 Martin Villalba
Martin Villalba Delphine Gitenay
Delphine Gitenay Sara Zemiti
Sara Zemiti Jean-François Rossi
Jean-François Rossi Mauricio Campos-Mora
Mauricio Campos-Mora- 1IRMB, Univ Montpellier, INSERM, CHU Montpellier, CNRS, Montpellier, France
- 2Institut du Cancer Avignon-Provence Sainte Catherine, Avignon, France
- 3IRMB, Univ Montpellier, INSERM, CHU Montpellier, Montpellier, France
Mammalian cells adapt to their environment by reshaping their metabolism. Increased fatty acid oxidation (FAO) enables metastatic cells to enhance their motility and colonize new niches, where the fatty acid transporter CD36 functions as both marker and driver of this process. The MAPK ERK5 regulates CD36 expression, FAO, and the epithelial-mesenchymal transition (EMT), a critical initial step in metastasis. Contrary to popular belief, metastasis is a highly inefficient process, in part due to natural killer (NK) cell immune surveillance. This cytotoxic lymphocyte lineage detects inhibitory and activating ligands on target cells to determine their fate. During EMT, the expression of specific ligands on metastatic cells triggers their recognition by NK cells. Interestingly, several of these ligands are regulated by ERK5. We hypothesize that ERK5 may serve as a central link between FAO, metastasis, and immune surveillance. Here, we review current knowledge and available evidence regarding ERK5 expression in tumor cells and its role in cancer cell migration and metastasis and speculate in the potential role of ERK5 in immune recognition and the clearance of metastasis by NK cells.
1 Metastatic cells rely on fatty acid oxidation
Metabolism is altered in cancer cells. Since Otto Warburg described aerobic glycolysis (1) many more metabolic alterations have been uncovered, including the upregulation of mitochondrial respiration alongside glycolysis in certain tumors (2). Thus, cancer cells adapt to changing environment by adjusting their metabolism to new conditions-for example, when they migrate and colonize new tissues. This migration, known as metastasis, is highly inefficient because metastatic cells must overcome numerous obstacles to establish colonies in distant tissues (3). Once metastatic lesions are formed, current treatments often fail to provide lasting responses. During their journey to a new “home”, tumor cells encounter a challenging metabolic environment and respond by increasing fatty acid (FA) consumption and generating energy through FA β-oxidation (FAO) (4). Consequently, lipid oxidation, which fuels β-oxidation and mitochondrial respiration, is linked to tumor progression, invasion and the metastatic process (5–7).
Interestingly, to sustain FAO, metastatic cells must increase FA import and activate various genes required for FAO, particularly CD36 (4, 6, 8). Our group has recently shown that this FA transporter is induced by extracellular-regulated kinase 5 (ERK5) (9), also known as big mitogen-activated protein kinase 1 (BMK1) due to its large size (110 kDa), and encoded by the mitogen-activated protein kinase 7 gene (MAPK7). This MAPK plays multiple roles in cancer, including metastasis (10, 11). Tumor cells express higher levels of ERK5 and/or ERK5 mRNA compared to their normal counterparts (12), and its expression levels may have prognostic value (13, 14). In fact, ERK5 is involved in several of the so-called hallmarks of cancer (10). Figure 1 summarizes its roles in these biological processes.
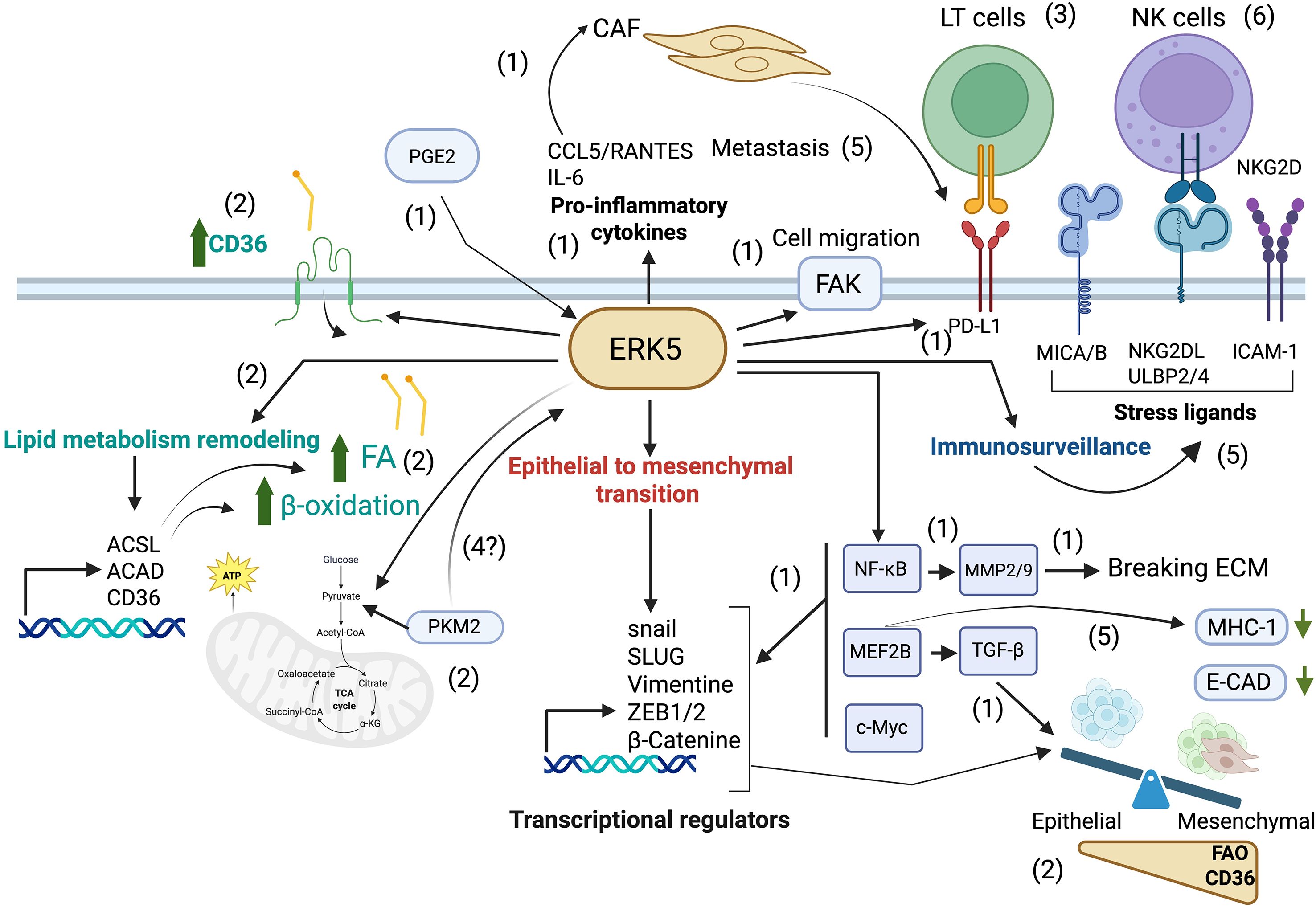
Figure 1. Pivotal role of ERK5 in metastasis. The epithelial-to-mesenchymal transition (EMT) is a critical step in metastasis. ERK5 promotes the accumulation of NF-κB, which activates MMP9, a matrix metalloproteinase that facilitates tumor cell detachment and invasion (1), and phosphorylates FAK favoring tumor cell migration (1). ERK5 also upregulates MEF2B, leading to the activation of TGF-β, a potent EMT inducer (1), and further promotes EMT by activating transcriptional regulators including SNAIL, SLUG, and ZEB/2, resulting in the downregulation of E-cadherin, a hallmark epithelial marker (1). In addition, ERK5 enhances the secretion of pro-inflammatory cytokines such as IL-6 and CCL5, which in turn activate cancer-associated fibroblasts [CAFs; (1)]. After gaining a mesenchymal phenotype, metastatic cells depend on metabolic reprogramming, e.g. to fatty acid β-oxidation [FAO, (2)], to adapt and colonize new environments. ERK5 supports the increased lipid requirements of metastatic cells by promoting fatty acid (FA) accumulation (2) through upregulation of the CD36 receptor (2), and by sustaining FAO (2) via enhanced expression of metabolic enzymes such as ACSLs and ACADs (2), which increases OXPHOS in mitochondria (2). During their journey, metastatic cells can interact with immune cells. By modulating tumor cell metabolism, ERK5 can modulate tumor immune recognition by cytotoxic lymphocytes (3). It is tempting to speculate that mitochondrial changes can signal ERK5 activation through PKM2 favoring tumor cell detachment (4), but also immune recognition by modulating expression of stress ligands (5), ICAM-1 (5) and MHC-I (5). CAFs also contribute to immune evasion by upregulating PD-L1 expression, thereby engaging the PD-1 receptor on T lymphocytes and suppressing their cytotoxic activity (5). By modulating all these molecules, ERK5 regulates NK cell-mediated immune surveillance (6), supporting a central role for ERK5 in the metastatic process. Created in BioRender. Zemiti, S. (2025) https://BioRender.com/.mn9cduu.
2 ERK5 regulates FAO
ERK5, described as a stress kinase (15), regulates cell metabolism (16–19), enables cells to manage oxidative stress (19–22), and mediates lipid metabolism in various cellular contexts (9, 23, 24). Knockdown of the MEK5/ERK5 axis in small-cell lung cancer (SCLC) cells not only reduced proliferation and tumor growth but also altered lipid metabolism-related pathways, including those involved in cholesterol homeostasis and FA metabolism (23). Additionally, transcriptomic analysis revealed connections between ERK5 signaling and pyruvate metabolism, as well as the citrate cycle (23). ERK5 enhances intracellular lipid availability by upregulating fatty acid transporters (9), supports FA activation by inducing the expression of long-chain acyl-CoA synthetase (ACSL) enzymes, and sustains FAO through the expression of Acyl-CoA dehydrogenases (ACADs) (9).
The final steps of FAO require oxidative phosphorylation (OXPHOS) via the redox potential in mitochondria. Recent evidence shows that mitochondrial complex I (CI) promotes kidney cancer metastasis (7), consistent with earlier findings that mutations in the ND2 subunit of mitochondrial complex I increase metastatic potential (25). Interestingly, mitochondrial CI also triggers an antioxidant response through ERK5 (19). In a related context, ACSL4 is a pro-hematogenous metastasis factor that facilitates metastatic extravasation by enhancing membrane fluidity and cellular invasiveness (26). Another ACSL family member, ACSL1, also promotes metastasis in endometrial cancer (27). As noted above, ERK5 induces ACSL expression (9). Therefore, ERK5 facilitates transcriptome remodeling to support FAO, which may be linked to pro-metastatic intracellular signaling.
3 ERK5 regulates epithelial-to-mesenchymal transition and metastasis
Increasing evidence supports the role of FA incorporation and metabolism in cancer and tumor cell migration (28–31). Consistent with its role in FAO, ERK5 activity enhances cell motility (10, 32), cell migration and invasiveness (32–34), EMT, and metastasis (11, 32, 35, 36). MAPK7 gene silencing is associated with reduced migration, invasion, and proliferation in osteosarcoma, oral squamous cell carcinoma (OSCC), and prostate cancer cells (37–40). In breast cancer cells, the MEK/ERK5 pathway is essential for suppressing the estrogen receptor, which is necessary to induce EMT and metastasis (41, 42).
Mechanistically, ERK5 activation upregulates focal adhesion kinase (FAK) expression and promotes its activation through direct phosphorylation, thereby facilitating cell migration and metastasis (32, 43–45). Conversely, MAPK7 knockout in triple-negative breast cancer (TNBC) and prostate cancer cells disrupts the interaction between FAK and proline-rich tyrosine kinase 2 (PYK2), reducing cell attachment to substrates such as vitronectin and fibronectin (46).
In melanoma xenografts, lung and lymph node metastases were drastically reduced upon transfection with dominant-negative isoforms of MEK5 and ERK5 (32). Similarly, MAPK7 knockout in TNBC cell lines impaired cell migration by diminishing mesenchymal features, including the expression of matrix-associated genes, integrins, and pro-angiogenic factors (47). Pharmacological inhibition of ERK5 reduced prostaglandin E2 (PGE2)-induced proliferation, migration, invasion, and stemness in non-small cell lung cancer (NSCLC) cells, highlighting the critical role of ERK5 in PGE2-induced tumorigenesis (48).
Another essential step in cell invasion is the degradation of extracellular matrix (ECM). In this context, ERK5 enhances the release of ECM metalloproteinases (MMPs), facilitating ECM breakdown and local tumor invasion (47, 49). ERK5 signaling has been shown to increase MMP2 and MMP9 gene expression (37), while ERK5 reduction reduced MMP9 mRNA and protein expression in lung metastases of osteosarcoma (49). A recent study using single-cell RNA sequencing to compare gene expression in primary osteosarcoma and circulating tumor cells (CTCs) supported the importance of the ERK5/MMP9 axis as a key driver of bone tumor metastasis (50). Furthermore, in a xenograft mouse model, MAPK7 silencing reduced tumor growth and significantly decreased metastatic spread to the lungs, which was associated with reduced MMP9 mRNA expression and secretion by osteosarcoma cells (50).
In breast cancer cells, overexpression of a constitutively active form of Signal transducer and activator of transcription-3 (STAT3) increased the expression of the EMT markers SNAIL, SLUG and vimentin via the MEK5/ERK5 pathway, while downregulating the epithelial marker E-cadherin (51). Similarly, expression of EMT-related transcription factors such as SNAIL, SLUG, and ZEB1/2 was significantly downregulated in pancreatic cancer xenografts from mice treated with XMD8-92, an ERK5 inhibitor that also reduced tumor growth; although the specificity of this inhibitor for ERK5 remains controversial (52). Other downstream targets of the ERK5 pathway, such as MEF2, activator protein-1 (AP-1), c-Myc (33) and NF-κB (53, 54), play critical roles in regulating EMT transcription factors like SNAIL, SLUG, and β-catenin (33, 55). Notably, MEF2B silencing prevents TGFβ-dependent EMT induction in murine epithelial mammary gland cells (11). In colon cancer cells, constitutive ERK5 activation results in nuclear accumulation of NF-κB, which upregulates vimentin expression and enhances cell migration (56). Accordingly, treatment with the MEK5/ERK5 inhibitor XMD8–92 decreases NF-κB transcriptional activity and reduces the expression of pluripotency transcription factors in colon cancer cells (57). Taken together, these findings confirm the direct and indirect roles of ERK5 in inducing EMT and metastasis across various malignancies, underscoring the potential of targeting the MEK5/ERK5 signaling pathway to develop more effective therapies aimed at preventing or limiting tumor metastasis.
4 Tumor cell metabolism modulates recognition by immune cells
Although “avoiding immune destruction” and “deregulating cellular energetics” were not initially recognized as hallmarks of cancer (58), they were later included in subsequent discussions (59). In the meantime, the link between tumor metabolism and immune escape became well established (18), and the potential for using metabolism-modulating drugs to enhance immunotherapy was proposed (60). Currently, there is significant interest in uncovering the mechanisms through which changes in tumor metabolism contribute to immune escape (61), with the aim of developing new immunotherapies (62). This topic has been extensively reviewed in the literature, e.g., (63, 64), and will not be further discussed here.
5 Metastatic metabolism and immune recognition
This issue was recently highlighted in an editorial (65). Interestingly, it was noted that pyruvate kinase isoform M2 (PKM2), a key rate-limiting enzyme catalyzing the conversion of phosphoenolpyruvate to pyruvate, plays a significant role in metastasis (66), partially via the ERK1/2 pathway (67). In that study, the authors did not explore whether PKM2 could also modulate ERK5 activity, which is essential for oxidative phosphorylation (OXPHOS) and the antioxidant response (19, 21, 68). Pyruvate produced by PKM2 is consumed in the mitochondria by mitochondrial CI, which regulates ERK5 activity through fumarate accumulation (19). Given that ERK5 promotes EMT and metastasis (as discussed above), it is tempting to hypothesize that PKM2 contributes to metastasis through CI and ERK5 activation. However, to our knowledge, the metabolic remodeling to FAO by metastatic cells has not yet been linked to immune cell recognition (64).
6 ERK5 is implicated in tumor immune surveillance
The interaction between different types of cells determines cancer growth and progression in the tumor microenvironment (TME). In this context, ERK5 is involved in several processes that stabilize the TME by immune system effector mechanisms, particularly those targeting tumor cells and pro-tumorigenic cell populations (12). Decreasing ERK5 in malignant mesothelioma cells injected into mice resulted in a significant reduction of tumor growth, due to downregulation of the pro-inflammatory cytokine CCL5/RANTES and other angiogenesis-related factors (69). Additionally, ERK5 is a crucial regulator of the pleiotropic cytokine IL-6 in various lung cancer cell lines and cancer-associated fibroblasts (CAFs), which modulate IL-12p70 secretion by dendritic cells (DCs), ultimately impairing their ability to induce a type 1 anti-tumor immune response (70). However, the precise mechanism by which ERK5 regulates IL-6 secretion remains to be elucidated. Furthermore, CAFs can also induce ERK5 phosphorylation and activation in colorectal cancer cells, leading to increased PD-L1 expression and promoting cell proliferation and survival. Nonetheless, in vivo experiments are needed to confirm these findings and clarify the connection between ERK5 and PD-L1 in the TME context (71). Tumor-associated macrophages (TAMs), derived from circulating monocytes recruited to the tumor site, exhibit an angiogenic and immunosuppressive phenotype that contributes to tumor growth and cancer progression (72, 73). ERK5 is overexpressed in TAMs from various tumor types, including breast, pancreatic, and lung cancer (74). Notably, tumor growth was reduced in mice with TAMs deficient in ERK5 signaling, primarily due to impaired activation of STAT3 signaling. These TAMs were unable to support tumor growth and angiogenesis in vivo (75), suggesting that the relevance of the ERK5/STAT3 axis is not limited to migrating tumor cells and EMT processes.
Moreover, ERK5 influences the expression of molecules involved in immune cell recognition (17, 76, 77), such as MHC-I (76, 77). Interestingly, forcing tumor cells to undergo respiration, such as through dichloroacetate (DCA) treatment, induces ERK5 expression and, consequently, MHC-I expression (17). Metabolism-regulated MHC-I expression can either enhance or reduce tumor cell recognition by cytotoxic lymphocytes (78). Other metabolic drugs also affect tumor cell immune surveillance. For instance, metformin, which inhibits glycolysis, induces MHC-I expression (79), facilitating recognition by natural killer (NK) cells and cytotoxic T lymphocytes (CTLs). This effect is mediated through the expression of Natural Killer G2-D (NKG2D) ligands (NKG2DL) and, primarily, intercellular adhesion molecule-1 (ICAM-1). Similarly, DCA induces NKG2DL and ICAM-1 expression, but this effect requires the presence of wild-type p53 (80). Finally, ERK5 modulates cancer cell sensitivity to extrinsic apoptosis triggered by death-receptor agonists (81), a mechanism used by cytotoxic lymphocytes to eliminate target cells.
Altogether, these observations suggest that ERK5 regulates multiple aspects of cell interactions within the TME, influencing immune recognition of tumor cells, immune cell activation, and the strength of the anti-tumor response.
7 Natural killer cells mediate metastasis immune surveillance
This subject has recently been reviewed (82, 83), and we will provide here only a summary of the field. EMT is one of the fundamental steps in the detachment of cancer cells from the primary tumor to initiate the metastatic process (84). During this process, metastatic cells are extensively exposed to immune cells, primarily cytotoxic lymphocytes, which patrol the blood and lymph. NK cells rapidly encounter metastatic tumor cells, leading to ERK activation and subsequent apoptosis of metastatic tumor cells (85).
NK cells play a key role in metastasis-specific immunosurveillance (86–89), and their numbers and activity correlate with the quantity of circulating tumor cells and metastases in various cancers (83, 90). Patients with different cancers who exhibit low levels of peripheral or infiltrating NK cells at tumor sites tend to have a higher number of metastatic lesions (82).
NK cells identify target cells by recognizing ligands on their plasma membranes. Although the mechanism by which NK cells recognize and kill metastatic cells is not fully understood, metastatic cells frequently lose MHC-I, which reduces inhibitory signaling and makes them more susceptible to NK cell-mediated killing (82). E-Cad, an epithelial marker, is downregulated by the transcriptional repressor Snail, which is induced during the EMT. In a model of TGFβ-induced EMT, E-Cad is recognized by Killer Cell Lectin-Like Receptor G1 (KLRG1), an inhibitory receptor on NK cells. Meanwhile, CADM1 is recognized by CRTAM, a receptor that activates NK cells (88). Thus, by decreasing E-Cad and increasing CADM1, metastatic cells can be more effectively recognized by NK cells. This partially explains metastatic cell recognition; however, losing the inhibitory interaction between E-Cad and KLRG1 alone is insufficient to reactivate NK cells’ cytotoxic functions (88).
Other ligands of NK cell activating receptors also increase during EMT such as PVR, UL16 binding protein2 (ULBP2), ULBP4, MHC class I polypeptide–related sequences A (MICA) and B (MICB), along with a decrease in the inhibitory ligand MHC-I (88, 91). The activating NKG2D receptor binds to MICA and MICB and various ULBPs, leading to target killing (92). In contrast, the inhibitory Killer-cell immunoglobulin-like receptor (KIR) recognizes MHC-I (93). Recognition of these ligands is crucial for NK cells’ anti-metastatic functions, and enhancing NK cell cytotoxicity within tissues can further strengthen the elimination of metastatic cells (94). Therefore, regardless of the ligands expressed on metastatic cells, strategies that boost NK cell function can improve their immune surveillance capabilities in this context.
NK cells are likely more effective at recognizing and/or eliminating individual metastatic cells or small metastatic clusters (83, 95). In contrast, circulating tumor cell (CTC) clusters are less sensitive to NK cells (83). Although large clusters exhibit reduced expression of NK cell-activating ligands (95), which can explain their decreased sensitivity to NK cells, the size of the cluster may also inhibit NK cell infiltration. For instance, clusters show elevated expression of epithelial and cell–cell adhesion genes (95), which likely prevents NK cell infiltration.
Another notable connection between ERK5 and the tumor microenvironment is its role in promoting EMT and a FAO/OXPHOS metabolism (10, 11), which may lead to increased extracellular reactive oxygen species (ROS), that suppress NK cell function. However, the presence of IL-15 protects NK cells from the detrimental effects of ROS (96), suggesting that activated NK cells may be shielded from ROS-induced damage.
8 Is ERK5 the link between metastasis and their recognition by NK cells?
We have described that ERK5 regulates immune cell recognition, EMT and FAO. Based on this, we hypothesized that ERK5 signaling could help explain why metastatic cells are promptly recognized by NK cells. Our unpublished findings show that rewiring cancer cell metabolism towards FAO using DCA activates ERK5, induces EMT gene expression (both in vitro and in vivo), and promotes tumor cell migration and invasion (97). Furthermore, DCA triggered the expression of a specific pattern of ligands in tumor cells, which facilitated NK cell recognition. As a result, NK cells infiltrated 3D tumor spheroids and exerted their cytotoxic effects (97). FAO drives EMT and the metastatic process, at least partially, through ERK5 activation. During EMT, various mechanisms could induce the expression of ligands recognized by NK cells, thereby enhancing immune surveillance and partially explaining the inefficiency of the metastatic process (86, 90). Thus, EMT and increased immune surveillance are linked, representing an Achilles’ heel for metastatic cells by limiting their ability to colonize new environments.
This insight could be leveraged in clinical settings by using specific metabolic drugs to promote the expression of NK cell-activating ligands on the tumor cell surface (18, 64), such as Intercellular Adhesion Molecule-1 (ICAM-1) or NKG2DL (80, 98), in combination with cytotoxic lymphocyte-based therapies to treat metastases.
ERK5 inhibition has also been shown to induce cellular senescence, making senescent cells susceptible to elimination by natural killer (NK) cells (68). This suggests that ERK5 may play a role not only in NK cell recognition of metastatic cells but also in the detection of senescent cells. These findings highlight a broader connection between ERK5 expression on target cells and NK cell-mediated immune surveillance.
9 Targeting ERK5 roles to fight metastasis
Although we will not detail ERK5 inhibitors (ERK5i) here, it is important to note that inhibition of ERK5 kinase activity does not necessarily block overall ERK5 function, given its kinase-independent roles (99, 100). Some ERK5i even paradoxically enhance C-terminal transactivation domain (TAD) activity and nuclear localization (101). This prompted the development of ERK5 degraders, such as the PROTAC INY-06-061, which selectively eliminates ERK5 protein. However, acute degradation failed to reproduce the anti-proliferative and anti-inflammatory effects observed with genetic ablation or earlier inhibitors (102). These findings dampened enthusiasm for ERK5 as an oncology target (102), suggesting that highly selective ERK5i lack intrinsic cytotoxicity, and should lack important in vivo toxic effects.
The ERK5 connections described here, particularly its role in immune surveillance, could represent interesting pharmaceutical targets to enhance immune cell activity. For example, immune checkpoint inhibitors (ICIs) are not directly cytotoxic but promote tumor cell destruction by the immune system, albeit with significant adverse events (103). Similarly, interfering with ERK5-mediated mechanisms could improve cancer treatment, especially against metastasis. Furthermore, engraftment of allogeneic, pre-activated NK cells may also prevent metastatic cell dissemination by eliminating them in the bloodstream. These approaches could provide new therapeutic options for patients at risk.
10 Conclusion
The spread of metastatic cells to distant locations is the leading cause of cancer-related deaths. NK cell-mediated immune surveillance plays a crucial role in regulating metastatic dissemination and represents a promising therapeutic target (90). Unveiling the mechanisms that link ERK5 to FAO and/or the expression of immune cell ligands and NK cell recognition could open new avenues for clinical treatments. The survival of metastatic cells depends on their ability to reprogram metabolism in order to colonize new environments, a process that involves ERK5. However, ERK5 activation concurrently induces the expression of a ligand repertoire recognized by NK cells, ultimately resulting in the elimination of these metastatic cells: we propose that these dual functions of ERK5 constitute a pharmacologically targetable Achilles’ heel of metastatic cells.
Author contributions
MV: Writing – original draft, Writing – review & editing. DG: Writing – original draft. SZ: Writing – review & editing. JR: Writing – review & editing. MC: Writing – original draft.
Funding
The authors declare financial support was received for the research and/or publication of this article. This work was supported by the Grant LabEx MAbImprove: ANR-10-LABX-53 (MV). We acknowledge the support of Immun4Cure IHU “Institute for innovative immunotherapies in autoimmune diseases” (France 2030/ANR-23-IHUA-0009).
Acknowledgments
This work was supported by the Grant LabEx MAbImprove: ANR-10-LABX-53 (MV). We acknowledge the support of Immun4Cure IHU “Institute for innovative immunotherapies in autoimmune diseases” (France 2030/ANR-23-IHUA-0009).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hsu PP and Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. (2008) 134:703–7. doi: 10.1016/j.cell.2008.08.021
2. Carracedo A, Cantley LC, and Pandolfi PP. Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer. (2013) 13:227–32. doi: 10.1038/nrc3483
3. Massagué J and Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. (2016) 529:298–306. doi: 10.1038/nature17038
4. Wang J and Li Y. CD36 tango in cancer: signaling pathways and functions. Theranostics. (2019) 9:4893–908. doi: 10.7150/thno.36037
5. Li Z and Kang Y. Lipid metabolism fuels cancer’s spread. Cell Metab. (2017) 25:228–30. doi: 10.1016/j.cmet.2017.01.016
6. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS-O, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. (2017) 541:41–5. doi: 10.1038/nature20791
7. Bezwada D, Perelli L, Lesner NP, Cai L, Brooks B, Wu Z, et al. Mitochondrial complex I promotes kidney cancer metastasis. Nature. (2024) 633:923–31. doi: 10.1038/s41586-024-07812-3
8. Ramakrishnan G, Terry AR, Nogueira V, Magdy A, and Hay N. Deletion of AMP-activated protein kinase impairs metastasis and is rescued by ROS scavenging or ectopic CD36 expression. Cell Rep. (2025) 44:115183. doi: 10.1016/j.celrep.2024.115183
9. Khan AUH, Salehi H, Alexia C, Valdivielso JM, Bozic M, Lopez-Mejia IC, et al. Glucose starvation or pyruvate dehydrogenase activation induce a broad, ERK5-mediated, metabolic remodeling leading to fatty acid oxidation. Cells. (2022) 11:1392. doi: 10.3390/cells11091392
10. Stecca B and Rovida E. Impact of ERK5 on the hallmarks of cancer. Int J Mol Sci. (2019) 20:E1426. doi: 10.3390/ijms20061426
11. Pavan S, Meyer-Schaller N, Diepenbruck M, Kalathur RKR, Saxena M, and Christofori G. A kinome-wide high-content siRNA screen identifies MEK5-ERK5 signaling as critical for breast cancer cell EMT and metastasis. Oncogene. (2018) 37:4197–213. doi: 10.1038/s41388-018-0270-8
12. Monti M, Celli J, Missale F, Cersosimo F, Russo M, Belloni E, et al. Clinical significance and regulation of ERK5 expression and function in cancer. Cancers (Basel). (2022) 14:348. doi: 10.3390/cancers14020348
13. Salinas-Sánchez AS, Serrano-Oviedo L, Nam-Cha SY, Roche-Losada O, Sánchez-Prieto R, and Giménez-Bachs JM. Prognostic value of the VHL, HIF-1α, and VEGF signaling pathway and associated MAPK (ERK1/2 and ERK5) pathways in clear-cell renal cell carcinoma. A long-term study. Clin Genitourin Cancer. (2017) 15:e923–33. doi: 10.1016/j.clgc.2017.05.016
14. Miranda M, Rozali E, Khanna KK, and Al-Ejeh F. MEK5-ERK5 pathway associates with poor survival of breast cancer patients after systemic treatments. Oncoscience. (2015) 2:99–101. doi: 10.18632/oncoscience.135
15. Abe J, Kusuhara M, Ulevitch RJ, Berk BC, and Lee JD. Big mitogen-activated protein kinase 1 (BMK1) is a redox-sensitive kinase. J Biol Chem. (1996) 271:16586–90. doi: 10.1074/jbc.271.28.16586
16. Guillén-Pérez YM, Ortiz-Ruiz MJ, Márquez J, Pandiella A, and Esparís-Ogando A. ERK5 interacts with mitochondrial glutaminase and regulates its expression. Int J Mol Sci. (2024) 25:3273. doi: 10.3390/ijms25063273
17. Charni S, de Bettignies G, Rathore MG, Aguilo JI, van den Elsen PJ, Haouzi D, et al. Oxidative phosphorylation induces de novo expression of the MHC class I in tumor cells through the ERK5 pathway. J Immunol. (2010) 185:3498–503. doi: 10.4049/jimmunol.1001250
18. Villalba M, Rathore MG, Lopez-Royuela N, Krzywinska E, Garaude J, and Allende-Vega N. From tumor cell metabolism to tumor immune escape. Int J Biochem Cell Biol. (2013) 45:106–13. doi: 10.1016/j.biocel.2012.04.024
19. Khan AUH, Allende-Vega N, Gitenay D, Garaude J, Vo DN, Belkhala S, et al. Mitochondrial Complex I activity signals antioxidant response through ERK5. Sci Rep. (2018) 8:7420. doi: 10.1038/s41598-018-23884-4
20. Abe Ji, Takahashi M, Ishida M, Lee JD, and Berk BC. c-Src is required for oxidative stress-mediated activation of big mitogen-activated protein kinase 1. J Biol Chem. (1997) 272:20389–94. doi: 10.1074/jbc.272.33.20389
21. Khan AU, Rathore MG, Allende-Vega N, Vo DN, Belkhala S, Orecchioni S, et al. Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen species (ROS) Production. EBioMedicine. (2016) 3:43–53. doi: 10.1016/j.ebiom.2015.11.045
22. Zhao J, Kyotani Y, Itoh S, Nakayama H, Isosaki M, and Yoshizumi M. Big mitogen-activated protein kinase 1 protects cultured rat aortic smooth muscle cells from oxidative damage. J Pharmacol Sci. (2011) 116:173–80. doi: 10.1254/jphs.11015FP
23. Cristea S, Coles GL, Hornburg D, Gershkovitz M, Arand J, Cao S, et al. The MEK5-ERK5 kinase axis controls lipid metabolism in small-cell lung cancer. Cancer Res. (2020) 80:1293–303. doi: 10.1158/0008-5472.CAN-19-1027
24. Khan AUH, Allende-Vega N, Gitenay D, Gerbal-Chaloin S, Gondeau C, Vo D-N, et al. The PDK1 inhibitor dichloroacetate controls cholesterol homeostasis through the ERK5/MEF2 pathway. Sci Rep. (2017) 7:10654. doi: 10.1038/s41598-017-10339-5
25. Marco-Brualla J, Al-Wasaby S, Soler R, Romanos E, Conde B, Justo-Méndez R, et al. Mutations in the ND2 subunit of mitochondrial complex I are sufficient to confer increased tumorigenic and metastatic potential to cancer cells. Cancers (Basel). (2019) 11. doi: 10.3390/cancers11071027
26. Wang Y, Hu M, Cao J, Wang F, Han JR, Wu TW, et al. ACSL4 and polyunsaturated lipids support metastatic extravasation and colonization. Cell. (2024) 188(2):412–429.e27. doi: 10.1016/j.cell.2024.10.047
27. Zhou Y, Li Y, Chen G, Guo X, Gao X, Meng J, et al. ACSL1-mediated fatty acid β-oxidation enhances metastasis and proliferation in endometrial cancer. Front Biosci (Landmark Ed). (2024) 29:66. doi: 10.31083/j.fbl2902066
28. Loo SY, Toh LP, Xie WH, Pathak E, Tan W, Ma S, et al. Fatty acid oxidation is a druggable gateway regulating cellular plasticity for driving metastasis in breast cancer. Sci Adv. (2021) 7:eabh2443. doi: 10.1126/sciadv.abh2443
29. Zeng F, Yao M, Wang Y, Zheng W, Liu S, Hou Z, et al. Fatty acid β-oxidation promotes breast cancer stemness and metastasis via the miRNA-328-3p-CPT1A pathway. Cancer Gene Ther. (2022) 29:383–95. doi: 10.1038/s41417-021-00348-y
30. Wang Y-N, Zeng Z-L, Lu J, Wang Y, Liu Z-X, He M-M, et al. CPT1A-mediated fatty acid oxidation promotes colorectal cancer cell metastasis by inhibiting anoikis. Oncogene. (2018) 37:6025–40. doi: 10.1038/s41388-018-0384-z
31. Wright HJ, Hou J, Xu B, Cortez M, Potma EO, Tromberg BJ, et al. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc Natl Acad Sci U S A. (2017) 114:E6556–65. doi: 10.1073/pnas.1703791114
32. Jiang W, Cai F, Xu H, Lu Y, Chen J, Liu J, et al. Extracellular signal regulated kinase 5 promotes cell migration, invasion and lung metastasis in a FAK-dependent manner. Protein Cell. (2020) 11:825–45. doi: 10.1007/s13238-020-00701-1
33. Bhatt AB, Wright TD, Barnes V, Chakrabarty S, Matossian MD, Lexner E, et al. Diverse and converging roles of ERK1/2 and ERK5 pathways on mesenchymal to epithelial transition in breast cancer. Trans Oncol. (2021) 14:101046. doi: 10.1016/j.tranon.2021.101046
34. Wang H, Dai YY, Zhang WQ, Hsu PC, Yang YL, Wang YC, et al. DCLK1 is correlated with MET and ERK5 expression, and associated with prognosis in Malignant pleural mesothelioma. Int J Oncol. (2017) 51:91–103. doi: 10.3892/ijo.2017.4021
35. Zhai L, Ma C, Li W, Yang S, and Liu Z. miR-143 suppresses epithelial-mesenchymal transition and inhibits tumor growth of breast cancer through down-regulation of ERK5. Mol Carcinog. (2016) 55:1990–2000. doi: 10.1002/mc.22445
36. Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, et al. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene. (2003) 22:1381–9. doi: 10.1038/sj.onc.1206154
37. Ramsay AK, McCracken SRC, Soofi M, Fleming J, Yu AX, Ahmad I, et al. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br J Cancer. (2011) 104:664–72. doi: 10.1038/sj.bjc.6606062
38. Tesser-Gamba F, Lopes LJ da S, Petrilli AS, and Toledo SRC. MAPK7 gene controls proliferation, migration and cell invasion in osteosarcoma. Mol Carcinog. (2016) 55:1700–13. doi: 10.1002/mc.22420
39. Dong X, Lv B, Li Y, Cheng Q, Su C, and Yin G. MiR-143 regulates the proliferation and migration of osteosarcoma cells through targeting MAPK7. Arch Biochem Biophys. (2017) 630:47–53. doi: 10.1016/j.abb.2017.07.011
40. Ding L, Fu Y, Zhu N, Zhao M, Ding Z, Zhang X, et al. OXTRHigh stroma fibroblasts control the invasion pattern of oral squamous cell carcinoma via ERK5 signaling. Nat Commun. (2022) 13:5124. doi: 10.1038/s41467-022-32787-y
41. Antoon JW, Martin EC, Lai R, Salvo VA, Tang Y, Nitzchke AM, et al. MEK5/ERK5 signaling suppresses estrogen receptor expression and promotes hormone-independent tumorigenesis. PLoS One. (2013) 8:e69291. doi: 10.1371/journal.pone.0069291
42. Dompe N, Klijn C, Watson SA, Leng K, Port J, Cuellar T, et al. A CRISPR screen identifies MAPK7 as a target for combination with MEK inhibition in KRAS mutant NSCLC. PLoS One. (2018) 13:e0199264. doi: 10.1371/journal.pone.0199264
43. Sawhney RS, Liu W, and Brattain MG. A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signaling. J Cell Physiol. (2009) 219:152–61. doi: 10.1002/jcp.21662
44. Xu Q, Zhang J, Telfer BA, Zhang H, Ali N, Chen F, et al. The extracellular-regulated protein kinase 5 (ERK5) enhances metastatic burden in triple-negative breast cancer through focal adhesion protein kinase (FAK)-mediated regulation of cell adhesion. Oncogene. (2021) 40:3929–41. doi: 10.1038/s41388-021-01798-2
45. Pozzato C, Outeiro-Pinho G, Galiè M, Ramadori G, and Konstantinidou G. ERK5 suppression overcomes FAK inhibitor resistance in mutant KRAS-driven non-small cell lung cancer. EMBO Mol Med. (2024) 16:2402–26. doi: 10.1038/s44321-024-00138-7
46. Ali M, Mutahir Z, and Riaz A. CRISPR/Cas9 engineering of ERK5 identifies its FAK/PYK2 dependent role in adhesion-mediated cell survival. Biochem Biophys Res Commun. (2019) 513:179–85. doi: 10.1016/j.bbrc.2019.03.145
47. Hoang VT, Matossian MD, Ucar DA, Elliott S, La J, Wright MK, et al. ERK5 is required for tumor growth and maintenance through regulation of the extracellular matrix in triple negative breast cancer. Front Oncol. (2020) 10:1164. doi: 10.3389/fonc.2020.01164
48. Filippelli A, Ciccone V, Del Gaudio C, Simonis V, Frosini M, Tusa I, et al. ERK5 mediates pro-tumorigenic phenotype in non-small lung cancer cells induced by PGE2. Biochim Biophys Acta Mol Cell Res. (2024) 1871:119810. doi: 10.1016/j.bbamcr.2024.119810
49. Yue B, Ren Q-X, Su T, Wang L-N, and Zhang L. ERK5 silencing inhibits invasion of human osteosarcoma cell via modulating the Slug/MMP-9 pathway. Eur Rev Med Pharmacol Sci. (2014) 18:2640–7.
50. Green D, Eyre H, Singh A, Taylor JT, Chu J, Jeys L, et al. Targeting the MAPK7/MMP9 axis for metastasis in primary bone cancer. Oncogene. (2020) 39:5553–69. doi: 10.1038/s41388-020-1379-0
51. Liu F, Zhang H, and Song H. Upregulation of MEK5 by Stat3 promotes breast cancer cell invasion and metastasis. Oncol Rep. (2017) 37:83–90. doi: 10.3892/or.2016.5256
52. Sureban SM, May R, Weygant N, Qu D, Chandrakesan P, Bannerman-Menson E, et al. XMD8–92 inhibits pancreatic tumor xenograft growth via a DCLK1-dependent mechanism. Cancer Lett. (2014) 351:151–61. doi: 10.1016/j.canlet.2014.05.011
53. Garaude J, Cherni S, Kaminski S, Delepine E, Chable-Bessia C, Benkirane M, et al. ERK5 activates NF-kappaB in leukemic T cells and is essential for their growth in vivo. J Immunol. (2006) 177:7607–17. doi: 10.4049/jimmunol.177.11.7607
54. Diéguez-Martínez N, Espinosa-Gil S, Yoldi G, Megías-Roda E, Bolinaga-Ayala I, Viñas-Casas M, et al. The ERK5/NF-κB signaling pathway targets endometrial cancer proliferation and survival. Cell Mol Life Sci. (2022) 79:524. doi: 10.1007/s00018-022-04541-6
55. Mirzaei S, Saghari S, Bassiri F, Raesi R, Zarrabi A, Hushmandi K, et al. NF-κB as a regulator of cancer metastasis and therapy response: A focus on epithelial-mesenchymal transition. J Cell Physiol. (2022) 237:2770–95. doi: 10.1002/jcp.30759
56. Simões AES, Pereira DM, Gomes SE, Brito H, Carvalho T, French A, et al. Aberrant MEK5/ERK5 signalling contributes to human colon cancer progression via NF-κB activation. Cell Death Dis. (2015) 6:e1718. doi: 10.1038/cddis.2015.83
57. Pereira DM, Gomes SE, Borralho PM, and Rodrigues CMP. MEK5/ERK5 activation regulates colon cancer stem-like cell properties. Cell Death Discov. (2019) 5:68. doi: 10.1038/s41420-019-0150-1
58. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. (2000) 100:57–70. doi: 10.1016/S0092-8674(00)81683-9
59. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
60. Villalba M, Lopez-Royuela N, Krzywinska E, Rathore MG, Hipskind RA, Haouas H, et al. Chemical metabolic inhibitors for the treatment of blood-borne cancers. Anti-cancer Agents medicinal Chem. (2014) 14:223–32. doi: 10.2174/18715206113136660374
61. Domblides C, Lartigue L, and Faustin B. Control of the antitumor immune response by cancer metabolism. Cells. (2019) 8:104. doi: 10.3390/cells8020104
62. Xu Y, He L, Fu Q, and Hu J. Metabolic reprogramming in the tumor microenvironment with immunocytes and immune checkpoints. Front Oncol. (2021) 11:759015. doi: 10.3389/fonc.2021.759015
63. Liang B, Deng L, and Zhou X. Targeting hypoxia-related metabolism molecules: How to improve tumour immune and clinical treatment? Biomedicine Pharmacotherapy. (2022) 156:113917. doi: 10.1016/j.biopha.2022.113917
64. Chuang Y-M, Tzeng S-F, Ho P-C, and Tsai C-H. Immunosurveillance encounters cancer metabolism. EMBO Rep. (2024) 25:471–88. doi: 10.1038/s44319-023-00038-w
65. Dong Q, Nelson PJ, and Zhao Y. Editorial: cancer cell metabolism and immunomodulation in the context of tumor metastasis. Front Oncol. (2021) 11:803213. doi: 10.3389/fonc.2021.803213
66. Yang Y-C, Chien M-H, Liu H-Y, Chang Y-C, Chen C-K, Lee W-J, et al. Nuclear translocation of PKM2/AMPK complex sustains cancer stem cell populations under glucose restriction stress. Cancer Lett. (2018) 421:28–40. doi: 10.1016/j.canlet.2018.01.075
67. Guo W, Zhang Z, Li G, Lai X, Gu R, Xu W, et al. Pyruvate kinase M2 promotes prostate cancer metastasis through regulating ERK1/2-COX-2 signaling. Front Oncol. (2020) 10:544288. doi: 10.3389/fonc.2020.544288. [Accessed November 30, 2022].
68. Tusa I, Menconi A, Tubita A, and Rovida E. Pathophysiological impact of the MEK5/ERK5 pathway in oxidative stress. Cells. (2023) 12:1154. doi: 10.3390/cells12081154
69. Shukla A, Miller JM, Cason C, Sayan M, MacPherson MB, Beuschel SL, et al. Extracellular signal-regulated kinase 5: a potential therapeutic target for Malignant mesotheliomas. Clin Cancer Res. (2013) 19:2071–83. doi: 10.1158/1078-0432.CCR-12-3202
70. Riegel K, Yurugi H, Schlöder J, Jonuleit H, Kaulich M, Kirschner F, et al. ERK5 modulates IL-6 secretion and contributes to tumor-induced immune suppression. Cell Death Dis. (2021) 12:969. doi: 10.1038/s41419-021-04257-8
71. Zhang M, Shi R, Guo Z, and He J. Cancer-associated fibroblasts promote cell growth by activating ERK5/PD-L1 signaling axis in colorectal cancer. Pathol Res Pract. (2020) 216:152884. doi: 10.1016/j.prp.2020.152884
72. Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. (2004) 4:71–8. doi: 10.1038/nrc1256
73. Martori C, Sanchez-Moral L, Paul T, Pardo JC, Font A, Ruiz de Porras V, et al. Macrophages as a therapeutic target in metastatic prostate cancer: A way to overcome immunotherapy resistance? Cancers (Basel). (2022) 14:440. doi: 10.3390/cancers14020440
74. Mantovani A and Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. (2015) 212:435–45. doi: 10.1084/jem.20150295
75. Giurisato E, Xu Q, Lonardi S, Telfer B, Russo I, Pearson A, et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc Natl Acad Sci U S A. (2018) 115:E2801–10. doi: 10.1073/pnas.1707929115
76. Charni S, Aguilo JI, Garaude J, de Bettignies G, Jacquet C, Hipskind RA, et al. ERK5 Knockdown generates mouse leukemia cells with low MHC class i levels that activate NK cells and block tumorigenesis. J Immunol. (2009) 182:3398–405. doi: 10.4049/jimmunol.0803006
77. Jiang H, Wang P, Li X, Wang Q, Deng Z-B, Zhuang X, et al. Restoration of miR17/20a in solid tumor cells enhances the natural killer cell antitumor activity by targeting Mekk2. Cancer Immunol Res. (2014) 2:789–99. doi: 10.1158/2326-6066.CIR-13-0162
78. Catalán E, Charni S, Jaime P, Aguiló J-I, Enríquez J-A, Naval J, et al. MHC-I modulation due to metabolic changes regulates tumor sensitivity to CTL and NK cells. Oncoimmunology. (2015) 4:e985924. doi: 10.4161/2162402X.2014.985924
79. Oliveras-Ferraros C, Cufi S, Vazquez-Martin A, Menendez OJ, Bosch-Barrera J, Martin-Castillo B, et al. Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle. (2012) 11:865–70. doi: 10.4161/cc.11.5.19252
80. Belkahla S, Brualla JM, Fayd’herbe de Maudave A, Falvo P, Allende-Vega N, Constantinides M, et al. The metabolism of cells regulates their sensitivity to NK cells depending on p53 status. Sci Rep. (2022) 12:3234. doi: 10.1038/s41598-022-07281-6
81. Espinosa-Gil S, Ivanova S, Alari-Pahissa E, Denizli M, Villafranca-Magdalena B, Viñas-Casas M, et al. MAP kinase ERK5 modulates cancer cell sensitivity to extrinsic apoptosis induced by death-receptor agonists. Cell Death Dis. (2023) 14:715. doi: 10.1038/s41419-023-06229-6
82. Yu Y. The function of NK cells in tumor metastasis and NK cell-based immunotherapy. Cancers (Basel). (2023) 15:2323. doi: 10.3390/cancers15082323
83. Masmoudi D, Villalba M, and Alix-Panabières C. Natural killer cells: the immune frontline against circulating tumor cells. J Exp Clin Cancer Res. (2025) 44:118. doi: 10.1186/s13046-025-03375-x
84. Roche J. The epithelial-to-mesenchymal transition in cancer. Cancers (Basel). (2018) 10:52. doi: 10.3390/cancers10020052
85. Ichise H, Tsukamoto S, Hirashima T, Konishi Y, Oki C, Tsukiji S, et al. Functional visualization of NK cell-mediated killing of metastatic single tumor cells. Elife. (2022) 11:e76269. doi: 10.7554/eLife.76269
86. López-Soto A, Gonzalez S, Smyth MJ, and Galluzzi L. Control of metastasis by NK cells. Cancer Cell. (2017) 32:135–54. doi: 10.1016/j.ccell.2017.06.009
87. Nakamura K and Smyth MJ. Immunoediting of cancer metastasis by NK cells. Nat Cancer. (2020) 1:670–1. doi: 10.1038/s43018-020-0081-z
88. Chockley PJ, Chen J, Chen G, Beer DG, Standiford TJ, and Keshamouni VG. Epithelial-mesenchymal transition leads to NK cell–mediated metastasis-specific immunosurveillance in lung cancer. J Clin Invest. (2018) 128:1384–96. doi: 10.1172/JCI97611
89. Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ, et al. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun. (2014) 5:4539. doi: 10.1038/ncomms5539
90. Lorenzo-Herrero S, López-Soto A, Sordo-Bahamonde C, Gonzalez-Rodriguez AP, Vitale M, and Gonzalez S. NK cell-based immunotherapy in cancer metastasis. Cancers (Basel). (2018) 11:E29. doi: 10.3390/cancers11010029
91. Sartor MA, Mahavisno V, Keshamouni VG, Cavalcoli J, Wright Z, Karnovsky A, et al. ConceptGen: a gene set enrichment and gene set relation mapping tool. Bioinformatics. (2010) 26:456–63. doi: 10.1093/bioinformatics/btp683
92. Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer. (2019) 18:29. doi: 10.1186/s12943-019-0956-8
93. Villalba M, Alexia C, Bellin-Robert A, Fayd’herbe de Maudave A, and Gitenay D. Non-genetically improving the natural cytotoxicity of natural killer (NK) cells. Front Immunol. (2019) 10:3026. doi: 10.3389/fimmu.2019.03026
94. Imianowski CJ, Whiteside SK, Lozano T, Evans AC, Benson JD, Courreges CJF, et al. BACH2 restricts NK cell maturation and function, limiting immunity to cancer metastasis. J Exp Med. (2022) 219:e20211476. doi: 10.1084/jem.20211476
95. Lo HC, Xu Z, Kim IS, Pingel B, Aguirre S, Kodali S, et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat Cancer. (2020) 1:709–22. doi: 10.1038/s43018-020-0068-9
96. Yang Y, Neo SY, Chen Z, Cui W, Chen Y, Guo M, et al. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J Clin Invest. (2020) 130:5508–22. doi: 10.1172/JCI137585
97. Zemiti S, Villalba M, and Gitenay D. Metabolic rewiring of cancer cells induces metastasis via ERK5 but triggers recognition by NK cells. BIORXIV/2025/672105. doi: 10.1101/2025.08.26.672105
98. Allende-Vega N, Marco Brualla J, Falvo P, Alexia C, Constantinides M, de Maudave AF, et al. Metformin sensitizes leukemic cells to cytotoxic lymphocytes by increasing expression of intercellular adhesion molecule-1 (ICAM-1). Sci Rep. (2022) 12:1341. doi: 10.1038/s41598-022-05470-x
99. Cook SJ and Lochhead PA. ERK5 signalling and resistance to ERK1/2 pathway therapeutics: the path less travelled? Front Cell Dev Biol. (2022) 10:839997. doi: 10.3389/fcell.2022.839997
100. Miller DC, Harnor SJ, Martin MP, Noble RA, Wedge SR, and Cano C. Modulation of ERK5 activity as a therapeutic anti-cancer strategy. J Med Chem. (2023) 66:4491–502. doi: 10.1021/acs.jmedchem.3c00072
101. Cook SJ, Tucker JA, and Lochhead PA. Small molecule ERK5 kinase inhibitors paradoxically activate ERK5 signalling: be careful what you wish for…. Biochem Soc Trans. (2020) 48:1859–75. doi: 10.1042/BST20190338
102. Potts MB. How to kill an ERKsome target: PROTACs deliver the deathblow. Cell Chem Biol. (2022) 29:1569–71. doi: 10.1016/j.chembiol.2022.11.002
Keywords: Erk5, CD36, fatty acid oxidation (FAO), epithelial-mesenchymal transition (EMT), natural killer (Nk) cell, immune surveillance
Citation: Villalba M, Gitenay D, Zemiti S, Rossi J-F and Campos-Mora M (2025) Is the MAPK ERK5 the nexus from FAO to NK cell-mediated metastasis immune surveillance? Front. Immunol. 16:1641865. doi: 10.3389/fimmu.2025.1641865
Received: 05 June 2025; Accepted: 22 September 2025;
Published: 08 October 2025.
Edited by:
Chiara Porta, University of Eastern Piedmont, ItalyReviewed by:
Jose M Lizcano, Autonomous University of Barcelona, SpainElisabetta Rovida, University of Florence, Italy
Copyright © 2025 Villalba, Gitenay, Zemiti, Rossi and Campos-Mora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Villalba, bWFydGluLnZpbGxhbGJhQGluc2VybS5mcg==