 Shanshan Zhang
Shanshan Zhang Ziqi Jin
Ziqi Jin Li Jiang2
Li Jiang2 Yibin Zhang
Yibin Zhang Jing Lu
Jing Lu- 1Department of Traditional Chinese Internal Medicine, Changchun University of Chinese Medicine, Changchun, Jilin, China
- 2Department of Encephalopathy, The Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, Jilin, China
- 3Research Center of Traditional Chinese Medicine, The Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, Jilin, China
Intracerebral hemorrhage (ICH), a common neurological disorder with a high rate of disability, involves complex immunoinflammatory mechanisms, particularly those related to secondary inflammatory injury. Neutrophils, as the earliest subtype of leukocytes recruited after stroke, play a pivotal role in secondary brain injury. Traditionally, neutrophils were thought to mediate tissue damage primarily via phagocytosis, chemotaxis, and degranulation. However, recent studies have shown that neutrophils also contribute to the pathogenesis of intracerebral hemorrhage by releasing neutrophil extracellular traps (NETs), which exacerbate blood-brain barrier disruption, amplify local inflammy -30ation, and promote neuronal injury. This review systematically examines the interactions between the central and peripheral immune systems following ICH. It focuses on the bidirectional regulatory relationship between microglia and neutrophils, and their coordinated roles in inflammation, blood-brain barrier disruption, neurological dysfunction, and cognitive impairment. In addition, this review summarizes recent potential therapeutic strategies targeting the formation and clearance of NETs, including peptidylarginine deiminase 4 inhibitors, reactive oxygen species inhibitors, histone inhibitors, and DNases. These interventions may offer theoretical insights into novel therapeutic targets for mitigating secondary injury following ICH.
1 Introduction
Stroke ranks among the foremost causes of mortality and disability globally, with its prevalence steadily rising in recent years, particularly in developing countries and aging populations. According to the latest statistics, in 2018, the mortality rate for cerebrovascular diseases in China was 149.49 per 100,000, with 1.57 million deaths (1). By 2020, the mortality rate for strokes had increased to 343.4 per 100,000 (2), with approximately 2.3 million deaths (3). In Europe and the United States, over 1.1 million and 790,000 new stroke cases (4), respectively, are reported annually, with a substantial proportion occurring in individuals with a history of stroke (5). The combination of high incidence, mortality, and disability rates has made stroke a pressing global public health challenge (6).
ICH, one of the most devastating type of stroke, encompasses parenchymal hemorrhage and subarachnoid hemorrhage (SAH), accounting for 15-30% of all stroke cases (7). The one-month mortality rate following onset can be as high as 40% (8). Currently, there are no effective neuroprotective treatments that significantly improve functional outcomes after ICH. Therefore, elucidating the pathological mechanisms underlying secondary injury following ICH and identifying potential therapeutic targets have become key priorities in both clinical and basic research. Following ICH, alongside the primary damage inflicted by the hematoma, secondary injuries such as neuroinflammation, immune cell infiltration, cerebral edema, and oxidative stress also occur (9–13). Among these, neuroinflammation is widely recognized as a central component of secondary injury (14).
Microglia, the resident immune cells of the central nervous system (CNS), promptly react to injury signals subsequent to ICH. Their activation states and polarization phenotypes play a decisive role in determining the extent of neuroinflammation and subsequent tissue repair (15, 16). Meanwhile, accumulating evidence indicates that involvement of the peripheral immune system—particularly the early infiltration of neutrophils into the brain—plays a critical role in amplifying the inflammatory response (17, 18).
During the immune response, neutrophils are rapidly recruited and activated, releasing nuclear and granular contents that form extensive web-like DNA structures known as NETs (19–21). NETs are composed of extracellular double-stranded DNA combined with various components, including histones, neutrophil elastase, myeloperoxidase (MPO), and cathepsins (22). In addition to their role in antimicrobial defense, NETs have emerged as a novel mechanism contributing to neuroinflammation and tissue damage (23). Studies have shown that NETs not only disrupt the blood-brain barrier (BBB) and increase its permeability but also promote the release of inflammatory mediators, thereby amplifying the local immune response (24, 25). Although the independent roles of microglia and neutrophils in the pathophysiology of ICH have been extensively reported, their interactions, particularly the molecular mechanisms linking NETs with central immune cells, remain poorly understood. Notably, most current studies focus on ischemic brain injury, leaving the immune response patterns specific to hemorrhagic stroke relatively underexplored.
This review systematically summarizes the interactions between the central and peripheral immune systems following ICH. It focuses on the signaling crosstalk and pathological synergy between microglia and neutrophils, particularly the bidirectional regulatory role of neutrophil NETs in this process. By outlining recent advances in this field, we aim to provide a theoretical foundation and reference for understanding the mechanisms of neuroinflammation in ICH, identifying potential biomarkers, and developing targeted therapeutic strategies.
2 Immune response following intracerebral hemorrhage
2.1 Interactions between the central and peripheral immune systems
Due to its antigen-induced immune tolerance and the physical isolation provided by the BBB and the blood-cerebrospinal fluid barrier, the CNS has long been considered an immune-privileged site. The presence of peripheral immune components within the central CNS was traditionally considered a pathological feature under healthy conditions. However, research over the past two decades has gradually challenged this view (26). Growing evidence indicates that the CNS and the peripheral immune system are not entirely distinct entities, but instead form an interactive network through complex crosstalk (27, 28). As illustrated in Figure 1. The immune system maintains immunosurveillance homeostasis by detecting not only pathogenic signals but also cues from damaged tissues, particularly in sterile injuries such as traumatic brain injury (TBI), spinal cord injury, and ICH (29–31). Consequently, following ICH, the peripheral immune system performs a vital function in secondary injury and directly influences long-term outcomes.
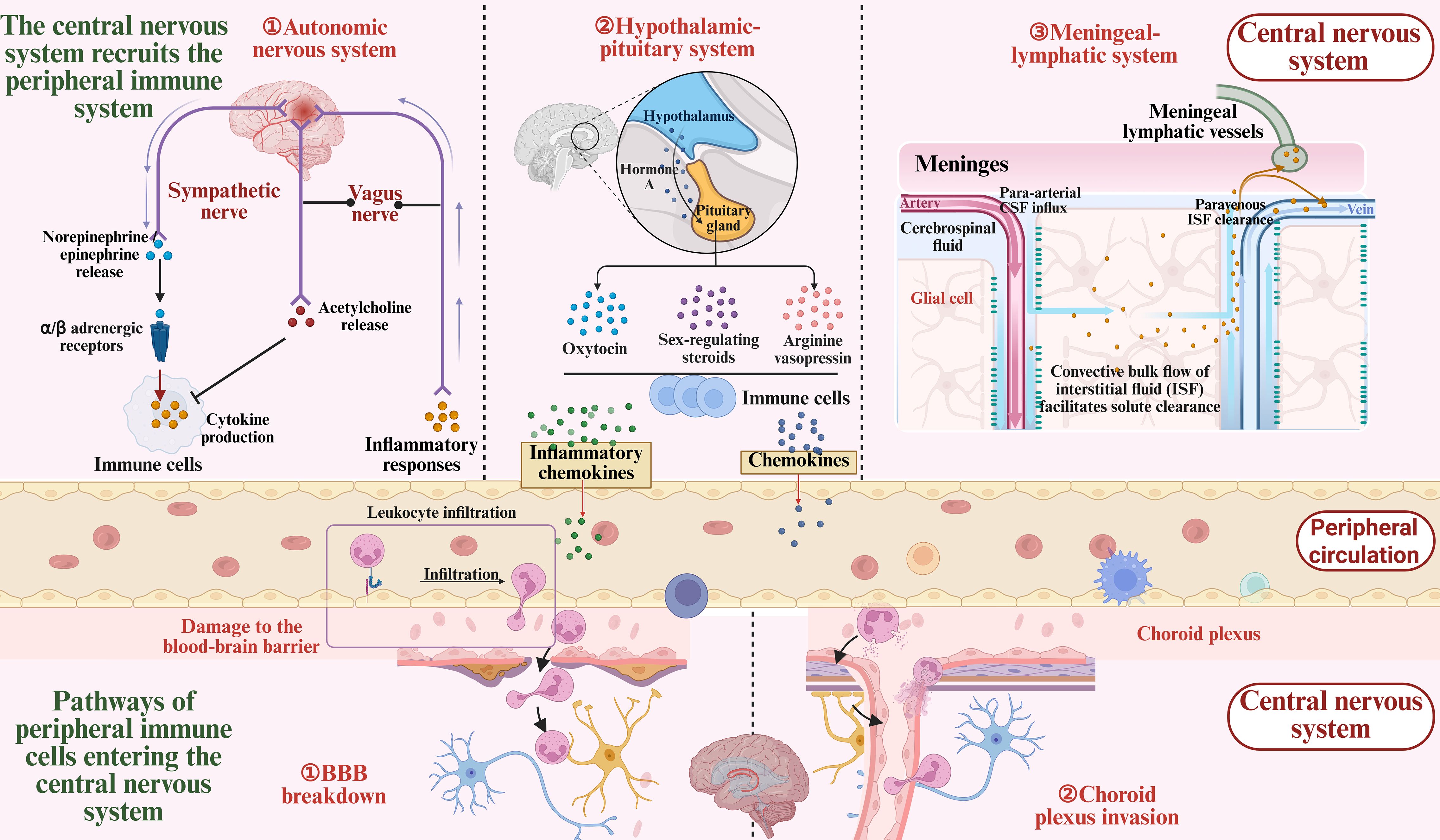
Figure 1. Bidirectional communication between the central and peripheral immune systems after ICH. Following intracerebral hemorrhage, the central nervous system (CNS) rapidly responds by engaging in bidirectional communication with the peripheral immune system via the autonomic nervous system, neuroendocrine system, andmeningeal lymphatic vasculature, while releasing signaling molecules such as inflammatory cytokines and chemokines. Meanwhile, peripheral immune cells infiltrate the CNS through the compromised blood-brain barrier and the choroid plexus, contributing to the local inflammatory response.
ICH occurs due to vascular rupture, resulting in hematoma formation within the brain. The primary injury is characterized by mechanical compression of brain tissue, including elevated intracranial pressure and brain herniation. The subsequent detrimental effects are termed secondary brain injury, which encompasses neuroinflammation, cerebral edema, BBB disruption, and other pathological alterations. Inflammation is one of the earliest defense responses following ICH, but it also contributes to the exacerbation of brain injury. Both central nervous system and peripheral immune cells play critical roles in orchestrating the inflammatory response surrounding the hematoma (32). The release of inflammatory mediators, activation and migration of immune cells, tissue destruction, cerebral edema formation, and neuronal repair together constitute this complex physiological and pathological process (33). In fact, the initial inflammatory response in the CNS may represent a component of the innate immune response, marked by the generation of damage-associated molecular patterns (DAMPs). These molecules activate resident innate immune cells, including microglia and astrocytes, which subsequently phagocytose dead cells and release cytokines and chemokines to initiate neuroinflammation. More precisely, following injury, the CNS activates and recruits components of the peripheral immune system through multiple pathways, including the autonomic nervous system, the neuroendocrine system, and the meningeal lymphatic vasculature. These immune components can access the CNS via various routes such as the BBB, where they interact with resident CNS cells to exert both detrimental and beneficial effects (34, 35). As illustrated in Figure 1.
The sympathetic nervous system is integral to the stress response after stroke (36). It exerts its effects by releasing norepinephrine and epinephrine, which activate α- and β-adrenergic receptors expressed on immune cells. The expression levels of these receptors are closely associated with the activation state and functional status of immune cells (37, 38). Through activation of these receptors, the CNS can modulate immune cell migration and cytokine secretion (39–41). However, these effects can be both beneficial and detrimental (42). For example, activation of β-adrenergic receptors can enhance CD4+ T cell proliferation and cytokine production, but may also suppress macrophage responses to lipopolysaccharide, thereby increasing the complexity of the immune response (43–45). In addition, the vagus nerve can sense inflammation via its afferent arc and suppress immune responses by releasing acetylcholine, particularly by inhibiting macrophage-derived TNF-α, thus mitigating inflammation (46–48).
More profound immune regulation is also mediated by the neuroendocrine system. The hypothalamic-pituitary axis modulates immune responses not only through the release of oxytocin and arginine vasopressin (AVP) (49–51), but also by regulating the immunological functions of sex steroids such as testosterone and estrogen, thereby influencing immune cell activity (52, 53). Studies have shown that oxytocin and AVP exert anti-inflammatory effects (54–57), whereas sex hormones exhibit dual roles in either suppressing or enhancing immune responses (53). In addition, thyroid hormones play an important role in immune responses by promoting lymphocyte proliferation and immunoactivation, a phenomenon associated with the reduced immune function observed after thyroidectomy (58, 59). Nonetheless, the precise mechanisms and regulatory factors underlying these neuroendocrine effects remain to be elucidated.
The meningeal lymphatic system also plays a critical role in the immune response following stroke (60). Recent studies have identified functional lymphatic vessels in the meninges that connect to the peripheral immune system, facilitating the clearance of immune waste from brain tissue and guiding immune cell trafficking (61–63). This process is particularly important after stroke. Meningeal lymphatic vessels not only mediate the transport of immune cells from the CNS meninges and cerebrospinal fluid (60), but also direct neuron-specific antigens released after injury to the peripheral immune system, thereby initiating immune responses. For example, in patients with acute stroke, neuron-derived antigens have been detected in deep cervical lymph nodes, where they are presented to T cells by antigen-presenting cells, triggering autoimmune responses (64, 65). Following ICH, NETs induce damage to lymphatic endothelial cells and promote lymphatic thrombosis through CX3CR1 signaling, ultimately contributing to secondary brain injuries such as hydrocephalus (66).
Peripheral immune cells access the CNS primarily via two routes: the classical BBB pathway and the choroid plexus pathway. In the early phase after injury, immune cells are recruited to the cerebral vasculature and, through interactions with endothelial cells, traverse the BBB into the brain parenchyma via adhesion molecules (67–70). Following intracerebral hemorrhage, increased BBB permeability facilitates immune cell infiltration (71), which drives the progression of the inflammatory response. Research indicates that this process is initiated within minutes after injury, as immune cells gradually infiltrate the central nervous system through vascular inflammation and endothelial activation, leading to the amplification of the inflammatory response. The choroid plexus, serving as a critical interface between the CNS and the peripheral immune system, also plays a significant role in post-stroke immune responses (72, 73). The choroid plexus is not only responsible for cerebrospinal fluid production but also contributes to the circulation and trafficking of immune cells. Studies have shown that peripheral immune cells can enter the ventricles via the choroid plexus and subsequently infiltrate the brain parenchyma, thereby modulating the immune status of the CNS. This finding highlights an alternative pathway for immune cell entry into the CNS (74), particularly in the context of primary central nervous system injury, where the peripheral immune system contributes to the secondary immune response through the choroid plexus.
In summary, stroke-induced peripheral immune responses involve multiple complex mechanisms, including regulation by the autonomic nervous system, neuroendocrine modulation, immune cell trafficking through meningeal lymphatic vessels, and alterations in BBB permeability. This intricate immune process affects not only the acute phase of stroke but may also significantly influence long-term neural repair and recovery. Therefore, a deeper understanding of these mechanisms is essential for the formulation of effective immunomodulatory strategies.
2.2 Immunological functions of microglia in the central nervous system
Microglia are among the most important immune cells in the central nervous system, comprising approximately 5% to 10% of the total CNS cell population (75). They not only perform immune surveillance by secreting cytokines but also exhibit phagocytic activity (76), enabling them to clear dead cells and cellular debris. As such, they are often referred to as the “macrophages of the brain.” (77). Following stroke, microglia play a pivotal role in the central nervous system response. They are involved not only in the immune response to brain injury but also in the regulation of neuroinflammation, as well as in neural repair and remodeling.
In healthy brain tissue, microglia exist in a “resting” state, primarily maintaining homeostasis by monitoring the microenvironment and clearing dead cells (15). However, following ICH, the release of hemoglobin and iron from the hematoma induces microglial activation (78). Activated microglia then migrate to the injury site and shift toward a pro-inflammatory M1 phenotype, releasing large amounts of cytokines, chemokines, and oxidative molecules, such as IL-1β, IL-6, and TNF-α (79, 80). The excessive immune response of microglia following ICH is not only neurotoxic but also exacerbates brain injury by increasing BBB permeability and promoting the infiltration of peripheral immune cells (81, 82). Studies have shown that excessively activated microglia disrupt the BBB by releasing large amounts of pro-inflammatory cytokines and matrix metalloproteinases (MMPs), thereby exacerbating damage to the surrounding neural tissue (82). Moreover, the release of these inflammatory mediators can induce the polarization of astrocytes into the neurotoxic A1 phenotype, thereby amplifying the inflammatory response. These pro-inflammatory cytokines also promote the infiltration of additional immune cells, creating a vicious cycle that further exacerbates inflammation and neuronal injury (80). As illustrated in Figure 2. Therefore, modulating the activation state of microglia is critical for improving stroke outcomes and guiding therapeutic interventions.
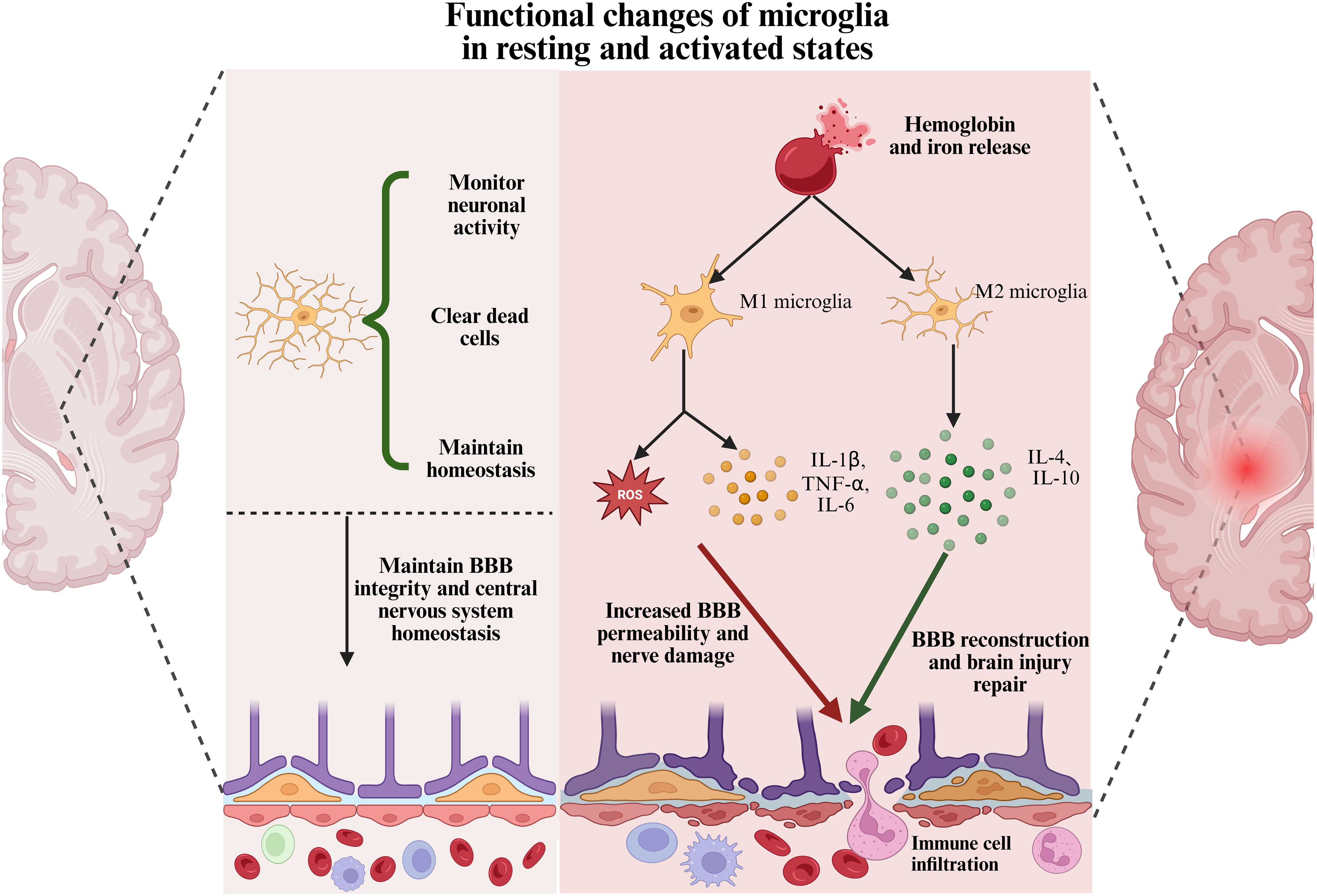
Figure 2. Functional changes of microglia in resting and activated states. In healthy brain tissue, microglia exist in a resting state with a ramified morphology. They monitor neuronal activity, clear apoptotic cells, and maintain central nervous system homeostasis. After ICH, microglia are activated by damage-associated molecules such as hemoglobin and iron. They adopt a pro-inflammatory M1 phenotype and release cytokines, including IL-1β, TNF-α, IL-6, and reactive oxygen species. This contributes to BBB disruption, immune cell infiltration, and neuronal damage. Subsequently, microglia transition to an anti-inflammatory M2 phenotype, secreting factors such as IL-4 and IL-10. This shift supports blood-brain barrier repair and promotes neurological recovery.
While microglial activation enhances the local immune response, it also exerts neuroprotective effects by phagocytosing damaged cells and cellular debris to clear the injury site (83). Within seven days following ICH, microglia transition from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype. This transition facilitates hematoma clearance, tissue repair, and anti-inflammatory responses through the release of cytokines such as IL-4 and IL-10. Furthermore, M2-polarized microglia promote the differentiation of Th2 and regulatory T cells, which further suppress M1 and Th1 phenotypes (18). As M2-type microglia differentiate, they contribute to BBB reconstruction and the reduction of cerebral edema, thereby alleviating mass effect. This process helps disrupt the cycle of escalating inflammation and fosters an environment conducive to brain tissue repair (80). As shown in Figure 2. Therefore, it is widely believed that facilitating the transition of microglia toward the M2 phenotype after intracerebral hemorrhage may alleviate brain injury and improve neurological outcomes in patients.
In recent years, there has been heightened focus on the functional polarization of microglia following a stroke, especially regarding the equilibrium between their pro-inflammatory and anti-inflammatory states. Studies have shown that activating signaling pathways such as Nrf2 (84), PPARγ/RAD21 (85), and TGF-β/ALK5 (86), or inhibiting inflammatory pathways such as TLR4/NF-κB and CDK5/DRP1 (87, 88), can promote microglial polarization toward the M2 phenotype, enhance phagocytic capacity, reduce neuroinflammation, and accelerate hematoma clearance and neural repair. In addition, miRNAs such as miRNA-182-5p and miRNA-27a suppress M1 microglial activation by targeting inflammatory mediators like TLR4 (89, 90). Meanwhile, miRNA-144 and miRNA-222 regulate pathways involving mTOR, autophagy, or apoptosis, thereby further modulating microglial inflammatory responses (91–93). These findings underscore the essential importance of regulating microglial functional states, particularly by inhibiting M1 activation and promoting M2 polarization, in the treatment and prognosis of stroke (94).
Although the “microglial polarization” and M1/M2 dichotomy are still widely used in the literature, studies have shown that this model oversimplifies the situation, particularly in in vivo environments (95, 96). In recent years, single-cell transcriptomics and spatial omics have revealed that microglia exhibit a continuous spectrum of subpopulations under injury and disease conditions, surpassing traditional classifications (97, 98). Among them, disease-associated microglia are characterized by TREM2–APOE signaling and lipid metabolism reprogramming, playing a role in phagocytosis and inflammation regulation (99). White matter-associated microglia and interferon-responsive microglia reflect specific responses to aging, demyelination, or viral-like stimuli (100, 101). However, there is limited evidence regarding the phenotypic characteristics of these microglial cells in the context of intracerebral hemorrhage, and the understanding of their dynamic changes and functional roles remains incomplete.
Microglia are crucial in the immune response after a stroke. Adjusting the equilibrium between pro-inflammatory and anti-inflammatory states can effectively alleviate brain injury and promote neural repair. Therefore, developing therapeutic strategies that target microglial function, particularly through the regulation of their activation states, may offer novel approaches for immunotherapy in stroke.
2.3 Neutrophils in intracerebral hemorrhage
Neutrophils, the predominant leukocytes in the peripheral immune system, are among the initial cells recruited to the central nervous system after intracerebral hemorrhage (33), where they are pivotal in the immune response (33, 102, 103). Studies have shown that the BBB becomes permeable as early as 3 minutes after brain injury (71). Neutrophil infiltration can be observed within 30 minutes and peaks at 2 to 3 days post-injury (104).
As shown in Figure 3, the marked increase in BBB permeability after brain injury facilitates the transendothelial migration of neutrophils. This process is primarily driven by DAMPs, which rapidly activate endothelial cells and enhance the expression of various cytokines, including vascular cell adhesion molecule-1, a key mediator of peripheral leukocyte adhesion and migration (105, 106). This activation can persist up to five days post-injury (107). Numerous studies have shown that targeting cell adhesion molecules, such as VLA-4, can reduce neutrophil adhesion to endothelial cells, significantly attenuating microglial activation and neuronal damage in the brain parenchyma (108). In addition, neutrophil-derived MMP-9 plays a critical role in this process. Numerous studies have demonstrated that, in subarachnoid hemorrhage models, neutrophil-released MMP-9 and IL-6 further disrupt the integrity of the BBB, promoting the infiltration of peripheral immune cells and perpetuating a vicious cycle of inflammation (109–112). Moreover, activated neutrophils not only exert direct cytotoxic effects on brain tissue, but also exacerbate local inflammation through interactions with other immune cells such as microglia and macrophages. Studies have shown that neutrophils can activate microglia, promoting the release of pro-inflammatory cytokines and amplifying the inflammatory response, ultimately worsening neuronal injury (113).
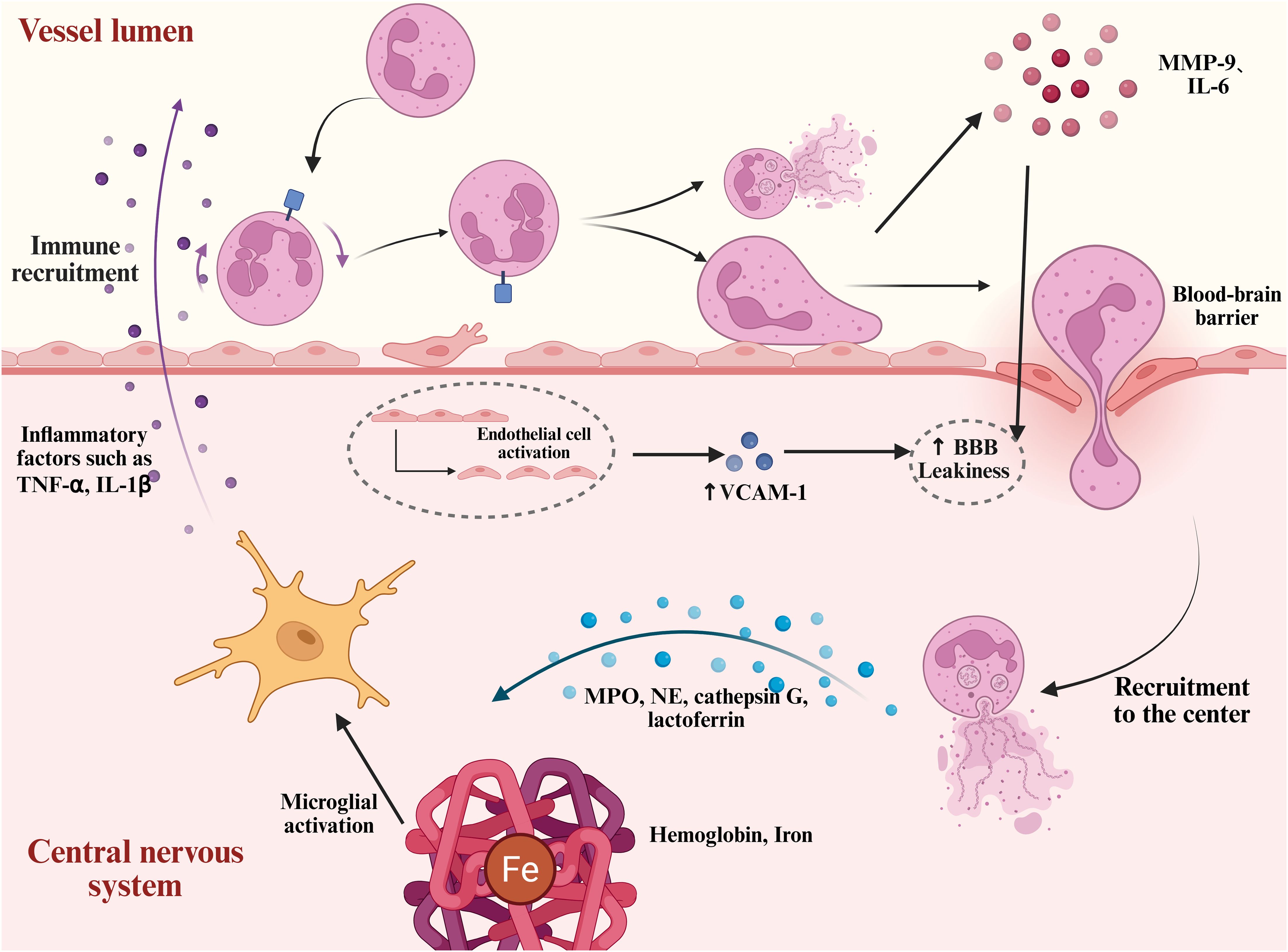
Figure 3. Neutrophil mobilization and neurotoxic mechanisms after intracerebral hemorrhage. After ICH, hemoglobin, iron, and other damage-associated molecules induce endothelial cell activation. This process is accompanied by the upregulation of adhesion molecules such as VCAM-1, which enhances neutrophil adhesion to the endothelium. Subsequently, neutrophils disrupt and traverse the BBB, migrating into the central nervous system. They release pro-inflammatory granules such as neutrophil elastase, cathepsin G, and lactoferrin. In addition, neutrophils form NETs, which, together with inflammatory mediators such as MMP-9 and IL-6, exacerbate BBB disruption and neuronal injury.
Neutrophils are considered a key component of the innate immune system’s initial response to microbial invasion (114). Neutrophil migration is not merely a simple chemotactic process; it also involves dynamic changes in their functional states. Before stimulation, naïve neutrophils exhibit a spherical morphology characterized by pronounced membrane ruffles. Upon stimulation with IL-8, phorbol myristate acetate, or lipopolysaccharide, they exhibit morphological alterations marked by a flattened morphology and the development of membrane protrusions (21). Following ICH, neutrophils transition from a resting to an activated state during their recruitment to the central nervous system. This activation involves the assembly of NADPH oxidase components (NOX), massive production of reactive oxygen species (ROS), cytoskeletal rearrangement, and initiation of the degranulation process (115). Neutrophil degranulation releases a variety of contents, including MPO, neutrophil elastase (NE), cathepsin G, and lactoferrin, which contribute to the clearance of necrotic tissue and pathogens. However, these granules also exhibit significant neurotoxicity. In particular, when combined with extracellular DNA to form NETs, they are considered key contributors to secondary neuronal injury (116). As shown in Figure 3.
3 NETs in intracerebral hemorrhage
3.1 Mechanisms of NETs formation and physiological functions
3.1.1 Formation and regulation of NETs
Takei was the first to observe that neutrophils can form NETs to kill bacteria (117). Brinkmann et al. further elucidated this process in 2004, during which they also introduced the term “NETosis.” (21) With continued research, it has become increasingly evident that the release of extracellular DNA is not invariably linked to cell death (22, 118, 119). In 2018, the Nomenclature Committee on Cell Death proposed substituting the term “NETosis” with “NETs” to more accurately describe this complex biological process (118). The formation of NETs is a highly regulated and programmed event, controlled by various endogenous and exogenous signals. Based on whether it is dependent on cell death or NOX activity, NETs formation can be categorized into three distinct mechanisms. As shown in Figure 4.
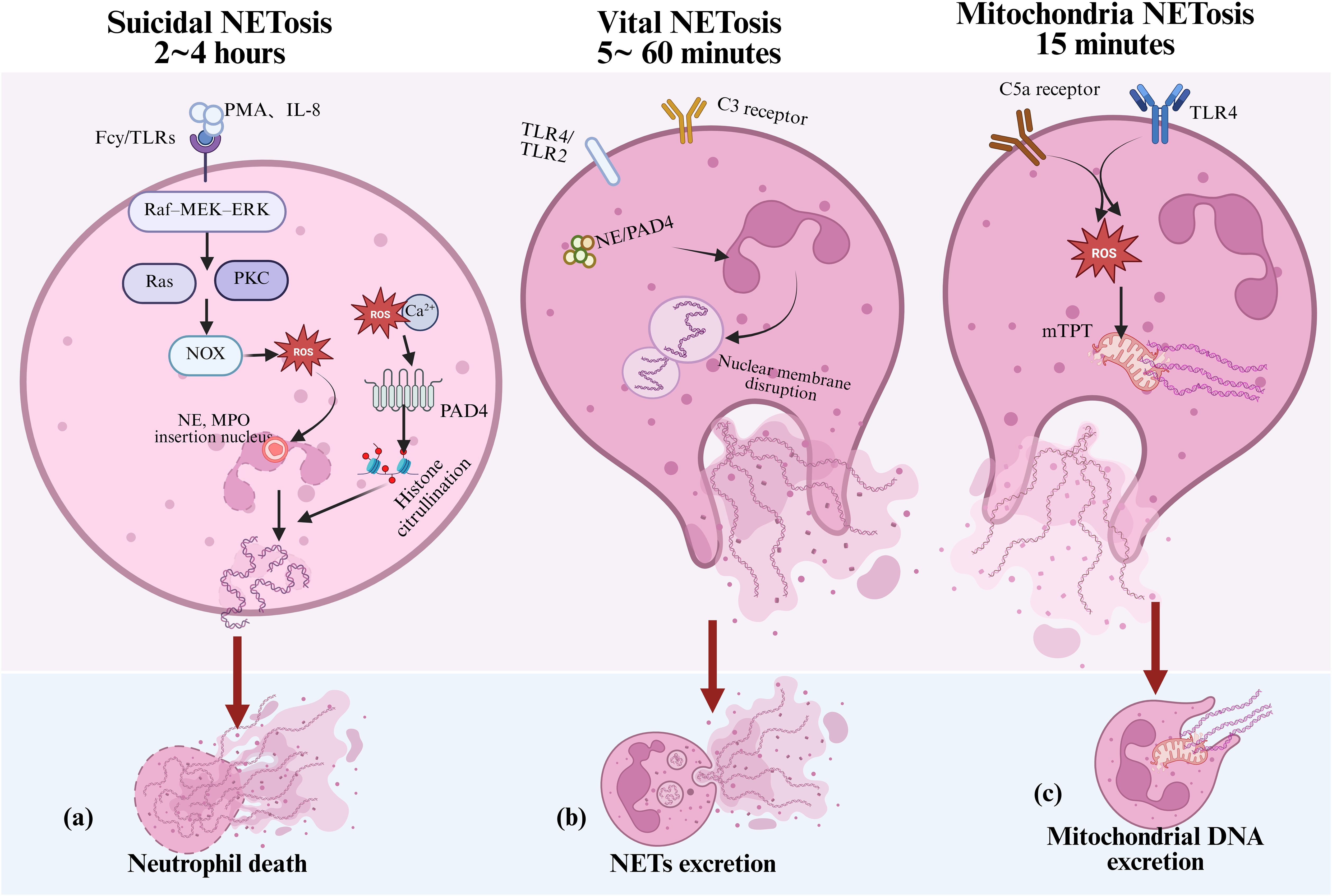
Figure 4. Mechanisms of NETs formation in inflammation. Under inflammatory stimulation, neutrophils can form NETs through three distinct mechanisms, contributing to both immune defense and tissue damage: (a) suicidal NETosis, which depends on NADPH oxidase activity and the accumulation of ROS; (b) vital NETosis, which is independent of NADPH oxidase and typically triggered by TLRs or complement component C3; and (c) mitochondrial NETosis, which is induced by factors such as LPS and C5a, leading to the release of mitochondrial DNA to form NETs. NETs, neutrophil extracellular traps; ROS, reactive oxygen species; NE, neutrophil elastase; MPO, myeloperoxidase; PAD4, Peptidylarginine deiminase 4; TLRs, toll-like receptors; LPS, lipopolysaccharide; mtDNA, mitochondrial DNA; mPTP, mitochondrial permeability transition pore; PKC, protein kinase C.
Suicidal NETosis is the earliest identified and most well-characterized form of NET formation. It depends on the activation of NOX and the production of ROS, typically occurring 2 to 4 hours after neutrophil activation. Specifically, stimuli such as phorbol myristate acetate and IL-8 activate Fcγ receptors or Toll-like receptors (TLRs), initiating the Raf–MEK–ERK signaling cascade. This sequentially activates Ras and protein kinase C (PKC), ultimately driving NOX activity and the production of ROS (120). The accumulation of ROS plays a central role in NETosis. On one hand, ROS promotes the translocation of NE and MPO into the nucleus, where they cooperatively mediate chromatin decondensation (121). On the other hand, extracellular calcium influx, together with ROS, activates peptidylarginine deiminase 4 (PAD4), which catalyzes the citrullination of histones H3, H4, and H2A. This modification weakens the electrostatic interactions between histones and DNA, leading to chromatin relaxation (122, 123). Ultimately, nuclear envelope rupture allows decondensed DNA to combine with granule proteins and form the characteristic NET structure, which is then released into the extracellular space. As shown in Figure 4, this process is typically accompanied by programmed neutrophil death.
However, studies have also shown that neutrophils can rapidly release NETs without undergoing cell death, a process termed “vital NETosis.” This form of NET release, as shown in Figure 4, is characterized by its independence from NADPH oxidase activity and is primarily induced by stimuli such as TLR2/TLR4 and complement component C3 (124). Vital NETosis typically transpires within 5 to 60 minutes after neutrophil activation and involves nuclear envelope vesiculation and the extrusion of DNA. During this process, neutrophils retain key immune functions such as phagocytosis and chemotaxis (125).
Further studies have identified a third type of NET formation termed “mitochondrial NETosis,” (126) which is induced by stimuli such as lipopolysaccharide, complement component C5a, and granulocyte-macrophage colony-stimulating factor (GM-CSF). As shown in Figure 4. This process typically occurs within 15 minutes after neutrophil activation. This process does not depend on nuclear DNA release. Instead, it is driven by the opening of the mitochondrial permeability transition pore (mPTP) and the generation of mitochondrial reactive oxygen species, which trigger the release of mitochondrial DNA and the formation of NETs without causing cell death (126). The three NET formation mechanisms occur at different time points following neutrophil activation, may or may not depend on NADPH oxidase activity, and can be associated with or independent of neutrophil death. However, the precise mechanisms underlying NET formation remain controversial and require further investigation.
3.1.2 Physiological functions of NETs
As a major effector mechanism of neutrophils, NETs were initially thought to play a key role in host defense, particularly against bacterial and viral infections (127). The DNA web-like structures, together with antimicrobial components such as MPO, NE, and histones, form a local physical barrier that restricts the spread of bacteria and pathogens (21), as shown in Figure 5. Studies have confirmed that NETs exert direct microbicidal effects against a variety of pathogens, including bacteria, fungi, and parasites (128). However, accumulating evidence indicate that NETs are involved not only in pathogen defense but also in the modulation of various aspects of the immune response (129). Antimicrobial components within NETs, such as NE, can target and degrade various bacterial virulence factors (130), thereby reducing their pathogenicity (131). MPO contributes to bacterial killing through the release of ROS (132). In addition, the DNA backbone of NETs carries a natural negative charge, which enables it to chelate cations and disrupt microbial membrane integrity, thereby exerting direct antimicrobial effects (133). When exogenous DNase is used to degrade NETs in mice, the structural integrity of NETs is disrupted, leading to a significant increase in bacterial load. This further confirms the barrier function of NETs in innate immunity (134). In addition to their direct antimicrobial functions, NETs also regulate immune responses in sterile inflammatory environments. NETs aggregation can degrade cytokines and chemokines, thereby interfering with neutrophil recruitment and activation, and ultimately suppressing the inflammatory response (23). Moreover, NETs are believed to play a role in the onset of autoimmune responses under certain conditions, potentially through mechanisms such as antigen citrullination.
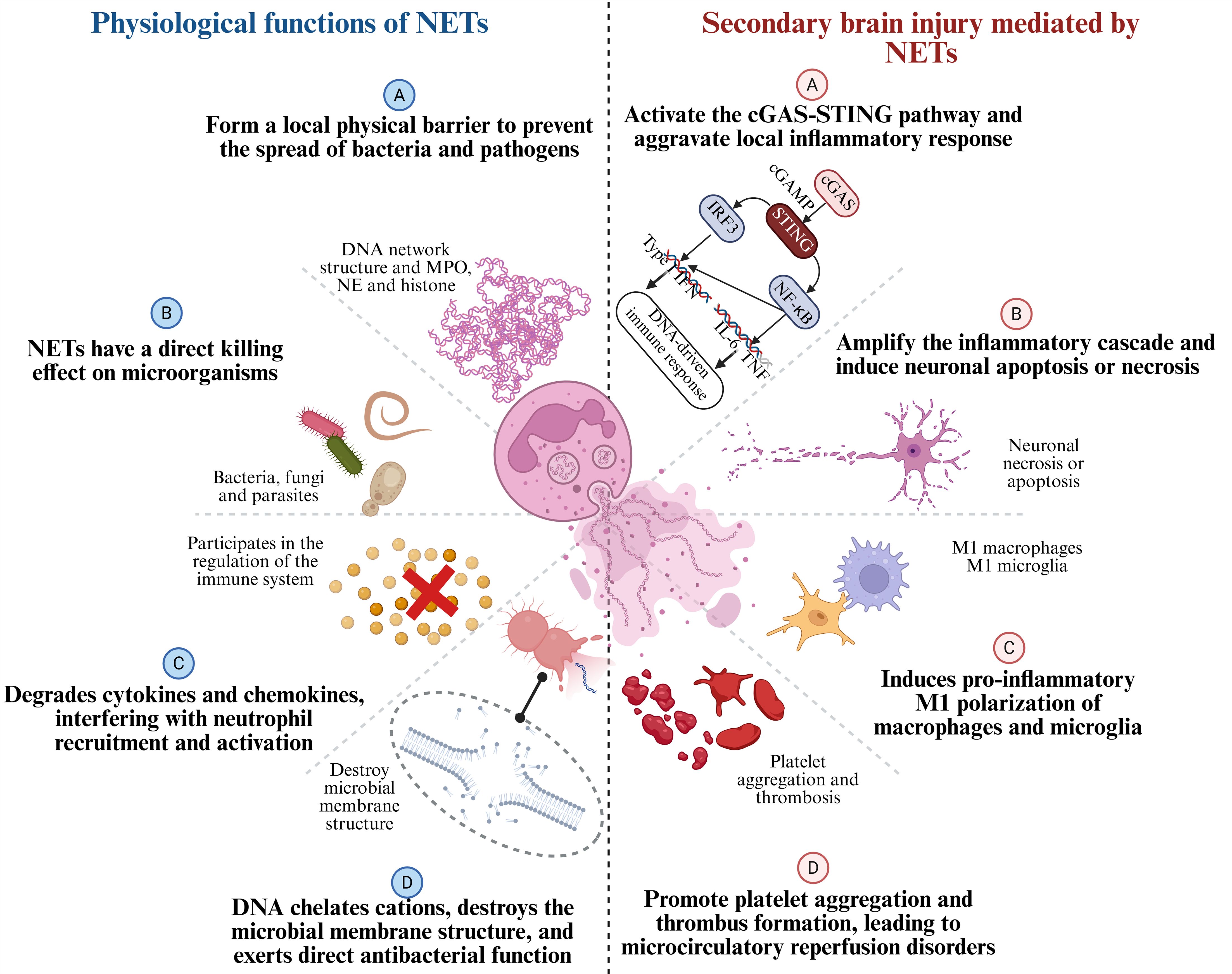
Figure 5. Physiological functions of NETs and their roles in secondary brain injury after ICH.NETs are composed of chromatin, histones, NE, and MPO. On the left, the physiological functions of NETs are shown: (A) forming a local physical barrier to prevent the spread of pathogens; (B) exerting direct killing effects on microorganisms; (C) degrading cytokines and chemokines to regulate neutrophil recruitment and activation; and (D) chelating cations, disrupting microbial membranes, and exerting direct antibacterial activity. On the right, after ICH, the detrimental effects of NETs as pro-inflammatory factors are shown: (A) activating the cGAS-STING pathway and aggravating local inflammation; (B) amplifying the inflammatory cascade and inducing neuronal apoptosis/necrosis; (C) promoting pro-inflammatory M1 polarization of macrophages and microglia; and (D) enhancing platelet aggregation and thrombosis, leading to microcirculatory disturbances.
Notably, under the immunologically active conditions of stroke, NETs exhibit a complex dual role in physiological function (17). While NETs contribute to immune defense, their excessive formation can lead to increased immune cell recruitment, amplify the inflammatory response, and exacerbate post-stroke tissue damage through the release of various cytokines and enzymes. Therefore, precise regulation of NET formation may represent a promising therapeutic approach in stroke immunomodulation.
3.2 NETs-mediated secondary brain injury
In recent years, research on NETs has provided novel insights into the pathogenic mechanisms of neutrophils. NETs are web-like structures composed of decondensed chromatin, histones, MPO, NE, and other associated proteins. Their formation process is known as NETosis. Significant accumulation of NETs has been observed in both ICH models and clinical specimens. A postmortem study by Laurent Puy and colleagues was the first to demonstrate the presence of abundant NETs in the human brain following ICH (135). These NETs were predominantly localized within and around the hematoma, and their release exhibited temporal dynamics—typically initiating within 72 hours after hemorrhage and persisting over the following 8 to 15 days. Jin et al. reported the presence of NETs in patients with ICH (136). They detected abundant NETs in hematoma, plasma, and drainage fluid samples from ICH patients, suggesting that NETs play a significant role in the pathophysiology of hemorrhagic stroke.
The formation of NETs depends on the activation of multiple signaling pathways, including NOX2-mediated production of ROS and activation of TLR4 (137), both of which have been identified as critical triggers for NET generation. Moreover, following ICH, interactions between platelets and neutrophils have also been shown to promote the formation of NETs. The binding of P-selectin to P-selectin glycoprotein ligand-1 (PSGL-1) on neutrophils further promotes the formation of NETs (138). Recent studies have demonstrated that blocking PSGL-1 reduces plasma levels of MPO-DNA complexes (139), inhibits NET formation, and alleviates secondary brain injury. In addition, elevated platelet counts can enhance high mobility group box 1 (HMGB1)-mediated NET formation (140). Notably, heme, a key oxidative stress byproduct following ICH, has also been shown to promote the release of NETs by inducing ROS and activating NOX2 (141).
Although NETs formation plays an important physiological role in acute infection defense, as shown in Figure 5, NETs themselves also exert multiple direct pathogenic effects. Components of NETs, including NE, MPO, cathepsin G, and citrullinated histone H3 (CitH3), can amplify inflammatory cascades in the extracellular space, leading to neuronal apoptosis or necrosis. The DNA backbone within NETs acts as a potent immunostimulatory molecule that activates the cGAS-STING pathway, exacerbates local inflammatory responses, and induces pro-inflammatory M1 polarization of macrophages and microglia (102). Additionally, by promoting platelet aggregation and thrombosis, NETs may contribute to microcirculatory reperfusion impairment in the later stages of stroke, representing a potential barrier to neurological recovery. As we and others have previously demonstrated, several well-characterized signaling proteins have been demonstrated to modulate the formation of NETs, including JNK, extracellular signal-regulated kinases 1/2, Akt, and Src (142–145). Moreover, phorbol 12-myristate 13-acetate, a PKC activator, has been extensively utilized as a potent inducer of NETs in fundamental research (146–148). The participation of various signaling proteins suggests that the mechanism underlying NETs formation is highly complex. Notably, inhibiting the formation of NETs has emerged as a promising neuroprotective strategy following ICH. In animal studies, the use of PAD4 inhibitors or systemic administration of Deoxyribonuclease I (DNase I) effectively reduces NET levels, alleviates vascular inflammation, decreases hemorrhage volume, and significantly improves neurological recovery (149). More importantly, NETs may also serve as a potential biomarker for stroke prognosis, as elevated plasma NET levels in acute stroke patients have been reported to positively correlate with neurological deficit scores (150). With continued advances in understanding the mechanisms of NETs formation, therapeutic strategies targeting NETs hold promise as an innovative strategy for the management of ICH.
3.3 Mechanisms of NET-induced programmed cell death in ICH
Although NETs contribute to pathogen clearance during stroke, their “double-edged sword” effect in the immune response should not be overlooked. Aberrant activation and excessive formation of NETs, as shown in Figure 6, can trigger intense immune reactions and initiate multiple forms of programmed cell death—including pyroptosis, apoptosis, and ferroptosis—ultimately contributing to secondary neuronal injury (151).
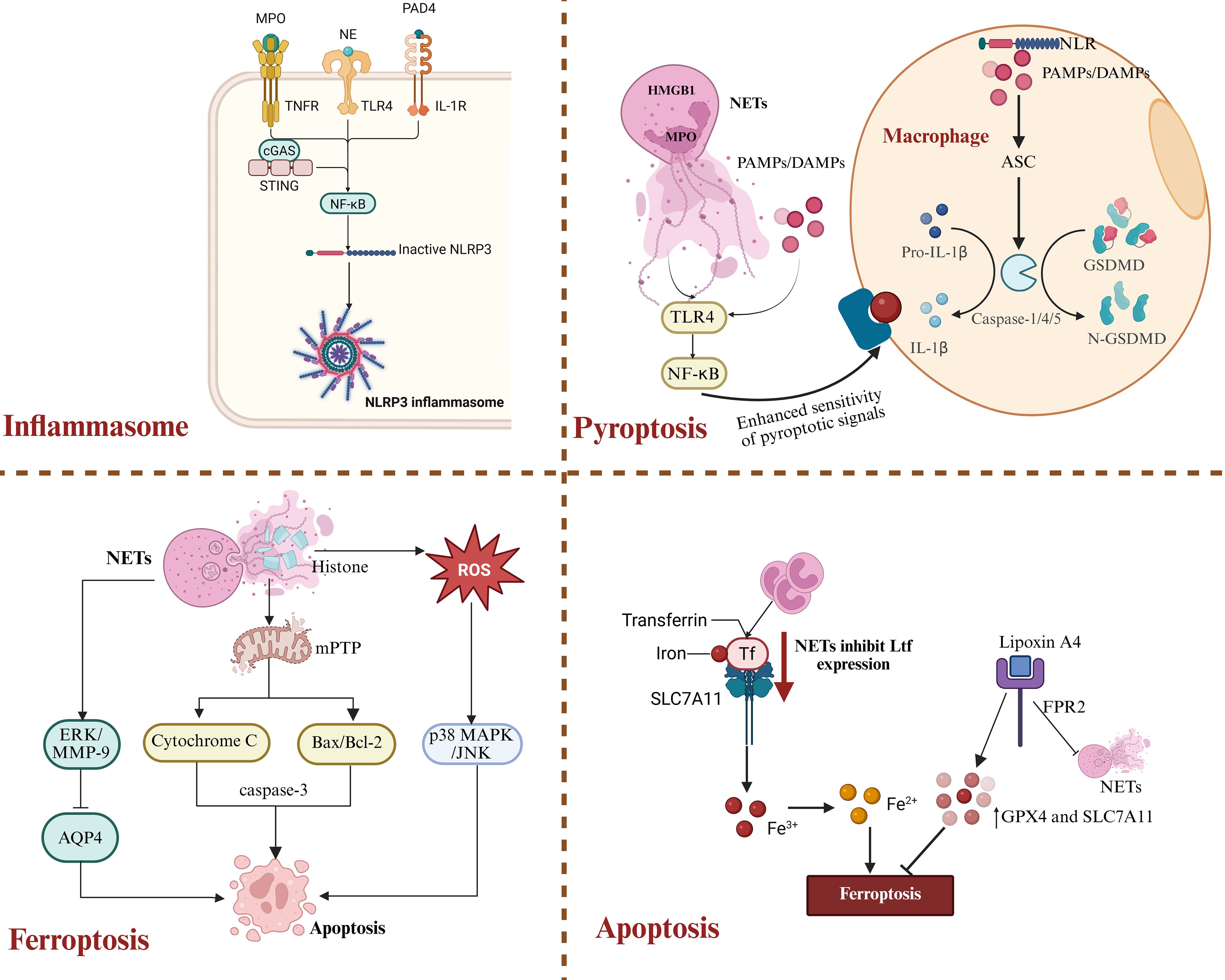
Figure 6. NETs-driven inflammatory cell death pathways after ICH. NETs play multiple roles in regulating neuronal cell death after ICH. By activating the NLRP3 inflammasome and various signaling pathways, NETs can induce pyroptosis, ferroptosis, and apoptosis, thereby exacerbating secondary brain injury.
Histones, MPO, NE, and PAD4, which are abundantly present in NETs, are considered classical DAMPs. These components can be recognized by mononuclear phagocyte lineages in the brain and activate the NLRP3 inflammasome pathway. Multiple studies have confirmed that the lesion area of ICH is often associated with high expression of caspase-1, IL-1β, and MPO (152, 153). In addition, extracellular DNA can activate the type I interferon pathway via the cGAS-STING axis, promoting pro-inflammatory polarization of microglia and exacerbating local neuroinflammatory responses (102). NETs induce activation of the TLR4/MyD88/NF-κB signaling axis through the specific binding of their histones to TLR4, thereby driving NF-κB dependent transcription of the IL-1β precursor (151). Moreover, the inflammasome contributes to atherosclerosis through this mechanism by inducing an interferon-α mediated neuroimmune cascade that amplifies immune cell recruitment within the plaque (154). This positive feedback loop involving the inflammasome, NETs, and TLR4 further impedes neural repair in the damaged area. As illustrated in Figure 6.
Pyroptosis is a highly inflammatory form of programmed cell death that primarily depends on caspase-1, -4, or -5-mediated cleavage of Gasdermin D, which leads to the formation of membrane pores and promotes the release of pro-inflammatory cytokines such as IL-1β. Upon sensing pathogen-associated molecular patterns (PAMPs) or DAMPs, NOD-like receptors (NLRs) recruit caspase-1 via the adaptor protein ASC, resulting in caspase-1 oligomerization and the induction of pyroptosis (155, 156). In addition, non-canonical inflammasome activation occurs when cytosolic lipopolysaccharide is sensed by caspase-4 (157, 158), leading to its activation, Gasdermin D pore formation, and subsequent activation of caspase-1 through the NLRP3 inflammasome. This mechanism has been confirmed in experiments using human-derived immune cells (159, 160). Components of NETs, such as MPO and high mobility group box 1, enhance macrophage sensitivity to pyroptotic signals and trigger their inflammatory, lytic cell death (154). Notably, in ICH models, cleavage products of Gasdermin D-N are frequently detected in the peritoneal region associated with NETs, indicating a prominent role of NETs in pyroptosis induction (161). Moreover, NETs-induced pyroptosis depends on the activation of the TLR4-NF-κB signaling axis (162). This process is not only mediated by NLRP3-driven caspase-1 activation but is also intricately linked to increased levels of ROS and activation of the HMGB1-TLR4 pathway (163, 164). DAMPs released during pyroptosis, in turn, promote the formation of NETs, establishing a self-amplifying “pyroptosis-NETs” inflammatory loop (165).
Ferroptosis, a form of programmed cell death characterized by iron dependency and the accumulation of lipid peroxides, has increasingly been recognized as a key contributor following ICH. Studies have shown that ICH-induced neutrophil infiltration reduces the transcription and expression of lactoferrin, thereby weakening its interaction with SLC7A11 and indirectly exacerbating neuronal ferroptosis (166). Animal model studies further demonstrate that Lipoxin A4 significantly inhibits the formation of NETs via an FPR2-dependent mechanism, thereby upregulating the expression of GPX4 and SLC7A11 and ultimately suppressing ferroptosis (167). Additionally, in fluoride-induced brain inflammation, NETs have been shown to disrupt calcium homeostasis, thereby inducing neutrophil self-death and promoting a feedback loop of NETs release (168). The interaction between NETs and ferroptosis has not yet been fully elucidated; however, their coupling under hypoxic conditions in the early stages of ICH has been demonstrated in multiple animal models, highlighting a promising emerging target for further investigation.
In addition to pyroptosis and ferroptosis, NETs can also induce neuronal apoptosis. Extracellular histones released from NETs directly disrupt the integrity of the cell membrane, trigger the opening of mitochondrial permeability transition pores, lead to cytochrome c release and Bax/Bcl-2 imbalance, and ultimately activate the caspase-3-dependent apoptotic pathway (169). Moreover, NETs-induced ROS stress activates the p38 MAPK and JNK signaling pathways, further amplifying cell death signals (170). However, the role of NETs in inducing apoptosis following ICH has not yet been systematically elucidated. Recent studies have shown that NETs disrupt the BBB by activating the ERK/MMP-9 signaling pathway and suppressing the expression of AQP4, leading to perihematomal edema and neuronal apoptosis. This represents one of the key mechanisms underlying secondary brain injury after ICH. In summary, NETs aggravate secondary injury to the central nervous system by triggering multiple forms of programmed cell death.
4 Regulatory role of microglia in ICH
Following ICH, microglia, the resident immune cells of the central nervous system, are among the first to be activated. Their functional and phenotypic changes perform a pivotal function in both secondary neuronal injury and tissue repair. Through interactions with astrocytes, oligodendrocytes, and peripheral immune cells, microglia help establish a complex immunoregulatory network that exerts both detrimental and reparative effects on tissue injury and recovery after ICH.
4.1 Dual roles of microglia in the CNS immune network
In the early stages of ICH, components such as hemoglobin and heme rapidly activate microglia via the TLR4, driving their polarization toward the pro-inflammatory M1 phenotype and resulting in the release of large amounts of DAMPs (81, 82). Molecules such as HMGB1, heat shock proteins, and nucleic acid fragments can bind to pattern recognition receptors (PRRs) on the surface of microglia, such as TLR4 and TLR2. This interaction triggers intracellular signaling via the MyD88 and TRIF pathways, leading to the activation of transcription factors such as NF-κB and the subsequent initiation of pro-inflammatory gene expression (171, 172). Moreover, microglial recognition of PRRs such as NLRP3 further amplifies the inflammatory cascade, thereby contributing to a pro-inflammatory microenvironment (171). Additionally, thrombin enhances the pro-inflammatory activation of microglia through protease-activated receptor-1. This process increases BBB permeability, facilitates the infiltration of inflammatory cells and harmful molecules into brain tissue, and exacerbates cerebral edema and neuronal injury. However, this inflammatory response may also confer short-term protective effects by aiding in the clearance of necrotic tissue and limiting lesion expansion. As shown in Figure 7.
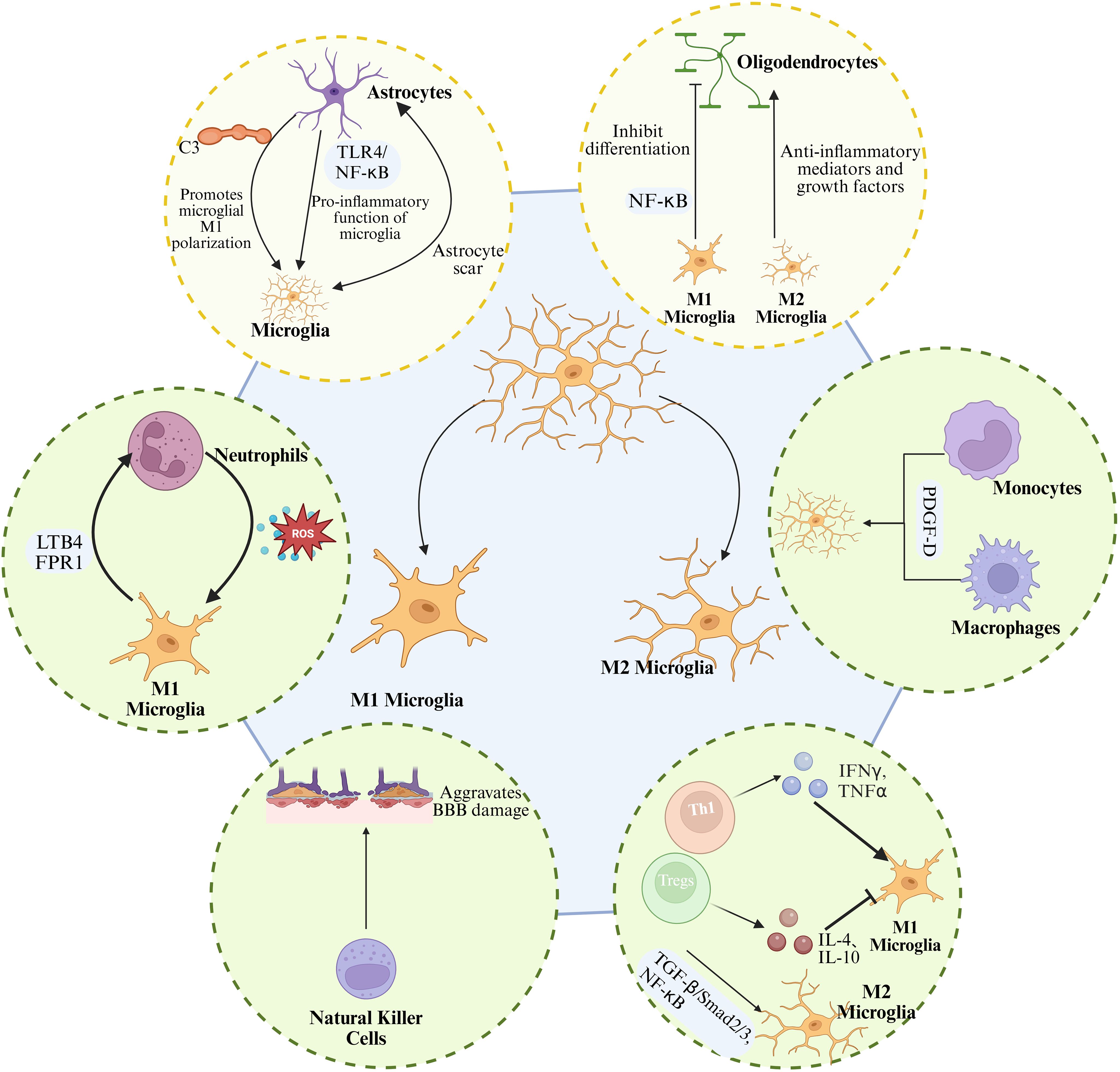
Figure 7. Microglial interactions with CNS and peripheral immune cells after ICH. In the CNS, microglia interact with astrocytes through multiple signaling pathways to regulate inflammation, glial scar formation, and neural repair. Moreover, microglial polarization influences oligodendrocyte differentiation and myelin regeneration. In the periphery, microglia release chemokines to interact with neutrophils, macrophages, natural killer (NK) cells, and T lymphocytes, establishing a complex network of inflammatory amplification and immune regulation.
Over time, activated microglia progressively shift toward the M2 phenotype, releasing anti-inflammatory cytokines such as IL-10 and TGF-β, as well as neurotrophic factors. This phenotypic transition contributes to hematoma clearance, vascular regeneration, and axonal repair, thereby reducing inflammation and improving neurological outcomes. This phenotypic shift is not only influenced by changes in the inflammatory microenvironment but also regulated by signaling pathways such as JAK1/STAT6, Nrf2, and PPARγ. For example, activation of Nrf2 enhances the phagocytic capacity of microglia and increases the expression of neurotrophic factors (84). In addition, upregulation of the PPARγ (85) and TGF-β/ALK-5 pathways (86), or suppression of key inflammatory mediators such as the TLR4/NF-κB pathway (87) and the CDK5)DRP1 axis (88), contributes to M2 polarization and inhibition of pro-inflammatory responses. Moreover, the regulatory role of microRNAs is also significant (173). For instance, miR-144 targets the mTOR pathway to modulate autophagic activity in microglia, contributing to hemoglobin-induced inflammatory responses (91–93). Meanwhile, miR-27a and miR-182-5p attenuate the release of pro-inflammatory cytokines by inhibiting downstream signaling of the TLR4 pathway (89, 90).
Within the central immune network, interactions between microglia and astrocytes are particularly intimate. A study by Shi et al. demonstrated that IL-15 exacerbates cerebral edema and neuronal injury after ICH by enhancing inflammatory signaling between astrocytes and microglia (174). In addition, the complement system serves as a critical communication pathway between astrocytes and microglia. A1-type astrocytes secrete complement component C3, which activates microglia through the C3/C3aR axis, promoting M1 polarization and exacerbating inflammation and white matter injury (175, 176). Moreover, overexpression of aquaporin-2 in astrocytes can induce inflammatory cascades by activating the TLR4/NF-κB signaling pathway, thereby enhancing the pro-inflammatory activity of microglia (177). Notably, microglia play roles not only in regulating inflammation but also in coordinating with astrocytes to modulate glial scar formation, thereby influencing brain tissue repair (178). Research indicated that astrocytes secrete functional mitochondria and neurotrophic factors, including brain-derived neurotrophic factor, which can be transferred to microglia. This transfer promotes microglial polarization toward the M2 phenotype, enhances their phagocytic and antioxidant capacity, reduces neurotoxicity, and facilitates the remodeling of neural networks (179).
White matter injury is a common consequence of ICH, characterized by oligodendrocyte death and demyelination. Microglia also play a critical role in both the injury and repair of white matter (175). Under the oxidative stress induced by ICH, M1-polarized microglia inhibit the differentiation of oligodendrocyte precursor cells into mature oligodendrocytes via NF-κB signaling, thereby impairing remyelination. In contrast, M2 microglia promote OPC maturation and remyelination by releasing anti-inflammatory mediators and growth factors. Therapeutic strategies targeting this pathological process, such as the application of nanoceria, have been shown to alleviate myelin damage and enhance remyelination.
4.2 Crosstalk and reciprocal regulation between microglia and peripheral immune cells
In the context of peripheral immune responses, microglia promote the infiltration of peripheral immune cells into brain tissue by releasing chemokines and cytokines, with neutrophils playing a particularly prominent role in secondary brain injury, as shown in Figure 7. Neutrophils are actively recruited in response to microglia-derived leukotriene and signals mediated by formyl peptide receptor 1. Upon infiltration, they release ROS and pro-inflammatory cytokines, which in turn further activate microglia and exacerbate tissue damage (180, 181). Meanwhile, the infiltration of macrophages and monocytes enhances microglial activation (182–184) and modulates their pro-inflammatory responses through pathways such as platelet-derived growth factor D (185). Natural killer (NK) cells and T lymphocytes also participate in the functional regulation of microglia. NK cells exacerbate BBB disruption by damaging endothelial cells and inducing pro-inflammatory activation of microglia (186). T cells, particularly Th1 cells, promote M1 polarization by secreting IFN-γ (187), whereas regulatory T cells and Th2 cells suppress the M1 phenotype and enhance M2 functions by releasing anti-inflammatory cytokines such as IL-4 and IL-10 (188). After ICH, an increase in regulatory T cells is positively correlated with microglial M2 polarization. Tregs limit inflammatory damage and promote hematoma clearance through the TGF-β/Smad2/3 signaling pathway and by inhibiting NF-κB activation (79). Importantly, peripheral immune cells also regulate the M1/M2 polarization of microglia by releasing various inflammatory mediators. IL-4 and IL-10 promote the shift from the M1 to M2 phenotype through the JAK1/STAT6 signaling pathway (189).
In summary, microglia play a central role in regulating central immune responses after ICH through dynamic phenotypic transitions. They not only orchestrate the early inflammatory response but also engage in complex interactions with peripheral immune cells to shape both the pathological progression and repair outcomes. Elucidating the mechanisms by which microglia operate within this complex network, particularly their functional crosstalk with neutrophils, will provide a theoretical basis for developing novel immunotherapeutic strategies and may improve neurological outcomes following ICH.
5 Bidirectional regulation between NETs and microglia: from stroke to neurodegenerative diseases
Immune responses triggered by neurological disorders involve not a single immune cell population, but rather a complex network of cellular interactions. Within this network, the bidirectional regulation between NETs and microglia plays a critical role in shaping post-stroke immune responses. The interaction between NETs and microglia not only regulates local immune responses during the acute phase, but may also profoundly influence neurorepair, BBB integrity, and functional recovery in stroke and other neurological disorders.
After ICH, microglial phenotypes and functions exhibit significant temporal changes: during the acute phase (within approximately 72 hours), a pro-inflammatory or interferon-responsive phenotype predominates, gradually transitioning to a phenotype characterized by phagocytosis and repair, such as DAM-like (18, 32, 97, 190). Correspondingly, NETs are closely associated with the acute phase of ICH and disease progression, promoting perihematomal edema and vascular permeability disruption via the ERK-MMP9/AQP4 axis. Clearance or inhibition of NETs has been shown to reduce vascular and brain tissue damage (66, 191). In lineage studies, disease-associated microglia have been found to exhibit both anti-inflammatory or phagocytic and pro-inflammatory characteristics (192). Their activation depends on the triggering receptor TREM2 on the microglial surface, which facilitates phagocytosis of apoptotic neurons and induces the generation of a small number of pro-inflammatory factors (193). In animal models of ICH, TREM2 is activated around the hematoma after intracerebral hemorrhage, reducing neuroinflammation and inhibiting neuronal apoptosis (194). However, in-depth studies on the DAM concept have largely focused on neurodegenerative diseases like Alzheimer’s, and related evidence in ICH remains limited. Existing studies suggest that microglia with different phenotypes may influence the generation and clearance of NETs through distinct mechanisms at different stages of ICH. During the acute phase, pro-inflammatory/IFN-responsive phenotypes are more likely to synergize with neutrophil activation and NET formation, contributing to lesion clearance but potentially exacerbating secondary damage. In contrast, in the subacute to chronic phase, microglia that shift toward repair or DAM-like phenotypes may limit the persistence of NETs by enhancing phagocytosis and anti-inflammatory signaling, thereby promoting tissue remodeling and functional recovery (192). Although the proposed dichotomy between M1 and M2 phenotypes is considered overly simplistic, this classification remains important in studying microglial function.
However, current research on the interactions between NETs and microglia in ICH remains limited. Most available evidence is derived from models of ischemic stroke, TBI, and neurodegenerative disorders such as Alzheimer’s disease (AD). Despite variations in disease contexts, current studies consistently reveal a highly conserved crosstalk mechanism between NETs and microglia in central inflammatory responses. Therefore, systematically elucidating their synergistic interactions, particularly in the regulation of inflammation, BBB disruption, and neurological dysfunction, will help uncover the key pathological mechanisms underlying secondary neural damage following ICH. As presented in Table 1.
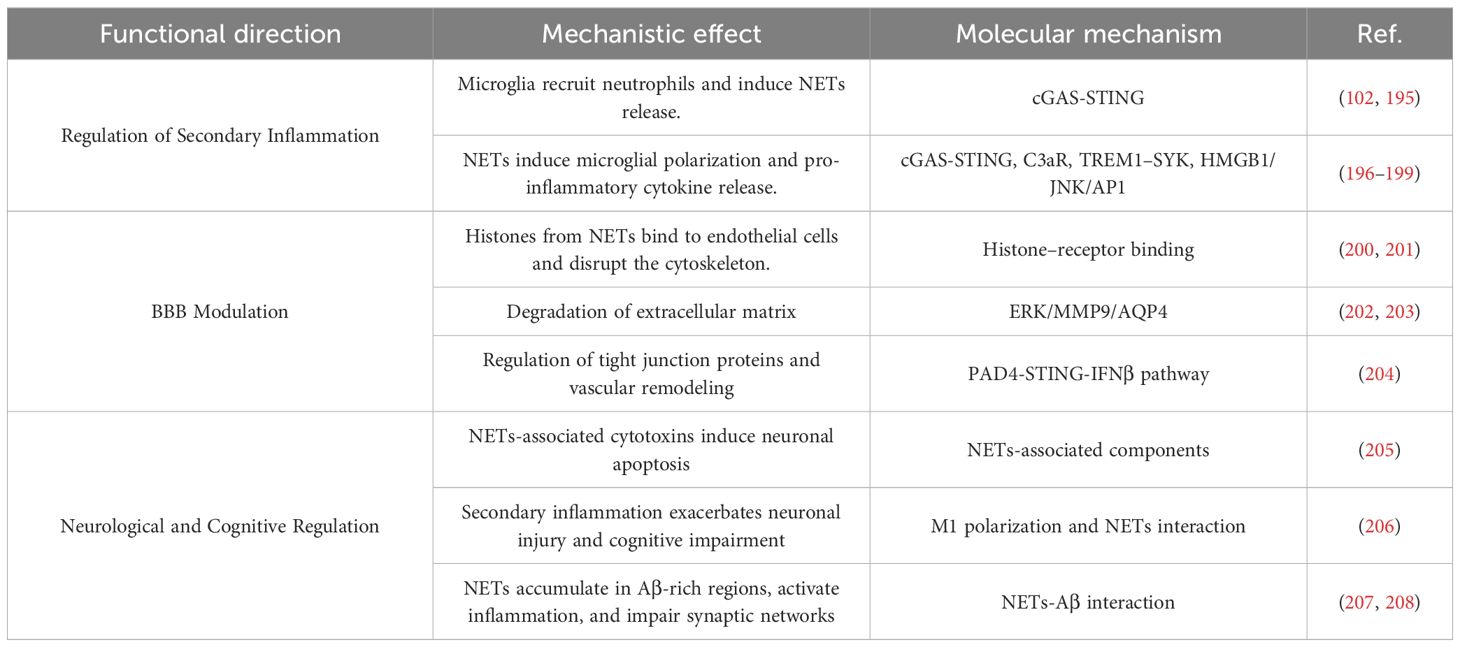
Table 1. Mechanisms of NET–microglia interaction.
5.1 Coordinated regulation of the inflammatory response
Inflammation is a principal pathological characteristic of ICH, with secondary inflammatory responses significantly influencing stroke progression, the degree of neuronal damage, and subsequent neurological recovery. Microglia promote the release of NETs, which in turn further activate microglia. Together, they facilitate the recruitment of local immune cells and influence the pathological progression of the disease. As illustrated in Table 1. NETs and microglia, as core participants in the immune response, form a self-amplifying inflammatory loop through bidirectional regulation. Microglia promote neutrophil recruitment and NET release, while NETs, in turn, activate microglia, driving pro-inflammatory responses and further infiltration of immune cells, thus establishing a self-sustaining positive feedback loop of inflammation (209).
Existing direct evidence in ICH shows that this bidirectional interaction is rapidly initiated during the acute phase. Hematoma degradation products, DAMPs, and endothelial signals exposed by blood-brain barrier disruption (e.g., ICAM-1, P-selectin) can drive extensive neutrophil infiltration into the brain parenchyma (82, 102, 210). After entering the brain parenchyma, neutrophils form NETs in the local inflammatory environment, releasing extracellular DNA, histones, and proteases. These molecules directly interact with microglial pattern recognition receptors, inducing the release of pro-inflammatory factors and chemokines such as IL-1β, IL-6, MCP-1, and CXCL-1 (200), which further enhance neutrophil recruitment and NET formation (210). Pharmacological studies also support this shared regulatory pattern. Degradation of NETs (e.g., DNase I, PAD4 inhibitor GSK484) can reduce the proportion of pro-inflammatory microglia and the levels of inflammatory factors (210). In contrast, inhibiting microglial activation (e.g., C3aR antagonist, TREM1 inhibitor LP17, Mincle–Syk blocker albumin) not only reduces local inflammatory responses but also decreases neutrophil recruitment and NET formation (4, 7, 211).
Research on this mechanism is continuously advancing, as shown in Figure 8. Studies have found that NETs can drive pro-inflammatory polarization and inflammation amplification in microglia through multiple signaling pathways. NETs contribute to neuroinflammation and BBB damage in tPA-related intracerebral hemorrhage by promoting the activation of the cGAS-STING pathway in microglia (102). In ICH, C3aR-mediated microglial activation promotes the expression of the NLRP3 inflammasome through the activation of the PKC/p38MAPK cascade. It also facilitates the infiltration of neutrophils into the perihematomal region, forming inflammation cell clusters marked by MPO, which further disrupt the blood-brain barrier and exacerbate cerebral edema (212). Additionally, in SAH, the activation of the TREM1–SYK axis drives microglial polarization toward a pro-inflammatory phenotype and activates the PAD4–NETs pathway, promoting neutrophil formation of NETs. HMGB1 released from NETs can exacerbate the inflammatory cascade through the JNK/AP1 pathway (201). This bidirectional regulation of central and peripheral immunity intensifies neuroinflammatory responses and secondary brain damage following SAH (136, 213).
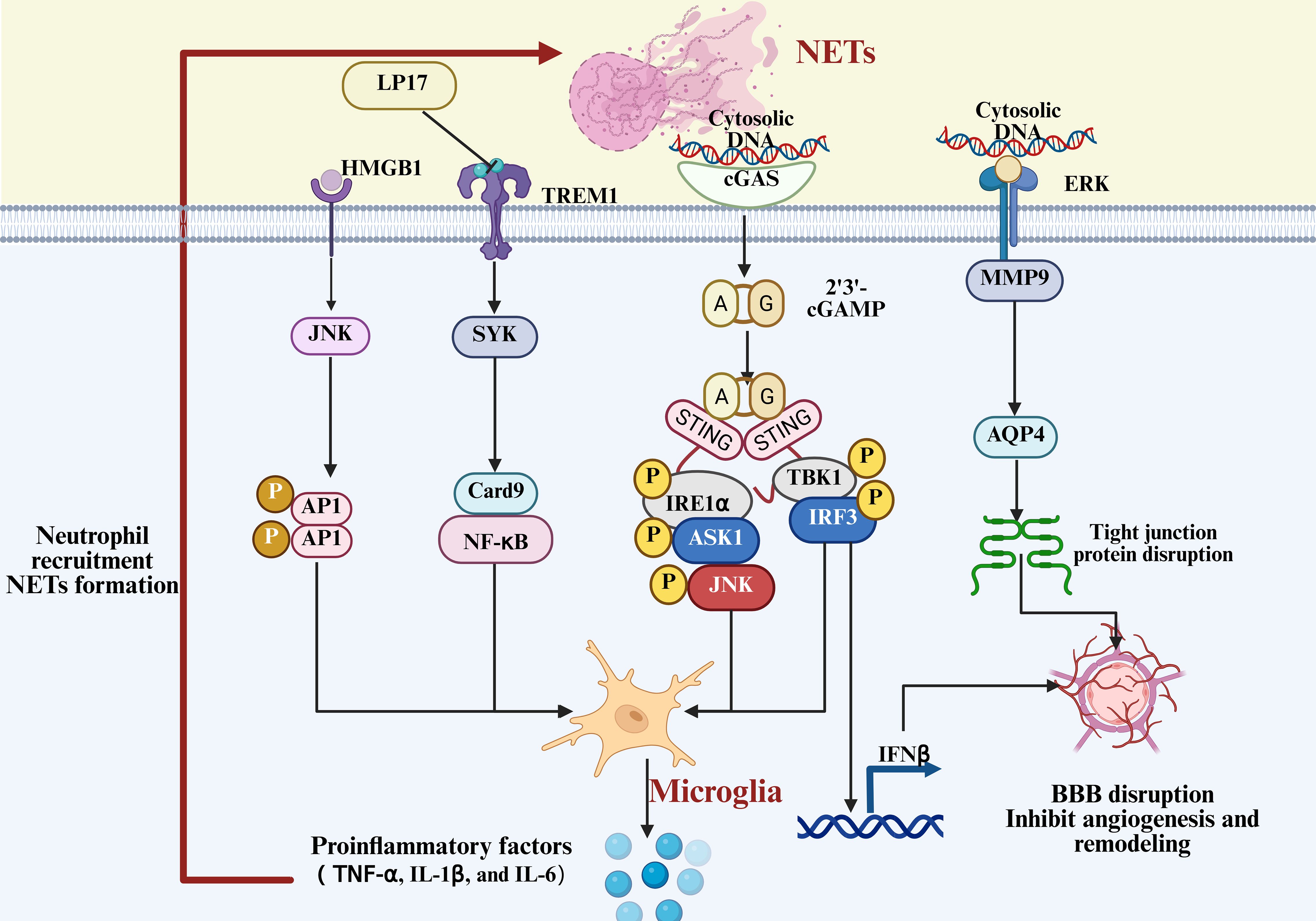
Figure 8. Mechanism of NET-induced microglial activation and BBB disruption. Neutrophil extracellular traps (NETs) activate microglia through multiple signaling cascades, amplifying neuroinflammatory responses and exacerbating blood-brain barrier disruption.
Interestingly, the bidirectional interaction between the two is also prominent in studies of other neurological disorders. Studies have found that ginsenoside Rg1-loaded vesicles enhance microglial clearance of neutrophils, significantly reducing NET release and promoting neovascularization and brain tissue repair (214). Further studies have found that the intelligent nanomaterial system C-Lipo/CA, containing a PAD4 inhibitor, can suppress NETosis and the cGAS-STING pathway, alleviating neuroinflammation and modulating the inflammatory phenotype of microglia (195). Further studies in TBI have found that NETs regulate microglial pro-inflammatory responses through the STING-dependent IRE1α/ASK1/JNK signaling pathway (215). In addition to stroke and TBI, Zenaro et al. were the first to report the existence of neutrophil-microglia crosstalk and NETs in both the vasculature and parenchyma in AD (216). Although these findings originate from non-ICH models, the revealed inflammation amplification pattern provides insights into the potential mechanisms in ICH.
In conclusion, although substantial evidence supports the bidirectional regulation between NETs and microglia in various neurological disorders, relevant studies in ICH remain scarce, particularly clinical research. ICH has a unique pathological context, including hematoma-driven inflammation and iron-mediated toxicity, and its immune regulatory mechanisms may differ significantly from those in ischemic stroke and TBI. Future studies should integrate ICH-specific animal models and clinical samples to further clarify the true role of NETs and microglia interactions in the progression of ICH and their therapeutic potential. Additionally, this inflammation amplification not only affects the release of cytokines but also further disrupts the structure and function of the blood-brain barrier, becoming a key mechanism in post-stroke secondary injury.
5.2 Synergistic disruption of the BBB
The BBB is a multilayered structure consisting of brain microvascular endothelial cells, the basement membrane, astrocytic end feet, and tight junction proteins. It serves as a critical defense barrier of the CNS, protecting against the infiltration of exogenous toxins and immune cells. After ICH, the structural integrity of the BBB is rapidly compromised during the acute phase, allowing harmful molecules and peripheral immune cells to enter the brain parenchyma, exacerbating secondary neural damage. Among them, the bidirectional regulation between NETs and microglia is one of the key mechanisms underlying BBB damage. NETs and microglia synergistically contribute to a compound injury pathway affecting the structure and function of the BBB, as shown in Table 1, through mutual amplification of inflammatory mediators, intercellular chemotaxis, and cross-activation of signaling pathways (217). Following stroke, microglia are rapidly activated into a pro-inflammatory M1 phenotype, releasing cytokines such as IL-1β and IL-6 that directly disrupt tight junctions of endothelial cells (218). In parallel, they promote astrocyte activation and glial scar formation, indirectly impairing the reconstruction and functional recovery of the BBB (219, 220). In addition, chemokines such as CXCL1 released by microglia facilitate the recruitment of neutrophils into the central nervous system. While modulating NETs formation, they also indirectly affect the integrity of the BBB via NETs, as shown in Figure 8.
Research has demonstrated that following ICH, excessive formation of NETs is not merely a reactive product of immune cells and pathological signals, but also contributes to BBB disruption through multiple mechanisms upon recruitment to the central nervous system. Histones released by NETs bind to receptors on the surface of endothelial cells, increasing membrane permeability and disrupting the cytoskeleton, thereby impairing endothelial barrier function (196). Additionally, elastase and proteases such as MMP-9 in NETs can degrade the extracellular matrix and tight junction proteins, downregulating AQP4, thereby further loosening the BBB structure (66, 197). At the same time, NETs can form dense complexes with fibrinogen and plasminogen, obstructing fibrinolysis and indirectly prolonging hematoma compression and mechanical damage to the BBB (136, 198). Moreover, under hypoxic conditions, the adhesion between NETs and endothelial cells is enhanced, further exacerbating endothelial injury and increasing vascular permeability (174, 199). In contrast, inhibition of key NETs-forming enzymes such as PAD4 reduces post-stroke BBB breakdown and enhances vascular plasticity.
It is important to note that the role of NETs and microglia in BBB damage is not a unidirectional process. NETs induce microglial inflammatory polarization through the TLR4/RAGE pathway. In turn, microglia stimulate the release of NETs via signaling axes such as the C3aR complement system (212), TREM1–SYK (201), and Mincle–Syk (201), amplifying the inflammatory response and endothelial barrier damage. After BBB damage, it also reciprocally promotes the infiltration of peripheral immune cells, allowing the positive feedback loop between NETs and microglia to continue. In therapeutic exploration, several drugs have shown potential in animal models to simultaneously protect the BBB and regulate NETs–microglia interactions. For example, Adjudin reduces neutrophil infiltration and MMP-9 levels while promoting the polarization of microglia to an anti-inflammatory phenotype (221). Methylene blue, on the other hand, upregulates IL-10 through the Akt/GSK-3β/MEF2D signaling pathway, inhibiting peripheral immune cell infiltration and microglial activation (211). Therefore, protecting the BBB should be considered one of the core downstream targets for targeting NETs–microglia interactions.
However, direct evidence for ICH remains very limited, despite a substantial body of research in ischemic stroke and neurodegenerative diseases. In ischemic stroke, NETs regulate the PAD4-STING-IFNβ pathway, promoting BBB damage and inhibiting neovascularization and vascular remodeling (207). Edaravone, in contrast, upregulates tight junction protein expression by reducing NET formation (222). Furthermore, NETs have been shown to drive synergistic damage to both the intestinal and blood-brain barriers through the DDIT4/IL-1β pathway, leading to the development of colitis and AD (223).
In the immune response, microglia act not only as initiators of inflammatory amplification but also as potential stabilizers that sustain barrier disruption. NETs and microglia engage in bidirectional inflammatory crosstalk, creating a sustained pro-inflammatory positive feedback loop that not only exacerbates local inflammation but also directly accelerates the deterioration of BBB structure and function. This synergistic mechanism has been reported in ischemic stroke, TBI, and various neurodegenerative diseases, but direct evidence in ICH remains limited and mostly confined to animal models. Given the unique pathological context of ICH, such as hematoma-driven inflammation and iron-mediated neurotoxicity, these extrapolated mechanisms may not fully reflect the actual disease progression. Future research must systematically verify the specific role of NETs–microglia interactions in BBB damage and their potential for intervention, using ICH-specific models and clinical samples.
5.3 Regulation of neural injury and cognition
NETs and microglia may play a crucial role in neuroinjury and cognitive dysfunction following ICH. Cognitive dysfunction is a key factor in long-term disability and decreased quality of life in ICH survivors, affecting various aspects such as attention, executive function, memory, and information processing speed. In recent years, immune-inflammatory responses, particularly the interaction between NETs and microglia, have been considered one of the key mechanisms driving cognitive decline following ICH. Histones and proteases released from NETs can directly damage neurons, inducing apoptosis and necrosis, and initiating an inflammatory cascade that further exacerbates neuronal loss. Moreover, NETs can further induce M1 polarization of microglia, forming a positive feedback loop between microglia and NETs. Thus, the interaction between NETs and microglia, as shown in Table 1, contributes to neuronal injury and cognitive impairment in central nervous system disorders through multiple mechanisms, exacerbating central nervous system inflammation and neuronal loss.
Existing animal studies on ICH show that NETs-mediated microglial pro-inflammatory activation is closely associated with early brain injury and poor functional outcomes (25). Acerboside D modulates neutrophil activity and NET formation, reducing inflammation and improving neurological function, motor coordination, and learning and memory abilities. The mechanism may involve upregulation of NTSR1 expression, activation of the cAMP/PKAc signaling pathway, and inhibition of PAD4 and citH3 activity, thereby suppressing NET formation (224). Additionally, in the ICH experimental model, neutrophil depletion improves brain perfusion and alleviates neurological deficits and long-term prognosis (113). Although these results suggest that the NETs–microglia axis may be involved in cognitive impairment following ICH, direct and systematic studies validating the relationship between their interaction and cognitive function remain extremely limited.
Due to the limited direct evidence linking ICH to cognitive impairment, some studies have drawn insights from findings in other neurological disorders. In ischemic stroke models, NETs have been found to contribute to delayed neuronal injury (108). The main mechanisms include disrupting the BBB (225), promoting thrombosis, and acting as an activation platform for inflammasomes (226), which synergistically amplify the inflammatory response, thereby exacerbating neurological dysfunction (203). Notably, the interaction between neutrophils and microglia is not limited to stroke models. In patients with cerebral small vessel disease, plasma NETs biomarkers are elevated and negatively correlate with overall cognition, executive function, and information processing speed (227). A similar phenomenon has also been observed in an AD mouse model (216, 228, 229). Although these studies provide clues for inferring the potential mechanisms of ICH, the pathological characteristics of ICH differ significantly from those of ischemic or degenerative diseases.
With growing research interest, the interaction between microglia and neutrophils has emerged as a focal point. Neumann et al. were the pioneers in presenting in vivo evidence of physical interaction between microglia and infiltrating neutrophils (108, 230). Emerging intervention strategies in neuronal injury and cognition, such as the gut-brain axis and nanozyme therapy, have become widely studied in various neurological disease models (202, 231, 232). However, their effectiveness in ICH and related cognitive impairment has yet to be directly validated. However, in a high-intensity inflammatory environment, microglia not only struggle to clear the large amounts of NETs formed but may also be activated by them, transitioning to a pro-inflammatory state, further promoting the recruitment of peripheral immune cells and exacerbating neuronal injury and cognitive dysfunction (136, 195, 201).
Although studies in ICH models have shown the role of NETs and microglia in neuroinflammation and functional impairment, direct evidence linking them to cognitive function remains lacking. In contrast, diseases like ischemic stroke and AD have provided insights into the underlying mechanisms. However, ICH has a unique pathological context, and extensive in vitro and in vivo research is needed to further clarify the interactions between NETs and microglia after ICH and evaluate their feasibility as intervention targets.
6 Therapeutic strategies targeting NETs After ICH
Aberrant formation of NETs plays a critical role in secondary inflammation after ICH by amplifying inflammatory signaling, disrupting the BBB, and inducing neuronal cell death. NETs formation is closely associated with PAD4 mediated histone citrullination and ROS production mediated by NADPH oxidase. In addition, NETs are primarily composed of a DNA backbone, histones, MPO, NE, and other cytotoxic proteins and enzymes. Therefore, inhibiting NET formation and promoting NET clearance represent key therapeutic strategies targeting NETs in the context of ICH, as summarized in Table 2.
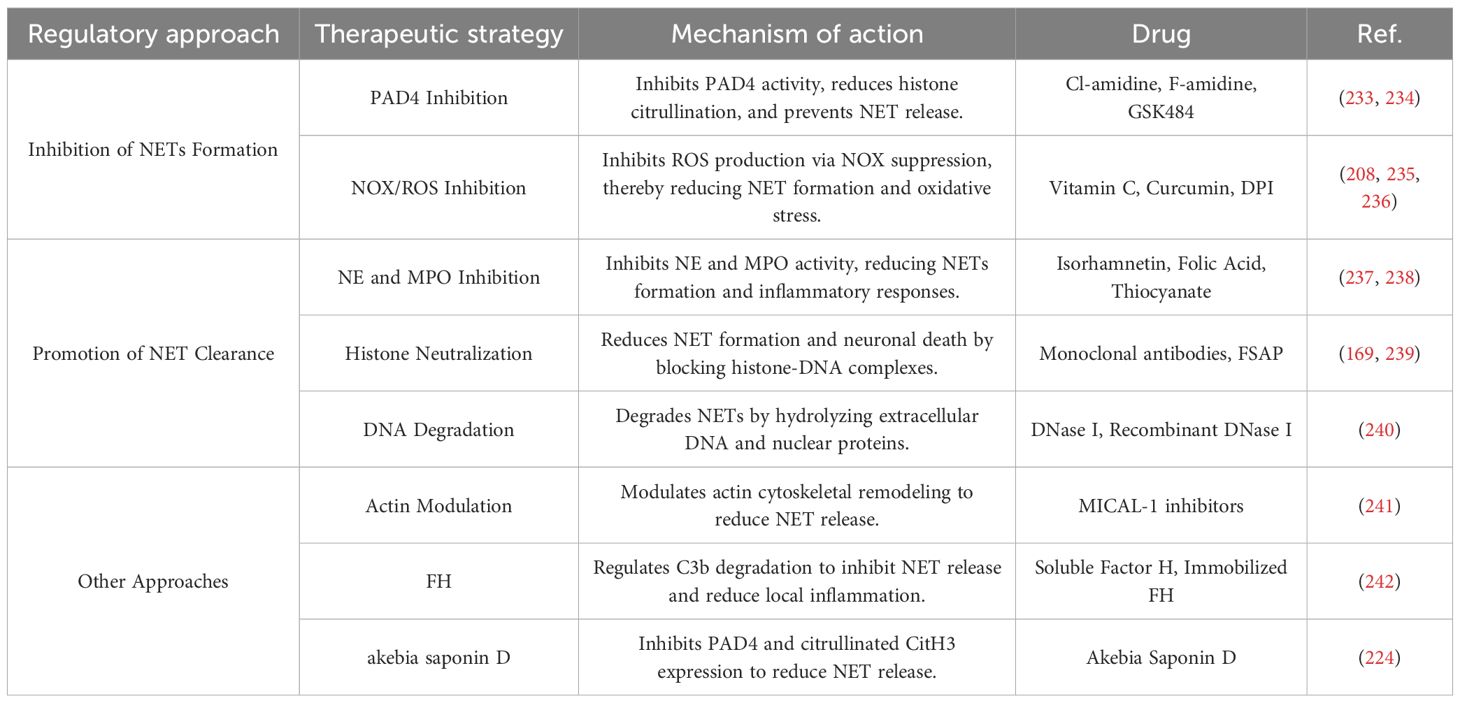
Table 2. Therapeutic strategies targeting NETs after ICH.
6.1 Inhibition of NETs formation
6.1.1 PAD4 inhibitors
PAD4 is a crucial enzyme in the synthesis of NETs. It catalyzes histone citrullination, leading to chromatin decondensation and promoting the release of nuclear DNA (233). In the context of ICH, PAD4 expression is significantly upregulated, particularly in neutrophils surrounding the hematoma. PAD4 activation promotes NETs release, thereby exacerbating neuroinflammation, BBB disruption, and vascular reperfusion impairment. Conversely, PAD4-/- mice exhibit reduced levels of citrullinated histone H3 and attenuated BBB damage (102). Therefore, targeting PAD4 has emerged as a promising strategy to inhibit NETs formation and mitigate secondary injury following ICH.
Studies have shown that the enzymatic activity of PAD4 is significantly enhanced upon binding to Ca²+. Irreversible PAD4 inhibitors targeting this calcium-dependent mechanism, such as Cl-amidine and F-amidine, effectively suppress NETs release (243, 244). GSK484, a selective and reversible PAD4 inhibitor, has been shown to effectively inhibit NET formation in vitro (122). In SAH models, GSK484 alleviates secondary brain edema and neuronal injury by suppressing inflammation (186), and also inhibits thrombosis by blocking NETs formation (234).
Notably, in addition to conventional small-molecule inhibitors, novel compounds have also been shown to modulate PAD4 activity. Studies have shown that miR-155 enhances PMA-induced PAD4 mRNA expression and promotes NETs formation, whereas its antagonist, antagomiR-155, reduces PAD4 expression, thereby attenuating NETs release and tissue damage (204). Collectively, these findings confirm that PAD4 gene knockout or pharmacological inhibition reduces NETs formation and alleviates secondary neural injury.
6.1.2 NOX and ROS inhibitors
ROS are key drivers of NETs formation, with NOX being the primary source of ROS production. Following ICH, neutrophils rapidly infiltrate the lesion site. Activation of NOX and the resulting ROS burst not only drive NETs formation but also directly contribute to BBB disruption, neuronal apoptosis, and intensified local oxidative stress. Therefore, regulating the production of ROS and NOX is critical for inhibiting NETs formation.
As a classic natural antioxidant, vitamin C not only scavenges ROS but also crosses the BBB in the form of dehydroascorbic acid (235), exerting neuroprotective effects (205, 245). Multiple clinical and preclinical studies have demonstrated that increased plasma vitamin C levels are significantly associated with a correlated risk of stroke (206, 246, 247). In animal models, vitamin C suppresses NETs formation and alleviates central inflammation by reducing ROS production (235, 245). Additionally, PKC has been shown to regulate NETosis by blocking ROS production in upstream signaling pathways (248, 249). The natural polyphenolic antioxidant curcumin exerts neuroprotective and anti-inflammatory effects by activating the Nrf2 pathway and inhibiting ROS-mediated NETs release (208).
As a key enzyme in ROS production, NOX has attracted increasing attention in NETs-targeted therapies. Diphenyleneiodonium, a non-specific NOX inhibitor, significantly reduces NETosis levels (236). In stroke models, attenuates MMP-2 and MMP-9 activity in brain tissue, alleviates cerebral edema, and mitigates BBB disruption (250, 251). Activation of NOX2 has been identified as a key mediator of NETosis and secondary brain injury. The use of NOX inhibitors has also shown beneficial effects in regulating vascular reperfusion and promoting brain tissue repair. In recent years, hypertonic saline has emerged as a promising non-pharmacological intervention with the potential to modulate NETosis. By altering the extracellular osmotic environment, it inhibits PMA- and LPS-induced ROS production, thereby interfering with both NOX-dependent and NOX-independent NETosis and promoting neutrophil apoptosis (252). This mechanism not only suppresses tissue damage caused by excessive NETs release but also contributes to the restoration of immune homeostasis.
In summary, interventions targeting the NOX/ROS pathway exert protective effects after ICH by reducing NETs formation, alleviating oxidative stress, stabilizing the BBB, and improving neurological function. These multifaceted benefits provide a strong pharmacological basis and translational potential for NETs-related therapies in ICH.
6.2 Promotion of NETs clearance
Current research on NETs formation has made considerable progress; however, most strategies target the early stages of NETs generation and remain insufficient to fully control NETs-mediated chronic inflammation and tissue damage. Therefore, further exploration of mechanisms for clearing established NETs may offer novel therapeutic approaches for targeting NETs-related pathological processes in central nervous system disorders.
6.2.1 NE and MPO inhibitors
NE and MPO are not only essential components of NETs but also play critical roles in the inflammatory response (8, 152). Inhibiting their enzymatic activity can effectively mitigate NETs-mediated inflammation. In stroke therapy, sivelestat sodium, a commonly used NE inhibitor, has been extensively studied and applied in clinical practice. Additionally, compounds such as trigonelline, isorhamnetin, and eupafolin have also been identified as effective inhibitors of MPO (237). Studies have shown that thiocyanate, selenocyanate, and various nitrogen oxides can act as alternative substrates for MPO and directly scavenge hypochlorous acid released by neutrophils, thereby inhibiting the formation of NETs (238). However, although these inhibitors have shown some efficacy in certain inflammatory conditions, research on the protective effects of NE and MPO inhibitors in ICH remains limited. Based on current research progress, the development of inhibitory strategies targeting NE and MPO may represent a promising direction for future therapeutic interventions in ICH.
6.2.2 Histone inhibitors
Histones exhibit significant cytotoxicity and, when bound to DNA, form histone-DNA complexes that constitute the structural backbone of NETs. Blocking histone-DNA complexes with monoclonal antibodies effectively inhibits NETs formation and reduces neuronal death. A study by Xu Jun et al. demonstrated that activated protein C can cleave histones and neutralize their cytotoxicity (169). Thrombomodulin-α promotes the generation of activated protein C in vitro, thereby reducing histone-induced thrombin production and endothelial cell death. A study by Simona Grasso et al. found that factor VII activating protease degrades histones, suppresses their cytotoxic effects on endothelial cells, reduces NETs formation, and mitigates NETosis-associated tissue damage (239).
6.2.3 DNases
DNase I is frequently employed as the principal enzyme for the degradation of NETs in both animal models and in vitro studies (136). Its mechanism of action involves specifically recognizing and hydrolyzing extracellular double-stranded DNA and associated nucleoproteins, thereby dismantling the NETs structure. This process not only suppresses NETs formation but also attenuates NETs-induced disruption of the BBB (253, 254). DNase I is naturally abundant in the bloodstream and can continuously eliminate circulating NETs (240). Research indicates that DNase I-mediated degradation of NETs enhances the efficacy of tissue-type plasminogen activator in hematoma clearance. In animal models of ICH, combined administration of DNase I and tissue-type plasminogen activator significantly reduces cerebral edema, decreases neuronal death, and improves neurological recovery (198).
Notably, the monoclonal antibody 2C5 specifically recognizes NETs and may serve as a targeting ligand for diagnostic and therapeutic applications (255). Nina Filipczak and colleagues further conjugated the monoclonal antibody 2C5 to functionalized DNase I-loaded nanomicelles, enabling specific recognition and targeted degradation of NETs (256), thereby significantly enhancing therapeutic efficacy. Multiple animal studies have demonstrated that DNase I inhibits the cGAS-STING pathway. This inhibition reduces NETs-mediated immune activation, alleviates secondary inflammatory responses after ICH, improves BBB integrity, and mitigates neuronal injury (102, 257). These findings highlight its strong potential for neuroprotection and ICH treatment.
To date, no clinical interventions have directly addressed the role of NETs in ICH. Nevertheless, clinical trials of DNase I have been registered in patients with ischemic stroke and have advanced to early exploratory phases (NCT05203224, NCT05880524) (254). Meanwhile, animal studies indicate that DNase I confers neuroprotective effects without increasing the risk of hemorrhage. In addition, systematic reviews and clinical data demonstrate substantial NETs formation in the peripheral blood of patients with TBI, with levels closely correlating with injury severity. These findings further support the critical role of NETs in secondary inflammation following acute brain injury (258). Although DNase I has not yet been clinically applied in intracerebral hemorrhage (ICH), its use as a NET-degrading strategy has demonstrated favorable safety and partial efficacy in clinical studies of cystic fibrosis (259), COVID-19–related ARDS (241), and autoimmune diseases (242). These cross-disease clinical findings, together with preclinical results from the stroke field, collectively support the feasibility of DNase I as a NET-targeted therapy for ICH.
6.3 Other approaches
Studies have shown that actin and associated cytoskeletal proteins are pivotal in regulating the formation of NETs. NETs extrusion is accompanied by local cortical F-actin depolymerization, a process primarily driven by F-actin oxidation mediated by the monooxygenase MICAL-1 and facilitated by the cooperative action of G-actin, binding proteins and gelsolin (260). Myosin accumulates with cortical F-actin at the cell periphery, where actomyosin interactions generate mechanical forces that facilitate the release of NETs (261). Inhibition of MICAL-1 oxidative activity or suppression of myosin ATPase activity significantly reduces NETs formation. Additionally, variations in gelsolin levels within neutrophils influence the structural type of NETs, suggesting that cytoskeletal composition not only regulates the efficiency of NETs release but also determines their morphological characteristics (262). Metzler et al. found that inhibition of actin dynamics impairs the nuclear translocation of neutrophil elastase, thereby disrupting the formation of NETs (259).
Factor H (FH) is a principal regulator of the alternative complement pathway. Studies have shown that FH not only modulates C3b degradation by colocalizing with CD11b on the neutrophil surface but also plays multiple roles in regulating neutrophil function. Soluble factor H promotes neutrophil migration, whereas immobilized FH induces cell spreading and enhances the release of IL-8. Although factor H alone does not induce NET formation, immobilized FH significantly suppresses NET release and associated ROS production under stimulation with PMA or fibronectin combined with β-glucan, potentially attenuating local inflammation and tissue damage (263).
In addition, akebia saponin D, a compound isolated from traditional Chinese medicine, has been shown to reduce NETs release following ICH by activating the NTSR1/cAMP/PKAc/p-CREB signaling pathway and suppressing the expression of the key NETs enzyme PAD4 and its downstream product citrullinated histone H3. This intervention effectively attenuates brain tissue damage and the release of proinflammatory cytokines (224). As a free radical scavenger, edaravone can eliminate singlet oxygen and significantly inhibit NETs formation in vitro (264). Its compound formulation, edaravone dexborneol, has been shown in patients with acute ischemic stroke to reduce serum NETs markers while improving blood–brain barrier integrity and neurological function (222). However, most of the supporting evidence derives from ischemic stroke, and direct clinical validation in ICH populations remains lacking. Nonetheless, these findings provide a biological rationale and indirect support for targeting NETs in ICH.
6.4 Challenges in clinical translation
Currently, immunomodulatory strategies for hemorrhagic stroke are being investigated at multiple levels. Clinical studies have shown that immunoregulatory agents exhibit therapeutic potential in animal models. For instance, fingolimod has been reported to improve neurological recovery in SAH models by inhibiting neutrophil adhesion to the vascular endothelium (265). Minocycline has been shown to reduce the infiltration of microglia and macrophages after ICH, alleviate cerebral edema, and downregulate the expression of TNF-α and MMP-12 (266). In addition, small molecules targeting neutrophil activation, such as the FPR1 inhibitor T-0080, can mitigate brain tissue injury by interfering with the IL-1β pathway and thereby reducing microglia-mediated recruitment of neutrophils (267, 268). In recent years, NETs have emerged as key mediators of neuroinflammation and secondary injury, representing potential therapeutic targets. NETs inhibitors, including PAD4 inhibitors, NADPH oxidase inhibitors, and recombinant DNase I, have demonstrated anti-inflammatory, neuroprotective, and BBB preserving effects in various animal models of intracerebral hemorrhage. However, their clinical translation still faces numerous challenges.
First, the limited permeability of the BBB is one of the primary obstacles. Although localized BBB disruption occurs in the early phase after ICH, overall permeability remains low. Moreover, BBB integrity exhibits high temporal variability across different time windows, substantially affecting both the efficacy and safety of therapeutic agents (269, 270). Second, the formation of NETs exhibits marked temporal dependence, yet studies precisely defining their peak formation and optimal intervention window are lacking, adding to the complexity of treatment and intervention strategies (271). Further complicating the issue, the role of NETs remains controversial due to their potential dual effects. On the one hand, studies have suggested that NETs can, under certain pathological conditions, clear hemorrhage-associated debris and limit microbial invasion, thereby exerting protective effects (127). On the other hand, animal studies have demonstrated that NETs can disrupt the BBB by activating pathways such as MMP9 and AQP4, leading to cerebral edema and secondary injury (66), and may even contribute to hydrocephalus formation by impairing lymphatic clearance of cerebrospinal fluid (66). This inconsistency poses significant challenges for NET-targeted therapies, further complicating their clinical application.
Notably, most current studies on NETs remain confined to in vitro experiments or animal models, with limited support from clinical trials. To advance the clinical translation of NET-targeted strategies, several emerging technologies in recent years have offered new opportunities for mechanistic elucidation and target validation. Single-cell RNA sequencing can be employed to characterize the dynamic expression of NET-related genes across distinct immune cell subsets after ICH (190). Advances in in vivo NET imaging techniques have also provided real-time assessment tools for targeted intervention strategies (272). These cutting-edge approaches hold promise for advancing the clinical translation of NET-related therapies. However, the current challenges remain major barriers to bridging the gap between basic research and clinical application, particularly given that most studies have yet to progress to the clinical stage. Future efforts should focus on overcoming these obstacles.
7 Perspectives and conclusion
Secondary inflammatory responses following ICH are a key mechanism driving ongoing brain tissue damage and functional impairment. Central nervous system injury signals to the periphery via the autonomic nervous system, neuroendocrine pathways, and meningeal lymphatic vessels. This activates the peripheral immune system and promotes the infiltration of neutrophils and other immune cells into the CNS through a disrupted BBB or cerebrospinal fluid pathways. Among these processes, the bidirectional interaction between NETs and microglia plays a central role in amplifying immune responses, disrupting the BBB, and contributing to neuronal injury. Central nervous system injury also signals to the periphery via the autonomic nervous system, neuroendocrine pathways, and meningeal lymphatic vessels. These signals activate the peripheral immune system and promote the infiltration of neutrophils and other immune cells into the CNS through a disrupted BBB or cerebrospinal fluid pathways. Through the release of inflammatory mediators, coordinated activation of signaling pathways, and indirect modulation of astrocytes and other immune effector cells, NETs and microglia establish a self-amplifying and reciprocal network of immune-mediated damage that not only exacerbates brain injury but also impedes post-stroke cognitive recovery. Therefore, the NETs-microglia axis may represent a critical therapeutic target for secondary neuroinflammation following ICH, with substantial potential for clinical translation.
Although basic research has made notable progress in NETs-targeted therapies, clinical translation still faces three major challenges. First, limited BBB permeability hampers the effective delivery of most NETs inhibitors or degrading enzymes to the lesion sites within the CNS, thereby reducing their therapeutic efficacy. Second, immune defense functions must be balanced, as NETs play a vital physiological role in host antimicrobial defense. Excessive inhibition may increase the risk of infection or even promote tumorigenesis. Third, the optimal therapeutic time window remains unclear. The function of NETs varies dynamically across different phases of intracerebral hemorrhage, and the lack of systematic research to define the best intervention timing introduces additional risks and uncertainties.
Future research should focus on stratified identification of NETs-related pathways and improved precision in target selection. It is essential to develop therapeutic strategies capable of crossing the BBB and maintaining immune homeostasis while enabling controllable modulation of central inflammatory injury. Such approaches may offer viable solutions for the precision treatment of secondary neurological damage following intracerebral hemorrhage. Additionally, further exploration of the interaction between NETs and microglia in animal models of aging or common comorbidities, such as metabolic and cardiovascular diseases, is needed. This will help systematically investigate their impact on inflammation progression and neurological recovery, enhancing the translational potential of the research findings.
Author contributions
SZ: Visualization, Writing – original draft. ZJ: Writing – review & editing. LJ: Validation, Writing – review & editing. YZ: Validation, Writing – review & editing, Supervision. TW: Conceptualization, Writing – review & editing, Investigation. PX: Validation, Writing – review & editing, Resources. YC: Funding acquisition, Writing – review & editing. DZ: Writing – review & editing, Funding acquisition. JL: Writing – review & editing, Funding acquisition.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was financially supported by the Joint Fund of the National Natural Science Foundation of China (Grant No. U24A20779), and by the Jilin Provincial Department of Science and Technology (Grant Nos. YDZJ202401062ZYTS and YDZJ202401081ZYTS).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1643524/full#supplementary-material
SUPPLEMENTARY IMAGE 1 | Mechanistic illustration of the crosstalk between peripheral NETs and microglia after intracerebral hemorrhage. Proposed mechanism after intracerebral hemorrhage, showing the crosstalk between peripheral neutrophil extracellular traps (NETs) and microglia, their role in inflammatory signaling, and potential therapeutic strategies targeting NETs.
Abbreviations
ICH, Intracerebral hemorrhage; NETs, neutrophil extracellular traps; SAH, subarachnoid hemorrhage; CNS, central nervous system; MPO, myeloperoxidase; BBB, blood-brain barrier; TBI, traumatic brain injury; DAMPs, damage-associated molecular patterns; AVP, arginine vasopressin; MMPs, matrix metalloproteinases; NOX, NADPH oxidase components; ROS, reactive oxygen species; NE, neutrophil elastase; TLRs, Toll-like receptors; PKC, protein kinase C; PAD4, peptidylarginine deiminase 4; GM-CSF, granulocyte-macrophage colony-stimulating factor; mPTP, mitochondrial permeability transition pore; PSGL-1, P-selectin glycoprotein ligand-1; HMGB1, high mobility group box 1; CitH3, histone H3; DNase I, Deoxyribonuclease I; PAMPs, pathogen-associated molecular patterns; NLRs, NOD-like receptors; PRRs, pattern recognition receptors; NK, Natural killer; AD, Alzheimer’s disease; BTK, Bruton’s tyrosine kinase; FH, Factor H.
References
1. Wang YJ, Li ZX, Gu HQ, Zhai Y, Zhou Q, Jiang Y, et al. China Stroke Statistics: an update on the 2019 report from the National Center for Healthcare Quality Management in Neurological Diseases, China National Clinical Research Center for Neurological Diseases, the Chinese Stroke Association, National Center for Chronic and Non-communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention and Institute for Global Neuroscience and Stroke Collaborations. Stroke Vasc Neurol. (2022) 7:415–50. doi: 10.1136/svn-2021-001374
2. Tu WJ, Zhao Z, Yin P, Cao L, Zeng J, Chen H, et al. Estimated burden of stroke in China in 2020. JAMA Netw Open. (2023) 6:e231455. doi: 10.1001/jamanetworkopen.2023.1455
3. Tu WJ and Wang LD. Special writing group of China stroke surveillance report. China stroke surveillance report 2021. Mil Med Res. (2023) 10:33. doi: 10.1186/s40779-023-00463-x
4. Wafa HA, Wolfe CDA, Emmett E, Roth GA, Johnson CO, and Wang Y. Burden of stroke in Europe: thirty-year projections of incidence, prevalence, deaths, and disability-adjusted life years. Stroke. (2020) 51:2418–27. doi: 10.1161/STROKEAHA.120.029606
5. Barthels D and Das H. Current advances in ischemic stroke research and therapies. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165260. doi: 10.1016/j.bbadis.2018.09.012
6. Feigin VL, Brainin M, Norrving B, Martins S, Sacco RL, Hacke W, et al. World Stroke Organization (WSO): global stroke fact sheet 2022. Int J Stroke. (2022) 17:18–29. doi: 10.1177/17474930211065917
7. Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, and Hanley DF. Spontaneous intracerebral hemorrhage. N Engl J Med. (2001) 344:1450–60. doi: 10.1056/NEJM200105103441907
8. Montaño A, Hanley DF, and Hemphill JC 3rd. Hemorrhagic stroke. Handb Clin Neurol. (2021) 176:229–48. doi: 10.1016/B978-0-444-64034-5.00019-5
9. Zhao X, Sun G, Zhang H, Ting SM, Song S, Gonzales N, et al. Polymorphonuclear neutrophil in brain parenchyma after experimental intracerebral hemorrhage. Transl Stroke Res. (2014) 5:554–61. doi: 10.1007/s12975-014-0341-2
10. Kim M, Byun J, Chung Y, Lee SU, Park JE, Park W, et al. Reactive oxygen species scavenger in acute intracerebral hemorrhage patients: A multicenter, randomized controlled trial. Stroke. (2021) 52:1172–81. doi: 10.1161/STROKEAHA.120.032266
11. Fu X, Zeng H, Zhao J, Zhou G, Zhou H, Zhuang J, et al. Inhibition of dectin-1 ameliorates neuroinflammation by regulating microglia/macrophage phenotype after intracerebral hemorrhage in mice. Transl Stroke Res. (2021) 12:1018–34. doi: 10.1007/s12975-021-00889-2
12. Yu X, Zhou G, Shao B, Zhou H, Xu C, Yan F, et al. Gut microbiota dysbiosis induced by intracerebral hemorrhage aggravates neuroinflammation in mice. Front Microbiol. (2021) 12:647304. doi: 10.3389/fmicb.2021.647304
13. Fu X, Zhou G, Wu X, Xu C, Zhou H, Zhuang J, et al. Inhibition of P2X4R attenuates white matter injury in mice after intracerebral hemorrhage by regulating microglial phenotypes. J Neuroinflammation. (2021) 18:184. doi: 10.1186/s12974-021-02239-3
14. Guo Y, Dai W, Zheng Y, Qiao W, Chen W, Peng L, et al. Mechanism and regulation of microglia polarization in intracerebral hemorrhage. Molecules. (2022) 27:7080. doi: 10.3390/molecules27207080
15. Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron. (2022) 110:3458–83. doi: 10.1016/j.neuron.2022.10.020
16. Castellani G, Croese T, Peralta Ramos JM, and Schwartz M. Transforming the understanding of brain immunity. Science. (2023) 380:eabo7649. doi: 10.1126/science.abo7649
17. Liu YW, Li S, and Dai SS. Neutrophils in traumatic brain injury (TBI): friend or foe? J Neuroinflamm. (2018) 15:146. doi: 10.1186/s12974-018-1173-x
18. Tschoe C, Bushnell CD, Duncan PW, Alexander-Miller MA, and Wolfe SQ. Neuroinflammation after intracerebral hemorrhage and potential therapeutic targets. J Stroke. (2020) 22:29–46. doi: 10.5853/jos.2019.02236
19. Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
20. Savchenko AS, Borissoff JI, Martinod K, De Meyer SF, Gallant M, Erpenbeck L, et al. VWF-mediated leukocyte recruitment with chromatin decondensation by PAD4 increases myocardial ischemia/reperfusion injury in mice. Blood. (2014) 123:141–8. doi: 10.1182/blood-2013-07-514992
21. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
22. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
23. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. (2014) 20:511–7. doi: 10.1038/nm.3547
24. Yin N, Wang W, Pei F, Zhao Y, Liu C, Guo M, et al. A neutrophil hijacking nanoplatform reprograming NETosis for targeted microglia polarizing mediated ischemic stroke treatment. Adv Sci (Weinh). (2024) 11:e2305877. doi: 10.1002/advs.202305877
25. Hanhai Z, Bin Q, Shengjun Z, Jingbo L, Yinghan G, Lingxin C, et al. Neutrophil extracellular traps, released from neutrophil, promote microglia inflammation and contribute to poor outcome in subarachnoid hemorrhage. Aging (Albany NY). (2021) 13:13108–23. doi: 10.18632/aging.202993
26. Louveau A, Harris TH, and Kipnis J. Revisiting the mechanisms of CNS immune privilege. Trends Immunol. (2015) 36:569–77. doi: 10.1016/j.it.2015.08.006
27. Schiller M, Ben-Shaanan TL, and Rolls A. Neuronal regulation of immunity: why, how and where? Nat Rev Immunol. (2021) 21:20–36. doi: 10.1038/s41577-020-0387-1
28. An C, Shi Y, Li P, Hu X, Gan Y, Stetler RA, et al. Molecular dialogs between the ischemic brain and the peripheral immune system: dualistic roles in injury and repair. Prog Neurobiol. (2014) 115:6–24. doi: 10.1016/j.pneurobio.2013.12.002
29. Claassen J and Park S. Spontaneous subarachnoid haemorrhage. Lancet. (2022) 400:846–62. doi: 10.1016/S0140-6736(22)00938-2
30. Cordonnier C, Demchuk A, Ziai W, and Anderson CS. Intracerebral haemorrhage: current approaches to acute management. Lancet. (2018) 392:1257–68. doi: 10.1016/S0140-6736(18)31878-6
31. Russo MV and McGavern DB. Inflammatory neuroprotection following traumatic brain injury. Science. (2016) 353:783–5. doi: 10.1126/science.aaf6260
32. Lan X, Han X, Li Q, Yang QW, and Wang J. Modulators of microglial activation and polarization after intracerebral haemorrhage. Nat Rev Neurol. (2017) 13:420–33. doi: 10.1038/nrneurol.2017.69
33. Wang J and Doré S. Inflammation after intracerebral hemorrhage. J Cereb Blood Flow Metab. (2007) 27:894–908. doi: 10.1038/sj.jcbfm.9600403
34. Xue M and Yong VW. Neuroinflammation in intracerebral haemorrhage: immunotherapies with potential for translation. Lancet Neurol. (2020) 19:1023–32. doi: 10.1016/S1474-4422(20)30364-1
35. Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, and Shi FD. Global brain inflammation in stroke. Lancet Neurol. (2019) 18:1058–66. doi: 10.1016/S1474-4422(19)30078-X
36. Nance DM and Sanders VM. Autonomic innervation and regulation of the immune system (1987-2007). Brain Behav Immun. (2007) 21:736–45. doi: 10.1016/j.bbi.2007.03.008
37. Elenkov IJ, Wilder RL, Chrousos GP, and Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. (2000) 52:595–638. doi: 10.1016/S0031-6997(24)01470-4
38. Scanzano A and Cosentino M. Adrenergic regulation of innate immunity: a review. Front Pharmacol. (2015) 6:171. doi: 10.3389/fphar.2015.00171
39. Maestroni GJ. Dendritic cell migration controlled by alpha 1b-adrenergic receptors. J Immunol. (2000) 165:6743–7. doi: 10.4049/jimmunol.165.12.6743
40. Szelényi J, Kiss JP, and Vizi ES. Differential involvement of sympathetic nervous system and immune system in the modulation of TNF-alpha production by alpha2- and beta-adrenoceptors in mice. J Neuroimmunol. (2000) 103:34–40. doi: 10.1016/s0165-5728(99)00234-9
41. Kohm AP and Sanders VM. Norepinephrine and beta 2-adrenergic receptor stimulation regulate CD4+T and B lymphocyte function in vitro and in vivo. Pharmacol Rev. (2001) 53:487–525. doi: 10.1016/S0031-6997(24)01510-2
42. Bellinger DL, Millar BA, Perez S, Carter J, Wood C, ThyagaRajan S, et al. Sympathetic modulation of immunity: relevance to disease. Cell Immunol. (2008) 252:27–56. doi: 10.1016/j.cellimm.2007.09.005
43. Loza MJ, Peters SP, Foster S, Khan IU, and Penn RB. beta-Agonist enhances type 2 T-cell survival and accumulation. J Allergy Clin Immunol. (2007) 119:235–44. doi: 10.1016/j.jaci.2006.09.019
44. Chelmicka-Schorr E, Kwasniewski MN, and Czlonkowska A. Sympathetic nervous system modulates macrophage function. Int J Immunopharmacol. (1992) 14:841–6. doi: 10.1016/0192-0561(92)90082-v
45. Swanson MA, Lee WT, and Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4+T cells exposed to norepinephrine. J Immunol. (2001) 166:232–40. doi: 10.4049/jimmunol.166.1.232
46. Bernik TR, Friedman SG, Ochani M, DiRaimo R, Susarla S, Czura CJ, et al. Cholinergic antiinflammatory pathway inhibition of tumor necrosis factor during ischemia reperfusion. J Vasc Surg. (2002) 36:1231–6. doi: 10.1067/mva.2002.129643
47. Pavlov VA, Ochani M, Yang LH, Gallowitsch-Puerta M, Ochani K, Lin X, et al. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit Care Med. (2007) 35:1139–44. doi: 10.1097/01.CCM.0000259381.56526.96
48. Guarini S, Altavilla D, Cainazzo MM, Giuliani D, Bigiani A, Marini H, et al. Efferent vagal fibre stimulation blunts nuclear factor-kappab activation and protects against hypovolemic hemorrhagic shock. Circulation. (2003) 107:1189–94. doi: 10.1161/01.CIR.0000050627.90734.ED
49. Churchland PS and Winkielman P. Modulating social behavior with oxytocin: how does it work? What does it mean? Horm Behav. (2012) 61:392–9. doi: 10.1016/j.yhbeh.2011.12.003
50. Neumann ID. Brain oxytocin: a key regulator of emotional and social behaviours in both females and males. J Neuroendocrinol. (2008) 20:858–65. doi: 10.1111/j.1365-2826.2008.01726.x
51. Boone M and Deen PM. Physiology and pathophysiology of the vasopressinregulated renal water reabsorption. Pflugers Arch. (2008) 456:1005–24. doi: 10.1007/s00424-008-0498-1
52. Tanriverdi F, Silveira LF, MacColl GS, and Bouloux PM. The hypothalamic-pituitary-gonadal axis: immune function and autoimmunity. J Endocrinol. (2003) 176:293–304. doi: 10.1677/joe.0.1760293
53. Taneja V. Sex hormones determine immune response. Front Immunol. (2018) 9:1931. doi: 10.3389/fimmu.2018.01931
54. Jankowski M, Bissonauth V, Gao L, Gangal M, Wang D, Danalache B, et al. Anti-inflammatory effect of oxytocin in rat myocardial infarction. Basic Res Cardiol. (2010) 105:205–18. doi: 10.1007/s00395-009-0076-5
55. Palin K, Moreau ML, Sauvant J, Orcel H, Nadjar A, Duvoid-Guillou A, et al. Interleukin-6 activates arginine vasopressin neurons in the supraoptic nucleus during immune challenge in rats. Am J Physiol Endocrinol Metab. (2009) 296:E1289–99. doi: 10.1152/ajpendo.90489.2008
56. Oliveira-Pelegrin GR, Saia RS, Carnio EC, and Rocha MJ. Oxytocin affects nitric oxide and cytokine production by sepsis-sensitized macrophages. Neuroimmunomodulation. (2013) 20:65–71. doi: 10.1159/000345044
57. Boyd JH, Holmes CL, Wang Y, Roberts H, and Walley KR. Vasopressin decreases sepsis-induced pulmonary inflammation through the V2R. Resuscitation. (2008) 79:325–31. doi: 10.1016/j.resuscitation.2008.07.006
58. Pawlikowski M, Stepien H, and Komorowski J. Hypothalamic-pituitary-thyroid axis and the immune system. Neuroimmunomodulation. (1994) 1:149–52. doi: 10.1159/000097154
59. Barreiro Arcos ML, Gorelik G, Klecha A, Genaro AM, and Cremaschi GA. Thyroid hormones increase inducible nitric oxide synthase gene expression downstream from PKC-zeta in murine tumor T lymphocytes. Am J Physiol Cell Physiol. (2006) 291:C327–36. doi: 10.1152/ajpcell.00316.2005
60. Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. (2015) 523:337–41. doi: 10.1038/nature14432
61. Kipnis J. Multifaceted interactions between adaptive immunity and the central nervous system. Science. (2016) 353:766–71. doi: 10.1126/science.aag2638
62. Louveau A, Herz J, Alme MN, Salvador AF, Dong MQ, Viar KE, et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat Neurosci. (2018) 21:1380–91. doi: 10.1038/s41593-018-0227-9
63. Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature. (2018) 560:185–91. doi: 10.1038/s41586-018-0368-8
64. Planas AM, Gómez-Choco M, Urra X, Gorina R, Caballero M, and Chamorro Á. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J Immunol. (2012) 188:2156–63. doi: 10.4049/jimmunol.1102289
65. Tsuchida T, Parker KC, Turner RV, McFarland HF, Coligan JE, and Biddison WE. Autoreactive CD8+ T-cell responses to human myelin protein-derived peptides. Proc Natl Acad Sci U S A. (1994) 91:10859–63. doi: 10.1073/pnas.91.23.10859
66. Zhang Q, Chen Y, Li Y, Feng Z, Liang L, Hao X, et al. Neutrophil extracellular trap-mediated impairment of meningeal lymphatic drainage exacerbates secondary hydrocephalus after intraventricular hemorrhage. Theranostics. (2024) 14:1909–38. doi: 10.7150/thno.91653
67. Nimmerjahn A, Kirchhoff F, and Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. (2005) 308:1314–8. doi: 10.1126/science.1110647
68. Vink R, Gabrielian L, and Thornton E. The role of substance P in secondary pathophysiology after traumatic brain injury. Front Neurol. (2017) 8:304. doi: 10.3389/fneur.2017.00304
69. Kubes P and Ward PA. Leukocyte recruitment and the acute inflammatory response. Brain Pathol. (2000) 10:127–35. doi: 10.1111/j.1750-3639.2000.tb00249.x
70. Engelhardt B and Ransohoff RM. The ins and outs of T-lymphocyte trafficking to the CNS: anatomical sites and molecular mechanisms. Trends Immunol. (2005) 26:485–95. doi: 10.1016/j.it.2005.07.004
71. Povlishock JT, Becker DP, Sullivan HG, and Miller JD. Vascular permeability alterations to horseradish peroxidase in experimental brain injury. Brain Res. (1978) 153:223–39. doi: 10.1016/0006-8993(78)90404-3
72. Ma Q, Ineichen BV, Detmar M, and Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun. (2017) 8:1434. doi: 10.1038/s41467-017-01484-6
73. Absinta M, Ha SK, Nair G, Sati P, Luciano NJ, Palisoc M, et al. Human and nonhuman primate meninges harbor lymphatic vessels that can be visualized noninvasively by MRI. Elife. (2017) 6:e29738. doi: 10.7554/eLife.29738
74. Saghazadeh A and Rezaei N. The role of timing in the treatment of spinal cord injury. BioMed Pharmacother. (2017) 92:128–39. doi: 10.1016/j.biopha.2017.05.048
75. Bian Z, Gong Y, Huang T, Lee CZW, Bian L, Bai Z, et al. Deciphering human macrophage development at single-cell resolution. Nature. (2020) 582:571–6. doi: 10.1038/s41586-020-2316-7
76. Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, et al. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. (2013) 33:1864–74. doi: 10.1038/jcbfm.2013.146
77. Eldahshan W, Fagan SC, and Ergul A. Inflammation within the neurovascular unit: Focus on microglia for stroke injury and recovery. Pharmacol Res. (2019) 147:104349. doi: 10.1016/j.phrs.2019.104349
78. Han X, Lan X, Li Q, Gao Y, Zhu W, Cheng T, et al. Inhibition of prostaglandin E2 receptor EP3 mitigates thrombin-induced brain injury. J Cereb Blood Flow Metab. (2016) 36:1059–74. doi: 10.1177/0271678X15606462
79. Deng S, Jin P, Liu S, He Y, Sherchan P, Zhang JH, et al. Recruitment of regulatory T cells with rCCL17 promotes M2 microglia/macrophage polarization through TGFβ/TGFβR/Smad2/3 pathway in a mouse model of intracerebral hemorrhage. Exp Neurol. (2023) 367:114451. doi: 10.1016/j.expneurol.2023.114451
80. Zhou Y, Wang Y, Wang J, Anne Stetler R, and Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. (2014) 115:25–44. doi: 10.1016/j.pneurobio.2013.11.003
81. Urday S, Kimberly WT, Beslow LA, Vortmeyer AO, Selim MH, Rosand J, et al. Targeting secondary injury in intracerebral haemorrhage–perihaematomal oedema. Nat Rev Neurol. (2015) 11:111–22. doi: 10.1038/nrneurol.2014.264
82. Atangana E, Schneider UC, Blecharz K, Magrini S, Wagner J, Nieminen-Kelhä M, et al. Intravascular inflammation triggers intracerebral activated microglia and contributes to secondary brain injury after experimental subarachnoid hemorrhage (eSAH). Transl Stroke Res. (2017) 8:144–56. doi: 10.1007/s12975-016-0485-3
83. Zhang Z, Zhang Z, Lu H, Yang Q, Wu H, and Wang J. Microglial polarization and inflammatory mediators after intracerebral hemorrhage. Mol Neurobiol. (2017) 54:1874–86. doi: 10.1007/s12035-016-9785-6
84. Liang C, Liu L, Bao S, Yao Z, Bai Q, Fu P, et al. Neuroprotection by Nrf2 via modulating microglial phenotype and phagocytosis after intracerebral hemorrhage. Heliyon. (2023) 9:e13777. doi: 10.1016/j.heliyon.2023.e13777
85. Jiao Y, Ren S, Wang L, and Wu G. PPARγ/RAD21 alleviates peripheral secondary brain injury in rat cerebral hemorrhage model through promoting M2 polarization of microglial cells. Int Immunopharmacol. (2023) 114:109572. doi: 10.1016/j.intimp.2022.109572
86. Wen H, Tan J, Tian M, Wang Y, Gao Y, and Gong Y. TGF-β1 ameliorates BBB injury and improves long-term outcomes in mice after ICH. Biochem Biophys Res Commun. (2023) 654:136–44. doi: 10.1016/j.bbrc.2023.03.007
87. Zhang Y, Lu W, and Xu N. Effects of butyphthalide on microglia polarization after intracerebral hemorrhage and the underlying mechanisms. Zhong Nan Da Xue Xue Bao Yi Xue Ban. (2022) 47:717–29. doi: 10.11817/j.issn.1672-7347.2022.210527
88. He M, Wang X, Liu Z, Cui Q, Chen Y, Geng W, et al. CDK5 mediates proinflammatory effects of microglia through activated DRP1 phosphorylation in rat model of intracerebral hemorrhage. Dis Markers. (2022) 2022:1919064. doi: 10.1155/2022/1919064
89. Lv YN, Ou-Yang AJ, and Fu LS. MicroRNA-27a negatively modulates the inflammatory response in lipopolysaccharide-stimulated microglia by targeting TLR4 and IRAK4. Cell Mol Neurobiol. (2017) 37:195–210. doi: 10.1007/s10571-016-0361-4
90. Wang J, Xu Z, Chen X, Li Y, Chen C, Wang C, et al. MicroRNA-182-5p attenuates cerebral ischemia-reperfusion injury by targeting Toll-like receptor 4. Biochem Biophys Res Commun. (2018) 505:677–84. doi: 10.1016/j.bbrc.2018.09.165
91. Wang Z, Yuan B, Fu F, Huang S, and Yang Z. Hemoglobin enhances miRNA-144 expression and autophagic activation mediated inflammation of microglia via mTOR pathway. Sci Rep. (2017) 7:11861. doi: 10.1038/s41598-017-12067-2
92. Bai Q, Xue M, and Yong VW. Microglia and macrophage phenotypes in intracerebral haemorrhage injury: therapeutic opportunities. Brain. (2020) 143:1297–314. doi: 10.1093/brain/awz393
93. Yu A, Zhang T, Zhong W, Duan H, Wang S, Ye P, et al. miRNA-144 induces microglial autophagy and inflammation following intracerebral hemorrhage. Immunol Lett. (2017) 182:18–23. doi: 10.1016/j.imlet.2017.01.002
94. Chang CF, Goods BA, Askenase MH, Beatty HE, Osherov A, DeLong JH, et al. Divergent functions of tissue-resident and blood-derived macrophages in the hemorrhagic brain. Stroke. (2021) 52:1798–808. doi: 10.1161/STROKEAHA.120.032196
95. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. (2016) 19:987–91. doi: 10.1038/nn.4338
96. Hu X, Leak RK, Shi Y, Suenaga J, Gao Y, Zheng P, et al. Microglial and macrophage polarization — new prospects for brain repair. Nat Rev Neurol. (2015) 11:56–64. doi: 10.1038/nrneurol.2014.207
97. Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. (2019) 566:388–92. doi: 10.1038/s41586-019-0924-x
98. Tan YL, Yuan Y, and Tian L. Microglial regional heterogeneity and its role in the brain. Mol Psychiatry. (2020) 25:351–67. doi: 10.1038/s41380-019-0609-8
99. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, DvirSzternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. (2017) 169:1276–1290.e17. doi: 10.1016/j.cell.2017.05.018
100. Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, et al. Single-cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell-state changes. Immunity. (2019) 50:253–271.e6. doi: 10.1016/j.immuni.2018.11.004
101. Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, et al. The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep. (2019) 27:1293–1306.e6. doi: 10.1016/j.celrep.2019.03.099
102. Wang R, Zhu Y, Liu Z, Chang L, Bai X, Kang L, et al. Neutrophil extracellular traps promote tPA-induced brain hemorrhage via cGAS in mice with stroke. Blood. (2021) 138:91–103. doi: 10.1182/blood.2020008913
103. Jassam YN, Izzy S, Whalen M, McGavern DB, and El Khoury J. Neuroimmunology of traumatic brain injury: time for a paradigm shift. Neuron. (2017) 95:1246–65. doi: 10.1016/j.neuron.2017.07.010
104. Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, and Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. (2015) 35:888–901. doi: 10.1038/jcbfm.2015.45
105. Göb E, Reymann S, Langhauser F, Schuhmann MK, Kraft P, Thielmann I, et al. Blocking of plasma kallikrein ameliorates stroke by reducing thromboinflammation. Ann Neurol. (2015) 77:784–803. doi: 10.1002/ana.24380
106. De Meyer SF, Denorme F, Langhauser F, Geuss E, Fluri F, and Kleinschnitz C. Thromboinflammation in stroke brain damage. Stroke. (2016) 47:1165–72. doi: 10.1161/STROKEAHA.115.011238
107. Villringer K, Sanz Cuesta BE, Ostwaldt AC, Grittner U, Brunecker P, Khalil AA, et al. DCE-MRI blood-brain barrier assessment in acute ischemic stroke. Neurology. (2017) 88:433–40. doi: 10.1212/WNL.0000000000003566
108. Neumann J, Riek-Burchardt M, Herz J, Doeppner TR, König R, Hütten H, et al. Very-late-antigen-4 (VLA-4)-mediated brain invasion by neutrophils leads to interactions with microglia, increased ischemic injury and impaired behavior in experimental stroke. Acta Neuropathol. (2015) 129:259–77. doi: 10.1007/s00401-014-1355-2
109. Mehta V, Russin J, Spirtos A, He S, Adamczyk P, Amar AP, et al. Matrix metalloproteinases in Cerebral Vasospasm following Aneurysmal Subarachnoid Hemorrhage. Neurol Res Int. (2013) 2013:943761. doi: 10.1155/2013/943761
110. Guo Z, Sun X, He Z, Jiang Y, Zhang X, and Zhang JH. Matrix metalloproteinase-9 potentiates early brain injury after subarachnoid hemorrhage. Neurol Res. (2010) 32:715–20. doi: 10.1179/016164109X12478302362491
111. Fassbender K, Hodapp B, Rossol S, Bertsch T, Schmeck J, Schutt S, et al. Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J Neurol Neurosurg Psychiatry. (2001) 70:534–7. doi: 10.1136/jnnp.70.4.534
112. Peeyush Kumar T, McBride DW, Dash PK, Matsumura K, Rubi A, and Blackburn SL. Endothelial cell dysfunction and Injury in Subarachnoid Hemorrhage. Mol Neurobiol. (2019) 56:1992–2006. doi: 10.1007/s12035-018-1213-7
113. Zeineddine HA, Hong SH, Peesh P, Dienel A, Torres K, Thankamani Pandit P, et al. Neutrophils and neutrophil extracellular traps cause vascular occlusion and delayed cerebral ischemia after subarachnoid hemorrhage in mice. Arterioscler Thromb Vasc Biol. (2024) 44:635–52. doi: 10.1161/ATVBAHA.123.320224
114. Borregaard N. Neutrophils, from marrow to microbes. Immunity. (2010) 33:657–70. doi: 10.1016/j.immuni.2010.11.011
115. Lee WL and Grinstein S. Immunology. The tangled webs that neutrophils weave. . Sci. (2004) 303:1477–8. doi: 10.1126/science.1095484
116. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
117. Takei H, Araki A, Watanabe H, Ichinose A, and Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. (1996) 59:229–40. doi: 10.1002/jlb.59.2.229
118. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. (2018) 25:486–541. doi: 10.1038/s41418-017-0012-4
119. Hamam HJ, Khan MA, and Palaniyar N. Histone acetylation promotes neutrophil extracellular trap formation. Biomolecules. (2019) 9:32. doi: 10.3390/biom9010032
120. Lu CH, Li KJ, Wu CH, Shen CY, Kuo YM, Hsieh SC, et al. The fcγRIII engagement augments PMA-stimulated neutrophil extracellular traps (NETs) formation by granulocytes partially via cross-talk between syk-ERK-NF-κB and PKC-ROS signaling pathways. Biomedicines. (2021) 9:1127. doi: 10.3390/biomedicines9091127
121. Papayannopoulos V, Metzler KD, Hakkim A, and Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. (2010) 191:677–91. doi: 10.1083/jcb.201006052
122. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD, et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat Chem Biol. (2015) 11:189–91. doi: 10.1038/nchembio.1735
123. Sørensen OE and Borregaard N. Neutrophil extracellular traps - the dark side of neutrophils. J Clin Invest. (2016) 126:1612–20. doi: 10.1172/JCI84538
124. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, and Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
125. Yipp BG and Kubes P. NETosis: how vital is it? Blood. (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
126. Yousefi S, Mihalache C, Kozlowski E, Schmid I, and Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. (2009) 16:1438–44. doi: 10.1038/cdd.2009.96
127. Monteleone M, Stanley AC, Chen KW, Brown DL, Bezbradica JS, von Pein JB, et al. Interleukin-1β Maturation triggers its relocation to the plasma membrane for gasdermin-D-dependent and -independent secretion. Cell Rep. (2018) 24:1425–33. doi: 10.1016/j.celrep.2018.07.027
128. Burgener SS and Schroder K. Neutrophil extracellular traps in host defense. Cold Spring Harb Perspect Biol. (2020) 12:a037028. doi: 10.1101/cshperspect.a037028
129. Dwivedi N and Radic M. Citrullination of autoantigens implicates NETosis in the induction of autoimmunity. Ann Rheum Dis. (2014) 73:483–91. doi: 10.1136/annrheumdis-2013-203844
130. Cedervall J, Hamidi A, and Olsson AK. Platelets, NETs and cancer. Thromb Res. (2018) 164 Suppl 1:S148–52. doi: 10.1016/j.thromres.2018.01.049
131. Weinrauch Y, Drujan D, Shapiro SD, Weiss J, and Zychlinsky A. Neutrophil elastase targets virulence factors of enterobacteria. Nature. (2002) 417:91–4. doi: 10.1038/417091a
132. Parker H, Albrett AM, Kettle AJ, and Winterbourn CC. Myeloperoxidase associated with neutrophil extracellular traps is active and mediates bacterial killing in the presence of hydrogen peroxide. J Leukoc Biol. (2012) 91:369–76. doi: 10.1189/jlb.0711387
133. Halverson TW, Wilton M, Poon KK, Petri B, and Lewenza S. DNA is an antimicrobial component of neutrophil extracellular traps. PloS Pathog. (2015) 11:e1004593. doi: 10.1371/journal.ppat.1004593
134. Liew PX and Kubes P. The neutrophil’s role during health and disease. Physiol Rev. (2019) 99:1223–48. doi: 10.1152/physrev.00012.2018
135. Puy L, Corseaux D, Perbet R, Deramecourt V, Cordonnier C, and Bérézowski V. Neutrophil extracellular traps (NETs) infiltrate haematoma and surrounding brain tissue after intracerebral haemorrhage: A post-mortem study. Neuropathol Appl Neurobiol. (2021) 47:867–77. doi: 10.1111/nan.12733
136. Jin J, Zhao X, Li W, Wang F, Tian J, Wang N, et al. Neutrophil extracellular traps: A novel therapeutic target for intracranial hemorrhage. Thromb Res. (2022) 219:1–13. doi: 10.1016/j.thromres.2022.08.024
137. Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. (2013) 304:L342–9. doi: 10.1152/ajplung.00151.2012
138. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, and Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126:242–6. doi: 10.1182/blood-2015-01-624023
139. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. doi: 10.1126/science.1256478
140. Kim SJ and Jenne CN. Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin Immunol. (2016) 28:546–54. doi: 10.1016/j.smim.2016.10.013
141. Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, and Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. (2014) 123:3818–27. doi: 10.1182/blood-2013-10-529982
142. Li Y, Yuan R, Ren T, Yang B, Miao H, Liu L, et al. Role of Sciellin in gallbladder cancer proliferation and formation of neutrophil extracellular traps. Cell Death Dis. (2021) 12:30. doi: 10.1038/s41419-020-03286-z
143. Ma F, Yang S, Zhou M, Lu Y, Deng B, Zhang J, et al. NADPH oxidase-derived reactive oxygen species production activates the ERK1/2 pathway in neutrophil extracellular traps formation by Streptococcus agalactiae isolated from clinical mastitis bovine. Veterinary Microbiol. (2022) 268:109427. doi: 10.1016/j.vetmic.2022.109427
144. Shao BZ, Yao Y, Li JP, Chai NL, and Linghu EQ. The role of neutrophil extracellular traps in cancer. Front Oncol. (2021) 11:714357. doi: 10.3389/fonc.2021.714357
145. Wright HL, Lyon M, Chapman EA, Moots RJ, and Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol. (2020) 11:584116. doi: 10.3389/fimmu.2020.584116
146. An Z, Li J, Yu J, Wang X, Gao H, Zhang W, et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-kappaB signaling in macrophages. Cell Cycle. (2019) 18:2928–38. doi: 10.1080/15384101.2019.1662678
147. Shirakawa K, Kobayashi E, Ichihara G, Kitakata H, Katsumata Y, Sugai K, et al. H(2) inhibits the formation of neutrophil extracellular traps. JACC: Basic to Trans Sci. (2022) 7:146–61. doi: 10.1016/j.jacbts.2021.11.005
148. Domer D, Walther T, Moller S, Behnen M, and Laskay T. Neutrophil extracellular traps activate proinflammatory functions of human neutrophils. Front Immunol. (2021) 12:636954. doi: 10.3389/fimmu.2021.636954
149. Denorme F, Rustad JL, Portier I, Crandell JL, de Araujo CV, Cody MJ, et al. Neutrophil extracellular trap inhibition improves survival in neonatal mouse infectious peritonitis. Pediatr Res. (2023) 93:862–9. doi: 10.1038/s41390-022-02219-0
150. Vallés J, Lago A, Santos MT, Latorre AM, Tembl JI, Salom JB, et al. Neutrophil extracellular traps are increased in patients with acute ischemic stroke: prognostic significance. Thromb Haemost. (2017) 117:1919–29. doi: 10.1160/TH17-02-0130
151. Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, and Papayannopoulos V. Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of TLR4. Cell Rep. (2020) 31:107602. doi: 10.1016/j.celrep.2020.107602
152. Cheng Y, Chen B, Xie W, Chen Z, Yang G, Cai Y, et al. Ghrelin attenuates secondary brain injury following intracerebral hemorrhage by inhibiting NLRP3 inflammasome activation and promoting Nrf2/ARE signaling pathway in mice. Int Immunopharmacol. (2020) 79:106180. doi: 10.1016/j.intimp.2019.106180
153. Zeng J, Chen Y, Ding R, Feng L, Fu Z, Yang S, et al. Isoliquiritigenin alleviates early brain injury after experimental intracerebral hemorrhage via suppressing ROS- and/or NF-κB-mediated NLRP3 inflammasome activation by promoting Nrf2 antioxidant pathway. J Neuroinflammation. (2017) 14:119. doi: 10.1186/s12974-017-0895-5
154. Warnatsch A, Ioannou M, Wang Q, and Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. . Sci. (2015) 349:316–20. doi: 10.1126/science.aaa8064
155. Jin Z, Lu J, Xu H, Zhang Y, Zhang S, Zhang D, et al. Exploring the correlation between innate immune activation of inflammasome and regulation of pyroptosis after intracerebral hemorrhage: From mechanism to treatment. BioMed Pharmacother. (2024) 179:117382. doi: 10.1016/j.biopha.2024.117382
156. Broz P and Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. (2016) 16:407–20. doi: 10.1038/nri.2016.58
157. Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. (2013) 341:1246–9. doi: 10.1126/science.1240248
158. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
159. Bakele M, Joos M, Burdi S, Allgaier N, Pöschel S, Fehrenbacher B, et al. Localization and functionality of the inflammasome in neutrophils. J Biol Chem. (2014) 289:5320–9. doi: 10.1074/jbc.M113.505636
160. Mankan AK, Dau T, Jenne D, and Hornung V. The NLRP3/ASC/Caspase-1 axis regulates IL-1β processing in neutrophils. Eur J Immunol. (2012) 42:710–5. doi: 10.1002/eji.201141921
161. Jin P, Qi D, Cui Y, Lenahan C, Zhang JH, Tao X, et al. Aprepitant attenuates NLRC4-dependent neuronal pyroptosis via NK1R/PKCδ pathway in a mouse model of intracerebral hemorrhage. J Neuroinflammation. (2022) 19:198. doi: 10.1186/s12974-022-02558-z
162. Xu C, Jiang F, Mao Y, Wei W, Song J, Jia F, et al. Disulfiram attenuates cell and tissue damage and blood–brain barrier dysfunction after intracranial haemorrhage by inhibiting the classical pyroptosis pathway. Sci Rep. (2024) 14:21860. doi: 10.1038/s41598-024-67118-2
163. Wang T, Nowrangi D, Yu L, Lu T, Tang J, Han B, et al. Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflammation. (2018) 15:2. doi: 10.1186/s12974-017-1039-7
164. Lei C, Chen K, Gu Y, Li Y, Wang L, Zhu X, et al. HMGB1/TLR4 axis promotes pyroptosis after ICH by activating the NLRP3 inflammasome. J Neuroimmunol. (2024) 393:578401. doi: 10.1016/j.jneuroim.2024.578401
165. Fang H, Bo Y, Hao Z, Mang G, Jin J, and Wang H. A promising frontier: targeting NETs for stroke treatment breakthroughs. Cell Commun Signal. (2024) 22:238. doi: 10.1186/s12964-024-01563-4
166. Xiao Z, Shen D, Lan T, Wei C, Wu W, Sun Q, et al. Reduction of lactoferrin aggravates neuronal ferroptosis after intracerebral hemorrhagic stroke in hyperglycemic mice. Redox Biol. (2022) 50:102256. doi: 10.1016/j.redox.2022.102256
167. Wei N, Lu T, Gu J, and Cai H. Lipoxin A4 suppresses neutrophil extracellular traps formation through the FPR2-dependent regulation of METTL3 in ischemic stroke. Brain Res Bull. (2025) 220:111178. doi: 10.1016/j.brainresbull.2024.111178
168. Wang D, Yin K, Zhang Y, Lu H, Hou L, Zhao H, et al. Fluoride induces neutrophil extracellular traps and aggravates brain inflammation by disrupting neutrophil calcium homeostasis and causing ferroptosis. Environ pollut. (2023) 331:121847. doi: 10.1016/j.envpol.2023.121847
169. Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F, et al. Extracellular histones are major mediators of death in sepsis. Nat Med. (2009) 15:1318–21. doi: 10.1038/nm.2053
170. Lv X, Han Y, Li Y, Wang X, Zhang T, Wang X, et al. Nonylphenol displays immunotoxicity by triggering hemocyte extracellular traps in Manila clam via ROS burst, ERK pathway and glycolysis. Ecotoxicol Environ Saf. (2024) 285:117145. doi: 10.1016/j.ecoenv.2024.117145
171. Wang YC, Wang PF, Fang H, Chen J, Xiong XY, and Yang QW. Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke. (2013) 44:2545–52. doi: 10.1161/STROKEAHA.113.001038
172. Wang YC, Zhou Y, Fang H, Lin S, Wang PF, Xiong RP, et al. Toll-like receptor 2/4 heterodimer mediates inflammatory injury in intracerebral hemorrhage. Ann Neurol. (2014) 75:876–89. doi: 10.1002/ana.24159
173. Yang Z, Jiang X, Zhang J, Huang X, Zhang X, Wang J, et al. Let-7a promotes microglia M2 polarization by targeting CKIP-1 following ICH. Immunol Lett. (2018) 202:1–7. doi: 10.1016/j.imlet.2018.07.007
174. Shi SX, Li YJ, Shi K, Wood K, Ducruet AF, and Liu Q. IL (Interleukin)-15 bridges astrocyte-microglia crosstalk and exacerbates brain injury following intracerebral hemorrhage. Stroke. (2020) 51:967–74. doi: 10.1161/STROKEAHA.119.028638
175. Zheng J, Lu J, Mei S, Wu H, Sun Z, Fang Y, et al. Ceria nanoparticles ameliorate white matter injury after intracerebral hemorrhage: microglia-astrocyte involvement in remyelination. J Neuroinflammation. (2021) 18:43. doi: 10.1186/s12974-021-02101-6
176. Tang J, Jila S, Luo T, Zhang B, Miao H, Feng H, et al. C3/C3aR inhibition alleviates GMH-IVH-induced hydrocephalus by preventing microglia-astrocyte interactions in neonatal rats. Neuropharmacology. (2022) 205:108927. doi: 10.1016/j.neuropharm.2021.108927
177. Deng S, Chen X, Lei Q, and Lu W. AQP2 promotes astrocyte activation by modulating the TLR4/NFκB-p65 pathway following intracerebral hemorrhage. Front Immunol. (2022) 13:847360. doi: 10.3389/fimmu.2022.847360
178. Zheng J, Wu H, Wang X, Zhang G, Lu J, Xu W, et al. Temporal dynamics of microglia-astrocyte interaction in neuroprotective glial scar formation after intracerebral hemorrhage. J Pharm Anal. (2023) 13:862–79. doi: 10.1016/j.jpha.2023.02.007
179. Tashiro R, Bautista-Garrido J, Ozaki D, Sun G, Obertas L, Mobley AS, et al. Transplantation of astrocytic mitochondria modulates neuronal antioxidant defense and neuroplasticity and promotes functional recovery after intracerebral hemorrhage. J Neurosci. (2022) 42:7001–14. doi: 10.1523/JNEUROSCI.2222-21.2022
180. Amulic B, Cazalet C, Hayes GL, Metzler KD, and Zychlinsky A. Neutrophil function: from mechanisms to disease. Annu Rev Immunol. (2012) 30:459–89. doi: 10.1146/annurev-immunol-020711-074942
181. Moxon-Emre I and Schlichter LC. Neutrophil depletion reduces blood-brain barrier breakdown, axon injury, and inflammation after intracerebral hemorrhage. J Neuropathol Exp Neurol. (2011) 70:218–35. doi: 10.1097/NEN.0b013e31820d94a5
182. Shtaya A, Bridges LR, Williams R, Trippier S, Zhang L, Pereira AC, et al. Innate immune anti-inflammatory response in human spontaneous intracerebral hemorrhage. Stroke. (2021) 52:3613–23. doi: 10.1161/STROKEAHA.121.034673
183. Liu J, Li N, Zhu Z, Kiang KM, Ng ACK, Dong CM, et al. Vitamin D enhances hematoma clearance and neurologic recovery in intracerebral hemorrhage. Stroke. (2022) 53:2058–68. doi: 10.1161/STROKEAHA.121.037769
184. Ye F, Yang J, Holste KG, Koduri S, Hua Y, Keep RF, et al. Characteristics of activation of monocyte-derived macrophages versus microglia after mouse experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. (2023) 43:1475–89. doi: 10.1177/0271678X231173187
185. Yang P, Manaenko A, Xu F, Miao L, Wang G, Hu X, et al. Role of PDGF-D and PDGFR-β in neuroinflammation in experimental ICH mice model. Exp Neurol. (2016) 283:157–64. doi: 10.1016/j.expneurol.2016.06.010
186. Li Z, Li M, Shi SX, Yao N, Cheng X, Guo A, et al. Brain transforms natural killer cells that exacerbate brain edema after intracerebral hemorrhage. J Exp Med. (2020) 217:e20200213. doi: 10.1084/jem.20200213
187. Gao L, Lu Q, Huang LJ, Ruan LH, Yang JJ, Huang WL, et al. Transplanted neural stem cells modulate regulatory T, γδ T cells and corresponding cytokines after intracerebral hemorrhage in rats. Int J Mol Sci. (2014) 15:4431–41. doi: 10.3390/ijms15034431
188. Yang Z, Yu A, Liu Y, Shen H, Lin C, Lin L, et al. Regulatory T cells inhibit microglia activation and protect against inflammatory injury in intracerebral hemorrhage. Int Immunopharmacol. (2014) 22:522–5. doi: 10.1016/j.intimp.2014.06.037
189. He Y, Gao Y, Zhang Q, Zhou G, Cao F, and Yao S. IL-4 switches microglia/macrophage M1/M2 polarization and alleviates neurological damage by modulating the JAK1/STAT6 pathway following ICH. Neuroscience. (2020) 437:161–71. doi: 10.1016/j.neuroscience.2020.03.008
190. Zhang P, Gao C, Guo Q, Yang D, Zhang G, Lu H, et al. Single-cell RNA sequencing reveals the evolution of the immune landscape during perihematomal edema progression after intracerebral hemorrhage. J Neuroinflammation. (2024) 21:140. doi: 10.1186/s12974-024-03113-8
191. Tang J, Yue J, Tao Y, Zhao G, Yi X, Zhang M, et al. Neutrophil extracellular traps induce brain edema around intracerebral hematoma via ERK-mediated regulation of MMP9 and AQP4. Transl Stroke Res. (2024) 16(5):1461–73. doi: 10.1007/s12975-024-01318-w
192. Rangaraju S, Dammer EB, Raza SA, Rathakrishnan P, Xiao H, Gao T, et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in alzheimer’s disease. Mol Neurodegener. (2018) 13:24. doi: 10.1186/s13024-018-0254-8
193. Arcuri C, Mecca C, Bianchi R, Giambanco I, and Donato R. The pathophysiological role of microglia in dynamic surveillance, phagocytosis and structural remodeling of the developing cns. Front Mol Neurosci. (2017) 10:191. doi: 10.3389/fnmol.2017.00191
194. Chen S, Peng J, Sherchan P, Ma Y, Xiang S, Yan F, et al. TREM2 activatio attenuates neuroinflammation and neuronal apoptosis via PI3K/akt pathway after intracerebral hemorrhage in mice. J Neuroinflamm. (2020) 17:168. doi: 10.1186/s12974-020-01853-x
195. Sun S, Lv W, Li S, Zhang Q, He W, Min Z, et al. Smart liposomal nanocarrier enhanced the treatment of ischemic stroke through neutrophil extracellular traps and cyclic guanosine monophosphate-adenosine monophosphate synthase-stimulator of interferon genes (cGAS-STING) pathway inhibition of ischemic penumbra. ACS Nano. (2023) 17:17845–57. doi: 10.1021/acsnano.3c03390
196. Shi Y, Zhang L, Pu H, Mao L, Hu X, Jiang X, et al. Rapid endothelial cytoskeletal reorganization enables early blood-brain barrier disruption and long-term ischaemic reperfusion brain injury. Nat Commun. (2016) 7:10523. doi: 10.1038/ncomms10523
197. Chen S, Pan J, Gong Z, Wu M, Zhang X, Chen H, et al. Hypochlorous acid derived from microglial myeloperoxidase could mediate high-mobility group box 1 release from neurons to amplify brain damage in cerebral ischemia-reperfusion injury. J Neuroinflammation. (2024) 21:70. doi: 10.1186/s12974-023-02991-8
198. Tan Q, Guo P, Zhou J, Zhang J, Zhang B, Lan C, et al. Targeting neutrophil extracellular traps enhanced tPA fibrinolysis for experimental intracerebral hemorrhage. Transl Res. (2019) 211:139–46. doi: 10.1016/j.trsl.2019.04.009
199. Cao Y, Shi M, Liu L, Zuo Y, Jia H, Min X, et al. Inhibition of neutrophil extracellular trap formation attenuates NLRP1-dependent neuronal pyroptosis via STING/IRE1α pathway after traumatic brain injury in mice. Front Immunol. (2023) 14:1125759. doi: 10.3389/fimmu.2023.1125759
200. Xie Y, Guo H, Wang L, Xu L, Zhang X, Yu L, et al. Human albumin attenuates excessive innate immunity via inhibition of microglial Mincle/Syk signaling in subarachnoid hemorrhage. Brain Behav Immun. (2017) 60:346–60. doi: 10.1016/j.bbi.2016.11.004
201. Wu X, Zeng H, Xu C, Chen H, Fan L, Zhou H, et al. TREM1 regulates neuroinflammatory injury by modulate proinflammatory subtype transition of microglia and formation of neutrophil extracellular traps via interaction with SYK in experimental subarachnoid hemorrhage. Front Immunol. (2021) 12:766178. doi: 10.3389/fimmu.2021.766178
202. Zhu Z, Jin L, Wang Q, Shi H, Cheng K, and Mao Z. Inhalable ce nanozyme-backpacked phage aims at ischemic cerebral injury by M1-microglia hitchhiking. Adv Mater. (2025) 15:e2419903. doi: 10.1002/adma.202419903
203. Perez-de-Puig I, Miró-Mur F, Ferrer-Ferrer M, Gelpi E, Pedragosa J, Justicia C, et al. Neutrophil recruitment to the brain in mouse and human ischemic stroke. Acta Neuropathol. (2015) 129:239–57. doi: 10.1007/s00401-014-1381-0
204. Hawez A, Al-Haidari A, Madhi R, Rahman M, and Thorlacius H. MiR-155 regulates PAD4-dependent formation of neutrophil extracellular traps. Front Immunol. (2019) 10:2462. doi: 10.3389/fimmu.2019.02462
205. Cherubini A, Ruggiero C, Morand C, Lattanzio F, Dell’aquila G, Zuliani G, et al. Dietary antioxidants as potential pharmacological agents for ischemic stroke. Curr Med Chem. (2008) 15:1236–48. doi: 10.2174/092986708784310431
206. Chen GC, Lu DB, Pang Z, and Liu QF. Vitamin C intake, circulating vitamin C and risk of stroke: a meta-analysis of prospective studies. J Am Heart Assoc. (2013) 2:e000329. doi: 10.1161/JAHA.113.000329
207. Kang L, Yu H, Yang X, Zhu Y, Bai X, Wang R, et al. Neutrophil extracellular traps released by neutrophils impair revascularization and vascular remodeling after stroke. Nat Commun. (2020) 11:2488. doi: 10.1038/s41467-020-16191-y
208. Ye S, Li S, Ma Y, Hu D, and Xiao F. Curcumin hinders PBDE-47-induced neutrophil extracellular traps release via Nrf2-associated ROS inhibition. Ecotoxicol Environ Saf. (2021) 225:112779. doi: 10.1016/j.ecoenv.2021.112779
209. Kraft P, Schwarz T, Göb E, Heydenreich N, Brede M, Meuth SG, et al. The phosphodiesterase-4 inhibitor rolipram protects from ischemic stroke in mice by reducing blood-brain-barrier damage, inflammation and thrombosis. Exp Neurol. (2013) 247:80–90. doi: 10.1016/j.expneurol.2013.03.026
210. Zeng H, Fu X, Cai J, Sun C, Yu M, Peng Y, et al. Neutrophil Extracellular Traps may be a Potential Target for Treating Early Brain Injury in Subarachnoid Hemorrhage. Transl Stroke Res. (2022) 13:112–31. doi: 10.1007/s12975-021-00909-1
211. Xu H, Li J, Wang Z, Feng M, Shen Y, Cao S, et al. Methylene blue attenuates neuroinflammation after subarachnoid hemorrhage in rats through the Akt/GSK-3β/MEF2D signaling pathway. Brain Behav Immun. (2017) 65:125–39. doi: 10.1016/j.bbi.2017.04.020
212. Qi D, Wei P, Cui Y, Lenahan C, Tao X, and Jin P. Inhibition of C3a/C3aR by SB290157 attenuates neuroinflammation via PKC/P38/NLRP3 signaling pathway after intracerebral hemorrhage. Neurocrit Care. (2025) 43:44–58. doi: 10.1007/s12028-025-02226-z
213. Qu X, Hou X, Zhu K, Chen W, Chen K, Sang X, et al. Neutrophil extracellular traps facilitate sympathetic hyperactivity by polarizing microglia toward M1 phenotype after traumatic brain injury. FASEB J. (2023) 37:e23112. doi: 10.1096/fj.202300752R
214. Hu K, Ye J, Fan P, Zheng R, Wang S, Peng Y, et al. Targeting and reprogramming microglial phagocytosis of neutrophils by ginsenoside Rg1 nanovesicles promotes stroke recovery. Bioact Mater. (2025) 47:181–97. doi: 10.1016/j.bioactmat.2025.01.017
215. Shi G, Liu L, Cao Y, Ma G, Zhu Y, Xu J, et al. Inhibition of neutrophil extracellular trap formation ameliorates neuroinflammation and neuronal apoptosis via STING-dependent IRE1α/ASK1/JNK signaling pathway in mice with traumatic brain injury. J Neuroinflammation. (2023) 20:222. doi: 10.1186/s12974-023-02903-w
216. Zenaro E, Pietronigro E, Della Bianca V, Piacentino G, Marongiu L, Budui S, et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat Med. (2015) 21:880–6. doi: 10.1038/nm.3913
217. Rodríguez C, Sobrino T, Agulla J, Bobo-Jiménez V, Ramos-Araque ME, Duarte JJ, et al. Neovascularization and functional recovery after intracerebral hemorrhage is conditioned by the Tp53 Arg72Pro single-nucleotide polymorphism. Cell Death Differ. (2017) 24:144–54. doi: 10.1038/cdd.2016.109
218. Yenari MA, Xu L, Tang XN, Qiao Y, and Giffard RG. Microglia potentiate damage to blood-brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke. (2006) 37:1087–93. doi: 10.1161/01.STR.0000206281.77178.ac
219. Wang Y, Tian M, Tan J, Pei X, Lu C, Xin Y, et al. Irisin ameliorates neuroinflammation and neuronal apoptosis through integrin αVβ5/AMPK signaling pathway after intracerebral hemorrhage in mice. J Neuroinflammation. (2022) 19:82. doi: 10.1186/s12974-022-02438-6
220. Katarzyna Greda A and Nowicka D. Hyaluronidase inhibition accelerates functional recovery from stroke in the mouse brain. J Neurochem. (2021) 157:781–801. doi: 10.1111/jnc.15279
221. Su Q, Su C, Zhang Y, Guo Y, Liu Y, Liu Y, et al. Adjudin protects blood-brain barrier integrity and attenuates neuroinflammation following intracerebral hemorrhage in mice. Int Immunopharmacol. (2024) 132:111962. doi: 10.1016/j.intimp.2024.111962
222. Huang Y, Zhang X, Zhang C, Xu W, Li W, Feng Z, et al. Edaravone dexborneol downregulates neutrophil extracellular trap expression and ameliorates blood-brain barrier permeability in acute ischemic stroke. Mediators Inflamm. (2022) 2022:3855698. doi: 10.1155/2022/3855698
223. Li SH, Huang QH, Yang QQ, Huang Q, Wang DX, Yang J, et al. The shared mechanism of barrier dysfunction in ulcerative colitis and Alzheimer’s disease: DDIT4/IL1β neutrophil extracellular traps drive macrophages-mediated phagocytosis. Int Immunopharmacol. (2025) 149:114188. doi: 10.1016/j.intimp.2025.114188
224. Gu L, Ye L, Chen Y, Deng C, Zhang X, Chang J, et al. Integrating network pharmacology and transcriptomic omics reveals that akebia saponin D attenuates neutrophil extracellular traps-induced neuroinflammation via NTSR1/PKAc/PAD4 pathway after intracerebral hemorrhage. FASEB J. (2024) 38:e23394. doi: 10.1096/fj.202301815R
225. Li C, Xing Y, Zhang Y, Hua Y, Hu J, and Bai Y. Neutrophil extracellular traps exacerbate ischemic brain damage. Mol Neurobiol. (2022) 59:643–56. doi: 10.1007/s12035-021-02635-z
226. Chen SH, Scott XO, Ferrer Marcelo Y, Almeida VW, Blackwelder PL, Yavagal DR, et al. Netosis and inflammasomes in large vessel occlusion thrombi. Front Pharmacol. (2021) 11:607287. doi: 10.3389/fphar.2020.607287
227. Shi Y, Mao H, Miao W, Deng J, Gao Q, Zeng S, et al. Potential association of neutrophil extracellular traps with cognitive impairment in cerebral small vessel disease. J Gerontol A Biol Sci Med Sci. (2023) 78:1999–2006. doi: 10.1093/gerona/glad184
228. Pietronigro EC, Della Bianca V, Zenaro E, and Constantin G. NETosis in alzheimer’s disease. Front Immunol. (2017) 8:211. doi: 10.3389/fimmu.2017.00211
229. Smyth LCD, Murray HC, Hill M, van Leeuwen E, Highet B, Magon NJ, et al. Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol Commun. (2022) 10:38. doi: 10.1186/s40478-022-01347-2
230. Neumann J, Sauerzweig S, Rönicke R, Gunzer F, Dinkel K, Ullrich O, et al. Microglia cells protect neurons by direct engulfment of invading neutrophil granulocytes: a new mechanism of CNS immune privilege. J Neurosci. (2008) 28:5965–75. doi: 10.1523/JNEUROSCI.0060-08.2008
231. Bosch ME, Dodiya HB, Michalkiewicz J, Lee C, Shaik SM, Weigle IQ, et al. Sodium oligomannate alters gut microbiota, reduces cerebral amyloidosis and reactive microglia in a sex-specific manner. Mol Neurodegener. (2024) 19:18. doi: 10.1186/s13024-023-00700-w
232. Shen J, Guo H, Liu S, Jin W, Zhang ZW, Zhang Y, et al. Aberrant branched-chain amino acid accumulation along the microbiota-gut-brain axis: Crucial targets affecting the occurrence and treatment of ischaemic stroke. Br J Pharmacol. (2023) 180:347–68. doi: 10.1111/bph.15965
233. Rohrbach AS, Slade DJ, Thompson PR, and Mowen KA. Activation of PAD4 in NET formation. Front Immunol. (2012) 3:360. doi: 10.3389/fimmu.2012.00360
234. Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun. (2019) 10:1322. doi: 10.1038/s41467-019-09160-7
235. Huang J, Agus DB, Winfree CJ, Kiss S, Mack WJ, McTaggart RA, et al. Dehydroascorbic acid, a blood-brain barrier transportable form of vitamin C, mediates potent cerebroprotection in experimental stroke. Proc Natl Acad Sci U S A. (2001) 98:11720–4. doi: 10.1073/pnas.171325998
236. Patel S, Kumar S, Jyoti A, Srinag BS, Keshari RS, Saluja R, et al. Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide. (2010) 22:226–34. doi: 10.1016/j.niox.2010.01.001
237. Zhao Z, Pan Z, Zhang S, Ma G, Zhang W, Song J, et al. Neutrophil extracellular traps: A novel target for the treatment of stroke. Pharmacol Ther. (2023) 241:108328. doi: 10.1016/j.pharmthera.2022.108328
238. Hallberg LAE, Barlous K, and Hawkins CL. Antioxidant strategies to modulate NETosis and the release of neutrophil extracellular traps during chronic inflammation. Antioxidants (Basel). (2023) 12:478. doi: 10.3390/antiox12020478
239. Grasso S, Neumann A, Lang IM, Etscheid M, von Köckritz-Blickwede M, and Kanse SM. Interaction of factor VII activating protease (FSAP) with neutrophil extracellular traps (NETs). Thromb Res. (2018) 161:36–42. doi: 10.1016/j.thromres.2017.11.012
240. von Köckritz-Blickwede M, Chow OA, and Nizet V. Fetal calf serum contains heat-stable nucleases that degrade neutrophil extracellular traps. Blood. (2009) 114:5245–6. doi: 10.1182/blood-2009-08-240713
241. Porter JC, Inshaw J, Solis VJ, Denneny E, Evans R, Temkin MI, et al. Anti-inflammatory therapy with nebulized dornase alfa for severe COVID-19 pneumonia: a randomized unblinded trial. Elife. (2024) 12:RP87030. doi: 10.7554/eLife.87030
242. Davis JC Jr, Manzi S, Yarboro C, Rairie J, Mcinnes I, Averthelyi D, et al. Recombinant human Dnase I (rhDNase) in patients with lupus nephritis. Lupus. (1999) 8:68–76. doi: 10.1191/096120399678847380
243. Seol SI, Oh SA, Davaanyam D, and Lee JK. Blocking peptidyl arginine deiminase 4 confers neuroprotective effect in the post-ischemic brain through both NETosis-dependent and -independent mechanisms. Acta Neuropathol Commun. (2025) 13:33. doi: 10.1186/s40478-025-01951-y
244. He W, Xi Q, Cui H, Zhang P, Huang R, Wang T, et al. Forsythiaside B ameliorates coagulopathies in a rat model of sepsis through inhibition of the formation of PAD4-dependent neutrophil extracellular traps. Front Pharmacol. (2022) 13:1022985. doi: 10.3389/fphar.2022.1022985
245. Li B, Xu L, Wang Z, Shi Q, Cui Y, Fan W, et al. Neutrophil extracellular traps regulate surgical brain injury by activating the cGAS-STING pathway. Cell Mol Neurobiol. (2024) 44:36. doi: 10.1007/s10571-024-01470-9
246. Kangisser L, Tan E, Bellomo R, Deane AM, and Plummer MP. Neuroprotective properties of vitamin C: A scoping review of pre-clinical and clinical studies. J Neurotrauma. (2021) 38:2194–205. doi: 10.1089/neu.2020.7443
247. Kurl S, Tuomainen TP, Laukkanen JA, Nyyssönen K, Lakka T, Sivenius J, et al. Plasma vitamin C modifies the association between hypertension and risk of stroke. Stroke. (2002) 33:1568–73. doi: 10.1161/01.str.0000017220.78722.d7
248. Gray RD, Lucas CD, MacKellar A, Li F, Hiersemenzel K, Haslett C, et al. Activation of conventional protein kinase C (PKC) is critical in the generation of human neutrophil extracellular traps. J Inflammation (Lond). (2013) 10:12. doi: 10.1186/1476-9255-10-12
249. Vorobjeva N, Dagil Y, Pashenkov M, Pinegin B, and Chernyak B. Protein kinase C isoforms mediate the formation of neutrophil extracellular traps. Int Immunopharmacol. (2023) 114:109448. doi: 10.1016/j.intimp.2022.109448
250. Kim DE, Suh YS, Lee MS, Kim KY, Lee JH, Lee HS, et al. Vascular NAD(P)H oxidase triggers delayed cerebral vasospasm after subarachnoid hemorrhage in rats. Stroke. (2002) 33:2687–91. doi: 10.1161/01.str.0000033071.99143.9e
251. Shin HK, Lee JH, Kim KY, Kim CD, Lee WS, Rhim BY, et al. Impairment of autoregulatory vasodilation by NAD(P)H oxidase-dependent superoxide generation during acute stage of subarachnoid hemorrhage in rat pial artery. J Cereb Blood Flow Metab. (2002) 22:869–77. doi: 10.1097/00004647-200207000-00012
252. Nadesalingam A, Chen JHK, Farahvash A, and Khan MA. Hypertonic saline suppresses NADPH oxidase-dependent neutrophil extracellular trap formation and promotes apoptosis. Front Immunol. (2018) 9:359. doi: 10.3389/fimmu.2018.00359
253. Whiteley WN, Slot KB, Fernandes P, Sandercock P, and Wardlaw J. Risk factors for intracranial hemorrhage in acute ischemic stroke patients treated with recombinant tissue plasminogen activator: a systematic review and meta-analysis of 55 studies. Stroke. (2012) 43:2904–9. doi: 10.1161/STROKEAHA.112.665331
254. Di G, Vázquez-Reyes S, Díaz B, Peña-Martinez C, García-Culebras A, Cuartero MI, et al. Daytime DNase-I administration protects mice from ischemic stroke without inducing bleeding or tPA-induced hemorrhagic transformation, even with aspirin pretreatment. Stroke. (2025) 56:527–32. doi: 10.1161/STROKEAHA.124.049961
255. Mendes LP, Rostamizadeh K, Gollomp K, Myerson JW, Marcos-Contreras OA, Zamora M, et al. Monoclonal antibody 2C5 specifically targets neutrophil extracellular traps. MAbs. (2020) 12:1850394. doi: 10.1080/19420862.2020.1850394
256. Filipczak N, Li X, Saawant GR, Yalamarty SSK, Luther E, and Torchilin VP. Antibody-modified DNase I micelles specifically recognize the neutrophil extracellular traps (NETs) and promote their degradation. J Control Release. (2023) 354:109–19. doi: 10.1016/j.jconrel.2022.12.062
257. Hao X, Zeng Z, Liang L, Feng Z, Li W, Xiong B, et al. The role of neutrophil extracellular traps in early microthrombosis and brain injury after subarachnoid hemorrhage in mice. Transl Stroke Res. (2023) 14:752–65. doi: 10.1007/s12975-022-01074-9
258. Mi L, Min X, Shi M, Liu L, Zhang Y, Zhu Y, et al. Neutrophil extracellular traps aggravate neuronal endoplasmic reticulum stress and apoptosis via TLR9 after traumatic brain injury. Cell Death Dis. (2023) 14(6):374. doi: 10.1038/s41419-023-05898-7
259. Yang C and Montgomery M. Dornase alfa for cystic fibrosis. Cochrane Database Syst Rev. (2021) 3:CD001127. doi: 10.1002/14651858.CD001127.pub5
260. Papayannopoulos V. Actin powers the neutrophil traps. Blood. (2022) 139:3104–5. doi: 10.1182/blood.2022015562
261. Sprenkeler EGG, Tool ATJ, Henriet SSV, van Bruggen R, and Kuijpers TW. Formation of neutrophil extracellular traps requires actin cytoskeleton rearrangements. Blood. (2022) 139:3166–80. doi: 10.1182/blood.2021013565
262. Mannherz HG, Budde H, Jarkas M, Hassoun R, Malek-Chudzik N, Mazur AJ, et al. Reorganization of the actin cytoskeleton during the formation of neutrophil extracellular traps (NETs). Eur J Cell Biol. (2024) 103:151407. doi: 10.1016/j.ejcb.2024.151407
263. Schneider AE, Sándor N, Kárpáti É, and Józsi M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol. (2016) 72:37–48. doi: 10.1016/j.molimm.2016.02.011
264. Zhang Z, Luo Z, Bi A, Yang W, An W, Dong X, et al. Compound edaravone alleviates lipopolysaccharide (LPS)-induced acute lung injury in mice. Eur J Pharmacol. (2017) 811:1–11. doi: 10.1016/j.ejphar.2017.05.047
265. Xu HL, Pelligrino DA, Paisansathan C, and Testai FD. Protective role of fingolimod (FTY720) in rats subjected to subarachnoid hemorrhage. J Neuroinflammation. (2015) 12:16. doi: 10.1186/s12974-015-0234-7
266. Wu J, Yang S, Hua Y, Liu W, Keep RF, and Xi G. Minocycline attenuates brain edema, brain atrophy and neurological deficits after intracerebral hemorrhage. Acta Neurochir Suppl. (2010) 106:147–50. doi: 10.1007/978-3-211-98811-4_26
267. Han D, Liu H, Gao Y, and Feng J. Targeting brain-spleen crosstalk after stroke: New insights into Stroke Pathology and Treatment. Curr Neuropharmacol. (2021) 19:1590–605. doi: 10.2174/1570159X19666210316092225
268. Li X and Chen G. CNS-peripheral immune interactions in hemorrhagic stroke. J Cereb Blood Flow Metab. (2023) 43:185–97. doi: 10.1177/0271678X221145089
269. Keep RF, Andjelkovic AV, Xiang J, Stamatovic SM, Antonetti DA, Hua Y, et al. Brain endothelial cell junctions after cerebral hemorrhage: Changes, mechanisms and therapeutic targets. J Cereb Blood Flow Metab. (2018) 38:1255–75. doi: 10.1177/0271678X18774666
270. Jia P, Peng Q, Fan X, Zhang Y, Xu H, Li J, et al. Immune-mediated disruption of the blood-brain barrier after intracerebral hemorrhage: Insights and potential therapeutic targets. CNS Neurosci Ther. (2024) 30:e14853. doi: 10.1111/cns.14853
271. Ohashi SN, DeLong JH, Kozberg MG, Mazur-Hart DJ, van Veluw SJ, Alkayed NJ, et al. Role of inflammatory processes in hemorrhagic stroke. Stroke. (2023) 54:605–19. doi: 10.1161/STROKEAHA.122.037155
Keywords: intracerebral hemorrhage, neutrophil extracellular traps, microglia, central and peripheral immune systems, mechanism analysis
Citation: Zhang S, Jin Z, Jiang L, Zhang Y, Wu T, Xu P, Cui Y, Zhang D and Lu J (2025) Unveiling the inflammatory messengers after intracerebral hemorrhage: the crosstalk between peripheral NETs and microglia. Front. Immunol. 16:1643524. doi: 10.3389/fimmu.2025.1643524
Received: 09 June 2025; Accepted: 26 August 2025;
Published: 17 September 2025.
Edited by:
Ma. Cecilia Opazo, Universidad de Las Américas, ChileReviewed by:
Wen-Jun Tu, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaHeling Chu, Shanghai Jiao Tong University, China
Huaqiu Zhang, Huazhong University of Science and Technology, China
Copyright © 2025 Zhang, Jin, Jiang, Zhang, Wu, Xu, Cui, Zhang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jing Lu, amluZ2x1MTk4OTA0QDE2My5jb20=; Dongmei Zhang, MTAzNjg3ODkyMUBxcS5jb20=
†These authors have contributed equally to this work