 Xiaoming Zhao
Xiaoming Zhao Chen Zhang1
Chen Zhang1 Yilei Zhang
Yilei Zhang Yingang Zhang
Yingang Zhang- 1Department of Orthopaedics of the First Affiliated Hospital, Xi’an Jiaotong University, Xi’an, Shaanxi, China
- 2The Institute of Molecular and Translational Medicine, Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Xi’an Jiaotong University Health Science Center, Xi’an, Shaanxi, China
Disulfidptosis is a novel form of programmed cell death triggered by cystine metabolic disorders and disulfide stress, initially studied primarily in the context of tumors. In recent years, its role in the occurrence and development of orthopedic diseases has gained increasing attention. This review systematically explores the dual regulatory mechanisms of disulfidptosis in degenerative orthopedic diseases, such as intervertebral disc degeneration, osteoporosis, and osteoarthritis, as well as in malignant bone tumors like osteosarcoma, along with their immunometabolic basis. The research findings indicate that in degenerative lesions, microenvironmental stresses such as ischemia and hypoxia exacerbate tissue degeneration by promoting abnormal accumulation of disulfide bonds and damaging the cytoskeleton. In osteosarcoma, tumor-associated oxidative stress can induce metabolism-dependent cell death, providing new opportunities for targeted therapy. The article further summarizes key signaling pathways and molecular regulatory networks, discussing the potential value of targeted intervention strategies in slowing disease progression and achieving precision treatment.
1 Introduction
In recent years, the field of programmed cell death has witnessed a breakthrough with the formal definition of disulfidptosis as a novel form of cell death. In 2023, Liu et al. (1) first revealed this new phenomenon: when cells with high expression of the cystine transporter SLC7A11 encounter glucose deprivation, the regeneration of NADPH is hindered, leading to an imbalance in cystine reduction. The abnormal accumulation of disulfides induces cross-linking and contraction of cytoskeletal proteins, ultimately resulting in unique cellular disintegration. This discovery not only enriches the theoretical framework of cell death but also opens new perspectives for studying the pathological mechanisms of orthopedic degenerative diseases and bone tumors, due to the close association of disulfidptosis with metabolic stress and oxidative damage in various diseases (2–10), and its potential to modulate immune responses in the bone microenvironment (10–14).
Within the spectrum of orthopedic diseases, degenerative conditions (such as intervertebral disc degeneration, osteoporosis, and osteoarthritis) and malignant bone tumors exhibit distinct pathologies, yet they share the core feature of metabolic microenvironment imbalance. The former is caused by nutritional supply disruptions or age-related oxidative stress, leading to homeostatic disturbances in chondrocytes, osteoblasts, and nucleus pulposus cells (15–17). In contrast, the latter maintains malignant proliferation and chemotherapy resistance through tumor cell metabolic reprogramming pathways (18–21). Notably, the triggering conditions for disulfidptosis (glucose deprivation and high expression of SLC7A11) significantly overlap with the microenvironment of orthopedic diseases: regions of degenerative changes are often accompanied by local ischemia and hypoxia (22), while osteosarcoma cells commonly exhibit abnormal activation of SLC7A11 to cope with oxidative stress (23). This intersection of pathological features suggests that disulfidptosis may play a “double-edged sword” role in the progression of orthopedic diseases—potentially accelerating the death of normal cells, leading to tissue degeneration, while also being strategically induced to eliminate malignant cells.
This review systematically analyzes the bidirectional regulatory role of disulfidptosis in orthopedic diseases for the first time: on one hand, in degenerative conditions such as intervertebral disc degeneration and osteoporosis, the nutrient-deprived microenvironment may exacerbate cell loss by activating the disulfidptosis pathway; on the other hand, the dependency of malignant tumors like osteosarcoma on SLC7A11 can be transformed into a therapeutic target, enabling selective killing through the induction of disulfidptosis. By integrating recent basic research with preclinical data, this article aims to reveal the key molecular switches involved in disulfidptosis during disease progression and assess the translational potential of targeted intervention strategies with immunomodulatory properties, providing a theoretical framework for precision medicine in orthopedic diseases.
2 Core molecular mechanisms of disulfidptosis
Disulfidptosis is a form of programmed cell death induced by intracellular disulfide stress, with its core mechanism involving cystine metabolism imbalance, abnormal cross-linking of cytoskeletal proteins, and disruption of redox homeostasis (24). Current research has identified its key driving factors and unique characteristics that distinguish it from other forms of programmed cell death, demonstrating mechanistic heterogeneity in both tumor and non-tumor diseases (8, 25). A schematic representation of the mechanisms of disulfidptosis is shown in Figure 1.
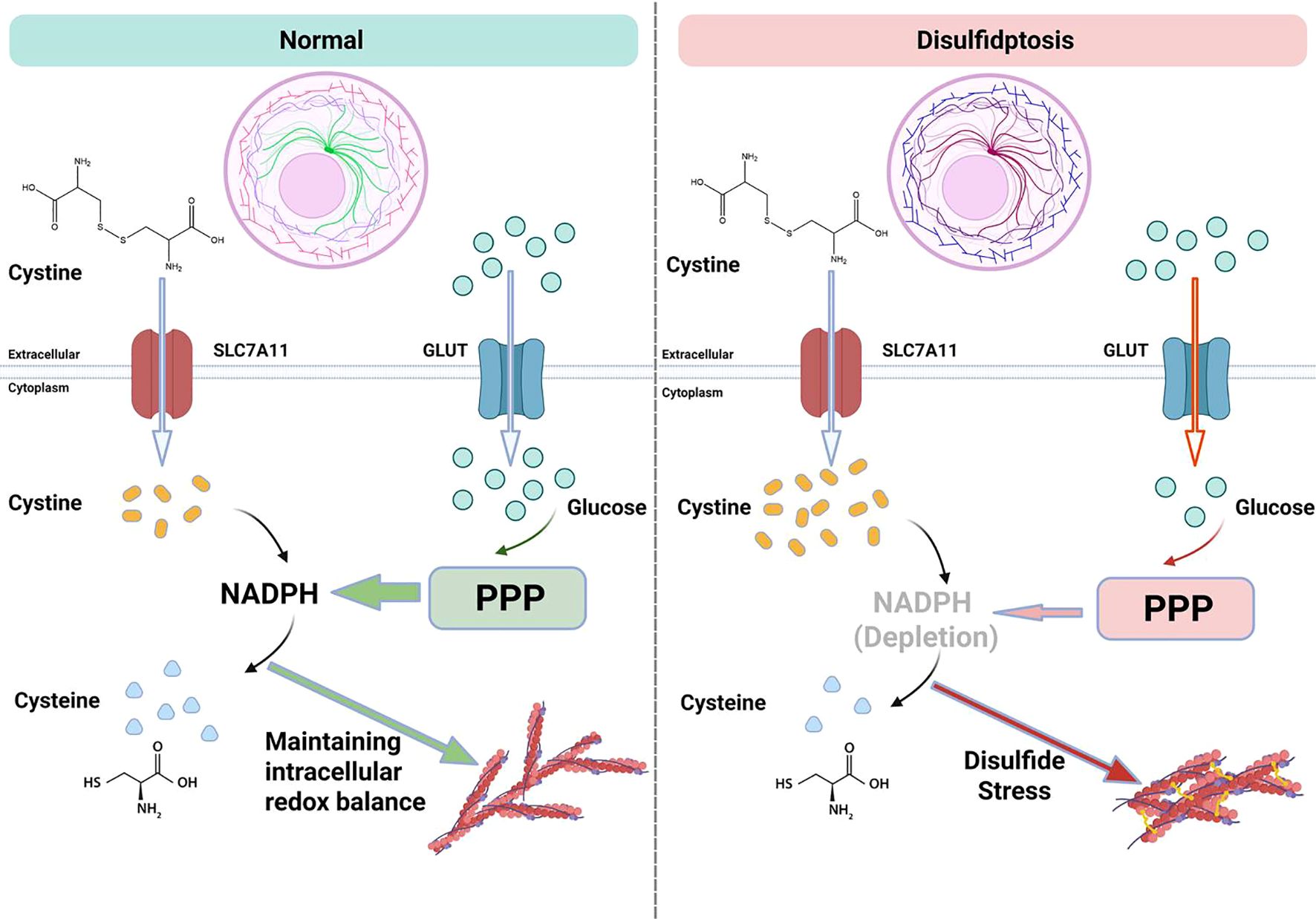
Figure 1. Schematic diagram of disulfidptosis mechanism.
2.1 Pathological associations of disulfidptosis in orthopedic diseases
The pathological relevance of disulfidptosis in orthopedic diseases is rooted in its unique metabolic microenvironment and cellular characteristics. The pathological processes of degenerative orthopedic diseases are often accompanied by local ischemia, hypoxia, nutritional supply disruptions, and age-related oxidative stress. This metabolic stress environment aligns closely with the triggering conditions for disulfidptosis (glucose deprivation and high expression of SLC7A11) (15, 22). Importantly, bone tissue cells (such as chondrocytes and osteoblasts) have a high dependence on cytoskeletal stability, making them particularly sensitive to abnormal disulfide cross-linking. For instance, the integrity of the cytoskeleton in osteoblasts and chondrocytes is crucial for mediating mechanosensitive signaling that regulates their basic functions. Nucleus pulposus cells maintain their morphology against intervertebral pressure through an F-actin network, while osteosarcoma cells rely on cytoskeletal remodeling to achieve invasion and metastasis. This dual vulnerability—metabolic and mechanical—gives the core mechanism of disulfidptosis (cystine metabolism imbalance leading to cytoskeletal collapse) special pathological significance in orthopedic diseases. The following sections will systematically analyze the specific and universal patterns of its molecular mechanisms.
2.2 Key driving factors
The initiation of disulfidptosis depends on the functional impairment of the cystine transporter SLC7A11/xCT. SLC7A11 mediates the influx of cystine, contributing to the synthesis of glutathione (GSH) and maintaining cellular antioxidant capacity. However, when cells are in a state of glucose deprivation, the pentose phosphate pathway (PPP) is suppressed, leading to insufficient NADPH production. As a result, cystine cannot be reduced to cysteine and accumulates abnormally within the cell, creating disulfide stress. This process is particularly pronounced in cells with high expression of SLC7A11, such as osteosarcoma cells and nucleus pulposus cells from degenerated intervertebral discs.
The unreduced cystine and its derivatives (such as cysteamine) form abnormal disulfide cross-links with the thiol groups of cytoskeletal proteins, including actin (ACTB) and non-muscle myosin heavy chain (MYH9). This leads to increased rigidity of the cytoskeletal network and loss of contractile function, ultimately resulting in plasma membrane rupture and cellular disintegration. Studies have shown that knocking down SLC7A11 or using disulfide reducing agents can effectively block this cell death process (7). Conversely, the inhibition of thioredoxin reductase activity further exacerbates the reduction deficiency of cystine (26), creating a vicious cycle of “cystine influx-NADPH depletion”.
2.3 Differences from other forms of programmed cell death
Disulfidptosis exhibits significant differences in molecular mechanisms and morphological characteristics compared to other forms of programmed cell death (27, 28). While both disulfidptosis and ferroptosis share SLC7A11-mediated cystine metabolism and glutathione (GSH) depletion mechanisms, disulfidptosis does not rely on lipid peroxidation and cannot be blocked by ferroptosis inhibitors, indicating its independence from ferroptosis signaling pathways. In apoptosis and necroptosis, typical events such as caspase activation or MLKL phosphorylation are not involved in the regulation of disulfidptosis. Moreover, disulfidptosis lacks characteristic morphological changes such as apoptotic body formation or plasma membrane pore formation. The uniqueness of disulfidptosis lies in its death signal being directly derived from mechanical damage to the cytoskeleton induced by disulfide stress, a mechanism that has been validated in chondrocytes of osteoarthritis and osteoclast precursor cells in osteoporosis.
2.4 Mechanistic heterogeneity in tumor and non-tumor diseases
The regulation of disulfidptosis exhibits essential differences between tumor and non-tumor diseases. In tumors, its triggering often arises from acute metabolic stress induced by therapeutic interventions. Tumor cells, due to high expression of SLC7A11, develop “cystine addiction,” where NADPH depletion leads to reduced cystine metabolism and disulfide-mediated cytoskeletal collapse (29–31). This mechanism is utilized for the targeted killing of malignant cells and may synergize with immunotherapy. In contrast, in non-tumor diseases, chronic microenvironments induce compensatory cystine metabolism imbalance through epigenetic modifications (e.g., NFATc1 activation of SLC7A11), creating conditions that promote pro-inflammatory immune cell infiltration (32–34). Terminally differentiated cells, with limited metabolic plasticity, struggle to cope with sustained NADPH deficiency, ultimately leading to progressive functional loss. Therefore, intervention strategies must accurately differentiate between disease types—tumor treatments should focus on activating disulfidptosis signaling while modulating the immunosuppressive microenvironment, while non-tumor diseases require the inhibition of this pathway to maintain cellular homeostasis to restore immune homeostasis.
3 Role of disulfidptosis in orthopedic diseases
Disulfidptosis, as a metabolism-dependent form of cell death, exhibits a significant “pro-death - anti-death” bidirectional regulatory characteristic in orthopedic diseases. In bone tumors, excessive activation of disulfidptosis can effectively eliminate malignant cells, while in degenerative diseases, the activation of disulfidptosis exacerbates the functional decline of cells. This bidirectional regulation is closely related to the disruption of the immune metabolic microenvironment. In orthopedic degenerative diseases (such as intervertebral disc degeneration, osteoporosis, osteoarthritis, etc.), chronic metabolic stress (ischemia, hypoxia, oxidative stress) is highly coupled with the key triggering conditions of disulfidptosis through the inflammation factor-metabolic enzyme network, accelerating the functional loss of terminally differentiated cells. Anti-death mechanisms can protect these cells from functional decline. In contrast, in bone tumors (such as osteosarcoma), malignant proliferation relies on cystine metabolic reprogramming and immune evasion, providing a new strategy for targeting and inducing disulfidptosis to eliminate tumor cells. This heterogeneity in immune metabolic regulation results in disulfidptosis displaying entirely opposite pathological effects in orthopedic diseases. The following sections will systematically analyze the specific mechanisms of disulfidptosis in orthopedic diseases through key molecular networks and disease-specific regulatory features (Table 1).
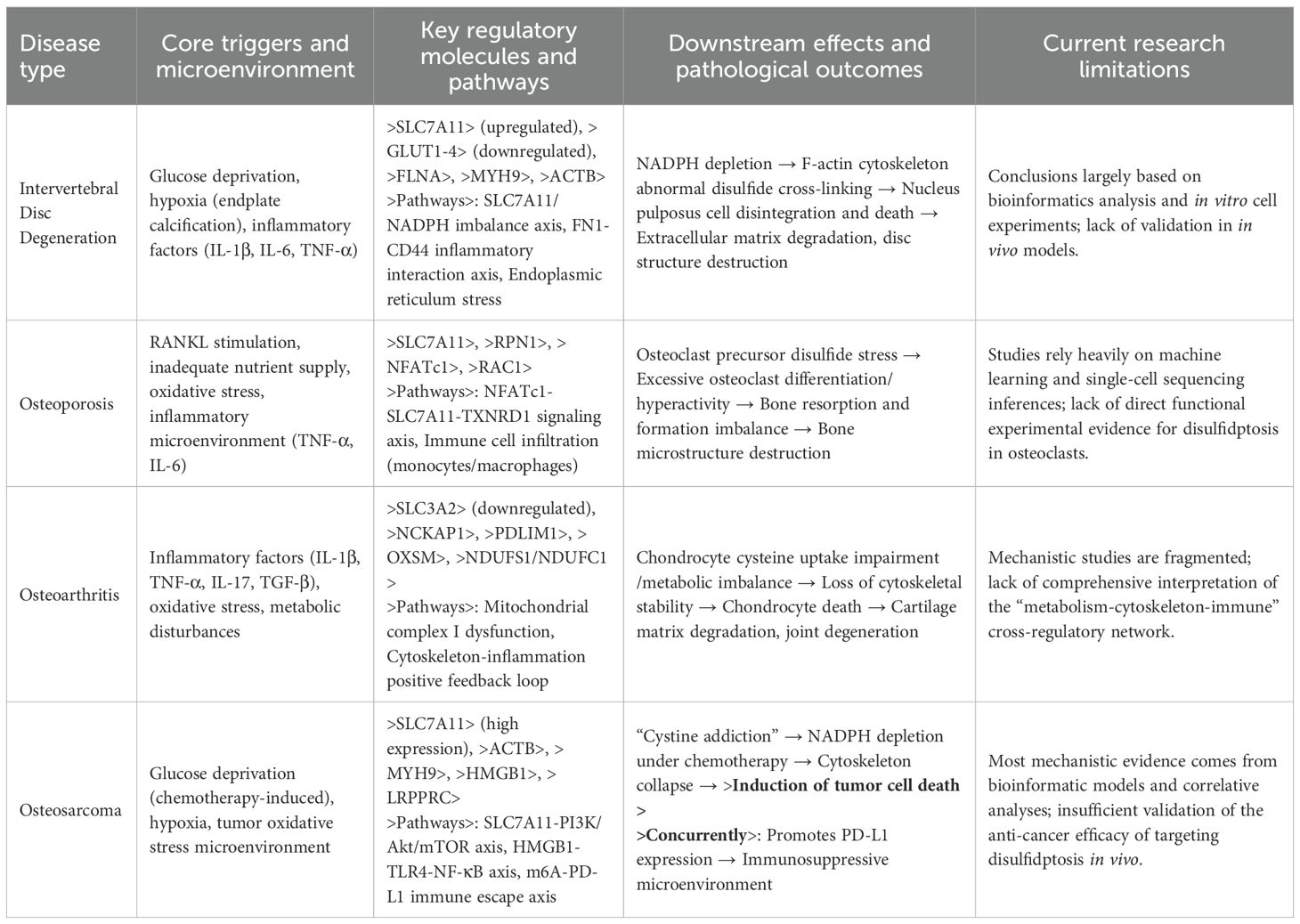
Table 1. Molecular mechanisms and microenvironmental triggers of disulfidptosis in orthopedic diseases.
3.1 Intervertebral disc degeneration
3.1.1 Pathological characteristics and potential association with disulfidptosis
The core pathological features of intervertebral disc degeneration (IVDD) include the degradation of the nucleus pulposus (NP) extracellular matrix, homeostatic imbalance, and disturbances in the nutritional microenvironment (35–40). Due to the avascular nature of intervertebral discs, nutrient supply relies on endplate diffusion, and calcification or aging of the endplates can impede the transport of glucose and oxygen, exacerbating metabolic stress in NP cells (41–43). As the only cellular component within the intervertebral disc, NP cell survival is highly dependent on glucose metabolism to maintain redox homeostasis. Research indicates that glucose deprivation can directly induce NP cell death; however, conventional apoptosis or necrosis inhibitors fail to fully rescue cell survival, suggesting the involvement of a novel cell death mechanism (44). Disulfidptosis, as a newly identified form of programmed cell death, is triggered by disulfide stress resulting from intracellular NADPH depletion and abnormal cross-linking of cytoskeletal proteins. This mechanism aligns closely with the restricted glucose metabolism and redox imbalance observed in IVDD. The high expression of the cystine transporter SLC7A11 in NP cells during degeneration may accelerate cystine uptake, further depleting NADPH reserves and triggering the key pathways of disulfidptosis.
3.1.2 Mechanisms of disulfidptosis in intervertebral disc degeneration
Recent studies have confirmed the role of disulfidptosis in IVDD for the first time (44). (1) SLC7A11-NADPH Imbalance Drives Disulfide Stress: SLC7A11 is significantly upregulated in degenerated nucleus pulposus tissue, promoting excessive uptake of cystine and depleting NADPH as it is reduced to cysteine. Glucose deprivation inhibits the pentose phosphate pathway (PPP), blocking NADPH production and leading to an increased NADP+/NADPH ratio, along with the abnormal accumulation of disulfides in cytoskeletal proteins such as actin (e.g., FLNA, MYH9). This results in cytoskeletal collapse and cell death. (2) Downregulation of Glucose Transporters (GLUTs) Exacerbates Metabolic Crisis: Single-cell sequencing analysis revealed a significant negative correlation between SLC7A11 and SLC2A1-4 (GLUT1-4) in the degenerative chondrocyte subpopulation (C4). The reduced expression of GLUTs further limits glucose influx, amplifying the effects of NADPH depletion. (3) Synergistic Effects of Endoplasmic Reticulum Stress and Oxidative Stress: The degenerative C4 subpopulation is enriched in genes related to endoplasmic reticulum stress, the p53 pathway, and oxidative stress, suggesting that disulfidptosis may accelerate NP cell loss by activating the unfolded protein response (UPR) and pro-apoptotic signals. Experimental evidence shows that the glucose analog 2-DG can reverse NADP+/NADPH imbalance by supplementing NADPH, significantly inhibiting disulfidptosis in NP cells. This provides a new strategy for targeting metabolic reprogramming to intervene in IVDD. Research on intervertebral disc degeneration is currently relatively limited, with verification methods primarily based on bioinformatics analysis and basic cellular experiments. The reliability of the conclusions and the underlying mechanisms require further exploration in the future.
3.2 Osteoporosis
3.2.1 Pathological characteristics and potential association with disulfidptosis
Osteoporosis (OP) is characterized by reduced bone density, compromised bone microstructure, and an increased risk of fractures, with the core pathological mechanism involving a dynamic imbalance between bone resorption and formation (45–51). Research indicates that metabolic disorders, immune dysregulation, and subsequent cell death within the bone microenvironment collectively contribute to the pathogenesis of OP (52–58). In recent years, a novel form of cell death—disulfidptosis—has been increasingly recognized for its role in osteoporosis. Disulfidptosis is a form of programmed cell death triggered by the abnormal accumulation of intracellular disulfides, characterized by SLC7A11-mediated excessive cystine uptake in a glucose-deprived environment, leading to NADPH depletion and disulfide stress, ultimately resulting in abnormal cross-linking of cytoskeletal proteins and cell death. The pathological environment of osteoporosis, with its unique microenvironmental characteristics of low glucose and low oxygen partial pressure, provides conditions conducive to the occurrence of disulfidptosis (59, 60). Bone-related cells, such as bone marrow mesenchymal stem cells (BM-MSCs) and osteoclast precursors, exhibit upregulated expression of SLC7A11 and redox imbalance under the stimulation of inflammatory factors, further increasing their susceptibility to disulfidptosis. Notably, the infiltration of immune cells and the release of inflammatory factors (e.g., TNF-α, IL-6) present in the bone microenvironment of osteoporosis patients not only directly participate in the regulation of bone metabolism but may also form a complex interactive network with the occurrence and progression of disulfidptosis by affecting cystine metabolism and redox balance (61, 62). Disulfidptosis may intertwine with traditional pathological mechanisms of osteoporosis, participating in the disease process through a unique “metabolic-immune-cytoskeletal” regulatory axis.
3.2.2 Mechanisms of disulfidptosis in osteoporosis
Recent studies have revealed the significant role of disulfidptosis in the progression of osteoporosis. In terms of metabolic regulation and osteoclast activation, research by Zhong et al. (61) found that disulfidptosis plays a critical role in osteoclast differentiation through the NFATc1-SLC7A11-TXNRD1 signaling axis. The study showed that during RANKL-induced osteoclast differentiation, NFATc1 significantly upregulates the expression of SLC7A11, enhancing the capacity of osteoclast precursors to uptake cystine. When TXNRD1 activity is inhibited, dysregulation of intracellular cystine metabolism leads to abnormal accumulation of disulfides, ultimately triggering characteristic F-actin contraction and cell death. This form of cell death is specific and cannot be reversed by conventional cell death inhibitors but can be effectively intervened by thiol compounds. Animal experiments confirmed that targeting this pathway can significantly improve bone resorption markers and bone microstructure.
In terms of remodeling the immune microenvironment, disulfidptosis participates in the immune metabolic imbalance of osteoporosis by regulating immune cell function. Wang et al. (59) conducted single-cell analysis of clinical data, revealing that osteoporosis patients can be categorized into different disulfidptosis subtypes, with high-scoring subtypes significantly associated with the infiltration of specific immune cell subsets (such as monocytes and T follicular helper cells). These immune cells activate the osteoclast differentiation pathway by secreting pro-inflammatory factors. Additionally, abnormal expression of PGRMC2 has been found to be associated with osteoporosis risk, potentially participating in the regulation of the bone immune microenvironment by influencing the differentiation process of monocytes into macrophages. Genetic analysis further supports the protective role of PGRMC2 in osteoporosis.
In terms of cytoskeletal stability and bone homeostasis, Zhang et al. (60) found that disulfidptosis regulators participate in the imbalance of bone remodeling by influencing cytoskeletal dynamics. Abnormal expression of key cytoskeletal proteins, such as FLNA and ACTB, leads to decreased stability of the actin network, promoting osteoclast differentiation. Conversely, excessive activation of regulators like RAC1 accelerates the formation of osteoclast bone resorption structures. Additionally, research by Pan et al. (62) revealed that abnormal expression of RPN1 recruits specific immune cells through pro-inflammatory signaling pathways, while targeted inhibition of RPN1 can effectively improve the abnormal bone microstructure in animal models.
These three mechanisms are interwoven, collectively forming a complete framework for the involvement of disulfidptosis in the pathological processes of osteoporosis: metabolic reprogramming alters cell fate determination, remodeling of the immune microenvironment affects local inflammatory states, and regulation of cytoskeletal stability directly participates in the modulation of bone resorption function. Nevertheless, the conclusions of this section primarily rely on single-cell sequencing, machine learning, and in vitro studies, lacking further validation through in vivo experiments and clinical trials, which will be a focus of future research.
3.3 Osteoarthritis
3.3.1 Pathological features and potential association with disulfidptosis
Osteoarthritis (OA) is characterized by degeneration of articular cartilage, synovial inflammation, and imbalance in chondrocyte homeostasis (63–68). Chondrocytes are critical for maintaining the function of articular cartilage, and their abnormal death (such as apoptosis and necroptosis) is closely related to the progression of OA (69–75).
Recently discovered disulfidptosis, a novel form of programmed cell death, is triggered by the abnormal accumulation of disulfides within cells, leading to cross-linking of cytoskeletal proteins and collapse of the actin network. This mechanism aligns well with the loss of cytoskeletal stability observed in chondrocytes during OA (76, 77). In the OA microenvironment, oxidative stress, inflammatory factors (such as IL-1β and TNF-α), and abnormalities in glucose metabolism may activate SLC7A11, leading to excessive cystine uptake and exacerbating intracellular disulfide stress, thereby triggering disulfidptosis. Moreover, dysfunction of mitochondrial respiratory chain complexes (such as dysregulation of NDUFS1 and NDUFC1 expression) may further amplify energy metabolism disturbances and redox imbalances, creating a vicious cycle that promotes the occurrence of disulfidptosis.
3.3.2 Mechanisms of disulfidptosis in osteoarthritis
Recent studies utilizing multi-omics analysis have revealed the critical role of disulfidptosis-related genes (DRGs) in osteoarthritis (OA). Wei et al. (76)integrated six OA datasets and single-cell sequencing, discovering that DRGs such as SLC3A2 and NDUFC1 are specifically highly expressed in OA chondrocytes and significantly correlated with the activation of pro-inflammatory pathways like IL-17 and TGF-β. Experimental validation showed that SLC3A2 expression is downregulated in OA models, and its deficiency exacerbates oxidative stress by inhibiting cystine transport, leading to the upregulation of extracellular matrix degradation markers (MMP3, ADAMTS5). This suggests that SLC3A2 may influence OA progression by regulating the balance between disulfidptosis and autophagy.
Cao et al. (77)employed machine learning to identify SLC3A2 and PDLIM1 as core DRGs. They found that PDLIM1 is abnormally highly expressed during late-stage chondrocyte differentiation, disrupting cytoskeletal integrity by competitively binding to α-actinin 4 (ACTN4) while inhibiting autophagy-related pathways (such as the mTOR-ULK1 axis), exacerbating the inflammatory response in chondrocytes. Hu et al. (78) further investigated that OA samples with different disulfidptosis scores exhibit distinct characteristics of immune cell infiltration. The C1 subtype (high score) shows activation of CD8+ T cells and a decrease in M0 macrophages, while the C2 subtype (low score) is characterized by an increase in Th17 cell infiltration and high expression of pro-inflammatory factors (IL-6, TNF-α). The imbalance in the proportion of these immune cell subpopulations is significantly correlated with the expression levels of key regulatory factors of disulfidptosis (such as NCKAP1, OXSM), indicating that disulfidptosis may participate in the inflammatory process of OA by regulating the activation and function of immune cells. These studies collectively suggest that disulfidptosis is involved in the pathological mechanisms of OA through a multidimensional network of “metabolism - oxidative stress - immune regulation,” providing new directions for targeted intervention.
3.4 Osteosarcoma
3.4.1 Pathological features and immunometabolic crosstalk in disulfidptosis
Osteosarcoma, as a highly heterogeneous malignant bone tumor, is characterized by rapid proliferation, a tendency for early metastasis, and chemotherapy resistance (79, 80). Disulfidptosis, as a novel form of programmed cell death, involves a core mechanism of cystine metabolic imbalance mediated by SLC7A11, leading to abnormal accumulation of intracellular disulfides, which in turn triggers cross-linking of cytoskeletal proteins and membrane rupture. This process is highly compatible with the oxidative stress microenvironment of osteosarcoma (81): osteosarcoma cells, due to rapid proliferation, often exist in states of hypoxia and nutrient deficiency, potentially exacerbating cystine-dependent metabolic abnormalities through overexpression of SLC7A11, thereby creating conditions that promote disulfidptosis. Additionally, the high expression of actin-related genes (such as ACTB and MYH9) in osteosarcoma tissues may further increase the sensitivity of the cytoskeleton to abnormal disulfides, providing a molecular basis for the occurrence of disulfidptosis.
3.4.2 Mechanisms of disulfidptosis in osteosarcoma
Recent studies have gradually revealed the regulatory networks and clinical significance of disulfidptosis-related genes (DRGs) in osteosarcoma. At the molecular mechanism level, abnormal activation of the SLC7A11-PI3K/Akt/mTOR signaling axis plays a critical role in the progression of osteosarcoma. Research by Xu et al. (82) indicates that SLC7A11 upregulates HK2 expression and glycolytic activity through mTORC1, while activated PI3K promotes the phosphorylation of GSK3β, inhibiting β-catenin degradation, thus forming a positive feedback loop of “cystine metabolism-glycolysis-Wnt signaling.” Clinical data show that patients with high expression of SLC7A11 exhibit increased sensitivity to mTOR inhibitors, providing important evidence for targeted therapy.
The interaction between microenvironmental factors and cytoskeletal stability is also crucial in the regulation of disulfidptosis. Wang et al. (83) found that exogenous HMGB1 upregulates ACTB expression via the TLR4/MyD88/NF-κB pathway, leading to the depolymerization of the F-actin network and disruption of cell membrane stability, accompanied by changes in histone modifications. This finding reveals the bridging role of the HMGB1-ACTB-TLR4 axis in connecting microenvironmental stimuli with cytoskeletal damage, also providing a new target for therapeutic intervention.
In terms of immune regulation, the prognostic model constructed by Chen et al. (84) shows that LRPPRC, as an m6A reader, recognizes a specific sequence in PD-L1 mRNA and enhances its translation efficiency by recruiting YTHDF1, leading to the functional suppression of CD8+ T cells. Additionally, LRPPRC may enhance the stability of PD-L1 mRNA through m6A modification, creating a malignant cycle of immune suppression and metabolic disorder. Meanwhile, MYH9 regulates the secretion of immune factors via the Hippo-YAP pathway, potentially leading to Treg infiltration and NK cell functional suppression, thus forming an immunosuppressive microenvironment. These findings reveal the important role of disulfidptosis-related genes in tumor immune evasion.
4 Dual role of disulfidptosis in targeted therapeutic strategies for orthopedic diseases
Previous studies have shown that Targeted therapies for orthopedic diseases exhibit bidirectional regulatory characteristics, necessitating precise differentiation in intervention strategies for tumor versus non-tumor conditions (85–90). Regarding disulfidptosis-targeted therapeutic strategies, in malignant tumors such as osteosarcoma, therapeutic strategies focus on activating disulfidptosis signaling pathways to induce tumor-specific cell death while concurrently overcoming immunosuppressive barriers, and enhance treatment efficacy especially when combined with immune checkpoint inhibitors (91–96). Conversely, in degenerative diseases like intervertebral disc degeneration, osteoporosis, and osteoarthritis, it is crucial to inhibit the excessive activation of disulfidptosis pathways to protect cellular functions, and mitigate pro-inflammatory immune responses and delay tissue degeneration (97–102).
This “pro-death vs. anti-death” dual logic underscores the core value of targeting disulfidptosis pathways in orthopedic precision medicine with integrated immunomodulation. By tailoring interventions based on the fundamental differences in disease nature, therapeutic goals can be optimized through specific immune-aware molecular mechanisms that simultaneously address metabolic dysfunction and immune microenvironment remodeling. A schematic representation of the “pro-death vs. anti-death” therapeutic strategy is shown in Figure 2, and Table 2 systematically summarizes the main therapeutic principles and disulfidptosis-related precision treatment strategies for the four types of diseases.
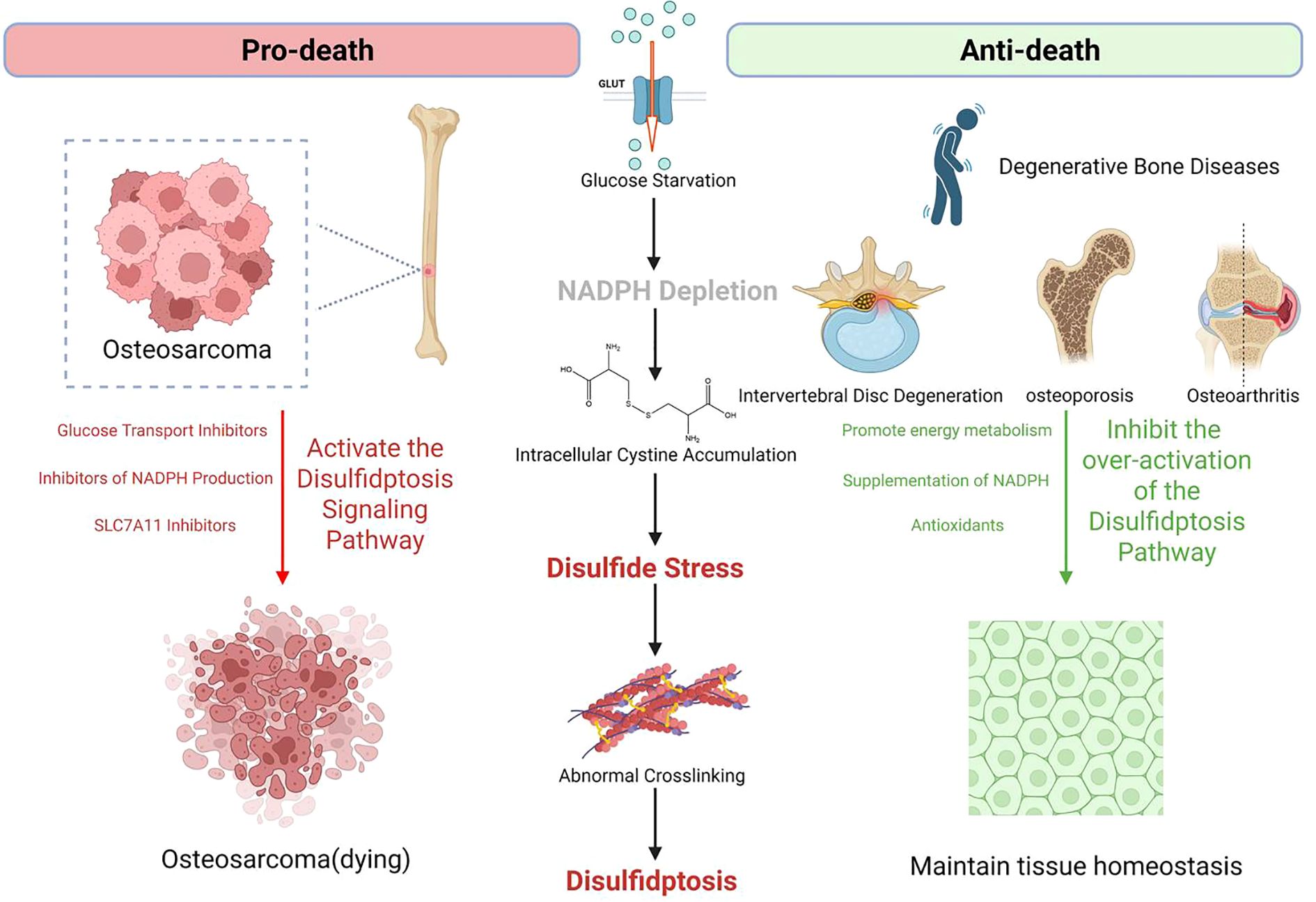
Figure 2. Bidirectional targeting of disulfidptosis in degenerative and malignant orthopedic disorders.
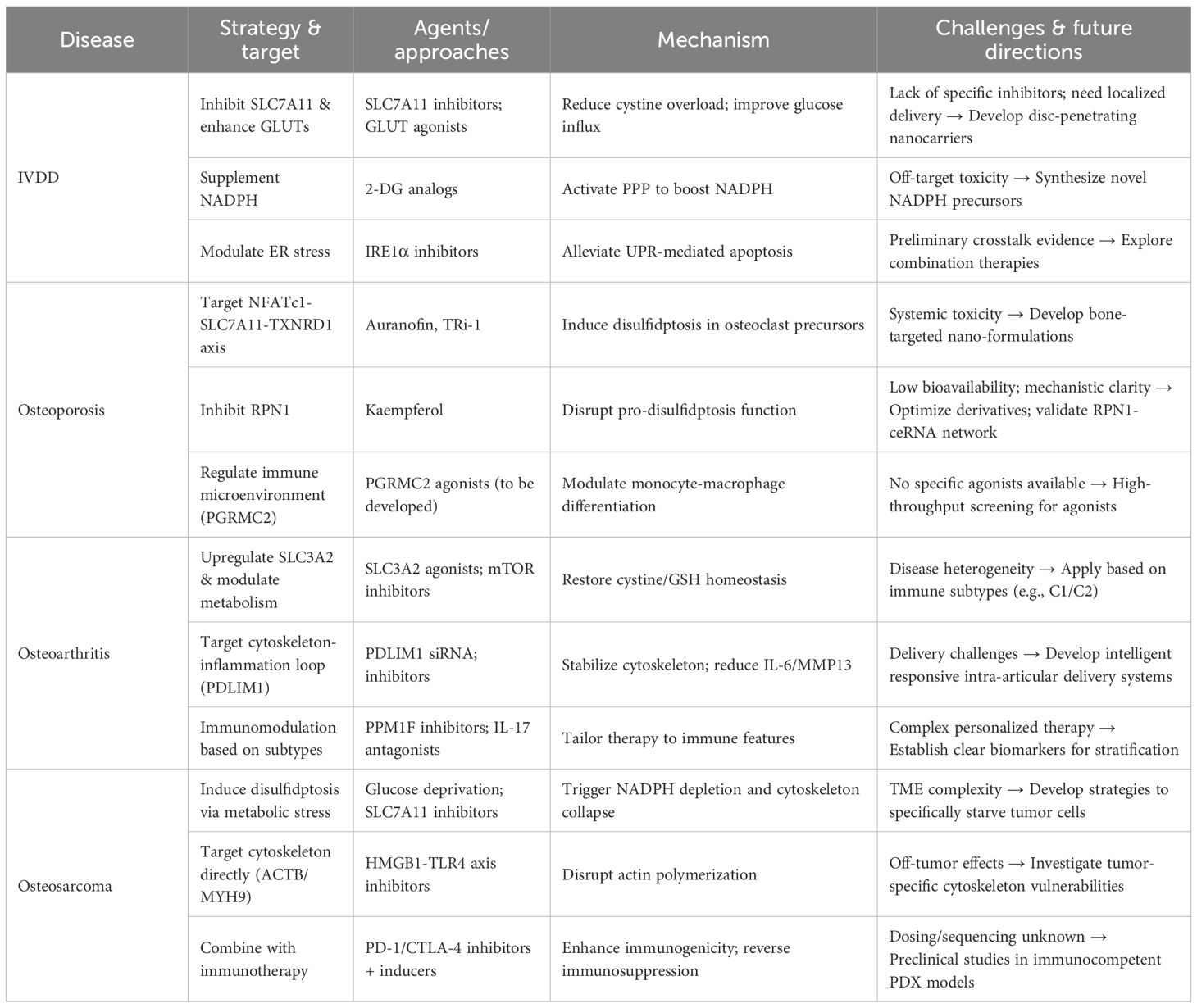
Table 2. Disulfidptosis-targeted therapeutic strategies for orthopedic diseases.
4.1 Intervertebral disc degeneration
In the targeted therapeutic strategies for disulfidptosis in intervertebral disc degeneration (IVDD), research focuses on regulating SLC7A11-mediated redox imbalance to maintain cellular homeostasis. Literature indicates (44)that glucose deficiency in IVDD drives NADPH depletion by upregulating SLC7A11, leading to disulfidptosis. The key to inhibiting this pathway lies in restoring NADPH balance. Experimental evidence shows that 2-deoxy-D-glucose (2-DG) can activate the pentose phosphate pathway to replenish NADPH, significantly reducing the NADP+/NADPH ratio and rescuing cells from glucose deprivation-induced death. Additionally, inhibiting SLC7A11 or enhancing the function of glucose transporters (such as GLUT1/SLC2A1) can block excessive cystine uptake and subsequent oxidative stress. Modulating endoplasmic reticulum stress pathways (such as the unfolded protein response) may also synergistically alleviate disulfidptosis. These strategies provide potential therapeutic targets for delaying intervertebral disc degeneration by precisely inhibiting disulfidptosis signals, such as developing SLC7A11 inhibitors or GLUT agonists to maintain redox homeostasis in nucleus pulposus cells.
4.2 Osteoporosis
In targeted therapeutic strategies for disulfidptosis in osteoporosis, research focuses on the precise intervention of key regulatory factors. Zhong et al. (61)found that NFATc1 activates SLC7A11 transcriptionally, driving osteoclast precursor cells’ sensitivity to thioredoxin reductase 1 (TXNRD1) inhibitors (such as Auranofin). Inhibiting TXNRD1 activity leads to cystine accumulation and disulfidptosis mediated by F-actin contraction, significantly reducing bone resorption.
Pan et al. (62)revealed that kaempferol could target and inhibit the aberrantly high expression of disulfidptosis-related gene RPN1, reversing bone microstructure damage in ovariectomized rats, suggesting the potential therapeutic value of flavonoids in regulating RPN1. Wang et al. (33) further discovered that PGRMC2 influences bone metabolism by promoting the differentiation of monocytes into macrophages; its downregulation exacerbates osteoporosis progression, while restoring PGRMC2 levels can improve bone homeostasis. Zhang et al. (60) constructed a diagnostic model based on genes like SLC7A11 and RAC1, proposing a combination therapy that involves regulating the immune microenvironment of monocyte infiltration to inhibit disulfidptosis and maintain the osteoblast/osteoclast balance. These studies provide molecular mechanisms and translational evidence for developing precision treatments targeting disulfidptosis in osteoporosis.
4.3 Osteoarthritis
Recent research advancements have led to breakthrough findings in targeted therapeutic strategies for disulfidptosis in osteoarthritis (OA). Wei et al. (76) integrated single-cell sequencing with machine learning algorithms, discovering that SLC3A2 is significantly downregulated in the EC subpopulation of OA chondrocytes. Its deficiency exacerbates cartilage degeneration by activating IL-17 and TGF-β inflammatory pathways. In vivo experiments confirmed that upregulating SLC3A2 effectively inhibits disulfidptosis-related F-actin network collapse.
Cao et al. (77) further revealed a bidirectional regulatory mechanism between SLC3A2 and PDLIM1: SLC3A2 maintains redox balance by promoting the cystine/glutathione axis, while aberrant high expression of PDLIM1 in late-stage OA disrupts the autophagy-cytoskeleton balance. Silencing PDLIM1 with siRNA resulted in a 47.3% reduction in inflammatory factors IL-6 and MMP13. Notably, Hu et al. (78) constructed a multi-omics diagnostic model showing that the NCKAP1-OXSM-SLC3A2 regulatory network is closely related to the OA immune microenvironment. Targeted inhibition of PPM1F (a magnesium-dependent phosphatase) increased chondrocyte survival by 32%, with mechanisms involving the restoration of mitochondrial complex I function and reduction of abnormal disulfide accumulation (p< 0.01). These findings provide a theoretical basis for developing specific small molecule inhibitors of disulfidptosis, such as SLC3A2 agonists or PDLIM1 antagonists.
4.4 Osteosarcoma
In the research on targeted therapeutic strategies for disulfidptosis in osteosarcoma, multiple studies have revealed key regulatory targets and potential therapeutic directions. Xu et al. (82) found that osteosarcoma cells with high expression of SLC7A11 are prone to disulfidptosis under glucose deprivation, suggesting that inhibiting glucose transport or pharmacologically targeting the SLC7A11 pathway could selectively induce tumor cell death.
Wang et al. (83) utilized single-cell sequencing to discover that HMGB1 regulates ACTB expression and participates in the disulfidptosis process. They confirmed that silencing ACTB significantly reduces osteosarcoma cell viability, while exogenous HMGB1 treatment enhances cell death sensitivity through the TP53/NF-κB signaling axis. Chen et al. (84) constructed a prognostic model indicating that MYH9 and LRPPRC are key risk genes, with their inhibitors potentially activating the disulfidptosis pathway by disrupting cytoskeletal stability. Notably, Zhang et al. (81) pointed out that the activation of disulfidptosis is associated with the remodeling of the immune microenvironment, where patients in the low-risk group demonstrated significantly increased sensitivity to PD-1/CTLA-4 inhibitors. This suggests that combined immune checkpoint blockade may produce a synergistic anti-tumor effect, with drug sensitivity analysis identifying targeted agents such as lapatinib, bortezomib, fruquintinib, and MG-132. Currently, targeted strategies primarily focus on metabolic interventions, key gene regulation, and the development of cytoskeletal-targeting drugs. These avenues provide a multidimensional therapeutic framework for precisely activating disulfidptosis in osteosarcoma.
5 Conclusion and outlook
This study systematically reveals the key mechanisms of disulfidptosis in orthopedic diseases and its potential for clinical translation. By integrating multi-omics data and experimental validation, we found that disulfidptosis exhibits distinctly different regulatory patterns in degenerative diseases such as intervertebral disc degeneration and osteoporosis compared to malignant tumors like osteosarcoma. In degenerative diseases, SLC7A11-mediated cystine metabolic imbalance leads to NADPH depletion, triggering abnormal cross-linking of cytoskeletal proteins and cell death; whereas in malignant tumors, targeted activation of the disulfidptosis pathway can selectively kill tumor cells. This finding provides new molecular targets and therapeutic strategies for precision diagnosis and treatment of orthopedic diseases.
Despite the significant progress made in this study, there are still several areas that require improvement: First, the limitations in sample sources may affect the generalizability of the research conclusions. The current study is primarily based on public databases and a limited number of clinical samples (44, 59, 76), and future efforts should expand the sample size and include a broader range of ethnic groups to validate the reliability of the results. Second, regarding mechanism studies, the interactions between disulfidptosis and other forms of programmed cell death, such as apoptosis, ferroptosis, autophagy, and necroptosis, have not been fully elucidated (103–107). In particular, the “molecular switch” function of SLC7A11 in different death pathways requires further exploration. Additionally, the lack of animal models limits the depth of in vivo validation, necessitating the development of genetically engineered animal models that more closely resemble human disease characteristics.
Future research should focus on several key directions: In translational medicine, there is an urgent need to develop a ctDNA-based LRPPRC mutation monitoring panel and an imaging radiomics early warning system, while optimizing the drug delivery technology of pH-responsive nanoparticles to improve targeted delivery efficiency. Clinical translational studies should establish SLC7A11 conditional knockout animal models and humanized PDX models to provide a reliable platform for treatment assessment. Mechanistic studies should concentrate on elucidating the spatiotemporal specificity of NFATc1-SLC7A11 transcriptional regulation and clarifying the molecular switch involved in TXNRD1 inhibitor-induced cytoskeletal remodeling. Additionally, exploring the metabolic dialogue mechanisms in the bone marrow microenvironment and developing disulfidptosis-inducing strategies targeting the tumor metabolic microenvironment will provide new insights for achieving precision therapy.
Author contributions
XZ: Conceptualization, Funding acquisition, Resources, Validation, Writing – original draft, Writing – review & editing. SW: Investigation, Supervision, Writing – review & editing. CZ: Investigation, Supervision, Writing – review & editing. JG: Resources, Writing – review & editing. LQ: Formal Analysis, Resources, Writing – review & editing. YGZ: Funding acquisition, Resources, Supervision, Visualization, Writing – review & editing. YLZ: Funding acquisition, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research received funding from the National Natural Science Foundation of China (No. 81371987 to Yingang Zhang and No. 82372847 to Yilei Zhang) and the Shaanxi Province Key Research and Development Project (No. 2023-YBSF-341 to Xiaoming Zhao).
Acknowledgments
We thank AJE for the linguistic editing and proofreading of the manuscript. The figures were created using Biorender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
2-DG, 2-Deoxy-D-glucose; ACTB, Actin Beta; ADAMTS5, A Disintegrin And Metalloproteinase with Thrombospondin Motifs 5; Akt, Protein Kinase B; BM-MSCs, Bone Marrow Mesenchymal Stem Cells; ceRNA, Competing Endogenous RNA; CTLA-4, Cytotoxic T-Lymphocyte-Associated Protein 4; ECM, Extracellular Matrix; ER, Endoplasmic Reticulum; FLNA, Filamin A; GLUT, Glucose Transporter; GSH, Glutathione; HMGB1, High Mobility Group Box 1; IL-1β, Interleukin-1 Beta; IL-6, Interleukin-6; IL-17, Interleukin-17; IVDD, Intervertebral Disc Degeneration; MMP3, Matrix Metalloproteinase 3; mTOR, Mammalian Target of Rapamycin; MYH9, Myosin Heavy Chain 9; NADPH, Nicotinamide Adenine Dinucleotide Phosphate; NF-κB, Nuclear Factor Kappa B; NFATc1, Nuclear Factor of Activated T Cells 1; NK cells, Natural Killer Cells; OA, Osteoarthritis; OP, Osteoporosis; OVX, Ovariectomized; PD-1, Programmed Cell Death Protein 1; PD-L1, Programmed Death-Ligand 1; PI3K, Phosphatidylinositol 3-Kinase; PMOP, Postmenopausal Osteoporosis; PPP, Pentose Phosphate Pathway; RAC1, Rac Family Small GTPase 1; RPN1, Ribophorin 1; SLC7A11, Solute Carrier Family 7 Member 11; TGF-β, Transforming Growth Factor Beta; Th17, T Helper 17 Cells; TNF-α, Tumor Necrosis Factor Alpha; TXNRD1, Thioredoxin Reductase 1; ULK1, Unc-51 Like Autophagy Activating Kinase 1; UPR, Unfolded Protein Response.
References
1. Liu X, Nie L, Zhang Y, Yan Y, Wang C, Colic M, et al. Actin cytoskeleton vulnerability to disulfide stress mediates disulfidptosis. Nat Cell Biol. (2023) 25:404–14. doi: 10.1038/s41556-023-01091-2
2. Liu X, Zhuang L, and Gan B. Disulfidptosis: disulfide stress-induced cell death. Trends Cell Biol. (2024) 34:327–37. doi: 10.1016/j.tcb.2023.07.009
3. Zheng P, Zhou C, Ding Y, and Duan S. Disulfidptosis: a new target for metabolic cancer therapy. J Exp Clin Cancer Res. (2023) 42:103. doi: 10.1186/s13046-023-02675-4
4. Wang X, Zheng C, Yao H, Guo Y, Wang Y, He G, et al. Disulfidptosis: six riddles necessitating solutions. Int J Biol Sci. (2024) 20:1042–4. doi: 10.7150/ijbs.90606
5. Mao C, Wang M, Zhuang L, and Gan B. Metabolic cell death in cancer: ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell. (2024) 15:642–60. doi: 10.1093/procel/pwae003
6. Zhao D, Meng Y, Dian Y, Zhou Q, Sun Y, Le J, et al. Molecular landmarks of tumor disulfidptosis across cancer types to promote disulfidptosis-target therapy. Redox Biol. (2023) 68:102966. doi: 10.1016/j.redox.2023.102966
7. Wang J, Wang M, Wu S, Zhu Y, Fan K, Chen Y, et al. Tumor suppressor BAP1 suppresses disulfidptosis through the regulation of SLC7A11 and NADPH levels. Oncogenesis. (2024) 13:31. doi: 10.1038/s41389-024-00535-0
8. Wang J, Chen J, Fan K, Wang M, Gao M, Ren Y, et al. Inhibition of endoplasmic reticulum stress cooperates with SLC7A11 to promote disulfidptosis and suppress tumor growth upon glucose limitation. Adv Sci (Weinh). (2025) 12:e2408789. doi: 10.1002/advs.202408789
9. Zheng T, Liu Q, Xing F, Zeng C, and Wang W. Disulfidptosis: a new form of programmed cell death. J Exp Clin Cancer Res. (2023) 42:137. doi: 10.1186/s13046-023-02712-2
10. Gu Q, An Y, Xu M, Huang X, Chen X, Li X, et al. Disulfidptosis, A novel cell death pathway: molecular landscape and therapeutic implications. Aging Dis. (2024) 16:917–45. doi: 10.14336/AD.2024.0083
11. Zhou S, Liu J, Wan A, Zhang Y, and Qi X. Epigenetic regulation of diverse cell death modalities in cancer: a focus on pyroptosis, ferroptosis, cuproptosis, and disulfidptosis. J Hematol Oncol. (2024) 17:22. doi: 10.1186/s13045-024-01545-6
12. Chen H, Yang W, Li Y, Ma L, and Ji Z. Leveraging a disulfidptosis-based signature to improve the survival and drug sensitivity of bladder cancer patients. Front Immunol. (2023) 14:1198878. doi: 10.3389/fimmu.2023.1198878
13. Hu G, Yao H, Wei Z, Li L, Yu Z, Li J, et al. A bioinformatics approach to identify a disulfidptosis-related gene signature for prognostic implication in colon adenocarcinoma. Sci Rep. (2023) 13:12403. doi: 10.1038/s41598-023-39563-y
14. Xie J, Deng X, Xie Y, Zhu H, Liu P, Deng W, et al. Multi-omics analysis of disulfidptosis regulators and therapeutic potential reveals glycogen synthase 1 as a disulfidptosis triggering target for triple-negative breast cancer. MedComm (2020). (2024) 5:e502. doi: 10.1002/mco2.502
15. Zhao XM, Chen AF, Lou XX, and Zhang YG. Comparison of three common intervertebral disc discectomies in the treatment of lumbar disc herniation: A systematic review and meta-analysis based on multiple data. J Clin Med. (2022) 11:6604. doi: 10.3390/jcm11226604
16. Zhao X, Ma H, Han H, Zhang L, Tian J, Lei B, et al. Precision medicine strategies for spinal degenerative diseases: Injectable biomaterials with in situ repair and regeneration. Mater Today Bio. (2022) 16:100336. doi: 10.1016/j.mtbio.2022.100336
17. Chen X, Zhang A, Zhao K, Gao H, Shi P, Chen Y, et al. The role of oxidative stress in intervertebral disc degeneration: Mechanisms and therapeutic implications. Ageing Res Rev. (2024) 98:102323. doi: 10.1016/j.arr.2024.102323
18. Shoaib Z, Fan TM, and Irudayaraj JMK. Osteosarcoma mechanobiology and therapeutic targets. Br J Pharmacol. (2022) 179:201–17. doi: 10.1111/bph.15713
19. Yu S and Yao X. Advances on immunotherapy for osteosarcoma. Mol Cancer. (2024) 23:192. doi: 10.1186/s12943-024-02105-9
20. Mohr A, Marques Da Costa ME, Fromigue O, Audinot B, Balde T, Droit R, et al. From biology to personalized medicine: Recent knowledge in osteosarcoma. Eur J Med Genet. (2024) 69:104941. doi: 10.1016/j.ejmg.2024.104941
21. Wu A, Yang ZK, Kong P, Yu P, Li YT, Xu JL, et al. Exploring osteosarcoma based on the tumor microenvironment. Front Immunol. (2024) 15:1423194. doi: 10.3389/fimmu.2024.1423194
22. Wei J, Ou Z, Tong B, Liao Z, and Yang C. Engineered extracellular vesicles as therapeutics of degenerative orthopedic diseases. Front Bioeng Biotechnol. (2023) 11:1162263. doi: 10.3389/fbioe.2023.1162263
23. Zou Q, Zhou X, Lai J, Zhou H, Su J, Zhang Z, et al. Targeting p62 by sulforaphane promotes autolysosomal degradation of SLC7A11, inducing ferroptosis for osteosarcoma treatment. Redox Biol. (2025) 79:103460. doi: 10.1016/j.redox.2024.103460
24. Li T, Song Y, Wei L, Song X, and Duan R. Disulfidptosis: a novel cell death modality induced by actin cytoskeleton collapse and a promising target for cancer therapeutics. Cell Commun Signal. (2024) 22:491. doi: 10.1186/s12964-024-01871-9
25. Huang L, Li Z, Lv Y, Zhang X, Li Y, Li Y, et al. Unveiling disulfidptosis-related biomarkers and predicting drugs in Alzheimer’s disease. Sci Rep. (2024) 14:20185. doi: 10.1038/s41598-024-70893-7
26. Tang M, Dirks K, Kim SY, Qiu Z, Gao Y, Sun D, et al. Inhibition of thioredoxin reductase 1 sensitizes glucose-starved glioblastoma cells to disulfidptosis. Cell Death Differ. (2025) 32:598–612. doi: 10.1038/s41418-024-01440-0
27. Moyer A, Tanaka K, and Cheng EH. Apoptosis in cancer biology and therapy. Annu Rev Pathol. (2025) 20:303–28. doi: 10.1146/annurev-pathmechdis-051222-115023
28. Wang Z, Yan Q, Wang Z, Hu Z, Wang C, Zhang X, et al. Ferroptosis and its implications in bone-related diseases. PeerJ. (2024) 12:e18626. doi: 10.7717/peerj.18626
29. Zhao S, Wang L, Ding W, Ye B, Cheng C, Shao J, et al. Crosstalk of disulfidptosis-related subtypes, establishment of a prognostic signature and immune infiltration characteristics in bladder cancer based on a machine learning survival framework. Front Endocrinol (Lausanne). (2023) 14:1180404. doi: 10.3389/fendo.2023.1180404
30. Kang Z, Wan ZH, Gao RC, Chen DN, Zheng QS, Xue XY, et al. Disulfidptosis-related subtype and prognostic signature in prostate cancer. Biol Direct. (2024) 19:97. doi: 10.1186/s13062-024-00544-4
31. Zhang K, Zhu Z, Zhou J, Shi M, Wang N, Yu F, et al. Disulfidptosis-related gene expression reflects the prognosis of drug-resistant cancer patients and inhibition of MYH9 reverses sorafenib resistance. Transl Oncol. (2024) 49:102091. doi: 10.1016/j.tranon.2024.102091
32. Wang Q, Xiao Z, Hou Z, and Li D. Effect of disulfidptosis-related genes SLC3A2, SLC7A11 and FLNB polymorphisms on risk of autoimmune thyroiditis in a Chinese population. Int Immunopharmacol. (2024) 129:111605. doi: 10.1016/j.intimp.2024.111605
33. Liu J, Gao J, Xing S, Yan Y, Yan X, Jing Y, et al. Bioinformatics analysis of signature genes related to cell death in keratoconus. Sci Rep. (2024) 14:12749. doi: 10.1038/s41598-024-63109-5
34. Xiong Z, Fang Y, Lu S, Sun Q, Sun Y, Yang P, et al. Exploring the relevance of disulfidptosis to the pathophysiology of ulcerative colitis by bioinformatics analysis. J Inflammation Res. (2024) 17:2757–74. doi: 10.2147/JIR.S454668
35. Zhang H, Shi S, Huang X, Gong C, Zhang Z, Zhao Z, et al. Identification of core genes in intervertebral disc degeneration using bioinformatics and machine learning algorithms. Front Immunol. (2024) 15:1401957. doi: 10.3389/fimmu.2024.1401957
36. Ye Y, Wan L, Hu J, Li X, and Zhang K. Combined single-cell RNA sequencing and mendelian randomization to identify biomarkers associated with necrotic apoptosis in intervertebral disc degeneration. Spine J. (2025) 25:165–83. doi: 10.1016/j.spinee.2024.09.011
37. Tu H, Gao Q, Zhou Y, Peng L, Wu D, Zhang D, et al. The role of sirtuins in intervertebral disc degeneration: Mechanisms and therapeutic potential. J Cell Physiol. (2024) 239:e31328. doi: 10.1002/jcp.31328
38. Qiu C, Cheng L, Di D, Xiang Z, Wang C, Li J, et al. TNFα-reliant FSP1 up-regulation promotes intervertebral disc degeneration via caspase 3-dependent apoptosis. Genes Dis. (2024) 12:101251. doi: 10.1016/j.gendis.2024.101251
39. Yang W, Yang Y, Wang Y, Gao Z, Zhang J, Gao W, et al. Metformin prevents the onset and progression of intervertebral disc degeneration: New insights and potential mechanisms (Review). Int J Mol Med. (2024) 54:71. doi: 10.3892/ijmm.2024.5395
40. Xue P, Lv L, Liu L, Xu Y, Zhou C, and Wang Y. Unveiling the role of CXCL8/CXCR2 in intervertebral disc degeneration: A path to promising therapeutic strategies. J Orthop Translat. (2024) 49:119–34. doi: 10.1016/j.jot.2024.08.022
41. Li Q, Long X, Wang R, Pengying N, Cai L, Wang L, et al. Correlation between degeneration of cervical intervertebral disc and degeneration of paravertebral muscle. Front Endocrinol (Lausanne). (2024) 15:1391970. doi: 10.3389/fendo.2024.1391970. Erratum in: Front Endocrinol (Lausanne). 2024,15:1454208.
42. Ran R, Zhang SB, Shi YQ, Dong H, Song W, Dong YB, et al. Spotlight on necroptosis: Role in pathogenesis and therapeutic potential of intervertebral disc degeneration. Int Immunopharmacol. (2024) 138:112616. doi: 10.1016/j.intimp.2024.112616
43. Dou Y, Zhang Y, Liu Y, Sun X, Liu X, Li B, et al. Role of macrophage in intervertebral disc degeneration. Bone Res. (2025) 13:15. doi: 10.1038/s41413-024-00397-7
44. Wu S, Wang J, Wang M, Zhou K, Huang D, Zhang Y, et al. Glucose deprivation-induced disulfidptosis in human nucleus pulposus cells: a novel pathological mechanism of intervertebral disc degeneration. Biol Direct. (2024) 19:81. doi: 10.1186/s13062-024-00528-4
45. Chai S, Yang Y, Wei L, Cao Y, Ma J, Zheng X, et al. Luteolin rescues postmenopausal osteoporosis elicited by OVX through alleviating osteoblast pyroptosis via activating PI3K-AKT signaling. Phytomedicine. (2024) 128:155516. doi: 10.1016/j.phymed.2024.155516
46. Li J, Chen X, Lu L, and Yu X. The relationship between bone marrow adipose tissue and bone metabolism in postmenopausal osteoporosis. Cytokine Growth Factor Rev. (2020) 52:88–98. doi: 10.1016/j.cytogfr.2020.02.003
47. Luo ZH, Ma JX, Zhang W, Tian AX, Gong SW, Li Y, et al. Alterations in the microenvironment and the effects produced of TRPV5 in osteoporosis. J Transl Med. (2023) 21:327. doi: 10.1186/s12967-023-04182-8
48. Wang Y, Wang Q, Xu Q, Li J, and Zhao F. Single-cell RNA sequencing analysis dissected the osteo-immunology microenvironment and revealed key regulators in osteoporosis. Int Immunopharmacol. (2022) 113:109302. doi: 10.1016/j.intimp.2022.109302
49. Wang Y, Che L, Chen X, He Z, Song D, Yuan Y, et al. Repurpose dasatinib and quercetin: Targeting senescent cells ameliorates postmenopausal osteoporosis and rejuvenates bone regeneration. Bioact Mater. (2023) 25:13–28. doi: 10.1016/j.bioactmat.2023.01.009
50. Weng Z, Ye J, Cai C, Liu Z, Liu Y, Xu Y, et al. Inflammatory microenvironment regulation and osteogenesis promotion by bone-targeting calcium and magnesium repletion nanoplatform for osteoporosis therapy. J Nanobiotechnol. (2024) 22:314. doi: 10.1186/s12951-024-02581-7
51. Huo S, Tang X, Chen W, Gan D, Guo H, Yao Q, et al. Epigenetic regulations of cellular senescence in osteoporosis. Ageing Res Rev. (2024) 99:102235. doi: 10.1016/j.arr.2024.102235
52. Jin Z, Xu H, Sun X, Yan B, and Wang L. Targeting SAT1 prevents osteoporosis through promoting osteoclast apoptosis. BioMed Pharmacother. (2024) 175:116732. doi: 10.1016/j.biopha.2024.116732
53. Ruan B, Dong J, Wei F, Huang Z, Yang B, Zhang L, et al. DNMT aberration-incurred GPX4 suppression prompts osteoblast ferroptosis and osteoporosis. Bone Res. (2024) 12:68. doi: 10.1038/s41413-024-00365-1
54. Jiang Z, Wang H, Qi G, Jiang C, Chen K, and Yan Z. Iron overload-induced ferroptosis of osteoblasts inhibits osteogenesis and promotes osteoporosis: An in vitro and in vivo study. IUBMB Life. (2022) 74:1052–69. doi: 10.1002/iub.2656
55. Jing Z, Li Y, Zhang H, Chen T, Yu J, Xu X, et al. Tobacco toxins induce osteoporosis through ferroptosis. Redox Biol. (2023) 67:102922. doi: 10.1016/j.redox.2023.102922
56. Xu Z, Zhang Z, Zhou H, Lin S, Gong B, Li Z, et al. Bazi Bushen attenuates osteoporosis in SAMP6 mice by regulating PI3K-AKT and apoptosis pathways. J Cell Mol Med. (2024) 28:e70161. doi: 10.1111/jcmm.70161
57. Lo HJ, Tsai CH, and Huang TW. Apoptosis-associated genetic mechanisms in the transition from rheumatoid arthritis to osteoporosis: A bioinformatics and functional analysis approach. APL Bioeng. (2024) 8:046107. doi: 10.1063/5.0233961
58. Wu Q, Liang H, Wang C, Chen Y, Yu C, Luo J, et al. Tetrahydroberberine prevents ovariectomy-induced bone loss by inhibiting RANKL-induced osteoclastogenesis and promoting osteoclast apoptosis. J Agric Food Chem. (2024) 72:20383–95. doi: 10.1021/acs.jafc.4c02982
59. Wang Y, Xiao H, Chen Y, Sheng X, Feng Z, Peng B, et al. PGRMC2 influences the onset of postmenopausal osteoporosis through disulfidptosis in monocytes: Evidence from experimental validation and Mendelian randomization. Heliyon. (2024) 10:e36570. doi: 10.1016/j.heliyon.2024.e36570
60. Zhang P, Li B, Chen H, Ge Z, Shang Q, Liang D, et al. RNA sequencing-based approaches to identifying disulfidptosis-related diagnostic clusters and immune landscapes in osteoporosis. Aging (Albany NY). (2024) 16:8198–216. doi: 10.18632/aging.205813
61. Zhong Z, Zhang C, Ni S, Ma M, Zhang X, Sang W, et al. NFATc1-mediated expression of SLC7A11 drives sensitivity to TXNRD1 inhibitors in osteoclast precursors. Redox Biol. (2023) 63:102711. doi: 10.1016/j.redox.2023.102711
62. Pan C, Zhang C, Lin Z, Liang Z, Cui Y, Shang Z, et al. Disulfidptosis-related Protein RPN1 may be a Novel Anti-osteoporosis Target of Kaempferol. Comb Chem High Throughput Screen. (2024) 27:1611–28. doi: 10.2174/0113862073273655231213070619
63. Shao Y, Zhang H, Guan H, Wu C, Qi W, Yang L, et al. PDZK1 protects against mechanical overload-induced chondrocyte senescence and osteoarthritis by targeting mitochondrial function. Bone Res. (2024) 12:41. doi: 10.1038/s41413-024-00344-6
64. Wang Q, Qi B, Shi S, Jiang W, Li D, Jiang X, et al. Melatonin alleviates osteoarthritis by regulating NADPH oxidase 4-induced ferroptosis and mitigating mitochondrial dysfunction. J Pineal Res. (2024) 76:e12992. doi: 10.1111/jpi.12992
65. Wu Y, Hu H, Wang T, Guo W, Zhao S, and Wei R. Characterizing mitochondrial features in osteoarthritis through integrative multi-omics and machine learning analysis. Front Immunol. (2024) 15:1414301. doi: 10.3389/fimmu.2024.1414301
66. Yao Q, Wu X, Tao C, Gong W, Chen M, Qu M, et al. Osteoarthritis: pathogenic signaling pathways and therapeutic targets. Signal Transduct Target Ther. (2023) 8:56. doi: 10.1038/s41392-023-01330-w
67. Fan X, Sun AR, Young RSE, Afara IO, Hamilton BR, Ong LJY, et al. Spatial analysis of the osteoarthritis microenvironment: techniques, insights, and applications. Bone Res. (2024) 12:7. doi: 10.1038/s41413-023-00304-6
68. Hu W, Chen Y, Dou C, and Dong S. Microenvironment in subchondral bone: predominant regulator for the treatment of osteoarthritis. Ann Rheum Dis. (2021) 80:413–22. doi: 10.1136/annrheumdis-2020-218089
69. Tang S, Zhang C, Oo WM, Fu K, Risberg MA, Bierma-Zeinstra SM, et al. Osteoarthritis. Nat Rev Dis Primers. (2025) 11:10. doi: 10.1038/s41572-025-00594-6
70. Musumeci G, Castrogiovanni P, Trovato FM, Weinberg AM, Al-Wasiyah MK, Alqahtani MH, et al. Biomarkers of chondrocyte apoptosis and autophagy in osteoarthritis. Int J Mol Sci. (2015) 16:20560–75. doi: 10.3390/ijms160920560
71. Liu Z, Wang T, Sun X, and Nie M. Autophagy and apoptosis: regulatory factors of chondrocyte phenotype transition in osteoarthritis. Hum Cell. (2023) 36:1326–35. doi: 10.1007/s13577-023-00926-2
72. Wang BW, Jiang Y, Yao ZL, Chen PS, Yu B, and Wang SN. Aucubin protects chondrocytes against IL-1β-induced apoptosis in vitro and inhibits osteoarthritis in mice model. Drug Des Devel Ther. (2019) 13:3529–38. doi: 10.2147/DDDT.S210220
73. Yang J, Hu S, Bian Y, Yao J, Wang D, Liu X, et al. Targeting cell death: pyroptosis, ferroptosis, apoptosis and necroptosis in osteoarthritis. Front Cell Dev Biol. (2022) 9:789948. doi: 10.3389/fcell.2021.789948
74. Zhao M, Song L, Liu Z, Li Y, and Wang Y. Necroptosis and its role in the pathogenesis and treatment of temporomandibular joint osteoarthritis: A narrative review. Arch Oral Biol. (2025) 176:106300. doi: 10.1016/j.archoralbio.2025.106300
75. Piao L, Wu D, Rui C, Yang Y, Liu S, Liu J, et al. The Bcr-Abl inhibitor DCC-2036 inhibits necroptosis and ameliorates osteoarthritis by targeting RIPK1 and RIPK3 kinases. BioMed Pharmacother. (2023) 161:114528. doi: 10.1016/j.biopha.2023.114528
76. Wei M, Shi X, Tang W, Lv Q, Wu Y, and Xu Y. Identification of a novel disulfidptosis-related gene signature in osteoarthritis using bioinformatics analysis and experimental validation. Sci Rep. (2025) 15:1339. doi: 10.1038/s41598-025-85569-z
77. Cao S, Wei Y, Yue Y, Wang D, Xiong A, Yang J, et al. Bioinformatics identification and experimental verification of disulfidptosis-related genes in the progression of osteoarthritis. Biomedicines. (2024) 12:1840. doi: 10.3390/biomedicines12081840
78. Hu K, Ou Y, Xiao L, Gu R, He F, Peng J, et al. Identification and construction of a disulfidptosis-mediated diagnostic model and associated immune microenvironment of osteoarthritis from the perspective of PPPM. J Inflammation Res. (2024) 17:3753–70. doi: 10.2147/JIR.S462179
79. Lian H, Zhang J, Hou S, Ma S, Yu J, Zhao W, et al. Immunotherapy of osteosarcoma based on immune microenvironment modulation. Front Immunol. (2025) 15:1498060. doi: 10.3389/fimmu.2024.1498060
80. Mao P, Feng Z, Liu Y, Zhang K, Zhao G, Lei Z, et al. The role of ubiquitination in osteosarcoma development and therapies. Biomolecules. (2024) 14:791. doi: 10.3390/biom14070791
81. Zhang K, Lu S, Jiang M, Zou X, Chen C, Lan Y, et al. Development of a risk model and genotyping patterns based on disulfidptosis-related lncRNAs to predict prognosis and immune landscape in osteosarcoma. Front Biosci (Landmark Ed). (2024) 29:193. doi: 10.31083/j.fbl2905193
82. Xu J, Guo K, Sheng X, Huang Y, Wang X, Dong J, et al. Correlation analysis of disulfidptosis-related gene signatures with clinical prognosis and immunotherapy response in sarcoma. Sci Rep. (2024) 14:7158. doi: 10.1038/s41598-024-57594-x
83. Wang L, Liu Y, Tai J, Dou X, Yang H, Li Q, et al. Transcriptome and single-cell analysis reveal disulfidptosis-related modification patterns of tumor microenvironment and prognosis in osteosarcoma. Sci Rep. (2024) 14:9186. doi: 10.1038/s41598-024-59243-9
84. Chen P and Shen J. A disulfidptosis-related gene signature associated with prognosis and immune cell infiltration in osteosarcoma. Bioeng (Basel). (2023) 10:1121. doi: 10.3390/bioengineering10101121
85. Song S, Guo Y, Yang Y, and Fu D. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharmacol Ther. (2022) 237:108168. doi: 10.1016/j.pharmthera.2022.108168
86. Li Z, Li D, Chen R, Gao S, Xu Z, and Li N. Cell death regulation: A new way for natural products to treat osteoporosis. Pharmacol Res. (2023) 187:106635. doi: 10.1016/j.phrs.2022.106635
87. Swahn H, Mertens J, Olmer M, Myers K, Mondala TS, Natarajan P, et al. Shared and compartment-specific processes in nucleus pulposus and annulus fibrosus during intervertebral disc degeneration. Adv Sci (Weinh). (2024) 11:e2309032. doi: 10.1002/advs.202309032
88. Zhang S, Li H, Li H, Zhao L, and Dong Z. Study on the effect of laser marker in the treatment of osteoarthritis of the knee joint and the accuracy of reconstruction of lower extremity alignment. Altern Ther Health Med. (2024) 30:368–76.
89. Ren Z, Yuan JY, Zhang J, Tan Y, Chen WQ, Zhang ZT, et al. Genetic analysis of seven pateints with Hereditary Multiple Osteochondromas (HMO). Am J Transl Res. (2022) 14:6303–12.
90. Li S, Zhang H, Liu J, and Shang G. Targeted therapy for osteosarcoma: a review. J Cancer Res Clin Oncol. (2023) 149:6785–97. doi: 10.1007/s00432-023-04614-4
91. Zhu Y, Wang X, Feng L, Zhao R, Yu C, Liu Y, et al. Intermetallics triggering pyroptosis and disulfidptosis in cancer cells promote anti-tumor immunity. Nat Commun. (2024) 15:8696. doi: 10.1038/s41467-024-53135-2
92. Liang J, Wang X, Yang J, Sun P, Sun J, Cheng S, et al. Identification of disulfidptosis-related subtypes, characterization of tumor microenvironment infiltration, and development of a prognosis model in breast cancer. Front Immunol. (2023) 14:1198826. doi: 10.3389/fimmu.2023.1198826
93. Ni L, Yang H, Wu X, Zhou K, and Wang S. The expression and prognostic value of disulfidptosis progress in lung adenocarcinoma. Aging (Albany NY). (2023) 15:7741–59. doi: 10.18632/aging.204938
94. Zheng H, Cheng J, Zhuang Z, Li D, Yang J, Yuan F, et al. A disulfidptosis-related lncRNA signature for analyzing tumor microenvironment and clinical prognosis in hepatocellular carcinoma. Front Immunol. (2024) 15:1412277. doi: 10.3389/fimmu.2024.1412277
95. Yang L, Cao ZJ, Zhang Y, Zhou JK, and Tian J. Disulfidptosis-related classification patterns and tumor microenvironment characterization in skin cutaneous melanoma. Melanoma Manag. (2024) 10:MMT65. doi: 10.2217/mmt-2023-0006
96. Hadian K and Stockwell BR. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat Rev Drug Discov. (2023) 22:723–42. doi: 10.1038/s41573-023-00749-8
97. Liu Y, Sun Y, Chen A, Chen J, Zhu T, Wang S, et al. Involvement of disulfidptosis in the pathophysiology of autism spectrum disorder. Life Sci. (2025) 369:123531. doi: 10.1016/j.lfs.2025.123531
98. Yang Y, Liu Q, Lu X, Wang Y, Tian X, Yang J, et al. Exploring a novel risk model based on core disulfidptosis-related genes in periodontitis: Bioinformatics analyses and experimental validation. FASEB J. (2025) 39:e70368. doi: 10.1096/fj.202401986R
99. Zhao L, Zhang J, Song Q, Dai C, Qin Y, and Li A. Comprehensive analysis of disulfidptosis-related genes and the immune microenvironment in heart failure. Front Cell Dev Biol. (2025) 12:1516898. doi: 10.3389/fcell.2024.1516898
100. Shi H, Zhou C, and Zhao Y. Establishment of a diagnostic model of endometriosis based on disulfidptosis-related genes. J Obstet Gynaecol Res. (2024) 50:1201–7. doi: 10.1111/jog.15945
101. Zhu Y, Kong L, Han T, Yan Q, and Liu J. Machine learning identification and immune infiltration of disulfidptosis-related Alzheimer’s disease molecular subtypes. Immun Inflammation Dis. (2023) 11:e1037. doi: 10.1002/iid3.1037
102. Liu Y, Zhu T, Wang J, Cheng Y, Zeng Q, You Z, et al. Analysis of network expression and immune infiltration of disulfidptosis-related genes in chronic obstructive pulmonary disease. Immun Inflammation Dis. (2024) 12:e1231. doi: 10.1002/iid3.1231
103. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. (2018) 20:1181–92. doi: 10.1038/s41556-018-0178-0
104. Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, et al. mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun. (2021) 12:1589. doi: 10.1038/s41467-021-21841-w
105. Dejas L, Santoni K, Meunier E, and Lamkanfi M. Regulated cell death in neutrophils: From apoptosis to NETosis and pyroptosis. Semin Immunol. (2023) 70:101849. doi: 10.1016/j.smim.2023.101849
106. Liu S, Yao S, Yang H, Liu S, and Wang Y. Autophagy: Regulator of cell death. Cell Death Dis. (2023) 14:648. doi: 10.1038/s41419-023-06154-8
Keywords: disulfidptosis, bone diseases, oxidative stress, immunometabolism, targeted therapy
Citation: Zhao X, Zhang C, Qu L, Gao J, Wu S, Zhang Y and Zhang Y (2025) Immunometabolic regulation of disulfidptosis in orthopedic diseases: mechanistic heterogeneity and therapeutic targets. Front. Immunol. 16:1647931. doi: 10.3389/fimmu.2025.1647931
Received: 16 June 2025; Accepted: 06 October 2025;
Published: 28 October 2025.
Edited by:
Gaocai Li, Huazhong University of Science and Technology, ChinaReviewed by:
Qi Xue, Connecticut Agricultural Experiment Station, United StatesRan Mo, Yale University, United States
Copyright © 2025 Zhao, Zhang, Qu, Gao, Wu, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yilei Zhang, emhhbmd5aWxlaUB4anR1LmVkdS5jbg==; Yingang Zhang, enlpbmdhbmdAbWFpbC54anR1LmVkdS5jbg==