 Jianbiao Xu1
Jianbiao Xu1 Lei Zou
Lei Zou- 1Department of General Surgery, The First People’s Hospital of Yunnan Province, The Affiliated Hospital of Kunming University of Science and Technology, Kunming, Yunnan, China
- 2Department of Radiology, The First People’s Hospital of Yunnan Province, The Affiliated Hospital of Kunming University of Science and Technology, Kunming, Yunnan, China
- 3Department of Hepatobiliary and Pancreatic Surgery, The First People’s Hospital of Yunnan Province, The Affiliated Hospital of Kunming University of Science and Technology, Kunming, Yunnan, China
- 4Department of Epidemiology and Health Statistics, School of Public Health, Kunming Medical University, Kunming, Yunnan, China
Pancreatic ductal adenocarcinoma (PDAC) remains a devastating malignancy characterized by profound lethality, aggressive local invasion, dismal prognosis, and significant resistance to existing therapies. Two critical biological features underpin the challenges in treating PDAC: extensive perineural invasion (PNI), the process by which cancer cells infiltrate and migrate along nerves, and a profoundly immunosuppressive, or “cold,” tumor microenvironment (TME). PNI is not only a primary route for local tumor dissemination and recurrence but also a major contributor to the severe pain often experienced by patients. Concurrently, the PDAC TME is typified by a dense desmoplastic stroma, hypoxia, and an abundance of immunosuppressive cells—including cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs)—while lacking sufficient infiltration of effector T cells, rendering it largely unresponsive to immunotherapies like checkpoint inhibitors. Although historically studied as separate entities, accumulating evidence reveals a deep-seated and complex bidirectional crosstalk between the neural components involved in PNI and the immune and stromal cells constituting the TME. Key cellular mediators, such as CAFs and TAMs, and shared signaling pathways, including the CXCL12/CXCR4 axis, TGF-β signaling, and neurotrophin pathways (e.g., NGF/TrkA), appear to act as critical nodes, coordinating the progression of PNI while simultaneously shaping and maintaining the immunosuppressive TME. This review synthesizes the current understanding of these intricate neuro-immune interactions in PDAC. We delineate the molecular and cellular mechanisms governing this crosstalk and explore how targeting these shared regulatory networks presents novel therapeutic opportunities, potentially disrupting PNI while concurrently “heating” the cold TME to overcome immunotherapy resistance. Elucidating this interplay is crucial not only for a deeper comprehension of PDAC’s invasive and metastatic mechanisms but also for uncovering new therapeutic vulnerabilities to improve patient outcomes.
1 Introduction
1.1 Pancreatic ductal adenocarcinoma: an unmet clinical challenge
Pancreatic ductal adenocarcinoma (PDAC) represents a major global health burden, characterized by a steadily increasing incidence and a mortality rate that closely mirrors its incidence (1, 2). It currently ranks as a leading cause of cancer-related deaths worldwide and is projected to become the second leading cause in Western countries by 2030 (3). The prognosis for PDAC patients remains exceptionally poor, with the overall 5-year survival rate being approximately 10-13%, a figure that has seen only marginal improvement despite decades of research (4).
PDAC typically arises from precursor lesions within the pancreatic ducts, such as pancreatic intraepithelial neoplasia (PanIN), or through acinar-to-ductal metaplasia (ADM) (5). Its development is driven by a characteristic sequence of genetic alterations, most notably activating mutations in the KRAS oncogene (present in >90% of cases) and inactivating mutations in tumor suppressor genes like CDKN2A (p16), TP53, and SMAD4 (6). Established risk factors include smoking, chronic pancreatitis, obesity, family history, and notably, type 2 diabetes (7).
The clinical management of PDAC is challenging. Due to the lack of specific early symptoms and reliable screening methods, the majority of patients (~80%) are diagnosed at advanced stages, precluding potentially curative surgical resection (3). Even for the minority who undergo surgery, recurrence rates are exceedingly high (8). Furthermore, PDAC exhibits significant intrinsic and acquired resistance to conventional treatments, including chemotherapy and radiotherapy (9). Contemporary approaches, such as targeted therapies and immunotherapies, have yielded limited success in unselected patient populations (10, 11).
1.2 Perineural invasion and the immunosuppressive TME: key features of PDAC aggressiveness
Two biological characteristics are major contributors to the aggressive nature and resistance to therapy of PDAC: perineural invasion (PNI) and the unique tumor microenvironment (TME). PNI, the infiltration of cancer cells along and within nerve structures, is an almost universal histological hallmark of PDAC, observed in 70-100% of cases, often even in early precursor lesions (12). This neurotropic behavior is an active invasion pathway facilitating local tumor spread, contributing significantly to post-surgical recurrence, and generating the debilitating pain associated with the disease (13).
Parallel to PNI, the PDAC TME presents a formidable barrier to treatment. It is characterized by an extensive desmoplastic reaction—a dense fibrotic stroma rich in extracellular matrix (ECM) components that can constitute up to 90% of the tumor mass (14). This stroma creates a hypoxic, hypovascular, and high-pressure environment that impedes the delivery of therapeutic agents and the infiltration of immune cells. Crucially, the PDAC TME is profoundly immunosuppressive, often described as immunologically “cold” (15). It is heavily infiltrated by immunosuppressive cell populations, including various subtypes of cancer-associated fibroblasts (CAFs), M2-polarized tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs). Conversely, it typically lacks significant infiltration of cytotoxic CD8+ T lymphocytes (CTLs). This landscape is a primary reason for the failure of immune checkpoint inhibitors (ICIs) in most PDAC patients (15, 16).
1.3 The emerging significance of neuro-immune crosstalk in PDAC
Historically, research into PDAC progression often focused on PNI and TME immunology in relative isolation. However, a growing body of evidence indicates that these phenomena are closely interconnected through complex, bidirectional signaling pathways—a concept referred to as neuro-immune crosstalk (5). Nerves and their associated signaling molecules (neurotransmitters, neurotrophins) can directly influence the function of immune and stromal cells, contributing to the immunosuppressive milieu (17). Conversely, components of the TME, including cancer cells, CAFs, and immune cells like TAMs, secrete factors that actively promote nerve growth, remodeling, and invasion, thereby facilitating PNI (18, 19).
This review is centered on the hypothesis that this neuro-immune crosstalk is a fundamental aspect of PDAC biology, where shared mediators mechanistically link PNI and the establishment of the cold, immunosuppressive TME (20). We aim to synthesize the current understanding of these interactions, dissect the key mechanisms, and evaluate the potential of targeting this axis as a novel therapeutic strategy. By disrupting pathways that simultaneously drive nerve invasion and immune suppression, it may be possible to inhibit local spread, alleviate pain, and “heat up” the TME, rendering it more susceptible to immunotherapy and improving outcomes for patients with PDAC (21).
2 Perineural invasion in PDAC pathogenesis
2.1 Definition, prevalence, and pathological features
Perineural invasion (PNI) is defined histologically as the presence of cancer cells in close proximity to nerves, specifically within the epineural, perineural, or endoneural spaces of the nerve sheath (8). A commonly used criterion requires cancer cells to track along or surround at least 33% of the nerve’s circumference, or to be present within any of the three nerve sheath layers (8, 22). Some studies further distinguish between invasion confined to the perineurial space (PNI) and deeper invasion into the endoneurium, affecting Schwann cells and axons directly, termed endoneurial or intraneural invasion (ENI/INI) (23). This distinction may hold prognostic significance, as ENI has been associated with more severe pain and potentially worse outcomes compared to PNI alone (24).
While PNI occurs in various solid tumors, its prevalence in PDAC is exceptionally high, ranging from 70% to nearly 100% in surgical specimens, far surpassing rates seen in cancers of the prostate, head and neck, or colorectum (8). It is suggested that PNI could be detected in virtually all PDAC cases if sufficient pathological sections are examined (16). Importantly, PNI is not merely a feature of advanced disease; it is frequently observed in early-stage PDAC and even within precursor PanIN lesions, indicating it is an early event in pancreatic carcinogenesis (5). Pathologically, PNI in PDAC is often associated with “neural remodeling,” characterized by nerve hypertrophy (increased size), increased nerve density, neurogenic inflammation, and signs of neuronal damage (25).
2.2 Molecular and cellular mechanisms driving PNI
PNI is an active biological process involving reciprocal communication between cancer cells, neural cells (neurons and Schwann cells), and various components of the TME within a “perineural niche” (26). The process involves several key steps:
Mutual chemotaxis: Nerves and cancer cells attract each other. Cancer cells release neurotrophins like Nerve Growth Factor (NGF), Brain-Derived Neurotrophic Factor (BDNF), Glial Cell Line-Derived Neurotrophic Factor (GDNF), and artemin (ARTN), which bind to receptors on nerve cells, promoting neurite outgrowth toward the tumor (8). Conversely, neural structures release factors such as NGF, GDNF, ARTN, and CXCL12 (SDF-1), which act as chemoattractants for cancer cells expressing corresponding receptors (21). Pancreatic stellate cells (PSCs) contribute via tenascin C (27). This reciprocal signaling establishes a chemotactic gradient guiding cancer cell migration toward nerves.
Extracellular Matrix (ECM) remodeling: To invade, cancer cells secrete matrix metalloproteinases (MMPs), particularly MMP2 and MMP9, to degrade the surrounding ECM (8). The activation of these MMPs is driven by signaling pathways initiated by factors like GDNF (via RET-PI3K/AKT and RAS/ERK pathways), L1CAM (via STAT3), and galectin-1 (LGALS1) from PSCs (via SRC signaling) (8, 28). Macrophages, recruited by Schwann cell-derived CCL2, release Cathepsin B, which degrades collagen IV in the perineurium (29). CAFs are major contributors to this process by producing collagen and MMPs (30).
Adhesion and invasion: Cancer cells adhere to the nerve sheath. This process is mediated by specific adhesion molecules, such as Mucin 1 (MUC1) on cancer cells, binding to Myelin-Associated Glycoprotein (MAG) on nerves, and interactions involving NCAM1 and β1 integrin (8). Invasion is further promoted by factors released from damaged nerves (e.g., PAP/REG3A), signaling via SDC3/PTN, metabolic support (e.g., serine from axons), and factors from Schwann cells (e.g., TGF-β enhancing invasiveness, CCL2 recruiting macrophages) (20). Schwann cells can even create “tracks” (TASTs) that guide cancer cells as they migrate (31).
Immune evasion within the perineural niche: Cancer cells evade local immune surveillance, a process aided by neurotransmitters like acetylcholine (ACh) (5) and norepinephrine (NE) (8) that suppress CD8+ T cell function. M2-polarized TAMs and CAFs also contribute to creating an immunosuppressive environment (32). CAFs contribute by promoting angiogenesis and secreting factors like IL-6 (33).
Nerve remodeling and regeneration: triggers nerve remodeling and regeneration, which paradoxically facilitates further cancer invasion (34). Damage to axons induces the release of factors like neuregulin 1 (NRG1), which activates Schwann cells to proliferate and migrate, creating pathways for tumor cells (8). This process is further stimulated by signals from CAFs, such as SLIT2 and Eph-B, and immune cells, including IL-6, which enhance Schwann cell migration and neuronal plasticity (35). Axon guidance molecules like SEMA3D (20) and the migration of neural precursor cells (36) also contribute to this environment.
Therefore, PNI is not a passive process but an active, orchestrated invasion driven by complex reciprocal signaling between cancer cells, nerves, and the TME, as illustrated in Figure 1. This molecular interplay must be the focus of future therapeutic strategies.
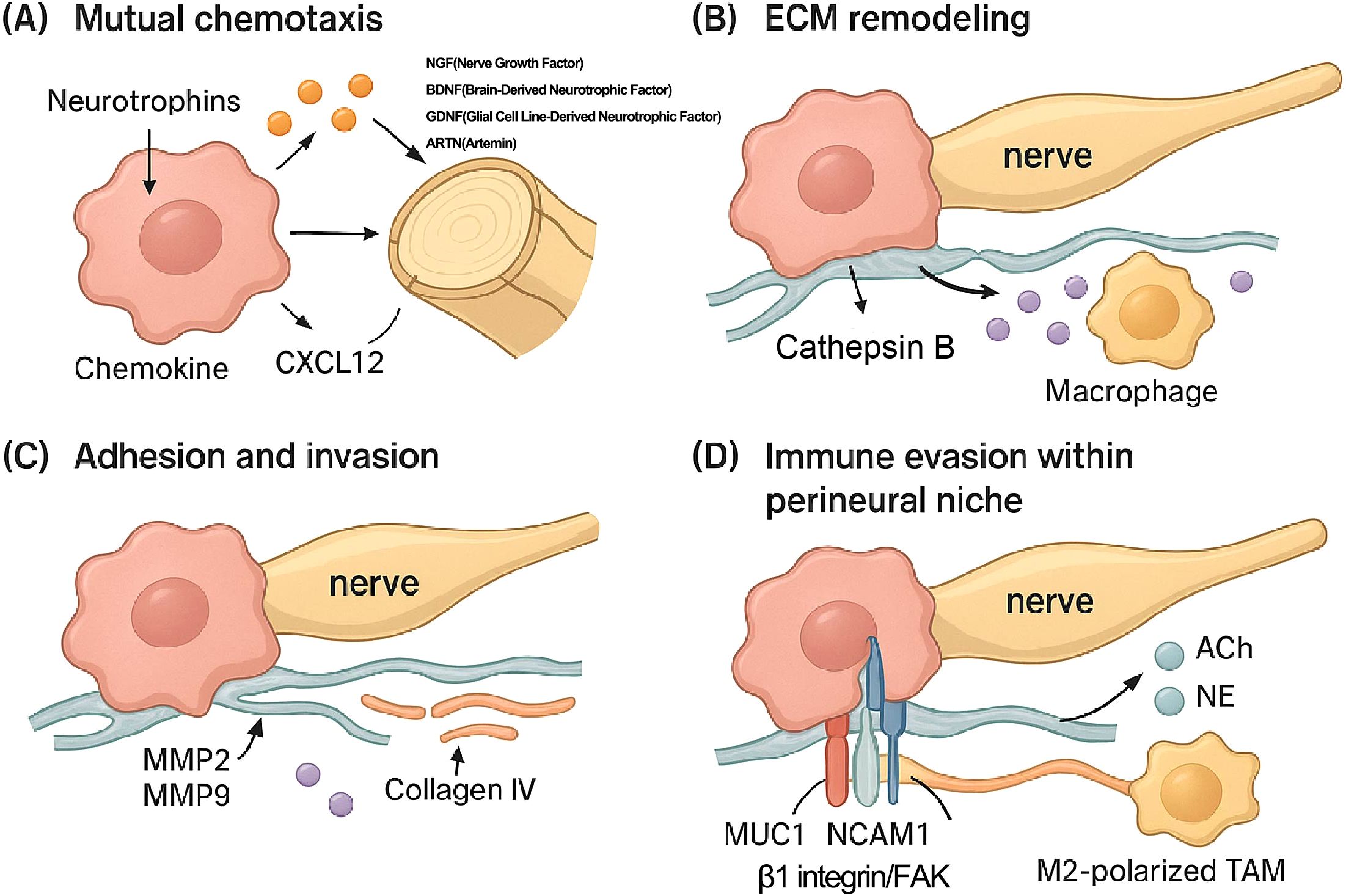
Figure 1. Molecular and cellular mechanisms driving perineural invasion (PNI). This figure details the key molecular and cellular steps of PNI in PDAC. (A) Mutual Chemotaxis: Cancer cells secrete neurotrophins (NGF, Nerve Growth Factor; BDNF, Brain-Derived Neurotrophic Factor; GDNF, Glial Cell Line-Derived Neurotrophic Factor; ARTN, Artemin) that promote neurite outgrowth. Conversely, nerves and associated cells release chemoattractants like CXCL12, guiding cancer cell migration toward the nerve. (B) ECM Remodeling: Macrophages recruited to the perineural niche secrete proteases, such as Cathepsin B, which degrade extracellular matrix (ECM) components of the nerve sheath (e.g., collagen IV), facilitating cancer cell entry. (C) Invasion: Cancer cells secrete matrix metalloproteinases (MMP2, MMP9) to further degrade the ECM and invade the perineural space. (D) Adhesion and Immune Evasion: Within the perineural niche, neurotransmitters (ACh, acetylcholine; NE, norepinephrine) released from nerves suppress local immune responses. M2-polarized tumor-associated macrophages (TAMs) further contribute to this immune suppression. Adhesion of the cancer cell to the nerve is critically mediated by molecules expressed on the cancer cell surface, such as Mucin 1 (MUC1), NCAM1, and the β1 integrin/focal adhesion kinase (FAK) signaling complex.
2.3 Clinical impact: PNI, pain, recurrence, and prognosis
The clinical consequences of PNI are significant. It is a primary mechanism underlying the severe, often difficult to manage, abdominal and back pain experienced by a majority (up to 80%) of PDAC patients (12). This pain is often neuropathic in nature, resulting from direct nerve damage, inflammation within the perineural niche, and sensitization of nerve endings by mediators released from cancer and immune cells (16). Beyond pain, PNI serves as a critical pathway for tumor dissemination, facilitating local spread and contributing significantly to the high rates of local and regional recurrence observed even after surgery (5). The presence of residual cancer cells within nerve sheaths after resection is thought to be a major factor in treatment failure (37). Consistent with its role in invasion and recurrence, PNI is widely recognized as an independent negative prognostic factor in PDAC (12), correlated with shorter overall survival (OS) and disease-free survival (DFS) (38).
3 The immunologically “cold” tumor microenvironment of PDAC
The TME of PDAC is a complex ecosystem comprising cellular and non-cellular components that profoundly influence tumor biology (1) and establish a profoundly immunosuppressive state (15), as illustrated in Figure 2.
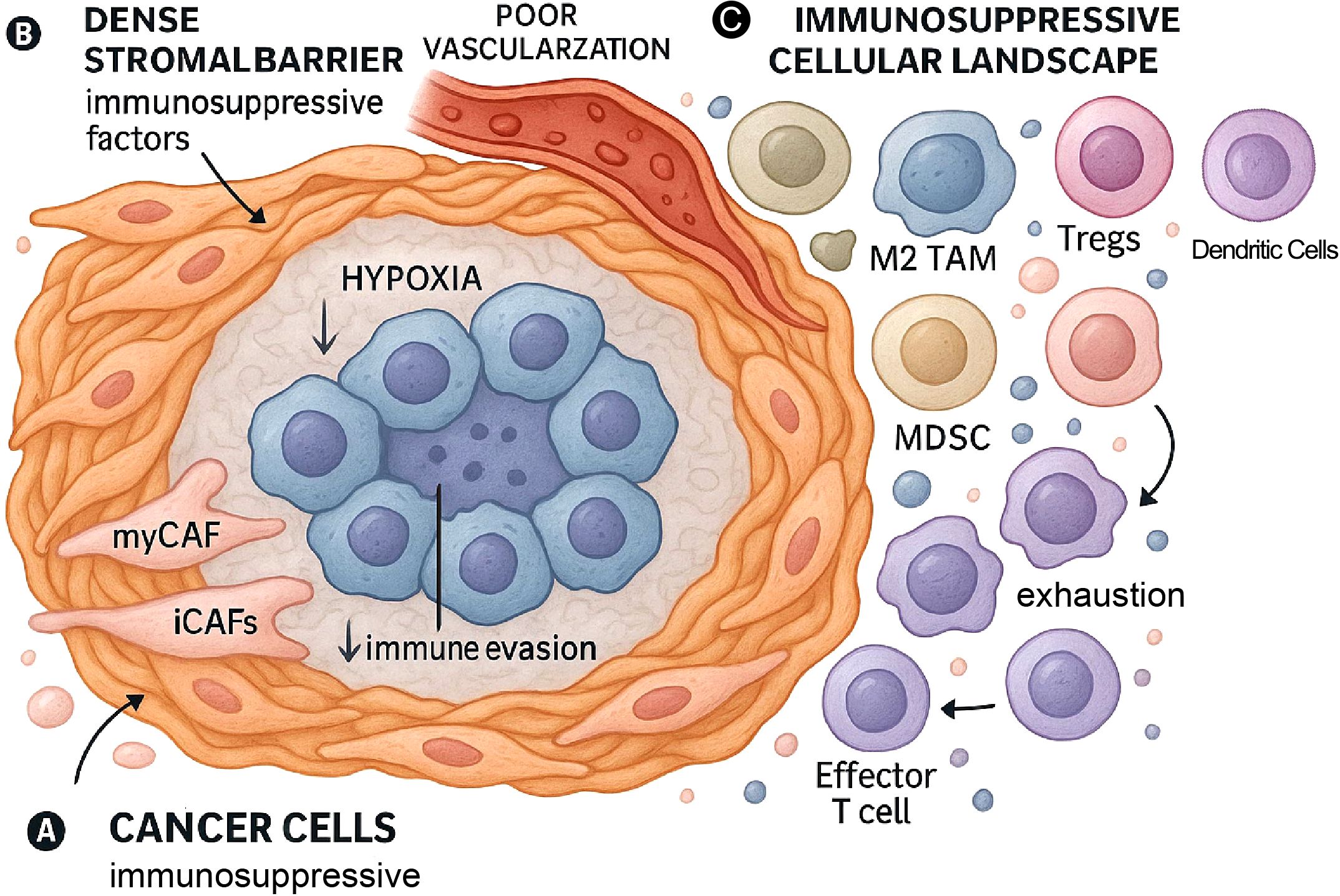
Figure 2. The immunologically “cold” tumor microenvironment (TME) of PDAC. This figure illustrates the key components that establish the immunosuppressive TME in PDAC. (A) Cancer Cells: Tumor cells themselves contribute to the immunosuppressive environment through various mechanisms. (B) Dense Stromal Barrier & Hypoxia: The TME is characterized by a dense desmoplastic stroma, largely produced by cancer-associated fibroblasts (CAFs). This stroma forms a physical barrier that, along with poor vascularization, leads to hypoxia and impedes immune cell infiltration. (C) Immunosuppressive Cellular Landscape: The TME is dominated by immunosuppressive cells, including phenotypically diverse CAFs, M2-polarized TAMs that exist on a spectrum of activation states, myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and dysfunctional dendritic cells (DCs). This environment is characterized by a scarcity of functional effector T cells.
3.1 Cellular and non-cellular composition: architects of immunosuppression
The immunosuppressive landscape of the PDAC TME is orchestrated by several key components:
Dense stroma (Desmoplasia): PDAC is notorious for its extensive desmoplastic reaction, a dense fibrotic stroma that can account for the vast majority of the tumor volume (14). This stroma is primarily composed of excessive ECM proteins, such as collagen, produced by CAFs (33). The dense matrix creates a physical barrier that hinders the infiltration of effector immune cells (15) and limits the efficacy of therapeutic agents (39).
Hypoxia: The dense stroma and compromised vasculature lead to significant regions of hypoxia (low oxygen) within the TME. Hypoxia activates hypoxia-inducible factor (HIF) signaling pathways (40), which not only drive tumor cell adaptation but also contribute significantly to immunosuppression (41).
Immunosuppressive cellular infiltrate: The cellular landscape is dominated by cells that actively suppress anti-tumor immunity:
Cancer-associated fibroblasts (CAFs): These are abundant stromal cells that act as key orchestrators of the TME (21). Beyond producing the ECM, CAFs actively contribute to immune suppression by secreting factors like CXCL12, which can sequester T cells in the stroma, and Transforming Growth Factor-β (TGF-β), a potent immunosuppressive cytokine (14). CAFs exhibit significant heterogeneity, with subtypes like inflammatory CAFs (iCAFs) (19) and myofibroblastic CAFs (myCAFs) having distinct roles in immune modulation and T-cell exclusion (42). The existence of potentially tumor-restraining CAF subsets further complicates therapeutic targeting (21, 43).
Tumor-associated macrophages (TAMs): TAMs are typically the most abundant immune cells within the PDAC TM (44). The traditional M1 (anti-tumor) vs. M2 (pro-tumor) dichotomy is now considered an oversimplification of their complex biology. Emerging evidence highlights a spectrum of activation states and significant functional heterogeneity within TAM populations (45). For instance, the “M2-like” phenotype encompasses multiple distinct subsets (e.g., M2a, M2b, M2c, M2d), and misinterpreting this diversity can impede the development of effective therapies. In PDAC, TAMs are predominantly polarized toward a pro-tumor, immunosuppressive state. They suppress T-cell activity via IL-10, TGF-β, and PD-L1 expression, and deplete essential amino acids like arginine via arginase-1 (Arg1) (46). The polarization and function of TAMs are dynamically regulated by various signals within the TME, including cytokines, metabolic cues, exosomes, and non-coding RNAs (47, 48).
Myeloid-derived suppressor cells (MDSCs): These are a heterogeneous population of immature myeloid cells that potently inhibit the cytotoxic functions of both T cells (15) and NK cells (42).
Regulatory T cells (Tregs): PDAC tumors are often enriched with CD4+FoxP3+ Tregs (15), which actively suppress the proliferation and effector functions of conventional T cells (49).
Dendritic cells (DCs): As professional antigen-presenting cells (APCs), DCs are essential for priming anti-tumor T-cell responses. However, their function is severely compromised in the PDAC TME. The dense stroma can limit their migration, while immunosuppressive factors from CAFs and TAMs inhibit DC maturation and antigen-presenting capacity. This impaired DC function is a key reason for poor T-cell priming and contributes significantly to the “cold” immune landscape (48–50).
Paucity of effector immune cells: A key feature of the “cold” PDAC TME is the scarcity or dysfunction of anti-tumor effector immune cells, particularly cytotoxic CD8+ T lymphocytes (CTLs) (15). This is attributed to physical exclusion, active immunosuppression, and poor immunogenicity due to a low tumor mutational burden (TMB) (51). The few T cells that do infiltrate often display markers of exhaustion (e.g., high PD-1, TIM-3, LAG-3 expression) (52).
3.2 Mechanisms underpinning immune evasion and immunotherapy resistance
The immune evasion of pancreatic ductal adenocarcinoma (PDAC) is driven by a multi-layered suppressive TME (21). This barrier consists of a dense stroma that physically blocks T-cell infiltration (53) and a variety of immunosuppressive cells that actively neutralize immune responses (54). This issue is exacerbated by the tumor’s intrinsically low immunogenicity due to a low mutation burden (51), which promotes T-cell exhaustion (55) and is enhanced by the cancer cells’ own escape tactics (56).
Consequently, PDAC is characterized as an immunologically “cold” tumor, demonstrating profound resistance to ICIs (15). Objective response rates to ICI monotherapy are minimal, typically below 2%, with significant efficacy confined to the rare, more immunogenic subset of tumors with high microsatellite instability (MSI-H/dMMR). The TME is an actively constructed barrier driving multi-therapy resistance (47); therefore, effective treatment strategies must aim to dismantle this suppressive architecture (21), not just stimulate immunity (50). This effort is complicated by substantial TME heterogeneity (56), suggesting that broad targeting may fail and highlighting the need for personalized therapeutic strategies (57). As shown in Figure 3, emerging evidence also links this cold TME to perineural invasion through shared molecular and cellular mediators.
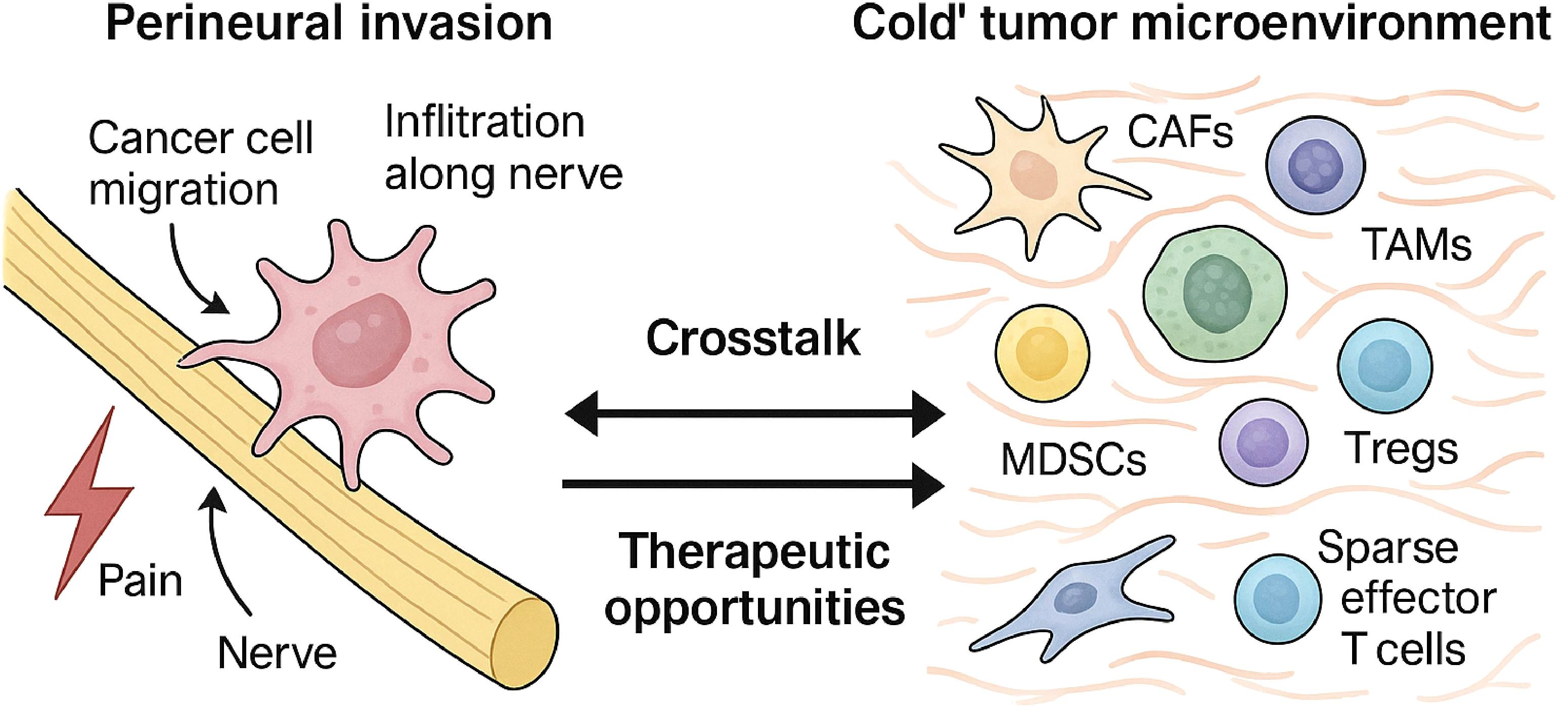
Figure 3. Bidirectional crosstalk between perineural invasion and the immunosuppressive TME. This figure illustrates the profound interconnection between PNI and the TME in PDAC. The left panel depicts PNI, where cancer cells invade and migrate along nerve structures. The right panel illustrates the immunosuppressive TME, populated by CAFs, TAMs, and Tregs, with a notable lack of effector T cells. The central arrows signify the critical bidirectional crosstalk driven by shared molecular mediators—including the CXCL12/CXCR4 axis, TGF-β signaling, and neurotrophin pathways (e.g., NGF/TrkA)—that mechanistically link PNI and immune suppression, creating a self-reinforcing cycle that promotes tumor aggression.
4 The intricate crosstalk between nerves and the immune microenvironment in PDAC
The realization that PNI and the cold TME are co-conspirators in PDAC progression stems from the growing understanding of the bidirectional communication between neural elements and the immune/stromal components within the tumor (5). This neuro-immune crosstalk involves direct cell interactions and soluble mediators, creating a complex regulatory network.
4.1 Neural regulation of the TME: impact of nerves, neurotransmitters, and neurotrophins
Nerves innervating the tumor are not passive structures but actively modulate the TME:
Neurotransmitter modulation of immune cells: Neurons release neurotransmitters that bind to receptors on immune cells, directly influencing their behavior (58).
Norepinephrine (NE): Released by sympathetic nerves, NE generally acts via β-adrenergic receptors to suppress anti-tumor immunity by inhibiting CTL activity (8) and promoting M2 TAM polarization (58).
Acetylcholine (ACh): Released by parasympathetic (vagal) nerves, ACh can impair CD8+ T cell recruitment and function via nicotinic ACh receptors (5).
GABA (Gamma-Aminobutyric Acid): Generally considered inhibitory, GABA can suppress T cell and macrophage activation (58).
Vasoactive Intestinal Peptide (VIP): This neuropeptide can inhibit T cell anti-tumor activity and promote the development of immunosuppressive Treg and Th2 cells (5).
Other Neurotransmitters: Serotonin and dopamine also have complex modulatory effects on T cells and macrophages within the TME (58).
Neurotrophin modulation of immune cells: Neurotrophins, primarily known for their roles in PNI, also interact with the immune system (59):
Nerve growth factor (NGF): NGF and its receptor TrkA are expressed by various immune cells (6). NGF is a key mediator of neurogenic inflammation (60) and can influence T-cell responses (61).
Brain-derived neurotrophic factor (BDNF): BDNF and its receptor TrkB are also implicated in immune modulation, with BDNF being produced by T cells, suggesting autocrine/paracrine loops (62).
Collectively, these neural signals contribute significantly to establishing and maintaining the immunosuppressive TME (17).
4.2 TME regulation of neural processes: promoting PNI and neural remodeling
The crosstalk is bidirectional. PDAC cells secrete neurotrophic factors (NGF, BDNF, GDNF) that stimulate neurite outgrowth (8) and attract nerve fibers (63). Stromal and immune cells also contribute significantly:
CAFs and PSCs secrete factors like SLIT2, Tenascin C, and IL-6 that promote neurite outgrowth and Schwann cell migration (8).
TAMs, recruited by Schwann cell-derived CCL2, secrete GDNF, which promotes PNI (5). Mast cells can secrete IL-6, contributing to Schwann cell plasticity (8). Macrophage-derived MIF acting via CD74 can also increase GDNF levels (64). TAMs also release proteases like Cathepsin B that degrade the protective nerve sheath, facilitating invasion (29, 65).
This TME-driven promotion of nerve growth and invasion creates a positive feedback loop that amplifies immunosuppression and stimulates tumor growth (34).
4.3 Metabolic interplay in the neuro-immune-cancer axis
Beyond signaling crosstalk, metabolic interactions are emerging as another crucial layer of complexity in the neuro-immune-cancer axis within the PDAC TME (6). Metabolic reprogramming is a fundamental hallmark of cancer, enabling cells to meet the bioenergetic and biosynthetic demands of rapid proliferation in a harsh, nutrient-deprived, and hypoxic environment (66). This reprogramming is heavily influenced by interactions within the TME.
Nerve-cancer metabolic crosstalk: Evidence suggests nerves can directly fuel PDAC progression during PNI. Axons and DRG can secrete serine, an amino acid utilized by PDAC cells for proliferation, particularly under nutrient stress conditions encountered during invasion (8). Glutamate released from nerve endings can activate NMDARs on PDAC cells, triggering downstream signaling (CaMKII/ERK/METTL3) that upregulates hexokinase 2 (HK2), promoting glycolysis (the Warburg effect) and PNI. Furthermore, signaling pathways crucial for PNI, like NGF/TrkA, can directly impact cancer cell metabolism by upregulating glucose transporters like GLUT1, further enhancing glycolytic flux (60).
Immune-cancer metabolic crosstalk: The TME’s metabolic landscape is significantly shaped by immune cells. Immunosuppressive cells like TAMs and MDSCs contribute to the hypoxic and acidic conditions that drive metabolic shifts in cancer cells (66). Moreover, there is intense metabolic competition within the TME. Immunosuppressive cells actively deplete nutrients essential for effector T cell function. For example, MDSCs and TAMs express high levels of Arg1 and iNOS, which consume arginine, an amino acid critical for T cell activation, proliferation, and survival, thereby contributing to T cell dysfunction (44).
Nerve-immune metabolic links: While less explored, potential metabolic interactions between nerves and immune cells within the TME likely exist. Neural signals might influence the metabolic state of immune cells, or vice versa. For instance, metabolic pathways involved in endocannabinoid and polyamine metabolism have been implicated in PNI and associated pain, potentially linking metabolic state to neuro-inflammation (12). The investigation of how nerve-derived metabolites or neurotransmitter signaling impacts immune cell metabolism (e.g., glycolysis vs. oxidative phosphorylation balance in T cells or TAMs) represents an important area for future research.
These findings indicate that the crosstalk governing PDAC progression involves not only complex signaling networks but also intricate metabolic dependencies and competition between cancer cells, nerves, and immune cells. This metabolic interplay likely influences PNI, immune suppression, and overall tumor growth, adding another dimension to the challenge of targeting the TME (20).
The neuro-immune crosstalk in PDAC thus establishes a self-reinforcing cycle driving tumor aggression. Nerves release signals that suppress anti-tumor immunity and promote pro-tumor immune cells, while the TME, including cancer cells, CAFs, and TAMs, secretes factors that stimulate nerve growth and invasion. This increased innervation further amplifies the immunosuppressive signals, creating a vicious loop (5). Breaking this cycle likely necessitates therapeutic strategies that simultaneously target both the neural signaling components and the mechanisms of immune suppression. Furthermore, the specific roles of different nerve types (sympathetic, parasympathetic, sensory) appear complex and context-dependent (17), suggesting that neuro-modulatory therapies must be carefully tailored based on the specific pathways involved and the stage of the disease.
5 Shared mediators and pathways orchestrating PNI and immune suppression
The crosstalk between PNI and the cold TME is orchestrated by specific molecular pathways and cellular mediators that function at the interface of these two processes.
5.1 The CXCL12/CXCR4 axis
The CXCL12/CXCR4 chemokine axis is pivotal in PDAC pathogenesis, driving both perineural invasion (PNI) and immune evasion (21). In the tumor microenvironment, CXCL12 secreted by stromal cells like CAFs/PSCs and potentially nerves (8) attracts CXCR4-overexpressing PDAC cells, guiding their migration toward neural structures and facilitating PNI (21). This interaction is clinically significant, as high CXCL12/CXCR4 expression is an independent negative prognostic factor associated with PNI (38), while the atypical receptor ACKR3 also promotes invasion (67). Concurrently, this axis creates an “immune-excluded” phenotype by using stromal CXCL12 to sequester CXCR4-positive T cells, preventing their infiltration into tumor nests (68). The axis’s immunomodulatory role is complex, as high CXCR4 expression, despite influencing the trafficking of immune cells like macrophages (66), also correlates with elevated inhibitory checkpoints such as PD-1/PD-L1, fostering a suppressed immune state (38). Thus, the CXCL12/CXCR4 axis functions as a critical node linking stromal activation with cancer cell invasion and profound immune dysfunction.
5.2 Transforming growth factor-β signaling
TGF-β is a pleiotropic cytokine that critically promotes both PNI and immune suppression within the PDAC TME (47). For PNI, TGF-β from stromal sources like Schwann cells and CAFs directly enhances PDAC cell invasive capacity (8), while also driving desmoplastic ECM remodeling and inducing an EMT program that increases cell motility (56). Simultaneously, TGF-β is one of the most potent immunosuppressive cytokines in the TME, secreted by cancer and stromal cells to inhibit the function of cytotoxic T cells and NK cells (56), promote regulatory T cells (Tregs), and polarize macrophages toward a suppressive M2 phenotype (69). The complexity of its role is highlighted by the “TGF-β paradox,” where its function switches from tumor-suppressive to pro-oncogenic during disease progression (56), a process influenced by factors like the frequent loss of SMAD4 in PDAC (70). Furthermore, non-canonical signaling pathways can promote aggressive phenotypes and PD-L1 upregulation (71). Thus, TGF-β signaling serves as a central node, fostering key mechanisms of PNI while orchestrating a profoundly immunosuppressive microenvironment.
5.3 Neurotrophin signaling pathways (NGF/TrkA, BDNF/TrkB)
These pathways are fundamental for the mutual chemotaxis that initiates PNI (8). Activation of Trk signaling promotes cancer cell proliferation, migration, and invasion (72). The NGF/TrkA pathway is also a key mediator of PNI-associated pain (12). These neurotrophins and their receptors are also expressed on various immune cells (59), participating in the complex neuro-immune dialogue (73).
5.4 Cancer-associated fibroblasts: heterogeneity and dual roles
CAFs are central, pleiotropic cells in the PDAC TME, actively driving both PNI and immune suppression (74). They facilitate PNI by producing and remodeling the dense ECM to create invasive tracks (8), with subtypes like αSMA-high myCAFs providing direct physical support (75), and by secreting numerous pro-invasive and neurotropic factors, including TGF-β, HGF, and SLIT2. As major architects of immunosuppression (76), CAFs construct a physical barrier, release potent inhibitory cytokines like TGF-β and IL-6 (77), recruit cells such as MDSCs and Tregs, and directly impair T cells via mechanisms like CXCL12 secretion (68). This dual functionality is governed by significant CAF heterogeneity: iCAFs are linked to inflammation and immunosuppression, myCAFs primarily contribute to ECM deposition, and apCAFs may induce T cell tolerance (75), with the existence of tumor-restraining subtypes further underscoring the complexity (78). Thus, CAFs represent a critical cellular hub that physically and chemically engineers the TME to promote invasion while orchestrating profound immunosuppression.
5.5 Tumor-associated macrophages: heterogeneity and dual roles
TAMs are abundant and functionally diverse cells that bridge PNI and immune suppression, often adopting an M2-like phenotype upon recruitment by factors such as CCL2 or CSF-1 (44). They actively facilitate PNI by degrading nerve barriers with enzymes like Cathepsin B, releasing pro-invasive factors like GDNF (8), and interacting with CAFs through mechanisms like LIF signaling to contribute to neural remodeling (79). Concurrently, M2-polarized TAMs are cornerstone immunosuppressive cells in the PDAC TME, potently inhibiting T and NK cell activity through secretion of IL-10 and TGF-β, expression of PD-L1, and recruitment of Tregs via chemokines like CCL22 (80). This convergence of PNI and immune suppression is driven by such shared cellular mediators and signaling nodes, with pleiotropic cells like TAMs and CAFs and pathways like CXCL12/CXCR4 and TGF-β mechanistically linking both processes (21). Therefore, targeting these central players offers a compelling ‘double hit’ strategy to simultaneously disrupt PNI and alleviate TME immunosuppression (Table 1).

Table 1. Key molecular pathways and cellular mediators linking PNI and immune suppression in PDAC.
6 Therapeutic strategies targeting the PNI-immune TME axis
Targeting the shared mediators of the PNI-TME crosstalk offers a promising strategy to simultaneously disrupt tumor invasion and “heat up” the cold TME (21). As illustrated in Figure 4, targeting the shared molecular pathways and cellular mediators that orchestrate the PNI-TME crosstalk offers promising strategies to simultaneously disrupt tumor invasion and ‘heat up’ the cold TME.
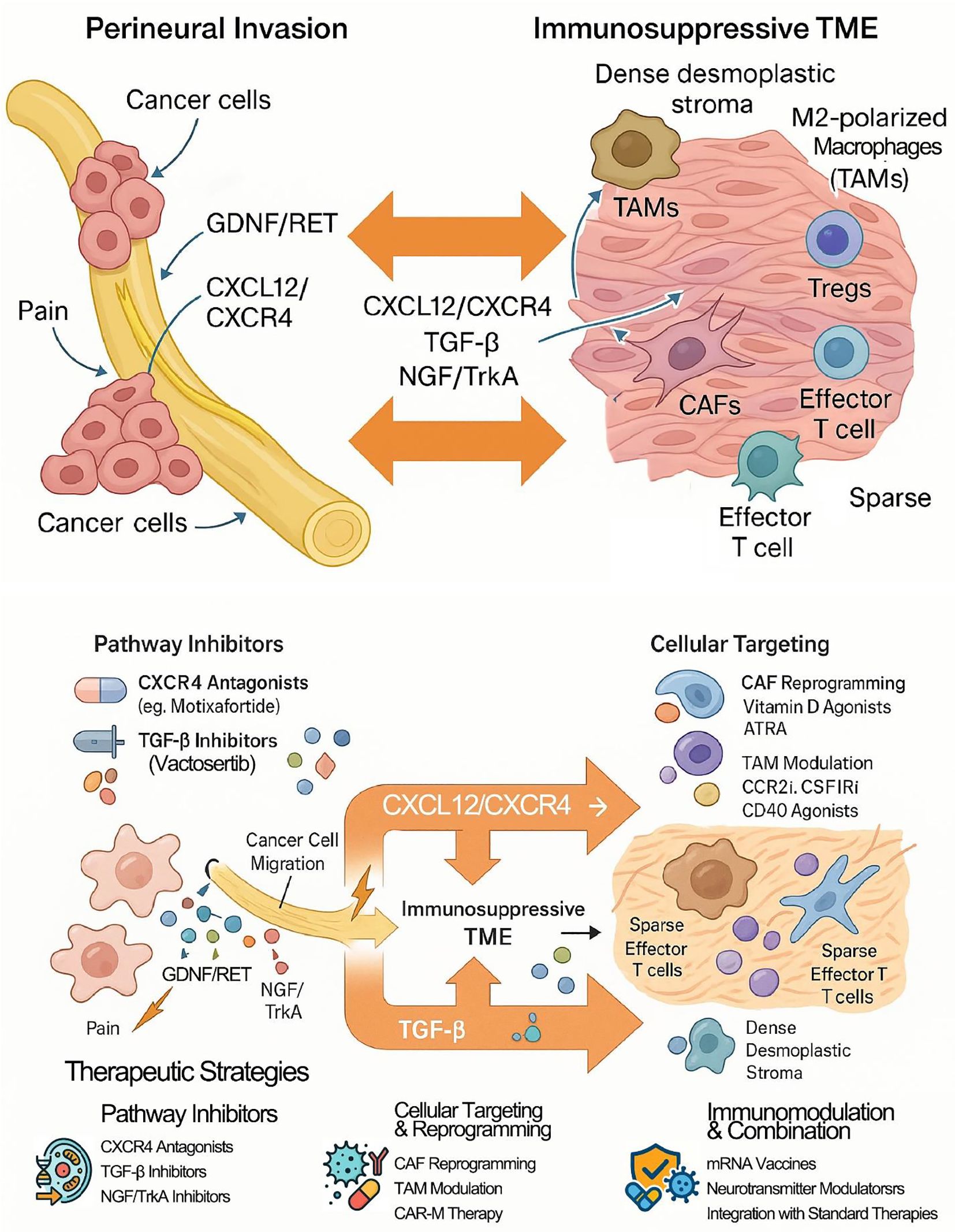
Figure 4. Neuro-immune crosstalk and therapeutic targets in pancreatic cancer. In pancreatic cancer (PDAC), perineural invasion (PNI) and the immunosuppressive tumor microenvironment (TME) form a vicious cycle driven by crosstalk through shared cells (e.g., CAFs, TAMs) and signaling pathways (e.g., CXCL12/CXCR4, TGF-β) that promotes tumor progression. Therapeutic strategies aim to disrupt this cycle by inhibiting key pathways, reprogramming stromal cells, and integrating emerging immunotherapies like mRNA vaccines. The ultimate goal is to simultaneously inhibit PNI while converting the immunologically "cold" TME into a "hot," therapy-responsive state.
6.1 Targeting shared molecular pathways
CXCR4 antagonists: Blocking the CXCL12/CXCR4 axis with agents like Plerixafor or Motixafortide aims to inhibit PNI and enhance T cell infiltration (85). The Phase IIa COMBAT trial (NCT02826486), combining Motixafortide with pembrolizumab and chemotherapy, showed promising signals of activity in metastatic PDAC (86).
TGF-β inhibitors: Inhibiting TGF-β signaling is an attractive strategy to reduce fibrosis and alleviate immunosuppression (70). However, the clinical development of some agents has faced challenges. For example, a Phase Ib trial combining the TGFBR1 inhibitor galunisertib with durvalumab showed limited clinical activity (87). The sentence structure describing the trial outcome has been corrected for clarity: The combination was tolerable, but clinical activity was limited (1 partial response, 7 stable diseases out of 32 patients), with a median PFS of 1.87 months and mOS of 5.7 months (87). It is noteworthy that the development of galunisertib for oncology indications was later discontinued. Other TGF-β inhibitors, such as the small molecule inhibitor Vactosertib or monoclonal antibodies targeting TGF-β, are under investigation in various cancers, representing alternative approaches to target this pathway (88).
Trk inhibitors: Targeting neurotrophin signaling, particularly NGF/TrkA, is under preclinical investigation as a strategy to inhibit PNI and alleviate associated pain (59). In mouse models, combining Trk inhibition with gemcitabine increased survival (89). Pan-Trk inhibitors like Larotrectinib are approved for rare TRK fusion-positive cancers, but preclinical studies using specific TrkA inhibitors or NGF-neutralizing antibodies have shown they can reduce the PNI potential of PDAC cells and inhibit neurite outgrowth (72).
6.2 Modulating key cellular players
Targeting CAFs: Given their heterogeneity, strategies are shifting from broad depletion to selective targeting or reprogramming (75). Approaches include targeting Fibroblast Activation Protein (FAP) or reprogramming CAFs toward a quiescent state using agents like Vitamin D receptor agonists (e.g., Calcipotriol) or all-trans retinoic acid (ATRA) (90) (91). Clinical trials investigating these approaches are ongoing (e.g., NCT03520790). More refined approaches aim to deplete subsets like Pdgfrb+ CAFs or adipose marker-expressing (ASC-like) CAFs, with the latter showing potential to enhance ICI efficacy in preclinical models (42). Targeting key signaling pathways within CAFs (e.g., JAK/STAT, HGF/c-Met) or inhibiting their ECM-modifying functions (e.g., LOX inhibitors) are also under investigation (92).
Targeting TAMs: Modulating the abundant and largely immunosuppressive TAM population is a key strategy to reprogram the TME (44). Approaches include inhibiting monocyte recruitment by blocking receptors like CCR2 and CSF1R (84), with a clinical trial combining the CCR2 antagonist PF-04136309 with FOLFIRINOX showing encouraging results in locally advanced PDAC (93); depleting existing TAMs via agents targeting CSF1R or other macrophage-specific markers (84); reprogramming M2 to M1 phenotypes using agents like TLR or CD40 agonists (94); and enhancing phagocytosis by blocking “don’t eat me” signals such as the CD47-SIRPα interaction (95). Targeting TAMs is frequently explored in combination with ICIs or other immunotherapies, aiming to reduce a major source of immunosuppression within the TME (44). Additionally, novel strategies such as CAR-macrophage (CAR-M) therapy are emerging, which engineer macrophages to directly target and phagocytose tumor cells (96, 97).
6.3 Neurotransmitter modulation strategies
Targeting neurotransmitter signaling is an emerging avenue:
β-blockers: Antagonizing NE signaling with drugs like propranolol has shown preclinical potential to reduce PNI and increase survival when combined with chemotherapy in KPC mouse models (a genetically engineered mouse model expressing oncogenic Kras and mutant Trp53, specifically KrasLSL-G12D/+; Trp53LSL-R172H/+; Pdx1-Cre) (89) (58). However, clinical data on the impact of β-blockers on cancer outcomes have been inconsistent, highlighting the need for a better understanding of specific β-AR subtype roles and patient selection (62).
VIP antagonists: Preclinical studies showed that blocking VIP signaling could synergize with anti-PD-1 therapy, enhancing T cell activation and recruitment (5).
6.4 Emerging strategies: mRNA neoantigen vaccines
A significant frontier in overcoming the poor immunogenicity of PDAC is personalized cancer vaccination. Recent breakthroughs with mRNA vaccine technology have shown remarkable promise. A notable study demonstrated that a personalized mRNA neoantigen vaccine (autogene cevumeran) could induce a substantial population of durable, polyfunctional CD8+ T cells targeting tumor-specific neoantigens in PDAC patients (98, 99). These vaccine-induced T cells persisted for up to two years and were associated with delayed tumor recurrence. This approach directly addresses the lack of pre-existing T-cell responses that limit ICI efficacy. By generating a potent de novo T-cell response, mRNA vaccines could potentially “heat up” the cold TME, making it more susceptible to checkpoint inhibition and other immunotherapies. Integrating such vaccination strategies with therapies that target the stromal and neural barriers of the TME represents a powerful future direction for combination treatments.
6.5 Combination therapies: disrupting PNI and “heating” the TME
Due to the complex nature of pancreatic cancer resistance, driven by PNI, stromal barriers, and immunosuppression, combination therapies are crucial for clinical advancement (100). Key strategies involve pairing agents that disrupt nerve-cancer signaling pathways, such as inhibitors of CXCR4, TGF-β, or Trk, with immunotherapies to amplify T cell responses (89). Another approach combines agents that modulate stromal cells like CAFs or TAMs with immune checkpoint inhibitors (ICIs) to dismantle physical and cellular barriers to immunity (44). Preclinical studies have also demonstrated durable responses by combining CXCR1/2 inhibition with T cell activating agents, or by targeting neurotransmitter pathways alongside ICIs (101).
These targeted combinations are frequently integrated with standard chemotherapy or radiotherapy. This integration aims to leverage the immunogenic cell death induced by conventional treatments, which can release tumor antigens and synergize with immunotherapy, despite the potential for inducing resistance (68). Examples include adding CXCR4 or CCR2 inhibitors to standard chemotherapy regimens, with or without ICIs (86) (93). The ultimate goal of these multifaceted strategies is to transform the immunologically “cold” and resistant tumor microenvironment into an inflamed, “hot” state that is susceptible to immune-mediated destruction, while simultaneously inhibiting PNI to control local invasion and recurrence (21).
6.6 Preclinical and clinical evidence landscape
While preclinical studies have generated considerable enthusiasm, translation into significant clinical benefit has remained challenging (102). Key clinical trials investigating these strategies include:
CXCR4 inhibition: The COMBAT trial (NCT02826486) showed promising signals of activity for Motixafortide combined with immunotherapy and chemotherapy (86).
TGF-β inhibition: The trial of Galunisertib plus Durvalumab (NCT02734160) was tolerable but had limited efficacy in pre-treated metastatic patients, and the drug’s development was not pursued for this indication (87).
CCR2 inhibition: Combining the CCR2 antagonist PF-04136309 with FOLFIRINOX (NCT01413022) showed potential benefit in locally advanced PDAC (93).
CAF/stroma targeting: Trials involving hyaluronidase inhibitors (PEGPH20) failed to show benefit in Phase III, potentially due to a lack of patient selection based on hyaluronan levels (97). Trials with Vitamin D analogues are ongoing (103).
ICI combinations: Numerous trials combining ICIs with other agents have yielded modest results overall, except for the rare subset of MSI-H PDAC (100). An ongoing Phase III trial (JCOG1908E) is assessing chemo-radiotherapy with or without durvalumab in locally advanced PDAC (104).
Major hurdles impeding clinical success include the profound heterogeneity of the PDAC TME, the lack of validated predictive biomarkers, difficulties in achieving adequate drug delivery, and determining optimal combination strategies (100). Strategies focusing on reprogramming rather than simple elimination, guided by precise biomarkers reflecting the TME state, may be more successful (Table 2).
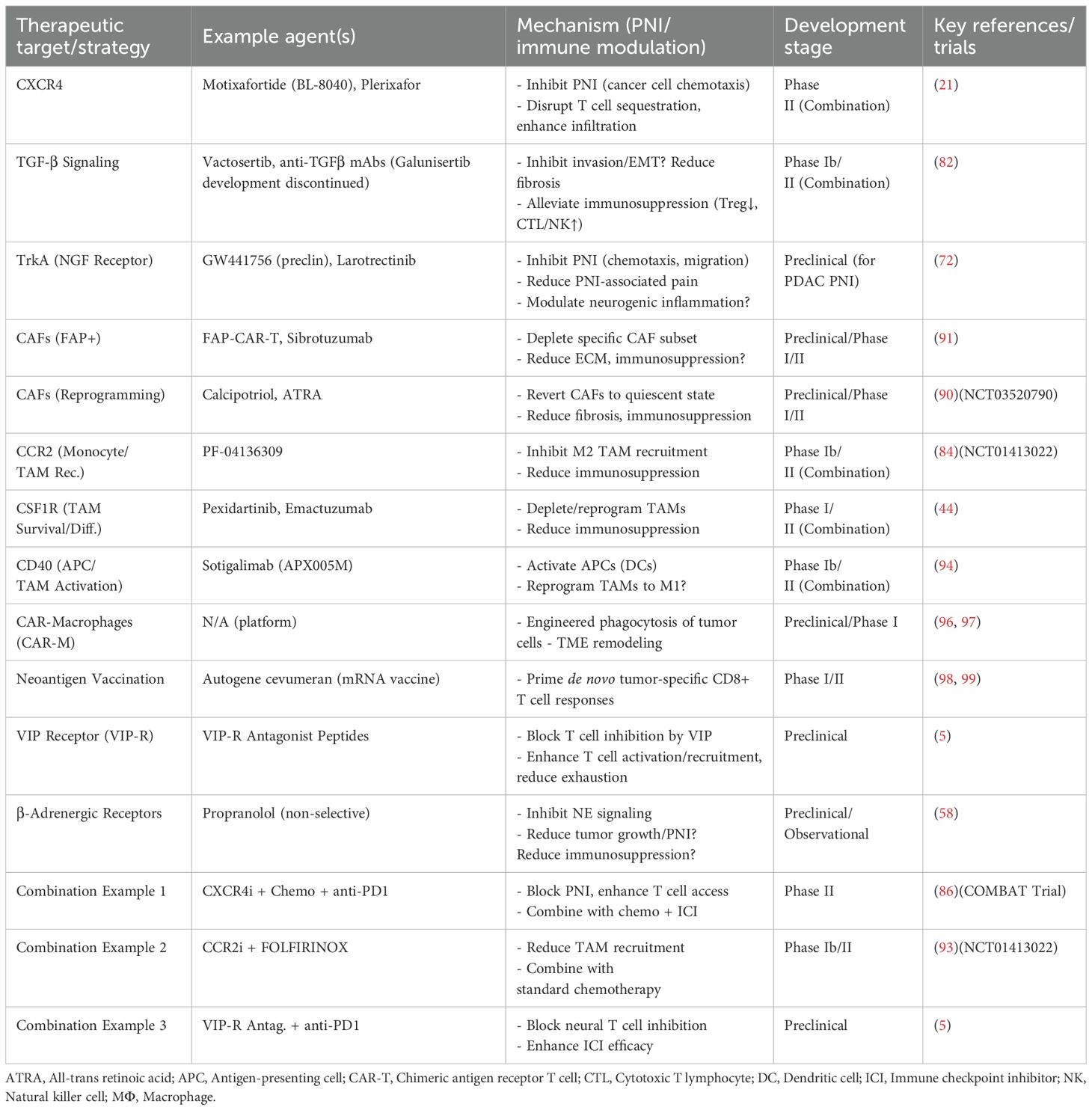
Table 2. Selected therapeutic strategies targeting the PNI-immune TME axis in PDAC.
7 Conclusion and future perspectives
7.1 Synthesizing the central role of PNI-TME crosstalk
The evidence reviewed herein strongly supports the conclusion that PNI and the immunologically cold TME are not independent pathological features of PDAC, but are deeply interwoven through extensive and bidirectional crosstalk (20). This neuro-immune axis is fundamental to PDAC’s aggressive biology. Shared signaling pathways and pleiotropic cellular players, particularly CAFs and the functionally diverse TAMs, act concertedly to orchestrate PNI while establishing profound immunosuppression (21). This interplay drives invasion, metastasis, therapy resistance, and critically underlies the failure of immunotherapies in most patients (12).
7.2 Key challenges and unanswered questions
Despite significant progress, substantial challenges remain:
Heterogeneity: The remarkable heterogeneity of PDAC—particularly within CAF and TAM populations—poses a major obstacle (75). Understanding how this impacts PNI-TME crosstalk and therapy response is crucial.
Biomarkers: There is an urgent need for validated biomarkers that can accurately reflect the state of the entire neuro-immune axis, not just cancer cell-centric markers, to guide patient stratification for TME-targeted therapies (2).
Mechanism nuances: A deeper understanding of the context-dependent roles of different nerve types, neurotransmitters, and specific CAF/TAM subsets is required (17). The contribution of metabolic crosstalk also warrants further investigation (6).
Therapeutic translation: Significant hurdles remain in translating promising preclinical findings into effective clinical therapies. These include optimizing drug delivery through the dense stroma, designing rational and tolerable combination regimens with appropriate sequencing, managing potential toxicities associated with targeting pathways with physiological roles, and improving clinical trial design to account for TME heterogeneity (39).
7.3 Future research directions and therapeutic outlook
Addressing these challenges requires a multi-pronged approach focused on deeper mechanistic understanding and smarter therapeutic design:
Advanced modeling and analysis: Continued use of sophisticated preclinical models combined with cutting-edge analytical tools (e.g., single-cell multi-omics, spatial transcriptomics) is essential to dissect PNI-TME interactions (8).
Precision targeting: Future therapies should move toward selectively targeting or reprogramming specific detrimental cell subsets (e.g., iCAFs, immunosuppressive TAMs), guided by robust biomarkers (76).
Rational combination therapies: The focus must be on designing mechanism-based, synergistic combinations. This includes strategically combining agents that target different nodes of the PNI-TME axis—for instance, pairing stromal modulators with ICIs, or integrating novel approaches like personalized mRNA vaccines to generate T-cell responses and CAR-M therapy to directly engage tumor cells and remodel the microenvironment (98).
Early intervention: Investigating the efficacy of targeting this axis in neoadjuvant or adjuvant settings may offer a crucial window of opportunity to prevent disease progression and recurrence.
In conclusion, while PDAC remains a formidable clinical challenge, the growing appreciation of the critical crosstalk between perineural invasion and the immunosuppressive TME offers new avenues for therapy. By continuing to unravel the complexities of this neuro-immune axis and developing rational therapeutic strategies to disrupt its detrimental effects, there is significant potential to improve the prognosis for patients with this disease (21).
Author contributions
JX: Data curation, Software, Writing – original draft, Investigation, Conceptualization. HY: Writing – review & editing, Software, Visualization. JW: Writing – review & editing, Visualization. YJ: Formal Analysis, Writing – review & editing. WC: Validation, Writing – review & editing. LL: Writing – review & editing, Methodology. LZ: Supervision, Writing – review & editing, Funding acquisition, Project administration.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by grants from the Science and Technology Department of Yunnan Province (grant numbers: 202101AY070001-032), the Beijing Medical Award Foundation (YXJL-2-23-0227-0081), and the Talent Program of the First People’s Hospital of Yunnan Province (KHYJ-6-2022-001).
Acknowledgments
We appreciate the technical assistance provided by the Cancer Research Center of Kunming Medical University.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ACh, Acetylcholine; ADM, Acinar-to-Ductal Metaplasia; APC, Antigen-Presenting Cell; ARTN, Artemin; BDNF, Brain-Derived Neurotrophic Factor; CAF, Cancer-Associated Fibroblast; CAR-M, Chimeric Antigen Receptor Macrophage; CTL, Cytotoxic T Lymphocyte; DC, Dendritic Cell; ECM, Extracellular Matrix; EMT, Epithelial-Mesenchymal Transition; FAP, Fibroblast Activation Protein; GABA, Gamma-Aminobutyric Acid; GDNF, Glial Cell Line-Derived Neurotrophic Factor; HIF, Hypoxia-Inducible Factor; ICI, Immune Checkpoint Inhibitor; MDSC, Myeloid-Derived Suppressor Cell; MMP, Matrix Metalloproteinase; NE, Norepinephrine; NGF, Nerve Growth Factor; OS, Overall Survival; PanIN, Pancreatic Intraepithelial Neoplasia; PDAC, Pancreatic Ductal Adenocarcinoma; PNI, Perineural Invasion; TAM, Tumor-Associated Macrophage; TGF-β, Transforming Growth Factor-β; TME, Tumor Microenvironment; TMB, Tumor Mutational Burden; Treg, Regulatory T cell; VIP, Vasoactive Intestinal Peptide.
References
1. Taherian M, Wang H, and Wang H. Pancreatic ductal adenocarcinoma: molecular pathology and predictive biomarkers. Cells. (2022) 11:3068. doi: 10.3390/cells11193068
2. Luo W, Wang J, Chen H, Ye L, Qiu J, Liu Y, et al. Epidemiology of pancreatic cancer: New version, new vision. Chin J Cancer Res. (2023) 35:438–50. doi: 10.21147/j.issn.1000-9604.2023.05.03
3. Bugazia D, Al-Najjar E, Esmail A, Abdelrahim S, Abboud K, Abdelrahim A, et al. Pancreatic ductal adenocarcinoma: the latest on diagnosis, molecular profiling, and systemic treatments. Front Oncol. (2024) 14:1386699. doi: 10.3389/fonc.2024.1386699
4. Lloyd EG, Henríquez JA, and Biffi G. Modelling the micro- and macro- environment of pancreatic cancer: from patients to pre-clinical models and back. Dis Model Mech. (2024) 17:dmm050624. doi: 10.1242/dmm.050624
5. Huang FF, Cui WH, Ma LY, Chen Q, and Liu Y. Crosstalk of nervous and immune systems in pancreatic cancer. Front Cell Dev Biol. (2023) 11:1309738. doi: 10.3389/fcell.2023.1309738
6. Dong M, Cao L, Cui R, and Xie Y. The connection between innervation and metabolic rearrangements in pancreatic cancer through serine. Front Oncol. (2022) 12:992927. doi: 10.3389/fonc.2022.992927
7. Argentiero A, Andriano A, Caradonna IC, de Martino G, and Desantis V. Decoding the intricate landscape of pancreatic cancer: insights into tumor biology, microenvironment, and therapeutic interventions. Cancers. (2024) 16:2438. doi: 10.3390/cancers16132438
8. Sun Y, Jiang W, Liao X, and Wang D. Hallmarks of perineural invasion in pancreatic ductal adenocarcinoma: new biological dimensions. Front Oncol. (2024) 14:1421067. doi: 10.3389/fonc.2024.1421067
9. Jain A and Bhardwaj V. Therapeutic resistance in pancreatic ductal adenocarcinoma: Current challenges and future opportunities. World J Gastroenterol. (2021) 27:6527–50. doi: 10.3748/wjg.v27.i39.6527
10. Luo J. KRAS mutation in pancreatic cancer. Semin Oncol. (2021) 48:10–8. doi: 10.1053/j.seminoncol.2021.02.003
11. Farhangnia P, Khorramdelazad H, Nickho H, and Delbandi A-A. Current and future immunotherapeutic approaches in pancreatic cancer treatment. J Hematol Oncol. (2024) 17:40. doi: 10.1186/s13045-024-01561-6
12. Lakis V, Chan NL, Lyons R, Blackburn N, Nguyen TH, Chang C, et al. Spatial transcriptomics reveals novel mechanisms involved in perineural invasion in pancreatic ductal adenocarcinomas. Cancers. (2025) 17:852. doi: 10.3390/cancers17050852
13. Demir IE, Ceyhan GO, Liebl F, D'Haese JG, Maak M, Friess H, et al. Neural invasion in pancreatic cancer: the past, present and future. Cancers. (2010) 2:1513–27. doi: 10.3390/cancers2031513
14. Shinkawa T, Ohuchida K, and Nakamura M. Heterogeneity of cancer-associated fibroblasts and the tumor immune microenvironment in pancreatic cancer. Cancers. (2022) 14:3994. doi: 10.3390/cancers14163994
15. Giurini EF, Ralph O, Pappas SG, and Gupta KH. Looking beyond checkpoint inhibitor monotherapy: uncovering new frontiers for pancreatic cancer immunotherapy. Mol Cancer Ther. (2025) 24:18–32. doi: 10.1158/1535-7163.Mct-24-0311
16. Gasparini G, Pellegatta M, Crippa S, Lena MS, Belfiori G, Doglioni C, et al. Nerves and pancreatic cancer: new insights into a dangerous relationship. Cancers (Basel). (2019) 11:893. doi: 10.3390/cancers11070893
17. Cheng J, Wang R, and Chen Y. Neuroimmune interactions in pancreatic cancer. Biomedicines. (2025) 13:609. doi: 10.3390/biomedicines13030609
18. Jurcak N and Zheng L. Signaling in the microenvironment of pancreatic cancer: Transmitting along the nerve. Pharmacol Ther. (2019) 200:126–34. doi: 10.1016/j.pharmthera.2019.04.010
19. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. (2021) 20:131. doi: 10.1186/s12943-021-01428-1
20. Li J, Kang R, and Tang D. Cellular and molecular mechanisms of perineural invasion of pancreatic ductal adenocarcinoma. Cancer Commun (Lond). (2021) 41:642–60. doi: 10.1002/cac2.12188
21. Roberto M, Arrivi G, Di Civita MA, Barchiesi G, Pilozzi E, Marchetti P, et al. The role of CXCL12 axis in pancreatic cancer: New biomarkers and potential targets. Front Oncol. (2023) 13:1154581. doi: 10.3389/fonc.2023.1154581
22. Tan X, Sivakumar S, Bednarsch J, Wiltberger G, Kather JN, Niehues J, et al. Nerve fibers in the tumor microenvironment in neurotropic cancer-pancreatic cancer and cholangiocarcinoma. Oncogene. (2021) 40:899–908. doi: 10.1038/s41388-020-01578-4
23. Ni B, Yin Y, Li Z, Wang J, Wang X, and Wang K. Crosstalk between peripheral innervation and pancreatic ductal adenocarcinoma. Neurosci Bull. (2023) 39:1717–31. doi: 10.1007/s12264-023-01082-1
24. Selvaggi F, Melchiorre E, Casari I, Cinalli S, Cinalli M, Aceto GM, et al. Perineural invasion in pancreatic ductal adenocarcinoma: from molecules towards drugs of clinical relevance. Cancers. (2022) 14:5793. doi: 10.3390/cancers14235793
25. Yuan H, Zhang Y, Liu F, Wu Y, Huang X, Liu X, et al. Exploring the biological mechanism and clinical value of perineural invasion in pancreatic cancer. Cancer Lett. (2025) 613:217515. doi: 10.1016/j.canlet.2025.217515
26. Chen SH, Zhang BY, Zhou B, Zhu CZ, Sun LQ, and Feng YJ. Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. Am J Cancer Res. (2019) 9:1–21.
27. Wang Z, Dong S, and Zhou W. Pancreatic stellate cells: Key players in pancreatic health and diseases (Review). Mol Med Rep. (2024) 30:109. doi: 10.3892/mmr.2024.13233
28. Ge W, Wang Y, Quan M, Mao T, Bischof EY, Xu H, et al. Activation of the PI3K/AKT signaling pathway by ARNTL2 enhances cellular glycolysis and sensitizes pancreatic adenocarcinoma to erlotinib. Mol Cancer. (2024) 23:48. doi: 10.1186/s12943-024-01965-5
29. Bakst RL, Xiong H, Chen CH, Deborde S, Lyubchik A, Zhou Y, et al. Inflammatory monocytes promote perineural invasion via CCL2-mediated recruitment and cathepsin B expression. Cancer Res. (2017) 77:6400–14. doi: 10.1158/0008-5472.Can-17-1612
30. Nissen NI, Karsdal M, and Willumsen N. Collagens and Cancer associated fibroblasts in the reactive stroma and its relation to Cancer biology. J Exp Clin Cancer Res. (2019) 38:115. doi: 10.1186/s13046-019-1110-6
31. Deborde S, Gusain L, Powers A, Marcadis A, Yu Y, Chen CH, et al. Reprogrammed schwann cells organize into dynamic tracks that promote pancreatic cancer invasion. Cancer Discov. (2022) 12:2454–73. doi: 10.1158/2159-8290.Cd-21-1690
32. Lankadasari MB, Mukhopadhyay P, Mohammed S, and Harikumar KB. TAMing pancreatic cancer: combat with a double edged sword. Mol Cancer. (2019) 18:48. doi: 10.1186/s12943-019-0966-6
33. Qin Q, Yu R, Eriksson JE, Tsai HI, and Zhu H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma therapy: Challenges and opportunities. Cancer Lett. (2024) 591:216859. doi: 10.1016/j.canlet.2024.216859
34. Wang J, Chen Y, Li X, and Zou X. Perineural invasion and associated pain transmission in pancreatic cancer. Cancers. (2021) 13:4594. doi: 10.3390/cancers13184594
35. Cai Z, Yao H, Chen J, Ahmed AA, Li C, Hu X, et al. Schwann cells in pancreatic cancer: Unraveling their multifaceted roles in tumorigenesis and neural interactions. Cancer Lett. (2024) 587:216689. doi: 10.1016/j.canlet.2024.216689
36. Jurcak NR, Rucki AA, Muth S, Thompson E, Sharma R, Ding D, et al. Axon guidance molecules promote perineural invasion and metastasis of orthotopic pancreatic tumors in mice. Gastroenterology. (2019) 157:838–50.e6. doi: 10.1053/j.gastro.2019.05.065
37. Zhang M, Zheng M, Dai L, Zhang WL, Fan HY, Yu XH, et al. CXCL12/CXCR4 facilitates perineural invasion via induction of the Twist/S100A4 axis in salivary adenoid cystic carcinoma. J Cell Mol Med. (2021) 25:7901–12. doi: 10.1111/jcmm.16713
38. D'Alterio C, Giardino A, Scognamiglio G, Butturini G, Portella L, Guardascione G, et al. CXCR4-CXCL12-CXCR7 and PD-1/PD-L1 in pancreatic cancer: CXCL12 predicts survival of radically resected patients. Cells. (2022) 11:3340. doi: 10.3390/cells11213340
39. Liu X, Shao Y, Li Y, Chen Z, Shi T, Tong Q, et al. Extensive review of nanomedicine strategies targeting the tumor microenvironment in PDAC. Int J Nanomedicine. (2025) 20:3379–406. doi: 10.2147/ijn.S504503
40. Tao J, Yang G, Zhou W, Qiu J, Chen G, Luo W, et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J Hematol Oncol. (2021) 14:14. doi: 10.1186/s13045-020-01030-w
41. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther. (2021) 6:362. doi: 10.1038/s41392-021-00670-9
42. Yamashita K and Kumamoto Y. CAFs-associated genes (CAFGs) in pancreatic ductal adenocarcinoma (PDAC) and novel therapeutic strategy. Int J Mol Sci. (2024) 25:6003. doi: 10.3390/ijms25116003
43. Zhang H, Yue X, Chen Z, Liu C, Wu W, Zhang N, et al. Define cancer-associated fibroblasts (CAFs) in the tumor microenvironment: new opportunities in cancer immunotherapy and advances in clinical trials. Mol Cancer. (2023) 22:159. doi: 10.1186/s12943-023-01860-5
44. Xiang X, Wang J, Lu D, and Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal transduction targeted Ther. (2021) 6:75. doi: 10.1038/s41392-021-00484-9
45. Wang Q and Ma W. Revisiting TAM polarization: beyond M1-and M2-type TAM toward clinical precision in macrophage-targeted therapy. Exp Mol Pathol. (2025) 143:104982. doi: 10.1016/j.yexmp.2025.104982
46. Toledo B, Zhu Chen L, Paniagua-Sancho M, Marchal JA, Perán M, Giovannetti E, et al. Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy. J Hematol Oncol. (2024) 17:44. doi: 10.1186/s13045-024-01559-0
47. Ramesh RPG, Yasmin H, Ponnachan P, Al-Ramadi B, Kishore U, and Joseph AM. Phenotypic heterogeneity and tumor immune microenvironment directed therapeutic strategies in pancreatic ductal adenocarcinoma. Front Immunol. (2025) 16:1573522. doi: 10.3389/fimmu.2025.1573522
48. Shao S, Miao H, and Ma W. Unraveling the enigma of tumor-associated macrophages: challenges, innovations, and the path to therapeutic breakthroughs. Front Immunol. (2023) 14:1295684. doi: 10.3389/fimmu.2023.1295684
49. Ajina R and Weiner LM. T-cell immunity in pancreatic cancer. Pancreas. (2020) 49:1014–23. doi: 10.1097/mpa.0000000000001621
50. Yu B, Shao S, and Ma W. Frontiers in pancreatic cancer on biomarkers, microenvironment, and immunotherapy. Cancer Lett. (2025) 610:217350. doi: 10.1016/j.canlet.2024.217350
51. Upadhrasta S and Zheng L. Strategies in developing immunotherapy for pancreatic cancer: recognizing and correcting multiple immune "Defects" in the tumor microenvironment. J Clin Med. (2019) 8:1472. doi: 10.3390/jcm8091472
52. Seifert L, Plesca I, Müller L, Sommer U, Heiduk M, von Renesse J, et al. LAG-3-expressing tumor-infiltrating T cells are associated with reduced disease-free survival in pancreatic cancer. Cancers. (2021) 13:1297. doi: 10.3390/cancers13061297
53. Karim M, Hasan MM, Kim SH, Azam Z, Wahab R, Islam T, et al. Stromal fibrin shapes immune infiltration landscape of pancreatic ductal adenocarcinoma. Biomaterials. (2025) 320:123280. doi: 10.1016/j.biomaterials.2025.123280
54. Zhang J, Li R, and Huang S. The immunoregulation effect of tumor microenvironment in pancreatic ductal adenocarcinoma. Front Oncol. (2022) 12:951019. doi: 10.3389/fonc.2022.951019
55. Saka D, Gökalp M, Piyade B, Cevik NC, Arik Sever E, Unutmaz D, et al. Mechanisms of T-cell exhaustion in pancreatic cancer. Cancers. (2020) 12:2274. doi: 10.3390/cancers12082274
56. Martinez-Bosch N, Vinaixa J, and Navarro P. Immune evasion in pancreatic cancer: from mechanisms to therapy. Cancers. (2018) 10:6. doi: 10.3390/cancers10010006
57. Ijichi H. Multiphasic heterogeneity of fibroblasts in the microenvironment of pancreatic ductal adenocarcinoma: dissection and the sum of the dynamics. Cancers. (2022) 14:4880. doi: 10.3390/cancers14194880
58. Liang Y, Li H, Gan Y, and Tu H. Shedding light on the role of neurotransmitters in the microenvironment of pancreatic cancer. Front Cell Dev Biol. (2021) 9:688953. doi: 10.3389/fcell.2021.688953
59. Blondy S, Christou N, David V, Verdier M, Jauberteau MO, Mathonnet M, et al. Neurotrophins and their involvement in digestive cancers. Cell Death Dis. (2019) 10:123. doi: 10.1038/s41419-019-1385-8
60. Trentini F, Agnetti V, Manini M, Giovannetti E, and Garajová I. NGF-mediated crosstalk: unraveling the influence of metabolic deregulation on the interplay between neural and pancreatic cancer cells and its impact on patient outcomes. Front Pharmacol. (2024) 15:1499414. doi: 10.3389/fphar.2024.1499414
61. Saloman JL, Singhi AD, Hartman DJ, Normolle DP, Albers KM, and Davis BM. Systemic depletion of nerve growth factor inhibits disease progression in a genetically engineered model of pancreatic ductal adenocarcinoma. Pancreas. (2018) 47:856–63. doi: 10.1097/mpa.0000000000001090
62. Benzaquen D, Lawrence YR, Taussky D, Zwahlen D, Oehler C, and Champion A. The crosstalk between nerves and cancer-A poorly understood phenomenon and new possibilities. Cancers. (2024) 16:1875. doi: 10.3390/cancers16101875
63. Silverman DA, Martinez VK, Dougherty PM, Myers JN, Calin GA, and Amit M. Cancer-associated neurogenesis and nerve-cancer cross-talk. Cancer Res. (2021) 81:1431–40. doi: 10.1158/0008-5472.Can-20-2793
64. Gregory E, Powers I, Jamshidi-Parsian A, Griffin R, and Song Y. Pancreatic tumor-derived extracellular vesicles stimulate schwann cell phenotype indicative of perineural invasion via IL-8 signaling. bioRxiv: preprint server Biol. (2023) 4:45–58. doi: 10.1101/2023.06.26.546629
65. Feng Y, Ye Z, Song F, He Y, and Liu J. The role of TAMs in tumor microenvironment and new research progress. Stem Cells Int. (2022) 2022:5775696. doi: 10.1155/2022/5775696
66. Anu RI, Shiu KK, and Khan KH. The immunomodulatory role of IDO1-Kynurenine-NAD(+) pathway in switching cold tumor microenvironment in PDAC. Front Oncol. (2023) 13:1142838. doi: 10.3389/fonc.2023.1142838
67. Righetti A, Giulietti M, Šabanović B, Occhipinti G, Principato G, and Piva F. CXCL12 and its isoforms: different roles in pancreatic cancer? J Oncol. (2019) 2019:9681698. doi: 10.1155/2019/9681698
68. Hartupee C, Nagalo BM, Chabu CY, Tesfay MZ, Coleman-Barnett J, West JT, et al. Pancreatic cancer tumor microenvironment is a major therapeutic barrier and target. Front Immunol. (2024) 15:1287459. doi: 10.3389/fimmu.2024.1287459
69. Batlle E and Massagué J. Transforming growth factor-β Signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
70. Ahmed S, Bradshaw AD, Gera S, Dewan MZ, and Xu R. The TGF-β/smad4 signaling pathway in pancreatic carcinogenesis and its clinical significance. J Clin Med. (2017) 6:5. doi: 10.3390/jcm6010005
71. Hussain SM, Kansal RG, Alvarez MA, Hollingsworth TJ, Elahi A, Miranda-Carboni G, et al. Role of TGF-β in pancreatic ductal adenocarcinoma progression and PD-L1 expression. Cell Oncol (Dordr). (2021) 44:673–87. doi: 10.1007/s13402-021-00594-0
72. Bapat AA, Munoz RM, Von Hoff DD, and Han H. Blocking nerve growth factor signaling reduces the neural invasion potential of pancreatic cancer cells. PloS One. (2016) 11:e0165586. doi: 10.1371/journal.pone.0165586
73. Benzaquen D, Lawrence YR, Taussky D, Zwahlen D, Oehler C, and Champion A. The crosstalk between nerves and cancer—A poorly understood phenomenon and new possibilities. Cancers (Basel) (2024) 16:1875. doi: 10.3390/cancers16101875
74. Vega JA, García-Suárez O, Hannestad J, Pérez-Pérez M, and Germanà A. Neurotrophins and the immune system. J Anat. (2003) 203:1–19. doi: 10.1046/j.1469-7580.2003.00203.x
75. Sherman MH and Beatty GL. Tumor microenvironment in pancreatic cancer pathogenesis and therapeutic resistance. Annu Rev Pathol. (2023) 18:123–48. doi: 10.1146/annurev-pathmechdis-031621-024600
76. Wang M, Xue W, Yuan H, Wang Z, and Yu L. Nano-drug delivery systems targeting CAFs: A promising treatment for pancreatic cancer. Int J Nanomedicine. (2024) 19:2823–49. doi: 10.2147/ijn.S451151
77. Vega JA, García-Suárez O, Hannestad J, Pérez-Pérez M, and Germanà A. Pancreatic ductal adenocarcinoma: characteristics of tumor microenvironment and barriers to treatment. J Adv Pract Oncol. (2020) 11:693–8. doi: 10.6004/jadpro.2020.11.7.4
78. Peiffer R, Boumahd Y, Gullo C, Crake R, Letellier E, Bellahcène A, et al. Cancer-associated fibroblast diversity shapes tumor metabolism in pancreatic cancer. Cancers (Basel). (2022) 15:61. doi: 10.3390/cancers15010061
79. Xiong C, Zhu Y, Xue M, Jiang Y, Zhong Y, Jiang L, et al. Tumor-associated macrophages promote pancreatic ductal adenocarcinoma progression by inducing epithelial-to-mesenchymal transition. Aging (Albany NY). (2021) 13:3386–404. doi: 10.18632/aging.202264
80. Gao C, Shen Q, and Yin T. Cellular crosstalk mediating immune evasion in pancreatic cancer microenvironment. Ann Pancreatic Cancer. (2019) 2(639):1–14. doi: 10.21037/apc.2019.06.04
81. Kocher F, Puccini A, Untergasser G, Martowicz A, Zimmer K, Pircher A, et al. Multi-omic characterization of pancreatic ductal adenocarcinoma relates CXCR4 mRNA expression levels to potential clinical targets. Clin Cancer Res. (2022) 28:4957–67. doi: 10.1158/1078-0432.Ccr-22-0275
82. Smith C, Zheng W, Dong J, Wang Y, Lai J, Liu X, et al. Tumor microenvironment in pancreatic ductal adenocarcinoma: Implications in immunotherapy. World J Gastroenterol. (2022) 28:3297–313. doi: 10.3748/wjg.v28.i27.3297
83. Principe DR, DeCant B, Mascariñas E, Wayne EA, Diaz AM, Akagi N, et al. TGFβ Signaling in the pancreatic tumor microenvironment promotes fibrosis and immune evasion to facilitate tumorigenesis. Cancer Res. (2016) 76:2525–39. doi: 10.1158/0008-5472.Can-15-1293
84. Petty AJ, Owen DH, Yang Y, and Huang X. Targeting tumor-associated macrophages in cancer immunotherapy. Cancers (Basel). (2021) 13:5318. doi: 10.3390/cancers13215318
85. Daniel SK, Seo YD, and Pillarisetty VG. The CXCL12-CXCR4/CXCR7 axis as a mechanism of immune resistance in gastrointestinal Malignancies. Semin Cancer Biol. (2020) 65:176–88. doi: 10.1016/j.semcancer.2019.12.007
86. Bockorny B, Semenisty V, Macarulla T, Borazanci E, Wolpin BM, Stemmer SM, et al. BL-8040, a CXCR4 antagonist, in combination with pembrolizumab and chemotherapy for pancreatic cancer: the COMBAT trial. Nat Med. (2020) 26:878–85. doi: 10.1038/s41591-020-0880-x
87. Melisi D, Oh DY, Hollebecque A, Calvo E, Varghese A, Borazanci E, et al. Safety and activity of the TGFβ receptor I kinase inhibitor galunisertib plus the anti-PD-L1 antibody durvalumab in metastatic pancreatic cancer. J Immunother Cancer. (2021) 9:e002068. doi: 10.1136/jitc-2020-002068
88. Gola M, Sejda A, Godlewski J, Cieślak M, and Starzyńska A. Neural component of the tumor microenvironment in pancreatic ductal adenocarcinoma. Cancers (Basel). (2022) 14:5246. doi: 10.3390/cancers14215246
89. Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, et al. β2 adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell. (2018) 33:75–90.e7. doi: 10.1016/j.ccell.2017.11.007
90. Yamazaki M and Ishimoto T. Targeting cancer-associated fibroblasts: eliminate or reprogram? Cancer Sci. (2025) 116:613–21. doi: 10.1111/cas.16443
91. Liu J, Wang Y, Mu C, Li M, Li K, Li S, et al. Pancreatic tumor eradication via selective Pin1 inhibition in cancer-associated fibroblasts and T lymphocytes engagement. Nat Commun. (2022) 13:4308. doi: 10.1038/s41467-022-31928-7
92. Deiana C, Margherita A, Giovanni B, and And Giovannetti E. The trend toward more target therapy in pancreatic ductal adenocarcinoma. Expert Rev Anticancer Ther. (2024) 24:525–65. doi: 10.1080/14737140.2024.2357802
93. Joshi S and Durden DL. Combinatorial approach to improve cancer immunotherapy: rational drug design strategy to simultaneously hit multiple targets to kill tumor cells and to activate the immune system. J Oncol. (2019) 2019:5245034. doi: 10.1155/2019/5245034
94. Firoozan S, Satpathy S, Shakiba M, and King DA. Recent advances in immunotherapy for pancreatic cancer: a narrative review. Digestive Med Res. (2024) 7:1–19. doi: 10.21037/dmr-24-2
95. Gautam SK, Batra SK, and Jain M. Molecular and metabolic regulation of immunosuppression in metastatic pancreatic ductal adenocarcinoma. Mol Cancer. (2023) 22:118. doi: 10.1186/s12943-023-01813-y
96. Li N, Geng S, Dong ZZ, Jin Y, Ying H, Li HW, et al. A new era of cancer immunotherapy: combining revolutionary technologies for enhanced CAR-M therapy. Mol Cancer. (2024) 23:117. doi: 10.1186/s12943-024-02032-9
97. Morva A, Arroyo AB, Andreeva L, Tapia-Abellán A, and Luengo-Gil G. Unleashing the power of CAR-M therapy in solid tumors: a comprehensive review. Front Immunol. (2025) 16:1615760. doi: 10.3389/fimmu.2025.1615760
98. Li J, Chen P, and Ma W. The next frontier in immunotherapy: potential and challenges of CAR-macrophages. Exp Hematol Oncol. (2024) 13:76. doi: 10.1186/s40164-024-00549-9
99. Sethna Z, Guasp P, Reiche C, Milighetti M, Ceglia N, Patterson E, et al. RNA neoantigen vaccines prime long-lived CD8+ T cells in pancreatic cancer. Nature. (2025) 2(639):1–10. doi: 10.1038/s41586-024-08508-4
100. Maru SY and Jaffee EM. Pancreatic cancer is feeling the heat. J Immunother Cancer. (2024) 12:e010124. doi: 10.1136/jitc-2024-010124
101. Gulhati P, Schalck A, Jiang S, Shang X, Wu CJ, Hou P, et al. Targeting T cell checkpoints 41BB and LAG3 and myeloid cell CXCR1/CXCR2 results in antitumor immunity and durable response in pancreatic cancer. Nat Cancer. (2023) 4:62–80. doi: 10.1038/s43018-022-00500-z
102. Upadhrasta S and Zheng L. Strategies in developing immunotherapy for pancreatic cancer: recognizing and correcting multiple immune “Defects” in the tumor microenvironment. JCM. (2019) 8:1472. doi: 10.3390/jcm8091472
103. Menezes S, Okail MH, Jalil SMA, Kocher HM, and Cameron AJM. Cancer-associated fibroblasts in pancreatic cancer: new subtypes, new markers, new targets. J Pathol. (2022) 257:526–44. doi: 10.1002/path.5926
Keywords: pancreatic ductal adenocarcinoma, perineural invasion, tumor microenvironment, immunosuppression, neuro-immune crosstalk, cancer-associated fibroblasts, tumor-associated macrophages, CXCL12/CXCR4
Citation: Xu J, Yao H, Wang J, Jin Y, Chang W, Li L and Zou L (2025) Perineural invasion and the “cold” tumor microenvironment in pancreatic cancer: mechanisms of crosstalk and therapeutic opportunities. Front. Immunol. 16:1650117. doi: 10.3389/fimmu.2025.1650117
Received: 19 June 2025; Accepted: 01 August 2025;
Published: 20 August 2025.
Edited by:
Wenxue Ma, University of California, San Diego, United StatesReviewed by:
Kevin Bode, German Cancer Research Center (DKFZ), GermanyHuan He, The Scripps Research Institute, United States
Copyright © 2025 Xu, Yao, Wang, Jin, Chang, Li and Zou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Zou, OVkxMjIwMzdAa3VzdC5lZHUuY24=