 James A. Pearson
James A. Pearson Stephanie J. Hanna
Stephanie J. Hanna- Diabetes Research Group, Division of Infection and Immunity, School of Medicine, Cardiff University, Cardiff, United Kingdom
Type 1 diabetes (T1D) is an autoimmune disease characterized by the destruction of insulin-producing β-cells in the pancreatic islets. The pathogenesis, involving complex interactions between genetic susceptibility and environmental factors, is mediated by T cells driven by multiple stimuli including cytokines. Interleukin-32 (IL-32), a predominantly proinflammatory cytokine, has emerged as a potential contributor to T1D pathogenesis. In this review we discuss current knowledge of IL-32 and its role in T1D pathogenesis, examining expression patterns in PBMCs and islets, possible functional mechanisms, and the potential for IL-32 as a biomarker. We will also consider how immunotherapies currently in clinical trials aiming to slow T1D progression may impact IL-32.
1 Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by destruction of insulin-producing β-cells in the pancreatic islets by autoantigen-specific T cells. The pathogenesis of T1D involves a complex interaction of genetic risk factors, environmental triggers, and immune dysregulation. T1D is currently treated with exogenous insulin, but in recent years a number of immunotherapies have entered clinical trials with the aim of slowing the loss of β-cells (1). Therefore, it has become crucial to understand how to both target and monitor the immune system in T1D. Among the various inflammatory mediators implicated in T1D, cytokines play a crucial role in orchestrating immune responses and β-cell destruction (2). Whilst many of these cytokines have been well characterized, until recently relatively little was known about IL-32. This review aims to summarize the role of IL-32 in the immune system, with a focus on its impact on T1D progression.
1.1 IL-32 structure
Interleukin-32 (IL-32), first identified as natural killer cell transcript 4 (NK4), is a (generally) proinflammatory cytokine that has gained attention for its potential role in various inflammatory and autoimmune diseases, such as rheumatoid arthritis and inflammatory bowel disease (3). IL-32 has 35 known splice variants (https://useast.ensembl.org/Homo_sapiens/Gene/Splice?g=ENSG00000008517;r=16:3065297-3082192) of which 29 produce proteins, varying in length from 131 to 234 amino acids. All of these, except IL-32γ, lack a typical secretory peptide sequence, and indeed bear little structural resemblance to other cytokines (3, 4). Orthologues have been found in primates but not in rodents, whilst in other mammals, putative homologues have very low sequence alignment with human IL-32 (5). The lack of a rodent model has likely contributed to the lack of knowledge of IL-32; however, in vitro and in vivo human studies are driving our better understanding of IL-32 in both health and disease settings.
1.2 Induction of IL-32 expression
IL-32 is highly expressed in CD4+ T cell subsets including Tregs, Th1, Th17, Th17.1, Tfh and Th2 cells, as well as activated and memory CD8+ T cells, with lower expression in naïve T cells (3, 6). In many cases, the isoforms produced have not been assessed, although the majority of PBMC subsets appear to produce at least the α, β, γ and δ isoforms (reviewed in (3)). Recent work suggests that CD4+ T cells predominantly produce IL-32β and are the major source of IL-32β found in the serum (7). IL-32 is also highly expressed in NK cells; however, expression is generally low in both monocytes and naïve B cells (3). IL-32 is often observed as upregulated in disease states, including autoimmunity, in these cells (3, 6, 8, 9).
The expression of IL-32 is induced in T cells, other leukocytes and cells such as epithelial cells and fibroblasts by various proinflammatory cytokines including IL-1β, TNFα, IFNγ, IL-12, IL-18 and IL-23 (reviewed in (3)). In NK cells IL-2 is a strong inducer of IL-32 with a somewhat weaker effect in T cells (10). Recently, it has been shown that IL-32 (particularly IL-32β) is produced in response to IL-2 and secreted via membrane pores and exosomes in response to TCR stimulation (7). In T cells, IL-32 can be induced by in vitro stimulation of T cells using anti-CD3 antibodies or PMA and ionomycin (10). This induction by pro-inflammatory cytokines and T cell activation has important implications for the role of IL-32 in T1D.
IL-32 can also be induced in response to hypoxia via HIF1α and cysteamine dioxygenase (ADO) (11). As both IL-1β and IL-18 can induce IL-32, it is not surprising that innate immune receptors such as Toll-like receptors (TLRs) (12, 13) and inflammasomes e.g. NLPR3 (14), which induce IL-1β and IL-18, are associated with increased IL-32 induction. Recent research has drawn attention to both the role of hypoxia (15) and TLR signaling (16) in T1D development, suggesting an additional role for IL-32 in T1D pathogenesis.
1.3 IL-32 functions
A classical receptor for IL-32 has not been identified; however it can bind proteinase 3 (PR3), a neutrophil granule serine protease (17), and can bind integrins (αVβ3, αVβ6 but not αVβ8) through an RGD domain (18, 19). Although, as mentioned above, most isoforms lack a secretory domain, IL-32 can be measured in the serum and therefore is likely to function both intra- and extracellularly.
The most widely studied isoforms (α, β, γ, δ), are thought to have distinct biological functions (5) with all four inducing IL-6 production from PBMC, but only the latter three capable of inducing TNFα (20). The isoform-specific actions of IL-32 have been summarized previously (3). All IL-32 isoforms can induce IL-8 production, although IL-32γ and IL-32θ were the most potent (18). IL-32γ is thought to be more proinflammatory, inducing TNFα and IL-6 in rheumatoid arthritis synovial fibroblasts whilst the IL-32β isoform is thought to reduce inflammation (21). Pro-inflammatory cytokines such as TNFα and IFNγ are known to play important roles in T1D development and thus it can be seen that IL-32 may contribute to T1D pathology (1).
Whilst lentiviral knock-down of IL-32 expression reduced CD8+ T cell production of IFNγ, it also reduced expression of FoxP3 by Tregs in vitro (22). Conversely, in vitro culture of PBMCs with IL-32α led to Treg cell death and downregulation of FoxP3 expression (9). These apparently contradictory studies are likely explainable by the role of different IL-32 isoforms acting intracellularly or extracellularly. Therefore, care must therefore be taken when considering monitoring or targeting IL-32 therapeutically in T1D to analyze the various isoforms and assess their differential functions.
IL-32 can induce apoptosis in a wide variety of cell types including T cells (10). Again it is likely that this function is enacted by specific isoforms, as in cell lines IL-32γ and IL-32β, but not IL-32α, induced caspase-8-dependent cell death (23).
IL-32 can aid mitochondrial metabolism (via interactions with the electron transport chain and promotion of oxidative phosphorylation), and promotes proliferation, and differentiation of plasma cells (11) (this effect was driven by intracellular IL-32β and various IL-32 isoforms added to the extracellular media had no effect, highlighting the complexity of studying IL-32). As oxidative phosphorylation is also key in driving activation of T cells and decisions between pathogenic and regulatory T cells (24), the role IL-32 has in this setting should be further investigated.
IL-32, particularly the IL-32γ isoform, has been shown to activate Langerhans cells in the skin and induce CD80, HLA-DR and CXCL10 production (25). This is of particular interest in T1D, where CXCL10 is raised in the peripheral blood (26) and CXCR3 expressed in the islets aids recruitment of autoantigen-specific T cells and thus is a target for immunotherapy (27). In DCs, IL-32γ can upregulate the expression of the chemokines CCL2, CCL4, and CCL5, with upregulation of CCL5 in particular leading to increased chemotaxis of T cells (28). Further, IL-32 can induce monocytes to differentiate into macrophage-like cells (29) and IL-32θ in particular can induce monocytes to differentiate to inflammatory M1 macrophages that produce IL-1β, TNFα and inducible nitric oxide synthase (30). In T1D due to the high numbers of macrophages that infiltrate the islets (31) and their role in driving CD8+ T cell destruction of the β-cells (32), IL-32 could therefore be important in modulating these autoimmune responses.
2 IL-32 in the serum and its role in other autoimmune and immune system-related diseases
Serum levels of IL-32 are elevated in a number of autoimmune conditions including Graves’ disease (where the percentage of IL-32α+ T cells was also increased) (33). In psoriasis IL-32 expression is increased in infiltrating Tregs, Th cells and cytotoxic T cells, although there are conflicting reports of whether IL-32 is increased in the serum (34, 35). IL-32α is elevated in the serum in myasthenia gravis (36) (reviewed (3)). In ankylosing spondylitis IL-32γ was elevated in the synovial fluid and in rheumatoid arthritis IL-32 was raised in synovial biopsies, correlating with pro-inflammatory cytokine levels and decreased with anti-TNFα therapy (37, 38). In vitro experiments suggested this effect was predominantly driven by the IL-32γ isoform. Similarly in Behçet’s disease IL-32 is raised in the serum and in the CSF of neuro-Behcet’s (14, 39). In IBD IL-32α was elevated in the mucosa (40) (reviewed (3)), whilst in atopic dermatitis, serum levels of IL-32 correlated to severity and were reduced with successful treatment (41), potentially implicating IL-32 as a useful biomarker for disease progression and favorable therapy responses.
Serum levels of IL-32 can also be affected by SNPs in the IL32 gene https://www.ebi.ac.uk/gwas/genes/IL32 (42) and the presence of a C allele or CC genotype in SNP rs45499297(T/C) has been implicated in the elevated serum levels of IL-32 seen in some people with multiple sclerosis and an earlier age of onset (14, 43), as well as a risk factor for the development of multiple sclerosis (44). In people with rheumatoid arthritis the SNP rs4786370 (T/C) CC genotype in the IL32 promoter was associated with a favorable lipoprotein profile (45) but also with higher IL-32 and pro-inflammatory cytokine production by PBMC (46). People with SLE are reported to have lower levels of IL-32 in the serum and the presence of the IL32 SNP rs28372698 (A/T) TT genotype was associated with SLE susceptibility (47). SNPs rs10431961(C/T) presence of T allele and rs7188573 (T/C) presence of C allele in the IL32 region were associated with juvenile idiopathic arthritis risk as well as extent of IL32 methylation (48). Although in a recent GWAS of T1D IL32 SNPs were not identified as significantly contributing to overall T1D risk (49), the effects of specific SNPs on age of development, endotype or response to immunotherapies have not been specifically investigated nor have IL-32 levels in the serum been studied in T1D.
3 IL-32 expression in PBMC in T1D
Kallionpää et al. performed longitudinal bulk RNAseq on peripheral blood samples from children as they progressed to diabetes and found that high IL-32 expression in CD4+, CD8+, CD4−CD8− cells, and PBMC fractions from peripheral blood was strongly associated with seroconversion and progression to T1D (50). Utilizing scRNAseq the authors identified highly activated and differentiated T cells and NK cells as a major source of IL-32 (50). In a further study, a locus at the promoter of IL32 was hypomethylated in CD8+ T cells of children who progressed to T1D compared to controls. This hypomethylation is thought to increase IL-32 expression (51).
Furthermore, when Honardoost et al. compared the upregulated genes in the PBMC of people living with T1D to healthy controls, using upregulated genes identified by Kallionpää et al. (50) and Fasolino et al. (31), they again identified IL-32 as overexpressed in those with T1D, specifically in the CD4+ T cells, CD8+ T cells, Tregs, MAIT, VD2+ γδT cells and NK cells (52).
It has been demonstrated that IL32 is highly expressed by CD4+ T cells from people living with T1D, as assessed by single cell sequencing, in response to neo- and native epitopes of diabetes autoantigens (53). Furthermore, Okamura et al. incubated PBMCs from people living with T1D with a pool of insulin peptides (mainly 15mers) for 2 hours in vitro and found that this stimulation significantly increased the expression of IL32 in the NK cell subset (54). In contrast, there is also a report of IL32 downregulation in peripheral blood CD8+ T cells in people with T1D compared to controls (55). It should be noted that in these studies, the isoform(s) of IL-32 produced at the protein level have not been determined and as described above these isoforms can activate different pro and anti-inflammatory pathways. Therefore, determination of the isoforms upregulated in T1D should be an urgent priority.
4 IL-32 expression in the pancreatic islets in T1D
Fasolino et al. performed scRNAseq of human pancreatic islet cells and in an analysis of differentially expressed genes (DEG) in immune cells between healthy and T1D pancreas samples identified IL-32 as highly differentially expressed (31). In further research, the same group used machine learning to classify whether pancreatic islet cells from autoantibody positive donors were more similar to cells from healthy controls or from people with T1D. They found that IL-32 was a key signature of islet cells from an autoantibody positive donor classified as similar to T1D (56).
Kallionpää et al. demonstrated that exposure to IL-1β and IFNγ induced IL-32 expression in a pancreatic β-cell line (EndoC-βH1), while treatment of the β-cell line with IL-32γ did not affect their survival or ability to produce insulin (50). Similarly, Dettmer et al. also found that stem-cell derived β-cells and the EndoC-βH1 β cell line upregulate IL-32 expression in response to pro-inflammatory cytokines IL-1β, TNFα and IFNγ (57). Finally, in a human IL-32γ-expressing transgenic mouse model, when mice expressed human IL-32γ, streptozotocin-induced diabetes was accelerated (58), highlighting IL-32 as an important biomarker for T1D progression.
5 IL-32 up- and downstream of drug targets for T1D
Taken together, the paragraphs above demonstrate the importance of IL-32 in many of the pathological processes in T1D. Due to the absence of IL-32 in mice, particularly in the well-studied Non-Obese Diabetic (NOD) mouse model of T1D, it remains unknown as to whether in humans, IL-32 function is necessary or sufficient for T1D initiation or progression. Nevertheless, it can be hypothesized that downregulating the expression or function of pro-inflammatory isoforms of IL-32 would be an attractive option in T1D immunotherapy. Yet, as a predominantly intracellular protein with an unclear secretory mechanism, many isoforms, and several putative binding partners but no identified specific receptor (5), it is perhaps unsurprising that no candidate drugs that target IL-32 are in clinical development. However, we can consider its role in specific pathways targeted in recent immunotherapy clinical trials (Figure 1, Table 1).
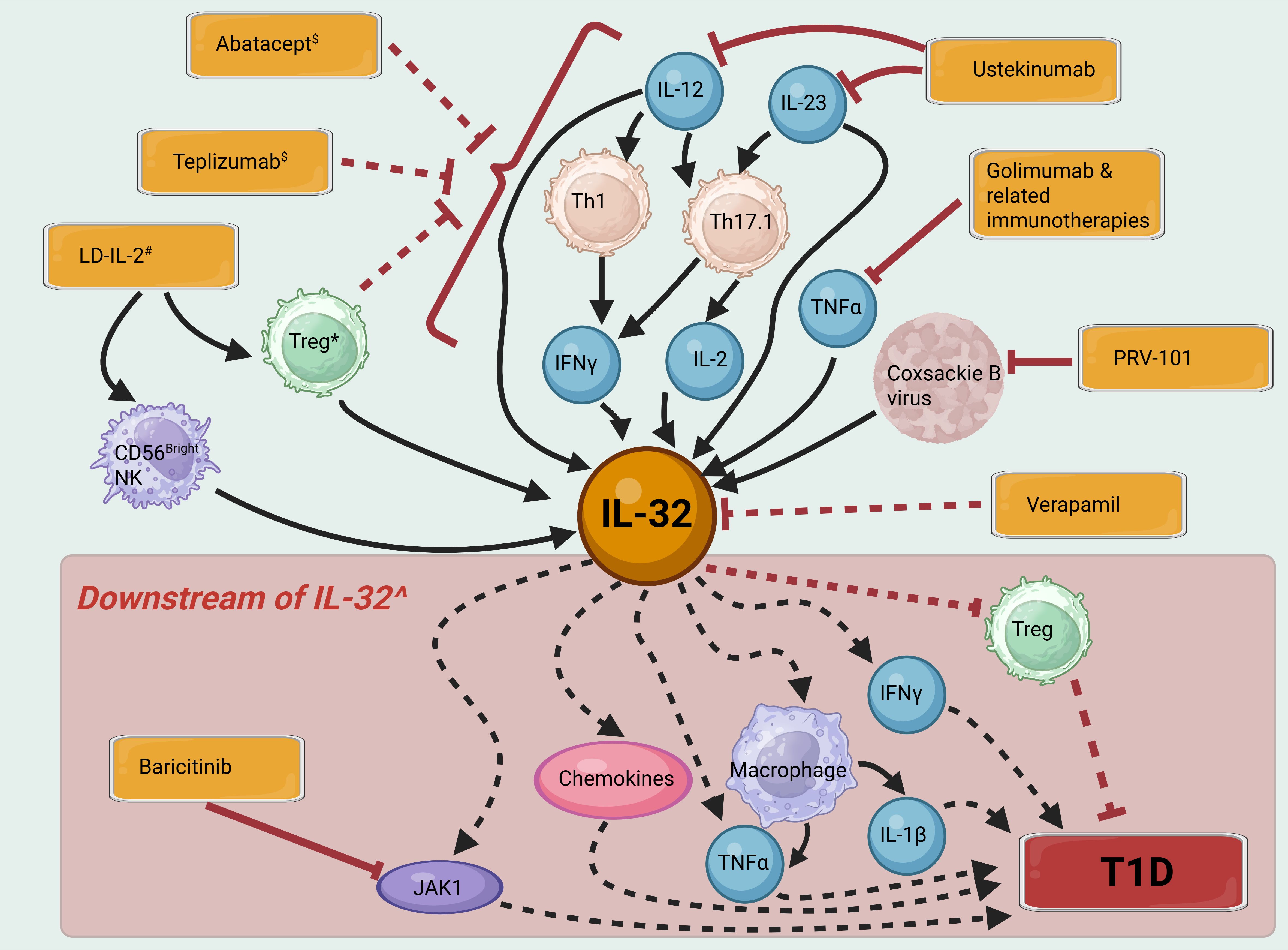
Figure 1. IL-32 signaling pathways and their potential contribution to T1D pathogenesis. Immunotherapies and vaccines are shown in orange boxes. Solid black arrows indicate activation of a pathway, dashed black arrows indirect activation, solid red bars inhibition and dashed red bars indirect inhibition. $ Immunotherapy with Abatacept and Teplizumab aims to induce exhaustion and depletion of a range of effector T cell subsets. # Low dose (LD)-IL-2 may increase IL-32 in some cell subsets, whilst depleting cell subsets producing more proinflammatory isoforms of IL-32. * Whilst Tregs are themselves a source of IL-32, their inhibitory effects on proinflammatory immune cell populations may be expected to have the overall effect of reducing IL-32 levels. ^ Most isoforms of IL-32 are thought to contribute in varying proportions to the pro-inflammatory actions shown here, however IL-32β may have some opposing, anti-inflammatory effects. Created in BioRender. H, S. (2025) https://BioRender.com/5r3h56d.

Table 1. Immunotherapies and vaccines for T1D that influence the IL-32 pathway.
5.1 Prevention of viral infection
Coxsackie B infection is hypothesized to contribute to T1D development (68) and a vaccine, PRV-101, for coxsackie B is in clinical development with a view to preventing T1D (69). Expression of IL-32 in pancreatic islets is increased by Coxsackie B infection (50). Furthermore, reporter cell lines infected with enterovirus strains isolated from Network for Pancreatic Organ Donors with Diabetes (nPOD) pancreases exhibited increased IL-32 expression compared to those infected with control enterovirus strains (64). Therefore, a potential mechanism of action of the vaccine may be through suppression of IL-32 production and thus should be investigated.
5.2 Immunotherapies that alter cytokine signaling
We have recently demonstrated that Ustekinumab, which blocks IL-12 and IL-23 signaling, can slow the loss of insulin production from β-cells in new-onset T1D (66). IL-32 is induced by IL-12 signaling in NK cells and by IL-12, IL-23, IL-2 and IFNγ signaling in T cells ((7) and reviewed (5)). Therefore, Ustekinumab’s blockade of IL-12 and IL-23 signaling, coupled with the downstream decrease in dual IL-17/IFNγ-secreting Th17.1 cells, (particularly those that co-express IL-2), may reduce β-cell loss partly through suppression of IL-32 induction.
TNFα and IL-32 have both been shown to induce each other in a positive feedback loop in arthritis in PBMCs, lymphoid tissue and DCs (62) again suggesting that the effects of Golimumab and other immunotherapies targeting TNFα to slow T1D progression may involve suppression of IL-32 (70, 71).
As discussed above, IL-2 is thought to induce IL-32 expression. However, treatment with low-dose IL-2 is thought to preserve β-cell function primarily through the specific expansion of Treg cells and reduction of IL-21-producing CD4+ T cells, without activating effector subsets of T cells and NK cells (63, 72). An examination of IL-32 expression in participants receiving low-dose IL-2 showed a significant upregulation of IL-32 a month after the final dose of IL-2 in CD56bright NK cells after stimulation with PMA and ionomycin (63). This population of IL-32-expressing CD56bright NK cells induced by low-dose IL-2 is thought to have immunoregulatory properties and therefore the impact of increased IL-32 expression is not clear but should be examined in future studies. If the IL-32 isoforms upregulated are predominantly pro-inflammatory (e.g. IL-32γ) this may reduce the immunoregulatory effect of the CD56bright NK cells, whereas if the IL-32 isoforms have anti-inflammatory actions (e.g. IL-32β) this may potentiate the immunoregulatory properties of the CD56bright NK cells (21, 63).
5.3 Inhibition of kinases
Inhibition of JAK1 can reverse T1D in NOD mice (73). In humans, the JAK1/2 inhibitor Baricitinib was successful in slowing progression of T1D in a Phase 2 trial [BANDIT (74)], this research is being continued in the recently launched JAKPOT T1D study (NCT05743244) and T1DPlus (ISRCTN45965456). IL-32 has been demonstrated to upregulate JAK1 expression and increase activation of the JAK1 signaling pathway (61), therefore inhibition with Baricitinib may act to reduce the effects of overexpression of IL-32 in T1D.
5.4 Modulation of T cell subsets
Teplizumab, an anti-CD3 monoclonal antibody, currently the sole licensed immunotherapy for delaying the onset of T1D in the USA (75) acts via inhibition of CD3 activation during TCR signaling, leading to modification of T cell subset abundance and the partial exhaustion of CD8+ T cells (65). In scRNAseq analysis, IL32 gene expression was significantly downregulated in CD4+ T cells from individuals treated with Teplizumab compared to placebo controls at 18 months (65).
Abatacept, a fusion protein of the extracellular domain of CTLA4 fused to the Fc region of IgG1,has been shown to decrease Th17 and Th1 cell abundance and proliferation in in rheumatoid arthritis (59, 60) and its efficacy is linked to baseline levels of Th17.1 cells. Therefore it may reduce IL-32 expression in T1D through decreased IFNγ signaling; however, as Abatacept also decreases abundance of Tregs the overall balance of its effect on IL-32 levels should also be determined (76).
5.5 β-cell preservation with verapamil
Verapamil has been shown in the CLVer study to slow C-peptide loss in T1D (77), a result that is being followed up in Vera-T1D [awaiting publication of results (78)]. Verapamil will also be given to all participants in T1DPlus (in addition to immunotherapies in different arms of the trial). In human islet samples in vitro treatment with verapamil has been shown to reduce IL-32 expression and it is hypothesized that this is one of the main pathways through which it exerts its protective effects on β-cells (67). Further investigation is needed to assess the effect of verapamil on IL-32 in immune cells.
6 Conclusion
In summary there is an increasing body of evidence which positions IL-32 as a key cytokine involved in the autoimmune pathogenesis of T1D. IL-32 upregulation occurs in peripheral blood immune cells early in the disease process and is a feature of the autoantigen-specific T cell response; however, the expression and functions of the different isoforms, particularly IL-32β which may have anti-inflammatory actions, remain poorly defined.
Many T1D immunotherapies are predicted to impact IL-32 production and signaling. Therefore, there is a strong case to develop IL-32 as a biomarker to not only monitor T1D progression but also to evaluate the effectiveness of immunotherapies in clinical trials. Whilst this could be through analysis of IL-32 at the mRNA or protein level in PBMC subsets, levels of IL-32 in the serum of people with T1D should also be assessed. Monitoring IL-32 as a novel biomarker may help identify the immunotherapy that individuals would respond best to, while also ensuring those at greatest risk of developing T1D are more closely monitored or offered disease-modifying therapy.
Because IL-32 is not expressed in most other mammals and is not targeted by any immunotherapies in clinical development, the field lacks definitive evidence that direct inhibition of IL-32 function would be sufficient to prevent either the initiation of β-cell autoimmunity or the progression of T1D in humans. However, development of monoclonal antibodies to target extracellular IL-32 or adaption of siRNA approaches (79) to knockdown intracellular IL-32 expression would allow this question to be addressed in both T1D and other autoimmune diseases where current evidence suggests a crucial role for IL-32.
Data availability statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Author contributions
JP: Funding acquisition, Writing – original draft, Writing – review & editing. SH: Conceptualization, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by a Medical Research Council Career Development Award (MR/T010525/1) and on behalf of the “Steve Morgan Foundation Type 1 Diabetes Grand Challenge” by Breakthrough T1D UK (formerly JDRF), and SMF (grant numbers 2-SRA-2024-1474-M-N and 2-SRA-2024-1473-M-N).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Gen AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Tatovic D and Dayan C. Clinical immunologic interventions for the treatment of type 1 diabetes: challenges, choice, and timing of immunomodulators. Cold Spring Harb Perspect Med. (2025), a041597. doi: 10.1101/cshperspect.a041597
2. Benaglio P, Zhu H, Okino M-L, Yan J, Elgamal R, Nariai N, et al. Type 1 diabetes risk genes mediate pancreatic beta cell survival in response to proinflammatory cytokines. Cell Genomics. (2022) 2:100214. doi: 10.1016/j.xgen.2022.100214
3. De Albuquerque R, Komsi E, Starskaia I, Ullah U, and Lahesmaa R. The role of Interleukin-32 in autoimmunity. Scand J Immunol. (2021) 93:e13012. doi: 10.1111/sji.13012
4. Sohn DH, Nguyen TT, Kim S, Shim S, Lee S, Lee Y, et al. Structural characteristics of seven IL-32 variants. Immune Netw. (2019) 19:e8. doi: 10.4110/in.2019.19.e8
5. Aass KR, Kastnes MH, and Standal T. Molecular interactions and functions of IL-32. J Leukocyte Biol. (2021) 109:143–59. doi: 10.1002/JLB.3MR0620-550R
6. Dey NS, Dey S, Brown N, Senarathne S, Campos Reis L, Sengupta R, et al. IL-32-producing CD8+ memory T cells define immunoregulatory niches in human cutaneous leishmaniasis. J Clin Invest. (2025) 135:e182040. doi: 10.1172/JCI182040
7. Sanna FC, Benešová I, Pervan P, Krenz A, Wurzel A, Lohmayer R, et al. IL-2 and TCR stimulation induce expression and secretion of IL-32β by human T cells. Front Immunol. (2024) 15:1437224. doi: 10.3389/fimmu.2024.1437224
8. Soussi S, Maione AS, Lefèvre L, Pizzinat N, Iacovoni J, Gonzalez-Fuentes I, et al. Analysis of effector/memory regulatory T cells from arrhythmogenic cardiomyopathy patients identified IL-32 as a novel player in ACM pathogenesis. Cell Death Dis. (2025) 16:87. doi: 10.1038/s41419-025-07364-y
9. Galván-Peña S, Leon J, Chowdhary K, Michelson DA, Vijaykumar B, Yang L, et al. Profound Treg perturbations correlate with COVID-19 severity. Proc Natl Acad Sci USA. (2021) 118:e2111315118. doi: 10.1073/pnas.2111315118
10. Goda C, Kanaji T, Kanaji S, Tanaka G, Arima K, Ohno S, et al. Involvement of IL-32 in activation-induced cell death in T cells. Int Immunol. (2006) 18:233–40. doi: 10.1093/intimm/dxh339
11. Aass KR, Mjelle R, Kastnes MH, Tryggestad SS, Van Den Brink LM, Aass Roseth I, et al. Intracellular IL-32 regulates mitochondrial metabolism, proliferation, and differentiation of Malignant plasma cells. iScience. (2022) 25:103605. doi: 10.1016/j.isci.2021.103605
12. Aass KR, Tryggestad SS, Mjelle R, Kastnes MH, Nedal TMV, Misund K, et al. IL-32 is induced by activation of toll-like receptors in multiple myeloma cells. Front Immunol. (2023) 14:1107844. doi: 10.3389/fimmu.2023.1107844
13. Zhang L, Che C, Lin J, Liu K, Li D-Q, and Zhao G. TLR-mediated induction of proinflammatory cytokine IL-32 in corneal epithelium. Curr Eye Res. (2013) 38:630–8. doi: 10.3109/02713683.2012.763102
14. Hamzaoui K, Borhani-Haghighi A, Dhifallah IB, and Hamzaoui A. Elevated levels of IL-32 in cerebrospinal fluid of neuro-Behcet disease: Correlation with NLRP3 inflammasome. J Neuroimmunol. (2022) 365:577820. doi: 10.1016/j.jneuroim.2022.577820
15. Fagundes RR, Zaldumbide A, and Taylor CT. Role of hypoxia-inducible factor 1 in type 1 diabetes. Trends Pharmacol Sci. (2024) 45:798–810. doi: 10.1016/j.tips.2024.07.001
16. Huang J, Peng J, Pearson JA, Efthimiou G, Hu Y, Tai N, et al. Toll-like receptor 7 deficiency suppresses type 1 diabetes development by modulating B-cell differentiation and function. Cell Mol Immunol. (2021) 18:328–38. doi: 10.1038/s41423-020-00590-8
17. Novick D, Rubinstein M, Azam T, Rabinkov A, Dinarello CA, and Kim S-H. Proteinase 3 is an IL-32 binding protein. Proc Natl Acad Sci USA. (2006) 103:3316–21. doi: 10.1073/pnas.0511206103
18. Shim S, Lee S, Hisham Y, Kim S, Nguyen TT, Taitt AS, et al. Comparison of the seven interleukin-32 isoforms’ Biological activities: IL-32θ Possesses the most dominant biological activity. Front Immunol. (2022) 13:837588. doi: 10.3389/fimmu.2022.837588
19. Heinhuis B, Koenders MI, van den Berg WB, Netea MG, Dinarello CA, and Joosten LAB. Interleukin 32 (IL-32) contains a typical α-helix bundle structure that resembles focal adhesion targeting region of focal adhesion kinase-1. J Biol Chem. (2012) 287:5733–43. doi: 10.1074/jbc.M111.288290
20. Choi J, Bae S, Hong J, Azam T, Dinarello CA, Her E, et al. Identification of the most active interleukin-32 isoform. Immunology. (2009) 126:535–42. doi: 10.1111/j.1365-2567.2008.02917.x
21. Heinhuis B, Koenders MI, Van De Loo FA, Netea MG, Van Den Berg WB, and Joosten LAB. Inflammation-dependent secretion and splicing of IL-32γ in rheumatoid arthritis. Proc Natl Acad Sci USA. (2011) 108:4962–7. doi: 10.1073/pnas.1016005108
22. Han L, Chen S, Chen Z, Zhou B, Zheng Y, and Shen L. Interleukin 32 promotes foxp3+ Treg cell development and CD8+ T cell function in human esophageal squamous cell carcinoma microenvironment. Front Cell Dev Biol. (2021) 9:704853. doi: 10.3389/fcell.2021.704853
23. Heinhuis B, Plantinga TS, Semango G, Küsters B, Netea MG, Dinarello CA, et al. Alternatively spliced isoforms of IL-32 differentially influence cell death pathways in cancer cell lines. CARCIN. (2016) 37:197–205. doi: 10.1093/carcin/bgv172
24. Shin B, Benavides GA, Geng J, Koralov SB, Hu H, Darley-Usmar VM, et al. Mitochondrial oxidative phosphorylation regulates the fate decision between pathogenic th17 and regulatory T cells. Cell Rep. (2020) 30:1898–1909.e4. doi: 10.1016/j.celrep.2020.01.022
25. Gonnet J, Perrin H, Hutton AJ, Boccara D, Bonduelle O, Mimoun M, et al. Interleukin-32 promotes detachment and activation of human Langerhans cells in a human skin explant model. Br J Dermatol. (2018) 179:145–53. doi: 10.1111/bjd.16721
26. Powell WE, Hanna SJ, Hocter CN, Robinson E, Davies J, Dunseath GJ, et al. Loss of CXCR3 expression on memory B cells in individuals with long-standing type 1 diabetes. Diabetologia. (2018) 61:1794–803. doi: 10.1007/s00125-018-4651-x
27. Christen U, Pouzol L, Tunis M, Sassi A, Tondello C, Bayer M, et al. Combination treatment of a novel CXCR3 antagonist ACT-777991 with an anti-CD3 antibody synergistically increases persistent remission in experimental models of type 1 diabetes. Clin Exp Immunol. (2023) 214:131–43. doi: 10.1093/cei/uxad083
28. Son MH, Jung MY, Choi S, Cho D, and Kim TS. IL-32γ induces chemotaxis of activated T cells via dendritic cell-derived CCL5. Biochem Biophys Res Commun. (2014) 450:30–5. doi: 10.1016/j.bbrc.2014.05.052
29. Netea MG, Lewis EC, Azam T, Joosten LAB, Jaekal J, Bae S-Y, et al. Interleukin-32 induces the differentiation of monocytes into macrophage-like cells. Proc Natl Acad Sci USA. (2008) 105:3515–20. doi: 10.1073/pnas.0712381105
30. Park H-M, Park J-Y, Kim N-Y, Kim H, Kim H-G, Son D-J, et al. Recombinant human IL-32θ Induces polarization into M1-like macrophage in human monocytic cells. Immune Netw. (2024) 24:e27. doi: 10.4110/in.2024.24.e27
31. Fasolino M, Schwartz GW, Patil AR, Mongia A, Golson ML, Wang YJ, et al. Single-cell multi-omics analysis of human pancreatic islets reveals novel cellular states in type 1 diabetes. Nat Metab. (2022) 4:284–99. doi: 10.1038/s42255-022-00531-x
32. Srivastava N, Hu H, Peterson OJ, Vomund AN, Stremska M, Zaman M, et al. CXCL16-dependent scavenging of oxidized lipids by islet macrophages promotes differentiation of pathogenic CD8+ T cells in diabetic autoimmunity. Immunity. (2024) 57:1629–1647.e8. doi: 10.1016/j.immuni.2024.04.017
33. Yao Q, Wang B, Jia X, Li Q, Yao W, and Zhang J. Increased human interleukin-32 expression is related to disease activity of graves’ Disease. Front Endocrinol. (2019) 10:613. doi: 10.3389/fendo.2019.00613
34. Al-Shobaili HA, Farhan J, Zafar U, and Rasheed Z. Functional role of human interleukin-32 and nuclear transcription factor-kB in patients with psoriasis and psoriatic arthritis. Int J Health Sci (Qassim). (2018) 12:29–34.
35. Frost B, Schmidt M, Klein B, Loeffler-Wirth H, Krohn K, Reidenbach T, et al. Single-cell transcriptomics reveals prominent expression of IL-14, IL-18, and IL-32 in psoriasis. Eur J Immunol. (2023) 53:2250354. doi: 10.1002/eji.202250354
36. Na S-J, So S-H, Lee KO, and Choi Y-C. Elevated serum level of interleukin-32α in the patients with myasthenia gravis. J Neurol. (2011) 258:1865–70. doi: 10.1007/s00415-011-6036-7
37. Heinhuis B, Koenders MI, Van Riel PL, Van De Loo FA, Dinarello CA, Netea MG, et al. Tumour necrosis factor alpha-driven IL-32 expression in rheumatoid arthritis synovial tissue amplifies an inflammatory cascade. Ann Rheumatic Dis. (2011) 70:660–7. doi: 10.1136/ard.2010.139196
38. Joosten LAB, Netea MG, Kim S-H, Yoon D-Y, Oppers-Walgreen B, Radstake TRD, et al. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc Natl Acad Sci USA. (2006) 103:3298–303. doi: 10.1073/pnas.0511233103
39. Ha Y, Park J, Kang M, Lee S, Park Y, and Lee S. Increased serum interleukin-32 levels in patients with Behçet’s disease. Int J Rheum Dis. (2018) 21:2167–74. doi: 10.1111/1756-185X.13072
40. Shioya M, Nishida A, Yagi Y, Ogawa A, Tsujikawa T, Kim-Mitsuyama S, et al. Epithelial overexpression of interleukin-32alpha in inflammatory bowel disease. Clin Exp Immunol. (2007) 149:480–6. doi: 10.1111/j.1365-2249.2007.03439.x
41. Wallimann A and Schenk M. IL-32 as a potential biomarker and therapeutic target in skin inflammation. Front Immunol. (2023) 14:1264236. doi: 10.3389/fimmu.2023.1264236
42. Dhindsa RS, Burren OS, Sun BB, Prins BP, Matelska D, Wheeler E, et al. Rare variant associations with plasma protein levels in the UK Biobank. Nature. (2023) 622:339–47. doi: 10.1038/s41586-023-06547-x
43. Morsaljahan Z, Rafiei A, Valadan R, Abedini M, Pakseresht M, and Khajavi R. Association between interleukin-32 polymorphism and multiple sclerosis. J Neurological Sci. (2017) 379:144–50. doi: 10.1016/j.jns.2017.05.045
44. Parray Z, Zargar MH, Asimi R, Dar WR, Yaqoob A, Raina A, et al. Interleukin 32 gene promoter polymorphism: A genetic risk factor for multiple sclerosis in Kashmiri population. Gene. (2022) 824:146261. doi: 10.1016/j.gene.2022.146261
45. Damen MSMA, Agca R, Holewijn S, De Graaf J, Dos Santos JC, Van Riel PL, et al. IL-32 promoter SNP rs4786370 predisposes to modified lipoprotein profiles in patients with rheumatoid arthritis. Sci Rep. (2017) 7:41629. doi: 10.1038/srep41629
46. Damen MSMA, Schraa K, Tweehuysen L, Den Broeder AA, Netea MG, Popa CD, et al. Genetic variant in IL-32 is associated with the ex vivo cytokine production of anti-TNF treated PBMCs from rheumatoid arthritis patients. Sci Rep. (2018) 8:14050. doi: 10.1038/s41598-018-32485-0
47. Wang Y, Zhou B, Zhao Y, Yu X, Liu Y, and Zhang L. Association of plasma IL-32 levels and gene polymorphisms with systemic lupus erythematosus in chinese han population. Dis Markers. (2016) 2016:1–6. doi: 10.1155/2016/2460206
48. Meyer B, Chavez RA, Munro JE, Chiaroni-Clarke RC, Akikusa JD, Allen RC, et al. DNA methylation at IL32 in juvenile idiopathic arthritis. Sci Rep. (2015) 5:11063. doi: 10.1038/srep11063
49. Crouch DJM, Inshaw JRJ, Robertson CC, Ng E, Zhang J, Chen W, et al. Bayesian effect size ranking to prioritise genetic risk variants in common diseases for follow-up studies. Genet Epidemiol. (2025) 49:e22608. doi: 10.1002/gepi.22608
50. Kallionpää H, Somani J, Tuomela S, Ullah U, de Albuquerque R, Lönnberg T, et al. Early detection of peripheral blood cell signature in children developing β-cell autoimmunity at a young age. Diabetes. (2019) 68:2024–34. doi: 10.2337/db19-0287
51. Starskaia I, Laajala E, Grönroos T, Härkönen T, Junttila S, Kattelus R, et al. Early DNA methylation changes in children developing beta cell autoimmunity at a young age. Diabetologia. (2022) 65:844–60. doi: 10.1007/s00125-022-05657-x
52. Honardoost MA, Adinatha A, Schmidt F, Ranjan B, Ghaeidamini M, Arul Rayan N, et al. Systematic immune cell dysregulation and molecular subtypes revealed by single-cell RNA-seq of subjects with type 1 diabetes. Genome Med. (2024) 16:45. doi: 10.1186/s13073-024-01300-z
53. Arif S, Pujol-Autonell I, Kamra Y, Williams E, Yusuf N, Domingo-Vila C, et al. Mapping T cell responses to native and neo-islet antigen epitopes in at risk and type 1 diabetes subjects. Front Immunol. (2021) 12:675746. doi: 10.3389/fimmu.2021.675746
54. Okamura T, Kitagawa N, Kitagawa N, Sakai K, Sumi M, Kobayashi G, et al. Single-cell analysis reveals islet autoantigen’s immune activation in type 1 diabetes patients. J Clin Biochem Nutr. (2025) 76:64–84. doi: 10.3164/jcbn.24-86
55. Okamura T, Hamaguchi M, Tominaga H, Kitagawa N, Hashimoto Y, Majima S, et al. Characterization of peripheral blood TCR in patients with type 1 diabetes mellitus by BD rhapsodyTM VDJ CDR3 assay. Cells. (2022) 11:1623. doi: 10.3390/cells11101623
56. Patil AR, Schug J, Liu C, Lahori D, Descamps HC, Naji A, et al. Modeling type 1 diabetes progression using machine learning and single-cell transcriptomic measurements in human islets. Cell Rep Med. (2024) 5:101535. doi: 10.1016/j.xcrm.2024.101535
57. Dettmer R, Niwolik I, Cirksena K, Yoshimoto T, Tang Y, Mehmeti I, et al. Proinflammatory cytokines induce rapid, NO-independent apoptosis, expression of chemotactic mediators and interleukin-32 secretion in human pluripotent stem cell-derived beta cells. Diabetologia. (2022) 65:829–43. doi: 10.1007/s00125-022-05654-0
58. Jhun H, Choi J, Hong J, Lee S, Kwak A, Kim E, et al. IL-32γ overexpression accelerates streptozotocin (STZ)-induced type 1 diabetes. Cytokine. (2014) 69:1–5. doi: 10.1016/j.cyto.2014.05.002
59. Scarsi M, Zanotti C, Chiarini M, Imberti L, Piantoni S, Frassi M, et al. Reduction of peripheral blood T cells producing IFN-γ and IL-17 after therapy with abatacept for rheumatoid arthritis. Clin Exp Rheumatol. (2014) 32:204–10.
60. Maeda S, Osaga S, Maeda T, Takeda N, Tamechika S, Naniwa T, et al. Circulating Th17.1 cells as candidate for the prediction of therapeutic response to abatacept in patients with rheumatoid arthritis: An exploratory research. PloS One. (2019) 14:e0215192. doi: 10.1371/journal.pone.0215192
61. Chang J, Zhou B, Wei Z, and Luo Y. IL-32 promotes the occurrence of atopic dermatitis by activating the JAK1/microRNA-155 axis. J Transl Med. (2022) 20:207. doi: 10.1186/s12967-022-03375-x
62. Shoda H, Fujio K, Yamaguchi Y, Okamoto A, Sawada T, Kochi Y, et al. Interactions between IL-32 and tumor necrosis factor alpha contribute to the exacerbation of immune-inflammatory diseases. Arthritis Res Ther. (2006) 8:R166. doi: 10.1186/ar2074
63. Zhang J-Y, Hamey F, Trzupek D, Mickunas M, Lee M, Godfrey L, et al. Low-dose IL-2 reduces IL-21+ T cell frequency and induces anti-inflammatory gene expression in type 1 diabetes. Nat Commun. (2022) 13:7324. doi: 10.1038/s41467-022-34162-3
64. Poma AM, Genoni A, Broccolo F, Denaro M, Pugliese A, Basolo F, et al. Immune transcriptome of cells infected with enterovirus strains obtained from cases of type 1 diabetes. Microorganisms. (2020) 8:1031. doi: 10.3390/microorganisms8071031
65. Lledó-Delgado A, Preston-Hurlburt P, Currie S, Clark P, Linsley PS, Long SA, et al. Teplizumab induces persistent changes in the antigen-specific repertoire in individuals at risk for type 1 diabetes. J Clin Invest. (2024) 134:e177492. doi: 10.1172/JCI177492
66. Tatovic D, Marwaha A, Taylor P, Hanna SJ, Carter K, Cheung WY, et al. Ustekinumab for type 1 diabetes in adolescents: a multicenter, double-blind, randomized phase 2 trial. Nat Med. (2024) 30:2657–66. doi: 10.1038/s41591-024-03115-2
67. Xu G, Grimes TD, Grayson TB, Chen J, Thielen LA, Tse HM, et al. Exploratory study reveals far reaching systemic and cellular effects of verapamil treatment in subjects with type 1 diabetes. Nat Commun. (2022) 13:1159. doi: 10.1038/s41467-022-28826-3
68. Carré A, Vecchio F, Flodström-Tullberg M, You S, and Mallone R. Coxsackievirus and type 1 diabetes: diabetogenic mechanisms and implications for prevention. Endocrine Rev. (2023) 44:737–51. doi: 10.1210/endrev/bnad007
69. Hyöty H, Kääriäinen S, Laiho JE, Comer GM, Tian W, Härkönen T, et al. Safety, tolerability and immunogenicity of PRV-101, a multivalent vaccine targeting coxsackie B viruses (CVBs) associated with type 1 diabetes: a double-blind randomised placebo-controlled Phase I trial. Diabetologia. (2024) 67:811–21. doi: 10.1007/s00125-024-06092-w
70. Rigby MR, Hayes B, Li Y, Vercruysse F, Hedrick JA, and Quattrin T. Two-year follow-up from the T1GER study: continued off-therapy metabolic improvements in children and young adults with new-onset T1D treated with golimumab and characterization of responders. Diabetes Care. (2023) 46:561–9. doi: 10.2337/dc22-0908
71. Bazile C, Abdel Malik MM, Ackeifi C, Anderson RL, Beck RW, Donath MY, et al. TNF-α inhibitors for type 1 diabetes: exploring the path to a pivotal clinical trial. Front Immunol. (2024) 15:1470677. doi: 10.3389/fimmu.2024.1470677
72. Rosenzwajg M, Salet R, Lorenzon R, Tchitchek N, Roux A, Bernard C, et al. Low-dose IL-2 in children with recently diagnosed type 1 diabetes: a Phase I/II randomised, double-blind, placebo-controlled, dose-finding study. Diabetologia. (2020) 63:1808–21. doi: 10.1007/s00125-020-05200-w
73. Ge T, Jhala G, Fynch S, Akazawa S, Litwak S, Pappas EG, et al. The JAK1 selective inhibitor ABT 317 blocks signaling through interferon-γ and common γ Chain cytokine receptors to reverse autoimmune diabetes in NOD mice. Front Immunol. (2020) 11:588543. doi: 10.3389/fimmu.2020.588543
74. Waibel M, Wentworth JM, So M, Couper JJ, Cameron FJ, MacIsaac RJ, et al. Baricitinib and β-cell function in patients with new-onset type 1 diabetes. N Engl J Med. (2023) 389:2140–50. doi: 10.1056/NEJMoa2306691
75. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An anti-CD3 antibody, teplizumab, in relatives at risk for type 1 diabetes. N Engl J Med. (2019) 381:603–13. doi: 10.1056/NEJMoa1902226
76. Russell WE, Bundy BN, Anderson MS, Cooney LA, Gitelman SE, Goland RS, et al. Abatacept for delay of type 1 diabetes progression in stage 1 relatives at risk: A randomized, double-masked, controlled trial. Diabetes Care. (2023) 46:1005–13. doi: 10.2337/dc22-2200
77. Forlenza GP, McVean J, Beck RW, Bauza C, Bailey R, Buckingham B, et al. Effect of verapamil on pancreatic beta cell function in newly diagnosed pediatric type 1 diabetes: A randomized clinical trial. JAMA. (2023) 329:990. doi: 10.1001/jama.2023.2064
78. Wych J, Brunner M, Stenson R, Chmura PJ, Danne T, Mander AP, et al. Investigating the effect of verapamil on preservation of beta-cell function in adults with newly diagnosed type 1 diabetes mellitus (Ver-A-T1D): protocol for a randomised, double-blind, placebo-controlled, parallel-group, multicentre trial. BMJ Open. (2024) 14:e091597. doi: 10.1136/bmjopen-2024-091597
Keywords: IL-32, type 1 diabetes, immunotherapy, T cells, beta cells, β-cells
Citation: Pearson JA and Hanna SJ (2025) Mini review: Interleukin-32 as a key mediator of type 1 diabetes pathogenesis. Front. Immunol. 16:1641698. doi: 10.3389/fimmu.2025.1641698
Received: 05 June 2025; Accepted: 11 August 2025;
Published: 04 September 2025.
Edited by:
Soohyun Kim, Konkuk University, Republic of KoreaReviewed by:
Makoto Miyara, Hôpitaux Universitaires Pitié Salpêtrière, FranceSaerok Shim, Konkuk University, Republic of Korea
Copyright © 2025 Pearson and Hanna. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stephanie J. Hanna, SGFubmFTSkBjYXJkaWZmLmFjLnVr