 Ziyu Wang1†
Ziyu Wang1† Ertao Jia
Ertao Jia Hongling Geng
Hongling Geng- 1The Fifth Clinical College of Guangzhou University of Chinese Medicine, Guangzhou, China
- 2Department of Rheumatology, Shenzhen Hospital, The University of Hong Kong, Shenzhen, China
- 3Department of Rheumatism, Guangdong Provincial Second Hospital of Traditional Chinese Medicine, Guangzhou, China
- 4Department of Gynecology, Guangdong Provincial Hospital of Chinese Medicine, Guangzhou, China
Neuromyelitis optica spectrum disorder (NMOSD) is an immune-mediated inflammatory demyelinating disease affecting the optic nerve and spinal cord. NMOSD frequently coexists with other autoimmune diseases. However, its concurrence with mixed connective tissue disease (MCTD) is rather rare and often overlooked. This study reports the first case in China of aquaporin-4 immunoglobulin G (AQP4-IgG) seropositive NMOSD preceding MCTD with long-term follow-up. Between 2016 and 2024, the patient successively developed left lower limb numbness, hiccups, vomiting, facial numbness, Raynaud’s phenomenon, finger swelling, digital sclerosis, and synovitis. Acute-phase management involved pulse steroid therapy, while remission maintenance utilized azathioprine, mycophenolate mofetil, rituximab, and inebilizumab for relapse prevention. This paper presents this case and reviews other cases of NMOSD combined with MCTD, aiming to contribute to the clinical understanding and management of this rare condition.
Introduction
Neuromyelitis optica spectrum disorder (NMOSD), formerly known as Devic’s disease, is a rare antibody-mediated central nervous system disorder. Typical manifestations include longitudinally extensive transverse myelitis, severe optic neuritis, and/or intractable vomiting and hiccups (postictal area syndrome) (1). Research indicates that aquaporin-4 immunoglobulin G (AQP4-IgG) is the causative factor of NMOSD. The pathological mechanism involves the binding of AQP4-IgG to anti-aquaporin-4 (AQP4) water channels on astrocyte foot processes, triggering complement cascade activation, resulting in astrocyte damage and subsequent secondary injury to oligodendrocytes and neurons (2).According to the revised criteria published by Wingerchuck et al., a positive AQP4 antibody with the presence of a single core clinical feature (optic neuritis, acute myelitis, or brainstem syndrome) is sufficient for diagnosis once other diseases have been ruled out (3). NMOSD may be associated with various types of autoimmune diseases. Among systemic autoimmune diseases, Sjögren’s Syndrome (SjS) and Systemic Lupus Erythematosus (SLE) were the most commonly associated with mixed connective tissue disease (MCTD). In contrast, Systemic Sclerosis (SSc) and MCTD are less common (4).
This paper reported a case of a middle-aged female with numbness of the left lower limb as the initial symptom and a positive serum AQP4-IgG, diagnosed with NMOSD. Seven years after the onset, the patient developed Raynaud’s phenomenon, swollen fingers, sclerodactyly, synovitis, alopecia, and localized cutaneous sclerosis with depigmentation on the forehead. Combined with positive anti–U1-ribonucleoprotein (RNP) antibodies and an Anti-Nuclear Antibody (ANA) titer >1:1000, the patient was ultimately diagnosed with MCTD concurrent with NMOSD. Furthermore, we also reviewed additional case reports on NMOSD coexisting with MCTD.
Case presentation
The 48-year-old female patient presented with progressive numbness in the left lower limb in June 2016, which was particularly prominent in the left foot. There was no weakness, blurred vision, dizziness, diplopia, crooked mouth, girdle sensation of the body, or disorders of urination and defecation. Among the ancillary diagnostic studies, magnetic resonance imaging (MRI) demonstrated a focal abnormal signal intensity in the spinal cord at the C7-T1 level, suggestive of a demyelinating lesion (Figure 1). The lumbar puncture indicated normal cerebrospinal fluid (CSF) pressure, with no significant abnormalities in the routine, biochemical, bacterial, cryptococcal, or tuberculosis examinations. Oligoclonal bands (OB) were negative; however, serum AQP4-IgG, as determined by cell-based assay (CBA), was positive with a titer of 1:100. Ultimately, she was diagnosed with NMOSD and received intravenous administration of methylprednisolone (MP) 0.5 grams/day for 5 days. Subsequently, the treatment regimen was adjusted to oral prednisone (Pred) 35 mg/day and azathioprine (AZA) 25 mg/day. Due to hepatic impairment, the Pred dosage was reduced to 5 mg/day, and the AZA dosage was increased to 50 mg/day.
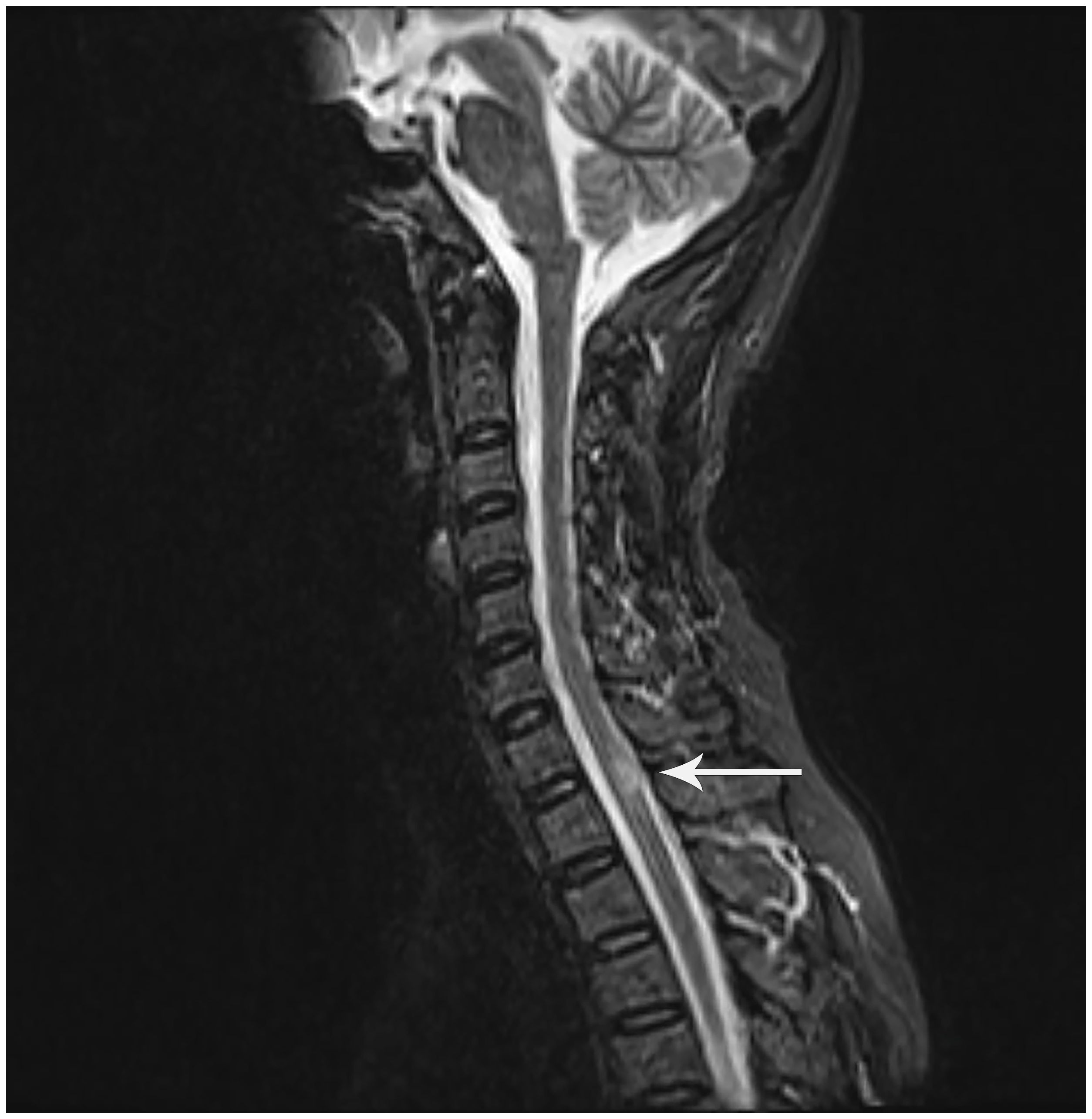
Figure 1. Sagittal T2-weighted MRI of the cervicothoracic spine: A hyperintense lesion at C7-T1 (white arrowhead). Although this lesion does not meet classical LETM criteria (≥3 vertebral segments), NMOSD diagnostic guidelines (Wingerchuk et al., 2015) waive the lesion length requirement for AQP4-IgG-seropositive patients. This clinically compatible lesion correlates with left lower limb numbness.
In April 2018, the patient experienced recurrent hiccups and vomiting, prompting consideration of a recurrence of NMOSD, which improved with intravenous MP. During the hospitalization, a follow-up MRI examination was performed. Comparative analysis with the 2016 study demonstrated no abnormalities in spinal cord signal intensity, morphological configuration, or anatomical alignment. Subsequently, rituximab (RTX) therapy was initiated for relapse prevention. The patient received two doses, achieving sustained disease stability. However, due to personal reasons, the patient was unable to continue the injections.
In April 2023, the patient presented with swelling of the right eyelid and face, which gradually spread to the right periauricular and external auditory canal with numbness of the right oral mucosa. The numbness eventually progressed to the left side of the face, but it was less severe than on the right side. Physical examination showed bilateral facial hyperalgesia with an onion-skin distribution, which was more prominent on the right side. Multimodal evoked potentials (somatosensory, brainstem auditory, and visual) and MRI (cranial, orbital, cervical, and thoracic) were unremarkable. Serum AQP4-IgG by CBA was positive (titer 1:100). Comprehensive laboratory investigations, including routine biochemistry, immunology, infectious serology, and hematology, showed no significant abnormalities. Given the possibility of NMOSD relapse, the patient was treated with intravenous MP at 1 g/day and maintenance therapy with mycophenolate mofetil (MMF) at 750 mg twice daily. Gabapentin was subsequently used for symptomatic treatment. Subsequently, the patient developed alopecia, Raynaud’s phenomenon, swollen fingers, frontal circumscribed scleroderma with hypopigmentation, sclerodactyly, and synovitis (Figure 2). The ENA profile showed anti–U1-RNP antibodies were positive and the ANA titer was >1:1000 with a speckled pattern (Table 1). Based on all examination results, the patient was diagnosed as NMOSD coexisting with MCTD and treated accordingly for NMOSD. The patient was successively treated with pregabalin, gabapentin, and oxcarbazepine for facial numbness, but the therapeutic effect was poor, and dizziness occurred.
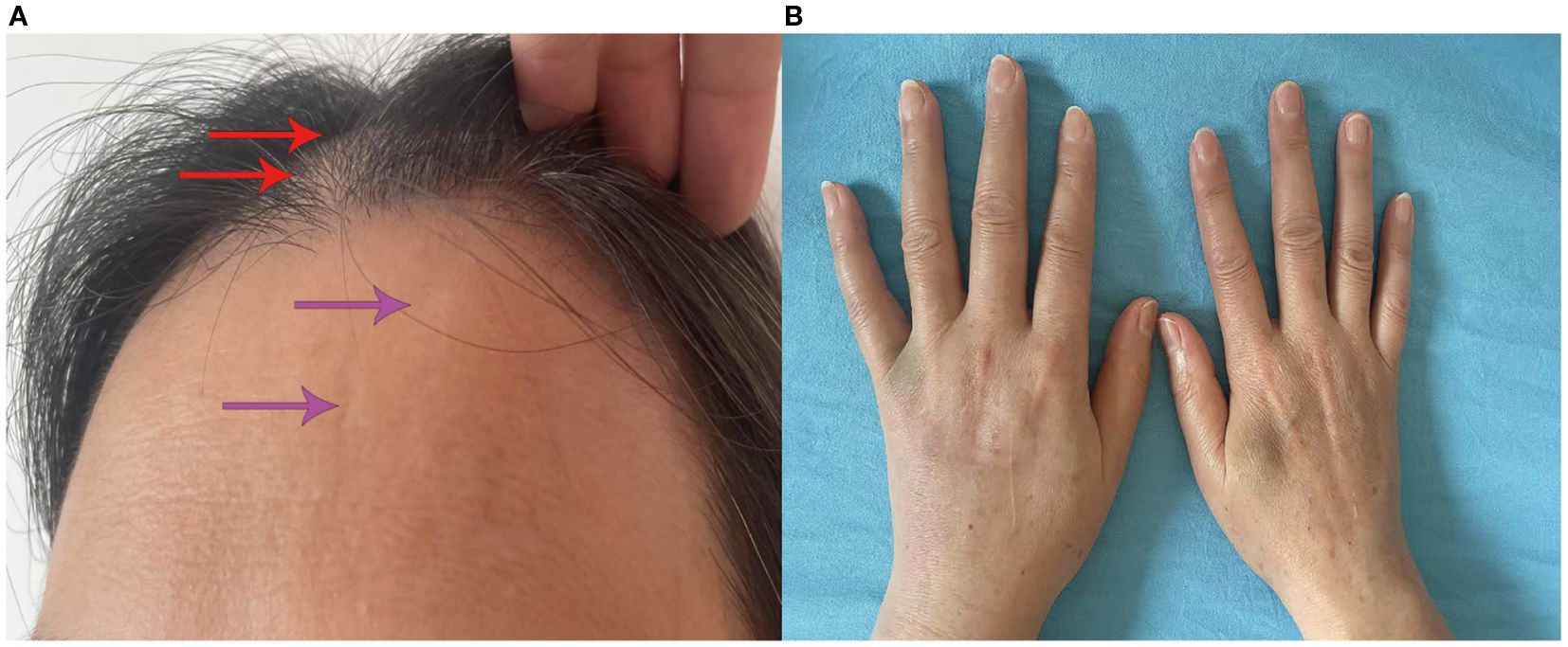
Figure 2. Clinical manifestations of the patient. (A) Alopecia (red arrowhead); localized cutaneous sclerosis with depigmentation on the forehead (purple arrowhead). (B) Finger swelling.
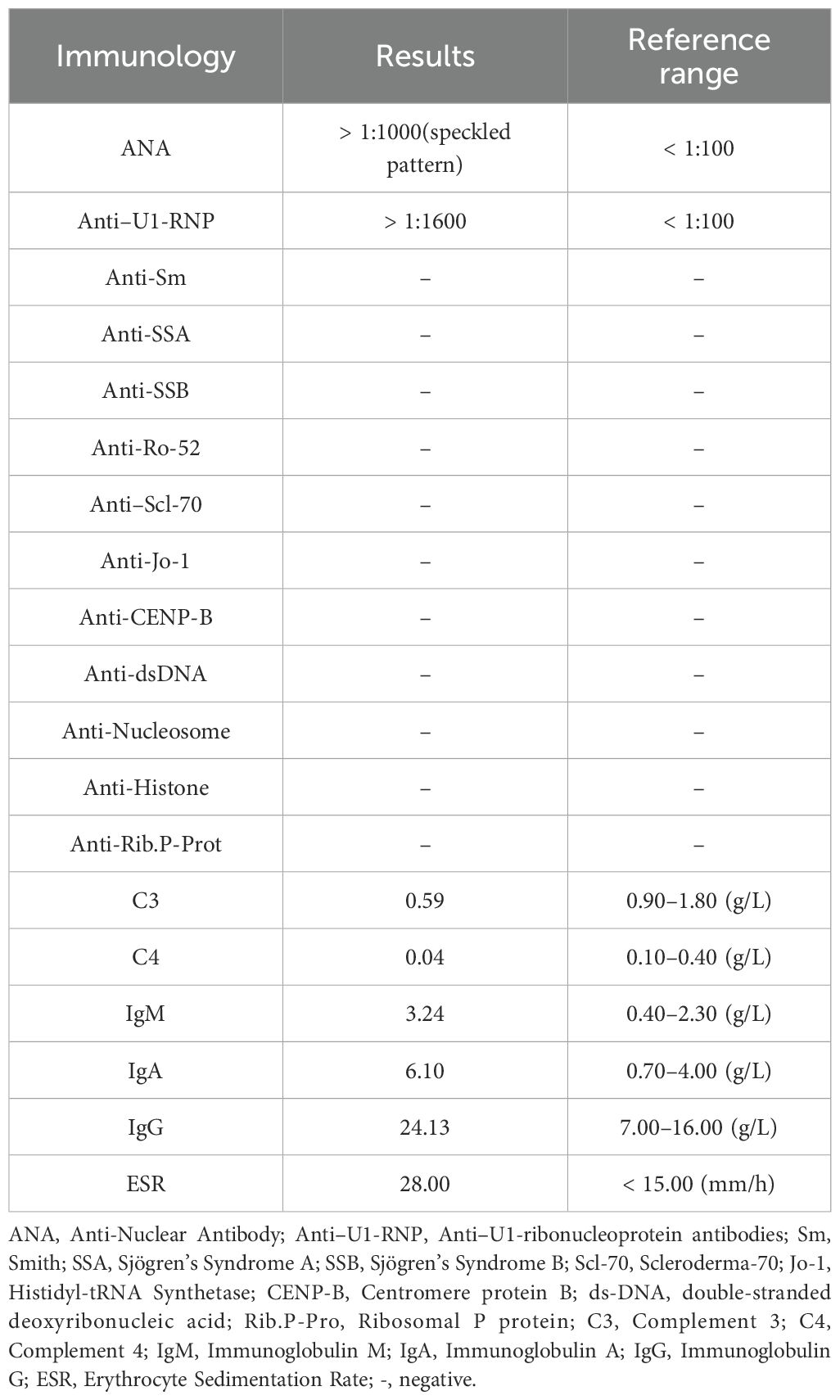
Table 1. Summary of the immunological work up.
In 2024, the patient started treatment with inebilizumab (IBZ) and has received three injections so far. The patient still has a sense of numbness, tightness, and discomfort mainly on the right side of the face, accompanied by stiffness and swelling of the fingers of both hands and tenderness at the proximal interphalangeal joint of the right middle finger.
Discussion
The case reported here involves a 48-year-old female patient with a 9-year history of NMOSD, and the AQP4 antibody was positive. Throughout treatment, the patient experienced multiple relapses of NMOSD, presenting with symptoms such as numbness in the left lower limb, hiccups, and numbness on the right side of the face. Subsequently, the patient also developed symptoms of Raynaud’s phenomenon, finger swelling, alopecia, localized cutaneous sclerosis with depigmentation on the forehead, sclerodactyly, and synovitis. Considering the positive anti–U1-RNP antibodies and an ANA titer exceeding 1:1000, the patient was diagnosed with MCTD. Following immunotherapy, the patient achieved clinical stability with only residual facial numbness and finger swelling persisting.
Management of NMOSD involves acute-phase treatment and disease-modifying therapy (DMT) during remission. This case details the patient’s immunotherapy course from 2016 to 2024 (Figure 3). In June 2016, the patient experienced the first acute attack of NMOSD. In accordance with the criteria, high-dose intravenous MP was administered, followed by oral AZA combined with low-dose Pred for relapse prevention. In April 2018, NMOSD relapsed. After alleviation of acute symptoms, the patient was switched to RTX as DMT. Previous studies have confirmed that RTX is effective in AQP4-IgG-positive NMOSD (5). However, RTX is used off-label in China, and the patient discontinued RTX due to excessive long-term financial burden. The patient relapsed again in April 2023. Studies have shown that MMF may be a favorable therapeutic option for patients with relapse or adverse reactions during AZA treatment (6). Considering the patient’s financial status and the suboptimal efficacy of previous AZA treatment, MMF was prescribed as DMT. IBZ, a first-line maintenance treatment, has been included in China’s national medical insurance. The N-MOmentum study demonstrated that switching from RTX to IBZ results in further reduction in relapse risk (7). After comprehensive consideration, the patient was switched to IBZ in 2024. Following the third dose, the patient’s condition was stable, with only mild residual facial numbness and finger swelling. Regular follow-up assessments every 3 months showed that the patient’s symptoms continued to improve without further relapses. When formulating immunotherapy regimens, clinicians must integrate disease activity, treatment frequency, reimbursement policies, and financial constraints. This approach enhances patient compliance, facilitates long-term standardized management, and reduces the risk of disease recurrence.
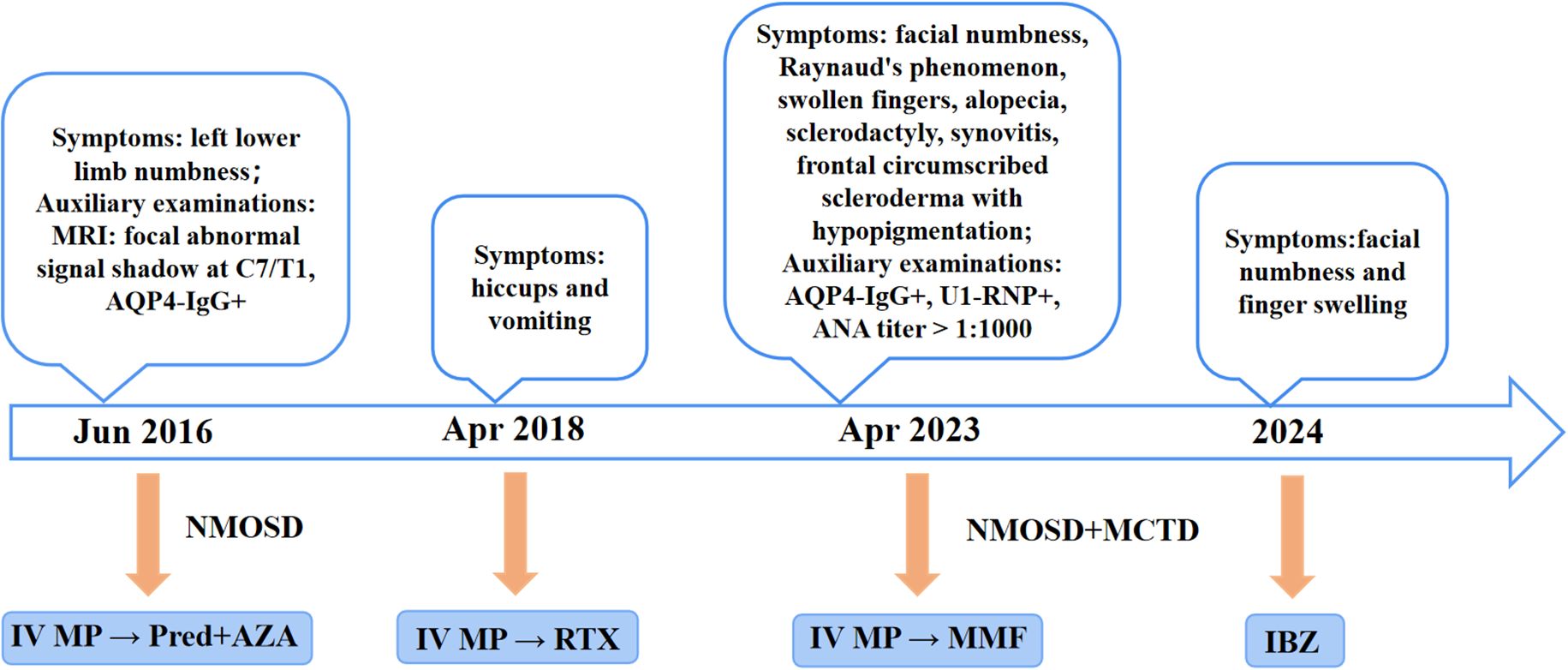
Figure 3. The treatment regimen for the patient, from the onset of the condition until the end of the follow-up period, is shown at different time points. IV, Intravenous; MP, Methylprednisolone; Pred, Prednisone; AZA, Azathioprine; RTX, Rituximab; MMF, Mycophenolate mofetil; IBZ, Inebilizumab.
Although case reports of connective tissue diseases (CTD) associated with NMOSD have been documented in the literature (8–11), those of MCTD coexisting with NMOSD remain relatively rare. Additionally, we systematically reviewed all published case reports of NMOSD combined with MCTD (Table 2, see Additional file 1).
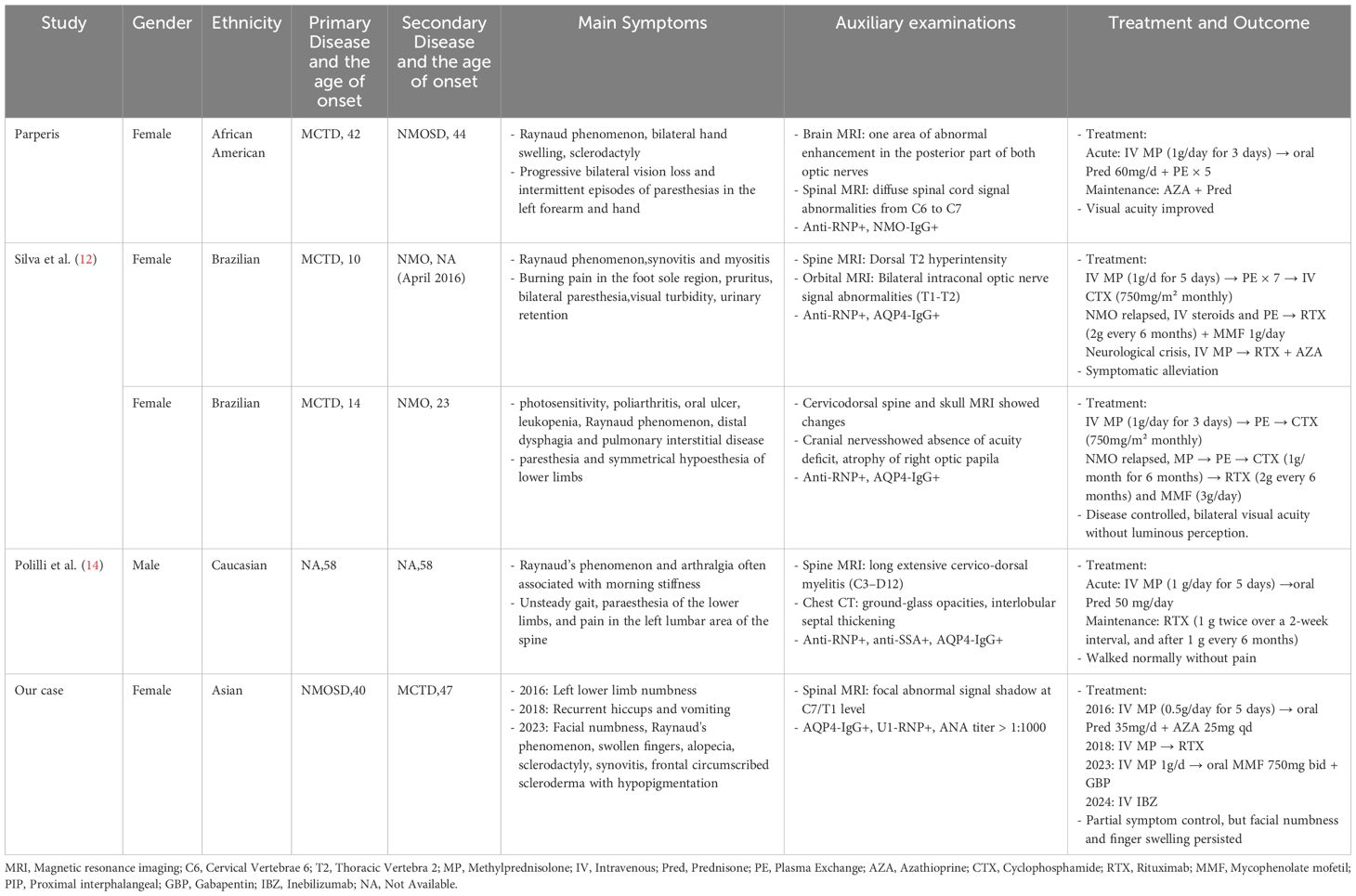
Table 2. Systematic review of cases with neuromyelitis optica spectrum disorder complicated by mixed connective tissue disease.
Silva et al. (12) reported two cases of Brazilian female patients who both presented with AQP4 antibody-positive NMOSD on the basis of MCTD. However, the clinical manifestations of the two cases differed. Case 1 was a 19-year-old patient with a 9-year history of MCTD. After experiencing viral prodromal symptoms, the patient developed symptoms such as sensory abnormalities, blurred vision, urinary retention, and muscle weakness. During immunotherapy, the patient’s neurological and visual symptoms showed periodic relief. Nevertheless, the condition relapsed after the third administration of cyclophosphamide. Subsequently, the treatment was discontinued due to an allergic reaction to MMF, leading to another relapse. Fortunately, the patient’s condition improved after subsequent treatment. Case 2 was a 29-year-old patient with a 15-year history of MCTD. After presenting with acute lower limb sensory abnormalities, symmetrical sensory loss, headache, and urinary retention, the patient was diagnosed with NMOSD based on examination results. During the treatment period, the patient discontinued treatment due to pregnancy. But unfortunately, the fetus was lost at the sixth month of gestation, and NMOSD relapsed, resulting in bilateral total amaurosis and lower limb sensory abnormalities. Regrettably, despite subsequent treatment, the patient’s vision did not recover, and the optic nerves were completely atrophic.
Parperis (13) reported a case of a 44-year-old African–American female patient with a 2-year history of MCTD. The patient had previously received treatment with prednisone but failed to continue regular treatment thereafter. The patient’s visual acuity gradually declined, with the left eye being more severely affected than the right, accompanied by intermittent paresthesia in the left upper limb and hand. MRI revealed abnormal lesions in the bilateral optic nerves and diffuse abnormal signals in the C6–C7 segments. In conjunction with positive serum NMO antibody (NMO-IgG) results, the patient was diagnosed with neuromyelitis optica (NMO). After immunotherapy, the patient’s visual acuity in both eyes improved. However, following discharge, the visual acuity of the left eye continued to deteriorate, and then prophylactic treatment with AZA and Pred was initiated.
Polilli et al. (14) reported a 58-year-old Caucasian man who presented to the Emergency Department with low back pain and inability to walk. He previously manifested repeated episodes of Raynaud’s phenomenon and arthralgia. Neurological examination revealed pyramidal signs with asymmetric and progressive paraparesis associated with hypoesthesia and bladder dysfunction. Spinal MRI revealed the presence of a long, extensive cervico-dorsal myelitis. Laboratory testing showed positivity for anti–U1-RNP, anti-Sjögren’s Syndrome A (SSA), and AQP4-IgG antibodies. Symptoms resolved following high-dose MP therapy. Subsequent RTX therapy was administered, enabling the patient to achieve normal ambulation, with the latest cervico-dorsal spine MRI demonstrating negative findings.
The coexistence of NMOSD and CTD is well recognized (15). However, the exact relationship between NMOSD and rheumatic diseases has not yet been fully elucidated. Research has revealed that the coexistence of NMOSD and CTD is more prevalent in female rheumatology patients diagnosed with SjS who present with neurological symptoms, as well as in patients with neurological diseases suspected of having SjS. Additionally, NMOSD was found to have a lower incidence in SLE patients while being exceedingly rare in MCTD patients (16).
At present, there is no research that clearly defines the sequence of onset between NMOSD and MCTD/CTD. CTD may occur prior to NMOSD, or NMOSD may precede CTD, or both may occur simultaneously (8–11, 17, 18). To date, only a limited number of cases with NMOSD coexisting with MCTD have been documented. A review of these cases reveals that Silva et al. And Parperisal. reported cases where MCTD served as the initial disease. Polilli et al. described a case with simultaneous occurrence of NMOSD and MCTD, although the chronological relationship between these disorders remains debated. Notably, the patient in the current report had a history of recurrent Raynaud’s phenomenon and arthralgia, often with morning stiffness, and the spinal MRI at presentation showed spinal cord abnormalities; however, pre-diagnostic clinical details were unavailable. In contrast, this study reports the first case in China of AQP4-IgG-seropositive NMOSD preceding MCTD, with a long-term follow-up period from 2016 to 2024.
The pathogenesis of NMOSD is complex. Genetic and environmental factors play crucial roles in the occurrence and development of this disease (19). In 2004, Lennon and colleagues first discovered NMO-IgG and the following year confirmed its target as AQP4 on astrocytes (20). AQP4-IgG is highly specific for the diagnosis of NMOSD. However, in some suspected NMOSD patients with negative AQP4-IgG test results or an unknown AQP4-IgG serological status, the diagnostic work faces significant challenges. The misapplication of the diagnostic criteria for seronegative NMOSD often leads to misdiagnosis (21). Given that some NMOSD patients may be antibody-negative, strict clinical follow-up is required for suspected cases to ensure diagnostic accuracy (22).
The spinal lesion in this case did not meet classical imaging criteria for longitudinally extensive transverse myelitis (LETM). However, diagnostic guidelines permit exemption from spinal lesion length requirements in AQP4-IgG seropositive patients (3). The temporospatial correlation between progressive left lower limb numbness and a focal demyelinating lesion at C7-T1 satisfied acute myelitis criteria. Lumbar puncture demonstrated absent OB with normal CSF parameters, providing critical laboratory evidence against multiple sclerosis and supporting NMOSD. The recurrent hiccups and vomiting in 2018, consistent with area postrema syndrome—a core NMOSD clinical phenotype—further confirmed the diagnosis.
MCTD is a rare autoimmune disorder first described by Sharp et al. in 1972, characterized by the serological hallmark of anti–U1-RNP antibodies. Specific diagnostic criteria for MCTD have not yet been established by the American College of Rheumatology or the European League Against Rheumatism. Currently, the diagnostic criteria proposed by Sharp, Alarcon-Segovia, Kahn, and Kasukawa are commonly utilized (23). Among these, the Kasukawa criteria demonstrated the maximum sensitivity (77.5%), whereas the Alarcon-Segovia and Kahn criteria showed the best specificity (24). MCTD presents an extensive range of clinical manifestations. During the disease progression, it can exhibit any typical symptoms and signs of SLE, SSc, polymyositis (PM), or rheumatoid arthritis (RA). Many patients may not meet the classification criteria for MCTD early in the disease, or they only meet the diagnostic criteria of one of these autoimmune diseases (25, 26). Therefore, the diagnosis of MCTD often poses significant challenges.
This patient met core serological criteria with positive anti–U1-RNP antibodies and high-titer ANA (1:1000). Raynaud’s phenomenon and swollen fingers were present, while digital sclerosis and synovitis represented key clinical features. Based on the Kasukawa criteria, mixed connective tissue disease (MCTD) was diagnosed. Key differentiating factors included, versus SSc, the absence of anti–Scl-70/anticentromere antibodies and limited (non-diffuse) skin sclerosis; versus systemic SLE, the absence of malar rash, proteinuria, or anti-dsDNA/anti-Sm antibodies, with non-specific alopecia; and versus SjS, the absence of sicca symptoms or anti-SSA/SSB antibodies. The hallmark features confirming MCTD were high-titer anti–U1-RNP antibodies and overlapping clinical manifestations.
NMOSD is characterized by recurrence and disability, and the accumulation of disability is mainly driven by acute clinical relapses (27, 28). Kaplan-Meier survival analysis revealed that patients with NMOSD combined with CTD had a significantly earlier time to first relapse compared to those without CTD. Except for recurrence events, patients with initial NMOSD accompanied by CTD are similar to those without CTD in terms of demographic data and clinical features (29). Meanwhile, research has indicated that NMOSD patients with CTD have longer segments of spinal cord injury compared to those without CTD (30). Notably, these cases generally exhibit relapsing tendencies and varying degrees of disability. Particularly, this 29-year-old Brazilian female patient interrupted her treatment due to pregnancy, which eventually led to irreversible optic nervous system disability, and this undoubtedly had a severe impact on her daily life. Other cases lack follow-up data, with their recurrence status and current conditions unknown. Conversely, our case, after long-term follow-up, shows stable condition and continuous improvement of symptoms.
When NMOSD coexists with MCTD, the clinical manifestations become more diverse. Rheumatologists and neurologists should fully consider the potential association between NMOSD and CTD during clinical diagnosis and treatment (15). Early identification of the presence of these two diseases is crucial for the timely initiation of immunosuppressive therapy and the prevention of systemic disability.
Conclusions
In conclusion, this study reports the first case in China of AQP4-IgG seropositive NMOSD preceding MCTD with long-term follow-up. Additionally, a review of other cases of NMOSD combined with MCTD was conducted. However, due to the rarity of this clinical condition and the scarcity of relevant literature, it is quite challenging to comprehensively document the disease course and fully confirm the correct diagnostic and treatment approaches.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Guangdong Provincial Second Hospital of Traditional Chinese Medicine. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
ZW: Writing – original draft. LW: Writing – review & editing. ZZ: Writing – review & editing. CZ: Investigation, Writing – review & editing. EJ: Supervision, Writing – review & editing. HG: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, and/or publication of this article.
Acknowledgments
The authors thank the patient for sharing her medical case, which provided valuable real-world insights for this study. We also acknowledge Deepseek for its assistance in language refinement, grammar improvement, and clarity enhancement of the manuscript. After completion of editing, we have verified the accuracy of the content.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that Generative AI was used in the creation of this manuscript. This manuscript utilized DeepSeek for language refinement, grammar improvement, and clarity enhancement. After completion of editing, we have verified the accuracy of the content.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Huda S, Whittam D, Bhojak M, Chamberlain J, Noonan C, and Jacob A. Neuromyelitis optica spectrum disorders. Clin Med (Lond). (2019) 19:169–76. doi: 10.7861/clinmedicine.19-2-169
2. Ratelade J and Verkman AS. Neuromyelitis optica: aquaporin-4 based pathogenesis mechanisms and new therapies. Int J Biochem Cell Biol. (2012) 44:1519–30. doi: 10.1016/j.biocel.2012.06.013
3. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. (2015) 85:177–89. doi: 10.1212/WNL.0000000000001729
4. Shahmohammadi S, Doosti R, Shahmohammadi A, Mohammadianinejad SE, Sahraian MA, Azimi AR, et al. Autoimmune diseases associated with Neuromyelitis Optica Spectrum Disorders: A literature review. Mult Scler Relat Disord. (2019) 27:350–63. doi: 10.1016/j.msard.2018.11.008
5. Dong GY, Meng YH, and Xiao XJ. A meta-analysis on efficacy and safety of rituximab for neuromyelitis optica spectrum disorders. Med (Baltimore). (2022) 101:e30347. doi: 10.1097/MD.0000000000030347
6. Pathomrattanapiban C, Tisavipat N, Jitprapaikulsan J, Prayoonwiwat N, Rattanathamsakul N, and Siritho S. The efficacy and safety of mycophenolate mofetil in Thai neuromyelitis optica spectrum disorder patients. Mult Scler Relat Disord. (2022) 63:103882. doi: 10.1016/j.msard.2022.103882
7. Flanagan EP, Levy M, Katz E, Cimbora D, Drappa J, Mealy MA, et al. Inebilizumab for treatment of neuromyelitis optica spectrum disorder in patients with prior rituximab use from the N-MOmentum Study. Mult Scler Relat Disord. (2022) 57:103352. doi: 10.1016/j.msard.2021.103352
8. Liu L, Tang L, Zhang L, Li X, Huang P, Xiong J, et al. The first case report of preschool-onset SS/SLE coexisting with NMOSD of Chinese origin. Front Immunol. (2022) 13:887041. doi: 10.3389/fimmu.2022.887041
9. Li S, Gao Y, He Y, and Zhang Z. A case report of AQP4-IgG-seropositive refractory neuromyelitis optica spectrum disorder patient with Sjögren’s syndrome and pancytopenia treated with inebilizumab. Front Neurol. (2024) 15:1371515. doi: 10.3389/fneur.2024.1371515
10. Li F, Sui X, Pan X, Liu C, Xie L, Zhao H, et al. Neuromyelitis optica spectrum disorder with ultra-longitudinally extensive transverse myelitis: A case report and literature review. Heliyon. (2024) 10:e39687. doi: 10.1016/j.heliyon.2024.e39687
11. Shidahara K, Hayashi K, Sada KE, Hiramatsu S, Morishita M, Watanabe H, et al. Refractory neuromyelitis optica spectrum disorder in systemic lupus erythematosus successfully treated with rituximab. Lupus. (2018) 27:1374–7. doi: 10.1177/0961203318760994
12. Silva SA, Cunha PS, Brito AS, Souza RB, and Ribeiro SLE. Devic’s syndrome and mixed connective tissue disease: an unusual association. Acta Reumatol Port. (2018) 43:146–50.
13. Parperis K. A rare case of mixed connective tissue disease complicated with neuromyelitis optica. J Clin Rheumatol. (2012) 18:261–2. doi: 10.1097/RHU.0b013e318262df28
14. Polilli E, Volpe P, Esposito JE, Di Risio A, Di Carmine C, Di Iorio G, et al. AQP4 antibody-seropositive neuromyelitis optica spectrum disorder in a patient with mixed connective tissue disease: a case report. AME Case Rep. (2025) 9:30. doi: 10.21037/acr-23-48
15. Cruz RA, Chaudhary S, Guevara M, and Meltzer E. Neuromyelitis optica spectrum disorders (NMOSD) and connective tissue disease (CTD): an update for the rheumatologist. Curr Rheumatol Rep. (2021) 23:33. doi: 10.1007/s11926-021-01000-2
16. Esposito JE, Annoni G, D'Amato M, Graziosi A, Troilo F, Di Risio A, et al. Systemic connective tissue disease and neuromyelitis optica spectrum disorder coexistence: A systematic review and meta-analysis. J Integr Neurosci. (2024) 23:35. doi: 10.31083/j.jin2302035
17. Kopp CR, Prasad CB, Naidu S, Sharma V, Misra DP, Agarwal V, et al. Overlap syndrome of anti-aquaporin-4 positive neuromyelitis optica spectrum disorder and systemic lupus erythematosus: A systematic review of individual patient data. Lupus. (2023) 32:1164–72. doi: 10.1177/09612033231191180
18. Martín-Nares E, Hernandez-Molina G, and Fragoso-Loyo H. Aquaporin-4-IgG positive neuromyelitis optica spectrum disorder and systemic autoimmune diseases overlap syndrome: a single-center experience. Lupus. (2019) 28:1302–11. doi: 10.1177/0961203319877255
19. Holroyd KB, Manzano GS, and Levy M. Update on neuromyelitis optica spectrum disorder. Curr Opin Ophthalmol. (2020) 31:462–8. doi: 10.1097/ICU.0000000000000703
20. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, and Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. (2005) 202:473–7. doi: 10.1084/jem.20050304
21. Zara P, Dinoto A, Carta S, Floris V, Turilli D, Budhram A, et al. Non-demyelinating disorders mimicking and misdiagnosed as NMOSD: a literature review. Eur J Neurol. (2023) 30:3367–76. doi: 10.1111/ene.15983
22. de Souza Moraes A, Brum DG, Ierich JCM, Higa AM, Assis ASJ, Miyazaki CM, et al. A highly specific and sensitive nanoimmunosensor for the diagnosis of neuromyelitis optica spectrum disorders. Sci Rep. (2019) 9:16136. doi: 10.1038/s41598-019-52506-w
23. Kubo S and Tanaka Y. Evolution of diagnostic criteria and new insights into clinical testing in mixed connective tissue disease; anti-survival motor neuron complex antibody as a novel marker of severity of the disease. Immunol Med. (2024) 47:52–7. doi: 10.1080/25785826.2024.2338593
24. Ferrara CA, La Rocca G, Ielo G, Libra A, and Sambataro G. Towards early diagnosis of mixed connective tissue disease: updated perspectives. Immunotargets Ther. (2023) 12:79–89. doi: 10.2147/ITT.S390023
25. Shan X and Ge Y. Interstitial lung disease in patients with mixed connective tissue disease: A retrospective study. Int J Gen Med. (2024) 17:2091–9. doi: 10.2147/IJGM.S464704
26. Hao Y, Xin M, Wang S, Ma D, and Feng J. Myelopathy associated with mixed connective tissue disease: clinical manifestation, diagnosis, treatment, and prognosis. Neurol Sci. (2019) 40:1785–97. doi: 10.1007/s10072-019-03935-y
27. Wingerchuk DM and Lucchinetti CF. Neuromyelitis optica spectrum disorder. N Engl J Med. (2022) 387:631–9. doi: 10.1056/NEJMra1904655
28. Jarius S, Aktas O, Ayzenberg I, Bellmann-Strobl J, Berthele A, Giglhuber K, et al. Update on the diagnosis and treatment of neuromyelits optica spectrum disorders (NMOSD) - revised recommendations of the Neuromyelitis Optica Study Group (NEMOS). Part I: Diagnosis and differential diagnosis. J Neurol. (2023) 270:3341–68. doi: 10.1007/s00415-023-11634-0
29. Yao Y, Yang X, Zhou Y, Xie H, Duan R, Jing L, et al. Comparative analysis of clinical and imaging data of first-attack neuromyelitis optica spectrum disorders with and without connective tissue disease. Front Neurol. (2022) 13:969762. doi: 10.3389/fneur.2022.969762
Keywords: neuromyelitis optica spectrum disorder, mixed connective tissue disease, AQP4, case report, overlap syndrome
Citation: Wang Z, Wu L, Zhang Z, Zhang C, Jia E and Geng H (2025) Overlap syndrome of anti-aquaporin-4 positive neuromyelitis optica spectrum disorder and mixed connective tissue disease: a case report. Front. Immunol. 16:1644259. doi: 10.3389/fimmu.2025.1644259
Received: 10 June 2025; Accepted: 20 August 2025;
Published: 10 September 2025.
Edited by:
Gian Marco Ghiggeri, Giannina Gaslini Institute (IRCCS), ItalyReviewed by:
Simone Parisi, University Hospital of the City of Health and Science of Turin, ItalyThanos Tsaktanis, University of Erlangen Nuremberg, Germany
Copyright © 2025 Wang, Wu, Zhang, Zhang, Jia and Geng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ertao Jia, c2FpbGluZzE5ODBAMTI2LmNvbQ==; Hongling Geng, Z2hsOTIwQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship