 Yangyang Zhao
Yangyang Zhao Chunlei Wu2†
Chunlei Wu2†- 1Department of Blood Transfusion, Affiliated Hospital of North Sichuan Medical College, Nanchong, Sichuan, China
- 2Department of Blood Transfusion, Beijing Anzhen Nanchong Hospital of Capital Medical University & Nanchong Central Hospital, Nanchong, Sichuan, China
- 3Department of Medical Laboratory, Chengdu Qingbaijiang District People’s Hospital, Chengdu, Sichuan, China
- 4Department of Cardiovascular Surgery, Affiliated Hospital of North Sichuan Medical College, Nanchong, Sichuan, China
- 5Graduate School of Comprehensive Human Science, University of Tsukuba, Tsukuba, Japan
Circulating cf-mtDNA has emerged as a dual-functional entity in human pathophysiology, serving not only as a disease biomarker but also as a potent innate immune activator through its molecular pattern recognition. Extracellular mtDNA engages PRRs, triggering dysregulated pro-inflammatory signaling in multiple cell lineages. Elevated mtDNA in circulation correlates with pathogenesis of autoimmune disorders, infectious diseases, critical illnesses, neurological disorders, and hematological abnormalities. Therapeutic strategies combining mtDNA monitoring with inhibitors targeting its release mechanisms and downstream pathways offer novel immunomodulatory strategies. This review systematically examines the therapeutic nexus of blood-derived mtDNA in immune activation and disease progression. Here we aim to elucidate the function of mtDNA in disease pathobiology while highlighting mitochondria’s central position in human systemic homeostasis.
1 Introduction
Mitochondria serve as the primary sites for energy production in eukaryotic cells and crucial reservoirs for effector molecules regulating fundamental cellular and physiological processes (1, 2). Various components of mitochondria and their metabolic byproducts released from damaged mitochondria exhibit immunogenicity, eliciting immune responses characterized by the presence of damage-associated molecular patterns (DAMPs) (3, 4). Specifically, mitochondrial DAMPs (mtDAMPs) encompass entities such as mitochondrial DNA (mtDNA), cardiolipin, N-formyl peptides (NFP), reactive oxygen species (ROS), adenosine triphosphate (ATP), and mitochondrial transcription factor A (TFAM) (5, 6). Recently, mtDNA has emerged as one of the most extensively researched DAMPs.
Mitochondria evolved from bacterial ancestors and retained a circular chromosome termed mtDNA. In vertebrates, this maternally inherited circular mtDNA is capable of self-replication (1, 7). It encodes 11 subunits of the electron transport chain (ETC) and 2 subunits of ATP synthase, which critical for the oxidative phosphorylation (OXPHOS) (8, 9). Circulating cell-free DNA (cfDNA) has emerged as a novel biomarker with diverse applications across various fields, including oncology, toxicology, cardiovascular diseases (CVD), and organ transplantation (10–14). The primary sources of circulating cfDNA are nuclear DNA (nDNA) and mtDNA (15). Healthy individuals possess approximately 50,000 times more copies of mitochondrial genomes in plasma compared to nuclear genomes, constituting 10% to 25% total circulating cfDNA (16). Circulating mtDNA demonstrates superior immunogenicity compared to Circulating nDNA (17, 18). While optimal mtDNA can maintain mitochondrial genome stability and facilitating its repair mechanisms, supraphysiological concentrations induce cellular damage and trigger immunity (19). As noted by Trumpff et al., the enhanced stability of cf-mtDNA as a biomarker stems from dual protective mechanisms that physical shielding via encapsulation within intact mitochondrial membranes or lipid vesicles, or through protein binding (e.g., TFAM-DNA adduct) (19). And intrinsic resistance to circulating nucleases conferred by its circular double-stranded topology and lack of histone association (20).
mtDNA exhibits a dual role as a tissue damage biomarker and DAMP, activating innate immunity via pattern recognition receptors (PRRs). Current research focuses on enhancing our understanding of the regulation of mtDNA in circulation and its influence on disease severity. A critical gap remains in pleiotropic injury mechanisms of systematizing mtDNA. Nevertheless, a comprehensive review of the mechanisms through which mtDNA induces damage have yet to be thoroughly examined.
2 Circulating mtDNA: from intracellular transport to extracellular release
2.1 mtDNA is released from mitochondria into the cytoplasm
Structurally, mitochondrial compartmentalization relies on its dual-membrane architecture—an inner mitochondrial membrane (IMM) and an outer mitochondrial membrane (OMM)—that sequester mtDAMPs from cytosolic PRRs (21). mtDNA release from the mitochondrial matrix into the cytosol must traverse both membranes (22) (Figure 1). Current researches indicate several major pathways regulate in the permeability of mitochondrial membranes, including the mitochondrial permeability transition pore (mPTP), Bcl-2-associated X protein (BAX)/Bcl-2 homologous antagonist/killer (BAK), voltage-dependent anion channel (VDAC), and gasdermin D (GSDMD). Activation of these channels induces mitochondrial depolarization, disruption of OXPHOS system, mitochondrial membrane permeabilization, and mtDNA extrusion (23–25). BAX and BAK are proapoptotic pore-forming proteins that mediate mitochondrial outer membrane permeabilization (MOMP), a process enabling release of cytochrome c and mtDNA to trigger apoptosis while impairing mitochondrial respiration (5, 25). The VDAC, a β-barrel membrane protein located at the OMM, involves the exchange of materials between mitochondria and the cytoplasm, maintains intracellular calcium homeostasis, and regulates apoptosis and necrosis (26, 27). During pyroptosis, caspase-mediated cleavage generates GSDMD-N-terminal fragments (GSDMD-NT) that target cardiolipin-containing mitochondrial membranes. This interaction disrupts mitochondrial phospholipid bilayers, facilitating mtDAMPs release prior to plasma membrane rupture in a cell lysis-independent manner (23, 28, 29). Furthermore, pyroptosis disturbs mitochondrial homeostasis and induces MOMP through membrane depolarization, ionic imbalance, and suppressed mitophagy (5). The mPTP, a non-specific channel located in the IMM, activates under stress conditions including Ca2+ influx, oxidative stress, BAX/BAK oligomerization, and VDAC (25, 30, 31) (Figure 1).
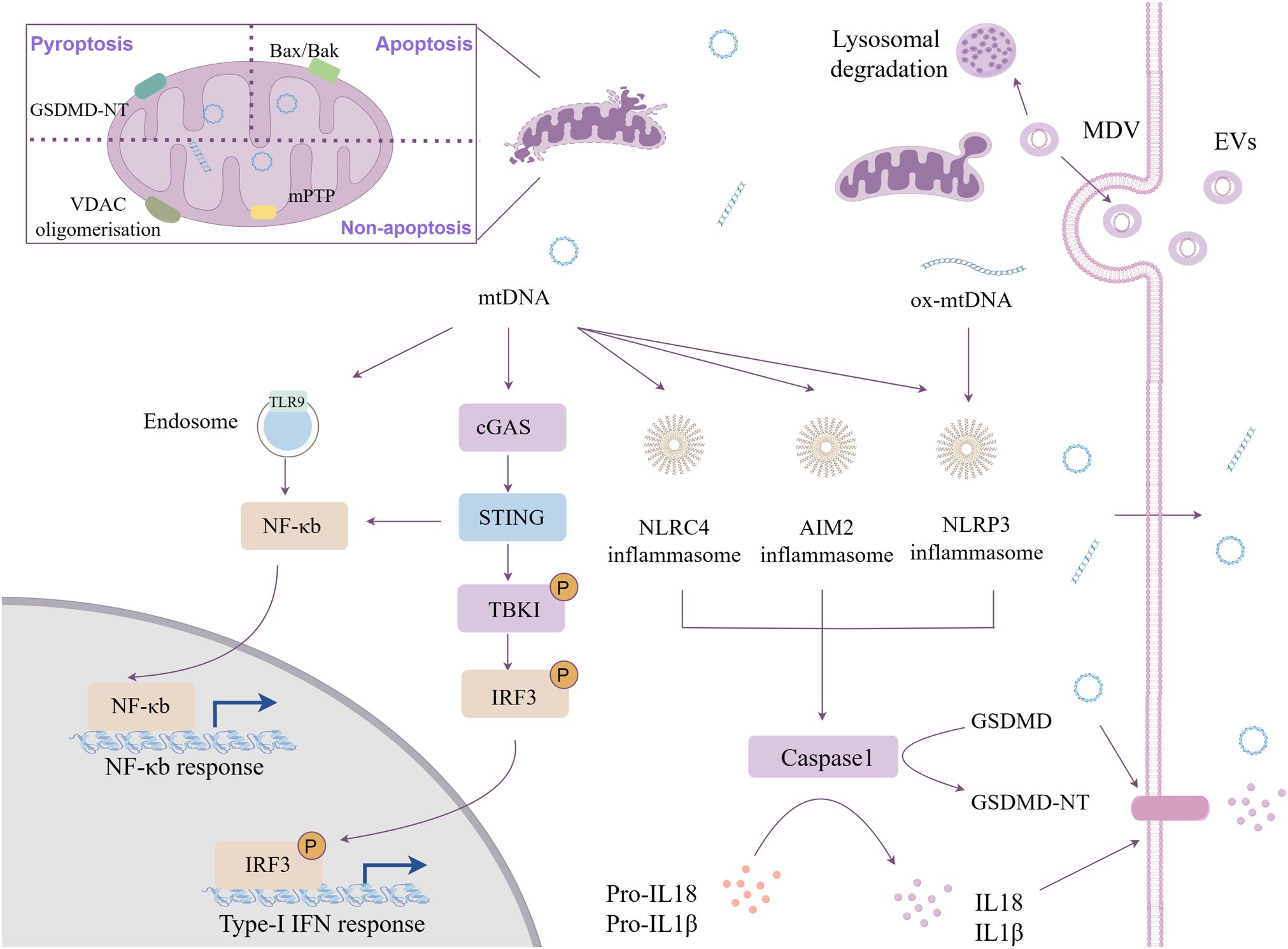
Figure 1. Cytoplasmic mtDNA mediated innate inflammation. The release of mtDNA into the cytoplasm occurs through the mitochondrial inner and outer membrane permeability transition pore, activating multiple pattern recognition receptors. Besides, mtDNA be packaged into mitochondrial-derived vesicles and transferred to recipient cells through extracellular vesicle mechanism.
2.2 Cytosolic mtDNA engages multiple pattern recognition receptors
Augmented immunogenicity of mtDNA attributable to its unique molecular architecture: high copy number, circular hypomethylated structure, and lacking free DNA termini (2, 29, 32). Simultaneously, these characteristics render mtDNA readily identifiable and capable of binding, which can elicit substantial biological effects. It is recognized by several PRRs: cyclic guanosine monophosphate adenosine monophosphate (cGAMP) synthase (cGAS), nucleotide oligomerization domain (NOD)-like receptors (NLRs), and Toll-like receptor 9 (TLR9) (33–35) (Figure 1).
cGAS: cGAS functions as a crucial cytosolic DNA sensor, activated in sequence-agnostic through hybridization with double-stranded DNA (dsDNA) (36). cGAS-mtDNA binding catalyzes the synthesis of the second messenger cyclic GMP-AMP (cGAMP) (37). This molecule subsequently regulates the downstream stimulator of interferon genes (STING) pathway, wherein STING1 activation ultimately recruits TANK-binding kinase 1 (TBK1) (37, 38). Then, TBK1 catalyzes the phosphorylation of interferon regulatory factor 3 (IRF3) and promotes nuclear factor kappa-B (NF-κB) signaling, leading to the production of type I interferons (IFNs) and other pro-inflammatory cytokines (37–39).
Inflammasomes: Beyond cGAS-mediated pathways, cytosolic mtDNA engages absent in melanoma 2 (AIM2) via C-terminal hematopoietic interferon-inducible nuclear (HIN) domain and NLRP3/NLRC4 inflammasomes, facilitating the secretion of proinflammatory cytokines (29, 40, 41).
TLR9: mtDNA retains evolutionarily conserved CpG motifs resembling bacterial nucleic acids, serving as potent TLR9 agonists through their hypomethylated DNA architecture (42). Cytosolic mtDNA containing CpG motifs will stimulate endolysosomal TLR-9 to recruit myeloid differentiation primary response 88 (MyD88), activating transcription factors such as NF-κB, driving pro-inflammatory cytokine and chemokine cascades (43, 44).
Transcriptional factor a mitochondrial (TFAM)-bound mtDNA resists ROS-mediated oxidation through its compact nucleoid structure within mitochondria (45, 46). In contrast, newly synthesized naked mtDNA, lacking TFAM shielding and other protective mechanisms, is prone to oxidative modification by OXPHOS-derived mtROS due to spatial proximity to the OXPHOS (30, 47). This susceptibility can result in the leakage of mtDNA into the cytoplasm (48, 49). Unlike nuclear DNA, mtDNA lacks histone protection and effective DNA repair mechanisms, rendering it more vulnerable to damage (50). The oxidized form of mtDNA (ox-mtDNA) acts as a potent DAMP that drives sustained pattern recognition receptor activation and amplifies sterile inflammation (30). However, the molecular mechanisms underlying mtDNA oxidation and fragmentation under various stress conditions require further investigation. Subsequent research demonstrates that ox-mtDNA released into the cytosol during mitochondrial dysfunction serves as an effective activator of the NLRP3 inflammasome and TLR9 (51, 52). The NLRP3 inflammasome appears to preferentially respond to oxidized DNA, while AIM2 is proposed to primarily recognize non-oxidized DNA (24, 47). Topoisomerase deficiency, which serve as genetic and pharmacological triggers of mitochondrial genome instability, induces left-handed Z-form mtDNA accumulation (53). This form of mtDNA is more readily recognized by the nucleic acid sensor Z-DNA binding protein 1 (ZBP1) (53). Recent studies reveal ZBP1 coordinates with cGAS and receptor-interacting protein kinase 1/3 (RIPK1/3) to sustain the type I interferon (IFN) signaling pathway activated by mtDNA instability (53, 54). Cytosolic DNA sensors differentiate mtDNA types. How distinct mtDNA forms engage specific PRRs to drive unique inflammatory responses remains unclear.
2.3 mtDNA is released from the cytoplasm to the extracellular milieu
Under pathological conditions, mtDNA escapes into the cytosol and extracellular space through three primary pathways: 1) regulated cell death (RCD): cfDNA is released into the circulation after apoptosis, necroptosis, and pyroptosis (28, 55, 56). Both necroptosis and pyroptosis result in the rupture of the cytoplasmic membrane; necroptosis is mediated by the oligomerization of mixed lineage kinase domain-like proteins (MLKLs), while pyroptosis is mediated by GSDMD (28, 56). Mitochondrial permeability transition (MPT)-driven necrosis is a necrotic variant of regulated cell death that can eventually result in the complete disintegration of mitochondrial membranes (57). This process is characterized by a rapid depletion of ATP and oxidative damage to macromolecules, occurring independently of caspase activation (58); 2) the efflux of mtDAMPs due to defects in mitochondrial quality control (MQC) (59); and 3) the active secretion of mitochondrial-derived vesicles (MDVs) (55, 60). Additionally, the leakage of mtDNA into the cytosol, whether in circular or fragmented form, can result from mitochondrial damage caused by oxidative stress and damage to membrane structures (61). The accumulation of mtDNA in the cytosol activates intracellular inflammatory signaling pathways, which subsequently alter the physiological state of cells and cause cell death (60). This process further contributes to the release of mtDNA into the extracellular environment. Mitophagy as part of the MQC could inhibit mtDNA leakage by facilitating the disposal of dysfunctional mitochondria and limit potential pro-inflammatory effect (60). Following the activation of PTEN-induced putative kinase 1 (PINK1)- and parkin (PRKN)-mediated mitophagy, the ubiquitination of mitochondrial proteins that induce oxidative damage takes place. Subsequently, these ubiquitinated mitochondria are engulfed by autophagosomes, which then coalesce with lysosomes to facilitate the systematic degradation and recycling of mitochondrial components (62). Sublethal MOMP activates mitophagy, enabling the processing of dysfunctional mitochondria through lysosomal degradation (24, 63).
2.4 Multiple forms of cf-mtDNA
Current evidence indicates that circulating cf-mtDNA exists in heterogeneous forms. These range from distinct compositional states, including naked DNA, lipid vesicle-encapsulated DNA, intact mitochondria, and neutrophil extracellular traps (NETs), to different modes of circulation, such as free diffusion or vesicle- mediated transport (Figure 2).
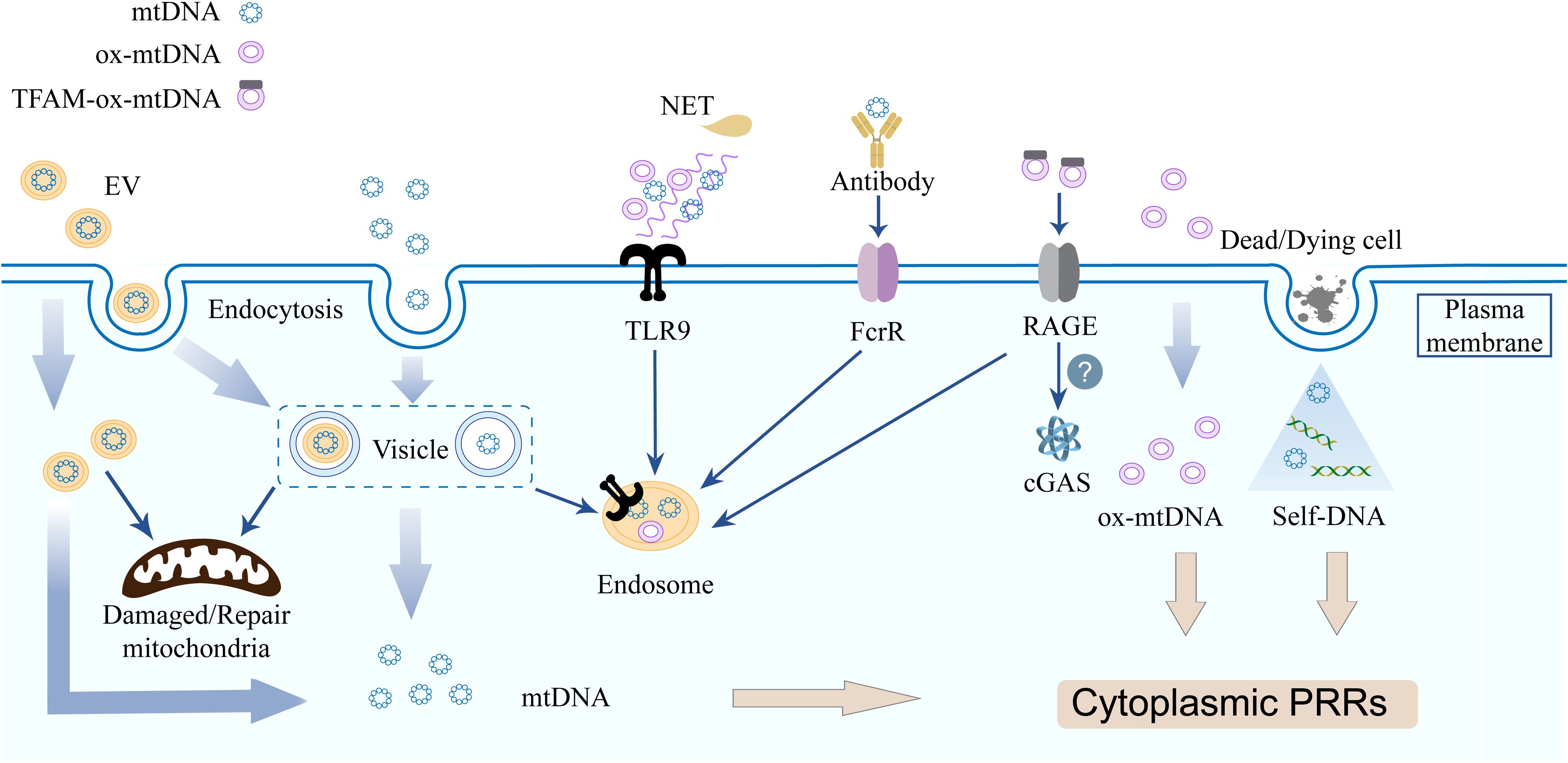
Figure 2. Cell free mitochondrial nucleic acids triggers inflammation via multiple distinct pathways. Innate immune cells acquire cf-mtDNA via endocytosis (EVs/free forms) or phagocytosis. mtDNA from deading/dying cells released during infection bind PRRs. TLR9 recognizes NET-DNA, mtDNA, and ox-mtDNA. TFAM-associated ox-mtDNA internalized by pDC via RAGE receptors, the IFN-generating sensor in recipient cells is unclear.
Naked as well as protein-bound mtDNA molecules are released into the extracellular microenvironment, where they functions as an autocrine, paracrine, or endocrine immune stimulus (24). TLR9 is primarily expressed in innate immune cells, B lymphocytes, and certain non-immune cell lineages. Both mtDNA and bacterial DNA possess low-methylated CpG dinucleotide motifs that can be recognized by TLR9 (64). In addition to being recognized directly by TLR9 on the cell membrane, extracellular mtDNA activates TLR9 occurring via two principal routes: phagocytosis of mtDNA-containing cellular debris or cells, and internalized through autophagic delivery of mitochondrial fragments within the endolysosomal compartment (65–67). Furthermore, the dissemination of mtDNA may occur through direct fusion between the mitochondrial membrane and the cell membrane; this fusion event has been associated with elevated levels of ox-mtDNA (51, 68).
Extracellular vesicles (EVs) represent a significant source of cf-mtDNA (69). EVs are lipid bilayer membrane vesicles found in body fluids, essential for intercellular communication, maintaining internal balance, and promoting pathological processes (70, 71). EVs, as crucial transport carriers for mtDNA and play a significant role in the sorting of mitochondrial components via the dual MDVs. The Snx9-dependent MDVs facilitates the transport of intact IMM and matrix proteins to EVs, whereas the Parkin-dependent MDVs is responsible for targeting oxidatively damaged mitochondrial components for lysosomal degradation (62). Generally, EVs contain original cell membrane structures and cytoplasmic components, playing a crucial role in intercellular signaling (72, 73). EVs have the capacity to transfer mtDNA to another cells, modifying the mitochondrial function or metabolic state of the recipient cells. This mechanism may be involved in tissue repair, immune regulation, and various pathological processes. Innate immune cells can uptake either EVs or cell free nucleotide through endocytosis, as well as mtDNA from cells and cell debris through phagocytosis (29, 74). TLR9 may detect EVs containing mtDNA (mtDNA-EVs) through endolysosomal compartments in a cell-autonomous manner (29). Specifically, PINK1 plays a crucial role in the packaging of mtDNA into EVs through a scaffolding mechanism that is independent of its kinase activity and traditional mitophagy processes. Furthermore, PINK1 can promote the release of mtDNA-containing EVs in breast cancer cells. This process results in the autocrine and paracrine activation of TLR9, which subsequently enhances the degradation of the extracellular matrix and the invasive capabilities of the cancer cells (75). Activated T cells release mtDNA-EVs, which stimulate the cGAS/STING pathway in dendritic cells (76). In a distinct clinical context, EVs have garnered significant attention across various domains, including drug delivery and regenerative medicine, owing to their numerous advantages such as high biocompatibility, low cytotoxicity, low immunogenicity, and a rich content of growth factors (77, 78). Mitochondrial transplantation via EVs ameliorates dysfunction in Leigh syndrome models (79). The nanoscale dimensions of EVs present significant challenges for current analytical systems in verifying or regulating their effects. This situation underscores the need for more advanced analytical techniques capable of detecting EV subpopulations of varying sizes, which would enhance the sensitivity and specificity of EV detection. Furthermore, critical unknowns include EV-delivered mtDNA integration/expression in recipient cells and its signaling mechanisms.
NETs are DNA mesh structures that function to capture and eliminate pathogens; however, excessive formation of these structures contributes to diverse pathologies (80). Initially, it was widely believed that NETs were primarily composed of nDNA, emerging evidence demonstrates the presence of mtDNA within NETs under specific stimulating conditions (81). Neutrophils release mtDNA through a process known as NETosis, and mtDNA can trigger the formation of NETs (15, 81, 82). The NETosis described by Yousefi et al. revealed that mtDNA that is released rather than nDNA during this process (81). Beyond active extrusion via NETosis, extracellular mtDNA accumulation may also passive diffusion from other leukocytes and necrotic cells (83, 84). Accumulating evidence links dysregulated NETosis to multiple inflammatory and autoimmune conditions, including atherosclerosis, psoriasis, rheumatoid arthritis (RA), gout, anti-neutrophil cytoplasmic antibody-associated vasculitis, and systemic lupus erythematosus (SLE) (85, 86). Characteristically, NET-derived DNA complexes amplify TLR7/9 signaling in plasmacytoid dendritic cells (pDCs) or pancreatic ductal epithelial cells, driving cytokine production. These cytokines reciprocally enhance NETosis, establishing a self-perpetuating inflammatory loop (87, 88).
The diagnostic research concerning mtDNA encounters numerous challenges and issues. A significant concern is the absence of a comprehensive classification system to differentiate between extracellular mtDNA (ex-mtDNA) and various biological forms of intact mitochondria (89). Caicedo et al. have delineated four categories of ex-mtDNA (89). These distinct forms of ex-mtDNA demonstrate varying levels and configurations under different health and disease states. Factors such as sample type, collection methods, processing techniques (including anticoagulant selection and centrifugation parameters), and storage conditions significantly influence detection accuracy, particularly in relation to the prevention of false positives resulting from platelet activation (16, 90). Therefore, the establishment of a standardized classification system and detection protocol is essential for enhancing the clinical diagnostic utility of mtDNA.
3 The effect of cf-mtDNA in blood circulation
3.1 Autoimmunity
Autoimmune diseases represent a complex array of disorders driven by intricate interactions between genetic predisposition and environmental triggers. Recent studies have increasingly highlighted the pathogenic significance of mtDNA in facilitating autoimmune diseases. Under physiological conditions, circulating cf-mtDNA is regulated by the activity of DNase, which maintains low levels to prevent aberrant immune activation. Clinically, impaired DNase activity has been implicated in the pathogenesis of multiple autoimmune disorders, highlighting its potential protective function (91). Mutations in the mitochondrial genome or prolonged exposure to pro-inflammatory cytokines promote mitochondrial dysfunction, leading to the release of mitochondrial components that initiate innate immune activation (92). Furthermore, the presence of antibodies targeting mitochondrial nucleic acids (mtDNA and mitochondrial RNA) in autoimmune diseases demonstrats active cross-talk between mitochondrial constituents and adaptive immunity (93–95). Pathological amplification of type I IFN response via self-DNA (including mtDNA) sensing underlies severe autoinflammatory manifestations, as observed in Aicardi-Goutières syndrome and infantile STING-associated vasculopathy (96, 97).
SLE: SLE is an autoimmune disorder characterized by high IFN-mediated multisystemic damage, with “DNA overload” emerging as a pivotal pathogenic driver (98, 99). mtDNA functions as a potent type I IFN pathway agonist to facilitate disease progression. Clinically, elevated serum anti-mtDNA IgG antibodies correlate with disease activity and predict nephritis (93). Plasma cf-mtDNA predominantly originates from naked mitochondria, with platelets serving as a major source (100). In SLE patients, the release of mtDNA is linked to platelet degranulation mediated by platelet FcγRIIA and the fibrinogen receptor α2bβ3 (101). When hydrolyzed by secretory phospholipase A2 group IIA, naked mitochondria release pro-inflammatory lipid mediators and mtDNA to enhance neutrophil activation (102). Furthermore, GSDMD not only facilitates the release of ex-DNA but requires ox-mtDNA-mediated GSDMD-N oligomerization, establishing a self-reinforcing cycle of lytic cell death that promotes the release of extracellular DNA and pro-inflammatory PCD in neutrophils in SLE (103). TLR9 and RAGE are involved in the uptake of extracellular TFAM-associated ox-mtDNA nucleoids by pDCs, thereby stimulating IFN production (51)(Figure 2). The pharmacological inhibition of VDAC-mediated mtDNA release, as well as the application of the mtROS scavenger MitoTEMPO, has been shown to reduce disease severity in murine models of lupus (68, 104).
RA: Elevated circulating and synovial cf-mtDNA in RA patients correlates with disease activity and serves as an early diagnostic biomarker (11, 91, 105). Crucially, mtDNA drives RA pathogenesis through multiple pro-inflammatory mechanisms: Platelet-derived microparticles carrying mitochondria contribute to immune complex formation and stimulate monocytes to release IL-1β and TNF-α (106, 107); mtDNA-exposed synovial neutrophils upregulate receptor activator of nuclear factor kappa-B ligand, promoting joint erosion (108). Supporting its pathogenic centrality, impaired mtDNA clearance triggers RA-like arthritis in mice (17, 109), anti-inflammatory therapies reduce cfDNA (91), and TLR9 inhibition hydroxychloroquine (HCQ) shows clinical efficacy (110). Expanding beyond canonical pathways of lysosomal pH elevation, TLR signaling inhibition, and cytokine regulation, HCQ suppresses TLR-mediated inflammation via the RNF13-LAMP-1 axis (111, 112). Additionally, racemic HCQ inhibits fibroblast-like synoviocyte function by blocking the PI3K/AKT pathway, thereby ameliorating synovitis (113). These findings collectively demonstrate the capacity of HCQ to modulate RA pathogenesis through multitargeted synergistic mechanisms.
Systemic sclerosis (SSc): In SSc, elevated plasma mtDNA levels correlate with TLR9 and cGAS pathway stimulation, inducing pathogenic IFN and IL-6 production that parallels declines in lung function (forced vital capacity) (114, 115). Critically, the increased of mtROS is driven by the inhibition of PINK1/Parkin-mediated mitophagy in type II alveolar epithelial cells, causing ox-mtDNA damage and suppressing DNA repair mechanisms (116).
3.2 Neurological disorders
In the field of neurological disorders, circulating cf-mtDNA serves as a potential biomarker for mitochondrial damage, neuroinflammation, and stress responses, exhibiting abnormal concentrations in multiple conditions including Parkinson’s disease (PD), Alzheimer’s disease (AD), multiple sclerosis (MS), and bipolar disorder (BD). However, significant inconsistencies exist across studies due to variables such as sample source (peripheral blood vs. cerebrospinal fluid), disease subtype heterogeneity, and methodological differences in detection (117–119). This issue has been elaborated in detail in the systematic review by Risi et al. (120).
cf-mtDNA not only correlates with neuroinflammatory progression in depression, dementia, and amyotrophic lateral sclerosis (ALS) but also directly contributes to pathogenesis through specific molecular mechanisms (121–123). Notably, current research on the pathogenic mechanisms of cf-mtDNA remains limited, with studies predominantly concentrated on its biomarker utility.
Serum cf-mtDNA was significantly elevated in ALS, particularly in SOD1 mutation carriers, and positively correlate with IL-6 levels and disease progression rate, indicating synergistic roles of mitochondrial dysfunction and neuroinflammation in pathogenesis (122). In narcolepsy type 1, elevated cerebrospinal fluid cf-mtDNA inversely correlates with hypocretin-1 concentration and associates with sleep architecture abnormalities. Concurrent changes in IL-6/IL-18 further implicate neuroinflammation in disease pathology (124). In BD, cf-mtDNA positively correlates with C-reactive protein (CRP), suggesting involvement in disease progression via inflammatory pathway activation and interaction with metabolic syndrome-associated low-grade inflammation (125). Together these studies suggest a potential link but not causality between cf-mtDNA and neuroinflammation pathologies.
Mechanistically, Tripathi et al. demonstrated that chronic restraint stress significantly increases serum cf-mtDNA in mice. They established the centrality of the cf-mtDNA-TLR9 signaling axis in mediating social behavior deficits, triggering neuroinflammation in the prefrontal cortex and ultimately driving behavioral abnormalities (126). In the chronic intermittent ethanol exposure mouse model, high numbers of mtDNA-EVs could promote in disease progression, exacerbating neuroinflammation and compromising the integrity of the blood-brain barrier (127). It’s also worth mentioning that the cGAS-STING pathway activated by mtDNA plays dual roles in neurodegeneration: while transient activation confers neuroprotection, excessive or chronic CNS stimulation drives neuroinflammation and neurodegeneration (43).
3.3 Infectious diseases and critical illnesses
Circulating cf-mtDNA has been consistently associated with the onset, severity, and prognosis of diverse diseases across multiple studies, demonstrating significant diagnostic potential in infectious diseases and critical illnesses. Concurrently, the inflammatory roles of cf-mtDNA in disease pathogenesis are actively being elucidated. Under pathogenic stimuli such as viruses or bacteria, mtDNA can be released from damaged mitochondria into the cytoplasm and subsequently enter the systemic circulation via active or passive release mechanisms (128, 129). In chronic inflammatory diseases, persistently elevated circulating cf-mtDNA is linked to progressive cellular stress and death (22, 59). During acute disease or injury, a marked increase in circulating cf-mtDNA can trigger acute systemic inflammatory response syndrome (SIRS) (130).
Cardiac dysfunction: Circulating cf-mtDNA levels correlate significantly with 30-day mortality in cardiogenic shock and decompensated heart failure patients, but lack prognostic value in cardiac arrest (where uric acid demonstrates superior predictive utility). This disease-specific association positions mtDNA as a targeted prognostic biomarker for cardiac dysfunction-related critical illness (131).
Severe fever with thrombocytopenia syndrome (SFTS): SFTS is an infectious disease caused by the tick-borne SFTS virus, with a high case fatality rate ranging from 10% to 50%. Studies have demonstrated significantly elevated circulating cf-mtDNA in SFTS patients, which strongly correlate with adverse clinical outcomes. Mechanistically, endothelial cell-derived mtDNA promotes B-cell activation, migration, and differentiation via the TLR9 pathway, enhancing B-cell susceptibility to SFTSV infection, thereby facilitating viral replication and exacerbating disease progression (132).
COVID-19: Circulating cf-mtDNA levels distinguish COVID-19 clinical subtypes (133–135). Critical cases: Non-survivors exhibit 76% lower mtDNA abundance and shorter fragments vs. survivors; elevated cf-mtDNA correlates with ICU admission/death risk (positive predictive value for mortality: 83.3%). Long COVID: Reduced cf-mtDNA with mitochondrial structural abnormalities indicate persistent mitochondrial dysfunction. Asymptomatic individuals show higher cf-mtDNA than symptomatic patients, who conversely exhibit elevated cf-nDNA (135, 136). During respiratory failure, serum cf-mtDNA levels are elevated and positively correlate with oxygen therapy requirement (137). In COVID-19-associated myocarditis models, cf-mtDNA activates TLR/NF-κB signaling, exacerbating myocardial injury via pro-inflammatory cytokine release in myocarditis models (128).
Sepsis: Sepsis, a life-threatening systemic inflammatory response to infection, manifests as multiorgan dysfunction with high mortality rates (138). circulating cf-mtDNA from damaged tissues, which functions as a DAMP to hyperactivate innate immunity, intensifying systemic inflammation and impairing organ function during the progression of sepsis (139). both the administration of mitochondrial fragments enriched with mtDNA in murine models and in vitro and in vivo mtDNA injections can induce comparable inflammatory cascade observed in clinical sepsis (139, 140). Clinical investigations demonstrate that circulating cf-mtDNA increased markedly in sepsis patients correlate with the onset of acute kidney injury (AKI), acute lung injury (ALI), and acute respiratory distress syndrome (ARDS) (139, 141, 142). In sepsis-induced AKI, mtDNA can enhance mitochondrial oxidative stress driving a self-reinforcing pathological loop (139). Clinical validation studies demonstrate strong correlations between circulating cf-mtDNA levels and AKI severity markers. Depletion of mtDNA mitigates acute tubular cell injury, indicating that position mtDNA-centric therapeutics as promising investigational approaches for AKI (143). Notably, hemodialysis patients with elevated circulating cf-mtDNA exhibit higher risks of adverse clinical outcomes (144). In the proinflammatory microenvironment in end-stage renal disease, the immunogenicity of circulating cf-mtDNA may adversely affect patient health, though lacking all-cause mortality association (145). In sepsis-associated ALI and ARDS, circulating cf-mtDNA activates a robust STING pathway in macrophages, disrupting autophagic flux by impairing lysosomal acidification in an IFN-dependent manner, thereby exacerbating lung endothelial barrier disruption and propagate a cytokine storm (141).
3.4 CVD
The concentration, copy number, and methylation profiles of circulating cf-mtDNA serve as critical indicators of disease status, progression stage, and prognostic risk, demonstrating significant clinical potential across cardiovascular and related disorders including diabetic macroangiopathy, abdominal aortic aneurysm (AAA), and atrial fibrillation (AF) (146–149). Additionally, cf-mtDNA abnormalities are closely linked to mitochondrial functional decline and accelerated biological aging: In chronic kidney disease (CKD), low mtDNA copy number (mtDNA-cn) coupled with high cf-mtDNA levels associates significantly with vascular calcification and epigenetic age acceleration (150).
Among heart failure (HF) and type 2 diabetes mellitus (T2DM) patients, heightened cf-mtDNA correlates with mitochondrial dysfunction and metabolic stress, exhibiting positive associations with systemic inflammatory markers—indicating its role as a mediator of metabolic-inflammatory crosstalk in disease progression (151). Individuals diagnosed with T2DM exhibit elevated circulating cf-mtDNA, driven by chronic hyperglycemia-induced mtROS and mitochondrial dysfunction (152, 153). A weak correlation has been identified between plasma IL-1β levels and circulating cf-mtDNA. Mechanically, circulating cf-mtDNA activates the AIM2 inflammasome in macrophages, triggering caspase-1-dependent IL-1β/IL-18 maturation and secretion (152). Additionally, cerebral vessel remodeling and impaired cerebrovascular reactivity may be associated with variations of mtDNA and inflammation particularly in early diabetic kidney disease, yet the precise mechanisms underlying mtDNA-driven cerebrovascular remodeling remain incompletely characterized (154). Mitochondrial debris from tubular and glomerular cells enters systemic circulation in diabetic kidney disease (155). In blood and urine, mtDNA levels were evaluated as a specific signature in relation to inflammatory response within the diabetic kidney at the glomerular and tubular (156). In maintenance hemodialysis (MHD) patients, exogenous cf-mtDNA upregulates TLR9, ICAM-1 and TNF-α in cardiac microvascular endothelial cells, intensifying microvascular inflammation and CVD progression (157).
AAA: In AAA, cf-mtDNA derived from patient peripheral blood mononuclear cells (PBMCs) stimulates macrophages to potentiate AIM2/IFI16 inflammasome assembly, upregulating apoptosis-associated speck-like protein (ASC) and IL-1β expression, thereby inducing ASC speck formation and exacerbating chronic aortic wall inflammation (146).
Myocardial ischemia/reperfusion (MI/R): During MI/R injury, cf-mtDNA activates the NLRP3 inflammasome in a TLR9-dependent manner, mediating splenic monocyte inflammatory responses that amplify IL-1β release and augment infarct size (158).
Metabolic complications with CVD: Patients with metabolic syndrome (MetS) exhibit elevated circulating ox-mtDNA and upregulated TLR9 expression in peripheral blood mononuclear cells. In vitro stimulation of THP-1 monocytes with cfDNA or ox-mtDNA activates TLR9/NF-κB signaling, driving proinflammatory cytokine secretion in MetS-associated cardiovascular disease (159). Obesity induces elevation of cf-mtDNA in cerebrospinal fluid, particularly found in major target organs in hypertension, such as the heart, kidneys, and brain (160, 161). Mechanistically, cf-mtDNA in the cerebrospinal fluid activates the sympathetic nervous system to cause hypertension via the TGFβ signaling pathway. This neuroimmune crosstalk increases sympathetic output to the cardiovascular system (160).
In summary, while substantial progress has been made in elucidating the roles and mechanisms of cf-mtDNA in CVD, its clinical utility requires further validation through large-scale prospective studies.
3.5 blood system diseases
Sickle cell disease (SCD): SCD exhibits pathologically elevated circulating cf-mtDNA levels, driven by abnormal mitochondrial retention in erythrocytes and hemolysis-mediated release during vaso-occlusive crises (VOC) (82). Hypomethylated mtDNA triggers the formation of NETs, exacerbating chronic inflammation and organ damage during VOC (82). The retention of functional mitochondria in mature erythrocytes accelerates cellular senescence, driving membrane fragility and hemolysis (162, 163). Accelerated intravascular lysis of sickle RBCs facilitates mitochondrial extrusion into circulation (82). Hemolysis-derived mtDNA can be transferred to antigen-presenting cells (APCs) by other RBCs, engaging TLR9 to potentiate inflammatory cytokines secretion. In SCD, mitochondria-positive RBCs exhibit reduced circulatory half-life, rendering them susceptible to phagocytosis (164). Under physiological conditions, aging or damaged erythrocytes undergo cleared by splenic macrophages without eliciting an immune response; however, in SCD, immunogenic APC subpopulations more effectively facilitate the removal of aging RBCs increasing the possibility of producing autoantibodies (165, 166). Consequently, circulating cf-mtDNA as a byproduct of hemolysis has the potential to activate coagulation and inflammatory pathways. This activation may contribute to self-perpetuating cycle of VOC and end-organ damage (82).
Multiple myeloma (MM): Patients with MM exhibit significantly elevated cf-mtDNA originating from malignant plasma cells in both peripheral blood and bone marrow. These mtDAMPs promote MM progression by activating the STING pathway in bone marrow macrophages, inducing chemokines release and enhancing the retention of MM cells within the bone marrow niche (167).
Anemia: RBCs bind and eliminate circulating cf-mtDNA via surface TLR9 receptors in the blood, which subsequently stimulate macrophages to phagocytize these complexes and provoke inflammatory responses (65, 168, 169). This clearance mechanism partially alleviates pulmonary inflammation in ARDS and sepsis patients (65, 169). Nonetheless, pathologically amplified RBC-mtDNA interactions accelerate erythrocyte clearance, contributing to anemia in sepsis, COVID-19, and hematologic malignancies (65, 170, 171).
As a potent proinflammatory mediator, mtDNA provides critical insights into inflammatory disease pathogenesis (Table 1). Its dual role as both a DAMP and a biomarker bridges mitochondrial dysfunction with systemic inflammation. Nevertheless, significant gaps remain in our understanding of the specific mechanisms by which circulating cf-mtDNA operates in different diseases. This includes the regulatory mechanisms governing mtDNA release, its interactions with other cellular signaling pathways, and the development of precise therapeutic strategies aimed at targeting circulating cf-mtDNA.
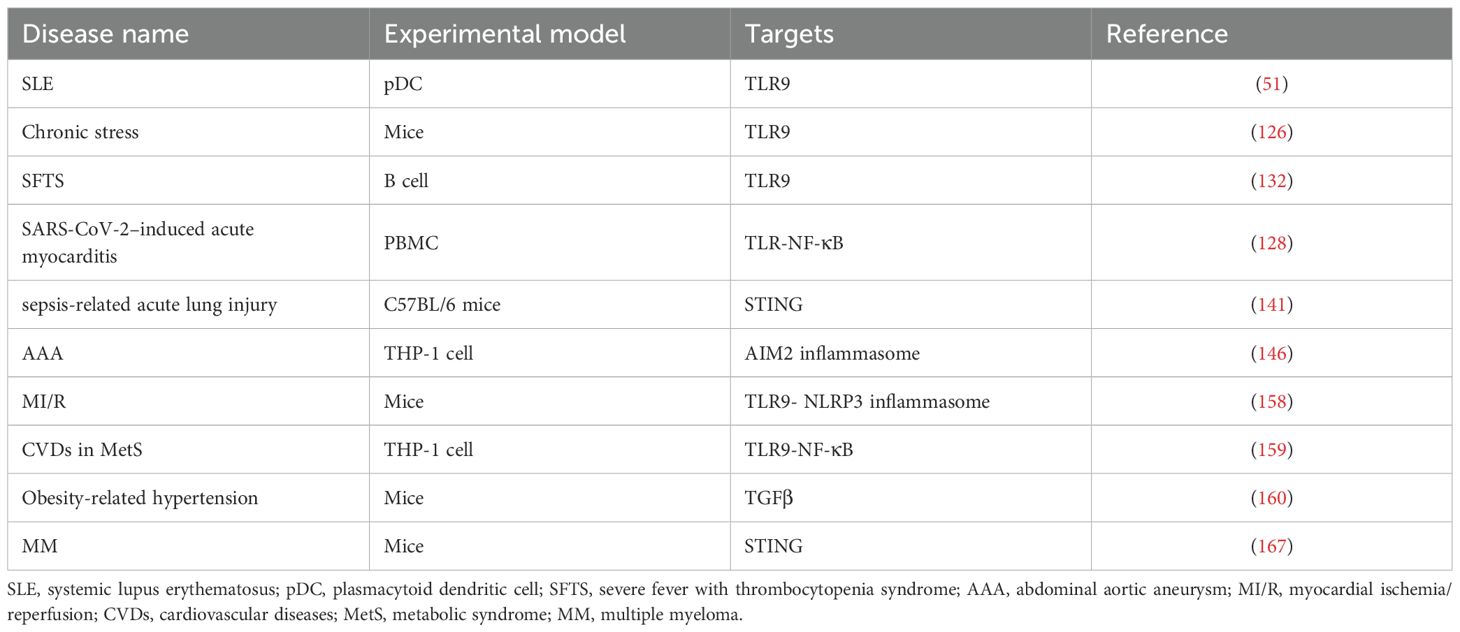
Table 1. Prominent examples of pathologies directly linked to circulating cf-mtDNA.
4 Pharmacology
mtDNA-driven immunity dysregulation constitutes a pathogenic axis in diverse human diseases, spanning hyperinflammatory conditions to inefficient inflammatory states (35). Pharmacological agents that target mitochondrial function have the potential to modulate inflammatory processes, offering novel therapeutic strategies for mtDNA- driven inflammatory diseases, particularly in scenarios where conventional treatments exhibit limited efficacy. The mitochondrial uncoupler BAM15 can reduce mortality and mitigate kidney injury in sepsis models by breaking the mtDNA-TLR9 feedforward loop that amplifies tissue injury (139).
Mitochondrial Pore Opening Inhibitors: Targeted mitochondrial membrane stability emerges as a strategic intervention to prevent pathological mtDNA release. Pharmacological agents such as Cyclosporine A (a Cyclosporine D inhibitor), VBIT-4 (a VDAC inhibitor), and Venetoclax (a BCL-2 inhibitor) is anticipated to facilitate the targeted inhibition of pore opening in the IMM or OMM limiting the release of mtDNA (172–174). Notably, Cyclosporine A is approved for use in humans, primarily for the treatment of autoimmune diseases and the prevention of transplant rejection (58). The application of this is mainly based on its interaction with the cytoplasmic protein PPIF, which inhibits calcineurin and subsequently suppresses lymphocyte activity. Yet the partial immunosuppressive effects of Cyclosporine A through MPT inhibition and the attenuation of mitochondrial-driven inflammatory responses, a hypothesis that warrants further investigation (24). VBIT-4 alleviates symptoms resembling SLE in lupus-prone murine models (104). Some antioxidants sustains mitochondrial function by preserving mitochondrial membrane integrity and reducing pathological mtROS (175).
Cell Death and Autophagy: The impairment of mitochondrial function and structural integrity associated with RCD drives pathogenic mtDNA release. Disulfiram blocks the formation of GSDMD pores, reducing ox-mtDNA release and alleviating symptoms in lupus models (103). Hypocrellin A, a component in ethnic medicinal fungus, targets NLRP3 NACHT domain to inhibit the assembly and activation of the inflammasome (176). Mitophagy facilitates the degradation of dysfunctional mitochondria via lysosomal pathways, limiting the release of mtDNA and mtROS and inhibiting PRR signaling and subsequent inflammatory responses (177, 178). Parkin-dependent mitophagy generates mitochondria-derived vesicles from mtDAMP, suppressing paracrine inflammation (179). At present, the specific pharmacological modulators of mitophagy that are available for clinical use remain unclear.
Targeted Therapies for PRRs and Their Signaling Pathways: Precision modulation of PRR signaling cascades activated by mtDNA emerges as a strategic frontier in autoimmune therapeutics. Notably, the cGAS-STING signaling pathway has garnered considerable attention, with several inhibitors already available, including the competitive inhibitor of IRF3 activation, known as MITA/STING activation, tetrahydroisoquinoline, and the cGAS cyclic peptide inhibitor XQ2B (180, 181). Hydroxychloroquine, functioning as a TLR-9 inhibitor, has demonstrated efficacy in the treatment of RA, SLE, and various other connective tissue disorders (110, 182). Over the past two decades, monoclonal antibodies (mAbs) targeting type I IFN pathway have undergone extensive evaluation in clinical trials. Among these, belimumab and anifrolumab have received clinical approval for the treatment of SLE (98). Nevertheless, > 50% of patients remain refractory to achieve the anticipated outcomes of disease improvement and reduced flare-ups when treated with these two mAbs, highlighting unmet needs for more effective therapeutic options (98).
Targeting the mtDNA inflammatory pathway offers therapeutic promise for refractory inflammatory diseases, yet several mechanisms and safety concerns must be addressed. Mechanistic ambiguities in drug pharmacology, such as the potential dual immunosuppressive effects of cyclosporine A. Safety trade-offs between immune modulation and host defense; for instance, inhibiting the STING pathway necessitates careful consideration of infection risk. Furthermore, the dose-dependent effects of drugs, gender differences, and long-term safety require extensive clinical trials for validation. Research on cross-pathway interactions is vital, as the relationships between mitochondrial membrane permeabilization, mitophagy, and EV release are complex. Future multidisciplinary studies should clarify mechanisms, optimize drug design, and advance personalized treatment, ultimately enabling precision immune interventions.
5 Conclusion
This review elucidates that elevated circulating cf-mtDNA in blood serve not only as a indicator of cellular metabolic status, but also participate in the pathogenesis of various diseases. A critical observation is that mtDNA can activate multiple signaling pathways, such as the cGAS-STING pathway, inflammasomes, and TLR9, thereby initiating innate immune responses, RCD, and alterations in lipid metabolism. This perspective offers novel insights into the emergence and progression of specific clinical symptoms, while simultaneously presenting opportunities for the development of innovative therapeutic strategies. Although existing studies have examined the mechanisms of mtDNA release and the immune signaling pathways mediated by mtDNA, additional clarification of the key molecular complexities and refinement of experimental strategies remain necessary. Furthermore, future research should prioritize the development of mitochondrial-targeted pharmacological agents capable of regulating mitochondrial membrane integrity, mitigating oxidative stress, and sustaining mitochondrial autophagy functions, as well as inhibitors of PRR immune signaling pathways.
Author contributions
YZ: Writing – original draft, Writing – review & editing. CW: Writing – original draft. XL: Writing – original draft. MY: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was funded by Nanchong Federation of Social Science Associations (No. NC22B032), grant from China Scholarship Council (No. 202208510018), the Foundation of the Affiliated Hospital of North Sichuan Medical College (No. ZX-51130001-2023-163), the Foundation of North Sichuan Medical College (No. CBY22-QNA22), the Foundation of Chengdu Municipal Health Commission (No. 2023280).
Acknowledgments
We would like to thank the drawing tools provided by Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Friedman JR and Nunnari J. Mitochondrial form and function. Nature. (2014) 505:335–43. doi: 10.1038/nature12985
2. Picard M and Shirihai OS. Mitochondrial signal transduction. Cell Metab. (2022) 34:1620–53. doi: 10.1016/j.cmet.2022.10.008
3. Harrington JS, Ryter SW, Plataki M, Price DR, and Choi AMK. Mitochondria in health, disease, and aging. Physiol Rev. (2023) 103:2349–422. doi: 10.1152/physrev.00058.2021
4. Jin HS, Suh H-W, Kim S-J, and Jo E-K. Mitochondrial control of innate immunity and inflammation. Immune Network. (2017) 17:77. doi: 10.4110/in.2017.17.2.77
5. Vringer E and Tait SWG. Mitochondria and cell death-associated inflammation. Cell Death Differentiation. (2022) 30:304–12. doi: 10.1038/s41418-022-01094-w
6. Yoon J, Kim S, Lee M, and Kim Y. Mitochondrial nucleic acids in innate immunity and beyond. Exp Mol Med. (2023) 55:2508–18. doi: 10.1038/s12276-023-01121-x
7. Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, et al. Sequence and organization of the human mitochondrial genome. Nature. (1981) 290:457–65. doi: 10.1038/290457a0
8. Bayona-Bafaluy MP, Fernández-Silva P, and Enrıíquez JA. The thankless task of playing genetics with mammalian mitochondrial DNA: a 30-year review. Mitochondrion. (2002) 2:3–25. doi: 10.1016/s1567-7249(02)00044-2
9. Wallace DC. Why do we still have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. (2007) 76:781–821. doi: 10.1146/annurev.biochem.76.081205.150955
10. Jamshidi A, Liu MC, Klein EA, Venn O, Hubbell E, Beausang JF, et al. Evaluation of cell-free DNA approaches for multi-cancer early detection. Cancer Cell. (2022) 40:1537–1549.e1512. doi: 10.1016/j.ccell.2022.10.022
11. Lehmann J, Giaglis S, Kyburz D, Daoudlarian D, and Walker UA. Plasma mtDNA as a possible contributor to and biomarker of inflammation in rheumatoid arthritis. Arthritis Res Ther. (2024) 26:97. doi: 10.1186/s13075-024-03329-2
12. Leotta C, Hernandez L, Tothova L, Arefin S, Ciceri P, Cozzolino MG, et al. Levels of Cell-Free DNA in Kidney Failure Patients before and after Renal Transplantation. Cells. (2023) 12:2774. doi: 10.3390/cells12242774
13. Raszeja-Wyszomirska J, Macech M, Kolanowska M, Krawczyk M, Nazarewski S, Wójcicka A, et al. Free-circulating nucleic acids as biomarkers in patients after solid organ transplantation. Ann Transplant. (2023) 28:e939750. doi: 10.12659/aot.939750
14. Yuwono NL, Warton K, and Ford CE. The influence of biological and lifestyle factors on circulating cell-free DNA in blood plasma. eLife. (2021) 10:e69679. doi: 10.7554/eLife.69679
15. Yang L, Yang D, Yang Q, Cheng F, and Huang Y. Extracellular DNA in blood products and its potential effects on transfusion. Bioscience Rep. (2020) 40:BSR20192770. doi: 10.1042/bsr20192770
16. Roch B, Pisareva E, Mirandola A, Sanchez C, Pastor B, Tanos R, et al. Impact of platelet activation on the release of cell-free mitochondria and circulating mitochondrial DNA. Clinica Chimica Acta. (2024) 553:117711. doi: 10.1016/j.cca.2023.117711
17. Collins LV, Hajizadeh S, Holme E, Jonsson I-M, and Tarkowski A. Endogenously oxidized mitochondrial DNA induces in vivo and in vitro inflammatory responses. J Leukoc Biol. (2004) 75:995–1000. doi: 10.1189/jlb.0703328
18. Otandault A, Abraham J-D, Al Amir Dache Z, Khalyfa A, Jariel-Encontre I, Forné T, et al. Hypoxia differently modulates the release of mitochondrial and nuclear DNA. Br J Cancer. (2020) 122:715–25. doi: 10.1038/s41416-019-0716-y
19. Trumpff C, Michelson J, Lagranha CJ, Taleon V, Karan KR, Sturm G, et al. Stress and circulating cell-free mitochondrial DNA: A systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion. (2021) 59:225–45. doi: 10.1016/j.mito.2021.04.002
20. Newell C, Hume S, Greenway SC, Podemski L, Shearer J, and Khan A. Plasma-derived cell-free mitochondrial DNA: A novel non-invasive methodology to identify mitochondrial DNA haplogroups in humans. Mol Genet Metab. (2018) 125:332–7. doi: 10.1016/j.ymgme.2018.10.002
21. Harapas CR, Idiiatullina E, Al-Azab M, Hrovat-Schaale K, Reygaerts T, Steiner A, et al. Organellar homeostasis and innate immune sensing. Nat Rev Immunol. (2022) 22:535–49. doi: 10.1038/s41577-022-00682-8
22. Hu M and Shu H. Mitochondrial DNA-triggered innate immune response: mechanisms and diseases. Cell Mol Immunol. (2023) 20:1403–12. doi: 10.1038/s41423-023-01086-x
23. de Torre-Minguela C, Gómez A, Couillin I, and Pelegrín P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2021) 35:e21757. doi: 10.1096/fj.202100085R
24. Marchi S, Guilbaud E, Tait SWG, Yamazaki T, and Galluzzi L. Mitochondrial control of inflammation. Nat Rev Immunol. (2022) 23:159–73. doi: 10.1038/s41577-022-00760-x
25. McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science. (2018) 359:eaao6047. doi: 10.1126/science.aao6047
26. Varughese JT, Buchanan SK, and Pitt AS. The role of voltage-dependent anion channel in mitochondrial dysfunction and human disease. Cells. (2021) 10:1737. doi: 10.3390/cells10071737
27. Yan J, Liu W, Feng F, and Chen L. VDAC oligomer pores: A mechanism in disease triggered by mtDNA release. Cell Biol Int. (2020) 44:2178–81. doi: 10.1002/cbin.11427
28. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. (2016) 535:153–8. doi: 10.1038/nature18629
29. VanPortfliet JJ, Chute C, Lei Y, Shutt TE, and West AP. Mitochondrial DNA release and sensing in innate immune responses. Hum Mol Genet. (2024) 33:R80–91. doi: 10.1093/hmg/ddae031
30. Xian H, Watari K, Sanchez-Lopez E, Offenberger J, Onyuru J, Sampath H, et al. Oxidized DNA fragments exit mitochondria via mPTP- and VDAC-dependent channels to activate NLRP3 inflammasome and interferon signaling. Immunity. (2022) 55:1370–1385.e1378. doi: 10.1016/j.immuni.2022.06.007
31. Y C-H, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell. (2020) 183:636–649.e618. doi: 10.1016/j.cell.2020.09.020
32. Menger KE, Rodríguez-Luis A, Chapman J, and Nicholls TJ. Controlling the topology of mammalian mitochondrial DNA. Open Biol. (2021) 11:210168. doi: 10.1098/rsob.210168
33. Kim J, Kim H-S, and Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. (2023) 55:510–9. doi: 10.1038/s12276-023-00965-7
34. Newman LE and Shadel GS. Mitochondrial DNA release in innate immune signaling. Annu Rev Biochem. (2023) 92:299–332. doi: 10.1146/annurev-biochem-032620-104401
35. Riley JS and Tait SWG. Mitochondrial DNA in inflammation and immunity. EMBO Rep. (2020) 21:e49799. doi: 10.15252/embr.201949799
36. Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. (2013) 498:332–7. doi: 10.1038/nature12305
37. Zhang C, Shang G, Gui X, Zhang X, Bai X-c, and Chen ZJ. Structural basis of STING binding with and phosphorylation by TBK1. Nature. (2019) 567:394–8. doi: 10.1038/s41586-019-1000-2
38. Sun L, Wu J, Du F, Chen X, and Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
39. Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science. (2015) 347:aaa2630. doi: 10.1126/science.aaa2630
40. Jabir MS, Hopkins L, Ritchie ND, Ullah I, Bayes HK, Li D, et al. Mitochondrial damage contributes toPseudomonas aeruginosaactivation of the inflammasome and is downregulated by autophagy. Autophagy. (2015) 11:166–82. doi: 10.4161/15548627.2014.981915
41. Xiao TS. The nucleic acid-sensing inflammasomes. Immunol Rev. (2015) 265:103–11. doi: 10.1111/imr.12281
42. Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, et al. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat Immunol. (2004) 5:190–8. doi: 10.1038/ni1028
43. Marques E, Kramer R, and Ryan DG. Multifaceted mitochondria in innate immunity. NPJ Metab Health Dis. (2024) 2:6. doi: 10.1038/s44324-024-00008-3
44. Schnare M, Holt† AC, Takeda K, Akira S, and Medzhitov R. Recognition of CpG DNA is mediated by signaling pathways dependent on the adaptor protein MyD88. Curr Biol. (2000) 10:1139–42. doi: 10.1016/s0960-9822(00)00700-4
45. Nunnari J and Suomalainen A. Mitochondria: in sickness and in health. Cell. (2012) 148:1145–59. doi: 10.1016/j.cell.2012.02.035
46. Zhao M, Wang Y, Li L, Liu S, Wang C, Yuan Y, et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics. (2021) 11:1845–63. doi: 10.7150/thno.50905
47. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X-j, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
48. Li Y, Yang Q, Chen H, Yang X, Han J, Yao X, et al. TFAM downregulation promotes autophagy and ESCC survival through mtDNA stress-mediated STING pathway. Oncogene. (2022) 41:3735–46. doi: 10.1038/s41388-022-02365-z
49. Liu H, Zhen C, Xie J, Luo Z, Zeng L, Zhao G, et al. TFAM is an autophagy receptor that limits inflammation by binding to cytoplasmic mitochondrial DNA. Nat Cell Biol. (2024) 26:878–91. doi: 10.1038/s41556-024-01419-6
50. Gu X, Chen Y, Cao K, Tu M, Liu W, and Ju J. Therapeutic landscape in systemic lupus erythematosus: mtDNA activation of the cGAS-STING pathway. Int Immunopharmacol. (2024) 133:112114. doi: 10.1016/j.intimp.2024.112114
51. Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, et al. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. (2016) 213:697–713. doi: 10.1084/jem.20151876
52. Shimada K, Crother Timothy R, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
53. Lei Y, VanPortfliet JJ, Chen Y-F, Bryant JD, Li Y, Fails D, et al. Cooperative sensing of mitochondrial DNA by ZBP1 and cGAS promotes cardiotoxicity. Cell. (2023) 186:3013–3032.e3022. doi: 10.1016/j.cell.2023.05.039
54. Al Khatib I, Deng J, Lei Y, Torres-Odio S, Rojas GR, Newman LE, et al. Activation of the cGAS-STING innate immune response in cells with deficient mitochondrial topoisomerase TOP1MT. Hum Mol Genet. (2023) 32:2422–40. doi: 10.1093/hmg/ddad062
55. Heitzer E, Auinger L, and Speicher MR. Cell-free DNA and apoptosis: how dead cells inform about the living. Trends Mol Med. (2020) 26:519–28. doi: 10.1016/j.molmed.2020.01.012
56. Murao A, Aziz M, Wang H, Brenner M, and Wang P. Release mechanisms of major DAMPs. Apoptosis. (2021) 26:152–62. doi: 10.1007/s10495-021-01663-3
57. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differentiation. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
58. Bonora M, Giorgi C, and Pinton P. Molecular mechanisms and consequences of mitochondrial permeability transition. Nat Rev Mol Cell Biol. (2021) 23:266–85. doi: 10.1038/s41580-021-00433-y
59. Picca A, Lezza AMS, Leeuwenburgh C, Pesce V, Calvani R, Bossola M, et al. Circulating mitochondrial DNA at the crossroads of mitochondrial dysfunction and inflammation during aging and muscle wasting disorders. Rejuvenation Res. (2018) 21:350–9. doi: 10.1089/rej.2017.1989
60. Lyu Y, Wang T, Huang S, and Zhang Z. Mitochondrial damage-associated molecular patterns and metabolism in the regulation of innate immunity. J Innate Immun. (2023) 15:665–79. doi: 10.1159/000533602
61. Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. (2012) 485:251–5. doi: 10.1038/nature10992
62. Todkar K, Chikhi L, Desjardins V, El-Mortada F, Pépin G, and Germain M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat Commun. (2021) 12:1971. doi: 10.1038/s41467-021-21984-w
63. Bernardini JP, Brouwer JM, Tan IKL, Sandow JJ, Huang S, Stafford CA, et al. Parkin inhibits BAK and BAX apoptotic function by distinct mechanisms during mitophagy. EMBO J. (2018) 38:e99916. doi: 10.15252/embj.201899916
64. Boguszewska K and Szewczuk M. Kaźmierczak-barańska, J., & Karwowski, B. T. The similarities between human mitochondria and bacteria in the context of structure, genome, and base excision repair system. Molecules (Basel Switzerland). (2020) 25:2857. doi: 10.3390/molecules25122857
65. Lam LKM, Murphy S, Kokkinaki D, Venosa A, Sherrill-Mix S, Casu C, et al. DNA binding to TLR9 expressed by red blood cells promotes innate immune activation and anemia. Sci Transl Med. (2021) 13:eabj1008. doi: 10.1126/scitranslmed.abj1008
66. Mukhopadhyay P, Bueno M, Zank D, Buendia-Roldán I, Fiedler K, Mays BG, et al. PINK1 attenuates mtDNA release in alveolar epithelial cells and TLR9 mediated profibrotic responses. PloS One. (2019) 14:e0218003. doi: 10.1371/journal.pone.0218003
67. Rodríguez‐Nuevo A, Díaz‐Ramos A, Noguera E, Díaz‐Sáez F, Duran X, Muñoz JP, et al. Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. EMBO J. (2018) 37:e96553. doi: 10.15252/embj.201796553
68. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
69. Keuren JF, Magdeleyns EJ, Govers‐Riemslag JW, Lindhout T, and Curvers J. Effects of storage-induced platelet microparticles on the initiation and propagation phase of blood coagulation. Br J haematology. (2006) 134:307–13. doi: 10.1111/j.1365-2141.2006.06167.x
70. Obeid S, Sung P-S, Le Roy B, Chou M-L, Hsieh S-L, Elie-Caille C, et al. NanoBioAnalytical characterization of extracellular vesicles in 75-nm nanofiltered human plasma for transfusion: A tool to improve transfusion safety. Nanomedicine: Nanotechnology Biol Med. (2019) 20:101977. doi: 10.1016/j.nano.2019.02.026
71. Tkach M and Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. (2016) 164:1226–32. doi: 10.1016/j.cell.2016.01.043
72. Pelletier M, Breton Y, Allaeys I, Becker Y, Benson T, and Boilard E. Platelet extracellular vesicles and their mitochondrial content improve the mitochondrial bioenergetics of cellular immune recipients. Transfusion. (2023) 63:1983–96. doi: 10.1111/trf.17524
73. Peruzzotti-Jametti L, Bernstock JD, Willis CM, Manferrari G, Rogall R, Fernandez-Vizarra E, et al. Neural stem cells traffic functional mitochondria via extracellular vesicles. PloS Biol. (2021) 19:e3001166. doi: 10.1371/journal.pbio.3001166
74. Cai Y, Li S, Yang Y, Duan S, Fan G, Bai J, et al. Intestinal epithelial damage-derived mtDNA activates STING-IL12 axis in dendritic cells to promote colitis. Theranostics. (2024) 14:4393–410. doi: 10.7150/thno.96184
75. Rabas N, Palmer S, Mitchell L, Ismail S, Gohlke A, Riley JS, et al. PINK1 drives production of mtDNA-containing extracellular vesicles to promote invasiveness. J Cell Biol. (2021) 220:e202006049. doi: 10.1083/jcb.202006049
76. Torralba D, Baixauli F, Villarroya-Beltri C, Fernández-Delgado I, Latorre-Pellicer A, Acín-Pérez R, et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat Commun. (2018) 9:2658. doi: 10.1038/s41467-018-05077-9
77. Johnson J, Law SQK, Shojaee M, Hall AS, Bhuiyan S, Lim MBL, et al. First-in-human clinical trial of allogeneic, platelet-derived extracellular vesicles as a potential therapeutic for delayed wound healing. J Extracellular Vesicles. (2023) 12:e12332. doi: 10.1002/jev2.12332
78. Meliciano A, Salvador D, Mendonça P, Louro AF, and Serra M. Clinically expired platelet concentrates as a source of extracellular vesicles for targeted anti-cancer drug delivery. Pharmaceutics. (2023) 15:953. doi: 10.3390/pharmaceutics15030953
79. Sofou K, De Coo IFM, Isohanni P, Ostergaard E, Naess K, De Meirleir L, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J rare Dis. (2014) 9:52. doi: 10.1186/1750-1172-9-52
80. De Gaetano A, Solodka K, Zanini G, Selleri V, Mattioli AV, Nasi M, et al. Molecular mechanisms of mtDNA-mediated inflammation. Cells. (2021) 10:2898. doi: 10.3390/cells10112898
81. Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med. (2008) 14:949–53. doi: 10.1038/nm.1855
82. Tumburu L, Ghosh-Choudhary S, Seifuddin FT, Barbu EA, Yang S, Ahmad MM, et al. Circulating mitochondrial DNA is a proinflammatory DAMP in sickle cell disease. Blood. (2021) 137:3116–26. doi: 10.1182/blood.2020009063
83. Berthelot J-M, Le Goff B, Neel A, Maugars Y, and Hamidou M. NETosis: At the crossroads of rheumatoid arthritis, lupus, and vasculitis. Joint Bone Spine. (2017) 84:255–62. doi: 10.1016/j.jbspin.2016.05.013
84. Thiam HR, Wong SL, Wagner DD, and Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
85. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M, et al. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
86. Koenig A and Buskiewicz-Koenig IA. Redox activation of mitochondrial DAMPs and the metabolic consequences for development of autoimmunity. Antioxidants Redox Signaling. (2022) 36:441–61. doi: 10.1089/ars.2021.0073
87. Li M, Liu Y, Wang J, Wang Y, Yang Y, and Yang A. Neutrophil extracellular DNA traps activate the TLR9 signaling pathway of pancreatic ductal epithelial cells in patients with type 2 autoimmune pancreatitis. Int Immunopharmacol. (2025) 144:113673. doi: 10.1016/j.intimp.2024.113673
88. Lin CMA, Isaacs JD, and Cooles FAH. Role of IFN-α in rheumatoid arthritis. Curr Rheumatol Rep. (2023) 26:37–52. doi: 10.1007/s11926-023-01125-6
89. Caicedo A, Benavides-Almeida A, Haro-Vinueza A, Peña-Cisneros J, Pérez-Meza ÁA, Michelson J, et al. Decoding the nature and complexity of extracellular mtDNA: Types and implications for health and disease. Mitochondrion. (2024) 75:101848. doi: 10.1016/j.mito.2024.101848
90. Michelson J, Rausser S, Peng A, Yu T, Sturm G, Trumpff C, et al. MitoQuicLy: A high-throughput method for quantifying cell-free DNA from human plasma, serum, and saliva. Mitochondrion. (2023) 71:26–39. doi: 10.1016/j.mito.2023.05.001
91. Macáková K, Illésová J, Mlynáriková V, Lesayová A, Konečná B, and Vlková B. The dynamics of extracellular DNA associates with treatment response in patients with rheumatoid arthritis. Sci Rep. (2022) 12:21099. doi: 10.1038/s41598-022-23954-8
92. Becker YLC, Duvvuri B, Fortin PR, Lood C, and Boilard E. The role of mitochondria in rheumatic diseases. Nat Rev Rheumatol. (2022) 18:621–40. doi: 10.1038/s41584-022-00834-z
93. Becker Y, Loignon R-C, Julien A-S, Marcoux G, Allaeys I, Lévesque T, et al. Anti-mitochondrial autoantibodies in systemic lupus erythematosus and their association with disease manifestations. Sci Rep. (2019) 9:4530. doi: 10.1038/s41598-019-40900-3
94. Becker Y, Marcoux G, Allaeys I, Julien A-S, Loignon R-C, Benk-Fortin H, et al. Autoantibodies in systemic lupus erythematosus target mitochondrial RNA. Front Immunol. (2019) 10:1026. doi: 10.3389/fimmu.2019.01026
95. Becker YLC, Julien A-S, Godbout A, Boilard É, and Fortin PR. Pilot study of anti-mitochondrial antibodies in antiphospholipid syndrome. Lupus. (2020) 29:1623–9. doi: 10.1177/0961203320944481
96. Dragoni F, Garau J, Sproviero D, Orcesi S, Varesio C, De Siervi S, et al. Characterization of mitochondrial alterations in aicardi–goutières patients mutated in RNASEH2A and RNASEH2B genes. Int J Mol Sci. (2022) 23:14482. doi: 10.3390/ijms232214482
97. Gul E, Sayar EH, Gungor B, Eroglu FK, Surucu N, Keles S, et al. Type I IFN–related NETosis in ataxia telangiectasia and Artemis deficiency. J Allergy Clin Immunol. (2018) 142:246–57. doi: 10.1016/j.jaci.2017.10.030
98. Casey KA, Smith MA, Sinibaldi D, Seto NL, Playford MP, Wang X, et al. Modulation of cardiometabolic disease markers by type I interferon inhibition in systemic lupus erythematosus. Arthritis Rheumatol. (2021) 73:459–71. doi: 10.1002/art.41518
99. Chan RWY, Jiang P, Peng X, Tam L-S, Liao GJW, Li EKM, et al. Plasma DNA aberrations in systemic lupus erythematosus revealed by genomic and methylomic sequencing. Proc Natl Acad Sci. (2014) 111:E5302–11. doi: 10.1073/pnas.1421126111
100. Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med. (2018) 25:75–81. doi: 10.1038/s41591-018-0254-9
101. Melki I, Allaeys I, Tessandier N, Lévesque T, Cloutier N, Laroche A, et al. Platelets release mitochondrial antigens in systemic lupus erythematosus. Sci Transl Med. (2021) 13:eaav5928. doi: 10.1126/scitranslmed.aav5928
102. Boudreau LH, Duchez A-C, Cloutier N, Soulet D, Martin N, Bollinger J, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. (2014) 124:2173–83. doi: 10.1182/blood-2014-05-573543
103. Miao N, Wang Z, Wang Q, Xie H, Yang N, Wang Y, et al. Oxidized mitochondrial DNA induces gasdermin D oligomerization in systemic lupus erythematosus. Nat Commun. (2023) 14:872. doi: 10.1038/s41467-023-36522-z
104. Kim J, Gupta R, Blanco L, Yang S, Shteinfer-Kuzmine A, Wang K, et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Sci (New York N.Y.). (2019) 366:1531–6. doi: 10.1126/science.aav4011
105. Dong C, Liu Y, Sun C, Liang H, Dai L, Shen J, et al. Identification of specific joint-inflammatogenic cell-free DNA molecules from synovial fluids of patients with rheumatoid arthritis. Front Immunol. (2020) 11:662. doi: 10.3389/fimmu.2020.00662
106. Knijff‐Dutmer EAJ, Koerts J, Nieuwland R, Kalsbeek‐Batenburg EM, and Van De Laar MAFJ. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis Rheumatism. (2002) 46:1498–503. doi: 10.1002/art.10312
107. Villar-Vesga J, Grajales C, Burbano C, Vanegas–García A, Muñoz–Vahos CH, Vásquez G, et al. Platelet-derived microparticles generated in vitro resemble circulating vesicles of patients with rheumatoid arthritis and activate monocytes. Cell Immunol. (2019) 336:1–11. doi: 10.1016/j.cellimm.2018.12.002
108. Contis A, Mitrovic S, Lavie J, Douchet I, Lazaro E, Truchetet ME, et al. Neutrophil-derived mitochondrial DNA promotes receptor activator of nuclear factor κB and its ligand signalling in rheumatoid arthritis. Rheumatol (Oxford). (2017) 56:1200–5. doi: 10.1093/rheumatology/kex041
109. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature. (2006) 443:998–1002. doi: 10.1038/nature05245
110. Han J, Li X, Luo X, He J, Huang X, Zhou Q, et al. The mechanisms of hydroxychloroquine in rheumatoid arthritis treatment: Inhibition of dendritic cell functions via Toll like receptor 9 signaling. Biomedicine Pharmacotherapy. (2020) 132:110848. doi: 10.1016/j.biopha.2020.110848
111. Bansal P, Goyal A, Cusick AT, Lahan S, Dhaliwal HS, Bhyan P, et al. Hydroxychloroquine: a comprehensive review and its controversial role in coronavirus disease 2019. Ann Med. (2021) 53:117–34. doi: 10.1080/07853890.2020.1839959
112. Liu W, Wang Y, Liu S, Zhang X, Cao X, and Jiang M. E3 ubiquitin ligase RNF13 suppresses TLR lysosomal degradation by promoting LAMP-1 proteasomal degradation. Adv Sci (Weinh). (2024) 11:e2309560. doi: 10.1002/advs.202309560
113. Wang X, He J, Zhong J, He J, Wang D, Bai L, et al. Hydroxychloroquine inhibits the PI3K/AKT pathway in synovial fibroblasts of rheumatoid arthritis and alleviates collagen-induced arthritis in mice. Clin Exp Rheumatol. (2025) 43:829–40. doi: 10.55563/clinexprheumatol/tczujg
114. Ghincea A, Woo S, Yu S, Pivarnik T, Fiorini V, Herzog EL, et al. Mitochondrial DNA-sensing pathogen recognition receptors in systemic sclerosis-associated interstitial lung disease: a review. Curr Treat Options Rheumatol. (2023) 9:204–20. doi: 10.1007/s40674-023-00211-1
115. Ryu C, Walia A, Ortiz V, Perry C, Woo S, Reeves BC, et al. Bioactive plasma mitochondrial DNA is associated with disease progression in scleroderma-associated interstitial lung disease. Arthritis Rheumatol. (2020) 72:1905–15. doi: 10.1002/art.41418
116. Jablonski RP, Kim SJ, Cheresh P, Williams DB, Morales‐Nebreda L, Cheng Y, et al. SIRT3 deficiency promotes lung fibrosis by augmenting alveolar epithelial cell mitochondrial DNA damage and apoptosis. FASEB J. (2017) 31:2520–32. doi: 10.1096/fj.201601077R
117. Ding B, Zhang X, Wan Z, Tian F, Ling J, Tan J, et al. Characterization of mitochondrial DNA methylation of alzheimer’s disease in plasma cell-free DNA. Diagnostics (Basel). (2023) 13:2351. doi: 10.3390/diagnostics13142351
118. Shao S, Zou Y, Kennedy KG, Dimick MK, Andreazza AC, Young LT, et al. Pilot study of circulating cell-free mitochondrial DNA in relation to brain structure in youth bipolar disorder. Int J Bipolar Disord. (2024) 12:21. doi: 10.1186/s40345-024-00334-x
119. Ying C, Li Y, Zhang H, Pang S, Hao S, Hu S, et al. Probing the diagnostic values of plasma cf-nDNA and cf-mtDNA for Parkinson’s disease and multiple system atrophy. Front Neurosci. (2024) 18:1488820. doi: 10.3389/fnins.2024.1488820
120. Risi B, Imarisio A, Cuconato G, Padovani A, Valente EM, and Filosto M. (mtDNA) as fluid biomarker in neurodegenerative disorders: A systematic review. Eur J Neurol. (2025) 32:e70014. doi: 10.1111/ene.70014
121. Daniels TE, Zitkovsky EK, Laumann LE, Kunicki ZJ, Price DJ, Peterson AL, et al. Circulating cell-free mitochondrial DNA and depressive symptoms among low-active adults who smoke. Psychosom Med. (2024) 86:37–43. doi: 10.1097/psy.0000000000001254
122. Li J, Gao C, Wang Q, Liu J, Xie Z, Zhao Y, et al. Elevated serum circulating cell-free mitochondrial DNA in amyotrophic lateral sclerosis. Eur J Neurol. (2024) 31:e16493. doi: 10.1111/ene.16493
123. Nidadavolu LS, Feger D, Chen D, Wu Y, Grodstein F, Gross AL, et al. Associations between circulating cell-free mitochondrial DNA, inflammatory markers, and cognitive and physical outcomes in community dwelling older adults. Immun Ageing. (2023) 20:24. doi: 10.1186/s12979-023-00342-y
124. Moresco M, Tropeano CV, Romagnoli M, Neccia G, Rapone A, Pizza F, et al. Elevated circulating cell-free mitochondrial DNA level in cerebrospinal fluid of narcolepsy type 1. Brain Commun. (2025) 7:fcaf125. doi: 10.1093/braincomms/fcaf125
125. Zachos KA, Choi J, Godin O, Chernega T, Kwak HA, Jung JH, et al. Mitochondrial biomarkers and metabolic syndrome in bipolar disorder. Psychiatry Res. (2024) 339:116063. doi: 10.1016/j.psychres.2024.116063
126. Tripathi A, Bartosh A, Whitehead C, and Pillai A. Activation of cell-free mtDNA-TLR9 signaling mediates chronic stress-induced social behavior deficits. Mol Psychiatry. (2023) 28:3806–15. doi: 10.1038/s41380-023-02189-7
127. Togre NS, Mekala N, Bhoj PS, Mogadala N, Winfield M, Trivedi J, et al. Neuroinflammatory responses and blood–brain barrier injury in chronic alcohol exposure: role of purinergic P2 × 7 Receptor signaling. J Neuroinflamm. (2024) 21:1971. doi: 10.1186/s12974-024-03230-4
128. Lu RXZ, Rafatian N, Zhao Y, Wagner KT, Beroncal EL, Li B, et al. Cardiac tissue model of immune-induced dysfunction reveals the role of free mitochondrial DNA and the therapeutic effects of exosomes. Sci Adv. (2024) 10:eadk0164. doi: 10.1126/sciadv.adk0164
129. Xiao L, Xu H, Liu S, Cheng Z, Kong Y, and Tian L. Preliminary study on the participation of TLR9 on erythrocyte surface combined with mtDNA in the monitoring of infectious diseases. Front Med (Lausanne). (2024) 11:1498627. doi: 10.3389/fmed.2024.1498627
130. Stover Cordula M, Gu X, Yao Y, Wu G, Lv T, Luo L, et al. The plasma mitochondrial DNA is an independent predictor for post-traumatic systemic inflammatory response syndrome. PloS One. (2013) 8:e72834. doi: 10.1371/journal.pone.0072834
131. Lenz M, Zilberszac R, Hengstenberg C, Wojta J, Richter B, Heinz G, et al. Nonlinear associations of uric acid and mitochondrial DNA with mortality in critically ill patients. J Clin Med. (2025) 14:4455. doi: 10.3390/jcm14134455
132. Zhang YF, Cui N, Yang T, Wang JX, Chen JH, Yang X, et al. Endothelial cell-released mitochondrial DNA promotes B cell differentiation and virus replication during severe fever with thrombocytopenia syndrome virus infection. J Virol. (2025) 99:e0132324. doi: 10.1128/jvi.01323-24
133. Mahmoodpoor A, Mohammadzadeh M, Asghari R, Tagizadeh M, Iranpour A, Rezayi M, et al. Prognostic potential of circulating cell free mitochondrial DNA levels in COVID-19 patients. Mol Biol Rep. (2023) 50:10249–55. doi: 10.1007/s11033-023-08841-3
134. Szögi T, Borsos BN, Masic D, Radics B, Bella Z, Bánfi A, et al. Novel biomarkers of mitochondrial dysfunction in Long COVID patients. Geroscience. (2025) 47:2245–61. doi: 10.1007/s11357-024-01398-4
135. Zhang H, Li L, Luo Y, Zheng F, Zhang Y, Xie R, et al. Fragmentomics of plasma mitochondrial and nuclear DNA inform prognosis in COVID-19 patients with critical symptoms. BMC Med Genomics. (2024) 17:243. doi: 10.1186/s12920-024-02022-2
136. Shoraka S, Mohebbi SR, Hosseini SM, and Zali MR. Comparison of plasma mitochondrial DNA copy number in asymptomatic and symptomatic COVID-19 patients. Front Microbiol. (2023) 14:1256042. doi: 10.3389/fmicb.2023.1256042
137. Tanaka A, Wakayama K, Fukuda Y, Ohta S, Homma T, Ando K, et al. Increased levels of circulating cell-free DNA in COVID-19 patients with respiratory failure. Sci Rep. (2024) 14:17399. doi: 10.1038/s41598-024-68433-4
138. Kristina ER, Sarah Charlotte J, Kareha MA, Katya Anne S, Derrick T, Daniel Rhodes K, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet. (2020) 395:200–11. doi: 10.1016/S0140-6736(19)32989-7
139. Tsuji N, Tsuji T, Yamashita T, Hayase N, Hu X, Yuen PST, et al. BAM15 treats mouse sepsis and kidney injury, linking mortality, mitochondrial DNA, tubule damage, and neutrophils. J Clin Invest. (2023) 133:e152401. doi: 10.1172/jci152401
140. Tsuji N, Tsuji T, Ohashi N, Kato A, Fujigaki Y, Yasuda H, et al. Role of mitochondrial DNA in septic AKI via toll-like receptor 9. J Am Soc Nephrol. (2016) 27:2009–20. doi: 10.1681/asn.2015040376
141. Liu Q, Wu J, Zhang X, Li X, Wu X, Zhao Y, et al. Circulating mitochondrial DNA-triggered autophagy dysfunction via STING underlies sepsis-related acute lung injury. Cell Death Dis. (2021) 12:673. doi: 10.1038/s41419-021-03961-9
142. Malik AN. Mitochondrial DNA – novel mechanisms of kidney damage and potential biomarker. Curr Opin Nephrol Hypertension. (2023) 32:528–36. doi: 10.1097/mnh.0000000000000922
143. Liu J, Jia Z, and Gong W. Circulating mitochondrial DNA stimulates innate immune signaling pathways to mediate acute kidney injury. Front Immunol. (2021) 12:680648. doi: 10.3389/fimmu.2021.680648
144. Burdmann EA, Cao H, Ye H, Sun Z, Shen X, Song Z, et al. Circulatory mitochondrial DNA is a pro-inflammatory agent in maintenance hemodialysis patients. PloS One. (2014) 9:e113179. doi: 10.1371/journal.pone.0113179
145. Kim K, Jung SW, Cho W-H, Moon H, Jeong KH, Kim JS, et al. Associations between cell-free mitochondrial DNA and inflammation, and their clinical implications for patients on hemodialysis: A prospective multicenter cohort study. Blood Purification. (2021) 50:214–21. doi: 10.1159/000510088
146. Dihlmann S, Kaduk C, Passek KH, Spieler A, Böckler D, and Peters AS. Exploring circulating cell-free DNA as a biomarker and as an inducer of AIM2-inflammasome-mediated inflammation in patients with abdominal aortic aneurysm. Sci Rep. (2025) 15:20196. doi: 10.1038/s41598-025-06220-5
147. Dong S, Wang H, Zhou X, Zhang Z, Zhan X, Jin Z, et al. Specific changes in the levels of circulating cell-free mitochondrial DNA in the serum of patients with diabetic microangiopathy. Lab Med. (2025). doi: 10.1093/labmed/lmaf034
148. Miyamoto S, Tokuyama T, Okubo Y, Okamura S, Miyauchi S, Furutani M, et al. Decreased plasma cell-free mitochondrial DNA may be a new biomarker of tachycardia-induced cardiomyopathy in patients with atrial fibrillation. Int J Cardiol. (2024) 417:132579. doi: 10.1016/j.ijcard.2024.132579
149. Pool L, van Wijk SW, van Schie MS, Taverne Y, de Groot NMS, and Brundel B. Quantifying DNA lesions and circulating free DNA: diagnostic marker for electropathology and clinical stage of AF. JACC Clin Electrophysiol. (2025) 11:321–32. doi: 10.1016/j.jacep.2024.10.008
150. Schwarz A, Qureshi AR, Hernandez L, Wennberg L, Wernerson A, Kublickiene K, et al. Reduced mtDNA copy number links to vascular calcification and restores after transplantation. Cells. (2025) 14:917. doi: 10.3390/cells14120917
151. Mengozzi A, Armenia S, De Biase N, Punta LD, Cappelli F, Duranti E, et al. Circulating mitochondrial DNA signature in cardiometabolic patients. Cardiovasc Diabetol. (2025) 24:106. doi: 10.1186/s12933-025-02656-1
152. Bae JH, Jo S, Kim SJ, Lee JM, Jeong JH, Kang JS, et al. Circulating cell-free mtDNA contributes to AIM2 inflammasome-mediated chronic inflammation in patients with type 2 diabetes. Cells. (2019) 8:328. doi: 10.3390/cells8040328
153. Deng X, Yang G, Zheng X, Yang Y, Qin H, Liu ZX, et al. Plasma mtDNA copy numbers are associated with GSTK1 expression and inflammation in type 2 diabetes. Diabetic Med. (2019) 37:1874–8. doi: 10.1111/dme.14132
154. Petrica L, Gadalean F, Muntean DM, Jianu DC, Vlad D, Dumitrascu V, et al. Mitochondrial DNA and inflammation are associated with cerebral vessel remodeling and early diabetic kidney disease in patients with type 2 diabetes mellitus. Biomolecules. (2024) 14:499. doi: 10.3390/biom14040499
155. Wei PZ, Kwan BC-H, Chow KM, Cheng PM-S, Luk CC-W, Li PK-T, et al. Urinary mitochondrial DNA level is an indicator of intra-renal mitochondrial depletion and renal scarring in diabetic nephropathy. Nephrol Dialysis Transplant. (2018) 33:784–8. doi: 10.1093/ndt/gfx339
156. Petrica L, Vlad A, Gadalean F, Muntean DM, Vlad D, Dumitrascu V, et al. Mitochondrial DNA changes in blood and urine display a specific signature in relation to inflammation in normoalbuminuric diabetic kidney disease in type 2 diabetes mellitus patients. Int J Mol Sci. (2023) 24:9803. doi: 10.3390/ijms24129803
157. Fan Z, Feng Y, Zang L, Guo Y, and Zhong XY. Association of circulating MtDNA with CVD in hemodialysis patients and in vitro effect of exogenous MtDNA on cardiac microvascular inflammation. BMC Cardiovasc Disord. (2023) 23:74. doi: 10.1186/s12872-023-03104-2
158. Xie D, Guo H, Li M, Jia L, Zhang H, Liang D, et al. Splenic monocytes mediate inflammatory response and exacerbate myocardial ischemia/reperfusion injury in a mitochondrial cell-free DNA-TLR9-NLRP3-dependent fashion. Basic Res Cardiol. (2023) 118:44. doi: 10.1007/s00395-023-01014-0
159. Ye W, Wen C, Zeng A, and Hu X. Increased levels of circulating oxidized mitochondrial DNA contribute to chronic inflammation in metabolic syndrome, and MitoQ-based antioxidant therapy alleviates this DNA-induced inflammation. Mol Cell Endocrinol. (2023) 560:111812. doi: 10.1016/j.mce.2022.111812
160. Alé A, Zhang Y, Han C, and Cai D. Obesity-associated extracellular mtDNA activates central TGFβ pathway to cause blood pressure increase. Am J Physiology-Endocrinology Metab. (2017) 312:E161–74. doi: 10.1152/ajpendo.00337.2016
161. Konstantinidis K. & Kitsis, R. N Escaped DNA inflames heart. Nat. (2012) 485:179–80. doi: 10.1038/485179a
162. Esperti S, Nader E, Stier A, Boisson C, Carin R, Marano M, et al. Increased retention of functional mitochondria in mature sickle red blood cells is associated with increased sickling tendency, hemolysis and oxidative stress. Haematologica. (2023) 108:3086–94. doi: 10.3324/haematol.2023.282684
163. Gallivan A, Alejandro M, Kanu A, Zekaryas N, Horneman H, Hong LK, et al. Reticulocyte mitochondrial retention increases reactive oxygen species and oxygen consumption in mouse models of sickle cell disease and phlebotomy-induced anemia. Exp Hematol. (2023) 122:55–62. doi: 10.1016/j.exphem.2023.02.005
164. Wood BL, Gibson DF, and Tait JF. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: flow-cytometric measurement and clinical associations. Blood. (1996) 88:1873–80. doi: 10.1182/blood.V88.5.1873.1873
165. Moriconi C, Dzieciatkowska M, Roy M, D'Alessandro A, Roingeard P, Lee J, et al. Retention of functional mitochondria in mature red blood cells from patients with sickle cell disease. Br J haematology. (2022) 198:574–86. doi: 10.1111/bjh.18287
166. Richards AL, Hendrickson JE, Zimring JC, and Hudson KE. Erythrophagocytosis by plasmacytoid dendritic cells and monocytes is enhanced during inflammation. Transfusion. (2016) 56:905–16. doi: 10.1111/trf.13497
167. Jibril A, Hellmich C, Wojtowicz EE, Hampton K, Maynard R, De Silva R, et al. Plasma cell-derived mtDAMPs activate the macrophage STING pathway, promoting myeloma progression. Blood. (2023) 141:3065–77. doi: 10.1182/blood.2022018711
168. Anderson HL, Brodsky IE, and Mangalmurti NS. The evolving erythrocyte: red blood cells as modulators of innate immunity. J Immunol. (2018) 201:1343–51. doi: 10.4049/jimmunol.1800565
169. Hotz MJ, Qing D, Shashaty MGS, Zhang P, Faust H, Sondheimer N, et al. Red blood cells homeostatically bind mitochondrial DNA through TLR9 to maintain quiescence and to prevent lung injury. Am J Respir Crit Care Med. (2018) 197:470–80. doi: 10.1164/rccm.201706-1161OC
170. Renoux C, Fort R, Nader E, Boisson C, Joly P, Stauffer E, et al. Impact of COVID-19 on red blood cell rheology. Br J haematology. (2021) 192:e108–11. doi: 10.1111/bjh.17306
171. Yue L, Li Y-H, Ma R-L, Niu J-W, Cui H-T, Sun Y, et al. Circulating mitochondrial DNA is associated with anemia in newly diagnosed hematologic Malignancies. Leukemia lymphoma. (2022) 64:178–87. doi: 10.1080/10428194.2022.2133537
172. Belosludtsev K N, Ilzorkina AI, Matveeva LA, Chulkov AV, Semenova AA, Dubinin MV, et al. Effect of VBIT-4 on the functional activity of isolated mitochondria and cell viability. Biochim Biophys Acta (BBA) - Biomembranes. (2024) 1866:184329. doi: 10.1016/j.bbamem.2024.184329
173. Diepstraten ST, Anderson MA, Czabotar PE, Lessene G, Strasser A, and Kelly GL. The manipulation of apoptosis for cancer therapy using BH3-mimetic drugs. Nat Rev Cancer. (2022) 22:45–64. doi: 10.1038/s41568-021-00407-4
174. Liu H, Fan H, He P, Zhuang H, Liu X, Chen M, et al. Prohibitin 1 regulates mtDNA release and downstream inflammatory responses. EMBO J. (2022) 41:e111173. doi: 10.15252/embj.2022111173
175. Chen P, Yao L, Yuan M, Wang Z, Zhang Q, Jiang Y, et al. Mitochondrial dysfunction: A promising therapeutic target for liver diseases. Genes Dis. (2024) 11:101115. doi: 10.1016/j.gendis.2023.101115
176. Yan L-j, Qi S, Wu C, Jin R, Hu C, Wang A-l, et al. Hypocrellin A from an ethnic medicinal fungus protects against NLRP3-driven gout in mice by suppressing inflammasome activation. Acta pharmacologica Sin. (2024) 46:1016–29. doi: 10.1038/s41401-024-01434-1
177. Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo‐San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. (2017) 36:1811–36. doi: 10.15252/embj.201796697
178. Lindqvist LM, Frank D, McArthur K, Dite TA, Lazarou M, Oakhill JS, et al. Autophagy induced during apoptosis degrades mitochondria and inhibits type I interferon secretion. Cell Death Differentiation. (2017) 25:784–96. doi: 10.1038/s41418-017-0017-z
179. Cadete VJJ, Deschênes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, et al. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol. (2016) 594:5343–62. doi: 10.1113/jp272703
180. Siu T, Altman MD, Baltus GA, Childers M, Ellis JM, Gunaydin H, et al. Discovery of a novel cGAMP competitive ligand of the inactive form of STING. ACS Medicinal Chem Lett. (2019) 10:92–7. doi: 10.1021/acsmedchemlett.8b00466
181. Wang X, Wang Y, Cao A, Luo Q, Chen D, Zhao W, et al. Development of cyclopeptide inhibitors of cGAS targeting protein-DNA interaction and phase separation. Nat Commun. (2023) 14:6132. doi: 10.1038/s41467-023-41892-5
Keywords: immunity, mitochondrial DNA, cell-free DNA, blood circulation, mitochondria, extracellular vesicles
Citation: Zhao Y, Wu C, Liang X and Yang M (2025) Cell-free mitochondrial DNA as a pro-inflammatory agent in blood circulation: mechanisms, therapeutic implications, and clinical challenges in immune dysregulation. Front. Immunol. 16:1640748. doi: 10.3389/fimmu.2025.1640748
Received: 04 June 2025; Accepted: 08 September 2025;
Published: 01 October 2025.
Edited by:
Antonio Riva, Foundation for Liver Research, United KingdomReviewed by:
Vishal C. Patel, King’s College London, United KingdomMayank Garg, Ashoka University, India
Copyright © 2025 Zhao, Wu, Liang and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yangyang Zhao, Wnl5NjMxNzAwNTQzQDE2My5jb20=
†These authors have contributed equally to this work