 Fei Du1,2*Linlin Xiao1
Fei Du1,2*Linlin Xiao1 Wang Guojun1Qian Dai1
Wang Guojun1Qian Dai1 Junxin Li3Xin Zhao1Qimin Zhang1Lan Yang1Yujie Liu1Yidan Hu4Bo Wen1Jingqiu Zhou1Jie Dai1Wenhao Zhang1
Junxin Li3Xin Zhao1Qimin Zhang1Lan Yang1Yujie Liu1Yidan Hu4Bo Wen1Jingqiu Zhou1Jie Dai1Wenhao Zhang1 Zhuo Zhang2,5*
Zhuo Zhang2,5*- 1Department of Pharmacy, The Fourth Affiliated Hospital of Southwest Medical University, Meishan, Sichuan, China
- 2Department of Pharmacology, School of Pharmacy, Southwest Medical University, Luzhou, Sichuan, China
- 3Department of Pharmacy, Zigong Fourth People’s Hospital, Zigong, China
- 4Department of Medical Administration, The Fourth Affiliated Hospital of Southwest Medical University, Meishan, Sichuan, China
- 5Key Laboratory of Luzhou City for Aging Medicine, School of Pharmacy, Southwest Medical University, Luzhou, China
Immunotherapy has rapidly emerged as a transformative advancement in cancer treatment, becoming essential for managing diverse malignancies. Despite the remarkable clinical efficacy of immunotherapies, including immune checkpoint inhibitors (ICIs) and chimeric antigen receptor (CAR)-T cells, across various tumor types, patient responses remain heterogeneous, with some tumors developing resistance through immune evasion strategies. Presently, the investigation of cell death mechanisms is gaining momentum as a promising avenue for immunotherapy optimization. Recent studies underscore that integrating cell death pathways with immunotherapy can significantly amplify anti-tumor immune responses. Ammonia, a metabolic byproduct within the tumor microenvironment (TME), has garnered increasing interest. Specifically, emerging research suggests that ammonia, accumulating in effector T cells as a result of glutamine metabolism, induces cell death. This distinct form of cell death, termed “ammonia death,” diverges from previously characterized mechanisms. This review examines the metabolic role of glutamine in various TME cells, explores the potential regulatory links between glutamine metabolism and ammonia-induced cell death, and evaluates the feasibility of targeting ammonia-induced cell death to enhance anti-tumor immunity and improve immunotherapy outcomes.
1 Introduction
Immunotherapy, an established strategy that leverages the immune system to combat tumors, has seen substantial advancements in recent years (1). Among the various therapeutic approaches, ICIs and CAR-T cell therapy have emerged as the most clinically impactful, significantly improving the prognosis for various cancers (2, 3). However, challenges such as inconsistent clinical efficacy and resistance continue to impede further optimization of immunotherapy (4, 5). Consequently, the exploration of novel immunotherapeutic strategies, particularly those integrating immunotherapy with other modalities, has become a central focus in cancer immunotherapy research. Metabolic reprogramming is a hallmark of cancer, with glutamine serving as a key nutrient for both tumor proliferation and immune cell function (6). This intersection positions ammonia metabolism as a critical therapeutic node.
Glutamine, a key amino acid abundant in blood and muscle, constitutes approximately 20% of circulating amino acids (6). While traditionally considered a non-essential amino acid due to its ability to be synthesized by most mammalian tissues, glutamine becomes conditionally essential under pathological conditions, where increased demand necessitates exogenous intake (7, 8). It plays a critical role as a metabolic nutrient in the TME, fueling energy production and serving as a precursor for biosynthetic processes essential for rapid cell proliferation (9, 10). Recent evidence suggests that reprogramming glutamine metabolism is not only vital for immune cell activation and proliferation but also critical for tumor cell invasion and TME remodeling (11, 12). Despite efforts to target amino acid metabolism in cancer therapy, clinical success has thus far been limited (13). Emerging studies, however, suggest that metabolic byproducts of glutamine also play pivotal roles within the TME (14, 15).
Ammonia, a byproduct of amino acid metabolism in the TME, has gained increasing attention in recent years. Notably, recent research indicates that during immune responses, ammonia produced from glutamine metabolism in activated effector T cells serves as a nitrogen source for energy production and biosynthesis. However, excessive ammonia accumulation induces a novel form of cell death in effector T cells, termed Ammonia death (15, 16).
Distinct from traditional cell death mechanisms such as apoptosis, necrosis, and pyroptosis, ammonia-induced cell death (termed “Ammonia death”) is a novel form of cell death triggered by overload of glutamine-derived ammonia, leading to the demise of effector T cells. It is characterized by mitochondrial swelling, impaired autophagic flux, and lysosomal alkalinization (15). Studies have demonstrated that inhibiting Ammonia death significantly extends the lifespan of effector T cells, thereby enhancing the effectiveness of tumor immunotherapy (15, 16). Targeting Ammonia death presents a promising novel strategy for cancer immunotherapy.
This review consolidates the metabolic features of glutamine in tumor and immune cells within the TME, explores the regulatory relationship between glutamine metabolism and Ammonia death, and evaluates current strategies targeting Ammonia death to boost anti-tumor immunity and immunotherapy. The article aims to provide novel insights and theoretical support for future targeted immunotherapy strategies aimed at overcoming the immune-suppressive effects of Ammonia death.
2 The metabolic characteristics of glutamine in the TME
2.1 Tumor cells
The Warburg effect is a hallmark metabolic characteristic of tumor cells, wherein they preferentially rely on glycolysis to metabolize glucose and produce substantial amounts of lactate, even in the presence of oxygen. This metabolic shift inhibits the conversion of glucose into acetyl-CoA, thereby preventing its entry into the tricarboxylic acid (TCA) cycle (17, 18). To compensate for this altered metabolism, tumor cells accelerate intracellular glutamine metabolism, ensuring a continuous supply of metabolic intermediates (nitrogen and carbon sources) to sustain the TCA cycle. This phenomenon is referred to as “anaplerosis” (19, 20). Glutamine undergoes metabolism through various enzymatic processes, producing byproducts that integrate into the TCA cycle. It enters the cytoplasm via solute carrier (SLC) proteins (21) and is converted into glutamate by glutaminase (GLS) (22). Glutamate is then converted to α-ketoglutarate (α-KG) by glutamate dehydrogenase (GLUD) or transaminases, facilitating the replenishment of the TCA cycle (22–24). Furthermore, glutamine metabolism supports the production of nicotinamide adenine dinucleotide phosphate (NADPH) and glutathione (GSH), contributing to the maintenance of cellular redox homeostasis (25, 26).
Glutamine metabolism is pivotal for tumor cell survival and proliferation. Studies indicate that many tumor cells upregulate SLC family transporters to enhance glutamine uptake and stimulate the activity of enzymes involved in glutamine metabolism, thus sustaining proliferative demands (27, 28). Among these, SLC1A5 (also known as ASCT2) serves as the primary transporter for glutamine, with its overexpression strongly linked to the growth, metastasis, and drug resistance of several cancers, including breast, lung, liver, and gastric cancers (29–31). Other transporters, such as SLC7A5, SLC38A2, and SLC7A11, also play contributory roles in glutamine and glutamate transport (32–34).
As a rate-limiting enzyme in the “anaplerosis” pathway, GLS catalyzes the breakdown of glutamine into free ammonia and glutamate (22). Studies have demonstrated that GLS1 is overexpressed in various tumor types and is closely associated with advanced tumor stages, high invasiveness, metastatic potential, and poor clinical prognosis (35, 36). Similarly, GLUD1, a key enzyme in glutamine catabolism, is highly expressed in many tumors (37). Research indicates that GLUD1 converts glutamate into α-KG, releasing substantial amounts of free ammonia in the process (38, 39). Under the influence of GLUD1 and enzymes such as glutamine synthetase (GS), the ammonia is incorporated into amino acid synthesis pathways, thereby promoting tumor cell proliferation and migration (40, 41). Alternatively, glutamate can be converted into α-KG by transaminases, such as glutamate-pyruvate transaminase 2 (GPT2), without producing ammonia. GPT2 catalyzes the reaction between glutamate and pyruvate, generating α-KG and alanine, which accelerates liver cancer cell proliferation (42). GPT2 is highly expressed in various cancers and correlates with patient prognosis (43, 44) (Figure 1).
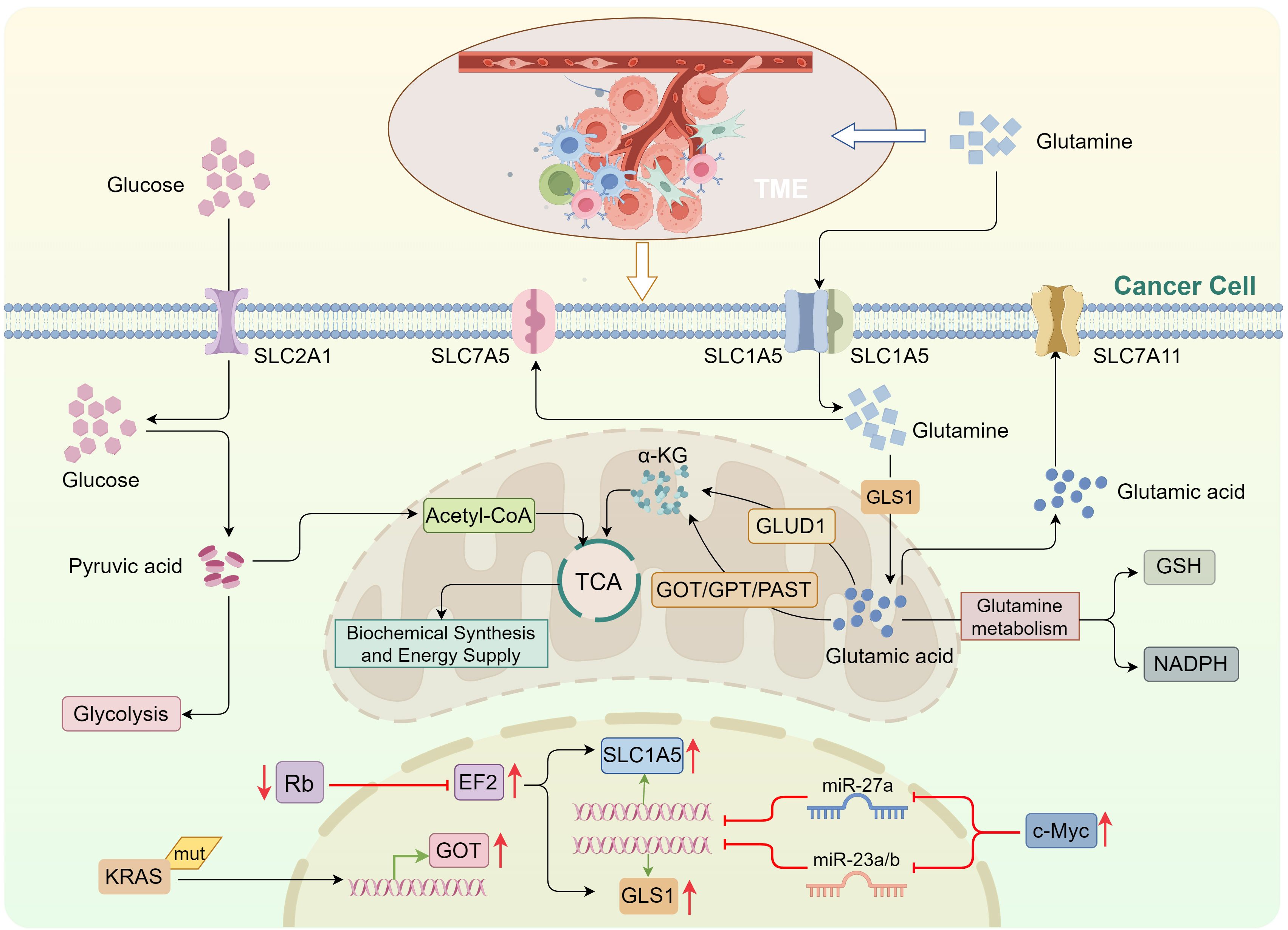
Figure 1. Glutamine metabolism in tumor cells replenishes the tricarboxylic acid cycle. Tumor cells primarily rely on glycolysis to metabolize glucose, impairing its conversion into acetyl-CoA for entry into the TCA cycle. To compensate, tumor cells enhance intracellular glutamine metabolism. Upon entering the cell via solute carrier (SLC) transporters, glutamine is converted into glutamic acid by GLS. Glutamic acid is then converted into α-KG, which enters the TCA cycle through two distinct pathways: via GLUD, and via transaminases, including GOT, GPT, and PSAT. c-Myc suppresses miR-27a and miR-23a/b expression, upregulating SLC1A5 and GLS1, respectively. E2F activation also promotes the expression of SLC1A5 and GLS1. KRAS mutations can induce GOT expression. TCA, tricarboxylic acid; GLS, glutaminase; GLUD, glutamate dehydrogenase; GOT, glutamate oxaloacetate transaminase; GPT, glutamate pyruvate transaminase; PSAT, phosphoserine aminotransferase; α-KG, α-ketoglutarate.
The oncogene c-Myc is a major regulator of glutamine utilization in tumor cells (45). c-Myc enhances glutamine uptake by binding to the promoter regions of glutamine transporters or repressing the expression of miR-27a, which promotes SLC1A5 upregulation (46, 47). Furthermore, c-Myc upregulates GLS1 expression by transcriptionally inhibiting miR-23a/b, thereby enhancing glutamine metabolism (48). c-Myc also influences glutamine metabolism indirectly by modulating the expression of miR-15a and miR-16 (49, 50). Recent studies have highlighted that c-Myc can also regulate glutamine metabolism through the upregulation of long non-coding RNAs (lncRNAs) such as H19 and MALAT1 (51, 52). Similarly, KRAS mutations in tumor cells promote glutamine metabolism by inducing the expression of glutamate oxaloacetate transaminase (GOT), contributing to the maintenance of tumor cell proliferation and survival (53, 54). Conversely, the loss of tumor suppressor proteins from the Rb family activates E2F transcriptional activity, directly upregulating the expression of SLC1A5 and GLS1, thus facilitating glutamine uptake and utilization (55) (Figure 1).
2.2 T cells
Immune cells are essential components of the TME, playing pivotal roles in tumor initiation, progression, immune evasion, and metastasis (56, 57). Increasing evidence suggests that while immune cells are tasked with mounting anti-tumor responses, they also contribute to tumor progression through various mechanisms (57, 58). Within the TME, tumor cells competitively consume glutamine, depriving immune cells of this vital nutrient. This results in restricted glutamine metabolism in immune cells, diminishing their anti-tumor efficacy and promoting immune evasion (57) (Figure 2). The metabolic profiles of immune cells significantly vary based on their differentiation and functional states.
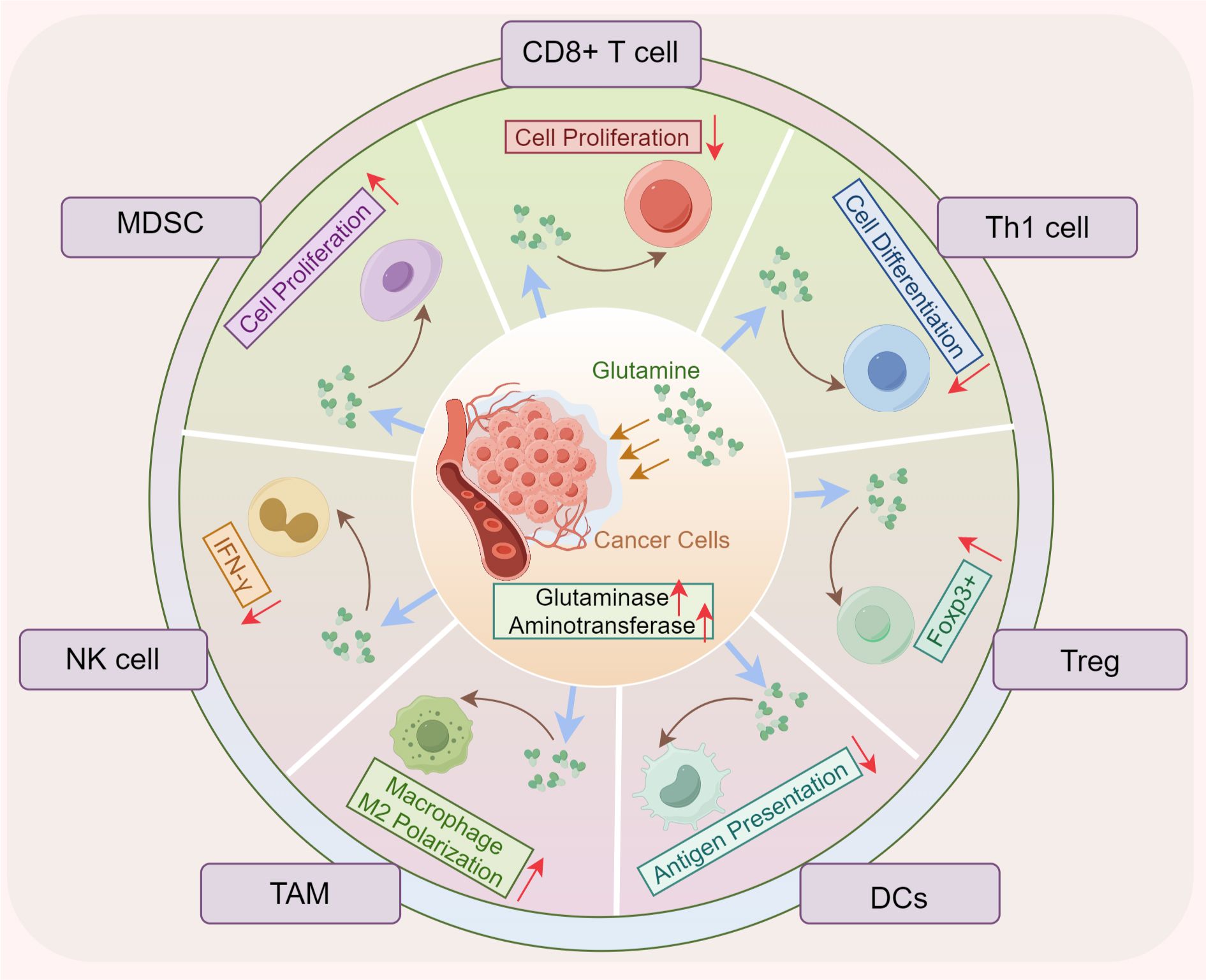
Figure 2. Glutamine metabolism promotes tumor immune evasion. The enhanced uptake of glutamine by tumor cells reduces its availability for infiltrating immune cells, impairing the proliferation and differentiation of CD8+ and CD4+ T cells, reducing antigen presentation by DCs, and diminishing natural killer (NK) cell antitumor activity. In contrast, the proliferation and polarization of MDSCs and TAMs are paradoxically increased. DCs, dendritic cells; Treg, regulator T cells; NK, natural killer; MDSC, myeloid-derived suppressor cells; FOXP3, forkhead box P3; TAM, tumor-associated macrophages.
2.2.1 CD8+ T cells
CD8+ T cells, key immune effector cells in the TME, are involved in tumor clearance, as well as in responding to viral infections and other pathological conditions (56). Glutamine metabolism is vital for the activation, proliferation, effector functions, and formation of immune memory in CD8+ T cells (59, 60). Intriguingly, in vitro studies have demonstrated that glutamine deprivation can promote the differentiation of memory CD8+ T cells, facilitating the development of long-lasting immune memory (61, 62). Conversely, in vivo inhibition of glutamine metabolism helps prevent CD8+ T cell exhaustion, sustaining their long-term effector functions (63). Recent research has elucidated the mechanisms underlying these effects: CD8+ T cells regulate isocitrate dehydrogenase 2 (IDH2) activity in the mitochondria, reducing glutamine through carboxylation and generating metabolites such as succinate. These metabolites not only provide energy for T cells but also “lock” them into a terminal effector differentiation state through epigenetic mechanisms, including histone modifications. Thus, inhibiting glutamine metabolism enhances memory T cell formation (62, 64). Moreover, inhibiting glutamine metabolism alleviates the long-term nutrient limitations and immune suppressive pressures within the TME, aiding in metabolic adaptation and preventing CD8+ T cell exhaustion (65). Additionally, reduced glutamine metabolism can lower the expression of immune checkpoint molecules (e.g., PD-1) on CD8+ T cells, thereby mitigating immune evasion within the TME (32).
2.2.2 CD4+ T cells
Upon antigenic stimulation, CD4+ T cells undergo metabolic reprogramming, activating pathways critical for their proliferation and differentiation (66, 67). Glutamine plays a central role in this process. Studies indicate that CD4+ T cells comprise distinct subpopulations, and modulation of glutamine metabolism profoundly influences their differentiation and effector functions (68, 69). In glutamine-deprived conditions, naïve CD4+ T cells preferentially differentiate into Foxp3+ regulatory T (Treg) cells, known for their immunosuppressive activity (70). Conversely, supplementation with exogenous α-KG or its analogs enhances mTORC1 activation in naïve CD4+ T cells, upregulating the transcription factor T-bet and driving Th1 differentiation (71). Glutamine metabolism thus promotes cytokine production, including interferon-γ (IFN-γ) and IL-2, through the activation of mTORC1 and c-Myc, contributing to potent anti-tumor responses (72). Additionally, glutamine metabolism regulates CD4+ T cell differentiation via epigenetic mechanisms. The enzyme IDH1/2 generates 2-hydroxyglutarate (2-HG), which inhibits α-KG’s metabolic functions (73). Inhibition of glutamate conversion to α-KG prevents 2-HG production in Th17 cells, thereby affecting DNA demethylase activity and reducing FOXP3 gene locus methylation, promoting Th17 to Treg cell differentiation (74). Notably, GLS deficiency impairs naïve T cell activity and Th17 differentiation, while enhancing T-bet expression to foster Th1 and CD8+ T cell differentiation and function (75).
2.3 Other cells
In addition to tumor cells and T cells, the TME also contains various other cell types that play important roles in tumor initiation, progression, immune evasion, and response to therapy.
As an indispensable component of the TME, tumor-associated macrophages (TAMs) also exhibit a high dependence on glutamine metabolism (58). Research has demonstrated that glutamine not only supports the TCA cycle in TAMs but also influences their polarization (76, 77). M2-like TAMs, compared to their M1-like counterparts, exhibit elevated expression of glutamine transporters and metabolic enzymes. Inhibition of glutamine synthetase can trigger the conversion of M2-like TAMs to the more inflammatory M1-like phenotype, thus impeding tumor metastasis (77, 78). TAMs, particularly M2 macrophages, rely on glutamine metabolism to sustain cell proliferation and immune suppression (79). Recent findings indicate that chemotherapy-responsive macrophages secrete interleukin (IL)-18, which upregulates the expression of L-type amino acid transporter 2 (LAT2) in osteosarcoma cells. This enhances leucine and glutamine uptake, activating mTORC1 and promoting c-Myc-mediated transcription of CD47, which inhibits macrophage phagocytosis and facilitates immune evasion (80). Another study highlights that competitive uptake of extracellular glutamine by clear cell renal carcinoma activates hypoxia-inducible factor 1α (HIF-1α) in tumor-infiltrating macrophages, inducing IL-23 secretion (81). IL-23 subsequently activates Treg proliferation and stimulates the release of immunosuppressive cytokines, including IL-10 and transforming growth factor (TGF)-β, thus suppressing cytotoxic lymphocyte effector functions (81).
Myeloid-derived suppressor cells (MDSCs) are a diverse subset of bone marrow-derived cells, typically generated in response to stress stimuli such as tissue injury and inflammation (82, 83). MDSCs play a pivotal role in maintaining immune tolerance and preventing excessive immune responses (82). In the TME, competition for glutamine between tumor cells and myeloid cell SLC1A5 transporters significantly promotes MDSC generation and recruitment, while inhibiting the formation of anti-tumor inflammatory TAMs, thus impairing anti-tumor immunity (79). This occurs as glutamine scarcity in the TME induces endoplasmic reticulum stress in myeloid cells, activating the IRE1α/XBP1 signaling pathway, which upregulates the expression of GPR109A, driving their immunosuppressive polarization (79). Additionally, glutamine metabolism regulates MDSC suppressive function via the glutamate-NMDA receptor axis (84).
Dendritic cells (DCs) are essential to the immune system, particularly in antigen presentation (85). Recent studies have emphasized the central role of glutamine metabolism in DC function, particularly in type 1 conventional dendritic cells (cDC1s) (33). In the TME, tumor cells and cDC1s compete for glutamine uptake through the SLC38A2 transporter (33, 86). This glutamine deprivation in cDC1s diminishes their antigen presentation capacity via the FLCN-Tfeb signaling pathway, ultimately impairing anti-tumor T cell responses (33).
Natural killer (NK) cells, key effectors in the immune defense against viruses and tumors, rely on glutamine for proper function (30, 87). Research shows that SLC7A5 serves as the primary glutamine transporter in activated NK cells. Glutamine uptake activates mTORC1, increasing c-Myc expression and promoting NK cell proliferation (30). However, when tumor cells competitively deplete glutamine, activated NK cells undergo glutamine scarcity, leading to reduced glycolysis and oxidative phosphorylation, which suppresses IFN-γ production (30, 88). Although glutamine is crucial for maintaining c-Myc expression, it is not a primary metabolic fuel for NK cells. Studies suggest that while glutamine metabolism inhibitors can significantly impact tumor cells, they do not suppress NK cell metabolism or functional responses (30).
3 Glutamine metabolism and ammonia death
3.1 Ammonia accumulation dynamics: metabolic sources vs. clearance pathways
Ammonia metabolism in the TME has garnered significant attention in recent years, emerging as a critical area of study in both immunology and oncology (14). As a major metabolic byproduct, ammonia plays multifaceted roles in the TME, influencing tumor progression, immune evasion, and therapeutic responses (89, 90). Beyond serving as a nitrogen source for nucleic acid and protein synthesis, ammonia accumulates in the TME through various metabolic pathways (14). Rapid tumor cell growth and proliferation in the TME often coincide with shifts in metabolic pathways, with ammonia accumulation tightly linked to cellular metabolic reprogramming, protein degradation, and the urea cycle (14, 91). Ammonia concentrations in the TME are elevated compared to normal tissues, with distinct regional distribution patterns (14).
Both tumor and immune cells exhibit increased amino acid demands to sustain their proliferation and survival (92, 93). The primary source of ammonia within cells is the deamination of amino acids (94). Glutamine, a key nitrogen and energy donor for tumor cells, is imported into the cytoplasm via the transporters SLC1A5, SLC38A1, and SLC38A2. It is subsequently transferred to the mitochondrial inner membrane via a variant of SLC1A5 (29, 95). Within the mitochondria, glutamine is converted into glutamate by glutaminases (GLS1, GLS2, GAC), releasing free ammonia (22, 96). The glutamate produced can either return to the cytoplasm via SLC25A18 and SLC25A22 transporters for the biosynthesis of GSH and non-essential amino acids (e.g., aspartate, alanine, arginine) or undergo complete oxidation or conversion into α-KG by mitochondrial transaminases (e.g., GPT2, GOT2) (97, 98). Pathway variations in the conversion of glutamine to α-KG occur under different pathological conditions. In liver and kidney diseases, such as hepatitis, cirrhosis, and renal failure, glutamate primarily converts to α-KG through transaminase pathways (99). In contrast, within the TME, factors like hypoxia and oxidative stress activate the GLUD pathway as a result of metabolic reprogramming. The oxidative metabolism of glutamate through GLUD activity generates substantial adenosine triphosphate (ATP), with ammonia and carbon dioxide as byproducts (93), significantly contributing to ammonia buildup in the TME (Figure 3). Isotope tracing studies have confirmed that the majority of ammonia in the TME is derived from the amino group of glutamines (15), further underscoring that ammonia accumulation is predominantly driven by glutamine metabolism.
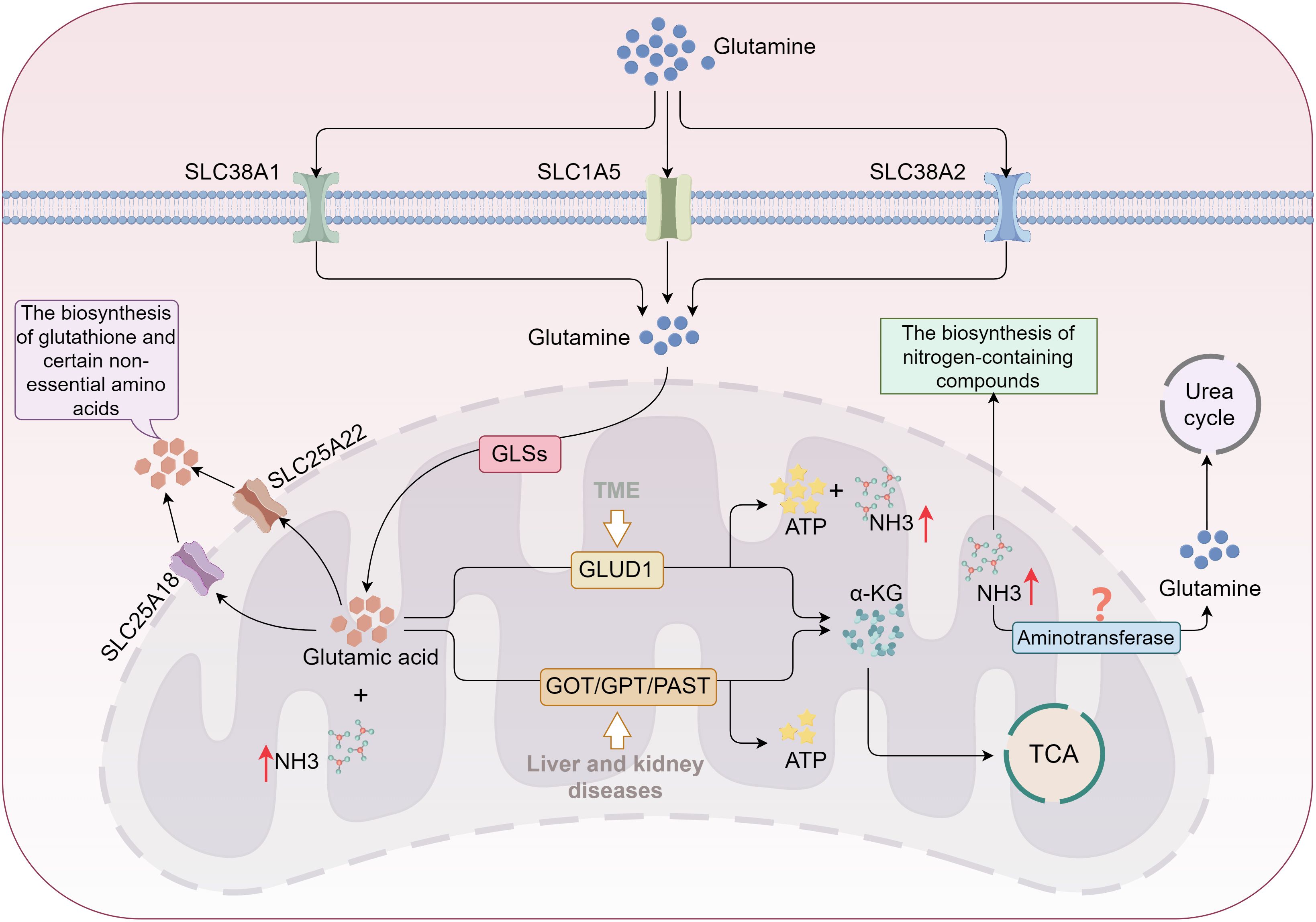
Figure 3. Glutamine metabolism promotes ammonia accumulation in tumor cells. Glutamine is transported into cells via SLC1A5 and SLC38A1/2, then converted into glutamic acid by GLS. A portion of glutamic acid is directed toward biosynthesis in the cytoplasm, while most is converted into α-KG and enters the TCA cycle through two distinct pathways. In kidney and liver diseases, glutamic acid primarily follows the transaminase pathway; however, in the TME, it favors the GLUD1 pathway, resulting in significant ammonia production. The TME downregulates enzymes involved in the urea cycle, destabilizing the tumor cells’ ability to clear ammonia and leading to its accumulation. TCA, tricarboxylic acid; GLS, glutaminase; α-KG, α-ketoglutarate; GLUD, glutamate dehydrogenase; GOT, glutamate oxaloacetate transaminase; GPT, glutamate pyruvate transaminase; PSAT, phosphoserine aminotransferase..
Ammonia clearance is essential for maintaining cellular homeostasis and supporting growth and proliferation. Under normal conditions, the urea cycle (ornithine cycle) serves as the primary mechanism for ammonia detoxification, particularly in hepatocytes (100). In liver cells, urease-related enzymes in the mitochondria—such as carbamoyl-phosphate synthetase I, ornithine transcarbamylase, and argininosuccinate synthetase—convert ammonia into urea, which is then transported to the kidneys via the bloodstream and excreted in urine (100). Extracellular tissues can also incorporate ammonia into glutamine via transaminases, with glutamine then transported through the circulatory system to the liver to participate in the urea cycle (101). While the urea cycle is not commonly present in most tissue cells, many cells retain some capacity to clear ammonia (101). Non-hepatic cells can utilize alternative synthetic pathways for nitrogen-containing compounds, such as purines and pyrimidines, to clear ammonia, although these mechanisms are less efficient and cannot completely eliminate ammonia (102). In the TME, despite various ammonia clearance mechanisms, rapidly proliferating cells, particularly tumor and immune effector cells, have a heightened demand for α-KG derived from glutamine breakdown to fuel the TCA cycle, leading to substantial free ammonia production (14). Research indicates that tumor cells downregulate the expression of urea cycle enzymes (e.g., ornithine aminotransferase), reducing ammonia conversion into urea (103). This disruption in ammonia production and clearance balance contributes to the accumulation of ammonia. Furthermore, although lysosomes, as crucial organelles for degrading and recycling cellular metabolic waste, can transport ammonia into the lumen via the Rhesus glycoprotein C (RHCG) to reduce the concentration of free ammonia in cells, this clearance pathway is biologically limited. Prolonged and sustained ammonia stimulation may instead impair lysosomal degradative function (15, 104). Consequently, ammonia accumulation is not a transient phenomenon; rather, ammonia-induced cellular damage is more closely associated with chronic conditions, such as cancer (Figure 3).
3.2 Mechanisms of ammonia death
Ammonia is generally regarded as a cytotoxin, and elevated blood levels lead to hyperammonemia, impairing cellular function (105, 106). In the TME, excessive ammonia accumulation triggers endoplasmic reticulum (ER) stress, activating ER stress sensors such as IRE1 and PERK. This activation promotes reactive oxygen species (ROS) production and induces oxidative stress, resulting in significant cellular and tissue toxicity (107).
Ammonia accumulation is closely associated with protein synthesis and degradation processes (90, 106, 108). Excess ammonia disrupts cellular protein metabolism. The mTOR pathway, a key regulator of cell growth, proliferation, and metabolism (90), is modulated by ammonia levels. Under normal conditions, ammonia activates amino acid sensors, enhancing mTORC1 activity and promoting protein synthesis. However, at high concentrations, ammonia and increased oxidative stress initiate negative feedback on mTORC1. Through modulation of upstream factors like AMPK, ammonia inhibits mTOR activity, thereby reducing protein synthesis (106). Regarding protein degradation, ammonia primarily influences this process via the ubiquitin-proteasome system (UPS) and autophagy (108). Excess ammonia enhances the activity of ubiquitin ligases, such as E3 ubiquitin ligase, facilitating protein degradation. It can also activate cellular stress responses, including plasma membrane and ER stress (e.g., UPR), accelerating the removal of damaged proteins (108).
Ammonia accumulation has a dual effect on autophagy (15, 109, 110). At low to moderate levels, ammonia activates the AMPK signaling pathway, enhancing autophagy to clear ROS-induced damage and dysfunctional proteins, helping cells adapt and survive (110). However, at high levels of ammonia, it may excessively activate the mTORC1 pathway or interfere with the autophagy initiation complex, inhibiting autophagic function. In this scenario, the failure to effectively clear damaged proteins and organelles leads to cellular dysfunction and eventual cell death (110).
In 2024, Professor Huang’s team uncovered a novel form of T cell death, termed “Ammonia death” (15, 16). This study, for the first time, explored ammonia-induced cell death from a metabolic perspective, offering a new explanation for the rapid exhaustion of effector T cells (Teffs) following anti-tumor activity. Ammonia death is a distinct form of cell death driven by lysosomal and mitochondrial damage due to ammonia accumulation, characterized by lysosomal alkalization and mitochondrial swelling (15, 16). In contrast to traditional cell death mechanisms such as apoptosis and T cell exhaustion, the accumulation of ammonia from glutamine metabolism is identified as the primary trigger for “Ammonia death” (15, 16). During tumor immune responses, rapidly proliferating T cells become highly dependent on glutamine metabolism, leading to the release of large quantities of ammonia in the mitochondria (15, 16). However, when ammonia accumulates excessively in lysosomes, it raises the lysosomal pH, which not only damages the lysosomal membrane but also halts the storage of ammonia in the lysosome, resulting in its release back into the cytoplasm (15). As lysosomes lose their capacity to store ammonia, it accumulates around the mitochondria, triggering mitochondrial dysfunction and initiating oxidative stress (15, 104). This dual damage to lysosomes and mitochondria due to excessive ammonia accumulation ultimately promotes cell death (15). Furthermore, autophagy is one of the crucial mechanisms for maintaining intracellular homeostasis, preserving normal cellular function by clearing damaged or unnecessary cellular components (15). However, due to lysosomal impairment, excess ammonia accumulates in mitochondria, leading to increased mitochondrial mass. Although autophagosomes can form in the cytoplasm, autophagic flux appears to be interrupted. The blockade of autophagy prevents the efficient clearance of damaged mitochondria, which further exacerbates cellular dysfunction and ultimately accelerates cell death (15, 16) (Figure 4).
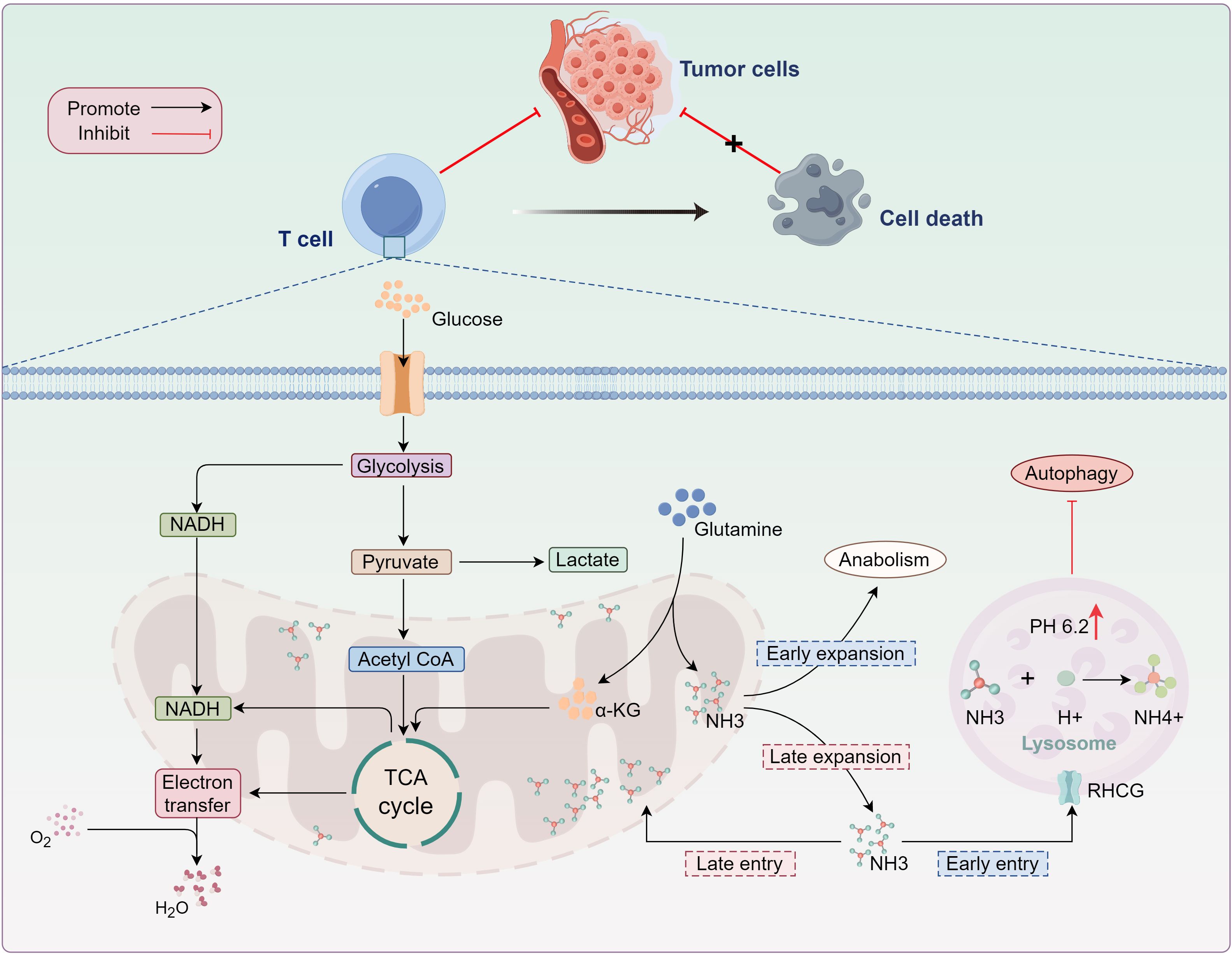
Figure 4. Ammonia-induced T-cell death. As glutamine metabolism progresses, ammonia (NH3) produced during glutamine decomposition exits the mitochondria and enters the lysosome, where it reacts with H+ to form NH4+. This reaction results in lysosomal alkalinization, terminating ammonia storage in the lysosome. Excess ammonia accumulation in the cytoplasm disrupts mitochondrial function. While damaged mitochondria are typically cleared via autophagy, lysosomal alkalinization inhibits enzymatic activity, obstructing the removal of dysfunctional mitochondria and ultimately inducing effector T-cell death. TCA, tricarboxylic acid; α-KG, α-ketoglutarate; NADPH, nicotinamide adenine dinucleotide phosphate; RHCG, rhesus glycoprotein C..
3.3 Potential relationship between ammonia death and others
While Ammonia death differs in morphology and mechanism from other cell death forms, excessive ammonia promotes the generation of ROS, inducing oxidative stress (15, 16). This suggests that ammonia-induced Ammonia death is not an isolated mode of cell death but may be interconnected with other well-established cell death processes, such as apoptosis, autophagic cell death, and necrosis.
Apoptosis, a form of programmed cell death, is typically executed through an intrinsic self-destruction pathway that involves caspase family activation (mainly caspases 3 and 7), alterations in the cell membrane, and DNA fragmentation (111). Mitochondrial dysfunction caused by ammonia accumulation creates an oxidative stress environment, a critical condition for activating the caspase pathway (112). At low ammonia concentrations, cells typically undergo a sub-lethal apoptotic response, a mechanism designed to eliminate mildly damaged cells (113). However, at higher concentrations, ammonia induces significant mitochondrial damage, compromising mitochondrial membrane integrity and promoting the release of cytochrome c and other pro-apoptotic molecules (such as SMAC/DIABLO) into the cytoplasm. This activates the apoptosome, leading to caspase-9 and caspase-3 activation, which triggers the apoptotic program (112).
Autophagy is a process in which cells degrade and recycle damaged organelles and macromolecules, typically activated in response to nutrient deprivation or stress (114). The relationship between Ammonia death and autophagy is complex. As mentioned earlier, varying ammonia concentrations yield different effects on autophagy (109, 115). In the early stages of autophagy, ammonia triggers oxidative stress and mitochondrial damage, stimulating the autophagic machinery to clear damaged mitochondria and oxidative damage. However, as ammonia accumulates to higher concentrations, it may disrupt the initiation and degradation stages of autophagy (e.g., inhibiting autophagosome-lysosome fusion), impairing autophagic flux and promoting autophagic cell death (109, 115).
Cell necrosis is an acute, non-programmed form of cell death typically triggered by membrane rupture, leading to the release of cellular contents and subsequent inflammation (116). The link between ammonia-induced cell death and necrosis is likely due to the local accumulation of ammonia at high concentrations, which disrupts cell membrane integrity and initiates necrotic responses such as cell swelling, membrane rupture, and the leakage of intracellular components into the extracellular space (117, 118). This leakage contributes to local inflammation and tissue damage.
Pyroptosis, a programmed cell death process driven by iron ions and lipid peroxidation, has been implicated in various pathological conditions (119). While direct evidence connecting ammonia-induced cell death with pyroptosis is currently lacking, several studies suggest that ammonia accumulation exacerbates oxidative stress, potentially activating inflammasomes and thereby promoting pyroptotic cell death (119, 120).
In conclusion, ammonia-induced cell death represents a distinct mode of cellular demise caused by ammonia accumulation, overlapping with and interacting with apoptosis, autophagy, necrosis, and other forms of cell death. Its unique characteristic is the metabolic reprogramming of the cell and the specialized response to ammonia buildup, positioning ammonia death as a promising target for future immunotherapeutic strategies.
4 Ammonia death in TME
4.1 Immune cells with ammonia death
Upon antigen stimulation, CD8+ naïve T cells differentiate into effector cells, with over 95% undergoing rapid cell death following antigen clearance in a process known as T cell contraction (121). This death of effector T cells is essential for maintaining immune homeostasis and preventing autoimmune responses. However, the precise mechanisms underlying effector T cell death have remained unclear. Professor Huang Bo’s team demonstrated that the ketone-body-gluconeogenesis-glycogen metabolic pathway activates the pentose phosphate pathway to reduce intracellular ROS, promoting the formation and maintenance of CD8+ T cell memory (122, 123). Furthermore, they showed that the urea cycle plays an active role in reducing intracellular ammonia levels, which supports the long-term survival of memory T cells (104).
Recent studies have highlighted ammonia’s critical role in effector CD8+ T cell death (15). While glutamine metabolism provides essential energy for CD8+ T cells during immune responses, contributing carbon and nitrogen for biosynthesis, its metabolic byproduct, ammonia, gradually accumulates within the cells, ultimately leading to their demise (15, 16). To prevent ammonia-induced cell death and extend the survival of CD8+ T cells, at least three strategies have been proposed. First, since glutamine metabolism is the precursor to ammonia accumulation, inhibitors such as 6-diazo-5-oxo-L-norleucine (DON) or CB-839, a non-competitive allosteric inhibitor of GLS1, can significantly reduce intracellular ammonia levels (35, 124). Both DON and CB839 have been shown to promote Teff survival, upregulate CD25, IFN-γ, and TNF-α, and enhance the formation of memory precursor effector cells (KLRG1-CD127+) (125–127). Second, removing excess ammonia from the cell provides an alternative approach to mitigate ammonia-induced cell death. Ammonia scavengers, such as phenylbutyrate (a prodrug of phenylacetic acid), reduce ammonia levels by promoting its conversion into phenylacetyl glutamine, which is excreted in urine, thus mitigating its toxic effects (128). Additionally, upregulating the key urea cycle enzyme, carbamoyl-phosphate synthetase 1 (CPS1), which is expressed at low levels in CD8+ T cells, can significantly lower intracellular ammonia concentrations (15, 129). Third, because ammonia-induced damage to lysosomes and mitochondria directly triggers cell death, accelerating the clearance of damaged organelles or enhancing autophagy offers a promising strategy to rescue T cells. Restoring lysosomal function, for example, by activating the transcription factor TFEB using compounds like Torin1 or Metformin to upregulate lysosomal gene expression, promotes lysosome biogenesis (130–132). When lysosomal function is severely impaired, lysosome-independent degradation pathways, such as those activated by Bortezomib, a proteasome modulator, can enhance ubiquitination and proteasomal activity, compensating for defective lysosomal function and facilitating the removal of damaged proteins and organelles, thus improving immunotherapy efficacy (133) (Table 1). In summary, targeting ammonia-induced cell death can significantly prolong the survival of CD8+ T cells in the TME, enhancing their anti-tumor activity. This approach offers a promising direction for future cancer immunotherapy.
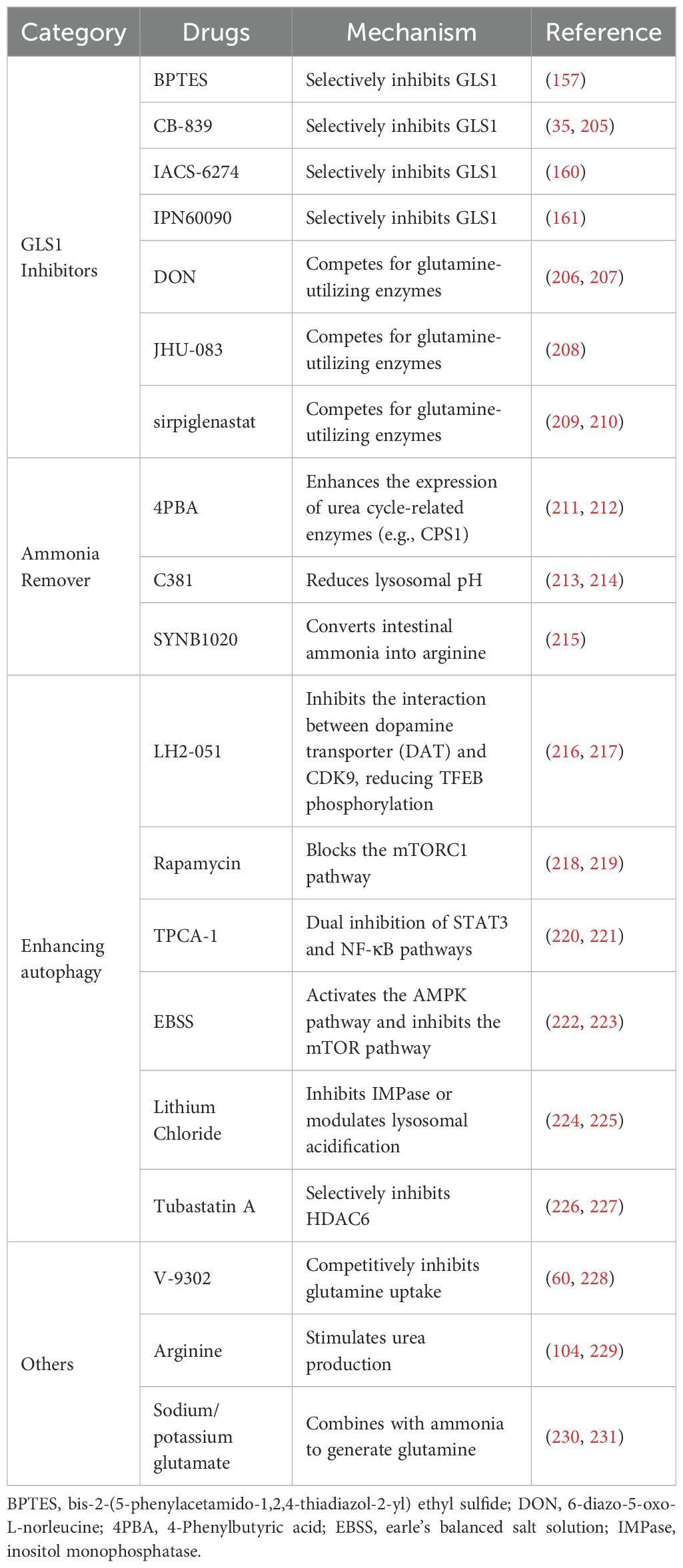
Table 1. Potential strategies for inhibiting ammonia death.
Although current experimental research on ammonia-induced cell death primarily focuses on CD8+ T cells, the immune system operates as a complex network. As discussed in the previous section, glutamine metabolism, which leads to ammonia accumulation, is active in a variety of immune cells. This metabolic pathway facilitates ammonia buildup in these cells (68, 69, 76, 77, 79). Furthermore, research indicates that CPS1, the key enzyme in the urea cycle, is predominantly expressed in hepatocytes and certain renal cells, with low or absent expression in most immune cells due to the urea cycle’s inactivity in these cells (134, 135). This limitation further contributes to ammonia accumulation in other immune cells. Notably, effector CD4+ T cells, including Th1, Th2, and Th17 cells, play vital roles in short-term immune responses. Following activation and execution of their effector functions, the majority of effector CD4+ T cells undergo programmed cell death (apoptosis), making them unsuitable for long-term immunity (136, 137). Therefore, it is reasonable to speculate that ammonia-induced cell death may not be a form of cell death unique to CD8+ T cells. Although CD8+ T cells may be more susceptible to ammonia under certain tumor microenvironments or immunosuppressive conditions, other immune cells (such as CD4+ T cells and macrophages)may also undergo cell death due to ammonia overload.
4.2 Tumor cells with ammonia death
Research on ammonia-induced cell death has primarily focused on immune cells, particularly CD8+ T cells (15, 16). Inhibiting ammonia-induced cell death appears to significantly extend the lifespan of CD8+ T cells, thereby enhancing long-term immune efficacy (15). However, whether tumor cells undergo ammonia-induced cell death has yet to be investigated. Current evidence suggests that the primary mechanism triggering ammonia-induced cell death involves the excessive accumulation of ammonia, a byproduct of glutamine metabolism, which leads to lysosomal and mitochondrial damage (15, 16). As a metabolic byproduct, ammonia plays a critical role not only in immune cells but also in tumor cells (138, 139).
In the TME, the rapid proliferation of tumor cells is often associated with increased metabolic activity and competition for nutrients, including glutamine, from the surrounding environment. Consequently, tumor cells typically exhibit higher glutamine metabolic activity than immune cells (140). Studies indicate that, under normal conditions, ammonia produced by glutamine metabolism can promote tumor progression. For instance, ammonia serves as a key activator of lipogenesis, which is a prominent marker of cancer progression and metastasis (141). Lipogenesis is regulated by Sterol Regulatory Element-Binding Proteins (SREBPs) (141, 142). Cheng et al. demonstrated that glutamine is essential for SREBP activation and lipogenesis. While glucose is required for the N-glycosylation-mediated stabilization of SCAP, glutamine is necessary for the nuclear translocation of SREBP (90). Further studies revealed that ammonia, rather than glutamate or α-KG, is the primary activator of SREBP cleavage (90). Interestingly, gene inhibition and CB-839, an inhibitor of GLS activity, can eliminate SREBP activation, suggesting that ammonia derived from GLS is a critical mediator of lipogenesis (90, 142). Additionally, studies have shown that in certain tumors, blocking glutamine uptake leads to cell death, a phenomenon known as “glutamine addiction” (143, 144). Consequently, recent studies have increasingly focused on inhibiting glutamine metabolism in tumor cells as a strategy to suppress tumor progression.
Given the established mechanisms of ammonia-induced cell death in CD8+ T cells, inducing ammonia-induced cell death in tumor cells through excessive ammonia accumulation presents a promising avenue for cancer immunotherapy research. CPS1 catalyzes the reaction between ammonia and carbon dioxide to form carbamoyl phosphate, the first step in urea synthesis (134, 145). This process helps maintain metabolic balance by clearing ammonia from cells and mitigating oxidative damage (134, 145). Research indicates that CPS1 is predominantly expressed in liver, kidney, and a small number of intestinal epithelial cells (134, 135, 146). In the TME, CPS1 expression varies across cancer types and is highly expressed in liver, pancreatic, kidney, colorectal, breast cancers, and non-small cell lung cancer (135, 146–148). Several small-molecule CPS1 inhibitors have been identified, showing potential in inhibiting CPS1 activity and inducing tumor cell death under experimental conditions (135, 146, 149–151). However, these inhibitors remain in the early stages of research and have yet to be extensively validated in clinical cancer treatments. Despite this, the common outcome observed across these studies is that CPS1 inhibition leads to a significant increase in intracellular ammonia levels. Therefore, inhibiting CPS1 expression in tumor cells may cause excessive ammonia accumulation, but whether this accumulation can induce ammonia-induced cell death in tumor cells requires further validation through comprehensive experimental studies.
5 Clinical relevance of ammonia death
5.1 Targeting glutamine metabolism-related pathways
Based on current research progress, anti-tumor strategies targeting glutamine metabolism have established a multi-level intervention system, primarily encompassing three major directions: transport inhibition, key enzyme blockade, and combination therapies. At the level of glutamine transport, SLC1A5, as a key transporter, has emerged as an important target. Preclinical studies have confirmed that IMD-0354 significantly suppresses the growth of melanoma and hepatocellular carcinoma (HCC) through competitive inhibition of SLC1A5 (152, 153). Similarly, V-9302, which also targets this transporter, has demonstrated anti-tumor activity in models of triple-negative breast cancer (TNBC), HCC, and melanoma. Its mechanism of action involves blocking glutamine uptake by tumor cells, thereby disrupting metabolic homeostasis (60, 154, 155). JHU083, a novel SLC1A5 inhibitor, has been shown to be effective against various solid tumors in animal experiments, particularly exhibiting potential in regulating the metabolic reprogramming of TAMs (76, 156).
Multiple inhibitors targeting glutaminase (GLS), a key enzyme in glutaminolysis, are currently in various stages of development. BPTES, a selective GLS inhibitor, has been shown to suppress the growth of multiple solid tumors in preclinical studies (157, 158). CB-839 (also known as Telaglenastat), one of the more advanced candidates, has entered clinical trials for the treatment of various solid tumors (156, 159). Other GLS inhibitors, such as IACS-6274 in head and neck squamous cell carcinoma (HNSCC) and IPN60090 in non-small cell lung cancer (NSCLC) and ovarian cancer, have demonstrated efficacy in preclinical models (160) (161). These agents inhibit the conversion of glutamine to glutamate, thereby blocking α-KG production, disrupting TCA cycle replenishment and biosynthesis, and ultimately leading to metabolic collapse in tumor cells.
Combination therapy strategies aim to overcome the limitations of monotherapy and enhance therapeutic efficacy. Preclinical studies have demonstrated that DRP-104, a glutamine mimetic prodrug, in combination with the MEK inhibitor trametinib, effectively suppresses pancreatic ductal adenocarcinoma by simultaneously inhibiting glutamine metabolism and the ERK signaling pathway (162, 163). Furthermore, progress has been made in targeting compensatory mechanisms employed by tumor cells under glutamine deprivation. For instance, IFRD1 knockout combined with CB-839 treatment effectively inhibited the “low-cost” survival of tumor cells under nutrient stress in HCC animal models by blocking the autophagic degradation pathway of histone H1.0 (164). Other strategies, such as targeting the RAS signaling pathway to suppress its mediated dysfunction in macrophage phagocytosis, remain at the theoretical exploration stage (165, 166). These multi-pathway, multi-target intervention strategies provide new research directions and therapeutic approaches for overcoming tumor metabolic heterogeneity and microenvironment-mediated drug resistance. (Table 2).
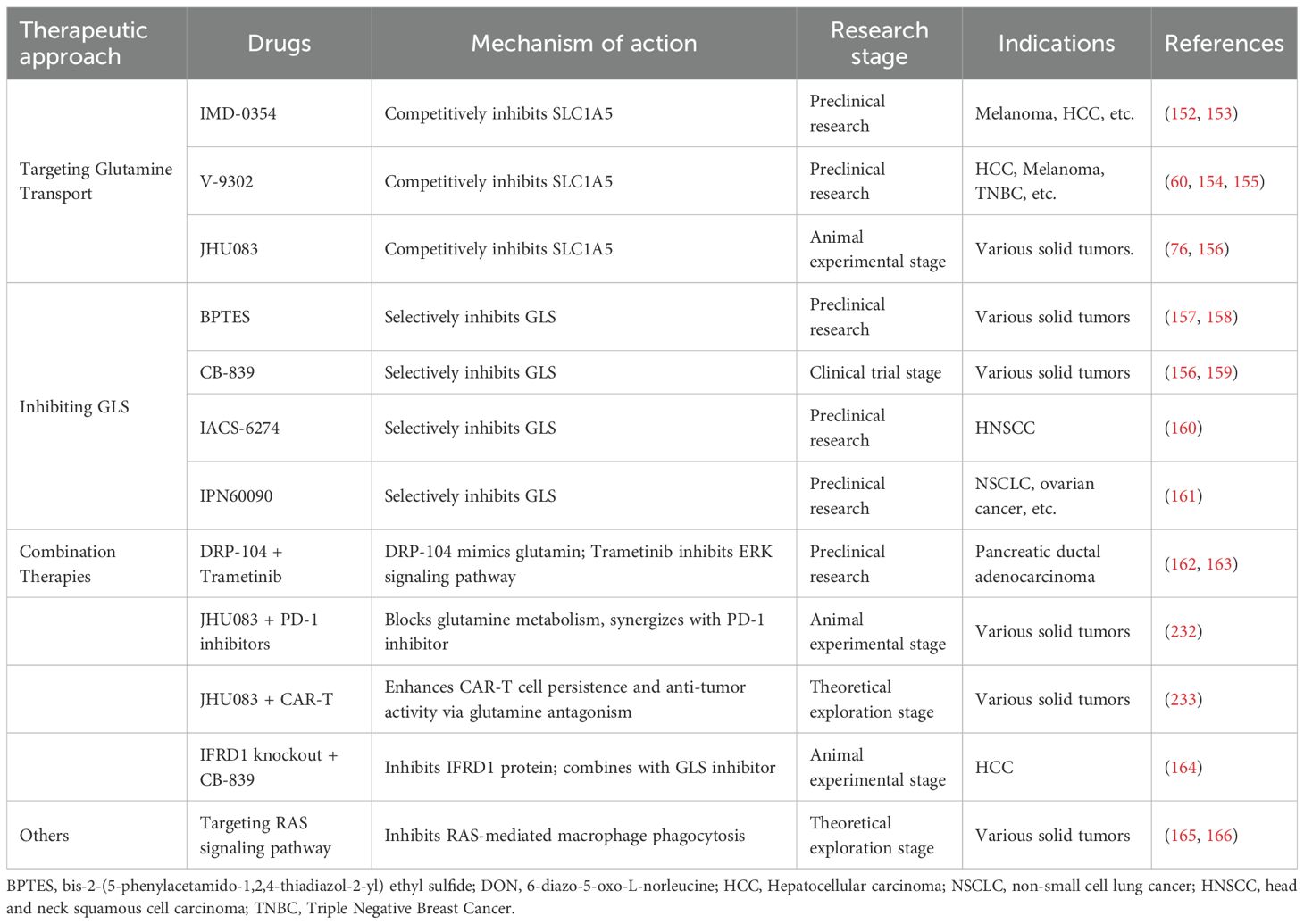
Table 2. Antitumor therapeutic strategies targeting glutamine metabolism-related pathways.
5.2 Targeting ammonia death to promote tumor immunotherapy
5.2.1 Ammonia death and adoptive T cell therapy
The concept of “ammonia death” in effector T cells, recently proposed by Professor Huang Bo’s team, offers valuable insights for improving T-cell-based cancer immunotherapy by enhancing the survival of Teff cells (15, 16). Their research demonstrates that targeting T-cell glutamine metabolism through drugs such as C381 or JHU083, or by adoptively transferring inducible GLS1 shRNA-expressing OT-I Teff cells into tumors, not only increases the number of OT-I Teff cells within the TME but also significantly reduces tumor growth and prolongs survival in animal models (15, 16). These findings suggest that blocking ammonia accumulation to improve effector T cell persistence may represent a promising new direction for advancing adoptive T-cell therapies aimed at tumors.
CAR-T immunotherapy, one of the most innovative approaches in adoptive cell therapy, has shown considerable success in treating hematological malignancies (167, 168). However, the short lifespan of effector T cells remains a major limitation of CAR-T therapy (168, 169). This limitation directly impacts the long-term outcomes of treatment, particularly in terms of tumor relapse and sustained immune surveillance (168, 169). Studies have shown that during the early phase, ammonia derived from glutamine metabolism serves as a nitrogen source for CD8+ T cell proliferation and indirectly upregulates RUNX2 (a key transcription factor for osteoblast differentiation; RUNX2 overexpression enhances the antitumor efficacy of murine CAR-T cells and improves the function of human CAR-T cells) via the mTOR pathway (170–172). In the late phase, however, excessive ammonia accumulation induces lysosomal alkalization, collapse of mitochondrial membrane potential, and ROS burst, triggering oxidative stress and mitochondrial dysfunction. This subsequently activates the HIF-1α signaling pathway, leading to upregulation of P4HA1 (a critical regulator of CD8+ T cell differentiation that is highly upregulated in tumor-draining lymph nodes (TDLNs) and hypoxic tumor microenvironments; inhibition of P4HA1 enhances the expansion of progenitor CD8+ T cells while alleviating exhaustion, thereby boosting systemic antitumor immunity). Concurrently, ammonia inhibits the catalytic function of P4HA1 by chelating iron and competing with 2-oxoglutarate (2OG), thereby accelerating T cell exhaustion (173–175). Moreover, intracellular ammonia overload appears to downregulate RUNX2 by promoting ROS accumulation (176–178) (Figure 5A).
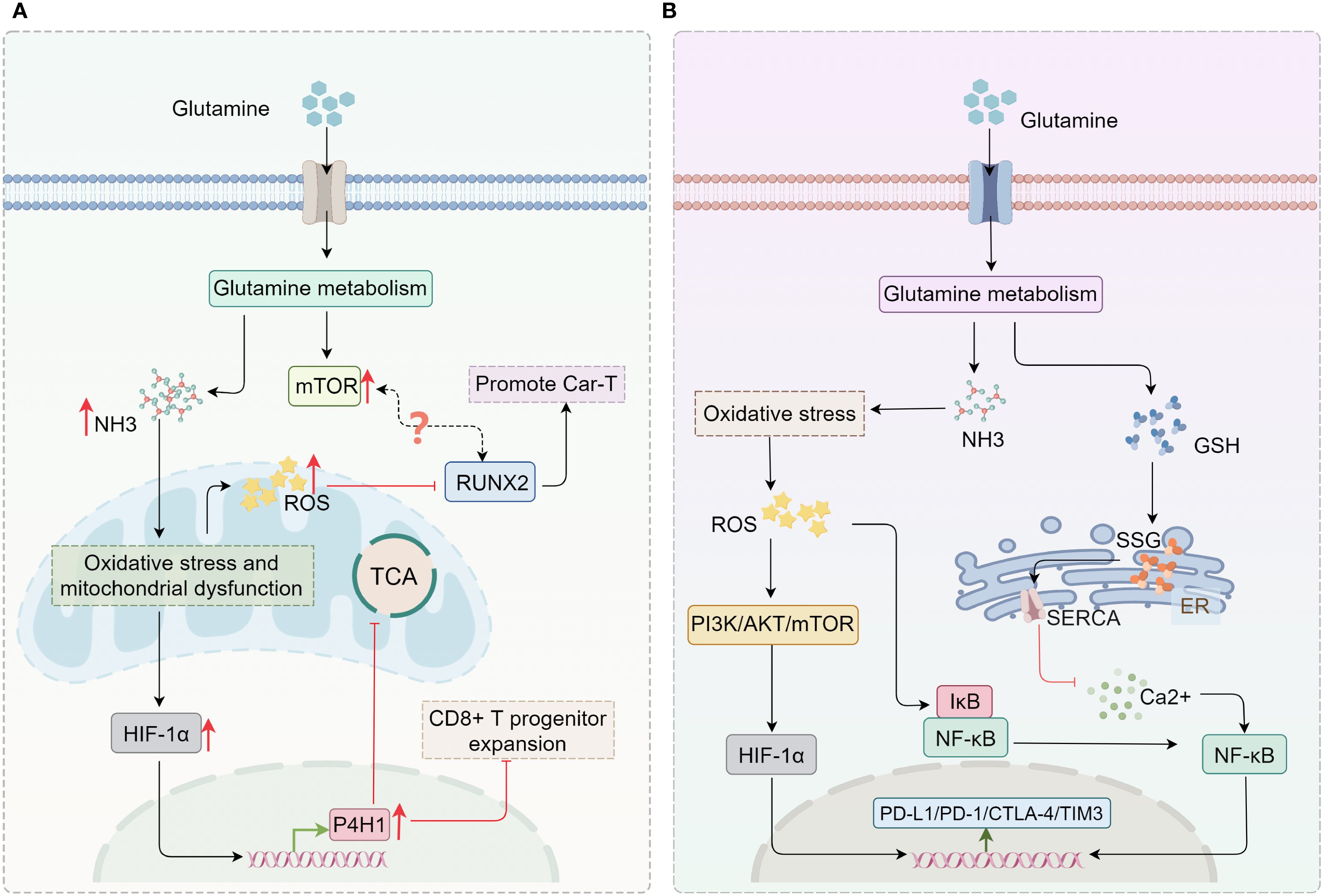
Figure 5. Potential association between ammonia accumulation and immunotherapy. (A) Ammonia accumulation resulting from glutamine metabolism can activate HIF-1α/P4HA1, thereby inhibiting the TCA cycle and the expansion of CD8+ T progenitor cells. Both ROS generated by oxidative stress and the mTOR pathway activated by glutamine metabolism can regulate RUNX2, further affecting CAR-T therapy. (B) Glutamine metabolism not only modulates the expression of immune checkpoints through the ROS-mediated PI3K/AKT pathway, but also influences the calcium/NF-κB signaling pathway by mediating GSH synthesis to regulate the expression of immune checkpoints. GSH, glutathione; ER, endoplasmic reticulum; SERCA, sarcoendoplasmic reticulum calcium ATPase; SSG, protein S-glutathionylation; TCA, tricarboxylic acid; P4HA1, prolyl 4-hydroxylase 1..
This time-dependent paradoxical effect of ammonia accumulation can be harnessed through precise interventional strategies to achieve therapeutic benefits. During the early stage of ammonia accumulation, maintaining an appropriate ammonia level supports RUNX2-mediated T cell function. For instance, transient early inhibition of ammonia production using glutaminase inhibitors (e.g., JHU083) can delay the exhaustion phenotype (15, 16). In the late phase, ammonia scavengers (e.g., C381, which lowers lysosomal pH) or overexpression of urea cycle enzymes (e.g., CPS1) can mitigate lysosomal damage and mitochondrial dysfunction, while inhibition of the HIF-1α/P4HA1 axis helps sustain T cell persistence (173, 179, 180). These findings suggest that suppressing ammonia accumulation in immune cells or targeting ammonia-induced cell death pathways may represent a promising strategy to prolong Teff survival and optimize CAR-T therapy. In this context, combining targeted ammonia metabolism intervention (e.g., using the GLS1 inhibitor CB-839) with CAR-T cell infusion not only blocks tumor cell utilization of ammonia but also enhances T cell adaptability through metabolic plasticity, thereby simultaneously alleviating the immunosuppressive microenvironment and prolonging CAR-T cell persistence (156, 159). This strategy integrates the dual roles of ammonia into a novel paradigm of “spatiotemporal regulation–metabolic intervention–immune synergy,” offering a new approach to overcome the limitations of CAR-T therapy.
5.2.2 Targeting ammonia death to enhance ICI efficacy
ICIs have emerged as revolutionary therapeutic strategies in tumor immunotherapy, primarily by activating T cells and alleviating immunosuppressive signals (3, 181). However, T cells may still experience exhaustion during prolonged immune responses (182). Thus, prolonging the survival of effector T cells is a critical strategy to enhance the long-term efficacy of ICIs. Additionally, immune evasion remains a major challenge in ICI therapy. Despite the impressive success of immune checkpoint inhibitors across various tumor types, tumor cells can circumvent immune system attacks through several mechanisms, undermining the therapeutic potential of ICIs (183, 184). For example, tumor cells can evade immune surveillance not only through the PD-L1/PD-1 pathway but also by suppressing T cell function through other immune checkpoints such as TIM-3 and CTLA-4 (183, 184).
Research suggests that ammonia accumulation can trigger intracellular metabolic disorders and oxidative stress, such as increased ROS production, which activates key signaling pathways, including NF-κB, PI3K/AKT/mTOR, and HIF-1α (178, 185, 186). These pathways are integral to cellular responses to metabolic stress, oxidative stress, and immune reactions. Their activation is closely associated with immune evasion, tumor progression, and the regulation of immune cell function (186, 187).
The interaction between ROS and NF-κB is a critical mechanism of immunosuppression in chronic inflammation and the tumor microenvironment (188). Ammonia accumulation in CD8+ T cells triggers PD-1 upregulation via ROS/NF-κB (189, 190), while scavenging ammonia augments PD-1 blockade response in vivo (191). Studies indicate that blocking glutamine metabolism in tumor cells can enhance the expression of PD-L1, enabling tumor cells to evade T cell-mediated immune killing (191, 192). Under glutamine-restricted conditions, intracellular GSH levels decrease, disrupting the activity of the sarco/endoplasmic reticulum calcium ATPase (SERCA) and activating the calcium/NF-κB signaling pathway. This activation leads to the translocation of NF-κB to the nucleus, where it binds directly to the PD-L1 promoter, promoting its transcription (191, 192). An analysis of clinical cohort data similarly demonstrated that the glutamine antagonist JHU083 promotes the proliferation of CD8+ T cells and enhances the efficacy of PD-1 inhibitors (193).
While blocking glutamine metabolism in combination with PD-1/PD-L1 inhibitors significantly boosts T cell anti-tumor activity in vitro and in vivo in tumor cells with primary resistance (low PD-L1 expression) (191), this approach may not be effective for immunotherapy in “hot” tumors that already exhibit high PD-L1 expression. Currently, detailed experimental studies on the regulatory role of ammonia accumulation in immune checkpoint expression in tumors are limited, and further research is needed. Nevertheless, evidence suggests that in TAMs and MDSCs, oxidative stress can activate IκB kinase (IKK) by promoting ROS production, leading to the degradation of IκB protein, the release of NF-κB, upregulation of PD-L1, and ultimately reduced anti-tumor T cell activity (194, 195). Similarly, under oxidative stress, ROS can inhibit PTEN, a negative regulator of the PI3K pathway, leading to the activation of the PI3K-AKT signaling axis (196, 197). AKT activation increases metabolic activity and transcriptional regulation via the mTOR pathway, which indirectly upregulates immune checkpoint molecules such as PD-1 and CTLA-4 (198, 199). HIF-1α, a key mediator of cellular responses to hypoxia and metabolic stress, is also activated by oxidative stress. By increasing ROS levels, oxidative stress can activate the PI3K/AKT and mTOR pathways, promoting the expression and stabilization of HIF-1α. The upregulation of HIF-1α induces T cells to express PD-1 and other exhaustion-related genes such as TIM-3 (200, 201) (Figure 5B). These findings suggest a potential link between ammonia accumulation and immune checkpoint expression, implying that ammonia accumulation can promote immune checkpoint expression in immune cells, leading to tumor drug resistance and immune evasion. Thus, targeting ammonia accumulation or ammonia death-related pathways in immune cells may provide a promising combination strategy to enhance the efficacy of ICIs.
6 Challenges and future directions of ammonia death in cancer therapy
Current strategies targeting ammonia-induced cell death face multiple challenges. Insufficient targeting specificity represents a core bottleneck: although glutaminase inhibitors (e.g., CB-839) can reduce ammonia accumulation in CD8+ T cells, their concurrent suppression of glutamine metabolism in tumor cells may exacerbate nutrient competition within the immune microenvironment, thereby impairing T-cell function (127, 202). Another major obstacle is the ammonia tolerance exhibited by tumor cells. Studies have shown that HCC and pancreatic cancer, among others, highly express CPS1, a urea cycle enzyme that promotes survival by enhancing ammonia detoxification (135, 203). Furthermore, the heterogeneity of the immune microenvironment further limits therapeutic efficacy: competitive uptake of glutamine by tumor cells via SLC38A2 significantly impairs the antigen-presenting capacity of dendritic cells (33), while MDSCs and TAMs exhibit enhanced immunosuppressive activity under glutamine-deficient conditions (79). Inadequate drug delivery efficiency is also a concern; small-molecule inhibitors such as V-9302, due to their lack of tumor targeting, may disrupt metabolic homeostasis in normal tissues (204).
Potential side effects also warrant serious consideration. Preclinical studies indicate that systemic glutamine deprivation may induce muscle atrophy and intestinal mucosal damage (7), while high-dose administration of the ammonia scavenger sodium phenylbutyrate carries a risk of neurotoxicity (128). Particularly noteworthy is that inhibiting ammonia clearance may inadvertently promote tumor progression: ammonia drives lipid synthesis by activating SREBP-1, and impaired ammonia clearance may accelerate tumor metastasis (90).
Future research should focus on four major breakthrough directions: (1). Spatiotemporally precise regulation: Develop tumor microenvironment-responsive prodrugs (e.g., DRP-104) to achieve localized induction of ammoniagenesis, thereby avoiding systemic toxicity. (2). Combined metabolic intervention: The combination of CPS1 inhibitors and GLS inhibitors may simultaneously disrupt tumor ammonia detoxification barriers and alleviate ammonia toxicity in CD8+ T cells. (3). Immunometabolic reprogramming: Enhancing SLC38A2 expression in type 1 conventional dendritic cells (cDC1s) may improve glutamine uptake and potentially reverse antigen presentation defects. (4). Dynamic monitoring techniques: Utilize positron emission tomography (PET) imaging with ¹¹C-labeled BPTES to monitor tumor glutaminase activity in real time for guiding precision drug administration. Special emphasis should be placed on elucidating the dual regulatory mechanisms of ammonia metabolism in CAR-T therapy: maintaining appropriate ammonia levels in the early phase to activate RUNX2-mediated T-cell function, while reducing lysosomal pH via agents such as C381 in the later phase to delay T-cell exhaustion (172).
7 Conclusion
Ammonia death, a newly identified form of T cell death, has been shown to play a key role in the demise of CD8+ T cells (15, 16). In the TME, activated CD8+ T cells undergo rapid proliferation and metabolic reprogramming to sustain their anti-tumor activity (59, 60). This reprogramming accelerates glutamine metabolism, producing large amounts of ammonia as a byproduct, which accumulates intracellularly and triggers cell death (15, 16). However, the TME is a highly complex regulatory network, encompassing not only CD8+ T cells but also other immune cells and tumor cells (57). This review examines glutamine metabolism in various immune and tumor cells within the TME and finds that elevated glutamine metabolic activity is not exclusive to CD8+ T cells. The activation of other immune cells also involves glutamine metabolism, and in particular, evidence suggests that glutamine metabolism in tumor cells may be even more pronounced than in immune cells. Therefore, regulating ammonia death-related pathways could potentially benefit other immune cells, while ammonia accumulation may also influence tumor cells themselves. These interactions warrant further investigation and exploration.
Inhibiting pathways associated with ammonia-induced cell death can enhance CD8+ T-cell survival, significantly improving the effectiveness of adoptive cell therapy (15). However, the precise role of ammonia in this process remains unclear. While previous research suggests that ammonia accumulation may indirectly influence potential CAR-T therapy targets and modulate the expression of immune checkpoints through the activation of oxidative stress, experimental validation is still needed. Elucidating the mechanisms of ammonia conversion and clearance within the TME could pave the way for new therapeutic strategies aimed at mitigating ammonia-induced damage in immune cells while promoting ammonia-induced cell death in tumor cells.
Current targeting strategies continue to confront a triple paradox: First, while inhibition of glutamine metabolism enhances memory differentiation of T cells (60), it may exacerbate nutrient competition within the tumor microenvironment. Second, although ammonia scavengers prolong T cell survival (104), they may promote tumor lipid synthesis via activation of the SREBP-1 pathway (90). Third, the “double-edged sword effect” of ammonia in CAR-T therapy—supporting T cell priming at early stages while accelerating exhaustion in later phases (172). Overcoming these challenges requires spatiotemporally dynamic regulatory strategies: maintaining appropriate ammonia levels during early treatment to promote T cell function, while controlling toxicity in later stages through lysosomal pH modulators (e.g., C381) or urea cycle enzyme activators.
Future research should focus on four dimensions: (1). In-depth mechanistic investigation: Clarifying the molecular switches of ammonia-induced death in other immune cells such as CD4+ T cells and TAMs. (2). Remodeling the metabolic microenvironment: Developing bifunctional nanocarriers targeting tumor ammonia metabolism (e.g., PD-L1 antibody conjugates loaded with JHU083). (3). Clinical translation exploration: Validating the efficacy of combinations such as DRP-104 with immune checkpoint blockers in specific subgroups, for instance, KEAP1-mutant lung cancer (163). (4). Technological innovation: Utilizing single-cell metabolomics to delineate spatiotemporal landscapes of ammonia transport (e.g., regulatory mechanisms of RHCG channels). Only through multi-omics integration and precise intervention can ammonia-induced cell death be transformed from a “stumbling block” into a “stepping stone” for immunotherapy, ultimately breaking through the current survival bottlenecks in tumor immunotherapy.
In conclusion, inhibiting ammonia death in effector CD8+ T-cells theoretically holds promise for improving immunotherapy efficacy. However, the underlying mechanisms of ammonia death in CD8+ T-cells remain under investigation, and mechanisms in other cell types are yet to be explored. Continued research into the immunological aspects of ammonia metabolism in the TME is essential for advancing tumor immunotherapy targeting ammonia death, with the goal of achieving innovative breakthroughs in cancer treatment and providing clinical benefits to cancer individuals.
Author contributions
FD: Conceptualization, Funding acquisition, Writing – review & editing, Writing – original draft. LX: Data curation, Writing – review & editing, Methodology. WG: Writing – review & editing, Formal analysis, Project administration. QD: Project administration, Writing – review & editing, Resources. JL: Validation, Visualization, Writing – review & editing, Methodology, Formal analysis. XZ: Writing – review & editing, Methodology, Project administration. QZ: Writing – review & editing, Investigation, Visualization. LY: Writing – review & editing, Methodology, Supervision. YL: Validation, Writing – review & editing, Software, Project administration. YH: Project administration, Investigation, Writing – original draft. BW: Writing – review & editing, Data curation. JZ: Validation, Writing – review & editing. JD: Writing – review & editing, Resources. WZ: Writing – review & editing, Formal analysis. ZZ: Methodology, Data curation, Writing – review & editing, Funding acquisition.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This project supported by Natural Fund of Sichuan Science and Technology Department (125104004-2022NSFSC0816/01).
Acknowledgments
We thank Bullet Edits Limited for the linguistic editing and proofreading of the manuscript. The figures were created using Figdraw.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
PD-1, programmed cell death protein 1; PD-L1, programmed cell death ligand 1; ICIs, immune checkpoint inhibitors; TME, tumor microenvironment; TCA, tricarboxylic acid; GLS, glutaminase; GLUD, glutamate dehydrogenase; GOT, glutamate oxaloacetate transaminase; GPT, glutamate pyruvate transaminase; PSAT, phosphoserine aminotransferase; α-KG, α-ketoglutarate; DCs, dendritic cells; CPS1, carbamoyl-phosphate synthetase 1; ROS, reactive oxygen species; Treg, regulator T cells; NK, natural killer; MDSC, myeloid-derived suppressor cells; FOXP3, forkhead box P3; TAM, tumor-associated macrophages; NADPH, nicotinamide adenine dinucleotide phosphate; RHCG, rhesus glycoprotein C.; P4HA1, prolyl 4-hydroxylase 1.
References
1. De Martino M, Rathmell JC, Galluzzi L, and Vanpouille-Box C. Cancer cell metabolism and antitumour immunity. Nat Rev Immunol. (2024) 24:654–69. doi: 10.1038/s41577-024-01026-4
2. Maalej KM, Merhi M, Inchakalody VP, Mestiri S, Alam M, Maccalli C, et al. CAR-cell therapy in the era of solid tumor treatment: current challenges and emerging therapeutic advances. Mol Cancer. (2023) 22:20. doi: 10.1186/s12943-023-01723-z
3. Kroemer G, Chan TA, Eggermont AMM, and Galluzzi L. Immunosurveillance in clinical cancer management. CA: Cancer J For Clin. (2024) 74:187–202. doi: 10.3322/caac.21818
4. Saw PE, Liu Q, Wong P-P, and Song E. Cancer stem cell mimicry for immune evasion and therapeutic resistance. Cell Stem Cell. (2024) 31:1101–12. doi: 10.1016/j.stem.2024.06.003
5. Khosravi G-R, Mostafavi S, Bastan S, Ebrahimi N, Gharibvand RS, and Eskandari N. Immunologic tumor microenvironment modulators for turning cold tumors hot. Cancer Commun (London England). (2024) 44:521–53. doi: 10.1002/cac2.12539
6. Olawuni B and Bode BP. Asparagine as a signal for glutamine sufficiency via asparagine synthetase: a fresh evidence-based framework in physiology and oncology. Am J Physiol Cell Physiol. (2024) 327:C1335–46. doi: 10.1152/ajpcell.00316.2024
7. Yoo HC, Yu YC, Sung Y, and Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. (2020) 52:1496–516. doi: 10.1038/s12276-020-00504-8
8. Wang D, Duan J-J, Guo Y-F, Chen J-J, Chen T-Q, Wang J, et al. Targeting the glutamine-arginine-proline metabolism axis in cancer. J Enzyme Inhibit Med Chem. (2024) 39:2367129. doi: 10.1080/14756366.2024.2367129
9. Gao L, Wei Z, Ying F, Huang L, Zhang J, Sun S, et al. Glutamine metabolism prognostic index predicts tumour microenvironment characteristics and therapeutic efficacy in ovarian cancer. J Cell Mol Med. (2024) 28:e18198. doi: 10.1111/jcmm.18198
10. Jin X-K, Zhang S-M, Liang J-L, Zhang S-K, Qin Y-T, Huang Q-X, et al. A PD-L1-targeting regulator for metabolic reprogramming to enhance glutamine inhibition-mediated synergistic antitumor metabolic and immune therapy. Adv Mater (Deerfield Beach Fla.). (2024) 36:e2309094. doi: 10.1002/adma.202309094
11. Li R, Liu X, Huang X, Zhang D, Chen Z, Zhang J, et al. Single-cell transcriptomic analysis deciphers heterogenous cancer stem-like cells in colorectal cancer and their organ-specific metastasis. Gut. (2024) 73:470–84.
12. Scirgolea C, Sottile R, De Luca M, Susana A, Carnevale S, Puccio S, et al. NaCl enhances CD8+ T cell effector functions in cancer immunotherapy. Nat Immunol. (2024) 25:1845–57. doi: 10.1038/s41590-024-01923-9
13. Jin J, Byun J-K, Choi Y-K, and Park K-G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. (2023) 55:706–15. doi: 10.1038/s12276-023-00971-9
14. Kim GW, Lee DH, Jeon YH, Yoo J, Kim SY, Lee SW, et al. Glutamine synthetase as a therapeutic target for cancer treatment. Int J Mol Sci. (2021) 22(4):1701. doi: 10.3390/ijms22041701
15. Zhang H, Liu J, Yuan W, Zhang Q, Luo X, Li Y, et al. Ammonia-induced lysosomal and mitochondrial damage causes cell death of effector CD8+ T cells. Nat Cell Biol. (2024) 26:1892–902. doi: 10.1038/s41556-024-01503-x
16. Li Z, Lin J, and Yin P. Ammonia death: a novel potential strategy to augment immunotherapy in cancer. Cancer Gene Ther. (2024) 31:1751–3. doi: 10.1038/s41417-024-00851-y
17. Fendt S-M. 100 years of the Warburg effect: A cancer metabolism endeavor. Cell. (2024) 187:3824–8. doi: 10.1016/j.cell.2024.06.026
18. Liao M, Yao D, Wu L, Luo C, Wang Z, Zhang J, et al. Targeting the Warburg effect: A revisited perspective from molecular mechanisms to traditional and innovative therapeutic strategies in cancer. Acta Pharm Sin B. (2024) 14(3):953–1008. doi: 10.1016/j.apsb.2023.12.003
19. Ziki RA and Colnot S. Glutamine metabolism, a double agent combating or fuelling hepatocellular carcinoma. JHEP Rep: Innovation In Hepatol. (2024) 6:101077. doi: 10.1016/j.jhepr.2024.101077
20. Martins F, Gonçalves LG, Pojo M, and Serpa J. Take advantage of glutamine anaplerosis, the kernel of the metabolic rewiring in Malignant gliomas. Biomolecules. (2020) 10(10):1370. doi: 10.3390/biom10101370
21. Song W, Li D, Tao L, Luo Q, and Chen L. Solute carrier transporters: the metabolic gatekeepers of immune cells. Acta Pharm Sin B. (2020) 10:61–78. doi: 10.1016/j.apsb.2019.12.006
22. Li S, Zeng H, Fan J, Wang F, Xu C, Li Y, et al. Glutamine metabolism in breast cancer and possible therapeutic targets. Biochem Pharmacol. (2023) 210:115464. doi: 10.1016/j.bcp.2023.115464
23. Chung C, Sweha SR, Pratt D, Tamrazi B, Panwalkar P, Banda A, et al. Integrated metabolic and epigenomic reprograming by H3K27M mutations in diffuse intrinsic pontine gliomas. Cancer Cell. (2020) 38(3):334–49.e9. doi: 10.1016/j.ccell.2020.07.008
24. Ciuffoli V, Feng X, Jiang K, Acevedo-Luna N, Ko KD, Wang AHJ, et al. Psat1-generated α-ketoglutarate and glutamine promote muscle stem cell activation and regeneration. Genes Dev. (2024) 38:151–67. doi: 10.1101/gad.351428.123
25. Ying M, You D, Zhu X, Cai L, Zeng S, and Hu X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. (2021) 46:102065. doi: 10.1016/j.redox.2021.102065
26. Yao X, Li W, Fang D, Xiao C, Wu X, Li M, et al. Emerging roles of energy metabolism in ferroptosis regulation of tumor cells. Adv Sci (Weinheim Baden-Wurttemberg Germany). (2021) 8:e2100997. doi: 10.1002/advs.202100997
27. Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S, et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm Sin B. (2022) 12:558–80. doi: 10.1016/j.apsb.2021.09.019
28. Ruan X, Yan W, Cao M, Daza RAM, Fong MY, Yang K, et al. Breast cancer cell-secreted miR-199b-5p hijacks neurometabolic coupling to promote brain metastasis. Nat Commun. (2024) 15:4549. doi: 10.1038/s41467-024-48740-0
29. Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, et al. A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab. (2020) 31(2):267–83.e12. doi: 10.1016/j.cmet.2019.11.020
30. Nachef M, Ali AK, Almutairi SM, and Lee S-H. Targeting SLC1A5 and SLC3A2/SLC7A5 as a potential strategy to strengthen anti-tumor immunity in the tumor microenvironment. Front In Immunol. (2021) 12:624324. doi: 10.3389/fimmu.2021.624324
31. Chen P, Jiang Y, Liang J, Cai J, Zhuo Y, Fan H, et al. SLC1A5 is a novel biomarker associated with ferroptosis and the tumor microenvironment: a pancancer analysis. Aging. (2023) 15:7451–75. doi: 10.18632/aging.204911
32. Huang R, Wang H, Hong J, Wu J, Huang O, He J, et al. Targeting glutamine metabolic reprogramming of SLC7A5 enhances the efficacy of anti-PD-1 in triple-negative breast cancer. Front In Immunol. (2023) 14:1251643. doi: 10.3389/fimmu.2023.1251643
33. Guo C, You Z, Shi H, Sun Y, Du X, Palacios G, et al. SLC38A2 and glutamine signalling in cDC1s dictate anti-tumour immunity. Nature. (2023) 620:200–8. doi: 10.1038/s41586-023-06299-8
34. Mathew M, Sivaprakasam S, Dharmalingam-Nandagopal G, Sennoune SR, Nguyen NT, Jaramillo-Martinez V, et al. Induction of oxidative stress and ferroptosis in triple-negative breast cancer cells by niclosamide via blockade of the function and expression of SLC38A5 and SLC7A11. Antioxid (Basel Switzerland). (2024) 13(3):291. doi: 10.3390/antiox13030291
35. Jiang B, Zhang J, Zhao G, Liu M, Hu J, Lin F, et al. Filamentous GLS1 promotes ROS-induced apoptosis upon glutamine deprivation via insufficient asparagine synthesis. Mol Cell. (2022) 82. doi: 10.1016/j.molcel.2022.03.016
36. Wang D, Li X, Gong G, Lu Y, Guo Z, Chen R, et al. An updated patent review of glutaminase inhibitors (2019-2022). Expert Opin On Ther Patents. (2023) 33:17–28. doi: 10.1080/13543776.2023.2173573
37. Wang L, Fang Z, Gao P, and Zheng J. GLUD1 suppresses renal tumorigenesis and development via inhibiting PI3K/Akt/mTOR pathway. Front In Oncol. (2022) 12:975517. doi: 10.3389/fonc.2022.975517
38. Zhan C-L, Lu Q-Y, Lee S-H, Li X-H, Kim J-D, Lee G-H, et al. IDH2 and GLUD1 depletion arrests embryonic development through an H4K20me3 epigenetic barrier in porcine parthenogenetic embryos. Zool Res. (2024) 45:1175–87. doi: 10.24272/j.issn.2095-8137.2024.219
39. Wang Q, Wu M, Li H, Rao X, Ao L, Wang H, et al. Therapeutic targeting of glutamate dehydrogenase 1 that links metabolic reprogramming and Snail-mediated epithelial-mesenchymal transition in drug-resistant lung cancer. Pharmacol Res. (2022) 185:106490. doi: 10.1016/j.phrs.2022.106490
40. Shang M, Cappellesso F, Amorim R, Serneels J, Virga F, Eelen G, et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature. (2020) 587:626–31. doi: 10.1038/s41586-020-2857-9
41. Moreira Franco YE, Alves MJ, Uno M, Moretti IF, Trombetta-Lima M, de Siqueira Santos S, et al. Glutaminolysis dynamics during astrocytoma progression correlates with tumor aggressiveness. Cancer Metab. (2021) 9:18. doi: 10.1186/s40170-021-00255-8
42. Ji L, Zhang Q, Cao Y, and Liu L. A prognostic risk model, tumor immune environment modulation, and drug prediction of ferroptosis and amino acid metabolism-related genes in hepatocellular carcinoma. Hum Cell. (2023) 36:1173–89. doi: 10.1007/s13577-023-00885-8
43. Li N, Xu X, Liu D, Gao J, Gao Y, Wu X, et al. The delta subunit of the GABAA receptor is necessary for the GPT2-promoted breast cancer metastasis. Theranostics. (2023) 13:1355–69. doi: 10.7150/thno.80544
44. Mitra D, Vega-Rubin-de-Celis S, Royla N, Bernhardt S, Wilhelm H, Tarade N, et al. Abrogating GPT2 in triple-negative breast cancer inhibits tumor growth and promotes autophagy. Int J Cancer. (2021) 148:1993–2009. doi: 10.1002/ijc.33456
45. Ganguly K, Bhatia R, Rauth S, Kisling A, Atri P, Thompson C, et al. Mucin 5AC serves as the nexus for β-catenin/c-myc interplay to promote glutamine dependency during pancreatic cancer chemoresistance. Gastroenterology. (2022) 162(1):253–68.e13. doi: 10.1053/j.gastro.2021.09.017
46. Xiang S, Zeng H, Xia F, Ji Q, Xue J, Ren R, et al. The dietary flavonoid isoliquiritigenin induced apoptosis and suppressed metastasis in melanoma cells: An in vitro and in vivo study. Life Sci. (2021) 264:118598. doi: 10.1016/j.lfs.2020.118598
47. Amaya ML, Inguva A, Pei S, Jones C, Krug A, Ye H, et al. The STAT3-MYC axis promotes survival of leukemia stem cells by regulating SLC1A5 and oxidative phosphorylation. Blood. (2022) 139:584–96. doi: 10.1182/blood.2021013201
48. Gao P, Tchernyshyov I, Chang T-C, Lee Y-S, Kita K, Ochi T, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. (2009) 458:762–5. doi: 10.1038/nature07823
49. Youness RA, Hafez HM, Khallaf E, Assal RA, Abdel Motaal A, and Gad MZ. The long noncoding RNA sONE represses triple-negative breast cancer aggressiveness through inducing the expression of miR-34a, miR-15a, miR-16, and let-7a. J Cell Physiol. (2019) 234:20286–97. doi: 10.1002/jcp.28629
50. Zhang X, Chen X, Lin J, Lwin T, Wright G, Moscinski LC, et al. Myc represses miR-15a/miR-16-1 expression through recruitment of HDAC3 in mantle cell and other non-Hodgkin B-cell lymphomas. Oncogene. (2012) 31:3002–8. doi: 10.1038/onc.2011.470
51. Boewe AS, Wrublewsky S, Hoppstädter J, Götz C, Kiemer AK, Menger MD, et al. C-Myc/H19/miR-29b axis downregulates nerve/glial (NG)2 expression in glioblastoma multiforme. Mol Ther Nucleic Acids. (2024) 35:102120. doi: 10.1016/j.omtn.2024.102120
52. Iaiza A, Tito C, Ianniello Z, Ganci F, Laquintana V, Gallo E, et al. METTL3-dependent MALAT1 delocalization drives c-Myc induction in thymic epithelial tumors. Clin Epigenet. (2021) 13:173. doi: 10.1186/s13148-021-01159-6
53. Hu S-S, Han Y, Tan T-Y, Chen H, Gao J-W, Wang L, et al. SLC25A21 downregulation promotes KRAS-mutant colorectal cancer progression by increasing glutamine anaplerosis. JCI Insight. (2023) 8(21):e167874. doi: 10.1172/jci.insight.167874
54. Bernfeld E and Foster DA. Glutamine as an essential amino acid for KRas-driven cancer cells. Trends In Endocrinol Metabol: TEM. (2019) 30:357–68. doi: 10.1016/j.tem.2019.03.003
55. Huber K, Giralt A, Dreos R, Michenthaler H, Geller S, Barquissau V, et al. E2F transcription factor-1 modulates expression of glutamine metabolic genes in mouse embryonic fibroblasts and uterine sarcoma cells. Biochim Et Biophys Acta Mol Cell Res. (2024) 1871:119721. doi: 10.1016/j.bbamcr.2024.119721
56. Vilbois S, Xu Y, and Ho P-C. Metabolic interplay: tumor macrophages and regulatory T cells. Trends In Cancer. (2024) 10:242–55. doi: 10.1016/j.trecan.2023.11.007
57. Vadakekolathu J and Rutella S. Escape from T-cell-targeting immunotherapies in acute myeloid leukemia. Blood. (2024) 143:2689–700. doi: 10.1182/blood.2023019961
58. Chu X, Tian Y, and Lv C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol Cancer. (2024) 23:150. doi: 10.1186/s12943-024-02064-1
59. Wang X, Fu S-Q, Yuan X, Yu F, Ji Q, Tang H-W, et al. A GAPDH serotonylation system couples CD8+ T cell glycolytic metabolism to antitumor immunity. Mol Cell. (2024) 84(4):760–75.e7. doi: 10.1016/j.molcel.2023.12.015
60. Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, et al. Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest. (2021) 131(4):760–75.e7. doi: 10.1172/JCI140100
61. Wenes M, Jaccard A, Wyss T, Maldonado-Pérez N, Teoh ST, Lepez A, et al. The mitochondrial pyruvate carrier regulates memory T cell differentiation and antitumor function. Cell Metab. (2022) 34(5):731–46.e9. doi: 10.1016/j.cmet.2022.03.013
62. Jaccard A, Wyss T, Maldonado-Pérez N, Rath JA, Bevilacqua A, Peng J-J, et al. Reductive carboxylation epigenetically instructs T cell differentiation. Nature. (2023) 621:849–56. doi: 10.1038/s41586-023-06546-y
63. Ma EH, Dahabieh MS, DeCamp LM, Kaymak I, Kitchen-Goosen SM, Oswald BM, et al. 13C metabolite tracing reveals glutamine and acetate as critical in vivo fuels for CD8 T cells. Sci Adv. (2024) 10:eadj1431.
64. Leung WK, Torres Chavez AG, French-Kim M, Shafer P, Mamonkin M, Hill LC, et al. Targeting IDH2R140Q and other neoantigens in acute myeloid leukemia. Blood. (2024) 143:1726–37. doi: 10.1182/blood.2023021979
65. Yang C, Wu S, Mou Z, Zhou Q, Dai X, Ou Y, et al. Exosome-derived circTRPS1 promotes Malignant phenotype and CD8+ T cell exhaustion in bladder cancer microenvironments. Mol Ther. (2022) 30:1054–70. doi: 10.1016/j.ymthe.2022.01.022
66. Lei X, de Groot DC, Welters MJP, de Wit T, Schrama E, van Eenennaam H, et al. CD4+ T cells produce IFN-I to license cDC1s for induction of cytotoxic T-cell activity in human tumors. Cell Mol Immunol. (2024) 21:374–92. doi: 10.1038/s41423-024-01133-1
67. Montauti E, Oh DY, and Fong L. CD4+ T cells in antitumor immunity. Trends In Cancer. (2024) 10:969–85. doi: 10.1016/j.trecan.2024.07.009
68. Endo K, Sawa T, Kitamura H, Umezawa K, Makabe H, and Tanaka S. Procyanidin B2 3,3″-di-O-gallate suppresses IFN-γ production in murine CD4+ T cells through the regulation of glutamine influx via direct interaction with ASCT2. Int Immunopharmacol. (2023) 115:109617. doi: 10.1016/j.intimp.2022.109617
69. Matheson LS, Petkau G, Sáenz-Narciso B, D'Angeli V, McHugh J, Newman R, et al. Multiomics analysis couples mRNA turnover and translational control of glutamine metabolism to the differentiation of the activated CD4+ T cell. Sci Rep. (2022) 12:19657. doi: 10.1038/s41598-022-24132-6
70. Hou Y-C, Wu J-M, Chen K-Y, Chen P-D, Lei C-S, Yeh S-L, et al. Effects of prophylactic administration of glutamine on CD4+ T cell polarisation and kidney injury in mice with polymicrobial sepsis. Br J Nutr. (2019) 122:657–65. doi: 10.1017/S0007114519000990
71. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine-dependent α-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signaling. (2015) 8:ra97. doi: 10.1126/scisignal.aab2610
72. Ai K, Li K, Jiao X, Zhang Y, Li J, Zhang Q, et al. IL-2-mTORC1 signaling coordinates the STAT1/T-bet axis to ensure Th1 cell differentiation and anti-bacterial immune response in fish. PloS Pathog. (2022) 18:e1010913. doi: 10.1371/journal.ppat.1010913
73. Wang C, McKeithan TW, Gong Q, Zhang W, Bouska A, Rosenwald A, et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. (2015) 126:1741–52. doi: 10.1182/blood-2015-05-644591
74. Xu T, Stewart KM, Wang X, Liu K, Xie M, Ryu JK, et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature. (2017) 548:228–33. doi: 10.1038/nature23475
75. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of th17 and th1 cell differentiation by glutaminase-dependent metabolism. Cell. (2018) 175(7):1780–95.e19. doi: 10.1016/j.cell.2018.10.001
76. Praharaj M, Shen F, Lee AJ, Zhao L, Nirschl TR, Theodros D, et al. Metabolic reprogramming of tumor-associated macrophages using glutamine antagonist JHU083 drives tumor immunity in myeloid-rich prostate and bladder cancers. Cancer Immunol Res. (2024) 12:854–75. doi: 10.1158/2326-6066.CIR-23-1105
77. Jeroundi N, Roy C, Basset L, Pignon P, Preisser L, Blanchard S, et al. Glycogenesis and glyconeogenesis from glutamine, lactate and glycerol support human macrophage functions. EMBO Rep. (2024) 25:5383–407. doi: 10.1038/s44319-024-00278-4
78. Chen S, Jiang Y, Zheng J, Li P, Liu M, Zhu Y, et al. Folate-targeted nanoparticles for glutamine metabolism inhibition enhance anti-tumor immunity and suppress tumor growth in ovarian cancer. J Controlled Release. (2025) 379:89–104. doi: 10.1016/j.jconrel.2024.12.073
79. Yang Y, Pei T, Liu C, Cao M, Hu X, Yuan J, et al. Glutamine metabolic competition drives immunosuppressive reprogramming of intratumour GPR109A+ myeloid cells to promote liver cancer progression. Gut. (2025) 74:255–69. doi: 10.1136/gutjnl-2024-332429
80. Wang Z, Li B, Li S, Lin W, Wang Z, Wang S, et al. Metabolic control of CD47 expression through LAT2-mediated amino acid uptake promotes tumor immune evasion. Nat Commun. (2022) 13:6308. doi: 10.1038/s41467-022-34064-4
81. Fu Q, Xu L, Wang Y, Jiang Q, Liu Z, Zhang J, et al. Tumor-associated macrophage-derived interleukin-23 interlinks kidney cancer glutamine addiction with immune evasion. Eur Urol. (2019) 75:752–63. doi: 10.1016/j.eururo.2018.09.030
82. Hegde S, Leader AM, and Merad M. MDSC: Markers, development, states, and unaddressed complexity. Immunity. (2021) 54:875–84. doi: 10.1016/j.immuni.2021.04.004
83. Freitas BFA, Verchere CB, and Levings MK. Advances in engineering myeloid cells for cell therapy applications. ACS Synthetic Biol. (2025) 14:10–20. doi: 10.1021/acssynbio.4c00589
84. Xia J, Zhang L, Zhu W, Tu J, Peng X, Deng Q, et al. Tumor cell-derived N-acetyl-aspartyl-glutamate reshapes the tumor microenvironment to facilitate breast cancer metastasis. Sci Bull. (2024) 70(7):1126–38.
85. Tai Y, Chen M, Wang F, Fan Y, Zhang J, Cai B, et al. The role of dendritic cells in cancer immunity and therapeutic strategies. Int Immunopharmacol. (2024) 128:111548. doi: 10.1016/j.intimp.2024.111548
86. Wang M, Sang J, Xu F, Wang S, Liu P, Ma J, et al. Microwave ablation combined with flt3L provokes tumor-specific memory CD8+ T cells-mediated antitumor immunity in response to PD-1 blockade. Adv Sci (Weinheim Baden-Wurttemberg Germany). (2024) p:e2413181.
87. Loftus RM, Assmann N, Kedia-Mehta N, O'Brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. (2018) 9:2341. doi: 10.1038/s41467-018-04719-2
88. Choi W-M, Ryu T, Lee J-H, Shim Y-R, Kim M-H, Kim H-H, et al. Metabotropic glutamate receptor 5 in natural killer cells attenuates liver fibrosis by exerting cytotoxicity to activated stellate cells. Hepatol (Baltimore Md). (2021) 74:2170–85. doi: 10.1002/hep.31875
89. Bell HN, Huber AK, Singhal R, Korimerla N, Rebernick RJ, Kumar R, et al. Microenvironmental ammonia enhances T cell exhaustion in colorectal cancer. Cell Metab. (2023) 35(1):134–49.e6. doi: 10.1016/j.cmet.2022.11.013
90. Cheng C, Geng F, Li Z, Zhong Y, Wang H, Cheng X, et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat Metab. (2022) 4:575–88. doi: 10.1038/s42255-022-00568-y
91. Yang L, Achreja A, Yeung T-L, Mangala LS, Jiang D, Han C, et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. (2016) 24:685–700. doi: 10.1016/j.cmet.2016.10.011
92. Yahsi B and Gunaydin G. Immunometabolism - the role of branched-chain amino acids. Front In Immunol. (2022) 13:886822. doi: 10.3389/fimmu.2022.886822
93. Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal: CCS. (2022) 20:114. doi: 10.1186/s12964-022-00909-0
94. Körner P. Hydrothermal degradation of amino acids. ChemSusChem. (2021) 14:4947–57. doi: 10.1002/cssc.202101487
95. Tambay V, Raymond V-A, Voisin L, Meloche S, and Bilodeau M. Reprogramming of glutamine amino acid transporters expression and prognostic significance in hepatocellular carcinoma. Int J Mol Sci. (2024) 25(14):7558. doi: 10.3390/ijms25147558
96. Mukha A, Kahya U, Linge A, Chen O, Löck S, Lukiyanchuk V, et al. GLS-driven glutamine catabolism contributes to prostate cancer radiosensitivity by regulating the redox state, stemness and ATG5-mediated autophagy. Theranostics. (2021) 11:7844–68. doi: 10.7150/thno.58655
97. Song J-Y, Jia Y, Han H, Yang X-H, Zhang J, Zhang Q, et al. Increased expression of SLC25A18 is associated with Alzheimer’s disease and is involved in Aβ42-induced mitochondrial dysfunction and apoptosis in neuronal cells. Mitochondrion. (2024) 78:101918. doi: 10.1016/j.mito.2024.101918
98. Bodineau C, Tomé M, Courtois S, Costa ASH, Sciacovelli M, Rousseau B, et al. Two parallel pathways connect glutamine metabolism and mTORC1 activity to regulate glutamoptosis. Nat Commun. (2021) 12:4814. doi: 10.1038/s41467-021-25079-4
99. Holeček M. Branched-chain amino acid supplementation in treatment of liver cirrhosis: Updated views on how to attenuate their harmful effects on cataplerosis and ammonia formation. Nutr (Burbank Los Angeles County Calif). (2017) 41:80–5. doi: 10.1016/j.nut.2017.04.003
100. Moedas MF, Simões RJM, and Silva MFB. Mitochondrial targets in hyperammonemia: Addressing urea cycle function to improve drug therapies. Biochem Pharmacol. (2024) 222:116034. doi: 10.1016/j.bcp.2024.116034
101. Hajaj E, Pozzi S, and Erez A. From the inside out: exposing the roles of urea cycle enzymes in tumors and their micro and macro environments. Cold Spring Harbor Perspect In Med. (2024) 14(4):a041538. doi: 10.1101/cshperspect.a041538
102. Kurmi K and Haigis MC. Nitrogen metabolism in cancer and immunity. Trends In Cell Biol. (2020) 30:408–24. doi: 10.1016/j.tcb.2020.02.005
103. Hajaj E, Sciacovelli M, Frezza C, and Erez A. The context-specific roles of urea cycle enzymes in tumorigenesis. Mol Cell. (2021) 81:3749–59. doi: 10.1016/j.molcel.2021.08.005
104. Tang K, Zhang H, Deng J, Wang D, Liu S, Lu S, et al. Ammonia detoxification promotes CD8+ T cell memory development by urea and citrulline cycles. Nat Immunol. (2023) 24:162–73. doi: 10.1038/s41590-022-01365-1
105. Roberts SC, Malik W, and Ison MG. Hyperammonemia syndrome in immunosuppressed individuals. Curr Opin In Infect Dis. (2022) 35:262–8. doi: 10.1097/QCO.0000000000000828
106. Kumar A, Bellar A, Mishra S, Sekar J, Welch N, and Dasarathy S. L-Isoleucine reverses hyperammonemia-induced myotube mitochondrial dysfunction and post-mitotic senescence. J Nutr Biochem. (2024) 123:109498. doi: 10.1016/j.jnutbio.2023.109498
107. Zhou S, Zhang X, Fu Q, Cheng Z, Ji W, and Liu H. The use of selenomethionine to reduce ammonia toxicity in porcine spleen by inhibiting endoplasmic reticulum stress and autophagy mediated by oxidative stress. Ecotoxicol Environ Saf. (2022) 242:113887. doi: 10.1016/j.ecoenv.2022.113887
108. Nguyen TV. USP15 antagonizes CRL4CRBN-mediated ubiquitylation of glutamine synthetase and neosubstrates. Proc Natl Acad Sci U S A. (2021) 118(40):e2111391118.
109. Soria LR and Brunetti-Pierri N. Ammonia and autophagy: An emerging relationship with implications for disorders with hyperammonemia. J Inherit Metab Dis. (2019) 42:1097–104. doi: 10.1002/jimd.12061
110. Cheong H, Lindsten T, and Thompson CB. Autophagy and ammonia. Autophagy. (2012) 8:122–3. doi: 10.4161/auto.8.1.18078
111. Bertheloot D, Latz E, and Franklin BS. Necroptosis, pyroptosis and apoptosis: an intricate game of cell death. Cell Mol Immunol. (2021) 18:1106–21. doi: 10.1038/s41423-020-00630-3
112. Shah SWA, Chen J, Han Q, Xu Y, Ishfaq M, and Teng X. Ammonia inhalation impaired immune function and mitochondrial integrity in the broilers bursa of fabricius: Implication of oxidative stress and apoptosis. Ecotoxicol Environ Saf. (2020) 190:110078. doi: 10.1016/j.ecoenv.2019.110078
113. Osama S, El-Sherei MM, Al-Mahdy DA, Bishr M, Salama O, and Raafat MM. Optimization and characterization of antileukemic L-asparaginase produced by Fusarium solani endophyte. AMB Express. (2023) 13:96. doi: 10.1186/s13568-023-01602-2
114. Liu S, Yao S, Yang H, Liu S, and Wang Y. Autophagy: Regulator of cell death. Cell Death Dis. (2023) 14:648. doi: 10.1038/s41419-023-06154-8
115. Zhang T-Y, Chen T, Hu W-Y, Li J-C, and Guo M-Y. Ammonia induces autophagy via circ-IFNLR1/miR-2188-5p/RNF182 axis in tracheas of chickens. BioFactors (Oxford England). (2022) 48:416–27. doi: 10.1002/biof.1795
116. Hadian K and Stockwell BR. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discov. (2023) 22:723–42. doi: 10.1038/s41573-023-00749-8
117. Tamnanloo F, Ochoa-Sanchez R, Oliveira MM, Lima C, Lépine M, Dubois K, et al. Multiple ammonia-induced episodes of hepatic encephalopathy provoke neuronal cell loss in bile-duct ligated rats. JHEP Rep: Innovation In Hepatol. (2023) 5:100904. doi: 10.1016/j.jhepr.2023.100904
118. Šebela M and Rašková M. Polyamine-derived aminoaldehydes and acrolein: cytotoxicity, reactivity and analysis of the induced protein modifications. Mol (Basel Switzerland). (2023) 28(21):7429. doi: 10.3390/molecules28217429
119. Liu J, Liu H, Tang H, Ran L, Wang D, Yang F, et al. Golgi apparatus regulated pyroptosis through the miR-32-5p/Golga7/NLRP3 axis in chicken splenic lymphocytes exposure to ammonia. Environ pollut (Barking Essex: 1987). (2024) 362:124923. doi: 10.1016/j.envpol.2024.124923
120. Chen D, Shen F, Liu J, Tang H, Teng X, Yang F, et al. Luteolin enhanced antioxidant capability and induced pyroptosis through NF-κB/NLRP3/Caspase-1 in splenic lymphocytes exposure to ammonia. Sci Total Environ. (2024) 919:170699. doi: 10.1016/j.scitotenv.2024.170699
121. Verma V, Jafarzadeh N, Boi S, Kundu S, Jiang Z, Fan Y, et al. MEK inhibition reprograms CD8+ T lymphocytes into memory stem cells with potent antitumor effects. Nat Immunol. (2021) 22:53–66. doi: 10.1038/s41590-020-00818-9
122. Ma R, Ji T, Zhang H, Dong W, Chen X, Xu P, et al. A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8+ T cells. Nat Cell Biol. (2018) 20:21–7. doi: 10.1038/s41556-017-0002-2
123. Zhang H, Tang K, Ma J, Zhou L, Liu J, Zeng L, et al. Ketogenesis-generated β-hydroxybutyrate is an epigenetic regulator of CD8+ T-cell memory development. Nat Cell Biol. (2020) 22:18–25. doi: 10.1038/s41556-019-0440-0
124. Huang J, Zhang X, Zhang H, Li Y, Huang H, Li Z, et al. Addressing clinical limitations of glutaminase inhibitors: novel strategies for osimertinib-resistant lung cancer by exploiting glutamine metabolic dependency. Adv Sci (Weinheim Baden-Wurttemberg Germany). (2024), e2411479.
125. Wu H, Tang X, Kim HJ, Jalali S, Pritchett JC, Villasboas JC, et al. Expression of KLRG1 and CD127 defines distinct CD8+ subsets that differentially impact patient outcome in follicular lymphoma. J For Immunother Cancer. (2021) 9(7):e002662. doi: 10.1136/jitc-2021-002662
126. Shen L, Xiao Y, Zhang C, Li S, Teng X, Cui L, et al. Metabolic reprogramming by ex vivo glutamine inhibition endows CAR-T cells with less-differentiated phenotype and persistent antitumor activity. Cancer Lett. (2022) 538:215710. doi: 10.1016/j.canlet.2022.215710
127. Best SA, Gubser PM, Sethumadhavan S, Kersbergen A, Negrón Abril YL, Goldford J, et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. (2022) 34(6):874–87.e6. doi: 10.1016/j.cmet.2022.04.003
128. Rockey DC, Vierling JM, Mantry P, Ghabril M, Brown RS, Alexeeva O, et al. Randomized, double-blind, controlled study of glycerol phenylbutyrate in hepatic encephalopathy. Hepatol (Baltimore Md). (2014) 59:1073–83. doi: 10.1002/hep.26611
129. Quan S, Li N, Lian S, Wang Y, Liu Y, Liu J, et al. SLC4A4 as a novel biomarker involved in immune system response and lung adenocarcinoma progression. Int Immunopharmacol. (2024) 140:112756. doi: 10.1016/j.intimp.2024.112756
130. Curnock R, Yalci K, Palmfeldt J, Jäättelä M, Liu B, and Carroll B. TFEB-dependent lysosome biogenesis is required for senescence. EMBO J. (2023) 42:e111241. doi: 10.15252/embj.2022111241
131. Ma T, Tian X, Zhang B, Li M, Wang Y, Yang C, et al. Low-dose metformin targets the lysosomal AMPK pathway through PEN2. Nature. (2022) 603:159–65. doi: 10.1038/s41586-022-04431-8
132. Chao X, Wang S, Zhao K, Li Y, Williams JA, Li T, et al. Impaired TFEB-mediated lysosome biogenesis and autophagy promote chronic ethanol-induced liver injury and steatosis in mice. Gastroenterology. (2018) 155(3):865–79.e12. doi: 10.1053/j.gastro.2018.05.027
133. Yamamoto S and Egashira N. Pathological mechanisms of bortezomib-induced peripheral neuropathy. Int J Mol Sci. (2021) 22(2):888. doi: 10.3390/ijms22020888
134. Nitzahn M and Lipshutz GS. CPS1: Looking at an ancient enzyme in a modern light. Mol Genet Metab. (2020) 131:289–98. doi: 10.1016/j.ymgme.2020.10.003
135. Zhang L, Zou Y, Lu Y, Li Z, and Gao F. Unraveling the therapeutic potential of carbamoyl phosphate synthetase 1 (CPS1) in human diseases. Bioorg Chem. (2023) 130:106253. doi: 10.1016/j.bioorg.2022.106253
136. Oh DY and Fong L. Cytotoxic CD4+ T cells in cancer: Expanding the immune effector toolbox. Immunity. (2021) 54:2701–11. doi: 10.1016/j.immuni.2021.11.015
137. Künzli M and Masopust D. CD4+ T cell memory. Nat Immunol. (2023) 24:903–14. doi: 10.1038/s41590-023-01510-4
138. Vallianou N, Christodoulatos GS, Karampela I, Tsilingiris D, Magkos F, Stratigou T, et al. Understanding the role of the gut microbiome and microbial metabolites in non-alcoholic fatty liver disease: current evidence and perspectives. Biomolecules. (2021) 12(1):56. doi: 10.3390/biom12010056
139. Ghosh N, Mahalanobish S, and Sil PC. Reprogramming of urea cycle in cancer: Mechanism, regulation and prospective therapeutic scopes. Biochem Pharmacol. (2024) 228:116326. doi: 10.1016/j.bcp.2024.116326
140. Dong C, Zhao Y, Han Y, Li M, and Wang G. Targeting glutamine metabolism crosstalk with tumor immune response. Biochim Et Biophys Acta Rev On Cancer. (2024) 1880:189257. doi: 10.1016/j.bbcan.2024.189257
141. Peng S, Wang Z, Tang P, Wang S, Huang Y, Xie Q, et al. PHF8-GLUL axis in lipid deposition and tumor growth of clear cell renal cell carcinoma. Sci Adv. (2023) 9:eadf3566. doi: 10.1126/sciadv.adf3566
142. Zhong Y, Geng F, Mazik L, Yin X, Becker AP, Mohammed S, et al. Combinatorial targeting of glutamine metabolism and lysosomal-based lipid metabolism effectively suppresses glioblastoma. Cell Rep Med. (2024) 5:101706. doi: 10.1016/j.xcrm.2024.101706
143. Wise DR and Thompson CB. Glutamine addiction: a new therapeutic target in cancer. Trends In Biochem Sci. (2010) 35:427–33. doi: 10.1016/j.tibs.2010.05.003
144. Li X, Peng X, Li Y, Wei S, He G, Liu J, et al. Glutamine addiction in tumor cell: oncogene regulation and clinical treatment. Cell Commun Signal: CCS. (2024) 22:12. doi: 10.1186/s12964-023-01449-x
145. Wu J, Liu J, Lapenta K, Desrouleaux R, Li M-D, and Yang X. Regulation of the urea cycle by CPS1 O-GlcNAcylation in response to dietary restriction and aging. J Mol Cell Biol. (2022) 14(3):mjac016.
146. Polletta L, Vernucci E, Carnevale I, Arcangeli T, Rotili D, Palmerio S, et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy. (2015) 11:253–70. doi: 10.1080/15548627.2015.1009778
147. Cortes J, Rugo HS, Cescon DW, Im S-A, Yusof MM, Gallardo C, et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. New Engl J Med. (2022) 387:217–26. doi: 10.1056/NEJMoa2202809
148. Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature. (2017) 546:168–72. doi: 10.1038/nature22359
149. Taguchi A, Fahrmann JF, and Hanash SM. A promising CPS1 inhibitor keeping ammonia from fueling cancer. Cell Chem Biol. (2020) 27:253–4. doi: 10.1016/j.chembiol.2020.03.002
150. Sun X-M, Wu X, Wei M-G, Zhu L-Z, Wu W-H, Zhou X-Y, et al. CPS1 augments hepatic glucagon response through CaMKII/FOXO1 pathway. Front In Pharmacol. (2024) 15:1437738. doi: 10.3389/fphar.2024.1437738
151. Yao S, Nguyen T-V, Rolfe A, Agrawal AA, Ke J, Peng S, et al. Small molecule inhibition of CPS1 activity through an allosteric pocket. Cell Chem Biol. (2020) 27(3):259–68.e5. doi: 10.1016/j.chembiol.2020.01.009
152. Feng Y, Pathria G, Heynen-Genel S, Jackson M, James B, Yin J, et al. Identification and characterization of IMD-0354 as a glutamine carrier protein inhibitor in melanoma. Mol Cancer Ther. (2021) 20:816–32. doi: 10.1158/1535-7163.MCT-20-0354
153. Wang T, Zhang J, Hou T, Yin X, and Zhang N. Selective targeting of tumor cells and tumor associated macrophages separately by twin-like core-shell nanoparticles for enhanced tumor-localized chemoimmunotherapy. Nanoscale. (2019) 11:13934–46. doi: 10.1039/C9NR03374B
154. Jin H, Wang S, Zaal EA, Wang C, Wu H, Bosma A, et al. A powerful drug combination strategy targeting glutamine addiction for the treatment of human liver cancer. ELife. (2020) 9:e56749. doi: 10.7554/eLife.56749
155. Ren H, Wu Z, Tan J, Tao H, Zou W, Cao Z, et al. Co-delivery nano system of MS-275 and V-9302 induces pyroptosis and enhances anti-tumor immunity against uveal melanoma. Adv Sci (Weinheim Baden-Wurttemberg Germany). (2024) 11:e2404375. doi: 10.1002/advs.202404375
156. Yang W-H, Qiu Y, Stamatatos O, Janowitz T, and Lukey MJ. Enhancing the efficacy of glutamine metabolism inhibitors in cancer therapy. Trends In Cancer. (2021) 7:790–804. doi: 10.1016/j.trecan.2021.04.003
157. Zhang Y, Kumata K, Xie L, Kurihara Y, Ogawa M, Kokufuta T, et al. The glutaminase-1 inhibitor [11C-carbony]BPTES: synthesis and positron emission tomography study in mice. Pharm (Basel Switzerland). (2023) 16(7):963. doi: 10.3390/ph16070963
158. Fu Z, Du H, Meng S, Yao M, Zhao P, Li X, et al. Tumor-targeted dual-starvation therapy based on redox-responsive micelle nanosystem with co-loaded LND and BPTES. Mater Today Bio. (2022) 16:100449. doi: 10.1016/j.mtbio.2022.100449
159. Wu H, Zhang J, Wang Q, Li Z, Li L, and Xie Y. Metformin combined with CB-839 specifically inhibits KRAS-mutant ovarian cancer. Sci Rep. (2025) 15:6072. doi: 10.1038/s41598-025-90963-8
160. Hoskin AJ, Holt AK, Legge DN, Collard TJ, Williams AC, and Vincent EE. Aspirin and the metabolic hallmark of cancer: novel therapeutic opportunities for colorectal cancer. Explor Target Anti-tumor Ther. (2023) 4:600–15. doi: 10.37349/etat
161. Soth MJ, Le K, Di Francesco ME, Hamilton MM, Liu G, Burke JP, et al. Discovery of IPN60090, a clinical stage selective glutaminase-1 (GLS-1) inhibitor with excellent pharmacokinetic and physicochemical properties. J Med Chem. (2020) 63:12957–77. doi: 10.1021/acs.jmedchem.0c01398
162. Rais R, Lemberg KM, Tenora L, Arwood ML, Pal A, Alt J, et al. Discovery of DRP-104, a tumor-targeted metabolic inhibitor prodrug. Sci Adv. (2022) 8:eabq5925. doi: 10.1126/sciadv.abq5925
163. Pillai R, LeBoeuf SE, Hao Y, New C, Blum JLE, Rashidfarrokhi A, et al. Glutamine antagonist DRP-104 suppresses tumor growth and enhances response to checkpoint blockade in KEAP1 mutant lung cancer. Sci Adv. (2024) 10:eadm9859. doi: 10.1126/sciadv.adm9859
164. Huang Y, Meng F, Zeng T, Thorne RF, He L, Zha Q, et al. IFRD1 promotes tumor cells “low-cost” survival under glutamine starvation via inhibiting histone H1.0 nucleophagy. Cell Discov. (2024) 10:57. doi: 10.1038/s41421-024-00668-x
165. Xie Y, Yang J, Zhu H, Yang R, and Fan Y. The efferocytosis dilemma: how neutrophil extracellular traps and PI3K/Rac1 complicate diabetic wound healing. Cell Commun Signal: CCS. (2025) 23:103. doi: 10.1186/s12964-025-02092-4
166. Audia S, Mahévas M, Nivet M, Ouandji S, Ciudad M, and Bonnotte B. Immune thrombocytopenia: recent advances in pathogenesis and treatments. HemaSphere. (2021) 5:e574. doi: 10.1097/HS9.0000000000000574
167. Brudno JN, Maus MV, and Hinrichs CS. CAR T cells and T-cell therapies for cancer: A translational science review. JAMA. (2024) 332:1924–35. doi: 10.1001/jama.2024.19462
168. Li Y-R, Lyu Z, Chen Y, Fang Y, and Yang L. Frontiers in CAR-T cell therapy for autoimmune diseases. Trends In Pharmacol Sci. (2024) 45:839–57. doi: 10.1016/j.tips.2024.07.005
169. Ohno R and Nakamura A. Advancing autoimmune Rheumatic disease treatment: CAR-T Cell Therapies - Evidence, Safety, and future directions. Semin In Arthritis Rheum. (2024) 67:152479. doi: 10.1016/j.semarthrit.2024.152479
170. Krishnan V. The RUNX family of proteins, DNA repair, and cancer. Cells. (2023) 12(8):1106. doi: 10.3390/cells12081106
171. Newton AH and Pask AJ. Evolution and expansion of the RUNX2 QA repeat corresponds with the emergence of vertebrate complexity. Commun Biol. (2020) 3:771. doi: 10.1038/s42003-020-01501-3
172. DeGolier KR, Danis E, D'Antonio M, Cimons J, Yarnell M, Kedl RM, et al. Antigen experience history directs distinct functional states of CD8+ CAR T cells during the antileukemia response. Nat Immunol. (2025) 26:68–81. doi: 10.1038/s41590-024-02034-1
173. Ma S, Ong L-T, Jiang Z, Lee WC, Lee PL, Yusuf M, et al. Targeting P4HA1 promotes CD8+ T cell progenitor expansion toward immune memory and systemic anti-tumor immunity. Cancer Cell. (2024) 43(2):213–231.e9.
174. Chang WH, Forde D, and Lai AG. Dual prognostic role of 2-oxoglutarate-dependent oxygenases in ten cancer types: implications for cell cycle regulation and cell adhesion maintenance. Cancer Commun (London England). (2019) 39:23. doi: 10.1186/s40880-019-0369-5
175. Weisshaar N, Ma S, Ming Y, Madi A, Mieg A, Hering M, et al. The malate shuttle detoxifies ammonia in exhausted T cells by producing 2-ketoglutarate. Nat Immunol. (2023) 24:1921–32. doi: 10.1038/s41590-023-01636-5
176. Sharma JR, Agraval H, and Yadav UCS. Cigarette smoke induces epithelial-to-mesenchymal transition, stemness, and metastasis in lung adenocarcinoma cells via upregulated RUNX-2/galectin-3 pathway. Life Sci. (2023) 318:121480. doi: 10.1016/j.lfs.2023.121480
177. Huang Y, Mo S, Jin Y, Zheng Z, Wang H, Wu S, et al. Ammonia-induced excess ROS causes impairment and apoptosis in porcine IPEC-J2 intestinal epithelial cells. Ecotoxicol Environ Saf. (2022) 243:114006. doi: 10.1016/j.ecoenv.2022.114006
178. Zheng P, Qin X, Feng R, Li Q, Huang F, Li Y, et al. Alleviative effect of melatonin on the decrease of uterine receptivity caused by blood ammonia through ROS/NF-κB pathway in dairy cow. Ecotoxicol Environ Saf. (2022) 231:113166. doi: 10.1016/j.ecoenv.2022.113166
179. Liu M-J, Guo H-Y, Liu B, Zhu K-C, Guo L, Liu B-S, et al. Gill oxidative damage caused by acute ammonia stress was reduced through the HIF-1α/NF-κb signaling pathway in golden pompano (Trachinotus ovatus). Ecotoxicol Environ Saf. (2021) 222:112504. doi: 10.1016/j.ecoenv.2021.112504
180. Xiong G, Stewart RL, Chen J, Gao T, Scott TL, Samayoa LM, et al. Collagen prolyl 4-hydroxylase 1 is essential for HIF-1α stabilization and TNBC chemoresistance. Nat Commun. (2018) 9:4456. doi: 10.1038/s41467-018-06893-9
181. Llovet JM, Pinyol R, Yarchoan M, Singal AG, Marron TU, Schwartz M, et al. Adjuvant and neoadjuvant immunotherapies in hepatocellular carcinoma. Nat Rev Clin Oncol. (2024) 21:294–311. doi: 10.1038/s41571-024-00868-0
182. Ni L. Potential mechanisms of cancer stem-like progenitor T-cell bio-behaviours. Clin Trans Med. (2024) 14:e1817. doi: 10.1002/ctm2.1817
183. Wu Y, Yi M, Niu M, Mei Q, and Wu K. Myeloid-derived suppressor cells: an emerging target for anticancer immunotherapy. Mol Cancer. (2022) 21:184. doi: 10.1186/s12943-022-01657-y
184. Du F, Yang L-H, Liu J, Wang J, Fan L, Duangmano S, et al. The role of mitochondria in the resistance of melanoma to PD-1 inhibitors. J Trans Med. (2023) 21:345. doi: 10.1186/s12967-023-04200-9
185. Han Q, Wang A, Fu Q, Zhou S, Bao J, and Xing H. Protective role of selenium on ammonia-mediated nephrotoxicity via PI3K/AKT/mTOR pathway: Crosstalk between autophagy and cytokine release. Ecotoxicol Environ Saf. (2022) 242:113918. doi: 10.1016/j.ecoenv.2022.113918
186. Wang A, Zhang X, Wang H, and Xing H. Recent evidence for toxic effects of NH3 exposure on lung injury: Protective effects of L-selenomethionine. Ecotoxicol Environ Saf. (2022) 242:113937. doi: 10.1016/j.ecoenv.2022.113937
187. Pan T, Sun S, Chen Y, Tian R, Chen E, Tan R, et al. Immune effects of PI3K/Akt/HIF-1α-regulated glycolysis in polymorphonuclear neutrophils during sepsis. Crit Care (London England). (2022) 26:29. doi: 10.1186/s13054-022-03893-6
188. Ming S, Tian J, Ma K, Pei C, Li L, Wang Z, et al. Oxalate-induced apoptosis through ERS-ROS-NF-κB signalling pathway in renal tubular epithelial cell. Mol Med (Cambridge Mass). (2022) 28:88. doi: 10.1186/s10020-022-00494-5
189. Zhang W, Chen L, Liu J, Chen B, Shi H, Chen H, et al. Inhibition of autophagy-related protein 7 enhances anti-tumor immune response and improves efficacy of immune checkpoint blockade in microsatellite instability colorectal cancer. J Exp Clin Cancer Res: CR. (2024) 43:114. doi: 10.1186/s13046-024-03023-w
190. Lopresti L, Capitani N, Tatangelo V, Tangredi C, Boncompagni G, Frezzato F, et al. p66Shc deficiency in CLL cells enhances PD-L1 expression and suppresses immune synapse formation. Front In Cell Dev Biol. (2024) 12:1297116. doi: 10.3389/fcell.2024.1297116
191. Matias MI, Dardalhon V, and Taylor N. Targeting glutamine metabolism and PD-L1: A novel anti-tumor pas de deux. Mol Cell. (2020) 80:555–7. doi: 10.1016/j.molcel.2020.11.005
192. Tang Y, Wang S, Li Y, Yuan C, Zhang J, Xu Z, et al. Simultaneous glutamine metabolism and PD-L1 inhibition to enhance suppression of triple-negative breast cancer. J Nanobiotechnol. (2022) 20:216. doi: 10.1186/s12951-022-01424-7
193. Chen J, Wang R, Liu Z, Fan J, Liu S, Tan S, et al. Unbalanced glutamine partitioning between CD8T cells and cancer cells accompanied by immune cell dysfunction in hepatocellular carcinoma. Cells. (2022) 11(23):3924. doi: 10.3390/cells11233924
194. Zhang W, Liu X, Cao S, Zhang Q, Chen X, Luo W, et al. Multifunctional redox-responsive nanoplatform with dual activation of macrophages and T cells for antitumor immunotherapy. ACS Nano. (2023) 17:14424–41. doi: 10.1021/acsnano.2c12498
195. Adeshakin AO, Liu W, Adeshakin FO, Afolabi LO, Zhang M, Zhang G, et al. Regulation of ROS in myeloid-derived suppressor cells through targeting fatty acid transport protein 2 enhanced anti-PD-L1 tumor immunotherapy. Cell Immunol. (2021) 362:104286. doi: 10.1016/j.cellimm.2021.104286
196. Kma L and Baruah TJ. The interplay of ROS and the PI3K/Akt pathway in autophagy regulation. Biotechnol Appl Biochem. (2022) 69:248–64. doi: 10.1002/bab.2104
197. Kang J, Wang Y, Guo X, He X, Liu W, Chen H, et al. N-acetylserotonin protects PC12 cells from hydrogen peroxide induced damage through ROS mediated PI3K/AKT pathway. Cell Cycle (Georgetown Tex). (2022) 21:2268–82. doi: 10.1080/15384101.2022.2092817
198. Chuangchot N, Jamjuntra P, Yangngam S, Luangwattananun P, Thongchot S, Junking M, et al. Enhancement of PD-L1-attenuated CAR-T cell function through breast cancer-associated fibroblasts-derived IL-6 signaling via STAT3/AKT pathways. Breast Cancer Res: BCR. (2023) 25:86. doi: 10.1186/s13058-023-01684-7
199. Wang S, Liu C, Yang C, Jin Y, Cui Q, Wang D, et al. PI3K/AKT/mTOR and PD−1/CTLA−4/CD28 pathways as key targets of cancer immunotherapy (Review). Oncol Lett. (2024) 28:567. doi: 10.3892/ol.2024.14700
200. Wang Y, Mang X, Li D, Wang Z, Chen Y, Cai Z, et al. Cold atmospheric plasma sensitizes head and neck cancer to chemotherapy and immune checkpoint blockade therapy. Redox Biol. (2024) 69:102991. doi: 10.1016/j.redox.2023.102991
201. Koh HS, Chang CY, Jeon S-B, Yoon HJ, Ahn Y-H, Kim H-S, et al. The HIF-1/glial TIM-3 axis controls inflammation-associated brain damage under hypoxia. Nat Commun. (2015) 6:6340. doi: 10.1038/ncomms7340
202. Madden MZ, Ye X, Chi C, Fisher EL, Wolf MM, Needle GA, et al. Differential effects of glutamine inhibition strategies on antitumor CD8 T cells. J Immunol (Baltimore Md: 1950). (2023) 211:563–75. doi: 10.4049/jimmunol.2200715
203. Chen S, Tang Q, Hu M, Song S, Wu X, Zhou Y, et al. Loss of carbamoyl phosphate synthetase 1 potentiates hepatocellular carcinoma metastasis by reducing aspartate level. Adv Sci (Weinheim Baden-Wurttemberg Germany). (2024) 11:e2402703. doi: 10.1002/advs.202402703
204. Chen R, Zheng S, Zhao X, Huang H, Xu Y, Qiu C, et al. Metabolic reprogramming of macrophages by a nano-sized opsonization strategy to restore M1/M2 balance for osteoarthritis therapy. J Controlled Release. (2025) 380:469–89. doi: 10.1016/j.jconrel.2025.02.005
205. Wu S, Fukumoto T, Lin J, Nacarelli T, Wang Y, Ong D, et al. Targeting glutamine dependence through GLS1 inhibition suppresses ARID1A-inactivated clear cell ovarian carcinoma. Nat Cancer. (2021) 2:189–200. doi: 10.1038/s43018-020-00160-x
206. Huang J, Zhang X, Zhang H, Li Y, Huang H, Li Z, et al. Addressing clinical limitations of glutaminase inhibitors: novel strategies for osimertinib-resistant lung cancer by exploiting glutamine metabolic dependency. Adv Sci (Weinheim Baden-Wurttemberg Germany). (2025) 12:e2411479. doi: 10.1002/advs.202411479
207. Okada T, Yamabe K, Jo M, Sakajiri Y, Shibata T, Sawada R, et al. Design and structural optimization of thiadiazole derivatives with potent GLS1 inhibitory activity. Bioorg Med Chem Lett. (2023) 93:129438. doi: 10.1016/j.bmcl.2023.129438
208. Tu H, Yin X, Wen J, Wu W, Zhai B, Li J, et al. Glutaminase 1 blockade alleviates nonalcoholic steatohepatitis via promoting proline metabolism. Biochem Biophys Res Commun. (2022) 634:1–9. doi: 10.1016/j.bbrc.2022.10.007
209. Encarnación-Rosado J, Sohn ASW, Biancur DE, Lin EY, Osorio-Vasquez V, Rodrick T, et al. Targeting pancreatic cancer metabolic dependencies through glutamine antagonism. Nat Cancer. (2024) 5:85–99. doi: 10.1038/s43018-023-00647-3
210. Yokoyama Y, Estok TM, and Wild R. Sirpiglenastat (DRP-104) induces antitumor efficacy through direct, broad antagonism of glutamine metabolism and stimulation of the innate and adaptive immune systems. Mol Cancer Ther. (2022) 21:1561–72. doi: 10.1158/1535-7163.MCT-22-0282
211. Yang Y, Gao C, Li Q, Liu Y, and Cao J. HMGA2-mediated glutamine metabolism is required for Cd-induced cell growth and cell migration. Toxicology. (2024) 507:153899. doi: 10.1016/j.tox.2024.153899
212. Wang L, Hu T, Shen Z, Zheng Y, Geng Q, Li L, et al. Inhibition of USP1 activates ER stress through Ubi-protein aggregation to induce autophagy and apoptosis in HCC. Cell Death Dis. (2022) 13:951. doi: 10.1038/s41419-022-05341-3
213. Vest RT, Chou C-C, Zhang H, Haney MS, Li L, Laqtom NN, et al. Small molecule C381 targets the lysosome to reduce inflammation and ameliorate disease in models of neurodegeneration. Proc Natl Acad Sci United States America. (2022) 119:e2121609119. doi: 10.1073/pnas.2121609119
214. Austin MC, Muralidharan C, Roy S, Crowder JJ, Piganelli JD, and Linnemann AK. Dysfunctional β-cell autophagy induces β-cell stress and enhances islet immunogenicity. Front In Immunol. (2025) 16:1504583. doi: 10.3389/fimmu.2025.1504583
215. Kurtz CB, Millet YA, Puurunen MK, Perreault M, Charbonneau MR, Isabella VM, et al. An engineered E. coli Nissle improves hyperammonemia and survival in mice and shows dose-dependent exposure in healthy humans. Sci Trans Med. (2019) 11(475):eaau7975.
216. Yin L, Zhou J, Li T, Wang X, Xue W, Zhang J, et al. Inhibition of the dopamine transporter promotes lysosome biogenesis and ameliorates Alzheimer’s disease-like symptoms in mice. Alzheimer’s Dementia. (2023) 19:1343–57. doi: 10.1002/alz.12776
217. Yin L, Zhou Y, Liu H, and Li Y. LYECs: lysosome-enhancing compounds as potential therapeutic approaches for Alzheimer disease. Autophagy. (2023) 19:1863–4. doi: 10.1080/15548627.2022.2131247
218. Ham DJ, Börsch A, Chojnowska K, Lin S, Leuchtmann AB, Ham AS, et al. Distinct and additive effects of calorie restriction and rapamycin in aging skeletal muscle. Nat Commun. (2022) 13:2025. doi: 10.1038/s41467-022-29714-6
219. Selvarani R, Mohammed S, and Richardson A. Effect of rapamycin on aging and age-related diseases-past and future. GeroScience. (2021) 43:1135–58. doi: 10.1007/s11357-020-00274-1
220. Liu L, Lu X, Fan Z, Deng J, Zhang S, Zhang L, et al. TPCA-1 compound, inhibiting testis-specific serine/threonine protein kinase 3 for potential male sterile in Bombyx mori. Pest Manage Sci. (2024) 80:6189–200. doi: 10.1002/ps.8347
221. Wang S, Li J, Hong S, Wang N, Xu S, Yang B, et al. Chemotherapy-elicited extracellular vesicle CXCL1 from dying cells promotes triple-negative breast cancer metastasis by activating TAM/PD-L1 signaling. J Exp Clin Cancer Res: CR. (2024) 43:121. doi: 10.1186/s13046-024-03050-7
222. Zhu J, Yan Y, Qiu X, Lin S, and Wen J. Endoscopic bariatric surgery for adults with overweight and obesity: a systematic review and network meta-analysis. Int J Obes. (2005) 49:237–45. doi: 10.1038/s41366-024-01678-1
223. Paquette M, El-Houjeiri L, Zirden C L, Puustinen P, Blanchette P, Jeong H, et al. AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy. (2021) 17:3957–75. doi: 10.1080/15548627.2021.1898748
224. Wong SK, Chin K-Y, and Ima-Nirwana S. The skeletal-protecting action and mechanisms of action for mood-stabilizing drug lithium chloride: current evidence and future potential research areas. Front In Pharmacol. (2020) 11:430. doi: 10.3389/fphar.2020.00430
225. De Meyer I, Martinet W, Van Hove CE, Schrijvers DM, Hoymans VY, Van Vaeck L, et al. Inhibition of inositol monophosphatase by lithium chloride induces selective macrophage apoptosis in atherosclerotic plaques. Br J Pharmacol. (2011) 162:1410–23. doi: 10.1111/j.1476-5381.2010.01152.x
226. Liu S, Zhang H-L, Li J, Ye Z-P, Du T, Li L-C, et al. Tubastatin A potently inhibits GPX4 activity to potentiate cancer radiotherapy through boosting ferroptosis. Redox Biol. (2023) 62:102677. doi: 10.1016/j.redox.2023.102677
227. Sui L, Huang R, Yu H, Zhang S, and Li Z. Inhibition of HDAC6 by tubastatin A disrupts mouse oocyte meiosis via regulating histone modifications and mRNA expression. J Cell Physiol. (2020) 235:7030–42. doi: 10.1002/jcp.29599
228. Wang X, Ding B, Liu W, Qi L, Li J, Zheng X, et al. Dual starvations induce pyroptosis for orthotopic pancreatic cancer therapy through simultaneous deprivation of glucose and glutamine. J Am Chem Soc. (2024) 146:17854–65. doi: 10.1021/jacs.4c03478
229. Missiaen R, Anderson NM, Kim LC, Nance B, Burrows M, Skuli N, et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab. (2022) 34(8):1151–67.e7. doi: 10.1016/j.cmet.2022.06.010
230. Xu X, Liu G-H, Zhao D, Chen J, Shao Y, Wang J, et al. Enhancement of anammox bacterial activity by sodium glutamate. Chemosphere. (2020) 244:125570. doi: 10.1016/j.chemosphere.2019.125570
231. Zhong Z, Wang C, Zhang H, Mi J, Liang JB, Liao X, et al. Sodium butyrate reduces ammonia emissions through glutamate metabolic pathways in cecal microorganisms of laying hens. Ecotoxicol Environ Saf. (2022) 233:113299. doi: 10.1016/j.ecoenv.2022.113299
232. Prange CJ, Sayed NYB, Feng B, Goepfert C, Trujillo DO, Hu X, et al. A redox-responsive prodrug for tumor-targeted glutamine restriction. J Controlled Release. (2024) 368:251–64. doi: 10.1016/j.jconrel.2024.02.031
Keywords: ammonia death, glutamine, CD8+ T cell, tumor microenvironment, immunotherapy
Citation: Du F, Xiao L, Guojun W, Dai Q, Li J, Zhao X, Zhang Q, Yang L, Liu Y, Hu Y, Wen B, Zhou J, Dai J, Zhang W and Zhang Z (2025) Glutamine metabolism and ammonia death: targeted modulation for enhanced cancer immunotherapy. Front. Immunol. 16:1643017. doi: 10.3389/fimmu.2025.1643017
Received: 07 June 2025; Accepted: 31 August 2025;
Published: 17 September 2025.
Edited by:
Hai Fang, Shanghai Jiao Tong University, ChinaReviewed by:
Zhi Li, Shanghai University of Traditional Chinese Medicine, ChinaXinmei Dong, BeiGene, China
Copyright © 2025 Du, Xiao, Guojun, Dai, Li, Zhao, Zhang, Yang, Liu, Hu, Wen, Zhou, Dai, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhuo Zhang, emh1b3poYW5nQHN3bXUuZWR1LmNu; Fei Du, MTg5ODkxNDYxMDFAMTYzLmNvbQ==