 Dorothy D. Yang
Dorothy D. Yang William Macmorland
William Macmorland James N. Arnold
James N. Arnold- School of Cancer and Pharmaceutical Sciences, King’s College London, London, United Kingdom
Chimeric antigen receptor (CAR) T-cell therapy is a transformative immunotherapeutic approach, yet its application in solid tumors is hindered by the immunosuppressive tumor microenvironment (TME). The TME restricts T-cell trafficking, impairs effector functions, and promotes exhaustion through soluble factors, metabolic stress, and suppressive cell populations. Recent efforts to enhance CAR T-cell efficacy have focused on armoring strategies that ‘reprogram’ and ‘boost’ T-cell responses within the TME. These include engineered expression of dominant-negative receptors or cytokine-releasing constructs (such as IL-12 and IL-18) to reshape the local immune milieu and improve T-cell effector function, synthetic Notch receptors for inducible gene expression, and chemokine receptor knock-ins to improve tumor infiltration. Additional approaches aim to modulate intrinsic metabolic pathways to improve CAR T-cell persistence under hypoxic or nutrient-deprived conditions. Armoring strategies that recruit bystander or endogenous immune cells also activate broader anti-tumor immunity that prevents antigen escape and may induce more durable anti-tumor responses. This review highlights the molecular and cellular mechanisms by which current armoring strategies enhance CAR T-cell functions in solid tumors, offering a perspective on improving immune cell engineering for overcoming the hurdles encountered in deploying these therapies against solid cancers.
Introduction
Chimeric antigen receptor (CAR) T-cell therapy has transformed the treatment of hematological malignancies, with seven CAR T-cell therapies approved by the US Food and Drug Administration for indications including relapsed or refractory B-cell lymphomas and multiple myeloma (1–3). This technology provides a method by which a patient’s T-cells are genetically engineered to express CARs that recognize tumor-associated antigens (TAAs). Upon antigen engagement, CARs trigger T-cell activation and cytotoxicity, leading to the targeted elimination of malignant cells (4, 5). In hematological cancers, CAR T-cells have demonstrated potent immune responses and potential for achieving long-term disease remission (2). However, CAR T-cell therapy has not yet achieved comparable clinical success in solid tumors. The immunosuppressive tumor microenvironment (TME), tumor antigen heterogeneity, and limited T-cell infiltration pose major barriers to efficacy (1, 4, 6, 7). Continued innovation in CAR design and function is therefore essential to overcome these challenges and expand the therapeutic impact of CAR T-cell therapies to solid malignancies.
The basic structure of a CAR comprises an extracellular antigen-binding domain that recognizes TAAs, a transmembrane domain, and an intracellular activation signaling domain. The extracellular domain most commonly consists of a single-chain variable fragment (scFv) that binds to the target antigen (1). The intracellular domain, derived from CD3ζ, delivers a pseudo-T-cell receptor (TCR) activation signal upon CAR engagement, initiating downstream killing pathways (4, 5).
Innovative generations of CAR designs have been developed to enhance the functionality and clinical benefits observed from the original design (Figure 1). Second generation CARs include a co-stimulatory domain (CsD), consisting most commonly of either 4-1BB or CD28, which has been demonstrated to improve CAR T-cell persistence, cytokine production, and potency (4, 8). Third generation CARs combine two co-stimulatory domains for a more potent response (4, 9). Subsequent generations have focused on further augmenting effector functions or persistence, for instance by co-expressing a biological payload such as a cytokine or chemokine alongside the CAR (4, 9, 10). These innovations are the focus of this review.
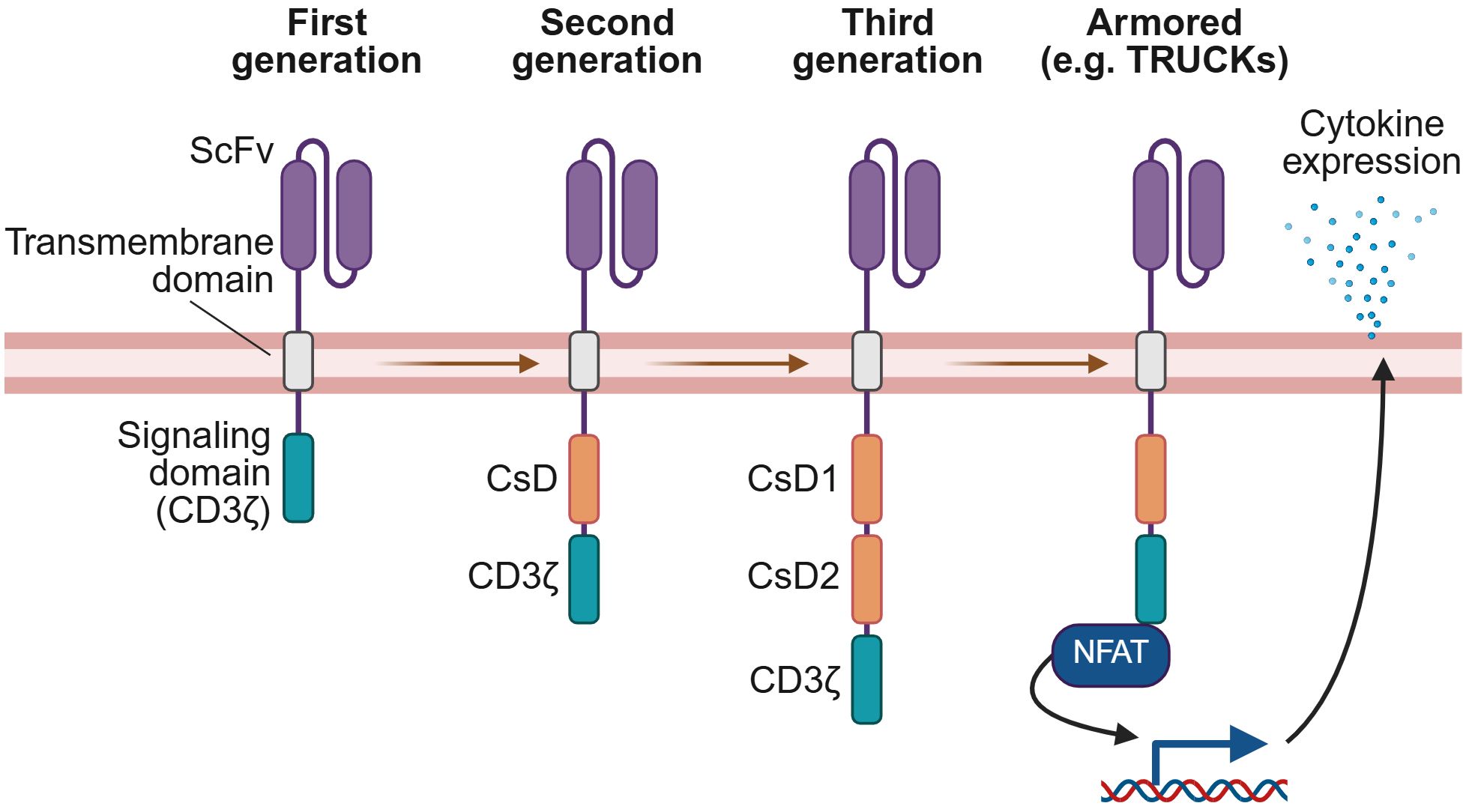
Figure 1. Generations of CAR technology. CARs have evolved from the original design of the first generation, with each subsequent generation incorporating new features to enhance CAR-elicited T-cell effector functions. First generation CARs comprise an extracellular domain with an antigen-specific scFv, a transmembrane domain, and an intracellular signaling domain derived from CD3ζ. Second generation CARs add a co-stimulatory domain (CsD), most commonly 4-1BB or CD28, while third generation CARs include two co-stimulatory domains. Armored CAR T-cells introduce co-expression of a biological payload that augments effector functions or persistence. For example, as illustrated here, TRUCKs are a type of armored CAR T-cell co-expressing a pro-inflammatory cytokine payload whose expression may be regulated by an NFAT-responsive promoter, linking release of the cytokine to CAR signaling. This figure was created using BioRender.com.
The primary distinction between solid and hematological cancers lies in the presence of a TME which can limit T-cell infiltration and functionality (Figure 2). Consequently, CAR T-cells face numerous obstacles that hinder successful clinical outcomes (6, 10, 11). One major challenge is the inefficient homing of T-cells to the tumor, which can result from reduced expression of chemokine ligands for effector T-cells, including CXCL9, CXCL10, and CXCL11, in the TME, impairing T-cell infiltration (12–14). Additionally, the movement of T-cells from blood vessels to the tumor site is hindered by the downregulation of extravasation mediators, including vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) (15–17). T-cells that successfully cross these barriers into the TME are then confronted by a collagen-rich stromal network formed by cancer-associated fibroblasts (CAFs), which can obstruct T-cell access to tumor cells (18). CAFs, along with immune cells such as regulatory T-cells (Tregs), myeloid-derived suppressor cells (MDSCs), and tumor-associated macrophages (TAMs), contribute further to the hostile environment by releasing immunosuppressive cytokines including interleukin (IL)-4, IL-10, and transforming growth factor (TGF)-β, which reduce T-cell function (6, 19). Upregulated expression of immune checkpoint proteins, such as PD-L1 and CTLA-4, on tumor cells and immune cells within the TME also activate immune-inhibitory signaling axes which curtail CAR T-cell activity and induce functional exhaustion (6, 19). Lastly, TAA heterogeneity, arising from genomic instability and clonal evolution, can represent a mechanism of antigen escape, as tumor cells alter antigen expression to evade immune recognition (20). This antigen variability further diminishes the anti-tumor efficacy of CAR T-cells, as not all tumor cells can be targeted and eliminated. Therefore, it is evident that to achieve clinical success in the treatment of solid cancers, CAR T-cells will need to be engineered to be capable of efficiently bypassing all of these obstacles within the solid TME.
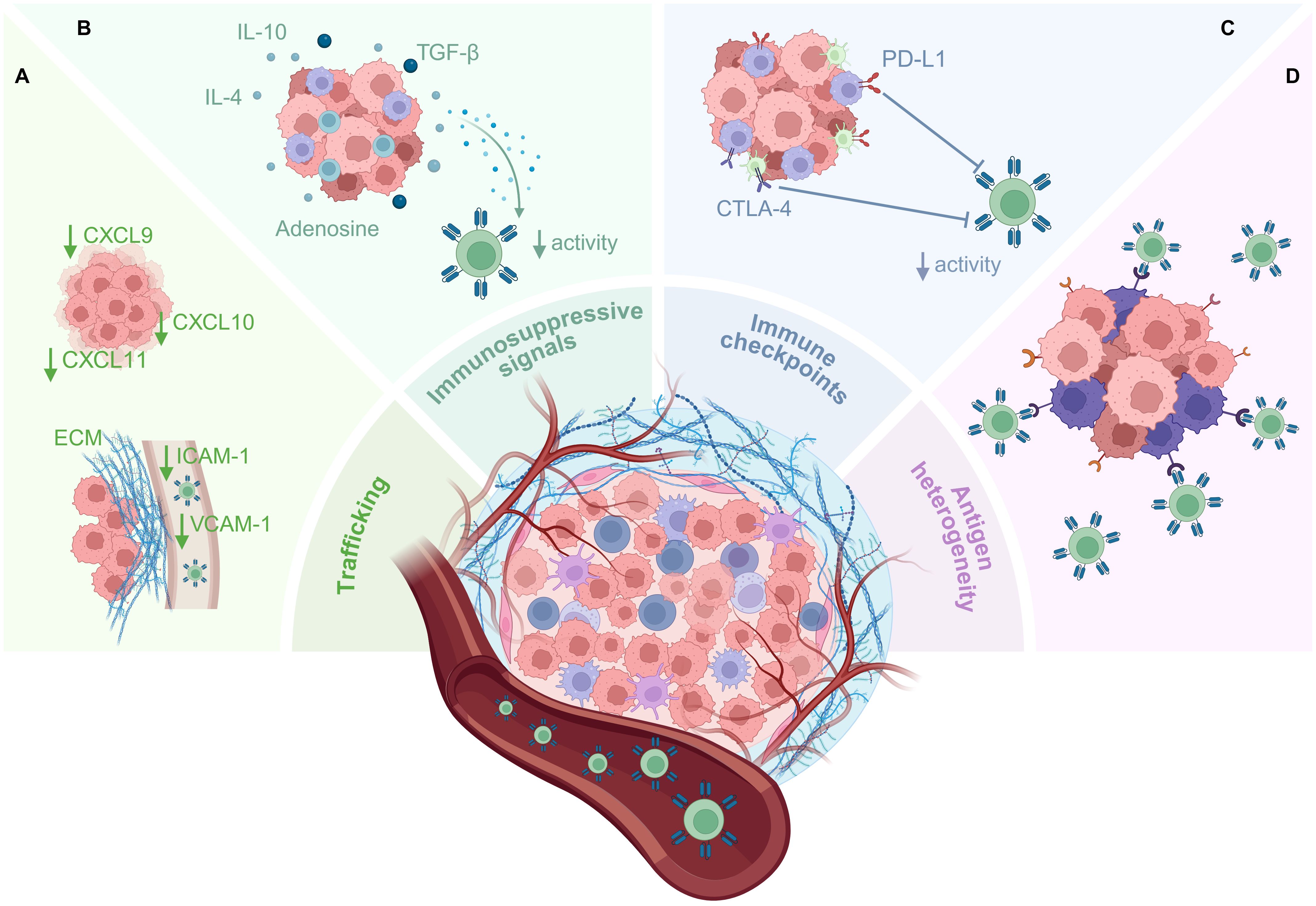
Figure 2. Barriers to CAR T-cell therapy in the solid TME. The solid TME poses multiple barriers to CAR T-cell infiltration and functionality which significantly impact anti-tumor efficacy. (A) Downregulation of chemokine ligands by the tumor can lead to inefficient trafficking of CAR T-cells to tumor sites, and downregulation of adhesion molecules such as VCAM-1 and ICAM-1 limits the migration of CAR T-cells from blood vessels into the tumor. CAR T-cells that do arrive at the tumor site are confronted by the stroma and ECM that surround solid tumors, limiting their infiltration into the tumor. (B) Immune and stromal cells within the TME, including CAFs, Tregs, MDSCs, and TAMs, can release immunosuppressive cytokines which reduce the function of CAR T-cells. Other immunosuppressive molecules, such as adenosine, also accumulate within the metabolically stressful milieu of the TME. (C) Inhibitory immune checkpoint molecules expressed by tumor and immune cells can activate immunosuppressive signaling axes within the CAR T-cells, inducing exhaustion/suppressing effector functions. (D) Target antigen heterogeneity allows some tumor cells to evade CAR T-cell killing, resulting in antigen escape and lack of tumor control. This figure was created using BioRender.com.
One strategy to overcome the barriers of the solid TME is to ‘armor’ CAR T-cells, typically by co-expressing a biological payload alongside the CAR that counteracts the immunosuppressive features of the TME (10, 11). A wide range of armoring approaches have been developed, many with pleiotropic effects that could fall into multiple functional categories. For the purposes of this review, we define armoring as the incorporation of a transgenic payload into CAR T-cells to enhance their anti-tumor activity. We broadly categorize armoring strategies into those that: 1) exploit cytokine signaling, 2) neutralize immune-inhibitory signals in the TME, 3) modulate metabolic pathways, 4) address antigen heterogeneity or antigen escape, and 5) improve CAR T-cell homing to tumors. As a comprehensive review of all armoring approaches is beyond the scope of this article, we particularly focus on recent or novel advances that have shown promising preclinical outcomes or have progressed to clinical evaluation (Table 1, Supplementary Table S1).
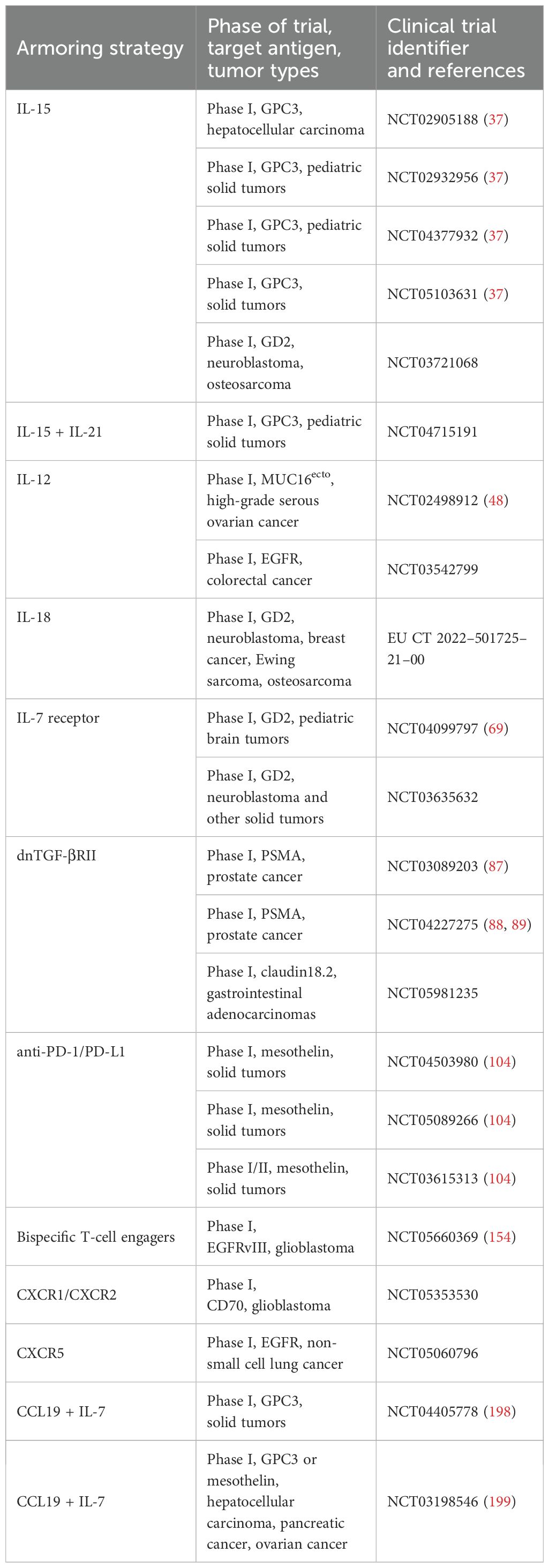
Table 1. Examples of armored CAR T-cells in clinical trials.
Exploiting cytokine signaling to enhance CAR T-cell potency and rewire the TME
T-cells Redirected for Universal Cytokine Killing (TRUCKs) represent some of the earliest examples of armored CAR T-cells (21–23). This term typically refers to CAR T-cells engineered to secrete pro-inflammatory cytokines that enhance anti-tumor activity by promoting CAR T-cell survival and effector functions, or by modulating the immunosuppressive TME (11, 24). More recently, novel armoring strategies have expanded beyond this classical definition, exploring alternative methods of inducing cytokine signaling within CAR T-cells, or incorporating cytokines that provide functional benefits unrelated to traditional inflammatory pathways (Figure 3).
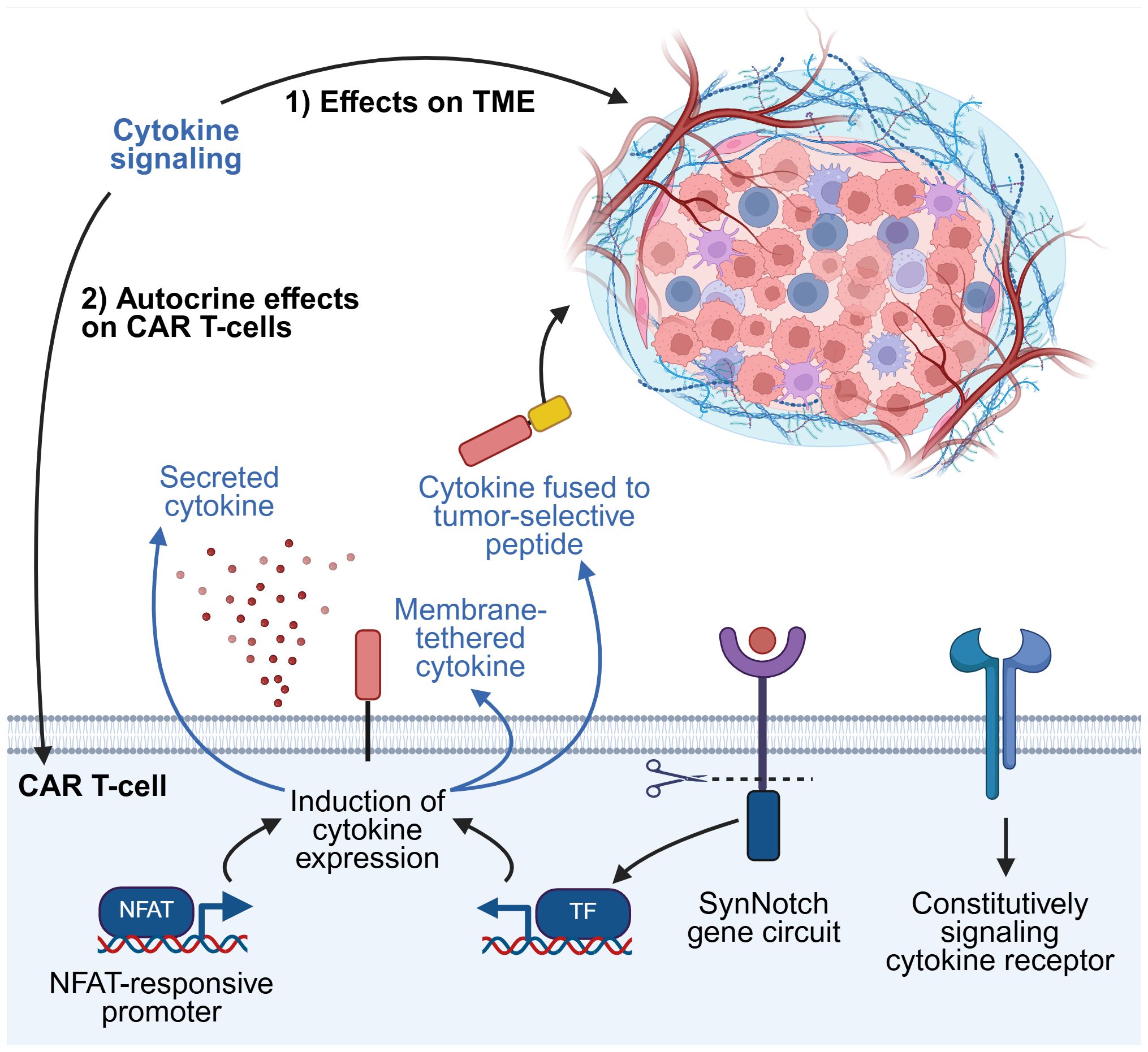
Figure 3. Strategies for exploiting cytokine signaling. Harnessing cytokine signaling pathways can induce beneficial effects by modulating the TME, the CAR T-cells themselves, or both. Cytokines can be secreted as soluble factors from CAR T-cells into the TME. Selected examples of other methods of regulating cytokine expression are illustrated here, including cytokines tethered to the CAR T-cell membrane or fused to tumor-selective peptides, modified cytokine receptors that signal constitutively but do not respond to native extracellular cytokine, and synthetic gene circuits. TF, transcription factor. This figure was created using BioRender.com.
Common γ-chain cytokines
The common γ-chain (γc) family of cytokines comprise IL-2, -4, -7, -9, -15, and -21, which all signal through receptors containing a shared γc subunit, and play crucial roles in lymphocyte proliferation, differentiation, and homeostasis (25). As such, this family of cytokines has received significant attention as potential armoring payloads for CAR T-cells.
Among these, IL-15 has emerged as a particularly promising candidate. IL-15-armored CAR T-cells have been demonstrated to exhibit enhanced proliferation, improved anti-tumor efficacy, sustained killing upon repeated tumor challenges in vitro and in vivo, prolonged survival in tumor-bearing mice, and promotion of central memory or stem cell memory-like phenotypes (26–30). In one study using a syngeneic melanoma mouse model, IL-15-armored CAR T-cells not only improved intrinsic T-cell function but also beneficially remodeled the TME, enhancing natural killer (NK) cell activation and reducing immunosuppressive ‘M2’ macrophage abundance (29). Another study found that CAR T-cells modified to fuse IL-15 to the scFv portion of the CAR could deplete immunosuppressive MDSCs in murine glioblastoma models, based on the observation that MDSCs in these models as well as human glioblastoma samples express the α subunit of the IL-15 receptor. This MDSC depletion was associated with reduced tumor growth and improved survival of the mice (31).
Despite these encouraging findings, concerns have been raised about the clinical translation of IL-15 armoring due to toxicities observed in trials of recombinant human IL-15, as well as some studies of IL-15-armored CAR T-cells in mice, including cytokine release syndrome (CRS), thrombocytopenia, and liver toxicity (32–36). To address safety concerns, several groups have developed regulatory mechanisms to try to prevent these toxicities. For example, Chen et al. incorporated an inducible caspase-9 (iC9) suicide gene into their IL-15-armored CAR construct. They demonstrated that administration of a chemical inducer of dimerization (CID) drug, which causes the iC9 protein to dimerize and activate apoptosis, enabled rapid elimination of the CAR T-cells (26).
IL-15-armored CAR T-cells, including those with the iC9 suicide gene, have now been evaluated in phase I trials. In one study, 4 out of 12 patients with glypican-3 (GPC3)-positive solid tumors achieved a partial response following treatment with IL-15-armored GPC3-targeting CAR T-cells, whereas none of the patients treated with unarmored CAR T-cells experienced an objective response (37). Notably, IL-15-CAR T-cell treatment was associated with a higher incidence of CRS that necessitated intervention with IL-1 or IL-6 blockade. In three patients with refractory CRS, activation of the iC9 switch successfully reduced circulating CAR T-cells and alleviated symptoms. Interestingly, this trial also reported an increase in effector memory and a decrease in central memory T-cell subsets among IL-15-armored CAR T-cells (37), an observation that contrasts with prior preclinical findings.
Another approach to circumvent the toxicities associated with constitutive IL-15 expression is to place IL-15 expression downstream of antigen-dependent T-cell activation. Ma et al. engineered CAR T-cells with IL-15 expression controlled by the interferon (IFN)-γ gene promoter, which activated upon TCR signaling. This antigen-dependent expression system led to selective IL-15 expression following tumor recognition and improved tumor control in a xenograft mouse model of gastric cancer (28).
The combination of two γc cytokines, IL-15 and IL-21, has also been tested in the preclinical setting, based on their known synergistic effects on cytotoxic lymphocytes (38, 39). CAR T-cells armored with both cytokines demonstrated superior anti-tumor activity in in vivo models compared to unarmored or single cytokine-armored CAR T-cells, and showed enhanced proliferation, increased frequencies of stem cell memory and central memory populations (40), and reduced exhaustion following repeated antigen stimulation (41).
IL-12 family of cytokines
The IL-12 family comprises heterodimeric cytokines with pleiotropic roles in immune regulation. Among these, IL-12 and IL-23, members of the family which are considered to have predominantly pro-inflammatory effects (42), have garnered particular interest as potential armoring payloads for CAR T-cells.
IL-12 consists of two subunits, a p35 and a p40, and is primarily produced by antigen-presenting cells (APCs) (42). It promotes IFN-γ secretion from T-cells and NK cells, which in turn can further stimulate IL-12 release by macrophages and other APCs (42, 43). As an armoring payload, IL-12 has been demonstrated to enhance CAR T-cell effector functions and reprogram endogenous immune cells in the TME toward a pro-inflammatory, anti-tumor state (23, 42, 44). Although early efforts to armor CAR T-cells with IL-12 showed improved anti-tumor activity, constitutive or systemic expression of IL-12 has been associated with significant toxicities (45–48). In a phase I trial of an IL-12-secreting CAR T-cell therapy, two-thirds of patients who also received lymphodepleting chemotherapy developed dose-limiting hemophagocytic lymphohistiocytosis or macrophage activation-like syndrome (48).
More recent efforts to develop IL-12-armored CAR T-cells have therefore also endeavored to improve their safety profile by restricting IL-12 expression to the TME. One solution has been to bind IL-12 to the membrane of CAR T-cells (49, 50), which Hu et al. combined with fusing the IL-12 to a peptide targeting cell surface vimentin, a protein overexpressed by various solid tumors (49). These studies demonstrated that, compared to unarmored CAR T-cells, membrane-bound IL-12-CAR T-cells released more IFN-γ, and demonstrated superior anti-tumor efficacy in murine models of solid tumors, including large established tumors, without inducing systemic toxicity. These effects were accompanied by changes in immune cell populations in the TME, including enhanced dendritic cell (DC) maturation (49, 50). Lee et al. confirmed these findings in a model of ovarian cancer peritoneal metastasis, where systemic IL-12 administration caused toxicity, but membrane-bound IL-12-armored CAR T-cells did not (50).
Hombach et al. inserted IL-12 into the extracellular domain of the CAR. This configuration modulated CAR T-cells toward an NK cell-like phenotype, enabling them to kill both antigen-positive and antigen-negative tumor cells. This was demonstrated both in vitro and in a mouse model of CEA-negative ovarian cancer, where IL-12-armored CAR T-cells were effective, while conventional unarmored CEA-targeted CAR T-cells were not (51).
Other methods of achieving tumor-restricted IL-12 expression include: fusing IL-12 to a collagen-binding domain to localize it to exposed collagen in tumor vasculature and stroma (52); fusing it to a tumor-specific scFv (53); and placing IL-12 expression under control of a nuclear factor of activated T-cells (NFAT)-responsive promoter to link cytokine secretion to CAR activation (54, 55).
IL-23 is a heterodimeric cytokine that shares the same p40 subunit as IL-12, but pairs this with a p19 subunit. Ma et al. discovered that due to upregulation of p19 expression in T-cells upon activation, transducing T-cells with the p40 subunit was sufficient to induce IL-23 secretion from T-cells. They demonstrated that IL-23-secreting CAR T-cells exhibited improved tumor killing and reduced functional exhaustion compared to unarmored CAR T-cells in syngeneic mouse models of solid tumors. Notably, IL-23 acted via autocrine signaling to enhance CAR T-cell function. When compared to other armoring strategies, IL-23-armored CAR T-cells did not induce weight loss in immunodeficient mice, suggesting a superior safety profile relative to IL-15 and IL-18-armoring (56).
In contrast, another study found that PSMA-targeted CAR T-cells engineered to co-express an IL-23-targeting monoclonal antibody (mAb) led to eradication of prostate cancer in a murine model, suggesting that IL-23 blockade, not supplementation, may be beneficial in some contexts (57). These findings illustrate that the benefits of armoring can be context-dependent, influenced by tumor type, the immune microenvironment, and the preclinical model being used. Moreover, IL-23 is a multifaceted cytokine with reported pro-tumoral and anti-tumor effects, which may reflect differences in cytokine concentration (56, 58). These observations underscore the importance of considering not only the CAR T-cell dose, but also the ‘dose’ of the armoring payload when evaluating therapeutic efficacy and safety, which are critical factors for clinical translation.
Other cytokines
Engineered IL-2 and IL-33
Although IL-2 is the prototypical T-cell growth factor and the first member of the γc family of cytokines to be discovered (25), its use as a payload has not gained significant traction due mainly to concerns about systemic toxicity. One group found that armoring CAR T-cells with superkine IL-2 (Super2), a variant of IL-2 that binds to the IL-2 receptor with higher affinity than the wild-type cytokine, and IL-33 significantly enhanced anti-tumor activity in vivo compared to armoring with either cytokine alone, without evidence of toxicity in the immunocompetent mouse models used (59). The rationale for armoring with IL-33 arose from studies that found that IL-33 activates intratumoral group 2 innate lymphoid cells (ILC2s) which can contribute to tissue-specific tumor immunity (59–61). The synergistic improvement in anti-tumor activity induced by Super2 and IL-33 armoring was independent of IFN-γ or perforin-mediated cytotoxicity. Instead, the combination appeared to function primarily by harnessing an endogenous immune response. This included increased tumor infiltration of endogenous T-cells and a shift toward pro-inflammatory ‘M1’ macrophage polarization, highlighting a potential strategy to remodel the TME and enhance CAR T-cell efficacy through immune orchestration rather than direct cytotoxic mechanisms (59).
IL-18
Like IL-12, IL-18 is a pro-inflammatory IFN-γ inducer, and has been found to have beneficial effects as an armoring payload. These include enhancing the proliferation and survival of CAR T-cells and modulating the TME to recruit and polarize endogenous immune cells to support the anti-tumor response (62–64). Jaspers et al. compared murine IL-18-armored, IL-12-armored and unarmored CAR T-cells in a syngeneic mouse metastatic model of small cell lung cancer. They found that only IL-18-armored CAR T-cell treatment induced tumor shrinkage, which was associated with improved survival. Moreover, phenotypic analysis of CAR T-cells post-injection into tumor-bearing mice revealed increased memory marker and reduced exhaustion marker expression by IL-18-secreting CAR T-cells compared to unarmored CAR T-cells (64).
As with other pro-inflammatory cytokine payloads, concerns about potential toxicities that may result from constitutive IL-18 signaling have led to efforts to regulate its expression (63, 65). Hull et al. demonstrated that constitutive expression of murine IL-18 by CAR T-cells induced lethal toxicity in mice, with postmortem blood analysis indicating CRS (65). To regulate IL-18 activity, they modified the cleavage site within pro-IL-18, the biologically inactive precursor, to one recognized by granzyme B, a cytotoxic protease released by T-cells. This design restricted IL-18 activation to the context of CAR T-cell activation and granzyme B release. They confirmed that this strategy enhanced CAR T-cell anti-tumor activity both in vitro and in vivo, without inducing significant toxicity in the same mouse model (65). Other strategies to link IL-18 expression to CAR T-cell activation have also been explored, including NFAT-responsive promoter systems, one of which is being evaluated in a first-in-human phase I trial against GD2-positive tumors (63, 66).
Lange et al. took a different approach by engineering a chimeric cytokine receptor consisting of the extracellular domain of the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor fused to the transmembrane and intracellular domains of the IL-18 receptor. Since GM-CSF is secreted by CAR T-cells upon antigen engagement, its binding to this chimeric receptor triggered activation-dependent IL-18 signaling within the CAR T-cells. This system was shown to induce tumor regression in two tumor xenograft models in which unarmored CAR T-cells were ineffective (67).
Modifying cytokine receptors
Apart from engineering CAR T-cells to secrete or express cytokines, another strategy to exploit cytokine signaling is to modify the cytokine receptors. This approach has been applied particularly to cytokines of the γc family and circumvents the off-target effects that can result from transgenic cytokine expression, which may activate signaling on bystander cells in addition to the CAR T-cells themselves. Shum et al. developed a constitutively signaling IL-7 receptor, termed C7R, which activated the IL-7 signaling axis in CAR T-cells but was unresponsive to extracellular IL-7. They demonstrated that C7R-expressing CAR T-cells were more resilient to repeated tumor challenges, and capable of controlling or clearing tumors in several in vivo models (68). A phase I clinical trial of C7R-CAR T-cells demonstrated tolerability, with mostly grade 1 inflammation-associated toxicities, apart from one case of grade 4 CRS, and reported partial responses in 2 out of 7 patients with diffuse midline glioma (69). Another approach to harnessing IL-7 signaling has been to incorporate a portion of the IL-7 receptor-α intracellular domain into the CAR structure itself, which was demonstrated to enhance T-cell proliferation and improve tumor control (70, 71).
An additional strategy to avoid off-target effects is the design of mutated, or orthogonal, cytokine and receptor pairs that interact exclusively with each other, and not with their native counterparts. This concept was first demonstrated using an orthogonal IL-2 and IL-2 receptor pair (72). Following on from this, Kalbasi et al. engineered T-cells to express a synthetic cytokine receptor ‘o9R’, which combines the extracellular domain of the orthogonal IL-2 receptor with the intracellular domain of the IL-9 receptor. Upon stimulation with orthogonal IL-2, o9R signaling induced features consistent with a stem cell memory phenotype in the T-cells, an attribute potentially beneficial for CAR T-cell therapy. CAR T-cells expressing o9R and receiving orthogonal IL-2 also exhibited greater anti-tumor activity in syngeneic mouse models compared to CAR T-cells expressing the orthogonal IL-2 receptor (73).
More recently, Bell et al. replaced the extracellular domains of several heterodimeric cytokine receptors, including those of γc cytokines, and IL-10 and IL-12 receptors, with heterodimerizing leucine zipper motifs. This modification led to constitutive signaling through these receptors. CAR T-cells expressing these leucine zipper-modified cytokine receptors showed improved cytotoxicity in response to multiple tumor challenges in vitro, and CAR T-cells with a modified IL-2 receptor demonstrated superior anti-tumor activity in vivo in mouse xenograft models of lung cancer and sarcoma (74).
Synthetic cytokine circuits
Designing synthetic circuits to restrict cytokine production specifically within the tumor is another strategy aimed at preventing systemic cytokine release and thereby reducing toxicity. Synthetic Notch (synNotch) receptors have been a pioneering example of synthetic gene circuits in the CAR field. These receptors consist of a synthetic antigen-recognizing extracellular domain, a transmembrane domain derived from the Notch signaling receptor, and a synthetic intracellular transcription factor. When the extracellular domain binds its cognate antigen, the synNotch receptor undergoes transmembrane cleavage, releasing the transcription factor to activate expression of chosen target genes placed downstream of a synthetic promoter (75). While the downstream payload can, in principle, be any gene of interest, synNotch-controlled cytokine release provides a compelling example of the utility of this system. Allen et al. demonstrated that CAR T-cells co-expressing a synNotch receptor designed to drive IL-2 production were able to eradicate solid tumors without inducing significant toxicity (76).
Combating immune-inhibitory signals in the TME
CAR T-cells face numerous immunosuppressive signals within the TME, which can lead to functional exhaustion and diminished effector activity, ultimately resulting in reduced or lost therapeutic efficacy. To address this, armoring CAR T-cells with payloads that block or counteract these immune-inhibitory signals represents a promising strategy to enhance their anti-tumor potential (Figure 4).
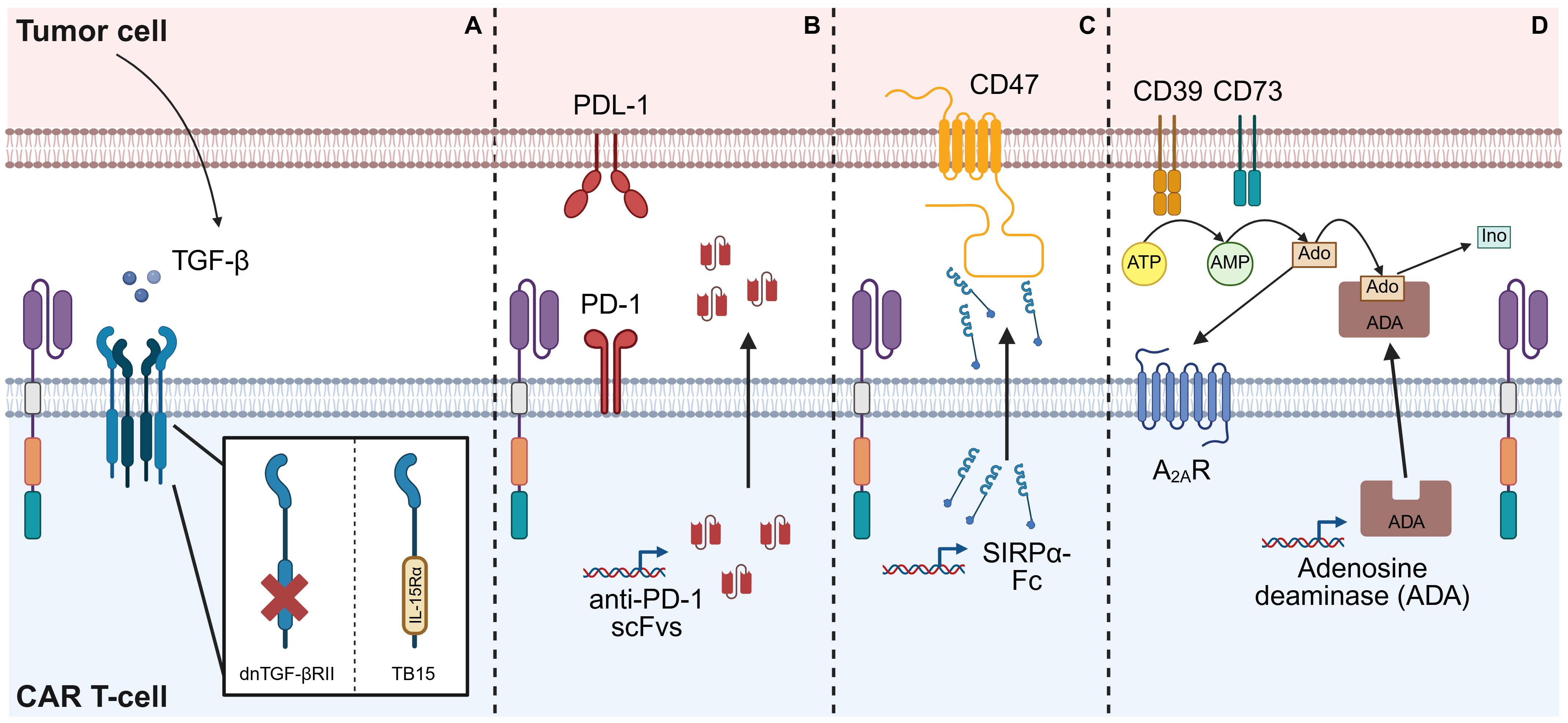
Figure 4. Strategies for combating immune-inhibitory signals in the TME. (A) TGF-β released within the TME can suppress CAR T-cell functions. Armoring a CAR T-cell with a ‘dominant-negative’ TGF-β receptor II (dnTGF-βRII), which features a truncated intracellular domain, prevents downstream signaling, thereby shielding the CAR T-cells from TGF-β-induced suppression. Alternatively, this intracellular domain can be replaced with that of a different receptor, such as IL-15 receptor-α, resulting in a chimeric receptor (TB15) which induces IL-15 signaling. (B) Binding of PD-L1 expressed on the surface of tumor cells to the PD-1 receptor on CAR T-cells suppresses CAR T-cell proliferation and effector functions. One method of preventing this is by engineering the CAR T-cells to secrete scFvs which neutralize PD-1 both on the CAR-T cells themselves and on adjacent cells, thereby blocking this inhibitory pathway. (C) CD47 expressed on the surface of tumor cells provide a ‘don’t eat me’ signal to APCs, protecting the tumor cells from phagocytosis. A strategy to overcome this is to engineer CAR T-cells to secrete molecules, such as a SIRPα-Fc fusion protein, that block CD47. (D) Adenosine (Ado) produced by breakdown of excess ATP within the TME can limit CAR T-cell functions by signaling through A2AR. Armoring CAR T-cells to release the enzyme adenosine deaminase (ADA) results in the catabolism of adenosine to inosine (Ino), thereby reducing the immunosuppressive effect of adenosine. This figure was created using BioRender.com.
TGF-β
Although TGF-β is a cytokine with pleiotropic effects, it has drawn particular interest in the CAR T-cell field for its tumor-promoting and immune-inhibitory roles in the TME of many solid tumors, especially its suppression of T-cell effector functions (77, 78). Kloss et al. aimed to counteract this by armoring CAR T-cells with a ‘dominant-negative’ TGF-β receptor II (dnTGF-βRII), a truncated receptor lacking the intracellular signaling domain, which acts as a decoy to sequester TGF-β and prevent signaling in both CAR T-cells and neighboring immune cells. These dnTGF-βRII-armored CAR T-cells were resistant to TGF-β-induced suppression and exhibited enhanced proliferation and improved anti-tumor efficacy in a xenograft mouse model of prostate cancer (79). Enhanced efficacy with dnTGF-βRII armoring has since been reported in preclinical models of prostate cancer (80), ovarian cancer (81, 82), pancreatic cancer (83, 84), esophagogastric cancers (84), and glioblastoma (85). Notably, Tran et al. showed that their ROR1-targeting CAR T-cells induced TGF-β expression in tumor cells that did not initially overexpress this cytokine, but that this was mitigated by dnTGF-βRII armoring, which restored anti-tumor activity (83). A further advanced version of this strategy replaces the intracellular domain of TGF-βRII with that of another receptor, such as IL-15 receptor-α, thereby simultaneously blocking TGF-β and inducing IL-15 signaling. This dual-function receptor, named TB15, was shown to enhance CAR T-cell function beyond dnTGF-βRII or IL-15 armoring alone (86).
A dnTGF-βRII-armored PSMA-targeting CAR T-cell has since been evaluated in a phase I trial against metastatic castration-resistant prostate cancer. Grade 2 or higher CRS occurred in 5 out of 13 patients and was mostly manageable with immunosuppressive therapy, although one patient developed grade 4 CRS and concurrent sepsis and subsequently died. Four patients experienced ≥30% reductions in prostate-specific antigen levels, although the best radiographic response was stable disease (87). In a second phase I trial, two of nine patients experienced fatal immune-mediated toxicities, leading to early trial termination (88, 89). Retrospective analyses could not conclusively explain these severe toxicities, although the investigators speculated that modifying the co-stimulatory domain by replacing 4-1BB might help mitigate cytokine release without impairing efficacy (89). These findings underscore the gap between promising preclinical outcomes and complex clinical realities, possibly due to the widespread use of immunodeficient mouse models, which fail to fully capture human immune responses and associated toxicities.
An alternative strategy for blocking TGF-β signaling involves engineering CAR T-cells with an extracellular TGF-β-binding scFv. One group found that this approach paradoxically converted TGF-β into a stimulatory signal that enhanced CAR T-cell proliferation and activated immunostimulatory pathways (90). This group subsequently developed a bispecific CAR, targeting both IL-13Rα2 and TGF-β, which improved survival in glioblastoma mouse models (91). Other approaches for inhibiting TGF-β signaling include SMAD7-expressing CAR T-cells (92, 93), and knockout of the endogenous TGF-βRII in CAR T-cells using CRISPR/Cas9 technology (94), both of which improved anti-tumor efficacy in vivo.
PD-1/PDL-1
A major factor limiting CAR T-cell efficacy within the TME is exhaustion, which can involve multiple signaling pathways, including activation of immune checkpoints that suppress CAR T-cell proliferation, cytotoxicity and other effector functions (95, 96).
PD-1, a cell surface receptor on T-cells, is one such inhibitory immune checkpoint; its sustained expression or upregulation is widely regarded as a hallmark of exhaustion (97). An armoring strategy that has gained significant interest involves engineering CAR T-cells that secrete anti-PD-1 scFvs, thereby blocking the PD-1/PD-L1 axis within the TME (95). This approach has been demonstrated to enhance CD107a expression (a marker of degranulation), as well as secretion of effector molecules, such as IFN-γ and granzyme B, by both CAR T-cells and endogenous tumor-specific T-cells, suggesting a broader restoration of anti-tumor immunity (98–100). Anti-PD-1 scFv-secreting CAR T-cells have demonstrated improved anti-tumor activity in preclinical in vivo models of non-small cell lung cancer (NSCLC) (98), breast cancer (101), ovarian cancer (99), and hepatocellular carcinoma (HCC) (100). Similarly, CAR T-cells that secrete antibodies that block PD-L1, the ligand for PD-1, have demonstrated enhanced efficacy in models of pancreatic cancer (102) and renal cell carcinoma (RCC) (103). In a humanized RCC mouse model, Wang et al. reported that tumors treated with anti-PD-L1 antibody-secreting CAR T-cells exhibited increased endogenous T-cell infiltration and a reduction in M2-like macrophages, indicating TME modulation by the secreted payload (103). Based on such promising preclinical results, several anti-PD-1 or anti-PD-L1-armored CAR T-cells have entered phase I clinical trials. One report described CAR T-cells with IFN-γ-induced secretion of anti-PD-1 nanobodies achieving partial responses in 6 out of 11 patients with mesothelin and PD-L1-positive mesotheliomas, as well as one complete response, and toxicities that were manageable with supportive care (104). Further reports from early phase clinical studies are awaited.
Combinations of PD-1 or PD-L1 blockade with additional mechanisms to enhance CAR T-cell function or modulate the TME have also been explored. For example, one group armored CAR T-cells to secrete a bispecific scFv targeting both PD-1 and TREM2 (105), an immunoglobulin receptor expressed on immunosuppressive TAMs and MDSCs (105, 106). These dual-targeting CAR T-cells outperformed those secreting scFvs against either PD-1 or TREM2 alone in vivo, and were associated with decreased M2 macrophages and MDSCs, and increased proportion of CD8+ T-cells (105). Another strategy combined PD-1 and TGF-β pathway inhibition via secretion of a bifunctional ‘trap’, a fusion of an anti-PD-1 scFv with the TGF-βRII ectodomain. In a xenograft model of prostate cancer, these dual-inhibition CAR T-cells suppressed tumor growth more effectively than unarmored or anti-PD-1-only armored CAR T-cells (107).
CD47
Another immune checkpoint, often referred to as the ‘don’t eat me’ signal, involves CD47, a molecule frequently overexpressed on tumor cells. CD47 binds to the signal regulatory protein α (SIRPα) receptor on APCs, thereby inhibiting their phagocytic function (108). One strategy to counter this has been to engineer CAR T-cells to secrete a fusion protein, SIRPα-Fc, which blocks CD47. Chen et al. provided in vitro evidence that SIRPα-Fc enhanced macrophage phagocytic activity. In vivo, SIRPα-Fc-armored CAR T-cells exhibited improved anti-tumor efficacy and prolonged survival across several mouse models of solid tumors. This armoring also modulated immune cell phenotypes, promoting a T-cell central memory phenotype and increasing M1 macrophages and CD11c+ DCs in the TME (109).
Alternative strategies to target this axis have also been reported. Martins et al. armored CAR T-cells with a signal regulatory protein γ (SIRPγ)-related protein that binds to CD47 with high affinity, while Xie et al. engineered CAR T-cells to secrete CD47-specific nanobodies. In both cases, blocking the CD47 checkpoint enhanced CAR T-cell function and anti-tumor activity, including in models with tumor antigen heterogeneity (110, 111).
Interestingly, another group observed that concomitantly administering an anti-CD47 mAb with CAR T-cells depleted the CAR T-cells themselves secondary to macrophage-mediated phagocytosis, revealing a significant potential shortcoming of utilizing CD47 blockade as an armoring strategy (112). This issue was overcome by engineering CAR T-cells to co-express a variant of CD47 (CD47E) that retained SIRPα interaction but was resistant to blockade by anti-CD47 antibodies. Co-administering CD47E-CAR T-cells and a CD47-blocking mAb thereby protected the CAR T-cells from macrophage-mediated phagocytosis, whilst unleashing phagocytosis against tumor cells. In multiple in vivo models, CD47E-CAR T-cells administered alongside a CD47-blocking mAb exhibited increased persistence, enhanced macrophage infiltration into the TME, and improved anti-tumor activity (112).
Adenosine
The solid TME is characterized by hypoxia, metabolic stress, and high cellular turnover, which leads to the accumulation of extracellular adenosine triphosphate (ATP). This ATP is converted to adenosine by CD39 and CD73, enzymes which are expressed on tumor cells and immunosuppressive cells within the TME. Activation of adenosine receptor 2A (A2AR) by adenosine represents another immune checkpoint that impairs anti-tumor immune responses, including limiting CAR T-cell effector functions (113–116).
Adenosine deaminase (ADA) is an enzyme that catabolizes adenosine into inosine, thereby preventing accumulation of adenosine. Qu et al. found that armoring CAR T-cells to secrete ADA increased the resistance of CAR T-cells to exhaustion, promoted intratumoral CAR T-cell expansion, and improved tumor control (117). Similarly, Hu et al. explored the benefits of armoring CAR T-cells with ADA, hypothesizing that this strategy would not only relieve adenosine-mediated immunosuppression, but also provide inosine as an alternative fuel to support CAR T-cell proliferation and function. To avoid supplying inosine to tumor cells, they engineered ADA-secreting CAR T-cells with the secreted ADA anchored to the CAR T-cell. This strategy resulted in enhanced CAR T-cell proliferation, higher inosine concentrations, greater T-cell infiltration into the TME, and improved anti-tumor activity (118).
Other strategies to combat adenosine-induced immunosuppression have also been explored, including knocking-down or deleting A2AR expression in CAR T-cells using short hairpin RNA (shRNA) (119) or CRISPR/Cas9 technology (120, 121). These approaches have been demonstrated to enhance CAR T-cell effector functions. As gene editing technologies like CRISPR/Cas9 continue to improve in safety and efficacy, the concept of armoring CAR T-cells may evolve beyond supplying one or two transgenic payloads to include more complex modifications to the CAR T-cell genome (122, 123).
Tumor stroma: CAFs and the ECM
Apart from immune-inhibitory signals from tumor and immune cells, the activities of other stromal cells in the TME, such as CAFs, can also contribute to the suppression of CAR T-cell effector functions. CAFs secrete soluble factors, including immunosuppressive cytokines, and play a key role in producing the collagen-rich extracellular matrix (ECM) which create a physical barrier to CAR T-cell infiltration (18, 19, 124). One strategy to counteract CAFs is to target fibroblast activation protein (FAP), a pan-CAF marker, which some groups have achieved by engineering FAP-targeting CAR T-cells (125–127). One study found that treating pancreatic tumors, characterized by their dense desmoplastic stroma, in mice with FAP-targeting CAR T-cells reduced the integrity of the desmoplastic matrix, thereby making the tumors more susceptible to infiltration by CAR T-cells targeted against mesothelin, a TAA (126). Wehrli et al. also aimed to address CAF-associated immunosuppression by armoring mesothelin-targeting CAR T-cells to secrete a bispecific molecule targeting both CD3 and FAP, thereby inducing CAR-mediated killing of antigen-expressing tumor cells and engaging T-cells to kill CAFs. These armored CAR T-cells were shown to be effective against multiple models of pancreatic cancer (128). Notably, the FAP-targeted scFv used in this study did not cross-react with murine FAP, a challenge that the authors navigated by using several preclinical models, including patient-derived organoids with patient-matched CAFs, to characterize the armored CAR T-cells (128). This highlights one challenge that affects the study of armored CAR T-cells, which is that of potential cross-species barriers between the armoring payload and host cells in preclinical mouse models.
Another approach to combating the tumor stroma is to target the ECM, which is composed of macromolecules including collagens, proteoglycans, and glycoproteins (129). One strategy is to armor CAR T-cells with enzymes that degrade ECM components. Both heparanase-expressing CAR T-cells (130) and hyaluronidase-expressing CAR T-cells (131) have demonstrated increased tumor infiltration and enhanced anti-tumor activity, including when hyaluronidase and IL-7 armoring were combined (132). Moreover, Zheng et al. engineered CAR T-cells to express a synNotch receptor that drives the expression of three ECM-degrading enzymes, matrix metalloproteinase (MMP) 9, MMP12, and heparanase. They demonstrated that these synNotch CAR T-cells were more resistant to exhaustion and apoptosis after tumor challenge and exhibited enhanced tumor infiltration and anti-tumor activity in vivo (133).
Optimizing CAR T-cell fitness by modulating metabolic pathways
Exploration of cellular metabolism and its pathways to identify novel targets for treating cancer has intensified over the past decade, although much remains to be elucidated in this complex field (134–136). Numerous groups have demonstrated that modulating CAR T-cell metabolism to overcome metabolic suppression in the TME can improve CAR T-cell survival and effector functions. Many of these strategies overlap with those designed to overcome immune suppression in the TME, as promoting immunostimulatory or blocking immune-inhibitory signals is often inextricably linked to the metabolic fitness of CAR T-cells. However, we review novel approaches for metabolic armoring separately here to facilitate discussion (Figure 5).
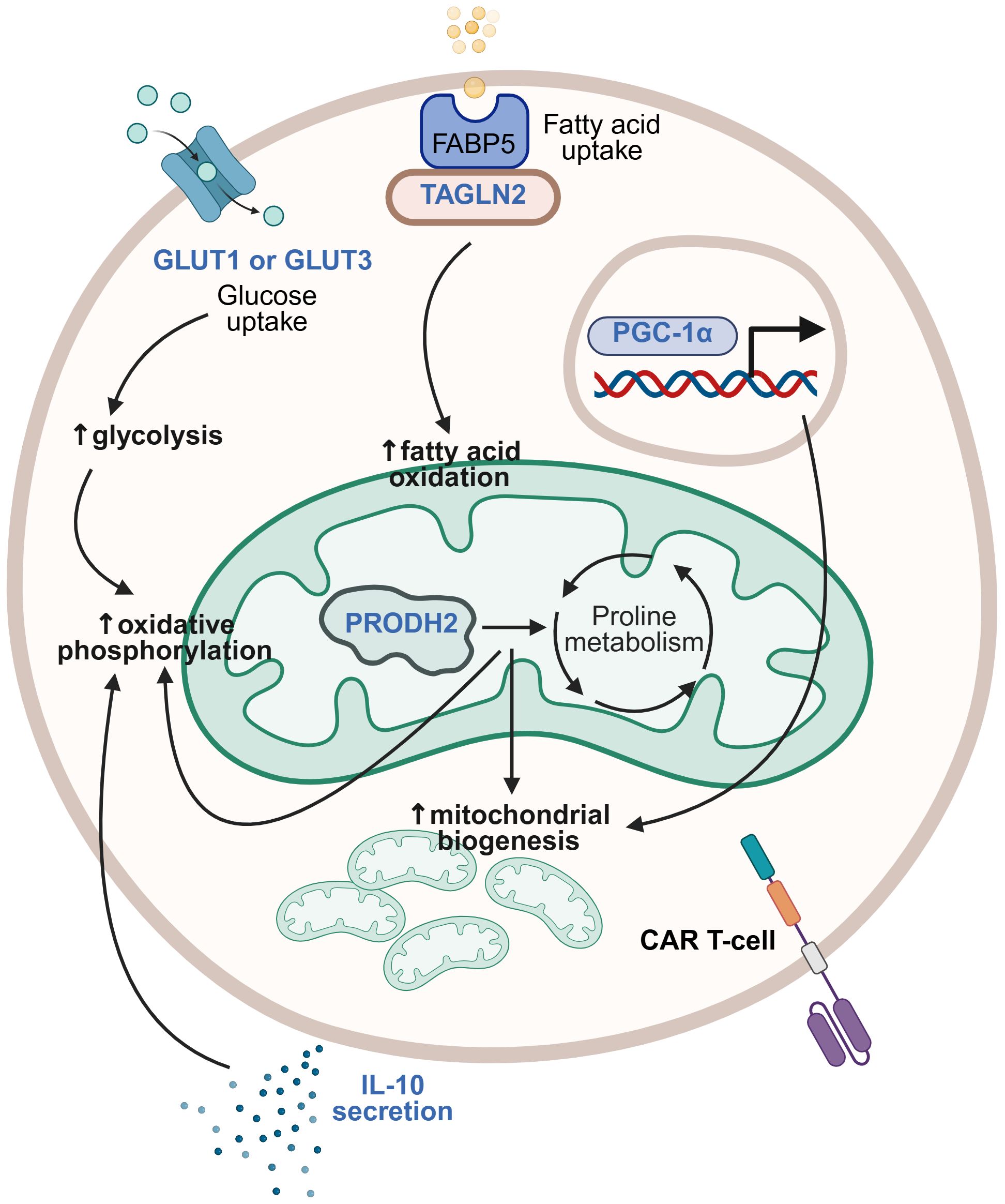
Figure 5. Strategies for modulating metabolic pathways. Different metabolic pathways may be modified in CAR T-cells to optimize their metabolic fitness. Metabolic armoring strategies that have been studied are illustrated here, with transgenic payloads (in blue) including glucose transporter (GLUT) 1 or GLUT3, transgelin 2 (TAGLN2), proline dehydrogenase 2 (PRODH2), IL-10, and PPARγ coactivator 1-α (PGC-1α) having been demonstrated to induce beneficial modulation of metabolic pathways including glycolysis, oxidative phosphorylation, and fatty acid oxidation, increased mitochondrial biogenesis, and associated improvement of CAR T-cell effector functions. This figure was created using BioRender.com.
The solid tumor TME is an environment of intense competition for nutrients, with the tumor cells typically prevailing. Glucose uptake and glycolysis in tumor cells are enhanced, whilst other cells in the TME, including immune cells, experience metabolic starvation and dysfunction (137, 138). Modulating glucose metabolism in CAR T-cells is therefore an appealing strategy. There are 14 human glucose transporters (GLUTs) that mediate glucose transport into cells, with GLUT1 being the most widely expressed across cell types, and GLUT3 being a high-affinity isoform expressed primarily in neurons (139). Shi et al. reported the benefits of overexpressing GLUT1 in CAR T-cells, including increased proliferative capacity and persistence in low-glucose conditions, increased proportions of stem cell-like memory T-cells after repeated tumor challenge, upregulation of genes involved in glycolysis and oxidative phosphorylation, and improved anti-tumor activity in vitro and in vivo in models of both leukemia and solid tumors, including RCC and glioblastoma. They did not observe the same with GLUT3 overexpression in CAR T-cells against leukemia models, although testing of GLUT3 armoring in solid tumor models was not reported (140). In contrast, Hu et al. observed that GLUT3-armored CAR T-cells demonstrated increased uptake of the glucose analog 2NBDG, greater release of effector cytokines, and enhanced tumor cell killing in vitro compared to GLUT1-armored CAR T-cells. Moreover, GLUT3-CAR T-cells demonstrated superior anti-tumor activity compared to unarmored CAR T-cells in both xenogeneic and syngeneic mouse models of solid tumors, including pancreatic, esophageal, and lung cancer (141). The seemingly disparate results regarding the relative benefits of GLUT1 versus GLUT3 overexpression may not actually be conflicting, but stem from the use of different tumor models or variations in CAR T-cell types. Regardless, the heterogeneity of solid tumors and of their preclinical models means that specific armoring strategies may not be universally beneficial across all solid tumor types.
Lipid metabolism has also been targeted to enhance CAR T-cell effector functions. The fatty acid-binding protein 5 (FABP5)-fatty acid β oxidation (FAO) axis in T-cells has emerged as a potential therapeutic target due to FABP5’s role in facilitating the transport of exogenous fatty acids to the mitochondria for FAO and energy generation. Hwang et al. identified transgelin 2 (TAGLN2), a cytoskeletal actin-binding protein, as a key FABP5-binding partner required for FABP5 localization to the plasma membrane for fatty acid uptake. They discovered that TAGLN2 expression was downregulated in T-cells from cancer patients, due to tumor-induced endoplasmic reticulum (ER) stress responses. By overexpressing TAGLN2 in CAR T-cells to circumvent stress responses and maintain CAR T-cell fitness, they demonstrated that TAGLN2-armored CAR T-cells improved control of metastatic disease progression and survival of mouse models of ovarian cancer (142).
Amino acid metabolism has also been implicated in tumorigenesis and homeostasis, with proline being one amino acid that plays diverse roles within the TME (143, 144). Using a CRISPR activation screen to identify genes that could enhance the effector functions of CD8+ T-cells, Ye et al. identified proline dehydrogenase 2 (PRODH2) as a promising enzyme target. Overexpression of PRODH2 in CAR T-cells led to increased proliferative capacity, upregulation of CAR T-cell effector molecules, including IFN-γ and granzyme B, and downregulation of apoptosis. PRODH2-armored CAR T-cells also demonstrated higher mitochondrial mass, were shifted toward oxidative phosphorylation metabolism, and exhibited enhanced anti-tumor efficacy in a xenograft mouse model of breast cancer (145).
Cytokine signaling, the manifold benefits of which were discussed earlier, can also contribute to CAR T-cell armoring by improving their metabolic fitness. One recent study focused particularly on the metabolic benefits of armoring with IL-10 (146), traditionally considered to be an anti-inflammatory cytokine but also now recognized for its complex roles in cancer immunity (147, 148). Zhao et al. demonstrated that IL-10-armored CAR T-cells exhibited decreased mitochondrial dysfunction, enhanced oxidative phosphorylation, and a stem cell-like memory phenotype, as well as increased proliferative capacity and cytotoxicity. Furthermore, these CAR T-cells showed enhanced tumor control in various solid tumor mouse models, including rejection of tumor rechallenges in long-term survivors (146).
Finally, Lontos et al. addressed mitochondrial dysfunction in exhausted CAR T-cells by targeting PPARγ coactivator 1-α (PGC-1α), a master regulator of mitochondrial biogenesis. By overexpressing an inhibition-resistant variant of PGC-1α in CAR T-cells, they demonstrated that these CAR T-cells had increased effector cytokine expression, higher expression of stem cell-like memory markers, and enhanced anti-tumor activity (149).
Taken together, these studies highlight the potential benefits of modifying various metabolic pathways in CAR T-cells. Whether these benefits will translate into clinical success is an exciting area for future research.
Overcoming antigen heterogeneity and antigen escape
Another variable that can impact CAR T-cell efficacy against solid tumors is the expression of antigen on tumor cells. A key challenge is the scarcity of bona fide TAAs that are entirely absent on healthy tissues and uniformly expressed across all tumor cells (150, 151). Additionally, antigen-negative tumor cells may become predominant through selective pressure, as CAR T-cells eliminate antigen-positive tumor cells. Tumors may develop mechanisms to downregulate the expression of the targeted antigen, resulting in a phenomenon known as ‘antigen escape’ in which CAR T-cells lose the ability to recognize and kill tumor cells (10, 150). One potential way to circumvent this issue is through recruitment of endogenous immune responses, particularly those mediated by innate immune cells that are not restricted to a single tumor antigen. This was previously discussed in the context of rewiring of the TME by cytokines such as IL-12, as well as the strategy of blocking CD47 to promote macrophage-mediated phagocytosis of tumor cells. Additional armoring strategies aimed at addressing antigen heterogeneity and escape are discussed below (Figure 6).
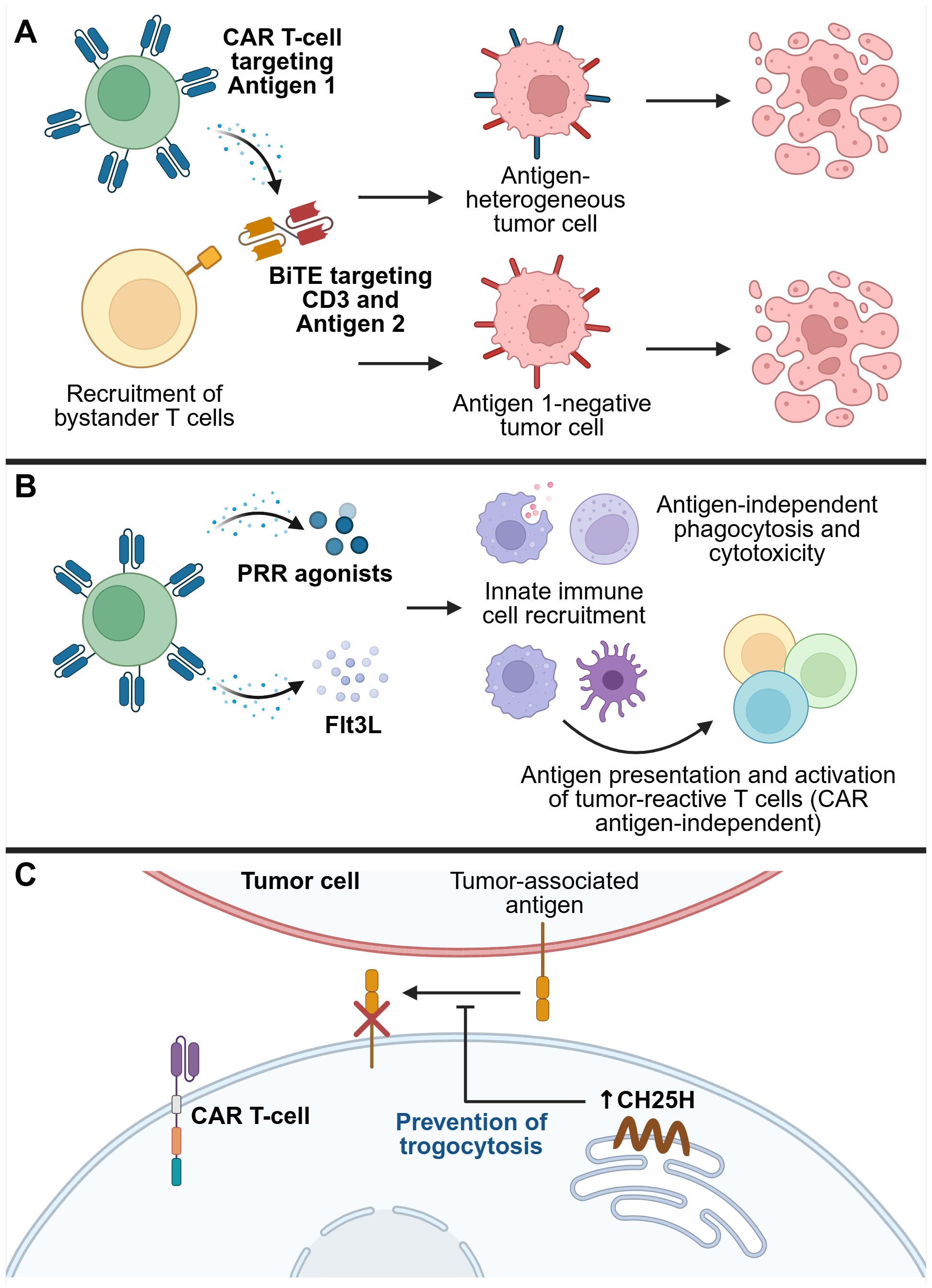
Figure 6. Strategies for overcoming tumor antigen heterogeneity and therapy escape mechanisms. (A) CAR T-cells secreting bispecific T-cell engagers (BiTEs) can both induce CAR-mediated tumor killing, and recruit bystander T-cells to kill tumor cells that do not express the CAR antigen. (B) Recruitment of endogenous innate immune cells, such as DCs, macrophages, and NK cells, activates their antigen-presenting, phagocytic, and cytotoxic capabilities to induce CAR antigen-independent tumor killing, as well as recruit the endogenous adaptive immune system, resulting in epitope spreading and broad anti-tumor immunity. (C) Restriction of trogocytosis can prevent the transfer of antigens from tumor cells to CAR T-cells, thereby preventing CAR T-cell fratricide and loss of efficacy. One mechanism that contributes to trogocytosis is upregulation of the transcription factor ATF3 by tumor exposure, which in turn suppresses the enzyme cholesterol 25-hydroxylase (CH25H). Overexpressing CH25H in CAR T-cells overcomes this issue, thereby reducing trogocytosis-mediated loss of antigen from tumor cells. This figure was created using BioRender.com.
Bispecific immune cell engagers
Bispecific T-cell engagers (BiTEs) are fusion proteins specific for two targets, one of which is an anti-CD3 scFv that engages T-cells, thereby enabling T-cell-mediated killing of cells expressing the second target (152). While BiTEs are being developed as standalone immunotherapeutics, engineering CAR T-cells to secrete BiTEs has also emerged as a strategy to overcome antigen heterogeneity. For example, Choi et al. engineered EGFRvIII-targeting CAR T-cells that secreted BiTEs against EGFR, and demonstrated in vitro that these armored CAR T-cells could recruit bystander T-cells to kill tumor cells. In orthotopic mouse models of glioblastoma with heterogeneous or negative EGFRvIII expression, these BiTE-armored CAR T-cells induced tumor regression (153). This strategy has since been tested in a first-in-human trial involving intraventricular delivery to three patients with recurrent glioblastoma, where it led to rapid tumor shrinkage. However, for two of the patients, the response was transient, underscoring the need for further optimization to enhance durability (154).
Similar strategies have been explored in other tumor types. These include: Muc16-targeting CAR T-cells armored with BiTEs against a peptide derived from the intracellular TAA Wilms tumor 1 (WT1), which demonstrated improved efficacy in ovarian cancer models with low Muc16 expression (155); and GPC3-targeting CAR T-cells that secrete BiTEs against B7-H3, which showed enhanced cytotoxicity against HCC cell lines with heterogeneous expression of GPC3 and B7-H3, although this was demonstrated only in vitro (156).
The repertoire of bispecific molecules that can be secreted by CAR T-cells continues to expand. Based on their findings of abundant CD16a-expressing innate immune cell infiltration in high-risk neuroblastoma TMEs, Pascual-Pasto et al. decided to engineer anti-glypican-2 (GPC2) CAR T-cells that secreted a bispecific innate immune cell engager (BiCE) targeting GD2 and CD16a, the latter serving to engage CD16a+ innate immune cells, including NK cells. They demonstrated that BiCE-armored CAR T-cells exhibited CAR-mediated cytotoxicity against GPC2-positive tumor cells and harnessed bystander NK cell activity against GD2-positive cells. This dual activity led to enhanced intratumoral NK cell retention and improved anti-tumor efficacy in patient-derived xenograft models of neuroblastoma in mice (157).
Targeting PRRs to activate endogenous innate immunity
Damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) are molecular motifs recognized by pattern recognition receptors (PRRs), which can activate innate immune responses in an antigen-independent manner (158). As such, they offer a potential strategy for circumventing tumor antigen heterogeneity.
Toll-like receptors (TLRs) are an important class of PRRs, each receptor recognizing distinct PAMPs in a specific subcellular compartment (158). TLR5, for example, recognizes flagellin, a protein subunit of bacterial flagella. Flagellin can initiate pro-inflammatory signaling pathways via TLR5, which is expressed on several innate immune cell types including macrophages and DCs, and has therefore attracted attention for its potential use as a vaccine adjuvant (159). Several groups have also explored its use as an armoring payload in CAR T-cells. More recently, Niu et al. engineered CAR T-cells that secreted flagellin under the control of an NFAT-responsive promoter. While flagellin armoring did not enhance the cytotoxicity of CAR T-cells in vitro, it significantly prolonged tumor control and survival in immunocompetent mouse models of solid tumors, suggesting that its benefit lies in activating endogenous immune responses. Supporting this, the authors observed increased intratumoral abundance of CD86+ macrophages (indicative of M1 macrophage polarization) and CD103+ DCs (associated with DC activation). Additionally, in a model of antigen-heterogeneous melanoma, flagellin-armored CAR T-cells suppressed tumor growth more effectively than their unarmored counterparts (160).
Another bacterial protein explored as a CAR T-cell armoring payload is neutrophil-activating protein (NAP) from the bacterium Helicobacter pylori, which has been shown to exert various immunomodulatory effects. These include acting as a neutrophil chemoattractant and promoting IL-12 and IL-23 expression via TLR2 on neutrophils and monocytes, thereby supporting Th1 responses (161, 162). Jin et al. demonstrated that armoring CAR T-cells with NFAT promoter-inducible NAP improved anti-tumor activity in multiple syngeneic tumor models, including those with antigen-heterogeneity. NAP-armored CAR T-cells increased tumor infiltration of neutrophils, M1 macrophages and NK cells, and promoted ‘epitope spreading’ among endogenous CD8+ T-cells, that is, activation of CD8+ T-cells recognizing antigens other than the CAR-targeted TAA, suggesting robust bystander immune activation (163).
Beyond protein-based PRR agonists, nucleic acid-sensing pathways have also been leveraged. The cytosolic PRR RIG-I is activated by RNA and induces type I IFNs, which play broad roles in innate and adaptive immunity (164). Johnson et al. engineered CAR T-cells to secrete RN7SL1, a non-coding RNA that activates RIG-I, via extracellular vesicles that are preferentially taken up by immune rather than tumor cells. They found that delivery of RN7SL1 by the CAR T-cells promoted pro-inflammatory features and restricted immunosuppressive features among endogenous myeloid cells, and promoted the development of endogenous CD8+ T-cells with an effector memory-like phenotype. When combined with immune checkpoint blockade, RN7SL1-CAR T-cells eradicated antigen-heterogeneous melanomas in immunocompetent mice and prevented tumor outgrowth upon rechallenge with antigen-negative tumor cells, suggesting the generation of CAR antigen-independent memory (165).
Finally, PRR agonists as immune adjuvants have been combined with CAR T-cells secreting immune cell growth factors as another method of harnessing the endogenous immune system. Lai et al. armored CAR T-cells with Fms-like tyrosine kinase 3 ligand (Flt3L), a hematopoietic growth factor that is essential for steady-state DC development (166, 167), and demonstrated associated intratumoral expansion of conventional type 1 DCs, which play an important role in cross-presenting antigens to and activating cytotoxic CD8+ T-cell responses. When combined with poly(I:C), an immunostimulatory TLR3 agonist, Flt3L-secreting CAR T-cells significantly suppressed tumor growth in both antigen-homogeneous and heterogeneous tumor models. Treated mice also rejected antigen-negative tumor rechallenges, again indicating induction of epitope spreading and a durable anti-tumor immune response (168).
Modulating trogocytosis
Trogocytosis is a phenomenon that has received increased attention in recent years within the field of immunology. It refers to the transfer of fragments of cell membrane, along with associated proteins and other molecules, between cells that are in direct contact with each other (169). Trogocytosis has been observed between tumor cells and CAR T-cells, leading to the transfer of TAAs from the tumor cells to the CAR T-cells. This can contribute to antigen escape and even CAR T-cell fratricide, where CAR T-cells inadvertently recognize and kill one another (170). In some cases, CAR molecules themselves can be transferred from CAR T-cells to tumor cells, further reducing anti-tumor efficacy (171). Lu et al. identified cholesterol metabolism as a key pathway involved in trogocytosis, potentially due to cholesterol’s role in modulating the biophysical properties of lipid membranes. They found that ATF3, a stress-responsive transcription factor, is upregulated in cytotoxic T lymphocytes (CTLs) exposed to tumor-derived signals. ATF3 suppresses expression of the enzyme cholesterol 25-hydroxylase (CH25H), which in turn promotes trogocytosis between CTLs and tumor cells, leading to T-cell dysfunction and tumor progression. By armoring CAR T-cells to co-express CH25H, they were able to reduce the extent of trogocytosis, preserve antigen expression on tumor cells, and enhance CAR T-cell infiltration and anti-tumor efficacy (172). Of note, other recent work has also examined how incorporation of different CAR transmembrane domains affect the probability of trogocytosis occurring, and how this can be used to modulate the effects of CAR T-cell therapy (173).
Improving CAR T-cell homing to tumors
Enhancing the ability of CAR T-cells to efficiently home to tumors represents another important challenge to overcome. Systemically administered CAR T-cells can become sequestered at non-tumor-bearing tissues, and a lack of chemotactic cues can allow tumor cells to evade recognition and elimination by circulating CAR T-cells (174, 175). One strategy to address these limitations involves armoring CAR T-cells with chemokines or chemokine receptors selected to improve trafficking of CAR T-cells to tumor sites (Figure 7).
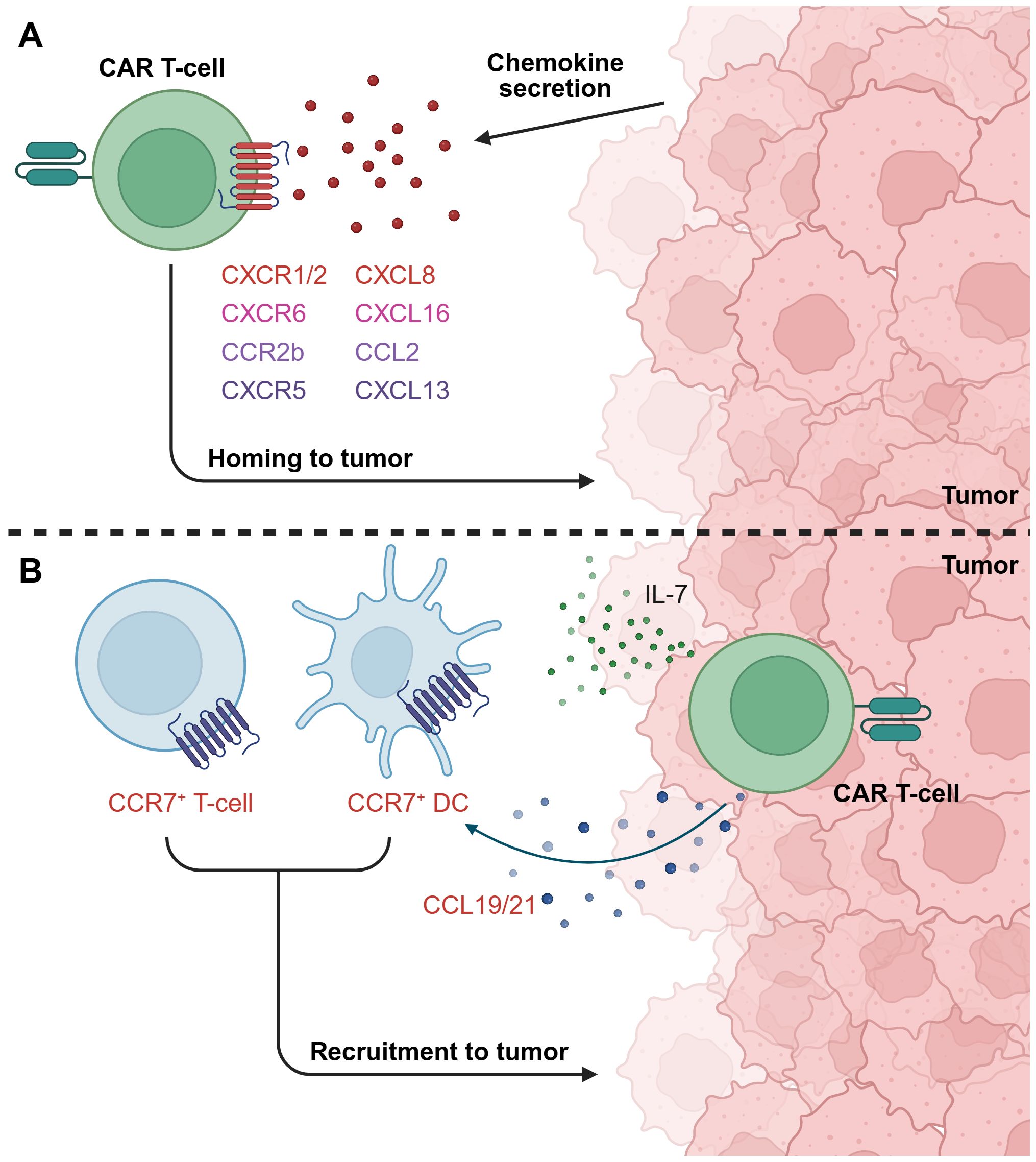
Figure 7. Exploiting chemokine/chemokine receptor signaling to improve CAR T-cell homing to tumors. (A) Armoring CAR T-cells with specific chemokine receptors, for example CXCR1/2, CXCR6, CCR2b, and CXCR5, can improve homing towards tumors that express the cognate chemokine ligand. (B) CAR T-cells can be engineered to secrete chemokines that recruit endogenous immune cells to support the anti-tumor response. For instance, illustrated here is a dual IL-7 and CCL19/21-armored CAR T-cell, which recruits T-cells and DCs expressing CCR7, the cognate receptor for CC19 and CCL21, to the TME. This figure was created using BioRender.com.
Chemokine/chemokine receptor axes
CXCL8, also known as IL-8, is a chemokine associated with poor prognosis in numerous solid tumor types and with the recruitment of immunosuppressive immune cells to the TME. Several groups have sought to target this by armoring CAR T-cells with CXCR1 or CXCR2, both of which are CXCL8 receptors, with the aim of exploiting tumor CXCL8 expression to improve CAR T-cell migration to tumors (12, 176, 177). Notably, CXCL8 expression has been shown to be upregulated by irradiation of some tumors, suggesting the benefits of combining radiotherapy with CAR T-cell therapy in specific contexts (176, 177). CXCR1/CXCR2-armoring has been demonstrated to improve CAR T-cell infiltration, persistence, and tumor control in vivo across several studies (176–180). However, findings by Talbot et al. suggested that the benefits of CXCR2-armoring were context-dependent, as this did not enhance tumor control in a rapidly growing primary orthotopic xenograft model of osteosarcoma, but did confer benefits in a metastatic model (179). Interestingly, multiple groups have reported potentially improved safety profiles of CXCR-armored CAR T-cells compared to unarmored counterparts, possibly due to more targeted trafficking to tumors and thus fewer off-tumor toxicities (178, 179). On the other hand, one group found that while CXCR2 armoring enhanced trafficking into the TME, CAR T-cell persistence, and therefore anti-tumor efficacy, remained limited by the immunosuppressive TME. They demonstrated that co-armoring with the pro-inflammatory cytokines IL-15 or IL-18 could overcome this limitation, resulting in significantly improved in vivo anti-tumor activity (181).
Lesch et al. found CXCL16 to be highly expressed by both tumor and myeloid stromal cells in murine pancreatic cancer models and human specimens. Armoring CAR T-cells with CXCR6, the receptor for CXCL16, led to increased anti-tumor efficacy and prolonged the survival of treated mice (182). They also noted elevated expression of very late antigen-4 (VLA-4), an integrin that facilitates transendothelial migration of lymphocytes, in CXCR6-transduced T-cells, suggesting that harnessing the CXCL16-CXCR6 axis may offer multiple benefits for improving CAR T-cell trafficking. Talbot et al., who also tested CXCR6-armored CAR T-cells against osteosarcoma, found similar benefits in a metastatic xenograft model, and again reported an improved safety profile compared to unarmored CAR T-cells (179).
The CCL2-CCR2 axis has also drawn attention, as some solid tumors, including pleural mesothelioma and NSCLC, can secrete high levels of CCL2 (183, 184). CAR T-cells transduced with CCR2b, the dominant isoform of CCR2, showed improved tumor infiltration and tumor control in mouse models, including successfully crossing the blood-brain barrier to control brain metastases from NSCLC (183–185). On a similar theme, Li et al. armored CAR T-cells with CXCR5, the receptor for CXCL13, based on the expression of CXCL13 in a high proportion of NSCLC samples. These CAR T-cells exhibited enhanced migration toward CXCL13-positive targets and improved in vivo anti-tumor activity (186). Moreover, the combination of chemokine receptor (including CCR2b and CXCR5) and IL-7 armoring has been demonstrated to boost CAR T-cell expansion and anti-tumor activity in immunodeficient mouse tumor models (187, 188).
Tian et al. identified the CCL4/CCL5-CCR5 axis as one that is significantly correlated with CD8+ T-cell infiltration in solid tumors. They engineered CAR T-cells to express both CCR5 and IL-12, which enhanced T-cell trafficking and reversed TAM-induced immunosuppression (189). Conversely, the CCL1-CCR8 axis has been identified as having a role in the recruitment of immunosuppressive Tregs into the TME. Cadilha et al. armored CAR T-cells with both CCR8, to drive recruitment of CAR T-cells into the TME, and dnTGF-βRII, reasoning that this would counteract any Treg-mediated immunosuppression. They demonstrated that these dual-armored CAR T-cells significantly improved tumor control and survival in vivo (190).
Finally, chemokine armoring has also been found to be beneficial for recruiting other immune cells to the TME. Dual armoring with IL-7 and CCL19 has received particular focus, as these cytokines are essential for organizing the T-cell zone in lymphoid organs, and for recruiting T-cells and DCs expressing CCR7, the receptor for CCL19 (191, 192). Adachi et al. applied this mechanism to facilitate T-cell and DC recruitment to tumors (193), a strategy that has since also been studied by other groups. IL-7 and CCL19 dual armoring has been found to result in enhanced proliferation of the CAR T-cells, reduced expression of exhaustion markers, development of a central memory phenotype, remodeling of the TME, and improved in vivo control of tumors and survival in preclinical mouse models, including survival superior to armoring with either cytokine alone (193–196). Similar findings have been reported with dual armoring with IL-7 and CCL21, another CCR7 ligand, which also promoted T-cell and DC recruitment, increased central memory phenotype T-cells, and improved tumor control in an antigen-heterogeneous tumor model (197). Early phase clinical trials of dual IL-7 and CCL19-armored CAR T-cell have reported preliminary evidence of anti-tumor activity and no grade >2 toxicities (198, 199), although further studies will be needed to confirm clinical benefit.
Taken together, this wide assortment of studies have demonstrated the utility of exploiting chemokine signaling pathways to improve recruitment of CAR T-cells and other immune cells to solid tumors. However, the benefits of individual strategies are likely to be context-dependent and must be tailored according to the chemokine/chemokine receptor expression profile of the specific tumor being targeted. While chemokine receptor armoring alone has yielded enhanced anti-tumor effects in some instances, other studies suggest that additional payloads, such as pro-inflammatory cytokines, may be necessary to overcome the immunosuppressive TME and fully realize therapeutic potential.
LIGHT
An armoring payload that potentially provides the benefits of both chemokine and pro-inflammatory signaling is LIGHT, also known as tumor necrosis factor superfamily member 14 (TNFSF14). Using RNA sequencing data from The Cancer Genome Atlas (TCGA) database, Zhang et al. identified a strong correlation between LIGHT expression and gene signatures of tertiary lymphoid structures (TLS) (200), structures in tumors that play key roles in recruiting immune cells and shaping immune responses (201). They demonstrated that CAR T-cells armored with LIGHT upregulated expression of chemokines CCL19, CCL21, and CXCL13 in both stromal cells and tumor cells, enhancing the ability of the stromal cells to recruit T-cells in migration assays. In vivo, LIGHT-armored CAR T-cells improved tumor infiltration, accelerated tumor regression, and prolonged survival in mice compared to their unarmored counterparts (200).
Separately, another group characterized the benefits of LIGHT-armoring from the perspective of overcoming tumor antigen heterogeneity (202). LIGHT binds to two receptors: lymphotoxin-β receptor (LTβR), which is broadly expressed on non-lymphoid cells, and herpesvirus entry mediator (HVEM), expressed on various immune cells. LIGHT–HVEM signaling has been shown to promote pro-inflammatory responses, including induction of Th1-type immunity and enhancement of NK and CD8+ T-cell activity (203, 204). In contrast, LIGHT–LTβR interactions can induce apoptosis in LTβR-expressing tumor cells, suggesting a mechanism for direct, antigen-independent tumor cell killing (205). Cai et al. demonstrated that LIGHT-armoring enhanced CAR T-cell proliferation and cytotoxic function in vitro, including against tumor cell lines with heterogeneous antigen expression. In vivo, LIGHT-armored CAR T-cells showed increased tumor infiltration and improved tumor control in immunodeficient mouse models, with no detectable toxicities observed in immunocompetent mice (202).
Discussion and future perspectives
CAR T-cells must surmount multiple barriers in order to effectively eradicate tumor cells, likely accounting for the limited clinical efficacy observed thus far in the treatment of solid tumors. Armoring CAR T-cells with a biological payload, such as molecules that enhance effector function, counteract immunosuppression within the TME, or recruit endogenous immune cells to support the anti-tumor response, has shown promise in numerous preclinical studies, often outperforming unarmored CAR T-cells in specific settings. However, the reproducibility and durability of these responses in the clinical setting remain uncertain. To date, no armored CAR T-cell product has progressed beyond early phase clinical trials, and only limited data from completed or ongoing phase I trials are available. Notably, some strategies, such as IL-12, IL-15 and dnTGF-βRII-armoring, have produced toxicity in phase I trials that were not fully anticipated by preclinical data, underscoring one of the major limitations in this field: the lack of a fully predictive preclinical model.
The large majority of CAR T-cell in vivo studies in mice thus far have been carried out in immunodeficient mice, predominantly NSG mice which lack T-cells, B-cells, and NK cells (206). While these models enable the study of human CAR T-cells, which may be perceived as a translational advantage, they fail to recapitulate key immune-mediated toxicities seen in the clinical setting. This is particularly problematic for armored CAR T-cells, where cross-species incompatibilities may prevent interaction of the payload with murine immune components. For instance, human IL-12, one of the most extensively studied cytokine payloads, does not cross-react with murine cells (207). Syngeneic immunocompetent mouse models do provide a method of investigating armored CAR T-cell therapy in organisms that have an intact immune system, and eliminate cross-species barriers. However, these models are not invulnerable to uncertainty or criticism either, given the biological differences between mice and humans. Humanized mouse models, or immunodeficient mice reconstituted with a humanized immune system, have been gaining attention, but difficulties remain in navigating the mismatch of the murine and human systems to establish stable models (206). Alternative platforms, such as canine (208), zebrafish (209), and patient-derived tumor organoid models (128, 210), offer additional insights, but are not widely adopted or accessible. As such, mouse models will likely remain the cornerstone of preclinical CAR T-cell evaluation in the near term. It will continue to be necessary to test armored CAR T-cells in multiple preclinical models to assess different aspects of safety and efficacy. Other challenges highlighted previously in this review, including the pleiotropic effects of different armoring payloads and the heterogeneity of solid tumors, also need to be navigated by judicious selection of a range of preclinical assays and models.
Another layer of complexity arises in defining what constitutes a “beneficial” effect of armoring on CAR T-cell phenotype and function. Many studies highlight increased proliferation, reduced expression of exhaustion markers, and enhanced effector molecule release as positive outcomes. Likewise, the enrichment of memory T-cell subsets, particularly central memory or stem cell-like memory subsets, is generally viewed as being beneficial, due to evidence from studies that demonstrated that these T-cells exhibit superior persistence and induce superior anti-tumor immunity compared to effector memory T-cells (211, 212). However, other studies have conversely found that more differentiated or effector memory-like T-cells could potentially have particular benefits for the therapy of solid tumors, as opposed to hematological malignancies (213–215). Ultimately, only clinical trials in human patients will be able to determine whether the effects on T-cell phenotype induced by armoring payloads in preclinical studies translate to durable benefits for patients with cancer.
As armoring strategies for enhancing the potency of CAR T-cells continue to advance, it is of paramount importance that progress is similarly made in developing mechanisms to ensure the safety of these therapies. As illustrated by a number of studies we have discussed, armoring strategies have evolved to become more refined in regulating expression of the payload, which is important for avoiding off-tumor effects. For example, harnessing the intrinsic biological effects of cytokine signaling is attractive as an armoring approach, but constitutive or uncontrolled cytokine signaling can cause severe toxicities. To address this, researchers have developed a range of expression control strategies, each with their strengths and pitfalls, including tethering cytokines to CAR T-cell membranes, targeting cytokines to tumors via tumor-selective peptides, designing chimeric cytokine receptors, and engineering synthetic promoters and gene circuits responsive to specific intracellular or environmental signals, such as the NFAT-activated promoter system and the synNotch receptor system. The NFAT-activated promoter approach has been widely employed as a means to link payload expression to CAR activation, but has also been found to allow ‘leaky’, or non-tumor-restricted, expression of the payload. For example, a first-in-human trial of tumor-infiltrating lymphocytes (TILs) armored with NFAT-inducible IL-12 was terminated early due to severe toxicities secondary to the secreted IL-12, peak serum levels of which were unpredictable and varied widely, including reaching potentially lethal levels (47). This highlights a limitation of inducible expression systems where the stimulus is CAR or TCR activation, rather than a bona fide tumor-specific stimulus. Efforts to reduce the leakiness of the NFAT promoter system have included utilizing affinity-tuned CARs. One group found that the threshold of CAR target antigen density for NFAT activation was inversely correlated with CAR affinity, such that restriction of NFAT activation to antigen-high tumors was more stringent with low-affinity CARs (216). Another recent study aimed to develop a tumor-inducible expression system by screening for endogenous genes that are differentially upregulated by CAR T-cells in the tumor compared to a non-tumor site, and identified NR4A2 and RGS16 as the two most promising candidates. Placing transgene expression under control of these two endogenous promoters through a CRISPR knock-in approach resulted in stringent tumor-restricted expression, and CAR T-cells armored in this manner with tumor-restricted IL-12 or IL-2 exhibited greater anti-tumor efficacy (217). Alternative approaches for linking transgene expression to a TME-intrinsic stimulus include hypoxia-induced expression, which exploits the hypoxic microenvironment of solid tumors (151, 218–220), and synNotch receptor systems, which utilize TAAs or other targets within the TME to induce transgene expression (75, 76, 133, 221). Nonetheless, many switchable technologies have been shown to be prone to leakiness which can result in off-tumor effects. Therefore, tailoring of different armored CAR T-cell therapies not just to tumor types, but to carefully profiled patient and disease-specific characteristics, is likely to become more important as a personalized approach to maximize safety and efficacy.
Finally, apart from engineering inducible expression systems to optimize tumor-restricted expression of the armoring payload, other strategies for improving the safety of armored CAR T-cells have been explored. One approach is to incorporate a module that blocks IL-1 or IL-6 signaling, key mediators of CRS, as the armoring payload itself (222). To avoid systemic immunosuppression, these neutralizing payloads, such as an IL-6 receptor α-blocking antibody, have been regulated using an NFAT promoter-based approach (54), or have been engineered as a chimeric cytokine receptor composed of the extracellular domains of the IL-6 receptor and the intracellular domains of a mutated IL-7 receptor, thereby combining IL-6 sequestration with constitutive IL-7 signaling (223). An alternative approach to alleviating toxicity is to incorporate suicide genes into CAR T-cells, which act as killing switches that rapidly eliminate the CAR T-cells when activated by the appropriate drug. Examples of suicide genes include iC9, which was previously discussed in the context of IL-15 armoring, as well as antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity switches, where administration of a mAb such as rituximab selectively eliminates CAR T-cells co-expressing epitopes from the corresponding antigen, in this case CD20 (224, 225). As armoring strategies become more advanced, however, involving more complex transgenes and genome engineering, remote approaches for modulating CAR T-cell function that do not require incorporation of another transgene are also appealing. For example, dasatinib is a tyrosine kinase inhibitor that reversibly inhibits CAR signaling, effectively suppressing CAR T-cell functions but allowing these functions to resume when the dasatinib is discontinued (224, 226, 227). This provides a further advantage compared to suicide switches, which permanently eliminate CAR T-cells, potentially impacting anti-tumor activity. Taken together, the range of safety mechanisms that have been developed thus far each offer different benefits, and the selection of the best approach will depend on the desired characteristics and outcomes for the specific CAR T-cell therapy.
Looking ahead, given the myriad barriers to effective CAR T-cell therapy, strategies to refine and combine armoring payload expression are likely to become ever more advanced. A recent example of a combination approach is the study by Erler et al. which described the generation of a multi-armored allogeneic CAR T-cell engineered using TALEN-mediated genome editing technology to achieve knockout of TGF-βRII and PD-1, and knock-in of activation-induced IL-12 (228). Although a comprehensive review of CAR T-cell engineering strategies was beyond the scope of this review, the field is clearly moving toward increasingly sophisticated designs. Future directions will likely involve combining multiple payloads, broadening the scope of payloads beyond secreted proteins, and leveraging advanced genome engineering and synthetic biology tools to tightly regulate CAR T-cell activity. Furthermore, new insights into TME-driven transcriptional and epigenetic reprogramming will continue to inform the rational selection of payloads for the design of next-generation CAR T-cells.
In summary, while armoring strategies have demonstrated significant promise in preclinical models, translation into effective and safe therapies for solid tumors will require a concerted focus on improving preclinical models, refining expression control and safety mechanisms, and deepening our understanding of CAR T-cell biology in the context of solid tumor immunology. The next decade will likely see the emergence of highly engineered, multi-functional CAR T-cells capable of addressing the complex challenges of solid tumors in the clinical setting.
Author contributions
DY: Writing – original draft, Writing – review & editing. WM: Writing – review & editing, Writing – original draft. JA: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. JA and WM are funded by Cancer Research UK grant DCRPGF\100009. DY is funded by the CRUK City of London Centre Award SEBCATP-2023\100006.
Conflict of interest
JA is co-founder and holds equity in Korecyte Bio and Aethox Therapeutics. JA and DY are named inventors on two submitted patents related to armored CAR T-cell products.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1643941/full#supplementary-material
References
1. Labanieh L and Mackall CL. CAR immune cells: design principles, resistance and the next generation. Nature. (2023) 614:635–48. doi: 10.1038/s41586-023-05707-3
2. Cappell KM and Kochenderfer JN. Long-term outcomes following CAR T cell therapy: what we know so far. Nat Rev Clin Oncol. (2023) 20:359–71. doi: 10.1038/s41571-023-00754-1
3. National Cancer Institute. CAR T Cells: Engineering Patients’ Immune Cells to Treat Their Cancers (2025). Available online at: https://www.cancer.gov/about-cancer/treatment/research/car-t-cells (Accessed June 6, 2025).
4. Sterner RC and Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. (2021) 11:69. doi: 10.1038/s41408-021-00459-7
5. Feins S, Kong W, Williams EF, Milone MC, and Fraietta JA. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am J Hematol. (2019) 94:S3–9. doi: 10.1002/ajh.25418
6. Zhang ZZ, Wang T, Wang XF, Zhang YQ, Song SX, and Ma CQ. Improving the ability of CAR-T cells to hit solid tumors: Challenges and strategies. Pharmacol Res. (2022) 175:106036. doi: 10.1016/j.phrs.2021.106036
7. Kosti P, Maher J, and Arnold JN. Perspectives on chimeric antigen receptor T-cell immunotherapy for solid tumors. Front Immunol. (2018) 9:1104. doi: 10.3389/fimmu.2018.01104
8. Weinkove R, George P, Dasyam N, and McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. (2019) 8:e1049. doi: 10.1002/cti2.1049
9. Mohanty R, Chowdhury CR, Arega S, Sen P, Ganguly P, and Ganguly N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol Rep. (2019) 42:2183–95. doi: 10.3892/or.2019.7335
10. Rafiq S, Hackett CS, and Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. (2020) 17:147–67. doi: 10.1038/s41571-019-0297-y
11. Lindo L, Wilkinson LH, and Hay KA. Befriending the hostile tumor microenvironment in CAR T-cell therapy. Front Immunol. (2020) 11:618387. doi: 10.3389/fimmu.2020.618387
12. Kohli K, Pillarisetty VG, and Kim TS. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. (2022) 29:10–21. doi: 10.1038/s41417-021-00303-x
13. Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. (2009) 69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281
14. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. (2015) 527:249–53. doi: 10.1038/nature15520
15. Piali L, Fichtel A, Terpe HJ, Imhof BA, and Gisler RH. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J Exp Med. (1995) 181:811–6. doi: 10.1084/jem.181.2.811
16. Kantari-Mimoun C, Barrin S, Vimeux L, Haghiri S, Gervais C, Joaquina S, et al. CAR T-cell entry into tumor islets is a two-step process dependent on IFNgamma and ICAM-1. Cancer Immunol Res. (2021) 9:1425–38. doi: 10.1158/2326-6066.CIR-20-0837
17. Griffioen AW, Damen CA, Blijham GH, and Groenewegen G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood. (1996) 88:667–73. doi: 10.1182/blood.V88.2.667.bloodjournal882667
18. Yamauchi M, Barker TH, Gibbons DL, and Kurie JM. The fibrotic tumor stroma. J Clin Invest. (2018) 128:16–25. doi: 10.1172/JCI93554
19. Kankeu Fonkoua LA, Sirpilla O, Sakemura R, Siegler EL, and Kenderian SS. CAR T cell therapy and the tumor microenvironment: Current challenges and opportunities. Mol Ther Oncolyt. (2022) 25:69–77. doi: 10.1016/j.omto.2022.03.009
20. Proietto M, Crippa M, Damiani C, Pasquale V, Sacco E, Vanoni M, et al. Tumor heterogeneity: preclinical models, emerging technologies, and future applications. Front Oncol. (2023) 13:1164535. doi: 10.3389/fonc.2023.1164535
21. Chmielewski M, Hombach AA, and Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev. (2014) 257:83–90. doi: 10.1111/imr.12125
22. Chmielewski M and Abken H. TRUCKs: the fourth generation of CARs. Expert Opin Biol Ther. (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
23. Chmielewski M, Kopecky C, Hombach AA, and Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. (2011) 71:5697–706. doi: 10.1158/0008-5472.CAN-11-0103
24. Thomas S and Abken H. CAR T cell therapy becomes CHIC: “cytokine help intensified CAR” T cells. Front Immunol. (2022) 13:1090959. doi: 10.3389/fimmu.2022.1090959
25. Leonard WJ, Lin JX, and O’Shea JJ. The gamma(c) family of cytokines: basic biology to therapeutic ramifications. Immunity. (2019) 50:832–50. doi: 10.1016/j.immuni.2019.03.028
26. Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, and Savoldo B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin Cancer Res. (2019) 25:2915–24. doi: 10.1158/1078-0432.CCR-18-1811
27. Gargett T, Ebert LM, Truong NTH, Kollis PM, Sedivakova K, Yu W, et al. GD2-targeting CAR-T cells enhanced by transgenic IL-15 expression are an effective and clinically feasible therapy for glioblastoma. J Immunother Cancer. (2022) 10:e005187. doi: 10.1136/jitc-2022-005187
28. Ma L, Zhang K, Xu J, Wang J, Jiang T, Du X, et al. Building a novel TRUCK by harnessing the endogenous IFN-gamma promoter for cytokine expression. Mol Ther. (2024) 32:2728–40. doi: 10.1016/j.ymthe.2024.06.017
29. Lanitis E, Rota G, Kosti P, Ronet C, Spill A, Seijo B, et al. Optimized gene engineering of murine CAR-T cells reveals the beneficial effects of IL-15 coexpression. J Exp Med. (2021) 218:e20192203. doi: 10.1084/jem.20192203
30. Shi H, Li A, Dai Z, Xue J, Zhao Q, Tian J, et al. IL-15 armoring enhances the antitumor efficacy of claudin 18.2-targeting CAR-T cells in syngeneic mouse tumor models. Front Immunol. (2023) 14:1165404. doi: 10.3389/fimmu.2023.1165404
31. Zannikou M, Duffy JT, Levine RN, Seblani M, Liu Q, Presser A, et al. IL15 modification enables CAR T cells to act as a dual targeting agent against tumor cells and myeloid-derived suppressor cells in GBM. J Immunother Cancer. (2023) 11:e006239. doi: 10.1136/jitc-2022-006239
32. Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. (2015) 33:74–82. doi: 10.1200/JCO.2014.57.3329
33. Conlon KC, Potter EL, Pittaluga S, Lee CR, Miljkovic MD, Fleisher TA, et al. IL15 by continuous intravenous infusion to adult patients with solid tumors in a phase I trial induced dramatic NK-cell subset expansion. Clin Cancer Res. (2019) 25:4945–54. doi: 10.1158/1078-0432.CCR-18-3468
34. Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, et al. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. (2019) 3:1970–80. doi: 10.1182/bloodadvances.2018028332
35. Sanchez-Moreno I, Lasarte-Cia A, Martin-Otal C, Casares N, Navarro F, Gorraiz M, et al. Tethered IL15-IL15Ralpha augments antitumor activity of CD19 CAR-T cells but displays long-term toxicity in an immunocompetent lymphoma mouse model. J Immunother Cancer. (2024) 12:e008572. doi: 10.1136/jitc-2023-008572
36. Ataca Atilla P, McKenna MK, Tashiro H, Srinivasan M, Mo F, Watanabe N, et al. Modulating TNFalpha activity allows transgenic IL15-Expressing CLL-1 CAR T cells to safely eliminate acute myeloid leukemia. J Immunother Cancer. (2020) 8:e001229. doi: 10.1136/jitc-2020-001229
37. Steffin D, Ghatwai N, Montalbano A, Rathi P, Courtney AN, Arnett AB, et al. Interleukin-15-armoured GPC3 CAR T cells for patients with solid cancers. Nature. (2025) 637:940–6. doi: 10.1038/s41586-024-08261-8
38. Strengell M, Matikainen S, Siren J, Lehtonen A, Foster D, Julkunen I, et al. IL-21 in synergy with IL-15 or IL-18 enhances IFN-gamma production in human NK and T cells. J Immunol. (2003) 170:5464–9. doi: 10.4049/jimmunol.170.11.5464
39. Zeng R, Spolski R, Finkelstein SE, Oh S, Kovanen PE, Hinrichs CS, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. (2005) 201:139–48. doi: 10.1084/jem.20041057
40. Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-specific CAR T cells coexpressing IL15 and IL21 have superior expansion and antitumor activity against hepatocellular carcinoma. Cancer Immunol Res. (2020) 8:309–20. doi: 10.1158/2326-6066.CIR-19-0293
41. Nguyen R, Doubrovina E, Mousset CM, Jin BY, Okada R, Zhang X, et al. Cooperative armoring of CAR and TCR T cells by T cell-restricted IL15 and IL21 universally enhances solid tumor efficacy. Clin Cancer Res. (2024) 30:1555–66. doi: 10.1158/1078-0432.CCR-23-1872
42. Vignali DA and Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. (2012) 13:722–8. doi: 10.1038/ni.2366
43. Bastos KR, Marinho CR, Barboza R, Russo M, Alvarez JM, and D’Imperio Lima MR. What kind of message does IL-12/IL-23 bring to macrophages and dendritic cells? Microbes Infect. (2004) 6:630–6. doi: 10.1016/j.micinf.2004.02.012
44. Kerkar SP, Goldszmid RS, Muranski P, Chinnasamy D, Yu Z, Reger RN, et al. IL-12 triggers a programmatic change in dysfunctional myeloid-derived cells within mouse tumors. J Clin Invest. (2011) 121:4746–57. doi: 10.1172/JCI58814
45. Leonard JP, Sherman ML, Fisher GL, Buchanan LJ, Larsen G, Atkins MB, et al. Effects of single-dose interleukin-12 exposure on interleukin-12-associated toxicity and interferon-gamma production. Blood. (1997) 90:2541–8.
46. Bortolanza S, Bunuales M, Otano I, Gonzalez-Aseguinolaza G, Ortiz-de-Solorzano C, Perez D, et al. Treatment of pancreatic cancer with an oncolytic adenovirus expressing interleukin-12 in Syrian hamsters. Mol Ther. (2009) 17:614–22. doi: 10.1038/mt.2009.9
47. Zhang L, Morgan RA, Beane JD, Zheng Z, Dudley ME, Kassim SH, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. (2015) 21:2278–88. doi: 10.1158/1078-0432.CCR-14-2085
48. O’Cearbhaill RE, Park JH, Halton EF, Diamonte CR, Mead E, Lakhman Y, et al. A phase I clinical trial of autologous chimeric antigen receptor (CAR) T cells genetically engineered to secrete IL-12 and to target the MUC16ecto antigen in patients (pts) with MUC16ecto+ recurrent high-grade serous ovarian cancer (HGSOC). Gynecol Oncol. (2020) 159:42. doi: 10.1016/j.ygyno.2020.06.089
49. Hu J, Yang Q, Zhang W, Du H, Chen Y, Zhao Q, et al. Cell membrane-anchored and tumor-targeted IL-12 (attIL12)-T cell therapy for eliminating large and heterogeneous solid tumors. J Immunother Cancer. (2022) 10:e003633. doi: 10.1136/jitc-2021-003633
50. Lee EHJ, Murad JP, Christian L, Gibson J, Yamaguchi Y, Cullen C, et al. Antigen-dependent IL-12 signaling in CAR T cells promotes regional to systemic disease targeting. Nat Commun. (2023) 14:4737. doi: 10.1038/s41467-023-40115-1
51. Hombach A, Barden M, Hannappel L, Chmielewski M, Rappl G, Sachinidis A, et al. IL12 integrated into the CAR exodomain converts CD8(+) T cells to poly-functional NK-like cells with superior killing of antigen-loss tumors. Mol Ther. (2022) 30:593–605. doi: 10.1016/j.ymthe.2021.10.011
52. Bhatia V, Kamat NV, Pariva TE, Wu LT, Tsao A, Sasaki K, et al. Targeting advanced prostate cancer with STEAP1 chimeric antigen receptor T cell and tumor-localized IL-12 immunotherapy. Nat Commun. (2023) 14:2041. doi: 10.1038/s41467-023-37874-2
53. Zhu Y, Wang K, Yue L, Zuo D, Sheng J, Lan S, et al. Mesothelin CAR-T cells expressing tumor-targeted immunocytokine IL-12 yield durable efficacy and fewer side effects. Pharmacol Res. (2024) 203:107186. doi: 10.1016/j.phrs.2024.107186
54. Smole A, Benton A, Poussin MA, Eiva MA, Mezzanotte C, Camisa B, et al. Expression of inducible factors reprograms CAR-T cells for enhanced function and safety. Cancer Cell. (2022) 40:1470–87 e7. doi: 10.1016/j.ccell.2022.11.006
55. Liu Y, Di S, Shi B, Zhang H, Wang Y, Wu X, et al. Armored inducible expression of IL-12 enhances antitumor activity of glypican-3-targeted chimeric antigen receptor-engineered T cells in hepatocellular carcinoma. J Immunol. (2019) 203:198–207. doi: 10.4049/jimmunol.1800033
56. Ma X, Shou P, Smith C, Chen Y, Du H, Sun C, et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat Biotechnol. (2020) 38:448–59. doi: 10.1038/s41587-019-0398-2
57. Wang D, Shao Y, Zhang X, Lu G, and Liu B. IL-23 and PSMA-targeted duo-CAR T cells in Prostate Cancer Eradication in a preclinical model. J Transl Med. (2020) 18:23. doi: 10.1186/s12967-019-02206-w
58. Yan J, Smyth MJ, and Teng MWL. Interleukin (IL)-12 and IL-23 and their conflicting roles in cancer. Cold Spring Harb Perspect Biol. (2018) 10:a028530. doi: 10.1101/cshperspect.a028530
59. Brog RA, Ferry SL, Schiebout CT, Messier CM, Cook WJ, Abdullah L, et al. Superkine IL-2 and IL-33 armored CAR T cells reshape the tumor microenvironment and reduce growth of multiple solid tumors. Cancer Immunol Res. (2022) 10:962–77. doi: 10.1158/2326-6066.CIR-21-0536
60. Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature. (2020) 579:130–5. doi: 10.1038/s41586-020-2015-4
61. Stier MT, Zhang J, Goleniewska K, Cephus JY, Rusznak M, Wu L, et al. IL-33 promotes the egress of group 2 innate lymphoid cells from the bone marrow. J Exp Med. (2018) 215:263–81. doi: 10.1084/jem.20170449
62. Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered tumor-targeted T cells mediate enhanced anti-tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. (2018) 23:2130–41. doi: 10.1016/j.celrep.2018.04.051
63. Chmielewski M and Abken H. CAR T cells releasing IL-18 convert to T-bet(high) foxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. (2017) 21:3205–19. doi: 10.1016/j.celrep.2017.11.063
64. Jaspers JE, Khan JF, Godfrey WD, Lopez AV, Ciampricotti M, Rudin CM, et al. IL-18-secreting CAR T cells targeting DLL3 are highly effective in small cell lung cancer models. J Clin Invest. (2023) 133:e166028. doi: 10.1172/JCI166028
65. Hull CM, Larcombe-Young D, Mazza R, George M, Davies DM, Schurich A, et al. Granzyme B-activated IL18 potentiates alphabeta and gammadelta CAR T cell immunotherapy in a tumor-dependent manner. Mol Ther. (2024) 32:2373–92. doi: 10.1016/j.ymthe.2024.05.013
66. Fischer-Riepe L, Kailayangiri S, Zimmermann K, Pfeifer R, Aigner M, Altvater B, et al. Preclinical development of CAR T cells with antigen-inducible IL18 enforcement to treat GD2-positive solid cancers. Clin Cancer Res. (2024) 30:3564–77. doi: 10.1158/1078-0432.CCR-23-3157
67. Lange S, Sand LGL, Bell M, Patil SL, Langfitt D, and Gottschalk S. A chimeric GM-CSF/IL18 receptor to sustain CAR T-cell function. Cancer Discov. (2021) 11:1661–71. doi: 10.1158/2159-8290.CD-20-0896
68. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. (2017) 7:1238–47. doi: 10.1158/2159-8290.CD-17-0538
69. Lin FY, Stuckert A, Tat C, White M, Ruggieri L, Zhang H, et al. Phase I trial of GD2.CART cells augmented with constitutive interleukin-7 receptor for treatment of high-grade pediatric CNS tumors. J Clin Oncol. (2024) 42:2769–79. doi: 10.1200/JCO.23.02019
70. Sakunrangsit N, Khuisangeam N, Inthanachai T, Yodsurang V, Taechawattananant P, Suppipat K, et al. Incorporating IL7 receptor alpha signaling in the endodomain of B7H3-targeting chimeric antigen receptor T cells mediates antitumor activity in glioblastoma. Cancer Immunol Immunother. (2024) 73:98. doi: 10.1007/s00262-024-03685-7
71. Zhai Y, Li G, Pan C, Yu M, Hu H, Wang D, et al. The development and potent antitumor efficacy of CD44/CD133 dual-targeting IL7Ralpha-armored CAR-T cells against glioblastoma. Cancer Lett. (2025) 614:217541. doi: 10.1016/j.canlet.2025.217541
72. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. (2018) 359:1037–42. doi: 10.1126/science.aar3246
73. Kalbasi A, Siurala M, Su LL, Tariveranmoshabad M, Picton LK, Ravikumar P, et al. Potentiating adoptive cell therapy using synthetic IL-9 receptors. Nature. (2022) 607:360–5. doi: 10.1038/s41586-022-04801-2
74. Bell M, Lange S, Sejdiu BI, Ibanez J, Shi H, Sun X, et al. Modular chimeric cytokine receptors with leucine zippers enhance the antitumour activity of CAR T cells via JAK/STAT signalling. Nat BioMed Eng. (2024) 8:380–96. doi: 10.1038/s41551-023-01143-w
75. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T cells with customized therapeutic response programs using synthetic notch receptors. Cell. (2016) 167:419–32 e16. doi: 10.1016/j.cell.2016.09.011
76. Allen GM, Frankel NW, Reddy NR, Bhargava HK, Yoshida MA, Stark SR, et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science. (2022) 378:eaba1624. doi: 10.1126/science.aba1624
77. Liu S, Ren J, and Ten Dijke P. Targeting TGFbeta signal transduction for cancer therapy. Signal Transduct Target Ther. (2021) 6:8. doi: 10.1038/s41392-020-00436-9
78. Chen W. TGF-beta regulation of T cells. Annu Rev Immunol. (2023) 41:483–512. doi: 10.1146/annurev-immunol-101921-045939
79. Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-beta receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. (2018) 26:1855–66. doi: 10.1016/j.ymthe.2018.05.003
80. Zanvit P, van Dyk D, Fazenbaker C, McGlinchey K, Luo W, Pezold JM, et al. Antitumor activity of AZD0754, a dnTGFbetaRII-armored, STEAP2-targeted CAR-T cell therapy, in prostate cancer. J Clin Invest. (2023) 133:e169655. doi: 10.1172/JCI169655
81. Li K, Xu J, Wang J, Lu C, Dai Y, Dai Q, et al. Dominant-negative transforming growth factor-beta receptor-armoured mesothelin-targeted chimeric antigen receptor T cells slow tumour growth in a mouse model of ovarian cancer. Cancer Immunol Immunother. (2023) 72:917–28. doi: 10.1007/s00262-022-03290-6
82. Zhu S, Hu J, Lin J, Wang C, and Wang E. Co-expression of dominant-negative TGF-beta receptor 2 enhances the therapeutic efficacy of novel TREM1/DAP12-BB-based CAR-T cells in solid tumours. Immunology. (2025) 174:310–21. doi: 10.1111/imm.13888
83. Tran TM, Chand Thakuri BK, Nurmukhambetova S, Lee JJ, Hu P, Tran NQ, et al. Armored TGFbetaRIIDN ROR1-CAR T cells reject solid tumors and resist suppression by constitutively-expressed and treatment-induced TGFbeta1. J Immunother Cancer. (2024) 12:e008261. doi: 10.1136/jitc-2023-008261
84. Barrett AM, Britton ZT, Carrasco RA, Breen S, Broggi MAS, Hatke AL, et al. Preclinical evaluation of AZD6422, an armored chimeric antigen receptor T cell targeting CLDN18.2 in gastric, pancreatic, and esophageal cancers. Clin Cancer Res. (2024) 30:5413–29. doi: 10.1158/1078-0432.CCR-24-1853
85. Li N, Rodriguez JL, Yin Y, Logun MT, Zhang L, Yu S, et al. Armored bicistronic CAR T cells with dominant-negative TGF-beta receptor II to overcome resistance in glioblastoma. Mol Ther. (2024) 32:3522–38. doi: 10.1016/j.ymthe.2024.07.020
86. Zheng S, Che X, Zhang K, Bai Y, and Deng H. Potentiating CAR-T cell function in the immunosuppressive tumor microenvironment by inverting the TGF-beta signal. Mol Ther. (2025) 33:688–702. doi: 10.1016/j.ymthe.2024.12.014
87. Narayan V, Barber-Rotenberg JS, Jung IY, Lacey SF, Rech AJ, Davis MM, et al. PSMA-targeting TGFbeta-insensitive armored CAR T cells in metastatic castration-resistant prostate cancer: a phase 1 trial. Nat Med. (2022) 28:724–34. doi: 10.1038/s41591-022-01726-1
88. McKean M, Carabasi MH, Stein MN, Schweizer MT, Luke JJ, Narayan V, et al. Safety and early efficacy results from a phase 1, multicenter trial of PSMA-targeted armored CAR T cells in patients with advanced mCRPC. J Clin Oncol. (2022) 40:094. doi: 10.1200/JCO.2022.40.6_suppl.094
89. Gladney W, Vultur A, Schweizer M, Fraietta J, Rech A, June C, et al. 335 Analyses of severe immune-mediated toxicity in patients with advanced mCRPC treated with a PSMA-targeted armored CAR T-cells. J ImmunoTher Cancer. (2022) 10:A353. doi: 10.1136/jitc-2022-SITC2022.0335
90. Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, and Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. (2018) 14:317–24. doi: 10.1038/nchembio.2565
91. Hou AJ, Shih RM, Uy BR, Shafer A, Chang ZL, Comin-Anduix B, et al. IL-13Ralpha2/TGF-beta bispecific CAR-T cells counter TGF-beta-mediated immune suppression and potentiate anti-tumor responses in glioblastoma. Neuro Oncol. (2024) 26:1850–66. doi: 10.1093/neuonc/noae126
92. Li G, Liao G, Xie J, Liu B, Li X, and Qiu M. Overexpression of SMAD7 improves the function of EGFR-targeted human CAR-T cells against non-small-cell lung cancer. Respirology. (2023) 28:869–80. doi: 10.1111/resp.14541
93. Liang S, Zheng R, Zuo B, Li J, Wang Y, Han Y, et al. SMAD7 expression in CAR-T cells improves persistence and safety for solid tumors. Cell Mol Immunol. (2024) 21:213–26. doi: 10.1038/s41423-023-01120-y
94. Tang N, Cheng C, Zhang X, Qiao M, Li N, Mu W, et al. TGF-beta inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. (2020) 5:e133977. doi: 10.1172/jci.insight.133977
95. Gumber D and Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. EBioMedicine. (2022) 77:103941. doi: 10.1016/j.ebiom.2022.103941
96. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. (2016) 126:3130–44. doi: 10.1172/JCI83092
97. Jiang Y, Li Y, and Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. (2015) 6:e1792. doi: 10.1038/cddis.2015.162
98. Ping Y, Li F, Nan S, Zhang D, Shi X, Shan J, et al. Augmenting the effectiveness of CAR-T cells by enhanced self-delivery of PD-1-neutralizing scFv. Front Cell Dev Biol. (2020) 8:803. doi: 10.3389/fcell.2020.00803
99. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol. (2018) 36:847–56. doi: 10.1038/nbt.4195
100. Yang C, You J, Pan Q, Tang Y, Cai L, Huang Y, et al. Targeted delivery of a PD-1-blocking scFv by CD133-specific CAR-T cells using nonviral Sleeping Beauty transposition shows enhanced antitumour efficacy for advanced hepatocellular carcinoma. BMC Med. (2023) 21:327. doi: 10.1186/s12916-023-03016-0
101. Harrasser M, Gohil SH, Lau H, Della Peruta M, Muczynski V, Patel D, et al. Inducible localized delivery of an anti-PD-1 scFv enhances anti-tumor activity of ROR1 CAR-T cells in TNBC. Breast Cancer Res. (2022) 24:39. doi: 10.1186/s13058-022-01531-1
102. Wang Y, Fang X, Li M, Ye J, Zhao S, Yu L, et al. Mesothelin CAR-T cells secreting PD-L1 blocking scFv for pancreatic cancer treatment. Cancer Genet. (2022) 268-269:103–10. doi: 10.1016/j.cancergen.2022.10.003
103. Wang Y, Cho JW, Kastrunes G, Buck A, Razimbaud C, Culhane AC, et al. Immune-restoring CAR-T cells display antitumor activity and reverse immunosuppressive TME in a humanized ccRCC mouse model. iScience. (2024) 27:108879. doi: 10.1016/j.isci.2024.108879
104. Liu Z, Xia Y, Li L, Sun Y, Lin Z, Rong L, et al. Abstract CT134: Non-viral mesothelin-targeted CAR-T cells armored with IFNg-induced secretion of PD-1 nanobody in treatment of Malignant mesothelioma in phase I clinical trial. Cancer Res. (2023) 83:CT134. doi: 10.1158/1538-7445.AM2023-CT134
105. Chen J, Zhu T, Jiang G, Zeng Q, Li Z, and Huang X. Target delivery of a PD-1-TREM2 scFv by CAR-T cells enhances anti-tumor efficacy in colorectal cancer. Mol Cancer. (2023) 22:131. doi: 10.1186/s12943-023-01830-x
106. Molgora M, Esaulova E, Vermi W, Hou J, Chen Y, Luo J, et al. TREM2 modulation remodels the tumor myeloid landscape enhancing anti-PD-1 immunotherapy. Cell. (2020) 182:886–900 e17. doi: 10.1016/j.cell.2020.07.013
107. Chen X, Yang S, Li S, Qu Y, Wang HY, Liu J, et al. Secretion of bispecific protein of anti-PD-1 fused with TGF-beta trap enhances antitumor efficacy of CAR-T cell therapy. Mol Ther Oncolyt. (2021) 21:144–57. doi: 10.1016/j.omto.2021.03.014
108. Logtenberg MEW, Scheeren FA, and Schumacher TN. The CD47-SIRPalpha immune checkpoint. Immunity. (2020) 52:742–52. doi: 10.1016/j.immuni.2020.04.011
109. Chen H, Yang Y, Deng Y, Wei F, Zhao Q, Liu Y, et al. Delivery of CD47 blocker SIRPalpha-Fc by CAR-T cells enhances antitumor efficacy. J Immunother Cancer. (2022) 10:e003737. doi: 10.1136/jitc-2021-003737
110. Martins TA, Kaymak D, Tatari N, Gerster F, Hogan S, Ritz MF, et al. Enhancing anti-EGFRvIII CAR T cell therapy against glioblastoma with a paracrine SIRPgamma-derived CD47 blocker. Nat Commun. (2024) 15:9718. doi: 10.1038/s41467-024-54129-w
111. Xie YJ, Dougan M, Ingram JR, Pishesha N, Fang T, Momin N, et al. Improved antitumor efficacy of chimeric antigen receptor T cells that secrete single-domain antibody fragments. Cancer Immunol Res. (2020) 8:518–29. doi: 10.1158/2326-6066.CIR-19-0734
112. Yamada-Hunter SA, Theruvath J, McIntosh BJ, Freitas KA, Lin F, Radosevich MT, et al. Engineered CD47 protects T cells for enhanced antitumour immunity. Nature. (2024) 630:457–65. doi: 10.1038/s41586-024-07443-8
113. Yegutkin GG and Boison D. ATP and adenosine metabolism in cancer: exploitation for therapeutic gain. Pharmacol Rev. (2022) 74:797–822. doi: 10.1124/pharmrev.121.000528
114. Vijayan D, Young A, Teng MWL, and Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer. (2017) 17:709–24. doi: 10.1038/nrc.2017.86
115. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. (2017) 127:929–41. doi: 10.1172/JCI89455
116. Seifert M, Benmebarek MR, Briukhovetska D, Markl F, Dorr J, Cadilha BL, et al. Impact of the selective A2(A)R and A2(B)R dual antagonist AB928/etrumadenant on CAR T cell function. Br J Cancer. (2022) 127:2175–85. doi: 10.1038/s41416-022-02013-z
117. Qu Y, Dunn ZS, Chen X, MacMullan M, Cinay G, Wang HY, et al. Adenosine deaminase 1 overexpression enhances the antitumor efficacy of chimeric antigen receptor-engineered T cells. Hum Gene Ther. (2022) 33:223–36. doi: 10.1089/hum.2021.050
118. Hu Y, Sarkar A, Song K, Michael S, Hook M, Wang R, et al. Selective refueling of CAR T cells using ADA1 and CD26 boosts antitumor immunity. Cell Rep Med. (2024) 5:101530. doi: 10.1016/j.xcrm.2024.101530
119. Liu G, Zhang Q, Liu G, Li D, Zhang L, Gu Z, et al. Disruption of adenosine 2A receptor improves the anti-tumor function of anti-mesothelin CAR T cells both in vitro and in vivo. Exp Cell Res. (2021) 409:112886. doi: 10.1016/j.yexcr.2021.112886
120. Giuffrida L, Sek K, Henderson MA, Lai J, Chen AXY, Meyran D, et al. CRISPR/Cas9 mediated deletion of the adenosine A2A receptor enhances CAR T cell efficacy. Nat Commun. (2021) 12:3236. doi: 10.1038/s41467-021-23331-5
121. Li N, Tang N, Cheng C, Hu T, Wei X, Han W, et al. Improving the anti-solid tumor efficacy of CAR-T cells by inhibiting adenosine signaling pathway. Oncoimmunology. (2020) 9:1824643. doi: 10.1080/2162402X.2020.1824643
122. Tao R, Han X, Bai X, Yu J, Ma Y, Chen W, et al. Revolutionizing cancer treatment: enhancing CAR-T cell therapy with CRISPR/Cas9 gene editing technology. Front Immunol. (2024) 15:1354825. doi: 10.3389/fimmu.2024.1354825
123. Dimitri A, Herbst F, and Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21:78. doi: 10.1186/s12943-022-01559-z
124. Chen M, Chen F, Gao Z, Li X, Hu L, Yang S, et al. CAFs and T cells interplay: The emergence of a new arena in cancer combat. BioMed Pharmacother. (2024) 177:117045. doi: 10.1016/j.biopha.2024.117045
125. Kakarla S, Chow KK, Mata M, Shaffer DR, Song XT, Wu MF, et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol Ther. (2013) 21:1611–20. doi: 10.1038/mt.2013.110
126. Xiao Z, Todd L, Huang L, Noguera-Ortega E, Lu Z, Huang L, et al. Desmoplastic stroma restricts T cell extravasation and mediates immune exclusion and immunosuppression in solid tumors. Nat Commun. (2023) 14:5110. doi: 10.1038/s41467-023-40850-5
127. Wang LC, Lo A, Scholler J, Sun J, Majumdar RS, Kapoor V, et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res. (2014) 2:154–66. doi: 10.1158/2326-6066.CIR-13-0027
128. Wehrli M, Guinn S, Birocchi F, Kuo A, Sun Y, Larson RC, et al. Mesothelin CAR T cells secreting anti-FAP/anti-CD3 molecules efficiently target pancreatic adenocarcinoma and its stroma. Clin Cancer Res. (2024) 30:1859–77. doi: 10.1158/1078-0432.CCR-23-3841
129. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, and Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. (2020) 11:5120. doi: 10.1038/s41467-020-18794-x
130. Caruana I, Savoldo B, Hoyos V, Weber G, Liu H, Kim ES, et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med. (2015) 21:524–9. doi: 10.1038/nm.3833
131. Zhao R, Cui Y, Zheng Y, Li S, Lv J, Wu Q, et al. Human hyaluronidase PH20 potentiates the antitumor activities of mesothelin-specific CAR-T cells against gastric cancer. Front Immunol. (2021) 12:660488. doi: 10.3389/fimmu.2021.660488
132. Xiong X, Xi J, Liu Q, Wang C, Jiang Z, Yue SY, et al. Co-expression of IL-7 and PH20 promote anti-GPC3 CAR-T tumour suppressor activity in vivo and in vitro. Liver Int. (2021) 41:1033–43. doi: 10.1111/liv.14771
133. Zheng R, Shen K, Liang S, Lyu Y, Zhang S, Dong H, et al. Specific ECM degradation potentiates the antitumor activity of CAR-T cells in solid tumors. Cell Mol Immunol. (2024) 21:1491–504. doi: 10.1038/s41423-024-01228-9
134. De Martino M, Rathmell JC, Galluzzi L, and Vanpouille-Box C. Cancer cell metabolism and antitumour immunity. Nat Rev Immunol. (2024) 24:654–69. doi: 10.1038/s41577-024-01026-4
135. Stine ZE, Schug ZT, Salvino JM, and Dang CV. Targeting cancer metabolism in the era of precision oncology. Nat Rev Drug Discov. (2022) 21:141–62. doi: 10.1038/s41573-021-00339-6
136. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
137. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
138. Su R, Shao Y, Huang M, Liu D, Yu H, and Qiu Y. Immunometabolism in cancer: basic mechanisms and new targeting strategy. Cell Death Discov. (2024) 10:236. doi: 10.1038/s41420-024-02006-2
139. Holman GD. Structure, function and regulation of mammalian glucose transporters of the SLC2 family. Pflugers Arch. (2020) 472:1155–75. doi: 10.1007/s00424-020-02411-3
140. Shi Y, Kotchetkov IS, Dobrin A, Hanina SA, Rajasekhar VK, Healey JH, et al. GLUT1 overexpression enhances CAR T cell metabolic fitness and anti-tumor efficacy. Mol Ther. (2024) 32:2393–405. doi: 10.1016/j.ymthe.2024.05.006
141. Hu W, Li F, Liang Y, Liu S, Wang S, Shen C, et al. Glut3 overexpression improves environmental glucose uptake and antitumor efficacy of CAR-T cells in solid tumors. J Immunother Cancer. (2025) 13:e010540. doi: 10.1136/jitc-2024-010540
142. Hwang SM, Awasthi D, Jeong J, Sandoval TA, Chae CS, Ramos Y, et al. Transgelin 2 guards T cell lipid metabolism and antitumour function. Nature. (2024) 635:1010–8. doi: 10.1038/s41586-024-08071-y
143. Kay EJ, Zanivan S, and Rufini A. Proline metabolism shapes the tumor microenvironment: from collagen deposition to immune evasion. Curr Opin Biotechnol. (2023) 84:103011. doi: 10.1016/j.copbio.2023.103011
144. Wei Z, Liu X, Cheng C, Yu W, and Yi P. Metabolism of amino acids in cancer. Front Cell Dev Biol. (2020) 8:603837. doi: 10.3389/fcell.2020.603837
145. Ye L, Park JJ, Peng L, Yang Q, Chow RD, Dong MB, et al. A genome-scale gain-of-function CRISPR screen in CD8 T cells identifies proline metabolism as a means to enhance CAR-T therapy. Cell Metab. (2022) 34:595–614 e14. doi: 10.1016/j.cmet.2022.02.009
146. Zhao Y, Chen J, Andreatta M, Feng B, Xie YQ, Wenes M, et al. IL-10-expressing CAR T cells resist dysfunction and mediate durable clearance of solid tumors and metastases. Nat Biotechnol. (2024) 42:1693–704. doi: 10.1038/s41587-023-02060-8
147. Salkeni MA and Naing A. Interleukin-10 in cancer immunotherapy: from bench to bedside. Trends Cancer. (2023) 9:716–25. doi: 10.1016/j.trecan.2023.05.003
148. Oft M. IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res. (2014) 2:194–9. doi: 10.1158/2326-6066.CIR-13-0214
149. Lontos K, Wang Y, Joshi SK, Frisch AT, Watson MJ, Kumar A, et al. Metabolic reprogramming via an engineered PGC-1alpha improves human chimeric antigen receptor T-cell therapy against solid tumors. J Immunother Cancer. (2023) 11:e006522. doi: 10.1136/jitc-2022-006522
150. Flugel CL, Majzner RG, Krenciute G, Dotti G, Riddell SR, Wagner DL, et al. Overcoming on-target, off-tumour toxicity of CAR T cell therapy for solid tumours. Nat Rev Clin Oncol. (2023) 20:49–62. doi: 10.1038/s41571-022-00704-3
151. Kosti P, Opzoomer JW, Larios-Martinez KI, Henley-Smith R, Scudamore CL, Okesola M, et al. Hypoxia-sensing CAR T cells provide safety and efficacy in treating solid tumors. Cell Rep Med. (2021) 2:100227. doi: 10.1016/j.xcrm.2021.100227
152. Lowe KL, Cole D, Kenefeck R, OK I, Lepore M, and Jakobsen BK. Novel TCR-based biologics: mobilising T cells to warm ‘cold’ tumours. Cancer Treat Rev. (2019) 77:35–43. doi: 10.1016/j.ctrv.2019.06.001
153. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol. (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
154. Choi BD, Gerstner ER, Frigault MJ, Leick MB, Mount CW, Balaj L, et al. Intraventricular CARv3-TEAM-E T cells in recurrent glioblastoma. N Engl J Med. (2024) 390:1290–8. doi: 10.1056/NEJMoa2314390
155. Mun SS, Meyerberg J, Peraro L, Korontsvit T, Gardner T, Malviya M, et al. Dual targeting ovarian cancer by Muc16 CAR T cells secreting a bispecific T cell engager antibody for an intracellular tumor antigen WT1. Cancer Immunol Immunother. (2023) 72:3773–86. doi: 10.1007/s00262-023-03529-w
156. Cao G, Zhang G, Liu M, Liu J, Wang Q, Zhu L, et al. GPC3-targeted CAR-T cells secreting B7H3-targeted BiTE exhibit potent cytotoxicity activity against hepatocellular carcinoma cell in the in vitro assay. Biochem Biophys Rep. (2022) 31:101324. doi: 10.1016/j.bbrep.2022.101324
157. Pascual-Pasto G, McIntyre B, Hines MG, Giudice AM, Garcia-Gerique L, Hoffmann J, et al. CAR T-cell-mediated delivery of bispecific innate immune cell engagers for neuroblastoma. Nat Commun. (2024) 15:7141. doi: 10.1038/s41467-024-51337-2
158. Li D and Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. (2021) 6:291. doi: 10.1038/s41392-021-00687-0
159. Hajam IA, Dar PA, Shahnawaz I, Jaume JC, and Lee JH. Bacterial flagellin-a potent immunomodulatory agent. Exp Mol Med. (2017) 49:e373. doi: 10.1038/emm.2017.172
160. Niu X, Zhang P, Dai L, Peng X, Liu Z, Tang Y, et al. Flagellin engineering enhances CAR-T cell function by reshaping tumor microenvironment in solid tumors. J Immunother Cancer. (2025) 13:e010237. doi: 10.1136/jitc-2024-010237
161. D’Elios MM, Amedei A, Cappon A, Del Prete G, and de Bernard M. The neutrophil-activating protein of Helicobacter pylori (HP-NAP) as an immune modulating agent. FEMS Immunol Med Microbiol. (2007) 50:157–64. doi: 10.1111/j.1574-695X.2007.00258.x
162. Amedei A, Cappon A, Codolo G, Cabrelle A, Polenghi A, Benagiano M, et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J Clin Invest. (2006) 116:1092–101. doi: 10.1172/JCI27177
163. Jin C, Ma J, Ramachandran M, Yu D, and Essand M. CAR T cells expressing a bacterial virulence factor trigger potent bystander antitumour responses in solid cancers. Nat BioMed Eng. (2022) 6:830–41. doi: 10.1038/s41551-022-00875-5
164. Rehwinkel J and Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. (2020) 20:537–51. doi: 10.1038/s41577-020-0288-3
165. Johnson LR, Lee DY, Eacret JS, Ye D, June CH, and Minn AJ. The immunostimulatory RNA RN7SL1 enables CAR-T cells to enhance autonomous and endogenous immune function. Cell. (2021) 184:4981–95 e14. doi: 10.1016/j.cell.2021.08.004
166. McKenna HJ, Stocking KL, Miller RE, Brasel K, De Smedt T, Maraskovsky E, et al. Mice lacking flt3 ligand have deficient hematopoiesis affecting hematopoietic progenitor cells, dendritic cells, and natural killer cells. Blood. (2000) 95:3489–97. doi: 10.1182/blood.V95.11.3489
167. Shortman K and Naik SH. Steady-state and inflammatory dendritic-cell development. Nat Rev Immunol. (2007) 7:19–30. doi: 10.1038/nri1996
168. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. (2020) 21:914–26. doi: 10.1038/s41590-020-0676-7
169. Bettadapur A, Miller HW, and Ralston KS. Biting off what can be chewed: trogocytosis in health, infection, and disease. Infect Immun. (2020) 88:10.1128/iai.00930-19. doi: 10.1128/IAI.00930-19
170. Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. (2019) 568:112–6. doi: 10.1038/s41586-019-1054-1
171. Zhai Y, Du Y, Li G, Yu M, Hu H, Pan C, et al. Trogocytosis of CAR molecule regulates CAR-T cell dysfunction and tumor antigen escape. Signal Transduct Target Ther. (2023) 8:457. doi: 10.1038/s41392-023-01708-w
172. Lu Z, McBrearty N, Chen J, Tomar VS, Zhang H, De Rosa G, et al. ATF3 and CH25H regulate effector trogocytosis and anti-tumor activities of endogenous and immunotherapeutic cytotoxic T lymphocytes. Cell Metab. (2022) 34:1342–58 e7. doi: 10.1016/j.cmet.2022.08.007
173. Barbera S, Schuiling MJA, Sanjaya NA, Pietila I, Saren T, Essand M, et al. Trogocytosis of chimeric antigen receptors between T cells is regulated by their transmembrane domains. Sci Immunol. (2025) 10:eado2054. doi: 10.1126/sciimmunol.ado2054
174. Hong Y, Walling BL, Kim HR, Serratelli WS, Lozada JR, Sailer CJ, et al. ST3GAL1 and betaII-spectrin pathways control CAR T cell migration to target tumors. Nat Immunol. (2023) 24:1007–19. doi: 10.1038/s41590-023-01498-x
175. Skovgard MS, Hocine HR, Saini JK, Moroz M, Bellis RY, Banerjee S, et al. Imaging CAR T-cell kinetics in solid tumors: Translational implications. Mol Ther Oncolyt. (2021) 22:355–67. doi: 10.1016/j.omto.2021.06.006
176. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
177. Lake JA, Woods E, Hoffmeyer E, Schaller KL, Cruz-Cruz J, Fernandez J, et al. Directing B7-H3 chimeric antigen receptor T cell homing through IL-8 induces potent antitumor activity against pediatric sarcoma. J Immunother Cancer. (2024) 12:e009221. doi: 10.1136/jitc-2024-009221
178. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-Cells Targeting the Integrin alphavbeta6 and Co-Expressing the Chemokine Receptor CXCR2 Demonstrate Enhanced Homing and Efficacy against Several Solid Malignancies. Cancers (Basel). (2019) 11:674. doi: 10.3390/cancers11050674
179. Talbot LJ, Chabot A, Ross AB, Beckett A, Nguyen P, Fleming A, et al. Redirecting B7-H3.CAR T cells to chemokines expressed in osteosarcoma enhances homing and antitumor activity in preclinical models. Clin Cancer Res. (2024) 30:4434–49. doi: 10.1158/1078-0432.CCR-23-3298
180. Liu G, Rui W, Zheng H, Huang D, Yu F, Zhang Y, et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur J Immunol. (2020) 50:712–24. doi: 10.1002/eji.201948457
181. Ruixin S, Yifan L, Chuanlong W, Min Z, Hong L, Guoxiu D, et al. Expressing IL-15/IL-18 and CXCR2 improve infiltration and survival of EGFRvIII-targeting CAR-T cells in breast cancer. Biochem Pharmacol. (2023) 212:115536. doi: 10.1016/j.bcp.2023.115536
182. Lesch S, Blumenberg V, Stoiber S, Gottschlich A, Ogonek J, Cadilha BL, et al. T cells armed with C-X-C chemokine receptor type 6 enhance adoptive cell therapy for pancreatic tumours. Nat BioMed Eng. (2021) 5:1246–60. doi: 10.1038/s41551-021-00737-6
183. Moon EK, Carpenito C, Sun J, Wang LC, Kapoor V, Predina J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res. (2011) 17:4719–30. doi: 10.1158/1078-0432.CCR-11-0351
184. Li H, Harrison EB, Li H, Hirabayashi K, Chen J, Li QX, et al. Targeting brain lesions of non-small cell lung cancer by enhancing CCL2-mediated CAR-T cell migration. Nat Commun. (2022) 13:2154. doi: 10.1038/s41467-022-29647-0
185. Wang Y, Wang J, Yang X, Yang J, Lu P, Zhao L, et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front Immunol. (2021) 12:628906. doi: 10.3389/fimmu.2021.628906
186. Li G, Guo J, Zheng Y, Ding W, Han Z, Qin L, et al. CXCR5 guides migration and tumor eradication of anti-EGFR chimeric antigen receptor T cells. Mol Ther Oncolyt. (2021) 22:507–17. doi: 10.1016/j.omto.2021.07.003
187. Li G, Zhang Q, Han Z, Zhu Y, Shen H, Liu Z, et al. IL-7 and CCR2b co-expression-mediated enhanced CAR-T survival and infiltration in solid tumors. Front Oncol. (2021) 11:734593. doi: 10.3389/fonc.2021.734593
188. Hui X, Farooq MA, Chen Y, Ajmal I, Ren Y, Xue M, et al. A novel strategy of co-expressing CXCR5 and IL-7 enhances CAR-T cell effectiveness in osteosarcoma. Front Immunol. (2024) 15:1462076. doi: 10.3389/fimmu.2024.1462076
189. Tian Y, Zhang L, Ping Y, Zhang Z, Yao C, Shen C, et al. CCR5 and IL-12 co-expression in CAR T cells improves antitumor efficacy by reprogramming tumor microenvironment in solid tumors. Cancer Immunol Immunother. (2025) 74:55. doi: 10.1007/s00262-024-03909-w
190. Cadilha BL, Benmebarek MR, Dorman K, Oner A, Lorenzini T, Obeck H, et al. Combined tumor-directed recruitment and protection from immune suppression enable CAR T cell efficacy in solid tumors. Sci Adv. (2021) 7:eabi5781. doi: 10.1126/sciadv.abi5781
191. Link A, Vogt TK, Favre S, Britschgi MR, Acha-Orbea H, Hinz B, et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat Immunol. (2007) 8:1255–65. doi: 10.1038/ni1513
192. Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, and McColl SR. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. (2013) 24:269–83. doi: 10.1016/j.cytogfr.2013.03.001
193. Adachi K, Kano Y, Nagai T, Okuyama N, Sakoda Y, and Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. (2018) 36:346–51. doi: 10.1038/nbt.4086
194. Sasaki T, Sakoda Y, Adachi K, Tokunaga Y, and Tamada K. Therapeutic effects of anti-GM2 CAR-T cells expressing IL-7 and CCL19 for GM2-positive solid cancer in xenograft model. Cancer Med. (2023) 12:12569–80. doi: 10.1002/cam4.5907
195. Lu LL, Xiao SX, Lin ZY, Bai JJ, Li W, Song ZQ, et al. GPC3-IL7-CCL19-CAR-T primes immune microenvironment reconstitution for hepatocellular carcinoma therapy. Cell Biol Toxicol. (2023) 39:3101–19. doi: 10.1007/s10565-023-09821-w
196. Ohta K, Sakoda Y, Adachi K, Shinozaki T, Nakajima M, Yasuda H, et al. Therapeutic efficacy of IL7/CCL19-expressing CAR-T cells in intractable solid tumor models of glioblastoma and pancreatic cancer. Cancer Res Commun. (2024) 4:2514–24. doi: 10.1158/2767-9764.CRC-24-0226
197. Luo H, Su J, Sun R, Sun Y, Wang Y, Dong Y, et al. Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clin Cancer Res. (2020) 26:5494–505. doi: 10.1158/1078-0432.CCR-20-0777
198. Nakajima TE, Kuboki Y, Fukahori M, Shimazu Y, Kondo S, Katsuya Y, et al. Updated results from first-in-human phase 1 dose-escalation trial of TAK-102, a GPC3-targeted armored CAR T cells, in patients with advanced solid tumors. J Clin Oncol. (2024) 42:2543. doi: 10.1200/JCO.2024.42.16_suppl.2543
199. Pang N, Shi J, Qin L, Chen A, Tang Y, Yang H, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol. (2021) 14:118. doi: 10.1186/s13045-021-01128-9
200. Zhang N, Liu X, Qin J, Sun Y, Xiong H, Lin B, et al. LIGHT/TNFSF14 promotes CAR-T cell trafficking and cytotoxicity through reversing immunosuppressive tumor microenvironment. Mol Ther. (2023) 31:2575–90. doi: 10.1016/j.ymthe.2023.06.015
201. Schumacher TN and Thommen DS. Tertiary lymphoid structures in cancer. Science. (2022) 375:eabf9419. doi: 10.1126/science.abf9419
202. Cai W, Tanaka K, Mi X, Rajasekhar VK, Khan JF, Yoo S, et al. Augmenting CAR T-cell functions with LIGHT. Cancer Immunol Res. (2024) 12:1361–79. doi: 10.1158/2326-6066.CIR-24-0246
203. Granger SW and Rickert S. LIGHT-HVEM signaling and the regulation of T cell-mediated immunity. Cytokine Growth Factor Rev. (2003) 14:289–96. doi: 10.1016/S1359-6101(03)00031-5
204. Fan Z, Yu P, Wang Y, Wang Y, Fu ML, Liu W, et al. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood. (2006) 107:1342–51. doi: 10.1182/blood-2005-08-3485
205. Shuptrine CW, Perez VM, Selitsky SR, Schreiber TH, and Fromm G. Shining a LIGHT on myeloid cell targeted immunotherapy. Eur J Cancer. (2023) 187:147–60. doi: 10.1016/j.ejca.2023.03.040
206. Duncan BB, Dunbar CE, and Ishii K. Applying a clinical lens to animal models of CAR-T cell therapies. Mol Ther Methods Clin Dev. (2022) 27:17–31. doi: 10.1016/j.omtm.2022.08.008
207. Zou JJ, Schoenhaut DS, Carvajal DM, Warrier RR, Presky DH, Gately MK, et al. Structure-function analysis of the p35 subunit of mouse interleukin 12. J Biol Chem. (1995) 270:5864–71. doi: 10.1074/jbc.270.11.5864
208. Zhang S, Black RG, Kohli K, Hayes BJ, Miller C, Koehne A, et al. B7-H3 specific CAR T cells for the naturally occurring, spontaneous canine sarcoma model. Mol Cancer Ther. (2022) 21:999–1009. doi: 10.1158/1535-7163.MCT-21-0726
209. Zhou Z, Li J, Hong J, Chen S, Chen M, Wang L, et al. Interleukin-15 and chemokine ligand 19 enhance cytotoxic effects of chimeric antigen receptor T cells using zebrafish xenograft model of gastric cancer. Front Immunol. (2022) 13:1002361. doi: 10.3389/fimmu.2022.1002361
210. Logun M, Wang X, Sun Y, Bagley SJ, Li N, Desai A, et al. Patient-derived glioblastoma organoids as real-time avatars for assessing responses to clinical CAR-T cell therapy. Cell Stem Cell. (2025) 32:181–90 e4. doi: 10.1016/j.stem.2024.11.010
211. Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. (2005) 102:9571–6. doi: 10.1073/pnas.0503726102
212. Busch DH, Frassle SP, Sommermeyer D, Buchholz VR, and Riddell SR. Role of memory T cell subsets for adoptive immunotherapy. Semin Immunol. (2016) 28:28–34. doi: 10.1016/j.smim.2016.02.001
213. Weninger W, Crowley MA, Manjunath N, and von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J Exp Med. (2001) 194:953–66. doi: 10.1084/jem.194.7.953
214. Csaplar M, Szollosi J, Gottschalk S, Vereb G, and Szoor A. Cytolytic activity of CAR T cells and maintenance of their CD4+ Subset is critical for optimal antitumor activity in preclinical solid tumor models. Cancers (Basel). (2021) 13:4301. doi: 10.3390/cancers13174301
215. Albelda SM. CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat Rev Clin Oncol. (2024) 21:47–66. doi: 10.1038/s41571-023-00832-4
216. Yang Y, Yang H, Alcaina Y, Puc J, Birt A, Vedvyas Y, et al. Inducible expression of interleukin-12 augments the efficacy of affinity-tuned chimeric antigen receptors in murine solid tumor models. Nat Commun. (2023) 14:2068. doi: 10.1038/s41467-023-37646-y
217. Chen AXY, Yap KM, Kim JS, Sek K, Huang YK, Dunbar PA, et al. Rewiring endogenous genes in CAR T cells for tumour-restricted payload delivery. Nature. (2025) 644:241–51. doi: 10.1038/s41586-025-09212-7
218. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. (2017) 7:39833. doi: 10.1038/srep39833
219. Javan B and Shahbazi M. Hypoxia-inducible tumour-specific promoters as a dual-targeting transcriptional regulation system for cancer gene therapy. Ecancermedicalscience. (2017) 11:751. doi: 10.3332/ecancer.2017.751
220. Zhou W, Miao J, Cheng Z, Wang Z, Wang J, Guo H, et al. Hypoxia-regulated secretion of IL-12 enhances antitumor activity and safety of CD19 CAR-T cells in the treatment of DLBCL. Mol Ther Oncolyt. (2023) 30:216–26. doi: 10.1016/j.omto.2023.08.009
221. Vogt KC, Silberman PC, Lin Q, Han JE, Laflin A, Gellineau HA, et al. Microenvironment actuated CAR T cells improve solid tumor efficacy without toxicity. Sci Adv. (2025) 11:eads3403. doi: 10.1126/sciadv.ads3403
222. Morris EC, Neelapu SS, Giavridis T, and Sadelain M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat Rev Immunol. (2022) 22:85–96. doi: 10.1038/s41577-021-00547-6
223. Yoshikawa T, Ito Y, Wu Z, Kasuya H, Nakashima T, Okamoto S, et al. Development of a chimeric cytokine receptor that captures IL-6 and enhances the antitumor response of CAR-T cells. Cell Rep Med. (2024) 5:101526. doi: 10.1016/j.xcrm.2024.101526
224. Lu L, Xie M, Yang B, Zhao WB, and Cao J. Enhancing the safety of CAR-T cell therapy: Synthetic genetic switch for spatiotemporal control. Sci Adv. (2024) 10:eadj6251. doi: 10.1126/sciadv.adj6251
225. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. (2014) 124:1277–87. doi: 10.1182/blood-2014-01-545020
226. Weber EW, Lynn RC, Sotillo E, Lattin J, Xu P, and Mackall CL. Pharmacologic control of CAR-T cell function using dasatinib. Blood Adv. (2019) 3:711–7. doi: 10.1182/bloodadvances.2018028720
227. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. (2019) 11:eaau5907. doi: 10.1126/scitranslmed.aau5907
Keywords: chimeric antigen receptor, armored, T-cells, immunotherapy, solid tumors, microenvironment, stroma, immunosuppression
Citation: Yang DD, Macmorland W and Arnold JN (2025) Current strategies for armoring chimeric antigen receptor T-cells to overcome barriers of the solid tumor microenvironment. Front. Immunol. 16:1643941. doi: 10.3389/fimmu.2025.1643941
Received: 09 June 2025; Accepted: 13 August 2025;
Published: 11 September 2025.
Edited by:
Astero Klampatsa, The Institute of Cancer Research, United KingdomReviewed by:
Yina Hsing Huang, Dartmouth College, United StatesKoichi Sasaki, Imperial College, United Kingdom
Copyright © 2025 Yang, Macmorland and Arnold. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James N. Arnold, amFtZXMuYXJub2xkQGtjbC5hYy51aw==