 Wenxin Shi1,2,3
Wenxin Shi1,2,3 Zhiqiang Zhang1,2Xiaotong Xu1,2Yanpeng Tian4Li Feng5Xianghua Huang1,2Yanfang Du1,2*
Zhiqiang Zhang1,2Xiaotong Xu1,2Yanpeng Tian4Li Feng5Xianghua Huang1,2Yanfang Du1,2* Zhongkang Li1,2*
Zhongkang Li1,2*- 1Department of Obstetrics and Gynecology, The Second Hospital of Hebei Medical University, Shijiazhuang, China
- 2Hebei Key Laboratory of Regenerative Medicine of Obstetrics and Gynecology, Shijiazhuang, China
- 3Department of Obstetrics and Gynecology, Hebei General Hospital, Shijiazhuang, China
- 4Department of Obstetrics and Gynecology, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, China
- 5Department of Obstetrics and Gynecology, The Fourth Hospital of Shijiazhuang, Shijiazhuang, China
Single-cell RNA sequencing (scRNA-seq) has emerged as an advanced biological technology capable of resolving the complexity of cancer landscapes at single-cell resolution. Spatial transcriptomics(ST), as an innovative complementary approach, effectively compensates for the lack of spatial information inherent in scRNA-seq data. This review explores the rapidly evolving integration of scRNA-seq and ST and their transformative role in deciphering the tumor microenvironment (TME). We highlight how these technologies jointly uncover cellular heterogeneity, stromal-immune interactions, and spatial niches driving tumor progression and therapy resistance. Moving beyond previous reviews, we emphasize emerging computational strategies for data integration—including deconvolution and mapping approaches—and evaluate their applications in characterizing immune evasion, fibroblast diversity, and cell-cell communication networks. Ultimately, this review provides a forward-looking perspective on how spatial multi-omics are poised to advance precision oncology through spatially-informed biomarkers and diagnostic tools. We conclude that the full clinical potential of these technologies relies on closing the gap between analytical innovation and robust clinical implementation.
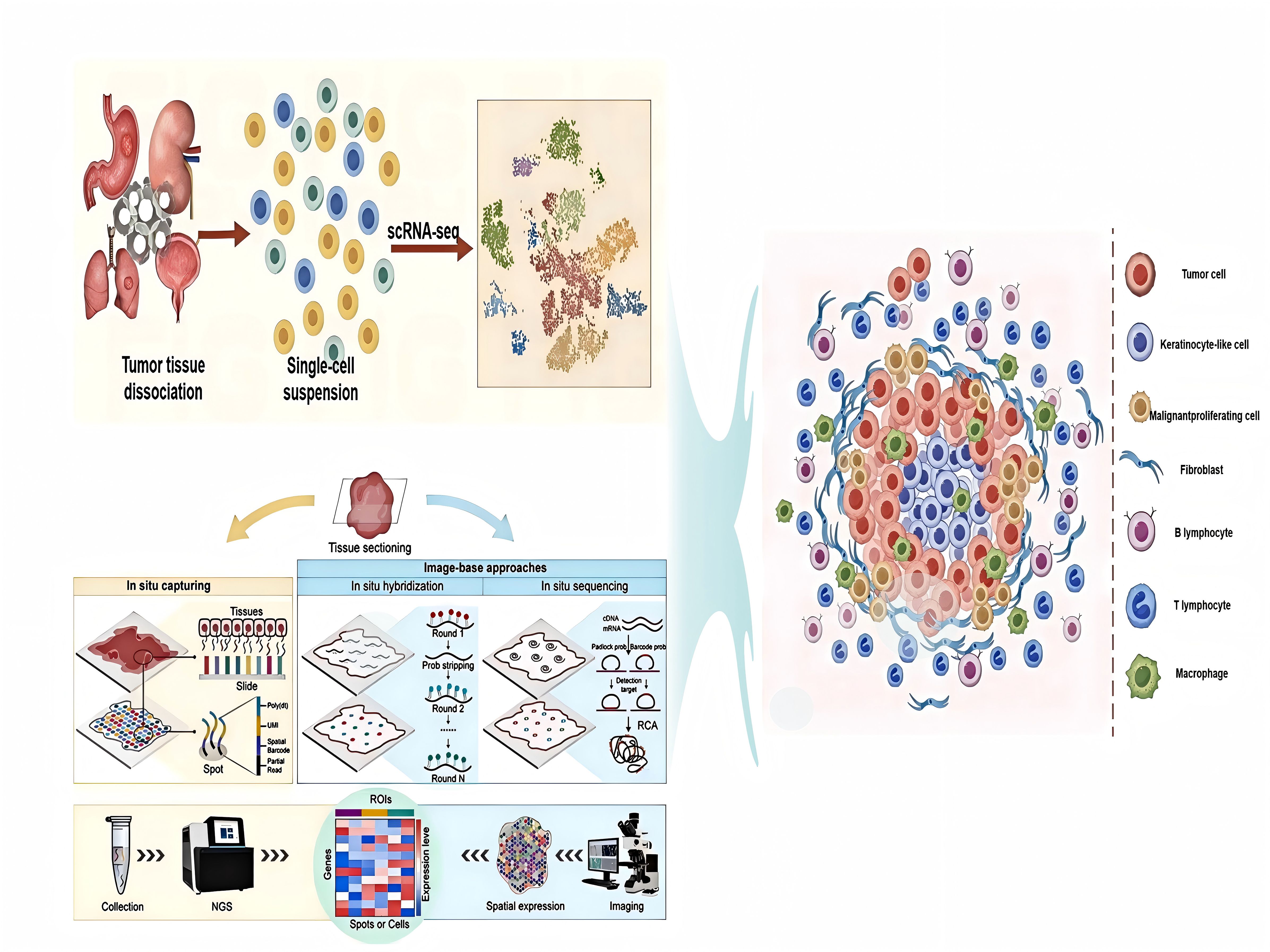
Graphical Abstract. The application of single-cell RNA sequencing (scRNA-seq) and spatial transcriptomic (ST) techniques in TME.
1 Introduction
Traditionally, tumors have been regarded as diseases primarily characterized by uncontrolled proliferation of malignant cells, and therapeutic strategies have predominantly focused on their direct eradication through chemotherapy and radiotherapy. However, this perspective has evolved significantly with the growing recognition of tumor complexity, wherein tumors are increasingly viewed as highly dynamic and heterogeneous ecosystems (1). The TME, in particular, represents a complex cellular and molecular landscape composed not only of malignant cells but also of diverse non-malignant components, including immune cells, cancer-associated fibroblasts (CAFs), vascular endothelial cells, pericytes, and tissue-resident stromal cells, all embedded within the extracellular matrix (ECM) (2). In certain tumor types, non-malignant cells may constitute the majority of the tumor mass (3). The cellular composition and functional states in the TME exhibit significant variability influenced by factors such as the anatomical origin of the tumor, genetic and epigenetic features of cancer cells, disease stage, and host-specific factors (4–6). Understanding the complex cellular interactions and spatial heterogeneity in the TME is crucial for enhancing our understanding comprehension of tumor biology and facilitating the development of more precise and effective anticancer therapies.
Despite its central role in cancer progression and therapeutic response, the TME presents significant analytical challenges. One primary limitation arises from the technical constraints of transcriptomic profiling methods. Conventional bulk RNA sequencing (RNA-seq) captures only average gene expression from heterogeneous cell populations, thereby obscuring intrinsic cellular heterogeneity in the TME and failing to identify rare but functionally critical subpopulations (7, 8). Tumor heterogeneity itself constitutes another substantial barrier (9–11). This heterogeneity exists both across patients (inter-tumor heterogeneity) and within individual tumors (intra-tumor heterogeneity), as cancer cells occupy various differentiation states while exhibiting divergent transcriptional profiles and mutational landscapes. Furthermore, non-malignant cell populations, including immune and stromal cells, exhibit extensive phenotypic and functional diversity. The complexity of mechanisms underlying therapy resistance further highlights the urgent need for deeper insights into the TME (12–15). Increasing evidence suggests that non-malignant cells actively contribute to resistance against chemotherapy, targeted therapies, and immunotherapies through multiple mechanisms. For instance, CAFs secrete ECM components and growth factors, establishing physical and biochemical barriers that hinder drug penetration (16, 17). Immunosuppressive cells such as regulatory T cells (Tregs) and M2-polarized macrophages suppress anti-tumor immunity by expressing immune checkpoint molecules (e.g., PD-1, CTLA-4) and releasing inhibitory cytokines such as IL-10 and TGF-β (18–20). Collectively, these findings underscore the necessity of comprehensively characterizing the TME—encompassing cellular composition, functional phenotypes, and spatial interaction networks—to inform the rational design of combination therapies (21, 22). To address these challenges, the integrating scRNA-seq with ST has emerged as a powerful strategy. This approach facilitates insights into the spatial and functional complexity of the TME.
scRNA-seq is a powerful technique enabling high-resolution gene expression profiling for the individual-cell level, enabling the identification and characterization of distinct cellular subpopulations with specialized functions (23). ST, a rapidly evolving complementary approach, maps gene expression within intact tissue sections, preserving critical spatial context and tissue architecture (24). Given the cellular complexity of the TME, no single technology can fully capture its spatial and functional heterogeneity. Although current ST platforms generally lack true single-cell resolution, their integration with scRNA-seq provides a comprehensive perspective on the TME. Combining scRNA-seq and ST overcomes these limitations by bridging cellular identity with spatial localization. For instance, multimodal intersection analysis (MIA) was introduced in 2020 to integrate scRNA-seq and ST data, aiming to map spatial associations cell-type relationships in pancreatic ductal adenocarcinoma (PDAC) (25). This study revealed that stress-associated cancer cells colocalize with inflammatory fibroblasts, the latter identified as major producers of interleukin-6 (IL-6), underscoring spatially organized tumor-stroma crosstalk in PDAC (25).
The integration of scRNA-seq and ST enables researchers to dissect the complexity and spatial organization of the TME with unprecedented resolution. This synergistic approach not only deepens our understanding of tumor biology but also accelerates the discovery of novel diagnostic and prognostic biomarkers, paving the way for more precise and effective therapeutic strategies. We conducted comprehensive searches in PubMed, Web of Science, and Scopus to ensure broad coverage of relevant studies. We used a combination of keywords related to single-cell sequencing, spatial transcriptomics, tumor microenvironment, cancer heterogeneity, and their respective applications in oncology. We defined explicit criteria for including studies based on relevance, study type (e.g., original research, key reviews), and publication status. Studies were excluded if they were not peer-reviewed, not published in English, or deemed outside the scope of this review. We focused primarily on literature published between January 2010 and June 2025 to capture the most recent and impactful advances in the field. (GA).
2 Advances in technologies for analyzing spatial distributions
ST is an emerging technology that enables spatially resolved gene expression profiling within intact tissue sections, preserving the native histological context. By combining high-resolution imaging and transcriptomic analysis, ST maps gene expression patterns with precise spatial localization, achieving subcellular resolution in some cases. Current ST methodologies can be broadly classified into two categories: image-based (I-B) and barcode-based (B-B) approaches (26, 27). Image-based methods, such as in situ hybridization (ISH) (28) and in situ sequencing (ISS) (29), utilize fluorescently labeled probes to directly detect RNA transcripts within tissues, allowing visualization of gene expression patterns while maintaining spatial integrity. In contrast, barcode-based approaches rely on spatially encoded oligonucleotide barcodes to capture RNA transcripts. In solid-phase transcriptome capture, RNAs hybridize to immobilized barcoded probes on slides before sequencing. Deterministic spatial barcoding assigns unique barcodes to each transcript, retaining positional information throughout sequencing (30–32). These complementary strategies facilitate comprehensive spatial transcriptome profiling, when integrated with single-cell techniques, they yield unprecedented resolution for investigating tissue architecture and tumor heterogeneity.
scRNA-seq of patient-derived tumors has uncovered diverse cellular subpopulations and revealed intricate intercellular communication networks within the TME (33–39). However, scRNA-seq requires tissue dissociation, leading to the loss of spatial context and limiting insights into tissue architecture and cell-cell interactions. To address this, several strategies have been developed to preserve or reconstruct spatial information. For example, combining ISH-based gene expression mapping with scRNA-seq data has proven effective for identifying rare cell types and subpopulations using targeted gene panels (40, 41). Recent advances have evolved ISH into high-plex RNA imaging (HPRI) techniques, including in situ sequencing, multiplexed error-robust fluorescence in situ hybridization (MERFISH) (42), and sequential fluorescence in situ hybridization (seqFISH) (43–45). However, these approaches are often limited to well-defined tissues and remain challenging when applied to heterogeneous solid tumors characterized with complex structures and diverse transcriptomic profiles.
Emerging methods, such as sci-Space, have been developed to address this limitation by generating spatially resolved transcriptomic maps at near-single-cell resolution across extensive tissue areas. In mouse embryonic development studies, sci-Space enabled the simultaneous capture of approximate spatial coordinates and complete transcriptomes from over 120,000 nuclei. However, its spatial resolution is currently limited to approximately 200 micrometers. Although there have been improvements in spot density and size, the resolution remains insufficient for precisely capturing interactions between neighboring cells. As a result, this approach typically yields composite transcriptomic profiles derived from small cell clusters or cellular fragments rather than genuine single-cell resolution. Currently, ST remains one of the most widely adopted approaches for high-throughput spatial gene expression analysis (46, 47). scRNA-seq is a high-throughput method for transcriptomic profiling at individual-cell resolution. By isolating individual cells, capturing their mRNA, and performing high-throughput sequencing, scRNA-seq reveals cellular heterogeneity typically masked in bulk RNA analyses. The advantages of scRNA-seq include: (i) identification of rare cell populations, including tumor stem cells and transitional cellular states, which are undetectable by bulk RNA-seq (48); (ii) classification of cells based on canonical markers, enabling precise identification of immune cell subsets and epithelial cell states (49); (iii) characterization of dynamic biological processes, such as differentiation trajectories and cellular transitions (50); and (iv) integration with multi-omics approaches, including single-cell ATAC-seq (chromatin accessibility) and CITE-seq (surface protein expression), providing multidimensional insights into cell states (51).
Despite these strengths, scRNA-seq also exhibits notable limitations. RNA capture efficiency per cell is relatively low (52). The method remains costly and technically challenging, necessitating careful optimization of sample processing protocols (53, 54). Critically, the mandatory tissue dissociation disrupts native spatial relationships, hindering analysis of cell–cell interactions within intact tissue architectures (55, 56). The comparison of scRNA-seq and ST is shown in Table 1.
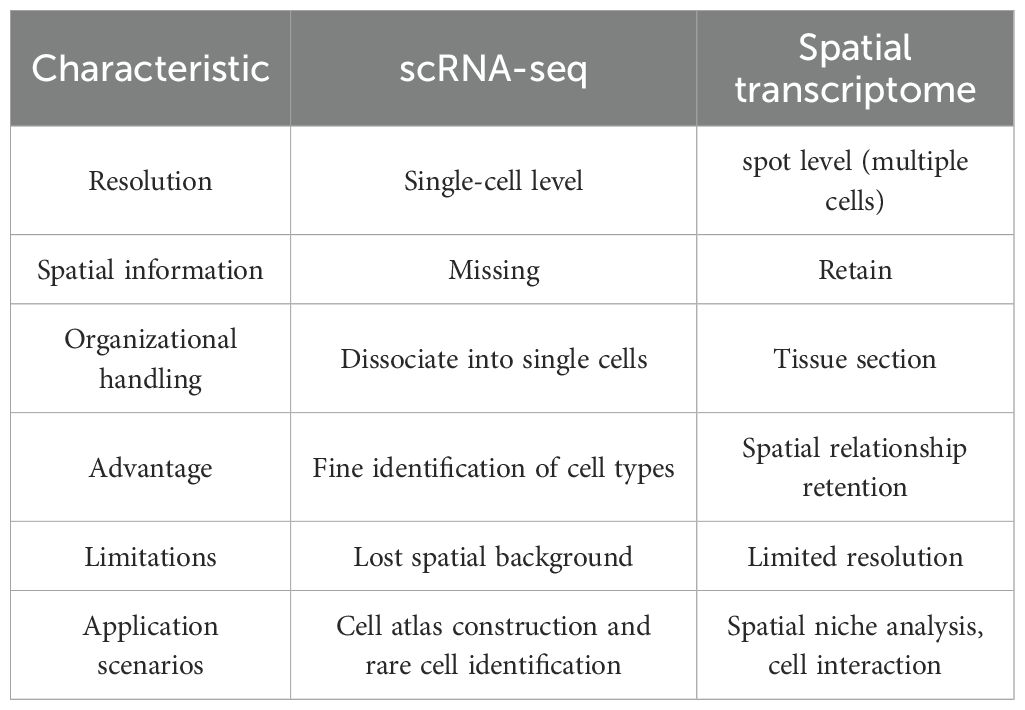
Table 1. Comparison between scRNA-seq and ST technology.
The integration of scRNA-seq and ST confers significant advantages for deciphering complex biological systems: (i) Comprehensive gene expression profiling: scRNA-seq enables high-resolution gene expression analyses, revealing cellular heterogeneity and transcriptional dynamics within tissues (8). It is essential for cell-type identification, developmental tracking, and elucidating disease mechanisms. (ii) Spatial context and tissue architecture: ST preserve native tissue spatial architecture, enabling localization of gene expression patterns, cellular distributions, and intercellular interactions (57). (iii) Complementary strengths: While scRNA-seq lacks spatial information, ST technologies face resolution and throughput limitations. Their integration overcomes their individual limitations, offering a comprehensive understanding of tissue biology (58–60).
Combining scRNA-seq and ST provides deeper insights into cellular interactions with their microenvironment (61–63), with critical implications for both diagnostics and therapeutics. This integrative strategy supports the identification of spatially informed biomarkers and therapeutic targets by linking gene expression patterns to precise tissue regions, thereby advancing personalized medicine and enhancing disease diagnosis (64). Currently, two major computational approaches are used to this integration: deconvolution and mapping. Deconvolution utilize single-cell reference datasets to computationally estimate the cellular composition within each spatial capture spot, determining proportions of various cell types. Mapping approaches assign scRNA-seq-defined cellular subtypes to cells within spatial maps or localize individual scRNA-seq profiles to specific tissue niches. The characteristics of different integration strategies are shown in Table 2. These analyses provide critical spatial context to inferred ligand–receptor interactions and other forms of intercellular communication derived from scRNA-seq data.
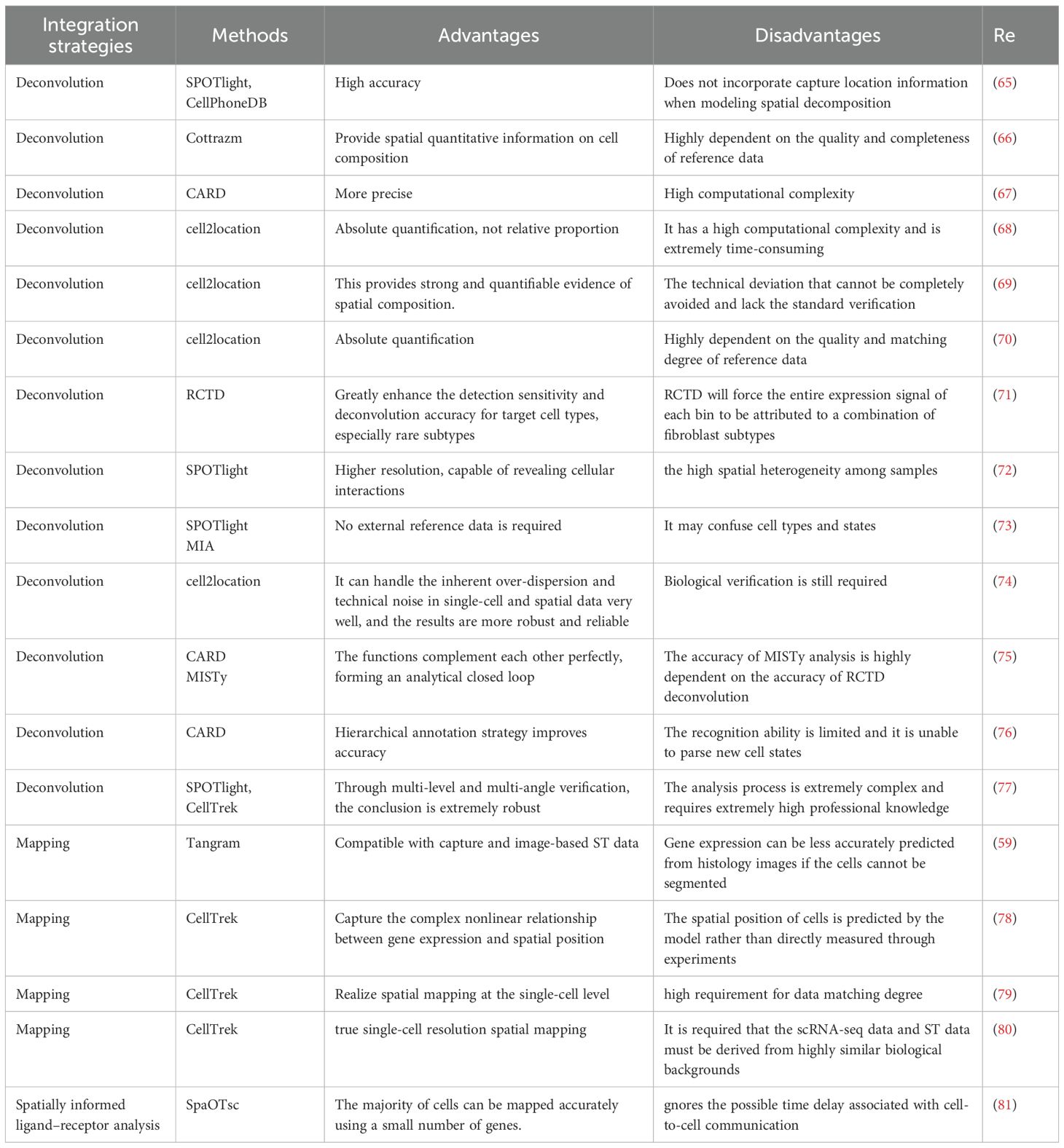
Table 2. seRNA-seq and spatial transcriptomic integration strategies.
3 Immunosuppressive tumor microenvironment
The immunosuppressive tumor microenvironment (ITME) is a specialized ecosystem in tumor tissues. The ITME suppresses anti-tumor immune responses via multiple mechanisms. This promotes immune escape, tumor growth, and therapy resistance. Complex cellular crosstalk drives ITME formation, representing a major challenge to immunotherapy. The interaction between immune-mediated tumor editing and cancer cell immune evasion influences disease progression and therapeutic outcomes (82, 83).
3.1 Cells in the tumor microenvironment
The TME comprises diverse cell types that collectively influence tumor behavior. ScRNA-seq and ST have revealed unprecedented heterogeneity and functional plasticity among these populations, uncovering their roles in immune evasion, metastasis, and treatment resistance.
3.1.1 CD8+ T cells
CD8+ T cells, also known as cytotoxic T lymphocytes (CTLs), serve as central effectors in anti-tumor immunity. They mediate tumor cell killing through cytolytic mechanisms, such as perforin and granzyme release, and secrete cytokines like IFN-γ to amplify immune responses. However, in the ITME, chronic antigen exposure, inhibitory signals, and metabolic disturbances often lead to CD8+T cell immune exhaustion (84–86). T cell exhaustion is a critical factor contributing to immune evasion and limited immunotherapy efficacy. Recent studies suggest that targeted transcriptional modulation (87), metabolic reprogramming (86, 88), and microenvironmental remodeling (89, 90) can restore CD8+ T cell functionality. These strategies offer promising directions for next-generation immunotherapies.
ST has become an essential tool for deciphering the functional states and spatial organization of CD8+ T cells in the TME. By mapping spatial proximity to other cell populations, ST can infer intercellular communication and elucidate how local cellular neighborhoods influence CD8+ T cell phenotypes (91–94).
3.1.2 CD4+ T cells
CD4+ T cells act as central coordinators of immune responses and differentiate into various functional subsets. In the TME, their activity is highly context-dependent, influenced by subset composition, cytokines and metabolites. scRNA-seq has revealed that CD4+ T cells can exert tumor-suppressive effects by producing TNF-α, while ST indicates spatial co-localization with CD8+ T cells, suggesting coordinated immune responses. These findings exemplify the complementary strengths of integrating scRNA-seq and ST (95). Future research should leverage these technologies to explore CD4+ T cell heterogeneity and spatial organization, facilitating precision immunotherapy.
CD4+ T cells mediate anti-tumor effects through both indirect and direct mechanisms (95). Dynamic changes in CD4+ T cell subsets correlate with tumor progression. For instance, scRNA-seq analyses of prostate cancer identified elevated regulatory T cell (Treg) activity scores in tumors relative to normal tissue, with tumor-infiltrating Tregs displaying increased expression of TNF receptor family genes. These findings suggest CD4+ T cells may promote both pro-inflammatory tumor progression and immunosuppressive niche formation via TNF signaling (96).
3.1.3 Tumor-associated macrophages
Macrophages represent essential innate immune components, mediating pathogen clearance and immune modulation. Within tumors, macrophages—termed tumor-associated macrophages (TAMs)—often exhibit immunosuppressive functions and promote tumor progression. TAMs exhibit remarkable plasticity, polarizing into pro-inflammatory, cytotoxic M1-like or immunosuppressive, tissue-remodeling M2-like phenotypes (97–99).
A recent study analyzed 97 paired samples from 24 colorectal cancer patients with liver metastases using scRNA-seq and spatial transcriptomics. It revealed extensive spatial remodeling in metastatic niches, driven largely by MRC1+CCL18+ M2-like macrophages (100). However, how the chemotherapy induces the functional changes of macrophages was not clear. Further experimental validation is required to validate that such state shift of macrophages is due to altered differentiation or population change. It showed intensified immunosuppression, highlighting the therapeutic potential of targeting M2-like TAMs (100). Similarly, a 2021 breast cancer study using scRNA-seq identified immunosuppressive macrophage subsets—lipid-associated macrophages (LAMs) and CXCL10+ macrophages—as key producers of suppressive cytokines. ST further demonstrated their proximity to PD-1+ lymphocytes (101). However, its number of cases per clinical subtype limited to estimate subtype-specific features.
In clear cell renal cell carcinoma (ccRCC), ST revealed distinct expression profiles between tumor cores and boundaries. Integrative analysis identified selective expression of IL-1β by macrophages at tumor edges. IL-1β expression correlated with epithelial–mesenchymal transition (EMT) induction and poor prognosis. IL-1β blockade reduced tumor burden in RCC murine models (102), while in another study, it was verified that IL-6 lowered lung cancer incidence (103), highlighting IL-1β as a promising therapeutic target (104).The limit is that the researchers chose mouse renal cell carcinoma lines as the tumor cell model. This cell line usually lacks mutations related to ccRCC (103).
3.2 Tumor cell–immune cell communication in the tumor microenvironment
Communication between tumor cells and immune cells in the TME, critically influences immune evasion or tumor eradication. scRNA-seq approaches have elucidated cell–cell interaction networks and identified pivotal immune cell signaling hubs (103). By inferring ligand–receptor interactions from scRNA-seq data, researchers can delineate intercellular communication pathways between cancer and TME, including those driving immunosuppression (105). Notably, epithelial cells engage strongly with myeloid cells and may demonstrate potential immunosuppressive communications with T cells.
Cell-cell interactions within the tumor microenvironment drive key processes including immune suppression, angiogenesis, and metastasis. Advances in single-cell and spatial multi-omics now enable systematic mapping of these communications, revealing ligand–receptor networks and functional cellular crosstalk. Targeting these interactions offers promising strategies for novel cancer immunotherapies.
3.2.1 T lymphocyte-cell interactions
Interactions between T lymphocytes and various tumor cells play a critical role in shaping the immune microenvironment. scRNA-seq analyses have revealed strong immunosuppression in tumors, characterized by increased infiltration of regulatory T cells (Tregs), which impair CD8+T cell cytotoxicity and promote tumor progression (106). ST further identified immune hotspots where Tregs are found in close proximity to effector T cells, suppressing anti-tumor responses within these regions (107, 108). Consistent with this, transcriptomic profiling shows elevated abundances of Tregs and exhausted CD8+T cells, underscoring the profound immunosuppression and immune infiltration features in the tumor microenvironment (109).
3.2.2 TAM-cell interactions
In TNBC tumors, macrophage subsets often co-express both M1 and M2 markers, suggesting their dual role in either suppressing or promoting tumor progression and metastasis (110). Specific subpopulations of tumor-associated macrophages (TAMs) are associated with T cell infiltration and immunosuppression, highlighting their critical influence on the immune landscape of TNBC (111). These TAMs can impair T cell function and dampen immune responses, thereby supporting immune evasion and fostering a tumor-permissive microenvironment (112, 113). Interestingly, macrophage infiltration also correlates with improved patient outcomes. Transcriptome studies indicate that a high density of CD163+macrophages is significantly associated with longer overall survival and TNBC-specific survival (114).
3.2.3 CAFs-cell interactions
It was showed that CAF phenotypes were a strong prognostic factor, and CAF phenotypes associated with good and poor patient prognosis. It was also discovered that different CAF types varied in their spatial distribution in the TME (Table 3). However, the interactions occurred at the edges of the cells was not investigated (115). In another study, the intercellular communication predominantly involved iCAFs, malignant epithelial cells, mCAFs, and pCAFs, each exhibiting distinct numbers and strengths of interactions. Although their study provided a detailed analysis of CAFs, it may not fully encapsulate all interactions and mechanisms (116). ST in breast cancer have revealed specific spatial enrichment between cancer-associated fibroblasts (CAFs) and T cell subsets (101). In multiple tumor types, certain CAF subsets are associated with T-cell exhaustion. For example, ecm-myCAF and TGF-β-myCAF in breast cancer, and a FAP+/PDGFRA-subset in lung cancer, have been linked to this immunosuppressive process (117, 118). Consistent with this, a separate lung cancer study also reported positive correlations between FAP+ CAFs and T-cell exhaustion markers (119). Spatial transcriptomics in head and neck cancer demonstrated co-localization of specific CAF subsets with exhausted T cells (120).
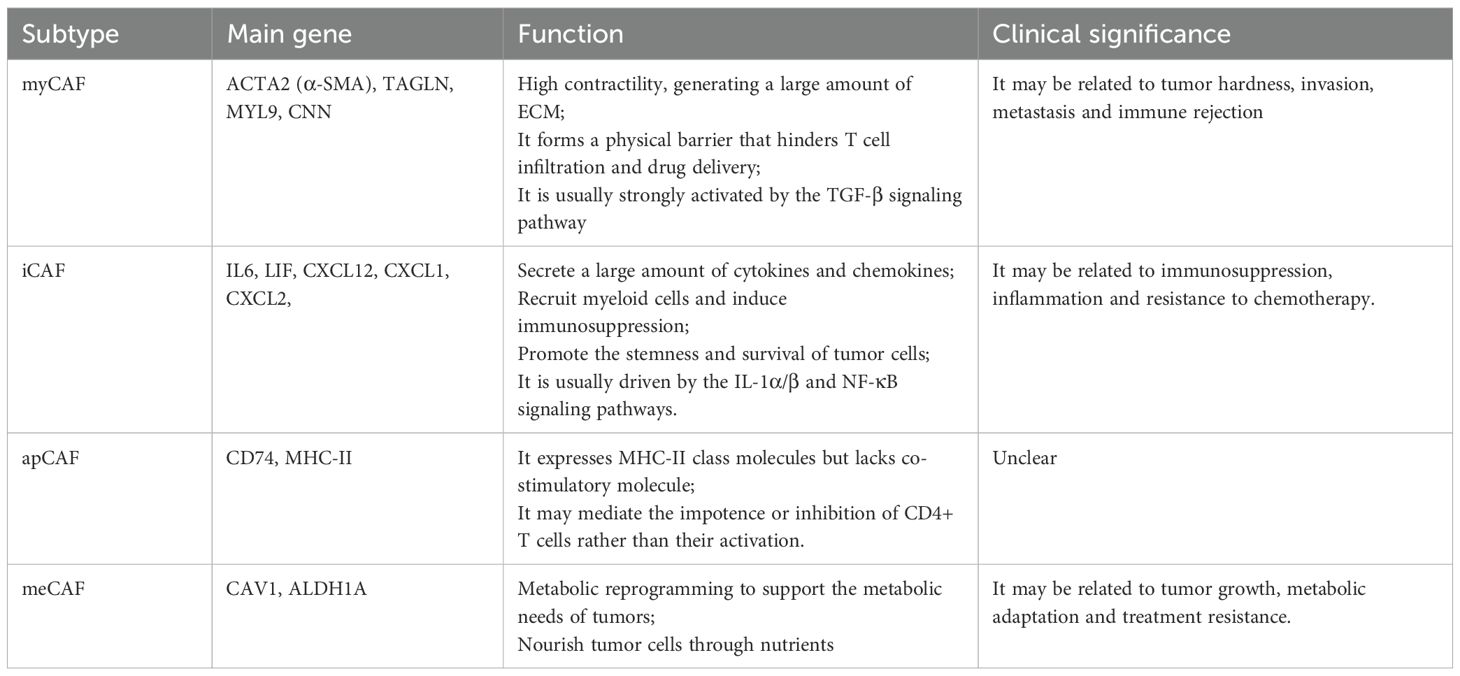
Table 3. Subtypes and comparisons of CAFs.
3.2.4 B lymphocyte-cell interactions
B cells influence the tumor microenvironment not only through antibody production, but also via cytokine secretion and direct cell-cell interactions. They exert regulatory effects on both tumor cells and other immune cells. For instance, ligand-receptor interactions can mediate direct contact between B cells and tumor cells (121). Such interactions may also suppress antibody-mediated immune responses (122). Together, these mechanisms help sustain an immunosuppressive microenvironment, promoting tumor proliferation and metastasis.
4 Functional heterogeneity of cancer-associated fibroblasts and their immunomodulatory roles
CAFs are a major stromal component in the TME, critically contributing to tumor initiation, progression, invasion, metastasis, and therapeutic resistance. CAFs typically originate from resident fibroblasts or precursor cells activated by tumor-derived signals. They exhibit high heterogeneity and secrete diverse cytokines, growth factors, and ECM components, collectively remodeling the TME to facilitate tumor development. CAFs significantly modulate tumor behavior (123–125). Their functional plasticity and diversity not only promote tumor progression but also represent potential therapeutic targets. SeRNA-seq has revealed substantial CAFs heterogeneity, identifying multiple transcriptionally distinct CAF subtypes within the TME (Table 3) (126–128).
4.1 Spatially resolved roles of CAFs in the TME
CAFs interact extensively with immune and tumor cells within the TME, significantly influencing tumor progression (129). A 2023 spatial transcriptomics study of 16 glioblastoma (GBM) patient samples demonstrated spatial proximity between CAFs, mesenchymal GBM stem cells, endothelial cells, and M2-like macrophages (130). Beyond immune modulation, CAFs shape the GBM vascular microenvironment (131). CAF-induced hypertrophic remodeling of tumor vasculature potentially underlies GBM resistance. ST revealed CAFs were preferentially localized in perivascular niches along with glioblastoma stem cells (GSCs), suggesting the interactions contributing to therapeutic resistance. These findings highlight CAF–GSC interactions as critical targets for therapeutic intervention in GBM (132).
4.2 CAFs in tumor metastasis
A 2022 study identified two major CAFs subtypes—iCAFs and myCAFs—in esophageal squamous cell carcinoma (ESCC), revealing the heterogeneity (133). Integrative scRNA-seq and ST analyses demonstrated the epithelial cells primarily localized in cancerous regions, whereas iCAFs were predominantly enriched in surrounding stroma. In contrast, myCAFs showed no distinct spatial preference. This spatial distribution suggested a pivotal role for iCAFs in tumor progression and metastasis.
4.3 CAFs remodeling in response to neoadjuvant chemotherapy
Neoadjuvant chemotherapy (NACT), administered before surgery or radiotherapy, reduces the tumor burden, enhances resection success, and eradicates micrometastases. Emerging evidence indicates NACT significantly reshapes CAF composition and function, influencing therapeutic outcomes. In rectal cancer, scRNA-seq demonstrated a distinct reorganization of CAFs following NACT, particularly characterized by an increase in myofibroblast populations after treatment. Elevated myCAFs facilitated ECM remodeling and immunosuppression, correlating with the poor prognosis (134, 135). However, the relationship between CAFs heterogeneity and NACT response remains incompletely characterized (136–139). Integrative scRNA-seq and ST analyses have begun to shed light on how NACT-induced remodeling affects therapeutic efficacy. In 2023, using combined scRNA-seq and STs, Qin et al. (140) identified a novel CAFs subpopulation termed positive-response–associated CAFs (pCAFs), which promoted anti-tumor immunity through spatial recruitment and immune cell interactions. Similar CAFs remodeling patterns were observed in pancreatic ductal adenocarcinoma (PDAC) (141). These findings indicate that NACT profoundly remodels both cancer cells and fibroblasts, leading to the formation of distinct immunological and stromal niches.
Collectively, these insights highlight the therapeutic potential of modulating specific CAFs subsets. Potential strategies include promoting immune-supportive pCAF differentiation, inhibiting tumor-promoting nCAF subpopulations, or targeting specific cytokines and ECM components driving therapy resistance. Nevertheless, the mechanisms underlying CAF heterogeneity are not yet fully understood. Systematic characterization of CAFs subsets and their context-specific functions will be essential for uncovering novel therapeutic targets.
5 Challenges and perspectives
Although scRNA-seq and ST have significantly enhanced our understanding of tumor biology, several challenges remain to be addressed (142). Tumors exhibit extensive somatic genetic heterogeneity (143), and their pathogenesis involves intricate regulatory mechanisms across multiple omics dimensions, including transcriptomics, epigenomics, proteomics, and metabolomics (144). With the rapid advancement of single-cell multi-omics technologies, research has increasingly transitioned from single-omics analyses to integrated approaches combining transcriptomic, genomic, epigenomic, and proteomic data. Such integrated multi-omics strategies have already provided valuable insights into several malignancies, including colorectal cancer (CRC) (145), lung cancer (146), and prostate cancer (147). The combination of single-cell multi-omics with ST is anticipated to offer a more comprehensive and spatially resolved understanding of tumor heterogeneity at single-cell resolution.
However, despite these technological advancements, clinical translation remains challenging. Several practical barriers remain for clinical transformation: (i) Cost-benefit trade-off: these technologies are currently expensive and have long experimental cycles; (ii) High requirements of infrastructure and data analysis capabilities; (iii) Lack of regulations and standardization. This requires collaborative efforts from regulators, industry, and academia (148, 149).
6 Outstanding questions
Achieving true single-cell resolution in spatial transcriptomics technologies and the associated computational challenges in analyzing such high-dimensional data. The necessary next step of integrating spatial multi-omics data, particularly spatial proteomics and metabolomics, to build a more comprehensive functional understanding of the tumor microenvironment. The urgent need for standardizing and validating analytical pipelines to ensure robustness, reproducibility, and ultimately, their successful translation into clinical settings for diagnostics and therapeutic decision-making.
7 Conclusion
In conclusion, the integration of single-cell and spatial transcriptomics technologies has fundamentally expanded our understanding of tumor heterogeneity and microenvironmental organization. However, to translate these insights into clinical impact, future work must focus on three critical frontiers. First, the integration of single-cell and spatial transcriptomics will be essential to move beyond transcriptional data and achieve a functional, multi-layered understanding of cellular phenotypes and interactions within their native context. Second, the prospective clinical validation of spatial biomarkers is urgently needed to establish their utility in patient stratification, prognosis, and therapy guidance. This will require rigorous standardization of analytical and reporting protocols to ensure reproducibility across platforms and cohorts. Finally, the development of advanced computational frameworks capable of unifying multi-omic spatial data—and ultimately enabling real-time mapping—will be crucial for informing diagnostic and even intraoperative decisions. With sustained development, these integrative approaches hold substantial promise for enhancing cancer diagnostics, guiding precision therapeutic strategies, and ultimately improving clinical outcomes for patients.
Author contributions
WS: Formal Analysis, Conceptualization, Writing – original draft. ZZ: Writing – review & editing, Investigation, Supervision. XX: Software, Writing – review & editing. YT: Formal Analysis, Project administration, Writing – review & editing. LF: Writing – review & editing, Investigation, Data curation. XH: Writing – review & editing, Validation, Methodology, Visualization. YD: Investigation, Formal Analysis, Writing – review & editing, Data curation. ZL: Writing – review & editing, Methodology, Conceptualization, Supervision, Resources, Formal Analysis.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. The Hengrui Hebei Innovation and Development Medical Cooperation Program 412 (Grant No. R202502019). The Natural Science Foundation of Hebei Province (Grant No. H2023206356); the Natural Science Foundation of Hebei Province (Grant No. H2024206427); the Central Government Guides Local Science and Technology Development Fund, specifically under the Science and Technology Innovation Base Project (Project No. 236Z7756G); the Excellent Clinical Medicine Talent Training Project funded by the Hebei Provincial Government in 2024 (Grant No. ZF2024040); the Excellent Clinical Medicine Talent Training Project funded by the Hebei Provincial Government in 2025 (Grant No. ZF2025102).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. de Visser KE and Joyce JA. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell. (2023) 41:374–403. doi: 10.1016/j.ccell.2023.02.016
2. Bantug GR and Hess C. The immunometabolic ecosystem in cancer. Nat Immunol. (2023) 24:2008–20. doi: 10.1038/s41590-023-01675-y
3. Saini H, Rahmani Eliato K, Veldhuizen J, Zare A, Allam M, Silva C, et al. The role of tumor-stroma interactions on desmoplasia and tumorigenicity within a microengineered 3D platform. Biomaterials. (2020) 247:119975. doi: 10.1016/j.biomaterials.2020.119975
4. Jing Y, Han Z, Zhang S, Liu Y, and Wei L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. (2011) 1:29. doi: 10.1186/2045-3701-1-29
5. Lorenzo-Martín LF, Broguiere N, Langer J, Tillard L, Nikolaev M, Coukos G, et al. Patient-derived mini-colons enable long-term modeling of tumor-microenviroment complexity. Nat Biotechnol. (2025) 43:727–36. doi: 10.1038/s41587-024-02301-4
6. Shah DD, Chorawala MR, Raghani NR, Patel R, Fareed M, Kashid VA, et al. Tumor microenvironment: recent advances in understanding and its role in modulating cancer therapies. Med Oncol. (2025) 42:117. doi: 10.1007/s12032-025-02641-4
7. Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ Myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. (2020) 10:232–53. doi: 10.1158/2159-8290
8. Longo SK, Guo MG, Ji AL, and Khavari PA. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet. (2021) 22:627–44. doi: 10.1038/s41576-021-00370-8
9. Yuan Y. Spatial heterogeneity in the tumor microenvironment. Cold Spring Harb Perspect Med. (2016) 6:a026583. doi: 10.1101/cshperspect.a026583
10. Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, et al. A single-cell atlas of the tumor and immune ecosystem of human breast cancer. Cell. (2019) 177:1330–1345.e18. doi: 10.1016/j.cell.2019.03.005
11. Ibrahim AM, Moss MA, Gray Z, Rojo MD, Burke CM, Schwertfeger KL, et al. Diverse macrophage populations contribute to the inflammatory microenvironment in premalignant lesions during localized invasion. Front Oncol. (2020) 10:569985. doi: 10.3389/fonc.2020.569985
12. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. (2018) 33:463–479.e10. doi: 10.1016/j.ccell.2018.01.011
13. Ghahremanifard P, Chanda A, Bonni S, and Bose P. TGF-β Mediated immune evasion in cancer-spotlight on cancer-associated fibroblasts. Cancers (Basel). (2020) 12:3650. doi: 10.3390/cancers12123650
14. Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
15. Elia I and Haigis MC. Metabolites and the tumour microenvironment: from cellular mechanisms to systemic metabolism. Nat Metab. (2021) 3:21–32. doi: 10.1038/s42255-020-00317-z
16. Cao Z, Quazi S, Arora S, Osellame LD, Burvenich IJ, Janes PW, et al. Cancer-associated fibroblasts as therapeutic targets for cancer: advances, challenges, and future prospects. J BioMed Sci. (2025) 32:7. doi: 10.1186/s12929-024-01099-2
17. Lu Q, Kou D, Lou S, Ashrafizadeh M, Aref AR, Canadas I, et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J Hematol Oncol. (2024) 17:16. doi: 10.1186/s13045-024-01535-8
18. Marangoni F, Zhakyp A, Corsini M, Geels SN, Carrizosa E, Thelen M, et al. Expansion of tumor-associated Treg cells upon disruption of a CTLA-4-dependent feedback loop. Cell. (2021) 184:3998–4015.e19. doi: 10.1016/j.cell.2021.05.027
19. Chen L, Huang H, Zheng X, Li Y, Chen J, Tan B, et al. IL1R2 increases regulatory T cell population in the tumor microenvironment by enhancing MHC-II expression on cancer-associated fibroblasts. J Immunother Cancer. (2022) 10:e004585. doi: 10.1136/jitc-2022-004585
20. Tauriello DVF, Sancho E, and Batlle E. Overcoming TGFβ-mediated immune evasion in cancer. Nat Rev Cancer. (2022) 22:25–44. doi: 10.1038/s41568-021-00413-6
21. Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat Commun. (2022) 13:1742. doi: 10.1038/s41467-022-29366-6
22. Fang H and Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. (2013) 73:4965–77. doi: 10.1158/0008-5472
23. Papalexi E and Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. (2018) 18:35–45. doi: 10.1038/nri.2017.76
24. Li Y, Zhang J, Gao X, and Zhang QC. Tissue module discovery in single-cell-resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding. Cell Syst. (2024) 15:578–592.e7. doi: 10.1016/j.cels.2024.05.001
25. Moncada R, Barkley D, Wagner F, Chiodin M, Devlin JC, Baron M, et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol. (2020) 38:333–42. doi: 10.1038/s41587-019-0392-8
26. Zhao E, Stone MR, Ren X, Guenthoer J, Smythe KS, Pulliam T, et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat Biotechnol. (2021) 39:1375–84. doi: 10.1038/s41587-021-00935-2
27. Burgess DJ. Spatial transcriptomics coming of age. Nat Rev Genet. (2019) 20:317. doi: 10.1038/s41576-019-0129-z
28. JLee JH, Daugharthy ER, Scheiman J, Kalhor R, Yang JL, Ferrante TC, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. (2014) 343:1360–3. doi: 10.1126/science.1250212
29. Hunter MV, Moncada R, Weiss JM, Yanai I, and White RM. Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface. Nat Commun. (2021) 12:6278. doi: 10.1038/s41467-021-26614-z
30. Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. (2016) 353:78–82. doi: 10.1126/science.aaf2403
31. Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. (2019) 363:1463–7. doi: 10.1126/science.aaw1219
32. Li B, Zhang W, Guo C, Xu H, Li L, Fang M, et al. Benchmarking spatial and single-cell transcriptomics integration methods for transcript distribution prediction and cell type deconvolution. Nat Methods. (2022) 19:662–70. doi: 10.1038/s41592-022-01480-9
33. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. (2014) 344:1396–401. doi: 10.1126/science.1254257
34. Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. (2016) 539:309–13. doi: 10.1038/nature20123
35. Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. (2016) 352:189–96. doi: 10.1126/science.aad0501
36. Venteicher AS, Tirosh I, Hebert C, Yizhak K, Neftel C, Filbin MG, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. (2017) 355:eaai8478. doi: 10.1126/science.aai8478
37. Darmanis S, Sloan SA, Croote D, Mignardi M, Chernikova S, Samghababi P, et al. Single-cell RNA-seq analysis of infiltrating neoplastic cells at the migrating front of human glioblastoma. Cell Rep. (2017) 21:1399–410. doi: 10.1016/j.celrep.2017.10.030
38. Chung W, Eum HH, Lee HO, Lee KM, Lee HB, Kim KT, et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat Commun. (2017) 8:15081. doi: 10.1038/ncomms15081
39. Horning AM, Wang Y, Lin CK, Louie AD, Jadhav RR, Hung CN, et al. Single-cell RNA-seq reveals a subpopulation of prostate cancer cells with enhanced cell-cycle-related transcription and attenuated androgen response. Cancer Res. (2018) 78:853–64. doi: 10.1158/0008-5472.CAN-17-1924
40. Achim K, Pettit JB, Saraiva LR, Gavriouchkina D, Larsson T, Arendt D, et al. High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nat Biotechnol. (2015) 33:503–9. doi: 10.1038/nbt.3209
41. Chen KH, Boettiger AN, Moffitt JR, Wang S, and Zhuang X. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. (2015) 348:aaa6090. doi: 10.1126/science.aaa6090
42. Satija R, Farrell JA, Gennert D, Schier AF, and Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. (2015) 33:495–502. doi: 10.1038/nbt.3192
43. Lubeck E, Coskun AF, Zhiyentayev T, Ahmad M, and Cai L. Single-cell in situ RNA profiling by sequential hybridization. Nat Methods. (2014) 11:360–1. doi: 10.1038/nmeth.2892
44. Shah S, Lubeck E, Zhou W, and Cai L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron. (2016) 92:342–57. doi: 10.1016/j.neuron
45. Eng CL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. (2019) 568:235–9. doi: 10.1038/s41586-019-1049-y
46. Diez-Roux G, Banfi S, Sultan M, Geffers L, Anand S, Rozado D, et al. A high-resolution anatomical atlas of the transcriptome in the mouse embryo. PloS Biol. (2011) 9:e1000582. doi: 10.1371/journal.pbio.1000582
47. Poovathingal S, Davie K, Borm LE, Vandepoel R, Poulvellarie N, Verfaillie A, et al. Nova-ST: Nano-patterned ultra-dense platform for spatial transcriptomics. Cell Rep Methods. (2024) 4:100831. doi: 10.1016/j.crmeth.2024.100831
48. Ortega-Batista A, Jaén-Alvarado Y, Moreno-Labrador D, Gómez N, García G, and Guerrero EN. Single-cell sequencing: genomic and transcriptomic approaches in cancer cell biology. Int J Mol Sci. (2025) 26:2074. doi: 10.3390/ijms26052074
49. Zilionis R, Engblom C, Pfirschke C, Savova V, Zemmour D, Saatcioglu HD, et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. (2019) 50:1317–1334.e10. doi: 10.1016/j.immuni.2019.03.009
50. Rohlenova K, Goveia J, García-Caballero M, Subramanian A, Kalucka J, Treps L, et al. Single-cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab. (2020) 31:862–877.e14. doi: 10.1016/j.cmet.2020.03.009
51. Zhuang X. Spatially resolved single-cell genomics and transcriptomics by imaging. Nat Methods. (2021) 18:18–22. doi: 10.1038/s41592-020-01037-8
52. Haque A, Engel J, Teichmann SA, and Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. (2017) 9:75. doi: 10.1186/s13073-017-0467-4
53. Lafzi A, Moutinho C, Picelli S, and Heyn H. Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies. Nat Protoc. (2018) 13:2742–57. doi: 10.1038/s41596-018-0073-y
54. Cheng M, Jiang Y, Xu J, Mentis AA, Wang S, Zheng H, et al. Spatially resolved transcriptomics: a comprehensive review of their technological advances, applications, and challenges. J Genet Genomics. (2023) 50:625–40. doi: 10.1016/j.jgg.2023.03.011
55. Strell C, Hilscher MM, Laxman N, Svedlund J, Wu C, Yokota C, et al. Placing RNA in context and space - methods for spatially resolved transcriptomics. FEBS J. (2019) 286:1468–81. doi: 10.1111/febs.14435
56. Bandyopadhyay S, Duffy MP, Ahn KJ, Sussman JH, Pang M, Smith D, et al. Mapping the cellular biogeography of human bone marrow niches using single-cell transcriptomics and proteomic imaging. Cell. (2024) 187:3120–3140.e29. doi: 10.1016/j.cell.2024.04.013
57. Qian J, Liao J, Liu Z, Chi Y, Fang Y, Zheng Y, et al. Reconstruction of the cell pseudo-space from single-cell RNA sequencing data with scSpace. Nat Commun. (2023) 14:2484. doi: 10.1038/s41467-023-38121-4
58. Kleshchevnikov V, Shmatko A, Dann E, Aivazidis A, King HW, Li T, et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat Biotechnol. (2022) 40:661–71. doi: 10.1038/s41587-021-01139-4
59. Biancalani T, Scalia G, Buffoni L, Avasthi R, Lu Z, Sanger A, et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat Methods. (2021) 18:1352–62. doi: 10.1038/s41592-021-01264-7
60. Liu Y, Yang M, Deng Y, Su G, Enninful A, Guo CC, et al. High-spatial-resolution multi-omics sequencing via deterministic barcoding in tissue. Cell. (2020) 183:1665–1681.e18. doi: 10.1016/j.cell.2020.10.026
61. Zhang L, Chen D, Song D, Liu X, Zhang Y, Xu X, et al. Clinical and translational values of spatial transcriptomics. Signal Transduct Target Ther. (2022) 7:111. doi: 10.1038/s41392-022-00960-w
62. Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. (2022) 185:1777–1792.e21. doi: 10.1016/j.cell.2022.04.003
63. Wu Y, Korobeynyk VI, Zamboni M, Waern F, Cole JD, Mundt S, et al. Multimodal transcriptomics reveal neurogenic aging trajectories and age-related regional inflammation in the dentate gyrus. Nat Neurosci. (2025) 28:415–30. doi: 10.1038/s41593-024-01848-4
64. Zhao T, Chiang ZD, Morriss JW, LaFave LM, Murray EM, Del Priore I, et al. Spatial genomics enables multi-modal study of clonal heterogeneity in tissues. Nature. (2022) 601:85–91. doi: 10.1038/s41586-021-04217-4
65. Wang F, Long J, Li L, Wu ZX, Da TT, Wang XQ, et al. Single-cell and spatial transcriptome analysis reveals the cellular heterogeneity of liver metastatic colorectal cancer. Sci Adv. (2023) 9:eadf5464. doi: 10.1126/sciadv.adf5464
66. Wang Y, Qiu X, Li Q, Qin J, Ye L, Zhang X, et al. Single-cell and spatial-resolved profiling reveals cancer-associated fibroblast heterogeneity in colorectal cancer metabolic subtypes. J Transl Med. (2025) 23:175. doi: 10.1186/s12967-025-06103-3
67. Ping S, Jia X, and Tian Y. Integration of scRNA-seq and ST-seq identifies hyperproliferative RRM2+ cells features and therapeutic targets in gastric cancer. J Transl Med. (2025) 23:795. doi: 10.1186/s12967-025-06847-y
68. Xiao M, Deng Y, Guo H, Ren Z, He Y, Ren X, et al. Single-cell and spatial transcriptomics profile the interaction of SPP1+ macrophages and FAP+ fibroblasts in non-small cell lung cancer. Transl Lung Cancer Res. (2025) 14:2646–69. doi: 10.21037/tlcr-2025-244
69. De Zuani M, Xue H, Park JS, Dentro SC, Seferbekova Z, Tessier J, et al. Single-cell and spatial transcriptomics analysis of non-small cell lung cancer. Nat Commun. (2024) 15:4388. doi: 10.1038/s41467-024-48700-8
70. Chen C, Guo Q, Liu Y, Hou Q, Liao M, Guo Y, et al. Single-cell and spatial transcriptomics reveal POSTN+ cancer-associated fibroblasts correlated with immune suppression and tumour progression in non-small cell lung cancer. Clin Transl Med. (2023) 13:e1515. doi: 10.1002/ctm2.1515
71. Chen J, Song Y, Huang J, Wan X, and Li Y. Integrated single-cell RNA sequencing and spatial transcriptomics analysis reveals the tumour microenvironment in patients with endometrial cancer responding to anti-PD-1 treatment. Clin Transl Med. (2024) 14:e1668. doi: 10.1002/ctm2.1668
72. Liu Y, Geng R, Zhao S, Yang J, Liu J, Zheng Y, et al. Single-cell and spatial transcriptomics explore purine metabolism-related prognostic risk model and tumor immune microenvironment modulation in ovarian cancer. Hum Mutat. (2025) 2025:5530325. doi: 10.1155/humu/5530325
73. Liu YM, Ge JY, Chen YF, Liu T, Chen L, Liu CC, et al. Combined single-cell and spatial transcriptomics reveal the metabolic evolvement of breast cancer during early dissemination. Adv Sci (Weinh). (2023) 10:e2205395. doi: 10.1002/advs.202205395
74. Xie Z, Peng S, Wang J, Huang Y, Zhou X, Zhang G, et al. Multi-omics analysis reveals the role of ribosome biogenesis in Malignant clear cell renal cell carcinoma and the development of a machine learning-based prognostic model. Front Immunol. (2025) 16:1602898. doi: 10.3389/fimmu.2025.1602898
75. Jiang L, Ren X, Yang J, Chen H, Zhang S, Zhou X, et al. Mitophagy and clear cell renal cell carcinoma: insights from single-cell and spatial transcriptomics analysis. Front Immunol. (2024) 15:1400431. doi: 10.3389/fimmu.2024.1400431
76. Hirz T, Mei S, Sarkar H, Kfoury Y, Wu S, Verhoeven BM, et al. Dissecting the immune suppressive human prostate tumor microenvironment via integrated single-cell and spatial transcriptomic analyses. Nat Commun. (2023) 14:663. doi: 10.1038/s41467-023-36325-2
77. Shi ZD, Sun Z, Zhu ZB, Liu X, Chen JZ, Hao L, et al. Integrated single-cell and spatial transcriptomic profiling reveals higher intratumour heterogeneity and epithelial-fibroblast interactions in recurrent bladder cancer. Clin Transl Med. (2023) 13:e1338. doi: 10.1002/ctm2.1338
78. Liu Y, Dong G, Yu J, and Liang P. Integration of single-cell and spatial transcriptomics reveals fibroblast subtypes in hepatocellular carcinoma: spatial distribution, differentiation trajectories, and therapeutic potential. J Transl Med. (2025) 23:198. doi: 10.1186/s12967-025-06192-0
79. Wu Y, Li S, Yu H, Zhang S, Yan L, Guan X, et al. Integrative single-cell and spatial transcriptomics analysis reveals ECM-remodeling cancer-associated fibroblast-derived POSTN as a key mediator in pancreatic ductal adenocarcinoma progression. Int J Biol Sci. (2025) 21:3573–96. doi: 10.7150/ijbs.108618
80. Yang J, Xu Q, and Lu Y. Decoding epithelial-fibroblast interactions in lung adenocarcinoma through single-cell and spatial transcriptomics. J Cancer Res Clin Oncol. (2025) 151:221. doi: 10.1007/s00432-025-06250-6
81. Cang Z and Nie Q. Inferring Spatial and signaling relationships between cells from single cell transcriptomic data. Nat Commun. (2020) 11:2084. doi: 10.1038/s41467-020-15968-5
82. O’Donnell JS, Teng MWL, and Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
83. Keren L, Bosse M, Marquez D, Angoshtari R, Jain S, Varma S, et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell. (2018) 174:1373–1387.e19. doi: 10.1016/j.cell.2018.08.039
84. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
85. Ping Y, Shan J, Qin H, Li F, Qu J, Guo R, et al. PD-1 signaling limits expression of phospholipid phosphatase 1 and promotes intratumoral CD8+ T cell ferroptosis. Immunity. (2024) 57:2122–2139.e9. doi: 10.1016/j.immuni
86. Tysoe O. FGF21 suppresses CD8+ T cell antitumour activity. Nat Rev Endocrinol. (2024) 20:191. doi: 10.1038/s41574-024-00964-2
87. Cheng J, Xiao Y, Peng T, Zhang Z, Qin Y, Wang Y, et al. ETV7 limits the antiviral and antitumor efficacy of CD8+ T cells by diverting their fate toward exhaustion. Nat Cancer. (2025) 6:338–56. doi: 10.1038/s43018-024-00892-0
88. Hu C, Qiao W, Li X, Ning ZK, Liu J, Dalangood S, et al. Tumor-secreted FGF21 acts as an immune suppressor by rewiring cholesterol metabolism of CD8+T cells. Cell Metab. (2024) 36:1168. doi: 10.1016/j.cmet.2024.03.013
89. Chen L, Sun R, Xu J, Zhai W, Zhang D, Yang M, et al. Tumor-derived IL33 promotes tissue-resident CD8+ T cells and is required for checkpoint blockade tumor immunotherapy. Cancer Immunol Res. (2020) 8:1381–92. doi: 10.1158/2326-6066
90. Park JA, Espinosa-Cotton M, Guo HF, Monette S, and Cheung NV. Targeting tumor vasculature to improve antitumor activity of T cells armed ex vivo with T cell engaging bispecific antibody. J Immunother Cancer. (2023) 11:e006680. doi: 10.1136/jitc-2023-006680
91. Li C, Guo H, Zhai P, Yan M, Liu C, Wang X, et al. Spatial and single-cell transcriptomics reveal a cancer-associated fibroblast subset in HNSCC that restricts infiltration and anti-tumor activity of CD8+ T cells. Cancer Res. (2024) 84:258–75. doi: 10.1158/0008-5472
92. Fan Q, Wang Y, Cheng J, Pan B, Zang X, Liu R, et al. Single-cell RNA-seq reveals T cell exhaustion and immune response landscape in osteosarcoma. Front Immunol. (2024) 15:1362970. doi: 10.3389/fimmu.2024.1362970
93. Barras D, Ghisoni E, Chiffelle J, Orcurto A, Dagher J, Fahr N, et al. Response to tumor-infiltrating lymphocyte adoptive therapy is associated with preexisting CD8+ T-myeloid cell networks in melanoma. Sci Immunol. (2024) 9:eadg7995. doi: 10.1126/sciimmunol.adg7995
94. Xun Z, Ding X, Zhang Y, Zhang B, Lai S, Zou D, et al. Reconstruction of the tumor spatial microenvironment along the Malignant-boundary-nonmalignant axis. Nat Commun. (2023) 14:933. doi: 10.1038/s41467-023-36560-7
95. Cachot A, Bilous M, Liu YC, Li X, Saillard M, Cenerenti M, et al. Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci Adv. (2021) 7:eabe3348. doi: 10.1126/sciadv.abe3348
96. Lubrano di Ricco M, Ronin E, Collares D, Divoux J, Grégoire S, Wajant H, et al. Tumor necrosis factor receptor family costimulation increases regulatory T-cell activation and function via NF-κB. Eur J Immunol. (2020) 50:972–85. doi: 10.1002/eji.201948393
97. Mantovani A, Sozzani S, Locati M, Allavena P, and Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. (2002) 23:549–55. doi: 10.1016/s1471-4906(02)02302-5
98. Chen D, Xie J, Fiskesund R, Dong W, Liang X, Lv J, et al. Chloroquine modulates antitumor immune response by resetting tumor-associated macrophages toward m1 phenotype. Nat Commun. (2018) 9:873. doi: 10.1038/s41467-018-03225-9
99. Pathria P, Louis TL, and Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. (2019) 40:310–27. doi: 10.1016/j.it.2019.02.003
100. Wu Y, Yang S, Ma J, Chen Z, Song G, Rao D, et al. Spatiotemporal immune landscape of colorectal cancer liver metastasis at single-cell level. Cancer Discov. (2022) 12:134–53. doi: 10.1158/2159-8290
101. Wu SZ, Al-Eryani G, Roden DL, Junankar S, Harvey K, Andersson A, et al. A single-cell and spatially resolved atlas of human breast cancers. Nat Genet. (2021) 53:1334–47. doi: 10.1038/s41588-021-00911-1
102. Li R, Ferdinand JR, Loudon KW, Bowyer GS, Laidlaw S, Muyas F, et al. Mapping single-cell transcriptomes in the intratumoral and associated territories of kidney cancer. Cancer Cell. (2022) 40:1583–1599.e10. doi: 10.1016/j.ccell.2022.11.001
103. Aggen DH, Ager CR, Obradovic AZ, Chowdhury N, Ghasemzadeh A, Mao W, et al. Blocking IL1 beta promotes tumor regression and remodeling of the myeloid compartment in a renal cell carcinoma model:multidimensional analyses. Clin Cancer Res. (2021) 27:608–21. doi: 10.1158/1078-0432.CCR-20-1610
104. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, and Glynn RJ. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet. (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
105. Lee JJ, Bernard V, Semaan A, Monberg ME, Huang J, Stephens BM, et al. Elucidation of tumor-stromal heterogeneity and the ligand-receptor interactome by single-cell transcriptomics in real-world pancreatic cancer biopsies. Clin Cancer Res. (2021) 27:5912–21. doi: 10.1158/1078-0432
106. Huang P, Zhou X, Zheng M, Yu Y, Jin G, and Zhang S. Regulatory T cells are associated with the tumor immune microenvironment and immunotherapy response in triple-negative breast cancer. Front Immunol. (2023) 14:1263537. doi: 10.3389/fimmu.2023.1263537
107. Zhang H, AbdulJabbar K, Moore DA, Akarca A, Enfield KSS, Jamal-Hanjani M, et al. Spatial positioning of immune hotspots reflects the interplay between B and T cells in lung squamous cell carcinoma. Cancer Res. (2023) 83:1410–25. doi: 10.1158/0008-5472.Can-22-2589
108. Zhang Y, Gong S, and Liu X. Spatial transcriptomics: A new frontier in accurate localization of breast cancer diagnosis and treatment. Front Immunol. (2024) 15:1483595. doi: 10.3389/fimmu.2024.1483595
109. Mao X, Zhou D, Lin K, Zhang B, Gao J, Ling F, et al. Single-cell and spatial transcriptome analyses revealed cell heterogeneity and immune environment alternations in metastatic axillary lymph nodes in breast cancer. Cancer Immunol Immunother. (2023) 72:679–95. doi: 10.1007/s00262-022-03278-2
110. Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, et al. Single-cell analyses reveal key immune cell subsets associated with response to pd-L1 blockade in triple-negative breast cancer. Cancer Cell. (2021) 39:1578–93.e8. doi: 10.1016/j.ccell.2021.09.010
111. Ding S, Qiao N, Zhu Q, Tong Y, Wang S, Chen X, et al. Single-Cell Atlas Reveals a Distinct Immune Profile Fostered by T cell-B cell Crosstalk in Triple Negative Breast Cancer. Cancer Commun (Lond). (2023) 43:661–84. doi: 10.1002/cac2.12429
112. Huang F, Wang F, Hu Q, Li Y, and Jiang D. Ptgr1-mediated immune evasion mechanisms in late-stage triple-negative breast cancer: mechanisms of M2 macrophage infiltration and cd8+ T cell suppression. Apoptosis. (2024) 29:2002–24. doi: 10.1007/s10495-024-01991-0
113. Liu F, Zhang J, Gu X, Guo Q, and Guo W. Single-cell transcriptome sequencing reveals spp1-cd44-mediated macrophage–tumor cell interactions drive chemoresistance in tnbc. J Cell Mol Med. (2024) 28:e18525. doi: 10.1111/jcmm.18525
114. Omilian AR, Cannioto R, Mendicino L, Stein L, Bshara W, Qin B, et al. Cd163(+) macrophages in the triple-negative breast tumor microenvironment are associated with improved survival in the women’s circle of health study and the women’s circle of health follow-up study. Breast Cancer Res. (2024) 26:75. doi: 10.1186/s13058-024-01831-8
115. Cords L, Engler S, Haberecker M, Rüschoff JH, Moch H, de Souza N, et al. Cancer-associated fibroblast phenotypes are associated with patient outcome in non-small cell lung cancer. Cancer Cell. (2024) 42:396–412.e5. doi: 10.1016/j.ccell
116. Zhang X, Ren B, Liu B, Wang R, Li S, Zhao Y, et al. Single-cell RNA sequencing and spatial transcriptomics reveal the heterogeneity and intercellular communication of cancer-associated fibroblasts in gastric cancer. J Transl Med. (2025) 23:344. doi: 10.1186/s12967-025-06376-8
117. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. (2020) 10:1330–51. doi: 10.1158/2159-8290.CD-19-1384
118. Pellinen T, Paavolainen L, MartÌn-BernabÈ A, Papatella Araujo R, Strell C, Mezheyeuski A, et al. Fibroblast subsets in non-small cell lung cancer: associations with survival, mutations, and immune features. J Natl Cancer Inst. (2023) 115:71–82. doi: 10.1093/jnci/djac17
119. Grout JA, Sirven P, Leader AM, Maskey S, Hector E, Puisieux I, et al. Spatial positioning and matrix programs of cancer-associated fibroblasts promote T-cell exclusion in human lung tumors. Cancer Discov. (2022) 12:2606–25. doi: 10.1158/2159-8290.CD-21-1714
120. Obradovic AZ, Dallos MC, Zahurak ML, Partin AW, Schaeffer EM, Ross AE, et al. T-cell infiltration and adaptive Treg resistance in response to androgen deprivation with or without vaccination in localized prostate cancer. Clin Cancer Res. (2020) 26:3182–92. doi: 10.1158/1078-0432.CCR-19-3372
121. Gazinska P, Milton C, Iacovacci J, Ward J, Buus R, Alaguthurai T, et al. Dynamic changes in the nk-, neutrophil-, and B-cell immunophenotypes relevant in high metastatic risk post neoadjuvant chemotherapy-resistant early breast cancers. Clin Cancer Res. (2022) 28:4494–508. doi: 10.1158/1078-0432.Ccr-22-0543
122. Toney NJ, Opdenaker LM, Cicek K, Frerichs L, Kennington CR, Oberly S, et al. Tumor-B-cell interactions promote isotype switching to an immunosuppressive igg4 antibody response through upregulation of il-10 in triple negative breast cancers. J Transl Med. (2022) 20:112. doi: 10.1186/s12967-022-03319-5
123. Ishimoto T, Miyake K, Nandi T, Yashiro M, Onishi N, Huang KK, et al. Activation of transforming growth factor beta 1 signaling in gastric cancer-associated fibroblasts increases their motility, via expression of rhomboid 5 homolog 2, and ability to induce invasiveness of gastric cancer cells. Gastroenterology. (2017) 153:191–204.e16. doi: 10.1053/j.gastro.2017.03.046
124. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
125. Zhi K, Shen X, Zhang H, and Bi J. Cancer-associated fibroblasts are positively correlated with metastatic potential of human gastric cancers. J Exp Clin Cancer Res. (2010) 29:66. doi: 10.1186/1756-9966-29-66
126. LeBleu VS and Kalluri R. A peek into cancer-associated fibroblasts: origins, functions and translational impact. Dis Model Mech. (2018) 11:dmm029447. doi: 10.1242/dmm.029447
127. Affo S, Nair A, Brundu F, Ravichandra A, Bhattacharjee S, Matsuda M, et al. Promotion of cholangiocarcinoma growth by diverse cancer-associated fibroblast subpopulations. Cancer Cell. (2021) 39:883. doi: 10.1016/j.ccell.2021.05.010
128. Martin-Serrano MA, Kepecs B, Torres-Martin M, Bramel ER, Haber PK, Merritt E, et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut. (2023) 72:736–48. doi: 10.1136/gutjnl-2021-326514
129. Zhang M, Yang H, Wan L, Wang Z, Wang H, Ge C, et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J Hepatol. (2020) 73:1118–30. doi: 10.1016/j.jhep.2020.05.039
130. Jain S, Rick JW, Joshi RS, Beniwal A, Spatz J, Gill S, et al. Single-cell RNA sequencing and spatial transcriptomics reveal cancer-associated fibroblasts in glioblastoma with protumoral Effect. J Clin Invest. (2023) 133:e147087. doi: 10.1172/JCI147087
131. Chen D, Zhang X, Li Z, and Zhu B. Metabolic regulatory crosstalk between tumor microenvironment and tumor-associated macrophages. Theranostics. (2021) 11:1016–30. doi: 10.7150/thno.51777
132. Wu JY, Huang TW, Hsieh YT, Wang YF, Yen CC, Lee GL, et al. Cancer-derived succinate promotes macrophage polarization and cancer metastasis via succinate receptor. Mol Cell. (2020) 77:213–227.e5. doi: 10.1016/j.molcel.2019.10.023
133. Guo W, Zhou B, Yang Z, Liu X, Huai Q, Guo L, et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-sequencing reveals tissue architecture in esophageal squamous cell carcinoma. EBioMedicine. (2022) 84:104281. doi: 10.1016/j.ebiom.2022.104281
134. Farin HF, Mosa MH, Ndreshkjana B, Grebbin BM, Ritter B, Menche C, et al. Colorectal cancer organoid-stroma biobank allows subtype-specific assessment of individualized therapy responses. Cancer Discov. (2023) 13:2192–211. doi: 10.1158/2159-8290
135. Huang Z, Cong Z, Luo J, Qiu B, Wang K, Gao C, et al. Association between cancer-associated fibroblasts and prognosis of neoadjuvant chemoradiotherapy in esophageal squamous cell carcinoma: a bioinformatics analysis based on single-cell RNA sequencing. Cancer Cell Int. (2025) 25:74. doi: 10.1186/s12935-025-03709-x
136. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. (2017) 214:579–96. doi: 10.1084/jem.20162024
137. Pelka K, Hofree M, Chen JH, Sarkizova S, Pirl JD, Jorgji V, et al. Spatially organized multicellular immune hubs in human colorectal cancer. Cell. (2021) 184:4734–4752.e20. doi: 10.1016/j.cell.2021.08.003
138. Righetti A, Giulietti M, Šabanović B, Occhipinti G, Principato G, and Piva F. CXCL12 and its isoforms: different roles in pancreatic cancer? J Oncol. (2019) 2019:9681698. doi: 10.1155/2019/9681698
139. Zhang Y, Guan XY, and Jiang P. Cytokine and chemokine signals of T-cell exclusion in tumors. Front Immunol. (2020) 11:594609. doi: 10.3389/fimmu.2020.594609
140. Qin P, Chen H, Wang Y, Huang L, Huang K, Xiao G, et al. Cancer-associated fibroblasts undergoing neoadjuvant chemotherapy suppress rectal cancer revealed by single-cell and spatial transcriptomics. Cell Rep Med. (2023) 4:101231. doi: 10.1016/j.xcrm.2023.101231
141. Hwang WL, Jagadeesh KA, Guo JA, Hoffman HI, Yadollahpour P, Reeves JW, et al. Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment. Nat Genet. (2022) 54:1178–91. doi: 10.1038/s41588-022-01134-8
142. Saviano A, Henderson NC, and Baumert TF. Single-cell genomics and spatial transcriptomics: Discovery of novel cell states and cellular interactions in liver physiology and disease biology. J Hepatol. (2020) 73:1219–30. doi: 10.1016/j.jhep.2020.06.004
143. Stuart T and Satija R. Integrative single-cell analysis. Nat Rev Genet. (2019) 20:257–72. doi: 10.1038/s41576-019-0093-7
144. Kang M, Ko E, and Mersha TB. A roadmap for multi-omics data integration using deep learning. Brief Bioinform. (2022) 23:bbab454. doi: 10.1093/bib/bbab454
145. Zhou Y, Bian S, Zhou X, Cui Y, Wang W, Wen L, et al. Single-cell multiomics sequencing reveals prevalent genomic alterations in tumor stromal cells of human colorectal cancer. Cancer Cell. (2020) 38:818–828.e5. doi: 10.1016/j.ccell.2020.09.015
146. Leader AM, Grout JA, Maier BB, Nabet BY, Park MD, Tabachnikova A, et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell. (2021) 39:1594–1609.e12. doi: 10.1016/j.ccell.2021.10.009
147. Han M, Li F, Zhang Y, Dai P, He J, Li Y, et al. FOXA2 drives lineage plasticity and KIT pathway activation in neuroendocrine prostate cancer. Cancer Cell. (2022) 40:1306–1323.e8. doi: 10.1016/j.ccell.2022.10.011
148. Zhao M, He W, Tang J, Zou Q, and Guo F. A hybrid deep learning framework for gene regulatory network inference from single-cell transcriptomic data. Brief Bioinform. (2022) 23:bbab568. doi: 10.1093/bib/bbab568
Keywords: spatial transcriptomics, single-cell RNA sequencing, tumor microenvironment, cancer heterogeneity, intercellular communication
Citation: Shi W, Zhang Z, Xu X, Tian Y, Feng L, Huang X, Du Y and Li Z (2025) Single-cell and spatial transcriptomics integration: new frontiers in tumor microenvironment and cellular communication. Front. Immunol. 16:1649468. doi: 10.3389/fimmu.2025.1649468
Received: 18 June 2025; Accepted: 16 September 2025;
Published: 02 October 2025.
Edited by:
Jiaheng Xie, Central South University, ChinaReviewed by:
Erdong Wei, University of Minnesota Twin Cities, United StatesAnand Kamal Singh, University of Texas MD Anderson Cancer Center, United States
Copyright © 2025 Shi, Zhang, Xu, Tian, Feng, Huang, Du and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhongkang Li, emhvbmdrYW5nbGlAaGVibXUuZWR1LmNu; Yanfang Du, ZHV5YW5mYW5nMTk3M0AxNjMuY29t