 Lei Shi1,2,3†
Lei Shi1,2,3† Qiheng Qian2†Jiding Xie3†Taoshuo Yang4Xinyu Zhao5Xiangqi Meng2*Jingang Dai3*Qiguan Jin1*
Qiheng Qian2†Jiding Xie3†Taoshuo Yang4Xinyu Zhao5Xiangqi Meng2*Jingang Dai3*Qiguan Jin1*- 1College of Physical Education, Yangzhou University, Yangzhou, Jiangsu, China
- 2Suzhou Hospital of Traditional Chinese Medicine, Suzhou, Jiangsu, China
- 3Experimental Research Center, China Academy of Chinese Medical Sciences, Beijing, China
- 4College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, United Kingdom
- 5Xuzhou Hospital of Traditional Chinese Medicine, Xuzhou, Jiangsu, China
Traumatic spinal cord injury (TSCI) is a devastating neurological condition with limited therapeutic options and a high likelihood of permanent disability. Among the multifaceted secondary injury mechanisms triggered by TSCI, pyroptosis—an inflammatory form of programmed cell death—has emerged as a key pathological process. In particular, microglial pyroptosis plays a pivotal role in exacerbating neuroinflammation and disrupting tissue homeostasis, thereby amplifying the secondary injury cascade. This review provides a comprehensive overview of the molecular pathways mediating microglial pyroptosis, including canonical (NLRP3–caspase-1–GSDMD), non-canonical (caspase-11–GSDMD), and atypical (caspase-3/8–GSDME/GSDMC) signaling. We also examine recent therapeutic strategies aimed at suppressing microglial pyroptosis—such as extracellular vesicle-based delivery systems, small-molecule compounds, and gene-targeted approaches—and assess their potential to enhance neurological and motor recovery following SCI. By elucidating both the pathological significance and therapeutic promise of microglial pyroptosis, this review offers novel perspectives on its translational potential as a target for spinal cord injury intervention.
1 Introduction
Traumatic spinal cord injury (TSCI) refers to denotes an abrupt, often irreversible disruption of spinal parenchyma precipitated by high-energy mechanical forces—such as falls, motor-vehicle collisions and sports trauma—which instantaneously destroy neurons, glia and the microvasculature, producing profound sensorimotor deficits and imposing substantial socioeconomic burdens (1, 2).
Pathologically, TSCI progresses through two distinct phases: primary injury and secondary injury (3). The primary injury arises directly from mechanical forces (e.g., compression, traction, laceration) that cause irreversible structural damage to spinal cord tissue (4). In contrast, secondary injury initiates rapidly after the primary insult and involves a complex, sustained cascade of pathophysiological events, including neuroinflammation, oxidative stress, excitotoxicity, apoptosis, pyroptosis, edema, and disruption of the blood-spinal cord barrier (BSCB). The progression of secondary injury can last from hours to weeks or longer, typically divided into acute (hours to 3 days), subacute (3 days to 2 weeks), and chronic (weeks to months) phases (5). Each phase may exacerbate the initial damage and impair long-term functional recovery. Notably, compared to the irreversible nature of primary structural damage, secondary injury exhibits greater plasticity and therapeutic potential, and timely interventions targeting secondary injury may reduce long-term neurological deficits.
Pyroptosis is a pro-inflammatory form of programmed cell death distinct from classical apoptosis (6). Its hallmark molecular features include the assembly of inflammasomes and activation of caspase-1, which cleaves substrates such as Gasdermin D (GSDMD) (7). The N-terminal fragment of GSDMD (GSDMD-NT) forms pores in the cell membrane, leading to rapid cell lysis and the release of cellular contents (8). Pyroptosis triggers the maturation and secretion of inflammatory mediators (e.g., IL-1β, IL-18), which induce intense local inflammation and exacerbate damage to adjacent cells.
Microglia, the resident immune cells of the central nervous system (CNS), play critical roles in immune surveillance, debris clearance, and synaptic pruning (9). Studies show that microglia are among the first cells to respond following spinal cord injury, undergoing morphological changes, migration, and phenotypic transformation to participate in inflammatory reactions at the injury site (10). Activated microglia exhibit a “double-edged sword” effect: they can promote repair by clearing necrotic debris and releasing neurotrophic factors, but may also aggravate the local inflammatory milieu through the secretion of pro-inflammatory cytokines such as TNF-α and IL-1β (11).
In the context of spinal cord injury, growing evidence highlights microglial pyroptosis as a pivotal event in secondary injury (12). As key contributors to post-injury inflammation, elevated pyroptosis in microglia is thought to worsen the neuroinflammatory environment (13). Numerous studies demonstrate significant upregulation of pyroptosis-related molecules (e.g., NLRP3 inflammasome components, cleaved GSDMD) in microglia after TSCI. Pyroptotic microglial death may also impair their beneficial roles in debris clearance and regenerative support. Thus, microglial pyroptosis is recognized as a critical link in the secondary injury cascade, profoundly impacting motor and neurological functional recovery.
2 Spinal cord injury and microglia
2.1 Spinal cord injury and secondary injury
During the acute phase of SCI, spinal cord ischemia, vasogenic edema and glutamate-mediated excitotoxicity inflict the primary insult, whereas neuroinflammation, mitochondrial dysfunction, overactive nitric-oxide-synthase (NOS), excessive apoptosis/necrosis, axonal degeneration and glial-scar formation synergistically hinder axonal remyelination and remodeling, ultimately dictating neurological prognosis (14). Minutes after trauma, an explosive inflammatory cascade releases damage-associated molecular patterns (DAMPs) that swiftly recruit and activate resident glia and peripheral immune cells within the CNS (15). Pro-inflammatory cytokines—IL-1β, IL-6 and TNF-α—rise steeply in tissue and cerebrospinal fluid within hours. Activated microglia and infiltrating macrophages are detectable in the parenchyma as early as 1 h, peak at 5–10 days and can persist for months (16). The diverse mediators released by these inflammatory cells collectively shape the secondary injury microenvironment, exacerbating pathological processes such as ischemia, edema, oxidative free radical accumulation, apoptosis, and pyroptosis (17). Timely curtailment of this cascade is therefore paramount for salvaging residual neural tissue and preserving function.
2.2 Activation of microglia and their associated roles in spinal cord injury
Microglia—the brain’s resident “sentinels” and “scavengers”—continually survey the parenchyma under homeostatic conditions (18). After SCI they are rapidly activated, becoming one of the earliest cellular responders (19). Within minutes-to-hours they enlarge, proliferate and migrate towards the lesion core. Activated microglia appear as early as 1 h, peak at 5–10 days and remain for weeks-to-months (20).
The activation state of microglia exhibits a dual nature: On one hand, excessive activation of microglia leads to the release of large amounts of pro-inflammatory mediators, exacerbating tissue damage (21). On the other hand, moderate activation facilitates debris clearance and secretion of neurotrophic factors, promoting tissue repair. Based on their activation states and functions, microglia are typically categorized into two phenotypes: the classically activated M1 phenotype and the alternatively activated M2 phenotype (22).
2.2.1 Activation states of microglia
M1 microglia predominate during the acute phase of SCI and exhibit pro-inflammatory and neurotoxic effects (23). They secrete high levels of inflammatory mediators, such as IL-6, IL-12, and IFN-γ (24, 25), which trigger inflammatory cascades in neighboring cells, leading to severe neuronal and glial cell death and demyelination. M1 microglia also generate excessive reactive oxygen species (ROS) and proteases, causing further tissue damage (26).
In contrast, M2 microglia exert anti-inflammatory and neuroprotective roles by releasing anti-inflammatory cytokines (e.g., IL-10, IL-4, TGF-β) and growth factors (27–29). These mediators suppress inflammation and promote tissue repair and axonal regeneration. However, recent studies emphasize that the M1/M2 classification represents a spectrum rather than a strict dichotomy (30). However, recent studies suggest that microglial classification represents a continuum rather than two extreme, polarized phenotypes. With the advancement of technologies such as single-cell sequencing and spatial transcriptomics, microglia are now classified into multiple functional subtypes based on molecular characteristics, each with distinct nomenclature (31).
Homeostatic microglia refer to the resident microglia that maintain CNS homeostasis under physiological conditions—traditionally described as the “resting” state. Their marker genes include P2RY12, TMEM119, CX3CR1, SIGLEC-H, and HEXB (30).
Interferon-responsive microglia exhibit gene signatures induced by type I interferon stimulation, typically observed in acute inflammation or viral infection. However, studies have shown that this phenotype also exists in healthy mice, with notable sex-specific differences—male mice predominantly exhibit the interferon-responsive profile (high expression of the male-specific gene Eif2s3y), whereas females retain a homeostatic phenotype (high expression of the female X-linked gene Xist) (32).
Disease-associated microglia (DAM) were first identified in neurodegenerative conditions, characterized by the upregulation of genes involved in phagocytosis and lipid metabolism, such as APOE, TREM2, CD11c/ITGAX, and CLEC7A, accompanied by downregulation of homeostatic genes. This subtype is associated with lipid dysregulation and impaired clearance function and is mainly observed in neurodegeneration, demyelinating diseases, and late-stage acute injuries.
Proliferative-region-associated microglia (PAMG) are detected in neurogenic niches during development and participate in clearing apoptotic cells and promoting neurogenesis. Wang et al. found that PAMGs appear predominantly in the early acute phase of SCI (within ~3 days), characterized by genes involved in cell proliferation and stress response. These cells can be further divided into two subclusters: PAMG1, which highly expresses cell cycle regulatory genes (e.g., Mcm3, Cdk1) to promote proliferation; and PAMG2, which upregulates genes related to oxidative stress and inflammation (e.g., Tlr2, Cd5l, Ifi204), suggesting a potential role in counteracting injury-induced oxidative environments.
Meanwhile, injury-associated microglia (IaMG) are prominently enriched during the subacute phase post-injury. These are mainly divided into IaMG1 and IaMG2, both expressing inflammation-related genes such as Stat1, Cst7, and Cybb. Notably, the IaMG2 subset also upregulates genes associated with angiogenesis and axon regeneration (e.g., Nrp2, Fn1, Cxcr4, Rab7b), indicating its potential role in tissue repair and axonal regrowth. This highlights that post-injury microglial subtypes are not functionally homogeneous (30).
In addition, other subtypes have been proposed based on disease models, such as glioma-associated microglia (GAM), post-stroke microglia, and Parkinson’s disease-associated microglia. Some studies have noted overlapping features and lineage connections among different subtypes (33), and evidence suggests that microglia in varying activation states can migrate between regions as disease progresses (33, 34). These findings complicate nomenclature and experimental interpretation but underscore the remarkable plasticity of microglia and their ability to transition across diverse states depending on temporal and microenvironmental cues. Understanding these subpopulations is essential for elucidating the mechanisms by which microglia contribute to injury repair.
2.2.2 Microglial efferocytosis after spinal cord injury
Microglial engulfment of dying cells and myelin debris is indispensable for establishing a pro-regenerative milieu in the CNS. This engulfment—termed efferocytosis—progresses through three coordinated steps: “find-me,” “eat-me,” and “digest” signals that sequentially attract, engage, and remove dying cells (35). Briefly, apoptotic cells emit chemo-attractants that bind dedicated receptors on phagocytes, triggering engulfment; the resulting phagosome then fuses with lysosomes, where the cargo is enzymatically degraded (36).
Efferocytosis therefore represents a pivotal checkpoint in inflammation resolution. By-products generated during digestion actively re-programme immune cells, steering them toward pro-resolving phenotypes and restoring tissue homeostasis (35). After brain injury, efferocytosis in the CNS is often suppressed. Notably, EphA4 overexpression in microglia inhibits the P-ERK/P-Stat6/MERTK signaling axis (37). By contrast, microglia enriched for MERTK display heightened efferocytosis, foster oligodendrocyte regeneration, and improve functional outcome in demyelinating models (38). Likewise, Gas6 limits pro-inflammatory microglial activation and curtails microglia–astrocyte crosstalk, thereby attenuating post-SCI inflammation and glial-scar formation (39).
Multiple studies have shown that enhancing microglial phagocytic capacity improves outcomes in ischemic stroke, subarachnoid hemorrhage, and related conditions, likely through mechanisms involving the reduction of neuronal injury and modulation of CNS inflammation (40, 41). Some researchers have proposed that microglial phagocytic capacity is closely tied to their activation state. During efferocytosis, microglia may also adopt a pro-resolving phenotype, secreting anti-inflammatory cytokines such as TGF-β and IL-10 to suppress secondary inflammation and maintain tissue homeostasis (42, 43). Additionally, some studies have employed a strategy combining neutrophil membrane-derived vesicles and a “Trojan Horse” system to promote nerve regeneration and modulate inflammation after SCI through efferocytosis. This effect is mediated by the reprogramming of immune cells and regulation of the immune cascade (44). Collectively, these data underscore efferocytosis as a central driver of immune resolution and tissue repair in SCI. Therapeutic reinforcement of microglial efferocytosis thus offers a compelling avenue for improving neurological outcome.
3 Pyroptosis pathways following spinal cord injury
3.1 Classical caspase-1-dependent pathway
In the canonical pathway, pyroptosis is initiated by multi-protein inflammasomes—most notably NLRP3—that sense danger-associated molecular patterns (DAMPs) liberated after primary mechanical trauma (45). A prototypical signal is extracellular ATP, which binds microglial P2X7 receptors, drives K+ efflux, and thereby activates the NLRP3 inflammasome in SCI (46). High mobility group box 1 (HMGB1), a nuclear protein under physiological conditions, is upregulated in damaged neurons and microglia following SCI and can bind to receptors such as TLR2/4, thereby promoting M1-type polarization of microglia and increasing the release of pro-inflammatory mediators (47). Cellular stress increases mitochondrial permeability; oxidized mtDNA escapes into the cytosol and directly couples to NLRP3, driving inflammasome assembly (48). In addition, SCI-induced cell damage can release other DAMPs such as heat shock proteins (e.g., HSP70, HSP90), S100 proteins, and related molecules. These too are recognized by pattern recognition receptors and contribute to sterile inflammation (49, 50)- (51). Collectively, ATP, HMGB1, and mtDNA represent well-characterized DAMPs in the context of SCI, corresponding to the release of metabolic, nuclear, and genetic materials, respectively. These molecules engage distinct receptors and pathways to drive NLRP3 inflammasome-mediated neuroinflammation.
NLRP3 inflammasome activation proceeds in two steps: the priming/transcriptional signal and the activating signal. The priming/transcriptional signal is initiated by DAMPs or other stimuli that activate transcriptional pathways such as NF-κB, resulting in upregulated transcription and translation of NLRP3 and its downstream pro-inflammatory cytokine precursors, including pro-IL-1β and pro-IL-18 (52). This step elevates the cellular abundance of inflammasome components and sensitizes the NLRP3 complex to activation, involving adapter proteins such as Myeloid differentiation primary response 88 (MyD88), Interleukin-1 receptor-associated kinase 1 (IRAK-1), TIR-domain-containing adaptor-inducing interferon-β (TRIF), and Fas-associated protein with death domain (FADD) (53, 54). The activating signal is closely tied to the aforementioned DAMPs and directly induces NLRP3 inflammasome assembly and activation of effector molecules such as caspase-1 (55). This second signal is often associated with ion fluxes, particularly potassium efflux and calcium influx, which are considered potential upstream events in NLRP3 activation (53, 56). In acute-to-subacute SCI, NLRP3–caspase-1 signaling surges in microglia and constitutes a linchpin of secondary degeneration (57). Excessive pyroptosis depletes protective microglia and floods the parenchyma with pro-inflammatory mediators, jeopardizing neuronal survival. Pharmacological or genetic inhibition of NLRP3 therefore constitutes a promising strategy to blunt neuroinflammation and foster recovery (58).
3.2 Non-canonical pyroptosis pathway mediated by Caspase-4/5/11
In the non-canonical route, human caspase-4/-5 (murine caspase-11) are directly engaged by cytosolic lipopolysaccharide (LPS), bypassing canonical inflammasome sensors (59). LPS docking to their CARD domains triggers rapid oligomerization and auto-activation of these caspases. The activated caspases cleave the linker region of Gasdermin D (GSDMD), releasing the N-terminal fragment (GSDMD-NT). This fragment inserts into the cell membrane, forming pores that trigger pyroptotic cell lysis (60).
While Caspase-4/5/11 do not directly process pro-IL-1β or pro-IL-18, the membrane pores formed by GSDMD-NT cause potassium ion efflux and other cellular disturbances (60, 61). These changes indirectly activate the NLRP3 inflammasome, leading to Caspase-1-dependent maturation and release of IL-1β and IL-18. Consequently, the non-canonical pathway often synergizes with the classical pathway, amplifying the inflammatory cascade (62). Caspase-11 can also cleave the large-pore channel pannexin-1, leading to massive ATP release from the cell. The extracellular ATP then activates the P2X7 receptor, which further triggers potassium efflux, thereby promoting the activation of the NLRP3 inflammasome (63). Mice lacking P2X7 or pannexin-1 exhibit greater resistance to LPS, indicating that this signaling axis is essential for caspase-11-dependent non-canonical pyroptosis. Such cross-talk intensifies neuroinflammation after SCI, underscoring the intricate tapestry of pyroptotic signaling in secondary pathology.
3.3 Atypical pyroptosis mediated by caspase-3/8
Mounting evidence indicates that the executioner caspases-3 and caspases-8, historically viewed as apoptotic proteases, can instigate pyroptosis via unconventional cleavage of specific gasdermins, thereby constituting inflammasome-independent “atypical” pathways (64).
Caspase-3 is recognized as the protease executing apoptosis (65). However, in cells with high GSDME expression, caspase-3 can cleave GSDME, releasing its N-terminal pore-forming domain. This shifts apoptosis toward pyroptosis-like lytic cell death (66). Research indicates that GSDME acts as a “molecular switch,” triggering membrane pore formation and inflammatory mediator release in caspase-3-activated cells (67). Post-SCI, elevated GSDME levels are observed, and its suppression reverses neuroinflammatory exacerbation (68). Microglia express GSDME under pathological conditions, undergoing caspase-3-dependent GSDME cleavage and pyroptotic death upon injury (69).
Caspase-8, a key enzyme in the extrinsic apoptosis pathway, has recently been shown to cleave Gasdermin C (GSDMC) under specific inflammatory conditions (e.g., high TNF-α and IFN-γ levels), inducing pyroptosis in cancer cells (70). Metabolite α-ketoglutarate (α-KG)-induced pyroptosis via death receptor DR6 and caspase-8-mediated GSDMC cleavage has also been reported, dependent on ROS elevation and acidic microenvironments (71). Additionally, caspase-8 can inefficiently cleave GSDMD or promote inflammasome activation, further linking it to pyroptosis (72). Muendlein et al (73) recently proposed the concept of “efferoptosis,” referring to a form of macrophage death termed “macrophage efferoptosis” induced by TNF during efferocytosis. In this process, TNF-activated macrophages undergo TRIF/caspase-8/GSDMD-dependent cell death after engulfing neutrophils. Notably, IL-1β maturation in this context does not rely on the NLRP3 inflammasome but instead occurs via direct cleavage by caspase-8. This suggests that a similar pathway may also be involved in microglial pyroptosis following SCI.
3.4 ROS-mediated pyroptosis
Excess reactive-oxygen species (ROS) generated after SCI constitute a pivotal trigger of inflammasome activation and ensuing pyroptosis. ROS potentiate NLRP3 oligomerization by inducing thiol oxidation, ionic flux and mitochondrial dysfunction (74, 75). Studies show that pathological events post-SCI, such as hemorrhage, hypoxia, and iron ion release, amplify ROS production (76). Excessive ROS triggers NLRP3 inflammasome-mediated pyroptosis. Additionally, ROS indirectly activate the inflammasome by disrupting lysosomal membranes (causing lysosomal enzyme leakage) and damaging mitochondria (releasing mitochondrial DNA and other DAMPs) (77). In microglia, uncontrolled ROS levels persistently stimulate caspase-1/GSDMD-dependent pyroptosis, releasing inflammatory mediators that exacerbate neurological damage.
Targeting oxidative stress via antioxidant therapies has emerged as a promising strategy to suppress pyroptosis and mitigate inflammation (78). For instance, Cynarin inhibits microglial pyroptosis in SCI models by enhancing Nrf2 antioxidant signaling, reducing ROS levels, and suppressing NLRP3 inflammasome assembly (79). This mechanism highlights the therapeutic potential of antioxidants in modulating pyroptosis and improving outcomes in SCI.
Mitochondrial Damage and Pyroptosis
3.5 Mitochondrial damage-mediated pyroptosis
As metabolic powerhouses, mitochondria are intimately linked to cell-death pathways; injury-induced dysfunction prompts excess ROS production and releases mtDNA, oxidized cardiolipin, and other DAMPs into the cytosol (80–82). These molecules act as DAMPs to activate inflammasomes such as NLRP3 or AIM2, triggering caspase-1-mediated pyroptosis (83, 84). Targeting this mechanism, enhancing mitophagy (selective autophagy of mitochondria) to clear damaged mitochondria has emerged as an effective strategy to suppress pyroptosis. For instance, the natural compound Betulinic acid promotes autophagy and mitophagy, clearing dysfunctional mitochondria and reducing ROS levels, thereby significantly inhibiting microglial pyroptosis during SCI (85). Similarly, Urolithin A alleviates microglial pyroptosis and inflammation by enhancing mitophagy in injured tissues (86). These findings underscore the importance of maintaining mitochondrial homeostasis to inhibit pyroptosis and mitigate secondary injury in SCI (Figure 1).
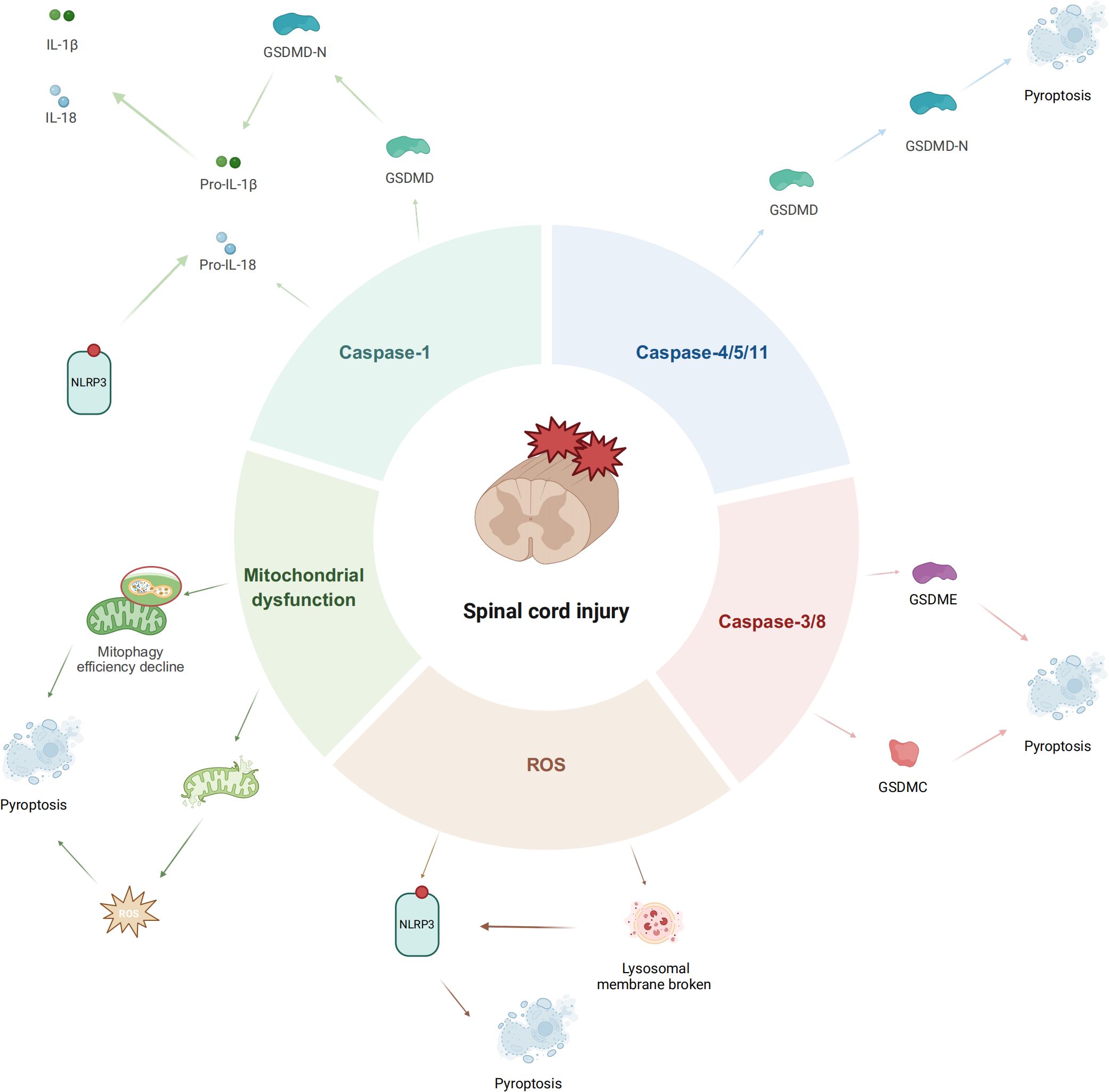
Figure 1. Diagram of pyroptosis pathway following spinal cord injury (created by Biorender).
4 Targeted modulation of microglial pyroptosis to promote neurological and motor recovery post-SCI
Currently, there are no clinically approved pyroptosis-targeted interventions for SCI. However, preclinical studies have demonstrated the critical importance of targeting microglial pyroptosis to improve neurological and motor functional recovery following SCI. Strategies such as cell transplantation, extracellular vesicles derived from other cell sources, synthetic drugs, natural compounds, and genetic modulation of key pyroptosis regulators have shown significant therapeutic potential (Shown Table 1 for details).
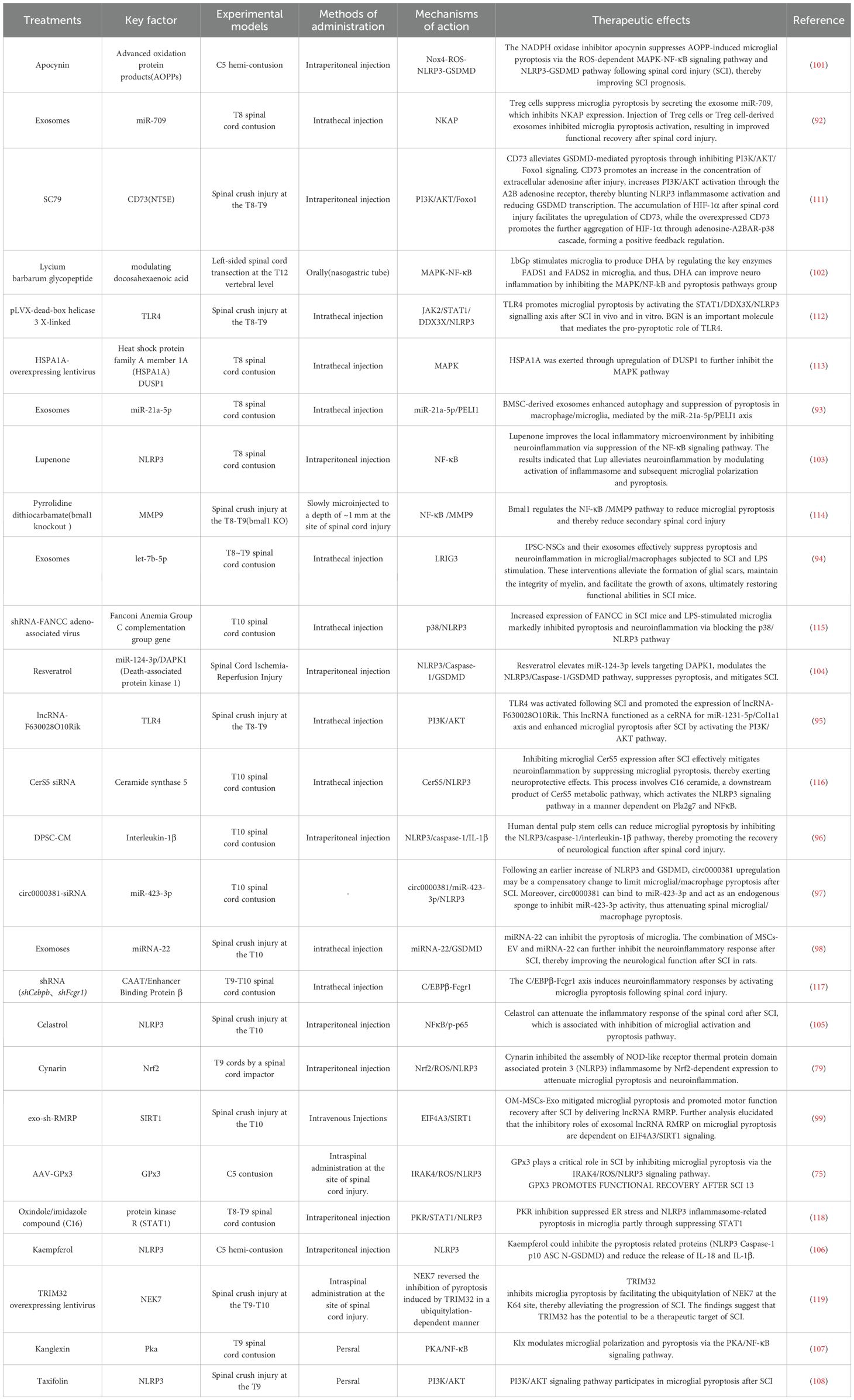
Table 1. Summary table of studies on targeted interventions in post-SCI microglial pyroptosis and their impact on prognosis.
4.1 Cell transplantation and extracellular vesicle-based interventions
In recent years, cell transplantation and extracellular vesicle (EV)-based drug delivery technologies have rapidly advanced in the field of regenerative medicine, emerging as third-generation “biological therapeutic” strategies following small-molecule drugs and genetic engineering (87). According to the International Society for Extracellular Vesicles (ISEV), EVs are lipid bilayer-enclosed particles (including exosomes and microvesicles) naturally released by cells, capable of carrying diverse bioactive cargo (88). In addition to miRNAs or circRNAs, EVs can deliver proteins, lipids, and other therapeutic factors that aid in spinal cord repair. Compared to traditional pharmaceuticals, these approaches can cross the blood-spinal cord barrier, achieve precise delivery to lesions, and remodel the damaged microenvironment through multi-target, network-based regulation, balancing high efficacy with controllability (89, 90). In animal studies and early clinical trials for neurological disorders, stem cells and their derived extracellular vesicle have demonstrated potential in promoting neuroprotection, inflammation modulation, axonal regeneration, and functional recovery (91). Notably, extracellular vesicle inherently offer advantages such as low immunogenicity, feasibility for large-scale production, and adaptability to engineering modifications, thereby enabling safe and repeatable administration.
Regulatory T cells (Tregs) suppress microglial pyroptosis by secreting exosomal miR-709, which down-regulates NKAP; administering either Tregs themselves or their extracellular vesicles blocks microglial pyroptotic activation and ultimately improves functional recovery after SCI (92). Extracellular vesicles derived from bone-marrow mesenchymal stem cells (BMSCs) deliver miR-21a-5p, which enhances PELI1-dependent autophagy and thereby inhibits microglial pyroptosis (93). Induced pluripotent stem-cell–derived neural stem cell (iPSC-NSC) extracellular vesicles can package and transfer let-7b-5p to modulate LRIG3 expression, reducing microglia/macrophage pyroptosis and boosting motor recovery in mice after SCI (94). lncRNA-F630028O10Rik, released in extracellular vesicles following TLR4 activation after SCI, heightens microglial pyroptosis through the PI3K/AKT pathway (95). Transplantation of human dental-pulp stem cells decreases microglial pyroptosis via the NLRP3/caspase-1/IL-1β axis, thereby promoting neurological recovery after SCI (96). While circ0000381 is up-regulated after SCI, miR-423-3p declines; silencing circ0000381 elevates miR-423-3p and increases microglia/macrophage pyroptosis (97). Mesenchymal-stem-cell extracellular vesicles loaded with miRNA-22 suppress microglial pyroptosis in rats following SCI (98). Exosomal lncRNA RMRP from olfactory-mucosa mesenchymal stem cells mitigates microglial pyroptosis and enhances motor recovery through the EIF4A3/SIRT1 pathway (99). In addition, miR-146a, up-regulated via Nrf2 after SCI, down-regulates GSDMD in microglia, thereby restraining their pyroptosis (100). Neutrophil membrane vesicles combined with a composite fiber scaffold reprogram microglial phenotype and metabolism during inflammation, regulating the innate immune cascade to reduce neuroinflammation and promote neural regeneration (44). This scaffold mimics an “efferocytosis-like” mechanism whereby the EVs are endocytosed by macrophages/microglia, reprogramming them towards a pro-regenerative phenotype and significantly promoting nerve fiber regeneration after SCI. his strategy exemplifies how combining biomaterial scaffolds with EV-mediated immune modulation can synergistically coordinate inflammatory resolution and tissue repair in SCI.
4.2 Pharmacological and small-molecule interventions
Small-molecule drugs and natural products are regarded as one of the most clinically translatable intervention strategies because their chemical structures are well-defined, their quality is controllable, and their routes of administration are flexible. In recent years, numerous bioactive constituents derived from medicinal herbs or diet have been shown to cross the blood–brain/spinal barriers, scavenge ROS, modulate the immune-inflammatory network and promote axonal regeneration—offering multiple-target advantages. Alongside technological advances, a series of newly synthesized small molecules have also exhibited excellent pharmacokinetic properties and selective microglial targeting, providing a rich pool of lead compounds for the precision treatment of nervous-system disorders.
The NADPH-oxidase inhibitor apocynin blocks AOPP-induced microglial pyroptosis after SCI via ROS-dependent MAPK–NF-κB and NLRP3–GSDMD pathways, thereby improving outcomes (101). Lycium barbarum glycopeptide (LbGp) up-regulates the key enzymes FADS1 and FADS2 in microglia to boost DHA production and, by suppressing the MAPK/NF-κB and pyroptosis cascades, mitigates neuro-inflammation and enhances recovery (102). Lupenone diminishes IκBα activation and p65 nuclear translocation; by modulating NF-κB it inhibits NLRP3-inflammasome activity, reduces microglial pyroptosis and alleviates motor deficits after SCI (103). Resveratrol elevates miR-124-3p, which targets DAPK1 and down-regulates the NLRP3/Caspase-1/GSDMD axis, thereby lowering microglial pyroptosis (104). Celastrol suppresses microglial pyroptosis after SCI through the NF-κB/p-p65 pathway (105). Cynarin attenuates microglial pyroptosis post-SCI by up-regulating Nrf2 (75). Lupeol activates mitophagy via the AMPK–mTOR–TFEB pathway and strengthens Na+/K+-ATPase activity, inhibiting microglial pyroptosis and slowing SCI progression. Kaempferol curbs ROS generation by inhibiting NADPH oxidase-4 and restrains microglial pyroptosis through the MAPK–NF-κB pathway (106). Kanglexin (Klx), an anthraquinone compound, enhances PKA phosphorylation while inhibiting NF-κB and IκBα phosphorylation, thus limiting NF-κB nuclear translocation and NLRP3-inflammasome-induced microglial pyroptosis (107). Taxifolin targets PI3K/Akt signaling, lessens neuro-inflammation, promotes axonal regeneration and lowers microglial pyroptosis, thereby improving functional outcomes after SCI (108).
4.3 Targeted gene intervention
With the rapid advances of gene-editing platforms such as CRISPR/Cas and TALEN, manipulating specific genes within the CNS has moved quickly from simple “proof-of-concept” studies to bona-fide functional interventions (106). Compared with conventional small-molecule or protein inhibitors, genome editing can silence or activate pathogenic/protective genes with high precision, efficiency and durability, providing a highly specific tool for modulating the inflammatory cascade and remodeling the micro-environment (109). When coupled with delivery vehicles that cross the blood–brain barrier—such as recombinant adeno-associated virus (rAAV) and lipid-nanoparticle (LNP) systems—gene editing has already shown longer-lasting efficacy and controllable safety profiles than pharmacological therapies in multiple models of neurodegenerative disease and SCI (110). Consequently, targeted gene intervention has become a major developmental direction for regulating microglial pyroptosis, mitigating secondary SCI, and treating other CNS disorders.
CD73 (ecto-5′-nucleotidase/NT5E) – an AMP-hydrolyzing ectoenzyme that converts extracellular ATP to adenosine. CD73 knock-down attenuates GSDMD-mediated pyroptosis by suppressing PI3K/AKT/Foxo1 signaling. After SCI, HIF-1α accumulation up-regulates CD73; in turn, CD73 over-expression amplifies HIF-1α via an adenosine–A2B receptor–p38 cascade, forming a positive-feedback loop (111). TLR4 – drives microglial pyroptosis after SCI through the STAT1/DDX3X/NLRP3 axis. Both TLR4 knockout and supplementation with biglycan (BGN) reverse this effect (112). HSPA1A (Heat-shock protein A member 1A) – a molecular chaperone highly induced after TSCI. Over-expression via lentiviral vectors up-regulates DUSP1 and inhibits MAPK signaling, thereby reducing microglial pyroptosis (113). Bmal1 – a core circadian-clock gene. Bmal1 limits microglial pyroptosis and secondary SCI by down-regulating the NF-κB/MMP9 pathway (114). FANCC (Fanconi-anemia complementation group C) – previously considered anti-inflammatory; its targeted inhibition lowers microglial pyroptosis via the p38/NLRP3 pathway (115). CerS5 (Ceramide-synthase 5) – silencing CerS5 in microglia alleviates neuroinflammation by suppressing pyroptosis. The mechanism involves the downstream product C16-ceramide, which activates the NLRP3 pathway through Pla2g7 and NF-κB (116). C/EBPβ (CCAAT/enhancer-binding protein β) – linked to inflammatory status in neurodegeneration; its knock-down diminishes microglia-mediated neuroinflammation by repressing Fcgr1 transcription (117). GPx3 (Glutathione-peroxidase 3) – an antioxidant enzyme. GPx3 silencing elevates ROS and increases IRAK4 and pro-inflammatory cytokines, thereby enhancing microglial pyroptosis (75). PKR (Protein-kinase R) – a type I ER-membrane kinase traditionally associated with ER stress. In SCI it modulates microglial pyroptosis via the STAT1 pathway (118). TRIM32 – an E3-ubiquitin ligase. TRIM32 inhibits microglial pyroptosis by promoting ubiquitination of NEK7 at lysine 64, slowing SCI progression (119).
5 Discussion and future directions
Compelling evidence now demonstrates that microglial pyroptosis orchestrates secondary degenerative cascades after SCI (13, 120). As resident immune sentinels of the central nervous system, microglia are rapidly recruited and activated within minutes of trauma, initiating a robust inflammatory response. During this lytic form of programmed cell death, microglia undergo rapid swelling and lysis, releasing inflammatory mediators (e.g., IL-1β, IL-18) and cellular contents. These mediators exacerbate local neuroinflammatory cascades, causing further damage to adjacent neurons and oligodendrocytes and amplifying secondary tissue damage (121). Conversely, multiple pre-clinical studies show that genetic or pharmacological suppression of microglial pyroptosis markedly attenuates neuroinflammation, limits cellular loss and accelerates locomotor recovery post-SCI (57, 75, 116).
Nano-sized extracellular vesicles have emerged as versatile carriers for anti-pyroptotic cargo. extracellular vesicles, with their small size and low immunogenicity, can penetrate the blood-spinal cord barrier and evade mononuclear phagocyte clearance (122, 123). Studies utilizing stem cell-derived exosomes as carriers for delivering anti-pyroptosis molecules have shown efficacy (124). The advantages of extracellular vesicles include targeted delivery and tissue permeability, but challenges remain in their high preparation/purification costs, complex processes, and lack of standardized quality control. Critically, batch-to-batch consistency in bioactivity and clarity of active components must be resolved before clinical translation.
A growing pharmacopeia of small-molecule inhibitors, antioxidant polyphenols and natural compounds can attenuate microglial pyroptosis in vivo (125, 126). Anti-inflammatory or antioxidant small molecules (e.g., Taxifolin, resveratrol, luteolin) have been shown to attenuate neuroinflammation and suppress microglial pyroptosis post-SCI. These drugs benefit from mature production processes and ease of administration, with some natural compounds exhibiting favorable biosafety (79, 105). Nevertheless, their pleiotropic targets and limited cell specificity raise concern regarding off-target immunosuppression, and systemic delivery must still overcome the blood–spinal cord barrier to achieve therapeutic concentrations while minimizing adverse effects.
Gene-based interventions, such as knockout or silencing of key nodes in pyroptosis pathways, provide robust evidence in animal studies (127, 128). Adeno-associated viruses (AAVs) or lipid nanoparticles delivering shRNA/siRNA have also emerged as tools to inhibit microglial pyroptosis (129, 130). Gene therapies offer high specificity and durable effects by targeting critical pyroptosis molecules. However, clinical translation faces hurdles, including immune responses to delivery vectors, safety/ethical concerns regarding gene editing, and ensuring cell-specific targeting without compromising systemic immunity (131, 132). Furthermore, these approaches are costly, technically demanding, and logistically challenging in acute injury scenarios.
Each intervention modality for modulating microglial pyroptosis carries distinct advantages and limitations. EV-based biological therapies (including cell transplants and EV carriers) enable targeted multi-factorial modulation of the injury microenvironment, with the ability to cross the BSCB and high biocompatibility; however, their production is costly and complex, and standardization of contents and potency remains challenging. Small-molecule drugs, by contrast, are easy to administer and can broadly suppress inflammation or oxidative stress; they benefit from well-established manufacturing and generally good safety profiles, but often lack cell-type specificity and must effectively penetrate into the spinal cord, raising concerns about off-target effects. Gene-editing and gene-silencing approaches (e.g. CRISPR/Cas9 or RNAi therapies) precisely target key pyroptosis-related genes with potentially long-lasting effects, yet they face significant hurdles including immune responses to viral or nanoparticle delivery vectors, ethical and safety considerations, and technical complexity in delivery to the injured CNS. In practice, the optimal approach may depend on the context: small molecules might be favored for acute, systemic intervention, whereas EV-based or gene therapies could offer more specific, sustained effects in subacute or chronic phases. Ultimately, a combination of these strategies may be required to achieve optimal neuroprotection and functional recovery after SCI.
Despite these advances, critical knowledge gaps persist. Foremost, the cell-type-specific contribution to the pyroptotic burden is poorly defined: infiltrating macrophages, astrocytes, oligodendrocytes and neurons may die via pyroptosis alongside microglia (2, 133). Most studies focus on inflammasome activation in mixed glial populations or whole spinal tissue, lacking resolution of pyroptosis dynamics in specific cell types (134). This obscures the relative contributions of microglial versus other cell pyroptosis to secondary injury. For instance, conflating microglia with monocyte-derived macrophages in analyses may mask functional differences. Advanced in vivo tracing and purified in vitro models are needed to dissect cell-specific mechanisms.
Second, functional distinctions among Gasdermin (GSDM) family members in SCI remain poorly understood (67). While GSDMD is widely recognized as the executor of inflammasome-mediated pyroptosis, recent studies suggest GSDME and other family members may mediate pyroptosis via alternative pathways (e.g., caspase-3 activation) (68, 135). In SCI, GSDMD-driven microglial pyroptosis is well-documented, but evidence for roles of GSDME, GSDMC, or other “non-canonical” pyroptosis pathways in neuronal or glial death is lacking. This gap limits our holistic understanding of pyroptosis networks in SCI.
Third, the optimal therapeutic window for pyroptosis inhibition requires clarification (136). Secondary injury spans acute, subacute, and chronic phases, with pyroptosis activity and tissue impacts likely varying across stages (137, 138). While inflammasome components and cleaved GSDMD surge in early injury (hours to days), long-term pyroptosis activity (weeks to chronic phases) remains inconsistently reported (139). Endogenous regulatory mechanisms may partially suppress pyroptosis but fail to halt progressive damage (140, 141). Timing interventions is thus critical: early blockade might disrupt essential immune clearance, whereas delayed action risks irreversible inflammatory cascades. Systematic temporal mapping of pyroptosis activity and intervention efficacy is needed to define optimal clinical windows.
In summary, converging advances in multi-omic analytics, bio-engineered delivery systems and genome editing are poised to transform our mechanistic understanding of microglial pyroptosis into clinically actionable therapies, with the potential to lessen the lifelong disability burden of SCI.
Author contributions
LS: Conceptualization, Writing – review & editing, Writing – original draft. QQ: Writing – original draft, Formal Analysis. JX: Resources, Writing – original draft. TY: Writing – review & editing, Writing – original draft, Software. XZ: Writing – original draft. XM: Writing – review & editing, Writing – original draft, Project administration. JD: Writing – review & editing, Writing – original draft, Project administration. QJ: Writing – original draft, Conceptualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the Beijing Natural Science Foundation (Project No. 7232308); the Youth Science and Technology Talent Training Program of China Academy of Chinese Medical Sciences (Project No. ZZ16-YQ-060); the Esoteric and Endangered Disciplines Research Program of the National Social Science Foundation of China (Project No. 23VJXG015), Suzhou Major Diseases Multicenter Clinical Research Project (Project No. DZXYJ202308), and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (Project No. KYCX25_3954).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cowan H, Lakra C, and Desai M. Autonomic dysreflexia in spinal cord injury. Bmj. (2020) 371:m3596. doi: 10.1136/bmj.m3596
2. Li C, Wu Z, Zhou L, Shao J, Hu x, Xu W, et al. Temporal and spatial cellular and molecular pathological alterations with single-cell resolution in the adult spinal cord after injury. Signal Transduct Target Ther. (2022) 7:65. doi: 10.1038/s41392-022-00885-4
3. Ryan F, Blex C, Ngo TD, Kopp MA, Michalke B, Venkataramani V, et al. Ferroptosis inhibitor improves outcome after early and delayed treatment in mild spinal cord injury. Acta Neuropathol. (2024) 147:106. doi: 10.1007/s00401-024-02758-2
4. Guha L and Kumar H. Drug repurposing for spinal cord injury: progress towards therapeutic intervention for primary factors and secondary complications. Pharmaceut Med. (2023) 37:463–90. doi: 10.1007/s40290-023-00499-3
5. Zavvarian MM, Modi AD, Sadat S, Hong J, and Fehlings MG. Translational relevance of secondary intracellular signaling cascades following traumatic spinal cord injury. Int J Mol Sci. (2024) 25:5708. doi: 10.3390/ijms25115708
6. Liu FS, Huang HL, Deng LX, Zhang QS, Wang XB, Li J, et al. Identification and bioinformatics analysis of genes associated with pyroptosis in spinal cord injury of rat and mouse. Sci Rep. (2024) 14:14023. doi: 10.1038/s41598-024-64843-6
7. Li X, Fu J, Guan M, Shi H, Pan W, and Lou X. Biochanin A attenuates spinal cord injury in rats during early stages by inhibiting oxidative stress and inflammasome activation. Neural Regener Res. (2024) 19:2050–6. doi: 10.4103/1673-5374.390953
8. Zhou H, Qian Q, Chen Q, Chen T, Wu C, Chen L, et al. Enhanced mitochondrial targeting and inhibition of pyroptosis with multifunctional metallopolyphenol nanoparticles in intervertebral disc degeneration. Small. (2024) 20:e2308167. doi: 10.1002/smll.202308167
9. Fu SP, Chen SY, Pang QM, Zhang M, Wu XC, Wan X, et al. Advances in the research of the role of macrophage/microglia polarization-mediated inflammatory response in spinal cord injury. Front Immunol. (2022) 13:1014013. doi: 10.3389/fimmu.2022.1014013
10. Ramos D and Cruz CD. Involvement of microglia in chronic neuropathic pain associated with spinal cord injury - a systematic review. Rev Neurosci. (2023) 34:933–50. doi: 10.1515/revneuro-2023-0031
11. Zhang C, Kang J, Zhang X, Zhang Y, Huang N, and Ning B. Spatiotemporal dynamics of the cellular components involved in glial scar formation following spinal cord injury. BioMed Pharmacother. (2022) 153:113500. doi: 10.1016/j.biopha.2022.113500
12. Al Mamun A, Wu Y, Monalisa I, Jia C, Zhou K, Munir F, et al. Role of pyroptosis in spinal cord injury and its therapeutic implications. J Adv Res. (2021) 28:97–109. doi: 10.1016/j.jare.2020.08.004
13. Oladapo A, Jackson T, Menolascino J, and Periyasamy P. Role of pyroptosis in the pathogenesis of various neurological diseases. Brain Behav Immun. (2024) 117:428–46. doi: 10.1016/j.bbi.2024.02.001
14. Hu X, Xu W, Ren Y, Wang Z, He X, Huang R, et al. Spinal cord injury: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. (2023) 8:245. doi: 10.1038/s41392-023-01477-6
15. Cantó-Santos J, Grau-Junyent JM, and Garrabou G. The impact of mitochondrial deficiencies in neuromuscular diseases. Antioxidants (Basel). (2020) 9:964. doi: 10.3390/antiox9100964
16. Zheng B and Tuszynski MH. Regulation of axonal regeneration after mammalian spinal cord injury. Nat Rev Mol Cell Biol. (2023) 24:396–413. doi: 10.1038/s41580-022-00562-y
17. Shi Z, Yuan S, Shi L, Li j, Ning B, Kong X, et al. Programmed cell death in spinal cord injury pathogenesis and therapy. Cell Prolif. (2021) 54:e12992. doi: 10.1111/cpr.12992
18. Borst K, Dumas AA, and Prinz M. Microglia: Immune and non-immune functions. Immunity. (2021) 54:2194–208. doi: 10.1016/j.immuni.2021.09.014
19. Vidal-Itriago A, Radford RAW, Aramideh JA, Maurel C, Scherer NM, Don EK, et al. Microglia morphophysiological diversity and its implications for the CNS. Front Immunol. (2022) 13:997786. doi: 10.3389/fimmu.2022.997786
20. Hasel P, Aisenberg WH, Bennett FC, and Liddelow SA. Molecular and metabolic heterogeneity of astrocytes and microglia. Cell Metab. (2023) 35:555–70. doi: 10.1016/j.cmet.2023.03.006
21. Devanney NA, Stewart AN, and Gensel JC. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol. (2020) 329:113310. doi: 10.1016/j.expneurol.2020.113310
22. Long Y, Li XQ, Deng J, Ye Q, Li D, Ma Y, et al. Modulating the polarization phenotype of microglia - A valuable strategy for central nervous system diseases. Ageing Res Rev. (2024) 93:102160. doi: 10.1016/j.arr.2023.102160
23. Javanmehr N, Saleki K, Alijanizadeh P, and Rezaei N. Microglia dynamics in aging-related neurobehavioral and neuroinflammatory diseases. J Neuroinflamm. (2022) 19:273. doi: 10.1186/s12974-022-02637-1
24. Li Q, Zhao Y, Guo H, Li Q, Yan C, Li Y, et al. Impaired lipophagy induced-microglial lipid droplets accumulation contributes to the buildup of TREM1 in diabetes-associated cognitive impairment. Autophagy. (2023) 19:2639–56. doi: 10.1080/15548627.2023.2213984
25. Choi BR, Johnson KR, Maric D, and McGavern DB. Monocyte-derived IL-6 programs microglia to rebuild damaged brain vasculature. Nat Immunol. (2023) 24:1110–23. doi: 10.1038/s41590-023-01521-1
26. Xia DY, Yuan JL, Jiang XC, Qi M, Lai NS, Wu LY, et al. SIRT1 promotes M2 microglia polarization via reducing ROS-mediated NLRP3 inflammasome signaling after subarachnoid hemorrhage. Front Immunol. (2021) 12:770744. doi: 10.3389/fimmu.2021.770744
27. Zhang L, Wei W, Ai X, Kilic E, Hermann DM, Venkataramani V, et al. Extracellular vesicles from hypoxia-preconditioned microglia promote angiogenesis and repress apoptosis in stroke mice via the TGF-β/Smad2/3 pathway. Cell Death Dis. (2021) 12:1068. doi: 10.1038/s41419-021-04363-7
28. Shemer A, Scheyltjens I, Frumer GR, Kim JS, Grozovski J, Ayanaw S, et al. Interleukin-10 prevents pathological microglia hyperactivation following peripheral endotoxin challenge. Immunity. (2020) 53:1033–49.e7. doi: 10.1016/j.immuni.2020.09.018
29. Guedes JR, Ferreira PA, Costa J, Laranjo M, Pinto MJ, Reis T, et al. IL-4 shapes microglia-dependent pruning of the cerebellum during postnatal development. Neuron. (2023) 111:3435–49.e8. doi: 10.1016/j.neuron.2023.09.031
30. Paolicelli RC, Sierra A, Stevens B, Tremblay ME, Aguzzi A, Ajami B, et al. Microglia states and nomenclature: A field at its crossroads. Neuron. (2022) 110:3458–83. doi: 10.1016/j.neuron.2022.10.020
31. Peng R, Zhang L, Xie Y, Guo S, Cao X, and Yang M. Spatial multi-omics analysis of the microenvironment in traumatic spinal cord injury: a narrative review. Front Immunol. (2024) 15:1432841. doi: 10.3389/fimmu.2024.1432841
32. Wang J, Xu L, Lin W, Yao Y, Li H, Shen G, et al. Single-cell transcriptome analysis reveals the immune heterogeneity and the repopulation of microglia by Hif1α in mice after spinal cord injury. Cell Death Dis. (2022) 13:432. doi: 10.1038/s41419-022-04864-z
33. Kracht L, Borggrewe M, Eskandar S, Brouwer N, Chuva de Sousa Lopes SM, Laman JD, et al. Human fetal microglia acquire homeostatic immune-sensing properties early in development. Science. (2020) 369:530–7. doi: 10.1126/science.aba5906
34. Skinnider MA, Rogalski J, Tigchelaar S, Manouchehri N, Prudova A, Jackson AM, et al. Proteomic portraits reveal evolutionarily conserved and divergent responses to spinal cord injury. Mol Cell Proteomics. (2021) 20:100096. doi: 10.1016/j.mcpro.2021.100096
35. Saas P, Vetter M, Maraux M, Bonnefoy F, and Perruche S. Resolution therapy: Harnessing efferocytic macrophages to trigger the resolution of inflammation. Front Immunol. (2022) 13:1021413. doi: 10.3389/fimmu.2022.1021413
36. Gerlach BD, Ampomah PB, Yurdagul A,JR, Liu C, Lauring MC, Wang X, et al. Efferocytosis induces macrophage proliferation to help resolve tissue injury. Cell Metab. (2021) 33:2445–63.e8. doi: 10.1016/j.cmet.2021.10.015
37. Soliman E, Leonard J, Basso EKG, Gershenson I, Ju J, Mills J, et al. Efferocytosis is restricted by axon guidance molecule EphA4 via ERK/Stat6/MERTK signaling following brain injury. J Neuroinflamm. (2023) 20:256. doi: 10.1186/s12974-023-02940-5
38. Nguyen LT, Aprico A, Nwoke E, Walsh AD, Blades F, Avneri R, et al. Mertk-expressing microglia influence oligodendrogenesis and myelin modelling in the CNS. J Neuroinflamm. (2023) 20:253. doi: 10.1186/s12974-023-02921-8
39. Chen J, Zeng X, Wang L, Zhang W, Li G, Cheng X, et al. Mutual regulation of microglia and astrocytes after Gas6 inhibits spinal cord injury. Neural Regener Res. (2025) 20:557–73. doi: 10.4103/nrr.Nrr-d-23-01130
40. Cai W, Dai X, Chen J, Zhang W, Li G, Cheng X, et al. STAT6/Arg1 promotes microglia/macrophage efferocytosis and inflammation resolution in stroke mice. JCI Insight. (2019) 4. doi: 10.1172/jci.insight.131355
41. Zhang G, Li Q, Tao W, Qin P, Chen J, Yang H, et al. Sigma-1 receptor-regulated efferocytosis by infiltrating circulating macrophages/microglial cells protects against neuronal impairments and promotes functional recovery in cerebral ischemic stroke. Theranostics. (2023) 13:543–59. doi: 10.7150/thno.77088
42. Chen Y, Kou Y, Ni Y, Yang H, Xu C, Fan H, et al. Microglia efferocytosis: an emerging mechanism for the resolution of neuroinflammation in Alzheimer's disease. J Neuroinflamm. (2025) 22:96. doi: 10.1186/s12974-025-03428-0
43. Liu X, Wang J, Jin J, Hu Q, Zhao T, Wang J, et al. S100A9 deletion in microglia/macrophages ameliorates brain injury through the STAT6/PPARγ pathway in ischemic stroke. CNS Neurosci Ther. (2024) 30:e14881. doi: 10.1111/cns.14881
44. Wu J, Tang J, Zhang L, Wang W, Li Z, Zhou L, et al. Biomimetic "Trojan horse" Fibers modulate innate immunity cascades for nerve regeneration. ACS Nano. (2025) 19:781–802. doi: 10.1021/acsnano.4c12036
45. Gu HY and Liu N. Mechanism of effect and therapeutic potential of NLRP3 inflammasome in spinal cord injury. Exp Neurol. (2025) 384:115059. doi: 10.1016/j.expneurol.2024.115059
46. Wang H, Zhang Y, Ma X, Wang W, Xu X, Huang M, et al. Spinal TLR4/P2X7 receptor-dependent NLRP3 inflammasome activation contributes to the development of tolerance to morphine-induced antinociception. J Inflammation Res. (2020) 13:571–82. doi: 10.2147/jir.S266995
47. Mo Y and Chen K. Review: The role of HMGB1 in spinal cord injury. Front Immunol. (2022) 13:1094925. doi: 10.3389/fimmu.2022.1094925
48. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. (2011) 12:222–30. doi: 10.1038/ni.1980
49. Lin QC, Wang J, Wang XL, Pan C, Jin SW, Char S, et al. Hippocampal HDAC6 promotes POCD by regulating NLRP3-induced microglia pyroptosis via HSP90/HSP70 in aged mice. Biochim Biophys Acta Mol Basis Dis. (2024) 1870:167137. doi: 10.1016/j.bbadis.2024.167137
50. Qu J, Tao XY, Teng P, Zhang Y, Guo CL, Hu L, et al. Blocking ATP-sensitive potassium channel alleviates morphine tolerance by inhibiting HSP70-TLR4-NLRP3-mediated neuroinflammation. J Neuroinflamm. (2017) 14:228. doi: 10.1186/s12974-017-0997-0
51. Ha JS, Choi HR, Kim IS, Kim EA, Cho SW, and Yang SJ. Hypoxia-induced S100A8 expression activates microglial inflammation and promotes neuronal apoptosis. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22031205
52. Thapa A, Adamiak M, Bujko K, Ratajczak J, Abdel-Latif AK, Kucia M, et al. Danger-associated molecular pattern molecules take unexpectedly a central stage in Nlrp3 inflammasome-caspase-1-mediated trafficking of hematopoietic stem/progenitor cells. Leukemia. (2021) 35:2658–71. doi: 10.1038/s41375-021-01158-9
53. Kodi T, Sankhe R, Gopinathan A, Nandakumar K, and Kishore A. New insights on NLRP3 inflammasome: mechanisms of activation, inhibition, and epigenetic regulation. J Neuroimmune Pharmacol. (2024) 19:7. doi: 10.1007/s11481-024-10101-5
54. He Y, Hara H, and Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. (2016) 41:1012–21. doi: 10.1016/j.tibs.2016.09.002
55. Chen J, Shen Y, Shao X, and Wu W. An emerging role of inflammasomes in spinal cord injury and spinal cord tumor. Front Immunol. (2023) 14:1119591. doi: 10.3389/fimmu.2023.1119591
56. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun. (2017) 8:202. doi: 10.1038/s41467-017-00227-x
57. Xiao X, Chen XY, Dong YH, Dong HR, Zhou LN, Ding YQ, et al. Pre-treatment of rapamycin transformed M2 microglia alleviates traumatic cervical spinal cord injury via AIM2 signaling pathway in vitro and in vivo. Int Immunopharmacol. (2023) 121:110394. doi: 10.1016/j.intimp.2023.110394
58. Zuo X, Ju C, Zhang Z, Wei X, Ma Y, Song Z, et al. Photobiomodulation regulates inflammation and autophagy in spinal cord injury through NLRP3/Caspase-1/IL-1β pathway by targeting TLR2. Mol Immunol. (2025) 182:1–10. doi: 10.1016/j.molimm.2025.03.014
59. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. (2014) 514:187–92. doi: 10.1038/nature13683
60. Shi X, Sun Q, Hou Y, Zeng H, Cao Y, Dong M, et al. Recognition and maturation of IL-18 by caspase-4 noncanonical inflammasome. Nature. (2023) 624:442–50. doi: 10.1038/s41586-023-06742-w
61. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. (2015) 526:666–71. doi: 10.1038/nature15541
62. Zhou Y, Chai Z, Pandeya A, Yang L, Zhang Y, Zhang G, et al. Caspase-11 and NLRP3 exacerbate systemic Klebsiella infection through reducing mitochondrial ROS production. Front Immunol. (2025) 16:1516120. doi: 10.3389/fimmu.2025.1516120
63. Yang D, He Y, Muñoz-Planillo R, Liu Q, and Núñez G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity. (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
64. Li Z, Rong Y, and Zhang Y. MiR-335 improves functional recovery in rats after spinal cord injury and protects PC12 cells against injury via the SPI-bax/caspase-3 axis. Spine (Phila Pa 1976). (2024) 49:583–93. doi: 10.1097/brs.0000000000004862
65. Zhao W, Li H, Hou Y, Jin Y, and Zhang L. Combined administration of poly-ADP-ribose polymerase-1 and caspase-3 inhibitors alleviates neuronal apoptosis after spinal cord injury in rats. World Neurosurg. (2019) 127:e346–e52. doi: 10.1016/j.wneu.2019.03.116
66. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. (2017) 547:99–103. doi: 10.1038/nature22393
67. Zhu C, Xu S, Jiang R, Yu Y, Bian J, and Zou Z. The gasdermin family: emerging therapeutic targets in diseases. Signal Transduct Target Ther. (2024) 9:87. doi: 10.1038/s41392-024-01801-8
68. Wu C, Wang L, Chen S, Shi L, Liu M, Tu P, et al. Iron induces B cell pyroptosis through Tom20-Bax-caspase-gasdermin E signaling to promote inflammation post-spinal cord injury. J Neuroinflamm. (2023) 20:171. doi: 10.1186/s12974-023-02848-0
69. Mckenzie BA, Fernandes JP, Doan MAL, Schmitt LM, Branton WG, and Power C. Activation of the executioner caspases-3 and -7 promotes microglial pyroptosis in models of multiple sclerosis. J Neuroinflamm. (2020) 17:253. doi: 10.1186/s12974-020-01902-5
70. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. (2020) 22:1264–75. doi: 10.1038/s41556-020-0575-z
71. Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. (2021) 31:980–97. doi: 10.1038/s41422-021-00506-9
72. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. (2019) 38. doi: 10.15252/embj.2019101638
73. Muendlein HI, Connolly WM, Leiriao J, Nolan MA, Judge J, Smirnova I, et al. TNF switches homeostatic efferocytosis to lytic caspase-8-dependent pyroptosis and IL-1β maturation. Sci Immunol. (2025) 10:eadq0043. doi: 10.1126/sciimmunol.adq0043
74. Zhou H, Li Z, Jing S, Wang B, Ye Z, Xiong W, et al. Repair spinal cord injury with a versatile anti-oxidant and neural regenerative nanoplatform. J Nanobiotechnology. (2024) 22:351. doi: 10.1186/s12951-024-02610-5
75. Liu Z, Shi J, Tu K, Ma H, Chen J, Xiang X, et al. GPx3 promotes functional recovery after spinal cord injury by inhibiting microglial pyroptosis through IRAK4/ROS/NLRP3 axis. Antioxid Redox Signal. (2025) 42:711–29. doi: 10.1089/ars.2024.0618
76. Ren Z, Liang W, Sheng J, Xun C, Xu T, Cao R, et al. Gal-3 is a potential biomarker for spinal cord injury and Gal-3 deficiency attenuates neuroinflammation through ROS/TXNIP/NLRP3 signaling pathway. Biosci Rep. (2019) 39. doi: 10.1042/bsr20192368
77. Liu Z, Tu K, Zou P, Liao C, Ding R, Huang Z, et al. Hesperetin ameliorates spinal cord injury by inhibiting NLRP3 inflammasome activation and pyroptosis through enhancing Nrf2 signaling. Int Immunopharmacol. (2023) 118:110103. doi: 10.1016/j.intimp.2023.110103
78. Yang S, Guan Y, Zheng C, Xia X, Ma X, and Jiang J. FOXO3-induced microRNA-128-3p promotes the progression of spinal cord injury in mice via regulating NLRP3 inflammasome-mediated pyroptosis. Front Immunol. (2025) 16:1526721. doi: 10.3389/fimmu.2025.1526721
79. Zhang B, Yu J, Bao L, Feng D, Qin Y, Fan D, et al. Cynarin inhibits microglia-induced pyroptosis and neuroinflammation via Nrf2/ROS/NLRP3 axis after spinal cord injury. Inflammation Res. (2024) 73:1981–94. doi: 10.1007/s00011-024-01945-x
80. Jiang W, He F, Ding G, and Wu J. Elamipretide reduces pyroptosis and improves functional recovery after spinal cord injury. CNS Neurosci Ther. (2023) 29:2843–56. doi: 10.1111/cns.14221
81. Zhou LY, Yao M, Tian ZR, Liu SF, Song YJ, Ye J, et al. Muscone suppresses inflammatory responses and neuronal damage in a rat model of cervical spondylotic myelopathy by regulating Drp1-dependent mitochondrial fission. J Neurochem. (2020) 155:154–76. doi: 10.1111/jnc.15011
82. Bai M, Cui Y, Sang Z, Gao S, Zhao H, and Mei X. Zinc ions regulate mitochondrial quality control in neurons under oxidative stress and reduce PANoptosis in spinal cord injury models via the Lgals3-Bax pathway. Free Radic Biol Med. (2024) 221:169–80. doi: 10.1016/j.freeradbiomed.2024.05.037
83. Rabchevsky AG, Michael FM, and Patel SP. Mitochondria focused neurotherapeutics for spinal cord injury. Exp Neurol. (2020) 330:113332. doi: 10.1016/j.expneurol.2020.113332
84. Slater PG, Domínguez-Romero ME, Villarreal M, Eisner V, and Larraín J. Mitochondrial function in spinal cord injury and regeneration. Cell Mol Life Sci. (2022) 79:239. doi: 10.1007/s00018-022-04261-x
85. Wu C, Chen H, Zhuang R, Zhang H, Wang Y, Hu X, et al. Betulinic acid inhibits pyroptosis in spinal cord injury by augmenting autophagy via the AMPK-mTOR-TFEB signaling pathway. Int J Biol Sci. (2021) 17:1138–52. doi: 10.7150/ijbs.57825
86. Chen K, Ying J, Zhu J, Chen L, Liu R, Jing M, et al. Urolithin A alleviates NLRP3 inflammasome activation and pyroptosis by promoting microglial mitophagy following spinal cord injury. Int Immunopharmacol. (2025) 148:114057. doi: 10.1016/j.intimp.2025.114057
87. Ran N, Li W, Zhang R, Lin C, Zhang J, Wei Z, et al. Autologous exosome facilitates load and target delivery of bioactive peptides to repair spinal cord injury. Bioact Mater. (2023) 25:766–82. doi: 10.1016/j.bioactmat.2022.07.002
88. Welsh JA, Goberdhan DCI, O'driscoll L, Buzas EI, Blenkiron C, Bussolati B, et al. Minimal information for studies of extracellular vesicles (MISEV2023): From basic to advanced approaches. J Extracell Vesicles. (2024) 13:e12404. doi: 10.1002/jev2.12404
89. Liu WZ, Ma ZJ, Li JR, and Kang XW. Mesenchymal stem cell-derived exosomes: therapeutic opportunities and challenges for spinal cord injury. Stem Cell Res Ther. (2021) 12:102. doi: 10.1186/s13287-021-02153-8
90. Debbi L, Guo S, Safina D, and Levenberg S. Boosting extracellular vesicle secretion. Biotechnol Adv. (2022) 59:107983. doi: 10.1016/j.bioteChadv.2022.107983
91. Xie Y, Sun Y, Liu Y, Zhao J, Liu Q, Xu J, et al. Targeted delivery of RGD-CD146(+)CD271(+) human umbilical cord mesenchymal stem cell-derived exosomes promotes blood-spinal cord barrier repair after spinal cord injury. ACS Nano. (2023) 17:18008–24. doi: 10.1021/acsnano.3c04423
92. Xiong W, Li C, Kong G, Zeng Q, Wang S, Yin G, et al. Treg cell-derived exosomes miR-709 attenuates microglia pyroptosis and promotes motor function recovery after spinal cord injury. J Nanobiotechnology. (2022) 20:529. doi: 10.1186/s12951-022-01724-y
93. Gu J, Wu J, Wang C, Xu Z, Jin Z, Yan D, et al. BMSCs-derived exosomes inhibit macrophage/microglia pyroptosis by increasing autophagy through the miR-21a-5p/PELI1 axis in spinal cord injury. Aging (Albany NY). (2024) 16:5184–206. doi: 10.18632/aging.205638
94. Liu J, Kong G, Lu C, Wang J, Li W, Lv Z, et al. IPSC-NSCs-derived exosomal let-7b-5p improves motor function after spinal cord Injury by modulating microglial/macrophage pyroptosis. J Nanobiotechnology. (2024) 22:403. doi: 10.1186/s12951-024-02697-w
95. Xu S, Wang J, Jiang J, Song J, Zhu W, Zhang F, et al. TLR4 promotes microglial pyroptosis via lncRNA-F630028O10Rik by activating PI3K/AKT pathway after spinal cord injury. Cell Death Dis. (2020) 11:693. doi: 10.1038/s41419-020-02824-z
96. Liu T, Ma Z, Liu L, Pei Y, Wu Q, Xu S, et al. Conditioned medium from human dental pulp stem cells treats spinal cord injury by inhibiting microglial pyroptosis. Neural Regener Res. (2024) 19:1105–11. doi: 10.4103/1673-5374.385309
97. Zhang Y, Zhang W, Liu T, Ma Z, Zhang W, Guan Y, et al. Upregulation of circ0000381 attenuates microglial/macrophage pyroptosis after spinal cord injury. Neural Regener Res. (2024) 19:1360–6. doi: 10.4103/1673-5374.386399
98. Sheng Y, Zhou X, Wang J, Shen H, Wu S, Guo W, et al. MSC derived EV loaded with miRNA-22 inhibits the inflammatory response and nerve function recovery after spinal cord injury in rats. J Cell Mol Med. (2021) 25:10268–78. doi: 10.1111/jcmm.16965
99. Wang C, Zhang J, Chen W, Gao L, He J, and Xia Y. Exosomal lncRNA RMRP-shuttled by Olfactory Mucosa-Mesenchymal Stem Cells Suppresses Microglial Pyroptosis to Improve Spinal Cord Injury via EIF4A3/SIRT1. Mol Neurobiol. (2025) 1–16. doi: 10.1007/s12035-025-04756-1
100. Zhang D, Mao F, Wang S, Wu H, Wang S, and Liao Y. Role of transcription factor nrf2 in pyroptosis in spinal cord injury by regulating GSDMD. Neurochem Res. (2023) 48:172–87. doi: 10.1007/s11064-022-03719-5
101. Liu Z, Yao X, Jiang W, Li W, Zhu S, Liao C, et al. Advanced oxidation protein products induce microglia-mediated neuroinflammation via MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. J Neuroinflamm. (2020) 17:90. doi: 10.1186/s12974-020-01751-2
102. Jiang Z, Zeng Z, He H, Li M, Lan Y, Hui J, et al. Lycium barbarum glycopeptide alleviates neuroinflammation in spinal cord injury via modulating docosahexaenoic acid to inhibiting MAPKs/NF-kB and pyroptosis pathways. J Transl Med. (2023) 21:770. doi: 10.1186/s12967-023-04648-9
103. Li F, Sun X, Sun K, Kong F, Jiang X, and Kong Q. Lupenone improves motor dysfunction in spinal cord injury mice through inhibiting the inflammasome activation and pyroptosis in microglia via the nuclear factor kappa B pathway. Neural Regener Res. (2024) 19:1802–11. doi: 10.4103/1673-5374.389302
104. Li D, Dai Y, Li Z, Bi H, Li H, Wang Y, et al. Resveratrol upregulates miR-124-3p expression to target DAPK1, regulating the NLRP3/caspase-1/GSDMD pathway to inhibit pyroptosis and alleviate spinal cord injury. J Cell Mol Med. (2025) 29:e70338. doi: 10.1111/jcmm.70338
105. Dai W, Wang X, Teng H, Li C, Wang B, Wang J, et al. Celastrol inhibits microglial pyroptosis and attenuates inflammatory reaction in acute spinal cord injury rats. Int Immunopharmacol. (2019) 66:215–23. doi: 10.1016/j.intimp.2018.11.029
106. Liu Z, Yao X, Sun B, Jiang W, Liao C, Dai X, et al. Pretreatment with kaempferol attenuates microglia-mediate neuroinflammation by inhibiting MAPKs-NF-κB signaling pathway and pyroptosis after secondary spinal cord injury. Free Radic Biol Med. (2021) 168:142–54. doi: 10.1016/j.freeradbiomed.2021.03.037
107. Yan R, Yuan Y, Shi C, Li Y, Li Y, Wang W, et al. Kanglexin attenuates spinal cord injury by modulating pyroptosis and polarization via the PKA/NF-κB signaling pathway. Int Immunopharmacol. (2025) 153:114401. doi: 10.1016/j.intimp.2025.114401
108. Hu Z, Xuan L, Wu T, Jiang N, Liu X, Chang J, et al. Taxifolin attenuates neuroinflammation and microglial pyroptosis via the PI3K/Akt signaling pathway after spinal cord injury. Int Immunopharmacol. (2023) 114:109616. doi: 10.1016/j.intimp.2022.109616
109. Wang B, Chang M, Zhang R, Wo J, Wu B, Zhang H, et al. Spinal cord injury target-immunotherapy with TNF-α autoregulated and feedback-controlled human umbilical cord mesenchymal stem cell derived exosomes remodelled by CRISPR/Cas9 plasmid. Biomater Adv. (2022) 133:112624. doi: 10.1016/j.msec.2021.112624
110. Chen Q, Chuai G, Zhang H, Tang J, Duan L, Guan H, et al. Genome-wide CRISPR off-target prediction and optimization using RNA-DNA interaction fingerprints. Nat Commun. (2023) 14:7521. doi: 10.1038/s41467-023-42695-4
111. Xu S, Wang J, Zhong J, Shao M, Jiang J, Song J, et al. CD73 alleviates GSDMD-mediated microglia pyroptosis in spinal cord injury through PI3K/AKT/Foxo1 signaling. Clin Transl Med. (2021) 11:e269. doi: 10.1002/ctm2.269
112. Wang J, Zhang F, Xu H, Yang H, Shao M, Xu S, et al. TLR4 aggravates microglial pyroptosis by promoting DDX3X-mediated NLRP3 inflammasome activation via JAK2/STAT1 pathway after spinal cord injury. Clin Transl Med. (2022) 12:e894. doi: 10.1002/ctm2.894
113. He X, Deng B, Zhang C, Zhang G, Yang F, Zhu D, et al. HSPA1A inhibits pyroptosis and neuroinflammation after spinal cord injury via DUSP1 inhibition of the MAPK signaling pathway. Mol Med. (2025) 31:53. doi: 10.1186/s10020-025-01086-9
114. Li D, Liu S, Lu X, Gong Z, Wang H, Xia X, et al. The circadian clock gene bmal1 regulates microglial pyroptosis after spinal cord injury via NF-κB/MMP9. CNS Neurosci Ther. (2024) 30:e70130. doi: 10.1111/cns.70130
115. Xia M, Li X, Ye S, Zhang Q, Zhao T, Li R, et al. FANCC deficiency mediates microglial pyroptosis and secondary neuronal apoptosis in spinal cord contusion. Cell Biosci. (2022) 12:82. doi: 10.1186/s13578-022-00816-4
116. Zhang W, Lu Y, Shen R, Wu Y, Liu C, Fang X, et al. Inhibiting ceramide synthase 5 expression in microglia decreases neuroinflammation after spinal cord injury. Neural Regener Res. (2025) 20:2955–68. doi: 10.4103/nrr.Nrr-d-23-01933
117. Li J, Yang Y, Zhao C, Zhao J, Wang X, Ye S, et al. Microglial C/EBPβ-Fcgr1 regulatory axis blocking inhibits microglial pyroptosis and improves neurological recovery. J Neuroinflamm. (2025) 22:29. doi: 10.1186/s12974-025-03362-1
118. Yang Z, Sheng M, Wang M, Cheng L, and Sun X. PKR inhibitor protects spinal cord injury through mitigating endoplasmic reticulum stress and pyroptosis. Neurochem Int. (2024) 172:105632. doi: 10.1016/j.neuint.2023.105632
119. Yu J, Feng D, Bao L, and Zhang B. TRIM32 inhibits NEK7 ubiquitylation-dependent microglia pyroptosis after spinal cord injury. Mol Biotechnol. (2025) 67:138–48. doi: 10.1007/s12033-023-00989-4
120. Zeng ZJ, Lin X, Yang L, Li Y, and Gao W. Activation of inflammasomes and relevant modulators for the treatment of microglia-mediated neuroinflammation in ischemic stroke. Mol Neurobiol. (2024) 61:10792–804. doi: 10.1007/s12035-024-04225-1
121. Wu X, Wan T, Gao X, Fu M, Duan Y, Shen X, et al. Microglia pyroptosis: A candidate target for neurological diseases treatment. Front Neurosci. (2022) 16:922331. doi: 10.3389/fnins.2022.922331
122. Wang T, Wang L, Zhang L, Long Y, Zhang Y, and Hou Z. Single-cell RNA sequencing in orthopedic research. Bone Res. (2023) 11:10. doi: 10.1038/s41413-023-00245-0
123. Ning B, Gao L, Liu RH, Liu Y, Zhang NS, and Chen ZY. microRNAs in spinal cord injury: potential roles and therapeutic implications. Int J Biol Sci. (2014) 10:997–1006. doi: 10.7150/ijbs.9058
124. Silvestro S and Mazzon E. MiRNAs as promising translational strategies for neuronal repair and regeneration in spinal cord injury. Cells. (2022) 11:2177. doi: 10.3390/cells11142177
125. Lu Y, Yang J, Wang X, Ma Z, Li S, Liu Z, et al. Research progress in use of traditional Chinese medicine for treatment of spinal cord injury. BioMed Pharmacother. (2020) 127:110136. doi: 10.1016/j.biopha.2020.110136
126. Wu X, Yan Y, and Zhang Q. Neuroinflammation and modulation role of natural products after spinal cord injury. J Inflammation Res. (2021) 14:5713–37. doi: 10.2147/jir.S329864
127. Yang R, Pan J, Wang Y, Xia P, Tai M, Jiang Z, et al. Application and prospects of somatic cell reprogramming technology for spinal cord injury treatment. Front Cell Neurosci. (2022) 16:1005399. doi: 10.3389/fncel.2022.1005399
128. Zeng CW. Stem cell-based approaches for spinal cord injury: the promise of iPSCs. Biol (Basel). (2025) 14:314. doi: 10.3390/biology14030314
129. Guo S, Perets N, Betzer O, Ben-Shaul S, Sheinin A, Michaelevski I, et al. Intranasal delivery of mesenchymal stem cell derived exosomes loaded with phosphatase and tensin homolog siRNA repairs complete spinal cord injury. ACS Nano. (2019) 13:10015–28. doi: 10.1021/acsnano.9b01892
130. Zeng H, Liu N, Yang YY, Xing HY, Liu XX, Li F, et al. Lentivirus-mediated downregulation of α-synuclein reduces neuroinflammation and promotes functional recovery in rats with spinal cord injury. J Neuroinflamm. (2019) 16:283. doi: 10.1186/s12974-019-1658-2
131. Rong Y, Wang J, Hu T, Shi Z, Lang C, Liu W, et al. Ginsenoside rg1 regulates immune microenvironment and neurological recovery after spinal cord injury through MYCBP2 delivery via neuronal cell-derived extracellular vesicles. Adv Sci (Weinh). (2024) 11:e2402114. doi: 10.1002/advs.202402114
132. Wang S, Li G, Liang X, Wu Z, Chen C, Zhang F, et al. Small extracellular vesicles derived from altered peptide ligand-loaded dendritic cell act as A therapeutic vaccine for spinal cord injury through eliciting CD4(+) T cell-mediated neuroprotective immunity. Adv Sci (Weinh). (2024) 11:e2304648. doi: 10.1002/advs.202304648
133. Li Y, He X, Kawaguchi R, Zhang Y, Wang Q, Monavarfeshani A, et al. Microglia-organized scar-free spinal cord repair in neonatal mice. Nature. (2020) 587:613–8. doi: 10.1038/s41586-020-2795-6
134. Brennan FH, Swarts EA, Kigerl KA, Mifflin KA, Guan Z, Noble BT, et al. Microglia promote maladaptive plasticity in autonomic circuitry after spinal cord injury in mice. Sci Transl Med. (2024) 16:eadi3259. doi: 10.1126/scitranslmed.adi3259
135. Neel DV, Basu H, Gunner G, Bergstresser MD, Giadone RM, Chung H, et al. Gasdermin-E mediates mitochondrial damage in axons and neurodegeneration. Neuron. (2023) 111:1222–40.e9. doi: 10.1016/j.neuron.2023.02.019
136. Tansley S, Uttam S, Ureña Guzmán A, Yaqubi M, Pacis A, Parisien M, et al. Single-cell RNA sequencing reveals time- and sex-specific responses of mouse spinal cord microglia to peripheral nerve injury and links ApoE to chronic pain. Nat Commun. (2022) 13:843. doi: 10.1038/s41467-022-28473-8
137. Mattei D, Ivanov A, Hammer J, Ugursu B, Schalbetter S, Richetto J, et al. Microglia undergo molecular and functional adaptations to dark and light phases in male laboratory mice. Brain Behav Immun. (2024) 120:571–83. doi: 10.1016/j.bbi.2024.07.007
138. Akhmetzyanova ER, Zhuravleva MN, Timofeeva AV, Tazetdinova LG, Garanina EE, Rizvanov AA, et al. Severity- and time-dependent activation of microglia in spinal cord injury. Int J Mol Sci. (2023) 24:8294. doi: 10.3390/ijms24098294
139. Bai Y, Pan Y, and Liu X. Mechanistic insights into gasdermin-mediated pyroptosis. Nat Rev Mol Cell Biol. (2025) 1–21. doi: 10.1038/s41580-025-00837-0
140. Wang S, Yang L, Wu Z, Li C, Wang S, Xiao Z, et al. Ferroptosis-related genes participate in the microglia-induced neuroinflammation of spinal cord injury via NF-κB signaling: evidence from integrated single-cell and spatial transcriptomic analysis. J Transl Med. (2025) 23:43. doi: 10.1186/s12967-025-06095-0
Keywords: traumatic spinal cord injury, pyroptosis, microglia, inflammation, NLRP3, GSDMD
Citation: Shi L, Qian Q, Xie J, Yang T, Zhao X, Meng X, Dai J and Jin Q (2025) Microglial pyroptosis as a therapeutic target after traumatic spinal cord injury: current progress and future directions. Front. Immunol. 16:1649790. doi: 10.3389/fimmu.2025.1649790
Received: 20 June 2025; Accepted: 05 August 2025;
Published: 22 August 2025.
Edited by:
Philippe Saas, Etablissement Français du Sang AuRA, FranceReviewed by:
Ziheng Pu, Daping Hospital, ChinaCopyright © 2025 Shi, Qian, Xie, Yang, Zhao, Meng, Dai and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangqi Meng, ZHJtZW5neGlhbmdxaUAxNjMuY29t; Jingang Dai, emhvbmd5aWRhaUBvdXRsb29rLmNvbQ==; Qiguan Jin, anFneXp1QHNpbmEuY29t
†These authors have contributed equally to this work