 Saleh Alrhmoun1,2*
Saleh Alrhmoun1,2* Roman Perik-Zavodskii1,2
Roman Perik-Zavodskii1,2 Marina Fisher1,2
Marina Fisher1,2 Julia Lopatnikova1,2
Julia Lopatnikova1,2 Olga Perik-Zavodskaia1
Olga Perik-Zavodskaia1 Julia Shevchenko1,2
Julia Shevchenko1,2 Kirill Nazarov1
Kirill Nazarov1 Julia Philippova1
Julia Philippova1 Vasily Kurilin1
Vasily Kurilin1 Olga Kichakova3
Olga Kichakova3 Evgenii Zavjalov3
Evgenii Zavjalov3 Elena Golikova2Petr Timashev2Petr Glybochko2
Elena Golikova2Petr Timashev2Petr Glybochko2 Sergey Sennikov1,2*
Sergey Sennikov1,2*- 1Laboratory of Molecular Immunology, Research Institute of Fundamental and Clinical Immunology, Novosibirsk, Russia
- 2Federal State Autonomous Educational Institution of Higher Education, I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation, Sechenov University, Moscow, Russia
- 3The Center for Genetic Resources of Laboratory Animals, ICG SB RAS, Novosibirsk, Russia
Introduction: T cell receptor-engineered T cell therapy has emerged as a promising approach in cancer immunotherapy, leveraging the ability of T cells to recognize tumor antigens presented on major histocompatibility complex molecules, offering a targeted approach for treating cancers. This study advances previous research conducted at the Laboratory of Molecular Immunology at RIFCI, where the full repertoire of HER2/neu-specific TCRs was identified. Specifically, here we are functionally validating a distinct TCR clonotype targeting the KIFGSLAFL peptide of HER2/neu protein presented by the HLA-A*02.
Methods: We employed an integrated approach combining in vitro cytotoxicity assays, single-cell RNA sequencing via BD Rhapsody, secretome profiling via LegendPlex, and in vivo HER2/neu-expressing xenograft models in SCID mice.
Results: Anti-HER2/neu TCR-T cells exhibited robust antigen-specific cytotoxicity in vitro, preferentially targeting tumor cells with high HER2/neu expression. Single-cell RNA sequencing revealed a unique double-positive (CD4+CD8+) T cell population emerging upon antigen engagement, characterized by a cytotoxic transcriptome with elevated granzyme B, granulysin, perforin, and TNF-α gene expression. Secretome profiling confirmed significantly enhanced production of effector molecules, including IL-2, granzyme B, TNF-α, and IFN-γ, supporting potent T cell activation and function. In vivo, anti-HER2/neu TCR-T cells achieved sustained and significant suppression of tumor growth in HER2/neu-expressing xenograft models, underscoring their therapeutic potential.
Discussion: These findings validate the broader utility of the previously identified HER2/neu-specific TCR repertoire and elucidate the molecular mechanisms driving its therapeutic efficacy, demonstrating the potential of TCR-T cells for treating solid tumors through robust cytotoxic activity and the emergence of a favorable CD4+CD8+ T cell population. This study offers critical mechanistic insights, establishing a foundation for advancing TCR-engineered therapies toward clinical use in HER2/neu-positive cancers.
1 Introduction
T cell receptor (TCR)-engineered T cell therapy represents a cutting-edge approach in cancer immunotherapy, leveraging genetic modification to equip T cells with TCRs that specifically recognize tumor antigens presented on major histocompatibility complex (MHC) molecules. This strategy enables precise targeting of malignant cells, offering a promising alternative to conventional treatments (1). Clinical trials targeting antigens such as NY-ESO-1 and MAGE-A4 have demonstrated significant therapeutic potential in solid tumors and hematological malignancies (2, 3). However, challenges, including inconsistent cytotoxic efficacy and tumor immune escape mechanisms, underscore the need for a deeper exploration of TCR T cell functionality to optimize clinical outcomes (4).
The success of TCR-T cell therapy relies on the cytotoxic potency and functional adaptability of engineered T cells across varied immunological settings. While in vitro assays offer valuable controlled insights into T-cell effector functions, in vivo studies reveal the intricate dynamics of the tumor microenvironment (TME) (5). Despite significant progress, the molecular factors governing TCR-T cell cytotoxicity, particularly the interplay between intrinsic T-cell signaling and extrinsic TME-driven suppression, remain insufficiently characterized, underscoring the need for integrated experimental strategies to address these unresolved challenges (6).
Recent advancements in single-cell RNA sequencing (scRNA-seq) have transformed the study of immune cell dynamics, enabling high-resolution analysis of transcriptional heterogeneity within T cell populations. These technological improvements allow for a more precise characterization of distinct T cell subsets, their functional states, and clonal evolution. Such insights are critical for understanding immune responses in both health and disease, and have already begun to inform the development of more effective immunotherapeutic strategies (7–13). By profiling TCR T cells at the moment of target cell engagement, scRNA-seq can elucidate the molecular pathways underpinning cytotoxic mechanisms, such as granzyme/perforin release, Fas/FasL interactions, and cytokine production (14). This approach holds immense potential to identify novel therapeutic targets and biomarkers that enhance the antitumor efficacy of TCR T cells.
In our previous work (15), we developed a comprehensive pipeline to identify and characterize naturally occurring TCRs specific to the HER2/neu tumor-associated antigen, aiming to enhance the development of TCR-T cell therapies for solid tumors. The process began with the generation of mature dendritic cells (DCs) from peripheral blood mononuclear cells, which were then pulsed with immunogenic peptides derived from HER2/neu. These antigen-loaded DCs effectively primed autologous CD8+ T cells, leading to a remarkable expansion of antigen-specific T cells. Following that, Flex-T™ technology (BioLegend, USA) was employed to isolate HER2/neu-specific CD8+ T cells based on their TCR-mediated recognition of the HER2/neu peptide–MHC complex, ensuring high purity of antigen-reactive clones. Subsequently, we employed scRNA-seq to analyze the expanded T cell repertoire, enabling the identification of over 100 unique TCR clonotypes specific to HER2/neu. To further characterize these clonotypes, we utilized the TCRscape tool (16), an open-source, Python-based bioinformatics toolkit optimized for BD Rhapsody™ multiomic datasets. This platform integrates full-length TCR sequence data with gene expression and surface-protein information, enabling multimodal clustering, clonotype quantification, and V(D)J gene usage profiling. Through this integrative analysis, TCRscape facilitates the identification of dominant TCR clonotypes, providing comprehensive insights into the structural and functional diversity of the antigen-specific T-cell repertoire. Overall, this workflow establishes a robust platform for the discovery and development of effective TCRs for adoptive cell therapy targeting HER2/neu and other tumor-associated/specific antigens. This study advances beyond the proof-of-concept stage to provide a comprehensive understanding of the antitumor immune response during tumor cell engagement. By functionally validating a distinct clonotype, we confirm the broader utility of the previously identified TCR repertoire and reveal its functionality, molecular mechanisms, and therapeutic efficacy through integrated in vitro and in vivo analyses, supported by scRNA-seq and secretome profiling. Importantly, the TCRs target the immunogenic KIFGSLAFL (KIF, spanning amino acids 369–377 of the HER2/neu protein) peptide, which has been shown to elicit strong CD8+ T-cell responses, confirming its immunogenicity (17). This peptide is presented on the HLA-A02 molecule, limiting its therapeutic application to individuals with this genotype. However, the widespread prevalence of HLA-A02 as the most frequent class I HLA genotype across nearly all human populations (18) makes it a practical choice for therapies that could help a large number of patients. These findings not only enhance our understanding of TCR-mediated antitumor immunity but also pave the way for the rational design of next-generation immunotherapies with improved therapeutic strategies.
In this work, we made the next step in our anti-HER2/neu T Cell Receptor analysis journey by employing a tumor cytotoxicity assay, single-cell RNA sequencing, bulk secretomics, and in vivo analysis of anti-HER2/neu TCR T Cells in a mouse model.
2 Materials and methods
2.1 Production of HER2/neu-specific TCR-T cells
One day before initiating the experiment, the wells of a 24-well plate were coated with Retronectin (25 µg/mL, Sci Store, Russia) and anti-CD3 antibodies (5 µg/mL, BioLegend) in ACDA buffer (415 µL per well) and incubated overnight. Peripheral blood was drawn from HLA-A*02-positive conditionally healthy donors into EDTA-coated vacuum tubes. Peripheral blood mononuclear cells (PBMCs) were then isolated by Ficoll™ (PanEco, Russia) density gradient centrifugation at 400 × g for 40 minutes at room temperature, with the buffy coat layer subsequently collected. All the donors signed an informed consent to participate in the study. From these PBMCs, CD3+ T cells were purified using the MojoSort™ Human CD3 Negative Magnetic Selection Kit (480134, BioLegend, USA) according to the manufacturer’s instructions. The isolated CD3+ T cells (0.75 × 10^6 cells/mL) were seeded into the pre-coated 24-well plates and activated with IL-2 (300 U/mL, Biotech LLC, Russia) for 48 hours.
On the day preceding transduction, an additional 24-well plate was prepared by coating the wells with Retronectin (25 µg/mL) in ACDA buffer (255 µL per well). Following the 48-hour activation period, the T cells were harvested, centrifuged at 350 g for 10 minutes, resuspended at a concentration of 4 × 105 cells/mL in RPMI-1640 serum-free medium, and counted. The cells were then split into two distinct groups: transduced T cells (anti-HER2/neu T cells) and non-transduced T cells (LV-neg T cells). Subsequently, 2 × 105 cells from each group were transferred into separate wells of the Retronectin-coated plate in a volume of 500 µL. For the transduced group, lentiviral particles encoding the anti-HER2/neu TCR (2 × 105 particles per well) at a Multiplicity Of Infection (MOI) = 1, as previously optimized (15), and protamine sulfate (5–8 µg/mL) (19) were added to facilitate T cell transduction. The plate was then centrifuged at 600 × g for 2 hours at 32°C. Post-centrifugation, 500 µL of warm complete RPMI-1640 medium containing IL-2 (final concentration 300 U/mL) was added, and the cells were incubated overnight. The next morning, the cells were transferred to 12-well plates with an equal volume of complete medium supplemented with IL-2 (300 U/mL). Cell growth was observed regularly, and every two days, partial media replacement was performed with the addition of fresh IL-2. Throughout the culture period, cell growth, conglomerate formation, and the cell culture medium state were visually monitored. Seven days post-transduction, the cells were collected, centrifuged at 350 × g for 10 minutes, and the cell count and viability were assessed in a counting chamber with trypan blue staining, transduction efficiency was also evaluated using Flex-T tetramers loaded with the KIFGSLAFL peptide as previously described (15), having positive cells stained simultaneously with two fluorochromes for their identification (20).
2.2 Assessment of TCR T cell cytotoxicity In Vitro
To evaluate TCR T cell cytotoxic activity against tumor cell lines, we employed the LDH assay (J2381, Promega, Madison, WI, USA). Tumor cells in the logarithmic growth phase were harvested using a 1:3 mixture of 0.25% trypsin (PanEco, Russia) and Versen solution (Vector, Russia). The harvested tumor cells were seeded into a 96-well flat-bottom plate at a density of 5,000 cells per well. Two to three hours later, transduced and non-transduced T cells were added at 50,000 cells per well, establishing an effector-to-target ratio of 10:1. The co-culture was incubated for 16–18 hours in a medium supplemented with 5% Fetal Calf Serum (FCS).
Forty-five minutes before the end of the incubation period, 10 µL of 10X lysing solution (J2381, Promega, Madison, WI, USA) was added per 100 µL of cell suspension to the control wells (tumor cells) to account for the maximum release of lactate dehydrogenase (LDH) from the cells. After completing the lysis, the plate was centrifuged at 250 × g for 4 minutes to gently pellet the cells. Subsequently, 50 µL aliquots from each well were transferred to a fresh 96-well flat-bottom plate for the immunoassay. To each well containing cell culture supernatants, 50 µL of reconstituted LDH enzyme-substrate mixture (J2381, Promega, Madison, WI, USA) was added. The plate was covered with foil or an opaque cover slip to shield it from light and incubated at room temperature for 30 minutes. The reaction was then terminated by adding a 1M acetic acid solution, and the optical density was measured immediately at 492 nm. Cytotoxic activity was calculated using the following formula:
The averaged per-sample cytotoxicity data were analyzed in GraphPad Prism version 10.2.3 using the Kruskal-Wallis test with Dunn’s correction for multiple comparisons and visualized using ggplot2 in R.
2.3 Single-cell analysis
2.3.1 Sample Tag cell staining and counting
Tumor cells in the logarithmic growth phase were harvested using a 1:3 mixture of 0.25% trypsin (PanEco, Russia) and Versen solution (Vector, Russia) and seeded into 96-well flat-bottom plates at a density of 40,000 cells per well. After 2–3 hours, transduced TCR-T cells and non-transduced T cells were added at 400,000 cells per well, achieving an effector-to-target (E:T) ratio of 10:1. The co-culture was incubated for 16–18 hours in medium supplemented with 5% fetal calf serum (FCS). Following incubation, both TCR-T cells and non-transduced T cells were collected via gentle pipetting. While this process may retain residual tumor cells, such contamination can be computationally excluded during downstream data analysis. Collected TCR-T cells (n = 4) and non-transduced T cells (n = 4) were then incubated for 20 minutes at room temperature with Sample Tag antibodies from the BD™ Single-Cell Multiplexing Kit (633781, BD Biosciences, USA) to barcode individual samples. After three washing cycles, cells were stained with Calcein (BD Biosciences, USA) as outlined in the BD Rhapsody Single-Cell Analysis System User Guide. Viable Calcein-positive cells were quantified using the Attune NxT flow cytometer (Thermo Fisher, USA), measured as events/μL. Subsequently, cells were pooled in equal proportions and resuspended in cold sample buffer to achieve a final concentration of 10 cells/μL and a volume of 620 µl for loading onto a BD Rhapsody Cartridge. The number of loaded cells was confirmed visually using the In Cell Analyzer 6000 (GE Healthcare, USA), calculated as the mean Calcein-positive cells across 5 fields of view (FOV), divided by 175 (microwells per FOV), and multiplied by 200,000 (total microwells per cartridge).
2.3.2 cDNA library preparation and sequencing
Single-cell capture and cDNA library preparation were performed using the BD Rhapsody Express Single-Cell Analysis System (BD Biosciences, USA), adhering to the manufacturer’s instructions (mRNA Targeted and Sample Tag Library Preparation Protocol) for targeted gene expression analysis with the BD Rhapsody™ Onco-BC Panel HS (633752, BD Biosciences, USA), a specialized panel targeting key oncology and immune-related genes in TCR-T cells, enabling precise analysis of their cytotoxic and activation states following tumor cell co-culture. Single cells were captured in the BD Rhapsody cartridge, followed by the addition of magnetic beads for poly-A-based mRNA capture, alongside Sample Tags for sample barcoding. After cell lysis, reverse transcription of the poly-A captured mRNA and Sample Tag was performed on the magnetic beads. Following that, the beads were treated with Exonuclease I and the on-bead cDNA was amplified using Sample Tag primer and the Onco-BC Panel HS (mRNA for short) primers. The resulting targeted mRNA PCR1 products and Sample Tag PCR1 products were then collected and subsequently purified using AMPure XP Beads (A63880, Beckman Coulter, USA) to remove primer dimers and small molecular weight by-products. The purified mRNA and Sample Tag PCR1 products were further amplified in a semi-nested PCR2 to increase the specificity of the transcript detection, and the resulting PCR2 products were purified using AMPure XP magnetic beads. Finally, the PCR2 product concentrations were assessed by Qubit 4 with the Qubit dsDNA High-Sensitivity Assay Kit (Q32854, Thermo Fisher, USA) and normalized to 5.5 ng/μL for the mRNA panel library and 3.0 ng/μL for the Sample Tag library to perform a final round of amplification using indexes for the Illumina sequencer to prepare the final libraries. The final libraries were then purified using AMPure XP Beads, quantified using Qubit 4, and quality was evaluated via Qsep1 capillary electrophoresis with the S2 Cartridge (Bioptic, China). Then the libraries were pooled (~94/6% mRNA/Sample Tag ratio, estimated read/cell: 20,000 (mRNA, deep sequencing read count quantity) and 1200 (Sample Tag)) to the final concentration of 5 nM. The final pooled libraries were sequenced (R1 = 51, R2 = 51, 700 million reads, SP flow cell) on a NovaSeq 6000 sequencer (Illumina, San Diego, California, USA).
2.3.3 Sequencing data processing
FASTQ files obtained from sequencing were processed using the BD Rhapsody pipeline v2.0 (BD Biosciences, USA). Low-quality read pairs were filtered based on read length, mean base quality score, and highest single-nucleotide frequency. High-quality R1 reads were analyzed to identify cell labels and unique molecular identifiers (UMIs). R2 reads were aligned to the BD Rhapsody™ Onco-BC Panel HS reference using Bowtie2. Reads sharing the same cell label, UMI, and gene were collapsed into single molecules, and UMI counts were adjusted using recursive substitution error correction (RSEC) and distribution-based error correction (DBEC) to correct for sequencing and PCR errors. Cell counts were determined via second derivative analysis to eliminate noise cell labels, where only cell labels beyond a single observed inflection point were considered valid. Sample demultiplexing and multiplet filtering were achieved using the Sample Tags. The pipeline identified single cells across the BD Rhapsody cartridges and produced gene expression matrices for each sample. Sequencing metrics confirmed high saturation and adequate depth for the targeted panel analysis.
2.3.4 Sequencing data analysis
We subjected the obtained gene expression matrices to the Seurat single-cell analysis pipeline, we performed quality control and data normalization via the LogNormalize() function, conducted PCA (principal component analysis) dimensionality reduction on the normalized data, performed Harmony batch correction and integration, used 20 Harmony-corrected principal components for UMAP dimensionality reduction, and found single-cell neighbors and clusters. We then identified T cell clusters using their canonical markers. We performed inter-cluster differential gene expression using the Wilcoxon signed-rank test. We set biological and statistical significance criteria as log2(FoldChange) > 1.0 or log2(FoldChange)< −1.0 and q-value< 0.05 using the FindMarkers() function.
2.4 Cell culturing
SK-Mel-37 cells were seeded at a density of 10,000 cells per well, while transduced and non-transduced T cells were added at 100,000 cells per well, in a total volume of 100 µL within a 96-well plate. The co-culture in complete RPMI-1640 medium was maintained at 37°C in a humidified atmosphere with 5% CO8. After 16–18 hours of co-culture, the conditioned medium was harvested by centrifuging the 96-well plate at 1,500 rpm for 10 minutes to pellet the cells. The supernatant was gently transferred into fresh 1.5 mL tubes, and bovine serum albumin (BSA) was added to a final concentration of 0.5%. The samples were subsequently stored at −80°C pending further analysis.
2.5 Cytokine quantification
Cytokine concentrations in the conditioned media were determined using the Human CD8/NK Panel with LegendPlex assay (Cat. No. 741187, BioLegend, USA) following the manufacturer’s instructions. Briefly, 25 µL of each sample was sequentially incubated with capture beads, detection antibodies, and streptavidin-phycoerythrin (SA-PE). The samples were analyzed on the Attune NxT flow cytometer (Thermo Fisher, USA).
The data from the LegendPlex assay were log2-transformed and Z-score standardized using Pandas. To evaluate differential cytokine production, multiple T-tests with false discovery rate (FDR) correction were performed. Statistical significance was established with a fold change > 1 or< −1 and a q-value< 0.01. Volcano plot and PCA plot were generated using BulkOmicsTools (21) to visualize differential cytokine expression, and a heatmap of cytokine concentrations was constructed using Bioinfokit (22) to depict secretion profiles.
2.6 In vivo TCR T cell cytotoxicity assessment
The immunodeficient mouse strain SCID (Severe combined immunodeficient) was used for the establishment of human tumor xenografts. Male and female mice, aged 8 weeks with Specific Pathogen Free (SPF) status, were housed in same-sex groups of 2–5 in individually ventilated cages (IVC) of the Opti Mice system (Animal Care Systems). Housing conditions were maintained at a temperature of 21–24°C, relative humidity of 30–50%, and a 12/12-hour light/dark cycle with lights on at 02:00. Mice had unlimited access to Ssniff diet (Germany) and reverse osmosis water supplemented with a mineral mixture.
Two to three weeks before the initiation of the experiment, SK-MEL-37 tumor cells were thawed and cultured for 4–5 passages in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS, Invitrogen) at 37°C in plastic culture flasks (TPP, Switzerland). On the day of transplantation, cells were detached using a trypsin/versene solution, centrifuged at 1,000 rpm for 5 minutes, and resuspended in serum-free medium. Xenografts were established by injecting 100 µL of the cell suspension subcutaneously into the right shoulder region of the mice.
Experimental therapy was initiated one week after tumor cell injection, by which time the average tumor volume had reached approximately 100 mm³. Mice were divided into three groups: a treatment group receiving anti-HER2/neu TCR-engineered T cells (HER2/neu), a control group receiving non-transduced T cells, and an untreated control group. Transduced and non-transduced T cells were administered locally in the vicinity of the developing tumor node, in a volume of 50 µL per injection, and the tumor volume was monitored for 45 days thereafter.
All procedures adhered to the principles of humane animal treatment as outlined in the European Community directive (86/609/EEC). Mice were monitored every 2–3 days over the 45-day experimental period, with assessments focusing on skin condition, motor activity, and behavioral changes. Tumor volumes in the different groups were measured at each assessment using calipers, and volumes were calculated using the formula (), where (a) represents the length and (b) the width. Mice exhibiting severe toxicity indicators (e.g., hunched posture, reduced activity), body weight loss exceeding 20%, or excessive xenograft volumes were humanely euthanized following ethical standards. Planned euthanasia was conducted via CO2 overdose followed by cervical dislocation at the end of the 45 days or earlier if necessary.
3 Results
3.1 In Vitro antitumor assessment demonstrates potent cytotoxicity and specific targeting of TCR-T cells against HER2/neu-expressing tumors
To assess the antigen-specific recognition and cytotoxic potential of anti-HER2/neu TCR-engineered T cells, we performed an in vitro tumor antigen stimulation experiment. T cells transduced with the HER2/neu-specific TCR, alongside non-transduced T cells (LV-neg), were co-cultured with four cancer cell lines expressing varying levels of HER2/neu. The selection of these cell lines was guided by previous reports describing their relative HER2 expression levels and was further confirmed by our own flow cytometric analysis, (See Supplementary Figure 1), using the PE anti-human CD340 (erbB2/HER-2) Antibody (Cat. No. 324406, BioLegend, USA). MDA-MB-231 cells, widely characterized as HER2-negative or HER2-low (23–25), showed low HER2 expression limited to a very small subset of the population in our analysis and therefore served as a negative control. HCT-116 cells, representing non-amplified colorectal carcinoma with baseline HER2 expression (26–28), showed uniform but moderate HER2 expression and therefore were classified as moderate expressers. As for the SK-MEL-5 and SK-MEL-37 melanoma lines, peer-reviewed publications explicitly reporting HER2 surface expression by flow cytometry or other methods are limited, public proteomic datasets and cell-line databases indicate detectable HER2 protein levels in SK-MEL-5 (29, 30), and some studies have shown that a subset of melanoma cell lines can express HER2 (29–31). Based on this contextual evidence we used our validated flow-cytometry measurements as the primary evidence for classifying these lines, as both SK-MEL-5 and SK-MEL-37 exhibited markedly higher mean fluorescence intensities and were therefore designated as high and very high HER2 expressers (HER2/neu+++ and HER2/neu++++, respectively).
Cytotoxicity was measured after 16–18 hours of co-culture using the LDH cytotoxicity assay, which quantifies the percentage of lysed target cells. The results revealed a statistically significant difference in cytotoxicity against SK-MEL-5 and SK-MEL-37 compared to HCT-116, MDA-MB-231, and the non-transduced T-cells co-cultured with these tumor cell lines (Figure 1). Specifically, anti-HER2/neu TCR T cells induced 61 ± 19% lysis of SK-MEL-5 and SK-MEL-37 cells, compared to the 16 ± 10% cell lysis for HCT-116, MDA-MB-231, and the non-transfected T-cells co-cultured with these tumor cell lines representing a 3.7-fold cell lysis difference (mean ± SD, n = 14 with 3 technical replicates).
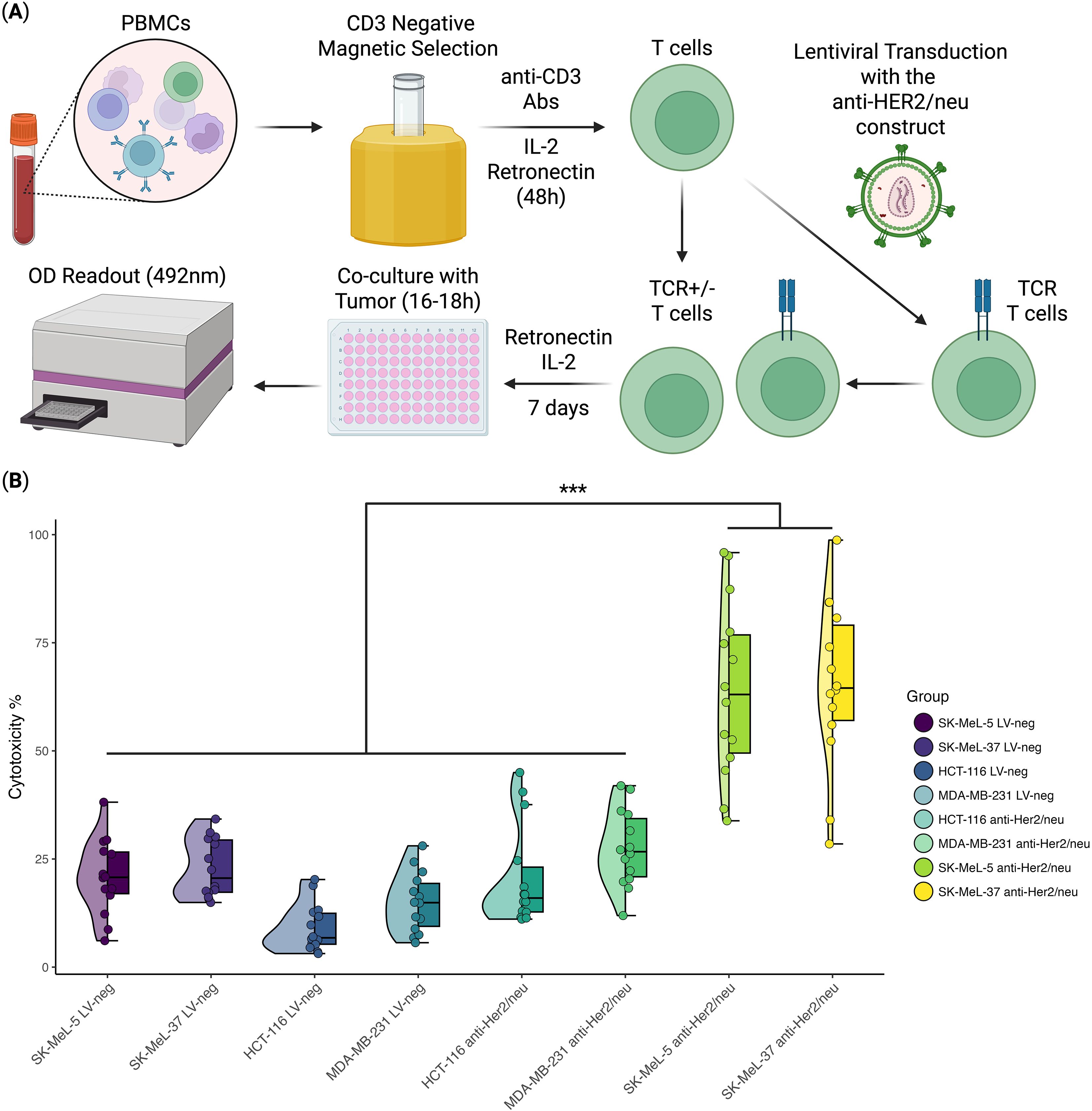
Figure 1. In Vitro anti-tumor cytotoxicity analysis of anti-HER2/neu TCR T cells (n = 14, with 3 technical repeats). (A) Schematic overview of the experimental design; (B) Violin/Box plot representing the percentage of tumor cell death in the HER2/neu-highly expressing cell lines (SK-MEL-5 and SK-MEL-37), HER2/neu-moderately expressing cell line (HCT-116), and the cell line with minimal HER2/neu expression (MDA-MB-231). The horizontal lines within the boxes represent median values. *** - Adjusted p-value< 0.0005.
3.2 scRNA-seq analysis reveals differentiation of HER2/neu-specific TCR-T cells into a cytotoxic CD4+CD8+ effector subset
To evaluate the molecular mechanisms involved in the implementation of the antitumor immune response and to identify the pathways of cytotoxicity realization of anti-HER2/neu TCR-engineered T cells at the moment of target cell engagement, we assessed their transcriptome in comparison to that of non-transduced T cells (LV-neg), following co-culture with HER2/neu-expressing SK-Mel-37 tumor cells (Figure 2). The scRNA-seq analysis revealed that all the transduced T cells differentiated into a distinct population of double-positive (CD4+CD8+) T cells (Figure 2B), a subset previously associated with enhanced effector functions in immunotherapy contexts (32–37). This double-positive population exhibited a pronounced cytotoxic phenotype, characterized by significantly elevated expression of key effector molecules like granzyme B (GZMB), granulysin (GNLY), perforin 1 (PRF1), and the pro-inflammatory cytokine tumor necrosis factor-alpha (TNF) (Figure 2C).
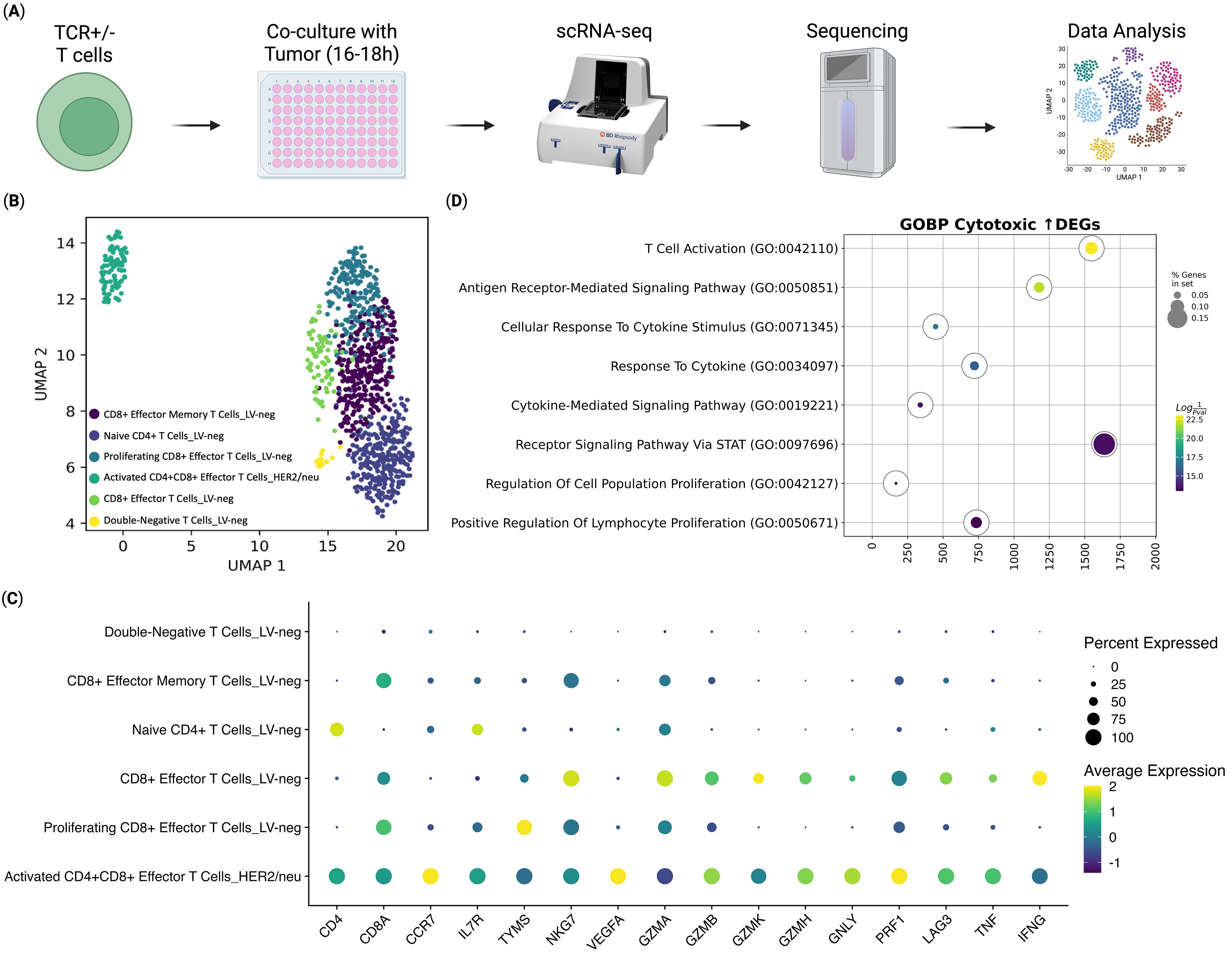
Figure 2. Transcriptomic Analysis of ani-HER2/neu T Cells Co-Cultured with Sk-Mel-37 Tumor Cells (n = 4). (A) Overview of the experiment, (B) UMAP Plot of the T cell clusters, (C) Dot plot of the gene expression per T cell cluster, (D) Gene Set Enrichment Analysis of differentially expressed genes between the anti-HER2/neu-transduced T cells and CD8+ Effector T cells from the LV-neg group.
In addition to their cytotoxic phenotype, the transduced T cells displayed molecular signatures indicative of successful antigen recognition and T-cell activation. Gene Set Enrichment Analysis (GSEA) of up-regulated genes of the transduced cells compared to the CD8 Effector T Cells from the non-transduced group, revealed significant upregulation of gene sets associated with T cell activation (Figure 2D). This included genes linked to antigen recognition and downstream signaling cascades, such as those involved in T cell receptor signaling and co-stimulatory pathways, confirming effective engagement with the HER2/neu antigen (38).
3.3 Secretome analysis highlights enhanced effector molecule secretion and broad immune activation in HER2/neu-specific TCR-T cells
To further characterize the functional properties of TCR-engineered T cells and to elucidate their effector molecule secretory profile, a critical indicator of their antigen-specific activation and antitumor potential, we analyzed their secretome compared to non-transduced T cells (LV-neg) following 16–18 hours of co-culture with HER2/neu-expressing SK-Mel-37 tumor cells, using the LEGENDplex multiplex cytokine profiling assay. Our analysis revealed a significant increase in the production of key effector molecules in anti-HER2/neu TCR T cells compared to LV-neg controls. Specifically, TCR T cells exhibited elevated secretion of interleukin-2 (IL-2), granzyme B, tumor necrosis factor-alpha (TNF-α), and interferon-gamma (IFN-γ), in anti-HER2/neu TCR T cells compared to LV-neg controls (Figure 3).
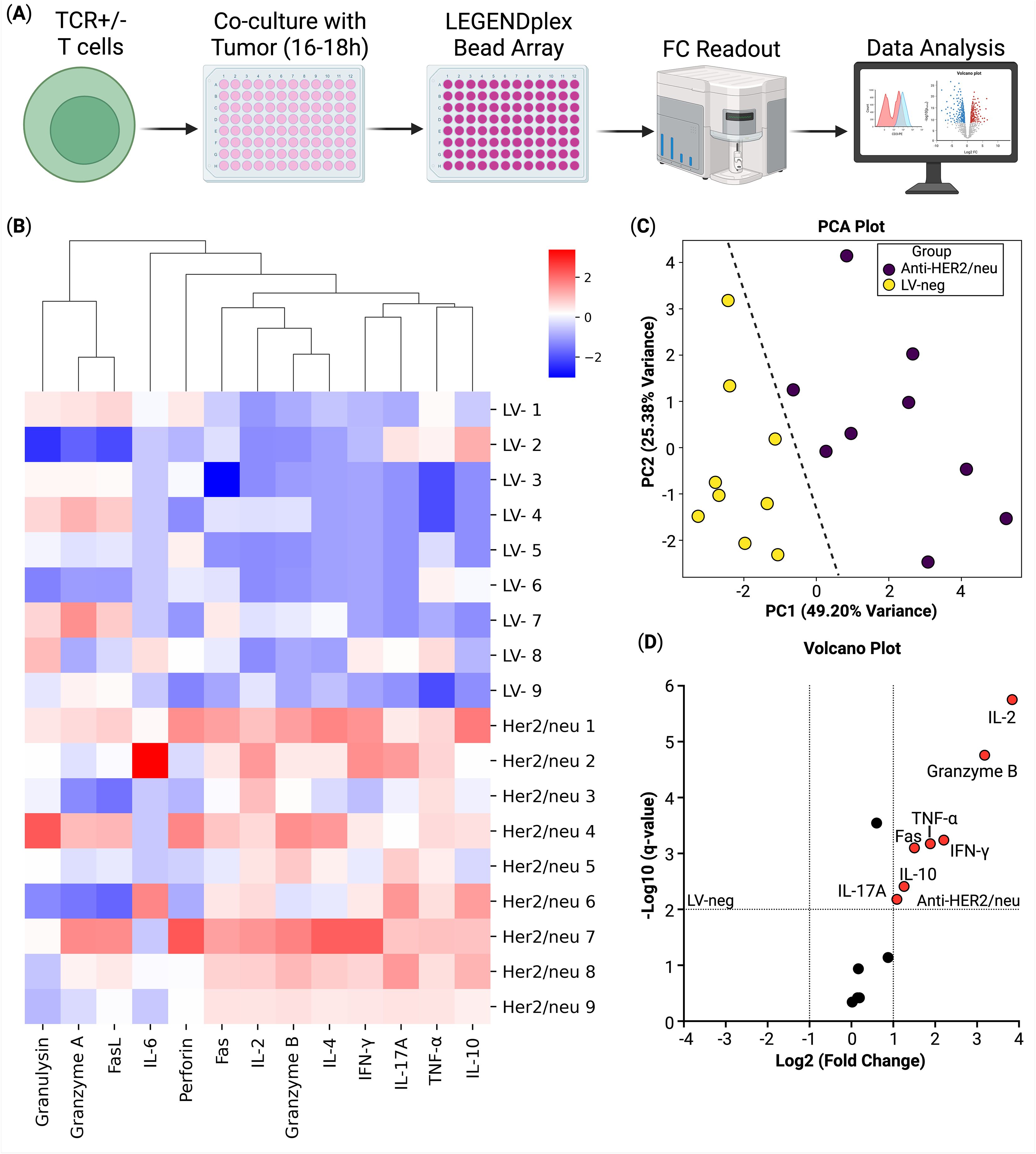
Figure 3. Secretome analysis of T cells co-cultured with Sk-Mel-37 tumor cells (n = 9). (A) Overview of the experiment, (B) Heatmap of cytokine production, (C) Principal component analysis of the samples, biogroups are split by a line; (D) Volcano plot of the differential cytokine secretion between the anti-HER2/neu T cells and the non-transduced T cells (LV-neg).
3.4 In Vivo antitumor assessment demonstrates sustained tumor suppression by HER2/neu-specific TCR-T cells in xenograft models
To evaluate the in vivo therapeutic efficacy of TCR-engineered T cells, we established a xenograft tumor model in SCID mice by injecting them with HER2/neu-expressing SK-Mel-37 tumor cells. Following tumor establishment, mice were divided into three groups: a treatment group receiving anti-HER2/neu TCR-engineered T cells (HER2/neu), a control group receiving non-transduced T cells (LV-neg), and an untreated control group (Control). Tumor volume was measured over 45 days after the administration of the treatment to assess the impact of TCR T cell therapy on tumor growth.
As the results show, untreated control mice (Control) and mice treated with non-transduced T cells (LV-neg) exhibited rapid tumor progression, with tumor volumes increasing steadily over time. By day 45, the mean tumor volume in the Control group reached approximately 350 mm³, while the LV-neg group showed a similar trajectory, with a mean tumor volume of around 450 mm³ (Figure 4B). In contrast, mice treated with anti-HER2/neu TCR T cells (HER2/neu) demonstrated significant tumor growth suppression throughout the observation period. Tumor volumes in the HER2/neu group remained relatively low, with a mean volume of less than 50 mm³ by day 45, representing a statistically significant reduction compared to both the Control and LV-neg groups (p< 0.05 and p< 0.0001, respectively, Kruskal-Wallis with Dunn’s post-hoc test; Figure 4B) with no statistically significant differences observed between the Control and LV-neg groups. Notably, the HER2/neu group showed minimal tumor growth after day 10, suggesting sustained antitumor activity mediated by the transduced T cells. This marked difference highlights the antigen-specific cytotoxicity of anti-HER2/neu TCR T cells, which effectively recognized and targeted HER2/neu-expressing tumor cells in vivo (2).
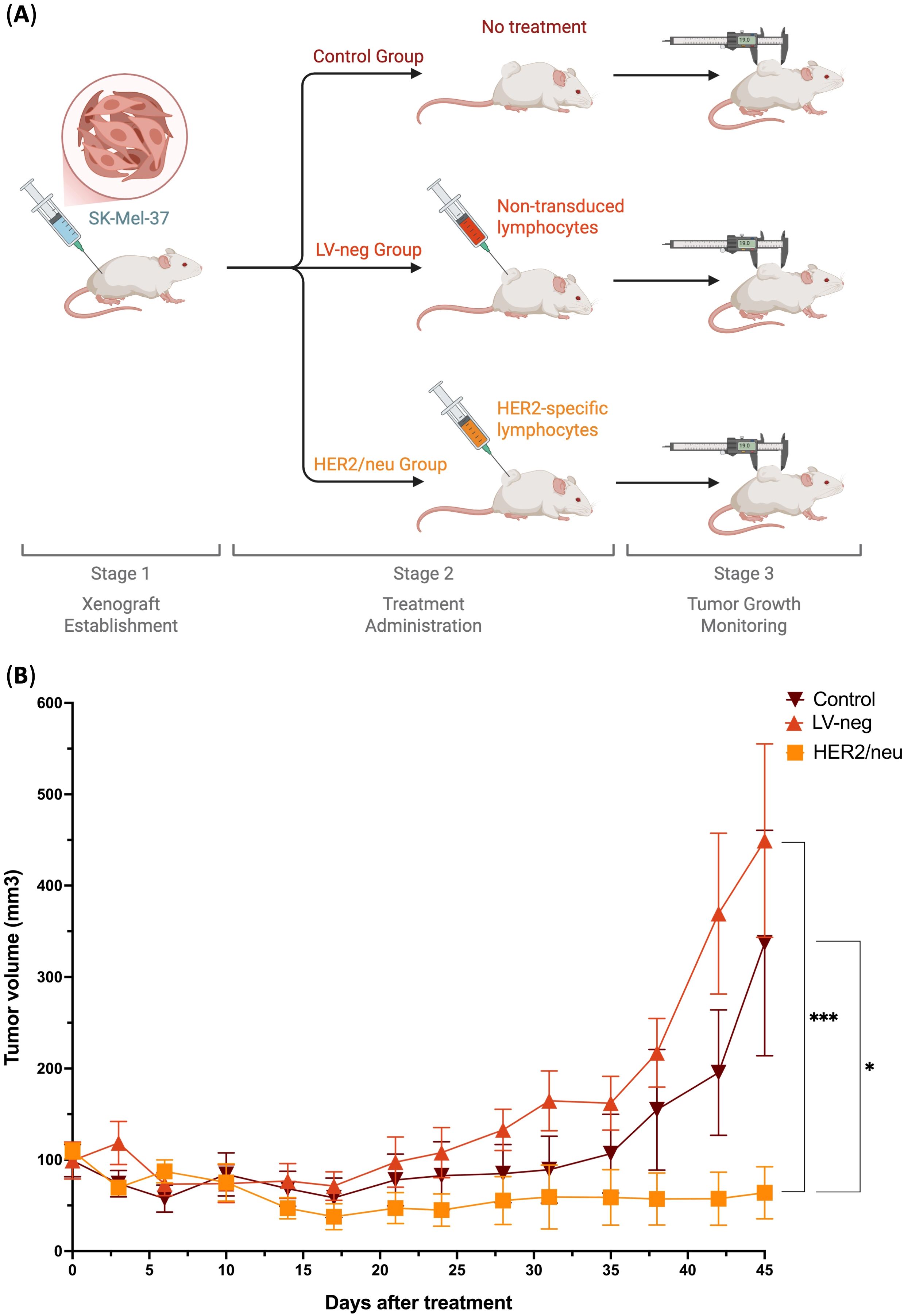
Figure 4. Tumor growth inhibition in a HER2/neu-expressing Xenograft Model by HER2/neu-specific transduced T cells (HER2/neu) compared to non-treated control (Control) and non-transduced T cells (LV-neg) (n = 8). (A) Overview of the experiment, (B) Tumor volume progression across different groups over 45 days. Data represented as Mean and SE; * - adjusted p-value< 0.05, *** - adjusted p-value< 0.0001.
4 Discussion
This study builds upon our previous work, where we analyzed the full repertoire of HER2/neu-specific TCRs (15). In the current investigation, we have advanced this research by functionally validating a distinct TCR clonotype through comprehensive in vitro and in vivo analyses, including single-cell RNA sequencing and secretome profiling. We demonstrate that these engineered TCR-T cells exhibit potent antigen-specific cytotoxicity and sustained tumor control. These findings not only confirm the broader applicability of the previously identified TCR repertoire but also provide deeper insights into the molecular mechanisms underpinning its therapeutic efficacy.
Our findings demonstrate that anti-HER2/neu TCR T cells exhibit antigen-specific cytotoxicity, preferentially targeting tumor cells with high HER2/neu expression while showing reduced activity against cells with lower expression, as evidenced by an in vitro cytotoxicity assay (See Figure 1). This selectivity correlates with the levels of HER2/neu expression on the target tumor cells, which can be 100–200-fold higher than in normal cells (39). These findings demonstrate the antigen specificity and selectivity of the engineered TCR, suggesting a potential therapeutic window for effectively targeting HER2/neu-overexpressing tumor cells while minimizing off-target effects.
At the molecular level, single-cell RNA sequencing revealed that transduced T cells differentiate into a unique double-positive (CD4+CD8+) population upon antigen engagement, exhibiting a pronounced cytotoxic transcriptome characterized by elevated expression of granzyme B, perforin, granulysin, and TNF-α (See Figure 2C). These molecules are critical components of the cytotoxic machinery, with granzyme B and perforin facilitating granule-mediated apoptosis of target cells, granulysin enhancing membrane disruption, and TNF-α contributing to both direct cytotoxicity and immune modulation. Collectively, these results highlight the successful generation of TCR-engineered T cells with enhanced cytotoxic potential and robust antigen-specific activation. The emergence of a double-positive T cell population, previously associated with enhanced effector functions in immunotherapy contexts (14, 40), may confer advantages in TCR T cell therapy by combining the helper functions of CD4+ T cells with the cytotoxic capabilities of CD8+ T cells that may contribute to superior antitumor activity. The activation of TCR signaling pathways and co-stimulatory molecule expression further validates the functionality of the engineered TCR T cells, effectively engaging their target antigens, triggering downstream signaling cascades critical for effector function (41). Furthermore, gene set enrichment analysis (GSEA) of differentially expressed genes revealed marked enrichment of pathways related to T cell activation, downstream signaling, and proliferation (See Figure 2D). These molecular insights provide a mechanistic and functional basis for the observed cytotoxicity, suggesting that the perforin-granzyme pathway and cytokine-mediated effects are key contributors to tumor cell killing (42).
Functional analysis of the secretome further corroborated the cytotoxic potential of anti-HER2/neu TCR T cells, showing significant upregulation of IL-2, granzyme B, TNF-α, and IFN-γ upon antigen-specific stimulation compared to non-transduced T cells (See Figure 3D). The elevated secretion of IL-2, a critical cytokine for T cell proliferation and survival, suggests enhanced autocrine and paracrine signaling in transduced T cells, which may sustain their effector functions in the tumor microenvironment (43). Similarly, the increased production of granzyme B, a serine protease integral to cytotoxic granule-mediated apoptosis, underscores the potent cytotoxic capacity of these T cells (42). The upregulation of TNF-α and IFN-γ, both hallmark cytokines of activated T cells, further indicates robust antigen-specific responses, as these molecules contribute to direct tumor cell killing and modulation of the immune microenvironment (44). These findings were corroborated by functional assays, which confirmed that the enhanced secretome correlated with increased cytotoxicity against antigen-expressing target cells (See Figure 1). The consistent enhancement of both transcriptomic and secretome profiles in the anti-HER2/neu TCR T cells suggests that these functional attributes are hallmarks of effective TCR T cells engineering. The robust secretion of these molecules in response to antigen encounter validates the specificity and functionality of the engineered TCRs, highlighting their ability to recognize and respond to HER2/neu antigen with potent effector activity (45).
The in vivo efficacy of anti-HER2/neu TCR T cells was demonstrated in a HER2/neu-expressing xenograft model, where treated mice exhibited significant and sustained tumor growth suppression compared to both non-transduced T cells and untreated controls. The sustained tumor control in the HER2/neu group aligns with our in vitro findings, where anti-HER2/neu TCR T cells exhibited enhanced secretion of effector molecules, as well as increased cytotoxicity against antigen-expressing target cells (See Figures 1, 3). The sustained suppression of tumor progression underscores the therapeutic potential of TCR T cell therapy for HER2/neu-positive cancers, providing a strong foundation for further preclinical and clinical investigations.
Collectively, our findings highlight the therapeutic potential of anti-HER2/neu TCR T cells for adoptive cell therapy. The integration of in vitro and in vivo data provides a comprehensive understanding of their functionality, from antigen recognition and molecular activation to effector function and tumor control. Compared to CAR T cell therapies, which are limited to surface antigens, TCR T cell therapies offer the advantage of targeting intracellular antigens presented by MHC molecules, broadening their applicability across cancer types, especially solid tumors (6, 15).
The current treatment landscape for HER2-positive tumors remains dominated by antibody-based and tyrosine kinase inhibitor (TKI) therapies, including trastuzumab, pertuzumab, and lapatinib (39, 46–48). While these agents have transformed outcomes in HER2-positive breast and gastric cancers, resistance frequently develops through HER2 mutations, receptor shedding, epitope masking, signaling pathway reactivation, or immune evasion mechanisms (46, 49). In this context, TCR-T cells demonstrate a fundamentally broader and more sensitive mode of antigen recognition compared to antibody-based strategies. TCRs detect intracellularly processed peptide–MHC complexes rather than relying on abundant surface proteins, which makes them able to identify tumor cells expressing even low levels of antigen or those that have undergone partial antigen loss. This unique property highlights the capacity of TCR-T therapy to address resistance mechanisms that limit the efficacy of monoclonal antibodies and even CAR-T cells, which depend on surface antigen density for recognition (1).
Previous studies have reported T-cell receptors (TCRs) specific for the HER2/neu-derived peptide KIFGSLAFL (EP3259284B1, WO2010012829A1) (50, 51). These patents describe alternative strategies for identifying, cloning, and functionally validating HER2-specific TCRs, each with distinct methodological principles. In contrast to these prior approaches, our workflow integrates several key methodological refinements that enhance the precision, specificity, and functional relevance of TCR discovery and validation.
The EP3259284B1 patent identified reactive T cells primarily through cytokine release assays after peptide stimulation. While effective in detecting peptide-reactive cells, this method can also capture bystander T cells activated indirectly by cytokines, which limits the accuracy of antigen-specific isolation (52, 53). The WO2010012829A1 patent, on the other hand, utilized PE-conjugated HLA-A2/peptide multimers to stain antigen-specific lymphocytes. Although this approach improves specificity compared to cytokine-based assays, single-color multimer staining may still lead to false positives, as demonstrated by our previous data (15), likely due to low-affinity or cross-reactive binding. In our study, antigen-specific T cells were isolated using Flex-T technology, in which the targeted peptide is presented within the MHC complex and conjugated to two distinct fluorophores (APC and PE). Only double-positive cells binding both MHC/peptide complexes were isolated by flow cytometry, ensuring that the recovered population was highly specific for the HER2/neu KIFGSLAFL peptide. This dual-label strategy significantly reduces nonspecific binding and yields a purer population of antigen-specific T cells than the methods used in the previous patents.
A major difference also lies in the strategy for TCR sequence identification. The EP3259284B1 patent used a 5′-RACE PCR kit to obtain TCR α and β sequences, while WO2010012829A1 relied on a subfamily-restricted PCR approach employing predefined sets of Vα and Vβ primers. The 5′-RACE PCR method, while primer-efficient, is prone to generating non-regular TCR sequences due to short DNA fragment artifacts and accumulated errors during library preparation, leading to biased quantification and incomplete repertoire coverage. Subfamily-restricted PCR, by design, exacerbates primer bias through predefined V-region sets, resulting in uneven amplification efficiencies, loss of rare V-J pairings, and skewed gene usage that underrepresents true repertoire diversity (54). In contrast, our TCR identification strategy leverages single-cell RNA sequencing, enabling the recovery of full-length paired α and β chains, including the complete V, D, J, and C segments (55), while simultaneously capturing the transcriptional profile of each clonotype. This eliminates the primer bias, allows the discovery of rare or unconventional TCR variants, and provides functional insight into each TCR-expressing cell. The integration of transcriptomic data enables rational selection of candidate TCRs based on activation, exhaustion, and differentiation states, resulting in a more informed and biologically relevant selection process.
Another key difference lies in the characterization of the resulting TCRs. The WO2010012829A1 patent does not define a single HER2-specific TCR but rather provides a combinatorial set of potential α/β pairings, making it difficult to determine the physiological relevance or safety profile of individual clonotypes. In contrast, our approach recovered a unique, naturally paired α/β TCR at the single-cell level, providing complete sequence information, preserving authentic pairing, and linking each clonotype to its corresponding transcriptional state. This ensures reproducibility, functional predictability, and accurate assessment of therapeutic potential.
Technically, we also advanced beyond the retroviral systems described in earlier patents by employing third-generation, self-inactivating lentiviral vectors, which offer improved biosafety, efficient transduction of non-dividing and dividing cells and sustained transgene expression (56, 57). Additionally, we incorporated the ERGO-II neural network for computational prediction and ranking of TCR–peptide affinity (58), introducing a precision layer absent in prior approaches.
In summary, these distinctions highlight that while earlier patents (EP3259284B1 and WO2010012829A1) established viable strategies for generating HER2-specific TCRs, our approach provides a more defined, precise, and functionally integrated system for the identification and engineering of antigen-specific T cells suitable for therapeutic application.
Nevertheless, several limitations should be acknowledged. First, our study primarily focuses on preclinical validation, future work must evaluate its safety and efficacy in clinical settings. This includes assessing the risk of cytokine release and off-target reactivity, which has been problematic in some TCR T-cell trials (4), not to mention the need to optimize T cell persistence and resistance to exhaustion in the tumor microenvironment (45). Second, while our study demonstrates robust antigen-specific cytotoxicity and cytokine release in vitro, it does not fully explore the long-term functional stability of the engineered T cells, such as their persistence, exhaustion dynamics, or responsiveness under repeated antigen stimulation. Future experiments assessing these aspects will be important to predict therapeutic durability and optimize TCR-T cell design for sustained antitumor activity. Third, while xenograft models provide proof-of-concept for antitumor efficacy, they do not fully recapitulate the complexity of human tumors, including the heterogeneity of tumor architecture, stromal and vascular components, and immune-tumor interactions. Patient-derived xenograft (PDX) models or humanized mouse models offer a more physiologically relevant platform, preserving tumor heterogeneity and enabling assessment of TCR-T cell persistence, trafficking, and function within a human-like immune microenvironment. Incorporating these models in future studies will be essential to evaluate therapeutic efficacy thereby providing a more comprehensive preclinical assessment of HER2-TCR-T cell therapy.
In conclusion, this study demonstrates that anti-HER2/neu TCR-engineered T cells exhibit robust antigen-specific cytotoxicity, molecular activation, and in vivo efficacy, positioning them as promising candidates for adoptive cell therapy. The integration of advanced single-cell sequencing, computational affinity modeling, and lentiviral engineering represents a significant step forward in the rational design of next-generation TCR-T therapies. These findings provide a strong foundation for further preclinical and translational development aimed at overcoming resistance mechanisms in HER2-positive malignancies and improving patient outcomes.
5 Patents
The clonotype investigated in this article is protected by the Patent of the Russian Federation with Application Number 2025105148.
Data availability statement
The data presented in the study are deposited in NCBI's Gene Expression Omnibus repository, accession number GSE311201.
Ethics statement
The studies involving humans were approved by local ethics committee of the Research Institute of Fundamental and Clinical Immunology. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. The animal study was approved by local ethics committee of the Research Institute of Fundamental and Clinical Immunology. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SA: Investigation, Writing – review & editing, Methodology, Supervision, Software, Writing – original draft, Validation, Visualization, Data curation, Formal analysis, Conceptualization. RP-Z: Data curation, Validation, Methodology, Visualization, Formal analysis, Conceptualization, Investigation, Software, Writing – review & editing, Writing – original draft, Supervision. MF: Methodology, Validation, Conceptualization, Investigation, Writing – review & editing, Formal analysis, Writing – original draft. JL: Validation, Data curation, Formal analysis, Methodology, Writing – review & editing, Investigation, Conceptualization, Supervision, Writing – original draft. OP-Z: Investigation, Software, Conceptualization, Writing – review & editing, Supervision, Data curation, Writing – original draft, Formal analysis, Validation, Methodology, Visualization. JS: Writing – original draft, Formal analysis, Conceptualization, Validation, Investigation, Data curation, Methodology, Supervision, Writing – review & editing. KN: Investigation, Writing – original draft, Methodology, Conceptualization, Writing – review & editing, Formal analysis, Validation. JP: Validation, Investigation, Conceptualization, Writing – review & editing, Formal analysis, Writing – original draft, Methodology. VK: Investigation, Methodology, Conceptualization, Validation, Writing – original draft, Formal analysis, Writing – review & editing. OK: Formal analysis, Validation, Investigation, Data curation, Methodology, Writing – review & editing. EZ: Formal analysis, Validation, Investigation, Data curation, Methodology, Writing – review & editing. EG: Supervision, Conceptualization, Resources, Writing – original draft, Writing – review & editing, Project administration, Funding acquisition. PT: Project administration, Conceptualization, Writing – original draft, Resources, Funding acquisition, Writing – review & editing, Supervision. PG: Writing – original draft, Conceptualization, Project administration, Funding acquisition, Writing – review & editing, Resources, Supervision. SS: Funding acquisition, Writing – original draft, Resources, Conceptualization, Methodology, Supervision, Project administration, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was funded by the Ministry of Higher Education and Science, State Assignment No.124112200103-3.
Acknowledgments
The authors express their sincere gratitude to the Academic Leadership Program Priority 2030 proposed by the Federal State Autonomous Educational Institution of Higher Education I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University) for providing essential financial and support that enabled this research.
Conflict of interest
Saleh Alrhmoun, Roman Perik-Zavodskii, Marina Fisher, Julia Lopatnikova, Julia Shevchenko, Elena Golikova, Petr Timashev, Petr Glybochko, and Sergey Sennikov are affiliated with the Federal State Budgetary Scientific Institution Research Institute of Fundamental and Clinical Immunology (RIFCI), which is the owner of a patent on a T-cell receptor recognizing the HER2/neu peptide (369–377, KIFGSLAFL) in complex with HLA-A*02. This patent describes a naturally-occuring T-cell receptor specific to the HER2/neu antigen and its use for adoptive cell therapy of HER2/neu-expressing tumors. The patent number is 2025105148.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2025.1646404/full#supplementary-material
References
1. Golikova EA, Alshevskaya AA, Alrhmoun S, Sivitskaya NA, and Sennikov SV. TCR-T cell therapy: current development approaches, preclinical evaluation, and perspectives on regulatory challenges. J Trans Med. (2024) 22:1–17. doi: 10.1186/S12967-024-05703-9
2. D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259T cells in synovial sarcoma. Cancer Discov. (2018) 8:944–57. doi: 10.1158/2159-8290.CD-17-1417
3. Hong D, Clarke J, Johanns T, Kebriaei P, Heymach J, Galal A, et al. 379 Initial safety, efficacy, and product attributes from the SURPASS trial with ADP-A2M4CD8, a SPEAR T-cell therapy incorporating an affinity optimized TCR targeting MAGE-A4 and a CD8α co-receptor. J Immunotherap Cancer. (2020) 8:A231.1–A231. doi: 10.1136/jitc-2020-SITC2020.0379
4. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1–reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
5. Marofi F, Motavalli R, Safonov VA, Thangavelu L, Yumashev AV, Alexander M, et al. CAR T cells in solid tumors: challenges and opportunities. Stem Cell Res Ther. (2021) 12:1–16. doi: 10.1186/S13287-020-02128-1
6. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, and Milone MC. CAR T cell immunotherapy for human cancer. Science. (2018) 359:1361–5. doi: 10.1126/science.aar6711
7. Papalexi E and Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. (2018) 18:35–45. doi: 10.1038/nri.2017.76
8. Perik-Zavodskii R, Perik-Zavodskaia O, Alrhmoun S, Volynets M, Shevchenko J, Nazarov K, et al. Single-cell multi-omics reveal stage of differentiation and trajectory-dependent immunity-related gene expression patterns in human erythroid cells. Front Immunol. (2024) 15:1431303. doi: 10.3389/fimmu.2024.1431303
9. Perik-Zavodskii R, Perik-Zavodskaia O, Shevchenko J, Denisova V, Alrhmoun S, Volynets M, et al. Immune transcriptome study of human nucleated erythroid cells from different tissues by single-cell RNA-sequencing. Cells. (2022) 11:3537. doi: 10.3390/cells11223537
10. Perik-Zavodskii R, Perik-Zavodskaia O, Alrhmoun S, Lopatnikova J, Alshevskaya A, Zhukova J, et al. Single-cell multi-omics reveals the TNF-α activation threshold for Classical Monocytes by studying healthy donors and rheumatoid arthritis patients. Front Immunol. (2025) 16:1572823. doi: 10.3389/fimmu.2025.1572823
11. Alsalloum A, Alrhmoun S, Perik-Zavosdkaia O, Fisher M, Volynets M, Lopatnikova J, et al. Decoding NY-ESO-1 TCR T cells: transcriptomic insights reveal dual mechanisms of tumor targeting in a melanoma murine xenograft model. Front Immunol. (2024) 15:1507218/BIBTEX. doi: 10.3389/FIMMU.2024.1507218
12. Sennikov S, Volynets M, Alrhmoun S, Perik-Zavodskii R, Perik-Zavodskaia O, Fisher M, et al. Modified Dendritic cell-based T-cell expansion protocol and single-cell multi-omics allow for the selection of the most expanded and in vitro-effective clonotype via profiling of thousands of MAGE-A3-specific T-cells. Front Immunol. (2024) 15:1470130/BIBTEX. doi: 10.3389/FIMMU.2024.1470130
13. Perik-Zavodskaia O, Perik-Zavodskii R, Alrhmoun S, Nazarov K, Shevchenko J, Zaitsev K, et al. Multi-omic analysis identifies erythroid cells as the major population in mouse placentas expressing genes for antigen presentation in MHC class II, chemokines, and antibacterial immune response. Front Immunol. (2025) 16:1644983/BIBTEX. doi: 10.3389/FIMMU.2025.1644983
14. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell. (2020) 181:442–459.e29. doi: 10.1016/j.cell.2020.03.048
15. Alrhmoun S, Fisher M, Lopatnikova J, Perik-Zavodskaia O, Volynets M, Perik-Zavodskii R, et al. Targeting precision in cancer immunotherapy: naturally-occurring antigen-specific TCR discovery with single-cell sequencing. Cancers (Basel). (2024) 16:4020. doi: 10.3390/cancers16234020
16. Perik-Zavodskii R, Perik-Zavodskaia O, Volynets M, Alrhmoun S, and Sennikov S. TCRscape: a single-cell multi-omic TCR profiling toolkit. Front Bioinf. (2025) 5:1641491/BIBTEX. doi: 10.3389/FBINF.2025.1641491
17. Kuznetsova M, Lopatnikova J, Shevchenko J, Silkov A, Maksyutov A, and Sennikov S. Cytotoxic activity and memory T cell subset distribution of in vitro-stimulated CD8+ T cells specific for HER2/neu epitopes. Front Immunol. (2019) 10:1017. doi: 10.3389/fimmu.2019.01017
18. Arrieta-Bolaños E, Hernández-Zaragoza DI, and Barquera R. An HLA map of the world: A comparison of HLA frequencies in 200 worldwide populations reveals diverse patterns for class I and class II. Front Genet. (2023) 14:866407. doi: 10.3389/fgene.2023.866407
19. Cornetta K and Anderson WF. Protamine sulfate as an effective alternative to polybrene in retroviral-mediated gene-transfer: implications for human gene therapy. J Virol Methods. (1989) 23:187–94. doi: 10.1016/0166-0934(89)90132-8
20. Lopatnikova JAJUA, Shevchenko YUA, Filippova JGJUG, Fisher MSMS, Obleukhova IAIA, Golikova EAEA, et al. Multimer-based technologies for the assessment of antigen-specific CD8+-lymphocytes. Immunologiya. (2024) 45:777–91. doi: 10.33029/1816-2134-2024-45-6-777-791
22. Bedre R. reneshbedre/bioinfokit: Bioinformatics data analysis and visualization toolkit. (2020). doi: 10.5281/ZENODO.3755192
23. Peng Y, Wu Z, Pang Z, Zhang L, Song D, Liu F, et al. Manufacture and evaluation of a HER2-positive breast cancer immunotoxin 4D5Fv-PE25. Microb Cell Fact. (2023) 22:1–11. doi: 10.1186/S12934-023-02115-0/TABLES/2
24. D’amico P, Reduzzi C, Qiang W, Zhang Y, Gerratana L, Zhang Q, et al. Single-cells isolation and molecular analysis: focus on HER2-low CTCs in metastatic breast cancer. Cancers (Basel). (2021) 14:79. doi: 10.3390/CANCERS14010079
25. Lin XL, Wang XL, Ma B, Jia J, Yan Y, Di LJ, et al. HER2-specific T lymphocytes kill both trastuzumab-resistant and trastuzumab-sensitive breast cell lines in vitro. Chin J Cancer Res. (2012) 24:143–50. doi: 10.1007/S11670-012-0143-6/METRICS
26. Takegawa N, Tsurutani J, Kawakami H, Yonesaka K, Kato R, Haratani K, et al. fam-] trastuzumab deruxtecan, antitumor activity is dependent on HER2 expression level rather than on HER2 amplification. Int J Cancer. (2019) 145:3414–24. doi: 10.1002/IJC.32408
27. Wang M, Xu T, Shen L, Li J, Zhu L, Wang C, et al. HER2 expression in circulating tumor cells as prognostic and predictive biomarkers for anti-HER2 therapy in previously treated metastatic colorectal cancer. Adv Ther (Weinh). (2023) 6:2300044. doi: 10.1002/ADTP.202300044
28. Wu X, Xu L, Li X, Zhou Y, Han X, Zhang W, et al. A HER2-targeting antibody-MMAE conjugate RC48 sensitizes immunotherapy in HER2-positive colon cancer by triggering the cGAS-STING pathway. Cell Death Dis. (2023) 14:1–10. doi: 10.1038/S41419-023-06073-8;TECHMETA
29. Datta KK, Periasamy P, Mohan SV, Ziegman R, and Gowda H. Temporal quantitative proteomics reveals proteomic and phosphoproteomic alterations associated with adaptive response to hypoxia in melanoma cells. Cancers (Basel). (2021) 13:2175. doi: 10.3390/CANCERS13092175
30. Gonçalves E, Poulos RC, Cai Z, Barthorpe S, Manda SS, Lucas N, et al. Pan-cancer proteomic map of 949 human cell lines. Cancer Cell. (2022) 40:835–849.e8. doi: 10.1016/j.ccell.2022.06.010
31. Ma J, Han H, Liu D, Li W, Feng H, Xue X, et al. HER2 as a promising target for cytotoxicity T cells in human melanoma therapy. PloS One. (2013) 8:e73261. doi: 10.1371/JOURNAL.PONE.0073261
32. Parel Y and Chizzolini C. CD4+ CD8+ double positive (DP) T cells in health and disease. Autoimmun Rev. (2004) 3:215–20. doi: 10.1016/j.autrev.2003.09.001
33. Clénet ML, Gagnon F, Moratalla AC, Viel EC, and Arbour N. Peripheral human CD4+CD8+ T lymphocytes exhibit a memory phenotype and enhanced responses to IL-2, IL-7 and IL-15. Sci Rep. (2017) 7:1–15. doi: 10.1038/S41598-017-11926-2;TECHMETA
34. Desfrançois J, Derré L, Corvaisier M, Le Mével B, Catros V, Jotereau F, et al. Increased frequency of nonconventional double positive CD4CD8 αβ T cells in human breast pleural effusions. Int J Cancer. (2009) 125:374–80. doi: 10.1002/IJC.24366
35. Desfrançois J, Moreau-Aubry A, Vignard V, Godet Y, Khammari A, Dréno B, et al. Double positive CD4CD8 αβ T cells: A new tumor-reactive population in human melanomas. PloS One. (2010) 5:e8437. doi: 10.1371/JOURNAL.PONE.0008437
36. Bagot M, Echchakir H, Mami-Chouaib F, Delfau-Larue MH, Charue D, and Bernheim A. Chouaib S, boumsell L, bensussan A. Isolation of tumor-specific cytotoxic CD4+ and CD4+CD8dim+ T-cell clones infiltrating a cutaneous T-cell lymphoma. Blood. (1998) 91:4331–41. doi: 10.1182/BLOOD.V91.11.4331
37. SChad SE, Chow A, Mangarin L, Pan H, Zhang J, Ceglia N, et al. Tumor-induced double positive T cells display distinct lineage commitment mechanisms and functions. J Exp Med. (2022) 219:e20212169. doi: 10.1084/JEM.20212169
38. Wherry EJ and Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
39. Alrhmoun S and Sennikov S. The role of tumor-associated antigen HER2/neu in tumor development and the different approaches for using it in treatment: many choices and future directions. Cancers. (2022) 14:6173. doi: 10.3390/CANCERS14246173
40. Nascimbeni M, Shin E-C, Chiriboga L, Kleiner DE, and Rehermann B. Peripheral CD4+CD8+ T cells are differentiated effector memory cells with antiviral functions. Blood. (2004) 104:478–86. doi: 10.1182/blood-2003-12-4395
41. Smith-Garvin JE, Koretzky GA, and Jordan MS. T cell activation. Annu Rev Immunol. (2009) 27:591–619. doi: 10.1146/annurev.immunol.021908.132706
42. Voskoboinik I, Whisstock JC, and Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. (2015) 15:388–400. doi: 10.1038/nri3839
43. Waldmann TA. The biology of interleukin-2 and interleukin-15: Implications for cancer therapy and vaccine design. Nat Rev Immunol. (2006) 6:595–601. doi: 10.1038/nri1901
44. Dunn GP, Ikeda H, Bruce AT, Koebel C, Uppaluri R, Bui J, et al. Interferon-γ and cancer immunoediting. Immunol Res. (2005) 32:231–46. doi: 10.1385/IR:32:1-3:231
45. Gattinoni L, Speiser DE, Lichterfeld M, and Bonini C. T memory stem cells in health and disease. Nat Med. (2017) 23:18–27. doi: 10.1038/nm.4241
46. Li X, Zhang X, Yin S, and Nie J. Challenges and prospects in HER2-positive breast cancer-targeted therapy. Crit Rev Oncol Hematol. (2025) 207:104624. doi: 10.1016/J.CRITREVONC.2025.104624
47. Canonici A, Ivers L, Conlon NT, Pedersen K, Gaynor N, Browne BC, et al. HER-targeted tyrosine kinase inhibitors enhance response to trastuzumab and pertuzumab in HER2-positive breast cancer. Invest New Drugs. (2019) 37:441–51. doi: 10.1007/S10637-018-0649-Y/METRICS
48. Stanowicka-Grada M and Senkus E. Anti-HER2 drugs for the treatment of advanced HER2 positive breast cancer. Curr Treat Options Oncol. (2023) 24:1633–50. doi: 10.1007/S11864-023-01137-5
49. Wang L, Wang Y, Li Y, Zhou L, Du J, Wang J, et al. Resistance mechanisms and prospects of trastuzumab. Front Oncol. (2024) 14:1389390/BIBTEX. doi: 10.3389/FONC.2024.1389390
50. WO2010012829A1 - Her2/neu specific t cell receptors - Google Patents . Available online at: https://patents.google.com/patent/WO2010012829A1/en (Accessed October 20, 2025).
51. Powell DJ. A fully-human t cell receptor specific for the 369–377 epitope derived from the her2/neu (erbb2) receptor protein (2016).
52. Boyman O. Bystander activation of CD4+ T cells. Eur J Immunol. (2010) 40:936–9. doi: 10.1002/EJI.201040466
53. Martin MD, Jensen IJ, Ishizuka AS, Lefebvre M, Shan Q, Xue HH, et al. Bystander responses impact accurate detection of murine and human antigen-specific CD8+ T cells. J Clin Invest. (2019) 129:3894–908. doi: 10.1172/JCI124443
54. Lin YH, Hung SJ, Chen YL, Lin CH, Kung TF, Yeh YC, et al. Dissecting efficiency of a 5’ rapid amplification of cDNA ends (5’-RACE) approach for profiling T-cell receptor beta repertoire. PloS One. (2020) 15:e0236366. doi: 10.1371/JOURNAL.PONE.0236366
55. Gao C, Zhang M, and Chen L. The comparison of two single-cell sequencing platforms: BD rhapsody and 10x genomics chromium. Curr Genomics. (2020) 21:602–9. doi: 10.2174/1389202921999200625220812
56. Schambach A, Zychlinski D, Ehrnstroem B, and Baum C. Biosafety features of lentiviral vectors. Hum Gene Ther. (2013) 24:132. doi: 10.1089/HUM.2012.229
57. Dufait I, Liechtenstein T, Lanna A, Bricogne C, Laranga R, Padella A, et al. Retroviral and lentiviral vectors for the induction of immunological tolerance. Scientifica (Cairo). (2012) 2012:694137. doi: 10.6064/2012/694137
Keywords: TCR-T cells, TCR-T, T cells, adoptive cell therapy, her2/neu, ErbB2, ScRNA-seq
Citation: Alrhmoun S, Perik-Zavodskii R, Fisher M, Lopatnikova J, Perik-Zavodskaia O, Shevchenko J, Nazarov K, Philippova J, Kurilin V, Kichakova O, Zavjalov E, Golikova E, Timashev P, Glybochko P and Sennikov S (2025) Anti-HER2/neu TCR-T Cells in Action: linking transcriptional signatures, secretomics, and In Vivo tumor suppression. Front. Immunol. 16:1646404. doi: 10.3389/fimmu.2025.1646404
Received: 13 June 2025; Accepted: 28 October 2025;
Published: 25 November 2025.
Edited by:
Xuefeng Wang, Soochow University, ChinaReviewed by:
Maria Turchaninova, Institute of Bioorganic Chemistry (RAS), RussiaFeifei Song, University of North Carolina at Chapel Hill, United States
Copyright © 2025 Alrhmoun, Perik-Zavodskii, Fisher, Lopatnikova, Perik-Zavodskaia, Shevchenko, Nazarov, Philippova, Kurilin, Kichakova, Zavjalov, Golikova, Timashev, Glybochko and Sennikov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Saleh Alrhmoun, c2FsZWguYWxyaG1vdW4xQGdtYWlsLmNvbQ==; Sergey Sennikov, c2Vubmlrb3ZAbmlpa2ltLnJ1