 Emily Slowikowski
Emily Slowikowski Céleste Willems
Céleste Willems Pedro Elias Marques
Pedro Elias Marques- Laboratory of Molecular Immunology, Department of Microbiology, Immunology and Transplantation, Rega Institute for Medical Research, Katholieke Universiteit Leuven (KU Leuven), Leuven, Belgium
Flaviviruses are capable of causing a myriad of diseases in humans, including viral encephalitis. This condition involves complex interactions between the virus, resident cells of the central nervous system and leukocytes recruited to the brain. We reviewed the mechanisms underlying leukocyte recruitment during flavivirus-induced encephalitis with a focus on the role of various chemoattractants and adhesion molecules. We discuss how these molecules orchestrate the migration of peripheral leukocytes into the brain parenchyma and how neurotropic flaviviruses induce this process. Moreover, we discuss evidence of leukocytes both controlling viral propagation and contributing to neuropathology, which poses a challenge for therapy development. This review summarizes our current understanding of the mechanisms behind leukocyte recruitment during encephalitis, addresses the gaps that remain in the field, and presents opportunities for therapeutic targeting unveiled by recent research on flaviviral encephalitis.
1 Introduction
Flaviviruses are a group of arthropod-borne RNA viruses estimated to infect up to 400 million people annually. Even though most flavivirus infections occur asymptomatically or cause mild febrile disease, these viruses can cause a wide spectrum of severe visceral, neurotropic and congenital diseases (1). Neurotropic flaviviruses have developed the capacity to overcome the protective barriers of the central nervous system (CNS), allowing them to cause severe inflammation of the meninges and/or the brain parenchyma, referred to respectively as meningitis and encephalitis, conditions that lead to neurodegeneration, edema and intracranial hypertension. Morbidity and mortality rates associated with CNS infections are high, especially in young children, the elderly and immunocompromised individuals. Neurological sequelae comprise headaches, movement disorders, altered consciousness, seizures and cognitive impairment, which can last up to long after the acute infection (2, 3). Clinically important neurotropic flaviviruses are Japanese encephalitis virus (JEV), West Nile virus (WNV) and tick-borne encephalitis virus (TBEV). Infections caused by flaviviruses such as Usutu virus (USUV), St. Louis encephalitis virus (SLEV) and Murray Valley encephalitis virus (MVEV) are rare. However, neglected flaviviruses pose a significant public health threat due to underestimation, lack of vaccines or antivirals, and their emerging potential for outbreaks (1). Other flaviviruses that are primarily associated with visceral or congenital diseases, such as dengue virus (DENV) and Zika virus (ZIKV), also possess the capacity to induce neurological disease (4–6).
Upon infection of the brain, resident cells such as neurons, microglia and astrocytes initiate an immune response by secretion of proinflammatory cytokines and chemokines. Consequently, peripheral leukocytes including monocytes and lymphocytes are recruited to the CNS, where they support viral clearance (7, 8), however, it has become clear that immune-mediated tissue damage and neuronal loss play a significant role in encephalitis. Moreover, neurotropic flaviviruses can infect and exploit leukocytes for neuroinvasion via a ‘trojan horse’ mechanism (9). The effector functions and consequences of cellular infiltration have been reviewed extensively elsewhere, as were the entry mechanisms of flaviviruses into the brain (10–14). This review focuses on the molecular mechanisms by which peripheral leukocytes migrate to the brain upon flavivirus infections. We discuss the leukocyte subsets that are recruited and the involvement of multiple chemoattractants and cellular adhesion molecules (CAMs).
2 Leukocyte entry sites of the brain
Specialized barriers separate physically the CNS from the periphery, posing a challenge for immune cells to reach the tissue. The blood-brain barrier (BBB) is undeniably the most well-known and studied barrier, consisting of a specialized endothelial layer that strictly controls the entry of solutes and immune cells from the blood into the brain parenchyma (Figure 1A). Nevertheless, leukocytes enter the brain via both paracellular and transcellular mechanisms. The paracellular route involves leukocyte migration between endothelial cells through junctional complexes. This process is facilitated by the interaction between leukocytes and endothelial cells through adhesion molecules such as intracellular adhesion molecule-1 (ICAM-1) and ICAM-2, as well as integrins like αLβ2 and αMβ2, which drive leukocyte crawling and their eventual passage through junctions (15). Tight junctions (TJ) between the endothelial cells strictly control paracellular transport and consist of transmembrane proteins such as occludin, claudins and junctional adhesion molecules (JAMs). This TJ backbone recruits several cytoplasmic scaffolding proteins, such as zona occludens, which contribute to TJ integrity. More specifically, claudin-5, the most abundant TJ protein in BBB endothelial cells, has been proposed to facilitate T-cell diapedeses across the BBB (16, 17). The regulation of TJ and BBB is profoundly dependent on the neurovascular unit, comprising astrocytes, perivascular microglia, pericytes and neurons (18, 19). Additionally, the endothelial and parenchymal basement membranes form another physical barrier to infiltrating leukocytes and are produced by endothelial cells and astrocytes, respectively.
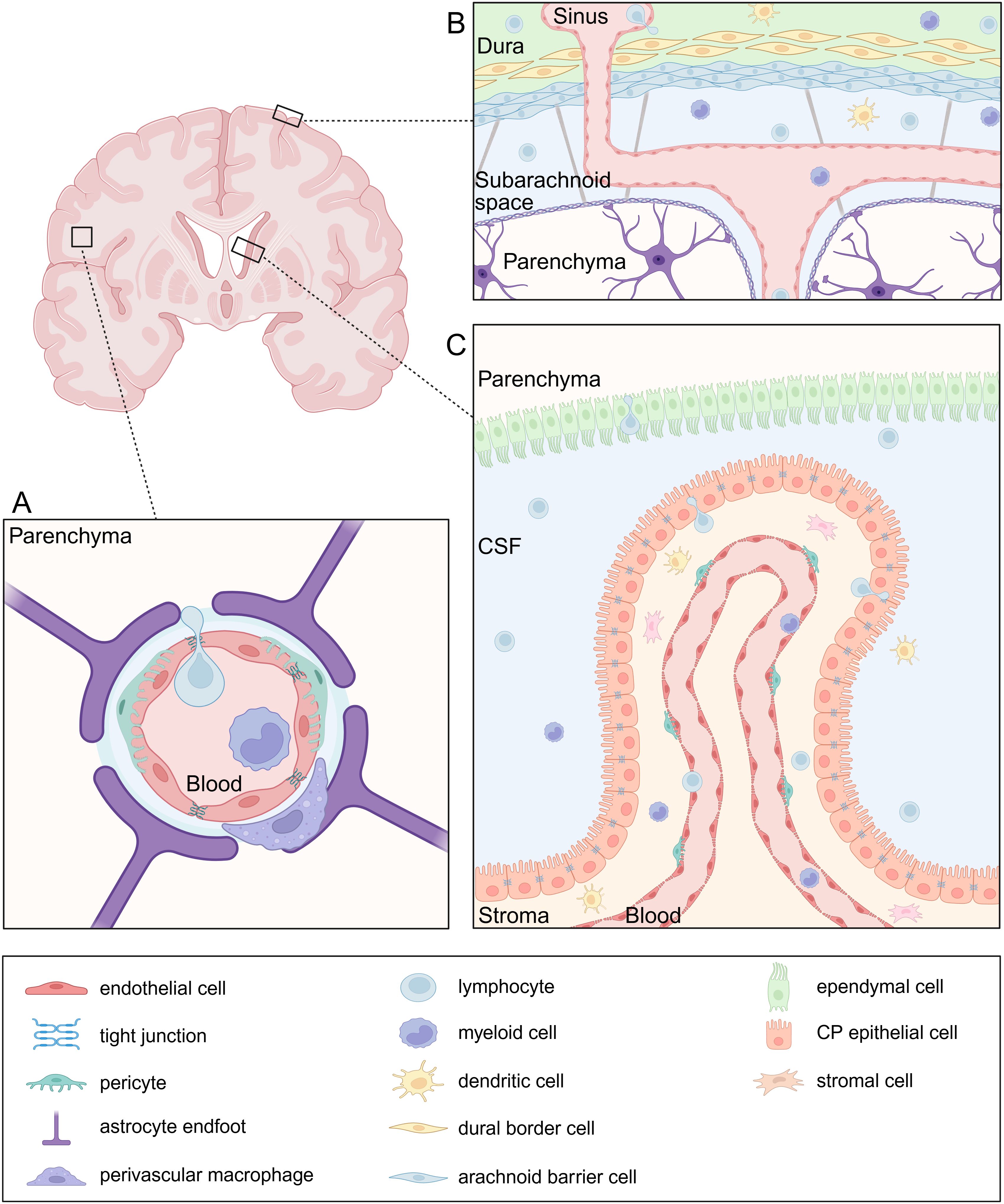
Figure 1. Neuroanatomy of leukocyte entry sites. (A) The blood-brain barrier (BBB) is composed of specialized endothelial cells connected by tight junctions (TJs), strictly regulating paracellular transport. Pericytes are embedded in the endothelial basement membrane which is distinct in composition from the parenchymal basement membrane. The latter forms the glia limitans together with astrocyte endfeet and serves as an additional barrier. At the level of the post-capillary venules, the basement membranes are separated by a perivascular space which holds antigen-presenting cells (APC) such as perivascular macrophages. (B) The meninges consist of three layers: the dura mater, arachnoid mater, and pia mater. Peripheral leukocytes infiltrate the dura through sinuses, which lack TJs. The arachnoid mater separates the dura from the cerebrospinal fluid (CSF)-filled subarachnoid space, which contains various immune cell populations. The pia mater together with the glia limitans form the last barrier to the parenchyma. (C) The blood-cerebrospinal fluid barrier (BCSFB) is located in the choroid plexus and is composed of an epithelial cell layer sealed by TJs and strictly regulating the entry of immune cells from the stroma into the CSF. APCs and T cells are abundant in the stroma which they can easily enter from the blood through a fenestrated endothelium.
The transcellular route involves leukocytes migrating directly through endothelial cells rather than between them. This route is characterized by the formation of vesiculotubular structures, intraendothelial structures or dynamic pores within endothelial cells (20). During transcellular diapedesis, endothelial cells form protrusions that surround the leukocyte, creating cup-like structures that facilitate the formation of temporary channels or pores, allowing leukocyte migration across the BBB while maintaining the TJ integrity (21). The molecular mechanisms directing paracellular and transcellular leukocyte diapedeses across the BBB are not yet understood. Research suggests that different leukocyte types may favor one route over the other based on their interactions with CAMs and the specific inflammatory context.
While the meninges are traditionally viewed as a protective barrier for the brain parenchyma, growing evidence highlights the role of meningeal immunity as a key contributor to CNS homeostasis and inflammation (Figure 1B). The meninges are composed of three layers, the outermost dura mater, the arachnoid mater and the innermost pia mater. Leukocytes access the dura from the circulation by extravasation through the dural sinuses which lack TJs. In addition, the skull bone marrow serves as a local source of leukocytes that migrate directly into the adjacent dura independently from the circulation. The dural immune cell population is heterogeneous and mainly consist of macrophages and lymphoid cells. The arachnoid mater separates the dura from the cerebrospinal fluid (CSF)-filled subarachnoid space, which houses a variety of immune cells. However, the blood vessels within the subarachnoid space restrict leukocyte extravasation (22, 23). Recently, arachnoid cuff exit points, which are gaps surrounding the bridging veins delineated by arachnoid barrier cells, have been identified as an additional route for leukocyte entry into the CNS. These exit points facilitate the direct efflux of CSF from the subarachnoid space to the dura and the entry of immune cells in reverse direction (24).
Another interface between blood and the CNS is the blood-cerebrospinal fluid barrier (BCSFB) (Figure 1C). CSF is produced by choroid plexus epithelial cells (CPE) within the four brain ventricles. The choroid plexus (CP) is lined by a fenestrated endothelium through which fluids, solutes and leukocytes easily pass in both homeostatic and encephalitic conditions. Moreover, the ependymal cells that separate the ventricles from the brain parenchyma contain solely loose junctions forming a leaky CSF-parenchyma interface. However, the CP epithelium itself serves as barrier and contains TJs that limit leukocyte passage (25–28). Importantly, the stroma contains a variety of innate and adaptive immune cells, including memory CD4+ T cells, which enter the CSF through the epithelium in physiological conditions and exert an important function of CNS immune surveillance (28, 29).
3 Chemokines and chemokine receptors
A hallmark of CNS infections is the infiltration of leukocytes into the brain parenchyma and the CSF, also referred to as pleocytosis. While viral CNS infections are characterized by lymphocytic predominance, several studies on WNV and TBEV infections point to neutrophils predominating in the earliest days following the onset of symptoms (30–33).
A key prerequisite for leukocyte recruitment is the formation of a chemokine gradient. Chemokines are small, secreted proteins that direct leukocyte migration by binding to glycosaminoglycans (GAGs) on endothelial cells and activating G protein-coupled receptors (GPCRs) on leukocytes. Based on the position of conserved amino-terminal cysteine residues, chemokines are structurally classified into four subgroups (CC, CXC, CX3C and XC) (34).
In the healthy CNS, several chemokines are constitutively expressed to support immune surveillance. However, during viral infection, their expression is strongly upregulated in neurons, astrocytes and microglia (Table 1). Elevated chemokine levels detected in the CSF of patients with viral encephalitis may serve as diagnostic or prognostic biomarkers (35–37). Likewise, the expression of chemokine receptors on infiltrating leukocytes is increased in response to neurotropic infections (Figure 2).
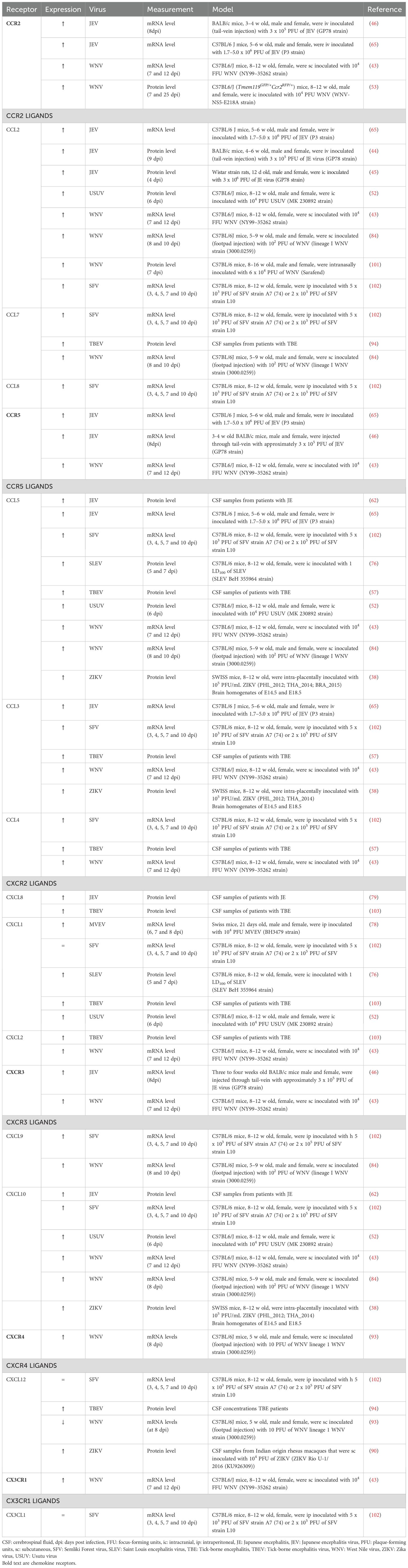
Table 1. Expression of chemokine receptors and their cognate ligands in the CNS upon neurotropic flavivirus infection in vivo.
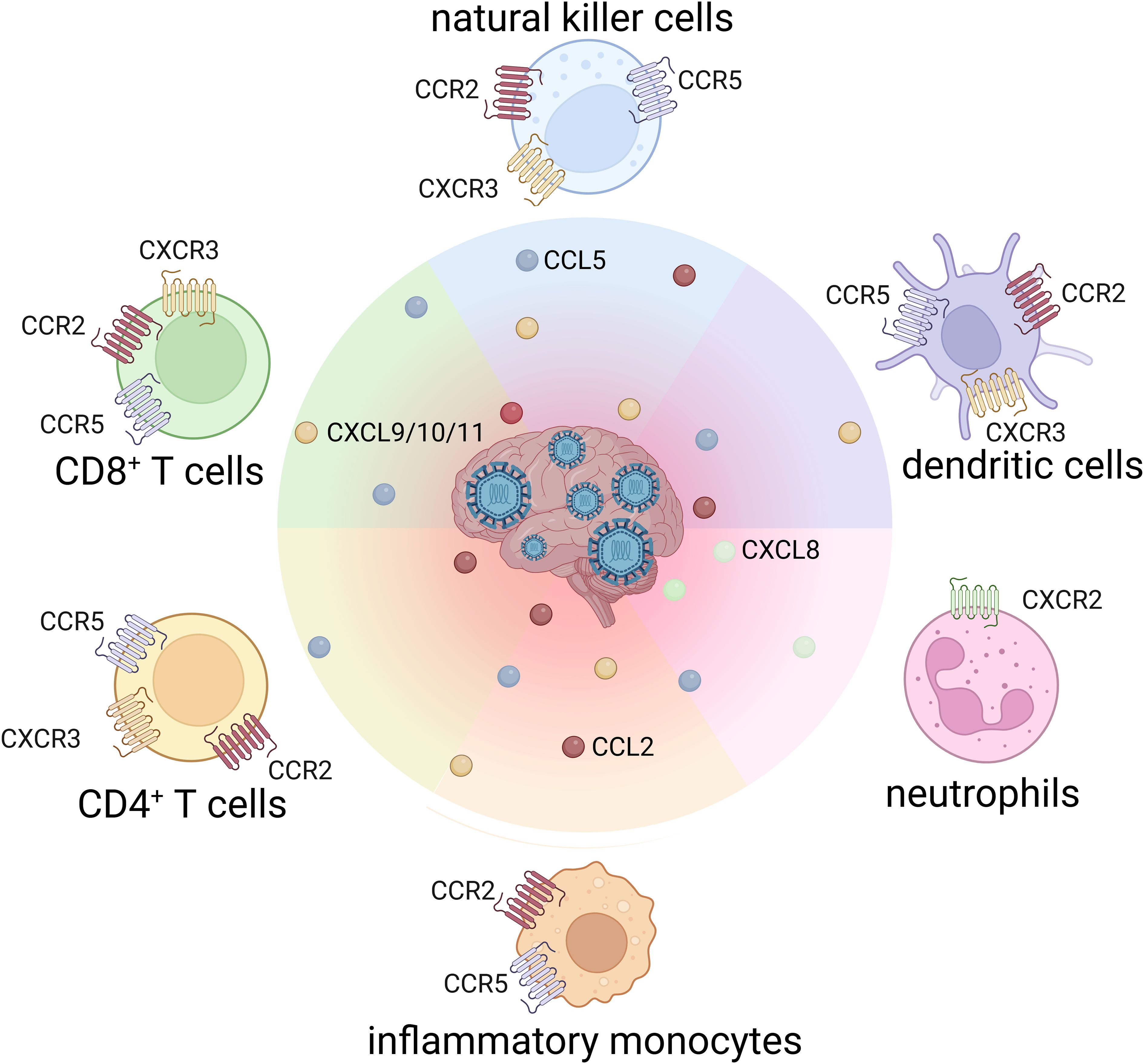
Figure 2. Chemokine receptor profiles on leukocytes driving CNS infiltration in flaviviral encephalitis.
Notably, the chemokine system is highly redundant, meaning that multiple chemokines signal through the same receptor and a single chemokine can activate different receptors. This redundancy ensures robust leukocyte recruitment but also complicates therapeutic targeting. On top of that, closely related neurotropic flavivirus can induce distinct patterns of chemokine expression and leukocyte infiltration (38, 39). In the following sections, we will discuss findings regarding chemokine receptors and their ligands during flaviviral encephalitis.
3.1 CCR2
CCR2 is a chemokine receptor that plays a crucial role in regulating monocyte egress from the bone marrow and their trafficking to sites of inflammation. CCR2 is highly expressed on inflammatory monocytes and is found on subsets of activated T cells, dendritic cells (DC) and natural killer (NK) cells (40, 41). The primary ligand of CCR2 is CCL2, which is significantly increased during flavivirus infections (42, 43). During JEV infection, elevated levels of CCL2 were observed in distinct brain regions including the cortex, striatum, thalamus, hippocampus, sub-ventricular zone and midbrain in both rat and mouse models. Additionally, JEV-infected mice showed increased CCR2 expression compared to mock controls (44–47). Monocytes and macrophages are the main sources of CCL2, but many cell types produce this chemokine, including endothelial cells, fibroblasts, epithelial cells, smooth muscle cells, mesangial cells, neurons, astrocytes and microglia (48).
CCR2-/- mice are often used to study functions of monocytes in vivo. For instance, CCR2 deficiency in WNV-infected mice increased mortality and viral load in the brain due to the lack of accumulating monocytes in the periphery during the early stages of WNV infection (49). Although some studies suggest that CCR2 is dispensable for monocyte trafficking from the blood to the brain, other research indicates that CCR2-expressing monocytes infiltrate the CNS more efficiently than CCR2-deficient monocytes (50, 51). Nevertheless, CCR2 deficiency alone was sufficient to completely block inflammatory monocyte recruitment to the brain in mice intracranially infected with USUV (52).
A recent study suggested that CCR2 is not required for the recruitment of T cells and viral control in acute WNV infection. However, the receptor does play a role in the recovery phase by regulating interferon-γ (IFN-γ) production by CD8+ T cells (53). Nevertheless, in the context of JEV infection, numerous CD8+ T cells expressing CCR2 and CCR5 infiltrate into the CNS.
In the context of JEV infection, CCR2 plays an interesting role: CCR2 ablation increases resistance, while CCL2 ablation increases susceptibility. This indicates that blocking CCR2, but not CCL2, could be a potential strategy for prophylaxis and therapy against JEV infection. However, the recruitment of inflammatory monocytes is not solely dependent on the CCL2-CCR2 axis, as other CCR2 ligands, such as CCL7, CCL8, CCL12 and CCL13, may also contribute (40–42).
In addition to its role in cell migration, CCR2 potentially contributes to BBB disruption. Endothelial CCR2 signaling alters TJ proteins, resulting in increased endothelial permeability in vitro and in vivo. Moreover CCR2-driven BBB permeability can be augmented indirectly through the recruitment of leukocytes (54). Notably, in a mouse model of USUV encephalitis, CCR2 deficient mice were protected against BBB disruption (52). These studies indicate that the role of CCR2 and CCL2 in the progression of viral encephalitis appears to be complex and context-dependent.
3.2 CCR5
CCR5 has been extensively studied for its role as the co-receptor for human immunodeficiency virus (HIV), but it was also the first chemokine receptor recognized to play an important role in flavivirus encephalitis (55). In contrast to HIV, the loss-of-function CCR5-Δ32 genetic variant was identified as a risk factor in WNV and TBEV infections. This led to the hypothesis that CCR5 deficiency is a contributing factor to symptomatic flavivirus infection due to the lack of its function in early peripheral clearance and driving lymphocyte migration into the CNS, resulting in severe disease (43, 56–59). Besides driving lymphocyte migration, CCR5 is involved in the CNS recruitment of monocytes, NK cells and the homing of CD4+ regulatory T cells (43, 60, 61).
CCL5 is the primary ligand of CCR5, although CCL3 and CCL4 are also recognized as cognate ligands. Elevated levels of CCL3 and CCL5 were observed in embryonic brains following intra-placental infection with various ZIKV strains (38). Furthermore, elevated levels of CCL5 in CSF were also detected in patients with JEV infection, with higher levels observed in non-surviving patients (62).
In addition, increased CCL5 production was observed in patients with mild TBEV symptoms, even without intrathecal inflammation, whereas patients with TBEV-induced meningoencephalitis showed elevated CCL5 levels in the CSF, which correlated with pleocytosis (57). Moreover, recent findings have identified higher recruitment and expansion of CD4+ and CD8+ T cells and B cells to be associated with more severe disease and neurological damage. CD4+ T cells are linked to encephalopathy, myelitis and cerebellar syndrome, while CD8+ T cells and B cells are associated with myelitis and encephalopathy (64). During JEV infection, CCR5 expression was dramatically increased in infiltrated CD8+ T cells, along with increased production of tumor necrosis factor-α (TNF-α), IFN-γ and granzyme B, indicating their activation and potential role in cell killing and viral clearance (65). However, CD8+ T cells can also contribute to immunopathology by mounting excessive cytotoxic responses that lead to tissue damage, as suggested by improved survival in TBEV-infected SCID mice, which lack functional T cells (66). Likewise, excessive production of proinflammatory cytokines by CD4+ T cells might increase disease severity in the context of high viral titers. Nevertheless, CD4+ T cells are critical for survival, as their absence in mice resulted in persistent CNS infection and uniform mortality following WNV infection (67).
3.3 CCR7
Leukocytes infiltrating into the CNS during viral encephalitis utilize a distinct combination of chemotactic and homing receptors to achieve selective trafficking toward the site of inflammation. While CCR2 and CCR5 serve as chemotactic receptors, CCR7 is generally described as a homing chemokine receptor. CCR7 is expressed on DCs, monocytes, neutrophils, T and B cells. During homeostasis, CCR7 regulates the migration of T cells into secondary lymphoid organs, where its cognate ligands CCL19 and CCL21 are abundantly present. Furthermore, DCs upregulate CCR7 following activation, which allows their efficient entry into terminal lymphatics expressing CCL21, promoting efficient interaction between DC and T cells (68). During viral infections, effector immune cells release IFN-γ, which reduces CCL19 and CCL21 levels in the T cell zones of secondary lymphoid organs (69). Combined with lower CCR7 expression on mature/effector T cells, this leads to the release of these cells into circulation, allowing them to migrate towards the site of inflammation (68).
Importantly, CCR7 plays a crucial role in limiting fatal WNV encephalitis in mice, by controlling leukocyte infiltration into the CNS. Loss of CCR7 led to persistent leukocytosis and increased leukocyte recruitment to the brain. Despite an excess of virus-specific T cells, CCR7-deficient mice exhibited higher viral loads and increased mortality. In addition, increased trafficking of infected myeloid cells into the brain of CCR7-deficient mice resulted in higher titers of WNV in the CNS (70, 71). Notably, CCR7 has been implicated in viral transmigration across the BBB in the context of the Trojan horse entry mechanism. In vitro models that mimic the transmigration of monocytes through BBB showed an upregulation of CCR7 on ZIKV-, WNV- and USUV-infected monocytes (39, 72).
3.4 CXCR2
Neutrophils are a critical component of the innate immune response, however, they play an underrecognized role in the pathogenesis of neurotropic flavivirus infections (73). CXCL8, CXCL1, CXCL2 and CXCL5 drive CXCR2-dependent neutrophil migration from the bone marrow into the blood. Importantly, the CXCL1–CXCR2 axis is also essential for neutrophil transendothelial migration into the brain (74). Macrophages rapidly upregulate CXCL1 and CXCL2 expression, while neurons and astrocytes also produce CXCL1 in vivo following HSV-1 infection which is the most common cause of viral encephalitis, although not a flavivirus (75). In mouse models of neurotropic flavivirus infections such as SLEV and MVE, neutrophil infiltration into the CNS was preceded by increased expression of CXCL1 within the CNS (76–78). A similar pattern was observed in patients, with higher CSF levels of CXCL8 associated with more severe outcomes of JE (79). Beyond their traditional immune functions, CXCR2+ neutrophils can act as viral reservoirs, supporting WNV replication and facilitating viral dissemination early during infection (74, 75).
In addition to its chemotactic function, CXCR2 also mediates the activation of the CNS endothelium. Mice deficient in either CXCL1 or CXCR2 show reduced leukocyte and endothelial interactions and lower expression of CAMs such as P-selectin, VCAM-1, and ICAM-1 in the brain following lipopolysaccharide (LPS) injection. Furthermore, in vitro stimulation of brain microvascular endothelial cells with CXCL1 increases BBB permeability and promotes monocyte transendothelial migration (80). This was also observed upon JEV infection, which led to activation of brain microvascular endothelial cells and the release of CXCL1 (63).
3.5 CXCR3
CXCR3 is preferentially expressed on activated T cells, particularly the T helper 1 (Th1) subset, NK cells and B cells. CXCR3 is activated by three highly related chemokines: CXCL9, CXCL10 and CXCL11, which are induced by CNS challenges such as neurotropic infections. Within the CNS, microglia and astrocytes are the main source of CXCL9 and CXCL10 (81, 82). The CXCL10-CXCR3 axis is particularly important in the context of WNV infection in the CNS, as reviewed by Benzarti et al. (83). In a mouse model of WNV encephalitis, neurons were identified as an additional source of CXCL10, inducing recruitment of CD8+ T cells (84). Similarly, CXCL10 facilitates T cell entry into the CNS parenchyma during TBEV infection. Patients with meningitis, encephalitis or meningoencephalitis caused by TBEV showed increased concentrations of CXCL10 and CXCL11 in the CSF and elevated CXCL10 levels in blood, indicating their potential role as biomarkers of inflammatory processes in the CNS. Moreover, the regression of CXCL10 in the CSF was proposed as an indicator of patient recovery during TBEV infection (85, 86).
Another ligand that is able to activate CXCR3 is CXCL4, also known as platelet factor 4 due to its release in high concentrations upon platelet activation. Elevated levels of CXCL4 have been observed during JEV infection, where it interacts with CXCR3 or CXCR3B, a splice variant of CXCR3, on vascular endothelial cells, epithelial cells, fibroblasts and leukocytes (87). Recent research highlighted the therapeutic potential of targeting the CXCR3 pathway. Blocking CXCR3 with the antagonist AMG487 in JEV-infected mice elevated the expression of type 1 IFNs, hereby reducing viral replication and increasing secretion of proinflammatory cytokines and leukocyte infiltration (88). Similar mechanisms were seen when infecting CXCL4-deficient mice with JEV. The suppression of the inflammatory response in these mice was associated with reduced percentages of monocytes, macrophages and T cells, along with decreased levels of proinflammatory cytokines in the brain compared to wild type (WT) mice (89).
3.6 CXCR4
CXCR4 is a chemokine receptor that is widely expressed on both immune- and non-immune cells, including hematopoietic cells, endothelial cells, neurons and stem cells. CXCR4 and its exclusive ligand CXCL12 are involved in numerous physiological and pathological conditions, including neurogenesis, immune responses and vascularization (90, 91). The constitutive expression of CXCL12 along the basolateral surfaces of CNS endothelial cells and within the brain microvasculature maintains an immune-privileged environment. This expression retains T cells, which commonly express CXCR4, in the perivascular space after crossing the BBB, thereby hindering CD8+ T cells from effectively clearing WNV within the CNS parenchyma (92). The antagonism of CXCR4 improved survival from lethal infection with WNV through enhanced intraparenchymal migration of WNV-specific CD8+ T cells, leading to reduced viral load and decreased immunopathology at this site (93). CXCR4 is expressed on inflammatory monocytes, but the CXCL12-CXCR4 axis is not involved in monocyte recruitment in TBE. Nevertheless, CXCL12 was synthesized intrathecally and detected in the periphery before the onset of pleocytosis, suggesting that this chemokine may contribute to CNS inflammation in its earliest stages (94). However, CXCL12 expression in the brain during WNV encephalitis depended on the mouse model used (83). Nevertheless, in adult Indian rhesus macaques, subcutaneous ZIKV infection caused increased CXCL12 expression in the CNS, which persisted long after the initial infection was cleared. In addition, CXCL12 plays a crucial role in regulating lymphocyte trafficking across the BBB into the CNS and in promoting the repair of damaged neural tissue, including remyelination (90).
3.7 CX3CR1
The chemokine CX3CL1 (fractalkine) is constitutively expressed by healthy neurons and endothelial cells (95, 96). CX3CR1 is expressed on resident brain cells, including microglia and astrocytes, as well as several leukocytes, such as DCs, monocytes, NK cells and T cells. The function of CX3CL1 varies depending on its form: soluble CX3CL1 acts as a chemoattractant, whereas membrane-anchored CX3CL1 functions as a CAM that facilitates the interaction of microglia and infiltrating leukocytes during inflammation (97). Binding of CX3CL1 to CX3CR1 on microglia provides potent inhibitory signals that help maintain their quiescent state, thus serving as a key mechanism by which neurons regulate innate immunity in the CNS (98). In addition, the soluble CX3CL1-CX3CR1 axis induces survival pathways in monocytes (99).
Studies investigating the role of CX3CR1 in the pathogenesis of neurotropic flavivirus-induced encephalitis are limited. Nevertheless, in WNV-infected mice, CX3CR1 and its ligand were upregulated in the brain by 7 days post infection (43). However, in later stages of disease, CX3CR1 expression on microglia was downregulated and these cells underwent apoptosis (100). Additionally, CX3CR1+ DC activation following JEV infection triggers their migration to the draining lymph nodes, promoting robust antiviral NK cell activity and virus-specific T cell responses (98).
4 Other chemoattractants
In addition to chemokines, other chemoattractants contribute to leukocyte recruitment during viral encephalitis. These include bioactive lipids (e.g., leukotriene B4 (LTB4)), complement anaphylatoxins (e.g., C3a and C5a) and find-me signals (e.g., nucleotides released by dying cells). In contrast to chemokines, these molecules do not depend on GAG binding and bind to specific receptors in a non-redundant manner.
4.1 Bioactive lipids
Leukotrienes are lipid mediators derived of the arachidonic acid metabolism via the 5-lipoxygenase pathway. LTB4 is a potent chemoattractant that activates leukocytes by binding to its high affinity receptor leukotriene B4 receptor 1 (BLT1). LTB4 synthesis is induced in response to other cytokines, such as IL-1 and TNF-α, bacterial products and other chemoattractants (platelet-activating factor (PAF) and C5a). Notably, during viral infection, there is evidence that leukotriene production is upregulated (104). In a mouse model of JEV infection, treatment with fenofibrate, which is a peroxisome proliferator-activated receptor-α (PPAR-α) agonist, reduced the levels of LTB4 in the brain. Fenofibrata acts by inducing the expression of cytochrome P450 4F (Cyp4f) enzymes, which catabolize LTB4 through hydroxylation (105).
Another notable lipid mediator is PAF, a potent proinflammatory phospholipid that is biosynthesized from phosphatidylcholine. The production of PAF and the subsequent release from platelets and leukocytes (neutrophils, macrophages and B cells) is induced by several cytokines such as IL-1, IL-6, IL-12 and TNF-α. PAF has pleiotropic effects after binding to its PAF receptor (PAFR), activating several proinflammatory signaling pathways including NF-κB and protein kinase C (106, 107).
PAF and LTB4 are often concomitantly produced and have been described in the recruitment of neutrophils upon stimulation with diverse toll like receptor (TLR) ligands (108). Although the production of PAF has not been studied in neurotropic flaviviruses yet, a previous study showed that macrophages from individuals with prior DENV infection increased PAF production compared to those from non-infected individuals (109). Therefore, excessive activation of PAFR was hypothesized to worsen symptoms of dengue hemorrhagic fever and dengue shock syndrome. Indeed, in PAFR-deficient mice inoculated with DENV, there was a decrease in thrombocytopenia, hyperpermeability and systemic cytokine levels, as well as the delay of mortality compared to infected WT mice (110).
4.2 Complement anaphylatoxins
The complement system is a large network of soluble and membrane-associated proteins that play an essential role in the innate immune response. Complement fragments C3a and C5a mediate leukocyte recruitment through C3aR and C5aR, respectively. C3aR is expressed on monocytes, activated T cells, neutrophils, basophils, eosinophils and mast cells. In the CNS, C3aR is constitutively expressed on neurons and during inflammation on astrocytes and microglia as well. C3aR-mediated chemotaxis was shown for monocytes, eosinophils and mast cells, but not neutrophils (111). C5aR expression is much more abundant than C3aR, and was shown on neutrophils, monocytes, T cells, B cells, DCs and mast cells. Evidence for C5aR-induced chemotaxis is available for neutrophils, monocytes, DCs, T cells and B cells (111). C5aR signaling in neutrophils also mediates cell recruitment indirectly via the release of LTB4 and upregulation of CAMs (112).
Even though it is clear that complement is activated upon flavivirus infection, its function during disease remains unclear. CSF proteome analysis of JEV-infected patients showed elevated levels of C3 (113). Mice deficient in C1q, C3, C4, factor B or factor D that were infected subcutaneously with WNV all showed decreased survival and increased viral burden in the brain and spinal cord (114, 115). However, C5aR-deficient mice shared similar susceptibility to WNV infection as WT mice. Importantly, this study showed that complement activation is required for T cell migration to the brain after peripheral WNV infection. Aberrant recruitment of both CD4+ and CD8+ T cells in C4 and factor B deficient mice, but not in C1q deficient mice suggests that the lectin and alternative pathways of complement activation are required (115).
Interestingly, viruses have developed specific strategies to antagonize complement activation. For instance, WNV, DENV and YF non-structural protein 1 (NS1) inhibits classical and lectin pathway activation through direct binding of C4 and promoting its cleavage to C4b (116). Moreover, WNV NS1 was shown to further inhibit the alternative pathway of complement activation by binding regulatory factor H (117).
Adverse effects of complement proteins in the brain have also been described following flavivirus neuroinvasion. Murine C3 levels in the brain are significantly upregulated following intracranial ZIKV infection. Neutralization with C3-specific antibodies rescued ZIKV-induced synapse pathology in the hippocampus and memory impairment (118). Likewise, C3 expression is highly increased following intracranial WNV infection and C3 or C3aR deficient mice are protected from synaptic loss in the hippocampus (119).
4.3 Find-me signals
Neurotropic flavivirus infections lead to non-lytic cell death by apoptosis and autophagy of neurons and, in some cases, glial cells, contributing to neurological dysfunction (120, 121). Clearance of apoptotic cells is mediated by find-me signals, which are molecules released by dying cells that attract phagocytes. This group of molecules mainly comprises the lipids lysophosphatidylcholine, sphingosine 1-phosphate, the chemokine CX3CL1 (vide supra), and the nucleotides ATP and UTP (122, 123). In-depth understanding of the mechanisms by which these mediators function is still required. Nevertheless, recent studies have reported an increase in LPC levels in the plasma of TBE patients as well as WNV-infected mice (124, 125).
Necroptosis is another type of cell death and was recently found to be a cause for neuronal loss in JEV-infected brains (126, 127). Moreover, WNV induces expression of necroptosis and pyroptosis cell death markers in the brain of infected mice. These programmed lytic forms of cell death release damage-associated molecular patterns (DAMPs) which are highly inflammatory. For instance, high mobility group Box 1 (HMGB1) protein is an abundant nuclear component released upon membrane permeabilization and secreted by activated monocytes, macrophages, DCs, endothelial cells and platelets (128). Once released, HMGB1 binds to receptors for advanced glycation end products (RAGE), TLR2 and TLR4, which are expressed by many cell types including neurons, endothelial cells and leukocytes (129, 130). HMGB1 induces multiple signaling pathways, leading to cell migration directly or inducing the transcription of cytokines and chemokines, stimulating immune cells in an autocrine and paracrine feedback loop (131, 132). Moreover, HMGB1 promotes CXCR4-dependent cell migration by upregulating CXCL12 via NF-κB and forming HMGB1-CXCL12 heterocomplexes, which protects CXCL12 degradation and induces CXCR4 signaling beyond CXCL12 alone (133).
HMGB1 also plays a role in leukocyte adhesion. ZIKV-infected monocytes were shown to release HMGB1 and upregulate RAGE expression in both monocytes and human brain microvascular endothelial cells (hBMECs) in coculture. This suggests that monocyte migration across the BBB is RAGE-dependent, with HMGB1 contributing to TJ disruption (72).
Another chemokine-like molecule is macrophage migration-inhibitory factor (MIF), which is produced by various cell types including immune cells, neurons, fibroblasts and endothelial cells. MIF receptors include CD74, CXCR2, CXCR4 and CXCR7, which form receptor complexes in order to establish MIF signaling and subsequent leukocyte migration (134, 135). Even though chemoattractant function of MIF in flavivirus encephalitis has not been studied, its plasma and CSF levels were increased in patients suffering from TBEV and WNV infection (136, 137). MIF mRNA levels are upregulated in the brain of JEV-infected mice and hBMECs stimulated with JEV NS1/NS1’ (138, 139). MIF is considered pathogenic in flavivirus infections as MIF-deficient mice were protected in WNV infection and showed lower viral loads and inflammatory responses. The detrimental role of MIF is attributed to disruption of the BBB favoring viral neuroinvasion and induced expression of chemokines and CAMs on endothelial cells and monocytes (136). Importantly, another study reported an association between high-expression MIF alleles and severe encephalitis in patients with WNV encephalitis (140).
Potent chemoattractant formyl peptides such as N-Formylmethionyl-leucyl-phenylalanine (fMLF) originate from both bacteria and endogenous mitochondria and have been described in neurodegenerative diseases (141). However, there are no reports describing fMLF or other formyl peptides in the context of viral encephalitis and their potential contribution to leukocyte recruitment to the CNS.
5 Cellular adhesion molecules
Leukocyte recruitment into the CNS is crucial for the antiviral defense during viral encephalitis. In addition to chemoattractants, this process requires CAMs. These transmembrane glycoproteins mediate adhesion between leukocytes and the endothelium of the vasculature, facilitating leukocyte tethering, rolling, firm adhesion and transmigration across the BBB into the CNS. CAMs are generally divided in five subgroups: selectins, integrins, cadherins, members of the immunoglobulin superfamily and others such as mucins (142, 143). The initial attachment and rolling are mediated by binding of endothelial E- and P-selectins to their ligands on leukocytes, including L-selectin, PSGL-1, CD44, CD43 and E-selectin ligand-(ESL)1 (144). These transient interactions slow down leukocytes as they roll along the vessel wall but remain reversible unless followed by signals from chemokines presented on endothelial proteoglycans. Chemokine signaling triggers the inside-out activation of integrins, to switch from a bent form to an extended form that more easily binds ligands. Leukocyte integrins such as α4β1 (VLA-4) and the β2 integrins LFA-1 and Mac-1 can then bind to their respective endothelial ligands VCAM-1 and ICAM-1 (145). Once firmly adhered, leukocytes undergo flattening, likely to minimize shear forces from blood flow and crawl along the endothelium to find permissive sites for transendothelial migration. Transmigration, or diapedesis, can occur via paracellular or transcellular routes, and is supported by additional molecules such as PECAM-1, CD99 and JAMs. These interactions help guide leukocytes through the endothelial barrier and into the CNS parenchyma (Figure 3).
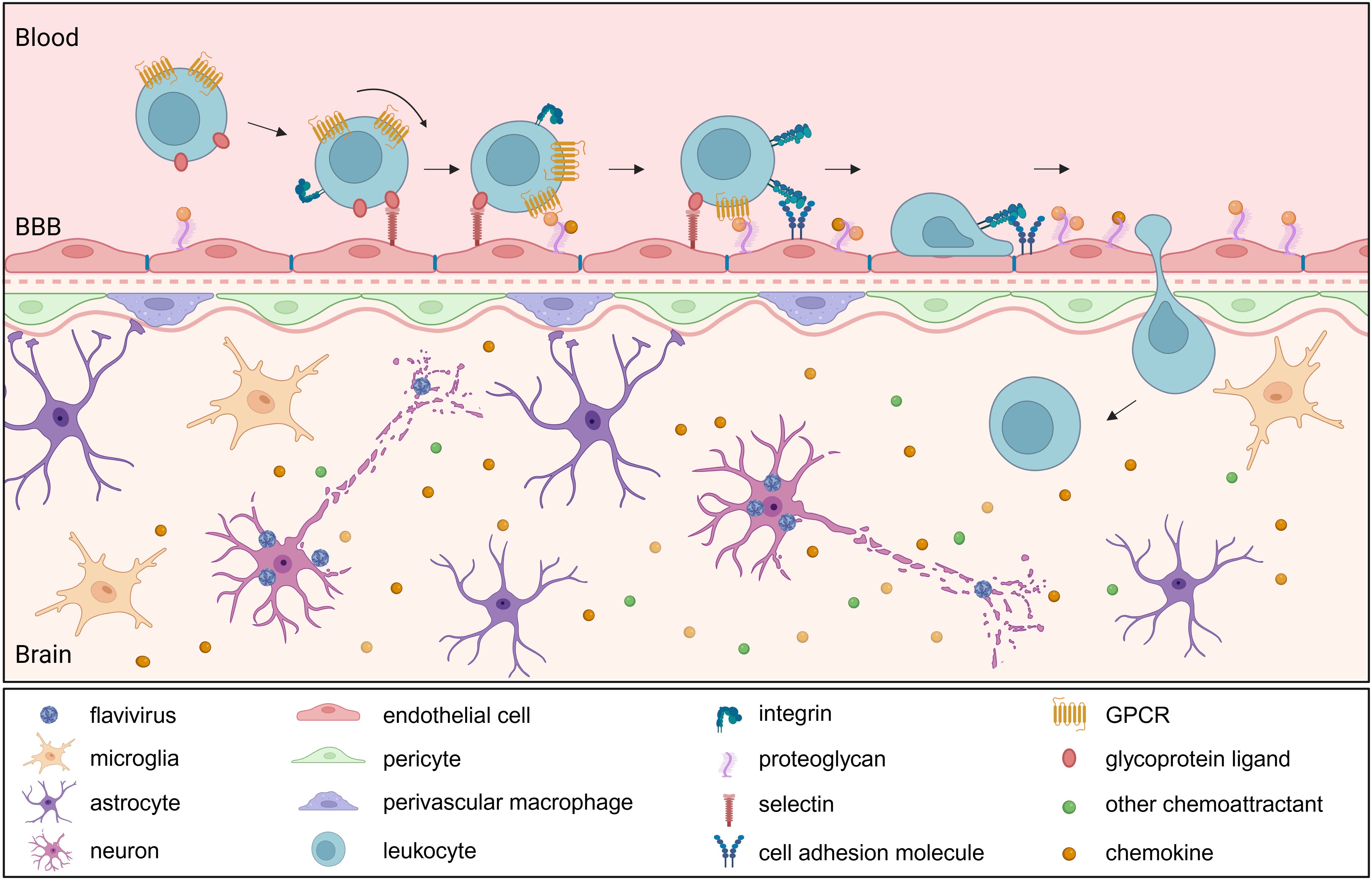
Figure 3. Leukocyte infiltration into the brain following neurotropic flavivirus infection. Upon neurotropic flavivirus infection, resident brain cells release chemokines (e.g., CCL2, CCL5), creating a gradient that drives leukocyte recruitment through binding to their cognate receptors (e.g., CCR2, CCR5). Activated endothelial cells upregulate adhesion molecules and present bound chemoattractants, initiating leukocyte tethering and rolling via selectin-ligand interactions. Chemoattractant receptor activation triggers integrin upregulation and activation, facilitating firm adhesion and subsequent transmigration. Once within the brain parenchyma, infiltrating immune cells amplify the inflammatory response. BBB, blood brain barrier; GPCR, G protein-coupled receptor.
Expression of CAMs on the endothelial cell surface is upregulated by cytokines such as TNF-α and IL-1β via NF-κB activation, in response to microbial infection or tissue damage (146–148). Increased CAM expression in response to neurotropic flavivirus infections has been demonstrated in multiple in vitro and in vivo models (Table 2).
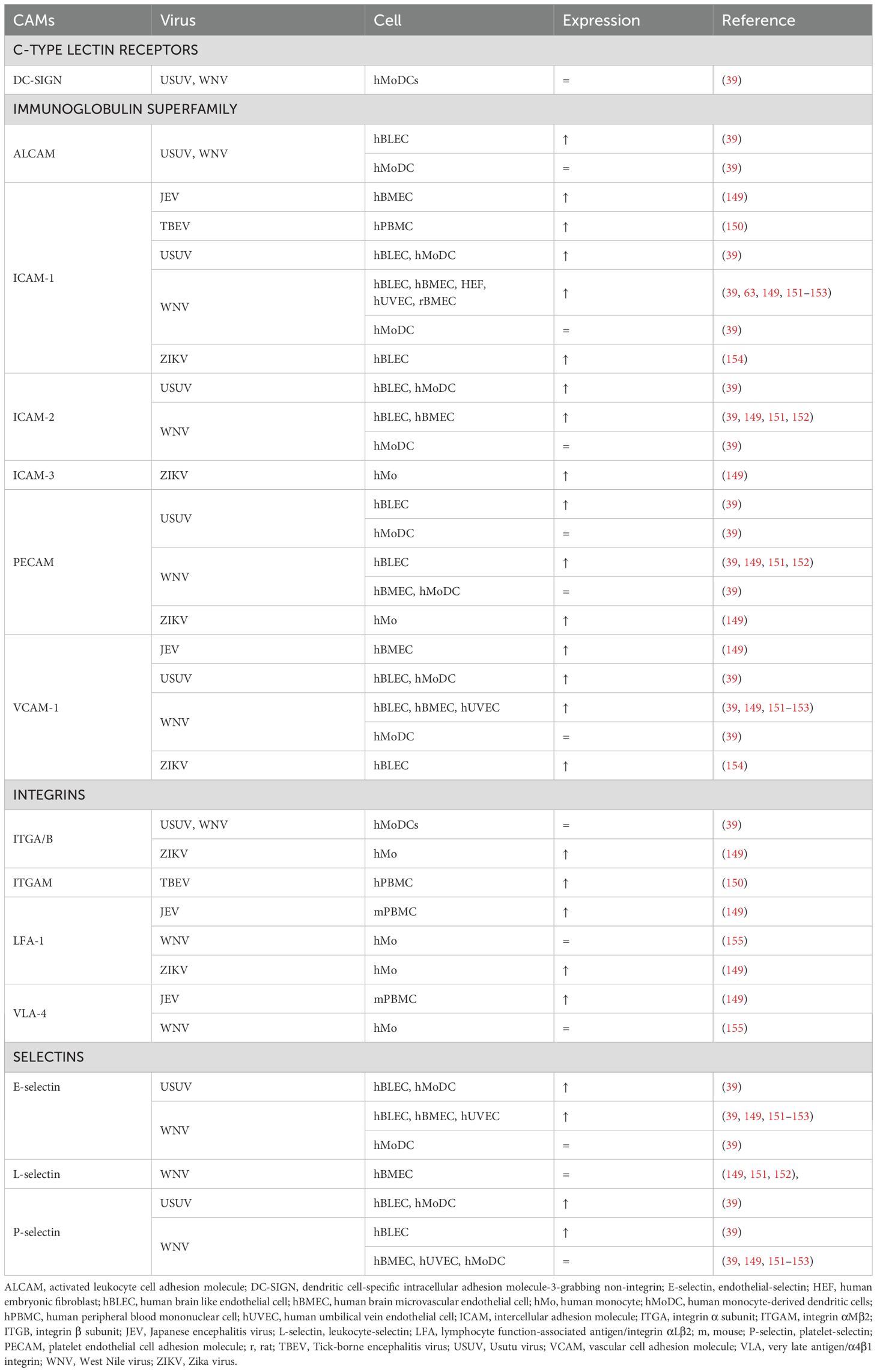
Table 2. Cell adhesion molecule expression in flaviviral encephalitis.
In vitro models have shown that human umbilical vein endothelial cells (hUVEC) infected with WNV upregulate E-selectin, ICAM-1, VCAM-1, but not P-selectin (153). Likewise, WNV-infected hBMECs showed increased mRNA levels of E-selectin, ICAM-1,VCAM-1, but not P-selectin, L-selectin and PECAM (151). However, a recent study comparing WNV and USUV infection on a hBBB model found that WNV, and to lesser extent USUV, increased mRNA levels of P-selectin, PECAM, ICAM-2 and ALCAM. Moreover, soluble E-selectin, P-selectin and ICAM-1 were detected in both apical and basolateral compartments (39).
In different mouse models of WNV, increased ICAM-1, VCAM-1 and E-selectin mRNA expression in the brain has been shown. Upregulation of these CAMs was correlated with peak WNV titers and leukocyte infiltration in the brain (151, 156). Moreover, ICAM-1 deficient mice were more resistant to lethal WNV infection, showing reduced viral load, leukocyte infiltration and neuronal damage (156). WNV is able to induce ICAM-1 expression in both a direct and an indirect cytokine-dependent manner (153). Indeed, a robust induction of CAM-inducing cytokines TNF-α and IL-1β is detected in the serum of patients with WNV fever and WNV encephalitis (157–159). The important role of ICAM-1 in leukocyte adhesion during flavivirus encephalitis is supported by research on JEV. Neutrophil and PBMC adhesion to rat brain microvascular endothelial cells (BMECs) was promoted by JEV infection. Moreover, pretreatment of the BMECs with ICAM-1 neutralizing antibodies almost completely inhibited leukocyte adhesion (63).
Moreover, endothelial-derived molecules indirectly promote leukocyte recruitment upon infection. For instance, hBMECs infected with JEV dramatically upregulate HMGB1 production, which enhances adhesion and transmigration of monocytes (149). RAGE functions as an endothelial adhesion receptor through direct interaction with the β2-integrin Mac-1 on innate leukocytes and in cooperation with ICAM-1 (160, 161). Moreover, HMGB1 activates endothelial RAGE and promotes expression of ICAM-1 and VCAM-1 (162). Additionally, HMGB1 also activates Mac-1 in a RAGE-dependent manner on neutrophils and upregulates the expression of LFA-1 and VLA-4 (149, 163). Lastly, MIF upregulates TNF-α-induced expression of endothelial E-selectin, P-selectin, ICAM-1 and VCAM-1and monocyte ICAM-1 and VCAM-1 (164, 165).
CAMs are absent in the choroid capillaries, aside from P-selectin, allowing leukocytes to enter the stroma freely. Interestingly, ICAM-1, VCAM-1 and mucosal addressin cell adhesion molecule 1 (MAdCAM-1) are expressed on the apical surface of CPEs, indicating their role in the final step of diapedesis and release in the CSF (27). Another CAM, epithelial V-like antigen is found on both apical and basal CPE surfaces and plays an important role for T cell adhesion, in vitro (166). Despite the recognized importance of leukocyte infiltration at the CP, the effects of flavivirus infection on CPE CAMs remains to be studied. Nevertheless, increased CAM expression by CPEs induced by TNF-α has been reported, suggesting a response in flavivirus encephalitis (26).
Flavivirus infection also increases the expression of complementary CAMs on leukocytes. However, the expression of integrins LFA-1 and VLA-4 in isolated monocytes, which are complementary to ICAM-1 and VCAM-1, respectively, did not increase upon WNV infection. Nevertheless, blocking VLA-4 reduced neutrophil, T-cell and especially monocyte counts in the brains of WNV-infected mice, alleviating encephalitic symptoms without affecting viral titers (155).
Monocytes and monocyte-derived dendritic cells (MoDCs) infected with WNV or USUV in vitro showed increased binding to both infected and non-infected human brain like endothelial cells (hBLECs), suggesting a direct effect of these viruses on leukocyte gene expression. The effect of USUV infection on binding ability and increased CAM expression of DCs was more pronounced compared to WNV infection (39). Likewise, ZIKV-infected monocytes showed increased efficiency in adherence, migration and transmigration assays compared to their mock-infected controls. Specifically, transmigration ability was improved by ZIKV-induced upregulation of LFA-1 (167, 168). Finally, the C-type lectin Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), which is abundantly expressed on immature DCs, has been identified as a key entry receptor for flaviviruses, interacting with glycosylation sites on the flavivirus E protein to facilitate viral entry (169).
6 Conclusion and future perspectives
Significant progress has been made in understanding how leukocytes infiltrate the CNS parenchyma during encephalitis (Box 1). However, many crucial questions persist regarding the precise mechanisms governing leukocyte recruitment into the inflamed CNS. For instance, it is still unknown which cells are producing which chemoattractants, and most lack confirmation at the protein level. The majority of the studies rely on measurements of mRNA levels of chemoattractants in whole tissue lysates. Much deeper insights would be obtained via single-cell/single-nucleus RNA sequencing and spatial transcriptomics, pinpointing the exact cellular sources for each of the molecules found during encephalitis. Importantly, RNA sequencing technologies can now be coupled to protein expression, such as CITE-seq (Cellular Indexing of Transcriptomes and Epitopes by sequencing), and techniques such as MILAN (multiple iterative labeling by antibody neodeposition) provide direct multiplexed protein identification in samples.
Box 1. Summary of key mechanisms driving leukocyte recruitment in the CNS
- Flaviviruses can enter the CNS via multiple routes, including the BBB, BCSFB and via infected leukocytes (Trojan horse mechanism).
- Once in the CNS, flavivirus infection triggers the release of chemoattractants.
- Chemokines guide leukocyte migration into the CNS by binding GAGs and activating specific GPCRs on leukocytes.
- Non-chemokine chemoattractants (e.g., LTB4, C5a, HMGB1) also contribute to leukocyte recruitment through distinct, non-redundant GPCR pathways.
- Different flaviviruses elicit both overlapping and virus-specific patterns of leukocyte recruitment and chemoattractant profiles.
- CAMs expressed on CNS endothelium (e.g., ICAM-1, VCAM-1, E-selectin) facilitate leukocyte adhesion, rolling and transmigration across barriers.
- Flavivirus infection modulates CAM expression on both endothelial cells and leukocytes.
It is evident that disrupting a single chemokine receptor or CAM is insufficient to completely block CNS inflammation and improve encephalitis outcomes. This is likely due to the redundant expression of multiple chemoattractants and CAMs, which can compensate for the absence of a single pathway. This highlights the importance of identifying and targeting the receptors primarily utilized by pathogenic leukocytes, while sparing those employed by resolving leukocytes. Future investigations are required to identify the most efficacious molecular targets to limit pathogenic leukocyte trafficking in encephalitis. Moreover, the distinct spatiotemporal and hierarchical contributions of chemoattractants are still elusive.
Besides attracting leukocytes, chemoattractants are also involved in cellular proliferation, which may significantly impact disease progression and recovery. Currently, this aspect of encephalitis is unknown. Multiple chemoattractants, including chemokines, also act on resident cell populations in the CNS, for instance, regulatory T cells and microglia. The impact of chemoattractant signalling locally, likely in early stages of encephalitis, is certainly worthy of investigation.
Author contributions
ES: Writing – original draft, Writing – review & editing. CW: Writing – review & editing, Writing – original draft. PM: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work is supported by FWO-Vlaanderen Junior Research Grants (G058421N and G025923N), a KU Leuven C1 grant (C14/23/143), an FWO-FAPESP grant (G0F8822N) and the Rega Foundation. CW holds a PhD fellowship from FWO-Vlaanderen 1132625N.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
BBB, Blood-brain barrier; BCSFB, Blood-cerebrospinal fluid barrier; BLT1, Leukotriene B4 receptor 1; CAMs, Cellular adhesion molecules; CNS, Central nervous system; CPE, Choroid plexus epithelial cells; CSF, Cerebrospinal fluid; DAMP, Damage-associated molecular pattern; DC, Dendritic cell; DC-SIGN, Dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin; DENV, Dengue virus; ESL1, E-selectin ligand-1; fMLF, N-Formylmethionyl-leucyl-phenylalanine; GAG, Glycosaminoglycans; GPCR, G protein-coupled receptors; hBLEC, Human brain like endothelial cells; hBMEC, Human brain microvascular endothelial cell; HIV, Human immunodeficiency virus; HMGB1, High mobility group box 1; hUVEC, Human umbilical vein endothelial cell; ICAM, Intercellular adhesion molecule; IFN, Interferon; JAMs, Junctional adhesion molecules; JEV, Japanese encephalitis virus; LFA1, Lymphocyte function-associated antigen 1; LPC, Lysophosphatidylcholine; LPS, Lipopolysaccharide; LTB4, Leukotriene B4; MadCAM1, Mucosal addressin cell adhesion molecule 1; MIF, Macrophage migration-inhibitory factor; MoDC, Monocyte-derived dendritic cell; MVEV, Murray Valley encephalitis virus; NK, Natural killer cell; NS1, Nonstructural protein 1; PAF, Platelet-activating factor; PPAR-α, Peroxisome proliferator-activated receptor α; RAGE, Receptors for advanced glycation end products; SLEV, St. Louis encephalitis virus; TBEV, Tick-borne encephalitis virus; Th1, T helper 1 cell; TJ, Tight junction; TLR, Toll-like receptor; TNF, Tumor necrosis factor; USUV, Usutu virus; VLA4, very late antigen/α4β1 integrin; WNV, West Nile virus; WT, Wild type; ZIKV, Zika virus.
References
1. Pierson TC and Diamond MS. The continued threat of emerging flaviviruses. Nat Microbiol. (2020) 5:796–812. doi: 10.1038/s41564-020-0714-0
2. Klein RS, Garber C, and Howard N. Infectious immunity in the central nervous system and brain function. Nat Immunol. (2017) 18:132–41. doi: 10.1038/ni.3656
3. Sadek JR, Pergam SA, Harrington JA, Echevarria LA, Davis LE, Goade D, et al. Persistent neuropsychological impairment associated with West Nile virus infection. J Clin Exp Neuropsychol. (2010) 32:81–7. doi: 10.1080/13803390902881918
4. Carod-Artal FJ, Wichmann O, Farrar J, and Gascón J. Neurological complications of dengue virus infection. Lancet Neurol. (2013) 12:906–19. doi: 10.1016/S1474-4422(13)70150-9
5. Carteaux G, Maquart M, Bedet A, Contou D, Brugières P, Fourati S, et al. Zika virus associated with meningoencephalitis. N Engl J Med. (2016) 374:1595–6. doi: 10.1056/NEJMc1602964
6. Artal FJC and Araujo AQC. Neurological complications in adults with Zika and chikungunya virus infection. Lancet Neurol. (2020) 19:799–801. doi: 10.1016/S1474-4422(20)30309-4
7. Ai S and Klein RS. Update on T cells in the virally infected brain: friends and foes. Curr Opin Neurol. (2020) 33:405–12. doi: 10.1097/WCO.0000000000000825
8. Terry RL, Getts DR, Deffrasnes C, Van Vreden C, Campbell IL, and King NJ. Inflammatory monocytes and the pathogenesis of viral encephalitis. J Neuroinflamm. (2012) 9:270. doi: 10.1186/1742-2094-9-270
9. Paul AM, Acharya D, Duty L, Thompson EA, Le L, Stokic DS, et al. Osteopontin facilitates West Nile virus neuroinvasion via neutrophil “Trojan horse” transport. Sci Rep. (2017) 7:4722. doi: 10.1038/s41598-017-04839-7
10. Klein RS, Garber C, Funk KE, Salimi H, Soung A, Kanmogne M, et al. Neuroinflammation during RNA viral infections. Annu Rev Immunol. (2019) 37:73–95. doi: 10.1146/annurev-immunol-042718-041417
11. Spiteri AG, Wishart CL, Pamphlett R, Locatelli G, and King NJC. Microglia and monocytes in inflammatory CNS disease: integrating phenotype and function. Acta Neuropathol (Berl). (2022) 143:179–224. doi: 10.1007/s00401-021-02384-2
12. Bai F, Thompson EA, Vig PJS, and Leis AA. Current understanding of west nile virus clinical manifestations, immune responses, neuroinvasion, and immunotherapeutic implications. Pathogens. (2019) 8:193. doi: 10.3390/pathogens8040193
13. Ashraf U, Ding Z, Deng S, Ye J, Cao S, and Chen Z. Pathogenicity and virulence of Japanese encephalitis virus: Neuroinflammation and neuronal cell damage. Virulence. (2021) 12:968–80. doi: 10.1080/21505594.2021.1899674
14. De Vries L and Harding AT. Mechanisms of neuroinvasion and neuropathogenesis by pathologic flaviviruses. Viruses. (2023) 15:261. doi: 10.3390/v15020261
15. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, and Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. (2006) 203:2569–75. doi: 10.1084/jem.20060925
16. Greene C, Hanley N, and Campbell M. Claudin-5: gatekeeper of neurological function. Fluids Barriers CNS. (2019) 16:3. doi: 10.1186/s12987-019-0123-z
17. Debayon P, Baena V, Ge S, Jiang X, Jellison ER, Kiprono T, et al. Appearance of claudin-5+ leukocytes in the central nervous system during neuroinflammation: a novel role for endothelial-derived extracellular vesicles. J Neuroinflamm. (2016) 13:292. doi: 10.1186/s12974-016-0755-8
18. Luissint AC, Artus C, Glacial F, Ganeshamoorthy K, and Couraud PO. Tight junctions at the blood brain barrier: physiological architecture and disease-associated dysregulation. Fluids Barriers CNS. (2012) 9:23. doi: 10.1186/2045-8118-9-23
19. Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, and Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis. (2010) 37:13–25. doi: 10.1016/j.nbd.2009.07.030
20. Lossinsky AS and Shivers RR. Structural pathways for macromolecular and cellular transport across the blood-brain barrier during inflammatory conditions. Rev Histol Histopathol. (2004) 19):535–64. doi: 10.14670/HH-19.535
21. Carman CV, Jun CD, Salas A, and Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol. (2003) 171:6135–44. doi: 10.4049/jimmunol.171.11.6135
22. Rua R and McGavern DB. Advances in meningeal immunity. Trends Mol Med. (2018) 24:542–59. doi: 10.1016/j.molmed.2018.04.003
23. Alves De Lima K, Rustenhoven J, and Kipnis J. Meningeal immunity and its function in maintenance of the central nervous system in health and disease. Annu Rev Immunol. (2020) 38:597–620. doi: 10.1146/annurev-immunol-102319-103410
24. Smyth LCD, Xu D, Okar SV, Dykstra T, Rustenhoven J, Papadopoulos Z, et al. Identification of direct connections between the dura and the brain. Nature. (2024) 627:165–73. doi: 10.1038/s41586-023-06993-7
25. Redzic Z and Segal M. The structure of the choroid plexus and the physiology of the choroid plexus epithelium. Adv Drug Delivery Rev. (2004) 56:1695–716. doi: 10.1016/j.addr.2004.07.005
26. Thompson D, Brissette CA, and Watt JA. The choroid plexus and its role in the pathogenesis of neurological infections. Fluids Barriers CNS. (2022) 19:75. doi: 10.1186/s12987-022-00372-6
27. Demeestere D, Libert C, and Vandenbroucke RE. Clinical implications of leukocyte infiltration at the choroid plexus in (neuro)inflammatory disorders. Drug Discov Today. (2015) 20:928–41. doi: 10.1016/j.drudis.2015.05.003
28. Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, and Engelhardt B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol (Berl). (2018) 135:337–61. doi: 10.1007/s00401-018-1807-1
29. De Graaf MT, Smitt PAES, Luitwieler RL, Van Velzen C, Van Den Broek PDM, Kraan J, et al. Central memory CD4+ T cells dominate the normal cerebrospinal fluid. Cytomet B Clin Cytom. (2011) 80B:43–50. doi: 10.1002/cyto.b.20542
30. Pepperell C, Rau N, Krajden S, Kern R, Humar A, Mederski B, et al. West Nile virus infection in 2002: morbidity and mortality among patients admitted to hospital in southcentral Ontario. CMAJ Can Med Assoc J J Assoc Med Can. (2003) 168:1399–405.
31. Jeha LE, Sila CA, Lederman RJ, Prayson RA, Isada CM, and Gordon SM. West Nile virus infection: A new acute paralytic illness. Neurology. (2003) 61:55–9. doi: 10.1212/01.WNL.0000073617.08185.0A
32. Crichlow R, Bailey J, and Gardner C. Cerebrospinal fluid neutrophilic pleocytosis in hospitalized west nile virus patients. J Am Board Fam Med. (2004) 17:470–2. doi: 10.3122/jabfm.17.6.470
33. LaSala PR and Holbrook M. Tick-borne flaviviruses. Clin Lab Med. (2010) 30:221–35. doi: 10.1016/j.cll.2010.01.002
34. Zlotnik A and Yoshie O. The chemokine superfamily revisited. Immunity. (2012) 36:705–16. doi: 10.1016/j.immuni.2012.05.008
35. Kothur K, Wienholt L, Brilot F, and Dale RC. CSF cytokines/chemokines as biomarkers in neuroinflammatory CNS disorders: A systematic review. Cytokine. (2016) 77:227–37. doi: 10.1016/j.cyto.2015.10.001
36. Soltani Khaboushan A, Pahlevan-Fallahy MT, Shobeiri P, Teixeira AL, and Rezaei N. Cytokines and chemokines profile in encephalitis patients: A meta-analysis. PloS One. (2022) 17:e0273920. doi: 10.1371/journal.pone.0273920
37. Zidovec-Lepej S, Bodulić K, Bogdanic M, Gorenec L, Savic V, Grgic I, et al. Proinflammatory chemokine levels in cerebrospinal fluid of patients with neuroinvasive flavivirus infections. Microorganisms. (2024) 12:657. doi: 10.3390/microorganisms12040657
38. Darmuzey M, Touret F, Slowikowski E, Gladwyn-Ng I, Ahuja K, Sanchez-Felipe L, et al. Epidemic Zika virus strains from the Asian lineage induce an attenuated fetal brain pathogenicity. Nat Commun. (2024) 15:10870. doi: 10.1038/s41467-024-55155-4
39. Constant O, Maarifi G, Barthelemy J, Martin MF, Tinto B, Savini G, et al. Differential effects of Usutu and West Nile viruses on neuroinflammation, immune cell recruitment and blood–brain barrier integrity. Emerg Microbes Infect. (2023) 12:2156815. doi: 10.1080/22221751.2022.2156815
40. Mitchell AJ, Roediger B, and Weninger W. Monocyte homeostasis and the plasticity of inflammatory monocytes. Cell Immunol. (2014) 291:22–31. doi: 10.1016/j.cellimm.2014.05.010
41. Tsou CL, Peters W, Si Y, Slaymaker S, Aslanian AM, Weisberg SP, et al. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J Clin Invest. (2007) 117:902–9. doi: 10.1172/JCI29919
42. Kim JH, Patil AM, Choi JY, Kim SB, Uyangaa E, Hossain FMA, et al. CCL 2, but not its receptor, is essential to restrict immune privileged central nervous system-invasion of Japanese encephalitis virus via regulating accumulation of CD 11b + Ly-6C hi monocytes. Immunology. (2016) 149:186–203. doi: 10.1111/imm.12626
43. Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, and Murphy PM. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med. (2005) 202:1087–98. doi: 10.1084/jem.20042530
44. Das S, Dutta K, Kumawat KL, Ghoshal A, Adhya D, and Basu A. Abrogated inflammatory response promotes neurogenesis in a murine model of Japanese encephalitis. PloS One. (2011) 6:e17225. doi: 10.1371/journal.pone.0017225
45. Srivastava R, Kalita J, Khan MY, and Misra UK. Status of proinflammatory and anti-inflammatory cytokines in different brain regions of a rat model of Japanese encephalitis. Inflammation Res. (2012) 61:381–9. doi: 10.1007/s00011-011-0423-5
46. Swarup V, Ghosh J, Das S, and Basu A. Tumor necrosis factor receptor-associated death domain mediated neuronal death contributes to the glial activation and subsequent neuroinflammation in Japanese encephalitis. Neurochem Int. (2008) 52:1310–21. doi: 10.1016/j.neuint.2008.01.014
47. Han YW, Choi JY, Uyangaa E, Kim SB, Kim JH, Kim BS, et al. Distinct dictation of Japanese encephalitis virus-induced neuroinflammation and lethality via triggering TLR3 and TLR4 signal pathways. PloS Pathog. (2014) 10:e1004319. doi: 10.1371/journal.ppat.1004319
48. Deshmane SL, Kremlev S, Amini S, and Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. (2009) 29:313–26. doi: 10.1089/jir.2008.0027
49. Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, and Murphy PM. Chemokine receptor ccr2 is critical for monocyte accumulation and survival in west nile virus encephalitis. J Immunol. (2011) 186:471–8. doi: 10.4049/jimmunol.1003003
50. Howe CL, LaFrance-Corey RG, Goddery EN, Johnson RK, and Mirchia K. Neuronal CCL2 expression drives inflammatory monocyte infiltration into the brain during acute virus infection. J Neuroinflamm. (2017) 14:238. doi: 10.1186/s12974-017-1015-2
51. Xu J, Ganguly A, Zhao J, Ivey M, Lopez R, Osterholzer JJ, et al. CCR2 signaling promotes brain infiltration of inflammatory monocytes and contributes to neuropathology during cryptococcal meningoencephalitis. mBio. (2021) 12:e01076–21. doi: 10.1128/mBio.01076-21
52. Slowikowski E, Willems C, Lemes RMR, Schuermans S, Berghmans N, Rocha RPF, et al. A central role for CCR2 in monocyte recruitment and blood–brain barrier disruption during Usutu virus encephalitis. J Neuroinflamm. (2025) 22:107. doi: 10.1186/s12974-025-03435-1
53. Ai S, Arutyunov A, Liu J, Hill JD, Jiang X, and Klein RS. CCR2 restricts IFN-γ production by hippocampal CD8 TRM cells that impair learning and memory during recovery from WNV encephalitis. J Neuroinflamm. (2024) 21:330. doi: 10.1186/s12974-024-03309-y
54. Stamatovic SM, Shakui P, Keep RF, Moore BB, Kunkel SL, Van Rooijen N, et al. Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab. (2005) 25:593–606. doi: 10.1038/sj.jcbfm.9600055
55. Marmor M, Sheppard HW, Donnell D, Bozeman S, Celum C, Buchbinder S, et al. Homozygous and heterozygous CCR5-Δ32 genotypes are associated with resistance to HIV infection. JAIDS J Acquir Immune Defic Syndr. (2001) 27:472–81. doi: 10.1097/00126334-200108150-00009
56. Kindberg E, Mickienė A, Ax C, Åkerlind B, Vene S, Lindquist L, et al. A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis. J Infect Dis. (2008) 197:266–9. doi: 10.1086/524709
57. Grygorczuk S, Osada J, Parczewski M, Moniuszko A, Świerzbińska R, Kondrusik M, et al. The expression of the chemokine receptor CCR5 in tick-borne encephalitis. J Neuroinflamm. (2016) 13:45. doi: 10.1186/s12974-016-0511-0
58. Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, Frank WA, et al. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. (2006) 203:35–40. doi: 10.1084/jem.20051970
59. Michlmayr D, Bardina SV, Rodriguez CA, Pletnev AG, and Lim JK. Dual function of ccr5 during langat virus encephalitis: reduction in neutrophil-mediated central nervous system inflammation and increase in T cell–mediated viral clearance. J Immunol. (2016) 196:4622–31. doi: 10.4049/jimmunol.1502452
60. Larena M, Regner M, and Lobigs M. The chemokine receptor CCR5, a therapeutic target for HIV/AIDS antagonists, is critical for recovery in a mouse model of Japanese encephalitis. PloS One. (2012) 7:e44834. doi: 10.1371/journal.pone.0044834
61. Yurchenko E, Tritt M, Hay V, Shevach EM, Belkaid Y, and Piccirillo CA. CCR5-dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med. (2006) 203:2451–60. doi: 10.1084/jem.20060956
62. Winter PM, Dung NM, Loan HT, Kneen R, Wills B, Thu LT, et al. Proinflammatory cytokines and chemokines in humans with Japanese encephalitis. J Infect Dis. (2004) 190:1618–26. doi: 10.1086/423328
63. Lai C, Ou Y, Chang C, Pan H, Chang C, Liao S, et al. Endothelial Japanese encephalitis virus infection enhances migration and adhesion of leukocytes to brain microvascular endothelia via MEK-dependent expression of ICAM1 and the CINC and RANTES chemokines. J Neurochem. (2012) 123:250–61. doi: 10.1111/j.1471-4159.2012.07889.x
64. Grygorczuk S, Osada J, Sulik A, Toczyłowski K, Dunaj-Małyszko J, Czupryna P, et al. Associations of the cerebrospinal fluid lymphocyte population with a clinical presentation of tick-borne encephalitis. Ticks Tick-Borne Dis. (2023) 14:102204. doi: 10.1016/j.ttbdis.2023.102204
65. Zhang F, Qi L, Li T, Li X, Yang D, Cao S, et al. PD1+CCR2+CD8+ T cells infiltrate the central nervous system during acute Japanese encephalitis virus infection. Virol Sin. (2019) 34:538–48. doi: 10.1007/s12250-019-00134-z
66. Růžek D, Salát J, Palus M, Gritsun TS, Gould EA, Dyková I, et al. CD8+ T-cells mediate immunopathology in tick-borne encephalitis. Virology. (2009) 384:1–6. doi: 10.1016/j.virol.2008.11.023
67. Sitati EM and Diamond MS. CD4 + T-cell responses are required for clearance of west nile virus from the central nervous system. J Virol. (2006) 80:12060–9. doi: 10.1128/JVI.01650-06
68. Choi H, Song H, and Jung YW. The roles of CCR7 for the homing of memory CD8+ T cells into their survival niches. Immune Netw. (2020) 20:e20. doi: 10.4110/in.2020.20.e20
69. Mueller SN, Hosiawa-Meagher KA, Konieczny BT, Sullivan BM, Bachmann MF, Locksley RM, et al. Regulation of homeostatic chemokine expression and cell trafficking during immune responses. Science. (2007) 317:670–4. doi: 10.1126/science.1144830
70. Bardina SV, Michlmayr D, Hoffman KW, Obara CJ, Sum J, Charo IF, et al. Differential roles of chemokines CCL2 and CCL7 in monocytosis and leukocyte migration during west nile virus infection. J Immunol. (2015) 195:4306–18. doi: 10.4049/jimmunol.1500352
71. Bardina SV, Brown JA, Michlmayr D, Hoffman KW, Sum J, Pletnev AG, et al. Chemokine receptor ccr7 restricts fatal west nile virus encephalitis. J Virol. (2017) 91:e02409–16. doi: 10.1128/JVI.02409-16
72. De Carvalho GC, Borget MY, Bernier S, Garneau D, Da Silva Duarte AJ, and Dumais N. RAGE and CCR7 mediate the transmigration of Zika-infected monocytes through the blood-brain barrier. Immunobiology. (2019) 224:792–803. doi: 10.1016/j.imbio.2019.08.007
73. Fontoura MA, Rocha RF, and Marques RE. Neutrophil recruitment and participation in severe diseases caused by flavivirus infection. Life. (2021) 11:717. doi: 10.3390/life11070717
74. Michael BD, Bricio-Moreno L, Sorensen EW, Miyabe Y, Lian J, Solomon T, et al. Astrocyte- and neuron-derived CXCL1 drives neutrophil transmigration and blood-brain barrier permeability in viral encephalitis. Cell Rep. (2020) 32:108150. doi: 10.1016/j.celrep.2020.108150
75. Bai F, Kong K, Dai J, Qian F, Zhang L, Brown CR, et al. A paradoxical role for neutrophils in the pathogenesis of west nile virus. J Infect Dis. (2010) 202:1804–12. doi: 10.1086/657416
76. Marques RE, Del Sarto JL, Rocha RPF, Gomes GF, Cramer A, Rachid MA, et al. Development of a model of Saint Louis encephalitis infection and disease in mice. J Neuroinflamm. (2017) 14:61. doi: 10.1186/s12974-017-0837-2
77. Matthews V, Robertson T, Kendrick T, Abdo M, Papadimitriou J, and Mcminn P. Morphological features of Murray Valley encephalitis virus infection in the central nervous system of swiss mice. Int J Exp Pathol. (2000) 81:31–40. doi: 10.1046/j.1365-2613.2000.00135.x
78. Andrews DM, Matthews VB, Sammels LM, Carrello AC, and McMinn PC. The severity of murray valley encephalitis in mice is linked to neutrophil infiltration and inducible nitric oxide synthase activity in the central nervous system. J Virol. (1999) 73:8781–90. doi: 10.1128/JVI.73.10.8781-8790.1999
79. Mathur A, Kulshreshtha R, and Singh A. Secretion of the chemokine interleukin-8 during Japanese encephalitis virus infection. J Med Microbiol. (2000) 49:607–12. doi: 10.1099/0022-1317-49-7-607
80. Wu F, Zhao Y, Jiao T, Shi D, Zhu X, Zhang M, et al. CXCR2 is essential for cerebral endothelial activation and leukocyte recruitment during neuroinflammation. J Neuroinflamm. (2015) 12:98. doi: 10.1186/s12974-015-0316-6
81. Abdelbaky HH, Mitsuhashi S, Watanabe K, Ushio N, Miyakawa M, Furuoka H, et al. Involvement of chemokine receptor CXCR3 in the defense mechanism against Neospora caninum infection in C57BL/6 mice. Front Microbiol. (2023) 13:1045106. doi: 10.3389/fmicb.2022.1045106
82. Phares TW, Stohlman SA, Hinton DR, and Bergmann CC. Astrocyte-derived CXCL10 drives accumulation of antibody-secreting cells in the central nervous system during viral encephalomyelitis. J Virol. (2013) 87:3382–92. doi: 10.1128/JVI.03307-12
83. Benzarti E, Murray KO, and Ronca SE. Interleukins, chemokines, and tumor necrosis factor superfamily ligands in the pathogenesis of west nile virus infection. Viruses. (2023) 15:806. doi: 10.3390/v15030806
84. Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, et al. Neuronal CXCL10 directs CD8 + T-cell recruitment and control of west nile virus encephalitis. J Virol. (2005) 79:11457–66. doi: 10.1128/JVI.79.17.11457-11466.2005
85. Lepej SŽ, Mišić-Majerus L, Jeren T, Rode OD, Remenar A, Šporec V, et al. Chemokines CXCL10 and CXCL11 in the cerebrospinal fluid of patients with tick-borne encephalitis. Acta Neurol Scand. (2007) 115:109–14. doi: 10.1111/j.1600-0404.2006.00726.x
86. Zajkowska J, Moniuszko-Malinowska A, Pancewicz S, Muszyńska-Mazur A, Kondrusik M, Grygorczuk S, et al. Evaluation of CXCL10, CXCL11, CXCL12 and CXCL13 chemokines in serum and cerebrospinal fluid in patients with tick borne encephalitis (TBE). Adv Med Sci. (2011) 56:311–7. doi: 10.2478/v10039-011-0033-z
87. Ojha A, Bhasym A, Mukherjee S, Annarapu GK, Bhakuni T, Akbar I, et al. Platelet factor 4 promotes rapid replication and propagation of Dengue and Japanese encephalitis viruses. EBioMedicine. (2019) 39:332–47. doi: 10.1016/j.ebiom.2018.11.049
88. Singh A, Ghosh R, and Guchhait P. CXCR3 antagonist rescues ER stress and reduces inflammation and JEV infection in mice brain. Cytokine. (2023) 172:156380. doi: 10.1016/j.cyto.2023.156380
89. Singh A, Ghosh R, Asuru TR, Prajapat SK, Joshi G, Gaur KK, et al. Inhibition of cellular activation induced by platelet factor 4 via the CXCR3 pathway ameliorates Japanese encephalitis and dengue viral infections. J Thromb Haemost. (2023) 22:S153878362300853X. doi: 10.1016/j.jtha.2023.11.015
90. Panganiban AT, Blair RV, Hattler JB, Bohannon DG, Bonaldo MC, Schouest B, et al. A Zika virus primary isolate induces neuroinflammation, compromises the blood-brain barrier and upregulates CXCL12 in adult macaques. Brain Pathol. (2020) 30:1017–27. doi: 10.1111/bpa.12873
91. Bianchi ME and Mezzapelle R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front Immunol. (2020) 11:2109. doi: 10.3389/fimmu.2020.02109
92. McCandless EE, Wang Q, Woerner BM, Harper JM, and Klein RS. CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol. (2006) 177:8053–64. doi: 10.4049/jimmunol.177.11.8053
93. McCandless EE, Zhang B, Diamond MS, and Klein RS. CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc Natl Acad Sci. (2008) 105:11270–5. doi: 10.1073/pnas.0800898105
94. Grygorczuk S, Czupryna P, Pancewicz S, Świerzbińska R, Dunaj J, Siemieniako A, et al. The increased intrathecal expression of the monocyte-attracting chemokines CCL7 and CXCL12 in tick-borne encephalitis. J Neurovirol. (2021) 27:452–62. doi: 10.1007/s13365-021-00975-z
95. Cardona SM, Mendiola AS, Yang YC, Adkins SL, Torres V, and Cardona AE. Disruption of fractalkine signaling leads to microglial activation and neuronal damage in the diabetic retina. ASN Neuro. (2015) 7:175909141560820. doi: 10.1177/1759091415608204
96. Tristão FSM, Lazzarini M, Martin S, Amar M, Stühmer W, Kirchhoff F, et al. CX3CR1 disruption differentially influences dopaminergic neuron degeneration in parkinsonian mice depending on the neurotoxin and route of administration. Neurotox Res. (2016) 29:364–80. doi: 10.1007/s12640-015-9557-5
97. Choi JY, Kim JH, Hossain FMA, Uyangaa E, Park SO, Kim B, et al. Indispensable role of CX3CR1+ Dendritic cells in regulation of virus-induced neuroinflammation through rapid development of antiviral immunity in peripheral lymphoid tissues. Front Immunol. (2019) 10:1467. doi: 10.3389/fimmu.2019.01467
98. Menasria R, Canivet C, Piret J, Gosselin J, and Boivin G. Protective role of CX3CR1 signalling in resident cells of the central nervous system during experimental herpes simplex virus encephalitis. J Gen Virol. (2017) 98:447–60. doi: 10.1099/jgv.0.000667
99. Rivas-Fuentes S, Salgado-Aguayo A, Arratia-Quijada J, and Gorocica-Rosete P. Regulation and biological functions of the CX3CL1-CX3CR1 axis and its relevance in solid cancer: A mini-review. J Cancer. (2021) 12:571–83. doi: 10.7150/jca.47022
100. Spiteri AG, Terry RL, Wishart CL, Ashhurst TM, Campbell IL, Hofer MJ, et al. High-parameter cytometry unmasks microglial cell spatio-temporal response kinetics in severe neuroinflammatory disease. J Neuroinflamm. (2021) 18:166. doi: 10.1186/s12974-021-02214-y
101. Getts DR, Terry RL, Getts MT, Müller M, Rana S, Shrestha B, et al. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. (2008) 205:2319–37. doi: 10.1084/jem.20080421
102. Michlmayr D, McKimmie CS, Pingen M, Haxton B, Mansfield K, Johnson N, et al. Defining the chemokine basis for leukocyte recruitment during viral encephalitis. J Virol. (2014) 88:9553–67. doi: 10.1128/JVI.03421-13
103. Grygorczuk S, Świerzbińska R, Kondrusik M, Dunaj J, Czupryna P, Moniuszko A, et al. The intrathecal expression and pathogenetic role of Th17 cytokines and CXCR2-binding chemokines in tick-borne encephalitis. J Neuroinflamm. (2018) 15:115. doi: 10.1186/s12974-018-1138-0
104. Sadik CD and Luster AD. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol. (2011) 91:207–15. doi: 10.1189/jlb.0811402
105. Sehgal N, Kumawat KL, Basu A, and Ravindranath V. Fenofibrate reduces mortality and precludes neurological deficits in survivors in murine model of Japanese encephalitis viral infection. PloS One. (2012) 7:e35427. doi: 10.1371/journal.pone.0035427
106. Brown SL, Jala VR, Raghuwanshi SK, Nasser MW, Haribabu B, and Richardson RM. Activation and regulation of platelet-activating factor receptor: role of gi and gq in receptor-mediated chemotactic, cytotoxic, and cross-regulatory signals. J Immunol. (2006) 177:3242–9. doi: 10.4049/jimmunol.177.5.3242
107. Ishizuka EK, Filgueiras LR, Rios FJ, Serezani CH, and Jancar S. PAFR activation of NF-κB p65 or p105 precursor dictates pro- and anti-inflammatory responses during TLR activation in murine macrophages. Sci Rep. (2016) 6:32092. doi: 10.1038/srep32092
108. Lefebvre JS, Marleau S, Milot V, Lévesque T, Picard S, Flamand N, et al. Toll-like receptor ligands induce polymorphonuclear leukocyte migration: key roles for leukotriene B 4 and platelet-activating factor. FASEB J. (2010) 24:637–47. doi: 10.1096/fj.09-135624
109. Yang KD, Lee CS, and Shaio MF. A higher production of platelet activating factor in ex vivo heterologously secondary dengue-2 virus infections. Acta Microbiol Immunol Hung. (1995) 42:403–7.
110. Souza DG, Fagundes CT, Sousa LP, Amaral FA, Souza RS, Souza AL, et al. Essential role of platelet-activating factor receptor in the pathogenesis of Dengue virus infection. Proc Natl Acad Sci. (2009) 106:14138–43. doi: 10.1073/pnas.0906467106
111. Vandendriessche S, Cambier S, Proost P, and Marques PE. Complement receptors and their role in leukocyte recruitment and phagocytosis. Front Cell Dev Biol. (2021) 9:624025. doi: 10.3389/fcell.2021.624025
112. Sadik CD, Miyabe Y, Sezin T, and Luster AD. The critical role of C5a as an initiator of neutrophil-mediated autoimmune inflammation of the joint and skin. Semin Immunol. (2018) 37:21–9. doi: 10.1016/j.smim.2018.03.002
113. Sengupta N, Mukherjee S, Tripathi P, Kumar R, Suryawanshi AR, and Basu A. Cerebrospinal fluid biomarkers of Japanese encephalitis. F1000Research. (2015) 4:334. doi: 10.12688/f1000research
114. Mehlhop E, Whitby K, Oliphant T, Marri A, Engle M, and Diamond MS. Complement activation is required for induction of a protective antibody response against west nile virus infection. J Virol. (2005) 79:7466–77. doi: 10.1128/JVI.79.12.7466-7477.2005
115. Mehlhop E and Diamond MS. Protective immune responses against West Nile virus are primed by distinct complement activation pathways. J Exp Med. (2006) 203:1371–81. doi: 10.1084/jem.20052388
116. Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, et al. Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med. (2010) 207:793–806. doi: 10.1084/jem.20092545
117. Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, et al. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci. (2006) 103:19111–6. doi: 10.1073/pnas.0605668103
118. Figueiredo CP, Barros-Aragão FGQ, Neris RLS, Frost PS, Soares C, Souza INO, et al. Zika virus replicates in adult human brain tissue and impairs synapses and memory in mice. Nat Commun. (2019) 10:3890. doi: 10.1038/s41467-019-11866-7
119. Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature. (2016) 534:538–43. doi: 10.1038/nature18283
120. Blackhurst BM and Funk KE. Molecular and cellular mechanisms underlying neurologic manifestations of mosquito-borne flavivirus infections. Viruses. (2023) 15:2200. doi: 10.3390/v15112200
121. Ghosh Roy S, Sadigh B, Datan E, Lockshin RA, and Zakeri Z. Regulation of cell survival and death during Flavivirus infections. World J Biol Chem. (2014) 5:93–105. doi: 10.4331/wjbc.v5.i2.93
122. Medina CB and Ravichandran KS. Do not let death do us part: ‘find-me’ signals in communication between dying cells and the phagocytes. Cell Death Differ. (2016) 23:979–89. doi: 10.1038/cdd.2016.13
123. Westman J, Grinstein S, and Marques PE. Phagocytosis of necrotic debris at sites of injury and inflammation. Front Immunol. (2020) 10:3030. doi: 10.3389/fimmu.2019.03030
124. Groth M, Łuczaj W, Dunaj-Małyszko J, Skrzydlewska E, and Moniuszko-Malinowska A. Differences in the plasma phospholipid profile of patients infected with tick-borne encephalitis virus and co-infected with bacteria. Sci Rep. (2022) 12:9538. doi: 10.1038/s41598-022-13765-2
125. Mingo-Casas P, Sanchez-Céspedes J, Blázquez AB, Casas J, Balsera-Manzanero M, Herrero L, et al. Lipid signatures of West Nile virus infection unveil alterations of sphingolipid metabolism providing novel biomarkers. Emerg Microbes Infect. (2023) 12:2231556. doi: 10.1080/22221751.2023.2231556
126. Bian P, Zheng X, Wei L, Ye C, Fan H, Cai Y, et al. MLKL mediated necroptosis accelerates JEV-induced neuroinflammation in mice. Front Microbiol. (2017) 8:303/full. doi: 10.3389/fmicb.2017.00303/full
127. Lim SM, Van Den Ham HJ, Oduber M, Martina E, Zaaraoui-Boutahar F, Roose JM, et al. Transcriptomic analyses reveal differential gene expression of immune and cell death pathways in the brains of mice infected with west nile virus and chikungunya virus. Front Microbiol. (2017) 8:1556. doi: 10.3389/fmicb.2017.01556
128. Rendon-Mitchell B, Ochani M, Li J, Han J, Wang H, Yang H, et al. IFN-γ Induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. (2003) 170:3890–7. doi: 10.4049/jimmunol.170.7.3890
129. Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. J Biol Chem. (1995) 270:25752–61. doi: 10.1074/jbc.270.43.25752
130. Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol. (1993) 143:1699–712.
131. Scaffidi P, Misteli T, and Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. (2002) 418:191–5. doi: 10.1038/nature00858
132. Degryse B, Bonaldi T, Scaffidi P, Müller S, Resnati M, Sanvito F, et al. The high mobility group (Hmg) boxes of the nuclear protein hmg1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J Cell Biol. (2001) 152:1197–206. doi: 10.1083/jcb.152.6.1197
133. Schiraldi M, Raucci A, Muñoz LM, Livoti E, Celona B, Venereau E, et al. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med. (2012) 209:551–63. doi: 10.1084/jem.20111739
134. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. (2007) 13:587–96. doi: 10.1038/nm1567
135. Jankauskas SS, Wong DWL, Bucala R, Djudjaj S, and Boor P. Evolving complexity of MIF signaling. Cell Signal. (2019) 57:76–88. doi: 10.1016/j.cellsig.2019.01.006
136. Arjona A, Foellmer HG, Town T, Leng L, McDonald C, Wang T, et al. Abrogation of macrophage migration inhibitory factor decreases West Nile virus lethality by limiting viral neuroinvasion. J Clin Invest. (2007) 117:3059–66. doi: 10.1172/JCI32218
137. Grygorczuk S, Parczewski M, Świerzbińska R, Czupryna P, Moniuszko A, Dunaj J, et al. The increased concentration of macrophage migration inhibitory factor in serum and cerebrospinal fluid of patients with tick-borne encephalitis. J Neuroinflamm. (2017) 14:126. doi: 10.1186/s12974-017-0898-2
138. Suzuki T, Ogata A, Tashiro K, Nagashima K, Tamura M, Yasui K, et al. Japanese encephalitis virus up-regulates expression of macrophage migration inhibitory factor (MIF) mRNA in the mouse brain. Biochim Biophys Acta BBA - Gene Struct Expr. (2000) 1517:100–6. doi: 10.1016/S0167-4781(00)00262-1
139. Zhang L, Nan X, Zhou D, Wang X, Zhu S, Li Q, et al. Japanese encephalitis virus NS1 and NS1′ protein disrupts the blood-brain barrier through macrophage migration inhibitory factor-mediated autophagy. J Virol. (2024) 98:e00116–24. doi: 10.1128/jvi.00116-24
140. Das R, Loughran K, Murchison C, Qian F, Leng L, Song Y, et al. Association between high expression macrophage migration inhibitory factor (MIF) alleles and West Nile virus encephalitis. Cytokine. (2016) 78:51–4. doi: 10.1016/j.cyto.2015.11.021
141. Cussell P, Gomez Escalada M, Milton N, and Paterson A. The N-formyl peptide receptors: contemporary roles in neuronal function and dysfunction. Neural Regener Res. (2020) 15:1191. doi: 10.4103/1673-5374.272566
142. Murray P, Frampton G, and Nelson P. Cell adhesion molecules. BMJ. (1999) 319:332–4. doi: 10.1136/bmj.319.7206.332
143. Carlos TM and Harlan JM. Leukocyte-endothelial adhesion molecules. Blood. (1994) 84:2068–101. doi: 10.1182/blood.V84.7.2068.2068
144. Yong K and Khwaja A. Leucocyte cellular adhesion molecules. Blood Rev. (1990) 4:211–25. doi: 10.1016/0268-960X(90)90001-9
145. McEver RP and Zhu C. Rolling cell adhesion. Annu Rev Cell Dev Biol. (2010) 26:363–96. doi: 10.1146/annurev.cellbio.042308.113238
146. Weller A, Isenmann S, and Vestweber D. Cloning of the mouse endothelial selectins. Expression of both E- and P-selectin is inducible by tumor necrosis factor alpha. J Biol Chem. (1992) 267:15176–83. doi: 10.1016/S0021-9258(18)42162-X
147. Gotsch U, Jäger U, Dominis M, and Vestweber D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-α in vivo. Cell Adhes Commun. (1994) 2:7–14. doi: 10.3109/15419069409014198
148. Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, and Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. (1995) 9:899–909. doi: 10.1096/fasebj.9.10.7542214
149. Zou SS, Zou QC, Xiong WJ, Cui NY, Wang K, Liu HX, et al. Brain microvascular endothelial cell-derived HMGB1 facilitates monocyte adhesion and transmigration to promote JEV neuroinvasion. Front Cell Infect Microbiol. (2021) 11:701820. doi: 10.3389/fcimb.2021.701820
150. Krylova NV, Smolina TP, and Leonova GN. Molecular mechanisms of interaction between human immune cells and far eastern tick-borne encephalitis virus strains. Viral Immunol. (2015) 28:272–81. doi: 10.1089/vim.2014.0083
151. Roe K, Orillo B, and Verma S. West nile virus-induced cell adhesion molecules on human brain microvascular endothelial cells regulate leukocyte adhesion and modulate permeability of the in vitro blood-brain barrier model. PloS One. (2014) 9:e102598. doi: 10.1371/journal.pone.0102598
152. Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, et al. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: Transmigration across the in vitro blood-brain barrier. Virology. (2009) 385:425–33. doi: 10.1016/j.virol.2008.11.047
153. Shen J, T-To SS, Schrieber L, and King NJ. Early E-selectin, VCAM-1, ICAM-1, and late major histocompatibility complex antigen induction on human endothelial cells by flavivirus and comodulation of adhesion molecule expression by immune cytokines. J Virol. (1997) 71:9323–32. doi: 10.1128/jvi.71.12.9323-9332.1997
154. Clé M, Desmetz C, Barthelemy J, Martin MF, Constant O, Maarifi G, et al. Zika virus infection promotes local inflammation, cell adhesion molecule upregulation, and leukocyte recruitment at the blood-brain barrier. mBio. (2020) 11:e01183–20. doi: 10.1128/mBio.01183-20
155. Getts DR, Terry RL, Getts MT, Müller M, Rana S, Deffrasnes C, et al. Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)-4-dependent recruitment of nitric oxide-producing macrophages. J Neuroinflamm. (2012) 9:246. doi: 10.1186/1742-2094-9-246
156. Dai J, Wang P, Bai F, Town T, and Fikrig E. ICAM-1 participates in the entry of west nile virus into the central nervous system. J Virol. (2008) 82:4164–8. doi: 10.1128/JVI.02621-07
157. Tobler LH, Cameron MJ, Lanteri MC, Prince HE, Danesh A, Persad D, et al. Interferon and interferon-induced chemokine expression is associated with control of acute viremia in west nile virus-infected blood donors. J Infect Dis. (2008) 198:979–83. doi: 10.1086/591466
158. Zidovec-Lepej S, Vilibic-Cavlek T, Barbic L, Ilic M, Savic V, Tabain I, et al. Antiviral cytokine response in neuroinvasive and non-neuroinvasive west nile virus infection. Viruses. (2021) 13:342. doi: 10.3390/v13020342
159. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1β Signaling promotes CNS-intrinsic immune control of west nile virus infection. PloS Pathog. (2012) 8:e1003039. doi: 10.1371/journal.ppat.1003039
160. Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, et al. The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins. J Exp Med. (2003) 198:1507–15. doi: 10.1084/jem.20030800
161. Frommhold D, Kamphues A, Hepper I, Pruenster M, Lukić IK, Socher I, et al. RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood. (2010) 116:841–9. doi: 10.1182/blood-2009-09-244293
162. Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. (2003) 101:2652–60. doi: 10.1182/blood-2002-05-1300
163. Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. (2007) 26:1129–39. doi: 10.1038/sj.emboj.7601552
164. Amin MA, Haas CS, Zhu K, Mansfield PJ, Kim MJ, Lackowski NP, et al. Migration inhibitory factor up-regulates vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 via Src, PI3 kinase, and NFκB. Blood. (2006) 107:2252–61. doi: 10.1182/blood-2005-05-2011
165. Cheng Q, McKeown SJ, Santos L, Santiago FS, Khachigian LM, Morand EF, et al. Macrophage migration inhibitory factor increases leukocyte–endothelial interactions in human endothelial cells via promotion of expression of adhesion molecules. J Immunol. (2010) 185:1238–47. doi: 10.4049/jimmunol.0904104
166. Wojcik E, Carrithers LM, and Carrithers MD. Characterization of epithelial V-like antigen in human choroid plexus epithelial cells: Potential role in CNS immune surveillance. Neurosci Lett. (2011) 495:115–20. doi: 10.1016/j.neulet.2011.03.051
167. Partiot E, Brychka D, and Gaudin R. Investigating human monocyte adhesion, migration and transmigration and their modulation by Zika virus. Eur J Cell Biol. (2024) 103:151453. doi: 10.1016/j.ejcb.2024.151453
168. Ayala-Nunez NV, Follain G, Delalande F, Hirschler A, Partiot E, Hale GL, et al. Zika virus enhances monocyte adhesion and transmigration favoring viral dissemination to neural cells. Nat Commun. (2019) 10:4430. doi: 10.1038/s41467-019-12408-x
Keywords: flavivirus, leukocyte, encephalitis, chemoattractant, chemokine, adhesion molecule
Citation: Slowikowski E, Willems C and Marques PE (2025) Leukocyte recruitment in flavivirus-induced encephalitis. Front. Immunol. 16:1650903. doi: 10.3389/fimmu.2025.1650903
Received: 20 June 2025; Accepted: 04 August 2025;
Published: 22 August 2025.
Edited by:
Juan Bautista De Sanctis, Palacký University Olomouc, CzechiaReviewed by:
Ermin Schadich, Palacký University, CzechiaAlexis Hipólito García, Central University of Venezuela, Venezuela
Copyright © 2025 Slowikowski, Willems and Marques. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pedro Elias Marques, cGVkcm8ubWFycXVlc0BrdWxldXZlbi5iZQ==
†These authors have contributed equally to this work