 May A. Alsayb1,2*
May A. Alsayb1,2*- 1Clinical Laboratory Sciences Department, College of Applied Medical Sciences, Taibah University, Madinah, Saudi Arabia
- 2Health and Life Research Center, Taibah University, Madinah, Saudi Arabia
Millions of people worldwide suffer from chronic and devastating autoimmune disorders, challenging contemporary medicine. These disorders develop when the immune system attacks its own tissues, causing inflammation and damage. Traditional treatments have focused on widespread immunosuppression, which can relieve symptoms but has serious adverse effects and does not address immunological dysregulation. This review discusses the current and future trends in immunotherapy for the management of autoimmune diseases, including advancements such as CAR T-cell therapy, bispecific antibodies, next-generation immune checkpoint modulators, targeted cytokine therapies, and microbiome-based interventions. The discussion is grounded in current scientific literature, focusing on mechanisms of action, recent breakthroughs, limitations, and potential future directions. Each of the related sections presents cutting-edge advancements, current challenges, and future opportunities for research and clinical translation.
1 Introduction
Autoimmune diseases represent a significant challenge in modern medicine, being chronic and debilitating conditions affecting millions of individuals worldwide. These diseases occur when the immune system targets the body’s own tissues, leading to inflammation and tissue damage (1). Among these conditions, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), type 1 diabetes (T1D), and multiple sclerosis (MS) affect a global population of millions and pose a considerable challenge to treatment in terms of efficacy, safety, and long-term disease control (2).
Conventional therapy for autoimmune diseases has primarily focused on broad immunosuppression, which can alleviate symptoms but often has significant side effects and does not address the underlying immune dysregulation (3–5). More recently, immunotherapy has emerged as a revolutionary treatment approach. Unlike conventional therapies, immunotherapy aims to modulate the immune system more precisely by enhancing its regulatory functions or specifically targeting the pathogenic immune cells and molecules involved in the disease process (6, 7). This approach not only improves efficacy but also can reduce the adverse effects of the immune response. The impact of recent breakthroughs in immunotherapy for autoimmune diseases can extend beyond symptomatic relief, offering the potential for long-term disease remission and even cure. As research progresses, the focus on personalized medicine, combination therapies, and improved drug delivery systems continues to shape the landscape of autoimmune disease management (8). This evolving paradigm holds promise for transforming patient outcomes and improving the quality of life of those afflicted by these chronic conditions.
This review focuses on recent breakthroughs in immunotherapy for autoimmune diseases, including the adaptation of chimeric antigen receptor (CAR)-T cell therapy, the development of bispecific antibodies (bsAbs), advancements in next-generation checkpoint inhibitors, targeted cytokine therapies, and microbiome-based interventions. Each of the related sections presents cutting-edge advancements, current challenges, and future opportunities for research and clinical translation.
2 Chimeric antigen receptor T-Cell therapy in autoimmune diseases
CAR T-cell therapy has shown promise against treatment-resistant autoimmune diseases (9). By genetically modifying a patient’s autologous T cells to express synthetic receptors targeting specific antigens, CAR T-cell therapy allows for the selective elimination of autoreactive immune cells, thereby “resetting” immune tolerance (Figure 1). Schett et al. (7) demonstrated this transformative potential when they treated 5 patients with refractory SLE with CD19-directed CAR T cells. The results were remarkable: all patients entered durable drug-free remission, with normalized complement levels, decreased anti-dsDNA titers, and no further disease flares during follow-up. These results demonstrate the major role of autoreactive B cells in lupus pathogenesis and validate CAR T-mediated B-cell depletion as a possible disease-modifying intervention.

Figure 1. CAR T cell production model. Leukapheresis of the patient is the first step in producing CAR T cells, followed by T cell activation and enrichment. To help introduce and occasionally permanently integrate the CAR transgene, activated T cells are transduced (e.g., by a lentiviral vector). After expansion in either static or dynamic culture, gene-modified T cells are cryopreserved and reinfused into the patient. This figure is adapted from (10). Created in BioRender. Alsayb, M. (2025) https://BioRender.com/1glnsej.
2.1 Breakthroughs
Recent studies show that CD19 CAR T-cell therapy can induce drug-free remission in refractory SLE and idiopathic inflammatory myopathies, with only mild, short-lived cytokine release syndrome as a well-tolerated side effect (11, 12). The therapy rapidly eliminates autoantibody-producing plasmablasts, and even after B-cell recovery, patients maintain remission with naïve, non-class-switched B cells over extended follow-up periods (13). Additionally, patients with systemic sclerosis experienced significant improvement in heart, joint, and skin manifestations, reinforcing the critical role of B-cell–mediated autoimmunity in these diseases and suggesting broader therapeutic potential for CD19 CAR T-cell therapy (14). Moreover, several preclinical and early clinical studies have looked into extending CAR T-cell therapy to other autoimmune conditions (15). Studies on CAR T-cell therapy for myasthenia gravis (MG) show that B-cell maturation antigen (BCMA)-targeted RNA-engineered CAR T cells led to clinical improvement and were deemed safe (16), while CD19-targeted CAR T cells achieved long-term disease stabilization without increasing infection risk, suggesting a safe and lasting treatment option for refractory MG (17, 18). Additionally, scientists at Xuzhou Medical University developed bispecific CAR T cells targeting CD19 and BCMA to reset immune responses in relapsed or treatment-resistant Chronic Inflammatory Demyelinating Polyneuropathy (CIDP), resulting in improved muscle function and reduced disability (19). Furthermore, CD19-directed CAR T cells ameliorated clinical disease in mouse models of MS and autoimmune encephalomyelitis, suggesting potential translational relevance for human autoimmune demyelinating diseases (20). Expanding the applications of CAR T-cell therapy, Lee et al. (21) demonstrated the use of desmoglein-3-specific CAR T cells in the preclinical treatment of pemphigus vulgaris, with the subsequent efficient elimination of autoreactive B cells. These milestones represent a paradigm shift: autoimmune disorders, previously treated only with lifelong immunosuppressants, may now be intervened upon, preferably through a single curative modality aimed specifically at the immunological cause (22). In essence, CAR T-cell therapy re-establishes the boundaries between oncology and immunology using precision immune-engineering. As for now, several clinical trials are currently ongoing, targeting different autoimmune diseases using CAR T-cell therapy (Table 1).
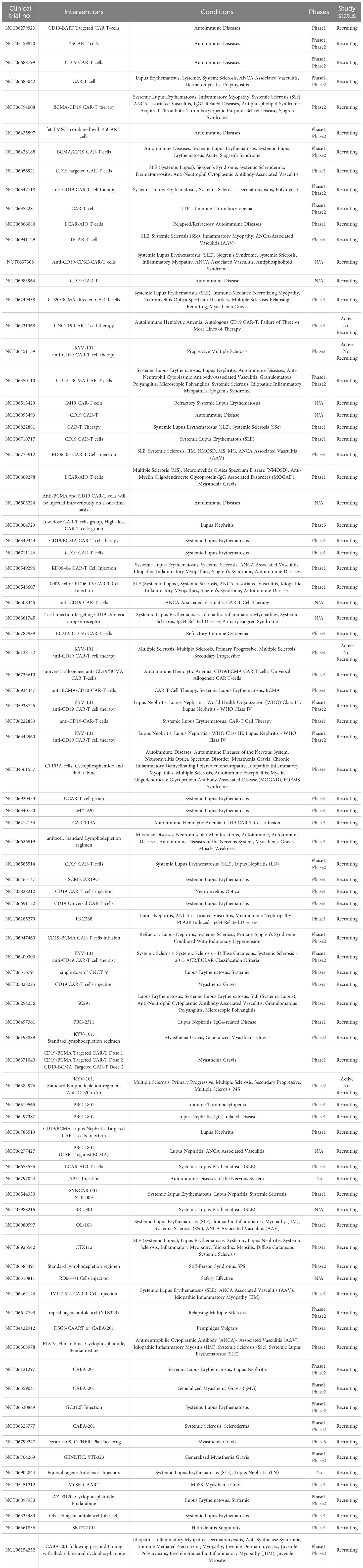
Table 1. CAR-T cell therapy currently in clinical trials for autoimmune diseases.
Biomarkers linked to endothelial cell activation, such as the ANG2:ANG1 ratio (23) and soluble adhesion molecules (sVCAM-1, sICAM-1) (24), have also been associated with predicting CAR-T therapy response. A rise in these biomarkers often reflects a endothelial dysfunction, which is connected poor prognostic effect of CAR-T therapy (24, 25). An increase in IL-6 level is likewise associated with Cytokine Release Syndrome (CRS) induction, acting as a negative prognostic marker (26). Additionally, an increase in the immune exhaustion markers (PD-1, CTLA-4, LAG-3, TIM-3) affects T cell activities and predicts poorer CAR T cell therapy outcomes. Whereas, higher levels of cytotoxicity biomarkers like granzyme and perforin are linked to successful target cell death and a better outlook for CAR T cell therapy (25, 27). An increase in C-Reactive Protein (CRP) levels, a non-specific marker for systemic inflammation, is associated with severe CRS and may serve as an indicator of poor treatment outcome (25, 26, 28, 29). However, it’s essential to note that CRP is a non-specific inflammatory marker and its elevation can be associated with other conditions such as infection, trauma, or autoimmune activity. Thus, CRP alone without consideration of other clinical parameters cannot provide a conclusive interpretation. Moreover, the chemokines represent positive prognostic biomarkers, enhancing tumor targeting by increasing the CAR T cell homing to tumor sites (25, 30). Therefore, these biomarkers offer significant insights into the mechanisms affecting CAR T cell therapy outcomes, facilitating improved prediction of treatment efficacy and potential adverse effects, which is vital for optimizing patient management and enhancing prognoses (25).
2.2 Challenges
The widespread use of CAR T-cell therapy is impeded by major clinical and translational challenges despite recent advances. As with all therapeutic agents, CAR T-cell therapy entails some risk of adverse events like CRS, immune effector cell-associated neurotoxicity syndrome, and B-cell aplasia (31). The ratio of therapeutic benefit to over-immunosuppression remains finely poised in autoimmune disorders; this is important considering that the patient cohort may often already be immunocompromised (32). The logistical complexity of CAR T-cell therapy, which entails leukapheresis, ex vivo T-cell modification, expansion, and reinfusion, makes the therapy extremely costly and time-consuming, raising ethical and economic concerns regarding access (6). Furthermore, patient-specific characteristics such as baseline immune profiles, autoantibody specificity, or markers of T-cell exhaustion can have strikingly variable influences on treatment outcomes. Currently, these factors are poorly characterized, creating uncertainty in the clinic and complicating trial design.
2.3 Future directions
One potential avenue involves generating universal or “off-the-shelf” CAR T cells using CRISPR/Cas9 genome-editing techniques to create allogeneic T cells that lack endogenous T-cell receptors and Human Leukocyte Antigen (HLA) to prevent graft-versus-host disease and host rejection (33, 34). Furthermore, modifying naturally occurring regulatory T cells (Tregs) to express CARs with specificity for a given antigen is being investigated as a new approach in CAR therapy. This approach, currently under investigation for autoimmune conditions such as type 1 diabetes (T1D) and Crohn’s disease, aims not to eliminate immune targets but to suppress pathological immune activation and restore tolerance as a novel immunomodulatory strategy (35). To enhance safety, engineered “suicide switches” such as inducible caspase-9 (iCasp9) are incorporated into CAR constructs, allowing for rapid ablation of the therapy in the event of severe toxicity (36, 37). Moreover, machine learning algorithms are utilized to categorize patients according to predictive biomarkers of therapeutic response or adverse events. These models utilize multi-omics information, encompassing genomes, transcriptomics, and proteomics, to inform tailored therapy decisions (38–40). However, these new paradigms will not be validated until they demonstrate their value with regard to numerous, heterogeneous populations with longitudinal follow-up as well.
3 Bispecific antibodies for immune modulation
bsAbs are engineered molecules that can simultaneously engage 2 distinct targets, providing a unique mechanism of action that modulates immune responses more precisely than traditional monoclonal antibodies (41). This dual-targeting capability allows bsAbs to precisely target disease-relevant cells or pathways, reducing off-target effects and minimizing systemic immunosuppression. By engaging multiple targets, bsAbs can mitigate the development of resistance mechanisms that often compromise the effectiveness of single-target therapies (42, 43). This versatility of bsAbs can enable either immune suppression or immune redirection and thereby represents a major step forward in immunomodulatory therapy.
3.1 Breakthroughs
Several in vivo preclinical studies have demonstrated the potential of a bsAb designed to target both CD20 on B cells and CD3 on T cells in treating various autoimmune diseases, with promising results in terms of efficacy and safety (44, 45). This bsAb aims to deplete autoreactive B cells while simultaneously modulating T cell activity. The efficacy of Mosunetuzumab was assessed utilizing a humanized CD20/CD3 mouse model and an immune reconstitution model in NSG mice transplanted with human CD34+ cells. These studies have demonstrated the proof of concept for the application of CD20/CD3 bispecific antibody therapy in the treatment of autoimmune disorders. Mosunetuzumab exhibited a promising safety profile in SLE patients, characterized by only minor side effects and the absence of dose-limiting toxicities. Initial clinical findings, including as decreases in disease activity and autoantibody levels, together with B-cell depletion and T-cell activation, warrant further exploration of Mosunetuzumab as a prospective treatment for SLE (46). Moreover, Imvotamab, a CD20/CD3 bispecific antibody, is being explored for the treatment of refractory autoimmune diseases like RA and SLE due to its ability to deplete B cells with reduced cytokine release (45).
Furthermore, A study demonstrated the first use of blinatumomab, a bispecific anti-CD3/CD19 antibody, as a B-cell depletion therapy for systemic sclerosis, revealing profound B-cell depletion with no increase in infection risk, along with an improvement in clinical manifestations (47). However, this reflects a single case study, and the finding remains preliminary; thus, further investigations and studies should explore and confirm these findings.
bsAbs offer mechanistic advantages over monoclonal antibodies by enabling simultaneous modulation of multiple immune pathways (48). For example, a bispecific construct that targets TNF-α and IL-17A has been demonstrated to suppress both TNF-α and IL-17A, resulting in decreased inflammation in animal models, thus supporting its potential role as a therapeutic agent for RA (49). In a rheumatoid arthritis clinical trial, ABT-122, a bispecific antibody targeting TNF-α and IL-17, was shown to reduce the chemokines CXCL9, CXCL10, CCL23, and E-selectin, which are involved in the recruitment of T cells and/or myeloid cells, suggesting that ABT-122 may influence the trafficking of immune cell populations (50, 51).
In the context of T1D, a bsAb targeting both cytotoxic T-lymphocyte–associated protein-4 (CTLA-4) on T cells and Glucose Transporter Type 2 (GLUT2) on pancreatic β cells has shown potential in preserving β-cell function. By binding to GLUT2 on pancreatic cells and interacting with CTLA-4 on autoreactive T cell, it can promote tolerance and delay the onset of diabetes without apparent adverse effects, thus providing a potential therapeutic strategy for T1D (52). This dual-targeting strategy aims to modulate the autoimmune response specifically against β cells while promoting immune tolerance, offering a novel approach to modifying disease progression in T1D. Additionally, a current phase 2 clinical trial is evaluating the safety and efficacy of SAR442970 a bsAb targeting both TNF-α and OX40L in preserving pancreatic β-cell function in individuals recently diagnosed with T1D (NCT06812988). SAR442970 act by inhibiting TNF-α and OX40L, while TNF-α promote inflammation, OX40L interacts with the OX40 receptor on T cells, promoting their activation and survival. By blocking these two pathways, SAR442970 aims to reduce excessive immune activation and inflammation. Additionally, a limited number of other clinical trials are also in progress (Table 2).
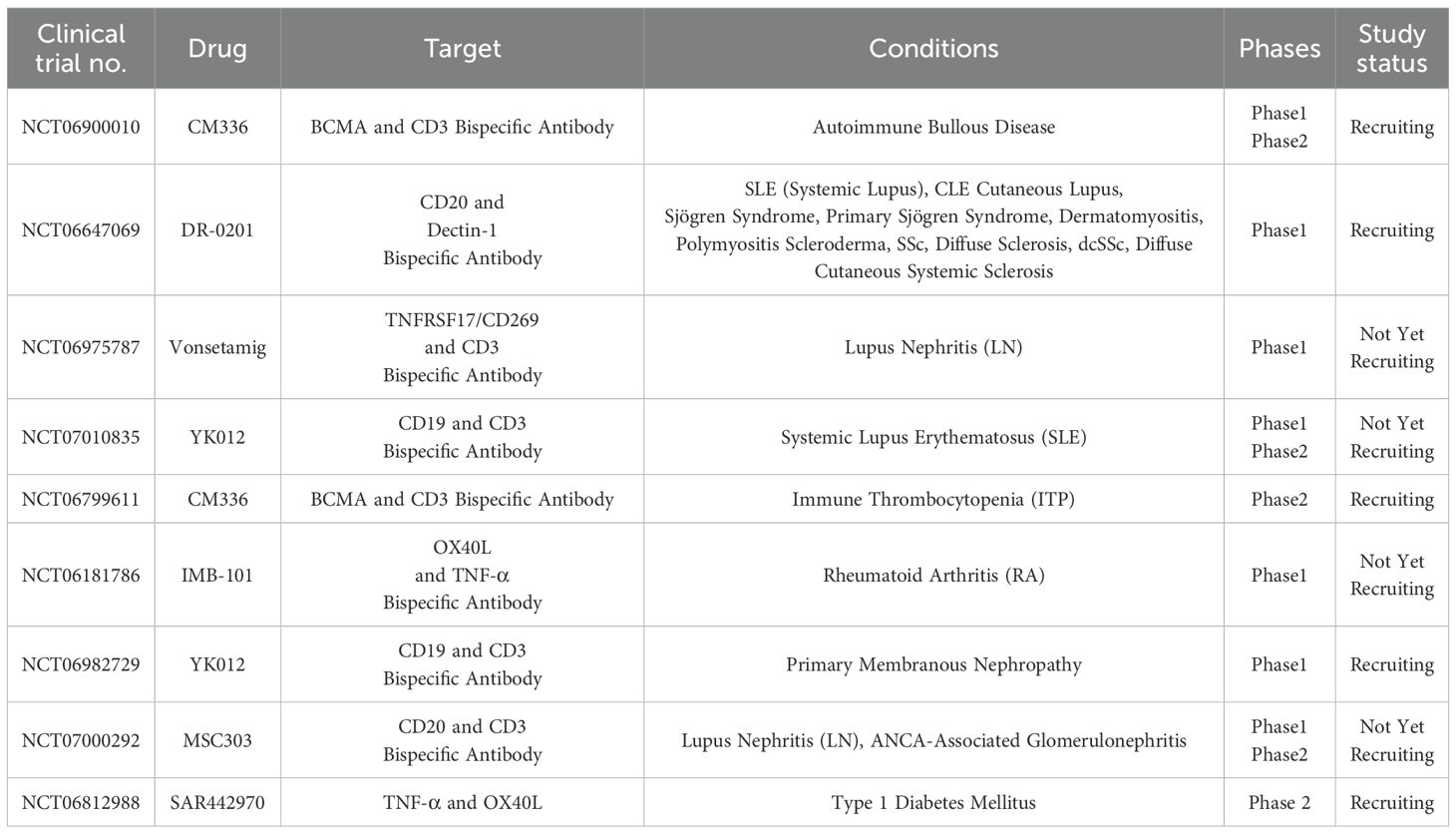
Table 2. Bispecific antibodies currently in clinical trials for autoimmune diseases.
3.2 Challenges
While bsAbs are designed to target CD3 and activate T cells toward autoreactive immune cells, uncoordinated CD3 engagement can overactivate T cells, resulting in excessive cytokine release (CRS) and tissue injury (53). It is noteworthy that while the most common adverse event associated with bsAbs is related to CRS, yet, it is typically milder than seen with CAR T-cell therapy (44). While early data on autoimmune conditions like SLE showed a favorable safety profile and temporary lymphocyte reduction, further studies are required to investigate the safety after long-term B cell depletion and its potential adverse events (44). Furthermore, safety and effectiveness of bsAbs depend on overcoming a number of significant challenges related to designing and manufacturing these bsAbs. For instance, the diverse molecular formats make manufacturing and purification more difficult, and their structural complexity lowers stability and raises the possibility of chain mispairing. It is also challenging to tune effector functions, ensure favorable pharmacokinetics, and achieve the proper binding balance for both targets (54). In addition, autoimmune diseases are massively heterogeneous; hence, the question of which pairs of antigens would form appropriate targets uniformly relevant across diverse populations of patients remains open. The lack of clear regulatory pathways for the use of bsAbs further adds to the complexities in introducing them into early human clinical studies. Despite the FDA’s guidance for bispecific antibodies in oncology, trial design is hampered by this regulatory gap, especially when it comes to defining safety margins, chronic dosing schedules, and patient comorbidities (55). Autoimmune patients often present with comorbidities and may be on concurrent immunosuppressive therapies. These factors complicate safety assessments and require tailored trial designs distinct from oncology protocols (42).
3.3 Future directions
Future innovation should be aimed at bsAbs that work on spatiotemporal control mechanisms for enhanced specificity and safety. With such approaches, conditionally active bsAbs may need inflammatory biomarkers or a particular tissue microenvironment to be activated (43). For example, pH-sensitive linkers or protease-activated formats are being researched to limit activity at the inflamed sites of joints or pancreatic islets (56). Such designs can provide safety against system toxicity as well as off-target effects for enhancement of the therapeutic index to a large extent. Furthermore, advancements in systems immunology and single-cell transcriptomics could allow for personalizing bsAb design according to patient immune signatures. Predictive models using artificial intelligence may better identify the possible antigen pairs for individual patients, thus paving the way for personalized bsAb therapy (57). Integrating these into modular bsAb platforms might allow for the fast adaptation of therapies for autoimmune conditions such as SLE, MS, and inflammatory bowel disease (IBD). There is limited follow-up data on safety, and the risk of immune activity, especially with non-human or chimeric domains, is still very much a concern (58). Future studies should investigate the establishment of biomarkers of durable tolerance induction and optimization criteria for dose regimens, as well as combinations with existing biologics or small molecules.
4 Next-generation checkpoint inhibitors
Inhibitory pathways, like programmed cell death-1 (PD-1), CTLA-4, and lymphocyte activation gene-3 (LAG-3), are crucial for maintaining the balance of tolerance to self and prevention of autoimmune pathology through the inhibition of overactive T cells (59, 60)., Immune checkpoint blockade reinvigorates exhausted T cells and enhances anti-tumor immune responses. In cancer, checkpoint inhibitors (e.g., anti–PD-1, anti–CTLA-4) block inhibitory receptors to unleash T cells against tumors. In autoimmunity, therapies may activate checkpoint pathways (e.g., CTLA-4 agonists like abatacept) to dampen overactive immune responses and restore homeostasis (61). Immune checkpoints such as LAG-3, T-cell immunoglobulin and mucin-domain containing-3 (Tim-3), T cell immunoglobulin and ITIM domain (TIGIT), and V-domain immunoglobulin suppressor of T cell activation (VISTA) are emerging targets for immune modulation and restoring immune tolerance and have been the focus of many studies on autoimmune disease (Figure 2).

Figure 2. Checkpoint schematic pathway. Mechanisms of LAG-3, Tim-3, TIGIT, and VISTA inhibitory pathways in T cells: (A) Upon ligand binding, LAG-3 inhibits early steps of the TCR pathway in a manner dependent on LAG-3’s cytoplasmic domain. The KIEELE motif is responsible for regulating downstream inhibitory signaling, while EP motifs interfere with T cell activation by blocking CD3/Lck interactions. (B) TIM-3 binds to Gal-9 and induces phosphorylation at Tyr256 and Tyr263, releasing Bat3, which regulates Lck tyrosine kinases, thereby inhibiting TCR signaling. (C) In T cells, TIGIT exhibits a variety of inhibitory mechanisms. 1) TIGIT directly lowers TCR expression and TCR signaling by binding to CD155 and delivering intracellular inhibitory signals. 2) TIGIT can outcompete CD226 in CD155 binding because it binds to CD155 with a significantly higher affinity than its co-stimulatory counterpart CD226; 3) TIGIT interferes with CD226 homodimerization to prevent CD226-mediated T cell activation. 4) TIGIT inhibits T cells indirectly by binding to CD155 on APCs, thereby increasing IL-10 production and decreasing IL-12 production. (D) VISTA can function as a receptor that preserves a tolerant or quiescent phenotype and as a ligand expressed by tumor cells that inhibits T-cell receptor-mediated activation by signaling through a putative receptor on T cells. VISTA can bind to PSGL-1 in acidic environments and to VSIG-3 in physiological environments, inhibiting T cell function and proliferation. This figure is adapted from (62, 63). Created in BioRender. Alsayb, M. (2025) https://BioRender.com/r2iqbmx.
4.1 Breakthroughs
LAG-3 is an immune checkpoint receptor that inhibits T-cell activation, proliferation, and contributes to immune homeostasis. In autoimmune-prone animal models, genetic deletion or pharmacologic blockade of LAG-3 exacerbates disease severity, underscoring its immunoregulatory role (64). Relapsing-remitting multiple sclerosis (RRMS) and T1D patients have a considerably low level of LAG-3+ CD4 and CD8 T cells. The low expression of LAG-3 has also been linked with T-cell resistance to apoptosis, promoting persistence of pathogenic T cells. These data suggest that LAG-3 agonists, by enhancing their inhibitory signaling, may be a promising target for restoring immune regulation in autoimmune conditions (65). Depending on the context of the disease, therapeutic approaches that target LAG-3 may involve either agonists or antagonists. Antagonists or depleting antibodies, like GSK2831781, are being studied to improve immune activity, while agonists may be investigated to suppress overactive immune responses in autoimmune diseases. GSK2831781, a monoclonal Ab targeting LAG-3 on activated T cells, can diminish LAG-3-expressing activated T cells in immuno-inflammatory conditions. Two clinical trials are assessing the safety and pharmacokinetics of GSK2831781 for the treatment of psoriasis (NCT03965533, NCT02195349). Another clinical trial was terminated in ulcerative colitis (NCT03893565) based on the assessment of clinical data, thus limiting the availability of this study’s safety and efficacy results. The preliminary data showed that GSK2831781 provides evidence of improvement in psoriasis; it has been shown to demonstrate the ability to downregulate the gene expression of IL-17A, IL-17F, IFNγ, and S100A12 (66). Additionally, the LAG-3 agonist IMP761 is being investigated in a Phase I clinical trial (NCT06637865) involving healthy volunteers but has not yet been tested in autoimmune patients (multiple sclerosis). These different approaches, agonism versus depletion, show how complicated LAG-3 biology is and how important it is to have disease-specific strategies for autoimmune therapy.
Tim-3, an immune checkpoint receptor that is highly expressed on immune cells and induces immunological tolerance by suppressing T cell activation and promoting apoptosis. Tim-3 and MHC-II dysregulation are associated with MS and other autoimmune diseases. Mechanistically, Tim-3 suppresses MHC-II–mediated autoantigen presentation and CD4+ T-cell activation by downregulating MHC-II expression in macrophages via the STAT1/CIITA signaling axis. In murine models of experimental autoimmune encephalomyelitis (EAE), overexpression of Tim-3 reduced MHC-II levels and ameliorated disease severity, while its inhibition led to increased MHC-II expression and worsened clinical outcomes. These findings suggest that targeted modulation of the Tim-3 and MHC-II pathway, potentially through Tim-3 agonists, may offer a novel therapeutic strategy for restoring immune tolerance in MS (67). A recent study showed that LPX3, a liposomal formulation that targets Tim-3 and Tim-4, can trigger immune tolerance without the need for an antigen. LPX3 demonstrated its potential to restore immune regulation by effectively penetrating lymph nodes, colocalizing with immune cells, and promoting regulatory T cells expansion. According to these results, LPX3 might be a possible treatment approach for autoimmune disorders that does not require the co-administration of particular antigens (68).
TIGIT is a newly identified co-inhibitory receptor, that modulates the immune system. CD226 promotes positive signals, whereas TIGIT transmits negative signals, forming a route comparable to the CD28/CTLA-4 signaling pathway (69). TIGIT can induce immunological tolerance by suppressing autoreactive T cells, increasing tolerogenic dendritic cells (DCs), and encouraging the production and suppressive capacity of Tregs (70). Preclinical studies using a TIGIT-Ig fusion protein, agonist antibodies, and other modalities have demonstrated protective effects in murine models of autoimmune diseases, such as SLE (71) and experimental autoimmune encephalomyelitis (72). Beyond T and NK cells, TIGIT also influences regulatory B cells, which help dampen immune responses by inhibiting T cell activation and reducing pro-inflammatory DC activity (73). Although the role of TIGIT in B cells is less well characterized, emerging evidence suggests it may be central to autoimmune regulation. These findings position TIGIT as a promising candidate for immune checkpoint therapy in autoimmunity (73).
In a variety of autoimmune and inflammatory diseases, VISTA, a novel negative checkpoint receptor, functions as a negative immune regulator, thereby preventing excessive immune activation (74). In autoimmune conditions, such as lupus, MS, and RA, VISTA is essential for suppressing autoreactive T-cell responses, controlling monocyte and macrophage activation, and reducing the production of pro-inflammatory cytokines such as IL-17, IFN-γ, and IL-23. Research indicates that the deletion of VISTA increases inflammation, heightens immune cell infiltration, and exacerbates disease severity in the SLE animal model, hence affirming its protective function (75). In contrast, the activation of VISTA using agonist antibodies resulted in a reduction in inflammatory markers and an enhancement of clinical outcomes in many models, including lupus (76). The specific mechanisms differ among diseases; for instance, VISTA can modulate Th1/Th17 responses in MS and Toll-like receptor signaling in psoriasis. It has been reported that the use of VISTA blocking antibody in the EAE mouse model has increased the infiltration of IFN-γ+ and IL17A + producing CD4+ T cells in the central nervous system (CNS), exhibiting an activation of T cell-mediated immunity and loss of peripheral tolerance, thus increasing the susceptibility to EAE (77). On the Other side, in a model of psoriasis, VISTA-deficient dendritic cells are unable to adequately regulate the TLR7 pathway, which exacerbates IL-23/IL-17-driven skin inflammation (78). Therefore, its primary role is to preserve immunological equilibrium by regulating both adaptive and innate immune cells, thus underscoring its potential as a biomarker for immunological dysregulation. Despite these promising preclinical findings, no VISTA-targeted intervention has been authorized or progressed to clinical trials for autoimmune disorders, highlighting the promising opportunities for translational research to exploit its immunoregulatory capabilities (77).
4.2 Challenges
There are major obstacles to the therapeutic promise of next-generation checkpoint agonists. Of primary concern is the risk of extended immunosuppression, which may lead to opportunistic infections or malignancies in recipients (79). While preclinical models support efficacy, translating that efficacy to human autoimmune disease remains fraught with complexity due to heterogeneity in disease phenotype, complicating the prediction of patient responses and the identification of accurate biomarkers, such as TIGIT+ cell populations, for therapeutic monitoring (80). The dual functions of checkpoints, such as VISTA, which may be activated or downregulated according to the kind and stage of disease, complicate therapeutic targeting and necessitate meticulous, context-specific strategies (77). Moreover, although preclinical models have shown promising findings, the incomplete comprehension of the underlying mechanisms and off-target effects, as well as the potential for exacerbating immune dysregulation, limits the translation of these findings into effective and safe clinical therapies. Additionally, the interaction between co-inhibitory and co-stimulatory pathways (e.g., TIGIT/CD226) is complex and not entirely understood, complicating the development of medicines that optimize efficacy without inducing undesirable immune activation (73, 81). Furthermore, extended checkpoint activation may initiate the opposite of tolerance by exhausting Tregs or shifting antigen presentation dynamics, going against the original intent of therapy. Manufacturing agonistic antibodies with the correct affinity for binding, epitope specificity, and isotype to engage inhibitory receptors without inducing any off-target consequences further complicates the matter (82). Current biomarker panels also do not adequately predict patient outcomes, hampering the ability to personalize checkpoint-based therapies.
4.3 Future directions
Next-generation approaches, focusing on the tissue-restricted or cell-targeted delivery of checkpoint modulators, are being developed to tackle the above issues. Nanoparticle-based systems and antibody-drug conjugates are being evaluated for the localized delivery of PD-1 or CTLA-4 agonism to inflamed tissues such as pancreatic islets or synovial membranes to minimize systemic exposure (83). Synthetic biology is being investigated to develop checkpoint agonists engineered with logic gates to activate only in the presence of cytopathic cytokines or antigens. Another promising direction is the combination of immune repertoire sequencing with machine learning algorithms to stratify patients based on their chances of responding to checkpoint agonists. Subpopulations of autoreactive T cells that could display selective sensitivity to PD-1 or LAG-3 signaling might already have been distinguished through single-cell transcriptomics (84). Such predictive immune-profiling would allow for better selection of therapeutic targets with the mitigation of off-target immunosuppression. Despite current constraints, immune-checkpoint-modulating drugs appear to be an increasingly rational and viable new therapeutic option for autoimmune diseases. However, daunting empirical gaps concerning the expected long-term effects of chronic checkpoint engagement, durability of immune tolerance, and optimal design of checkpoint combinational therapy should be the focus of future research (81). Therefore, apart from proving efficacy, future clinical trials must generate robust biomarkers for safety and durability that allow personalized checkpoint immunomodulation.
5 Targeted cytokine therapies
Cytokines are considered key mediators in the immune system, regulating the recruitment, activation, and differentiation of various immune cells (85). Aberrant cytokine expression sometimes stimulates the persistent inflammation and tissue damage characterizing autoimmune diseases. Hence, a central strategy for treating autoimmune diseases has become targeting the specific cytokines modulating disease progression (86). While some of these therapies are already in clinical use, current research is further improving their specificity and enhancing their therapeutic reach.
5.1 Breakthroughs
Monoclonal antibodies targeting pro-inflammatory cytokines have revolutionized the treatment of autoimmune diseases, offering precision without broad immunosuppression. Among the most widely used are IL-6 inhibitors, such as tocilizumab and sarilumab, which have demonstrated clinical efficacy in rheumatoid arthritis (RA), systemic juvenile idiopathic arthritis, and giant cell arteritis (87). By blocking key drivers of autoimmune inflammation such as IL-6 signaling, these agents suppress acute-phase reactants (CRP, SAA, fibrinogen, etc), modulate B-cell activity, and inhibit Th17 cell differentiation (88). Building on this approach, IL-17A blockers (secukinumab, ixekizumab) and IL-23 inhibitors (guselkumab, risankizumab) have shown remarkable success in psoriasis, psoriatic arthritis, and ankylosing spondylitis. These cytokines are central to the Th17 axis, and their inhibition disrupts inflammatory circuits while preserving broader immune function (89). Monoclonal antibodies inhibit this axis and disrupt key inflammatory circuits without globally inhibiting the immune system. IFN-γ neutralization, although infrequently used, is beneficial in diseases such as hemophagocytic lymphohistiocytosis and has been investigated in autoimmune uveitis (Box 1) (90). In contrast to blocking inflammation, enhancing anti-inflammatory cytokines such as IL-10 represents a complementary strategy. While recombinant IL-10 has faced challenges due to poor pharmacokinetics and systemic toxicity, gene therapy and fusion protein delivery systems are being developed to enable localized, sustained release, potentially restoring immune balance without adverse effects (91, 92). Currently, several clinical trials are highlighting the translational progress of targeted cytokine therapy in autoimmune disorders (Table 3).
Box 1. FN-γ neutralization in primary Hemophagocytic Lymphohistiocytosis (HLH).
Primary Hemophagocytic Lymphohistiocytosis (HLH) results from a defect in cytotoxic T lymphocytes and natural killer cells' abilities to kill infected cells by perforin-mediated cytotoxicity. The uncontrolled activation of CTL and NK cells increases cytokine production that, in turn, hyperactivates macrophages and induces cytokine storm, where IFN-γ plays a particularly key role in the development of HLH. Emapalumab is a monoclonal antibody that functions by neutralizing IFN-γ by blocking its activity. It helps to reduce the excessive inflammation and immune activation associated with HLH. This therapeutic approach highlights the potential of IFN-γ inhibitors (e.g., Emapalumab) in managing macrophage activation syndrome (MAS), a severe complication of autoimmune diseases (89).

Table 3. Cytokine based therapy currently in clinical trials for autoimmune diseases.
5.2 Challenges
While cytokine-targeted therapies have proved effective, they come with significant downsides. A major reason is cytokine redundancy, wherein, since most cytokines have overlapping functions, blockade of a single cytokine leads to the compensatory upregulation of parallel pathways (93). For example, some IL-17 inhibitors result in disease activity reoccurrence in patients due to increased levels of IL-22, GM-CSF, or both, but all maintain inflammatory circuits independently. On the other hand, the long-term inhibition of cytokines leads to certain symptoms and effects caused by immunosuppression, such as increased susceptibility to bacterial, fungal, and viral infections (94). Patients treated with IL-6 blockade, for instance, may fail to show symptoms of an infection because fever and CRP production are suppressed, impeding diagnosis and treatment (95). Some cytokines may also have double functions depending on the tissue context; for example, IL-17 has a role in mucosal defense against several Candida species, and its inhibition is associated with mucocutaneous candidiasis (96). The diverse nature of autoimmune diseases means that cytokine profiles vary greatly across patients and even over time in the same patient. Therefore, applying the same set of single-cytokine blockers in every instance is not a valid approach, indicating that better tools for stratification, as well as dynamic biomarkers, are required (97).
5.3 Future directions
The above barriers have led to multifunctional approaches to next-generation cytokine therapies. Dual-cytokine inhibitors that target 2 cytokines synergistically are an interesting tool. An example is bimekizumab, which inhibits IL-17A and IL-17F, leading to superior efficacy over IL-17A blockade alone in psoriasis and spondyloarthropathies (98). Co-inhibition of IL-12 and IL-23 (e.g., ustekinumab) in Crohn’s disease and psoriasis modulates both Th1 and Th17 pathways. Another innovation aimed at enhancing specificity and reducing immunogenicity involves cytokine traps or engineered receptor decoys that sequester cytokines (99). Examples are rilonacept, an IL-1 trap used in autoinflammatory syndromes, and newer constructs targeting IL-6 and GM-CSF. Some cytokine traps are designed with altered Fc regions or joined ligands to enhance their half-life and improve tissue targeting (100). Synthetic receptors for cytokines under development can be engineered to switch pro-inflammatory signals into tolerogenic ones or restrict cytokine activity to defined tissues or cell types (101). For example, IL-2 variants engineered to preferentially expand Tregs while sparing effector T cells and NK cells are in late-stage clinical development for T1D and lupus (102). Advances in systems immunology and machine learning will play a transformative role in identifying cytokine signatures that predict therapeutic response. By integrating transcriptomics, proteomics, and spatial mapping of immune networks, cytokine therapy regimens can be tailored to each patient and their effects dynamically monitored.
6 Microbiome-based therapy
Microbiome-based therapy has recently become a promising approach in immunotherapy. The human gut microbiome is a major regulator of immune function. The microorganisms that populate the gastrointestinal tract greatly influence immune development, tolerance, and responsiveness (103). Imbalances in gut microbiota (dysbiosis) have been increasingly correlated with the etiology of several autoimmune diseases, including SLE (104), IBD, MS, T1D, and RA, leading an interest in microbiome-related therapies that seek to restore microbial balance and, accordingly, immune homeostasis (105). Dysbiosis has been shown to be correlated with the production of autoantibodies (106) and its effects are mediated by Th17/Treg balance, gut barrier integrity, and dietary interactions. While it is closely linked to B-cell differentiation, autoantibody production, and systemic immune responses in autoimmune diseases like SLE, RA, Graves’ disease, and Hashimoto’s thyroiditis, a direct cause-and-effect relationship is not firmly established across all conditions. Additionally, microbial metabolites, such as SCFAs, have been identified as important modulators of T-cell function and immune responses (107). Thus, microbiome-based immunotherapy has been extensively investigated for autoimmune disease treatment.
6.1 Breakthroughs
Probiotics and prebiotics are interventions designed to modulate immune responses through dietary measures. Preclinical studies that include animal models and in vitro studies demonstrated the ability of probiotics, including Lactobacillus rhamnosus GG, Bifidobacterium longum, and Ruminococcaceae, and Lachnospiraceae, to induce changes in the expansion of Tregs, reduce IL-6 and TNF-α as pro-inflammatory cytokines production, and restore mucosal barrier integrity (108, 109). What these measures imply is the influence that commensal microbes can have on systemic immune responses. In animal studies, dietary fibers (such as inulin and fructo-oligosaccharides) improved regulatory immunological responses (such as the growth of Tregs) and decreased disease severity. They also raised the number of beneficial SCFA-producing bacteria (110). Acetate and butyrate contribute to suppressing pro-inflammatory responses, while butyrate and propionate are especially crucial for promoting Treg differentiation through epigenetic regulation. These metabolites demonstrate how important gut microbial products are in regulating the immune balance of the host (111).
Fecal microbiota transplantation (FMT) now shows great promise in autoimmune contexts, particularly in ulcerative colitis (UC) (112). Multiple randomized controlled clinical trials have illustrated that FMT can induce clinical and endoscopic remission in patients with UC; the response rates correlated with increased microbial diversity, along with the enrichment of beneficial taxa, such as Faecalibacterium prausnitzii and Akkermansia muciniphila (113, 114). Preclinical studies investigating immune response following the transfer of healthy microbiota demonstrated a reduction in inflammation in RA and MS animal models. Moreover, FMT from healthy donors increased insulin sensitivity and delayed the onset of T1D in an animal model. A similar positive outcome was demonstrated with SLE, showing a decrease in autoantibody production and improvement in kidney function (115). Hence, several up-to-date clinical trials are actively investigating the therapeutic potential of the microbiome in the management of autoimmune diseases (Table 4).
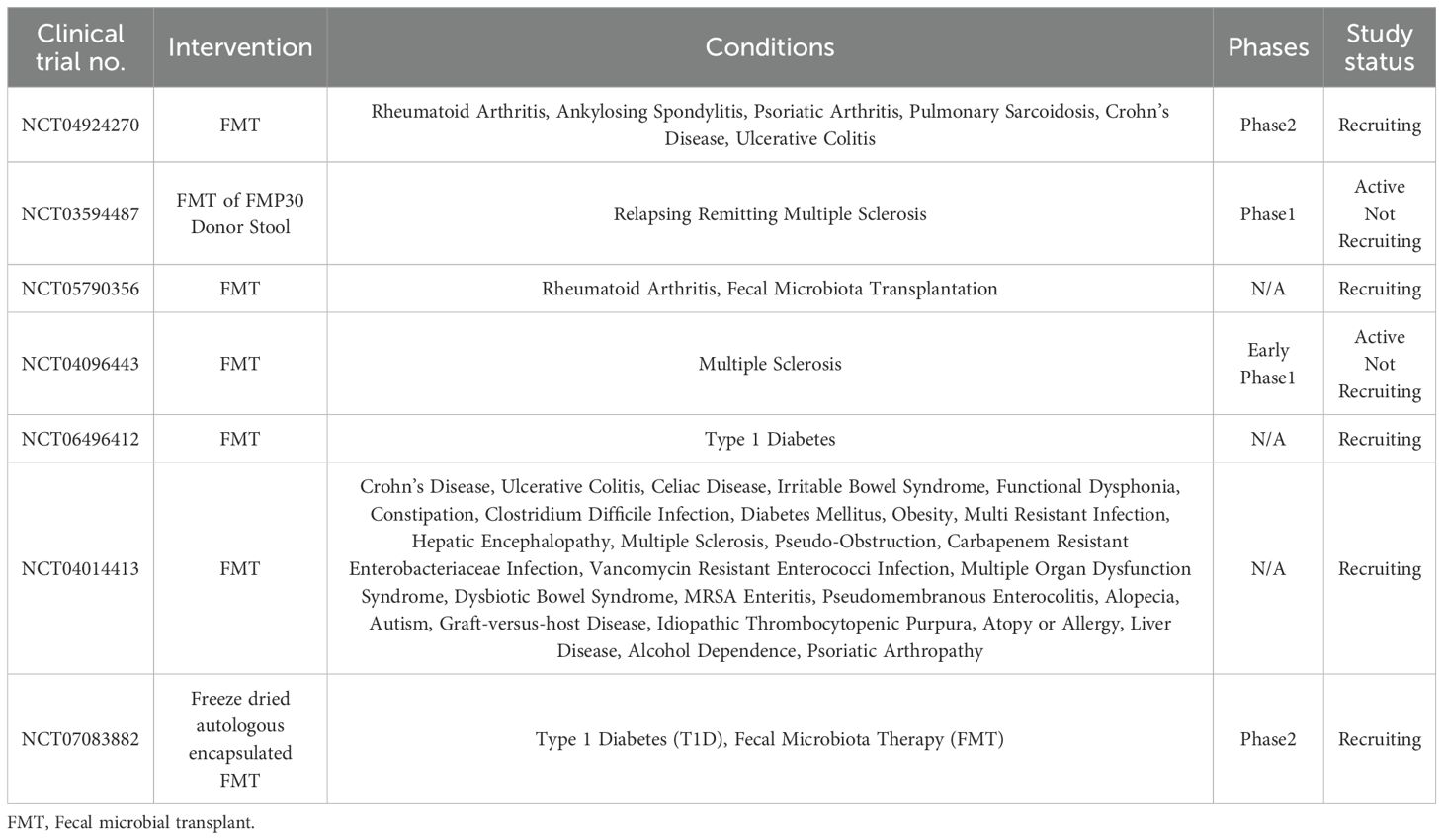
Table 4. Microbiome-based therapy currently in clinical trials for autoimmune diseases.
6.2 Challenges
Microbiome-based therapies face several major obstacles. The predominant obstacle constitutes individual variation, wherein every person has a unique microbial fingerprint shaped by genetics, environment, diet, and antibiotic use. Thus, some therapies may work for an individual or cohort but be useless, or worse, harmful, to another (116). Furthermore, FMT lacks standardization on donor selection, preparation, administration routes, and dosing frequency. This heterogeneity hampers reproducibility and raises safety concerns, like the potential transfer of opportunistic pathogens or undesirable metabolic characteristics (117, 118). Although screening protocols have improved, rare but severe adverse events, as well as the transmission of multidrug-resistant organisms, have triggered the FDA to intensify its controls. The mechanistic basis of microbiome-immune interactions is complex and not completely appreciated. Microbes affect immunity through different means, such as short-chain fatty acid production, changes in antigen-presenting cells, modulation of epithelial tight junctions, and interactions with pattern-recognition receptors (119). This issue is rendered even more complicated by the dynamic nature of the microbiome, which hinders persistence over time. Changes in diet, stress, sickness, or antibiotic consumption can drive a drastic shift in the community, compromising or entirely reversing previous benefits (120, 121).
6.3 Future directions
The future of microbiome therapeutics is in precision modulation interventions targeted to the individual’s specific microbial and immunological landscape. Next-generation sequencing and metagenomic profiling allow for the deep characterization of microbial communities and their functional potential and the identification of dysbiosis biomarkers and predictive response signatures (122). Currently, with the help of CRISPR-Cas systems, the idea is to edit bacterial genomes in the gut, thus allowing in situ reprogramming of the microbiome without the need to introduce foreign microbes into the environment. CRISPR-guided antimicrobials can kill pathogenic and pro-inflammatory strains while leaving beneficial commensals intact. However, gene insertion may enable the expression of immunostimulatory molecules by endogenous microorganisms (123). Synthetic biology is also accelerating the development of LBPs for sensing environmental cues and the controlled, tissue-specific release of therapeutic agents. Smart probiotics engineered to detect inflammation and respond by producing IL-10 or retinoic acid are part of this picture. Finally, computational modeling and machine learning are applied to simulate host-microbe interactions and predict treatment responses (124). By integrating all microbiome-initiated datasets on the genetic, dietary, and immune fronts, these tools will allow for the rational design of patient-specific microbiome therapies with optimized efficacy and safety.
7 Conclusion
Immunotherapy for autoimmune diseases is undergoing a transition, shifting from immunosuppression to immune modulation. This trend can be observed in the advanced fields of CAR T-cell therapy, bsAbs, checkpoint agonists, cytokine targeting, and microbiome interventions, which reflect a new system of rapid change in therapeutics. Each has distinct advantages but presents challenges to safety, cost, scale, or regulatory approval. The future integration of such therapies into certified care as they advance in preclinical and clinical studies will depend on interdisciplinary research, technological refinements, and personalized approaches concerning patients’ various immune profiles (Figure 3). The future of autoimmune disease treatment will hinge on a personalized, multifaceted approach that incorporates immune modulation, tolerance establishment, and tissue regeneration.
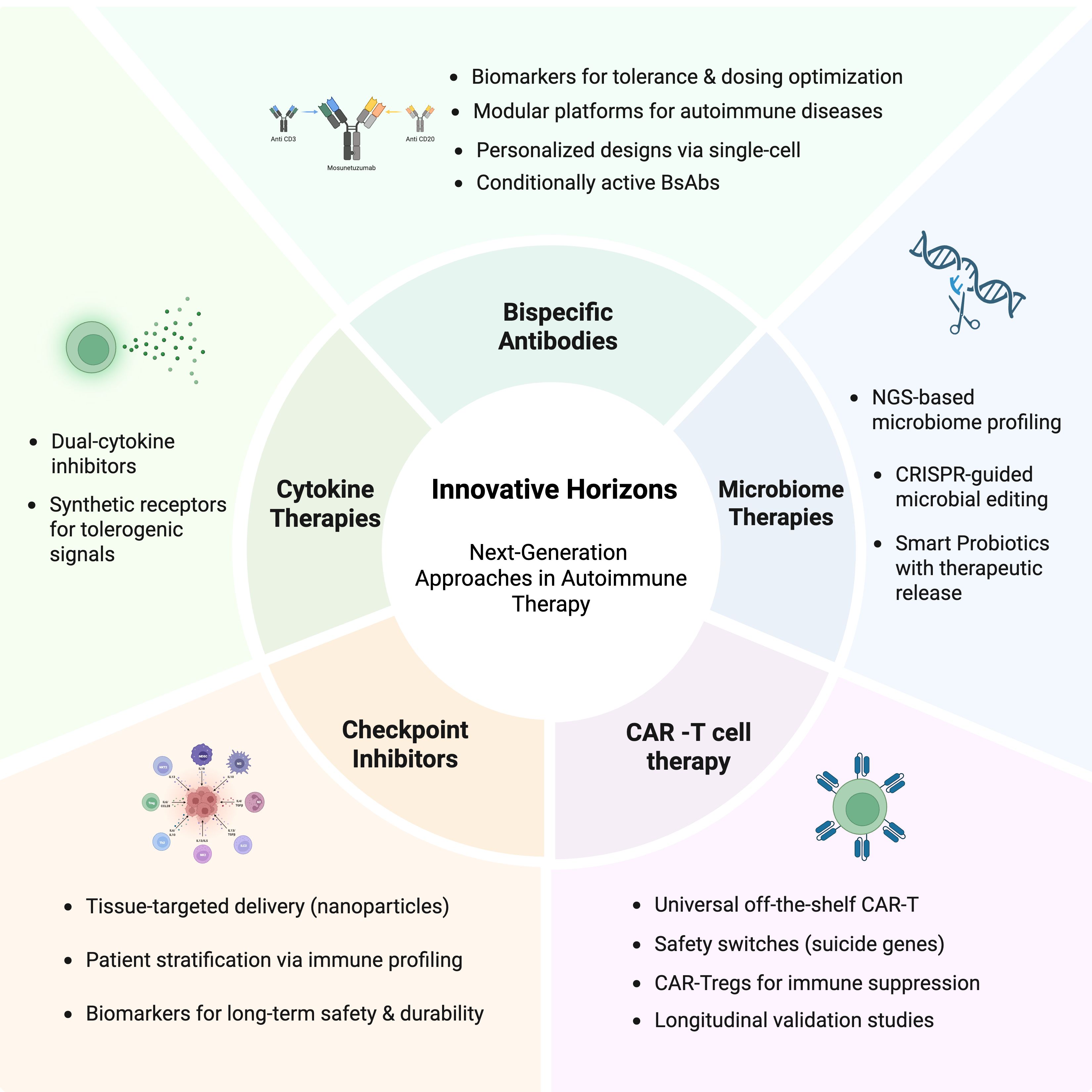
Figure 3. Innovative Horizons in Next-Generation Therapeutic Approaches for Autoimmune Diseases. This figure demonstrates the emerging schemes directed at immune modulation. These include CAR T-cell therapy, bispecific antibodies, next-generation immune checkpoint modulators, targeted cytokine therapies, and microbiome-based interventions, representing the foreground of therapeutic advancement in autoimmunity. Created in BioRender. Alsayb, M. (2025) https://BioRender.com/o15madg.
Author contributions
MA: Writing – review & editing, Writing – original draft.
Funding
The authors declare that no financial support was received for the research, and/or publication of this article.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Khalid S, Asif H, and Kabir M. Immunity and Autoimmune system-a review: Immunity and Autoimmune system. Pakistan J Health Sci. (2021) 2:3–7. doi: 10.54393/pjhs.v2i02.24
2. Wang L, Wang FS, and Gershwin ME. Human autoimmune diseases: a comprehensive update. J Internal Med. (2015) 278:369–95. doi: 10.1111/joim.12395
3. Cai Z, Wang S, and Li J. Treatment of inflammatory bowel disease: a comprehensive review. Front Med. (2021) 8:765474. doi: 10.3389/fmed.2021.765474
4. Konen FF, Mohn N, Witte T, Schefzyk M, Wiestler M, Lovric S, et al. Treatment of autoimmunity: The impact of disease-modifying therapies in multiple sclerosis and comorbid autoimmune disorders. Autoimmun Rev. (2023) 22:103312. doi: 10.1016/j.autrev.2023.103312
5. von Ahsen N and Chan A. Therapeutic monitoring of immunotherapies in autoimmune diseases. Curr Pharm Des. (2012) 18:4550–5. doi: 10.2174/138161212802502152
6. Liu Y, Dong M, Chu Y, Zhou L, You Y, Pang X, et al. Dawn of CAR-T cell therapy in autoimmune diseases. Chin Med J. (2024) 137:1140–50. doi: 10.1097/CM9.0000000000003111
7. Schett G, Mackensen A, and Mougiakakos D. CAR T-cell therapy in autoimmune diseases. Lancet. (2023) 402:2034–44. doi: 10.1016/S0140-6736(23)01126-1
8. Nasra S, Bhatia D, and Kumar A. Recent advances in nanoparticle-based drug delivery systems for rheumatoid arthritis treatment. Nanoscale Adv. (2022) 4:3479–94. doi: 10.1039/D2NA00229A
9. Guo Q, Li J, Wang J, Li L, Wei J, and Zhang L. The advent of chimeric antigen receptor T Cell therapy in recalibrating immune balance for rheumatic autoimmune disease treatment. Front Pharmacol. (2024) 15:1502298. doi: 10.3389/fphar.2024.1502298
10. Dias J, Garcia J, Agliardi G, and Roddie C. CAR-T cell manufacturing landscape-Lessons from the past decade and considerations for early clinical development. Mol Ther Methods Clin Dev. (2024) 32:101250. doi: 10.1016/j.omtm.2024.101250
11. Muller F, Taubmann J, Bucci L, Wilhelm A, Bergmann C, Volkl S, et al. CD19 CAR T-cell therapy in autoimmune disease - A case series with follow-up. N Engl J Med. (2024) 390:687–700. doi: 10.1056/NEJMoa2308917
12. Muller F, Boeltz S, Knitza J, Aigner M, Volkl S, Kharboutli S, et al. CD19-targeted CAR T cells in refractory antisynthetase syndrome. Lancet. (2023) 401:815–8. doi: 10.1016/S0140-6736(23)00023-5
13. Mahevas M, Michel M, Weill JC, and Reynaud CA. Long-lived plasma cells in autoimmunity: lessons from B-cell depleting therapy. Front Immunol. (2013) 4:494. doi: 10.3389/fimmu.2013.00494
14. Bergmann C, Muller F, Distler JHW, Gyorfi AH, Volkl S, Aigner M, et al. Treatment of a patient with severe systemic sclerosis (SSc) using CD19-targeted CAR T cells. Ann Rheum Dis. (2023) 82:1117–20. doi: 10.1136/ard-2023-223952
15. Zhou J, Lei B, Shi F, Luo X, Wu K, Xu Y, et al. CAR T-cell therapy for systemic lupus erythematosus: current status and future perspectives. Front Immunol. (2024) 15:1476859. doi: 10.3389/fimmu.2024.1476859
16. Granit V, Benatar M, Kurtoglu M, Miljkovic MD, Chahin N, Sahagian G, et al. Safety and clinical activity of autologous RNA chimeric antigen receptor T-cell therapy in myasthenia gravis (MG-001): a prospective, multicentre, open-label, non-randomised phase 1b/2a study. Lancet Neurol. (2023) 22:578–90. doi: 10.1016/S1474-4422(23)00194-1
17. Haghikia A, Schett G, and Mougiakakos D. B cell-targeting chimeric antigen receptor T cells as an emerging therapy in neuroimmunological diseases. Lancet Neurol. (2024) 23:615–24. doi: 10.1016/S1474-4422(24)00140-6
18. Zhang Y, Liu D, Zhang Z, Huang X, Cao J, Wang G, et al. Bispecific BCMA/CD19 targeted CAR-T cell therapy forces sustained disappearance of symptoms and anti-acetylcholine receptor antibodies in refractory myasthenia gravis: a case report. J Neurol. (2024) 271:4655–9. doi: 10.1007/s00415-024-12367-4
19. Zhang W, Liu D, Zhang T, Cao J, Wang G, Li H, et al. BCMA-CD19 bispecific CAR-T therapy in refractory chronic inflammatory demyelinating polyneuropathy. hLife. (2024) 2:434–8. doi: 10.1016/j.hlife.2024.05.005
20. Radic M, Neeli I, and Marion T. Prospects for CAR T cell immunotherapy in autoimmune diseases: Clues from Lupus. Expert Opin Biol Ther. (2022) 22:499–507. doi: 10.1080/14712598.2022.2026921
21. Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. (2020) 130:6317–24. doi: 10.1172/JCI138416
22. Fu Y, Feng C, Qin S, Xing Z, Liu C, Liu Z, et al. Breaking barriers: advancing cellular therapies in autoimmune disease management. Front Immunol. (2024) 15:1503099. doi: 10.3389/fimmu.2024.1503099
23. Jiang P, Zhang Z, Hu Y, Liang Z, Han Y, Li X, et al. Single-cell ATAC-seq maps the comprehensive and dynamic chromatin accessibility landscape of CAR-T cell dysfunction. Leukemia. (2022) 36:2656–68. doi: 10.1038/s41375-022-01676-0
24. Leahy AB, Newman H, Li Y, Liu H, Myers R, DiNofia A, et al. CD19-targeted chimeric antigen receptor T-cell therapy for CNS relapsed or refractory acute lymphocytic leukaemia: a post-hoc analysis of pooled data from five clinical trials. Lancet Haematol. (2021) 8:e711–e22. doi: 10.1016/S2352-3026(21)00238-6
25. Levstek L, Janzic L, Ihan A, and Kopitar AN. Biomarkers for prediction of CAR T therapy outcomes: current and future perspectives. Front Immunol. (2024) 15:1378944. doi: 10.3389/fimmu.2024.1378944
26. Liu Y, Jie X, Nian L, Wang Y, Wang C, Ma J, et al. A combination of pre-infusion serum ferritin, CRP and IL-6 predicts outcome in relapsed/refractory multiple myeloma patients treated with CAR-T cells. Front Immunol. (2023) 14:1169071. doi: 10.3389/fimmu.2023.1169071
27. Zhu X, Hu H, Xiao Y, Li Q, Zhong Z, Yang J, et al. Tumor-derived extracellular vesicles induce invalid cytokine release and exhaustion of CD19 CAR-T Cells. Cancer Lett. (2022) 536:215668. doi: 10.1016/j.canlet.2022.215668
28. Kawalekar OU, OC RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:712. doi: 10.1016/j.immuni.2016.02.023
29. Greenbaum U, Strati P, Saliba RM, Torres J, Rondon G, Nieto Y, et al. CRP and ferritin in addition to the EASIX score predict CAR-T-related toxicity. Blood Adv. (2021) 5:2799–806. doi: 10.1182/bloodadvances.2021004575
30. Cox MJ, Lucien F, Sakemura R, Boysen JC, Kim Y, Horvei P, et al. Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol Ther. (2021) 29:1529–40. doi: 10.1016/j.ymthe.2020.12.033
31. Grant SJ, Grimshaw AA, Silberstein J, Murdaugh D, Wildes TM, Rosko AE, et al. Clinical presentation, risk factors, and outcomes of immune effector cell-associated neurotoxicity syndrome following chimeric antigen receptor T cell therapy: a systematic review. Transplant Cell Ther. (2022) 28:294–302. doi: 10.1016/j.jtct.2022.03.006
32. Bach J-F. Immunosuppressive therapy of autoimmune diseases. Trends Pharmacol Sci. (1993) 14:213–6. doi: 10.1016/0165-6147(93)90211-2
33. Dimitri A, Herbst F, and Fraietta JA. Engineering the next-generation of CAR T-cells with CRISPR-Cas9 gene editing. Mol Cancer. (2022) 21:78. doi: 10.1186/s12943-022-01559-z
34. Qasim W. Genome-edited allogeneic donor “universal” chimeric antigen receptor T cells. Blood. (2023) 141:835–45. doi: 10.1182/blood.2022016204
35. Mohammadi V, Maleki AJ, Nazari M, Siahmansouri A, Moradi A, Elahi R, et al. Chimeric antigen receptor (CAR)-based cell therapy for type 1 diabetes mellitus (T1DM); current progress and future approaches. Stem Cell Rev Rep. (2024) 20:585–600. doi: 10.1007/s12015-023-10668-1
36. Diaconu I, Ballard B, Zhang M, Chen Y, West J, Dotti G, et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol Ther. (2017) 25:580–92. doi: 10.1016/j.ymthe.2017.01.011
37. Gargett T and Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. (2014) 5:235. doi: 10.3389/fphar.2014.00235
38. Yeo YY, Chang Y, Qiu H, Yiu SPT, Michel HA, Wu W, et al. Same-slide spatial multi-omics integration reveals tumor virus-linked spatial reorganization of the tumor microenvironment. bioRxiv. (2024). doi: 10.1101/2024.12.20.629650
39. Tao W, Sun Q, Xu B, and Wang R. Towards the prediction of responses to cancer immunotherapy: A multi-omics review. Life (Basel). (2025) 15:283. doi: 10.3390/life15020283
40. Rashid MM and Selvarajoo K. Advancing drug-response prediction using multi-modal and -omics machine learning integration (MOMLIN): a case study on breast cancer clinical data. Brief Bioinform. (2024) 25:bbae300. doi: 10.1093/bib/bbae300
41. Brinkmann U and Kontermann RE. Bispecific antibodies. Science. (2021) 372:916–7. doi: 10.1126/science.abg1209
42. Liu H, Saxena A, Sidhu SS, and Wu D. Fc engineering for developing therapeutic bispecific antibodies and novel scaffolds. Front Immunol. (2017) 8:38. doi: 10.3389/fimmu.2017.00038
43. Wei J, Yang Y, Wang G, and Liu M. Current landscape and future directions of bispecific antibodies in cancer immunotherapy. Front Immunol. (2022) 13:1035276. doi: 10.3389/fimmu.2022.1035276
44. Huang H and Wei X. Therapeutic potential of CD20/CD3 bispecific antibodies in the treatment of autoimmune diseases. Rheumatol Immunol Res. (2024) 5:209–16. doi: 10.1515/rir-2024-0029
45. Domingo-Gonzalez R, Baribaud I, Oyasu M, Hart K, Burns K, Pandey S, et al. POS1062 therapeutic potential of imvotamab, a CD20-targeted bispecific igm t cell engager, for the treatment of refractory autoimmune disease patients. Ann Rheumatic Dis. (2024) 83:935. doi: 10.1136/annrheumdis-2024-eular.3799
46. Chindalore V, Martins E, Pendergraft WF, Sheng XR, Schroeder A, Gearhart L, et al. POS1160 a phase ib, multicentre, open-label, dose-escalation study to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamics of subcutaneously administered mosunetuzumab in participants with systemic lupus erythematosus. Ann Rheumatic Dis. (2025) 84:1232–3. doi: 10.1016/j.ard.2025.06.510
47. Subklewe M, Magno G, Gebhardt C, Bucklein V, Szelinski F, Arevalo HJR, et al. Application of blinatumomab, a bispecific anti-CD3/CD19 T-cell engager, in treating severe systemic sclerosis: A case study. Eur J Cancer. (2024) 204:114071. doi: 10.1016/j.ejca.2024.114071
48. Sedykh SE, Prinz VV, Buneva VN, and Nevinsky GA. Bispecific antibodies: design, therapy, perspectives. Drug Design Dev Ther. (2018) 12:195–208. doi: 10.2147/DDDT.S151282
49. Zheng S, Shen F, Jones B, Fink D, Geist B, Nnane I, et al. Characterization of concurrent target suppression by JNJ-61178104, a bispecific antibody against human tumor necrosis factor and interleukin-17A. MAbs. (2020) 12:1770018. doi: 10.1080/19420862.2020.1770018
50. Fleischmann RM, Wagner F, Kivitz AJ, Mansikka HT, Khan N, Othman AA, et al. Safety, tolerability, and pharmacodynamics of ABT-122, a tumor necrosis factor- and interleukin-17-targeted dual variable domain immunoglobulin, in patients with rheumatoid arthritis. Arthritis Rheumatol. (2017) 69:2283–91. doi: 10.1002/art.40319
51. Genovese MC, Weinblatt ME, Aelion JA, Mansikka HT, Peloso PM, Chen K, et al. ABT-122, a bispecific dual variable domain immunoglobulin targeting tumor necrosis factor and interleukin-17A, in patients with rheumatoid arthritis with an inadequate response to methotrexate: A randomized, double-blind study. Arthritis Rheumatol. (2018) 70:1710–20. doi: 10.1002/art.40580
52. Bhattacharya P, Fan J, Haddad C, Essani A, Gopisetty A, Elshabrawy HA, et al. A novel pancreatic beta-cell targeting bispecific-antibody (BsAb) can prevent the development of type 1 diabetes in NOD mice. Clin Immunol. (2014) 153:187–98. doi: 10.1016/j.clim.2014.04.014
53. Li J, Piskol R, Ybarra R, Chen YJ, Li J, Slaga D, et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci Transl Med. (2019) 11:eaax8861. doi: 10.1126/scitranslmed.aax8861
54. Madsen AV, Pedersen LE, Kristensen P, and Goletz S. Design and engineering of bispecific antibodies: insights and practical considerations. Front Bioeng Biotechnol. (2024) 12:1352014. doi: 10.3389/fbioe.2024.1352014
55. Krah S, Kolmar H, Becker S, and Zielonka S. Engineering IgG-like bispecific antibodies—an overview. Antibodies. (2018) 7:28. doi: 10.3390/antib7030028
56. Byrne H, Conroy PJ, Whisstock JC, and O’Kennedy RJ. A tale of two specificities: bispecific antibodies for therapeutic and diagnostic applications. Trends Biotechnol. (2013) 31:621–32. doi: 10.1016/j.tibtech.2013.08.007
57. Kandari D and Bhatnagar R. Antibody engineering and its therapeutic applications. Int Rev Immunol. (2023) 42:156–83. doi: 10.1080/08830185.2021.1960986
58. Lewis SM, Wu X, Pustilnik A, Sereno A, Huang F, Rick HL, et al. Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol. (2014) 32:191–8. doi: 10.1038/nbt.2797
59. Li Y-Q, Chen X-M, Si G-F, and Yuan X-M. Progress of lymphocyte activation gene 3 and programmed cell death protein 1 antibodies for cancer treatment: A review. Biomol Biomed. (2024) 24:764. doi: 10.17305/bb.2024.10339
60. Ibis B, Aliazis K, Cao C, Yenyuwadee S, and Boussiotis VA. Immune-related adverse effects of checkpoint immunotherapy and implications for the treatment of patients with cancer and autoimmune diseases. Front Immunol. (2023) 14:1197364. doi: 10.3389/fimmu.2023.1197364
61. Sierro S, Romero P, and Speiser DE. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin Ther Targets. (2011) 15:91–101. doi: 10.1517/14712598.2011.540563
62. Ge Z, Peppelenbosch MP, Sprengers D, and Kwekkeboom J. TIGIT, the next step towards successful combination immune checkpoint therapy in cancer. Front Immunol. (2021) 12:699895. doi: 10.3389/fimmu.2021.699895
63. Yang M, Cui M, Sun Y, Liu S, and Jiang W. Mechanisms, combination therapy, and biomarkers in cancer immunotherapy resistance. Cell Commun Signal. (2024) 22:338. doi: 10.1186/s12964-024-01711-w
64. Hu S, Liu X, Li T, Li Z, and Hu F. LAG3 (CD223) and autoimmunity: Emerging evidence. J Autoimmun. (2020) 112:102504. doi: 10.1016/j.jaut.2020.102504
65. Jones BE, Maerz MD, Bahnson HT, Somasundaram A, McCarthy LH, Speake C, et al. Fewer LAG-3(+) T cells in relapsing-remitting multiple sclerosis and type 1 diabetes. J Immunol. (2022) 208:594–602. doi: 10.4049/jimmunol.2100850
66. Chocarro L, Blanco E, Arasanz H, Fernandez-Rubio L, Bocanegra A, Echaide M, et al. Clinical landscape of LAG-3-targeted therapy. Immunooncol Technol. (2022) 14:100079. doi: 10.1016/j.iotech.2022.100079
67. Tang L, Li G, Zheng Y, Hou C, Gao Y, Hao Y, et al. Tim-3 relieves experimental autoimmune encephalomyelitis by suppressing MHC-II. Front Immunol. (2021) 12:770402. doi: 10.3389/fimmu.2021.770402
68. Karzoun B, Ramadan A, Allababidi S, and Fathallah AM. Breaking the triad: immune tolerance induction without antigen co-presentation via tim agonist for the treatment of autoimmune diseases. Int J Mol Sci. (2025) 26:5531. doi: 10.3390/ijms26125531
69. Dougall WC, Kurtulus S, Smyth MJ, and Anderson AC. TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev. (2017) 276:112–20. doi: 10.1111/imr.12518
70. Muhammad F, Wang D, McDonald T, Walsh M, Drenen K, Montieth A, et al. TIGIT(+) A2Ar-Dependent anti-uveitic Treg cells are a novel subset of Tregs associated with resolution of autoimmune uveitis. J Autoimmun. (2020) 111:102441. doi: 10.1016/j.jaut.2020.102441
71. Liu S, Sun L, Wang C, Cui Y, Ling Y, Li T, et al. Treatment of murine lupus with TIGIT-Ig. Clin Immunol. (2019) 203:72–80. doi: 10.1016/j.clim.2019.04.007
72. Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T, et al. Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J Immunol. (2018) 200:3000–7. doi: 10.4049/jimmunol.1700407
73. Yue C, Gao S, Li S, Xing Z, Qian H, Hu Y, et al. TIGIT as a promising therapeutic target in autoimmune diseases. Front Immunol. (2022) 13:911919. doi: 10.3389/fimmu.2022.911919
74. Wang G, Tai R, Wu Y, Yang S, Wang J, Yu X, et al. The expression and immunoregulation of immune checkpoint molecule VISTA in autoimmune diseases and cancers. Cytokine Growth Factor Rev. (2020) 52:1–14. doi: 10.1016/j.cytogfr.2020.02.002
75. Ceeraz S, Sergent PA, Plummer SF, Schned AR, Pechenick D, Burns CM, et al. VISTA deficiency accelerates the development of fatal murine lupus nephritis. Arthritis Rheumatol. (2017) 69:814–25. doi: 10.1002/art.40020
76. Han X, Vesely MD, Yang W, Sanmamed MF, Badri T, Alawa J, et al. PD-1H (VISTA)-mediated suppression of autoimmunity in systemic and cutaneous lupus erythematosus. Sci Transl Med. (2019) 11:eaax1159. doi: 10.1126/scitranslmed.aax1159
77. Shekari N, Shanehbandi D, Kazemi T, Zarredar H, Baradaran B, and Jalali SA. VISTA and its ligands: the next generation of promising therapeutic targets in immunotherapy. Cancer Cell Int. (2023) 23:265. doi: 10.1186/s12935-023-03116-0
78. Li N, Xu W, Yuan Y, Ayithan N, Imai Y, Wu X, et al. Immune-checkpoint protein VISTA critically regulates the IL-23/IL-17 inflammatory axis. Sci Rep. (2017) 7:1485. doi: 10.1038/s41598-017-01411-1
79. Ocana-Guzman R, Osorio-Perez D, and Chavez-Galan L. Opportunistic infections and immune-related adverse events associated with administering immune checkpoint inhibitors: A narrative review. Pharm (Basel). (2023) 16:1119. doi: 10.3390/ph16081119
80. Lee DJ. The relationship between TIGIT+ regulatory T cells and autoimmune disease. Int Immunopharmacol. (2020) 83:106378. doi: 10.1016/j.intimp.2020.106378
81. Norde WJ, Hobo W, and Dolstra H. Role of Co-inhibitory molecules in tumor escape from CTL attack. Resistance Cancer Cells CTL-Mediated Immunother. (2015) 31–58. doi: 10.1007/978-3-319-17807-3_2
82. Seňavová J, Rajmonová A, Heřman V, Jura F, Veľasová A, Hamova I, et al. Immune checkpoints and their inhibition in T-cell lymphomas. Folia Biol. (2024) 70:123–51. doi: 10.14712/fb2024070030123
83. Alausa A, Victor UC, Fadahunsi OS, Owolabi N, Adeniji A, Olatinwo M, et al. Checkpoints and immunity in cancers: Role of GNG12. Pharmacol Res. (2022) 180:106242. doi: 10.1016/j.phrs.2022.106242
84. Metabolic modulation of immune checkpoints and novel therapeutic strategies in cancer. Semin Cancer Biol. (2022) 86(Pt 3):542–65. doi: 10.1016/j.semcancer.2022.02.010
85. Oberholzer A, Oberholzer C, and Moldawer LL. Cytokine signaling-regulation of the immune response in normal and critically ill states. Crit Care Med. (2000) 28:N3–N12. doi: 10.1097/00003246-200004001-00002
86. Altan-Bonnet G and Mukherjee R. Cytokine-mediated communication: a quantitative appraisal of immune complexity. Nat Rev Immunol. (2019) 19:205–17. doi: 10.1038/s41577-019-0131-x
87. Mahapatro M, Erkert L, and Becker C. Cytokine-mediated crosstalk between immune cells and epithelial cells in the gut. Cells. (2021) 10:111. doi: 10.3390/cells10010111
88. Salvador AF, de Lima KA, and Kipnis J. Neuromodulation by the immune system: a focus on cytokines. Nat Rev Immunol. (2021) 21:526–41. doi: 10.1038/s41577-021-00508-z
89. Lacy P and Stow JL. Cytokine release from innate immune cells: association with diverse membrane trafficking pathways. Blood J Am Soc Hematol. (2011) 118:9–18. doi: 10.1182/blood-2010-08-265892
90. Vallurupalli M and Berliner N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood. (2019) 134:1783–6. doi: 10.1182/blood.2019002289
91. Minshawi F, Lanvermann S, McKenzie E, Jeffery R, Couper K, Papoutsopoulou S, et al. The generation of an engineered interleukin-10 protein with improved stability and biological function. Front Immunol. (2020) 11:1794. doi: 10.3389/fimmu.2020.01794
92. Fioranelli M. Twenty-five years of studies and trials for the therapeutic application of IL-10 immunomodulating properties. From high doses administration to low dose medicine new paradigm. J Integr Cardiol. (2015) 1:2–6. doi: 10.15761/JIC.1000102
93. Reardon C, Murray K, and Lomax AE. Neuroimmune communication in health and disease. Physiol Rev. (2018) 98:2287–316. doi: 10.1152/physrev.00035.2017
94. Xie J, Tato CM, and Davis MM. How the immune system talks to itself: the varied role of synapses. Immunol Rev. (2013) 251:65–79. doi: 10.1111/imr.12017
95. Huang H, Patel DD, and Manton KG. The immune system in aging: roles of cytokines, T cells and NK cells. Front Biosci. (2005) 10:192–215. doi: 10.2741/1521
96. Davidson L, van den Reek JMPA, Bruno M, van Hunsel F, Herings RMC, Matzaraki V, et al. Risk of candidiasis associated with interleukin-17 inhibitors: A real-world observational study of multiple independent sources. Lancet Regional Health Europe. (2022) 13:100266. doi: 10.1016/j.lanepe.2021.100266
97. Fenton KA and Pedersen HL. Advanced methods and novel biomarkers in autoimmune diseases − a review of the recent years progress in systemic lupus erythematosus. Front Med. (2023) 10. doi: 10.3389/fmed.2023.1183535
98. Reich K, Warren RB, Lebwohl M, Gooderham M, Strober B, Langley RG, et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med. (2021) 385:142–52. doi: 10.1056/NEJMoa2102383
99. Dantzer R. Cytokine-induced sickness behaviour: a neuroimmune response to activation of innate immunity. Eur J Pharmacol. (2004) 500:399–411. doi: 10.1016/j.ejphar.2004.07.040
100. Qin S, Xie B, Wang Q, Yang R, Sun J, Hu C, et al. New insights into immune cells in cancer immunotherapy: from epigenetic modification, metabolic modulation to cell communication. MedComm. (2024) 5:e551. doi: 10.1002/mco2.551
101. Guo X, Sui R, and Piao H. Exosomes-mediated crosstalk between glioma and immune cells in the tumor microenvironment. CNS Neurosci Ther. (2023) 29:2074–85. doi: 10.1111/cns.14239
102. Alvarez F, Acuff NV, La Muraglia GM, Sabri N, ME M, JM M, et al. The IL-2 SYNTHORIN molecule promotes functionally adapted Tregs in a preclinical model of type 1 diabetes. JCI Insight. (2024) 9:e182064. doi: 10.1172/jci.insight.182064
103. Bengmark S. Gut microbiota, immune development and function. Pharmacol Res. (2013) 69:87–113. doi: 10.1016/j.phrs.2012.09.002
104. Ali AY, Zahran SA, Eissa M, Kashef MT, and Ali AE. Gut microbiota dysbiosis and associated immune response in systemic lupus erythematosus: impact of disease and treatment. Gut Pathog. (2025) 17:10. doi: 10.1186/s13099-025-00683-7
105. Yoo JY, Groer M, Dutra SVO, Sarkar A, and McSkimming DI. Gut microbiota and immune system interactions. Microorganisms. (2020) 8:1587. doi: 10.3390/microorganisms8101587
106. He L, Li X, Jiang S, Ou Y, Wang S, Shi N, et al. The influence of the gut microbiota on B cells in autoimmune diseases. Mol Med. (2025) 31:149. doi: 10.1186/s10020-025-01195-5
107. Bhutta NK, Xu X, Jian C, Wang Y, Liu Y, Sun J, et al. Gut microbiota mediated T cells regulation and autoimmune diseases. Front Microbiol. (2024) 15:1477187. doi: 10.3389/fmicb.2024.1477187
108. Takiishi T, Fenero CIM, and Câmara NOS. Intestinal barrier and gut microbiota: Shaping our immune responses throughout life. Tissue Barriers. (2017) 5:e1373208. doi: 10.1080/21688370.2017.1373208
109. Amdekar S, Singh V, Singh R, Sharma P, Keshav P, and Kumar A. Lactobacillus casei reduces the inflammatory joint damage associated with collagen-induced arthritis (CIA) by reducing the pro-inflammatory cytokines: Lactobacillus casei: COX-2 inhibitor. J Clin Immunol. (2011) 31:147–54. doi: 10.1007/s10875-010-9457-7
110. Zegarra-Ruiz DF, El Beidaq A, Iniguez AJ, Lubrano Di Ricco M, Manfredo Vieira S, Ruff WE, et al. A diet-sensitive commensal lactobacillus strain mediates TLR7-dependent systemic autoimmunity. Cell Host Microbe. (2019) 25:113–27 e6. doi: 10.1016/j.chom.2018.11.009
111. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. (2013) 504:446–50. doi: 10.1038/nature12721
112. Gefen R, Dourado J, Emile SH, Wignakumar A, Rogers P, Aeschbacher P, et al. Fecal microbiota transplantation for patients with ulcerative colitis: a systematic review and meta-analysis of randomized control trials. Tech Coloproctol. (2025) 29:103. doi: 10.1007/s10151-025-03113-7
113. Rees NP, Shaheen W, Quince C, Tselepis C, Horniblow RD, Sharma N, et al. Systematic review of donor and recipient predictive biomarkers of response to faecal microbiota transplantation in patients with ulcerative colitis. EBioMedicine. (2022) 81:104088. doi: 10.1016/j.ebiom.2022.104088
114. Haifer C, Luu LDW, Paramsothy S, Borody TJ, Leong RW, and Kaakoush NO. Microbial determinants of effective donors in faecal microbiota transplantation for UC. Gut. (2022) 5:2022-327742. doi: 10.1136/gutjnl-2022-327742
115. Yang R, Chen Z, and Cai J. Fecal microbiota transplantation: Emerging applications in autoimmune diseases. J Autoimmun. (2023) 141:103038. doi: 10.1016/j.jaut.2023.103038
116. Pickard JM, Zeng MY, Caruso R, and Núñez G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol Rev. (2017) 279:70–89. doi: 10.1111/imr.12567
117. Adak A and Khan MR. An insight into gut microbiota and its functionalities. Cell Mol Life Sci. (2019) 76:473–93. doi: 10.1007/s00018-018-2943-4
118. Zeng L, Yang K, He Q, Zhu X, Long Z, Wu Y, et al. Efficacy and safety of gut microbiota-based therapies in autoimmune and rheumatic diseases: a systematic review and meta-analysis of 80 randomized controlled trials. BMC Med. (2024) 22:110. doi: 10.1186/s12916-024-03303-4
119. Shahid A, Fatima M, Khan MSI, Ali U, Fareed SZ, and Qureshi MA. From microbes to immunity: a comprehensive review of microbiome modulation. J Health Rehabil Res. (2023) 3:801–7. doi: 10.61919/jhrr.v3i2.238
120. Rastogi S and Singh A. Gut microbiome and human health: Exploring how the probiotic genus Lactobacillus modulate immune responses. Front Pharmacol. (2022) 13:1042189. doi: 10.3389/fphar.2022.1042189
121. Wang X, Yuan W, Yang C, Wang Z, Zhang J, Xu D, et al. Emerging role of gut microbiota in autoimmune diseases. Front Immunol. (2024) 15:1365554. doi: 10.3389/fimmu.2024.1365554
122. Anwar H, Irfan S, Hussain G, Faisal MN, Muzaffar H, Mustafa I, et al. Gut microbiome: a new organ system in body. Parasitol Microbiol Res. (2019) 1:17–21. doi: 10.3389/fimmu.2024.1365554
123. Colella M, Charitos IA, Ballini A, Cafiero C, Topi S, Palmirotta R, et al. Microbiota revolution: How gut microbes regulate our lives. World J Gastroenterol. (2023) 29:4368. doi: 10.3748/wjg.v29.i28.4368
Keywords: autoimmunity, immunotherapy, CAR T-cells, bispecific antibodies, checkpoint modulators, cytokine therapy, microbiome interventions
Citation: Alsayb MA (2025) Innovations in immunotherapy for autoimmune diseases: recent breakthroughs and future directions. Front. Immunol. 16:1647066. doi: 10.3389/fimmu.2025.1647066
Received: 14 June 2025; Accepted: 29 August 2025;
Published: 17 September 2025.
Edited by:
Seng-Lai Tan, TFC Therapeutics, United StatesReviewed by:
Mattia Moratti, University of Rome Tor Vergata, ItalySelene Nunez-Cruz, University of Pennsylvania, United States
Copyright © 2025 Alsayb. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: May A. Alsayb, bXNheWJAdGFpYmFodS5lZHUuc2E=