 Ran Tang1,2†
Ran Tang1,2† Min Zhang
Min Zhang Xiaoyi Jia
Xiaoyi Jia- 1School of Pharmacy, Anhui University of Chinese Medicine, Hefei, Anhui, China
- 2Anhui Province Key Laboratory of Research & Development of Chinese Medicine, Hefei, Anhui, China
- 3Department of Rheumatology and Immunology, The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, Anhui, China
Neutrophil extracellular traps (NETs) are reticular fiber structures released by neutrophils in response to various stimuli. Although NETs have antibacterial defense functions, their excessive formation has been proven to accelerate the progression of autoimmune diseases. Increasing studies have shown that NETs play an important role in the pathogenesis of autoimmune diseases. The pathogenesis of recent advances in autoimmune disease research, with a focus on the role of NETs in the etiology and pathogenesis of these disorders, and summarizes the current treatment strategies targeting NETs, aiming to provide new directions for the treatment of autoimmune diseases.
1 Introduction
Neutrophils are the most abundant immune cells in the human body and constitute the first defense against pathogen invasion, playing a crucial role in host immunity (1). Typically, neutrophils degranulate by releasing antibacterial and proteolytic enzymes, then perform phagocytosis to kill invading microorganisms; however, when encountering large biological structures that cannot be engulfed (e.g., fungi and parasites), they undergo a distinct process to release DNA, histones, and granular proteins—such as neutrophil elastase (NE) and myeloperoxidase (MPO)—thus forming `neutrophil extracellular traps (NETs) (2). These NETs immobilize, kill, and degrade the pathogens extracellularly through the action of associated proteolytic enzymes.
NETs are involved in many autoimmune diseases and are thought to be crucial in the inflammatory process. Although NETs are beneficial during infection, they may play a harmful role in inflammatory, autoimmune, and other pathophysiological conditions (3–5). NETs promote inflammatory processes by releasing active molecules such as hazardous associated molecular patterns (DAMPs), histones, and extracellular active lyases, leading to further immune responses (6). Thus, NETs can also serve as a potential source of autoantigens that bind to associated autoantibodies produced by inflammatory autoimmune diseases. In autoimmune diseases, including gouty arthritis (GA), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), psoriasis, antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV), antiphospholipid syndrome (APS), and type 1 diabetes mellitus (T1DM), NETs exhibit aberrant accumulation and impaired clearance. Based on gradient density, neutrophils are classified into low-density neutrophils (LDNs) and normal-density neutrophils subgroups. LDNs is more likely to form NETs in patients with SLE and psoriasis, which may explain the link between the disease and NETs formation (7, 8). In addition, the composition of NETs may vary according to different stimuli, as well as the diseases associated with them (9). In some cases, NETs may also have anti-inflammatory features (10).
Therefore, this article will elucidate the role of NETs in autoimmune diseases from the perspective of their formation and function, and explore their potential as therapeutic targets, thereby providing new insights for the clinical treatment of autoimmune diseases.
2 Overview of NETs
2.1 Formation of NETs
The concept of NETs was first proposed by Brinkmann et al. in 2004, who found that neutrophils have a novel mode of death, which is different from cell necrosis and apoptosis (11). Through the breakdown of the neutrophil plasma membrane, a highly active mixture of nucleic acids and proteins is released outside the cell, forming this smooth filamentous structure with an DNA as a skeleton to which various protein particles are anchored called NETs (12). The formation of NETs is triggered by a variety of factors, including cytokines, bacteria, fungi, viruses and protozoa (13, 14). For example, stimulated by Phorbol-12-myristate-13-acetate (PMA), PMA can promote the assembly and activation of nicotinamide adenine dinucleotide phosphate oxidase (NOX) and induce the production of reactive oxygen species (ROS) without forming phagosomes (15, 16). The RAF-MEK-ERK pathway is located upstream of NOX, regulates the production of ROS. However, there are also NOX independent ROS, which are produced by mitochondria (17). ROS damage secretory granules and lysosome membranes, resulting in the release of NE and MPO. NE is first translocated to the nucleus, cutting some specific histones and promoting chromatin depolymerization. Subsequently, MPO also enters the nucleus and collaborates with NE to promote chromatin depolymerization (18). PMA can also activate peptidyl arginine deiminase 4 (PAD4) by binding to protein kinase C (PKC) to induce the release of intracellular calcium ions. PAD4 can dominate histone arginine residues to form citrulline residues, reduce the positive charge, weaken the electrostatic binding force with DNA, and thus depolymerize chromatin (19). It can be seen that the core process in the formation of NETs is chromatin deaggregation, which requires the participation of ROS, NE, MPO and PAD4. Depolymerized chromatin is released from the ruptured nucleus into the cytoplasm, and together with other substances in the cytoplasm such as MPO and NE, it is discharged into the extracellular space through membrane tearing, resulting in the death of neutrophils (20). The mechanism of NETs formation is shown in Figure 1.
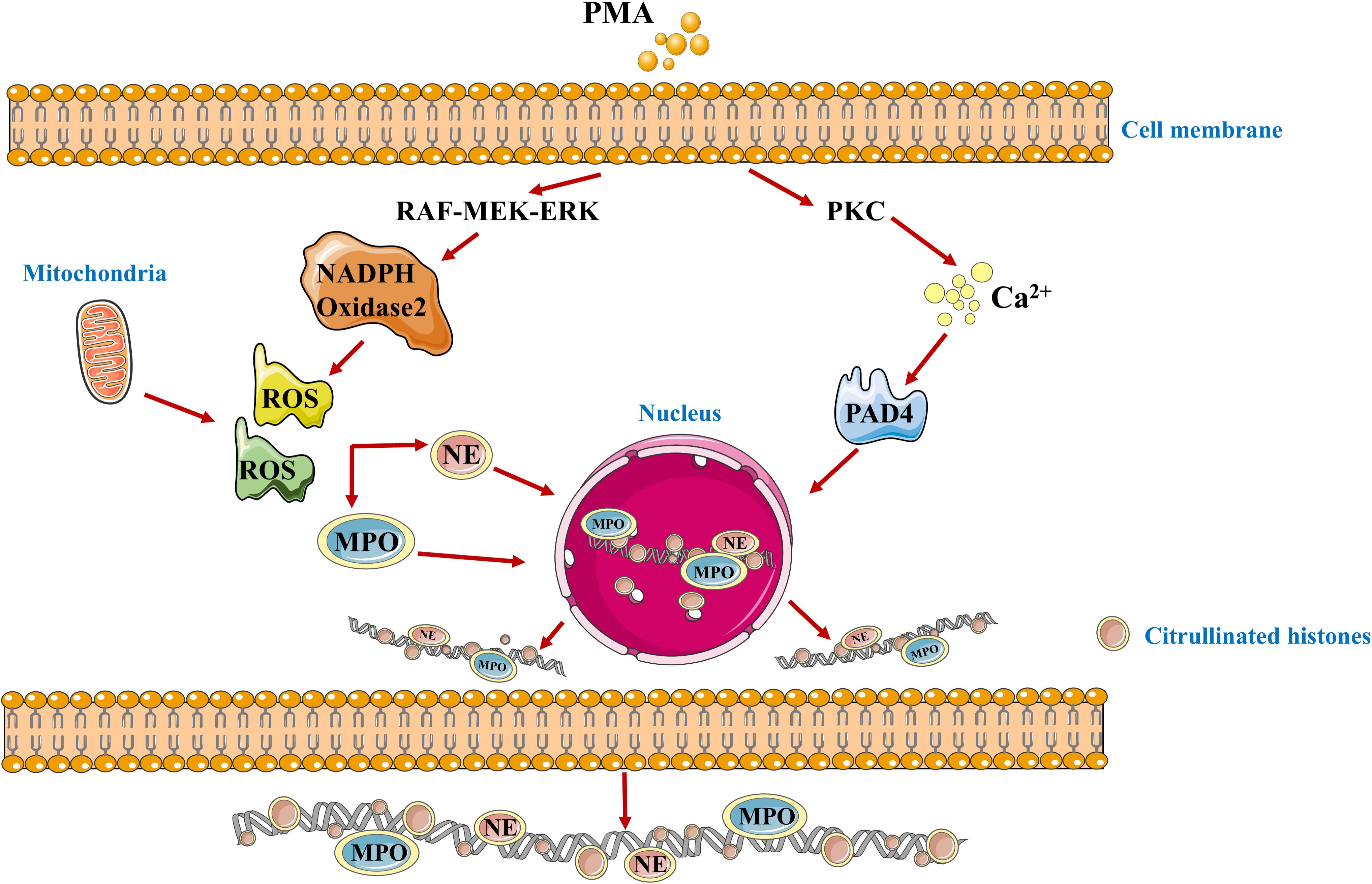
Figure 1. The schematic diagram of the formation mechanism of NETs. PMA, Phorbol-12-myristate-13-acetate; PKC, protein kinase C; ROS, Reactive oxygen species; PAD4, peptidyl arginine xdeiminase 4; MPO, myeloperoxidase; NE, neutrophilic elastase.
2.2 Functions of NETs
NETs are a double-edged sword. On the one hand, NETs have been shown to play a positive role in controlling bacterial infections (21, 22). Substances such as histone, NE, MPO, cathepsin G, lactoferrin, and antimicrobial peptides can protect wounds and prevent the spread of infection. Histone proteins play an important role in the decomposition of bacterial cell membrane (23). In anti-HIV, influenza, and novel coronavirus, viruses stimulate the formation of NETs via Toll Like Receptor 4 (TLR4), TLR7, and TLR8, NETs inhibit viral replication by capturing the virus or blocking the PKC pathway (24–26). In terms of anti-parasite, immune cells play an irreplaceable role in host defense. For example, neutrophils play a protective role in toxoplasmosis infected fibroblasts (27). Neutrophils can also produce antimicrobial factors to stop the spread of leishmania (28). On the other hand, excessive formation of NETs or inadequate clearance by the body may result in uncontrolled inflammatory response. NETs can regulate congenital and adaptive immune disorders through various mechanisms, and NETs can also amplify inflammatory responses, possibly worsening diseases and even organ damage (29, 30). Because some of the released proteins have non-specific effects, they will directly cause damage to other cells, form immune complexes, induce the production of autoantibodies, and result in pathological tissue damage (31). It has been found that there are a large number of circulating NETs in patients with sepsis, and their presence is associated with poor prognosis and multiple organ failure (32–34). NETs, through their pro-inflammatory and cytotoxic effects, can promote the progression of various diseases including autoimmune disorders, thrombotic conditions, cancer metastasis and progression, as well as severe COVID-19 (35). Histones have DAMPs that increase the release of pro-inflammatory cytokines and activate the Pyrin Domain Containing Protein 3 (NLRP3) inflammasome to further amplify the inflammatory response (36). Induce cytotoxicity and increase the production of ROS, cause endothelial dysfunction and induce organ damage.
Therefore, although NETs constitute a crucial antimicrobial defense mechanism, their uncontrolled release poses a significant threat to host tissues. Thus, the following section will focus on how the detrimental effects of NETs—resulting from either excessive formation or impaired clearance—play a critical role in the pathogenesis of various autoimmune diseases.
3 NETs and autoimmune diseases
Autoimmune diseases are caused by the breakdown of the balance between the body’s immune defense and its own antigens, resulting in immune response and damage to its own tissues. At this time, autoimmune cells unable to distinguish between “self” and “non-self” components, the immune system is abnormal, the body produces antibodies to attack itself, causing organ and tissue damage (37). Neutrophils play an irreplaceable role in autoimmune diseases, the excessive formation or insufficient clearance of NETs affects the course of autoimmune disease. Therefore, the study of NETs may be a new direction in the treatment of autoimmune diseases, and possibly the prevention of other diseases associated with the disease. The mechanism of neutrophils and NETs in autoimmune diseases is shown in Figure 2.
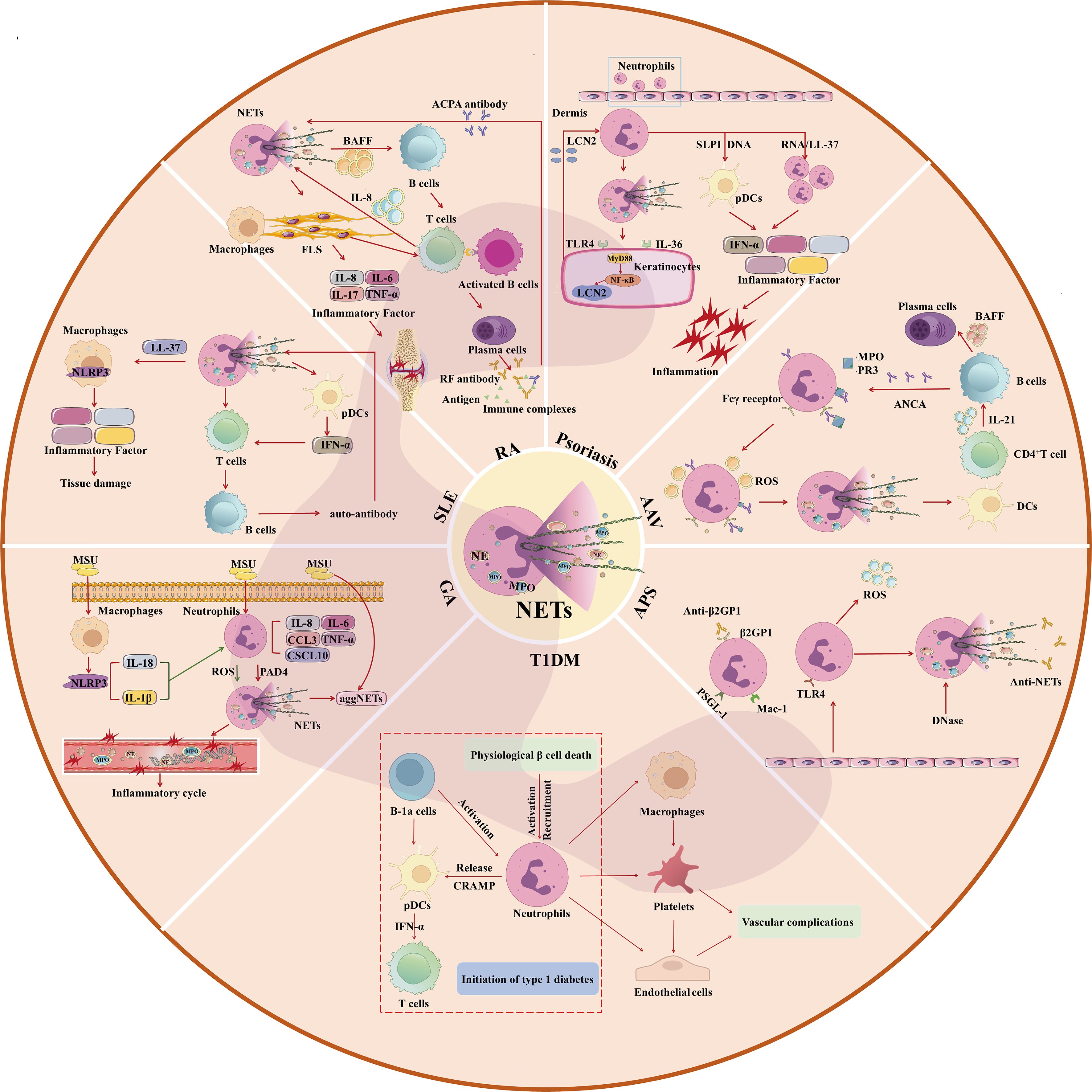
Figure 2. Mechanism of Neutrophils and NETs in Autoimmune Diseases (GA, SLE, RA, Psoriasis, AAV, APS, T1DM). MSU, Monosodium urate; aggNETs, aggregated NETs; NETs, neutrophil extracellular traps. pDCs, Plasmacytoid Dendritic Cells; IFN-α, interferon-α; TNF-α, Tumor Necrosis Factor-α; ACPA, anti-citrullinated protein antibody; RF, rheumatoid factor; BAFF, B Cell Activating Factor; LCN2, Lipocalin-2; MyD88, Myeloid differentiation factor-88; NF-κB, nuclear factor-kappaB; TLR4, Toll-like receptor 4; PR3, protease 3; ANCA, anti-neutrophil cytoplasmic antibody; DCs, Dendritic cells; MPO, myeloperoxidase; ROS, Reactive oxygen species; β2GP1, β2-glycoprotein-1; PSGL-1, P-selectin glycoprotein-1; DNase, deoxyribonuclease; TLR4, Toll-like receptor 4; MAC-1, Macrophage-1; CRAMP, cathelicidin-related antimicrobial peptide.
3.1 NETs and GA
GA, the most common inflammatory arthritis, is a multifactorial autoinflammatory disease uniquely characterized by the deposition of monosodium urate (MSU) crystals within joints, which triggers acute and painful synovitis (38, 39). During the onset of acute GA, the accumulation of MSU crystals can induce a large infiltration of inflammatory cells (such as neutrophils and monocytes) into the MSU crystal deposit site of the patient. The release of from these immune cells further triggers the release of various pro-inflammatory cytokines and chemokines, which upregulates selectin and integrin on the surface of endothelial cell lumen, and further enhances neutrophil recruitment (40). Upon contact with MSU crystals, neutrophils release a range of inflammatory mediators—including Tumor Necrosis Factor alpha (TNF-α), Interleukin-6 (IL-6), as well as neutrophil inducers (e.g., IL-8) and activators (such as CCL3 and CXCL10) (41). Furthermore, MSU crystals activate infiltrating neutrophils not only to secrete cytokines but also to form NETs through two principal mechanisms: a ROS-independent vital pathway involving calcium-mediated direct activation of PAD4, and a ROS-dependent suicidal pathway driven by calcium/NOX-generated ROS leading to PAD4 activation—both of which ultimately result in NETs release (42). Macrophage phagocytosis of MSU crystals activates the NLRP3 inflammasome, prompting release of IL-1β and IL-18, which recruit neutrophils that undergo oxidative burst (a rapid ROS-producing process) and form NETs through genomic DNA and granular protein release (43). These NETs components—notably histones and DNA—directly damage tissues such as vascular endothelium, inducing further release of inflammatory mediators like Adenosine Triphosphate and uric acid and thereby sustaining a feed-forward inflammatory cycle (44). NETs release proinflammatory mediators in the early stages of neutrophil recruitment or when the number of neutrophils is comparable to that of peripheral blood (40). Conversely, a high density of neutrophils—such as that found in the synovial fluid of GA or within highly infiltrated inflammatory tissues—promotes the formation of aggregated NETs (aggNETs) both in vivo and in vitro (45). These aggNETs can be covered on the surface of MSU crystals to isolate them from inflammatory mediators, thus promoting the formation of gout stones and indirectly alleviating the damage caused by MSU crystals to the body (46). Thus, in GA, the formation of NETs has a double-edged sword effect: on the one hand, NETs can package and isolate MSU crystals or adhere to apoptotic cells and clear them, inhibit inflammatory response and protect the body by controlling the production of cytokines by proteases, which shows that NETs play a role in promoting and alleviating inflammation. On the other hand, NETs release various damage-related molecular modes (histone or DNA-activated TLR or NLRP3 inflammatory bodies), which aggravates the inflammatory response by releasing pro-inflammatory factors or direct action, and excessive NETs will form aggNETs, promote the formation of gout, cause bone erosion, and induce chronic inflammatory reaction. Given the unique role of NETs in GA, targeting NETs is considered a highly attractive and promising therapeutic approach for treating GA.
3.2 NETs and SLE
SLE is a complex multi-system autoimmune disease involving epigenetic, genetic, ecological, and environmental factors (47). It is characterized by the presence of autoantibodies targeting nuclear and cytoplasmic antigens, with antinuclear antibody serving as a key serological marker (48). Patients with SLE produce a variety of autoantibodies, among which anti-dsDNA antibodies are highly specific and contribute to disease pathogenesis (49). NETs play a crucial role in SLE through multiple mechanisms. Anti-dsDNA antibodies can be components of NETs, and impaired NETs clearance—due to deoxyribonuclease (DNase) inhibition, DNase inhibitors, or anti-NET antibodies—leads to NETs accumulation, further elevating anti-dsDNA antibody titers and activating the complement system, thereby perpetuating a vicious cycle of inflammation (50).
Studies have indicated that levels of NETs are elevated in patients with SLE. NET-associated proteins, such as LL-37, can promote hyperactivation of the NLRP3 inflammasome in adjacent macrophages, leading to the release of large quantities of inflammatory cytokines and thereby causing severe tissue damage (51). In this process, released IL-18 further stimulates neutrophils to produce more NETs (52). Moreover, inflammasome-mediated activation of gasdermin D is also a key factor in NET formation (53, 54). Additionally, NETs can lower the activation threshold of T cells and promote T cell activation via T cell receptor signaling, thereby linking innate and adaptive immune responses (55). NETs in SLE are enriched in ox-mtDNA and citrullinated histones, which act as potent autoantigens. These structures activate Toll-like receptors and intracellular nucleic acid sensors, triggering Interferon Alpha (IFN-α) production by plasmacytoid dendritic cells (pDCs). IFN-α promotes dendritic cell maturation, T-cell activation, and autoantibody production by B cells, further stimulating NETs release (56, 57).
LDNs are a distinct subpopulation of neutrophils in patients with SLE (58). LDNs is significantly increased in the peripheral blood of SLE patients. LDNs from SLE patients demonstrate the capacity to activate T cells and induce the release of cytokines, including IFN-γ and TNF-α, a function not exhibited by their normal-density neutrophil counterparts (59). CD10+ LDNs exhibit a mature polymorphonuclear morphology, express high levels of type I interferon-stimulated genes (ISG15, MX1) and proinflammatory cytokines (IL-6, IL-8, TNF-α), and demonstrate enhanced NET-forming capacity (60). These NETs contain ox-mtDNA and citrullinated histone H4, which not only act as autoantigens driving anti-dsDNA antibody production via B-cell TLR9 activation but also stimulate the NLRP3 inflammasome in macrophages, promoting pyroptosis and IL-1β/IL-18 release (51). This cascade further potentiates Neutrophil Extracellular Traposis (NETosis), while IFN-α released from pDCs feedback enhances NETs formation and impairs endothelial repair, establishing a self-sustaining inflammatory loop.
Given the core role of NETs in the pathogenesis of SLE and their detectability in patient serum and tissues, NETs are expected to serve as biomarkers for disease activity, organ involvement, and treatment response.
3.3 NETs and RA
RA is a systemic inflammatory autoimmune disease characterized by joint inflammation and bone damage (61). It is characterized by persistent synovitis, systemic inflammation, the presence of autoantibodies, and the production of a large number of inflammatory cytokines, which can lead to articular cartilage and bone damage (62). The serological hallmark of RA is anti-citrullinated protein antibodies (ACPA) (63). Citrullinated histones are thought to be a persistent source of B cell antigens that promote the production of new ACPA (64).
It was found that the synovial fluid in RA patients was infiltrated by neutrophils, which were prone to form NETs (65, 66). When neutrophils are activated, a large number of histones are citrullinated by PAD4, which is a key step in chromatin decondensation and NETs release (67, 68). About 70% of the proteins in NETs are histones (69). Citrullinated antigens on NETs play a critical role in initiating and perpetuating autoimmunity and ACPA production (70). Therefore, ACPA-related immune responses and the formation of NETs play an important role in the pathogenesis of RA. Studies have shown that circulating neutrophils in patients with RA are more likely than those in healthy subjects to undergo NETosis (71, 72). As in other autoimmune diseases, NETs act as a source of extracellular autoantigens, leading to excessive innate and adaptive immune responses within the joint and subsequent tissue damage.
NETs promote synovial inflammation by stimulating the release of pro-inflammatory cytokines such as IL-6, IL-8, TNF-α, and IL-17 from macrophages and fibroblast-like synoviocytes (FLSs) (73). They also enhance cartilage damage through the internalization of arthritogenic peptides by FLSs via the RAGE-TLR9 pathway, upregulation of MHC class II, and activation of T cells and B cells, leading to ACPA production and inflammatory spread. Additionally, NET-derived enzymes such as NE, matrix metalloproteinase-8 (MMP8), and MMP9 contribute to cartilage matrix degradation (65, 74). Activated neutrophils release B-cell Activating Factor (BAFF) and activate B cells (75). Then, the activated B cells release cytokines to cascade with other immune cells, and B-cell-derived IL-8 recruits neutrophils to the synovial membrane (76). Additionally, B cells, with the help of T cells, promote the production of auto-antibodies. Some of these plasma cells produce a large number of auto-antibodies, including RF and ACPA; these formed immune complexes activate the complement pathway and promote inflammation, which is particularly abundant in RA (77, 78).
Studies have confirmed that the release of NETs exacerbates the occurrence and development of RA (79). ACPA, rheumatoid factor, and inflammatory cytokines (TNF-α, IL-17) can enhance the formation of NETs. In the pathogenesis of RA, NETs repeatedly stimulate the body to produce autoimmune responses through exposure to autoantigens, which aggravates NETosis, forming a vicious cycle and leading to a sustained inflammatory response. In vitro and in vivo experiments have shown that during the pathogenesis of RA, neutrophils undergo significant activation and death, inducing the formation of NETs and thus exacerbating their own apoptosis (80). Therefore, inhibiting the formation of NETs may provide a new direction for the treatment of RA.
3.4 NETs and Psoriasis
Psoriasis is a chronic inflammatory systemic disease with a genetic basis, characterized by symmetrical erythematous skin lesions covered with silvery-white scales (81, 82). While its exact cause remains unknown, neutrophils are among the earliest cells infiltrating nascent psoriatic plaques, and their epidermal accumulation is a hallmark of the disease (83).
Recent evidence highlights the significant role of NETs in psoriasis pathogenesis. Stimulating keratinocytes to produce high levels of various inflammatory mediators induces TLR4 expression (84). The endogenous neutrophil-derived TLR4 ligand then acts synergistically with IL-36 to induce the production of LCN2 through MyD88 and NF-kB activation signaling (85). In turn, upregulated LCN2 regulates the formation of NETs and neutrophil migration, enhancing and maintaining the inflammatory response (86). Circulating neutrophils in psoriasis patients exhibit a heightened tendency for spontaneous or stimulus-induced NETosis, which correlates with disease severity (87). These neutrophils, including an increased population of LDNs, are primed for NETs release (88). Notably, exosomes derived from keratinocytes treated with psoriasis-related cytokines (e.g., IL-17A, IL-22, IFN-γ, TNF-α) can activate normal neutrophils, leading to NETs formation via NF-κB and p38 mitogen-activated protein kinase (MAPK) signaling pathways (89).
Within psoriatic lesions, NETotic neutrophils are found in both the epidermis (e.g., within Munro’s microabscesses) and dermis. These neutrophils can produce IL-17 through NETs formation, contributing to further neutrophil recruitment and sustained inflammation (90). NETs also facilitate Th17 cell differentiation and induce the expression of antimicrobial peptides like human β-defensin-2 in keratinocytes (87, 91). Moreover, NET-derived components form complexes such as DNA/cathepsin G/secretory leukocyte protease inhibitor and RNA/LL-37, which activate pDCs and neighboring neutrophils, respectively (92, 93). These interactions promote the production of type I interferons and proinflammatory cytokines, amplifying the inflammatory cascade.Interestingly, dimethylfumarate—a therapeutic agent for psoriasis—inhibits neutrophil activation, including NETs formation, suggesting that targeting NETosis may be a viable treatment strategy.
3.5 NETs and AAV
AAV is a systemic necrotizing small vasculitis that includes Wegener’s granulomatosis, eosinophilic granuloma with vasculitis, and microvasculitis (94). ANCA is a specific antibody that targets MPO and Recombinant Proteinase 3 (PR3) (95). The study suggests that ANCA may be involved in the activation of NETs formation in patients with AAV. AAV occurs when ANCA binds to autoantigens PR3 and MPO, which are granular proteins found on the surface of neutrophils that are associated with GPA and MPA, respectively (96). NETs also contain the targeted antigen MPO (stored within neutrophilic granulocyte) or PR3 (expressed on the membrane of resting neutrophils) whose expression rises when neutrophils are activated by cytokines (97). Studies have shown that NETs is also modified by MPO and PR3 in vitro and in vivo immunofluorescence in AAV necrotic lesions (98). For example, the co-localization of DNA, MPO, and PR3 in the kidney tissue of patients with small vasculitis (SVV) glomerulonephritis indicates the presence of NETs and ANCA antigens in the inflammatory tissue (99). In patients with low NETs degradation activity, these NETs persist, particularly MPO and PR3. These antigens are presented to CD4+T cells via dendritic cells, producing ANCA (100). Neutrophils express MPO and PR3 on the plasma membrane, and PR3-ancas and MPO-ANCAs bind to them. At the same time, these crystallizable fragment (Fc) regions of ANCA bind to the Fc-γ receptor on neutrophils (101). This binding induces hyperactivation of neutrophils, resulting in abnormal cytokine production, while releasing ROS and lyases, which further form NETs and damage vascular endothelial cells. In addition to ANCA, BAFF are produced by activated neutrophils, and CD4+T cells (via IL-21) stimulate B cells, enabling continuous ANCA production (102). The study found that higher levels of MPO-DNA were detected in the serum of patients with active AAV compared to those with AAV in remission (103). However, another study did not find a difference in serum MPO-DNA levels between patients with active AAV and those with AAV in remission (104). This discrepancy may be related to differences in NETs clearance capacity among individual patients, such as reduced serum DNase I activity or impaired macrophage phagocytic function, which can lead to NETs accumulation that does not fully correlate with current clinical activity (105, 106). These findings indicate that while NETs are clearly present in patients with AAV, their utility as biomarkers for assessing disease activity remains to be determined. Furthermore, despite the potential of NET-associated biomarkers (e.g., MPO-DNA, citrullinated histones, and cell-free DNA) to aid in diagnosis, prognostic evaluation, and relapse prediction, further standardization and validation are still required.
NETs contribute to the progression of AAV through multiple mechanisms. On one hand, NETs are not only involved in the initiation of ANCA autoimmune responses but also directly cause vascular damage via histone-mediated cytotoxicity (107). On the other hand, the persistence of NETs is closely associated with an imbalance in their clearance. Studies have demonstrated that the endogenous degrading factor DNase1 can effectively degrade NETs, while intravenous immunoglobulin (IVIG) exhibits therapeutic potential by significantly inhibiting NET formation (108, 109). In therapeutic approaches, targeted strategies addressing NETs formation and clearance have emerged as research hotspots, including: C5a receptor antagonists (e.g., Avacopan), Syk inhibitors, PAD4 inhibitors, and recombinant DNase I. To sum up, NETs play an extremely important role in the pathogenesis of AAV. NETs can be used as the information of disease diagnosis and the target of future treatment. Effective intervention in the formation of NETs is expected to provide new ideas for the treatment of autoimmune vasculitis.
3.6 NETs and APS
APS is an autoimmune disorder associated with elevated levels of antiphospholipid antibodies (aPL), characterized by arterial, venous, or small vessel thrombosis or recurrent early pregnancy loss, fetal loss (110). aPL is a general term for antibodies to phospholipids and surface proteins, including lupus anticoagulants, anti-β2-glycoprotein-1, and anti-cardiolipin (111, 112). aPL is known to promote thrombosis by activating endothelial cells, monocytes, and platelets. Several mechanisms contribute to the release of NETs in APS. Anti-β2GP1 antibodies recognize β2GP1 bound to the surface of neutrophils, leading to the upregulation of adhesion molecules PSGL-1 and Mac-1 (113). This enhances their adherence to the endothelium. Subsequently, under the influence of this endothelial adhesion, TLR4 signaling, and potential interferon stimulation, APS neutrophils become activated and release ROS and NETs (114). Furthermore, anti-NETs antibodies present in APS may impair the clearance of NETs by inhibiting circulating DNase, preventing their effective degradation (115, 116). Studies have found that NETs play a key role in the involvement of platelets and neutrophils in the formation, stabilization, and growth of peripheral and coronary thrombosis (117). In patients with APS, increased NETs release is associated with autoimmunity and inflammation, driven by stimuli such as immune complexes, autoantibodies and complement activation (118). Seminal work by Yalavarthi et al. showed that IgG from APS patients stimulates NETosis in control neutrophils via mechanisms dependent on ROS and TLR4 signaling (119, 120). Moreover, impaired degradation of NETs—due to DNase inhibitors or anti-NET antibodies—further contributes to NETs persistence and thrombotic risk (121). Inhibition of NETs release may have potential benefits in patients with APS.
Experimental studies have demonstrated that serum and purified IgG isolated from patients with APS can induce neutrophils to release NETs (122). Furthermore, inhibition of ROS production or blockade of TLR4 signaling has been shown to reduce NET formation (123). In an animal model of APS, administration of patient-derived IgG was associated with increased thrombosis; conversely, degradation of NETs via DNase I treatment or depletion of neutrophils significantly attenuated thrombotic events (124). These findings suggest that modulating NETs formation or enhancing their clearance may represent a promising therapeutic strategy for APS.
3.7 NETs and T1D
T1DM is an autoimmune disease characterized by destruction of islet β cells, characterized by elevated blood glucose levels, often accompanied by absolute lack of endogenous insulin (125). Although the pathogenesis of T1DM is unknown, physiologic β cell death is a predisposing factor in the development of the disease through recruitment and activation of neutrophils, which penetrate the pancreas. In the pancreas, neutrophils can release CRAMP, pDCs can be induced to produce interferons alpha (126). The interaction between immune cells is necessary to induce a diabetic T cell response that subsequently leads to the development of T1DM (127). In addition, interactions between neutrophils and other non-immune cells, such as platelets in the blood or vascular endothelial cells, are thought to play an important role in the microvascular and macrovascular complications of diabetes. The number of circulating neutrophils decreased in T1DM patients and high-risk subjects before symptoms. Previous studies have shown that neutrophils infiltrate the pancreas before onset and form NETs within it, exhibiting strong pro-inflammatory biological activity (128). NETs is an important link for neutrophils to participate in the occurrence and development of T1DM, T1DM neutrophils express high levels of PAD4 and produce more NETs. In human T1DM, reduced circulating neutrophils and elevated NETs markers (e.g., NE, PR3, CitH3) correlate with autoantibody levels and beta cell loss (129). NETs and histones directly damage human islets in vitro, an effect reversible with polyanions like Mcbs (130).In the serum of children 10 days after T1DM onset, the levels of NETs, mtDNA and nuclear DNA in peripheral blood were higher than those in healthy children, and T1DM serum could induce normal neutrophils to form NETs (131). However, some studies have found that the levels of NE and PR3 in T1DM subjects decreased significantly, especially in the subjects within three years after diagnosis (132). The levels of NE and PR3 were correlated with the absolute neutrophil count. This may reflect disease stage-dependent changes in neutrophil activity. In the study of T1DM, non-obese diabetic (NOD) mice can spontaneously be T1DM, which is often used to study the pathogenesis and intervention of T1DM (133). You et al. found that the formation of PAD4 dependent NETs is involved in the aggravation of intestinal barrier dysfunction, the production of autoantibodies and the activation of intestinal autoimmune T cells in DSS-induced colitis and PAD4 knockout experiments in NOD mice, and then these cells migrated to pancreatic lymph nodes to cause injury (134). Neutrophils also promote early autoimmunity in NOD mice via pDC activation and IFN production (135). In female NOD mice, physiological β cell death induced the recruitment and activation of B-1a cells, neutrophils and plasma cell-like dendritic cells (pDC) to the pancreas (136). Activated Bmur1a cells secrete double-stranded DNA-specific IgG and activate neutrophils to form NETs. This DNA-specific IgG activates pDCs through Toll-like receptors, resulting in the production of IFN-α in islets and the formation of T1DM (137). Notably, PAD4 inhibition prevents diabetes in NOD mice, underscoring the role of NETosis (138). NETs impair wound healing and are more pronounced in T1DM, and inhibiting NETs may improve wound healing in diabetes and reduce NETs-driven chronic inflammation (139).
In T1DM, although the level of NETs is uncertain, the presence of NETs directly or indirectly activates innate and adaptive immune responses in the pancreas, damages islet β cells, and participates in the occurrence and development of T1DM. Accumulating evidence from humans and NOD models indicates NETs contribute to islet autoimmunity through cytotoxicity and immune activation. Therefore, the study of NETs may be one of the directions in the treatment of T1DM.
4 Drug intervention in NETs to treat autoimmune diseases
A variety of drugs have been used to treat autoimmune diseases in clinic, and their mechanism of action has been gradually explored. Studies have found that a variety of drugs may act on NETs to play a therapeutic.
4.1 NE and MPO inhibitors
NE and MPO are key synergistic molecules in the process of NETs formation, as well as core functional components of NETs structure. They collectively participate in immune defense and mediate the amplification of inflammation and tissue damage during the pathogenesis of diseases. Therefore, inhibitors targeting NE and MPO have emerged as potential therapeutic strategies, some of which have advanced into clinical research.
Among NE inhibitors, Sivelestat is a selective, reversible, and competitive small-molecule inhibitor that suppresses NE enzymatic activity by binding to its active site, thereby reducing NETs formation and mitigating inflammatory responses and tissue injury (140). Studies have shown that early administration of Sivelestat in diabetic mouse models significantly reduces the incidence of spontaneous insulitis and autoimmune diabetes (141). Furthermore, this compound has demonstrated therapeutic potential in various animal models of acute respiratory distress syndrome, sepsis, non-alcoholic steatohepatitis, and acute lung injury. Other NE inhibitors that have entered clinical stages include POL6014, PHP-303, Elafin, CHF6333, and alvelestat, all of which inhibit NE activity through a similar competitive mechanism (142–145). On the other hand, MPO inhibitors such as PF-1355 can significantly reduce MPO activity in mouse plasma, thereby inhibiting neutrophil recruitment and vascular edema, and have been used in basic research on immune complex-mediated vasculitis (146). In addition, ceruloplasmin has been shown to decrease plasma MPO activity in mice and inhibit the production of MPO-derived oxidants during inflammation, demonstrating protective effects (147). Recent studies have also indicated that ABAH reduces MPO-dependent hepatocyte death in a non-alcoholic steatohepatitis model, decreases MPO activity in a mouse model of acute stroke, and inhibits MPO activity in sputum from pulmonary cystic fibrosis (148–150). Similarly, compounds such as INV-315, PF-0628999, and AZM198 alleviate inflammatory responses by inhibiting MPO activity (151, 152).
In summary, NE and MPO inhibitors exhibit promising therapeutic effects in various disease models by regulating NETs formation and neutrophil-mediated inflammatory responses. Some compounds have progressed to clinical research stages, offering new directions for the treatment of related inflammatory and autoimmune diseases.
4.2 DNase I
DNA serves as the primary structural framework of NETs. DNase I is an enzyme capable of degrading DNA, effectively breaking down the DNA component within NETs, thereby reducing NETs formation (153). Although the early use of recombinant human DNase I (rhDNase I) in the treatment of SLE demonstrated a favourable safety profile, its clinical efficacy was limited; nevertheless, it has been approved for the treatment of cystic fibrosis (CF) (154). Recently, a novel bioenzyme with dual DNase1/DNase1L3 activity has shown significant effects in murine lupus models, effectively suppressing autoantibody production and resisting neutralization by autoantibodies in SLE patients (155). In RA patients, DNase I can also inhibit neutrophil NET generation and mitigate NET-induced thrombosis and endothelial damage (79). On the other hand, advances in production technology have provided crucial support for the clinical application of DNase I. Recent studies indicate that the use of a Pichia pastoris expression system enables successful recombinant production of active human DNase I (156). This breakthrough is expected to substantially reduce manufacturing costs and lay the foundation for large-scale applications in various NET-related diseases.
4.3 Targeted IFN preparations
Therapeutic targeting of IFN signaling can reduce NET-induced inflammation and autoimmune responses. For instance, both the JAK inhibitor tofacitinib and the type I IFN inhibitor anifrolumab have been demonstrated in clinical studies to lower NETs levels in SLE patients and improve their clinical symptoms (157, 158).
Current biologic agents for SLE treatment primarily consist of monoclonal antibodies that directly target either IFN-α or the type I IFN receptor (IFNAR). Sifalimumab and rontalizumab are two anti-IFN-α monoclonal antibodies. Among them, sifalimumab has been shown to significantly reduce SLE disease activity, whereas rontalizumab did not demonstrate notable efficacy—the underlying mechanisms remain unclear (159, 160). Anifrolumab, an anti-IFNAR monoclonal antibody, has been approved by the U.S. FDA and the European Union for the treatment of moderate to severe SLE (161). Additionally, QX006N is another monoclonal antibody targeting IFNAR1. It specifically binds to the SD3 domain of IFNAR1, creating steric hindrance that prevents the binding of type I IFN ligands and inhibits the assembly of the IFN/IFNAR1/IFNAR2 complex (162). This agent is currently under investigation for SLE therapy.
4.4 PAD4 inhibitor
PAD4 serves as a nuclear promoter that mediates citrullination of histone H3 in neutrophils, facilitating chromatin decondensation and promoting NET formation. Studies have shown that inhibiting PAD4 to suppress NETosis confers protective effects in mouse models of lupus, diabetes, and atherosclerosis without significant adverse events. Cl-amidine inhibits the citrullination of histone H3 by irreversibly binding to PAD4, thereby restraining NET formation (163).Research indicates that Cl-amidine alleviates endothelial dysfunction in SLE mice and reduces the deposition of immune complexes in renal tissues (164). Furthermore, Cl-amidine suppresses the production of NETs and inflammatory cytokines by reducing PAD4 levels in the joint tissues of arthritic mice, thereby ameliorating joint edema (165). Meanwhile, in vivo studies demonstrate that GSK484, a reversible PAD4 inhibitor, also inhibits NET release and exerts immunomodulatory effects. It enhances radiosensitivity in colorectal cancer by promoting DNA double-strand breaks and suppresses NET formation both in vivo and in vitro (166). Administration of GSK484 in CIA mice reduces the expression of synovial MPO, NE, and PAD4, decreases NET generation, attenuates arthritis severity, and inhibits macrophage infiltration, supporting its therapeutic potential (167). In various lupus models, PAD inhibitors can reduce NETs formation while protecting the vasculature, kidneys, and skin from damage. The selective PAD4 inhibitor JBI-589 blocks NET formation and PAD4-dependent citrullination; oral administration in mouse models reduces the incidence and severity of arthritis and inhibits ACPA production (168).
4.5 ROS remover
ROS are essential for the formation of NETs. A range of ROS scavengers have demonstrated therapeutic potential in autoimmune diseases. As a scavenger of ROS, N-acetylcysteine (NAC) has been observed to reduce NET generation upon treatment (169). In two clinical studies, NAC administration improved disease outcomes in SLE patients, though related mechanistic investigations remain at an early stage (170). Moreover, MitoTempo, a specific scavenger of mitochondrial ROS, prevented spontaneous NETosis and reduced disease severity in a lupus mouse model (171). Ethyl pyruvate attenuates NET formation and sepsis-induced intestinal injury by inhibiting ROS-mediated activation of MAPK/ERK1/2 and p38 MAPK (172). Additionally, other agents targeting ROS also exhibit efficacy. For instance, diphenyleneiodonium demonstrates significant anti-tumor activity in MYCN-amplified neuroblastoma by targeting MYCN-induced mitochondrial alterations and ROS production, thereby inducing apoptosis and suppressing tumor growth (173).
4.6 Traditional Chinese medicine compounds
Traditional Chinese medicine (TCM) represents a precious treasure endowed by nature and is increasingly demonstrating its therapeutic value. Numerous compounds derived from TCM that are currently under preclinical investigation have shown potential for targeting NETs. For instance, triptolide has been found to inhibit NET generation in vitro independently of cellular ROS levels (174). In a murine model of RA, it suppressed neutrophil autophagy, NET formation, tissue damage, and inflammation (175). Similarly, tetrandrine exhibited therapeutic effects in an RA model by modulating neutrophil-associated inflammatory responses and inhibiting NET formation (176). Quercetin was shown to reduce neutrophil infiltration, plasma cytokine levels, and autophagy-dependent NET formation (177). Andrographolide decreased joint levels of CXCL2, MPO, and NE, while also reducing neutrophil infiltration in ankle tissues (178). The classical TCM formula Simiao Yong’an Tang inhibited neutrophil migration, promoted apoptosis, and reduced ROS production and NET formation in vitro (179). Additionally, emodin alleviated arthritis in AA mice by diminishing neutrophil infiltration, inhibiting the release of pro-inflammatory cytokines (IL-6, IFN-γ, and TNF-α), suppressing autophagy-mediated NETosis, and promoting neutrophil apoptosis (180). Furthermore, our recent study demonstrated that Ermiao San and its primary active components (phellodendrine and atractylenolide-I) exert therapeutic effects against RA by suppressing PAD4 to reduce the formation of NETs (181).
5 Conclusion
The burgeoning field of NETosis has fundamentally redefined our understanding of autoimmune pathogenesis, establishing NETs not merely as inflammatory effectors but as central orchestrators that bridge innate and adaptive immunity. NETs contribute to autoimmunity through multiple mechanisms: they serve as a source of autoantigens, amplify inflammatory cascades, activate innate and adaptive immune pathways via Toll-like receptors, inflammasomes, and type I interferon responses, and directly cause tissue damage through cytotoxic components. Their involvement across various autoimmune diseases, including SLE, RA, APS, and T1DM, highlights a shared pathological mechanism rooted in dysregulated NET formation and clearance. Therapeutic strategies targeting NETs, such as inhibitors (e.g., PAD4 inhibitors, neutrophil elastase inhibitors, myeloperoxidase inhibitors, reactive oxygen species (ROS) inhibitors), DNase-based interventions, and biologics targeting interferon signaling pathways, have demonstrated significant potential in both preclinical and clinical studies. Additionally, multi-omics-driven biomarker discovery and exploration of the microbiome–NET axis hold promise for improving diagnosis, subtyping, and personalized treatment. The integration of advanced technologies—such as single-cell analysis, real-time NET imaging, and neutrophil engineering—will be crucial to translate these mechanistic insights into precise clinical interventions, ultimately revolutionizing the management of autoimmune diseases.
Author contributions
RT: Writing – original draft. JY: Writing – original draft, Writing – review & editing. ZQ: Writing – original draft. MZ: Writing – review & editing. XJ: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82074090, 81603362), and Natural Science Foundation of Anhui Province (No. 1808085MH298).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
AAV: antineutrophil cytoplasmic antibody -associated vasculitis
LL-37: Cathelicidin Antimicrobial Peptide
ACPA: anti-citrullinated protein antibodies
MAPK: mitogen-activated protein kinase
aggNETs: aggregated NETs
MMP8: matrix metalloproteinase-8
ANCA: antineutrophil cytoplasmic antibody
MMP9: matrix metalloproteinase-9
aPL: antiphospholipid antibodies
MPO: myeloperoxidase
aPL: antiphospholipid antibodies
MSU: monosodium urate
APS: antiphospholipid syndrome
NE: neutrophil elastase
BAFF: B-cell Activating Factor
NETosis: Neutrophil Extracellular Traposis
CCL3: C-C chemokine ligand 3
NETs: Neutrophil extracellular traps
CF: cystic fibrosis
NF-κB: Nuclear factor kappa-light-chain-enhancer of activated B cells
CRAMP: Dopamine-Cathelicidin-related antimicrobial peptide
NLRP3: the Pyrin Domain Containing Protein 3
CXCL10: C-X-C motif chemokine ligand 10
NOD: non-obese diabetic
DAMPs: hazardous associated molecular patterns
NOX: nicotinamide adenine dinucleotide phosphate oxidase
DNase: deoxyribonuclease
PAD4: peptidyl arginine deiminase 4
FLSs: fibroblast-like synoviocytes
pDCs: plasmacytoid dendritic cells
GA: gouty arthritis
PKC: protein kinase C
IFN-α: Interferon Alpha
PMA: Phorbol-12-myristate-13-acetate
IFN-γ: Interferon-gamma
PR3: Proteinase 3
IFN-γ: Interferon-gamma
PR3: Recombinant Proteinase 3
IL-17: Interleukin-17
RA: rheumatoid arthritis
IL-17A: Interleukin-17A
rhDNase I: recombinant human DNase I
IL-18: Interleukin-18
ROS: reactive oxygen species
IL-1β: Interleukin-1β
SLE: systemic lupus erythematosus
IL-21: Interleukin-21
T1DM: type 1 diabetes mellitus
IL-22: Interleukin-22
TLR4: Toll Like Receptor 4
IL-6: Interleukin-6
TLR7: Toll Like Receptor 7
IL-8: Interleukin-8
TLR8: Toll Like Receptor 8
IVIG: Intravenous immunoglobulin
TLR9: Toll Like Receptor 9
LDNs: low-density neutrophils
TNF-α: Tumor Necrosis Factor alpha
References
1. Nathan C. Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol. (2006) 6:173–82. doi: 10.1038/nri1785
2. Mutua V and Gershwin LJ. A review of neutrophil extracellular traps (NETs) in disease: potential anti-NETs therapeutics. Clin Rev Allergy Immunol. (2021) 61:194–211. doi: 10.1007/s12016-020-08804-7
3. Liu L and Sun B. Neutrophil pyroptosis: new perspectives on sepsis. Cell Mol Life Sci. (2019) 76:2031–42. doi: 10.1007/s00018-019-03060-1USA
4. Basyreva LY, Voinova EV, Gusev AA, Mikhalchik EV, Kuskov AN, Goryachaya AV, et al. Fluorouracil neutrophil extracellular traps formation inhibited by polymer nanoparticle shielding. Mater Sci Eng C Mater Biol Appl. (2020) 108:110382. doi: 10.1016/j.msec.2019.110382
5. Li T, Wang C, Liu Y, Li B, Zhang W, Wang L, et al. Neutrophil extracellular traps induce intestinal damage and thrombotic tendency in inflammatory bowel disease. J Crohns Colitis. (2020) 14:240–53. doi: 10.1093/ecco-jcc/jjz132
6. Müller-Redetzky H. Targeting neutrophil extracellular traps in acute lung injury: a novel therapeutic approach in acute respiratory distress syndrome? Anesthesiology. (2015) 122:725–7. doi: 10.1097/ALN.0000000000000604
7. Carmona-Rivera C and Kaplan MJ. Low-density granulocytes: a distinct class of neutrophils in systemic autoimmunity. Semin Immunopathol. (2013) 35:455–63. doi: 10.1007/s00281-013-0375-7
8. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol. (2011) 187:538–52. doi: 10.4049/jimmunol.1100450
9. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
10. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, et al. Neutrophil extracellular traps exert both pro- and anti-inflammatory actions in rheumatoid arthritis that are modulated by C1q and LL-37. J Autoimmun. (2019) 98:122–31. doi: 10.1016/j.jaut.2019.01.003
11. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
12. Zhang Z, Niu R, Zhao L, Wang Y, and Liu G. Mechanisms of neutrophil extracellular trap formation and regulation in cancers. Int J Mol Sci. (2023) 24:10265. doi: 10.3390/ijms241210265
13. Branzk N and Papayannopoulos V. Molecular mechanisms regulating NETosis in infection and disease. Semin Immunopathol. (2013) 35:513–30. doi: 10.1007/s00281-013-0384-6
14. Cheng OZ and Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Front Immunol. (2013) 4:1. doi: 10.3389/fimmu.2013.00001
15. Vazquez-Torres A, Xu Y, Jones-Carson J, Holden DW, Lucia SM, Dinauer MC, et al. Salmonella pathogenicity island 2-dependent evasion of the phagocyte NADPH oxidase. Science. (2000) 287:1655–8. doi: 10.1126/science.287.5458.1655
16. Li XJ, Marchal CC, Stull ND, Stahelin RV, and Dinauer MC. p47phox Phox homology domain regulates plasma membrane but not phagosome neutrophil NADPH oxidase activation. J Biol Chem. (2010) 285:35169–79. doi: 10.1074/jbc.M110.164475
17. Liu L, Mao Y, Xu B, Zhang X, Fang C, Ma Y, et al. Induction of neutrophil extracellular traps during tissue injury: Involvement of STING and Toll-like receptor 9 pathways. Cell Prolif. (2019) 52:e12579. doi: 10.1111/cpr.12579
18. De Volder J, Bontinck A, De Grove K, Dirven I, Haelterman V, Joos G, et al. Trajectory of neutrophilic responses in a mouse model of pollutant-aggravated allergic asthma. Environ pollut. (2023) 329:121722. doi: 10.1016/j.envpol.2023.121722
19. Wang Y, Wysocka J, Sayegh J, Lee YH, Perlin JR, Leonelli L, et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science. (2004) 306:279–83. doi: 10.1126/science.1101400
20. Burgener SS and Schroder K. Neutrophil extracellular traps in host defense. Cold Spring Harb Perspect Biol. (2020) 12:a037028. doi: 10.1101/cshperspect.a037028
21. Brinkmann V and Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol. (2012) 198:773–83. doi: 10.1083/jcb.201203170
22. Neumann A, Berends ET, Nerlich A, Molhoek EM, Gallo RL, Meerloo T, et al. The antimicrobial peptide LL-37 facilitates the formation of neutrophil extracellular traps. Biochem J. (2014) 464:3–11. doi: 10.1042/BJ20140778
23. HIRSCH JG. Bactericidal action of histone. J Exp Med. (1958) 108:925–44. doi: 10.1084/jem.108.6.925
24. Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. (2012) 12:109–16. doi: 10.1016/j.chom.2012.05.015
25. Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, et al. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol. (2011) 179:199–210. doi: 10.1016/j.ajpath.2011.03.013
26. Muraro SP, De Souza GF, Gallo SW, Da Silva BK, De Oliveira SD, Vinolo MAR, et al. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci Rep. (2018) 8:14166. doi: 10.1038/s41598-018-32576-y
27. Hunter CA and Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol. (2012) 10:766–78. doi: 10.1038/nrmicro2858
28. Ribeiro-Gomes FL, Moniz-de-Souza MC, Alexandre-Moreira MS, Dias WB, Lopes MF, Nunes MP, et al. Neutrophils activate macrophages for intracellular killing of Leishmania major through recruitment of TLR4 by neutrophil elastase. J Immunol. (2007) 179:3988–94. doi: 10.4049/jimmunol.179.6.3988
29. Goel RR and Kaplan MJ. Deadliest catch: neutrophil extracellular traps in autoimmunity. Curr Opin Rheumatol. (2020) 32:64–70. doi: 10.1097/BOR.0000000000000667
30. Kim TS, Silva LM, Theofilou VI, Greenwell-Wild T, Li L, Williams DW, et al. Neutrophil extracellular traps and extracellular histones potentiate IL-17 inflammation in periodontitis. J Exp Med. (2023) 220:e20221751. doi: 10.1084/jem.20221751
31. Song W, Ye J, Pan N, Tan C, and Herrmann M. Neutrophil extracellular traps tied to rheumatoid arthritis: points to ponder. Front Immunol. (2021) 11:578129. doi: 10.3389/fimmu.2020.578129
32. Li RHL and Tablin F. A comparative review of neutrophil extracellular traps in sepsis. Front Vet Sci. (2018) 5:291. doi: 10.3389/fvets.2018.00291
33. Li RHL, Johnson LR, Kohen C, and Tablin F. A novel approach to identifying and quantifying neutrophil extracellular trap formation in septic dogs using immunofluorescence microscopy. BMC Vet Res. (2018) 14:210. doi: 10.1186/s12917-018-1523-z
34. Medeiros SK, Sharma N, Dwivedi D, and Liaw PC. Investigation of the pathological effects of histones, DNA, and nucleosomes in a murine model of sepsis. Shock. (2023) 60:291–7. doi: 10.1097/SHK.000000000000216
35. Middleton EA, He XY, Denorme F, Campbell RA, Ng D, Salvatore SP, et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood. (2020) 136:1169–79. doi: 10.1182/blood.2020007008
36. Klopf J, Brostjan C, Eilenberg W, and Neumayer C. Neutrophil extracellular traps and their implications in cardiovascular and inflammatory disease. Int J Mol Sci. (2021) 22:559. doi: 10.3390/ijms22020559
37. Bieber K, Hundt JE, Yu X, Ehlers M, Petersen F, Karsten CM, et al. Autoimmune pre-disease. Autoimmun Rev. (2023) 22:103236. doi: 10.1016/j.autrev.2022.103236
38. Liu YR, Wang JQ, and Li J. Role of NLRP3 in the pathogenesis and treatment of gout arthritis. Front Immunol. (2023) 14:1137822. doi: 10.3389/fimmu.2023.1137822
39. Galozzi P, Bindoli S, Doria A, Oliviero F, and Sfriso P. Autoinflammatory features in gouty arthritis. J Clin Med. (2021) 10:1880. doi: 10.3390/jcm10091880
40. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, et al. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PloS One. (2011) 6:e29318. doi: 10.1371/journal.pone.0029318
41. Maueröder C, Kienhöfer D, Hahn J, Schauer C, Manger B, Schett G, et al. How neutrophil extracellular traps orchestrate the local immune response in gout. J Mol Med (Berl). (2015) 93:727–34. doi: 10.1007/s00109-015-1295-x
42. Lu CH, Shen CY, Li KJ, Wu CH, Chen YH, Kuo YM, et al. esolution of acute inflammation induced by monosodium urate crystals (MSU) through neutrophil extracellular trap-MSU aggregate-mediated negative signaling. J Inflammation (Lond). (2024) 21:50. doi: 10.1186/s12950-024-00423-9
43. Schorn C, Frey B, Lauber K, Janko C, Strysio M, Keppeler H, et al. Sodium overload and water influx activate the NALP3 inflammasome. J Biol Chem. (2011) 286:35–41. doi: 10.1074/jbc.M110.139048
44. Chen YH, Chen WY, Yu CL, Tsai CY, and Hsieh SC. Gouty arthritis involves impairment of autophagic degradation via cathepsin D inactivation-mediated lysosomal dysfunction that promotes apoptosis in macrophages. Biochim Biophys Acta Mol Basis Dis. (2023) 1869:166703. doi: 10.1016/j.bbadis.2023.166703
45. Tao H, Mo Y, Liu W, and Wang H. A review on gout: Looking back and looking ahead. Int Immunopharmacol. (2023) 117:109977. doi: 10.1016/j.intimp.2023.109977
46. Manger B, Lell M, Wacker J, Schett G, and Rech J. Detection of periarticular urate deposits with dual energy CT in patients with acute gouty arthritis. Ann Rheum Dis. (2012) 71:470–2. doi: 10.1136/ard.2011.154054
47. Ameer MA, Chaudhry H, Mushtaq J, Khan OS, Babar M, Hashim T, et al. An overview of systemic lupus erythematosus (SLE) pathogenesis, classification, and management. Cureus. (2022) 14:e30330. doi: 10.7759/cureus.30330
48. Meroni PL, Bizzaro N, Cavazzana I, Borghi MO, and Tincani A. Automated tests of ANA immunofluorescence as throughput autoantibody detection technology: strengths and limitations. BMC Med. (2014) 12:38. doi: 10.1186/1741-7015-12-38
49. Bossuyt X, De Langhe E, Borghi MO, and Meroni PL. Understanding and interpreting antinuclear antibody tests in systemic rheumatic diseases. Nat Rev Rheumatol. (2020) 16:715–26. doi: 10.1038/s41584-020-00522-w
50. Lou H, Wojciak-Stothard B, Ruseva MM, Cook HT, Kelleher P, Pickering MC, et al. Autoantibody-dependent amplification of inflammation in SLE. Cell Death Dis. (2020) 11:729. doi: 10.1038/s41419-020-02928-6
51. Kahlenberg JM, Carmona-Rivera C, Smith CK, and Kaplan MJ. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. (2013) 190:1217–26. doi: 10.4049/jimmunol.1202388
52. Bao C, Liu B, Zhu R, Xiao J, Li Z, Jiang H, et al. IFN-γ-/- mice resist actinobacillus pleuropneumoniae infection by promoting early lung IL-18 release and PMN-I accumulation. Infect Immun. (2021) 89:e00069–21. doi: 10.1128/IAI.00069-21
53. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, et al. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6676. doi: 10.1126/sciimmunol.aar6676
54. Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR, Kinoshita T, et al. Gasdermin D mediates the maturation and release of IL-1α downstream of inflammasomes. Cell Rep. (2021) 34:108887. doi: 10.1016/j.celrep.2021.108887
55. Tsai CY, Hsieh SC, Liu CW, Lu CS, Wu CH, Liao HT, et al. Cross-Talk among Polymorphonuclear Neutrophils, Immune, and Non-Immune Cells via Released Cytokines, Granule Proteins, Microvesicles, and Neutrophil Extracellular Trap Formation: A Novel Concept of Biology and Pathobiology for Neutrophils. Int J Mol Sci. (2021) 22:3119. doi: 10.3390/ijms22063119
56. Corsiero E, Bombardieri M, Carlotti E, Pratesi F, Robinson W, Migliorini P, et al. Single cell cloning and recombinant monoclonal antibodies generation from RA synovial B cells reveal frequent targeting of citrullinated histones of NETs. Ann Rheum Dis. (2016) 75:1866–75. doi: 10.1136/annrheumdis-2015-208356
57. Georgakis S, Gkirtzimanaki K, Papadaki G, Gakiopoulou H, Drakos E, Eloranta ML, et al. NETs decorated with bioactive IL-33 infiltrate inflamed tissues and induce IFN-α production in patients with SLE. JCI Insight. (2021) 6:e147671. doi: 10.1172/jci.insight.147671
58. McKenna E, Mhaonaigh AU, Wubben R, Dwivedi A, Hurley T, Kelly LA, et al. Neutrophils: need for standardized nomenclature. Front Immunol. (2021) 12:602963. doi: 10.3389/fimmu.2021.602963
59. Rahman S, Sagar D, Hanna RN, Lightfoot YL, Mistry P, Smith CK, et al. Low-density granulocytes activate T cells and demonstrate a non-suppressive role in systemic lupus erythematosus. Ann Rheum Dis. (2019) 78:957–66. doi: 10.1136/annrheumdis-2018-214620
60. Mistry P, Nakabo S, O'Neil L, Goel RR, Jiang K, Carmona-Rivera C, et al. Transcriptomic, epigenetic, and functional analyses implicate neutrophil diversity in the pathogenesis of systemic lupus erythematosus. Proc Natl Acad Sci USA. (2019) 116:25222–8. doi: 10.1073/pnas.1908576116
61. Zhao T, Wei Y, Zhu Y, Xie Z, Hai Q, Li Z, et al. Gut microbiota and rheumatoid arthritis: From pathogenesis to novel therapeutic opportunities. Front Immunol. (2022) 13:1007165. doi: 10.3389/fimmu.2022.1007165
62. Li K, Wang M, Zhao L, Liu Y, and Zhang X. ACPA-negative rheumatoid arthritis: From immune mechanisms to clinical translation. EBioMedicine. (2022) 83:104233. doi: 10.1016/j.ebiom.2022.104233
63. Wright HL, Lyon M, Chapman EA, Moots RJ, and Edwards SW. Rheumatoid arthritis synovial fluid neutrophils drive inflammation through production of chemokines, reactive oxygen species, and neutrophil extracellular traps. Front Immunol. (2021) 11:584116. doi: 10.3389/fimmu.2020.584116
64. Tedeschi SK, Cui J, Arkema EV, Robinson WH, Sokolove J, Lingampalli N, et al. Elevated BMI and antibodies to citrullinated proteins interact to increase rheumatoid arthritis risk and shorten time to diagnosis: A nested case-control study of women in the Nurses' Health Studies. Semin Arthritis Rheumatol. (2017) 46:692–8. doi: 10.1016/j.semarthrit.2016.09.001
65. Carmona-Rivera C, Carlucci PM, Moore E, Lingampalli N, Uchtenhagen H, James E, et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci Immunol. (2017) 2:eaag3358. doi: 10.1126/sciimmunol.aag3358
66. Hidalgo AI, Carretta MD, Alarcón P, Manosalva C, Müller A, Navarro M, et al. Pro-inflammatory mediators and neutrophils are increased in synovial fluid from heifers with acute ruminal acidosis. BMC Vet Res. (2019) 15:225. doi: 10.1186/s12917-019-1974-x
67. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. (2009) 184:205–13. doi: 10.1083/jcb.200806072
68. Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. PAD4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. (2012) 3:307. doi: 10.3389/fimmu.2012.00307
69. Thiam HR, Wong SL, Wagner DD, and Waterman CM. Cellular mechanisms of NETosis. Annu Rev Cell Dev Biol. (2020) 36:191–218. doi: 10.1146/annurev-cellbio-020520-111016
70. Wu S, Peng W, Liang X, and Wang W. Anti-citrullinated protein antibodies are associated with neutrophil extracellular trap formation in rheumatoid arthritis. J Clin Lab Anal. (2021) 35:e23662. doi: 10.1002/jcla.23662
71. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. (2013) 178):178ra40. doi: 10.1126/scitranslmed.3005580
72. Bach M, Moon J, Moore R, Pan T, Nelson JL, and Lood C. A neutrophil activation biomarker panel in prognosis and monitoring of patients with rheumatoid arthritis. Arthritis Rheumatol. (2020) 2:47–56. doi: 10.1002/art.41062
73. Tang J, Xia J, Gao H, Jiang R, Xiao L, Sheng H, et al. IL33-induced neutrophil extracellular traps (NETs) mediate a positive feedback loop for synovial inflammation and NET amplification in rheumatoid arthritis. Exp Mol Med. (2024) 6:2602–16. doi: 10.1038/s12276-024-01351-7
74. Carmona-Rivera C, Carlucci PM, Goel RR, James E, Brooks SR, Rims C, et al. Neutrophil extracellular traps mediate articular cartilage damage and enhance cartilage component immunogenicity in rheumatoid arthritis. JCI Insight. (2020) 13):e139388. doi: 10.1172/jci.insight.139388
75. Jung N, Bueb JL, Tolle F, and Bréchard S. Regulation of neutrophil pro-inflammatory functions sheds new light on the pathogenesis of rheumatoid arthritis. Biochem Pharmacol. (2019) 65:170–80. doi: 10.1016/j.bcp.2019.03.010
76. Wang T, Vasconcellos A, Marken J, Skopelja-Gardner S, Lood C, and Giltiay NV. Immune complex-driven neutrophil activation and BAFF release: a link to B cell responses in SLE. Lupus Sci Med. (2022) 1):e000709. doi: 10.1136/lupus-2022-000709
77. Karmakar U and Vermeren S. Crosstalk between B cells and neutrophils in rheumatoid arthritis. Immunology. (2021) 64:689–700. doi: 10.1111/imm.13412
78. Wang Q, Ma Y, Liu D, Zhang L, and Wei W. The roles of B cells and their interactions with fibroblast-like synoviocytes in the pathogenesis of rheumatoid arthritis. Int Arch Allergy Immunol. (2011) 55:205–11. doi: 10.1159/000321185
79. Wang N, Ma J, Song W, and Zhao C. An injectable hydrogel to disrupt neutrophil extracellular traps for treating rheumatoid arthritis. Drug Deliv. (2023) 0:2173332. doi: 10.1080/10717544.2023.2173332
80. Yang F, Luo X, Luo G, Zhai Z, Zhuang J, He J, et al. Inhibition of NET formation by polydatin protects against collagen-induced arthritis. Int Immunopharmacol. (2019) 7:105919. doi: 10.1016/j.intimp.2019.105919
81. Griffiths CE and Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. (2007) 70:263–71. doi: 10.1016/S0140-6736(07)61128-3
82. Cao T, Yuan X, Fang H, Chen J, Xue K, Li Z, et al. Neutrophil extracellular traps promote keratinocyte inflammation via AIM2 inflammasome and AIM2-XIAP in psoriasis. Exp Dermatol. (2023) 2:368–78. doi: 10.1111/exd.14711
83. Christophers E, Metzler G, and Röcken M. Bimodal immune activation in psoriasis. Br J Dermatol. (2014) 70:59–65. doi: 10.1111/bjd.12631
84. Choudhary V, Griffith S, Chen X, and Bollag WB. Pathogen-associated molecular pattern-induced TLR2 and TLR4 activation increases keratinocyte production of inflammatory mediators and is inhibited by phosphatidylglycerol. Mol Pharmacol. (2020) 7:324–35. doi: 10.1124/mol.119.118166
85. Shao S, Fang H, Dang E, Xue K, Zhang J, Li B, et al. Neutrophil extracellular traps promote inflammatory responses in psoriasis via activating epidermal TLR4/IL-36R crosstalk. Front Immunol. (2019) 0:746. doi: 10.3389/fimmu.2019.00746
86. Shao S, Cao T, Jin L, Li B, Fang H, Zhang J, et al. Increased lipocalin-2 contributes to the pathogenesis of psoriasis by modulating neutrophil chemotaxis and cytokine secretion. J Invest Dermatol. (2016) 36:1418–28. doi: 10.1016/j.jid.2016.03.002
87. Hu SC, Yu HS, Yen FL, Lin CL, Chen GS, and Lan CC. Neutrophil extracellular trap formation is increased in psoriasis and induces human β-defensin-2 production in epidermal keratinocytes. Sci Rep. (2016) 6:31119. doi: 10.1038/srep31119
88. Lin AM, Rubin CJ, Khandpur R, Wang JY, Riblett M, Yalavarthi S, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis. J Immunol. (2011) 187:490–500. doi: 10.4049/jimmunol.1100123
89. Jiang M, Fang H, Shao S, Dang E, Zhang J, Qiao P, et al. Keratinocyte exosomes activate neutrophils and enhance skin inflammation in psoriasis. FASEB J. (2019) 33:13241–53. doi: 10.1096/fj.201900642R
90. Saadh MJ, Ahmed HH, Kareem RA, Ballal S, Sharma S, Guntaj J, et al. Stem cell therapy: A new approach and effective treatment for psoriasis. Dermatol Pract Concept. (2025) 15:5333. doi: 10.5826/dpc.1503a5333
91. Peng G, Tsukamoto S, Ikutama R, Nguyen HLT, Umehara Y, JV T-P, et al. Human β-defensin-3 attenuates atopic dermatitis-like inflammation through autophagy activation and the aryl hydrocarbon receptor signaling pathway. J Clin Invest. (2022) 132:e156501. doi: 10.1172/JCI156501
92. Lande R, Mennella A, Palazzo R, Favaro R, Facheris P, Mancini F, et al. The nature of the post-translational modifications of the autoantigen LL37 influences the autoreactive T-helper cell phenotype in psoriasis. Front Immunol. (2025) 16:1546422. doi: 10.3389/fimmu.2025.1546422
93. Skrzeczynska-Moncznik J, Wlodarczyk A, Banas M, Kwitniewski M, Zabieglo K, Kapinska-Mrowiecka M, et al. DNA structures decorated with cathepsin G/secretory leukocyte proteinase inhibitor stimulate IFNI production by plasmacytoid dendritic cells. Am J Clin Exp Immunol. (2013) 2:186–94.
94. Sargin G. The evaluation of changing the eponym churg-strauss syndrome due to the 2012 revised international chapel hill consensus conference nomenclature of vasculitides. J Clin Med. (2024) 13:3424. doi: 10.3390/jcm13123424
95. Dereseviciene G, Dadoniene J, and Miltiniene D. Similarities and differences between patients diagnosed with ANCA-associated vasculitis who are positive and negative for ANCA: university clinic practice and expertise. Med (Kaunas). (2025) 61:1369. doi: 10.3390/medicina61081369
96. Pyo JY, Ahn SS, Song JJ, Park YB, and Lee SW. Reclassification of previously diagnosed GPA patients using the 2022 ACR/EULAR classification criteria. Rheumatol (Oxford). (2023) 62:1179–86. doi: 10.1093/rheumatology/keac267
97. Nakazawa D, Masuda S, Tomaru U, and Ishizu A. Pathogenesis and therapeutic interventions for ANCA-associated vasculitis. Nat Rev Rheumatol. (2019) 15:91–101. doi: 10.1038/s41584-018-0145-y
98. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, WL G, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. (2009) 15:623–5. doi: 10.1038/nm.1959
99. Zhang F, Zhang Q, Dang RN, Tian XX, Li LJ, Zhou QM, et al. Effects of environmentally-induced anxiety on autoimmunity in the MRL/lpr mouse. Lupus Sci Med. (2025) 12:e001528. doi: 10.1136/lupus-2025-001528
100. Fousert E, Toes R, and Desai J. Neutrophil extracellular traps (NETs) take the central stage in driving autoimmune responses. Cells. (2020) 9:915. doi: 10.3390/cells9040915
101. Wojcik I, Wuhrer M, Heeringa P, Stegeman CA, Rutgers A, and Falck D. Specific IgG glycosylation differences precede relapse in PR3-ANCA associated vasculitis patients with and without ANCA rise. Front Immunol. (2023) 14:1214945. doi: 10.3389/fimmu.2023.1214945
102. Banos A, Thomas K, Garantziotis P, Filia A, Malissovas N, Pieta A, et al. The genomic landscape of ANCA-associated vasculitis: Distinct transcriptional signatures, molecular endotypes and comparison with systemic lupus erythematosus. Front Immunol. (2023) 14:1072598. doi: 10.3389/fimmu.2023.1072598
103. Takeuchi S, Kawakami T, Okano T, Shida H, Nakazawa D, Tomaru U, et al. Elevated myeloperoxidase-DNA complex levels in sera of patients with igA vasculitis. Pathobiology. (2022) 89:23–8. doi: 10.1159/000519869
104. Wang H, Sha LL, Ma TT, Zhang LX, Chen M, and Zhao MH. Circulating level of neutrophil extracellular traps is not a useful biomarker for assessing disease activity in antineutrophil cytoplasmic antibody-associated vasculitis. PloS One. (2016) 11:e0148197. doi: 10.1371/journal.pone.0148197
105. Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, et al. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol. (2014) 25:990–7. doi: 10.1681/ASN.2013060606
106. Angeletti A, Volpi S, Bruschi M, Lugani F, Vaglio A, Prunotto M, et al. Neutrophil extracellular traps-DNase balance and autoimmunity. Cells. (2021) 10:2667. doi: 10.3390/cells10102667
107. Kumar SV, Kulkarni OP, Mulay SR, Darisipudi MN, Romoli S, Thomasova D, et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J am soc nephrol. (2015) 26:2399–413. doi: 10.1681/ASN.2014070673
108. Wang CY, TT L, Hu L, Xu CJ, Hu F, Wan L, et al. Neutrophil extracellular traps as a unique target in the treatment of chemotherapy-induced peripheral neuropathy. EBioMedicine. (2023) 90:104499. doi: 10.1016/j.ebiom.2023.104499
109. Uozumi R, Iguchi R, Masuda S, Nishibata Y, Nakazawa D, Tomaru U, et al. Pharmaceutical immunoglobulins reduce neutrophil extracellular trap formation and ameliorate the development of MPO-ANCA-associated vasculitis. Mod Rheumatol. (2020) 30:544–50. doi: 10.1080/14397595.2019.1602292
110. Salle V. Syndrome des antiphospholipides « séronégatif »: mythe ou réalité? [Seronegative antiphospholipid syndrome: Myth or reality]? Rev Med Interne. (2020) 41:265–74. doi: 10.1016/j.revmed.2020.02.005
111. Reshetnyak T, Nurbaeva K, Ptashnik I, Kudriaeva A, Belogurov A Jr, Lila A, et al. Markers of NETosis in patients with systemic lupus erythematosus and antiphospholipid syndrome. Int J Mol Sci. (2023) 24:9210. doi: 10.3390/ijms24119210
112. Grossi C, Capitani N, Benagiano M, Baldari CT, Della Bella C, Macor P, et al. Beta 2 glycoprotein I and neutrophil extracellular traps: Potential bridge between innate and adaptive immunity in anti-phospholipid syndrome. Front Immunol. (2023) 13:1076167. doi: 10.3389/fimmu.2022.1076167
113. Sule G, Kelley WJ, Gockman K, Yalavarthi S, Vreede AP, Banka AL, et al. Increased adhesive potential of antiphospholipid syndrome neutrophils mediated by β2 integrin mac-1. Arthritis Rheumatol. (2020) 72:114–24. doi: 10.1002/art.41057
114. Zuo Y, Yalavarthi S, Gockman K, Madison JA, Gudjonsson JE, Kahlenberg JM, et al. Anti-neutrophil extracellular trap antibodies and impaired neutrophil extracellular trap degradation in antiphospholipid syndrome. Arthritis Rheumatol. (2020) 72:2130–5. doi: 10.1002/art.41460
115. de Buhr N, Baumann T, Werlein C, Fingerhut L, Imker R, Meurer M, et al. Insights into immunothrombotic mechanisms in acute stroke due to vaccine-induced immune thrombotic thrombocytopenia. Front Immunol. (2022) 13:879157. doi: 10.3389/fimmu.2022.879157
116. Hakkim A, Fürnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V, et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA. (2010) 107:9813–8. doi: 10.1073/pnas.0909927107
117. González-Sierra M, Quevedo-Rodríguez A, Romo-Cordero A, González-Chretien G, Quevedo-Abeledo JC, de Vera-González A, et al. Relationship of blood inflammatory composite markers with cardiovascular risk factors and subclinical atherosclerosis in patients with rheumatoid arthritis. Life (Basel). (2023) 13:1469. doi: 10.3390/life13071469
118. Shahab F, Zachrisson H, Svensson C, Åström Aneq M, Sjöwall C, and Kylhammar D. Heterogeneous myocardial contraction detected by speckle tracking echocardiography in systemic lupus erythematosus is associated with complement protein C4: a cross-sectional study from a Swedish tertiary referral centre. Rheumatol Int. (2025) 45:183. doi: 10.1007/s00296-025-05939-8
119. Yalavarthi S, Gould TJ, Rao AN, Mazza LF, Morris AE, Núñez-Álvarez C, et al. Release of neutrophil extracellular traps by neutrophils stimulated with antiphospholipid antibodies: a newly identified mechanism of thrombosis in the antiphospholipid syndrome. Arthritis Rheumatol. (2015) 67:2990–3003. doi: 10.1002/art.39247
120. Tambralli A, Harbaugh A, NaveenKumar SK, Radyk MD, Rysenga CE, Sabb K, et al. Neutrophil glucose flux as a therapeutic target in antiphospholipid syndrome. J Clin Invest. (2024) 134:e169893. doi: 10.1172/JCI169893
121. Chen XQ, Tu L, Tang Q, Zou JS, Yun X, and Qin YH. DNase I targeted degradation of neutrophil extracellular traps to reduce the damage on IgAV rat. PloS One. (2023) 18:e0291592. doi: 10.1371/journal.pone.0291592
122. Kmeťová K, Lonina E, Yalavarthi S, Levine JS, Hoy CK, Sarosh C, et al. Interaction of the antiphospholipid syndrome autoantigen beta-2 glycoprotein I with DNA and neutrophil extracellular traps. Clin Immunol. (2023) 255:109714. doi: 10.1016/j.clim.2023.109714
123. Zhang K, Jiang N, Sang X, Feng Y, Chen R, and Chen Q. Trypanosoma brucei Lipophosphoglycan Induces the Formation of Neutrophil Extracellular Traps and Reactive Oxygen Species Burst via Toll-Like Receptor 2, Toll-Like Receptor 4, and c-Jun N-Terminal Kinase Activation. Front Microbiol. (2021) 12:713531. doi: 10.3389/fmicb.2021.713531
124. Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. (2017) 69:655–67. doi: 10.1002/art.39938
125. Atkinson MA, Eisenbarth GS, and Michels AW. Type 1 diabetes. Lancet. (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
126. Diana J and Lehuen A. Macrophages and β-cells are responsible for CXCR2-mediated neutrophil infiltration of the pancreas during autoimmune diabetes. EMBO Mol Med. (2014) 6:1090–104. doi: 10.15252/emmm.201404144
127. Huang J, Xiao Y, Xu A, and Zhou Z. Neutrophils in type 1 diabetes. J Diabetes Investig. (2016) 7:652–63. doi: 10.1111/jdi.12469
128. Klocperk A, Vcelakova J, Vrabcova P, Zentsova I, Petruzelkova L, Sumnik Z, et al. Elevated biomarkers of NETosis in the serum of pediatric patients with type 1 diabetes and their first-degree relatives. Front Immunol. (2021) 12:699386. doi: 10.3389/fimmu.2021.699386
129. Petrelli A, Popp SK, Fukuda R, Parish CR, Bosi E, and Simeonovic CJ. The contribution of neutrophils and NETs to the development of type 1 diabetes. Front Immunol. (2022) 13:930553. doi: 10.3389/fimmu.2022.930553
130. Popp SK, Vecchio F, Brown DJ, Fukuda R, Suzuki Y, Takeda Y, et al. Circulating platelet-neutrophil aggregates characterize the development of type 1 diabetes in humans and NOD mice. JCI Insight. (2022) 7:e153993. doi: 10.1172/jci.insight.153993
131. Skoglund C, Appelgren D, Johansson I, Casas R, and Ludvigsson J. Increase of neutrophil extracellular traps, mitochondrial DNA and nuclear DNA in newly diagnosed type 1 diabetes children but not in high-risk children. Front Immunol. (2021) 12:628564. doi: 10.3389/fimmu.2021.628564
132. Qin J, Fu S, Speake C, Greenbaum CJ, and Odegard JM. NETosis-associated serum biomarkers are reduced in type 1 diabetes in association with neutrophil count. Clin Exp Immunol. (2016) 184:318–22. doi: 10.1111/cei.12783
133. Berry G and Waldner H. Accelerated type 1 diabetes induction in mice by adoptive transfer of diabetogenic CD4+ T cells. J Vis Exp. (2013) 75):e50389. doi: 10.3791/50389
134. You Q, Shen Y, Wu Y, Li Y, Liu C, Huang F, et al. Neutrophil extracellular traps caused by gut leakage trigger the autoimmune response in nonobese diabetic mice. Front Immunol. (2022) 12:711423. doi: 10.3389/fimmu.2021.711423
135. Garciafigueroa Y, Phillips BE, Engman C, Trucco M, and Giannoukakis N. Neutrophil-associated inflammatory changes in the pre-diabetic pancreas of early-age NOD mice. Front Endocrinol (Lausanne). (2021) 12:565981. doi: 10.3389/fendo.2021.565981
136. Hu H, Vomund AN, Peterson OJ, Srivastava N, Li T, Kain L, et al. Crinophagic granules in pancreatic β cells contribute to mouse autoimmune diabetes by diversifying pathogenic epitope repertoire. Nat Commun. (2024) 15:8318. doi: 10.1038/s41467-024-52619-5
137. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med. (2013) 19:65–73. doi: 10.1038/nm.3042
138. Sodré FMC, Bissenova S, Bruggeman Y, Tilvawala R, DP C, Berthault C, et al. Peptidylarginine deiminase inhibition prevents diabetes development in NOD mice. Diabetes. (2021) 70:516–28. doi: 10.2337/db20-0421
139. Wong SL, Demers M, Martinod K, Gallant M, Wang Y, Goldfine AB, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. (2015) 21:815–9. doi: 10.1038/nm.3887
140. Ni Y, Hu BC, Wu GH, Shao ZQ, Zheng Y, Zhang R, et al. Interruption of neutrophil extracellular traps formation dictates host defense and tubular HOXA5 stability to augment efficacy of anti-Fn14 therapy against septic AKI. Theranostics. (2021) 11:9431–51. doi: 10.7150/thno.61902
141. Shu L, Shu L, Zhong L, Xiao Y, Wu X, Liu Y, et al. Neutrophil elastase triggers the development of autoimmune diabetes by exacerbating innate immune responses in pancreatic islets of non-obese diabetic mice. Clin Sci (Lond). (2020) 134:1679–96. doi: 10.1042/CS20200021
142. Barth P, Bruijnzeel P, Wach A, Sellier Kessler O, Hooftman L, Zimmermann J, et al. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J Cyst Fibros. (2020) 19:299–304. doi: 10.1016/j.jcf.2019.08.020
143. Watz H, Nagelschmitz J, Kirsten A, Pedersen F, van der Mey D, Schwers S, et al. Safety and efficacy of the human neutrophil elastase inhibitor BAY 85-8501 for the treatment of non-cystic fibrosis bronchiectasis: A randomized controlled trial. Pulm Pharmacol Ther. (2019) 56:86–93. doi: 10.1016/j.pupt.2019.03.009
144. Gramegna A, Amati F, Terranova L, Sotgiu G, Tarsia P, Miglietta D, et al. Neutrophil elastase in bronchiectasis. Respir Res. (2017) 18:211. doi: 10.1186/s12931-017-0691-x
145. Stockley R, De Soyza A, Gunawardena K, Perrett J, Forsman-Semb K, Entwistle N, et al. Phase II study of a neutrophil elastase inhibitor (AZD9668) in patients with bronchiectasis. Respir Med. (2013) 107:524–33. doi: 10.1016/j.rmed.2012.12.009
146. Ali M, Pulli B, Courties G, Tricot B, Sebas M, Iwamoto Y, et al. Myeloperoxidase inhibition improves ventricular function and remodeling after experimental myocardial infarction. JACC Basic Transl Sci. (2016) 1:633–43. doi: 10.1016/j.jacbts.2016.09.004
147. Chapman AL, Mocatta TJ, Shiva S, Seidel A, Chen B, Khalilova I, et al. Ceruloplasmin is an endogenous inhibitor of myeloperoxidase. J Biol Chem. (2013) 288:6465–77. doi: 10.1074/jbc.M112.418970
148. Pulli B, Ali M, Iwamoto Y, Zeller MW, Schob S, Linnoila JJ, et al. Myeloperoxidase-hepatocyte-stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid Redox Signal. (2015) 23:1255–69. doi: 10.1089/ars.2014.6108
149. Kim H, Wei Y, Lee JY, Wu Y, Zheng Y, Moskowitz MA, et al. Myeloperoxidase inhibition increases neurogenesis after ischemic stroke. J Pharmacol Exp Ther. (2016) 359:262–72. doi: 10.1124/jpet.116.235127
150. Hair PS, Sass LA, Krishna NK, and Cunnion KM. Inhibition of myeloperoxidase activity in cystic fibrosis sputum by peptide inhibitor of complement C1 (PIC1). PloS One. (2017) 12:e0170203. doi: 10.1371/journal.pone.0170203
151. Sadiq G, Sharma S, Stevens JS, Martinez-Bulit P, Hunnisett LM, Cameron C, et al. An integrated approach combining experimental, informatics and energetic methods for solid form derisking of PF-06282999. J Pharm Sci. (2025) 114:371–82. doi: 10.1016/j.xphs.2024.10.013
152. Chniguir A, Saguem MH, Dang PM, El-Benna J, and Bachoual R. Eugenol inhibits neutrophils myeloperoxidase in vitro and attenuates LPS-induced lung inflammation in mice. Pharm (Basel). (2024) 17:504. doi: 10.3390/ph17040504
153. Scozzi D, Liao F, Krupnick AS, Kreisel D, and Gelman AE. The role of neutrophil extracellular traps in acute lung injury. Front Immunol. (2022) 13:953195. doi: 10.3389/fimmu.2022.953195
154. Macanovic M, Sinicropi D, Shak S, Baughman S, Thiru S, and Lachmann PJ. The treatment of systemic lupus erythematosus (SLE) in NZB/W F1 hybrid mice; studies with recombinant murine DNase and with dexamethasone. Clin Exp Immunol. (1996) 106:243–52. doi: 10.1046/j.1365-2249.1996.d01-839.x
155. Stabach PR, Sims D, Gomez-Bañuelos E, Zehentmeier S, Dammen-Brower K, Bernhisel A, et al. A dual-acting DNASE1/DNASE1L3 biologic prevents autoimmunity and death in genetic and induced lupus models. JCI Insight. (2024) 9:e177003. doi: 10.1172/jci.insight.177003
156. Krischek JO, Mannherz HG, and Napirei M. Different results despite high homology: Comparative expression of human and murine DNase1 in Pichia pastoris. PloS One. (2025) 20:e0321094. doi: 10.1371/journal.pone.0321094
157. Hasni SA, Gupta S, Davis M, Poncio E, Temesgen-Oyelakin Y, Carlucci PM, et al. Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus. Nat Commun. (2021) 12:3391. doi: 10.1038/s41467-021-23361-z
158. Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-α Receptor monoclonal antibody, in moderate-to-severe systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:376–86. doi: 10.1002/art.39962
159. Greth W, Robbie GJ, Brohawn P, Hultquist M, and Yao B. Targeting the interferon pathway with sifalimumab for the treatment of systemic lupus erythematosus. Immunotherapy. (2017) 9:57–70. doi: 10.2217/imt-2016-0090
160. Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. (2016) 75:196–202. doi: 10.1136/annrheumdis-2014-206090
161. Kalunian KC, Furie R, Morand EF, Bruce IN, Manzi S, Tanaka Y, et al. A randomized, placebo-controlled phase III extension trial of the long-term safety and tolerability of anifrolumab in active systemic lupus erythematosus. Arthritis Rheumatol. (2023) 75:253–65. doi: 10.1002/art.42392
162. Chen X, Ke H, Li W, Yin L, Chen W, Chen T, et al. Structural basis for the recognition of IFNAR1 by the humanized therapeutic monoclonal antibody QX006N for the treatment of systemic lupus erythematosus. Int J Biol Macromol. (2024) 268:131721. doi: 10.1016/j.ijbiomac.2024.131721
163. Knight JS, Luo W, O'Dell AA, Yalavarthi S, Zhao W, Subramanian V, et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ Res. (2014) 114:947–56. doi: 10.1161/CIRCRESAHA.114.303312
164. Knight JS, Zhao W, Luo W, Subramanian V, AA O, Yalavarthi S, et al. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J Clin Invest. (2013) 123:2981–93. doi: 10.1172/JCI67390
165. Willis Willis VC, Gizinski AM, Banda NK, Causey CP, Knuckley B, Cordova KN, et al. N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of murine collagen-induced arthritis. J Immunol. (2011) 186:4396–404. doi: 10.4049/jimmunol.1001620
166. Wang B, Su X, Zhang B, and Pan S. GSK484, an inhibitor of peptidyl arginine deiminase 4, increases the radiosensitivity of colorectal cancer and inhibits neutrophil extracellular traps. J Gene Med. (2023) 25:e3530. doi: 10.1002/jgm.3530
167. Ye H, Yang Q, Guo H, Wang X, Cheng L, Han B, et al. Internalisation of neutrophils extracellular traps by macrophages aggravate rheumatoid arthritis via Rab5a. RMD Open. (2024) 10:e003847. doi: 10.1136/rmdopen-2023-003847
168. Gajendran C, Fukui S, Sadhu NM, Zainuddin M, Rajagopal S, Gosu R, et al. Alleviation of arthritis through prevention of neutrophil extracellular traps by an orally available inhibitor of protein arginine deiminase 4. Sci Rep. (2023) 13:3189. doi: 10.1038/s41598-023-30246-2
169. Lai ZW, Hanczko R, Bonilla E, Caza TN, Clair B, Bartos A, et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: a randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. (2012) 64:2937–46. doi: 10.1002/art.34502
170. Gupta S and Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. (2016) 12:402–13. doi: 10.1038/nrneph.2016.71
171. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. (2016) 22:146–53. doi: 10.1038/nm.4027
172. Wang X, Sun S, Duan Z, Yang C, Chu C, Wang K, et al. Protective effect of ethyl pyruvate on gut barrier function through regulations of ROS-related NETs formation during sepsis. Mol Immunol. (2021) 132:108–16. doi: 10.1016/j.molimm.2021.01.012
173. Yin X, Zhang J, Zhao W, Liu Z, and Wang J. Combined Levo-tetrahydropalmatine and diphenyleneiodonium chloride enhances antitumor activity in hepatocellular carcinoma. Pharmacol Res. (2022) 179:106219. doi: 10.1016/j.phrs.2022.106219
174. Guan H, Xie L, Ji Z, Song R, Qi J, and Nie X. Triptolide inhibits neutrophil extracellular trap formation. Ann Transl Med. (2021) 9:1384. doi: 10.21037/atm-21-3522
175. Huang G, Yuan K, Zhu Q, Zhang S, Lu Q, Zhu M, et al. Triptolide inhibits the inflammatory activities of neutrophils to ameliorate chronic arthritis. Mol Immunol. (2018) 101:210–20. doi: 10.1016/j.molimm.2018.06.012
176. Lu Q, Jiang H, Zhu Q, Xu J, Cai Y, Huo G, et al. Tetrandrine ameliorates rheumatoid arthritis in mice by alleviating neutrophil activities. Evid Based Complement Alternat Med. (2022) 2022:8589121. doi: 10.1155/2022/8589121
177. Yuan K, Zhu Q, Lu Q, Jiang H, Zhu M, Li X, et al. Quercetin alleviates rheumatoid arthritis by inhibiting neutrophil inflammatory activities. J Nutr Biochem. (2020) 84:108454. doi: 10.1016/j.jnutbio.2020.108454
178. Luo S, Li H, Liu J, Xie X, Wan Z, Wang Y, et al. Andrographolide ameliorates oxidative stress, inflammation and histological outcome in complete Freund's adjuvant-induced arthritis. Chem Biol Interact. (2020) 319:108984. doi: 10.1016/j.cbi.2020.108984
179. Jie SS, Sun HJ, Liu JX, Gao Y, Bai D, Zhu LL, et al. Simiao Yong'an decoction ameliorates murine collagen-induced arthritis by modulating neutrophil activities: An in vitro and in vivo study. J Ethnopharmacol. (2023) 305:116119. doi: 10.1016/j.jep.2022.116119
180. Zhu M, Yuan K, Lu Q, Zhu Q, Zhang S, Li X, et al. Emodin ameliorates rheumatoid arthritis by promoting neutrophil apoptosis and inhibiting neutrophil extracellular trap formation. Mol Immunol. (2019) 112:188–97. doi: 10.1016/j.molimm.2019.05.010
181. Tang R, Qin ZF, Yin JH, Wang JY, Su WR, Xuan ZH, et al. Er Miao San and its main components phellodendrine and atractylenolide-I exert anti-rheumatoid arthritis effects by inhibiting PAD4 and thereby reducing the formation of NETs. Fitoterapia. (2025) 185:106771. doi: 10.1016/j.fitote.2025.106771
Keywords: neutrophil, neutrophil extracellular traps, autoimmune disease, mechanism, targeted therapy
Citation: Tang R, Yin J, Qin Z, Zhang M and Jia X (2025) NETs: a new target for autoimmune disease. Front. Immunol. 16:1646527. doi: 10.3389/fimmu.2025.1646527
Received: 13 June 2025; Accepted: 10 October 2025;
Published: 23 October 2025.
Edited by:
Suneel Kumar, The State University of New Jersey, United StatesReviewed by:
Jessy J Alexander, University at Buffalo, United StatesMeraj Khan, The Hospital for Sick Children, Canada
Copyright © 2025 Tang, Yin, Qin, Zhang and Jia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Zhang, ZG9jemhhbmdtaW5AdXN0Yy5lZHUuY24=; Xiaoyi Jia, amlheHlAYWh0Y20uZWR1LmNu
†These authors have contributed equally to this work