytes is independent of the bacterial vaca and cag status. Cell Microbiol. (2003) 5:887–99. doi: 10.1046/j.1462-5822.2003.00328.x
44. Schwartz JT and Allen LA. Role of urease in megasome formation and helicobacter pylori survival in macrophages. J Leukoc Biol. (2006) 79:1214–25. doi: 10.1189/jlb.0106030
45. Qaria MA, Qumar S, Sepe LP, and Ahmed N. Cholesterol glucosylation-based survival strategy in helicobacter pylori. Helicobacter. (2021) 26:e12777. doi: 10.1111/hel.12777
46. Zheng PY and Jones NL. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining taco (Coronin 1) protein. Cell Microbiol. (2003) 5:25–40. doi: 10.1046/j.1462-5822.2003.00250.x
47. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
48. Yunna C, Mengru H, Lei W, and Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
49. Zitta K, Hummitzsch L, Lichte F, Fandrich F, Steinfath M, Eimer C, et al. Effects of temporal ifngamma exposure on macrophage phenotype and secretory profile: exploring gmp-compliant production of a novel subtype of regulatory macrophages (Mreg(Ifngamma0)) for potential cell therapeutic applications. J Transl Med. (2024) 22:534. doi: 10.1186/s12967-024-05336-y
50. Khan A, Zhang K, Singh VK, Mishra A, Kachroo P, Bing T, et al. Human M1 Macrophages Express Unique Innate Immune Response Genes after Mycobacterial Infection to Defend against Tuberculosis. Commun Biol. (2022) 5:480. doi: 10.1038/s42003-022-03387-9
51. Gong Z, Han S, Liang T, Zhang H, Sun Q, Pan H, et al. Mycobacterium tuberculosis effector ppe36 attenuates host cytokine storm damage via inhibiting macrophage M1 polarization. J Cell Physiol. (2021) 236:7405–20. doi: 10.1002/jcp.30411
52. Jiang L, Wang P, Song X, Zhang H, Ma S, Wang J, et al. Salmonella typhimurium reprograms macrophage metabolism via T3ss effector sope2 to promote intracellular replication and virulence. Nat Commun. (2021) 12:879. doi: 10.1038/s41467-021-21186-4
53. Zhichao L, Xin S, Chunhui Y, Jun W, Yun X, and Qinzhen C. Direct interaction between urease B of helicobacter pylori and tlr2 negatively regulates immune fucntions of macrophages. Chin J Microbiol Immunol. (2021) 41:507–15. doi: 10.3760/cma.j.cn112309-20200730-00380
54. Krakowiak MS, Noto JM, Piazuelo MB, Hardbower DM, Romero-Gallo J, Delgado A, et al. Matrix metalloproteinase 7 restrains helicobacter pylori-induced gastric inflammation and premalignant lesions in the stomach by altering macrophage polarization. Oncogene. (2015) 34:1865–71. doi: 10.1038/onc.2014.135
55. Kim SH, Sierra RA, McGee DJ, and Zabaleta J. Transcriptional profiling of gastric epithelial cells infected with wild type or arginase-deficient helicobacter pylori. BMC Microbiol. (2012) 12:175. doi: 10.1186/1471-2180-12-175
56. Hardbower DM, Asim M, Murray-Stewart T, Casero RA Jr., Verriere T, Lewis ND, et al. Arginase 2 deletion leads to enhanced M1 macrophage activation and upregulated polyamine metabolism in response to helicobacter pylori infection. Amino Acids. (2016) 48:2375–88. doi: 10.1007/s00726-016-2231-2
57. Lewis ND, Asim M, Barry DP, de Sablet T, Singh K, Piazuelo MB, et al. Immune evasion by helicobacter pylori is mediated by induction of macrophage arginase ii. J Immunol. (2011) 186:3632–41. doi: 10.4049/jimmunol.1003431
58. Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, and Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
59. Zhuang Y, Shi Y, Liu XF, Zhang JY, Liu T, Fan X, et al. Helicobacter pylori-infected macrophages induce th17 cell differentiation. Immunobiology. (2011) 216:200–7. doi: 10.1016/j.imbio.2010.05.005
60. Gobert AP, Verriere T, Asim M, Barry DP, Piazuelo MB, de Sablet T, et al. Heme oxygenase-1 dysregulates macrophage polarization and the immune response to helicobacter pylori. J Immunol. (2014) 193:3013–22. doi: 10.4049/jimmunol.1401075
61. Devi S, Rajakumara E, and Ahmed N. Induction of mincle by helicobacter pylori and consequent anti-inflammatory signaling denote a bacterial survival strategy. Sci Rep. (2015) 5:15049. doi: 10.1038/srep15049
62. Peng R, Xu C, Zhang L, Liu X, Peng D, Chen X, et al. M2 macrophages participate in ilc2 activation induced by helicobacter pylori infection. Gut Microbes. (2024) 16:2347025. doi: 10.1080/19490976.2024.2347025
63. Wenjing L, Kaiyun G, Junzi L, Yunxing H, Jie D, Na W, et al. Gastric cancer cell-derived exosome mir-382-5p induced by helicobacter pylori inhibits macrophage autophagy and promotes M2 polarization by targeting pten. Chin J Immunol. (2024) 40:1153–9. doi: 10.3969/j.issn.1000-484X.2024.06.007
64. Hato L, Vizcay A, Eguren I, Perez-Gracia JL, Rodriguez J, Gallego Perez-Larraya J, et al. Dendritic cells in cancer immunology and immunotherapy. Cancers (Basel). (2024) 16:981. doi: 10.3390/cancers16050981
65. Stopforth RJ and Ward ES. The role of antigen presentation in tumor-associated macrophages. Crit Rev Immunol. (2020) 40:205–24. doi: 10.1615/CritRevImmunol.2020034910
66. Pagliari M, Munari F, Toffoletto M, Lonardi S, Chemello F, Codolo G, et al. Helicobacter pylori affects the antigen presentation activity of macrophages modulating the expression of the immune receptor cd300e through mir-4270. Front Immunol. (2017) 8:1288. doi: 10.3389/fimmu.2017.01288
67. Codolo G, Toffoletto M, Chemello F, Coletta S, Soler Teixidor G, Battaggia G, et al. Helicobacter pylori dampens hla-ii expression on macrophages via the up-regulation of mirnas targeting ciita. Front Immunol. (2019) 10:2923. doi: 10.3389/fimmu.2019.02923
68. Coletta S, Battaggia G, Della Bella C, Furlani M, Hauke M, Faass L, et al. Adp-heptose enables helicobacter pylori to exploit macrophages as a survival niche by suppressing antigen-presenting hla-ii expression. FEBS Lett. (2021) 595:2160–8. doi: 10.1002/1873-3468.14156
69. Wang C, Wang X, Zhang D, Sun X, Wu Y, Wang J, et al. The macrophage polarization by mirnas and its potential role in the treatment of tumor and inflammation (Review). Oncol Rep. (2023) 50:190. doi: 10.3892/or.2023.8627
70. Gobert AP, Cheng Y, Wang JY, Boucher JL, Iyer RK, Cederbaum SD, et al. Helicobacter pylori induces macrophage apoptosis by activation of arginase ii. J Immunol. (2002) 168:4692–700. doi: 10.4049/jimmunol.168.9.4692
71. Cheng Y, Chaturvedi R, Asim M, Bussiere FI, Scholz A, Xu H, et al. Helicobacter pylori-induced macrophage apoptosis requires activation of ornithine decarboxylase by C-myc. J Biol Chem. (2005) 280:22492–6. doi: 10.1074/jbc.C500122200
72. Menaker RJ, Ceponis PJ, and Jones NL. Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Infect Immun. (2004) 72:2889–98. doi: 10.1128/IAI.72.5.2889-2898.2004
73. Chaturvedi R, Cheng Y, Asim M, Bussiere FI, Xu H, Gobert AP, et al. Induction of polyamine oxidase 1 by helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J Biol Chem. (2004) 279:40161–73. doi: 10.1074/jbc.M401370200
74. Asim M, Chaturvedi R, Hoge S, Lewis ND, Singh K, Barry DP, et al. Helicobacter pylori induces erk-dependent formation of a phospho-C-fos C-jun activator protein-1 complex that causes apoptosis in macrophages. J Biol Chem. (2010) 285:20343–57. doi: 10.1074/jbc.M110.116988
75. Tenguria S, Ansari SA, Khan N, Ranjan A, Devi S, Tegtmeyer N, et al. Helicobacter pylori cell translocating kinase (Ctka/jhp0940) is pro-apoptotic in mouse macrophages and acts as auto-phosphorylating tyrosine kinase. Int J Med Microbiol. (2014) 304:1066–76. doi: 10.1016/j.ijmm.2014.07.017
76. Alvi A, Ansari SA, Ehtesham NZ, Rizwan M, Devi S, Sechi LA, et al. Concurrent proinflammatory and apoptotic activity of a helicobacter pylori protein (Hp986) points to its role in chronic persistence. PloS One. (2011) 6:e22530. doi: 10.1371/journal.pone.0022530
77. Pathak SK, Tavares R, de Klerk N, Spetz AL, and Jonsson AB. Helicobacter pylori protein jhp0290 binds to multiple cell types and induces macrophage apoptosis via tumor necrosis factor (Tnf)-dependent and independent pathways. PloS One. (2013) 8:e77872. doi: 10.1371/journal.pone.0077872
78. Tavares R and Pathak SK. Helicobacter pylori secreted protein hp1286 triggers apoptosis in macrophages via tnf-independent and erk mapk-dependent pathways. Front Cell Infect Microbiol. (2017) 7:58. doi: 10.3389/fcimb.2017.00058
79. Sit WY, Cheng ML, Chen TJ, Chen CJ, Chen BN, Huang DJ, et al. Helicobacter pylori plda modulates tnfr1-mediated P38 signaling pathways to regulate macrophage responses for its survival. Gut Microbes. (2024) 16:2409924. doi: 10.1080/19490976.2024.2409924
80. Li C and Zhang Y. Helicobacter pylori vaca up-regulates secretion of macrophages by activating nuclear factor kb. Chin J Microbiol Immunol. (2009) 29:454–9. doi: 10.3321/j.issn:0001-6209.2008.03.013
81. Yao Y, Li G, Wu J, Zhang X, and Wang J. Inflammatory response of macrophages cultured with helicobacter pylori strains was regulated by mir-155. Int J Clin Exp Pathol. (2015) 8:4545–54.
82. Koch M, Mollenkopf HJ, Klemm U, and Meyer TF. Induction of microrna-155 is tlr- and type iv secretion system-dependent in macrophages and inhibits DNA-damage induced apoptosis. Proc Natl Acad Sci U.S.A. (2012) 109:E1153–62. doi: 10.1073/pnas.1116125109
83. Bejarano L, Jordao MJC, and Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Discov. (2021) 11:933–59. doi: 10.1158/2159-8290.CD-20-1808
84. Navashenaq JG, Shabgah AG, Banach M, Jamialahmadi T, Penson PE, Johnston TP, et al. The interaction of helicobacter pylori with cancer immunomodulatory stromal cells: new insight into gastric cancer pathogenesis. Semin Cancer Biol. (2022) 86:951–9. doi: 10.1016/j.semcancer.2021.09.014
85. Korkaya H and Orsulic S. Editorial: the tumor microenvironment: recent advances and novel therapeutic approaches. Front Cell Dev Biol. (2020) 8:586176. doi: 10.3389/fcell.2020.586176
86. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol. (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
87. Wang Z, Yang Y, Cui Y, Wang C, Lai Z, Li Y, et al. Tumor-associated macrophages regulate gastric cancer cell invasion and metastasis through tgfbeta2/nf-kappab/kindlin-2 axis. Chin J Cancer Res. (2020) 32:72–88. doi: 10.21147/j.issn.1000-9604.2020.01.09
88. Li J, Sun J, Zeng Z, Liu Z, Ma M, Zheng Z, et al. Tumour-associated macrophages in gastric cancer: from function and mechanism to application. Clin Transl Med. (2023) 13:e1386. doi: 10.1002/ctm2.1386
89. Moeini P and Niedzwiedzka-Rystwej P. Tumor-associated macrophages: combination of therapies, the approach to improve cancer treatment. Int J Mol Sci. (2021) 22:7239. doi: 10.3390/ijms22137239
90. Thrift AP and El-Serag HB. Burden of gastric cancer. Clin Gastroenterol Hepatol. (2020) 18:534–42. doi: 10.1016/j.cgh.2019.07.045
91. Gambardella V, Castillo J, Tarazona N, Gimeno-Valiente F, Martinez-Ciarpaglini C, Cabeza-Segura M, et al. The role of tumor-associated macrophages in gastric cancer development and their potential as a therapeutic target. Cancer Treat Rev. (2020) 86:102015. doi: 10.1016/j.ctrv.2020.102015
92. Yu B, de Vos D, Guo X, Peng S, Xie W, Peppelenbosch MP, et al. Il-6 facilitates cross-talk between epithelial cells and tumor- associated macrophages in helicobacter pylori-linked gastric carcinogenesis. Neoplasia. (2024) 50:100981. doi: 10.1016/j.neo.2024.100981
93. He Z and Zhang S. Tumor-associated macrophages and their functional transformation in the hypoxic tumor microenvironment. Front Immunol. (2021) 12:741305. doi: 10.3389/fimmu.2021.741305
94. Che Y, Geng B, Xu Y, Miao X, Chen L, Mu X, et al. Helicobacter pylori-induced exosomal met educates tumour-associated macrophages to promote gastric cancer progression. J Cell Mol Med. (2018) 22:5708–19. doi: 10.1111/jcmm.13847
95. Malfertheiner P, Megraud F, Rokkas T, Gisbert JP, Liou JM, Schulz C, et al. Management of helicobacter pylori infection: the maastricht vi/florence consensus report. Gut. (2022) 71:1724–62. doi: 10.1136/gutjnl-2022-327745
96. Chey WD, Howden CW, Moss SF, Morgan DR, Greer KB, Grover S, et al. Acg clinical guideline: treatment of helicobacter pylori infection. Am J Gastroenterol. (2024) 119:1730–53. doi: 10.14309/ajg.0000000000002968
97. Ierardi E, Losurdo G, Fortezza RF, Principi M, Barone M, and Leo AD. Optimizing proton pump inhibitors in helicobacter pylori treatment: old and new tricks to improve effectiveness. World J Gastroenterol. (2019) 25:5097–104. doi: 10.3748/wjg.v25.i34.5097
98. Mladenova I. Epidemiology of helicobacter pylori resistance to antibiotics (a narrative review). Antibiot (Basel). (2023) 12:1184. doi: 10.3390/antibiotics12071184
99. Savoldi A, Carrara E, Graham DY, Conti M, and Tacconelli E. Prevalence of antibiotic resistance in helicobacter pylori: A systematic review and meta-analysis in world health organization regions. Gastroenterology. (2018) 155:1372–82 e17. doi: 10.1053/j.gastro.2018.07.007
100. Liu M, Gao H, Miao J, Zhang Z, Zheng L, Li F, et al. Helicobacter pylori infection in humans and phytotherapy, probiotics, and emerging therapeutic interventions: A review. Front Microbiol. (2023) 14:1330029. doi: 10.3389/fmicb.2023.1330029
101. Fei X, Li N, Xu X, and Zhu Y. Macrophage biology in the pathogenesis of helicobacter pylori infection. Crit Rev Microbiol. (2024) 51:399–416. doi: 10.1080/1040841X.2024.2366944
102. He J, Hu W, Ouyang Q, Zhang S, He L, Chen W, et al. Helicobacter pylori infection induces stem cell-like properties in correa cascade of gastric cancer. Cancer Lett. (2022) 542:215764. doi: 10.1016/j.canlet.2022.215764
103. Lian DW, Xu YF, Deng QH, Lin XM, Huang B, Xian SX, et al. Effect of patchouli alcohol on macrophage mediated helicobacter pylori digestion based on intracellular urease inhibition. Phytomedicine. (2019) 65:153097. doi: 10.1016/j.phymed.2019.153097
104. Bittencourt MLF, Rodrigues RP, Kitagawa RR, and Goncalves RCR. The gastroprotective potential of silibinin against helicobacter pylori infection and gastric tumor cells. Life Sci. (2020) 256:117977. doi: 10.1016/j.lfs.2020.117977
105. Ardisson JS, Goncalves RCR, Rodrigues RP, and Kitagawa RR. Antitumour, immunomodulatory activity and in silico studies of naphthopyranones targeting inos, a relevant target for the treatment of helicobacter pylori infection. BioMed Pharmacother. (2018) 107:1160–5. doi: 10.1016/j.biopha.2018.08.098
106. Gebremariam HG, Qazi KR, Somiah T, Pathak SK, Sjolinder H, Sverremark Ekstrom E, et al. Lactobacillus gasseri suppresses the production of proinflammatory cytokines in helicobacter pylori-infected macrophages by inhibiting the expression of adam17. Front Immunol. (2019) 10:2326. doi: 10.3389/fimmu.2019.02326
107. Garcia-Castillo V, Marin-Vega AM, Ilabaca A, Albarracin L, Marcial G, Kitazawa H, et al. Characterization of the immunomodulatory and anti-helicobacter pylori properties of the human gastric isolate lactobacillus rhamnosus uco-25a. Biofouling. (2019) 35:922–37. doi: 10.1080/08927014.2019.1675153
108. Garcia-Castillo V, Zelaya H, Ilabaca A, Espinoza-Monje M, Komatsu R, Albarracin L, et al. Lactobacillus fermentum uco-979c beneficially modulates the innate immune response triggered by helicobacter pylori infection in vitro. Benef Microbes. (2018) 9:829–41. doi: 10.3920/BM2018.0019
109. Yang T, Wang R, Liu H, Wang L, Li J, Wu S, et al. Berberine regulates macrophage polarization through il-4-stat6 signaling pathway in helicobacter pylori-induced chronic atrophic gastritis. Life Sci. (2021) 266:118903. doi: 10.1016/j.lfs.2020.118903
110. Wang Z, Zhou X, Hu X, and Zheng C. Quercetin ameliorates helicobacter pylori-induced gastric epithelial cell injury by regulating specificity protein 1/lipocalin 2 axis in gastritis. J Appl Toxicol. (2024) 44:641–50. doi: 10.1002/jat.4566
111. Chen Z, Tang Z, Li W, Deng X, Yu L, Yang J, et al. Weizmannia coagulans bcf-01: A novel gastrogenic probiotic for helicobacter pylori infection control. Gut Microbes. (2024) 16:2313770. doi: 10.1080/19490976.2024.2313770
112. Somiah T, Gebremariam HG, Zuo F, Smirnova K, and Jonsson AB. Lactate causes downregulation of helicobacter pylori adhesin genes saba and laba while dampening the production of proinflammatory cytokines. Sci Rep. (2022) 12:20064. doi: 10.1038/s41598-022-24311-5
Keywords: Helicobacter pylori, macrophages, phagocytosis, inflammation, antigen presentation, immunomodulation
Citation: Xiang M, Li P, Yue X, Liu L, Wang L, Sun N, Wang K, Zhang Y and Wang H (2025) Dysregulated macrophage immunity in Helicobacter pylori infection: unveiling mechanistic insights and therapeutic implications. Front. Immunol. 16:1636768. doi: 10.3389/fimmu.2025.1636768
Received: 28 May 2025; Accepted: 14 July 2025;
Published: 04 August 2025.
Edited by:
Yoshio Yamaoka, Oita University, JapanReviewed by:
Paweł Krzyżek, Wroclaw Medical University, PolandAlessandro Palma, Sapienza University of Rome, Italy
Copyright © 2025 Xiang, Li, Yue, Liu, Wang, Sun, Wang, Zhang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuying Zhang, d2Z6emJ6eUAxMjYuY29t; Hongyan Wang, d2FuZ2h5QHNkc211LmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship