 Piaopiao Wu
Piaopiao Wu Wei You2
Wei You2 Wentong Liu
Wentong Liu Ni Luo
Ni Luo- 1Department of Gerontology, CR & WISCO General Hospital Affiliated to Wuhan University of Science and T echnology, Wuhan, Hubei, China
- 2School of Basic Medicine, Hubei University of Arts and Science, Xiangyang, China
X-linked agammaglobulinemia (XLA) is a rare primary immunodeficiency disorder caused by mutations in the Bruton tyrosine kinase (BTK) gene. This article presents a fatal case of a 20-year-old male with XLA complicated by septic shock due to Pseudomonas aeruginosa infection, highlighting two novel BTK insertion mutations in exon 15 (NM_000061.3 exon15:c.1561insG and c.1565insTAGAA). Concurrently, we provide a systematic review of XLA’s genetic basis, clinical manifestations, diagnostic challenges, and therapeutic advancements. The patient’s delayed diagnosis, lack of immunoglobulin replacement therapy, and fatal outcome underscore the importance of early genetic screening and standardized management. This case and review aim to enhance clinical awareness and emphasize the integration of genetic diagnostics into routine practice for primary immunodeficiencies.
Introduction
X-linked agammaglobulinemia (XLA) is a rare primary immunodeficiency disorder (PID) (1, 2). It was first described in 1952 by the American pediatrician Ogden Bruton, making it the first hereditary immunodeficiency identified in humans (3, 4). The disease is caused by mutations in the Bruton’s tyrosine kinase (BTK) gene located on the X chromosome (Xq22), leading to B-cell developmental arrest (5). This results in a significant reduction or absence of mature B cells and extremely low or undetectable levels of immunoglobulins (IgG, IgA, IgM) (6).
Affected individuals are typically male (females are usually carriers), and symptoms usually appear 6–12 months after birth, when maternal antibody protection wanes. Patients suffer from recurrent bacterial infections (e.g., otitis media, pneumonia, sinusitis), particularly from encapsulated bacteria (e.g., Streptococcus pneumoniae, Haemophilus influenzae) (7). Without timely treatment, complications such as chronic lung disease and sepsis may occur. Intravenous immunoglobulin (IVIG) replacement therapy and antibiotic prophylaxis are the mainstays of management. Advances in genetic diagnosis and early intervention have significantly improved patient outcomes, but lifelong monitoring is required.
In this study, we report a previously undocumented BTK mutation in an XLA patient who died from septic shock and multi-organ failure.
Case presentation
Chief complaints
A 20-year-old male was admitted with fever (peak temperature: 39°C), cough, bloody sputum, and hemoptysis for two days. He was initially diagnosed with septic shock and treated with antibiotics and vasopressors, but his condition did not improve. He was subsequently transferred to the ICU of a tertiary hospital.
History of past illness
The patient had a 7-year history of bronchiectasis and had been hospitalized multiple times in the past two years for severe pneumonia. He denied any history of hypertension, diabetes, coronary heart disease, hepatitis, tuberculosis, trauma, blood transfusions, or surgeries. He also had no history of long-term smoking, alcohol abuse, or drug allergies.
Personal and family history
His mother denied any specific family history.
Physical examination
On admission, his temperature was 38.7°C, heart rate 148 bpm, respiratory rate 39 breaths/min, and blood pressure 94/52 mmHg (maintained with vasopressors). Oxygen saturation was 94% (with oxygen support). The patient was conscious but appeared acutely ill and lethargic. Coarse breath sounds and dry rales were heard in both lungs. A scattered rash was present on his back and buttocks, with a stage II pressure ulcer (6 cm × 3.5 cm) on the left ischial tuberosity. The left lower limb showed scattered eczema and ulcerations. No lymphadenopathy was detected in the neck or supraclavicular fossae. There was no jaundice, cyanosis, or edema in the lower limbs.
Imaging examinations
Chest CT revealed patchy high-density shadows in the left lower lobe and right lung, with significant involvement of the right lower lobe and a small amount of right pleural effusion (Figure 1). Bedside thoracic ultrasound confirmed bilateral pleural effusion. Cardiac ultrasound indicated tachycardia, while abdominal, lower limb, and urinary tract ultrasounds showed no abnormalities.
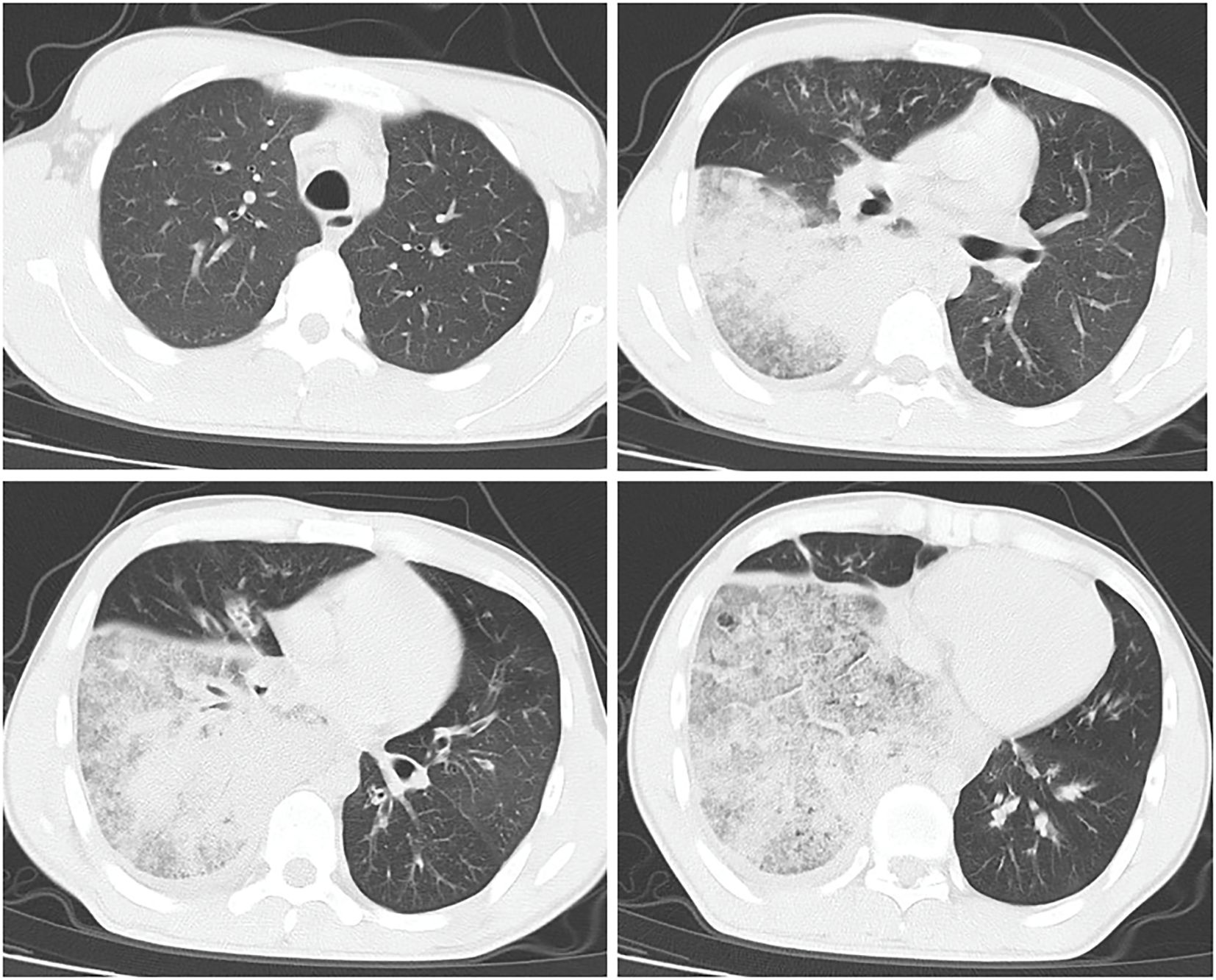
Figure 1. CT results of the lungs.
Laboratory examinations
Initial tests showed elevated white blood cells (12.92 × 109/L) and neutrophils (11.90 × 109/L), suggesting bacterial infection. By the second day, white blood cells surged to 24.98 × 109/L, with neutrophils accounting for 95.7% (23.91 × 109/L) and severe lymphopenia (0.47 × 109/L), indicating severe sepsis. Platelets dropped from 216 × 109/L to 73 × 109/L. Inflammatory markers were markedly elevated: CRP remained extremely high, PCT >200 ng/mL, and a cytokine storm was evident (IL-6 >5000 pg/mL, IL-10 >1000 pg/mL, TNF-α 44.3 pg/mL). Immunoglobulin levels were critically low: IgG <0.3 g/L, IgA <0.07 g/L, IgM <0.04 g/L, with reduced complement C3/C4. Rheumatologic and ANCA tests were negative, excluding autoimmune diseases. Metagenomic next-generation sequencing (mNGS) of bronchoalveolar lavage fluid identified Pseudomonas aeruginosa (Table 1).
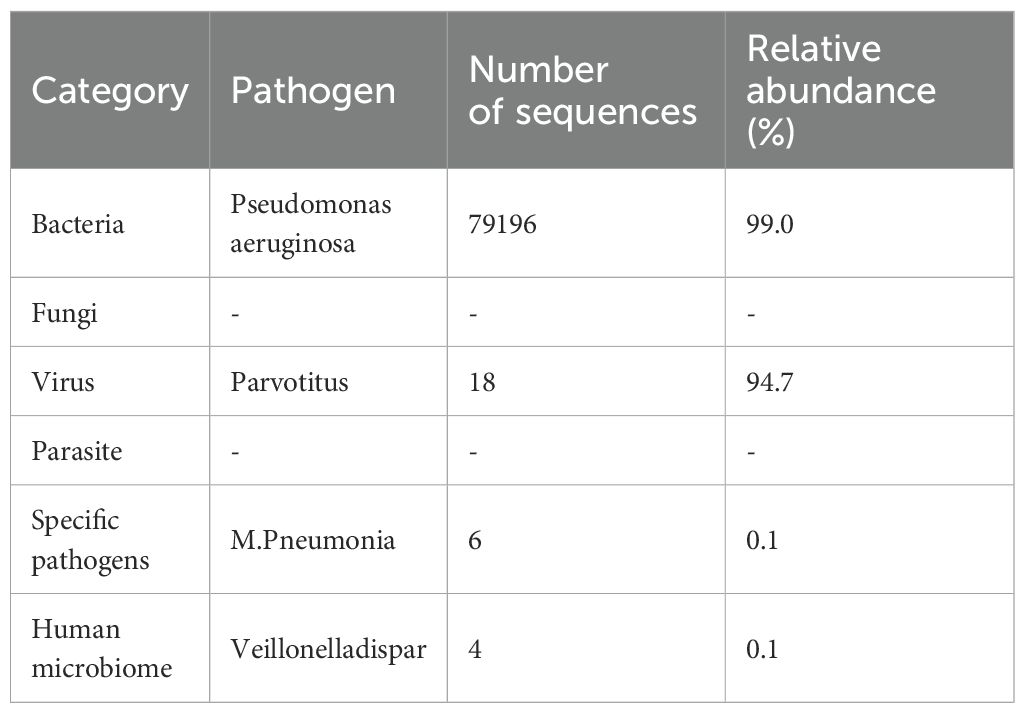
Table 1. NGS detection of alveolar lavage fluid.
Treatment
The patient was immediately treated with antibiotics (meropenem + linezolid), vasopressors (norepinephrine + metaraminol + dopamine), fresh frozen plasma for coagulopathy, IVIG (5 g), and glucocorticoids (dexamethasone). On the second day, he developed respiratory distress with oxygen saturation dropping to 75%, necessitating intubation. Treatment was adjusted to stronger antibiotics (meropenem + tigecycline), increased IVIG (30 g/day), and optimized vasopressor therapy. On the third day, recurrent hypotension and progressive bradycardia culminated in cardiac arrest. Resuscitation efforts failed, and the patient was declared clinically dead.
Final diagnosis
With family consent, whole-exome sequencing of peripheral blood cells revealed two previously unreported insertion mutations in exon 15 of the BTK gene on the X chromosome (Figure 2). Lymphocyte subset analysis showed near-absence of B cells (Table 2), confirming a diagnosis of XLA with a novel BTK mutation. The patient died from Pseudomonas aeruginosa infection leading to multiple organ failure.
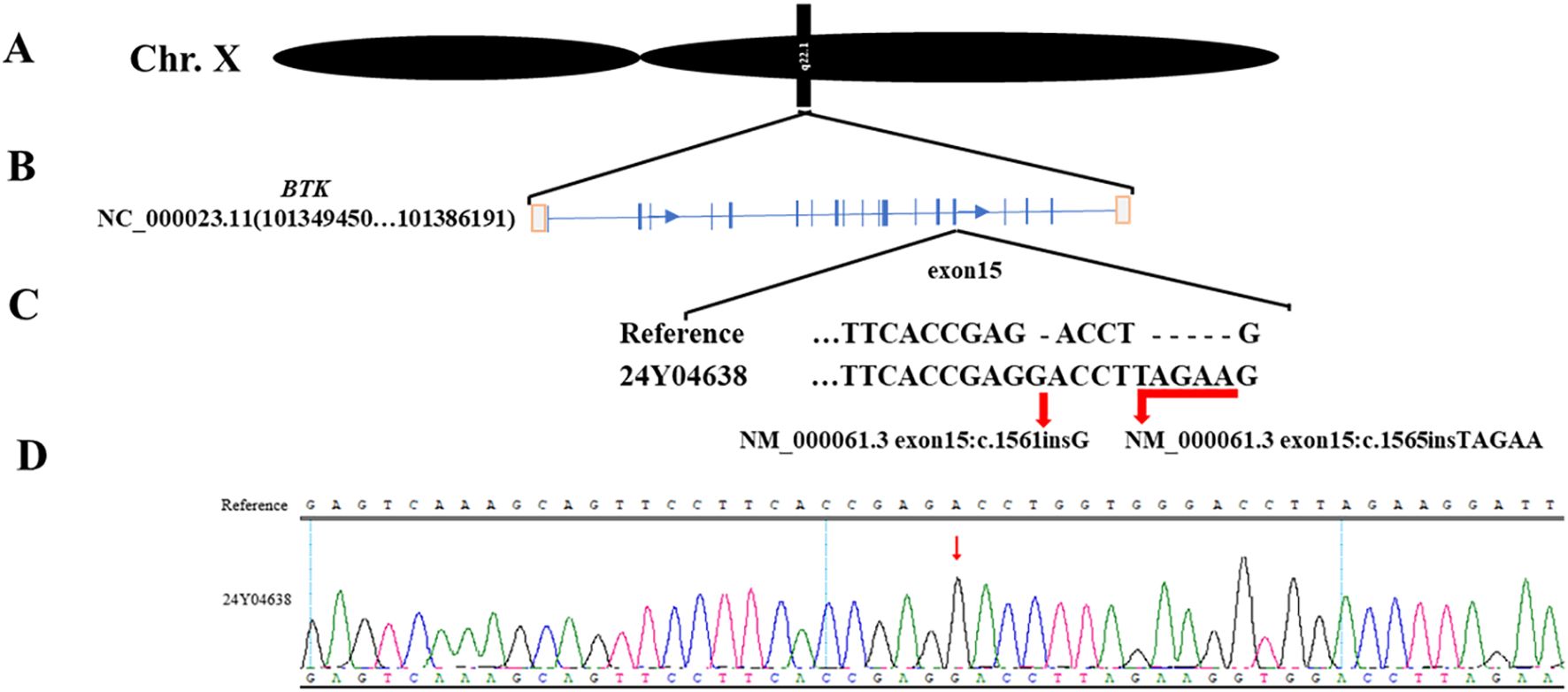
Figure 2. Mutation details. (A). The mutated gene is located on the X chromosome. (B). The two mutation sites are located on exon 15 of the BTK gene. (C). Whole-exome sequencing shows the information of the two mutation sites. (D). Peripheral blood sequencing verifies the mutation results.
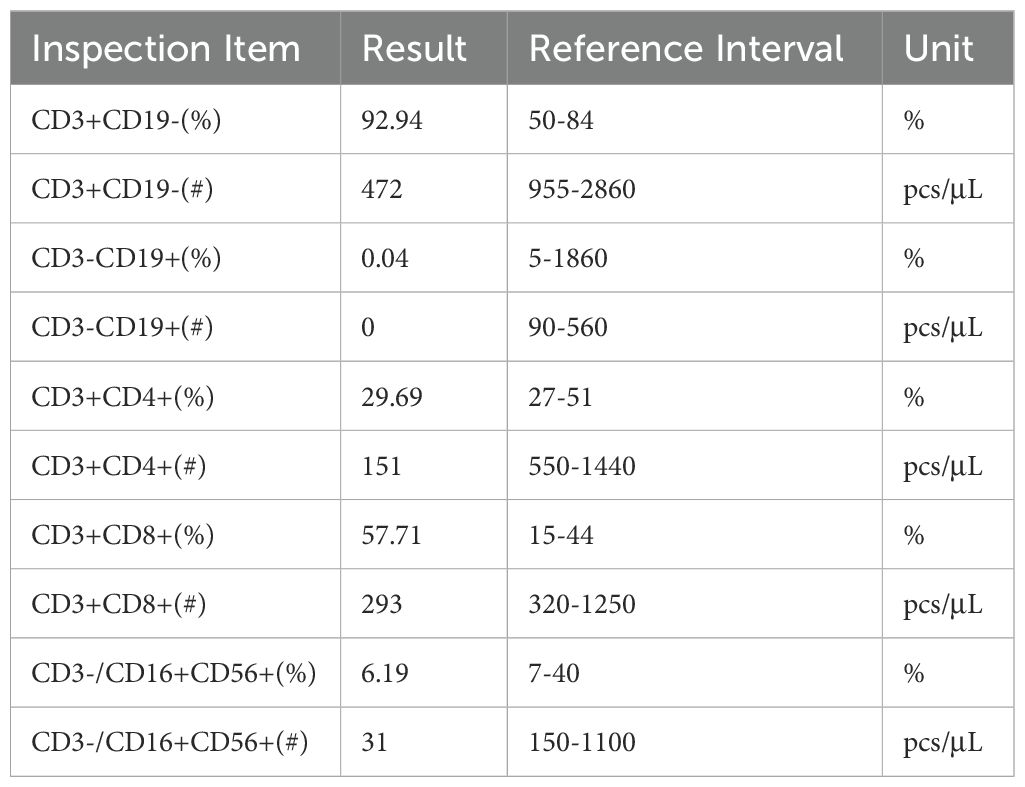
Table 2. Lymphocyte typing assay.
Discussion
XLA is a primary immunodeficiency disorder characterized by impaired B-cell development, leading to antibody deficiency and recurrent infections (8). Patients often present with severe bacterial infections, particularly in the ears, nose, throat, and respiratory tract. A 2022 study of the USIDNET registry involving 231 XLA patients with known BTK mutations identified common pathogenic microorganisms as Haemophilus influenzae (10.4%), Staphylococcus aureus (10.4%), Streptococcus pneumoniae (7.8%), and Pseudomonas aeruginosa (6.1%) (9). In this case, the patient had a history of multiple severe pneumonias and was admitted with septic shock, ultimately succumbing to multi-organ failure due to Pseudomonas aeruginosa infection. A 2016 study reported that the average age of onset for XLA was 2.15 ± 2.16 years, with a median of 1 year. Among 135 patients (77.59%), symptoms appeared before the age of 3, and only one case presented at 13 years (10). The average age at diagnosis was 7.09 ± 3.98 years (range: 0.17–19 years), with 87 patients (50.58%) diagnosed within the first 6 years. However, only 5 patients (2.91%) were diagnosed before 1 year of age. This patient exhibited bronchiectasis at 13 but was not properly evaluated. Over the past two years, recurrent pneumonias also failed to prompt an XLA diagnosis. The cornerstone of XLA treatment is regular immunoglobulin replacement therapy to prevent infections (9, 11). Unfortunately, this patient was not diagnosed early or given standardized immunoglobulin therapy, leading to recurrent infections and rapid disease progression. Additionally, septic shock and multi-organ failure further complicated treatment, resulting in a poor prognosis.
Mutations in the BTK gene, are the primary cause of XLA (12). BTK consists of five domains: catalytic kinase (SH1), SH2, Src homology (SH3), Pleckstrin homology (PH), and Tec homology (TH), spanning 19 exons (13, 14). The BTKbase, an international database documenting disease-causing variants in BTK associated with XLA, currently contains information on 2,310 DNA variants from 2,291 individuals, including 1,025 unique variants (15). Mutations predominantly occur in the kinase domain (49.4%), with missense mutations accounting for 41.5% and insertions for 1.8% (https://structure.bmc.lu.se/idbase/BTKbase/). Due to the wide variety of BTK mutations, correlating specific mutations with disease severity remains challenging, and no objective standard for symptom severity exists (16, 17).
In this case, whole-exome sequencing revealed two previously unreported insertion mutations in exon 15 of the BTK gene(NM_000061.3 exon15:c.1561insG, NM_000061.3 exon15:c.1565insTAGAA), located within the catalytic kinase (SH1) domain. These mutations likely impair BTK kinase function, leading to symptom onset at 13 and death at 20 due to Pseudomonas aeruginosa-induced multi-organ failure. Genetic testing is now widely used in clinical practice, and early diagnosis combined with standardized treatment can prevent disease progression (18). Dana O’Toole’s research indicates that respiratory infections are a common cause of death in XLA patients. Screening for B-cell developmental defects in newborns provides an opportunity to prevent pulmonary infections through early diagnosis and treatment initiation. This approach helps increase clinical suspicion of XLA in patients with recurrent infections and assists healthcare providers in anticipating infection patterns in XLA patients (9). Although experimental approaches such as virus-mediated oligonucleotide gene therapy in mice and hematopoietic stem cell gene editing are under investigation, there is currently no cure for XLA in clinical practice (19, 20).
Conclusion
X-linked agammaglobulinemia is a life-threatening immunodeficiency. Early diagnosis and standardized immunoglobulin replacement therapy are critical. This case underscores the need for heightened clinical awareness of primary immunodeficiencies, particularly in patients with recurrent infections. Prompt immunological and genetic testing can improve outcomes. Therefore, we advocate for the inclusion of severe B-cell developmental defects (such as XLA) in newborn screening programs to prevent such fatal outcomes.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethical Approval for Clinical Research Projects under Medical Ethics Committee of China Resources & WISCO General Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article. Written informed consent was obtained from the patient's mother for the publication of this case report.
Author contributions
WL: Writing – original draft, Funding acquisition, Conceptualization, Writing – review & editing. NL: Supervision, Writing – review & editing, Data curation, Writing – original draft, Conceptualization, Visualization. XJ: Conceptualization, Writing – review & editing, Formal analysis, Writing – original draft. YW: Writing – original draft. PW: Writing – original draft, Conceptualization, Formal analysis.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. This research was supported by the Research Project of Hubei Provincial Department of Education (Q20222601).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript. Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Conley ME, Brown P, Pickard AR, Buckley RH, Miller DS, Raskind WH, et al. Expression of the gene defect in X-linked agammaglobulinemia. N Engl J Med. (1986) 315:564–7. doi: 10.1056/NEJM198608283150907
2. Conley ME, Mathias D, Treadaway J, Minegishi Y, and Rohrer J. Mutations in btk in patients with presumed X-linked agammaglobulinemia. Am J Hum Genet. (1998) 62:1034–43. doi: 10.1086/301828
3. Smith CIE and Berglof A. X-linked agammaglobulinemia. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, and Amemiya A, editors. GeneReviews((R)). Seattle (WA (1993).
4. Ochs HD and Smith CI. X-linked agammaglobulinemia. A clinical and molecular analysis. Med (Baltimore). (1996) 75:287–99. doi: 10.1097/00005792-199611000-00001
5. Ponader S and Burger JA. Bruton’s tyrosine kinase: from X-linked agammaglobulinemia toward targeted therapy for B-cell Malignancies. J Clin Oncol. (2014) 32:1830–9. doi: 10.1200/JCO.2013.53.1046
6. Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F, et al. The gene involved in X-linked agammaglobulinaemia is a member of the Src family of protein-tyrosine kinases. 1993. J Immunol. (2012) 188:2948–55. doi: 10.1093/jimmunol/188.7.2948
7. Ramalho VD, Oliveira Junior EB, Tani SM, Roxo Junior P, and Vilela MM. Mutations of Bruton’s tyrosine kinase gene in Brazilian patients with X-linked agammaglobulinemia. Braz J Med Biol Res. (2010) 43:910–3. doi: 10.1590/S0100-879X2010007500079
8. Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S, et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol. (2009) 27:199–227. doi: 10.1146/annurev.immunol.021908.132649
9. O’Toole D, Groth D, Wright H, Bonilla FA, Fuleihan RL, Cunningham-Rundles C, et al. X-linked agammaglobulinemia: infection frequency and infection-related mortality in the USIDNET registry. J Clin Immunol. (2022) 42:827–36. doi: 10.1007/s10875-022-01237-1
10. Chen XF, Wang WF, Zhang YD, Zhao W, Wu J, and Chen TX. Clinical characteristics and genetic profiles of 174 patients with X-linked agammaglobulinemia: Report from Shanghai, China (2000-2015). Med (Baltimore). (2016) 95:e4544. doi: 10.1097/MD.0000000000004544
11. Quartier P, Debre M, De Blic J, de Sauverzac R, Sayegh N, Jabado N, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: a retrospective survey of 31 patients. J Pediatr. (1999) 134:589–96. doi: 10.1016/S0022-3476(99)70246-5
12. Khan F, Person H, Dekio F, Ogawa M, Ho HE, Dunkin D, et al. Crohn’s-like enteritis in X-linked agammaglobulinemia: A case series and systematic review. J Allergy Clin Immunol Pract. (2021) 9:3466–78. doi: 10.1016/j.jaip.2021.04.070
13. Carrillo-Tapia E, Garcia-Garcia E, Herrera-Gonzalez NE, Yamazaki-Nakashimada MA, Staines-Boone AT, Segura-Mendez NH, et al. Delayed diagnosis in X-linked agammaglobulinemia and its relationship to the occurrence of mutations in BTK non-kinase domains. Expert Rev Clin Immunol. (2018) 14:83–93. doi: 10.1080/1744666X.2018.1413349
14. Hagemann TL, Chen Y, Rosen FS, and Kwan SP. Genomic organization of the Btk gene and exon scanning for mutations in patients with X-linked agammaglobulinemia. Hum Mol Genet. (1994) 3:1743–9. doi: 10.1093/hmg/3.10.1743
15. Schaafsma GCP, Väliaho J, Wang Q, Berglöf A, Zain R, Smith CIE, et al. BTKbase, bruton tyrosine kinase variant database in X-linked agammaglobulinemia: looking back and ahead. Hum Mutat. (2023) 2023:1–12. doi: 10.1155/2023/5797541
16. Cardenas-Morales M and Hernandez-Trujillo VP. Agammaglobulinemia: from X-linked to autosomal forms of disease. Clin Rev Allergy Immunol. (2022) 63:22–35. doi: 10.1007/s12016-021-08870-5
17. Lopez-Granados E, Perez de Diego R, Ferreira Cerdan A, Fontan Casariego G, and Garcia Rodriguez MC. A genotype-phenotype correlation study in a group of 54 patients with X-linked agammaglobulinemia. J Allergy Clin Immunol. (2005) 116:690–7. doi: 10.1016/j.jaci.2005.04.043
18. Collins CJ, Yi F, Dayuha R, Whiteaker JR, Ochs HD, Freeman A, et al. Multiplexed proteomic analysis for diagnosis and screening of five primary immunodeficiency disorders from dried blood spots. Front Immunol. (2020) 11:464. doi: 10.3389/fimmu.2020.00464
19. Bestas B, Estupinan HY, Wang Q, Kharazi S, He C, K.M. D, et al. Cell-penetrating peptide-conjugated, splice-switching oligonucleotides mitigate the phenotype in BTK/Tec double deficient X-linked agammaglobulinemia model. RSC Chem Biol. (2025) 6:761–71. doi: 10.1039/D4CB00312H
Keywords: case report, BTK mutations, septic shock, delayed diagnosis, primary immunodeficiency
Citation: Wu P, You W, Liu W, Luo N and Xu J (2025) Fatal X-linked agammaglobulinemia complicated by septic shock: a case report and comprehensive review of novel BTK mutations. Front. Immunol. 16:1645337. doi: 10.3389/fimmu.2025.1645337
Received: 12 June 2025; Accepted: 30 July 2025;
Published: 15 August 2025.
Edited by:
Silvia Clara Giliani, University of Brescia, ItalyReviewed by:
Vassilios Lougaris, University of Brescia, ItalyJessica Palmieri, Ann & Robert H. Lurie Children’s Hospital of Chicago, United States
Copyright © 2025 Wu, You, Liu, Luo and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wentong Liu, MTE4NDdAaGJ1YXMuZWR1LmNu; Ni Luo, MjYxNTI0MjM5QHFxLmNvbQ==; Jiayue Xu, MTgwNDA2MDUxMDBAMTYzLmNvbQ==
†These authors have contributed equally to this work