 Xudong Cheng
Xudong Cheng Yian Wang2
Yian Wang2 Bryon Johnson
Bryon Johnson Ming You
Ming You- 1Department of Pharmacy, Suzhou Traditional Chinese Medicine (TCM) Hospital Affiliated to Nanjing University of Chinese Medicine, Suzhou, China
- 2Center for Cancer Prevention, Houston Methodist Neal Cancer Center, Houston Methodist Hospital, Weill Cornell Medicine, Houston, TX, United States
- 3Division of Hematology and Oncology, Department of Medicine, Medical College of Wisconsin, Milwaukee, WI, United States
Mitochondria, as regulators of cellular energy production and metabolism, play a crucial role in tumor growth and survival. Tumors are reprogrammed to accommodate rapid proliferation through the Warburg effect. This reprogramming leads to the accumulation of metabolites such as lactate and ketone bodies, thereby lowering the pH of the tumor microenvironment, inhibiting the activity of effector T cells and NK cells, while promoting the infiltration of regulatory T cells and MDSCs, forming an immunosuppressive microenvironment. ROS produced by mitochondria can affect immune cell function by modulating their signaling pathways. Mitochondria also release DAMPs, which activate the antigen-presenting capacity of dendritic cells and initiate anti-tumor immune responses. Currently, various methods have been employed, such as DLCs modifications and mitochondrial targeted delivery, which enable drugs to penetrate the lipid bilayer and enter the mitochondria, thereby helping to reduce immunosuppression in the tumor microenvironment. In this review, we will discuss the impact of mitochondria on tumor immunity, strategies to target tumor cell mitochondria, and progress on the discovery of mitochondria-targeted drugs to enhance tumor immunity, providing potential directions for developing new cancer therapeutic strategies.
1 Introduction
Mitochondria are essential intracellular organelles that primarily facilitate energy production and metabolic regulation (1). They generate adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS), thereby providing cells with the energy required for various functions (2). Moreover, mitochondria modulate diverse physiological processes including calcium homeostasis, redox balance, apoptosis and immune responses through mitochondrial DNA (mtDNA), reactive oxygen species (ROS), and metabolite signaling pathways (3, 4). Consequently, abnormal mitochondrial function may contribute to the pathogenesis of various diseases, including cancer.
Mitochondria play a particularly critical role in tumorigenesis and progression (5). Tumor cells frequently undergo metabolic reprogramming to support rapid proliferation, exemplified by the “Warburg effect,” wherein cells preferentially utilize glycolysis over OXPHOS despite adequate oxygen availability (6). This metabolic shift not only sustains tumor cell growth and survival but also modulates immune cell function within the tumor microenvironment (TME) (7). For instance, hypoxia in the TME can induce T-cell exhaustion, thereby impairing anti-tumor immune responses (8). Additionally, mitochondrial dysfunction is closely associated with immune escape mechanisms that promote tumor progression and metastasis (9).
Recent years have witnessed significant advancements in tumor immunotherapy, particularly with the advent of immune checkpoint inhibitors. Nevertheless, many patients exhibit either primary or acquired resistance to such therapies, a phenomenon closely linked to the immunosuppressive nature of the TME (10). Immune cells, such as tumor-associated macrophages (TAMs), rely on mitochondrial metabolic functions to maintain their immunosuppressive activity (11). Therefore, targeted therapies aimed at modulating mitochondrial function may overcome immune resistance by reprogramming immunosuppressive cells (12). For example, recent progress in mitochondrial-targeted metabolic reprogramming has demonstrated that enhancing T-cell bioenergetics can restore antitumor activity (13). Furthermore, mitochondrial-derived ROS modulates immune cell function via redox signaling; low ROS levels promote T-cell exhaustion, whereas normal ROS levels enhance antigen presentation by dendritic cells (DCs) (14). Given the critical role of mitochondria in tumor metabolism and immunotherapy, elucidating how mitochondria-targeting strategies influence tumor immunotherapy represents a promising area of research. In this review, we will explore the emerging role of mitochondria in tumor immunotherapy and discuss the recent advances in mitochondria-targeted drugs that enhance tumor immunity, thereby providing important directions for future therapeutic strategies.
2 Mitochondria and tumor immunity
Tumor cells adapt to increasing energy and biosynthetic demands by reprogramming relevant metabolic pathways (15). Nutrient depletion and overproduction of metabolic byproducts driven by tumor development in the TME help to establish an immunosuppressive TME by regulating the metabolic reprogramming of tumor-infiltrating immune cells and associated signaling activation to control the polarization of different types of immune cells, ultimately resulting in metabolic derangement-mediated deficiencies and decreased anti-tumor immune responses (16). Mitochondria, as intracellular organelles with diverse biological functions and highly variable, have key regulatory roles in metabolism and activating immune cells (17). Glucose, fatty acid and amino acid metabolism are abnormal during tumor development and progression (18) (Figure 1). ROS-induced mtDNA damage impairs mitochondrial OXPHOS, forcing tumor cells to rely on glycolysis for ATP production (19). Abnormal mitochondrial function in the TME is an important cause of cancer formation, progression and metastasis.
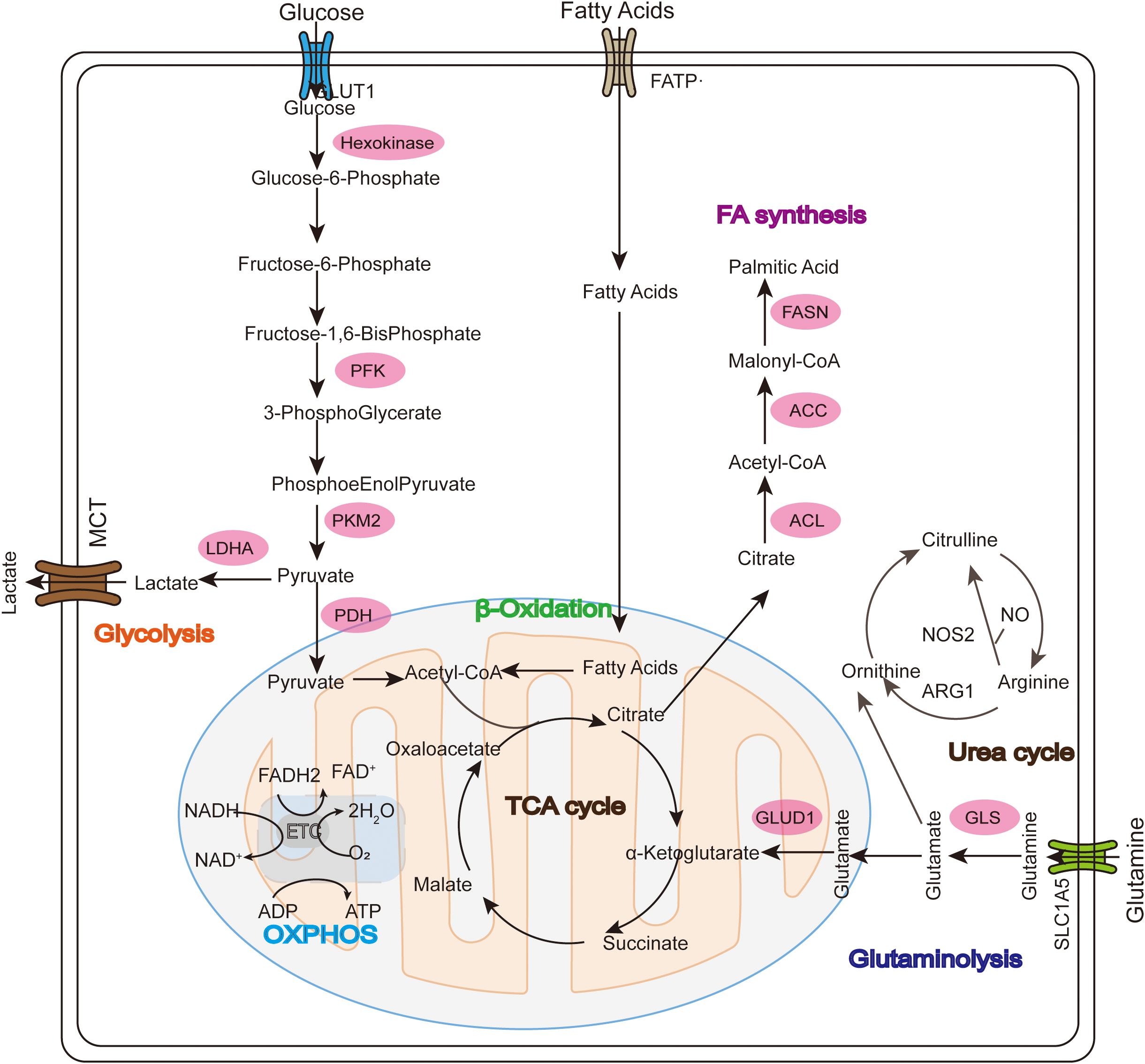
Figure 1. Glucose, fatty acid, and glutamine metabolism in mitochondria during tumor development. In the Warburg effect, unlike normal differentiated cells, glucose enters the cell through GLUT1 and mainly relies on mitochondrial oxidative phosphorylation to provide energy for the cell, while most tumor cells rely on aerobic glycolysis and are eventually oxidized to lactate instead of acetyl-CoA (ac-COA). The main substrate for lipid synthesis is cytoplasmic acetyl-CoA synthesized through a series of reactions. Fatty acid oxidation (FAO) allows long-chain FA to be converted into acetyl-CoA in the mitochondria and enter the TCA cycle to generate ATP and malic enzyme-dependent NADPH. Glutamate is then converted into α-ketoglutarate (α-KG) through two different pathways and can participate in the TCA cycle as a replenishing substrate. Glutamine sequentially catalyzes the formation of arginine (Arg) through citrulline (Cit), and then continues to decompose under the action of arginase (ARG) to produce urea and ornithine (Orn), thus forming the urea cycle.
2.1 Mitochondrial metabolic regulation of immune cells
T cells rely on OXPHOS and fatty acid oxidation in the resting state, but switch to aerobic glycolysis and fatty acid synthesis upon activation to support proliferation (20, 21) (Figure 2). During T cell activation, mitochondria accumulate in the immune synapse formed by T cells and antigen-presenting cells (APCs), and activation of the T cell receptor stimulates an increase in mitochondrial fission, which increases the number of mitochondria and cristae loosening, and generation of ROS and ATP, which are essential in maintaining calcium homeostasis and regulating its downstream-related signaling (22). During the transformation of CD8+ T cells from effector T cells to memory T cells, activation of Sirt3, a mitochondrial deacetylase, reduces protein acetylation, which enhances OXPHOS activity and generation and survival of memory T cells, resulting in increased anti-tumor immune activity (23). In contrast, competition of tumor cells for glucose and other nutrients in the TME suppresses the metabolism and function of immune cells (24). Hypoxia in the TME promotes mitochondrial structural damage and reduces ATP production by down-regulating MYC expression levels, which induces T-cell exhaustion (TExh) and anti-tumor dysfunction of CD8+ T cells. Tumor-infiltrating T cells are in a state of high oxidative stress for long periods of time due to glucose and oxygen-deficient environment-mediated metabolic insufficiency and impairment of mitochondrial function and quality (25). In addition, peroxisome proliferator-activated receptor γ coactivator-1α (PGC-1α), a key regulator of mitochondrial biogenesis, is upregulated in CD8+ T cells in the TME resulting in their dysfunction. This dysfunction can be reversed/rescued by enhancing cellular expression of PGC1α, which increases CD8+ T cell anti-tumor activity (26).
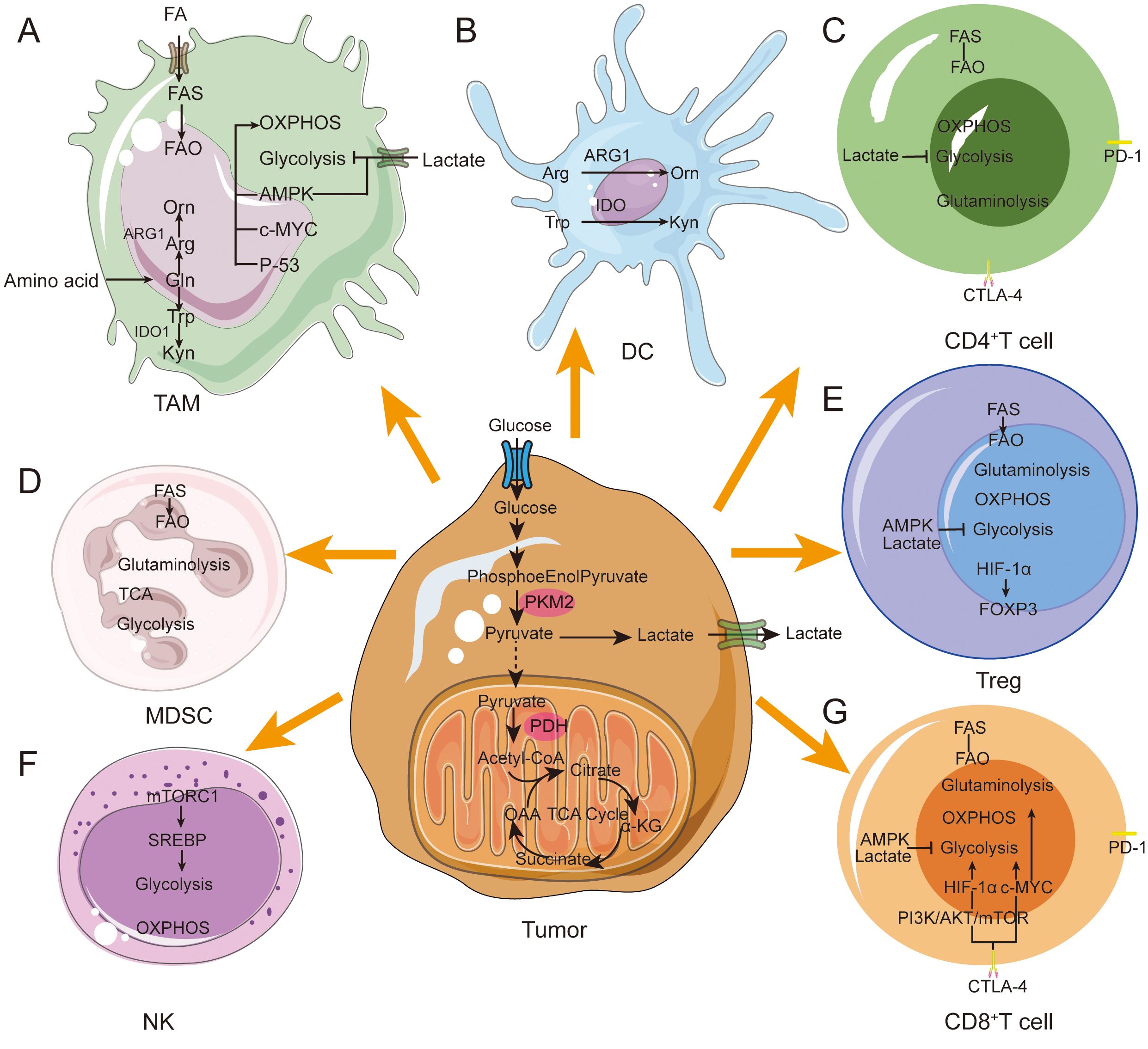
Figure 2. Metabolic reprogramming in immune cells during tumor progression. Immune cells in the TME achieve immunosuppressive and pro-tumor phenotypes through metabolic reprogramming. (A) M1 macrophages prefer glycolysis and secrete a large amount of lactate. M2 macrophages tend to show enhanced fatty acid oxidative phosphorylation ability. M2 macrophages mainly rely on FAO, OXPHOS and glutamine metabolism. (B) Immediately after DC activation, glycolysis increases rapidly to provide ATP. DCs express ARG1 and IDO enzymes. Hydrolyze arginine and tryptophan. (C) High lactate concentration blocks CD4+ T cell glycolysis. CD4+ T cells increase lipid uptake leading to a metabolic shift toward FAO. (D) MDSCs express ARG1 and IDO enzymes. Hydrolyze arginine and tryptophan. Tumor-infiltrating MDSCs exhibit enhanced glycolysis and OXPHOS. (E) In Tregs, FOXP3 expression inhibits glycolysis while promoting OXPHOS. FASN overexpression enhances lipid metabolism. (F) NK-mediated glycolysis via mTORC1 signaling. and OXPHOS. (G) High lactate concentrations block T cell glycolysis. Activated CD8+ T cells convert glucose and glutamine into biomass and rely on the Pl3K and AKT pathways. CD8+ T cells tend to FAO increase lipid uptake.
Natural killer (NK) cells are cytotoxic lymphocytes, and their cellular activity is significantly correlated with levels of glucose metabolism. When glucose levels are elevated, NK cell activity is significantly enhanced. After activation of NK cells, intracellular sterol regulatory element binding protein (SREBP) binds to and upregulates its mechanistic target rapamycin complex 1 (mTORC1) expression and enhances aerobic glycolysis and OXPHOS metabolism (27). The transcription factor cMyc can significantly increase NK cell metabolism; if the c-Myc protein is defective, NK cells will reduce their expression of key gluconeogenesis and mitochondrial enzymes, leading to impaired immune function (28). When NK cells transition to the memory stage, mitochondrial autophagy-related proteins Bnip3-Bnip3L promote their transition by inducing mitochondrial autophagy to remove damaged mitochondria and reduce the generation of ROS (29). Studies have shown that in a hypoxic TME, the mitochondrial morphology of tumor-infiltrating NK cells shows significant fragmentation and division compared to normal NK cells. These changes in mitochondrial morphology significantly reduce the ability of NK cells to mediate tumor immune surveillance (30).
Mitochondria also play a key role in macrophage polarization. In the early stage of tumor formation, pro-inflammatory cytokines such as toll-like receptor (TLR) agonists can promote the polarization of TAM to an M1 phenotype, and nitric oxide (NO) and ROS produced by M1 type macrophages can significantly inhibit the proliferation and induce tumor cell death (31). During tumor progression, interleukin (IL)-4 and colony stimulating factor 1 (CSF1) induce the polarization of TAM to M2 phenotype. M2 macrophages secrete epidermal growth factor (EGF), matrixmetalloprotein9 (MMP-9), and other proteins to inhibit anti-tumor immunity and promote tumor progression (32). M2 macrophages rely on the OXPHOS metabolic pathway for energy supply (33), which is linked to fatty acid oxidation (FAO) and characterized by high expression of CD36. CD36 promotes the mitochondrial OXPHOS process, resulting in mitochondrial fusion and lengthening (34). Additionally, M2 macrophages synthesize large amounts of arginase (ARG) and indoleamine2,3-dioxygenase1 (IDO1), which deplete arginine and tryptophan respectively, leading to immune dysfunction (35). FAO plays a key role in human M2 macrophage function by enhancing IL-1β secretion to promote cancer cell migration (36).
The metabolic shift from OXPHOS to glycolysis and dynamic changes in mitochondrial morphology lead to alterations in immune cell polarity and phenotype, which in turn affects the biology of immune cells. Therefore, studying the role of mitochondrial metabolism is important for understanding regulation of tumor immunity and developing new drugs that can promote tumor immunity.
2.2 Mitochondrial ROS in tumor immunity
Mitochondria are the main intracellular ROS-generating organelles, producing ROS through the electron transport chain (ETC) and OXPHOS during aerobic respiration (37). ROS have dual roles in tumorigenesis and progression. Low levels of ROS act as important cellular signaling molecules involved in multiple life activities such as gene expression, cell proliferation, differentiation, and stress responses. However, when the intracellular levels of ROS are too high, oxidative damage to nucleoplasm, mitochondrial DNA, proteins and lipids occurs, which ultimately leads to cellular damage. High levels of ROS facilitate tumorigenesis by promoting tumor cell proliferation, migration, invasion and angiogenesis, inflammatory responses and immune escape, helping tumor cells adapt to the harsh TME. In addition, ROS-mediated inflammatory responses can also change the composition of immune cells in the TME and enhance immune suppression (38). Therefore, maintaining a balance between intracellular ROS production and consumption is essential for maintaining cellular homeostasis and organismal health.
2.2.1 ROS and formation of a tumor immunosuppressive microenvironment
As highlighted in the previous section, ROS plays a central regulatory role in the TME and drives cancer development and progression (39). Tumor cells adapt to the high reactive oxygen environment and avoid cell death by inducing the secretion of inflammatory cytokines, stabilizing hypoxia-inducible factor-1α (HIF-1α), activating AMP-activated protein kinase (AMPK) signaling, and promoting the production of nicotinamide adenine dinucleotide phosphate (NADPH), which in turn promotes tumor metastasis and angiogenesis (40). In addition, ROS regulates the activation status of immune cells in the TME that affect cancer progression. High ROS levels oxidize major histocompatibility complex (MHC) class I molecules, which impairs antigen peptide loading and T-cell receptor(TCR)-MHC/peptide complex stability (41). Tumor cells and immunosuppressive cells in the microenvironment act synergistically to induce mitochondrial ROS (mtROS) generation, aiding in the establishment of immune tolerance (42). Lon protease in the mitochondrial quality control system induces ROS generation by interacting with multiple proteins, mediating activation of the NF-κB signaling axis and enhancing downstream signaling activity to promote tumorigenesis (43). Highly expressed HIF-1α promotes mtROS production by inducing Lon protease expression (44). Lon protease binds PYCR1, a key enzyme in proline metabolism, enhancing NADPH consumption and promoting electron leakage in the ETC, thereby elevating mtROS (45).
2.2.2 Effects of mtROS on immune cell activation in the TME
To avoid the deleterious effects of high ROS levels on immune cells, there exists a set of strict regulatory mechanisms in the organism to maintain a delicate balance between immune cell activity and ROS levels (46). Precise control of ROS levels in NK cells and T lymphocytes prevents their damage to other lymphocytes (Figure 3). In the tumor microenvironment, IL-15 has been shown to induce NK cells to enhance their resistance to oxidative stress and protect against ROS via the thioredoxin system (47). During anti-tumor immunity, activated T lymphocytes and NK cells recruit neutrophils and macrophages by increasing ROS production, ultimately killing tumor cells (48). On the other hand, elevated ROS inhibits prolyl hydroxylases (PHDs), stabilizing HIF-1α to drive myeloid-derived suppressor cell (MDSC) differentiation (49). For example, tumor-associated fibroblasts promote the transformation of peripheral monocytes into MDSCs by increasing their oxidative stress, thereby inhibiting the proliferation of CD8+ T cells and promoting tumor progression (50).
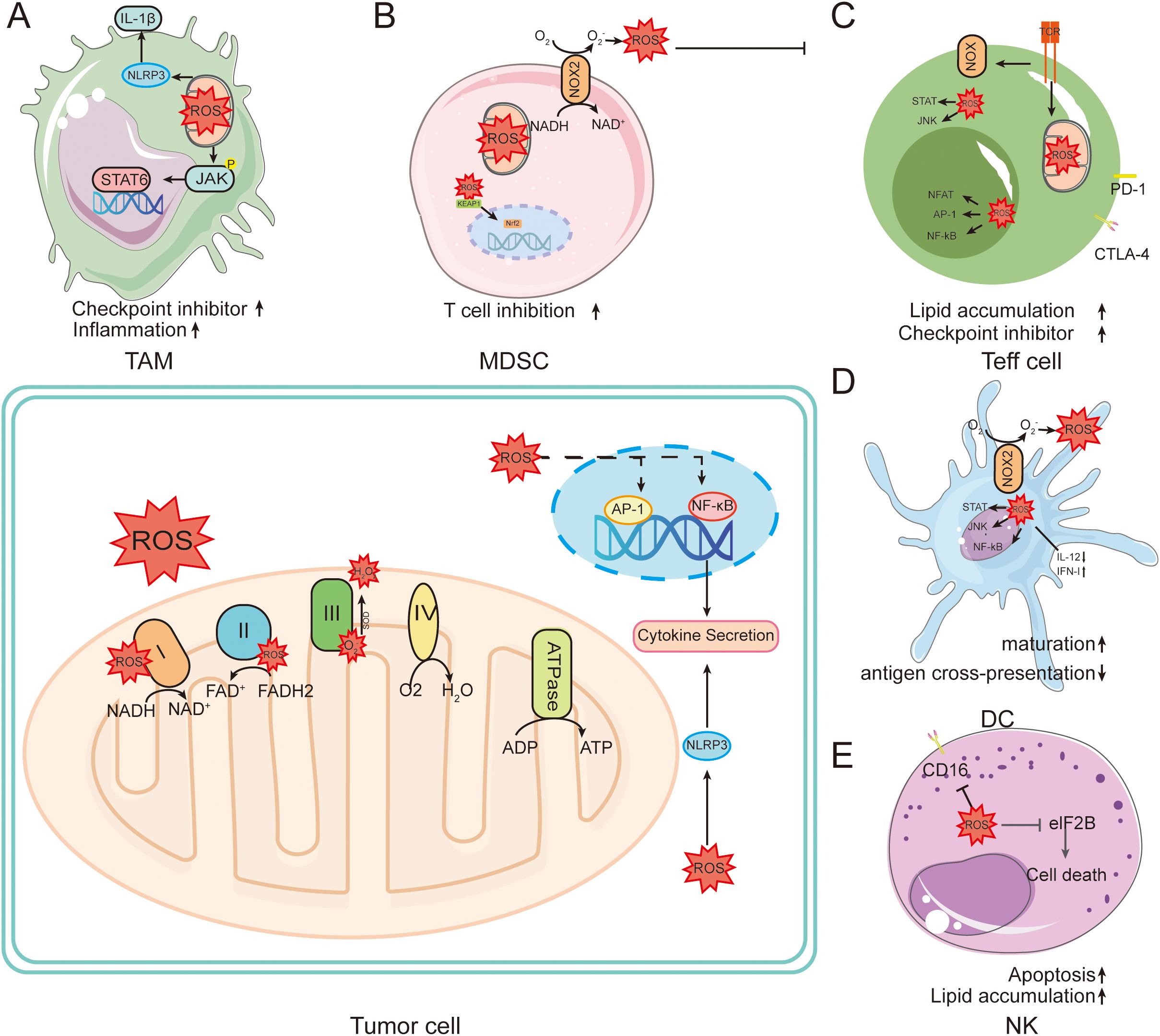
Figure 3. Role of ROS in tumors and immune cells. ROS are mainly generated in the electron transport chain on the inner membrane of mitochondria during oxidative phosphorylation. This leads to intracellular oxidative stress. (A) In macrophages, ROS induce powerful intracellular changes by activating signaling pathways such as NF-kB, AP-1, and NRF2. Activation of NF-kB and AP-1 leads to the production of key pro-inflammatory cytokines. (B) NOX family members in MDSC cells directly mediate the production of ROS, further promoting the inflammatory cascade. (C) TCR engagement can mediate the production of ROS through NOX family members, increasing the activity of NFAT, Myc, and mTOR. (D) ROS can affect DC maturation, antigen presentation, NF-κB activation, and induction of anti-inflammatory cytokines. (E) ROS induced in NK promotes NK cell apoptosis.
ROS plays an important regulatory role in T cell activation, promotion of T cell antigen-specific proliferation and apoptosis (51). Moderate levels of ROS are essential for the normal activation and differentiation of T lymphocytes, whereas high levels of ROS promote T cell apoptosis by up-regulating the apoptosis-related factor Fas and down-regulating expression of the anti-apoptotic protein Bcl-2 (52). In addition, extracellular ROS affects T cell activation by altering the immunogenicity of antigenic peptides in APCs (53). During immunogenic cell death (ICD), intracellular damage-associated molecular patterns (DAMPs) such as ATP, endoplasmic reticulum calmodulin, and high mobility group protein B1 leaks to the extracellular space, which in turn activates DCs by interacting with its receptors and triggers anti-tumor immune responses in T lymphocytes (54). Nanoparticle-delivered catalase scavenges extracellular H2O2, enhancing T cell infiltration and reversing immunosuppression (55). In addition, reduced glutathione deficiency in regulatory T cells (Treg) leads to abnormal serine metabolism and down-regulates transcription factor forkhead box P3 (Foxp3), which ultimately attenuates the immunosuppressive function of Treg (56). These studies suggest that ROS levels and sustained generation capacity have a key role in ICD.
ROS have long been considered to be harmful metabolites of mitochondria, but recent studies have shown that mtROS have a necessary signaling role in preventing excessive immune responses, and in particular ROS play a key role in regulating macrophage immune responses (57). Under normal conditions, ROS affects macrophage polarization by modulating relevant signaling pathways (58). ROS also have important regulatory roles in macrophages subsets. For example, M1-type macrophages generate ROS through NADPH-oxidase (NOX) 2 signaling, which activates NF-κB signaling and enhances cellular phagocytosis (59). In contrast, high levels of ROS have harmful effects on macrophages (60). During tumorigenesis, macrophages become an important immune cell population for maintaining immune homeostasis in the TME. Tumor cells remodel the peripheral and distal TME by secreting tumor-derived factors, which stimulate the activation of both monocytes and macrophages in the microenvironment and accelerate tumor progression (61). Although TAM can exhibit both pro-inflammatory M1-type and anti-inflammatory M2-type polarized forms, it is generally accepted that TAM exhibit similar functions to M2-type macrophages, promoting tumor growth, metastasis, angiogenesis, and immunosuppression by secreting cytokines, chemokines, and proteases (62). Mitochondrial Lon protease is upregulated in M2-type macrophages, suggesting that during tumorigenesis macrophages may regulate Lon expression through multiple signals, inducing mtROS generation and participating in the TAM differentiation process (63).
DCs differentiated from monocytes have potent antigen presentation properties, promote T-cell activation, and play an important role in initiating and regulating immune responses (64). DC maturation is regulated by different types of stimuli. When immature DCs are stimulated by the pro-inflammatory cytokine IL-6 or the TLR ligand lipopolysaccharide, they are transformed into mature DCs that express CD80, CD86 and IL-6, and initiate effector T cell responses (65). In contrast, when DCs are stimulated by the regulatory factors IL-10, transforming growth factor beta (TGF-β), vitamin D3 and corticosteroids, they are transformed into tolerogenic DCs, which ultimately contribute to impaired differentiation of effector T cells and activation of Treg (66). The TME establishes an immunosuppressive state by inducing the differentiation of regulatory DCs and MDSCs, thus helping the tumor to escape immune surveillance (67). In addition, TGF-β and IL-10 secreted by tumor cells and TAM inhibit DC-mediated antigen presentation and adaptive immune responses (68). Ultimately, the concentration of ROS in the TME has an important role in regulating the cytotoxic or immunosuppressive effects of immune cells.
Mitochondria are the main ROS-producing organelles in cells, producing ROS and OXPHOS through ETC, which have a dual role in tumors: low levels are important cell signaling molecules, high levels cause oxidative damage and promote tumor development, and ROS balance is key to cell homeostasis.
In tumors, ROS drives cancer progression, and tumor cells can adapt to a high ROS environment, and also regulate the activation state of TME immune cells, such as damaging the stability of T cell-related complexes, synergizing with immunosuppressive cells to help build immune tolerance, and Lon protease is also involved in promoting mtROS production. At the same time, ROS affects a variety of immune cells: regulates the activity of NK cells and T lymphocytes, promotes MDSC differentiation, affects macrophage polarization and function, regulates DC maturation and antigen presentation, and ROS concentration in the TME plays an important role in regulating immune cell function.
2.3 Role of mtDNA in anti-tumor immunity
Genomic mutations in mitochondria are an important part of the cancer mutant genome, and mtDNA dysfunction and gene mutations are closely related to cancer development (69). Mitochondrial gene copy number abnormalities, aberrant gene expression and altered mtDNA epigenetic modifications frequently affect cancer development and malignant transformation by regulating cellular metabolism, ROS production and cell-cell interactions. Furthermore, the location and level of mtDNA gene mutations can confer different degrees of competitive advantages to cancer cells (70). mtDNA leaked into the cytoplasm by mitochondria during stress is an important source of DAMPs, and cytoplasmic mtDNA binds to and activates different DNA pattern recognition receptors, inducing strong intrinsic immune responses (71) (Figure 4).
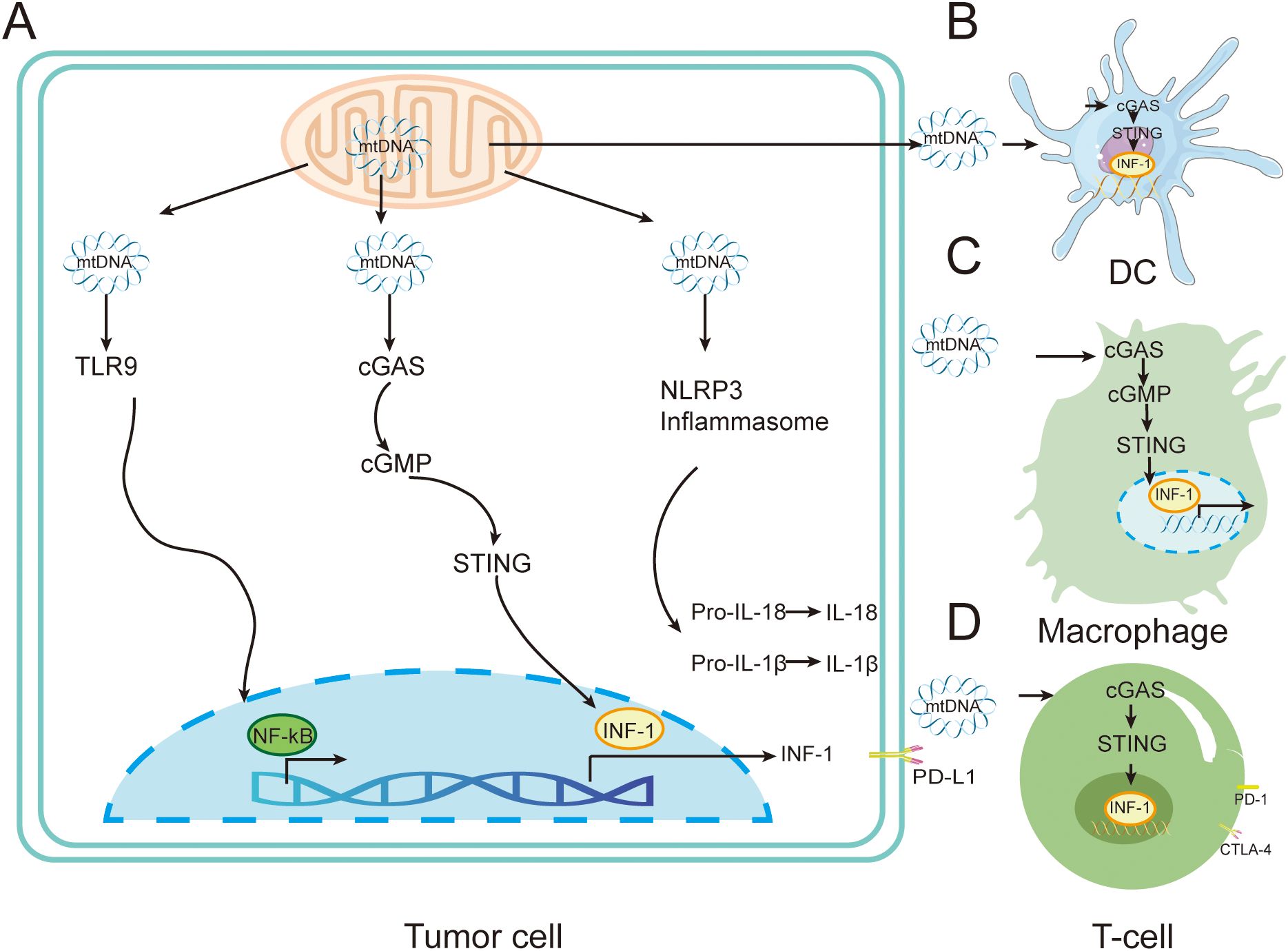
Figure 4. Immune regulation by mtDNA in tumors cells. (A) Mitochondria can release mtDNA in response to external or endogenous stress. The released mtDNA triggers various pro-inflammatory signaling pathways through TLR9, cGAS-STING, or through the cytoplasmic inflammasome NLRP3. (B) mtDNA released by tumor cells is transferred to DC cells, stimulating the activation of the cGAS-STING pathway and leading to the release of type I interferons. (C) mtDNA released by tumor cells is transferred to DC cells, stimulating the activation of the cGAS-STING pathway and leading to the release of type I interferons, inducing immunosuppressive M2 phenotype macrophages. (D) mtDNA released by tumor cells is transferred to T cells, stimulating the cGAS-STING pathway and leading to the release of type I interferons.
When mitochondrial stress occurs, mtDNA can be activated by BAX/BAK-dependent mitochondrial outer membrane permeabilization (MOMP) or mitochondrial permeability transition pore (mPTP), and mtDNA can bind to and activate different DNA pattern recognition receptors to induce strong intrinsic immune responses (72). After release into the cytoplasm, mtDNA can be recognized by pattern recognition receptors such as cGAS, TLR9 and NLRP3, which activate downstream inflammatory signaling pathways.
When mtDNA leakage occurs, the cytoplasmic localized receptor cGAS recognizes mtDNA and induces generation of the second messenger 2´3´-cGAMP. Subsequently, cGAS activates endoplasmic reticulum-localized protein STING and mediates the downstream activation of the type I interferon (INF-I) signaling pathway and the associated inflammatory response (73). Studies have shown that cGAS-STING signaling activation has an important regulatory role in tumor immunity. Tumor-specific adaptive immune responses, including cytotoxic T-cell (CD8+ T-cell) activation, are dependent on INF-I signaling from APCs. Activation of INF-I is largely mediated by the cGAS-STING signaling pathway (74). mtDNA can enhance the function of Treg through the cGAS-STING signaling pathway, thereby suppressing tumor immunity and promoting the development of T lymphoma (75). Fatty acid binding protein 5 (FABP5) is one of the proteins that maintains mitochondrial stability in T cells. Thus, FABP5 inhibitors impair mitochondrial integrity and promote the release of mitochondrial DNA, thereby inducing interleukin 10 (IL-10) production via activation of the cGAS-STING signaling pathway. IL-10 facilitates T-lymphoma development by promoting Treg in the TME to suppress the viability of other T-cells (76). Mitochondrial DNA upregulates programmed cell death ligand 1 (PD-L1) and IDO-1 via the cGAS-STING signaling pathway, thereby inhibiting T cell function (77). CD47 blockade disrupts SIRPα-CD47 signaling, preventing lysosomal degradation of phagocytosed mtDNA in DCs, thereby enhancing cGAS sensing (78). Ionizing radiation can damage the mitochondria of tumor cells such as colon cancer, lung cancer and T lymphoma, resulting in the release of mtDNA, which is phagocytosed by DC cells and activates the cGAS-STING signaling pathway, enhancing the ability of DC cells to deliver antigens to CD8+ T cells, and ultimately enhancing tumor immunity (79). The ataxia telangiectasia mutated (ATM) protein detects DNA double-strand breaks and promotes DNA damage repair. Pharmacological inhibition of ATM (e.g., KU-55933) reduces mitochondrial transcription factor A (TFAM) expression in melanoma and breast cancer cells, promotes mtDNA leakage into the cytoplasm, activates the cGAS-STING signaling pathway and downstream cytokine production, and enhances lymphocyte infiltration into the TME, resulting in anti-tumor therapeutic effects (80). Activation of cGAS-STING signaling can also activate a variety of immune cells including DCs, macrophages, NK cells, CD4+ and CD8+ T cells by triggering the relevant natural immune signals, leading to reduction or even complete disappearance of a variety of tumors in vivo (81).
TLR9 supports tumor cell growth and chemoresistance by recognizing the CpG structural domain of mtDNA which activates downstream MAPK and NF-κB signaling to promote the associated inflammatory responses (82). Notably, mtDNA leaking into the extracellular space can also be involved in the polarization and functional regulation of a variety of immune cells, including macrophages, DCs, and T lymphocytes, through the activation of TLR9 and cGAS-STING signaling in neighboring immune cells (83).
NLRP3, as a multicomponent protein complex in the cytoplasm, recognizes mtDNA leaking into the cytoplasm and activates downstream MAPK and NF-κB signaling (84). mtDNA activates NLRP3 inflammasome assembly via K+ efflux, leading to caspase-1-dependent IL-1β maturation (85).
Mitochondrial genome mutations constitute a significant portion of the mutation genome in cancer. Their functional impairments, mutations, copy number abnormalities, aberrant expressions, and alterations in epigenetic modifications can influence cancer development and malignant transformation by regulating cellular metabolism, ROS production, and intercellular interactions. Moreover, the specific locations and levels of these mutations can confer a competitive advantage to cancer cells. Under mitochondrial stress, mitochondrial DNA can be activated via specific mechanisms and released into the cytoplasm or extracellular space, where it can be recognized by receptors such as cGAS, TLR9, and NLRP3. Notably, cGAS activates STING upon recognition, mediating associated signaling pathways and inflammatory responses, which play a crucial role in tumor immunity—potentially inhibiting tumor immunity and promoting cancer, or enhancing tumor immunity and producing anti-tumor effects; it can also activate various immune cells to reduce tumors. TLR9 recognizes specific structural domains, supports tumor growth, enhances chemotherapy resistance, and contributes to the regulation of immune cell function. NLRP3, upon recognition, activates downstream signals, and mtDNA can induce the assembly of its inflammasome through potassium efflux.
2.4 Role of mitochondrial autophage in anti-tumor immunity
Tumor mitochondrial autophagy is a specialized autophagic process in tumor cells that selectively eliminates damaged or redundant mitochondria (86). This autophagy relies on pathways such as Parkin-PINK1, BNIP3/BNIP3L, and FUNDC1, serving as a key mechanism regulating mitochondrial homeostasis in tumor cells (87). Mitochondrial autophagy exhibits dual-sided effects: on one hand, it helps tumor cells adapt to hypoxic and nutrient-deprived microenvironments by eliminating damaged mitochondria, reducing ROS accumulation and mtDNA release, thereby maintaining metabolic balance for survival; on the other hand, its excessive activation reduces tumor cell immunogenicity, suppresses innate immune pathways like cGAS-STING, and facilitates immune evasion (88). Conversely, mitochondrial autophagy defects lead to mtDNA accumulation, activating immune responses resulting in the enhancement of antitumor immunity. Currently, targeted regulation of tumor mitochondrial autophagy has emerged as a promising therapeutic approach. By intervening in related pathways and synergizing with immune checkpoint blockade, it can improve tumor treatment efficacy.
Autophagy is a key mechanism supporting the maintenance of activated states and antitumor functions in CD8+ T cells. Following tumor antigen recognition by the T cell receptor (TCR), basal autophagy activity is triggered: it degrades intracellular surplus proteins to release amino acids and other metabolic substrates, providing energy for CD8+ T cell proliferation while maintaining organelle homeostasis. Simultaneously, it regulates immunological synapse formation, promotes TCR signaling activation, and drives CD8+ T cells from a quiescent to an effector state.
However, the hypoxic microenvironment of the TME can induce autophagy via HIF1α, downregulating MHC-I molecule expression and thereby reducing CD8+ T cell cytotoxicity (89). Similarly, CXCL1 mediates MHC-I degradation via autophagy in colorectal cancer (CRC), while the oncogene PACSIN1 promotes MHC-I lysosomal degradation through autophagy in gastric cancer, both inhibiting antigen presentation and CD8+ T cell infiltration to drive immune escape (90, 91). Furthermore, defects in autophagy-related genes (ATG) significantly impact CD8+ T cell function: Atg5/Atg7 deficiency enhances CD8+ T cell infiltration and IFN-γ secretion, while Atg4/Atg5 knockdown upregulates MHC-I expression and antigen presentation in lung cancer cells; while Atg7 deficiency suppresses tumor cells through metabolic promotes CD8+ T cell accumulation in the colonic lamina propria (92–94). Clinical studies reveal that LC3B expression in hypopharyngeal squamous cell carcinoma (HSCC) positively correlates with CD8+/CD39+ T cell infiltration, while LC3B deficiency in breast cancer reduces CD8+ T cell infiltration and increases FOXP3+ Treg/CD68+ macrophage numbers, suggesting autophagy influences tumor prognosis by regulating CD8+ T cell infiltration (95).
Notably, autophagy modulates CD8+ T cell function by regulating immune checkpoints and cytokines: in CRC, CTSS upregulates PD-L1 via autophagy and reduces CD8+ T cell infiltration (96). In acute myeloid leukemia (AML), C/EBPα DM alleviates CD8+ T cell immunosuppression by inhibiting autophagy-associated IL-1β secretion (97). Combining autophagy inducers with chemotherapeutic agents specifically activates CD8+ T cell-dependent anticancer immunity, suggesting that targeting autophagy may serve as a potential strategy to enhance CD8+ T cell antitumor function.
In the TME autophagy provides essential survival support for Tregs by degrading intracellular glycogen, damaged proteins, and releasing metabolic substrates from mitochondria. Simultaneously, mitochondrial autophagy clears hypoxia-induced damaged mitochondria, reduces ROS accumulation, and prevents premature Treg apoptosis. Autophagy stabilizes Foxp3 transcription factor expression in Tregs, promotes synthesis of inhibitory cytokines like IL-10 and TGF-β, and enhances their immunosuppressive effects on CD8+ T cells. CAFs in the TME can activate Treg expansion through antigen-dependent and autophagy-dependent pathways by forming immune synapses with Tregs (98). UNC-51-like kinase 1 (ULK1), as an autophagy-activating molecule, is a key candidate target for regulating Treg function (99). Clinical studies reveal abnormally elevated CD39+ Treg levels in patients with autophagy genetic defects, and these CD39+ Tregs show low expression of autophagy-related genes NEFL and PLAC8 (100). Furthermore, defects in the GTPase-activating regulator RGS1 disrupt Treg metabolism and autophagy via the FOXP3-c-MYC axis, diminishing their immunosuppressive capacity (101). This suggests autophagy is a core pathway for maintaining Treg function.
In the TME, autophagy regulates TAM polarization. In undifferentiated pleomorphic sarcoma (UPS), COLVI induces CD8+ T cell dysfunction by inhibiting T cell autophagy while promoting TAM M2 polarization and VEGF/TGF-β secretion, thereby facilitating tumor angiogenesis (102). Oxidative stress induces tumor cells to release KRAS(G12D), which is packaged into exosomes via autophagy-dependent ferroptosis and induces M2 polarization in macrophages through the AGER-STAT3 axis (103).
Conversely, under stimuli like chemotherapy drugs, autophagy induces M1 polarization in TAMs. Autophagy inhibition enhances pro-inflammatory effects associated with M1 polarization by regulating macrophage migration inhibitory factor (MIF) secretion via ROS; while mTOR signaling inhibition reduces M2-type TAMs and MDSCs in the TME by downregulating autophagy, simultaneously upregulating CD8+/CD4+ T cells (104). This suggests autophagy serves as a critical regulatory node in TAM polarization.
Hypoxia and immunosuppressive factors in the TME can impair DC function, whereas autophagy maintains DC activity through multiple mechanisms: on one hand, it degrades senescent mitochondria and damaged proteins within DCs, preserving energy homeostasis and reducing ROS-induced apoptosis to safeguard DC numbers; on the other hand, it degrades tumor antigens through autophagosome-lysosome fusion, generating antigenic peptides for presentation via MHC class I/II molecules. Crucially, it facilitates antigen shunting to the MHC class I pathway during cross-presentation, efficiently activating CD8+ T cells (105).
Autophagy also regulates DC maturation and cytokine secretion: activating autophagy promotes DC expression of co-stimulatory molecules like CD80 and CD86 while secreting IL-12, enhancing immune activation. Conversely, autophagy defects leave DCs in an immature state, even secreting TGF-β to exacerbate immune suppression (105). Furthermore, ROS-dependent endoplasmic reticulum stress in tumor cells suppresses DC surface calretinins exposure via autophagy, diminishing their maturation capacity and IL-6 secretion, thereby inhibiting CD4+/CD8+ T cell proliferation (106). High Mobility Group Box 1 (HMGB1) inhibits DC apoptosis via the JNK-autophagy axis, contributing to colon cancer cell immune evasion. This suggests autophagy is a core regulatory mechanism for DC-mediated antitumor immunity (106).
Autophagy enhances NK cell antitumor activity by regulating the synthesis and recognition of killing molecules. Disrupting the interaction between ATG7 and phosphorylated FOXO1 in the cytoplasmic solute of immature NK cells blocks autophagy, a process critical for NK cell maturation. Activating autophagy may support the maturation of NK cells and other ILCs exhibiting anticancer activity (107). In the B16-F10 melanoma model, the autophagy-critical gene Beclin1 induces substantial infiltration of functional NK cells into the tumor bed by activating the MAPK8/JNK-JUN/c-Jun signaling pathway, significantly inhibiting tumor growth (108). This further confirms autophagy's positive regulatory role in NK cell antitumor function. Targeting Becn1 to inhibit autophagy significantly restores the levels of serine protease GZMB/granzyme B within target cells under hypoxic conditions and induces tumor regression in vivo by promoting NK cell-mediated tumor cell killing (109, 110).
Autophagy defects impair MDSC lysosomal degradation, upregulate MHC-II molecule expression, thereby activating tumor-specific CD4+T cells and reducing tumor volume; conversely, in multiple myeloma (MM), MDSCs induce tumor cell autophagy via AMPK phosphorylation, upregulating MCL-1/BCL-2 expression to enhance MM cell survival (111). Mechanistically, glycolysis inhibits CCAAT enhancer-binding protein β (CEBPβ) subtype LAP expression via the AMPK-ULK1-autophagy axis, thereby regulating G-CSF/GM-CSF secretion to support MDSC development (111). While the lysosomal inhibitor LCL521 disrupts autophagy by activating cathepsin B/D, inducing endoplasmic reticulum stress in MDSCs and promoting their death, providing a basis for therapies targeting MDSC autophagy (112).
3 Mitochondrial targeting strategies
Mitochondria have great potential as therapeutic targets. Drug entry into cells requires passage through several lipid bilayers, especially the inner mitochondrial membrane which is highly selective for molecular traversal, which is the reason why mitochondria-targeted drugs are difficult to deliver (113). Currently, several methods have been developed to enable targeted drugs to break through the lipid bilayer into the mitochondria, such as delocalized lipophilic cation (DLCs) modification and mitochondria-targeted drug delivery technologies (114) (Figure 5).
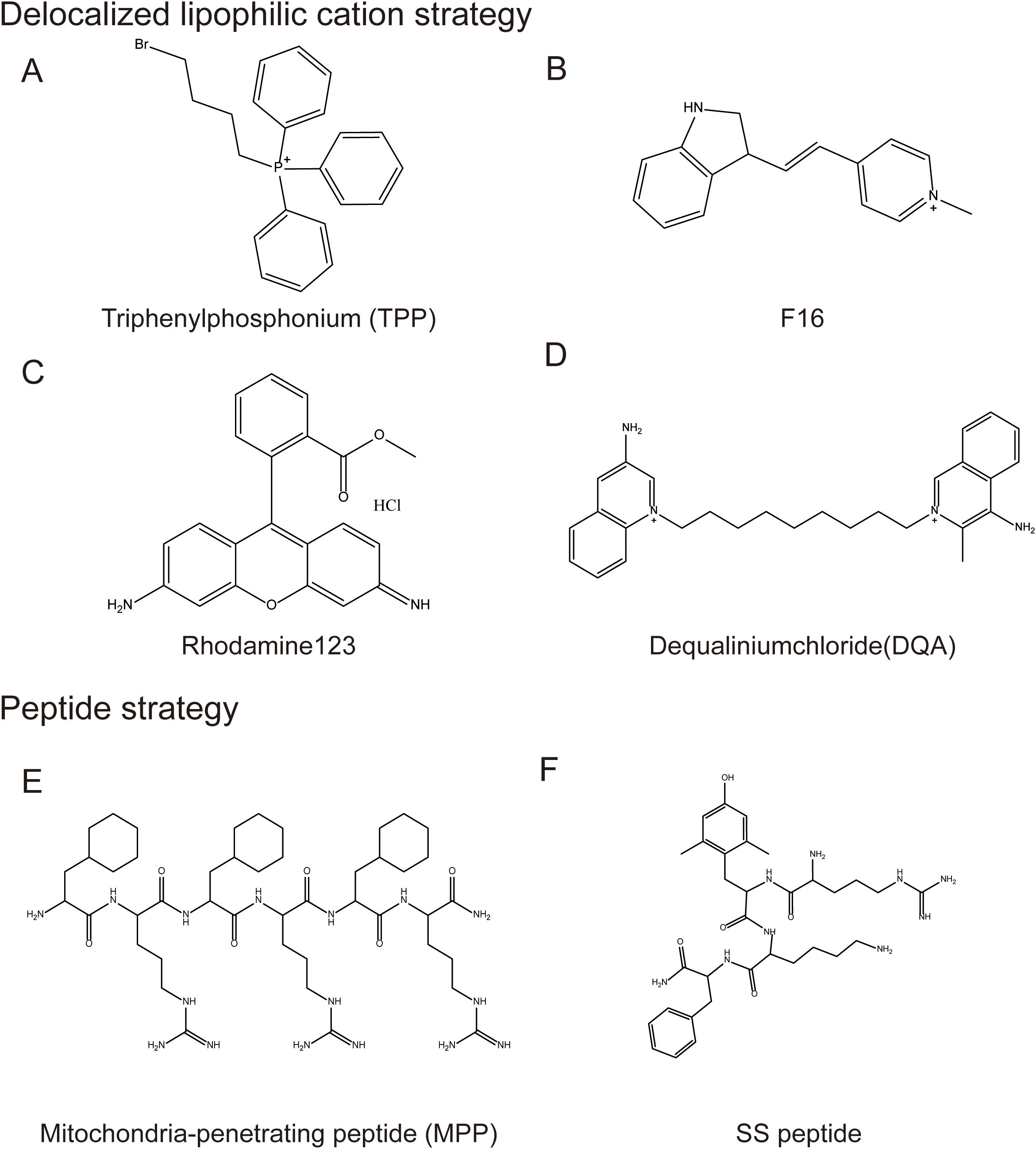
Figure 5. Mitochondrial-targeted structural modification strategies. Typical Mitochondrial-targeted structural modification strategies: 1. Delocalized lipophilic cation strategy, including (A) Triphenylphosphonium, (B) F16, (C) Rhodamine123, (D) Dequaliniumchloride; 2. Peptide strategy including (E) Mitochondria-penetrating peptide and (F) SS peptide.
3.1 Mitochondrial targeting based on DLC modification
DLCs are a class of compounds that can penetrate the lipid bilayer and accumulate in mitochondria, due to the high mitochondrial membrane potential of tumor cells. These compounds can be used for targeted delivery of drugs to mitochondria of tumor cells by covalently linking them with small molecules. DLCs identified in current studies include triphenylphosphine (TPP+) and its derivatives, F16, rhodamine analogs, and dequaliniumchloride (DQA).
3.1.1 TPP and its derivatives
The chemical structure of TPP contains three phenyl groups, which makes it highly lipid-soluble (115). At the same time, the positively charged phosphorus ions can be delocalized to the three benzene rings, allowing them to pass smoothly through the lipid bilayer (116). TPP drives drug accumulation within mitochondria due to the the negative mitochondrial membrane potential (ΔΨm) (117) (Figure 6).
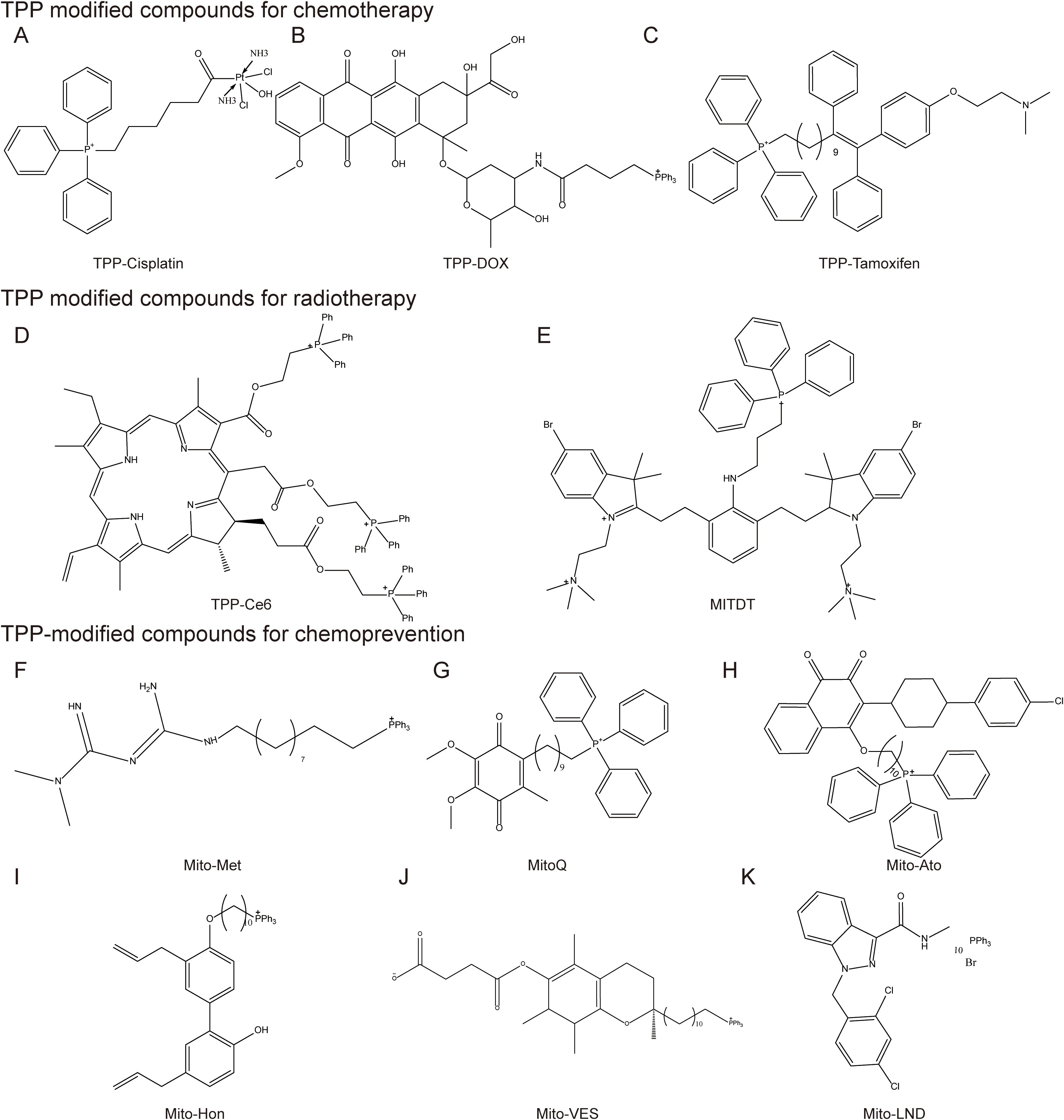
Figure 6. Mitochondrial-targeted triphenylphosphine compounds. TPP modified compounds for chemotherapy include (A) TPP-Cisplatin, (B) TPP-DOX, (C) TPP-Tamoxifen; TPP modified compounds for radiotherapy include (D) TPP-Ce6 (E) MITDT; TPP-modified compounds for chemoprevention, including (F) Mito-Met, (G) MitoQ, (H) Mito-Ato, (I) Mito-Hon, (J) Mito-VES, and (K) Mito-LND.
Chemotherapy drugs have long been the mainstay of therapy for many cancer types. However, severe side effects, low bioavailability, poor stability, and acquired drug resistance limit their clinical application. Mitochondria-targeted monofunctional platinum complexes can accumulate in the mitochondria, induce significant changes in mitochondrial ultrastructure and membrane, release cytochrome c(Cytc) from mitochondria, and disrupt mitochondrial OXPHOS and glycolysis (118). Paclitaxel (PTX) modified with TPP cations reduced the decrease in ΔΨm and significantly inhibited the growth of MCF-7 cells. Doxorubicin (DOX) resistance is a common problem in cancer treatment (119). Addition of TPP to DOX-PLGA/CPT nanoparticles leads to effective mitochondrial localization of DOX-PLGA/CPT, releases DOX to target mtDNA, induces tumor cell apoptosis and overcomes DOX resistance in MCF-7/ADR breast cancer cells (120) (Figure 7).
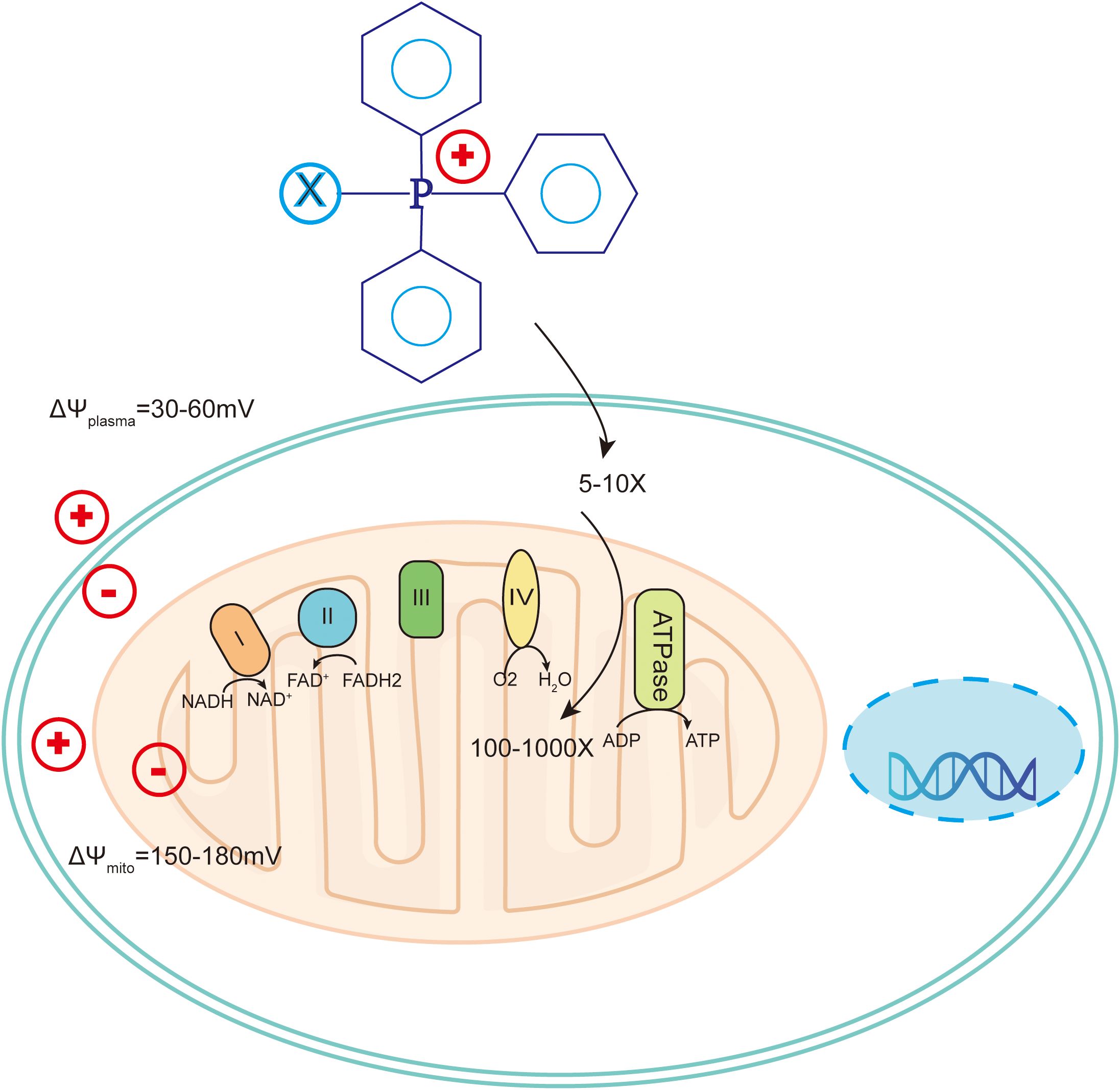
Figure 7. Mechanism of triphenylphosphine targeting to mitochondria in tumor cells. Cancer cells have more hyperpolarized membranes than normal cells, helping to drive uptake of TPP+-conjugated compounds by up to 100-1000-fold. .
Photodynamic therapy (PDT) is being used to treat some cancers, and it has become a promising approach for the treatment of malignant brain tumors. Mitochondria-targeted triphenylphosphine can enhance PDT efficacy in brain cancer. The TPP-conjugated photosensitizer chloramphenicol e6 (Ce6) selectively accumulates in mitochondria, colocalizes with 88% of mitochondria, and has potent cytotoxic activity, thereby significantly enhancing PDT efficacy (121, 122). The PDT effect of the mitochondria-targeting photodynamic therapeutic (MitDt) agent is amplified after laser irradiation because mitochondria are susceptible to ROS, triggering apoptotic anticancer effects (123). TPP-modified photosensitizer zinc phthalocyanine (ZnPc) selectively accumulates in mitochondria, showing excellent mitochondrial targeting for ROS-activated chemotherapy and PDT (124). TPP-modified liposomes encapsulating black phosphorus (BP) and calcium peroxide (CaO2) accumulate in tumor mitochondria and are activated by near-infrared (NIR) laser irradiation to generate abundant PO43- and Ca2+ to accelerate in situ mitochondrial mineralization, leading to mitochondrial dysfunction and cancer cell death (125).
Radiotherapy has been an important form of cancer treatment for many years. TPP can significantly increase the efficacy of radiation enhancers and improve the effect of radiotherapy. Smaller doses of radiation (4Gy vs standard 12Gy) can be given in combination with TPP-based PDT to control tumor growth, reducing radiation side effects (126). 4-hydroxy-2,2,6,6-tetramethyl-1-oxy-piperidin(Tempol) coupled with TTP enhances X-irradiation-induced germ cell death, reduces basal ΔΨm and inhibits X-ray-induced increase in ATP production (127).
Photothermal therapy (PTT) is a new non-invasive tumor treatment that uses photothermal agents (PTA) to convert light energy into heat energy to kill tumor cells under irradiation with external light sources such as NIR light. Micro-nanoparticles loaded with TPP and S-nitrosothiol can release NO generated by surface overheating and elicit PTT upon NIR laser irradiation. The released NO can also destroy collagen fibers by activating matrix metalloproteinases (MMPs), thereby loosening the dense extracellular matrix (ECM) to enhance immune cell infiltration. The highly toxic reactive nitrogen species (RNS) peroxynitrite (ONOO-) is produced, resulting in mitochondrial damage and induction of cell apoptosis (128). Nanoparticles with core-shell-disulfide-shell nanoparticles burst in the high GSH environment of tumors to achieve targeted drug release. The loaded DOX can quickly enter mitochondria, subsequently destroying mitochondrial DNA and leading to cell apoptosis. The synergistic effect of PTT and chemotherapy targeting mitochondria significantly enhances cancer treatment (129). The heat stress-damaged mitochondria produced can cause ICD in tumor cells, release damage-related factors, reactivate the immune response of macrophages against tumor cells, and effectively activate tumor-associated macrophages to fight against tumor cells (130).
TPP conjugates enhance the accumulation of chemopreventive agents in tumor cell mitochondria, enhancing efficacy and reducing toxicity to normal tissues (131). Metformin (Met), a commonly used hypoglycemic drug, has certain mitochondria-targeting effects and anti-tumor ability, but its clinical performance is not ideal. In pancreatic ductal adenocarcinoma (PDAC) cell lines, the IC50 concentration required to inhibit proliferation by Mito-Met is nearly 1,000 times lower than that of Met (132). Mito-Met induces superoxide (O2•-) production through complex I, inducing ROS-disrupting membrane potential, and activating calcineurin- and Cn-dependent retrograde signaling pathways in multiple cells (133). Atovaquone(ATO), an antimalarial drug, was discovered to have anti-tumor potential in the form of TPP-modified and PEGylated mitochondrial-targeted ATO (Mito-(PEG)n-ATO). Mito-ATO analogs inhibit mitochondrial complex I and complex III-induced OXPHOS in human pancreatic and brain cancer cells. Combined use with inhibitors of monocarboxylate transporters (MCT), Krebs cycle redox metabolism, or glutaminolysis, the Mito-ATO analogs can synergistically eliminate tumor cell proliferation (134). TPP mitochondrial targeting increases the drug's involvement in metabolic processes within mitochondria, inhibits tumor cell development, and promotes tumor cell death (135).
TPP has a high fat-soluble transportable positively charged phosphorus ion, which can drive the accumulation of drugs in mitochondria with the help of mitochondrial negative membrane potential. Drugs such as metformin and atropine can be used in chemotherapy to increase efficacy and overcome resistance, in radiotherapy to increase efficacy and reduce side effects, and in PTT to aid the initiation of treatment and destruction of ectoplasm, induction of apoptosis. TPP-derived compounds exhibit good antitumor activity, but there are few clinical studies to verify their anticancer efficacy, and further clinical studies are needed in the future (117). Although the TPP cation itself has low toxicity, some of the TPP-derived compounds administered systemically have non-specific toxicity, and the current strategy is mainly to modify the structure of TPP cations, encapsulate the modified compounds in liposomes, and reduce the toxicity of TPP cations, enhance its targeting selectivity to tumor cells and mitochondria to reduce toxicity (131).
3.1.2 F16
F16 is a delocalized DLC that can target and aggregate in the mitochondrial matrix of tumor cells. F16 induces mPTP opening by inhibiting the interaction between mitochondrial inner membrane adenine nucleotide translocase (ANT) and cyclophilin D. F16 can form conjugates with other substances to enhance anti-tumor effects. At the same time, the reduced availability of intracellular adenosine 5'-triphosphate induced by the uncoupling effect of F16 is the main factor in the enhanced cytotoxicity mediated by F16 (136). F16 conjugates show higher cytotoxicity at low doses, and F16 conjugates initiate cell cycle arrest at the G0/G1 phase leading to mitochondrial dysfunction and excessive production of ROS, thereby inducing apoptosis (137–139). The F16-modified compounds accumulate in cancer cell mitochondria to depolarize ΔΨm, increase ROS and attack mtDNA, effectively killing cancer cells and overcoming multi-drug resistance (140, 141). Fluorescent mitochondria-targeted organic arsenic accumulates in mitochondria and inhibits the activity of pyruvate dehydrogenase complex (PDHC), leading to ATP synthesis inhibition and heat production disorders. The inhibition of respiratory chain complexes accelerates mitochondrial dysfunction and causes cell apoptosis (142).
F16 can also be used for fluorescence imaging of mitochondria. CyM is a multifunctional organic biological probe that can facilitate NIR imaging and PDT in vivo and in vitro (143). There are two F16 isomers that can specifically display mitochondria in the green and red channels, respectively, due to their unique fluorescence properties, providing new ways of studying mitochondrial targeting by F16. The above studies suggest that F16 and its derivatives can be of great value in cancer treatment and tumor imaging. However, the clinical application of F16 is limited by its toxicity to normal cells (144). Therefore, scientists have focused on enhancing the selectivity of F16 and its derivatives for tumor cells, and future research will likely uncover new drugs that can specifically target tumor cell mitochondria and reduce toxic effects on normal cells.
In summary, F16 acts as a DLC that can be targeted to cluster in the mitochondrial matrix of tumor cells, and its conjugate can depolarize δψm, increase Ros to attack mtDNA to kill cancer cells, and overcome multiple drug resistance. F16 can also be used for mitochondrial fluorescence imaging.
3.1.3 Rhodamine
Rhodamine is an organic fluorescent dye based on xanthene that can be substituted with different 3- and 6-amino groups. It has a darker color and stronger fluorescence signal. Rhodamine can penetrate the cell membrane and selectively stain the mitochondria of living cells. Rhodamine dyes have photophysical properties such as high fluorescence quantum yield, high molar extinction coefficient and good water solubility, and low biological toxicity, making them attractive for wide use as biomarkers and fluorescent probes.
Rhodamine conjugates can be delivered to tumor mitochondria and functional proteins through organic cation transporters, improving their tumor inhibitory effects. Rhodamine B mitochondria-targeted multi-drug nanoparticles focus on mitochondrial stress-induced ICD to improve their therapeutic effect on treatment of ovarian cancer (145). Centella asiatica-rhodamine B conjugates are highly cytotoxic to human tumor cell lines, affect cell apoptosis, and can overcome resistance to chemotherapeutic drugs (146). Rhodamine B-conjugated oleanolic acid derivatives (RhodOA) reduce tumor cell viability, reduce cell migration and disrupt mitochondrial function (147). Hybrid peptide-fused rhodamine B increases anticancer activity by up to 37.5 fold, targeting the nucleus and triggering apoptosis to enhance anticancer cell activity (148). Enrichment of rhodamine B-modified catalase in cancer tissues can effectively inhibit mouse xenograft human lung tumors (149). Differences in ΔΨm and ATPase sensitivity in tumor cells contribute to the selective cytotoxicity of rhodamine123 against certain cell types in vitro (150). Mitochondrial targeting by rhodamine enhances the preferential cellular uptake of paclitaxel and SN-38 in cancer cells by 2–3 fold (151). Multi-walled carbon nanotubes (MWCNTs) with mitochondria-targeted fluorescent rhodamine-110 colocalize 80% with mitochondria and exhibit superior efficacy to drugs without PtBz (152). Ciacic acid-rhodamine 101 conjugates induce proliferation or growth arrest of MDA-MB-231 breast cancer cells at low doses and induce apoptosis at higher doses (153).
Rhodamine can significantly improve the effect of PDT on tumors and is by itself a potential PDT agent. The combination of rhodamine organic dyes and luminescent transition metal centers exhibits low cytotoxicity, increases tumor cell uptake, and enhances antitumor efficacy (154). Rhodamine 6G-based organic salts are stable under physiological conditions and show excellent fluorescence photostability. More importantly, they have tunable chemotherapeutic properties. Rhodamine fluorescent groups synthesized from Rh-6G and amines show pH-dependent anticancer bioactivity and trigger cell apoptosis through mitochondrial pathways, showing anticancer bioactivity in bladder cancer (155). Rhodamine 6G-based organic salts can produce nanoparticles that are toxic to cancer cells but not normal cells (156). The apoptotic index of Dasatinib (DST) contained nanoparticles is 7.5 times higher than that of free DST and is non-toxic to normal cells (157). Rhodamine-mediated novel supramolecular assemblies can efficiently capture phosphorescence energy transfer (PET) processes and have potential applications in delayed fluorescence cell imaging (158). Mitochondria-targeted silicon rhodamine-based photosensitizer (SiR-PXZ) can be rapidly taken up by mitochondria and effectively induce cancer cell apoptosis, showing excellent anti-tumor effects and potential value in photodynamic cancer therapy (159). Burst-specific PDT in mitochondria by the rhodamine derivative UCNP-GQD/TRITC induces a sharp drop in ΔΨm, thereby irreversibly initiating tumor cell apoptosis (160).
Rhodamine, with its ability to penetrate cell membranes and selectively stain mitochondria in living cells, is often used as a biomarker and fluorescent probe, its conjugates can target tumor mitochondria to inhibit tumor cells, overcome drug resistance and enhance anticancer drug uptake through a variety of mechanisms. Rhodamine can improve the effect of tumor PDT and is a potential PDT agent, showing potential value in fields such as photodynamic cancer therapy and delayed fluorescence cell imaging.
3.1.4 DQA
DQA is a DLC with two positive charge centers. It can selectively accumulate in mitochondria driven by transmembrane potential, allowing anti-tumor drugs to target mitochondria in tumor cells. DQA-coupled FMPSi-Cis@GO targets mitochondria in cancer cells and destroys their function (161). DQA-containing micelles deliver DOX to the mitochondria and nucleus of tumor cells, significantly inhibiting the growth of DOX-resistant tumors without obvious systemic toxicity (162). Amphiphilic polymer GC-DQA nanoparticles were synthesized as carriers to efficiently deliver curcumin to mitochondria (163). The emulsion of DQA and α-tocopheryl succinate (α-TOS) targeting mitochondria has good stability and can effectively target mitochondria and inhibit the growth of HeLa cells (164).
DQA modification destroys mitochondrial structure and induces cell death by generating ROS and dissipating ΔΨm. DQA causes loss of mitochondrial transmembrane potential, O2*- accumulation and ATP depletion in this tumor cell line, alters mitochondrial function and induces cell death (165). Hinokiflavone (HF) hybrid micelles increase ROS levels, reduce ΔΨm, and induce mitochondria-mediated apoptosis (166). DQA chloride vesicles (HPS-DQAsomes) of DOX increase cytotoxicity to MCF-7/ADR cell lines, can target the delivery of therapeutic agents to mitochondria and induce mitochondria-driven apoptosis (167). DQA- polyethylene glycol (PEG)-modified resveratrol liposomes DLS (Res) selectively accumulate in mitochondria, inducing cytotoxicity of cancer cells by generating ROS and dissipating ΔΨm (168).
DQA is an inhibitor of apoptotic proteins that can directly inhibit the activity of caspases, regulate apoptosis through multiple pathways, and promote the degradation of Bcl-2 as an E3 ligase, thereby exerting anti-tumor effects. DQA hybrid micelles enhance the uptake of paclitaxel by drug-resistant breast cancer cells. Induction of tumor cell apoptosis is related to the activation of pro-apoptotic proteins Bax, Cytc, caspases-3, 9 and the inhibition of Bcl-2 and Mcl-1 (169). Targeted lonidamine liposomes selectively accumulate in mitochondria of drug-resistant A549 lung cancer cells, dissipate ΔΨm, open mitochondrial permeability transition pores, and release Cytc through translocation. A cascade of caspases 9 and 3 reactivity is initiated, which activates the pro-apoptotic Bax protein and inhibits the anti-apoptotic Mcl-1 protein, thereby enhancing cytotoxicity by acting on mitochondrial signaling pathways (170). The development of targeted resveratrol liposomes modified with DQA-PEG (2000)-DSPE on the liposome surface significantly enhance cellular uptake and selectively accumulate in mitochondria. They induce apoptosis in non-resistant and resistant cancer cells by dissipating ΔΨm, releasing Cytc, and increasing the activity of caspases 9 and 3 (171).
DQA can be combined with other drugs to exert a wide range of anti-tumor activities through targeting of mitochondria and is a safe and economical cancer treatment. However, the existing combination of nanomaterials and drugs has not yet achieved breakthrough results, and further research is needed on the role of DQA in targeting mitochondria and its synergistic effect with other drugs.
Due to the high mitochondrial membrane potential of tumor cells, DLC can penetrate the lipid bilayer to accumulate mitochondria, and is often covalently linked to drugs for targeted delivery, mainly including TPP and its derivatives, F16, rhodamine, and DQA. TPP and its derivatives are highly lipid-soluble and positively charged, which can help the accumulation of mitochondrial cells of drugs, enhance the efficacy of chemotherapy, PDT, radiotherapy, PTT, etc., and can also improve the effect of chemopreventive agents. F16 targets the mitochondrial stroma and induces mPTP opening, and its conjugates enhance cytotoxicity and aid tumor imaging, but are toxic to normal cells. Rhodamine can stain mitochondria, and the conjugate can enhance tumor suppression and improve PDT effect. DQA has two positive charge centers that help drugs target mitochondria, disrupt mitochondrial function, induce cell death, and can be combined with other drugs to fight tumors.
3.2 Peptide targeting sequences
3.2.1 Mitochondria-penetrating peptide
MPPs are synthetic mitochondrial localization peptides composed of 4 to 16 amino acids, containing cationic and hydrophobic residues. Similar to DLC, MPPs can finely regulate the localization of mitochondria by changing lipophilicity and charge, and have a significant inhibitory effect on growth of tumor cells in vivo and in vitro (172).
MPP-modified DOX copolymers can promote cell apoptosis and inhibit tumor metastasis by destroying mitochondria, inhibit the growth of breast cancer 4T1 cells in vivo, and overcome tumor resistance (173). MPP-modified DOX significantly enhances drug accumulation in mitochondria by 11.6 fold, resulting in a significant increase in ROS generation and a decrease in the production of ATP that can inhibit drug efflux and the growth of drug-resistant cancer cells (174). Nanoparticles (NPs) consisting of membrane-permeable peptide amphiphiles (MMPA) and small interfering RNA (siRNA) can specifically accumulate in mitochondria and inhibit tumor growth by inhibiting ATP production and repolarizing TAMs (175). Nanocomplexes of mitochondrial-penetrating peptides mtCPP1 and PepFect14 affect biological functions in cytoplasm and mitochondria and can effectively target mitochondrial genes (176). Poly(lactide-co-glycolide) (PLGA) conjugates with 6-mer mitochondrial penetrating peptides (MPP) can be used for mitochondrial targets without cytotoxicity. DOX modified with mitochondrial penetrating peptides (MPP) delivers the drug to cancer cell mitochondria, mediating apoptosis and enhancing therapeutic outcomes for multidrug resistant tumors (177). DOX modified with MPP promotes apoptosis and inhibits tumor metastasis by disrupting mitochondria (178).
Compared with DLC, MPP may have more potential as a ligand targeting mitochondria due to its advantages including good biocompatibility and low toxicity. Molecules modified by MPP include RNA, DNA and proteins which can be exploited for human cancer treatment (179).
MPP can regulate lipophilicity and charge to achieve mitochondrial localization, promote apoptosis, inhibit tumor growth and metastasis, and overcome drug resistance. Compared with DLC, MPP has the advantages of good Biocompatibility and low toxicity, and has the potential to be a mitochondrial targeting ligand. Its modified RNA, DNA and protein molecules can be used for human cancer treatment.
3.2.2 Szeto-Schiller peptides
Szeto-Schiller (SS) peptides are usually composed of four positively charged amino acids and are a new small peptide targeting strategy. They can specifically target mitochondrial cardiolipin, enhance mitochondrial plasticity and re-establish optimal bioenergetics.
SS peptides can reduce mtROS production, inhibit the opening of mitochondrial permeability transition pores, and have significant effects in preventing oxidative stress or inhibiting mitochondrial ETC-induced cell apoptosis and necrosis (180). The peptide SS-31 specifically localizes in the mitochondrial inner membrane by interacting with cardiolipin and can be used in the treatment of patients with abnormal ΔΨm (181). The SS-31-modified DOX-loaded liposome delivery system LS-DOX can effectively cross the blood brain barrier (BBB) to target gliomas, and mitochondrial targeting of SS-31 can enhance cellular uptake (182). SS-31 selectively binds to cardiolipin through electrostatic and hydrophobic interactions. By interacting with cardiolipin, SS-31 prevents cardiolipin from converting Cytc to peroxidase while protecting its electron-carrying function. Therefore, SS-31 protects the structure of mitochondrial cristae and promotes OXPHOS (183). Similarly, with excellent mitochondrial targeting ability, SS-20 peptide modification is a promising strategy for mitochondria-targeted drug delivery systems (184). Conjugation of α-TOS with SS-20 achieves delivery to mitochondria, increases ROS generation, opens the mitochondrial permeability transition pore, reduces ΔΨm, and promotes cell apoptosis (185). SS peptides are therefore expected to be studied more extensively in the future as strategic molecules for targeting cancer.
Peptide targeting sequences mainly include mitochondrial penetrating peptides (MPPs) and Stuart Schiller (SS) peptides. MPP is composed of 4–16 amino acids, contains cations and hydrophobic residues, can regulate lipophilicity and charge to locate mitochondria, can enhance the accumulation of mitochondria, promote apoptosis of tumor cells, inhibit metastasis and drug resistance, and has good biocompatibility and low toxicity, and the modified molecule can be used in cancer treatment. SS peptides often contain four positively charged amino acids, which can target mitochondrial cardiolipids, reduce mtROS production, inhibit the opening of mitochondrial permeability transition pores, and protect mitochondrial structure and function.
3.3 Mitochondria-targeted drug delivery technology
Anti-tumor drugs can be loaded into various carriers such as liposomes, polymer nanoparticles, micelles, and solid lipid nanoparticles to work in conjunction with mitochondria-targeted modifications to achieve better anti-tumor effects (186) (Figure 8).
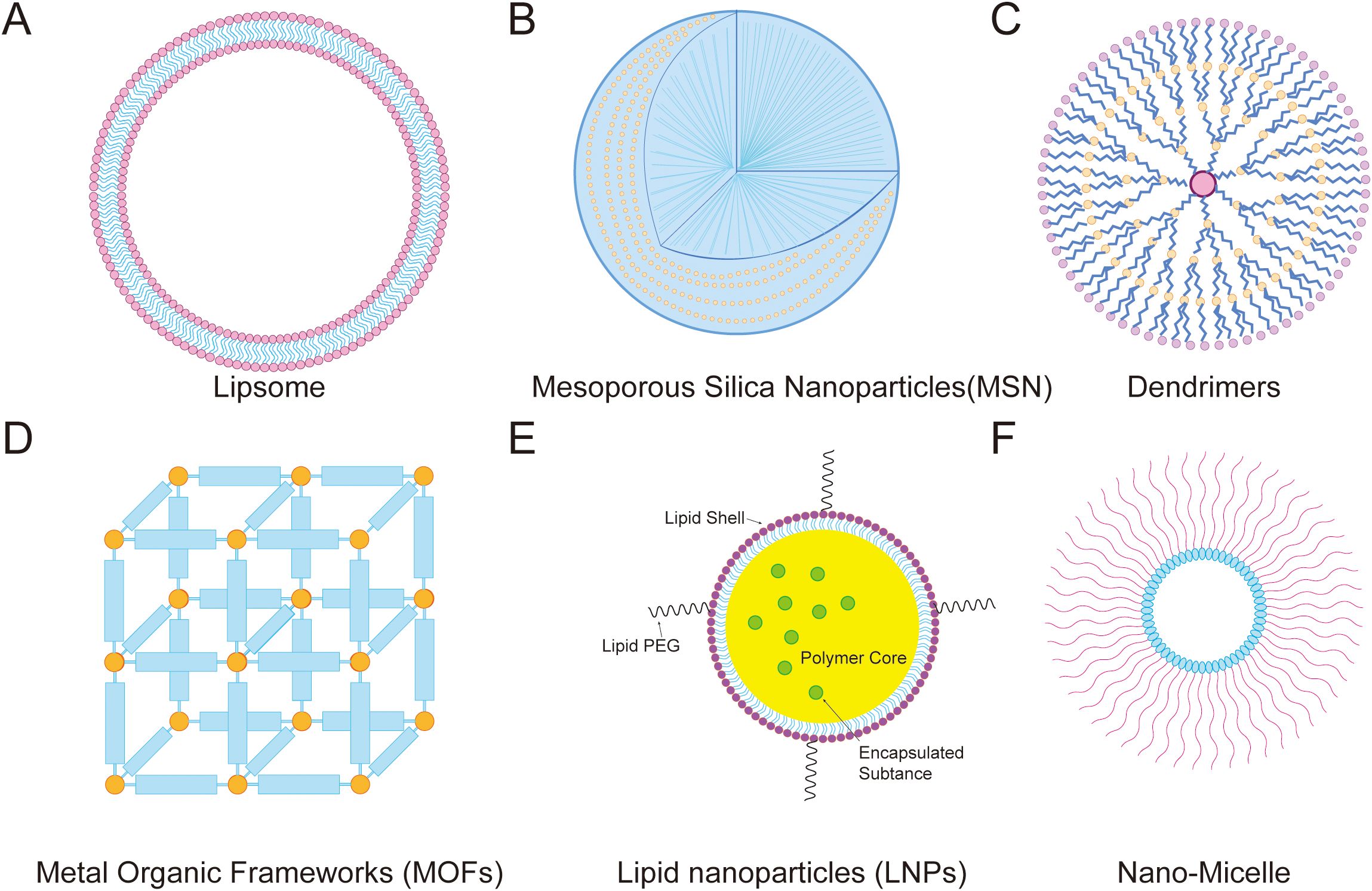
Figure 8. Mitochondrial-targeted nanodrug delivery strategies. (A) Lipsome, (B) Mesoporous Silica Nanoparticles(MSN), (C) Dendrimers, (D) Metal Organic Frameworks (MOFs), (E) Lipid nanoparticles (LNPs), and (F) Nano-Micelle.
3.3.1 Mitochondria-targeted liposomes
Liposomes are artificial membranes with bilayers, which are generally prepared by high-pressure homogenization, ethanol injection, rotary evaporation and ultrasound, and microfluidics. They can carry hydrophilic and lipophilic drugs, with the former distributed in the core compartment and the latter distributed in the bilayer membrane (187). Liposomes have attracted extensive attention due to their excellent drug delivery capabilities, biocompatibility, biodegradability, and ease of manufacture. Mitochondria-targeted liposomes have advantages in tumor-targeted therapies (188).
Liposomes enhance mitochondrial uptake of DOX and the chemosensitizer lonidamine (LND) by cancer cells, inhibiting tumor cell proliferation and inducing cell apoptosis. Lip-SPG significantly alters mitochondrial functions including reduced production of intracellular ATP, induction of ROS production, and enhancing ΔΨm depolarization (189). Milpoxetine (MPt)-loaded liposomes target mitochondria and trigger mtDNA replication blockage to induce mitophagy (190). DOX-loaded liposomes localize to mitochondria, and generate higher ROS levels (191).
Mitochondria-targeted photosensitizer liposomes exhibit high photodynamic therapy efficiency. Nanophotosensitizers can monitor abnormal mitochondrial morphology during photodynamic therapy under the guidance of fluorescence imaging. Liposome-encapsulated photosensitizers enhance cellular uptake, localize in mitochondria, and enhance anti-angiogenesis in PDT treatment (192). After TPP-modified liposomes are internalized by cells, a large amount of ROS can be generated upon laser irradiation, and a stimulatory effect on STING activation and enhanced infiltration of anti-tumor immune cells is observed, which can be used for PDT treatment (193).
3.3.2 Mitochondria-targeted mesoporous silica nanoparticles
MSNs are an ordered mesoporous material prepared by sol-gel methodologies including microwave-assisted technology, chemical etching technology, and template methods. The material has characteristics of good biocompatibility, high specific surface area, controllable size, and degradability. MSN can improve the targeting of drugs in tumor mitochondria through direct coupling with drugs and enhance the killing effect of tumors. MSNs can efficiently deliver DOX and α-TOS to tumor cell mitochondria, enhancing cancer cell killing effects (194, 195). MSN modified Bcl-2 conversion peptides enter mitochondria and bind to Bcl-2, exposing the BH3 domain and inducing apoptosis of DOX-resistant cells (196). MSN increases the accumulation of folate membrane cell receptors (folate) in tumor cells and targets mitochondria (197). MicroRNA-31 coupled to MSNs loaded with DOX increases active transport and promotes intracellular accumulation of drugs. MicroRNA-31 not only directs targeted mtEF4 to promote cell death, but also has a synergistic effect when used in combination with DOX (198). Phenylboronic acid (PBA)-labeled MSN carriers induce mitochondria-dependent apoptosis in MCF-7 cells through oxidative stress (199).
MSNs can also be combined with mitochondria targeting strategies such as DLC or MPP to further improve the targeting effect of drugs in tumor mitochondria. Pt-loaded MSNs achieve ROS burst in mitochondria, leading to cell apoptosis (200). Mesoporous connections of MSNs can deliver DOX to mitochondria and enhance copper consumption by producing H2O2 (201). After blocking surface pores through disulfide bonds, MSNs can target cancer cells with DOX, penetrate the cell membrane and quickly release anticancer drugs and mitochondria-targeted peptides, and induce significant synergistic anticancer effects (202).
MSNs can target the delivery of photosensitive and thermosensitive drugs to mitochondria, increase ROS, and enhance the efficacy and tumor imaging of new treatments such as tumor PDT. α-Tocopherol succinate and indocyanine green (IDG) MSNs reduce innate oxygen consumption by blocking the mitochondrial respiratory chain, leading to endogenous mitochondrial ROS burst and imaging-guided PDT (203). IR780-loaded MSNs nanoparticles can accumulate in tumors, destroy mitochondria and inhibit cellular respiration by decomposing H2O2, resulting in sustained reduction of hypoxia in tumor tissues, thereby enhancing the therapeutic effect of PDT (204). Redox-responsive drugs delivered by MSNs target mitochondria in living cells and induce apoptosis derived from mitochondrial membrane depolarization (205).
3.3.3 Mitochondria-targeted dendrimers
Dendrimers have a hyperbranched structure that can fill hydrophobic drug small molecules into polymer gaps and graft drugs onto polymer chains. Targeted dendrimer curcumin (TDC) colocalizes with mitochondria of cancer cells, inducing potent apoptosis and cell cycle arrest. It reduces ATP and glutathione and increases ROS levels in isolated mitochondria of rat hepatocytes (206). Poly(amidoamine) (PAMAM) is a common dendrimer targeting strategy with the ability to effectively regulate dendrimer targeting mitochondria. Active targeting of dendrimers induces P-glycoprotein (P-gp) overexpression and apoptosis in multidrug-resistant cells (207). TPP conjugated to PAMAM dendrimers, optimizes the density of surface TPP by adjusting the length of TPP-PEG linker, enhancing mitochondrial targeting ability and antitumor bioactivity (208).
3.3.4 Mitochondrial-targeted metal-organic frameworks
Metal-organic frameworks (MOFs) are a class of porous materials formed by the coordination of inorganic metal ions and organic ligands. Compared with other nano-drug carriers, MOFs have the advantages of high porosity, adjustable structure, controllable size, and easy modification. In addition, MOFs exhibit unique advantages: (1) easy preparation and good stability, assembled from non-toxic metals (Fe, Zn, Ca, Mg, etc.) and low-toxic carboxylic acids or phosphonic acids; (2) biodegradable, especially when exposed to water; (3) an internal microenvironment suitable for the delivery of drug molecules with different activities (209). These properties make MOFs ideal materials for biomedical applications, such as the delivery of drugs or imaging agents. Surface modification of materials further enriches the approach of using MOF as a drug delivery platform to treat diseases, such as PTT combined with chemotherapy, ultrasound therapy combined with chemotherapy, and other combination treatment strategies (210).
MOFs encapsulated in macrophage-cancer hybrid membranes (MCHMs) enhance the cancer homing targeting ability of nanoparticles (NPs), damage ΔΨm, and lead to cancer cell apoptosis (211). The ZCProP nanoplatform triggers cell ferroptosis through cuproposis and inhibits the anti-ferroptosis protein glutathione peroxidase 4 (212). MOFs can deliver oxymatrine (Om) and astragaloside IV (As) into the HCC microenvironment, and increase the oxygen consumption rate and proton efflux rate of tumor-infiltrating lymphocytes (TILs) by regulating the mitochondrial function of CAFs and TILs (213). The mitochondrial targeting drug of gallium-based organic frameworks produces ROS and releases L-Arg, which reacts with ROS to generate NO, downregulates HIF-1α expression to improve tumor hypoxia, and enhances immune responses by increasing calreticulin (CRT), high-mobility group box 1 (HMGB1), and T cell proliferation (214). The metal-organic framework GCZMT targets mitochondria and releases NO under MW irradiation, interfering with the cell's energy supply and inhibiting tumor cell growth. Upregulation of heat shock protein (HSP)70 expression can facilitate CD4+ and CD8+ T cell activation to promote anti-tumor immunity (215).
3.3.5 Other mitochondria-targeted nanoparticles
Lipid-polymer nanoparticles (LPNPs) are composed of a polymer core and a biocompatible lipid shell. LPNPs have a longer half-life compared with conventional liposomes. After lipid-polymer hybrid nanoparticles are taken up by cancer cells, the surface charge of LPNPs is restored due to the separation of PEG under intracellular reducing conditions, resulting in rapid and precise targeting of mitochondria (216). Nanomicelles are nanocarriers with a core-shell structure formed by self-assembly of amphiphilic copolymers in aqueous media, which have the advantages of simple preparation and small particle size. Micelles not only significantly improve drug solubility, but also increase drug accumulation in tumor sites through enhanced permeability and retention (EPR), and they improve the effect of chemotherapy and can partially reverse tumor drug resistance (217). Mitochondria-targeted polymeric micelles (OPDEA-PDCA) target mitochondria and induce mitochondrial oxidative stress via inhibition of pyruvate dehydrogenase kinase 1(PDHK1), leading to immunogenic pyroptosis in osteosarcoma cell lines (218). Charge-reversible nanocopolymers are copolymers that are modified with anions to shield the positive charge of the nanosystem, avoiding nonspecific binding with other proteins and subsequent elimination (219). In normal physiological environments (pH 7.4), the copolymers are neutral or negatively charged, which can reduce the uptake of nanomedicines by macrophages in the reticuloendothelial system while ensuring their stability in the blood circulation. However, when they reach the tumor site, their potential undergoes a charge reversal, and their affinity with the tumor cell surface is significantly enhanced, leading to the accumulation of nanoparticles in tumor cells (220). Tumor acidity triggers charge reversal and mitochondrial targeting activation of TPP-containing nanomedicines, which is a simple and effective strategy for delivering DOX to cancer cell mitochondria and overcoming DOX resistance in MCF-7/ADR breast tumor cells in vitro and in vivo (120).
In summary, mitochondrial-targeted drug delivery systems can deliver drugs to tumor sites and enhance the effectiveness of cancer treatment through various mechanisms, including disrupting mitochondrial energy metabolism, regulating ROS levels, modulating cell death-related proteins, damaging mitochondrial DNA, and regulating mitophagy. Liposomes can carry both hydrophilic and lipophilic drugs, enhancing drug uptake and inducing mitophagy, while their photosensitizer liposomes can also assist in photodynamic therapy (PDT). MSN boasts good biocompatibility and high surface area, allowing for direct drug conjugation or the integration of other targeted strategies, thus enhancing drug targeting and cytotoxic effects, and can also deliver photothermal sensitizers. Dendritic polymers are suitable for carrying hydrophobic drugs. MOFs are porous, structurally tunable, and biodegradable, making them apt for delivering drugs or imaging agents, and they can also be employed in combination therapies. Additionally, LPNP has a long half-life, nanomicelles can reverse drug resistance, and charge-reversible nanocopolymers offer precise targeting. These systems can improve cancer treatment outcomes through various approaches, making them a promising therapeutic strategy, reducing the high risks associated with traditional treatments such as surgery.
4 Mechanism of action of mitochondria-targeted therapy strategies in tumor immunity
Mitochondrial dysfunction plays an important role in tumor-induced immunosuppression. Mitochondrial-targeted drugs that restore mitochondrial function may overcome this dilemma and improve the efficacy of cancer therapies (221). Mechanisms of action for mitochondrial-targeted drugs in enhancing tumor immunity include targeting of mitochondrial metabolism, mitochondrial ROS, ICD, mtDNA and immune checkpoints.
4.1 Targeting mitochondrial metabolic pathways to improve tumor immunotherapy
4.1.1 Targeting glycolysis metabolic pathways
Glucose is one of the main players in tumor progression and a promoter of tumor invasion and metastasis. Glycolysis rapidly produces ATP, which provides sufficient energy for tumor cell proliferation. Under aerobic or hypoxic conditions, tumor cells have enhanced glycolytic activity and reduced mitochondrial respiration. Therefore, reversing the high glycolytic state of tumor cells to induce cell death is a possible approach to cancer treatment. Key glycolytic genes include glucose transporter 1 (GLUT1), hexokinase 2 (HK2), pyruvate kinase-M2 splicing isoform (PKM2) and lactate dehydrogenase (LDH-A) (Figure 9).
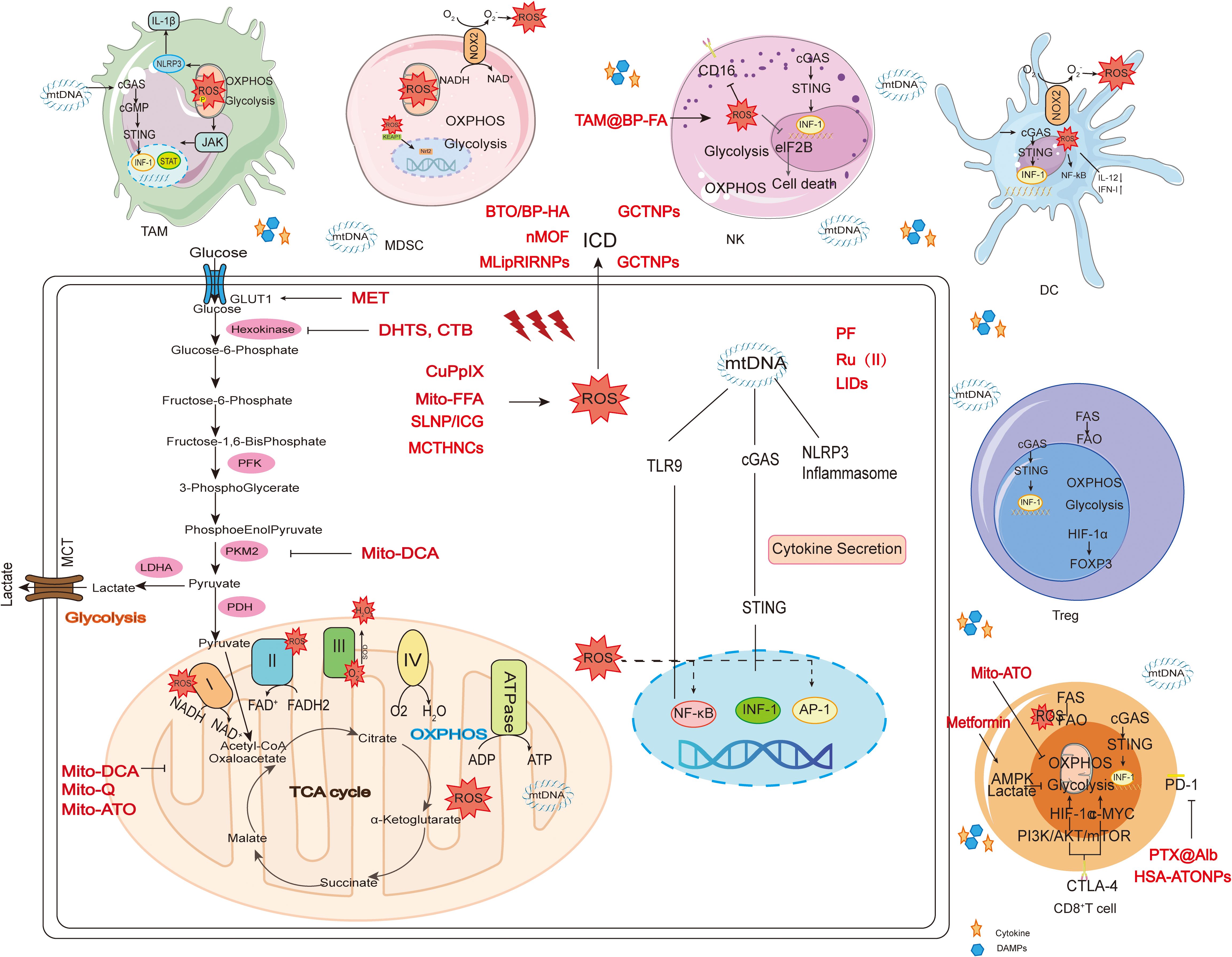
Figure 9. Employing mitochondria-targeted drugs to alter immune regulation. Mitochondrial targeting strategy drugs can affect tumor immunity through multiple pathways. Acting on glycolysis such as MET, DHTS, Mito-DCA; Acting on ROS such as CuPpIX, Mito-FFA, and SLNP/ICG; Acting on OXPHOS: Mito-DCA, Mito-Q, Mito-ATO; Acting on ICD such as nMOF, MLipRIRNPs, BTO/BP-HA; Acting on mtDNA such as PF, Ru (II), and LIDs; Acting on CD8+ T cell glucose metabolism such as metformin and Mito-ATO; Acting on PD-1 such as PTX@Alb and HSA-ATONPs.
Glucose uptake provides a key metabolic control point by targeting the Glut family of glucose transporters (120). Interference with HK1/2 and GLUT1 function in hematopoietic cells inhibits glycolysis, reduces ATP production, enhances the apoptotic effect, and preserves normal CD34+ bone marrow progenitors (222). Glycosylated poly(amidoamine)/celastrol (PAMAM/Cel) complexes are characterized by high photothermal conversion efficiency, hypoxia-sensitive PEG outer layer detachment, and alkali-sensitive drug release. The complexes show specific cellular uptake and accumulation in tumor cell mitochondria in hypoxic environments that overexpress GLUT1 (223). Met administration elevates mtROS and cell surface Glut-1, leading to IFN-γ production in CD8+ TILs in tumor cells (224).
Hexokinase-2 (HK2) is located at mitochondrial-endoplasmic reticulum (ER) contact sites called mitochondrial associated membranes (MAMs). HK2 expression is significantly elevated in most HCC cell lines and tumor tissues compared to normal cell lines and tissues (225). Since HK2 is the major rate-limiting enzyme in the aerobic glycolysis pathway, inhibiting HK2 is an effective strategy to block glycolysis. HK2 degraders cause mitochondrial damage and then induce GSDME-dependent pyroptosis and ICD, resulting in increased anti-tumor immunity (226). HK2-targeting peptides trigger mitochondrial Ca2+ overload, leading to Ca2+-dependent calpain activation, mitochondrial depolarization and cell death, and can also cause massive death of chronic lymphocytic leukemia B cells (227). Dihydrotanshinone I (DHTS) reverses metabolic reprogramming in colon cancer by inhibiting hexokinase activity and free fatty acids (FFA) via the PTEN/AKT/HIF1α-mediated signaling pathway (228).
Pyruvate kinase (PK) is a key enzyme that regulates the last step of glycolysis, catalyzing phosphoenolpyruvate (PEP) and ADP to generate pyruvate and ATP. Pyruvate kinase includes erythrocyte/liver pyruvate kinase (PKLR) and muscle pyruvate kinase (PKM), and PKM has two isoforms, PKM1 and PKM2. Mitochondra-targeted dichloroacetate (Mito-DCA) enhances delivery of DCA to mitochondria, leading to a shift from glycolysis to glucose oxidation, cell death through apoptosis, and increses DCs secretion of IL-12 (229). ATO treatment inhibits oxygen consumption and metabolically induces aerobic glycolysis and oxidative stress, and also induces apoptosis in CD44+/CD24low/- and ALDH+ cancer stem cells (230).
Another way to target glycolysis is to reduce levels of ATP. The mechanism of Met's anti-tumor effect may be that it directly reduces the production of ATP due to its function as an inhibitor of mitochondrial electron transfer chain complex I and an activator of AMPK (132). Metformin (Met) reduces tumor-infiltrating Treg (Ti-Treg), especially in the terminally differentiated CD103+KLRG1+ population, and also reduces expression of immune suppressive effector molecules such as CTLA4 and IL-10. Met inhibits the differentiation of naive CD4+ T cells into induced Treg (iTreg) by reducing forkhead box P3 (Foxp3) protein expression, which is determined by the increase of phosphorylated S6 (pS6), a downstream molecule of mTORC1. Rapamycin and compound C (AMPK inhibitor) restores the generation of iTreg, further indicating that mTORC1 and AMPK are involved (231).
A variety of strategies targeting key glycolytic genes (such as GLUT1, HK2, PK, etc.) can induce tumor cell death or enhance anti-tumor immunity by reducing ATP production and triggering mitochondrial damage. By inhibiting the mitochondrial electron transfer chain complex and activating AMPK, ATP production is directly reduced, which affects tumor immune cells and plays an anti-tumor role.
4.1.2 Targeting mitochondrial OXPHOS
The "Warburg effect" is necessary for tumor cells to undergo malignant transformation and escape immune attack. Reversing the Warburg effect by using drugs to intervene in the metabolic behavior of tumor cells so that their main energy production pathway changes from glycolysis to OXPHOS, may be a promising therapeutic strategy for treating such tumors. Upregulating the expression of genes related to mitochondrial biogenesis, thereby increasing mitochondrial activity and reprogramming cellular energy metabolism, produces an "anti-Warburg effect", and is known to regulate the differentiation of glioblastoma cells towards normal cell phenotype (118). Mitochondria-targeted OXPHOS inhibitors reduce hypoxia in tumor cells in a dose-dependent manner, potentially sensitizing hypoxic tumor cells to radiotherapy (232). Mitoquinone (Mito-Q) adsorbed to the inner mitochondrial membrane blocks ATP synthase, dissipates ΔΨm in HepG2 cells, and induces uncoupling of autophagy from OXPHOS in cancer cells (233). Functionalized Ir(III) complexes selectively localize in mitochondria and generate singlet oxygen and superoxide anion radicals upon two-photon irradiation, leading to the disruption of the mitochondrial respiratory chain, thereby interfering with mitochondrial OXPHOS and glycolytic metabolism, triggering cell death by combining ICD and ferritin autophagy (234).
Metabolic reprogramming of T cells in the TME impairs effector T cell responses against tumor cells. Mitochondrial-targeted drugs modulate the immune microenvironment via OXPHOS. Mitochondria-targeted hydroxyurea (Mito-HU) reduces mitochondrial complex I and complex III-induced oxygen consumption, effectively inhibits monocytic MDSCs and suppressive neutrophils, and stimulates T cell responses (235). Mito-CI reduces mitochondrial complex I oxygen consumption and Akt-FOXO signaling, blocks cell cycle progression, melanoma cell proliferation, and inhibits tumor progression. Anti-proliferative properties of mitochondria-targeted complex I inhibitors (Mito-CI) inhibit differentiation, viability, and suppressive function of bone marrow-derived MDSCs and increase activation of T cells (236). Mitochondria-targeted ATO inhibits the expression of mitochondrial complex components, OXPHOS, and glycolysis genes in granulocytic-MDSCs and Treg (237). The resulting reduction in intra-tumoral granulocytic-MDSCs (G-MDSCs) and Treg could contribute to the observed increase in tumor-infiltrating CD4+ T cells. In contrast, Mito-ATO significantly inhibits OXPHOS and glycolysis in G-MDSCs. These observations support the predicted higher OXPHOS and glycolysis in effector memory CD8+ T cells and lower OXPHOS and glycolysis in G-MDSCs after Mito-ATO treatment (238). AMPK is considered a major intracellular energy sensor and key regulator of mitochondrial biogenesis that can control metabolic reprogramming in immune cells and enhance anti-tumor immunity. The AMPK activator Met is considered a candidate drug to improve cancer treatment efficacy by interfering with tumor metabolic reprogramming (239). Met increases the numbers of CD8+ T cells and protects them from apoptosis and exhaustion (240).
Mitochondria-targeted OXPHOS inhibitors can induce tumor cell death or enhance their sensitivity to radiotherapy by affecting mitochondrial function and interfering with energy metabolism. Mitochondria-targeted drugs can also improve TME, stimulate T cell responses, protect CD8+ T cells and enhance anti-tumor immunity by regulating OXPHOS.
4.2 Targeting mtROS to improve tumor immunotherapy
Since ROS in tumor cell mitochondria have been shown to play an important role in immunotherapy, how mitochondrial targeted drugs can promote tumor cells to produce excessive ROS, damage mtDNA, release immunogenic intracellular substrates, and activate anti-tumor immunity are important areas of focus in anti-tumor research (Figure 9). Cu-modified protoporphyrin(CuPpIX) can be delivered to the mitochondria, inducing ROS burst in situ under ultrasound irradiation, which leads to severe mitochondrial dysfunction and amplies ICD to stimulate the body's immune response and enhance the infiltration of CD8+ T cells into tumors (241). The nanosystem delivers Ca@GOx to mitochondria, inducing mitochondrial Ca2+ overload and generating high levels of ROS, leading to pyroptosis and promoting tumor infiltration of CD8+ T cells (242). Indocyanine green (ICG) nanoparticles (SLNP/ICG) stimulate glioma cells to produce abundant ROS under NIR irradiation, activate mitochondria-mediated apoptosis pathways, and increase CD4+ and CD8+ T cell proliferation (243). The PDT photosensitizer IR700DX generates ROS upon light irradiation and promotes downstream p38 phosphorylation and CASP3-mediated gasderminE (GSDME) cleavage, which induces pyroptosis, triggers ICD, and enhances the anti-cancer efficacy of PD-1 blockade (244). The nanoplatform CS@KET/P780NPs induces apoptosis by enhancing ROS accumulation, which triggers ICD and a long-term antitumor response by releasing tumor-associated antigens (TAA) and DAMPs (245). The molecular photosensitizer FEPT, used in NIR-II, generates ROS and hyperthermia under laser irradiation, leading to mitochondrial dysfunction and light-induced apoptosis through the caspase-3 pathway, releasing immunogenic intracellular substrates, and thus promotes activation of anti-tumor immunity (246). Photosensitizer IR780 triggers ROS production through a Fenton-like reaction, induces ferroptosis of tumor cells, and simultaneously induces DC maturation, promotes cytotoxic T lymphocyte infiltration, decreases immune suppression in the tumor microenvironment, and activates a systemic immune response (247). Sonodynamic therapy (SDT) drugs stimulate ROS production and reduce ΔΨm, and induce antitumor immune responses by upregulating NK cell activity and reducing numbers of immunosuppressive macrophages (248). Mitochondria-targeted FFa (Mito-FFa) increases the production of mtROS that triggers endoplasmic reticulum (ER) stress, and also oxidizes mtDNA and promotes its leakage into the cytoplasm. This leads to cGAS-STING-dependent IFN-I secretion, improving tumor antigen uptake, DC maturation, and cross-priming of CD8+ T cells (249). Mitochondria-targeted liposomes induce massive lipid peroxidation and increase ROS in mitochondria, ultimately triggering ferroptosis in bladder cancer cells, promoting the release of intracellular DAMPs, inducing the release of mtDNA into the cytoplasm, activating the cGAS-STING pathway to secrete IFN-β, and increasing DC cross-presentation of antigens to T cells, and increasing macrophage phagocytosis (250).
Mitochondria-targeted drugs can activate the cGAS-STING pathway, promote DAMPs release, induce ferroptosis, and regulate immune cell activity by producing MTROS, reducing δψm, triggering endoplasmic reticulum stress or Lipid peroxidation, etc. The reduction of immunosuppressive cells will ultimately improve the efficacy of tumor immunotherapy.
4.3 Targeted mitochondrial-mediated ICD
ICD is a form of cell death that releases TAA and tumor-specific antigens (TSA), and provides "danger signals" to facilitate generation of an effective T cell response. It is characterized by the release and/or increased expression of DAMPs, precursor antigens, inflammatory cytokines, and inflammatory mediators. The main DAMPs include ATP, calreticulin (CRT), high mobility group box protein B1 (HMGB1), heat shock protein (HSP), type I interferon (IFN I) and Annexin 1 (ANXA1), which then activate and recruit APCs such as macrophages and DCs to activate T cells reactive to tumor antigens. DAMPs bind to pattern recognition receptors (PRRs) to induce a series of immune events (251). Mitochondria-targeted drugs trigger ICD by releasing ROS. Mitochondrial-targeting liposomal nanoparticles cause severe ferroptosis of tumor cells and trigger ICD through the accumulation of lipid peroxides, activating anti-tumor immunity (252).
The released DAMPs promote DC maturation, activate cytotoxic T lymphocytes, and reverse immune suppression in the tumor microenvironment. Polymeric nanoparticles induce mitochondrial dysfunction and amplify endoplasmic reticulum stress, leading to tumor cell apoptosis and ICD, which promotes DC maturation and increases numbers of tumor-infiltrating cytotoxic T lymphocytes (253). DOX-containing liposomes induce effective ICD in targeted cancer cells to promote DC maturation and stimulate T cell proliferation and activation, transforming the immunosuppressive tumor microenvironment (ITM) into an immune responsive environment (254). Toll-like receptor agonist R837 synergistically promotes DC maturation. Promoting DC activation by inducing ICDs resulted in more robust antitumor efficacy, which can inhibit metastatic disease progression and promote the development of durable antitumor memory responses (255).
DAMPs released by ICD effectively induce M1 polarization and migration of the polarized macrophages. TPP-modified CuET triggers ICD, and the released DAMPs induce macrophage M1 polarization and migration to activate an immune response consisting of CD8+ T cells and NK cells (256). SDT promotes the maturation of DCs and increases numbers of infiltrating immune cells. M2 macrophages are also re-polarized to an M1 phenotype and MDSCs are depleted to reverse immunosuppression and enhance immune responses (257).
Mitochondria-targeting drugs can trigger ICD by releasing ROS and other DAMPs, which can promote DC maturation, activate cytotoxic T lymphocyte, and reverse immunosuppression in the tumor microenvironment. They can also induce macrophage M1 polarization and migration, and SDT can also enhance the immune response by regulating the function of ICD-associated immune cells (such as macrophages, MDSCs).
4.4 Targeting mitochondria autophagy to increase tumor immunity
Mitochondrial autophagy, as a core mechanism regulating mitochondrial homeostasis, exerts bidirectional effects in tumor immunity by influencing immune cell function, tumor microenvironment metabolism, and innate immune pathways: It can maintain effector function by clearing damaged mitochondria from immune cells and enhance immune responses by activating the STING pathway. It also promotes antitumor effects by facilitating antigen presentation and activating effector immune cells. Conversely, it can promote immune escape by degrading MHC molecules, supporting immune-suppressive cells, and regulating immune checkpoints.
The autophagy inhibitor chloroquine (CQ) induces Ca²+ release through lysosomal Ca²+ channels, activating p38 and NF-κB to reprogram TAMs from M2 to M1 phenotypes, eliminating cancer cell resistance and achieving enhanced therapeutic effects (258). The autophagy inhibitor bafamilomycin confers ADCC resistance by altering cell death, modulating immunoregulatory factors in NK and/or cancer cells, and regulating HER2 kinetics (259). Spongy calcium carbonate (CaCO3) nanoparticles disrupt DC autophagy and antigen cross-presentation, enhancing DAMP release from tumor cells to improve DC maturation (260).
Autophagy activator like rapamycin enhances NK cell cytotoxicity by upregulating IL-27R expression, thereby restricting tumor growth (261). Metformin significantly activates AMPK signaling, reducing Th1 and Th17 cells while increasing Th2 and Treg cells (262). Combined with nelfinavir, it induces SIRT3/mROS-dependent autophagy and sensitizes NK cells against human cervical cancer cells (262). Temsirolimus (TEM) activates autophagy to suppress tumor-derived sEV PD-L1 secretion and increase the number and activation of CD4+and CD8+T cells, inducing systemic anticancer immunity (263). PD-1 blockade combined with endostar significantly suppressed tumor growth, leading to reduced IL-17 and TGF-β1 levels, increased IFN-γ secretion, decreased MDSC, and reversal of CD8+ T cell suppression. It improved the tumor microenvironment and activated autophagy (264). The nanomaterial MoO3-x nanowires (MoO3-x NWs) combined with PTT activated autophagy, induced DC maturation and antigen presentation, subsequently activating CD8+ T cell-mediated adaptive immunity. It promoted TAM polarization toward M1 macrophages, suppressed Treg cell infiltration at tumor sites, and alleviated immune suppression in the tumor microenvironment (265). Lanthanum nickel oxide (LNO) nanoenzyme-induced macrophage autophagy promotes macrophage M1 polarization (266).
Natural compounds exhibit bidirectional effects on autophagy, further influencing tumor immunity. (-)-Guaiol inhibits tumor growth by inducing autophagy, inducing ICD, enhancing DC activation, and boosting T cell infiltration (267). Berberine hydrochloride (Ber) increases autophagosome accumulation, elevates LC3-II and p62 levels in melanoma cells to enhance MHC-I-mediated antigen presentation, and improves CD8+ T cell infiltration (268). Rocaglamide (RocA) targets ULK1 to inhibit autophagy and restore NK cell GZMB levels, activating cGAS-STING signaling to promote NK cell infiltration (269). Naringenin can at least partially induce BC cell inhibition of autophagy and cell proliferation by modulating the FKBP4/NR3C1/NRF2 signaling pathway, while also enhancing DC differentiation and maturation (270).
4.5 Targeting immune checkpoints to increase tumor immunity
Immune checkpoints are a class of immune inhibitory molecules that are expressed on immune cells to regulate the degree of immune activation. These molecules are known to play an important role in preventing the occurrence of autoimmunity, but they can also be hijacked by cancers to inhibit tumor immune responses. Mitochondria-targeted treatments can be used in conjunction with immune checkpoint therapies to facilitate better induction of tumor-reactive T cells. The mitochondria-targeted drug Met attenuates upregulation of the immune checkpoints programmed cell death protein 1 (PD-1) and lymphocyte activation gene-3 (LAG-3), thereby increasing CD8+ T cell infiltration and survival in the harsh tumor microenvironment (271). Synergistic effects between ATO and anti-PD-L1, which blocks binding of PD-L1 to its receptor PD-1, leads to the activation of tumor-reactive CD8+ T cells and promotes the establishment of tumor-specific immune memory (272).
Mitochondria-targeted drugs can reverse tumor hypoxia, inhibit PD-L1 expression, thereby enhancing therapeutic effects. Albumin-bound paclitaxel (PTX@Alb) accumulates in 4T1 breast tumors and reduces the expression of PD-L1 and TGF-β, resulting in enhanced T cell infiltration of the tumors (273). Mitochondria-targeted ATO promotes CD8+ T cell recruitment by reducing tumor hypoxia, and ATO treatment enhances the efficacy of anti-PD-1 immunotherapy (274). MHI-TMX@ALB nanoparticle photosensitizers can reverse tumor hypoxia and inhibit PD-L1 protein expression in the tumor microenvironment, resulting in enhanced efficacy of photodynamic immunotherapy by increasing T cell infiltration (275).
Mitochondria-targeted drugs induce ICD, reverse the immunosuppressive TME, and promote immune checkpoint blockade therapy. MOF induces ICD after ultrasound exposure to promote DC activation. It can achieve in vivo synergy with anti-CTLA-4 immune checkpoint blockade to reverse the ITM (255). Nanodiagnostic therapeutics stimulate ICD, leading to the massive release of TAA and DAMPs, thereby improving the ITM and providing another treatment that can be combined with immune checkpoint blockade therapy (276). SDT leads to local production of ROS after ultrasound irradiation, damaging tumor cell mitochondria, downregulating PD-L1 expression, and promoting ICD (277). Zero-valent-iron nanoparticles (ZVI-NP) enhanced anti-tumor immunity by re-polarizing pro-tumor M2 macrophages to anti-tumor M1 macrophages, reducing the number of Treg, downregulating PD-1 and CTLA4 on CD8+ T cells to enhance their cytolytic activity against cancer cells, and reducing expression of PD-L1 on the tumor (278).
Mitochondrial targeted therapy can be combined with immune checkpoint therapy to synergistically activate tumor-reactive CD8+ T cells and establish immune memory. Mitochondria-targeted drugs can also synergize with immune checkpoint blockade therapy by reversing tumor hypoxia, inhibiting PD-L1 expression, or inducing ICD, reversing immunosuppression TME, and reducing tumor necrosis factor-α expression, enhance the effect of photodynamic immunotherapy, enhance T cell lytic activity, and then enhance tumor immunity.
4.6 Improving chimeric antigen receptor T cell therapy
Chimeric antigen receptor T (CAR-T) cell therapy is an exciting form of immunotherapy that is being used to primarily treat patients with hematologic cancers (leukemia, lymphoma, multiple myeloma), but also holds promise for treating solid tumors. CARs are constructed through genetic technology, where a gene encoding the CAR that recognizes antigen(s) on tumor cells is introduced into T cells. Upon CAR recognition of cognate antigen(s) on tumor cells, intracellular co-signaling domains in the CAR trigger T cell activation and killing of the tumor cells. Essentially, the CAR adds a new antigen binding receptor on the surface of T cells that is independent of MHC/antigen presentation. Despite impressive early clinical responses after CAR-T cell therapy, many patients still experience disease relapse, which is frequently due to loss of target antigen(s) on the tumor cells, so improvements to this form of cancer immunotherapy are warranted.
Increased mitochondrial biomass preserves bioenergetic potential to meet the metabolic demands of activated T cells. When T cells are modified to generate CAR-T cells, a unique mitochondrial adaptation is required to establish stemness and persistence of the CAR-T cells (279). Targeting mitochondrial metabolism to promote T cell memory formation and metabolic adaptation may represent an attractive strategy to improve CAR-T cell therapies and other immunotherapies (280).
CD8+ T cell migration depends on mitochondrial oxidation of glucose and glutamine and both ATP and ROS production. Drug interventions that increase mitochondrial activity can improve CAR-T cell recruitment to tumors, thereby better controlling tumor growth (281). Knockout of endogenous TCR in ARI-0001 CAR-T cells increases the percentage of energetic mitochondria (282). Nuclear receptor 4A (NR4A)1/2/3 triple knockout CAR-T cells show enhanced mitochondrial OXPHOS, increasing the persistence and stemness of CAR-T cells (283). Addition of the cosignaling molecule 4-1BB to the CAR construct promotes memory T cell respiratory capacity, increases fatty acid oxidation and enhances mitochondrial biogenesis, generating increased numbers of T cells with an effector-memory cell phenotype (284).
Upregulating T cell mitochondrial plasticity to increase the efficacy of adoptive cellular immunotherapies (ACI), including CAR-T cells, will generate T cells with strong metabolic adaptability and durable immune function, thereby preventing tumor metastasis and recurrence (285). Targeting methylation-controlled J protein (MCJ) in CD8+ CAR-T cells can increase mitochondrial metabolism and improve the anti-tumor activity of CAR-T therapy (286). CAR-T cells prevent staurosporine (STS)-induced apoptosis of human CD3+ T cells by interfering with the caspase pathway and improving their metabolic fitness and resistance to environmental stress (287).
Mitochondria are essential for CAR-T cells, and strategies targeting mitochondrial metabolism, such as increasing mitochondrial biomass, enhancing mitochondrial activity with OXPHOS, upregulating mitochondrial plasticity, and so on, have been proposed to reduce mitochondrial plasticity in CAR-T cells. They can improve the stemness, persistence, metabolic adaptability and anti-tumor activity of CAR-T cells, and help to improve the therapeutic effect.
4.7 Targeting mtDNA to improve tumor immunotherapy
Since tumor cell mitochondria are typically dysfunctional due to mutations in mtDNA, correcting mtDNA mutations is considered an effective strategy to restore mitochondrial function. Mitochondria-targeted drugs induce tumor mtDNA oxidation and specific release into the cytoplasm, activating the cGAS-STING pathway and affecting tumor immune-related responses. Mitochondrial lipid peroxidation and ROS promote the release of intracellular DAMPs, thereby facilitating the release of mtDNA into the cytoplasm, which activates the cGAS-STING pathway and increases cross-presentation of antigens by DCs and macrophages. Ultimately, this process induces CD8+ T cell infiltration into the TME to inhibit tumor growth (288).
Under NIR irradiation, mitochondria-targeted ROS are generated and mtDNA released to provide endogenous danger-associated molecules that activate the cGAS-STING pathway, promoting the maturation of DCs and the infiltration of cytotoxic T lymphocytes (289). Treatment with mitochondria-targeting gold(I) complexes generates large amounts of ROS and promotes DNA excretion. The ROS induces ICD and the released DNA activates the cGAS-STING pathway, generating a strong anti-cancer immune response (290). Indocyanine green and doxorubicin plus ultrasound enhances the nuclear delivery of doxorubicin, induces tumor mitochondrial DNA oxidation, activates cGAS-STING signaling, and triggers anti-tumor T cell immunity (291).
Mitochondrion-targeting drugs can induce tumor mtDNA oxidation and release to the cytoplasm, activate the cGAS-STING pathway, while mitochondrion Lipid peroxidation and ROS can also promote mtDNA release. This in turn promotes antigen cross-presentation, DC maturation, and CD8+ T cell infiltration to trigger anti-tumor immune responses.
In summary, mitochondrial targeting offers various strategies to enhance tumor immunotherapy. Targeting metabolic pathways can regulate immune cell function by inhibiting key enzymes in glycolysis (such as GLUT1 and HK2) or modulating OXPHOS to reverse the Warburg effect; targeting mtROS can induce excessive mitochondrial ROS, damage mitochondrial DNA, trigger immunogenic cell death (ICD), and activate anti-tumor immunity. Targeting mitochondria-mediated ICD can release damage-associated molecular patterns (DAMPs) that promote dendritic cell (DC) maturation and T cell activation; when combined with immune checkpoint therapy, it can reduce the expression of PD-1, reverse hypoxia, and enhance efficacy; improving CAR-T therapy requires increasing its mitochondrial activity and plasticity, enhancing stemness and durability; targeting mitochondrial DNA can induce its oxidative release, activate the cGAS-STING pathway, and promote immune cell infiltration. These strategies contribute to enhancing anti-tumor immunity (Table 1).
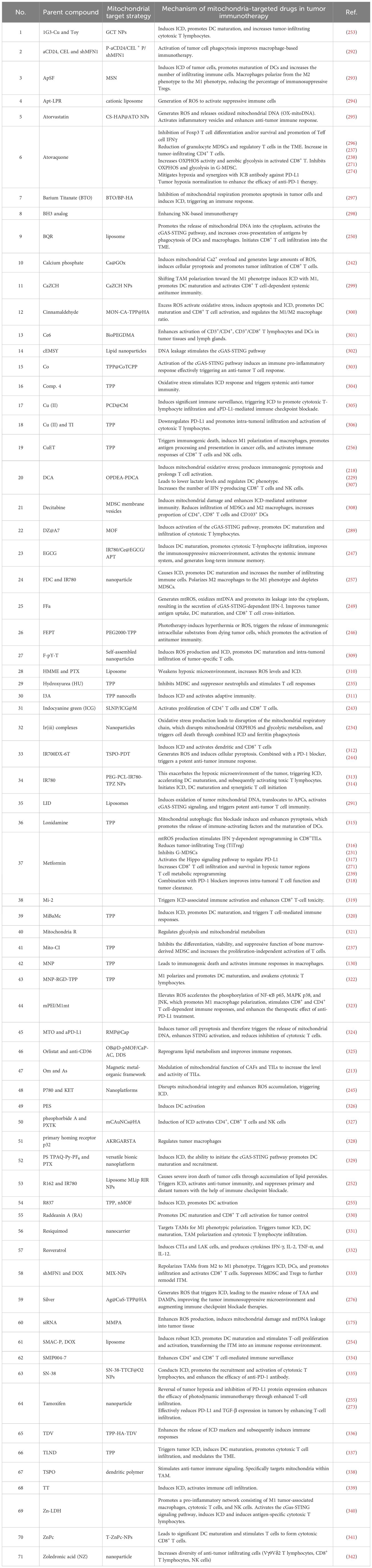
Table 1. Features of mitochondria-targeted drugs on cancer immunotherapy.
5 Conclusion
In conclusion, mitochondria play a crucial role in tumor immunity. Abnormalities in mitochondria, such as genomic mutations, and autophagy can regulate cancer progression by regulating cellular metabolism and ROS production and may further confer competitive advantage to cancer cells. Under mitochondrial stress, mtDNA is released into the cytoplasm or extracellular fluid where it can be recognized by PRRs cGAS, TLR9, and NLRP3. Among them, the cGAS-STING signaling pathway plays a dual role in tumor immunity: it can suppress anti-tumor immunity to promote cancer progression, or enhance tumor antigen presentation to exert an anti-tumor effect. TLR9 promotes tumor growth and increases chemoresistance by recognizing the specific domain of mtDNA, whereas activated NLRP3 triggers downstream signaling cascades and assembles inflammasomes via potassium ion efflux.
Due to the elevated mitochondrial membrane potential (δψm) in tumor cells, DLCs including TPP and its derivatives, F16, Rhodamine and DQA can selectively accumulate in mitochondria and deliver loaded drugs. Specifically, TPP and its derivatives enhance the efficacy of chemotherapy, PDT, radiotherapy, and PTT; F16-targeted mitochondrial matrix induces mitochondrial permeability transition; Rhodamine stains mitochondria and enhances tumor suppression, and DQA disrupts mitochondrial function to induce cancer cell death. In terms of peptide-targeting sequences, MPPs consisting of 4–16 cationic and hydrophobic residues, can localize to mitochondria, promote apoptosis in cancer cells, and inhibit apoptosis in cancer cells, and shows low toxicity and good biocompatibility. SS peptides, which typically contain 4 positively charged amino acids, specifically target mitochondrial cardiolipin to reduce mitochondrial ROS production, inhibit MPTP turn-on, and protect mitochondrial structure and function.
These mitochondria-targeting strategies can also enhance tumor immunity through multiple mechanisms: targeting metabolic pathways (e.g., inhibiting key glycolytic enzymes such as GLUT1 and HK2, or modulating OXPHOS to reverse the Warburg effect; targeting mtROS to trigger ICD; targeting autophagy; combining immune checkpoint therapy to downregulate PD-1 expression; improving CAR-T cell therapy; and targeting mtDNA to induce their oxidative release and activate the cGAS-STING pathway, thereby promoting the infiltration of anti-tumor immune cells). Collectively, these strategies contribute to enhancing anti-tumor immunity.
Although mitochondria-targeting strategies have shown significant research potential and application prospects in the field of tumor immunity, providing an innovative direction for anti-tumor therapy, the development of anti-tumor drugs is still in the early stage, however, there are still several key challenges and limitations clinical application. On the one hand, the vast majority of studies on mitochondrial targeting are still in the preclinical stage, mainly focusing on in vitro cell experiments and in vivo animal model validation. The relative paucity of human clinical research data, especially the lack of support from multicentre, large sample phase III trials, makes it difficult to fully validate the effectiveness and applicability of these strategies in clinical cancer treatment. Further studies in the future include the following aspects: 1 How to regulate the activity and biological function of mitochondria in the activation of different immune cell subsets? 2 How does the energy balance transfer to immune cells? How to promote the repair of mitochondrial dysfunction and metabolic deficiency in immune cells while inhibiting tumor cell metabolism? Effective means for evaluating the safety of mitochondria-targeting drugs has not been established, and the potential risk of toxic side effects needs to be focused on. For example, some targeted molecules may have nonspecific effects on the mitochondria of normal cells, interfere with energy metabolism and cellular homeostasis in normal tissues, or induce immune-related adverse reactions. The above issues need to be addressed through in-depth mechanistic studies, dosage form optimization, and future clinical translation studies to promote the safe and effective application of mitochondria-targeted drugs in clinical anti-tumor therapy.
Author contributions
XC: Investigation, Writing – original draft. YW: Conceptualization, Formal Analysis, Writing – review & editing. BJ: Supervision, Writing – review & editing. MY: Funding acquisition, Resources, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by National Institutes of Health (NIH): R01CA223804, R01CA232433, R01CA205633, and R01CA280746, Jiangsu Health International (regional) exchange support program, Jiangsu Province Chinese Medicine Key Talent Project, Project of Jiangsu Traditional Chinese Medicine (ZXFZ2024064), Project of Suzhou Science and Technology (SKYD2023263).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
ATG: Autophagy-related genes
ACI: adoptive cellular immunotherapy
AML: acute myeloid leukemia
AMP: adenosine monophosphate
AMPK: AMP-activated protein kinase
ANT: adenine nucleotide translocase
ANXA1: annexin 1
APCs: antigen-presenting cells
ARG: arginase
As: astragaloside IV
ATM: ataxia telangiectasia mutated
ATP: adenosine triphosphate
BNIP3: BCL2 Interacting Protein 3
BNIP3L: BCL2 Interacting Protein 3 Like
BPD-MA: benzoporphyrin derivative monoacid ring A
BTO: Barium Titanate
CAR-T cell: Chimeric antigen receptor T cell
CD8+ T: cytotoxic T-cell
CDT: Chemodynamic therapy
CEBPβ: CCAAT Enhancer Binding Protein Beta
CGAS: Cyclic GMP-AMP Synthase
CRC: Colorectal cancer
CRT: calreticulin
CSF1: colony stimulating factor 1
CTSS: Cathepsin S
CuPpIX: Cu-modified protoporphyrin
CXCL1: C-X-C Motif Chemokine Ligand 1
Cytc: cytochrome c
DAMPs: damage-associated molecular patterns
DC: dendritic cells
DLC: delocalized lipophilic cation
DOX: doxorubicin
DQA: dequaliniumchloride
ECM: extracellular matrix
EGF: epidermalgrowthfactor
EPR: enhanced permeability and retention
ER: endoplasmic reticulum
FABP5: Fatty acid binding protein 5
FAO: Fatty acid oxidation
FFA: free fatty acids
Foxp3: Forkhead box P3
FUNDC1: FUN14 Domain Containing 1
G-CSF: Granulocyte-Colony Stimulating Factor
GLUT1: glucose transporter 1
GM-CSF: granulocyte macrophage-colony stimulating factor
GZMB: Granzyme B
HF: Hinokiflavone
HIF-1α: hypoxia-inducible factor-1α
HK2: hexokinase 2
HMGB1: high-mobility group box 1
HSP: heat shock protein
ICD: immunogenic cell death
ICG: Indocyanine green
IDO1: indoleamine2,3-dioxygenase1
IFN-I: type I interferon
IL: interleukin
ITM: immunosuppressive tumor microenvironment
iTreg: induced Treg
KRAS: KRAS Proto-Oncogene, GTPase
LAG-3: lymphocyte activation gene 3
LC3B: Microtubule-associated protein 1 light chain 3B
LDH-A: lactate dehydrogenase
LND: lonidamine
LPNPs: Lipid-polymer nanoparticles
MCHMs: macrophage-cancer hybrid membranes
MCJ: methylation-controlled J protein
MCT: monocarboxylate transporters
MDSC: myeloid-derived suppressor cell
Met: Metformin
MHC: major histocompatibility complex
MHC-Ⅰ: Major Histocompatibility Complex, Class I
MIF: migration inhibitory factor
Mito-: TPP-modified PEGylated mitochondrial-targeted ATO
Mito-CI: mitochondria-targeted complex I inhibitors
Mito-HU: Mitochondria-targeted hydroxyurea
Mito-LND: Mitochondria-targeted mito-LND
Mito-Q: Mitoquinone
MMP-9: matrixmetalloprotein 9
MMPA: membrane-permeable peptide amphiphiles
MOFs: Metal-organic frameworks
MOMP: membrane permeabilization
MPP: Mitochondria-penetrating peptide
mPTP: mitochondrial permeability transition pore
mtDNA: mitochondrial DNA
mTORC1: rapamycin complex 1
mtROS: mitochondrial ROS
MWCNTs: Multi-walled carbon nanotubes
NADPH: nicotinamide adenine dinucleotide phosphate
NEFL: Neurofilament Light Chain
NIR: near-infrared
NK: natural killer
NO: nitric oxide
NOX: NADPH-oxidase
NPs: nanoparticles
NZ: Zoledronic acid
O2·-: superoxide
OCT: organic cation transporters
Om: oxymatrine
OX-mitoDNA: oxidized mitochondrial DNA
OXPHOS: oxidative phosphorylation
PACSIN1: Protein Kinase C And Casein Kinase Substrate In Neurons 1
parkin: E3 ubiquitin-protein ligase parkin
PBA: Phenylboronic acid
PD-1: programmed cell death protein 1
PDHC: pyruvate dehydrogenase complex
PD-L1: programmed cell death ligand 1
PDT: Photodynamic therapy
PEG: polyethylene glycol
PEP: phosphoenolpyruvate
PET: phosphorescence energy transfer
PGC1α: peroxide-activated receptor 1α
PHDs: prolyl hydroxylases
PINK1: PTEN-induced putative kinase 1
PK: Pyruvate kinase
PKM2: pyruvate kinase-M2 splicing isoform
PLAC8: Placenta Associated 8
PRRs: pattern recognition receptors
pS6: phosphorylated S6
PTA: photothermal agents
PTT: Photothermal therapy
PTX: Paclitaxel
RA: Raddeanin A
RGS1: Regulator Of G Protein Signaling 1
RhodOA: Rhodamine B conjugated oleanolic acid derivatives
RNS: reactive nitrogen
ROS: reactive oxygen species
SDT: sonodynamic therapy
siRNA: small interfering RNA
SiR-PXZ: silicon rhodamine-based photosensitizer
SREBP: sterol regulatory element-binding protein
SS: Szeto-Schiller
STING: Stimulator Of Interferon Response CGAMP Interactor
TAA: tumor-associated antigens
TAM: tumor-associated macrophages
TAMs: tumor-associated macrophages
TCPP: tumor-targeted cell membrane penetrating peptides
TCR: T cell receptor
TDC: Targeted dendritic curcumin
TExh: T-cell exhaustion
TGF-: transforming growth factor beta
TILs: tumor-infiltrating lymphocytes
Ti-Treg: tumor-infiltrating Treg
TLR: toll-like receptors
TME: tumor microenvironment
TPP+: triphenylphosphine
TPPLs: DSPE-PEG-TPP polymer liposomes
Treg: regulatory T-cells
TSA: tumor-specific antigens
ULK1: UNC-51-like kinase 1
UPS: undifferentiated pleomorphic sarcoma
ZnPc: zinc phthalocyanine
ZVI-NPs: Zero-valent-iron nanoparticle
α-TOS: α-tocopheryl succinate
ΔΨm: mitochondrial membrane potential
References
1. Desideri E, Vegliante R, and Ciriolo MR. Mitochondrial dysfunctions in cancer: Genetic defects and oncogenic signaling impinging on TCA cycle activity. Cancer Lett. (2015) 356:217–23. doi: 10.1016/j.canlet.2014.02.023
2. Glancy B and Balaban RS. Energy metabolism design of the striated muscle cell. Physiol Rev. (2021) 101:1561–607. doi: 10.1152/physrev.00040.2020
3. Chernorudskiy AL and Zito E. Regulation of calcium homeostasis by ER redox: A close-up of the ER/mitochondria connection. J Mol Biol. (2017) 429:620–32. doi: 10.1016/j.jmb.2017.01.017
4. Chen S, Liao Z, and Xu P. Mitochondrial control of innate immune responses. Front Immunol. (2023) 14:1166214. doi: 10.3389/fimmu.2023.1166214
5. Zong W-X, Rabinowitz JD, and White E. Mitochondria and cancer. Mol Cell. (2016) 61:667–76. doi: 10.1016/j.molcel.2016.02.011
6. Vander Heiden MG, Cantley LC, and Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
7. Xia L, Oyang L, Lin J, Tan S, Han Y, Wu N, et al. The cancer metabolic reprogramming and immune response. Mol Cancer. (2021) 20:28. doi: 10.1186/s12943-021-01316-8
8. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. (2021) 22:205–15. doi: 10.1038/s41590-020-00834-9
9. Wu Q, Tsai H-I, Zhu H, and Wang D. The entanglement between mitochondrial DNA and tumor metastasis. Cancers. (2022) 14:1862. doi: 10.3390/cancers14081862
10. O’Donnell JS, Teng MWL, and Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
11. Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduct Target Ther. (2023) 8:207. doi: 10.1038/s41392-023-01452-1
12. Mills EL, Kelly B, and O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol. (2017) 18:488–98. doi: 10.1038/ni.3704
13. Jackson CM, Pant A, Dinalankara W, Choi J, Jain A, Nitta R, et al. The cytokine meteorin-like inhibits anti-tumor CD8+ T cell responses by disrupting mitochondrial function. Immunity. (2024) 57:1864–1877.e9. doi: 10.1016/j.immuni.2024.07.003
14. Malla R, Surepalli N, Farran B, Malhotra SV, and Nagaraju GP. Reactive oxygen species (ROS): Critical roles in breast tumor microenvironment. Crit Rev Oncol Hematol. (2021) 160:103285. doi: 10.1016/j.critrevonc.2021.103285
15. Liu Y, Zhao Y, Song H, Li Y, Liu Z, Ye Z, et al. Metabolic reprogramming in tumor immune microenvironment: Impact on immune cell function and therapeutic implications. Cancer Lett. (2024) 597:217076. doi: 10.1016/j.canlet.2024.217076
16. Zhang H, Li S, Wang D, Liu S, Xiao T, Gu W, et al. Metabolic reprogramming and immune evasion: The interplay in the tumor microenvironment. biomark Res. (2024) 12:96. doi: 10.1186/s40364-024-00646-1
17. Weinberg SE, Sena LA, and Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. (2015) 42:406–17. doi: 10.1016/j.immuni.2015.02.002
18. Li Z and Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci CMLS. (2016) 73:377–92. doi: 10.1007/s00018-015-2070-4
19. Zeng Q, Lv C, Qi L, Wang Y, Hao S, Li G, et al. Sodium selenite inhibits cervical cancer progression via ROS-mediated suppression of glucose metabolic reprogramming. Life Sci. (2024) 357:123109. doi: 10.1016/j.lfs.2024.123109
20. Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. (2016) 24:104–17. doi: 10.1016/j.cmet.2016.06.007
21. Quintana A, Pasche M, Junker C, Al-Ansary D, Rieger H, Kummerow C, et al. Calcium microdomains at the immunological synapse: How ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J. (2011) 30:3895–912. doi: 10.1038/emboj.2011.289
22. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
23. He J, Shangguan X, Zhou W, Cao Y, Zheng Q, Tu J, et al. Glucose limitation activates AMPK coupled SENP1-Sirt3 signalling in mitochondria for T cell memory development. Nat Commun. (2021) 12:4371. doi: 10.1038/s41467-021-24619-2
24. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. (2016) 45:374–88. doi: 10.1016/j.immuni.2016.07.009
25. Arner EN and Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. (2023) 41:421–33. doi: 10.1016/j.ccell.2023.01.009
26. Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, et al. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell Mol Immunol. (2021) 18:1761–71. doi: 10.1038/s41423-020-0365-3
27. Walls J, Sinclair L, and Finlay D. Nutrient sensing, signal transduction and immune responses. Semin Immunol. (2016) 28:396–407. doi: 10.1016/j.smim.2016.09.001
28. Dong H, Adams NM, Xu Y, Cao J, Allan DSJ, Carlyle JR, et al. The IRE1 endoplasmic reticulum stress sensor activates natural killer cell immunity in part by regulating c-myc. Nat Immunol. (2019) 20:865–78. doi: 10.1038/s41590-019-0388-z
29. O’Sullivan TE, Johnson LR, Kang HH, and Sun JC. BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity. (2015) 43:331–42. doi: 10.1016/j.immuni.2015.07.012
30. Zheng X, Qian Y, Fu B, Jiao D, Jiang Y, Chen P, et al. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol. (2019) 20:1656–67. doi: 10.1038/s41590-019-0511-1
31. Yang M, Li J, Gu P, and Fan X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact Mater. (2021) 6:1973–87. doi: 10.1016/j.bioactmat.2020.12.010
32. Watson CK, Schloesser D, Fundel-Clemens K, Lerner C, Gabler S, Baskaran P, et al. Antifibrotic drug nintedanib inhibits CSF1R to promote IL-4-associated tissue repair macrophages. Am J Respir Cell Mol Biol. (2023) 68:366–80. doi: 10.1165/rcmb.2022-0021OC
33. Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, et al. Metabolic reprogramming in macrophage responses. biomark Res. (2021) 9:1. doi: 10.1186/s40364-020-00251-y
34. Farge T, Saland E, de Toni F, Aroua N, Hosseini M, Perry R, et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. (2017) 7:716–35. doi: 10.1158/2159-8290.CD-16-0441
35. Chimal-Ramírez GK, Espinoza-Sánchez NA, Chávez-Sánchez L, Arriaga-Pizano L, and Fuentes-Pananá EM. Monocyte differentiation towards protumor activity does not correlate with M1 or M2 phenotypes. J Immunol Res. (2016) 2016:6031486. doi: 10.1155/2016/6031486
36. Zhang Q, Song Q, Liu S, Xu Y, Gao D, Lu P, et al. Integrated transcriptomic and metabolomic analysis reveals the metabolic programming of GM-CSF- and M-CSF- differentiated mouse macrophages. Front Immunol. (2023) 14:1230772. doi: 10.3389/fimmu.2023.1230772
37. Dan Dunn J, Alvarez LA, Zhang X, and Soldati T. Reactive oxygen species and mitochondria: A nexus of cellular homeostasis. Redox Biol. (2015) 6:472–85. doi: 10.1016/j.redox.2015.09.005
38. Cheung EC and Vousden KH. The role of ROS in tumour development and progression. Nat Rev Cancer. (2022) 22:280–97. doi: 10.1038/s41568-021-00435-0
39. Kennel KB and Greten FR. Immune cell - produced ROS and their impact on tumor growth and metastasis. Redox Biol. (2021) 42:101891. doi: 10.1016/j.redox.2021.101891
40. Moloney JN and Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. (2018) 80:50–64. doi: 10.1016/j.semcdb.2017.05.023
41. Chen Y, Zhou Z, and Min W. Mitochondria, oxidative stress and innate immunity. Front Physiol. (2018) 9:1487. doi: 10.3389/fphys.2018.01487
42. Alissafi T, Kalafati L, Lazari M, Filia A, Kloukina I, Manifava M, et al. Mitochondrial oxidative damage underlies regulatory T cell defects in autoimmunity. Cell Metab. (2020) 32:591–604.e7. doi: 10.1016/j.cmet.2020.07.001
43. Sung Y-J, Kao T-Y, Kuo C-L, Fan C-C, Cheng AN, Fang W-C, et al. Mitochondrial lon sequesters and stabilizes p53 in the matrix to restrain apoptosis under oxidative stress via its chaperone activity. Cell Death Dis. (2018) 9:697. doi: 10.1038/s41419-018-0730-7
44. Su X-L, Su Z-R, and Xu W-H. The protease lon prolongs insect lifespan by responding to reactive oxygen species and degrading mitochondrial transcription factor A. Biochim Biophys Acta Mol Cell Res. (2024) 1871:119648. doi: 10.1016/j.bbamcr.2023.119648
45. Kuo C-L, Chou H-Y, Chiu Y-C, Cheng AN, Fan C-C, Chang Y-N, et al. Mitochondrial oxidative stress by lon-PYCR1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett. (2020) 474:138–50. doi: 10.1016/j.canlet.2020.01.019
46. Checa J and Aran JM. Reactive oxygen species: Drivers of physiological and pathological processes. J Inflammation Res. (2020) 13:1057–73. doi: 10.2147/JIR.S275595
47. Yang Y, Neo SY, Chen Z, Cui W, Chen Y, Guo M, et al. Thioredoxin activity confers resistance against oxidative stress in tumor-infiltrating NK cells. J Clin Invest. (2020) 130:5508–22. doi: 10.1172/JCI137585
48. Yin M and O’Neill LAJ. The role of the electron transport chain in immunity. FASEB J Off Publ Fed Am Soc Exp Biol. (2021) 35:e21974. doi: 10.1096/fj.202101161R
49. Wang L, Kuang Z, Zhang D, Gao Y, Ying M, and Wang T. Reactive oxygen species in immune cells: A new antitumor target. BioMed Pharmacother Biomedecine Pharmacother. (2021) 133:110978. doi: 10.1016/j.biopha.2020.110978
50. Xiang H, Ramil CP, Hai J, Zhang C, Wang H, Watkins AA, et al. Cancer-associated fibroblasts promote immunosuppression by inducing ROS-generating monocytic MDSCs in lung squamous cell carcinoma. Cancer Immunol Res. (2020) 8:436–50. doi: 10.1158/2326-6066.CIR-19-0507
51. Peng H-Y, Lucavs J, Ballard D, Das JK, Kumar A, Wang L, et al. Metabolic reprogramming and reactive oxygen species in T cell immunity. Front Immunol. (2021) 12:652687. doi: 10.3389/fimmu.2021.652687
52. Hildeman DA. Regulation of T-cell apoptosis by reactive oxygen species. Free Radic Biol Med. (2004) 36:1496–504. doi: 10.1016/j.freeradbiomed.2004.03.023
53. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity. (2013) 38:225–36. doi: 10.1016/j.immuni.2012.10.020
54. Li Z, Lai X, Fu S, Ren L, Cai H, Zhang H, et al. Immunogenic cell death activates the tumor immune microenvironment to boost the immunotherapy efficiency. Adv Sci Weinh Baden-Wurtt Ger. (2022) 9:e2201734. doi: 10.1002/advs.202201734
55. Deng H, Yang W, Zhou Z, Tian R, Lin L, Ma Y, et al. Targeted scavenging of extracellular ROS relieves suppressive immunogenic cell death. Nat Commun. (2020) 11:4951. doi: 10.1038/s41467-020-18745-6
56. Kurniawan H, FranChina DG, Guerra L, Bonetti L, -Baguet LS, Grusdat M, et al. Glutathione restricts serine metabolism to preserve regulatory T cell function. Cell Metab. (2020) 31:920–936.e7. doi: 10.1016/j.cmet.2020.03.004
57. Wang Y, Li N, Zhang X, and Horng T. Mitochondrial metabolism regulates macrophage biology. J Biol Chem. (2021) 297:100904. doi: 10.1016/j.jbc.2021.100904
58. Kang S and Kumanogoh A. The spectrum of macrophage activation by immunometabolism. Int Immunol. (2020) 32:467–73. doi: 10.1093/intimm/dxaa017
59. Tan H-Y, Wang N, Li S, Hong M, Wang X, and Feng Y. The reactive oxygen species in macrophage polarization: Reflecting its dual role in progression and treatment of human diseases. Oxid Med Cell Longev. (2016) 2016:2795090. doi: 10.1155/2016/2795090
60. Virág L, Jaén RI, Regdon Z, Boscá L, and Prieto P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol. (2019) 26:101261. doi: 10.1016/j.redox.2019.101261
61. Wu K, Lin K, Li X, Yuan X, Xu P, Ni P, et al. Redefining tumor-associated macrophage subpopulations and functions in the tumor microenvironment. Front Immunol. (2020) 11:1731. doi: 10.3389/fimmu.2020.01731
62. Cassetta L and Pollard JW. A timeline of tumour-associated macrophage biology. Nat Rev Cancer. (2023) 23:238–57. doi: 10.1038/s41568-022-00547-1
63. Kuo C-L, Ponneri Babuharisankar A, Lin Y-C, Lien H-W, Lo YK, Chou H-Y, et al. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: Foe or friend? J BioMed Sci. (2022) 29:74. doi: 10.1186/s12929-022-00859-2
64. Murphy TL and Murphy KM. Dendritic cells in cancer immunology. Cell Mol Immunol. (2022) 19:3–13. doi: 10.1038/s41423-021-00741-5
65. Blanco P, Palucka AK, Pascual V, and Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. (2008) 19:41–52. doi: 10.1016/j.cytogfr.2007.10.004
66. Morante-Palacios O, Fondelli F, Ballestar E, and Martínez-Cáceres EM. Tolerogenic dendritic cells in autoimmunity and inflammatory diseases. Trends Immunol. (2021) 42:59–75. doi: 10.1016/j.it.2020.11.001
67. Kozłowska A, Mackiewicz J, and Mackiewicz A. Therapeutic gene modified cell based cancer vaccines. Gene. (2013) 525:200–7. doi: 10.1016/j.gene.2013.03.056
68. Liang J, Liu X, Xie Q, Chen G, Li X, Jia Y, et al. Endostatin enhances antitumor effect of tumor antigen-pulsed dendritic cell therapy in mouse xenograft model of lung carcinoma. Chin J Cancer Res Chung-Kuo Yen Cheng Yen Chiu. (2016) 28:452–60. doi: 10.21147/j.issn.1000-9604.2016.04.09
69. Brandon M, Baldi P, and Wallace DC. Mitochondrial mutations in cancer. Oncogene. (2006) 25:4647–62. doi: 10.1038/sj.onc.1209607
70. Liu Y, Chen C, Wang X, Sun Y, Zhang J, Chen J, et al. An epigenetic role of mitochondria in cancer. Cells. (2022) 11:2518. doi: 10.3390/cells11162518
71. Koenig A and Buskiewicz-Koenig IA. Redox activation of mitochondrial DAMPs and the metabolic consequences for development of autoimmunity. Antioxid Redox Signal. (2022) 36:441–61. doi: 10.1089/ars.2021.0073
72. Riley JS, Quarato G, Cloix C, Lopez J, O’Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mtDNA release during apoptosis. EMBO J. (2018) 37:e99238. doi: 10.15252/embj.201899238
73. Tao G, Liao W, Hou J, Jiang X, Deng X, Chen G, et al. Advances in crosstalk among innate immune pathways activated by mitochondrial DNA. Heliyon. (2024) 10:e24029. doi: 10.1016/j.heliyon.2024.e24029
74. Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING-mediated DNA sensing maintains CD8+ T cell stemness and promotes antitumor T cell therapy. Sci Transl Med. (2020) 12:eaay9013. doi: 10.1126/scitranslmed.aay9013
75. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial integrity regulated by lipid metabolism is a cell-intrinsic checkpoint for treg suppressive function. Cell Metab. (2020) 31:422–437.e5. doi: 10.1016/j.cmet.2019.11.021
76. Liu J, Sun B, Guo K, Yang Z, Zhao Y, Gao M, et al. Lipid-related FABP5 activation of tumor-associated monocytes fosters immune privilege via PD-L1 expression on treg cells in hepatocellular carcinoma. Cancer Gene Ther. (2022) 29:1951–60. doi: 10.1038/s41417-022-00510-0
77. Cheng AN, Cheng L-C, Kuo C-L, Lo YK, Chou H-Y, Chen C-H, et al. Mitochondrial lon-induced mtDNA leakage contributes to PD-L1-mediated immunoescape via STING-IFN signaling and extracellular vesicles. J Immunother Cancer. (2020) 8:e001372. doi: 10.1136/jitc-2020-001372
78. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross-priming through signal regulatory protein α signaling. Immunity. (2017) 47:363–373.e5. doi: 10.1016/j.immuni.2017.07.016
79. Wang L, Luo R, Onyshchenko K, Rao X, Wang M, Menz B, et al. Adding liposomal doxorubicin enhances the abscopal effect induced by radiation/αPD1 therapy depending on tumor cell mitochondrial DNA and cGAS/STING. J Immunother Cancer. (2023) 11:e006235. doi: 10.1136/jitc-2022-006235
80. Hu M, Zhou M, Bao X, Pan D, Jiao M, Liu X, et al. ATM inhibition enhances cancer immunotherapy by promoting mtDNA leakage and cGAS/STING activation. J Clin Invest. (2021) 131:e139333. doi: 10.1172/JCI139333
81. Lv M, Chen M, Zhang R, Zhang W, Wang C, Zhang Y, et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res. (2020) 30:966–79. doi: 10.1038/s41422-020-00395-4
82. Bao W, Xia H, Liang Y, Ye Y, Lu Y, Xu X, et al. Toll-like receptor 9 can be activated by endogenous mitochondrial DNA to induce podocyte apoptosis. Sci Rep. (2016) 6:22579. doi: 10.1038/srep22579
83. Woo S-R, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MYK, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity. (2014) 41:830–42. doi: 10.1016/j.immuni.2014.10.017
84. Wu G, Zhu Q, Zeng J, Gu X, Miao Y, Xu W, et al. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-κB. J Thorac Dis. (2019) 11:4816–28. doi: 10.21037/jtd.2019.10.26
85. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin X-J, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature. (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
86. Yamazaki T, Bravo-San Pedro JM, Galluzzi L, Kroemer G, and Pietrocola F. Autophagy in the cancer-immunity dialogue. Adv Drug Delivery Rev. (2021) 169:40–50. doi: 10.1016/j.addr.2020.12.003
87. Debnath J, Gammoh N, and Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. (2023) 24:1–16. doi: 10.1038/s41580-023-00585-z
88. Gerada C and Ryan KM. Autophagy, the innate immune response and cancer. Mol Oncol. (2020) 14:1913–29. doi: 10.1002/1878-0261.12774
89. Zhou X, Cai M, Yang F, Huang L, Ling Y, Zhang Y, et al. Hypoxia-induced autophagy in pancreatic cancer counteracts the cytotoxicity of CD8+ T cells by inhibiting the expression of MHC-I. Genes Immun. (2025) 26:45–53. doi: 10.1038/s41435-024-00315-1
90. Kong J, Xu S, Zhang P, and Zhao Y. CXCL1 promotes immune escape in colorectal cancer by autophagy-mediated MHC-I degradation. Hum Immunol. (2023) 84:110716. doi: 10.1016/j.humimm.2023.09.002
91. Liu Z, Li X, Muhammad A, Sun Q, Zhang Q, Wang Y, et al. PACSIN1 promotes immunosuppression in gastric cancer by degrading MHC-I. Acta Biochim Biophys Sin. (2024) 56:1473–82. doi: 10.3724/abbs.2024059
92. Cao S, Hung Y-W, Wang Y-C, Chung Y, Qi Y, Ouyang C, et al. Glutamine is essential for overcoming the immunosuppressive microenvironment in Malignant salivary gland tumors. Theranostics. (2022) 12:6038–56. doi: 10.7150/thno.73896
93. Liu H, Lou J, Liu Y, Liu Z, Xie J, Sun J, et al. Intestinal epithelial cell autophagy deficiency suppresses inflammation-associated colon tumorigenesis. Mol Ther Nucleic Acids. (2022) 28:35–46. doi: 10.1016/j.omtn.2022.02.012
94. Song Y, Jiang Y-X, Guan J-Y, Jiang J-B, Xu M-S, Zhong X-Y, et al. Fangchinoline-mediated autophagy inhibition amplifies antigen presentation and PD-1 blockade efficacy in lung cancer. Acta Pharmacol Sin. (2025) 46:2751–64. doi: 10.1038/s41401-025-01541-7
95. Ladoire S, Enot D, Senovilla L, Ghiringhelli F, Poirier-Colame V, Chaba K, et al. The presence of LC3B puncta and HMGB1 expression in Malignant cells correlate with the immune infiltrate in breast cancer. Autophagy. (2016) 12:864–75. doi: 10.1080/15548627.2016.1154244
96. Taheri Baghmisheh S, Chen C-H, Yeh Y-M, Lin P-C, Chen P-C, Chan R-H, et al. Cathepsin S regulates antitumor immunity through autophagic degradation of PD-L1 in colorectal cancer cells. Cancer Immunol Immunother CII. (2025) 74:287. doi: 10.1007/s00262-025-04140-x
97. Wang J-D, Xu J-Q, Zhang X-N, Huang Z-W, Liu L-L, Zhang L, et al. Mutant C/EBPα p30 alleviates immunosuppression of CD8+ T cells by inhibiting autophagy-associated secretion of IL-1β in AML. Cell Prolif. (2022) 55:e13331. doi: 10.1111/cpr.13331
98. Varveri A, Papadopoulou M, Papadovasilakis Z, Compeer EB, Legaki A-I, Delis A, et al. Immunological synapse formation between T regulatory cells and cancer-associated fibroblasts promotes tumour development. Nat Commun. (2024) 15:4988. doi: 10.1038/s41467-024-49282-1
99. Park Y and Jang J. Prospect of ULK1 modulators in targeting regulatory T cells. Bioorganic Chem. (2022) 129:106141. doi: 10.1016/j.bioorg.2022.106141
100. Gerner MC, Ziegler LS, Schmidt RLJ, Krenn M, Zimprich F, Uyanik-Ünal K, et al. The TGF-b/SOX4 axis and ROS-driven autophagy co-mediate CD39 expression in regulatory T-cells. FASEB J Off Publ Fed Am Soc Exp Biol. (2020) 34:8367–84. doi: 10.1096/fj.201902664
101. Flynn AL, Gans J, Escobedo J, Zhu C, Florescu A-M, Shankara S, et al. RGS1 modulates autophagic and metabolic programs and is a critical mediator of human regulatory T cell function. J Immunol Baltim Md. (2023) 211:1656–68. doi: 10.4049/jimmunol.2200402
102. Fuller AM, Pruitt HC, Liu Y, Irizarry-Negron VM, Pan H, Song H, et al. Oncogene-induced matrix reorganization controls CD8+ T cell function in the soft-tissue sarcoma microenvironment. J Clin Invest. (2024) 134:e167826. doi: 10.1172/JCI167826
103. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. (2020) 16:2069–83. doi: 10.1080/15548627.2020.1714209
104. Zhang Z, Chen W-Q, Zhang S-Q, Bai J-X, Lau C-L, Sze SC-W, et al. The human cathelicidin peptide LL-37 inhibits pancreatic cancer growth by suppressing autophagy and reprogramming of the tumor immune microenvironment. Front Pharmacol. (2022) 13:906625. doi: 10.3389/fphar.2022.906625
105. Ghislat G and Lawrence T. Autophagy in dendritic cells. Cell Mol Immunol. (2018) 15:944–52. doi: 10.1038/cmi.2018.2
106. Garg AD, Dudek AM, and Agostinis P. Autophagy-dependent suppression of cancer immunogenicity and effector mechanisms of innate and adaptive immunity. Oncoimmunology. (2013) 2:e26260. doi: 10.4161/onci.26260
107. López-Soto A, Bravo-San Pedro JM, Kroemer G, Galluzzi L, and Gonzalez S. Involvement of autophagy in NK cell development and function. Autophagy. (2017) 13:633–6. doi: 10.1080/15548627.2016.1274486
108. Noman MZ, Berchem G, and Janji B. Targeting autophagy blocks melanoma growth by bringing natural killer cells to the tumor battlefield. Autophagy. (2018) 14:730–2. doi: 10.1080/15548627.2018.1427398
109. Viry E, Baginska J, Berchem G, Noman MZ, Medves S, Chouaib S, et al. Autophagic degradation of GZMB/granzyme B: A new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy. (2014) 10:173–5. doi: 10.4161/auto.26924
110. Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U.S.A. (2013) 110:17450–5. doi: 10.1073/pnas.1304790110
111. De Veirman K, Menu E, Maes K, De Beule N, De Smedt E, Maes A, et al. Myeloid-derived suppressor cells induce multiple myeloma cell survival by activating the AMPK pathway. Cancer Lett. (2019) 442:233–41. doi: 10.1016/j.canlet.2018.11.002
112. Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, et al. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget. (2016) 7:83907–25. doi: 10.18632/oncotarget.13438
113. Huang Y, Ji W, Zhang J, Huang Z, Ding A, Bai H, et al. The involvement of the mitochondrial membrane in drug delivery. Acta Biomater. (2024) 176:28–50. doi: 10.1016/j.actbio.2024.01.027
114. Cho H, Cho Y-Y, Shim MS, Lee JY, Lee HS, and Kang HC. Mitochondria-targeted drug delivery in cancers. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165808. doi: 10.1016/j.bbadis.2020.165808
115. Kalyanaraman B, Cheng G, Hardy M, and You M. OXPHOS-targeting drugs in oncology: New perspectives. Expert Opin Ther Targets. (2023) 27:939–52. doi: 10.1080/14728222.2023.2261631
116. Chen Z-P, Li M, Zhang L-J, He J-Y, Wu L, Xiao Y-Y, et al. Mitochondria-targeted drug delivery system for cancer treatment. J Drug Target. (2016) 24:492–502. doi: 10.3109/1061186X.2015.1108325
117. Zielonka J, Joseph J, Sikora A, Hardy M, Ouari O, Vasquez-Vivar J, et al. Mitochondria-targeted triphenylphosphonium-based compounds: Syntheses, mechanisms of action, and therapeutic and diagnostic applications. Chem Rev. (2017) 117:10043–120. doi: 10.1021/acs.chemrev.7b00042
118. Zhu Z, Wang Z, Zhang C, Wang Y, Zhang H, Gan Z, et al. Mitochondrion-targeted platinum complexes suppressing lung cancer through multiple pathways involving energy metabolism. Chem Sci. (2019) 10:3089–95. doi: 10.1039/c8sc04871a
119. Han X, Su R, Huang X, Wang Y, Kuang X, Zhou S, et al. Triphenylphosphonium-modified mitochondria-targeted paclitaxel nanocrystals for overcoming multidrug resistance. Asian J Pharm Sci. (2019) 14:569–80. doi: 10.1016/j.ajps.2018.06.006
120. Yu H, Li J-M, Deng K, Zhou W, Wang C-X, Wang Q, et al. Tumor acidity activated triphenylphosphonium-based mitochondrial targeting nanocarriers for overcoming drug resistance of cancer therapy. Theranostics. (2019) 9:7033–50. doi: 10.7150/thno.35748
121. Wang S, Guo F, Ji Y, Yu M, Wang J, and Li N. Dual-mode imaging guided multifunctional theranosomes with mitochondria targeting for photothermally controlled and enhanced photodynamic therapy in vitro and in vivo. Mol Pharm. (2018) 15:3318–31. doi: 10.1021/acs.molpharmaceut.8b00351
122. Nguyen Cao TG, Kang JH, Kang SJ, Truong Hoang Q, Kang HC, Rhee WJ, et al. Brain endothelial cell-derived extracellular vesicles with a mitochondria-targeting photosensitizer effectively treat glioblastoma by hijacking the blood–brain barrier. Acta Pharm Sin B. (2023) 13:3834–48. doi: 10.1016/j.apsb.2023.03.023
123. Noh I, Lee D, Kim H, Jeong C-U, Lee Y, Ahn J-O, et al. Enhanced photodynamic cancer treatment by mitochondria-targeting and brominated near-infrared fluorophores. Adv Sci Weinh Baden-Wurtt Ger. (2018) 5:1700481. doi: 10.1002/advs.201700481
124. Yue C, Yang Y, Zhang C, Alfranca G, Cheng S, Ma L, et al. ROS-responsive mitochondria-targeting blended nanoparticles: Chemo- and photodynamic synergistic therapy for lung cancer with on-demand drug release upon irradiation with a single light source. Theranostics. (2016) 6:2352–66. doi: 10.7150/thno.15433
125. Ma Z, Zeng P, Zhai T, Zhao Y, and Liang H. In situ mitochondrial biomineralization for drug-free cancer therapy. Adv Mater Deerfield Beach Fla. (2024) 36:e2310218. doi: 10.1002/adma.202310218
126. Deng W, McKelvey KJ, Guller A, Fayzullin A, Campbell JM, Clement S, et al. Application of mitochondrially targeted nanoconstructs to neoadjuvant X-ray-induced photodynamic therapy for rectal cancer. ACS Cent Sci. (2020) 6:715–26. doi: 10.1021/acscentsci.9b01121
127. Yasui H, Yamamoto K, Suzuki M, Sakai Y, Bo T, Nagane M, et al. Lipophilic triphenylphosphonium derivatives enhance radiation-induced cell killing via inhibition of mitochondrial energy metabolism in tumor cells. Cancer Lett. (2017) 390:160–7. doi: 10.1016/j.canlet.2017.01.006
128. Liu P, Wang Y, Liu Y, Tan F, Li J, and Li N. S-nitrosothiols loaded mini-sized au at silica nanorod elicits collagen depletion and mitochondrial damage in solid tumor treatment. Theranostics. (2020) 10:6774–89. doi: 10.7150/thno.42661
129. Zhang J, Hu F, Zhang J, Xie J, Wang Z, Lv L, et al. Physical-matched nanoplatelets boost heterogeneous thrombi targeting through self-adaptive deformation for thrombolysis and endothelial repairing. Small Weinh Bergstr Ger. (2025) 21:e2406262. doi: 10.1002/smll.202406262
130. Jiang H, Fu H, Guo Y, Hu P, and Shi J. Evoking tumor associated macrophages by mitochondria-targeted magnetothermal immunogenic cell death for cancer immunotherapy. Biomaterials. (2022) 289:121799. doi: 10.1016/j.biomaterials.2022.121799
131. Huang M, Myers CR, Wang Y, and You M. Mitochondria as a novel target for cancer chemoprevention: Emergence of mitochondrial-targeting agents. Cancer Prev Res Phila Pa. (2021) 14:285–306. doi: 10.1158/1940-6207.CAPR-20-0425
132. Cheng G, Zielonka J, Ouari O, Lopez M, McAllister D, Boyle K, et al. Mitochondria-targeted analogues of metformin exhibit enhanced antiproliferative and radiosensitizing effects in pancreatic cancer cells. Cancer Res. (2016) 76:3904–15. doi: 10.1158/0008-5472.CAN-15-2534
133. Chowdhury AR, Zielonka J, Kalyanaraman B, Hartley RC, Murphy MP, and Avadhani NG. Mitochondria-targeted paraquat and metformin mediate ROS production to induce multiple pathways of retrograde signaling: A dose-dependent phenomenon. Redox Biol. (2020) 36:101606. doi: 10.1016/j.redox.2020.101606
134. Cheng G, Hardy M, You M, and Kalyanaraman B. Combining PEGylated mito-atovaquone with MCT and krebs cycle redox inhibitors as a potential strategy to abrogate tumor cell proliferation. Sci Rep. (2022) 12:5143. doi: 10.1038/s41598-022-08984-6
135. Suárez-Rozas C, Jara JA, Cortés G, Rojas D, Araya-Valdés G, Molina-Berrios A, et al. Antimigratory effect of lipophilic cations derived from gallic and gentisic acid and synergistic effect with 5-fluorouracil on metastatic colorectal cancer cells: A new synthesis route. Cancers. (2024) 16:2980. doi: 10.3390/cancers16172980
136. Wang J, He H, Xiang C, Fan X-Y, Yang L-Y, Yuan L, et al. Uncoupling effect of F16 is responsible for its mitochondrial toxicity and anticancer activity. Toxicol Sci Off J Soc Toxicol. (2018) 161:431–42. doi: 10.1093/toxsci/kfx218
137. Belosludtsev KN, Ilzorkina AI, Belosludtseva NV, Sharapov VA, Penkov NV, Serov DA, et al. Comparative study of cytotoxic and membranotropic properties of betulinic acid-F16 conjugate on breast adenocarcinoma cells (MCF-7) and primary human fibroblasts. Biomedicines. (2022) 10:2903. doi: 10.3390/biomedicines10112903
138. Miao H, Cui W, Zhang T, Zhang Y, Zhang J, Lou H, et al. Mitochondrial targeting derivatives of honokiol enhanced selective antitumor activity in NCI-H446 cells and decreased in vivo toxicity in caenorhabditis elegans. Eur J Med Chem. (2024) 264:115996. doi: 10.1016/j.ejmech.2023.115996
139. Dubinin MV, Semenova AA, Nedopekina DA, Davletshin EV, Spivak AY, and Belosludtsev KN. Effect of F16-betulin conjugate on mitochondrial membranes and its role in cell death initiation. Membranes. (2021) 11:352. doi: 10.3390/membranes11050352
140. Peng YB, Zhao ZL, Liu T, Xie GJ, Jin C, Deng TG, et al. A multi-mitochondrial anticancer agent that selectively kills cancer cells and overcomes drug resistance. ChemMedChem. (2017) 12:250–6. doi: 10.1002/cmdc.201600538
141. Xing W, Liu G, Zhang Y, Zhang T, Lou H, and Fan P. Selective antitumor effect and lower toxicity of mitochondrion-targeting derivatization of triptolide. J Med Chem. (2024) 67:1093–114. doi: 10.1021/acs.jmedchem.3c01508
142. Liu Y-J, Fan X-Y, Zhang D-D, Xia Y-Z, Hu Y-J, Jiang F-L, et al. Dual inhibition of pyruvate dehydrogenase complex and respiratory chain complex induces apoptosis by a mitochondria-targeted fluorescent organic arsenical in vitro and in vivo. ChemMedChem. (2020) 15:552–8. doi: 10.1002/cmdc.201900686
143. Zhang L, Jiang F-L, Guo Q-L, Liu Y, and Jiang P. pH-sensitive bioprobe for multichannel mitochondrial imaging and photodynamic therapy. Anal Chem. (2022) 94:4126–33. doi: 10.1021/acs.analchem.2c00306
144. Xu J, He H, Zhou L-J, Liu Y-Z, Li D-W, Jiang F-L, et al. Pyridinium and indole orientation determines the mitochondrial uncoupling and anti-cancer efficiency of F16. Eur J Med Chem. (2018) 154:305–13. doi: 10.1016/j.ejmech.2018.05.036
145. Zhang W, Chen G, Chen Z, Yang X, Zhang B, Wang S, et al. Mitochondria-targeted polyprodrug nanoparticles induce mitochondrial stress for immunogenic chemo-photodynamic therapy of ovarian cancer. J Control Release Off J Control Release Soc. (2024) 371:470–83. doi: 10.1016/j.jconrel.2024.06.014
146. Kraft O, Hartmann A-K, Brandt S, Hoenke S, Heise NV, Csuk R, et al. Asiatic acid as a leading structure for derivatives combining sub-nanomolar cytotoxicity, high selectivity, and the ability to overcome drug resistance in human preclinical tumor models. Eur J Med Chem. (2023) 250:115189. doi: 10.1016/j.ejmech.2023.115189
147. Macasoi I, Pavel IZ, Moacă AE, Stefana A, VL D, Coricovac D, et al. Mechanistic investigations of antitumor activity of a rhodamine B−oleanolic acid derivative bioconjugate. Oncol Rep. (2020) 44:1169–83. doi: 10.3892/or.2020.7666
148. Yin H, Chen X-T, Chi Q-N, Ma Y-N, Fu X-Y, Du S-S, et al. The hybrid oncolytic peptide NTP-385 potently inhibits adherent cancer cells by targeting the nucleus. Acta Pharmacol Sin. (2023) 44:201–10. doi: 10.1038/s41401-022-00939-x
149. Shi J, Zhao D, Li X, Ding F, Tang X, Liu N, et al. The conjugation of rhodamine B enables carrier-free mitochondrial delivery of functional proteins. Org Biomol Chem. (2020) 18:6829–39. doi: 10.1039/d0ob01305f
150. Modica-Napolitano JS and Aprille JR. Basis for the selective cytotoxicity of rhodamine 123. Cancer Res. (1987) 47:4361–5.
151. Rahman KMM, Bist G, Kumbham S, Foster BA, Woo S, and You Y. Mitochondrial targeting improves the selectivity of singlet-oxygen cleavable prodrugs in NMIBC treatment. Photochem Photobiol. (2024) 100:1622–35. doi: 10.1111/php.13928
152. Yoong SL, Wong BS, Zhou QL, Chin CF, Li J, Venkatesan T, et al. Enhanced cytotoxicity to cancer cells by mitochondria-targeting MWCNTs containing platinum(IV) prodrug of cisplatin. Biomaterials. (2014) 35:748–59. doi: 10.1016/j.biomaterials.2013.09.036
153. Heise N, Becker S, Mueller T, Bache M, Csuk R, and Güttler A. Mitochondria-targeting 1,5-diazacyclooctane-spacered triterpene rhodamine conjugates exhibit cytotoxicity at sub-nanomolar concentration against breast cancer cells. Int J Mol Sci. (2023) 24:10695. doi: 10.3390/ijms241310695
154. Liu C, Zhou L, Wei F, Li L, Zhao S, Gong P, et al. Versatile strategy to generate a rhodamine triplet state as mitochondria-targeting visible-light photosensitizers for efficient photodynamic therapy. ACS Appl Mater Interfaces. (2019) 11:8797–806. doi: 10.1021/acsami.8b20224
155. Lu D, Yang T, Tang N, Li C, Song Y, Wang L, et al. A pH-dependent rhodamine fluorophore with antiproliferative activity of bladder cancer in vitro/vivo and apoptosis mechanism. Eur J Med Chem. (2022) 236:114293. doi: 10.1016/j.ejmech.2022.114293
156. Magut PKS, Das S, Fernand VE, Losso J, McDonough K, Naylor BM, et al. Tunable cytotoxicity of rhodamine 6G via anion variations. J Am Chem Soc. (2013) 135:15873–9. doi: 10.1021/ja407164w
157. Saren BN, Mahajan S, Aalhate M, Kumar R, Chatterjee E, Maji I, et al. Fucoidan-mediated targeted delivery of dasatinib-loaded nanoparticles amplifies apoptosis and endows cytotoxic potential in triple-negative breast cancer. Colloids Surf B Biointerfaces. (2024) 233:113631. doi: 10.1016/j.colsurfb.2023.113631
158. Shen F-F, Chen Y, Dai X, Zhang H-Y, Zhang B, Liu Y, et al. Purely organic light-harvesting phosphorescence energy transfer by β-cyclodextrin pseudorotaxane for mitochondria targeted imaging. Chem Sci. (2020) 12:1851–7. doi: 10.1039/d0sc05343k
159. Zhang H, Ren G, Hou W, Wang L, Sun Y, and Liu J. A silicon-rhodamine-based heavy-atom-free photosensitizer for mitochondria-targeted photodynamic therapy. Spectrochim Acta A Mol Biomol Spectrosc. (2024) 308:123688. doi: 10.1016/j.saa.2023.123688
160. Zhang D, Wen L, Huang R, Wang H, Hu X, and Xing D. Mitochondrial specific photodynamic therapy by rare-earth nanoparticles mediated near-infrared graphene quantum dots. Biomaterials. (2018) 153:14–26. doi: 10.1016/j.biomaterials.2017.10.034
161. Tran VA, Vo GV, Tan MA, Park J-S, An SSA, and Lee S-W. Dual stimuli-responsive multifunctional silicon nanocarriers for specifically targeting mitochondria in human cancer cells. Pharmaceutics. (2022) 14:858. doi: 10.3390/pharmaceutics14040858
162. Song Y, Liu D, Cheng Y, Liu M, Ye W, Zhang B, et al. Dual subcellular compartment delivery of doxorubicin to overcome drug resistant and enhance antitumor activity. Sci Rep. (2015) 5:16125. doi: 10.1038/srep16125
163. Mallick S, Song SJ, Bae Y, and Choi JS. Self-assembled nanoparticles composed of glycol chitosan-dequalinium for mitochondria-targeted drug delivery. Int J Biol Macromol. (2019) 132:451–60. doi: 10.1016/j.ijbiomac.2019.03.215
164. Thuy LT, Lee S, Dongquoc V, and Choi JS. Nanoemulsion composed of α-tocopherol succinate and dequalinium shows mitochondria-targeting and anticancer effects. Antioxid Basel Switz. (2023) 12:437. doi: 10.3390/antiox12020437
165. Galeano E, Nieto E, García-Pérez AI, Delgado MD, Pinilla M, and Sancho P. Effects of the antitumoural dequalinium on NB4 and K562 human leukemia cell lines. Mitochondrial implication Cell death. Leuk Res. (2005) 29:1201–11. doi: 10.1016/j.leukres.2005.03.014
166. Chen Y, Feng X, Li L, Song K, and Zhang L. Preparation and antitumor evaluation of hinokiflavone hybrid micelles with mitochondria targeted for lung adenocarcinoma treatment. Drug Delivery. (2020) 27:565–74. doi: 10.1080/10717544.2020.1748760
167. Shi M, Zhang J, Li X, Pan S, Li J, Yang C, et al. Mitochondria-targeted delivery of doxorubicin to enhance antitumor activity with HER-2 peptide-mediated multifunctional pH-sensitive DQAsomes. Int J Nanomedicine. (2018) 13:4209–26. doi: 10.2147/IJN.S163858
168. Kang JH and Ko YT. Enhanced subcellular trafficking of resveratrol using mitochondriotropic liposomes in cancer cells. Pharmaceutics. (2019) 11:423. doi: 10.3390/pharmaceutics11080423
169. Dian L-H, Hu Y-J, Lin J-Y, Zhang J-Y, Yan Y, Cui Y-N, et al. Fabrication of paclitaxel hybrid nanomicelles to treat resistant breast cancer via oral administration. Int J Nanomedicine. (2018) 13:719–31. doi: 10.2147/IJN.S150140
170. Li N, Zhang C-X, Wang X-X, Zhang L, Ma X, Zhou J, et al. Development of targeting lonidamine liposomes that circumvent drug-resistant cancer by acting on mitochondrial signaling pathways. Biomaterials. (2013) 34:3366–80. doi: 10.1016/j.biomaterials.2013.01.055
171. Wang X-X, Li Y-B, Yao H-J, Ju R-J, Zhang Y, Li R-J, et al. The use of mitochondrial targeting resveratrol liposomes modified with a dequalinium polyethylene glycol-distearoylphosphatidyl ethanolamine conjugate to induce apoptosis in resistant lung cancer cells. Biomaterials. (2011) 32:5673–87. doi: 10.1016/j.biomaterials.2011.04.029
172. Horton KL, Stewart KM, Fonseca SB, Guo Q, and Kelley SO. Mitochondria-penetrating peptides. Chem Biol. (2008) 15:375–82. doi: 10.1016/j.chembiol.2008.03.015
173. Zhou M, Li L, Li L, Lin X, Wang F, Li Q, et al. Overcoming chemotherapy resistance via simultaneous drug-efflux circumvention and mitochondrial targeting. Acta Pharm Sin B. (2019) 9:615–25. doi: 10.1016/j.apsb.2018.11.005
174. Zhang Y, Sun T, and Jiang C. Biomacromolecules as carriers in drug delivery and tissue engineering. Acta Pharm Sin B. (2018) 8:34–50. doi: 10.1016/j.apsb.2017.11.005
175. Xu R, Huang L, Liu J, Zhang Y, Xu Y, Li R, et al. Remodeling of mitochondrial metabolism by a mitochondria-targeted RNAi nanoplatform for effective cancer therapy. Small Weinh Bergstr Ger. (2024) 20:e2305923. doi: 10.1002/smll.202305923
176. Cerrato CP, Kivijärvi T, Tozzi R, Lehto T, Gestin M, and Langel Ü. Intracellular delivery of therapeutic antisense oligonucleotides targeting mRNA coding mitochondrial proteins by cell-penetrating peptides. J Mater Chem B. (2020) 8:10825–36. doi: 10.1039/d0tb01106a
177. Chen H, Wang Y, Yao Y, Qiao S, Wang H, and Tan N. Sequential delivery of cyclopeptide RA-V and doxorubicin for combination therapy on resistant tumor and in situ monitoring of cytochrome c release. Theranostics. (2017) 7:3781–93. doi: 10.7150/thno.20892
178. Yang J, Li Q, Zhou M, Li X, Huang Y, Yang N, et al. Concurrent impairment of nucleus and mitochondria for synergistic inhibition of cancer metastasis. Int J Pharm. (2021) 608:121077. doi: 10.1016/j.ijpharm.2021.121077
179. Wu J, Li J, Wang H, and Liu C-B. Mitochondrial-targeted penetrating peptide delivery for cancer therapy. Expert Opin Drug Delivery. (2018) 15:951–64. doi: 10.1080/17425247.2018.1517750
180. Szeto HH. Mitochondria-targeted cytoprotective peptides for ischemia-reperfusion injury. Antioxid Redox Signal. (2008) 10:601–19. doi: 10.1089/ars.2007.1892
181. Kuang X, Zhou S, Guo W, Wang Z, Sun Y, and Liu H. SS-31 peptide enables mitochondrial targeting drug delivery: A promising therapeutic alteration to prevent hair cell damage from aminoglycosides. Drug Delivery. (2017) 24:1750–61. doi: 10.1080/10717544.2017.1402220
182. Cen J, Dai X, Zhao H, Li X, Hu X, Wu J, et al. Doxorubicin-loaded liposome with the function of “killing two birds with one stone” against glioma. ACS Appl Mater Interfaces. (2023) 15:46697–709. doi: 10.1021/acsami.3c10364
183. Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. (2014) 171:2029–50. doi: 10.1111/bph.12461
184. Toyama S, Shimoyama N, Szeto HH, Schiller PW, and Shimoyama M. Protective effect of a mitochondria-targeted peptide against the development of chemotherapy-induced peripheral neuropathy in mice. ACS Chem Neurosci. (2018) 9:1566–71. doi: 10.1021/acschemneuro.8b00013
185. Liu Y, Li Q, Xiong X, Huang Y, and Zhou Z. Mitochondria-targeting and cell-penetrating peptides-co-modified HPMA copolymers for enhancing therapeutic efficacy of α-tocopheryl succinate. J Mater Chem B. (2018) 6:7674–83. doi: 10.1039/c8tb02621a
186. Buchke S, Sharma M, Bora A, Relekar M, Bhanu P, and Kumar J. Mitochondria-targeted, nanoparticle-based drug-delivery systems: Therapeutics for mitochondrial disorders. Life Basel Switz. (2022) 12:657. doi: 10.3390/life12050657
187. Shah S, Dhawan V, Holm R, Nagarsenker MS, and Perrie Y. Liposomes: Advancements and innovation in the manufacturing process. Adv Drug Delivery Rev. (2020) 154–155:102–22. doi: 10.1016/j.addr.2020.07.002
188. Khan MS, Jaswanth Gowda BH, Almalki WH, Singh T, Sahebkar A, and Kesharwani P. Unravelling the potential of mitochondria-targeted liposomes for enhanced cancer treatment. Drug Discov Today. (2024) 29:103819. doi: 10.1016/j.drudis.2023.103819
189. Peng Y, Lu J, Li R, Zhao Y, Hai L, Guo L, et al. Glucose and triphenylphosphonium co-modified redox-sensitive liposomes to synergistically treat glioma with doxorubicin and lonidamine. ACS Appl Mater Interfaces. (2021) 13:26682–93. doi: 10.1021/acsami.1c02404
190. Wang X, Wang M, Cai M, Shao R, Xia G, and Zhao W. Miriplatin-loaded liposome, as a novel mitophagy inducer, suppresses pancreatic cancer proliferation through blocking POLG and TFAM-mediated mtDNA replication. Acta Pharm Sin B. (2023) 13:4477–501. doi: 10.1016/j.apsb.2023.07.009
191. Ekmekcioglu A, Gok O, Oz-Arslan D, Erdal MS, Yagan Uzuner Y, and Muftuoglu M. Mitochondria-targeted liposomes for drug delivery to tumor mitochondria. Pharmaceutics. (2024) 16:950. doi: 10.3390/pharmaceutics16070950
192. Tian M, Chen W, Wu Y, An J, Hong G, Chen M, et al. Liposome-based nanoencapsulation of a mitochondria-stapling photosensitizer for efficient photodynamic therapy. ACS Appl Mater Interfaces. (2022) 14:12050–8. doi: 10.1021/acsami.1c23156
193. Xiao A, Yin L, Chen T, and Qian H. Lipo/TK-CDN/TPP/Y6 nanoparticles inhibit cutaneous melanoma formation. J Drug Target. (2024) 32:931–40. doi: 10.1080/1061186X.2024.2365243
194. Qu Q, Ma X, and Zhao Y. Targeted delivery of doxorubicin to mitochondria using mesoporous silica nanoparticle nanocarriers. Nanoscale. (2015) 7:16677–86. doi: 10.1039/c5nr05139h
195. Qu Q, Ma X, and Zhao Y. Anticancer effect of α-tocopheryl succinate delivered by mitochondria-targeted mesoporous silica nanoparticles. ACS Appl Mater Interfaces. (2016) 8:34261–9. doi: 10.1021/acsami.6b13974
196. Jia Q, Zheng X, Ge J, Liu W, Ren H, Chen S, et al. Synthesis of carbon dots from hypocrella bambusae for bimodel fluorescence/photoacoustic imaging-guided synergistic photodynamic/photothermal therapy of cancer. J Colloid Interface Sci. (2018) 526:302–11. doi: 10.1016/j.jcis.2018.05.005
197. López V, Villegas MR, Rodríguez V, Villaverde G, Lozano D, Baeza A, et al. Janus mesoporous silica nanoparticles for dual targeting of tumor cells and mitochondria. ACS Appl Mater Interfaces. (2017) 9:26697–706. doi: 10.1021/acsami.7b06906
198. Wang F, Zhang L, Bai X, Cao X, Jiao X, Huang Y, et al. Stimuli-responsive nanocarrier for co-delivery of MiR-31 and doxorubicin to suppress high MtEF4 cancer. ACS Appl Mater Interfaces. (2018) 10:22767–75. doi: 10.1021/acsami.8b07698
199. Kundu M, Sadhukhan P, Ghosh N, Ghosh S, Chatterjee S, Das J, et al. In vivo therapeutic evaluation of a novel bis-lawsone derivative against tumor following delivery using mesoporous silica nanoparticle based redox-responsive drug delivery system. Mater Sci Eng C Mater Biol Appl. (2021) 126:112142. doi: 10.1016/j.msec.2021.112142
200. Cai X, Luo Y, Song Y, Liu D, Yan H, Li H, et al. Integrating in situ formation of nanozymes with three-dimensional dendritic mesoporous silica nanospheres for hypoxia-overcoming photodynamic therapy. Nanoscale. (2018) 10:22937–45. doi: 10.1039/c8nr07679k
201. Hu T, Gong X, Liu X, Xu H, Zhou F, Tan S, et al. Smart design of a therapeutic nanoplatform for mitochondria-targeted copper-depletion therapy combined with chemotherapy. J Mater Chem B. (2023) 11:8433–48. doi: 10.1039/d3tb00979c
202. Cheng Y-J, Zeng X, Cheng D-B, Xu X-D, Zhang X-Z, Zhuo R-X, et al. Functional mesoporous silica nanoparticles (MSNs) for highly controllable drug release and synergistic therapy. Colloids Surf B Biointerfaces. (2016) 145:217–25. doi: 10.1016/j.colsurfb.2016.04.051
203. Dong P, Hu J, Yu S, Zhou Y, Shi T, Zhao Y, et al. A mitochondrial oxidative stress amplifier to overcome hypoxia resistance for enhanced photodynamic therapy. Small Methods. (2021) 5:e2100581. doi: 10.1002/smtd.202100581
204. Yang Z, Wang J, Ai S, Sun J, Mai X, and Guan W. Self-generating oxygen enhanced mitochondrion-targeted photodynamic therapy for tumor treatment with hypoxia scavenging. Theranostics. (2019) 9:6809–23. doi: 10.7150/thno.36988
205. He H, Meng S, Li H, Yang Q, Xu Z, Chen X, et al. Nanoplatform based on GSH-responsive mesoporous silica nanoparticles for cancer therapy and mitochondrial targeted imaging. Mikrochim Acta. (2021) 188:154. doi: 10.1007/s00604-021-04810-4
206. Kianamiri S, Dinari A, Sadeghizadeh M, Rezaei M, Daraei B, Bahsoun NE-H, et al. Mitochondria-targeted polyamidoamine dendrimer-curcumin construct for hepatocellular cancer treatment. Mol Pharm. (2020) 17:4483–98. doi: 10.1021/acs.molpharmaceut.0c00566
207. Anbazhagan R, Muthusamy G, Krishnamoorthi R, Kumaresan S, Rajendra Prasad N, Lai J-Y, et al. PAMAM G4.5 dendrimers for targeted delivery of ferulic acid and paclitaxel to overcome P-glycoprotein-mediated multidrug resistance. Biotechnol Bioeng. (2021) 118:1213–23. doi: 10.1002/bit.27645
208. Bielski ER, Zhong Q, Brown M, and da Rocha SRP. Effect of the conjugation density of triphenylphosphonium cation on the mitochondrial targeting of poly(amidoamine) dendrimers. Mol Pharm. (2015) 12:3043–53. doi: 10.1021/acs.molpharmaceut.5b00320
209. McKinlay AC, Morris RE, Horcajada P, Férey G, Gref R, Couvreur P, et al. BioMOFs: Metal-organic frameworks for biological and medical applications. Angew Chem Int Ed Engl. (2010) 49:6260–6. doi: 10.1002/anie.201000048
210. Wu M-X and Yang Y-W. Metal-organic framework (MOF)-based drug/cargo delivery and cancer therapy. Adv Mater Deerfield Beach Fla. (2017) 29:1606134. doi: 10.1002/adma.201606134
211. Li W, Dong M, Li Y, and Dong H. Macrophages-cancer membrane-encapsulated metal–organic frameworks with copper-depleting moiety for mitochondria-targeted therapeutics. Adv Healthc Mater. (2023) 12:2202986. doi: 10.1002/adhm.202202986
212. Deng J, Zhuang H, Shao S, Zeng X, Xue P, Bai T, et al. Mitochondrial-targeted copper delivery for cuproptosis-based synergistic cancer therapy. Adv Healthc Mater. (2024) 13:2304522. doi: 10.1002/adhm.202304522
213. Guo H, Liu Y, Li X, Wang H, Mao D, Wei L, et al. Magnetic metal-organic framework-based nanoplatform with platelet membrane coating as a synergistic programmed cell death protein 1 inhibitor against hepatocellular carcinoma. ACS Nano. (2023) 17:23829–49. doi: 10.1021/acsnano.3c07885
214. Guo W, Chen Z, Wu Q, Tan L, Ren X, Fu C, et al. Prepared MW-immunosensitizers precisely release NO to downregulate HIF-1α expression and enhance immunogenic cell death. Small Weinh Bergstr Ger. (2024) 20:e2308055. doi: 10.1002/smll.202308055
215. Guo W, Niu M, Chen Z, Wu Q, Tan L, Ren X, et al. Programmed upregulation of HSP70 by metal-organic frameworks nanoamplifier for enhanced microwave thermal-immunotherapy. Adv Healthc Mater. (2022) 11:2201441. doi: 10.1002/adhm.202201441
216. Zhou W, Yu H, Zhang L-J, Wu B, Wang C-X, Wang Q, et al. Redox-triggered activation of nanocarriers for mitochondria-targeting cancer chemotherapy. Nanoscale. (2017) 9:17044–53. doi: 10.1039/c7nr06130g
217. Guo Y, Yang X, Zhang Y, Luo F, Yang J, Zhang X, et al. Hyaluronic acid/dextran-based polymeric micelles co-delivering ursolic acid and doxorubicin to mitochondria for potentiating chemotherapy in MDR cancer. Carbohydr Polym. (2024) 332:121897. doi: 10.1016/j.carbpol.2024.121897
218. Jin J, Yuan P, Yu W, Lin J, Xu A, Xu X, et al. Mitochondria-targeting polymer micelle of dichloroacetate induced pyroptosis to enhance osteosarcoma immunotherapy. ACS Nano. (2022) 16:10327–40. doi: 10.1021/acsnano.2c00192
219. Li L, Yang Q, Zhou Z, Zhong J, and Huang Y. Doxorubicin-loaded, charge reversible, folate modified HPMA copolymer conjugates for active cancer cell targeting. Biomaterials. (2014) 35:5171–87. doi: 10.1016/j.biomaterials.2014.03.027
220. Liu C, Liu Q, Chen L, Li M, Yin J, Zhu X, et al. A pH-sensitive self-assembled and carrier-free nanoparticle based on charge reversal for enhanced synergetic chemo-phototherapy. Adv Healthc Mater. (2020) 9:2000899. doi: 10.1002/adhm.202000899
221. Kalyanaraman B. Exploiting the tumor immune microenvironment and immunometabolism using mitochondria-targeted drugs: Challenges and opportunities in racial disparity and cancer outcome research. FASEB J Off Publ Fed Am Soc Exp Biol. (2022) 36:e22226. doi: 10.1096/fj.202101862R
222. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. (2014) 20:61–72. doi: 10.1016/j.cmet.2014.05.004
223. Banella C, Catalano G, Travaglini S, Pelosi E, Ottone T, Zaza A, et al. Ascorbate plus buformin in AML: A metabolic targeted treatment. Cancers. (2022) 14:2565. doi: 10.3390/cancers14102565
224. Wei G, Chen J, Jing Z, Li Y, Li Z, Zheng W, et al. Glucose transporter 1 (GLUT1)-targeting and hypoxia-activated mitochondria-specific chemo-thermal therapy via a glycosylated poly(amido amine)/celastrol (PAMAM/cel) complex. J Colloid Interface Sci. (2022) 608:1355–65. doi: 10.1016/j.jcis.2021.10.129
225. Xu D, Jin J, Yu H, Zhao Z, Ma D, Zhang C, et al. Chrysin inhibited tumor glycolysis and induced apoptosis in hepatocellular carcinoma by targeting hexokinase-2. J Exp Clin Cancer Res CR. (2017) 36:44. doi: 10.1186/s13046-017-0514-4
226. Nishida M, Yamashita N, Ogawa T, Koseki K, Warabi E, Ohue T, et al. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8T lymphocytes. J Immunother Cancer. (2021) 9:e002954. doi: 10.1136/jitc-2021-002954
227. Lis P, Dyląg M, Niedźwiecka K, Ko YH, Pedersen PL, Goffeau A, et al. The HK2 dependent “warburg effect” and mitochondrial oxidative phosphorylation in cancer: Targets for effective therapy with 3-bromopyruvate. Mol Basel Switz. (2016) 21:1730. doi: 10.3390/molecules21121730
228. Aublin-Gex A, Jacolin F, Diaz O, Jacquemin C, Marçais A, Walzer T, et al. Tethering of hexokinase 2 to mitochondria promotes resistance of liver cancer cells to natural killer cell cytotoxicity. Eur J Immunol. (2024) 54:e2350954. doi: 10.1002/eji.202350954
229. Pathak RK, Marrache S, Harn DA, and Dhar S. Mito-DCA: A mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem Biol. (2014) 9:1178–87. doi: 10.1021/cb400944y
230. Fiorillo M, Lamb R, Tanowitz HB, Mutti L, Krstic-DeMonacos M, Cappello AR, et al. Repurposing atovaquone: Targeting mitochondrial complex III and OXPHOS to eradicate cancer stem cells. Oncotarget. (2016) 7:34084–99. doi: 10.18632/oncotarget.9122
231. Kunisada Y, Eikawa S, Tomonobu N, Domae S, Uehara T, Hori S, et al. Attenuation of CD4+CD25+ regulatory T cells in the tumor microenvironment by metformin, a type 2 diabetes drug. EBioMedicine. (2017) 25:154–64. doi: 10.1016/j.ebiom.2017.10.009
232. Beerkens APM, Boreel DF, Nathan JA, Neuzil J, Cheng G, Kalyanaraman B, et al. Characterizing OXPHOS inhibitor-mediated alleviation of hypoxia using high-throughput live cell-imaging. Cancer Metab. (2024) 12:13. doi: 10.1186/s40170-024-00342-6
233. Lyamzaev KG, Tokarchuk AV, Panteleeva AA, Mulkidjanian AY, Skulachev VP, and Chernyak BV. Induction of autophagy by depolarization of mitochondria. Autophagy. (2018) 14:921–4. doi: 10.1080/15548627.2018.1436937
234. Feng T, Tang Z, Karges J, Shu J, Xiong K, Jin C, et al. An iridium(III)-based photosensitizer disrupting the mitochondrial respiratory chain induces ferritinophagy-mediated immunogenic cell death. Chem Sci. (2024) 15:6752–62. doi: 10.1039/d4sc01214c
235. Cheng G, Hardy M, Topchyan P, Zander R, Volberding P, Cui W, et al. Mitochondria-targeted hydroxyurea inhibits OXPHOS and induces antiproliferative and immunomodulatory effects. iScience. (2021) 24:102673. doi: 10.1016/j.isci.2021.102673
236. AbuEid M, McAllister DM, McOlash L, Harwig MC, Cheng G, Drouillard D, et al. Synchronous effects of targeted mitochondrial complex I inhibitors on tumor and immune cells abrogate melanoma progression. iScience. (2021) 24(6):102653. doi: 10.1016/j.isci.2021.102653
237. Huang M, Xiong D, Pan J, Zhang Q, Wang Y, Myers CR, et al. Prevention of tumor growth and dissemination by in situ vaccination with mitochondria-targeted atovaquone. Adv Sci Weinh Baden-Wurtt Ger. (2022) 9:e2101267. doi: 10.1002/advs.202101267
238. Xiong D, Yin Z, Huang M, Wang Y, Hardy M, Kalyanaraman B, et al. Mitochondria-targeted atovaquone promotes anti-lung cancer immunity by reshaping tumor microenvironment and enhancing energy metabolism of anti-tumor immune cells. Cancer Commun Lond Engl. (2024) 44:448–52. doi: 10.1002/cac2.12500
239. Bahrambeigi S and Shafiei-Irannejad V. Immune-mediated anti-tumor effects of metformin; targeting metabolic reprogramming of T cells as a new possible mechanism for anti-cancer effects of metformin. Biochem Pharmacol. (2020) 174:113787. doi: 10.1016/j.bcp.2019.113787
240. Eikawa S, Nishida M, Mizukami S, Yamazaki C, Nakayama E, and Udono H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc Natl Acad Sci. (2015) 112:1809–14. doi: 10.1073/pnas.1417636112
241. Wang Y, Li H, Niu G, Li Y, Huang Z, Cheng S, et al. Boosting sono-immunotherapy of prostate carcinoma through amplifying domino-effect of mitochondrial oxidative stress using biodegradable cascade-targeting nanocomposites. ACS Nano. (2024) 18:5828–46. doi: 10.1021/acsnano.3c12511
242. Deng Y, Jia F, Jiang P, Chen L, Xing L, Shen X, et al. Biomimetic nanoparticle synchronizing pyroptosis induction and mitophagy inhibition for anti-tumor therapy. Biomaterials. (2023) 301:122293. doi: 10.1016/j.biomaterials.2023.122293
243. Zhang F, Wen C, Peng Y, Hu Z, Zheng S, Chen W, et al. Biomimetic lipid nanoparticles for homologous-targeting and enhanced photodynamic therapy against glioma. Eur J Pharm Sci Off J Eur Fed Pharm Sci. (2023) 190:106574. doi: 10.1016/j.ejps.2023.106574
244. Zhou Y, Zhang W, Wang B, Wang P, Li D, Cao T, et al. Mitochondria-targeted photodynamic therapy triggers GSDME-mediated pyroptosis and sensitizes anti-PD-1 therapy in colorectal cancer. J Immunother Cancer. (2024) 12:e008054. doi: 10.1136/jitc-2023-008054
245. Liu S, Tian H, Ming H, Zhang T, Gao Y, Liu R, et al. Mitochondrial-targeted CS at KET/P780 nanoplatform for site-specific delivery and high-efficiency cancer immunotherapy in hepatocellular carcinoma. Adv Sci Weinh Baden-Wurtt Ger. (2024) 11:e2308027. doi: 10.1002/advs.202308027
246. Yang S, Wu G-L, Li N, Wang M, Wu P, He Y, et al. A mitochondria-targeted molecular phototheranostic platform for NIR-II imaging-guided synergistic photothermal/photodynamic/immune therapy. J Nanobiotechnology. (2022) 20:475. doi: 10.1186/s12951-022-01679-0
247. Mu M, Chen B, Li H, Fan R, Yang Y, Zhou L, et al. Augmented the sensitivity of photothermal-ferroptosis therapy in triple-negative breast cancer through mitochondria-targeted nanoreactor. J Control Release Off J Control Release Soc. (2024) 375:733–44. doi: 10.1016/j.jconrel.2024.09.042
248. Wang J, Chen W, Du W, Zhang H, Ilmer M, Song L, et al. ROS generative black phosphorus-tamoxifen nanosheets for targeted endocrine-sonodynamic synergistic breast cancer therapy. Int J Nanomedicine. (2023) 18:2389–409. doi: 10.2147/IJN.S406627
249. Wang Y, Wang W, Gu R, Chen J, Chen Q, Lin T, et al. In situ vaccination with mitochondria-targeting immunogenic death inducer elicits CD8+ T cell-dependent antitumor immunity to boost tumor immunotherapy. Adv Sci Weinh Baden-Wurtt Ger. (2023) 10:e2300286. doi: 10.1002/advs.202300286
250. Ding Q, Tang W, Li X, Ding Y, Chen X, Cao W, et al. Mitochondrial-targeted brequinar liposome boosted mitochondrial-related ferroptosis for promoting checkpoint blockade immunotherapy in bladder cancer. J Control Release Off J Control Release Soc. (2023) 363:221–34. doi: 10.1016/j.jconrel.2023.09.024
251. Ahmed A and Tait SWG. Targeting immunogenic cell death in cancer. Mol Oncol. (2020) 14:2994–3006. doi: 10.1002/1878-0261.12851
252. Ren J, Zhou J, Liu H, Jiao X, Cao Y, Xu Z, et al. Ultrasound (US)-activated redox dyshomeostasis therapy reinforced by immunogenic cell death (ICD) through a mitochondrial targeting liposomal nanosystem. Theranostics. (2021) 11:9470–91. doi: 10.7150/thno.62984
253. Guo Y, Fan Y, Wang Z, Li G, Zhan M, Gong J, et al. Chemotherapy mediated by biomimetic polymeric nanoparticles potentiates enhanced tumor immunotherapy via amplification of endoplasmic reticulum stress and mitochondrial dysfunction. Adv Mater Deerfield Beach Fla. (2022) 34:e2206861. doi: 10.1002/adma.202206861
254. Kim J, Shim MK, Moon Y, Kim J, Cho H, Yun WS, et al. Cancer cell-specific and pro-apoptotic SMAC peptide-doxorubicin conjugated prodrug encapsulated aposomes for synergistic cancer immunotherapy. J Nanobiotechnology. (2024) 22:109. doi: 10.1186/s12951-024-02314-w
255. Luo J, Wang X, Shi Z, Zeng Y, He L, Cao J, et al. Enhancement of antitumor immunotherapy using mitochondria-targeted cancer cell membrane-biomimetic MOF-mediated sonodynamic therapy and checkpoint blockade immunotherapy. J Nanobiotechnology. (2022) 20:228. doi: 10.1186/s12951-022-01453-2
256. Lu Y, Fan X, Pan Q, He B, and Pu Y. A mitochondria-targeted anticancer copper dithiocarbamate amplifies immunogenic cuproptosis and macrophage polarization. J Mater Chem B. (2024) 12:2006–14. doi: 10.1039/d3tb02886k
257. Ji C, Si J, Xu Y, Zhang W, Yang Y, He X, et al. Mitochondria-targeted and ultrasound-responsive nanoparticles for oxygen and nitric oxide codelivery to reverse immunosuppression and enhance sonodynamic therapy for immune activation. Theranostics. (2021) 11:8587–604. doi: 10.7150/thno.62572
258. Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward M1 phenotype. Nat Commun. (2018) 9:873. doi: 10.1038/s41467-018-03225-9
259. Bede ÁM, Váróczy C, Polgár Z, Fazekas G, Hegedűs C, Kókai E, et al. The autophagy inhibitor bafilomycin inhibits antibody-dependent natural killer cell-mediated killing of breast carcinoma cells. Int J Mol Sci. (2025) 26:6273. doi: 10.3390/ijms26136273
260. An J, Zhang K, Wang B, Wu S, Wang Y, Zhang H, et al. Nanoenabled disruption of multiple barriers in antigen cross-presentation of dendritic cells via calcium interference for enhanced chemo-immunotherapy. ACS Nano. (2020) 14:7639–50. doi: 10.1021/acsnano.0c03881
261. Zhou W-J, Chang K-K, Wu K, Yang H-L, Mei J, Xie F, et al. Rapamycin synergizes with cisplatin in antiendometrial cancer activation by improving IL-27-stimulated cytotoxicity of NK cells. Neoplasia N Y N. (2018) 20:69–79. doi: 10.1016/j.neo.2017.11.003
262. Park M-J, Lee S-Y, Moon S-J, Son H-J, Lee S-H, Kim E-K, et al. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and tregs. Transl Res J Lab Clin Med. (2016) 173:115–30. doi: 10.1016/j.trsl.2016.03.006
263. Park S-S, Kim J-I, Lee C-H, Bae J-H, Park J-M, Choe E-J, et al. Temsirolimus enhances anti-cancer immunity by inducing autophagy-mediated degradation of the secretion of small extracellular vesicle PD-L1. Cancers. (2022) 14:4081. doi: 10.3390/cancers14174081
264. Wu J, Zhao X, Sun Q, Jiang Y, Zhang W, Luo J, et al. Synergic effect of PD-1 blockade and endostar on the PI3K/AKT/mTOR-mediated autophagy and angiogenesis in lewis lung carcinoma mouse model. BioMed Pharmacother Biomedecine Pharmacother. (2020) 125:109746. doi: 10.1016/j.biopha.2019.109746
265. He L, Chen Q, Lu Q, Yang M, Xie B, Chen T, et al. Autophagy-inducing MoO3-x nanowires boost photothermal-triggered cancer immunotherapy. Angew Chem Int Ed Engl. (2024) 63:e202404822. doi: 10.1002/anie.202404822
266. Baimanov D, Li S, Gao XJ, Cai R, Liu K, Li J, et al. A phosphatase-like nanomaterial promotes autophagy and reprograms macrophages for cancer immunotherapy. Chem Sci. (2024) 15:10838–50. doi: 10.1039/d4sc01690d
267. Yang X, Yang J, Gu X, Tao Y, Ji H, Miao X, et al. (-)-guaiol triggers immunogenic cell death and inhibits tumor growth in non-small cell lung cancer. Mol Cell Biochem. (2023) 478:1611–20. doi: 10.1007/s11010-022-04613-y
268. Xian J, Gao L, Ren Z, Jiang Y, Pan J, Ying Z, et al. Inhibition of autophagy by berbamine hydrochloride mitigates tumor immune escape by elevating MHC-I in melanoma cells. Cells. (2024) 13:1537. doi: 10.3390/cells13181537
269. Yao C, Ni Z, Gong C, Zhu X, Wang L, Xu Z, et al. Rocaglamide enhances NK cell-mediated killing of non-small cell lung cancer cells by inhibiting autophagy. Autophagy. (2018) 14:1831–44. doi: 10.1080/15548627.2018.1489946
270. Xiong H, Chen Z, Lin B, Xie B, Liu X, Chen C, et al. Naringenin regulates FKBP4/NR3C1/NRF2 axis in autophagy and proliferation of breast cancer and differentiation and maturation of dendritic cell. Front Immunol. (2021) 12:745111. doi: 10.3389/fimmu.2021.745111
271. Finisguerra V, Dvorakova T, Formenti M, Van Meerbeeck P, Mignion L, Gallez B, et al. Metformin improves cancer immunotherapy by directly rescuing tumor-infiltrating CD8 T lymphocytes from hypoxia-induced immunosuppression. J Immunother Cancer. (2023) 11:e005719. doi: 10.1136/jitc-2022-005719
272. Rodriguez-Berriguete G, Puliyadi R, MaChado N, Barberis A, Prevo R, McLaughlin M, et al. Antitumour effect of the mitochondrial complex III inhibitor atovaquone in combination with anti-PD-L1 therapy in mouse cancer models. Cell Death Dis. (2024) 15:32. doi: 10.1038/s41419-023-06405-8
273. Zhou Z, Luo W, Zheng C, Wang H, Hu R, Deng H, et al. Mitochondrial metabolism blockade nanoadjuvant reversed immune-resistance microenvironment to sensitize albumin-bound paclitaxel-based chemo-immunotherapy. Acta Pharm Sin B. (2024) 14:4087–101. doi: 10.1016/j.apsb.2024.05.028
274. Wang S, Zhou X, Zeng Z, Sui M, Chen L, Feng C, et al. Atovaquone-HSA nano-drugs enhance the efficacy of PD-1 blockade immunotherapy by alleviating hypoxic tumor microenvironment. J Nanobiotechnology. (2021) 19:302. doi: 10.1186/s12951-021-01034-9
275. Jiang X, Yi L, Li C, Wang H, Xiong W, Li Y, et al. Mitochondrial disruption nanosystem simultaneously depressed programmed death ligand-1 and transforming growth factor-β to overcome photodynamic immunotherapy resistance. ACS Nano. (2024) 18:3331–48. doi: 10.1021/acsnano.3c10117
276. Bai Y, Hua J, Zhao J, Wang S, Huang M, Wang Y, et al. A silver-induced absorption red-shifted dual-targeted nanodiagnosis-treatment agent for NIR-II photoacoustic imaging-guided photothermal and ROS simultaneously enhanced immune checkpoint blockade antitumor therapy. Adv Sci. (2024) 11:2306375. doi: 10.1002/advs.202306375
277. Zuo L, Nie W, Yu S, Zhuang W-R, Liang C, Li S, et al. Biomimetic nanovesicle with mitochondria-synthesized sonosensitizer and mitophagy inhibition for cancer sono-immunotherapy. Nano Lett. (2023) 23:3005–13. doi: 10.1021/acs.nanolett.3c00383
278. Hsieh C-H, Hsieh H-C, Shih F-S, Wang P-W, Yang L-X, Shieh D-B, et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics. (2021) 11:7072–91. doi: 10.7150/thno.57803
279. Jena B, Rushworth D, McNamara GT, and Cooper LJ. Mitochondrial biomass as a measure of fitness for T cells expressing chimeric antigen receptors. Blood. (2015) 126:3242. doi: 10.1182/blood.V126.23.3242.3242
280. Li W and Zhang L. Rewiring mitochondrial metabolism for CD8+ T cell memory formation and effective cancer immunotherapy. Front Immunol. (2020) 11:1834. doi: 10.3389/fimmu.2020.01834
281. Simula L, Fumagalli M, Vimeux L, Rajnpreht I, Icard P, Birsen G, et al. Mitochondrial metabolism sustains CD8+ T cell migration for an efficient infiltration into solid tumors. Nat Commun. (2024) 15:2203. doi: 10.1038/s41467-024-46377-7
282. Maldonado-Pérez N, Tristán-Manzano M, Justicia-Lirio P, Martínez-Planes E, Muñoz P, Pavlovic K, et al. Efficacy and safety of universal (TCRKO) ARI-0001 CAR-T cells for the treatment of B-cell lymphoma. Front Immunol. (2022) 13:1011858. doi: 10.3389/fimmu.2022.1011858
283. Nakagawara K, Ando M, Srirat T, Mise-Omata S, Hayakawa T, Ito M, et al. NR4A ablation improves mitochondrial fitness for long persistence in human CAR-T cells against solid tumors. J Immunother Cancer. (2024) 12:e008665. doi: 10.1136/jitc-2023-008665
284. Kawalekar OU, O’Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
285. Tian H, Zhang B, Li L, Wang G, Li H, and Zheng J. Manipulation of mitochondrial plasticity changes the metabolic competition between “foe” and “friend” during tumor Malignant transformation. Front Oncol. (2020) 10:1692. doi: 10.3389/fonc.2020.01692
286. Wu M-H, Valenca-Pereira F, Cendali F, Giddings EL, Pham-Danis C, Yarnell MC, et al. Deleting the mitochondrial respiration negative regulator MCJ enhances the efficacy of CD8+ T cell adoptive therapies in pre-clinical studies. Nat Commun. (2024) 15:4444. doi: 10.1038/s41467-024-48653-y
287. Court AC, Parra-Crisóstomo E, Castro-Córdova P, Abdo L, Aragão EAA, Lorca R, et al. Survival advantage of native and engineered T cells is acquired by mitochondrial transfer from mesenchymal stem cells. J Transl Med. (2024) 22:868. doi: 10.1186/s12967-024-05627-4
288. Pan Q, Fan X, Xie L, Wu D, Liu R, Gao W, et al. Nano-enabled colorectal cancer therapy. J Control Release Off J Control Release Soc. (2023) 362:548–64. doi: 10.1016/j.jconrel.2023.09.014
289. Guo X, Tu P, Wang X, Du C, Jiang W, Qiu X, et al. Decomposable nanoagonists enable NIR-elicited cGAS-STING activation for tandem-amplified photodynamic-metalloimmunotherapy. Adv Mater. (2024) 36:2313029. doi: 10.1002/adma.202313029
290. Li F, Wen Z, Wu C, Yang Z, Wang Z, Diao W, et al. Simultaneous activation of immunogenic cell death and cGAS-STING pathway by liver- and mitochondria-targeted gold(I) complexes for chemoimmunotherapy of hepatocellular carcinoma. J Med Chem. (2024) 67:1982–2003. doi: 10.1021/acs.jmedchem.3c01785
291. Wang C, Zhang R, He J, Yu L, Li X, Zhang J, et al. Ultrasound-responsive low-dose doxorubicin liposomes trigger mitochondrial DNA release and activate cGAS-STING-mediated antitumour immunity. Nat Commun. (2023) 14:3877. doi: 10.1038/s41467-023-39607-x
292. Zhao M, Li J, Chen F, Han Y, Chen D, and Hu H. Engineering nanoparticles boost TNBC therapy by CD24 blockade and mitochondrial dynamics regulation. J Control Release Off J Control Release Soc. (2023) 355:211–27. doi: 10.1016/j.jconrel.2023.01.075
293. Zhang X, He Q, Sun J, Gong H, Cao Y, Duan L, et al. Near-infrared-enpowered nanomotor-mediated targeted chemotherapy and mitochondrial phototherapy to boost systematic antitumor immunity. Adv Healthc Mater. (2022) 11:e2200255. doi: 10.1002/adhm.202200255
294. Xiong H, Song Z, Wang T, Huang K, Yu F, Sun W, et al. Photoswitchable dynamics and RNAi synergist with tailored interface and controlled release reprogramming tumor immunosuppressive niche. Biomaterials. (2025) 312:122712. doi: 10.1016/j.biomaterials.2024.122712
295. Yang Y, Yang J, Zhu N, Qiu H, Feng W, Chen Y, et al. Tumor-targeting hydroxyapatite nanoparticles for remodeling tumor immune microenvironment (TIME) by activating mitoDNA-pyroptosis pathway in cancer. J Nanobiotechnology. (2023) 21:470. doi: 10.1186/s12951-023-02231-4
296. Cheng G, Hardy M, Topchyan P, Zander R, Volberding P, Cui W, et al. Potent inhibition of tumour cell proliferation and immunoregulatory function by mitochondria-targeted atovaquone. Sci Rep. (2020) 10:17872. doi: 10.1038/s41598-020-74808-0
297. Fu Y, He Y, Wei X, Zhang X, Tu W, Xue W, et al. Sonocatalysis regulates tumor autophagy for enhanced immunotherapy. ACS Nano. (2024) 18:28793–809. doi: 10.1021/acsnano.4c08468
298. Pan R, Ryan J, Pan D, Wucherpfennig KW, and Letai A. Augmenting NK cell-based immunotherapy by targeting mitochondrial apoptosis. Cell. (2022) 185:1521–1538.e18. doi: 10.1016/j.cell.2022.03.030
299. Cheng F, He L, Wang J, Lai L, Ma L, Qu K, et al. Synergistic immunotherapy with a calcium-based nanoinducer: Evoking pyroptosis and remodeling tumor-associated macrophages for enhanced antitumor immune response. Nanoscale. (2024) 16:18570–83. doi: 10.1039/d4nr01497a
300. Zhu L, Li W, Liu C, Yue S, Qiao Y, Cui Y, et al. Glutathione-sensitive mesoporous nanoparticles loaded with cinnamaldehyde for chemodynamic and immunological therapy of cancer. J Mater Chem B. (2023) 11:8717–31. doi: 10.1039/d3tb01094e
301. Peng N, Yu H, Yu W, Yang M, Chen H, Zou T, et al. Sequential-targeting nanocarriers with pH-controlled charge reversal for enhanced mitochondria-located photodynamic-immunotherapy of cancer. Acta Biomater. (2020) 105:223–38. doi: 10.1016/j.actbio.2020.01.005
302. Zhang Y, Song X, Feng Y, Qian Y, Chen B, Zhang T, et al. The circRNA cEMSY induces immunogenic cell death and boosts immunotherapy efficacy in lung adenocarcinoma. Cancer Res. (2025) 85:497–514. doi: 10.1158/0008-5472.CAN-24-1484
303. Lei J, Zhang W, Ma L, He Y, Liang H, Zhang X, et al. Sonodynamic amplification of cGAS-STING activation by cobalt-based nanoagonist against bone and metastatic tumor. Biomaterials. (2023) 302:122295. doi: 10.1016/j.biomaterials.2023.122295
304. Xie L, Ding Y, Zhang X, Zhang Z, Zeng S, Wang L, et al. A cascade-targeted enzyme-instructed peptide self-assembly strategy for cancer immunotherapy through boosting immunogenic cell death. Small Methods. (2023) 7:e2201416. doi: 10.1002/smtd.202201416
305. Dai Y, Zhu L, Li X, Zhang F, Chen K, Jiao G, et al. A biomimetic cuproptosis amplifier for targeted NIR-II fluorescence/photoacoustic imaging-guided synergistic NIR-II photothermal immunotherapy. Biomaterials. (2024) 305:122455. doi: 10.1016/j.biomaterials.2023.122455
306. Li Y, Liu J, Weichselbaum RR, and Lin W. Mitochondria-targeted multifunctional nanoparticles combine cuproptosis and programmed cell death-1 downregulation for cancer immunotherapy. Adv Sci Weinh Baden-Wurtt Ger. (2024) 11:e2403520. doi: 10.1002/advs.202403520
307. Ohashi T, Akazawa T, Aoki M, Kuze B, Mizuta K, Ito Y, et al. Dichloroacetate improves immune dysfunction caused by tumor-secreted lactic acid and increases antitumor immunoreactivity. Int J Cancer. (2013) 133:1107–18. doi: 10.1002/ijc.28114
308. Lan Z, Liu W-J, Yin W-W, Yang S-R, Cui H, Zou K-L, et al. Biomimetic MDSCs membrane coated black phosphorus nanosheets system for photothermal therapy/photodynamic therapy synergized chemotherapy of cancer. J Nanobiotechnology. (2024) 22:174. doi: 10.1186/s12951-024-02417-4
309. Zheng D, Liu J, Xie L, Wang Y, Ding Y, Peng R, et al. Enzyme-instructed and mitochondria-targeting peptide self-assembly to efficiently induce immunogenic cell death. Acta Pharm Sin B. (2022) 12:2740–50. doi: 10.1016/j.apsb.2021.07.005
310. Guo X, Tu P, Zhu L, Cheng C, Jiang W, Du C, et al. Nanoenabled tumor energy metabolism disorder via sonodynamic therapy for multidrug resistance reversal and metastasis inhibition. ACS Appl Mater Interfaces. (2023) 15:309–26. doi: 10.1021/acsami.2c16278
311. Shen J, Sun C, Wang Z, Chu Z, Liu C, Xu X, et al. Sequential receptor-mediated mixed-charge nanomedicine to target pancreatic cancer, inducing immunogenic cell death and reshaping the tumor microenvironment. Int J Pharm. (2021) 601:120553. doi: 10.1016/j.ijpharm.2021.120553
312. Xie Q, Li Z, Liu Y, Zhang D, Su M, Niitsu H, et al. Translocator protein-targeted photodynamic therapy for direct and abscopal immunogenic cell death in colorectal cancer. Acta Biomater. (2021) 134:716–29. doi: 10.1016/j.actbio.2021.07.052
313. Ma B, Sheng J, Wang P, Jiang Z, and Borrathybay E. Combinational phototherapy and hypoxia-activated chemotherapy favoring antitumor immune responses. Int J Nanomedicine. (2019) 14:4541–58. doi: 10.2147/IJN.S203383
314. Jiang Q, Zhang C, Wang H, Peng T, Zhang L, Wang Y, et al. Mitochondria-targeting immunogenic cell death inducer improves the adoptive T-cell therapy against solid tumor. Front Oncol. (2019) 9:1196. doi: 10.3389/fonc.2019.01196
315. Ye Y, Ren K, Dong Y, Yang L, Zhang D, Yuan Z, et al. Mitochondria-targeting pyroptosis amplifier of lonidamine-modified black phosphorus nanosheets for glioblastoma treatments. ACS Appl Mater Interfaces. (2023) 15:26285–97. doi: 10.1021/acsami.3c01559
316. Mitochondrial reactive oxygen species trigger metformin-dependent antitumor immunity via activation of Nrf2/mTORC1/p62 axis in tumor-infiltrating CD8T lymphocytes - PubMed . Available online at (Accessed February 25, 2025).
317. Zhang J-J, Zhang Q-S, Li Z-Q, Zhou J-W, and Du J. Metformin attenuates PD-L1 expression through activating hippo signaling pathway in colorectal cancer cells. Am J Transl Res. (2019) 11:6965–76.
318. Scharping NE, Menk AV, Whetstone RD, Zeng X, and Delgoffe GM. Efficacy of PD-1 blockade is potentiated by metformin-induced reduction of tumor hypoxia. Cancer Immunol Res. (2017) 5:9–16. doi: 10.1158/2326-6066.CIR-16-0103
319. Yao Y, Yuan M, Shi M, Li W, Sha Y, Zhang Y, et al. Halting multiple myeloma with MALT1 inhibition: Suppressing BCMA-induced NF-κB and inducing immunogenic cell death. Blood Adv. (2024) 8:4003–16. doi: 10.1182/bloodadvances.2023012394
320. Wang D, Liu J, Wang C, Zhang W, Yang G, Chen Y, et al. Microbial synthesis of pRussian blue for potentiating checkpoint blockade immunotherapy. Nat Commun. (2023) 14:2943. doi: 10.1038/s41467-023-38796-9
321. Rencelj A, Gvozdenovic N, and Cemazar M. MitomiRs: Their roles in mitochondria and importance in cancer cell metabolism. Radiol Oncol. (2021) 55:379–92. doi: 10.2478/raon-2021-0042
322. Jiang H, Guo Y, Yu Z, Hu P, and Shi J. Nanocatalytic bacteria disintegration reverses immunosuppression of colorectal cancer. Natl Sci Rev. (2022) 9:nwac169. doi: 10.1093/nsr/nwac169
323. Zhang C-J, Li J-M, Xu D, Wang D-D, Qi M-H, Chen F, et al. Surface molecularly engineered mitochondria conduct immunophenotype repolarization of tumor-associated macrophages to potentiate cancer immunotherapy. Adv Sci Weinh Baden-Wurtt Ger. (2024) 11:e2403044. doi: 10.1002/advs.202403044
324. Sun W, Wang H, Qi Y, Li M, Zhang R, Gao Z, et al. Metal-phenolic vehicles potentiate cycle-cascade activation of pyroptosis and cGAS-STING pathway for tumor immunotherapy. ACS Nano. (2024) 18:23727–40. doi: 10.1021/acsnano.4c08613
325. Duan Y, Deng M, Liu B, Meng X, Liao J, Qiu Y, et al. Mitochondria targeted drug delivery system overcoming drug resistance in intrahepatic cholangiocarcinoma by reprogramming lipid metabolism. Biomaterials. (2024) 309:122609. doi: 10.1016/j.biomaterials.2024.122609
326. Granato M, Lacconi V, Peddis M, Lotti LV, Di Renzo L, Gonnella R, et al. HSP70 inhibition by 2-phenylethynesulfonamide induces lysosomal cathepsin D release and immunogenic cell death in primary effusion lymphoma. Cell Death Dis. (2013) 4:e730. doi: 10.1038/cddis.2013.263
327. Yu W, He X, Yang Z, Yang X, Xiao W, Liu R, et al. Sequentially responsive biomimetic nanoparticles with optimal size in combination with checkpoint blockade for cascade synergetic treatment of breast cancer and lung metastasis. Biomaterials. (2019) 217:119309. doi: 10.1016/j.biomaterials.2019.119309
328. Hunt H, Simón-Gracia L, Tobi A, Kotamraju VR, Sharma S, Nigul M, et al. Targeting of p32 in peritoneal carcinomatosis with intraperitoneal linTT1 peptide-guided pro-apoptotic nanoparticles. J Control Release Off J Control Release Soc. (2017) 260:142–53. doi: 10.1016/j.jconrel.2017.06.005
329. Wu C, Feng D, Xu H, He Z, and Hou J. Optimized bionic drug-delivery-inducing immunogenic cell death and cGAS-STING pathway activation for enhanced photodynamic-chemotherapy-driven immunotherapy in prostate cancer. ACS Appl Mater Interfaces. (2024) 16:43257–71. doi: 10.1021/acsami.4c07072
330. Yin M, Dong J, Sun C, Liu X, Liu Z, Liu L, et al. Raddeanin a enhances mitochondrial DNA-cGAS/STING axis-mediated antitumor immunity by targeting transactive responsive DNA-binding protein 43. Adv Sci Weinh Baden-Wurtt Ger. (2023) 10:e2206737. doi: 10.1002/advs.202206737
331. Guo Y, Li Y, Zhang M, Ma R, Wang Y, Weng X, et al. Polymeric nanocarrier via metabolism regulation mediates immunogenic cell death with spatiotemporal orchestration for cancer immunotherapy. Nat Commun. (2024) 15:8586. doi: 10.1038/s41467-024-53010-0
332. Gao X, Xu YX, Janakiraman N, Chapman RA, and Gautam SC. Immunomodulatory activity of resveratrol: Suppression of lymphocyte proliferation, development of cell-mediated cytotoxicity, and cytokine production. Biochem Pharmacol. (2001) 62:1299–308. doi: 10.1016/s0006-2952(01)00775-4
333. Zhao M, Li J, Liu J, Xu M, Ji H, Wu S, et al. Charge-switchable nanoparticles enhance cancer immunotherapy based on mitochondrial dynamic regulation and immunogenic cell death induction. J Control Release Off J Control Release Soc. (2021) 335:320–32. doi: 10.1016/j.jconrel.2021.05.036
334. Jain S, Hu C, Kluza J, Ke W, Tian G, Giurgiu M, et al. Metabolic targeting of cancer by a ubiquinone uncompetitive inhibitor of mitochondrial complex I. Cell Chem Biol. (2022) 29:436–450.e15. doi: 10.1016/j.chembiol.2021.11.002
335. Feng Q, Xu J, Zhuang C, Xiong J, Wang H, and Xiao K. Mitochondria-targeting and multiresponsive nanoplatform based on AIEgens for synergistic chemo-photodynamic therapy and enhanced immunotherapy. Biomacromolecules. (2023) 24:977–90. doi: 10.1021/acs.biomac.2c01416
336. Peng M, Dong H, Shao M, Zhang X, Sun J, Ding C, et al. Self-heating mitochondrion-induced free radical blast for immunogenic cell death stimulation and HCC immunotherapy. J Control Release Off J Control Release Soc. (2024) 366:694–711. doi: 10.1016/j.jconrel.2024.01.022
337. Chen H, Li T, Liu Z, Tang S, Tong J, Tao Y, et al. A nitric-oxide driven chemotactic nanomotor for enhanced immunotherapy of glioblastoma. Nat Commun. (2023) 14:941. doi: 10.1038/s41467-022-35709-0
338. Sharma A, Liaw K, Sharma R, Thomas AG, Slusher BS, Kannan S, et al. Targeting mitochondria in tumor-associated macrophages using a dendrimer-conjugated TSPO ligand that stimulates antitumor signaling in glioblastoma. Biomacromolecules. (2020) 21:3909–22. doi: 10.1021/acs.biomac.0c01033
339. Cullen JK, Yap P-Y, Ferguson B, Bruce ZC, Koyama M, Handoko H, et al. Tigilanol tiglate is an oncolytic small molecule that induces immunogenic cell death and enhances the response of both target and non-injected tumors to immune checkpoint blockade. J Immunother Cancer. (2024) 12:e006602. doi: 10.1136/jitc-2022-006602
340. Zhang L, Zhao J, Hu X, Wang C, Jia Y, Zhu C, et al. A peritumorally injected immunomodulating adjuvant elicits robust and safe metalloimmunotherapy against solid tumors. Adv Mater Deerfield Beach Fla. (2022) 34:e2206915. doi: 10.1002/adma.202206915
341. Marrache S, Tundup S, Harn DA, and Dhar S. Ex vivo generation of functional immune cells by mitochondria-targeted photosensitization of cancer cells. Methods Mol Biol Clifton NJ. (2015) 1265:113–22. doi: 10.1007/978-1-4939-2288-8_9
Keywords: mitochondria targeted, triphenylphosphonium, TME, cancer, immunotherapy
Citation: Cheng X, Wang Y, Johnson B and You M (2025) Mitochondria-targeted strategies in tumor immunity. Front. Immunol. 16:1646138. doi: 10.3389/fimmu.2025.1646138
Received: 12 June 2025; Accepted: 14 October 2025;
Published: 30 October 2025.
Edited by:
Xianhuo Wang, Tianjin Medical University Cancer Institute and Hospital, ChinaReviewed by:
Dipendra Khadka, Purbanchal University, NepalBei Zhang, Southern Methodist University, United States
Copyright © 2025 Cheng, Wang, Johnson and You. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ming You, bXlvdUBob3VzdG9ubWV0aG9kaXN0Lm9yZw==