 Wenzhao Wang1†Shengwen Li2†Yanjun Liu3†Xin Ding1Yongqi Yang1Shiyun Chen2
Wenzhao Wang1†Shengwen Li2†Yanjun Liu3†Xin Ding1Yongqi Yang1Shiyun Chen2 Jiafan Cao2
Jiafan Cao2 Frank Tacke4
Frank Tacke4 Wei Dong5*
Wei Dong5* Tian Lan1,5*
Tian Lan1,5*- 1Department of Pharmacology, College of Pharmacy, Harbin Medical University, State Key Laboratory of Frigid Zone Cardiovascular Diseases, Harbin, China
- 2School of Pharmacy, Guangdong Pharmaceutical University, Guangzhou, China
- 3Department of Pharmacy, The People’s Hospital of Miyi County, Panzhihua, China
- 4Department of Hepatology and Gastroenterology, Charité Universitätsmedizin Berlin, Campus Virchow Klinikum and Campus Charité Mitte, Berlin, Germany
- 5Department of Hepatopancreatobiliary Surgery, Harbin Medical University Cancer Hospital, Harbin, China
Liver fibrosis represents a universal pathological endpoint in chronic hepatic disorders, in which hepatic macrophages play a pivotal role through dynamic phenotypic modulation. These versatile immune cells undergo functional and phenotypic transformations mediated by diverse molecular mediators, with their heterogeneity arising from both cellular origin differences and disease-specific microenvironments. The development of technologies such as single-cell and spatial omics has broken through the traditional M1/M2 classification paradigm of macrophages, revealing the molecular signatures and functional distinctions of hepatic macrophages during liver injury, fibrogenesis, and regression. Hepatic macrophages are central to the pathogenesis of chronic liver injury and considered as potential targets for drug discovery. While numerous macrophage-targeting strategies for liver fibrosis intervention currently remain in preclinical development, advancing our comprehension of macrophage plasticity and subset-specific functions holds significant potential. A deeper understanding of macrophage heterogeneity could provide a new therapeutic strategy against liver fibrosis, ultimately improving clinical outcomes for patients with chronic liver diseases.
1 Introduction
Liver fibrosis emerges as a common pathological consequence of chronic liver diseases. It is characterized by an excessive accumulation of extracellular matrix (ECM) proteins, largely derived from activated hepatic stellate cells (HSCs), culminating in the formation of fibrotic scar tissue (1). During this process, hepatic macrophages serve a critical function (2). Hepatic macrophages, including both Kupffer cells (KCs) and recruited macrophages, constitute a heterogeneous population of immune cells characterized by remarkable functional and molecular diversity (3). Their strategic positioning at the interface of hepatic blood flow and the sub-sinusoidal space of Disse, coupled with their heightened sensitivity to microenvironmental factors and high phagocytic capabilities, enable KCs to perform a variety of roles. These include immune responses, protection against infections, and the modulation of metabolic processes (4). This heterogeneity manifests through distinct cytokine profiles, surface marker expression patterns, and transcriptomic signatures, which collectively define their phenotypic identity. Macrophages are also extremely plastic, as demonstrated by their ability to alter their phenotype to adapt to the liver microenvironment and perform different functions (5). Injury-induced inflammation prompts the recruitment of macrophages to the liver, where they secrete pro-inflammatory cytokines that activate HSCs, thereby initiating liver fibrosis (6). In contrast, their phenotypic transition leads to the breakdown of extracellular matrix components and the secretion of cytokines with anti-inflammatory properties (7).
Hepatic macrophages play an important role in maintaining the dynamic balance of the liver and the pathogenesis of both acute and chronic liver injury. They are involved in various processes related to liver disease, such as exacerbating injury, reducing inflammation, promoting tissue repair, and influencing fibrosis progression and regression, as well as tumor promotion and suppression (8). These discoveries are catalyzing the development of macrophage-centric therapeutic strategies, with emerging evidence underscoring their potential for improving clinical management of chronic liver diseases. Therefore, we summarize therapeutic approaches that target hepatic macrophages for liver fibrosis. With the current improved understanding of the complex heterogeneity and functional diversity of macrophages, therapies targeting macrophages may represent a promising avenue for the treatment of liver fibrosis.
2 Origin of hepatic macrophages
2.1 Kupffer cells
KCs originate from yolk sac-derived colony-stimulating factor 1 receptor (CSF1R)+ erythromyeloid progenitors (EMPs) (9). Hepatic Transforming Growth Factor-beta (TGF-β) and desmosterol synergistically regulate SMAD and Liver X receptor (LXR) signaling pathways to maintain KCs identity (10). In healthy livers, KCs are mainly confined to the hepatic sinusoids and do not migrate, whereas monocyte-derived macrophages can be found extravascularly (11). KCs-specific markers in mice include C-type lectin domain family 4 member F (CLEC4F), V-set and immunoglobulin domain containing 4 (VSIG4), C-type lectin domain family 2 (CLEC2), and Folate receptor 2 (FOLR2), whereas in humans no consensus has been reached (12). KCs express a wide range of pattern recognition receptors (PRRs), including toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors (NLRs), and retinoic acid-inducible gene I-like receptors (RLRs) (13). KCs help maintain liver homeostasis and play important modulatory roles in bacterial clearance, antigen presentation, and modulation of iron/lipid metabolism (14).
2.2 Monocyte-derived macrophages
In the healthy liver, monocyte-derived macrophages (MoMϕs) predominantly localize to the portal triad region, where they maintain iron homeostasis and regulate cholesterol metabolism (15). MoMϕs are Cluster of Differentiation 11b (CD11b)+, F4/80intermediate (int), Lymphocyte antigen 6 complex locus C (Ly6C)+ and CSF1R+, which are derived from bone marrow (BM) C-X3-C motif chemokine receptor 1 CX3CR1+ CD117+Lin- progenitor cells (16, 17). These MoMϕs are primarily recruited to the liver by chemokines, such as C-C motif chemokine ligand 2 (CCL2), CCL1, and their receptors C-C chemokine receptor type 2 (CCR2) and CCR8 (18). The murine system features two principal circulating monocyte subsets characterized by Ly-6C expression levels: pro-inflammatory Ly-6C high (Ly-6Chi) monocytes and patrolling Ly-6C low (Ly-6Clow) monocytes (19). In humans, monocytes are classified by their expression of CD14 and CD16 as classical (CD14hiCD16−), intermediate (CD14+CD16+) and nonclassical (CD14−CD16hi) monocytes, which to some extent correspond to Ly-6Chi and Ly-6Clow monocytes in mice respectively (20). Ly-6Chi monocytes are characterized by their expression of inflammatory chemokine receptors, pattern recognition receptors, and cytokines, whereas Ly-6Clow monocytes demonstrate a patrolling function within the liver and exhibit a higher expression of scavenging receptors (20). Notably, phenotypic plasticity exists between these subsets. Ly-6Chi MoMϕs can transition to a restorative Ly-6Clow phenotype through distinct mechanisms: phagocytic activity or exposure to interleukin-4 (IL-4) and IL-33 released by necrotic KCs (18). This phenotypic switching represents a critical adaptive mechanism in the process of hepatic fibrosis. Multiple lineage-tracing models have shown that MoMϕs are also the major population of immunosuppressive and liver metastasis-associated macrophages (LMAM) (21). Furthermore, MoMϕs can replace KCs when they are experimentally depleted due to liver injury, and these macrophages can subsequently acquire a phenotype that is almost identical to that of KCs (22, 23).
2.3 Peritoneal and splenic macrophages
Peritoneal macrophages (PMs), which are located in the peritoneal cavity, may migrate into the liver. PMs selectively express the transcription factor GATA6, which is not expressed by either liver-resident KCs or circulating monocytes (24). In the context of acute liver injury, silencing the pro-inflammatory protein High mobility group protein B1 (HMGB1) in liver-infiltrating PMs alleviates the liver injury phenotype in mice (25). However, it has been suggested that PMs do not deeply infiltrate the liver parenchyma during liver injury, which seems to contradict the conclusions of relevant studies (26, 27).
Splenic macrophages (SMs) exhibit regulatory roles in liver homeostasis and pathology. SMs express CD11b and CD115 but show low or no expression of CD90, B220, CD49b, NK1.1, and Ly-6G surface proteins (28). SMs enhance the secretion of CCL2 by hepatic macrophages, which in turn facilitates monocyte recruitment and the augmentation of liver fibrosis (29). In another study, a subtype of spleen‐derived monocytes identified as CD11b+CD43hiLy6Clo cells has been demonstrated to preferentially infiltrate fibrotic liver tissue and adopt macrophage characteristics, thereby exacerbating fibrogenesis (30). However, it remains controversial whether SMs can migrate to the liver. These hypotheses require more advanced imaging techniques or cell tracking methods to validate the migration pathways of PMs and SMs.
3 Heterogeneity and plasticity of macrophages
The dynamic process of macrophage polarization entails the acquisition of specialized phenotypes and functional capabilities by macrophages as a reaction to stimuli present in their immediate surroundings. In 2000, Mills et al. categorized macrophages into two distinct subtypes, M1 and M2, based on differences in their metabolism, secretion, and function (31). This classification was based on the differential responses of macrophages in vitro to stimuli (32). Moreover, these polarized states demonstrate bidirectional interconversion when exposed to specific microenvironmental stimuli (33). Pro-inflammatory macrophages are typically triggered by stimulation with lipopolysaccharide (LPS), interferon-γ (IFN-γ), tumor necrosis factor (TNF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and TLR ligands (34). Normally, pro-inflammatory macrophages are characterized by their robust secretion of pro-inflammatory cytokines, including TNF-α, interleukin-1 beta (IL-1β), and IL-12. These cytokines eventually drive the activation of HSCs and promote liver fibrosis progression (35). Additionally, pro-inflammatory macrophages generate substantial amounts of reactive oxygen species (ROS) and reactive nitrogen species (RNS), which collectively enable them to effectively kill invading pathogens, as well as phagocytose and clear senescent, damaged, and degenerated cells (36). In contrast, alternatively activated macrophages play a crucial role in defending against parasitic infections, participating in tissue remodeling and secreting immunomodulatory mediators such as IL-10, TGF-β, IL-4 and IL-13 (37). Among these cytokines, TGF-β plays a crucial role in HSCs activation and liver fibrosis (38) (Figure 1).
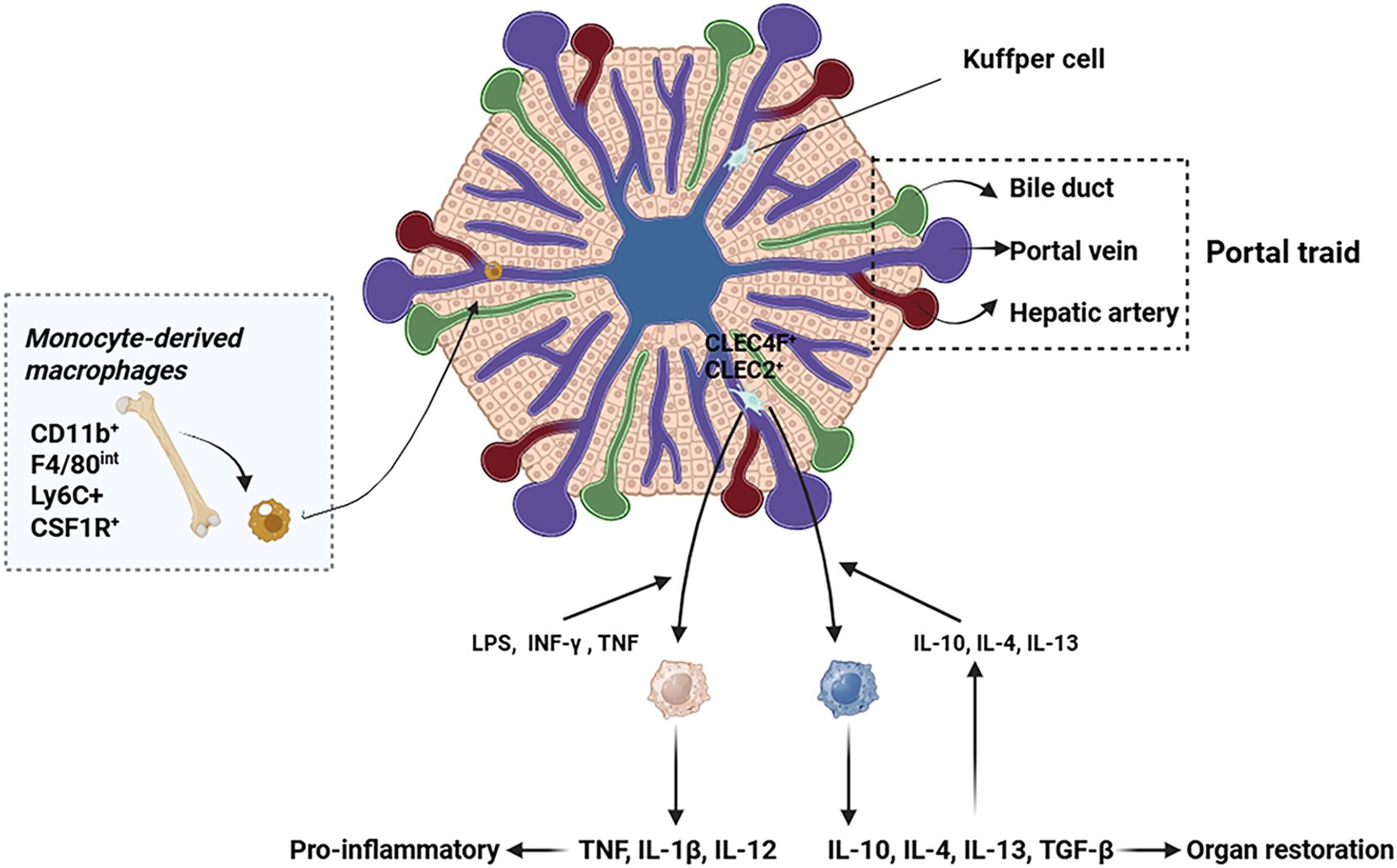
Figure 1. Origin and migration pathway of hepatic macrophages. This figure illustrates the heterogeneous origins and recruitment mechanisms of hepatic macrophages. KCs, the liver resident macrophages, are strategically positioned within the hepatic sinusoids. During liver injury, monocyte-derived macrophages are predominantly recruited to the liver via the systemic circulation.
Recent studies have shown that the traditional M1/M2 paradigm for classifying macrophages has been rendered obsolete by new technological breakthroughs, particularly in characterizing the complexity of hepatic macrophage populations (39). Hepatic macrophages are heterogeneous in the healthy liver, comprising distinct subsets with unique transcriptional profiles and, consequently, distinct functional roles (40). While the traditional M1/M2 classification remains useful for a broad understanding, it is insufficient to capture the full spectrum of macrophage functionality. Instead, distinct macrophage subpopulations exhibit unique biological characteristics across various disease contexts, and the functional differences among these subpopulations play crucial roles in disease progression and treatment response. Moreover, reliance on this general M1/M2 classification may impede the development of targeted therapies tailored to specific diseases (31). Therefore, despite its utility as a foundational framework, the M1/M2 paradigm’s limitations in explaining and treating complex diseases have prompted researchers to adopt more refined macrophage subpopulation analyses. This nuanced approach facilitates the identification of specific roles for different macrophage subtypes in various diseases, providing an essential foundation for the development of personalized targeted therapies.
4 Mechanisms of macrophage polarization
The dynamic regulation of immune cell responses by environmental stimuli manifests particularly through modifiable macrophage activity and functional plasticity. This adaptive “short-term memory” mechanism induces transient yet sustained modifications in macrophage phenotypes, thereby dynamically influencing their pathogenic contributions during disease progression (41). Macrophage polarization is controlled by a variety of molecular mechanisms, mainly including metabolic reprogramming, autophagy, iron metabolism, Signal Transducer and Activator of Transcription (STAT) and Notch signaling pathways (35, 42) (Figure 2).
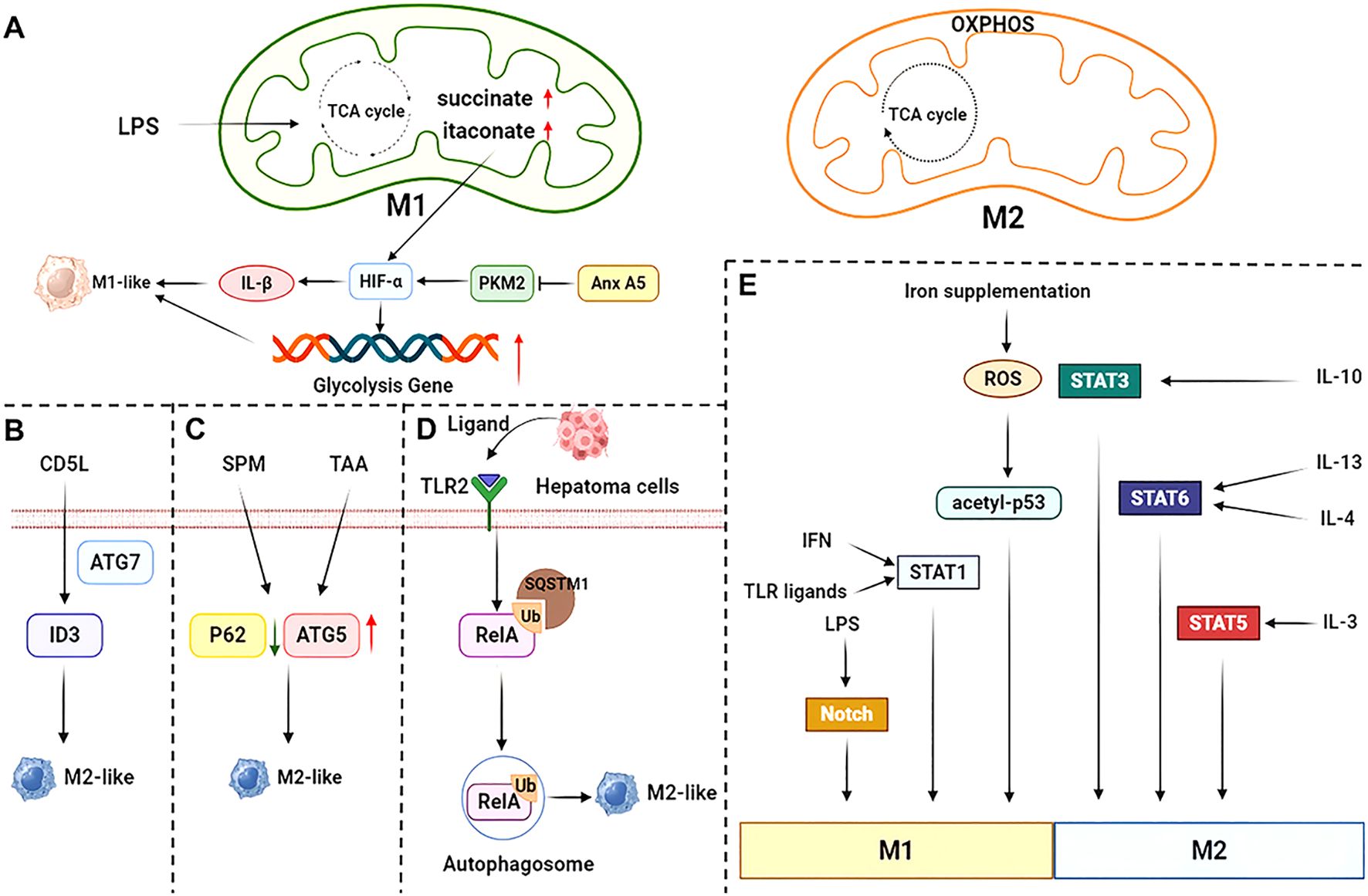
Figure 2. Mechanisms of macrophage polarization. (A) The tricarboxylic acid cycle is impaired in pro-inflammatory-polarized macrophages. Both Annexin A5 (Anx A5) and succinate upregulate HIF-α, which in turn promotes glycolytic gene expression and pro-inflammatory factor IL-1β expression. (B) CD5L induces ID3 expression and promotes alternatively activated polarization via the autophagy protein ATG7. (C) SPM-mediated KCs autophagy promotes alternatively activated polarization in TAA-induced liver injury through downregulation of P62 and upregulation of ATG5 expression. (D) Autophagy in hepatocellular carcinoma tissue is triggered by TLR2 ligand activation, which causes NF-κB RelA to be ubiquitinated and recognized by Sequestosome 1 (SQSTM1). This results in inhibition of the NF-κB pathway and consequently promotes alternatively activated polarization. (E) LPS, IFN, TLR ligands and iron supplementation promote pro-inflammatory polarization through different signaling pathways, while IL-10, IL-4, IL-13 and IL-3 promote alternatively activated polarization. SPM, spermine; TAA, thioacetamide.
4.1 Metabolic reprogramming
Metabolic adaptations play a pivotal role in macrophage activation and fibrogenic processes (35). In quiescent conditions, macrophages predominantly utilize tricarboxylic acid (TCA) cycle coupled with oxidative phosphorylation (OXPHOS) to generate Adenosine triphosphate (ATP), establishing energy equilibrium through mitochondrial respiration (43). During macrophage polarization toward pro-inflammatory phenotypes, glycolysis is the predominant metabolic pathway, while the TCA cycle is disrupted at two key points. These interruptions lead to the accumulation of itaconate and succinate, which are critical metabolites that contribute to the pro-inflammatory phenotype of macrophages (44). Excessive succinate stabilizes hypoxia inducible factor-1α (HIF-1α), which in turn activates the transcription of glycolytic genes, thereby maintaining glycolytic metabolism in pro-inflammatory macrophages (44). Moreover, HIF-1α exerts a regulatory influence on sphingosine 1-phosphate (S1P) metabolism, thereby modulating the migration, activation, differentiation, and polarization of macrophages (45). However, recent research suggests that the stabilization of HIF-1α takes place at a later stage in the process of inflammatory macrophage polarization. Furthermore, it indicates that the initial production of lactate through glycolysis is not governed by HIF-1α (46). Pro-inflammatory macrophages are distinguished by their enhanced glycolysis, elevated levels of glutathione, increased expression of ferritin, upregulated expression of cyclooxygenase (COX) 2, low expression of COX1, robust activity of inducible nitric oxide synthase (iNOS), and diminished activity of arginase 1 (Arg1) (47).
In contrast, alternatively activated macrophages exhibit a greater dependence on OXPHOS. Their TCA cycle remains intact, providing essential substrates for the electron transport chain (ETC). The coupling of mitochondrial OXPHOS with the TCA cycle is a slower process but generates significantly more ATP through the ETC (48). Although glycolysis produces less ATP compared to OXPHOS, its rapid rate of ATP generation is crucial for maintaining energy levels, especially under conditions demanding a rapid response (49). These macrophages are characterized by augmented fatty acid oxidation (FAO), reduced expression of ferritin, lower levels of glutathione, decreased production of COX2, heightened COX1 expression, weak iNOS activity, and enhanced Arg1 activity (47). Among the myriad metabolic alterations, the divergent metabolism of L-arginine represents one of the earliest described and most distinctive features used to differentiate between pro-inflammatory and alternatively activated macrophages. iNOS and Arg1 serve as quintessential effector molecules for pro-inflammatory and alternatively activated macrophages, respectively (50).
Pyruvate kinase M2 (PKM2) is a key determinant of macrophage glycolytic reprogramming and maintenance of pro-inflammatory polarization (51). Follistatin-like protein 1 (FSTL1) binds directly to PKM2 and promotes PKM2 phosphorylation and nuclear translocation (52). Conversely, Annexin A5 targeting to PKM2 causes glycolysis inhibition and activation of mitochondrial oxidative metabolism, thereby triggering macrophages to switch to an anti-inflammatory phenotype (53). In addition, growth differentiation factor 15 (GDF15) reprograms macrophage metabolic pathways, leading them to acquire an OXPHOS-dependent anti-inflammatory functional fate (54). Collectively, these distinct metabolic adaptations are not merely energetic adaptations, but constitute essential regulatory nodes that biochemically enforce macrophage polarization while dynamically coordinating immune functionality within specific microenvironmental niches.
4.2 Autophagy
Autophagy is essential for maintaining cellular homeostasis and significantly contributes to macrophage development, while also influencing their apoptosis via modulation of colony-stimulating factors (55, 56). Previous studies showed that cell division cycle 5-like (CD5L) regulates the up-regulation of inhibitor of DNA binding 3 (ID3) through the autophagy-related gene 7 (ATG7) and promotes an anti-inflammatory cytokine profile in response to TLR activation (57). In Thioacetamide (TAA)-induced KCs injury, spermine (SPM) pretreatment decreases P62 protein expression and increases ATG5 protein expression, thereby promoting anti-inflammatory polarization (58). Furthermore, TLR2-induced autophagosomal degradation of NF-κB RelA (P65) inhibits the NF-κB signaling pathway and drives alternatively activated macrophage polarization (59). In addition, it has been demonstrated that enhancing macrophage autophagy flux through ubiquitin-specific protease 19 (USP19) promotes the polarization of macrophages towards an anti-inflammatory phenotype (60). Fibroblast growth factor 21 (FGF21) significantly attenuates pro-inflammatory macrophage activation through autophagy-mediated degradation of HIF-1α (61). Additionally, inhibition of macrophage autophagy promotes M2-like polarization through ubiquitination-mediated degradation of TGF-β-activated kinase 1 and MAP3K7-binding protein 3 (TAB3), resulting in destabilization of the NF-κB signaling pathway (62). Collectively, these findings underscore the multifaceted role of autophagy in modulating macrophage polarization.
4.3 Iron metabolism
Iron homeostasis and the expression of iron-related genes strikingly shift during macrophage polarization, indicating a potential role for iron in macrophage activation. For example, in pro-inflammatory macrophages, the expression of Hepcidin antimicrobial peptide (Hamp) and FtH/FtL is highly upregulated, while Ferroportin (FPN) and IRP1/2 are downregulated (63). Upregulation of iron uptake and storage activates liver macrophages through the NF-κB pathway (64). Iron overload can polarize macrophages to the pro-inflammatory phenotype through the ROS/acetyl-p53 pathway (65). A recent study has shown that glycyrrhetic acid 3-O-mono-β-d-glucuronide (GAMG) induces ferroptosis of inflammatory macrophages through downregulation of solute carrier family 7 member 11 (SLC7A11) (66). However, exogenous iron supplementation and iron-rich ECM from human dermal fibroblasts induce the polarization of THP-1 cells and bone marrow-derived macrophages (BMDMs) into alternatively activated macrophages (67). These studies demonstrate the complexity of iron metabolism in macrophage polarization and function.
4.4 STAT signaling pathway
The STAT signaling pathway is a crucial mediator of cytokine signaling (e.g., IL-4, IL-6, IFN-γ) (68). Its core mechanism involves ligand binding to transmembrane receptors, which triggers JAK phosphorylation and subsequent STAT protein activation. Phosphorylated STAT proteins dimerize, translocate to the nucleus, and regulate target gene expression (69). In macrophage polarization, this pathway modulates the transition between pro-inflammatory and anti-inflammatory phenotypes through selective activation of distinct STAT isoforms (68). Specifically, IFN-γ and TLR-activated IRF-STAT1-signaling pathways orient macrophage function toward the pro-inflammatory phenotype, whereas IL-4 and IL-13 activate alternatively activated macrophages through STAT6 (70). Additionally, IL-10 and IL-3 activate STAT3 and STAT5, respectively, to promote alternatively activated macrophage polarization (71).
4.5 Notch signaling pathway
Notch signaling, known for its critical role in liver development, is also involved in liver regeneration, carcinogenesis, and metabolism (72). Macrophages express Notch ligands and receptors, indicating that Notch signaling participates in macrophage activation (73). LPS can upregulate Notch1 expression in macrophages via MyD88-dependent pathways, thereby induces the expression of its downstream genes (74). The Notch1 signaling pathway enhances the pro-inflammatory activation of hepatic macrophages by directly increasing the transcription of pro-inflammatory genes and by altering mitochondrial metabolism toward glucose oxidation, which leads to the production of mitochondrial reactive oxygen species (mtROS), further boosting the expression of pro-inflammatory genes (75).
5 The role of macrophages in liver fibrosis
Increasing evidence has shown that liver-resident macrophages and recruited monocyte-derived macrophages, which play an important role in liver fibrosis, are involved from initial liver injury and fibrosis formation to fibrosis regression (8). Among these cells, the existence of specialized subpopulations with distinct functional roles in health and disease has been documented (76).
Scar-associated macrophages (SAMs), which derive from BMDMs, accumulate in mouse fibrotic livers (77). There is a notable proliferation of the Scar-associated TREM2+ CD9+ macrophage subset. These cells are derived from circulating monocytes and play a role in enhancing the fibrotic response (78). In the initial phases of hepatic fibrosis, the activation of HSCs by macrophages through the release of inflammatory cytokines constitutes a pivotal mechanism that intensifies fibrosis (79). Macrophages accelerate fibrosis by secreting various cytokines, including TGF-β1, Vascular Endothelial Growth Factor (VEGF), and angiotensin II, which activate local tissue cells such as HSCs and myofibroblasts (80). Interestingly, activated HSCs further promote the transformation of macrophages into pro-inflammatory and pro-fibrogenic phenotypes. Activated HSCs attract monocytes/macrophages through the production of chemokines such as CCL2 and the infiltrating monocytes or macrophages can then further activate HSCs (81). For instance, sphingosine kinase 1 (SphK1) in KCs mediates CCL2 secretion, while SphK1 in HSCs upregulates CCR2 by downregulating miR-19b-3p (82). Furthermore, SphK1 aggravates liver fibrosis by promoting macrophage recruitment and M1/M2 polarization (83). The interaction between Jagged-1 on liver macrophages and Notch1 on HSCs drives Notch1-mediated HSCs activation and liver fibrosis (84). Additionally, MyD88 signaling in HSCs increases the secretion of CXCL10, which promotes macrophage polarization toward a pro-inflammatory phenotype and subsequent fibrosis (85).
Notably, in a carbon Tetrachloride (CCl4)-induced liver fibrosis model, macrophages exhibit contrasting functions in development and resolution of fibrosis: their elimination curbs development of fibrosis, while their absence following the cessation of injury hinders the resolution process, thereby worsening the fibrosis (86). In recent studies, the Ly6CloCD11BhiF4/80int macrophage population aggregates in the liver and constitute the main matrix metalloproteinase (MMP)-expressing macrophage subset during maximal fibrosis regression. It is crucial for degradation of tissue scar and originates from the infiltration of Ly-6Chi inflammatory monocytes (87). Collectively, these observations highlight that the dual regulation between macrophages and HSCs is a principal driver of fibrosis advancement.
6 The role of macrophages in different liver fibrosis induced by multiple disease
6.1 Metabolic dysfunction-associated steatotic liver disease
Metabolic dysfunction-associated steatotic liver disease (MASLD), formerly known as nonalcoholic fatty liver disease, is characterized by excessive hepatic lipid accumulation (88). The spectrum of MASLD extends from hepatic steatosis to metabolic dysfunction-associated steatohepatitis (MASH), which may progress to advanced liver fibrosis, cirrhosis, or even hepatocellular carcinoma (HCC) (89). Recent single-cell sequencing analyses have revealed a significant characteristic of both human and murine MASH, namely the formation of crown-like macrophage clusters. These clusters are observed encircling hepatocytes that are either dead or dying, which are characterized by substantial lipid accumulation. Additionally, these macrophage aggregates are found in close proximity to regions exhibiting fibrosis and to areas where HSCs have been activated (39, 40). Macrophages are important mediators of the inflammatory response that underlies the progression of MASLD to fibrosis.
Under normal physiological conditions, KCs are attached to the space of Disse within the hepatic sinusoids, and display thin, silk-like or flat, plate-like pseudopodia. In contrast, during the course of steatohepatitis, KCs tend to form clusters and lose their typical villus-like or digit-like extensions (90). Knockdown of Jun N-terminal kinase-1/2 (JNK-1/2) in KCs reverses liver fibrosis in choline-deficient, L-amino acid-defined (CDAA) diet-fed mice and reduces inflammatory responses (91). Dietary fat and cholesterol can suppress type 1 cytokine expression and oppositely upregulate the type 2 cytokines in murine KCs (92). The equilibrium among macrophage polarization states significantly influences the advancement of steatohepatitis. For instance, arginase-2 knockout mice develop spontaneous steatohepatitis, which can be mitigated by KCs depletion (93). Histidine-rich glycoprotein (HRG), produced by hepatocytes, induces macrophage pro-inflammatory polarization, whereas HRG knockout mice are protected from experimental steatohepatitis (94).
As MASLD progresses, resident KCs in the liver are gradually replaced by recruited macrophages (95). KCs expressing TREM2 localize to sites of inflammation, hepatic damage and fibrosis, and soluble TREM2 correlates with disease severity in humans (96). Additionally, TREM2 ligation inhibits TLR4-driven inflammation in KCs (97). Single-cell analysis has revealed that recruited macrophages exist in two subsets with distinct activation states, either CCR2+, CX3CR1+, Ly-6Chi monocytes or TREM2+,CD63+, CD9+ lipid-associated macrophages (LAMs) (98, 99). Genetic deletion of TREM2 in LAMs significantly impairs their tissue repair capacity, leading to exacerbated macrophage-mediated hepatic inflammation and accelerated fibrogenesis (100, 101). Given the crucial role of TREM2 in regulating both lipid metabolism and immune responses, therapeutic interventions targeting TREM2 modulation may offer promising novel strategies for the treatment of MASH (102). Another study has shown that hepatic LAMs express osteopontin (SPP1), a biomarker for patients with MASH, which is linked with the development of fibrosis (95). SPP1 has been reported to be upregulated in liver fibrosis and is tightly linked to dismal prognosis in end-stage hepatocellular carcinoma (103, 104). Studies have reported that myeloid-specific Glycoprotein Non-Metastatic Melanoma Protein B (GPNMB) knockout contributes to monocyte-derived macrophages occupation of the KCs niche and inhibits the formation of LAMs, thereby decreasing liver fibrosis (105). Another study suggested that the absence of Breast Regression Protein 39 (BRP39) reduces infiltration of LAMs, quelling liver inflammation and fibrosis (106). Notably, TREM2 also promotes lung fibrosis via protecting against macrophage apoptosis (107), while itaconate secreted by TREM2+ macrophages prevents apoptosis in cardiomyocytes and stimulates the growth of fibroblasts, which in turn enhances the process of cardiac tissue repair (108). Therefore, searching for new targets for LAMs is of great significance in the treatment of liver fibrosis in MASLD.
The genes elevated in MASLD have also been found to regulate macrophage polarization. In human and murine MASH, upregulated CD47 on necroptotic hepatocytes (necHC) and SIRPα on liver macrophages impair necHC uptake by liver macrophages, thereby promoting HSCs activation and fibrosis (109). Furthermore, macrophage-derived FGF12 and Tim3 have been shown to differentially activate HSCs through distinct mechanisms via the Monocyte Chemoattractant Protein-1 (MCP-1)/CCR2 axis and TGF-β secretion, respectively, all of which contribute to MASH pathogenesis (110, 111). Another study found that Niemann-Pick C1 (NPC1)-deficient macrophages exhibited inefficient efferocytosis in MASLD (112). HIF-1α, particularly in macrophages is increased in mice and patients with MASH, stimulating the release of inflammatory cytokines, which exacerbates both hepatic steatosis and inflammation (113). Simultaneously, it has been reported that macrophage HIF-2α mitigates insulin resistance and inflammation in adipose tissue by promoting an alternative activation polarization state (114). While PPARγ, rather than PPARδ, is essential for initiating the metabolic shift in response to IL-4, the deletion of either isoform has been demonstrated to hinder IL-4-triggered alternative macrophage activation, leading to insulin resistance and the development of hepatic steatosis (115, 116). Notably, a recent study identified a dopamine receptor D2 (DRD2) antagonist that selectively inhibits Yes-associated protein (YAP) in macrophages but not hepatocytes and thereby blocks the crosstalk between macrophages and the CTGF+VCAM1+ vascular niche, thereby promoting liver regeneration rather than fibrosis (117).
6.2 Alcoholic liver disease
Alcohol-associated liver disease (ALD) ranks among the most common liver conditions globally (118). Pericellular and perisinusoidal matrix accumulation with a chicken-wire appearance are also a characteristic fibrotic pattern in ALD (119). Alcohol consumption leads to malondialdehyde-acetaldehyde (MAA) adduct accumulation and stimulates KCs to produce IL-6, thereby accelerating hepatic inflammation and fibrosis in aldehyde dehydrogenase 2 (ALDH2) knockout mice (120). During ALD, the death of hepatocytes releases damage-associated molecular patterns, which in combination with necrotic cellular remnants and acetaldehyde—a byproduct of ethanol metabolism—induce the activation of KCs. This activation initiates hepatic inflammation through both innate and adaptive immune reactions (119, 121). Additionally, KCs produce nitric oxide (NO) and nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which further contribute to ALD (122).
Intestinal barrier dysfunction is an important contributor to ALD. Excessive alcohol consumption disrupts gut epithelial tight junctions, which increases intestinal permeability and facilitates the translocation of gut-derived LPS to the liver (123). During alcohol ingestion, high miR-212 expression suppresses zonula occludens-1 (ZO-1), a major component of tight junctions, causing disruption of gut integrity and permeability, thereby leading to LPS transport to the liver and subsequent activation of KCs (122, 124). Both KCs and activated HSCs contribute to fibrosis progression in alcohol-induced fibrosis through TLR4 (125). Silvia Affò et al. suggested that the upregulation of CCL20, mainly produced by macrophages, was strongly associated with LPS and silencing of CCL20 in mice reduces the expression of LPS-induced hepatic pro-inflammatory and pro-fibrogenic genes (126). In another study, monocyte-derived macrophages exhibit a pronounced inflammatory phenotype in a Notch-dependent manner (127).
6.3 Viral hepatitis
The global prevalence of viral hepatitis is predominantly attributed to five distinct hepatotropic viruses that are biologically unrelated, including hepatitis B virus (HBV), hepatitis C virus (HCV) among others (128). The estimated number of deaths due to viral hepatitis increased from 1.1 million in 2019 to 1.3 million in 2022, with 83% of deaths caused by HBV and 17% caused by HCV globally (129). Both adaptive and innate immunity are involved in the immune response to viral hepatitis, and the essential role of non-specific defense—especially the function of hepatic macrophages—has received wide attention (130).
A high HBV/HCV titer not only suppresses the polarization of pro-inflammatory macrophages, but also encourages their differentiation into a tolerogenic state (131). During immune activation in human and rodent infections, hepatitis B virus suppresses NF-κB pathway and ROS production in LPS-induced KCs, thereby inhibiting NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome activation and IL-1β production (132). In agreement with the finding, HCV core protein can inhibit the NF-κB pathway to greatly reduce the expression of CCL2 and CXCL10 in macrophages (133). Similarly, HBV splicing-generated protein (HBSP) impacts liver monocyte/macrophage recruitment through a down-regulation of hepatocyte CCL2 expression upon acute liver injury (134).
In human liver, primarily via stimulating macrophages, IFN-λ not only drives antiviral responses, but also promotes inflammation and fibrosis in viral diseases (135). Research corresponding to this statement has uncovered that IFN-λ3, but not IFN-λ4, is likely to be the major IFN-λ subclass mediating hepatic inflammation and fibrosis progression in HCV patients (136). However, exposure of human naive liver macrophages to HBV leads to an increased proportion of anti-inflammatory macrophages, which favors HBV development by releasing IL-10 (137). Moreover, HBV stimulates monocyte/macrophage secretion of TGF-β (138), while inhibiting the secretion of IL-12 induced by TLR2 to induce immune suppression (139). A recent study has confirmed that TLR2 is the direct binding receptor of hepatitis B e-antigen (HBeAg), which promotes the proliferation of HSCs in a macrophage-dependent manner (140). Consistently, prokineticin 2 (PK2), as a potential cytokine expressed in KCs, modulates the number of pro-inflammatory cells, thereby regulating their role in the progression of liver fibrosis after HBV infection (141). In addition, the activation of Stimulator of Interferon Genes (STING) signaling suppresses macrophage inflammasome activation by activating autophagic flux to alleviate HBV-induced liver fibrosis (142). The present investigation identifies MMP9+ macrophages as the pivotal drivers of end-stage hepatocellular carcinoma in patients with chronic HCV infection (143).
6.4 Cholestatic disease
Cholestatic diseases such as primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are characterized by the retention of bilirubin and bile salts in the liver and elevations of these metabolites in systemic circulation with a significant impact on organ function (144). The activation and recruitment of macrophages are mediated by ductular reactive cells (the epithelial cells characterized as a biliary phenotype) via the secretion of various factors (145). Exosomal lncRNA H19 derived from cholangiocytes enhances the pro-inflammatory polarization of KCs and promoted the recruitment and differentiation of BMDMs via inducing the expression and secretion of CCL2 and IL-6 in KCs (146). Flow cytometry analysis of non-parenchymal liver cells in PBC reveals massive infiltration of BMDMs in the liver, whereas the number of KCs decreases. These BMDMs exhibit high levels of TREM2 and SPP1 expression, which are characteristics of hepatic bile duct-associated macrophages. They are predominantly found surrounding the portal triad, a pattern that has been validated in patients with PSC (147). In contrast, SPP1+ macrophage infiltration in intrahepatic cholangiocarcinoma is associated with reduced tumor aggressiveness and improved patient survival (148). Another study has shown that high expression levels of IL-23 mRNA in CX3CR1hiCD11c+ BMDMs, inducing a significant intrahepatic increase in the frequency of hepatic IL-17A-producing CD4+ T cells and activity of the IL-23-IL17 axis, thereby aggravating PBC (149).
Moreover, KCs isolated from PBC mice showed increased surface RAE-1 protein expression and cytokine secretion, which subsequently activated NK cell-mediated target cell killing via Natural Killer Group 2 Member D (NKG2D)/Retinoic Acid Early Transcript 1 (RAE-1) recognition, increased inflammation, and fibrosis (150). IFN-γ further increased frequencies of inflammatory macrophages in the liver and aggravated liver fibrosis (151). In the absence of Protein Tyrosine Phosphatase 1B (PTP1B), which normally restricts the duration of pro-inflammatory signaling cascades, the activation and recruitment of hepatic macrophages are markedly enhanced after bile-duct ligation (BDL) (152). Macrophage phagocytosis of apoptotic cells was delayed by the induced high expression of CD16 in PBC BMDMs, promoting inflammation and fibrosis (153). In Mdr2-/- mice, CCL24-driven macrophages induce proliferation of HSCs and cholangiocytes to promote cholestasis and fibrosis (154).
7 Therapeutic approach for targeting macrophage in liver fibrosis
Hepatic macrophages, including KCs and other resident macrophages, play a crucial role in maintaining liver homeostasis and modulating the progression or regression of liver fibrosis. These cells are of significant therapeutic interest due to their central role in normal tissue homeostasis and their dual functions in promoting and inhibiting fibrosis. As the first line of defense against liver injury, hepatic macrophages orchestrate both pro-fibrotic and anti-fibrotic responses, making them attractive targets for therapeutic intervention. Although most macrophage-based therapies have been tested primarily in experimental animal models, some have been evaluated in clinical trials (155). Emerging translational strategies focus on multidimensional modulation of macrophage biology:
● Dampening KCs activation: Targeting the activation of KCs to reduce pro-inflammatory signaling and subsequent fibrogenesis.
● Inhibiting the recruitment of inflammatory cells (monocytes and macrophages) to the injured liver: Preventing the recruitment of inflammatory cells, such as monocytes and macrophages, to the injured liver to mitigate excessive inflammation.
● Shaping the heterogeneity of liver macrophages: Shaping the diverse phenotypes and functions of hepatic macrophages to promote an anti-fibrotic environment.
● Augmenting the differentiation into restorative macrophages: Shifting the hepatic microenvironment from inflammation toward resolution, as well as enhancing restorative differentiation pathways in macrophages by delivering phagocytic stimuli.
● Cell-based therapies involving autologous macrophages infusion: Utilizing autologous macrophage infusion to introduce macrophages with specific anti-inflammatory or pro-resolving properties.
● Targeting macrophages with nanostructures: Employing nanostructures to selectively target and modulate macrophage function in the liver.
7.1 Dampening KCs activation
Emricasan, a pan-caspases inhibitor, reduces inflammation and apoptosis by inhibiting KCs activation caused by NLRP3 inflammasome cascades (156, 157). However, it did not improve liver fibrosis in patients with MASH (158).
Apoptosis signal-regulating kinase 1 (ASK1) is a ubiquitously expressed redox-sensitive regulator of both JNK and p38-mediated inflammation and apoptosis (159). KCs are activated by p38 and JNK in liver fibrosis and blocking the inflammatory signaling pathway of KCs can reduce inflammation and fibrosis in NASH. Selonsertib, an ASK1 inhibitor, failed to demonstrate improvement in liver fibrosis in phase III trials (160).
The farnesoid X receptor (FXR) is a bile acid-activated nuclear receptor that is abundantly expressed in the liver and intestine (161). Direct activation of FXR enhances anti-inflammatory cytokines (162). Obeticholic acid (OCA), an effective FXR agonist, has been shown to prevent liver fibrosis by inhibiting KCs activation by blocking multiple inflammatory signaling pathways (163). In a Phase III trial, OCA significantly improved fibrosis in patients with MASH (164).
7.2 Inhibiting the recruitment of inflammatory cells to the injured liver
The recruitment of inflammatory cells to the injured liver is a critical step in the progression of liver inflammation and fibrosis. This process is largely dependent on the chemotactic effects of various chemokines secreted by activated hepatocytes, macrophages, and HSCs. Among these inflammatory cells, Ly-6Chi MoMϕs are particularly reliant on the signaling pathways involving CCL2/CCR2, CCL1/CCR8, and CCL25/CCR9 (165). Inhibition or elimination of macrophage recruitment via these signaling pathways can significantly ameliorate liver inflammation and global fibrosis in mice. Currently, strategies to interfere with chemokine signal transduction include the use of monoclonal antibodies, receptor antagonists, or small-molecule inhibitors to block chemokine-induced intracellular signaling (8). Among patients with steatohepatitis, the activation of KCs triggers the attraction of BMDMs via the CCR2/CCL2 and CCR5/CCL5 interaction pathways. This process promotes inflammation and contributes to the progression of fibrosis (166).
Notably, a dual inhibitor of CCR2/CCR5, known as Cenicriviroc (CVC), effectively blocks CCL2-mediated monocyte recruitment to the liver and exhibits anti-fibrotic effects in a mouse model of liver fibrosis (167). The phase III clinical trial demonstrated that a 12-month regimen of CVC 150 mg once daily failed to achieve histological improvement in liver fibrosis among MASH patients. However, CVC maintained a favorable safety profile and was well tolerated in this cohort with MASH and liver fibrosis (168).
In addition, medium chain fatty acid receptor G protein junction acceptor 84 (GPR84) has been identified as a mediator of myeloid immune cell infiltration under inflammatory conditions. Small-molecule antagonists (CpdA and CpdB) targeting GPR84 have been shown to obstruct macrophage recruitment to sites of injury in mice with both acute and chronic liver injury, thereby alleviating liver inflammation and fibrosis (169). Moreover, CCR9-deficient HSCs exhibit reduced fibrotic potential in vitro (170). Blocking the CCR9/CCL25 axis with a CCR9 antagonist represents an effective approach to mitigate the progression of hepatic fibrosis (171).
7.3 Shaping the heterogeneity of hepatic macrophages
Macrophage phenotypes exert contrasting functions, with pro-inflammatory macrophages typically associated with pro-inflammatory responses and alternatively activated macrophages with anti-inflammatory and tissue-repair functions. Consequently, therapeutic strategies aimed at promoting a switch from a pathogenic phenotype to a restorative phenotype hold promise for accelerating disease resolution and liver regeneration. This can be achieved using therapies that regulate macrophage polarization or reprogram macrophages into a restorative phenotype (155).
β-cryptoxanthin, a lutein carotenoid, has been shown to exert protective effects on markers of hepatic fat accumulation and inflammation (172). β-cryptoxanthin can directly attenuate LPS-induced pro-inflammatory macrophage activation while enhancing IL-4-induced alternatively activated macrophage activation, suggesting that β-cryptoxanthin may represent a promising therapeutic option for patients with liver fibrosis (173). Astaxanthin exhibits stronger antioxidant activity than β-carotene, and is particularly effective in reducing liver inflammation and inhibiting the activation of HSCs (174). It inhibits the activation of JNK/P38 and NF-κB signaling pathways by suppressing T-cell activity, macrophage recruitment, and KCs activation (175). Similarly, astaxanthin has been shown to decrease pro-inflammatory macrophages (176).
Glucagon-like peptide-1 (GLP-1) is a hormone secreted by the gut that lowers blood glucose levels by promoting glucose-dependent insulin secretion and inhibiting glucagon secretion. The glucose-lowering drug liraglutide, an analogue of GLP-1, has shown good efficacy in liver fibrosis (177). In vitro experiments showed that liraglutide counteracted the pro-inflammatory polarization of F4/80+ macrophages induced by palmitic acid (PA) in wild type mice, mediated through modulation of the cAMP–PKA–STAT3 signaling cascade (178). Moreover, corilagin, a gallotannin, mediating the reprogramming of alternatively activated macrophages to a pro-inflammatory phenotype by regulating the expression of Indoleamine 2,3-dioxygenase 1 (IDO1) in vitro, thereby alleviating liver fibrosis (179).
7.4 Augmenting the differentiation into restorative macrophages
Peroxisome proliferator-activated receptors (PPARs) play a key regulatory role in the liver, controlling insulin sensitivity, glucose and lipid metabolism, inflammation, and fibrosis (180). PPAR δ plays an anti-inflammatory role by promoting alternatively activated polarization of KCs and decreasing the expression of NLRP3, caspase-1 and IL-1β upon stimulation with saturated fatty acids and LPS. Elafibranor (GFT505), a dual PPARα/δ agonist, has been shown to reduce steatosis, inflammation, and fibrosis in several mouse models of steatohepatitis and decrease the gene expression of pro-inflammatory and pro-fibrotic markers (181). Lanifibranor, as a novel pan-PPAR agonist, decreases the pro-inflammatory activation of macrophages in the liver (182). In a phase 2b trial, it has been indicated that lanifibranor can alleviate liver fibrosis (183).
Gal-3 can directly trigger the NOD-like receptor thermal protein domain associated protein 3 (NLRP3) inflammasome in liver macrophages. Macrophage-derived pro-inflammatory cytokines ultimately result in the cascade of events leading to fibrosis (184). Gal-3 ablation protects mice from diet-induced steatohepatitis and reduces liver inflammation and fibrosis in HFD-fed mice (185). Gal-3 inhibitor GR-MD-02 shows potential efficacy in MASH with advanced fibrosis (186).
GPBAR-1 (TGR5) is a bile acid-activated receptor (BAR) expressed in various liver cells, including KCs, sinusoidal endothelial cells, and HSCs (187, 188). Bar501, a selective ligand of GPBAR-1, can effectively reduce bile duct inflammation, mitigate liver fibrosis and restore bile acid homeostasis (189).
7.5 Cell therapy with autologous macrophage infusion
More recently, mesenchymal stem cell (MSC) therapy has emerged as a promising alternative for treating liver diseases (190). MSCs possess the potential to differentiate into hepatocytes and exhibit immunomodulatory properties. They also secrete various trophic factors, including growth factors and cytokines, which have therapeutic implications. In addition, mesenchymal stem cells can inhibit the inflammatory response, reduce hepatocyte apoptosis, promote hepatocyte regeneration, attenuate liver fibrosis, and enhance liver function (191). However, depending on the route of MSC injection and the status of liver disease, MSCs may differentiate into myofibroblasts, thereby exacerbating liver fibrosis. Despite these potential risks, the therapeutic efficacy of MSCs in liver fibrosis has been demonstrated in both preclinical and clinical studies (192).
In addition to MSC therapy, cell therapy involving the transfer of autologous beneficial macrophages has also been explored. Macrophage cell therapy improves clinically relevant parameters in experimental chronic liver injury (193). BMDMs can recruit and modify endogenous macrophages to activate natural killer (NK) cells by modulating the hepatic microenvironment. Pro-inflammatory macrophages also increase the total number of NK cells and activated NK cells in the fibrotic liver, which promoted HSCs apoptosis through TRAIL release. (194).
7.6 Targeting macrophages with nanostructures
Nanodrugs have been demonstrated to improve inflammation and liver fibrosis by targeting macrophages. The polarization and reprogramming of macrophages can be differentially modulated by nanoparticles that vary in their physicochemical attributes, such as chemical makeup, size, and surface modification (195). A nanomedicine delivery system has been engineered to target KCs by exploiting receptors that are predominantly present on them, such as mannose and scavenger receptors. This system is intended to deliver a range of therapeutics, including anti-inflammatory medications, ROS scavengers, agents that modify the KCs phenotype, and small interfering RNA (siRNA) drugs aimed at inflammatory mediators, directly to KCs. This approach holds significant promise for the treatment of liver fibrosis (196). A polydatin-loaded micelle demonstrates highly efficient liver-targeted drug release in response to the fibrotic microenvironment (197). Moreover, researchers have developed a dual-drug-loaded lipid nanoparticle. It can effectively suppress macrophage pro-inflammatory signaling and degrade the ECM barrier (198). Therapeutic approaches for targeting macrophages in liver diseases are summarized in Table 1.
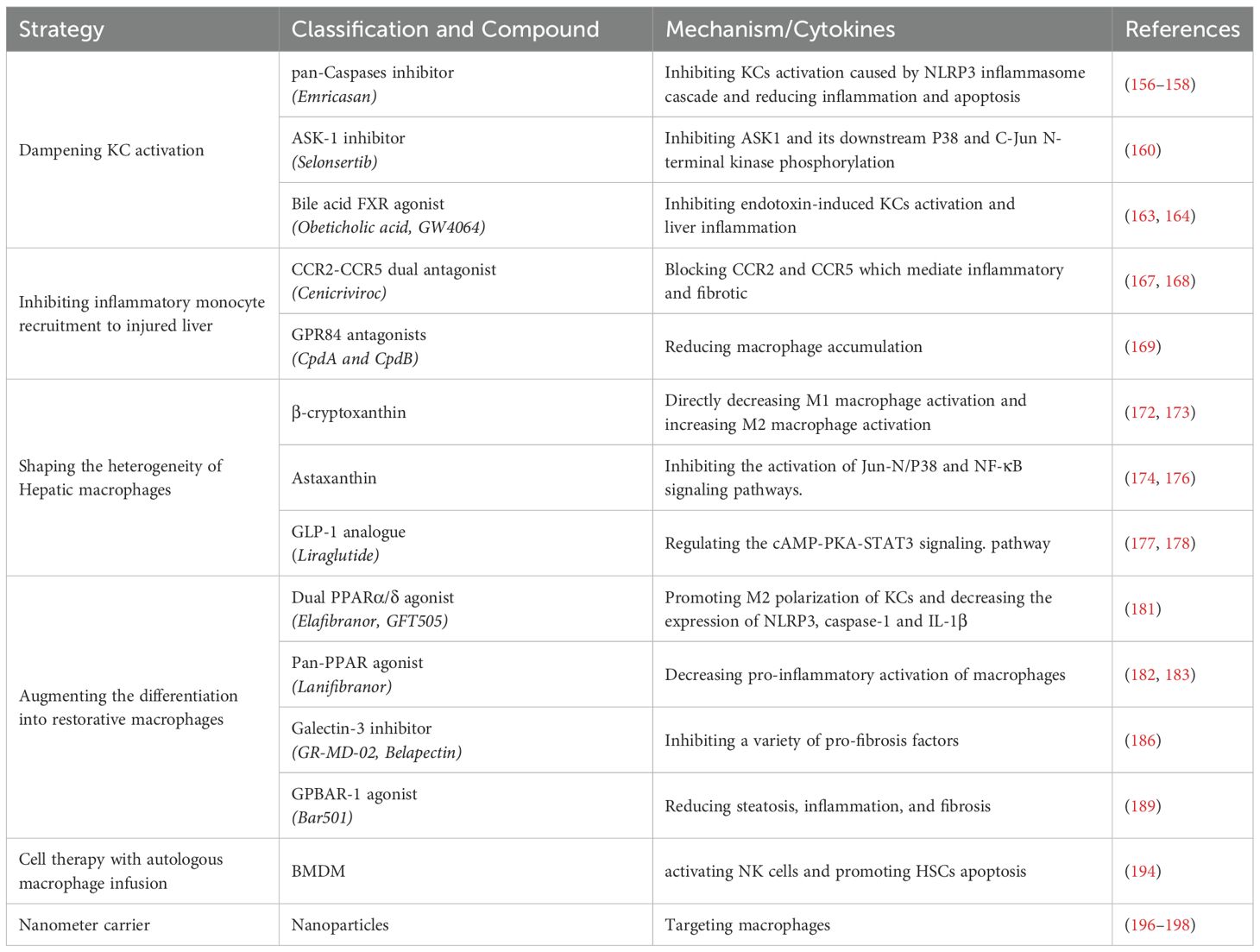
Table 1. Therapeutic approaches for targeting macrophages in liver diseases.
8 Conclusions and perspectives
In summary, translating the concept of macrophage heterogeneity into clinically effective therapy for liver fibrosis requires addressing two fundamental questions: (i) What are the precise functional roles and pathophysiological significance of distinct macrophage phenotypes across different disease stages? (ii) How can we achieve spatiotemporally precise reprogramming macrophage phenotypes to favor fibrosis resolution while minimizing off-target effects?
The current translational challenges primarily stem from interspecies discrepancies and human-specific complexities. Although murine models have provided foundational insights, they often fail to fully recapitulate the multidimensional heterogeneity of human macrophages, which is shaped by genetic polymorphisms, epigenetic modifications, demographic variables (age, sex, ethnicity), and dynamic host-microbiome interactions. This biological divergence contributes to the frequent discordance between preclinical efficacy and clinical trial outcomes. The functional plasticity of macrophages—acting as double-edged swords in disease initiation (pro-inflammatory), progression (pro-fibrotic), and resolution (pro-reparative)—demands phenotype-specific targeting strategies rather than global macrophage modulation. In addition, macrophage biology in the liver is complicated by phenotypic plasticity, overlapping markers, and inconsistent classification, making it difficult to distinguish between different populations and their functional roles. On the one hand, microenvironmental signals drive rapid phenotypic switching in both KCs and BMDMs, leading to shared surface markers and functional profiles. This bidirectional interconversion blurs the line between resident and recruited macrophages, challenging traditional identification methods. On the other hand, inconsistent nomenclature and unclear definitions further complicate the field. Many studies classify macrophage subsets with distinct names, yet there is significant overlap between datasets, raising questions about whether identified clusters represent true distinct populations or merely different activation states. Additionally, much of the existing research remains descriptive, lacking mechanistic insights into macrophage functions in health and disease.
Future studies would be focused on elucidating the precise roles of distinct macrophage phenotypes at each stage of liver fibrosis and developing targeted therapies that can precisely modulate macrophage function in a spatiotemporal manner. This approach holds promise for improving therapeutic outcomes and addressing the inherent complexities in liver fibrosis.
Author contributions
WW: Writing – original draft, Writing – review & editing. SL: Writing – original draft. YL: Writing – original draft. XD: Writing – original draft. YY: Writing – original draft. SC: Writing – original draft. JC: Writing – original draft. FT: Writing – review & editing. WD: Writing – review & editing. TL: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by grants from the National Natural Science Foundation of China (82373930), Guangdong-Hong Kong-Macao Joint Innovation Project of Guangdong Science and Technology Plan (2024A0505040025) and Natural Science Foundation Research Team Project of Heilongjiang Province (TD2024H003).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Friedman SL. Liver fibrosis – from bench to bedside. J Hepatology. (2003) 38:38–53. doi: 10.1016/S0168-8278(02)00429-4
2. Sun C and Matsukawa A. Role of macrophages in liver fibrosis. Acta Med Okayama. (2024) 7(1):1–8. doi: 10.18926/AMO/66664
3. Hammerich L and Tacke F. Hepatic inflammatory responses in liver fibrosis. Nat Rev Gastroenterol Hepatology. (2023) 20:633–46. doi: 10.1038/s41575-023-00807-x
4. Horn P and Tacke F. Liver macrophage diversity in health and disease. In: Monocytes and Macrophages in Development, Regeneration, and Disease. Results and Problems in Cell Differentiation (2024) 74:175–209. doi: 10.1007/978-3-031-65944-7_7
5. Tacke F and Zimmermann HW. Macrophage heterogeneity in liver injury and fibrosis. J Hepatology. (2014) 60:1090–6. doi: 10.1016/j.jhep.2013.12.025
6. Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. (2007) 117:539–48. doi: 10.1172/JCI30542
7. Yu W, Wang S, Wang Y, Chen H, Nie H, Liu L, et al. MicroRNA: role in macrophage polarization and the pathogenesis of the liver fibrosis. Front Immunol. (2023) 14:1147710. doi: 10.3389/fimmu.2023.1147710
8. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatology. (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
9. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2014) 518:547–51. doi: 10.1038/nature13989
10. Sakai M, Troutman TD, Seidman JS, Ouyang Z, Spann NJ, Abe Y, et al. Liver-derived signals sequentially reprogram myeloid enhancers to initiate and maintain kupffer cell identity. Immunity. (2019) 51:655–670.e8. doi: 10.1016/j.immuni.2019.09.002
11. David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB, et al. Combination of mass cytometry and imaging analysis reveals origin, location, and functional repopulation of liver myeloid cells in mice. Gastroenterology. (2016) 151:1176–91. doi: 10.1053/j.gastro.2016.08.024
12. Guilliams M and Scott CL. Liver macrophages in health and disease. Immunity. (2022) 55:1515–29. doi: 10.1016/j.immuni.2022.08.002
13. Kanneganti T-D, Lamkanfi M, and Núñez G. Intracellular NOD-like receptors in host defense and disease. Immunity. (2007) 27:549–59. doi: 10.1016/j.immuni.2007.10.002
14. Li W, Chang N, and Li L. Heterogeneity and function of kupffer cells in liver injury. Front Immunol. (2022) 13:940867. doi: 10.3389/fimmu.2022.940867
15. Krenkel O and Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. (2017) 17:306–21. doi: 10.1038/nri.2017.11
16. Fogg DK, Sibon C, Miled C, Jung S, Aucouturier P, Littman DR, et al. A clonogenic bone marrow progenitor specific for macrophages and dendritic cells. Science. (2006) 311:83–7. doi: 10.1126/science.1117729
17. Wu T, Zhang C, Shao T, Chen J, and Chen D. The role of NLRP3 inflammasome activation pathway of hepatic macrophages in liver ischemia–reperfusion injury. Front Immunol. (2022) 13:905423. doi: 10.3389/fimmu.2022.905423
18. Wen Y, Lambrecht J, Ju C, and Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. (2020) 18:45–56. doi: 10.1038/s41423-020-00558-8
19. Varol C, Mildner A, and Jung S. Macrophages: development and tissue specialization. Annu Rev Immunol. (2015) 33:643–75. doi: 10.1146/annurev-immunol-032414-112220
20. Ingersoll MA, Spanbroek R, Lottaz C, Gautier EL, Frankenberger M, Hoffmann R, et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood. (2010) 115:e10–9. doi: 10.1182/blood-2009-07-235028
21. Huang HY, Chen YZ, Zhao C, Zheng XN, Yu K, Yue JX, et al. Alternations in inflammatory macrophage niche drive phenotypic and functional plasticity of Kupffer cells. Nat Commun. (2024) 15:9337. doi: 10.1038/s41467-024-53659-7
22. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S, et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat Commun. (2016) 7:10321. doi: 10.1038/ncomms10321
23. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F, et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. J Hepatology. (2016) 65:758–68. doi: 10.1016/j.jhep.2016.05.037
24. Rosas M, Davies LC, Giles PJ, Liao CT, Kharfan B, Stone TC, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science. (2014) 334:645–8. doi: 10.1126/science.1251414
25. Oza D, Ivich F, Deprey K, Bittner K, Bailey K, Goldman S, et al. Treatment of acute liver injury through selective tropism of high mobility group box 1 gene-silenced large peritoneal macrophages. ACS Nano. (2025) 19:12102–18. doi: 10.1021/acsnano.4c18345
26. Jin H, Liu K, Tang J, Huang X, Wang H, Zhang Q, et al. Genetic fate-mapping reveals surface accumulation but not deep organ invasion of pleural and peritoneal cavity macrophages following injury. Nat Commun. (2021) 12:2863. doi: 10.1038/s41467-021-23197-7
27. Wang J and Kubes P. A reservoir of mature cavity macrophages that can rapidly invade visceral organs to affect tissue repair. Cell. (2016) 165:668–78. doi: 10.1016/j.cell.2016.03.009
28. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. (2009) 325:612–6. doi: 10.1126/science.1175202
29. Li L, Wei W, Li Z, Chen H, Li Y, Jiang W, et al. The spleen promotes the secretion of CCL2 and supports an M1 dominant phenotype in hepatic macrophages during liver fibrosis. Cell Physiol Biochem. (2018) 51:557–74. doi: 10.1159/000495276
30. Zhang S, Wan D, Zhu M, Wang G, Zhang X, Huang N, et al. CD11b+CD43hiLy6Clo splenocyte-derived macrophages exacerbate liver fibrosis via spleen–liver axis. Hepatology. (2023) 77:1612–29. doi: 10.1002/hep.32782
31. Guan F, Wang R, Yi Z, Luo P, Liu W, Xie Y, et al. Tissue macrophages: origin, heterogenity, biological functions, diseases and therapeutic targets. Signal Transduction Targeted Ther. (2025) 10:93. doi: 10.1038/s41392-025-02124-y
32. Zwicker C, Bujko A, and Scott CL. Hepatic macrophage responses in inflammation, a function of plasticity, heterogeneity or both? Front Immunol. (2021) 12:690813. doi: 10.3389/fimmu.2021.690813
33. Cheng K, Cai N, Zhu J, Yang X, Liang H, and Zhang W. Tumor-associated macrophages in liver cancer: From mechanisms to therapy. Cancer Commun. (2022) 42:1112–40. doi: 10.1002/cac2.12345
34. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. (2018) 18:545–58. doi: 10.1038/s41577-018-0029-z
35. Horn P and Tacke F. Metabolic reprogramming in liver fibrosis. Cell Metab. (2024) 36:1439–55. doi: 10.1016/j.cmet.2024.05.003
36. Peng Y, Zhou M, Yang H, Qu R, Qiu Y, Hao J, et al. Regulatory mechanism of M1/M2 macrophage polarization in the development of autoimmune diseases. Mediators Inflammation. (2023) 2023:1–20. doi: 10.1155/2023/8821610
37. Murray PJ and Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. (2011) 11:723–37. doi: 10.1038/nri3073
38. Roehlen N, Crouchet E, and Baumert TF. Liver fibrosis: mechanistic concepts and therapeutic perspectives. Cells. (2020) 9:875. doi: 10.3390/cells9040875
39. Ahamed F, Eppler N, Jones E, and Zhang Y. Understanding macrophage complexity in metabolic dysfunction-associated steatotic liver disease: transitioning from the M1/M2 paradigm to spatial dynamics. Livers. (2024) 4:455–78. doi: 10.3390/livers4030033
40. De Ponti FF, Liu Z, and Scott CL. Understanding the complex macrophage landscape in MASLD. JHEP Rep. (2024) 6:101196. doi: 10.1016/j.jhepr.2024.101196
41. Monticelli S and Natoli G. Short-term memory of danger signals and environmental stimuli in immune cells. Nat Immunol. (2013) 14:777–84. doi: 10.1038/ni.2636
42. Wang C, Ma C, Gong L, Guo Y, Fu K, Zhang Y, et al. Macrophage polarization and its role in liver disease. Front Immunol. (2021) 12:803037. doi: 10.3389/fimmu.2021.803037
43. Sun X, Li Y, Deng Q, Hu Y, Dong J, Wang W, et al. Macrophage polarization, metabolic reprogramming, and inflammatory effects in ischemic heart disease. Front Immunol. (2022) 13:934040. doi: 10.3389/fimmu.2022.934040
44. Viola A, Munari F, Sánchez-Rodríguez R, Scolaro T, and Castegna A. The metabolic signature of macrophage responses. Front Immunol. (2019) 10:1462. doi: 10.3389/fimmu.2019.01462
45. Hutami IR, Izawa T, Khurel-Ochir T, Sakamaki T, Iwasa A, and Tanaka E. Macrophage motility in wound healing is regulated by HIF-1α via S1P signaling. Int J Mol Sci. (2021) 22:8992. doi: 10.3390/ijms22168992
46. Bae S, Park PSU, Lee Y, Mun SH, Giannopoulou E, Fujii T, et al. MYC-mediated early glycolysis negatively regulates proinflammatory responses by controlling IRF4 in inflammatory macrophages. Cell Rep. (2021) 35:109264. doi: 10.1016/j.celrep.2021.109264
47. Li M, Yang Y, Xiong L, Jiang P, Wang J, and Li C. Metabolism, metabolites, and macrophages in cancer. J Hematol Oncol. (2023) 16:80. doi: 10.1186/s13045-023-01478-6
48. Thapa B LK. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. (2019) 52:360–72. doi: 10.5483/BMBRep.2019.52.6.140
49. El Kasmi KC and Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. Semin Immunol. (2015) 27:267–75. doi: 10.1016/j.smim.2015.09.001
50. Rath M, Müller I, Kropf P, Closs EI, and Munder M. Metabolism via arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. (2014) 5:532. doi: 10.3389/fimmu.2014.00532
51. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, et al. Pyruvate kinase M2 regulates hif-1α Activity and IL-1β Induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. (2015) 21:65–80. doi: 10.1016/j.cmet.2014.12.005
52. Rao J, Wang H, Ni M, Wang Z, Wang Z, Wei S, et al. FSTL1 promotes liver fibrosis by reprogramming macrophage function through modulating the intracellular function of PKM2. Gut. (2022) 71:2539–50. doi: 10.1136/gutjnl-2021-325150
53. Xu F, Guo M, Huang W, Feng L, Zhu J, Luo K, et al. Annexin A5 regulates hepatic macrophage polarization via directly targeting PKM2 and ameliorates NASH. Redox Biol. (2020) 36:101634. doi: 10.1016/j.redox.2020.101634
54. Li X, Huai Q, Zhu C, Zhang X, Xu W, Dai H, et al. GDF15 ameliorates liver fibrosis by metabolic reprogramming of macrophages to acquire anti-inflammatory properties. Cell Mol Gastroenterol Hepatology. (2023) 16:711–34. doi: 10.1016/j.jcmgh.2023.07.009
55. Vitaliti A, Reggio A, and Palma A. Macrophages and autophagy: partners in crime. FEBS J. (2024) 292:2957–72. doi: 10.1111/febs.17305
56. Germic N, Frangez Z, Yousefi S, and Simon H-U. Regulation of the innate immune system by autophagy: monocytes, macrophages, dendritic cells and antigen presentation. Cell Death Differentiation. (2019) 26:715–27. doi: 10.1038/s41418-019-0297-6
57. Sanjurjo L, Aran G, Téllez É, Amézaga N, Armengol C, López D, et al. CD5L promotes M2 macrophage polarization through autophagy-mediated upregulation of ID3. Front Immunol. (2018) 9:480. doi: 10.3389/fimmu.2018.00480
58. Zhou S, Gu J, Liu R, Wei S, Wang Q, Shen H, et al. Spermine alleviates acute liver injury by inhibiting liver-resident macrophage pro-inflammatory response through ATG5-dependent autophagy. Front Immunol. (2018) 9:948. doi: 10.3389/fimmu.2018.00948
59. Chang C, Su Y, Lee P, and Lei H. Targeting NFKB by autophagy to polarize hepatoma-associated macrophage differentiation. Autophagy. (2014) 9:619–21. doi: 10.4161/auto.23546
60. Liu T, Wang L, Liang P, Wang X, Liu Y, Cai J, et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol. (2020) 18:2431–42. doi: 10.1038/s41423-020-00567-7
61. Zhu J, Jin Z, Wang J, Wu Z, Xu T, Tong G, et al. FGF21 ameliorates septic liver injury by restraining proinflammatory macrophages activation through the autophagy/HIF-1α axis. J Advanced Res. (2025) 69:477–94. doi: 10.1016/j.jare.2024.04.004
62. Gao Z, Li XG, Feng S-R, Chen JF, Song K, Shi YH, et al. Autophagy suppression facilitates macrophage M2 polarization via increased instability of NF-κB pathway in hepatocellular carcinoma. Int Immunopharmacology. (2023) 123:110685. doi: 10.1016/j.intimp.2023.110685
63. Gan Z, Wang Q, Li J, Wang X, Wang Y, and Du H. Iron reduces M1 macrophage polarization in RAW264.7 macrophages associated with inhibition of STAT1. Mediators Inflammation. (2017) 2017:1–9. doi: 10.1155/2017/8570818
64. Xiong S, She H, Zhang A, Wang J, Mkrtchyan H, Dynnyk A, et al. Hepatic macrophage iron aggravates experimental alcoholic steatohepatitis. Am J Physiology-Gastrointestinal Liver Physiol. (2008) 295:G512–21. doi: 10.1152/ajpgi.90327.2008
65. Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, et al. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med. (2018) 7:4012–22. doi: 10.1002/cam4.1670
66. Pang Q, Zhou S, Wang Y, Pan H, Wang Z, Qin X, et al. GAMG alleviates liver fibrosis through inducing ferroptosis in inflammatory macrophages via the IRF1/SLC7A11 signaling pathway. Redox Biol. (2025) 80:103509. doi: 10.1016/j.redox.2025.103509
67. Wilkinson HN, Roberts ER, Stafford AR, Banyard KL, Matteucci P, Mace KA, et al. Tissue iron promotes wound repair via M2 macrophage polarization and the chemokine (C-C motif) ligands 17 and 22. Am J Pathology. (2019) 189:2196–208. doi: 10.1016/j.ajpath.2019.07.015
68. Chi Y, Jiang H, Yin Y, Zhou X, Shao Y, Li Y, et al. Macrophage signaling pathways in health and disease: from bench to bedside applications. MedComm. (2025) 6:e70256. doi: 10.1002/mco2.70256
69. Hu X, li J, Fu M, Zhao X, and Wang W. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduction Targeted Ther. (2021) 6:402. doi: 10.1038/s41392-021-00791-1
70. Biswas SK, Chittezhath M, Shalova IN, and Lim J-Y. Macrophage polarization and plasticity in health and disease. Immunologic Res. (2012) 53:11–24. doi: 10.1007/s12026-012-8291-9
71. Sica A, Invernizzi P, and Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. (2014) 59:2034–42. doi: 10.1002/hep.26754
72. Siebel C and Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. (2017) 97:1235–94. doi: 10.1152/physrev.00005.2017
73. Keewan E and Naser SA. The role of notch signaling in macrophages during inflammation and infection: implication in rheumatoid arthritis? Cells. (2020) 9:111. doi: 10.3390/cells9010111
74. Zhang Q, Wang C, Liu Z, Liu X, Han C, Cao X, et al. Notch signal suppresses toll-like receptor-triggered inflammatory responses in macrophages by inhibiting extracellular signal-regulated kinase 1/2-mediated nuclear factor κB activation. J Biol Chem. (2012) 287:6208–17. doi: 10.1074/jbc.M111.310375
75. Xu J, Chi F, and Tsukamoto H. Notch signaling and M1 macrophage activation in obesity-alcohol synergism. Clinics Res Hepatol Gastroenterology. (2015) 39:S24–8. doi: 10.1016/j.clinre.2015.05.016
76. Reggio A, Fuoco C, Deodati R, and Palma A. SPP1 macrophages across diseases: A call for reclassification? FASEB J. (2025) 39:e70448. doi: 10.1096/fj.202403227R
77. Yang Y, Li W, Liu C, Liu J, Yang L, Yue W, et al. Single-cell RNA seq identifies Plg-RKT-PLG as signals inducing phenotypic transformation of scar-associated macrophage in liver fibrosis. Biochim Biophys Acta (BBA) - Mol Basis Disease. (2023) 1869:166754. doi: 10.1016/j.bbadis.2023.166754
78. Ramachandran P, Dobie R, Wilson-Kanamori JR, Dora EF, Henderson BEP, Luu NT, et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature. (2019) 575:512–8. doi: 10.1038/s41586-019-1631-3
79. Barnes MA, McMullen MR, Roychowdhury S, Madhun NZ, Niese K, Olman MA, et al. Macrophage migration inhibitory factor is required for recruitment of scar-associated macrophages during liver fibrosis. J Leukocyte Biol. (2015) 97:161–9. doi: 10.1189/jlb.3A0614-280R
80. Sun YY, Li XF, Meng XM, Huang C, Zhang L, and Li J. Macrophage phenotype in liver injury and repair. Scandinavian J Immunol. (2017) 85:166–74. doi: 10.1111/sji.12468
81. Marra F and Tacke F. Roles for chemokines in liver disease. Gastroenterology. (2014) 147:577–94.e1. doi: 10.1053/j.gastro.2014.06.043
82. Lan T, Li C, Yang G, Sun Y, Zhuang L, Ou Y, et al. Sphingosine kinase 1 promotes liver fibrosis by preventing miR-19b-3p-mediated inhibition of CCR2. Hepatology. (2018) 68:1070–86. doi: 10.1002/hep.29885
83. Ding X, Zhang X, Cao J, Chen S, Chen Y, Yuan K, et al. Sphingosine kinase 1 aggravates liver fibrosis by mediating macrophage recruitment and polarization. Cell Mol Gastroenterol Hepatology. (2024) 18:101406. doi: 10.1016/j.jcmgh.2024.101406
84. Yang YM, Noureddin M, Liu C, Koichiro O, So YK, Divya R, et al. Hyaluronan synthase 2–mediated hyaluronan production mediates Notch1 activation and liver fibrosis. Sci Trans Med. (2019) 11(496):eaat9284. doi: 10.1126/scitranslmed.aat9284
85. Zhang J, Liu Y, Chen H, Yuan Q, Wang J, Niu M, et al. MyD88 in hepatic stellate cells enhances liver fibrosis via promoting macrophage M1 polarization. Cell Death Disease. (2022) 13:411. doi: 10.1038/s41419-022-04802-z
86. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. (2005) 115:56–65. doi: 10.1172/JCI200522675
87. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci. (2012) 109:E3186–95. doi: 10.1073/pnas.1119964109
88. Rinella ME, Lazarus JV, Ratziu V, Francque SM, Sanyal AJ, Kanwal F, et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Ann Hepatology. (2024) 29:101133. doi: 10.1016/j.aohep.2023.101133
89. Yin X, Guo X, Liu Z, and Wang J. Advances in the diagnosis and treatment of non-alcoholic fatty liver disease. Int J Mol Sci. (2023) 24:2844. doi: 10.3390/ijms24032844
90. Wang H, Li L, Li Y, Li Y, Sha Y, Wen S, et al. Intravital imaging of interactions between iNKT and kupffer cells to clear free lipids during steatohepatitis. Theranostics. (2021) 11:2149–69. doi: 10.7150/thno.51369
91. Kodama Y, Kisseleva T, Iwaisako K, Miura K, Taura K, De Minicis S, et al. c-jun N-terminal kinase-1 from hematopoietic cells mediates progression from hepatic steatosis to steatohepatitis and fibrosis in mice. Gastroenterology. (2009) 137:1467–1477.e5. doi: 10.1053/j.gastro.2009.06.045
92. McGettigan B, McMahan R, Orlicky D, Burchill M, Danhorn T, Francis P, et al. Dietary lipids differentially shape nonalcoholic steatohepatitis progression and the transcriptome of kupffer cells and infiltrating macrophages. Hepatology. (2019) 70:67–83. doi: 10.1002/hep.30401
93. Navarro LA, Wree A, Povero D, Berk MP, Eguchi A, Ghosh S, et al. Arginase 2 deficiency results in spontaneous steatohepatitis: A novel link between innate immune activation and hepatic de novo lipogenesis. J Hepatology. (2015) 62:412–20. doi: 10.1016/j.jhep.2014.09.015
94. Bartneck M, Fech V, Ehling J, Govaere O, Warzecha KT, Hittatiya K, et al. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology. (2016) 63:1310–24. doi: 10.1002/hep.28418
95. Remmerie A, Martens L, Thoné T, Castoldi A, Seurinck R, Pavie B, et al. Osteopontin expression identifies a subset of recruited macrophages distinct from kupffer cells in the fatty liver. Immunity. (2020) 53:641–657.e14. doi: 10.1016/j.immuni.2020.08.004
96. Guha Ray A, Odum OP, Wiseman D, and Weinstock A. The diverse roles of macrophages in metabolic inflammation and its resolution. Front Cell Dev Biol. (2023) 11:1147434. doi: 10.3389/fcell.2023.1147434
97. Perugorria MJ, Esparza-Baquer A, Oakley F, Labiano I, Korosec A, Jais A, et al. Non-parenchymal TREM-2 protects the liver from immune-mediated hepatocellular damage. Gut. (2019) 68:533–46. doi: 10.1136/gutjnl-2017-314107
98. Guilliams M, Bonnardel J, Haest B, Vanderborght B, Wagner C, Remmerie A, et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell. (2022) 185:379–396.e38. doi: 10.1016/j.cell.2021.12.018
99. Daemen S, Gainullina A, Kalugotla G, He L, Chan MM, Beals JW, et al. Dynamic shifts in the composition of resident and recruited macrophages influence tissue remodeling in NASH. Cell Rep. (2021) 34:108626. doi: 10.1016/j.celrep.2020.108626
100. Wang X, Qiu Z, Zhong Z, and Liang S. TREM2-expressing macrophages in liver diseases. Trends Endocrinol Metab. (2025). doi: 10.1016/j.tem.2025.04.009
101. De Ponti FF, Bujko A, Liu Z, Collins PJ, Schuermans S, Maueroder C, et al. Spatially restricted and ontogenically distinct hepatic macrophages are required for tissue repair. Immunity. (2025) 58:362–380.e10. doi: 10.1016/j.immuni.2025.01.002
102. Shi S, Zhou Y, Zhang H, and Zhang J. TREM2 in MASH: integrating lipid metabolism and immune response. Front Immunol. (2025) 16:1604837. doi: 10.3389/fimmu.2025.1604837
103. Palma A. The landscape of SPP1+ Macrophages across tissues and diseases: A comprehensive review. Immunology. (2025). doi: 10.1111/imm.13952
104. Fan G, Xie T, Li L, Tang L, Han X, and Shi Y. Single-cell and spatial analyses revealed the co-location of cancer stem cells and SPP1+ macrophage in hypoxic region that determines the poor prognosis in hepatocellular carcinoma. NPJ Precis Oncol. (2024) 8:75. doi: 10.1038/s41698-024-00564-3
105. Wang J, Wang H, Yang W, Zhao D, Liu D, Tang L, et al. GPNMB regulates the differentiation and transformation of monocyte-derived macrophages during MASLD. Int Immunopharmacology. (2025) 154:114554. doi: 10.1016/j.intimp.2025.114554
106. Kui L, Kim AD, Onyuru J, Hoffman HM, and Feldstein AE. BRP39 regulates neutrophil recruitment in NLRP3 inflammasome-induced liver inflammation. Cell Mol Gastroenterol Hepatology. (2024) 17:481–97. doi: 10.1016/j.jcmgh.2023.12.002
107. Cui H, Banerjee S, Xie N, Hussain M, Jaiswal A, Liu H, et al. TREM2 promotes lung fibrosis via controlling alveolar macrophage survival and pro-fibrotic activity. Nat Commun. (2025) 16:1761. doi: 10.1038/s41467-025-57024-0
108. Gong S, Zhai M, Shi J, Yu G, Lei Z, Shi Y, et al. TREM2 macrophage promotes cardiac repair in myocardial infarction by reprogramming metabolism via SLC25A53. Cell Death Differentiation. (2024) 31:239–53. doi: 10.1038/s41418-023-01252-8
109. Shi H, Wang X, Li F, and Gerlach BD. CD47-SIRPα axis blockade in NASH promotes necroptotic hepatocyte clearance by liver macrophages and decreases hepatic fibrosis. Sci Transl Med. (2022) 14:eabp8309. doi: 10.1126/scitranslmed.abp8309
110. Li S, Zhou B, Xue M, Zhu J, Tong G, Fan J, et al. Macrophage-specific FGF12 promotes liver fibrosis progression in mice. Hepatology. (2023) 77:816–33. doi: 10.1002/hep.32640
111. Li C, Fang L, Su X, Zhang J, Xiong H, Yu H, et al. Macrophage miR-4524a-5p/TBP promotes β-TrCP -TIM3 complex activation and TGFβ release and aggravates NAFLD-associated fibrosis. Cell Death Disease. (2025) 16:315. doi: 10.1038/s41419-025-07574-4
112. Guan D, Huang P, Liu X, Li Q, Zhang X, Liu N, et al. Deficiency of myeloid NPC1 exacerbates liver injury and fibrosis by impairing macrophage efferocytosis. J Advanced Res. (2025) 72:213–27. doi: 10.1016/j.jare.2024.11.020
113. Wang X, de Carvalho Ribeiro M, Iracheta-Vellve A, Lowe P, Ambade A, Satishchandran A, et al. Macrophage-specific hypoxia-inducible factor-1α Contributes to impaired autophagic flux in nonalcoholic steatohepatitis. Hepatology. (2019) 69:545–63. doi: 10.1002/hep.30215
114. Choe SS SK, Ka S, Lee YK, Chun JS, and Kim JB. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes. (2014) 63:3359–71. doi: 10.2337/db13-1965
115. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature. (2007) 447:1116–20. doi: 10.1038/nature05894
116. Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, et al. Adipocyte-derived th2 cytokines and myeloid PPARδ Regulate macrophage polarization and insulin sensitivity. Cell Metab. (2008) 7:485–95. doi: 10.1016/j.cmet.2008.04.002
117. Qing J, Ren Y, Zhang Y, Yan M, Zhang H, Wu D, et al. Dopamine receptor D2 antagonism normalizes profibrotic macrophage-endothelial crosstalk in non-alcoholic steatohepatitis. J Hepatology. (2022) 76:394–406. doi: 10.1016/j.jhep.2021.09.032
118. Åberg F, Jiang ZG, Cortez-Pinto H, and Männistö V. Alcohol-associated liver disease—Global epidemiology. Hepatology. (2024) 80:1307–22. doi: 10.1097/HEP.0000000000000899
119. Seitz HK, Bataller R, Cortez-Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver disease. Nat Rev Dis Primers. (2018) 4:16. doi: 10.1038/s41572-018-0014-7
120. Kwon H-J, Won Y-S, Park O, Chang B, Duryee MJ, Thiele GE, et al. Aldehyde dehydrogenase 2 deficiency ameliorates alcoholic fatty liver but worsens liver inflammation and fibrosis in mice. Hepatology. (2014) 60:146–57. doi: 10.1002/hep.27036
121. Gao B and Bataller R. Liver fibrosis in alcoholic liver disease. Semin Liver Disease. (2015) 35:146–56. doi: 10.1055/s-0035-1550054
122. Slevin E, Baiocchi L, Wu N, Ekser B, Sato K, Lin E, et al. Kupffer cells. Am J Pathology. (2020) 190:2185–93. doi: 10.1016/j.ajpath.2020.08.014
123. Liang S, Zhong Z, Kim SY, Uchiyama R, Roh YS, Matsushita H, et al. Murine macrophage autophagy protects against alcohol-induced liver injury by degrading interferon regulatory factor 1 (IRF1) and removing damaged mitochondria. J Biol Chem. (2019) 294:12359–69. doi: 10.1074/jbc.RA119.007409
124. Kumar V, Mansfield J, Fan R, MacLean A, Li J, and Mohan M. miR-130a and miR-212 Disrupt the Intestinal Epithelial Barrier through Modulation of PPARγ and Occludin Expression in Chronic Simian Immunodeficiency Virus–Infected Rhesus Macaques. J Immunol. (2018) 200:2677–89. doi: 10.4049/jimmunol.1701148
125. Inokuchi S, Tsukamoto H, Park E, Liu Z-X, Brenner DA, and Seki E. Toll-like receptor 4 mediates alcohol-induced steatohepatitis through bone marrow-derived and endogenous liver cells in mice. Alcoholism: Clin Exp Res. (2011) 35:1509–18. doi: 10.1111/j.1530-0277.2011.01487.x
126. Affò S, Morales-Ibanez O, Rodrigo-Torres D, Altamirano J, Blaya D, Dapito DH, et al. CCL20 mediates lipopolysaccharide induced liver injury and is a potential driver of inflammation and fibrosis in alcoholic hepatitis. Gut. (2014) 63:1782–92. doi: 10.1136/gutjnl-2013-306098
127. Xu J, Chi F, Guo T, Punj V, Lee WNP, French SW, et al. NOTCH reprograms mitochondrial metabolism for proinflammatory macrophage activation. J Clin Invest. (2015) 125:1579–90. doi: 10.1172/JCI76468
128. Xiang Z, Li J, Lu D, Wei X, and Xu X. Advances in multi-omics research on viral hepatitis. Front Microbiol. (2022) 13. doi: 10.3389/fmicb.2022.987324
129. Viral hepatitis elimination — time to act. Nat Rev Gastroenterol Hepatol. (2024) 21:529–. doi: 10.1038/s41575-024-00963-8
130. Li Y, Li S, Duan X, Yang C, Xu M, and Chen L. Macrophage phenotypes and hepatitis B virus infection. J Clin Trans Hepatology. (2020) 8:1–8. doi: 10.14218/JCTH.2020.00046
131. Dou L, Shi X, He X, and Gao Y. Macrophage phenotype and function in liver disorder. Front Immunol. (2020) 10:3112. doi: 10.3389/fimmu.2019.03112
132. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. J Hepatology. (2017) 66:693–702. doi: 10.1016/j.jhep.2016.12.018
133. Song X, Gao X, Wang Y, Raja R, Zhang Y, Yang S, et al. HCV core protein induces chemokine CCL2 and CXCL10 expression through NF-κB signaling pathway in macrophages. Front Immunol. (2021) 12:654998. doi: 10.3389/fimmu.2021.654998
134. Duriez M, Mandouri Y, Lekbaby B, Wang H, Schnuriger A, Redelsperger F, et al. Alternative splicing of hepatitis B virus: A novel virus/host interaction altering liver immunity. J Hepatology. (2017) 67:687–99. doi: 10.1016/j.jhep.2017.05.025
135. Read SA, Wijaya R, Ramezani-Moghadam M, Tay E, Schibeci S, Liddle C, et al. Macrophage coordination of the interferon lambda immune response. Front Immunol. (2019) 10:2674. doi: 10.3389/fimmu.2019.02674
136. Eslam M, McLeod D, Kelaeng KS, Mangia A, Berg T, Thabet K, et al. IFN-λ3, not IFN-λ4, likely mediates IFNL3–IFNL4 haplotype–dependent hepatic inflammation and fibrosis. Nat Genet. (2017) 49:795–800. doi: 10.1038/ng.3836
137. Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebossé F, Fresquet J, et al. Hepatitis B virus-induced modulation of liver macrophage function promotes hepatocyte infection. J Hepatology. (2019) 71:1086–98. doi: 10.1016/j.jhep.2019.06.032
138. Li H, Zheng H-W, Chen H, Xing Z-Z, You H, Cong M, et al. Hepatitis B virus particles preferably induce Kupffer cells to produce TGF-β1 over pro-inflammatory cytokines. Digestive Liver Disease. (2012) 44:328–33. doi: 10.1016/j.dld.2011.11.005
139. Wang S, Chen Z, Hu C, Qian F, Cheng Y, Wu M, et al. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand–induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J Immunol. (2013) 190:5142–51. doi: 10.4049/jimmunol.1201625
140. Xie X, Lv H, Liu C, Su X, Yu Z, Song S, et al. HBeAg mediates inflammatory functions of macrophages by TLR2 contributing to hepatic fibrosis. BMC Med. (2021) 19:247. doi: 10.1186/s12916-021-02085-3
141. Liu X-Q, Wei R-R, Wang C-C, Chen L-Y, Liu C, and Liu K. Relationship between PK2 and number of Kupffer cells duringthe progression of liver fibrosis in patients with HBV. Turkish J Med Sci. (2018) 48:52–61. doi: 10.3906/sag-1705-32
142. Li Y, He M, Wang Z, Duan Z, Guo Z, Wang Z, et al. STING signaling activation inhibits HBV replication and attenuates the severity of liver injury and HBV-induced fibrosis. Cell Mol Immunol. (2021) 19:92–107. doi: 10.1038/s41423-021-00801-w
143. Lu Y, Yang A, Quan C, Pan Y, Zhang H, Li Y, et al. A single-cell atlas of the multicellular ecosystem of primary and metastatic hepatocellular carcinoma. Nat Commun. (2022) 13:4594. doi: 10.1038/s41467-022-32283-3
144. Jansen PLM, Ghallab A, Vartak N, Reif R, Schaap FG, Hampe J, et al. The ascending pathophysiology of cholestatic liver disease. Hepatology. (2017) 65:722–38. doi: 10.1002/hep.28965
145. Azad AI, Krishnan A, Troop L, Li Y, Katsumi T, Pavelko K, et al. Targeted apoptosis of ductular reactive cells reduces hepatic fibrosis in a mouse model of cholestasis. Hepatology. (2020) 72:1013–28. doi: 10.1002/hep.31211
146. Li X, Liu R, Wang Y, Zhu W, Zhao D, Wang X, et al. Cholangiocyte-Derived Exosomal lncRNA H19 Promotes Macrophage Activation and Hepatic Inflammation under Cholestatic Conditions. Cells. (2020) 9:190. doi: 10.3390/cells9010190
147. De Muynck K, Heyerick L, De Ponti FF, Vanderborght B, Meese T, Van Campenhout S, et al. Osteopontin characterizes bile duct–associated macrophages and correlates with liver fibrosis severity in primary sclerosing cholangitis. Hepatology. (2024) 79:269–88. doi: 10.1097/HEP.0000000000000557
148. Song G, Shi Y, Meng L, Ma J, Huang S, Zhang J, et al. Single-cell transcriptomic analysis suggests two molecularly distinct subtypes of intrahepatic cholangiocarcinoma. Nat Commun. (2022) 13:2848. doi: 10.1038/s41467-022-30599-8
149. Reuveni D, Brezis MR, Brazowski E, Vinestock P, Leung PSC, Thakker P, et al. Interleukin 23 produced by hepatic monocyte-derived macrophages is essential for the development of murine primary biliary cholangitis. Front Immunol. (2021) 12:718841. doi: 10.3389/fimmu.2021.718841
150. Fu H-Y, Bao W-M, Yang C-X, Lai W-J, Xu J-M, Yu H-Y, et al. Kupffer cells regulate natural killer cells via the NK group 2, member D (NKG2D)/retinoic acid early inducible-1 (RAE-1) interaction and cytokines in a primary biliary cholangitis mouse model. Med Sci Monitor. (2020) 26:e923726. doi: 10.12659/MSM.923726
151. Ravichandran G, Neumann K, Berkhout LK, Weidemann S, Langeneckert AE, Schwinge D, et al. Interferon-γ-dependent immune responses contribute to the pathogenesis of sclerosing cholangitis in mice. J Hepatology. (2019) 71:773–82. doi: 10.1016/j.jhep.2019.05.023
152. García-Ruiz I, Blanes Ruiz N, Rada P, Pardo V, Ruiz L, Blas-García A, et al. Protein tyrosine phosphatase 1b deficiency protects against hepatic fibrosis by modulating nadph oxidases. Redox Biol. (2019) 26:101263. doi: 10.1016/j.redox.2019.101263
153. Allina J, Stanca CM, Garber J, Hu B, Sautes-Fridman C, Bach N, et al. Anti-CD16 autoantibodies and delayed phagocytosis of apoptotic cells in primary biliary cirrhosis. J Autoimmunity. (2008) 30:238–45. doi: 10.1016/j.jaut.2007.10.003
154. Greenman R, Segal-Salto M, Barashi N, Hay O, Katav A, Levi O, et al. CCL24 regulates biliary inflammation and fibrosis in primary sclerosing cholangitis. JCI Insight. (2023) 8:e162270. doi: 10.1172/jci.insight.162270
155. van der Heide D, Weiskirchen R, and Bansal R. Therapeutic targeting of hepatic macrophages for the treatment of liver diseases. Front Immunol. (2019) 10:2852. doi: 10.3389/fimmu.2019.02852
156. Wu X, Dong L, Lin X, and Li J. Relevance of the NLRP3 inflammasome in the pathogenesis of chronic liver disease. Front Immunol. (2017) 8:1728. doi: 10.3389/fimmu.2017.01728
157. Sumida Y and Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterology. (2017) 53:362–76. doi: 10.1007/s00535-017-1415-1
158. Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatology. (2020) 72:816–27. doi: 10.1016/j.jhep.2019.11.024
159. Yu Y and Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J Biol Chem. (2011) 286:15413–27. doi: 10.1074/jbc.M111.225946
160. Harrison SA, Wong VW, Okanoue T, Bzowej N, Vuppalanchi R, Younes Z, et al. Selonsertib for patients with bridging fibrosis or compensated cirrhosis due to NASH: Results from randomized phase III STELLAR trials. J Hepatology. (2020) 73:26–39. doi: 10.1016/j.jhep.2020.02.027
161. Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. (1999) 284:1365–8. doi: 10.1126/science.284.5418.1365
162. Anderson KM, Handler DA, and Gayer CP. Farnesoid X receptor-mediated changes to macrophage cytokine expression. J Am Coll Surgeons. (2021) 233:S185. doi: 10.1016/j.jamcollsurg.2021.07.375
163. Verbeke L, Mannaerts I, Schierwagen R, Govaere O, Klein S, Vander Elst I, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep. (2016) 6:33453. doi: 10.1038/srep33453
164. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. (2019) 394:2184–96. doi: 10.1016/S0140-6736(19)33041-7
165. Kholodenko IV and Yarygin KN. Hepatic macrophages as targets for the MSC-based cell therapy in non-alcoholic steatohepatitis. Biomedicines. (2023) 11:3056. doi: 10.3390/biomedicines11113056
166. Saldarriaga OA, Wanninger TG, Arroyave E, Gosnell J, Krishnan S, Oneka M, et al. Heterogeneity in intrahepatic macrophage populations and druggable target expression in patients with steatotic liver disease-related fibrosis. JHEP Rep. (2024) 6:100958. doi: 10.1016/j.jhepr.2023.100958
167. Sookoian SC, Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, et al. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One. (2016) 11:e0158156. doi: 10.1371/journal.pone.0158156
168. Anstee QM, Neuschwander-Tetri BA, Wai-Sun Wong V, Abdelmalek MF, Rodriguez-Araujo G, Landgren H, et al. Cenicriviroc lacked efficacy to treat liver fibrosis in nonalcoholic steatohepatitis: AURORA phase III randomized study. Clin Gastroenterol Hepatology. (2024) 22:124–134.e1. doi: 10.1016/j.cgh.2023.04.003
169. Puengel T, De Vos S, Hundertmark J, Kohlhepp M, Guldiken N, Pujuguet P, et al. The medium-chain fatty acid receptor GPR84 mediates myeloid cell infiltration promoting steatohepatitis and fibrosis. J Clin Med. (2020) 9:1140. doi: 10.3390/jcm9041140
170. Chu P, Nakamoto N, Ebinuma H, Usui S, Saeki K, Matsumoto A, et al. C-C motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology. (2013) 58:337–50. doi: 10.1002/hep.26351
171. Morikawa R, Nakamoto N, Amiya T, P-s C, Koda Y, Teratani T, et al. Role of CC chemokine receptor 9 in the progression of murine and human non-alcoholic steatohepatitis. J Hepatology. (2021) 74:511–21. doi: 10.1016/j.jhep.2020.09.033
172. Clugston RD. β-cryptoxanthin and fatty liver disease: new insights. Hepatobiliary Surg Nutr. (2023) 12:450–2. doi: 10.21037/hbsn-23-201
173. Ota T, Kaneko S, Ogawa K, Sugiura M, Kobori M, Nagata N, et al. Prevention and reversal of lipotoxicity-induced hepatic insulin resistance and steatohepatitis in mice by an antioxidant carotenoid, β-cryptoxanthin. Endocrinology. (2015) 156:987–99. doi: 10.1210/en.2014-1776
174. Ni Y, Nagashimada M, Zhuge F, Zhan L, Nagata N, Tsutsui A, et al. Astaxanthin prevents and reverses diet-induced insulin resistance and steatohepatitis in mice: A comparison with vitamin E. Sci Rep. (2015) 5:17192. doi: 10.1038/srep17192
175. Kitade H, Chen G, Ni Y, and Ota T. Nonalcoholic fatty liver disease and insulin resistance: new insights and potential new treatments. Nutrients. (2017) 9:387. doi: 10.3390/nu9040387
176. Kim B, Farruggia C, Ku CS, Pham TX, Yang Y, Bae M, et al. Astaxanthin inhibits inflammation and fibrosis in the liver and adipose tissue of mouse models of diet-induced obesity and nonalcoholic steatohepatitis. J Nutr Biochem. (2017) 43:27–35. doi: 10.1016/j.jnutbio.2016.01.006
177. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. (2016) 387:679–90. doi: 10.1016/S0140-6736(15)00803-X
178. Li Z, Feng P, Zhao Z, Zhu W, Gong J, and Du H. Liraglutide protects against inflammatory stress in non-alcoholic fatty liver by modulating Kupffer cells M2 polarization via cAMP-PKA-STAT3 signaling pathway. Biochem Biophys Res Commun. (2019) 510:20–6. doi: 10.1016/j.bbrc.2018.12.149
179. Wang Y, Huang S, Kong W, Wu C, Zeng T, Xie S, et al. Corilagin alleviates liver fibrosis in zebrafish and mice by repressing IDO1-mediated M2 macrophage repolarization. Phytomedicine. (2023) 119:155016. doi: 10.1016/j.phymed.2023.155016
180. Feige JN, Gelman L, Michalik L, Desvergne B, and Wahli W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog Lipid Res. (2006) 45:120–59. doi: 10.1016/j.plipres.2005.12.002
181. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al. Elafibranor, an agonist of the peroxisome proliferator–activated receptor–α and –δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. (2016) 150:1147–59. doi: 10.1053/j.gastro.2016.01.038
182. Lefere S, Puengel T, Hundertmark J, Penners C, Frank AK, Guillot A, et al. Differential effects of selective- and pan-PPAR agonists on experimental steatohepatitis and hepatic macrophages. J Hepatology. (2020) 73:757–70. doi: 10.1016/j.jhep.2020.04.025
183. Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, et al. A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. New Engl J Med. (2021) 385:1547–58. doi: 10.1056/NEJMoa2036205
184. Mackinnon AC, Tonev D, Jacoby B, Pinzani M, and Slack RJ. Galectin-3: therapeutic targeting in liver disease. Expert Opin Ther Targets. (2023) 27:779–91. doi: 10.1080/14728222.2023.2258280
185. Jeftic I, Jovicic N, Pantic J, Arsenijevic N, Lukic ML, and Pejnovic N. Galectin-3 ablation enhances liver steatosis, but attenuates inflammation and IL-33-dependent fibrosis in obesogenic mouse model of nonalcoholic steatohepatitis. Mol Med. (2015) 21:453–65. doi: 10.2119/molmed.2014.00178
186. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, et al. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. (2020) 158:1334–1345.e5. doi: 10.1053/j.gastro.2019.11.296
187. Biagioli M, Marchianò S, Di Giorgio C, Bordoni M, Urbani G, Bellini R, et al. Activation of GPBAR1 attenuates vascular inflammation and atherosclerosis in a mouse model of NAFLD-related cardiovascular disease. Biochem Pharmacol. (2023) 218:115900. doi: 10.1016/j.bcp.2023.115900
188. Zhuang L, Jia N, Zhang L, Zhang Q, Antwi SO, Sartorius K, et al. Gpbar-1/cAMP/PKA signaling mitigates macrophage-mediated acute cholestatic liver injury via antagonizing NLRP3-ASC inflammasome. Biochim Biophys Acta (BBA) - Mol Basis Disease. (2024) 1870:167266. doi: 10.1016/j.bbadis.2024.167266
189. Di Giorgio C, Urbani G, Marchianò S, Biagioli M, Bordoni M, Bellini R, et al. Liver GPBAR1 associates with immune dysfunction in primary sclerosing cholangitis and its activation attenuates cholestasis in abcb4–/– mice. Liver Int. (2025) 45:e16235. doi: 10.1111/liv.16235
190. Hu C, Zhao L, Zhang L, Bao Q, and Li L. Mesenchymal stem cell-based cell-free strategies: safe and effective treatments for liver injury. Stem Cell Res Ther. (2020) 11:377. doi: 10.1186/s13287-020-01895-1
191. Pinheiro D, Dias I, Ribeiro Silva K, Stumbo AC, Thole A, Cortez E, et al. Mechanisms underlying cell therapy in liver fibrosis: an overview. Cells. (2019) 8:1339. doi: 10.3390/cells8111339
192. Eom YW, Shim KY, and Baik SK. Mesenchymal stem cell therapy for liver fibrosis. Korean J Internal Med. (2015) 30:580–9. doi: 10.3904/kjim.2015.30.5.580
193. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. (2011) 53:2003–15. doi: 10.1002/hep.24315
194. Ma P, Gao C, Yi J, Zhao J, Liang S, Zhao Y, et al. Cytotherapy with M1-polarized macrophages ameliorates liver fibrosis by modulating immune microenvironment in mice. J Hepatology. (2017) 67:770–9. doi: 10.1016/j.jhep.2017.05.022
195. Miao X, Leng X, and Zhang Q. The current state of nanoparticle-induced macrophage polarization and reprogramming research. Int J Mol Sci. (2017) 18:336. doi: 10.3390/ijms18020336
196. Peng W, Cheng S, Bao Z, Wang Y, Zhou W, Wang J, et al. Advances in the research of nanodrug delivery system for targeted treatment of liver fibrosis. Biomedicine Pharmacotherapy. (2021) 137:111342. doi: 10.1016/j.biopha.2021.111342
197. Lin L, Gong H, Li R, Huang J, Cai M, Lan T, et al. Nanodrug with ROS and pH Dual-Sensitivity Ameliorates Liver Fibrosis via Multicellular Regulation. Advanced Science. (2020) 7:1903138. doi: 10.1002/advs.201903138
Keywords: macrophage, liver fibrosis, heterogeneity and plasticity, mechanisms, therapeutic approaches
Citation: Wang W, Li S, Liu Y, Ding X, Yang Y, Chen S, Cao J, Tacke F, Dong W and Lan T (2025) Macrophage heterogeneity in liver fibrosis. Front. Immunol. 16:1639455. doi: 10.3389/fimmu.2025.1639455
Received: 02 June 2025; Accepted: 08 August 2025;
Published: 04 September 2025.
Edited by:
Lara Campana, Resolution Therapeutics Ltd, United KingdomReviewed by:
Alessandro Palma, Sapienza University of Rome, ItalyFederico De Ponti, Ghent University, Belgium
Copyright © 2025 Wang, Li, Liu, Ding, Yang, Chen, Cao, Tacke, Dong and Lan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tian Lan, bGFudGlhbkBocmJtdS5lZHUuY24=; Wei Dong, ZG9uZ3dlaTIwMjQxMkAxNjMuY29t
†These authors have contributed equally to this work and share first authorship