 Fujie Liang
Fujie Liang Xiang Wang
Xiang Wang- Department of Urology, The First Affiliated Hospital of Guangxi Medical University, Nanning, China
Calcium Oxalate Nephrolithiasis is a globally prevalent urological disorder, with its pathogenesis involving multiple mechanisms such as inflammatory responses, oxidative stress, crystal-cell interactions, macrophage polarization, and fibrosis. In recent years, the multidimensional regulatory roles of interleukins (ILs) and chemokines in stone formation have garnered increasing attention. Pro-inflammatory interleukins, such as IL-1β, may promote crystal deposition, oxidative stress, and renal tubular epithelial cell injury by activating signaling pathways including NLRP3 inflammasome, NF-κB, and MAPK. In contrast, anti-inflammatory interleukins, by stimulating M2 macrophage polarization and suppressing crystal adhesion and oxidative damage, exhibit nephroprotective effects. Notably, IL-6 demonstrates unique bidirectional regulatory properties. Chemokines play critical roles in recruiting immune cells, amplifying inflammatory responses, modulating crystal-cell interactions, and sustaining the fibrosis-stone vicious cycle. The CXCL12/CXCR4 axis has emerged as a potential hub in regulating crystal autophagy and fibrotic progression. Additionally, miR-124-3p overexpression inhibits pro-inflammatory factor expression and promotes M2 macrophage polarization, while the IL-6/MCP-1 axis may reverse this suppression via a negative feedback network. This review integrates the multidimensional regulatory mechanisms of interleukins and chemokines in Calcium Oxalate Nephrolithiasis and proposes three novel hypotheses: the dynamic regulatory model of IL-6, the MCP-1-mediated fibrosis-stone vicious cycle, and the IL-6/MCP-1/miR-124-3p negative feedback loop.
1 Introduction
Kidney stones are a global health burden, with 80%–90% of cases being calcium oxalate (CaOx) stones (1). The incidence and recurrence rates of nephrolithiasis have risen due to modern lifestyle changes and global warming. In China, approximately 1 in 17 adults suffers from kidney stones, with males aged 31–60 exhibiting significantly higher prevalence than females (2–4). The formation of kidney stones is a multi-step pathophysiological process, triggered by a pathological imbalance in the urinary environment, influenced by cell damage, and regulated by genetic and metabolic factors (5). Under supersaturated conditions caused by excessive intake of solutes, dehydration, or metabolic abnormalities, compounds such as calcium, oxalate, or uric acid form microcrystalline nuclei that precipitate in the renal tubules. These crystals typically nucleate on Randall’s plaques, which are deposits of calcium phosphate located on the basement membrane of the renal papilla. Additionally, crystal-induced cell damage activates pathogenic pathways involving oxidative stress and mitochondrial dysfunction, leading to the release of extracellular vesicles and adhesion molecules (5, 6). Overall, these mechanisms promote crystal aggregation and subsequent stone growth.
There are mainly five types of kidney stones: calcium oxalate, calcium phosphate, uric acid, magnesium ammonium phosphate and cystine. Among them, calcium oxalate kidney stones are the most common. The formation of Calcium Oxalate Nephrolithiasis involves diverse physiological and chemical processes, including urine composition alterations, oxidative stress, inflammatory responses, macrophage infiltration, autophagy, gut microbiota dysbiosis, and renal fibrosis (7–11). The pathophysiological process of calcium oxalate kidney stones stems from the nucleation and deposition of calcium oxalate crystals in the renal tubules, mainly caused by the supersaturation of calcium oxalate in urine (12). This supersaturated state promotes the precipitation, growth and aggregation of crystals, eventually forming the initial center point of stone formation.These crystals attach to the epithelial cells of renal tubules, triggering oxidative stress and activating multiple signaling pathways such as NLRP3 inflammasome, TLR4/NF-κB, and p38 MAPK, resulting in the massive release of inflammatory cytokines and amplification of local inflammatory responses (12, 13). Meanwhile, the immunomodulatory mechanism induces macrophages to polarize towards the M1 phenotype, jointly promoting the occurrence and development of calcium oxalate kidney stones (14).
Emerging evidence suggests that inflammation is not merely a consequence but an active driver of CaOx stone formation (15, 16). Interleukins and chemokines are markedly upregulated in renal tubular epithelial cells and stone microenvironments, highlighting their critical roles in pathogenesis. While previous reviews focused on their roles in inflammation and oxidative stress (Table 1), this article systematically integrates their regulatory networks and elucidates their interplay with fibrosis, offering new insights into the dynamic interactions between inflammatory drivers and renal structural remodeling.
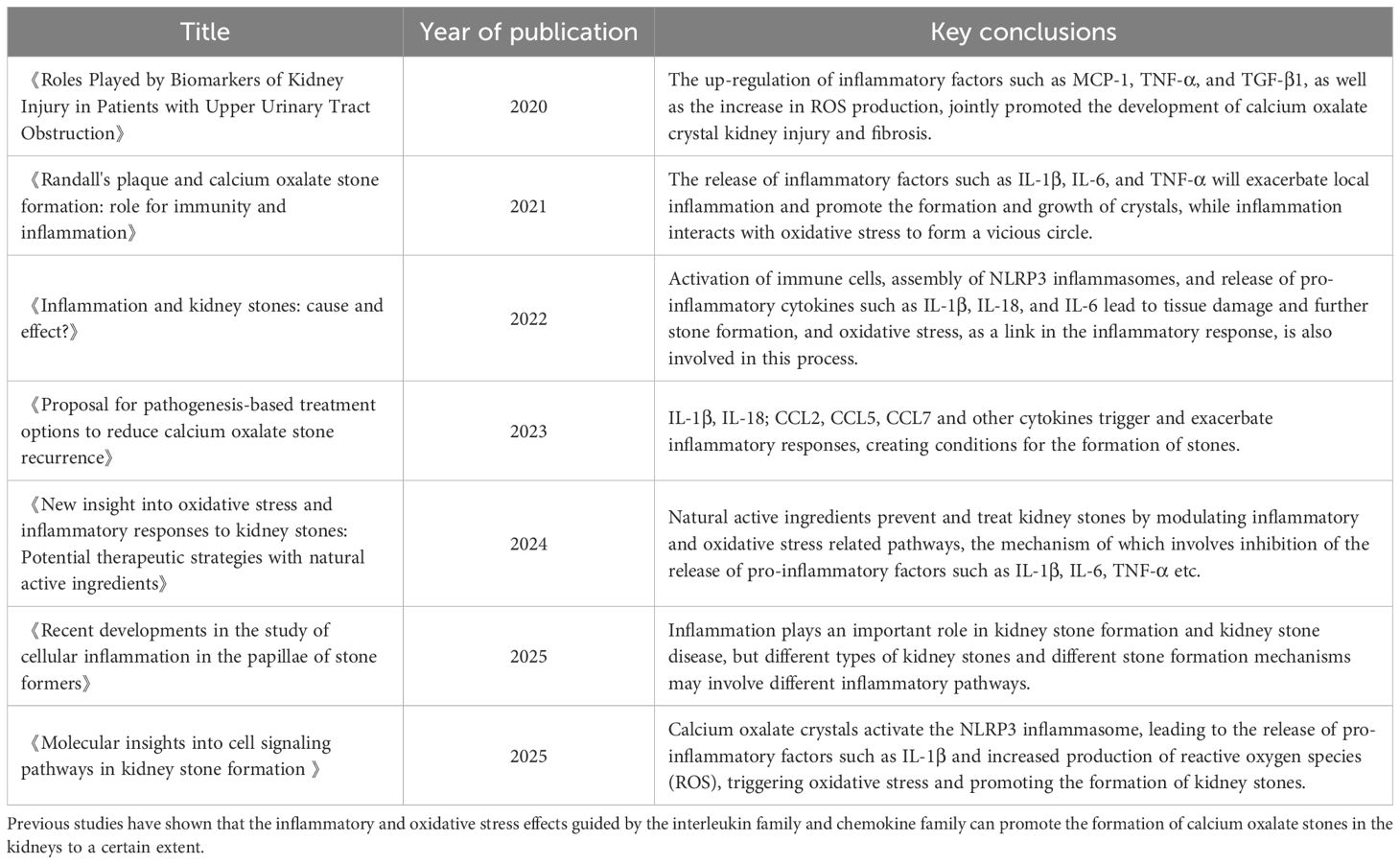
Table 1. Regulatory mechanisms of interleukin and chemokine families in Calcium Oxalate Nephrolithiasis: recent advances (9, 16, 87–91).
2 Dual regulatory roles of interleukins in calcium oxalate nephrolithiasis
Interleukins(ILs), secreted by immune and non-immune cells, are pleiotropic cytokines central to innate immunity, inflammation, and tissue homeostasis. Based on functional characteristics, ILs are classified into pro-inflammatory (e.g., IL-1β) and anti-inflammatory (e.g., IL-10) subgroups. Recent studies reveal their pivotal roles in CaOx stone formation through inflammation, oxidative stress, and crystal-cell interactions.
2.1 IL-1β/IL-6-mediated inflammation and oxidative stress promote CaOx stone formation
2.1.1 Inflammatory roles and molecular mechanisms of IL-1β and IL-6
At the regulatory level of pro-inflammatory cytokines, interleukin-1β (IL-1β) and interleukin-6 (IL-6), as core mediators of inflammatory responses, play pivotal regulatory roles in Calcium Oxalate Nephrolithiasis formation (Figure 1). Substantial evidence indicates significant upregulation of IL-1β and IL-6 expression levels during nephrolithiasis development, particularly in calcium oxalate stone formation (17). The high-oxalate microenvironment and calcium oxalate crystals activate the NLRP3 inflammasome, leading to caspase-1-dependent maturation and release of IL-1β, which subsequently drives inflammatory injury in renal tubular epithelial cells and promotes reactive oxygen species (ROS) generation (18, 19). Furthermore, emerging studies suggest that the elevated expression of IL-1β and IL-6 in Calcium Oxalate Nephrolithiasis may also depend on NF-κB and MAPK signaling pathway regulation (20, 21). Notably, the upregulated IL-1β expression demonstrates a significant positive correlation with enhanced calcium oxalate crystal adhesion capacity, suggesting its potential role in mediating intrarenal crystal deposition through strengthening crystal-cell interfacial interactions (17), thereby facilitating calcium oxalate stone pathogenesis.
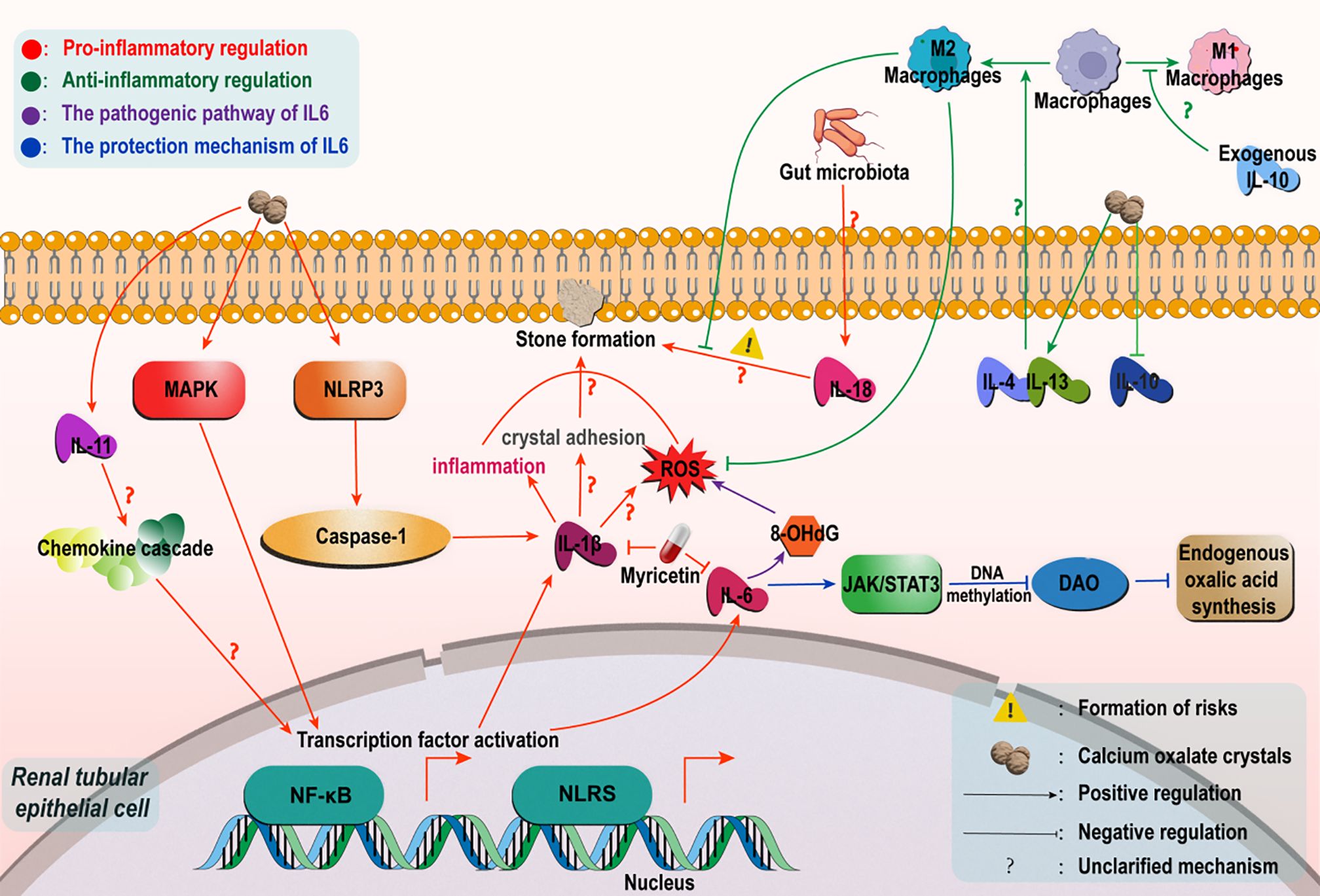
Figure 1. Regulatory mechanisms of the interleukin family in Calcium Oxalate Nephrolithiasis. In a high oxalic acid environment, calcium oxalate crystals mediate caspase-1-dependent IL-1βrelease by activating the NLRP3 inflammatome, trigger tubular inflammatory injury and ROS generation, and synergize with the NF-κB/MAPK pathway to enhance crystal adhesion and deposition. IL-6 has a biphasic effect: In the early stage, it inhibits DAO through the IL-6/JAK/STAT3 pathway to reduce oxalic acid synthesis; In the later stage, ROS/NF-κB is activated to intensify oxidative stress and adhesion. Among non-classical factors, IL-11 is involved in stone formation through chemokine/TLR/NLR signaling. Dysbiosis of the intestinal flora upregulates IL-18 via the gut-renal axis to increase susceptibility. In terms of anti-inflammation, IL-4/IL-13 promotes M2-polarized phagocytosis crystals in macrophages, and IL-10 can improve immune dysfunction induced by high oxalic acid.
Recent advances in therapeutic strategies have demonstrated that targeted inhibition of pro-inflammatory cytokines IL-1β and IL-6 expression can effectively intervene in calcium oxalate stone formation (Figure 1). For instance, myricetin exhibits anti-nephrolithic effects by suppressing IL-1β and IL-6 release, consequently alleviating renal stone formation and nephrotoxicity (22). Similarly, downregulation of IL-6 expression significantly inhibits oxalate-induced oxidative stress and inflammatory damage, thereby reducing lithogenic propensity (23). These collective findings substantiate that abnormal activation of the IL-1β/IL-6 signaling axis constitutes a critical mechanism underlying inflammatory injury and oxidative stress initiation during calcium oxalate calculogenesis. Concurrently, this pathway enhances crystal-cell adhesion interactions, creating a permissive microenvironment for calcium oxalate renal stone development.
2.1.2 Dual regulatory dynamics of IL-6: protective suppression versus pro-inflammatory aggravation
While both IL-6 and IL-1β are canonical pro-inflammatory cytokines, their mechanistic roles in Calcium Oxalate Nephrolithiasis exhibit distinct divergence (Table 2). Notably, IL-1β demonstrates consistent pro-lithogenic effects, whereas IL-6 displays paradoxical regulatory duality in calcium oxalate stone pathogenesis. Recent convergent validation across multi-species models (Drosophila, murine, and HK-2 cells) revealed that IL-6/JAK/STAT3 pathway activation epigenetically suppresses D-amino acid oxidase (DAO) transcription via DNA methylation modifications, thereby attenuating endogenous oxalate biosynthesis and mitigating hyperoxaluria risks. This underscores the evolutionary conservation and functional universality of the IL-6/JAK/STAT3 signaling axis (24). Paradoxically, in ethylene glycol-induced rat models of Calcium Oxalate Nephrolithiasis, elevated IL-6 levels exhibit significant positive correlation with oxidative stress marker 8-OHdG, implicating its role in promoting crystal deposition through ROS-mediated cascades (23) (Figure 1). To reconcile this functional dichotomy, we postulate a temporal regulatory hypothesis: During acute oxalate exposure, IL-6 may exert protective effects by suppressing DAO-driven oxalate synthesis via IL-6/JAK/STAT3 activation; conversely, in chronic lithogenic phases, sustained inflammatory microenvironments potentially drive IL-6-mediated ROS/NF-κB pathway activation, exacerbating oxidative stress and crystal adhesion. And the duration of calcium oxalate crystal exposure may be the underlying factor that triggers the transition between these two mechanisms. This paradigm highlights the necessity for stage-specific therapeutic targeting of IL-6 signaling. Future investigations should prioritize developing phase-defined IL-6 knockout models (e.g., conditional IL-6 depletion in acute oxalate challenge versus chronic stone-forming stages) to elucidate the microenvironment-dependent functional equilibrium governing IL-6’s dual roles.

2.2 Emerging roles of non-classical interleukins in calcium oxalate nephrolithiasis
Beyond IL-1β and IL-6, emerging evidence highlights the involvement of other interleukin family members in CaOx renal stone pathogenesis. Clinical data analyses reveal significantly elevated serum IL-11 levels in idiopathic Calcium Oxalate Nephrolithiasis patients. Mechanistic investigations propose its potential engagement in lithogenesis, possibly through orchestrating chemokine cascade interactions and activating Toll-like receptor (TLR)/NOD-like receptor (NLR) signaling pathways (25). Furthermore, investigations along the gut microbiota-kidney axis demonstrate that intestinal dysbiosis increases nephrolithiasis susceptibility via IL-18 upregulation (26), revealing novel inflammatory cytokine-mediated crosstalk in gut-renal pathophysiology (Figure 1). Nevertheless, research on non-classical pro-inflammatory interleukins remains nascent, with critical knowledge gaps persisting in three domains: (1) signaling specificity regarding detailed regulatory mechanisms of their activated pathways; (2) cytokine synergy involving combinatorial effects with other inflammatory mediators; and (3) systems-level integration of cross-talk between interleukin networks and metabolic reprogramming. Future studies should employ multi-omics strategies, particularly integrated metabolomic-proteomic analyses, to systematically map interleukin-mediated inflammatory networks and their spatial-temporal interactions in CaOx stone microenvironments. Such approaches will advance our mechanistic understanding beyond isolated cytokine observations toward comprehensive pathway deciphering.
2.3 Advances and challenges of anti-inflammatory factors in calcium oxalate kidney stone formation
The inflammatory microenvironment represents a dynamic equilibrium between pro-inflammatory and anti-inflammatory factors. In the field of anti-inflammatory regulation, IL-4 and IL-13 have been shown to stimulate macrophage polarization toward the M2 phenotype, enhancing their capacity to phagocytose calcium oxalate crystals and reducing oxidative stress damage in human renal tubular epithelial cells (HK-2) (27). Notably, emerging research highlights the critical protective role of IL-10. Experimental evidence demonstrates that oxalate-rich microenvironments significantly suppress IL-10 expression, while exogenous IL-10 supplementation effectively reverses oxalate-induced monocyte/macrophage dysfunction. Furthermore, a recent study demonstrated that IL-10-secreting constitutive RAW264.7 macrophages attenuate calcium oxalate crystal adhesion and deposition in both murine models of Calcium Oxalate Nephrolithiasis and in vitro models of COM-induced renal injury, mechanistically through downregulation of osteopontin (OPN) expression. This suggests that targeting the IL-10 signaling pathway may represent a novel therapeutic strategy for preventing and treating Calcium Oxalate Nephrolithiasis (28, 29).These findings elucidate the mechanisms underlying anti-inflammatory factor-mediated macrophage-epithelial cell interactions, providing a theoretical foundation for developing multi-target synergistic anti-lithiasis therapies (Figure 1).
Although these studies preliminarily reveal the regulatory roles of anti-inflammatory factors in calcium oxalate stone formation, systematic investigations into their involvement remain scarce compared to the well-characterized pro-inflammatory cytokine network. Other cytokines with potential anti-inflammatory properties, including IL-37 and IL-1RA, play key negative regulatory roles in the inflammatory response during gout crystal formation. It is worth noting that IL-37 promotes its own nuclear translocation by forming a complex with phosphorylated Smad3, thereby inhibiting the NLRP3–caspase-1–IL-1β signaling axis and mediating its anti-inflammatory effects. IL-1Ra mainly negatively regulates gout-associated inflammation by competitively inhibiting the IL-1β signaling pathway (30, 31). Currently, the roles of IL-37 and IL-1RA in the calcium oxalate crystal microenvironment remain unclear, and related studies are relatively limited. Inspired by their known functions in the gout inflammatory pathway, further investigation into the specific mechanisms of these two cytokines in calcium oxalate crystal-induced inflammation may provide new research directions and theoretical breakthroughs in this field.
3 Research status of chemokines in calcium oxalate nephrolithiasis
3.1 Overview of chemokines
Chemokines are small secreted proteins that interact with G protein-coupled receptors (GPCRs) on cell surfaces to stimulate cellular migration, particularly the directional movement of leukocytes. Based on the arrangement of cysteine (Cys) residues in their structures, chemokines are classified into four subfamilies: CC, CXC, CX3C, and XC. Beyond guiding cell migration, chemokines participate in diverse biological processes, including cell proliferation, survival, differentiation, and cytokine production. They play essential roles in immune system development, homeostasis maintenance, and immune response regulation (32, 33).
3.2 Structural and receptor-mediated mechanisms of MCP-1/CCL2 in inflammatory diseases
Monocyte chemoattractant protein-1 (MCP-1/CCL2), a key member of the CC chemokine subfamily, is a potent chemoattractant for monocytes. The MCP-1 gene is located on chromosome 17q11.2–q12 and encodes a 76-amino acid protein with a molecular weight of 13 kDa. Structurally, MCP-1 comprises a flexible N-terminal region, a long loop, and an α-helix, with the N-terminal domain being critical for receptor binding and activation. MCP-1 is expressed in various cell types, including endothelial cells, epithelial cells, and tumor cells. Its specific receptor, CCR2—a GPCR predominantly expressed on monocytes and T lymphocytes—activates downstream signaling pathways upon MCP-1 binding, recruiting immune cells such as monocytes/macrophages to inflammatory sites. This mechanism may contribute to the pathogenesis of diseases such as asthma, atherosclerosis, and breast cancer, potentially through amplified inflammatory signaling mediated by monocyte recruitment and cytokine release (34, 35). The MCP-1/CCR2 axis is also pivotal in renal pathologies. In nephritis, renal injury, kidney failure, and renal carcinoma, MCP-1 exacerbates inflammatory responses by promoting monocyte and macrophage infiltration, with its expression levels correlating strongly with the severity of renal damage (36). In diabetic nephropathy, MCP-1 acts as a critical inflammatory mediator, regulating inflammatory cascades and accelerating disease progression via oxidative stress activation (37–40).
3.3 Molecular mechanisms of MCP-1 in driving nephrolithiasis formation
3.3.1 Expression characteristics of MCP-1 in nephrolithiasis
Multiple clinical studies have demonstrated significantly elevated levels of MCP-1 and CCR2 in the urine and renal papillae of kidney stone patients (41, 42). Additionally, stone composition analysis revealed prominent MCP-1 expression in renal tissues of CaOx stone formers, suggesting its potential involvement in crystal adhesion and inflammatory microenvironment formation (43). In ethylene glycol-induced rat models of Calcium Oxalate Nephrolithiasis, MCP-1, CXCL1, and CXCL2 are released, activating TNF and IL-17 signaling pathways, which may play pivotal roles in stone pathogenesis (44, 45) (Figure 2). In murine CaOx stone models, intraperitoneal glyoxylate administration upregulated MCP-1 expression, accompanied by macrophage infiltration and tubular injury. Notably, CCR2 inhibition significantly reduced crystal deposition and ameliorated renal function (46) (Figure 3).
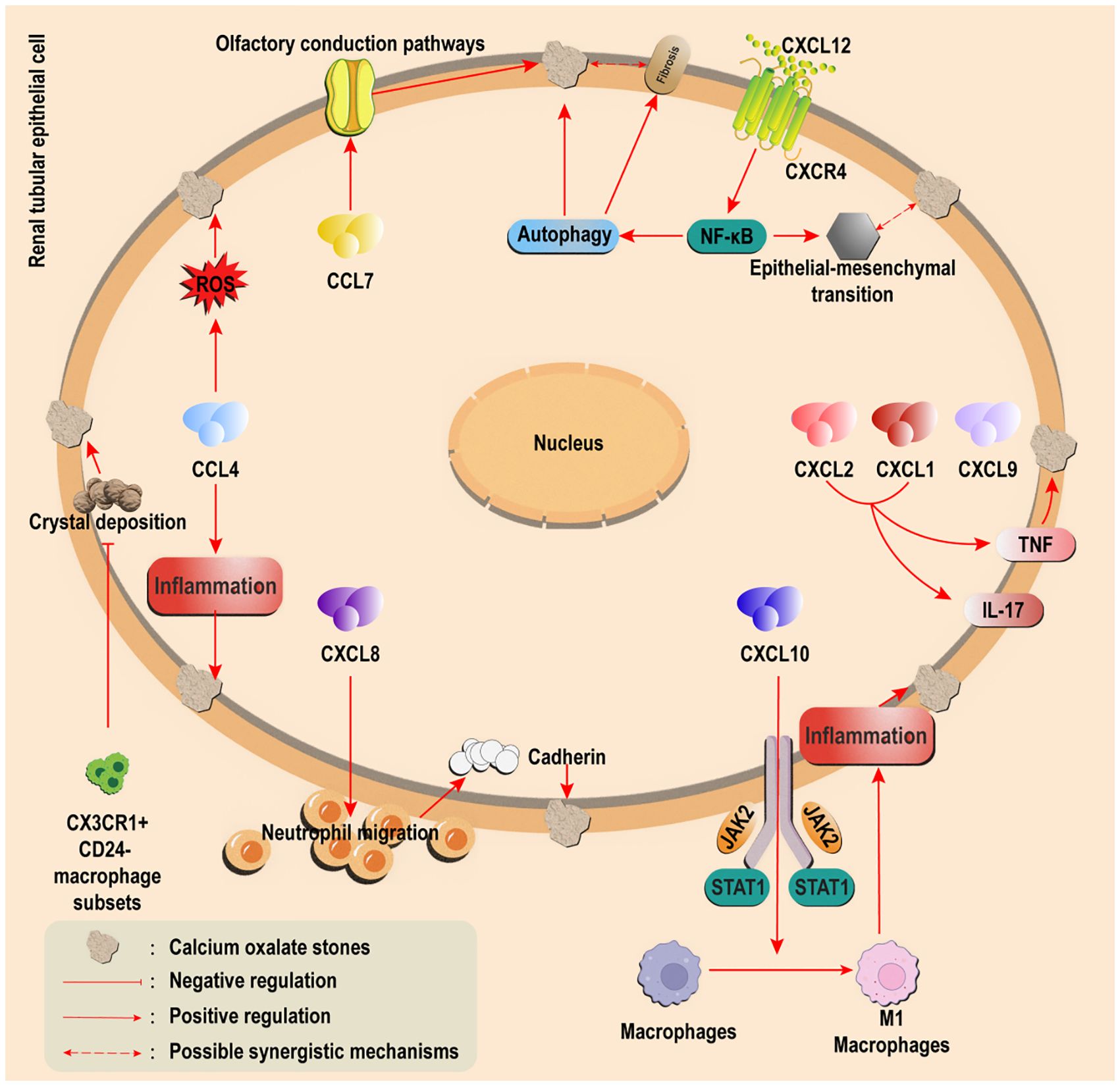
Figure 2. Regulatory effects of other chemokines on calcium oxalate stones. Overexpression of CCL5/7 (CCL7 or through the olfactory pathway) Pathogenic CXCL1/2/8/9/10 was upregulated (CXCL8 promotes inflammation and retention, and CXCL10 drives M1 polarization); Protective CXCL14 induces M2 polarization to promote crystal clearance; The CXCL12/CXCR4 axis drives autophagy/EMT via NF-κB and connects stones with fibrosis. CX3CR1+CD24− macrophages inhibit crystal adhesion.

Figure 3. Regulatory pathway of MCP-1 in Calcium Oxalate Nephrolithiasis formation. CaOx crystals induce high expression of MCP-1. MCP-1/CCR2 axis: 1) Drives the recruitment and activation of M1 macrophages, releases inflammatory factors/ROS, and damages renal tubules; 2) Enhance crystal adhesion; 3) Promote fibrosis. The fibrotic microenvironment promotes stones in reverse. This axis is the core hub and potential target.
3.3.2 Synergistic effects of MCP-1 in directly mediating inflammation and oxidative stress
Accumulating evidence indicates that MCP-1 promotes the pathological progression of nephrolithiasis by orchestrating inflammatory and oxidative stress mechanisms. Experimental studies demonstrate that selenium-modified corn silk polysaccharides significantly alleviate oxidative damage in renal tubular epithelial cells by suppressing MCP-1 expression induced by CaOx crystals (47). Similarly, febuxostat reduces ROS generation through modulation of the MCP-1 signaling pathway, thereby inhibiting CaOx crystal-induced oxidative stress (48). Additionally, compounds such as hydroxycitrate and metformin have been shown to downregulate MCP-1 expression, exerting anti-inflammatory and antioxidant effects that reduce renal crystal deposition (49, 50). These findings collectively suggest that MCP-1 facilitates CaOx crystallization by directly activating synergistic interactions between inflammatory responses and oxidative stress, thereby establishing a favorable microenvironment for stone formation (Figure 3).
3.3.3 Immunoregulatory mechanisms of macrophage polarization
M1-polarized macrophages are regarded as contributing factors to nephrolithiasis due to their role in exacerbating inflammatory injury and oxidative stress (14). Recent studies have further revealed that MCP-1 modulates stone progression by regulating immune cell phenotypes. In CaOx crystal stimulation models, MCP-1 binding to its receptor CCR2 significantly promotes macrophage polarization toward the M1 phenotype, a process closely associated with renal oxidative stress and apoptosis levels. Specific inhibition of CCR2 effectively reverses M1 polarization and alleviates crystal-induced inflammatory damage (46). Notably, the flavonoid compound Vitexin demonstrates marked renoprotective effects by suppressing MCP-1-mediated macrophage infiltration (51). These findings confirm that MCP-1 not only mediates immune cell chemotaxis but also amplifies inflammatory signaling by reshaping macrophage functional phenotypes, thereby fostering a vicious cycle that promotes stone development (Figure 3).
3.3.4 Molecular regulation of crystal adhesion
The adhesion of crystals to renal tubular epithelia represents a critical step in stone formation. Recent studies demonstrate a positive correlation between MCP-1 expression levels and the extent of crystal deposition. Lactiplantibacillus plantarum AR1089 significantly inhibits renal crystallization by downregulating MCP-1 and related adhesion molecule expression (52). Mechanistic investigations confirm that MCP-1 synergizes with molecules such as osteopontin (OPN) to enhance CaOx crystal adhesion to renal tubular cells (53). However, the potential cooperative mechanism of these two factors has not been specifically clarified. The current evidence is limited to their parallel overexpression during the formation of calcium oxalate stones and their promotion of formation. Furthermore, in vascular endothelial cell models, suppression of MCP-1 effectively reduces intercellular adhesion molecule activity, suggesting that MCP-1 may similarly modulate crystal adhesion properties in tubular epithelia (54). Emerging studies further indicate that MCP-1 may indirectly influence crystal deposition through immune cell-mediated mechanisms. Activation of M1-polarized macrophages promotes adhesion-related gene expression, creating a favorable microenvironment for crystal retention (55). These findings highlight the need to investigate how MCP-1-regulated M1 macrophage polarization modulates crystal-cell interactions, which may provide novel perspectives for understanding nephrolithiasis pathogenesis (Figure 3).
3.3.5 MCP-1 as a potential regulatory hub linking renal fibrosis and nephrolithiasis
Beyond directly mediating inflammatory responses, chemokines such as MCP-1 may indirectly influence stone formation by regulating fibrotic processes (Figure 3). The interplay between renal fibrosis and nephrolithiasis has emerged as a research focus, though their intrinsic relationship remains incompletely elucidated. This interaction involves multifactorial regulation. Studies indicate that fibronectin, a hallmark of renal fibrosis, exhibits expression levels positively correlated with fibrotic progression (56, 57). Meanwhile, the MCP-1/CCR2 axis promotes renal interstitial fibrosis by recruiting inflammatory cells and inducing macrophage-to-myofibroblast transdifferentiation (58, 59). Despite their shared involvement in fibrosis, the direct regulatory relationship between MCP-1 and fibronectin requires validation through co-expression analyses or genetic editing models. Notably, inhibition of the MCP-1 pathway concurrently ameliorates both fibrosis and stone formation (60, 61),underscoring its potential as a dual therapeutic target.
Intriguingly, fibronectin demonstrates dual roles: it may facilitate stone formation by promoting crystal aggregation and extracellular matrix invasion, while simultaneously inhibiting CaOx crystal nucleation and adhesion on renal tubular surfaces (62) (Figure 3). Current debates persist regarding the causality between fibrosis and stone formation. Some clinical studies suggest that fibrosis in stone patients may be driven by alternative pathways, such as STAT6 and ferroptosis signaling (63, 64). In ischemia-reperfusion injury models, tubular GM-CSF upregulates the MCP-1/CCR2 axis to drive advanced fibrosis (65), implying MCP-1 may act as a downstream effector of other mediators. Collectively, existing evidence indicates pathophysiological interactions between these processes: renal fibrosis promotes crystal formation by disrupting calcium ion metabolism, while fibrotic microenvironments strengthen crystal-cell interactions, thereby providing favorable conditions for stone development.
Future research should utilize conditional MCP-1 knockout mouse models to clarify the temporal sequence between fibrosis and stone formation. Furthermore, clinical analyses assessing the dynamic changes in MCP-1 levels and their correlation with fibrosis progression in stone patients are essential to validate the potential bridging role of the MCP-1/CCR2 signaling axis. The bidirectional regulatory properties of MCP-1 provide new research perspectives for unraveling the intricate network linking renal fibrosis and nephrolithiasis, while also opening up potential avenues for exploring pathological mechanisms and therapeutic strategies.
3.4 Multidimensional regulation by the chemokine family in the pathological progression of nephrolithiasis
Beyond MCP-1, other chemokines exhibit complex regulatory networks during CaOx stone formation (Figure 2). Although CCL4 has been implicated in mediating inflammatory injury and oxidative stress (66), its specific role in nephrolithiasis remains to be fully elucidated. Notably, CCL5 and CCL7 show marked overexpression in CaOx stone patients (41, 67), with CCL7 potentially regulating crystal formation through olfactory signaling pathways (68).
Within the CXC chemokine subfamily, CXCL1, CXCL2, CXCL8 (IL-8), CXCL9, and CXCL10 are significantly upregulated in CaOx stone models and play pivotal roles in pathogenesis (44, 69). Among these, CXCL8 is the most well-characterized: it enhances neutrophil migration to amplify inflammation and upregulates adhesion molecules such as cadherins on renal tubular epithelial cells, directly promoting crystal retention and deposition. Additionally, CXCL8-mediated cadherin modulation may remodel the stone-forming microenvironment (70). Other CXC members, including CXCL10, drive macrophage M1 polarization via JAK2/STAT1 pathway activation, exacerbating renal inflammation (71). In contrast, CXCL14—elevated in high-fat diet murine models—induces macrophage M2 polarization, enhancing phagocytic clearance of crystals on bladder epithelial surfaces and thereby inhibiting pathological stone progression (72), this finding highlights CXCL14 as a promising target for Calcium Oxalate Nephrolithiasis research.
The CXCL12/CXCR4 axis, where CXCL12 is the sole ligand for CXCR4, has recently emerged as a critical hub linking stone formation to fibrosis. By activating the NF-κB pathway, this axis drives autophagy and epithelial-mesenchymal transition (EMT), directly contributing to stone-related renal injury while establishing a molecular bridge between crystal deposition and fibrotic progression (73).Further investigation of this mechanism may clarify chemokines’ dual roles in secondary stone injury and reveal CXCL12’s underappreciated contributions to Calcium Oxalate Nephrolithiasis.
Finally, the CX3CR1+CD24− macrophage subset demonstrates unique protective functions by suppressing crystal adhesion mechanisms, offering novel insights into preventive strategies for CaOx stone formation (74).
4 The IL-6/MCP-1/miR-124-3p negative feedback regulatory network
In recent years, the regulatory roles of non-coding RNAs in inflammatory microenvironments have garnered increasing attention. Emerging studies reveal that miR-124-3p directly binds to the 3’-untranslated regions (3’-UTRs) of TRAF6 and STAT3, suppressing NLRP3 inflammasome activation and downstream inflammatory signaling, thereby significantly inhibiting the protein expression of key pro-inflammatory cytokines such as IL-6 and IL-1β (75, 76). Overexpression of miR-124-3p also attenuates NF-κB and p38 MAPK signaling pathway activity, reducing pro-inflammatory factor expression and alleviating inflammatory responses (77, 78). Notably, miR-124-3p has been shown to directly target and regulate interleukin family members (79). In Calcium Oxalate Nephrolithiasis, miR-124-3p expression is downregulated alongside MCP-1 upregulation. Mechanistically, miR-124-3p inhibits MCP-1 expression by targeting its 3’-UTR (80, 81). Furthermore, miR-124-3p is closely linked to macrophage polarization, suppressing macrophage migration in inflammatory environments and promoting their transition to the M2 phenotype (82, 83). These findings suggest that miR-124-3p may serve as a molecular hub connecting inflammatory responses to stone formation through multi-target regulation of inflammatory networks, offering new avenues for miRNA-based targeted therapies (Figure 4).

Figure 4. Possible mechanisms by which IL-1βIL-6/MCP-1 regulates calcium oxalate stone formation by regulating miR-124-3p. Under pathological conditions, overexpressed IL-6 and MCP-1 inhibit the anti-inflammatory molecule miR-124-3p. The reduction of miR-124-3p leads to its inability to effectively inhibit the key targets (TRAF6/STAT3/MCP-1) and downstream pathways (NF-κB/MAPK/NLRP3), and simultaneously weakens the ability to promote M2 polarization in macrophages. Together, it aggravates the inflammatory response, deteriorates the microenvironment, and promotes the deposition of CaOx crystals and the formation of stones. Form a self-perpetuating vicious cycle, highlighting the core hub position and therapeutic potential of miR-124-3p.
Building on the multi-target nature of miR-124-3p, we propose the “IL-6/MCP-1/miR-124-3p negative feedback loop” hypothesis: In CaOx crystal-rich environments, overexpressed IL-6 and MCP-1 suppress miR-124-3p expression via negative feedback. This dual mechanism (1) diminishes miR-124-3p’s inhibitory effects on STAT3/NF-κB/MAPK pathways, amplifying pro-inflammatory cytokine release and exacerbating inflammation; and (2) reverses anti-inflammatory macrophage polarization, thereby promoting CaOx stone formation. Future studies should employ miR-124-3p-specific knockout models to assess its impact on stone susceptibility and cytokine profiles. Additionally, this regulatory loop highlights the potential for synergistic therapies combining miR-124-3p mimics or exosome-based delivery systems with existing anti-inflammatory agents. In-depth investigation of this closed-loop regulatory network is expected to advance our understanding of the molecular mechanisms underlying Calcium Oxalate Nephrolithiasis and identify novel research directions.
5 Conclusions and perspectives
The critical role of inflammatory microenvironments in driving Calcium Oxalate Nephrolithiasis has been firmly established. This review comprehensively outlines the molecular mechanisms by which interleukin and chemokine families promote stone formation through inflammation, oxidative stress, crystal adhesion, and macrophage polarization. These insights not only refine our understanding of nephrolithiasis pathogenesis but also highlight potential therapeutic targets for clinical intervention. Emerging evidence further underscores the interplay between renal fibrosis and stone formation, with MCP-1 serving as a pivotal mediator. Building on this, we propose the “fibrosis-stone vicious cycle” hypothesis, wherein CaOx crystals activate the MCP-1/CCR2 axis to recruit pro-inflammatory M1 macrophages, which release fibrogenic factors like TGF-β1, accelerating renal fibrosis. Conversely, fibrotic microenvironments disrupt calcium metabolism and extracellular matrix dynamics, fostering crystal retention. Validating this hypothesis necessitates combinatorial therapies (e.g., CCR2 antagonists with anti-fibrotic agents) and longitudinal clinical studies tracking urinary MCP-1 levels alongside fibrosis biomarkers (e.g., α-SMA, collagen III). While this model offers a theoretical foundation for dual therapeutic targeting, its feasibility requires validation in advanced experimental systems such as organoids or humanized mice.
Therapeutic strategies targeting inflammatory pathways have gained momentum. Monotherapies against pro-inflammatory cytokines or supplementation of anti-inflammatory factors have demonstrated efficacy in modulating stone microenvironments, with preclinical studies showing promising anti-lithogenic effects. As previously highlighted, IL-10-engineered macrophage transplantation is an emerging therapeutic strategy that can effectively promote the clearance of calcium oxalate kidney stones and alleviate related renal tubular damage (29). In addition, chloroquine-containing nano-selenium and Burttet Hill (CA) suppress NLRP3 expression by inhibiting NF-κB pathway phosphorylation, thereby reducing secretion of IL-1β and IL-6 as well as OPN expression, which minimizes crystal deposition and kidney damage (84, 85). Roxadustat, a proline hydroxylase domain inhibitor,significantly lowers MCP-1 and OPN levels both in vivo and in vitro, inhibiting calcium oxalate stone formation (86). Furthermore, CCR2 antagonists alleviate the damage of HK-2 cells in vivo and in vitro by inhibiting CaOx-induced M1 polarization of macrophages and reducing the expression of inflammatory markers IL-1β, IL-6 and MCP-1, thereby reducing renal oxidative stress, inflammation and apoptosis (46). However, clinical evidence for these novel strategies remains limited, and human trials are still scarce. Future research should focus on prospective randomized controlled trials in kidney stone patients, evaluating efficacy and safety through stone recurrence rates and urinary inflammatory biomarkers. Such efforts will facilitate the translation of these approaches from bench to bedside. Future innovations may include involve dual-target systems (e.g., CCR2 antagonists combined with miR-124-3p agonists) to simultaneously suppress inflammation and enhance crystal clearance. Integrating gut microbiota modulation with cytokine-targeted therapies could further reduce recurrence risks, while biomarker discovery may enable early risk stratification and treatment monitoring.
Despite significant advances in understanding the roles of interleukins and chemokines in nephrolithiasis, critical challenges remain unresolved. The specific contributions of anti-inflammatory interleukins (e.g., IL-37, IL-1RA) and non-canonical pro-inflammatory interleukins (e.g., IL-11, IL-18) to stone formation are incompletely elucidated, particularly their interactions with the gut microbiota-kidney axis, which demand further exploration. Additionally, the synergistic effects of chemokines such as CXCL12/CXCR4 with inflammatory, oxidative stress, and fibrotic pathways remain ambiguous. Future studies should integrate multi-omics technologies to delineate their regulatory networks, supplemented by longitudinal clinical cohort analyses to strengthen causal evidence linking chemokines to fibrosis. Translational hurdles persist, as the safety and efficacy of existing targeted therapies require systematic validation. Further research should also focus on refining molecular mechanisms, including the detailed roles of anti-inflammatory factors like IL-10 in suppressing crystal deposition via macrophage phenotype modulation, as well as elucidating structure-activity relationships of chemokines at crystal-cell interfaces. Notably, while the development of multi-pathway combination therapies and elucidation of the regulatory roles of the miR-124-3p-centered inflammatory network in Calcium Oxalate Nephrolithiasis hold significant potential for advancing prevention and treatment, they concurrently present substantial scientific and translational challenges.
In summary, interleukins and chemokines orchestrate Calcium Oxalate Nephrolithiasis via multifaceted inflammatory and fibrotic networks. The proposed IL-6 dynamic regulation, fibrosis-stone vicious cycle, and IL-6/MCP-1/miR-124-3p feedback loop hypotheses provide novel frameworks to address current research bottlenecks. Future studies should employ interdisciplinary approaches to validate these models, explore the cooperative mechanisms between interleukins and chemokine families in depth, and advance precision therapeutic strategies targeting inflammatory microenvironments, thereby offering new directions for the prevention and management of nephrolithiasis.
Author contributions
FL: Writing – original draft, Writing – review & editing. FG: Writing – original draft. RL: Writing – original draft. XW: Funding acquisition, Writing – review & editing, Writing – original draft.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China (NO.82060134).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ye ZQ, Zeng GH, Yang H, Li JX, Tang K, Wang GM, et al. The status and characteristics of urinary stone composition in China. BJU Int. (2020) 125:801–9. doi: 10.1111/bju.14765
2. Zhang D, Li S, Zhang Z, Li N, Yuan X, Jia Z, et al. Urinary stone composition analysis and clinical characterization of 1520 patients in central China. Sci Rep. (2021) 11. doi: 10.1038/s41598-021-85723-3
3. Hong Y, Yu LP, Huang XB, An LZ, Xiong LL, Xu QQ, et al. Composition analysis of renal and ureteral calculi in a single center in northern China in the past decade. Medicine. (2024) 103. doi: 10.1097/md.0000000000037374
4. Abufaraj M, Xu TL, Cao C, Waldhoer T, Seitz C, D’Andrea D, et al. Prevalence and trends in kidney stone among adults in the USA: analyses of national health and nutrition examination survey 2007–2018 data. Eur Urol Focus. (2021) 7:1468–75. doi: 10.1016/j.euf.2020.08.011
5. Shastri S, Patel J, Sambandam KK, and Lederer ED. Kidney stone pathophysiology, evaluation and management: core curriculum 2023. Am J Kidney Diseases. (2023) 82:617–34. doi: 10.1053/j.ajkd.2023.03.017
6. Alelign T and Petros B. Kidney stone disease: an update on current concepts. Adv urology. (2018) 2018:3068365. doi: 10.1155/2018/3068365
7. Simmons KE, Nair HR, Phadke M, Motamedinia P, Singh D, Montgomery TA, et al. Risk factors for common kidney stones are correlated with kidney function independent of stone composition. Am J Nephrol. (2023) 54:329–36. doi: 10.1159/000531046
8. Suttapitugsakul S, Sassanarakkit S, Peerapen P, and Thongboonkerd V. Integrated proteomics reveals enrichment of oxidative stress and inflammatory proteins in the urine and stone matrix of calcium oxalate stone formers. Urolithiasis. (2025) 53. doi: 10.1007/s00240-025-01697-1
9. Khan SR, Canales BK, and Dominguez-Gutierrez PR. Randall’s plaque and calcium oxalate stone formation: role for immunity and inflammation. Nat Rev Nephrology. (2021) 17:417–33. doi: 10.1038/s41581-020-00392-1
10. Song BF, Li BJ, Ning JZ, Xia YQ, Ye ZH, Yuan TH, et al. Overexpression of sirtuin 1 attenuates calcium oxalate-induced kidney injury by promoting macrophage polarization. Int Immunopharmacol. (2023) 121. doi: 10.1016/j.intimp.2023.110398
11. Zhou JW, Meng LC, He ZQ, Song QL, Liu JW, Su XZ, et al. Melatonin exerts a protective effect in ameliorating nephrolithiasis via targeting AMPK/PINK1-Parkin mediated mitophagy and inhibiting ferroptosis in vivo and in vitro. Int Immunopharmacol. (2023) 124. doi: 10.1016/j.intimp.2023.110801
12. Liu JN, Huang JL, Gong B, Cheng ST, Liu YD, Chen YD, et al. Polydatin protects against calcium oxalate crystal-induced renal injury through the cytoplasmic/mitochondrial reactive oxygen species-NLRP3 inflammasome pathway. Biomedicine Pharmacotherapy. (2023) 167. doi: 10.1016/j.biopha.2023.115621
13. Tang K, Ye T, He Y, Ba XZ, Xia D, Peng EJ, et al. Ferroptosis, necroptosis, and pyroptosis in calcium oxalate crystal-induced kidney injury. Biochim Et Biophys Acta-Molecular Basis Dis. (2025) 1871. doi: 10.1016/j.bbadis.2025.167791
14. Okada T, Okada A, Aoki H, Onozato D, Kato T, Takase H, et al. Phagocytosis model of calcium oxalate monohydrate crystals generated using human induced pluripotent stem cell-derived macrophages. Urolithiasis. (2024) 52. doi: 10.1007/s00240-024-01553-8
15. Dejban PM, Wilson E, Jayachandran MP, Hernandez LH, Haskic ZE, Wellik L, et al. Inflammatory cells in nephrectomy tissue from patients without and with a history of urinary stone disease. Clin J Am Soc Nephrology. (2022) 17:414–22. doi: 10.2215/cjn.11730921
16. Capolongo G, Ferraro PM, and Unwin R. Inflammation and kidney stones: cause and effect? Curr Opin Urol. (2023) 33:129–35. doi: 10.1097/mou.0000000000001066
17. Nong WJ, Tong XY, and Ouyang JM. Comparison of endoplasmic reticulum stress and pyroptosis induced by pathogenic calcium oxalate monohydrate and physiologic calcium oxalate dihydrate crystals in HK-2 cells: insights into kidney stone formation. Cells. (2024) 13. doi: 10.3390/cells13242070
18. Thongboonkerd V, Yasui T, and Khan SR. Editorial: immunity and inflammatory response in kidney stone disease. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.795559
19. Lv P, Liu HR, Ye T, Yang XQ, Duan C, Yao XY, et al. XIST Inhibition Attenuates Calcium Oxalate Nephrocalcinosis-Induced Renal Inflammation and Oxidative Injury via the miR-223/NLRP3 Pathway. Oxid Med Cell Longevity. (2021) 2021. doi: 10.1155/2021/1676152
20. Tian Y, Zhao J, Chen L, Zhang C, Chu X, and Xia YG. Sanjin Paishi Decoction improves the imbalance of gut microbiota and regulates MAPK signaling pathway to inhibit calcium oxalate stones in rats. Int Urol Nephrology. (2023) 55:2421–9. doi: 10.1007/s11255-023-03641-x
21. Si YC, Liu LL, Cheng J, Zhao TT, Zhou Q, Yu JP, et al. Oral hydrogen-rich water alleviates oxalate-induced kidney injury by suppressing oxidative stress, inflammation, and fibrosis. Front Med. (2021) 8:713536. doi: 10.3389/fmed.2021.713536
22. Yang XJ, Zhang P, Jiang J, Almoallim HS, Alharbi SA, and Li YF. Myricetin attenuates ethylene glycol-induced nephrolithiasis in rats via mitigating oxidative stress and inflammatory markers. Appl Biochem Biotechnol. (2024) 196:5419–34. doi: 10.1007/s12010-023-04831-0
23. Tian Y, Ye ZY, Wang XR, Guan HT, Liu WF, Duan XL, et al. MOF-818 nanozyme suppresses calcium oxalate kidney stones by alleviating oxidative stress and inflammatory injury. Advanced Healthcare Materials. (2024) 14(8). doi: 10.1002/adhm.202401574
24. Ju YJ, Du MW, Wang ZY, Mu X, Miao YD, Guo ZM, et al. Kukoamine A alleviates nephrolithiasis by inhibiting endogenous oxalate synthesis via the IL-6/JAK/STAT3/DAO signaling pathway. Phytomedicine. (2024) 135. doi: 10.1016/j.phymed.2024.156145
25. Li J and Chen Y. Research on key pathogenesis and potential intervention targets of idiopathic renal calculi composed of calcium oxalate (CaOx) based on bioinformatics. Trans Andrology Urology. (2024) 13:1582–91. doi: 10.21037/tau-24-302
26. Guo L, Lan Q, Zhou M, and Liu F. From gut to kidney: microbiota modulates stone risk through inflammation-a mediated Mendelian randomization study. Mamm Genome. (2024) 36(1):250–61. doi: 10.1007/s00335-024-10094-9
27. Liu Q, Liu YL, Guan XF, Wu JH, He ZQ, Kang JN, et al. Effect of M2 macrophages on injury and apoptosis of renal tubular epithelial cells induced by calcium oxalate crystals. Kidney Blood Pressure Res. (2019) 44:777–91. doi: 10.1159/000501558
28. Kumar P, Laurence E, Crossman DK, Assimos DG, Murphy MP, and Mitchell T. Oxalate disrupts monocyte and macrophage cellular function via Interleukin-10 and mitochondrial reactive oxygen species (ROS) signaling. Redox Biol. (2023) 67. doi: 10.1016/j.redox.2023.102919
29. Zhao WL, Wang KL, Chen HR, Xu JN, Wen LQ, Wu Y, et al. Revolutionizing nephrocalcinosis treatment: IL-10 engineered macrophages as a novel therapeutic approach. Bioengineering Trans Med. (2025). doi: 10.1002/btm2.70047
30. Kim SK, Choe JY, Kim JW, Park KY, and Kim B. Anti-inflammatory effect of atorvastatin and rosuvastatin on monosodium urate-induced inflammation through IL-37/smad3-complex activation in an in vitro study using THP-1 macrophages. Pharmaceuticals. (2024) 17. doi: 10.3390/ph17070883
31. Gaal OI, Leask M, Nica V, Cabau G, Badii M, Hotea I, et al. Gout-associated SNP at the IL1RN-IL1F10 region is associated with altered cytokine production in PBMCs of patients with gout and controls. Arthritis Res Ther. (2024) 26. doi: 10.1186/s13075-024-03436-0
32. Vilgelm AE and Richmond A. Chemokines modulate immune surveillance in tumorigenesis, metastasis, and response to immunotherapy. Front Immunol. (2019) 10:333. doi: 10.3389/fimmu.2019.00333
33. Hughes CE and Nibbs RJB. A guide to chemokines and their receptors. FEBS J. (2018) 285:2944–71. doi: 10.1111/febs.14466
34. Singh S and Anshita D. Ravichandiran. MCP-1: Function, regulation, and involvement in disease. Int Immunopharmacol. (2021) 101. doi: 10.1016/j.intimp.2021.107598
35. Bianconi V, Sahebkar A, Atkin SL, and Pirro M. The regulation and importance of monocyte chemoattractant protein-1. Curr Opin Hematology. (2018) 25:44–51. doi: 10.1097/moh.0000000000000389
36. Liu Y, Xu K, Xiang Y, Ma B, Li H, Li Y, et al. Role of MCP-1 as an inflammatory biomarker in nephropathy. Front Immunol. (2024) 14:1303076. doi: 10.3389/fimmu.2023.1303076
37. Chen F, Wei G, Zhou Y, Ma X, and Wang Q. The mechanism of miR-192 in regulating high glucose-induced MCP-1 expression in rat glomerular mesangial cells. Endocrine Metab Immune Disord - Drug Targets. (2019) 19:1055–63. doi: 10.2174/1871530319666190301154640
38. Yu J, Da J, Yu F, Yuan J, and Zha Y. HMGN1 down-regulation in the diabetic kidney attenuates tubular cells injury and protects against renal inflammation via suppressing MCP-1 and KIM-1 expression through TLR4. J Endocrinological Invest. (2024) 47:1015–27. doi: 10.1007/s40618-023-02292-0
39. Tanase DM, Gosav EM, Anton MI, Floria M, Seritean Isac PN, Hurjui LL, et al. Oxidative stress and NRF2/KEAP1/ARE pathway in diabetic kidney disease (DKD): new perspectives. Biomolecules. (2022) 12. doi: 10.3390/biom12091227
40. Swaminathan SM, Rao IR, Bhojaraja MV, Attur RP, Nagri SK, Rangaswamy D, et al. Role of novel biomarker monocyte chemo-attractant protein-1 in early diagnosis & predicting progression of diabetic kidney disease: A comprehensive review. J Natl Med Assoc. (2024) 116:33–44. doi: 10.1016/j.jnma.2023.12.001
41. Sun AY, Hinck B, Cohen BR, Keslar K, Fairchild RL, and Monga M. Inflammatory cytokines in the papillary tips and urine of nephrolithiasis patients. J Endourology. (2018) 32:236–44. doi: 10.1089/end.2017.0699
42. Wang Y, Sun C, Li CY, Deng YL, Zeng GH, Tao ZW, et al. Urinary MCP-1 EuroHMGB1 increased in calcium nephrolithiasis patients and the influence of hypercalciuria on the production of the two cytokines. Urolithiasis. (2017) 45:159–75. doi: 10.1007/s00240-016-0902-9
43. Wang XL, Bhutani G, Vaughan LE, Enders FT, Haskic Z, Milliner D, et al. Urinary monocyte chemoattractant protein 1 associated with calcium oxalate crystallization in patients with primary hyperoxaluria. BMC Nephrol. (2020) 21. doi: 10.1186/s12882-020-01783-z
44. Wang BQ, Tan ZK, She WS, Wang X, Guan XF, Tao ZW, et al. Characterizing chemokine signaling pathways and hub genes in calcium oxalate-induced kidney stone formation: insights from rodent models. Biochem Genet. (2025). doi: 10.1007/s10528-025-11036-z
45. Hsieh SL, Yang SY, Lin CY, He XY, Tsai CH, Fong YC, et al. MCP-1 controls IL-17-promoted monocyte migration and M1 polarization in osteoarthritis. Int Immunopharmacol. (2024) 132. doi: 10.1016/j.intimp.2024.112016
46. Wang XP, Xie LG, and Liu CY. CCR2 antagonist attenuates calcium oxalate-induced kidney oxidative stress and inflammation by regulating macrophage activation. Exp Animals. (2024) 73:211–22. doi: 10.1538/expanim.23-0113
47. Zheng YY, Tong XY, Zhang DY, and Ouyang JM. Enhancement of antioxidative and anti-inflammatory activities of corn silk polysaccharides after selenium modification. J Inflammation Res. (2024) 17:7965–91. doi: 10.2147/jir.S467665
48. Miyazawa K, Nakai D, Nakamura Y, Tatsuno T, Inoue S, Nakazawa Y, et al. Effects of the xanthine oxidase inhibitor, febuxostat, on the expression of monocyte chemoattractant protein-1 and synchronous genes in MDCK cells treated with calcium oxalate monohydrate crystals. Int J Urology. (2021) 28:339–45. doi: 10.1111/iju.14450
49. Liu X, Yuan P, Sun XF, and Chen ZQ. Hydroxycitric acid inhibits renal calcium oxalate deposition by reducing oxidative stress and inflammation. Curr Mol Med. (2020) 20:527–35. doi: 10.2174/1566524020666200103141116
50. Yang X, Yang T, Li J, Yang R, Qi SY, Zhao Y, et al. Metformin prevents nephrolithiasis formation by inhibiting the expression of OPN and MCP-1 in vitro and in vivo. Int J Mol Med. (2019) 43:1611–22. doi: 10.3892/ijmm.2019.4084
51. Ding T, Zhao TT, Li YH, Liu ZX, Ding JR, Ji BY, et al. Vitexin exerts protective effects against calcium oxalate crystal-induced kidney pyroptosis in vivo and in vitro. Phytomedicine. (2021) 86. doi: 10.1016/j.phymed.2021.153562
52. Xu M, Qin Y, Xia Y, Wang G, Xiong Z, Song X, et al. Screening of oxalate-degrading probiotics and preventive effect of Lactiplantibacillus plantarum AR1089 on kidney stones. Food Funct. (2024) 15:10163–78. doi: 10.1039/d4fo03133d
53. Zuo L, Tozawa K, Okada A, Yasui T, Taguchi K, Ito Y, et al. A paracrine mechanism involving renal tubular cells, adipocytes and macrophages promotes kidney stone formation in a simulated metabolic syndrome environment. J Urology. (2014) 191:1906–12. doi: 10.1016/j.juro.2014.01.013
54. Li HL, Peng WH, Jian WX, Li YM, Li Q, Li WM, et al. ROCK inhibitor fasudil attenuated high glucose-induced MCP-1 and VCAM-1 expression and monocyte-endothelial cell adhesion. Cardiovasc Diabetol. (2012) 11. doi: 10.1186/1475-2840-11-65
55. Taguchi K, Okada A, Hamamoto S, Unno R, Moritoki Y, Ando R, et al. M1/M2-macrophage phenotypes regulate renal calcium oxalate crystal development. Sci Rep. (2016) 6. doi: 10.1038/srep35167
56. Zheng ZY, Ma TJ, Lian X, Gao JL, Wang WG, Weng WY, et al. Clopidogrel reduces fibronectin accumulation and improves diabetes-induced renal fibrosis. Int J Biol Sci. (2019) 15:239–52. doi: 10.7150/ijbs.29063
57. Xiao X, Huo E, Guo C, Zhou X, Hu X, Dong C, et al. Hypermethylation suppresses microRNA-219a-2 to activate the ALDH1L2/GSH/PAI-1 pathway for fibronectin degradation in renal fibrosis. Res square. (2023). doi: 10.21203/rs.3.rs-2986934/v1
58. Puthumana J, Thiessen-Philbrook H, Xu LY, Coca SG, Garg AX, Himmelfarb J, et al. Biomarkers of inflammation and repair in kidney disease progression. J Clin Invest. (2021) 131. doi: 10.1172/jci139927
59. He SY, Yao L, and Li J. Role of MCP-1/CCR2 axis in renal fibrosis: Mechanisms and therapeutic targeting. Medicine. (2023) 102. doi: 10.1097/md.0000000000035613
60. Deguchi R, Komori T, Yamashita S, Hisaoka T, Kajimoto M, Kohjimoto Y, et al. Suppression of renal crystal formation, inflammation, and fibrosis by blocking oncostatin M receptor β signaling. Sci Rep. (2024) 14. doi: 10.1038/s41598-024-80411-4
61. Li Y, Zhang J, Liu H, Yuan J, Yin Y, Wang T, et al. Curcumin ameliorates glyoxylate-induced calcium oxalate deposition and renal injuries in mice. Phytomedicine. (2019) 61. doi: 10.1016/j.phymed.2019.152861
62. Khamchun S, Sueksakit K, Chaiyarit S, and Thongboonkerd V. Modulatory effects of fibronectin on calcium oxalate crystallization, growth, aggregation, adhesion on renal tubular cells, and invasion through extracellular matrix. J Biol Inorganic Chem. (2019) 24:235–46. doi: 10.1007/s00775-019-01641-w
63. Yuan TH, Xia YQ, Pan SY, Li BJ, Ye ZH, Yan XZ, et al. STAT6 promoting oxalate crystal deposition-induced renal fibrosis by mediating macrophage-to-myofibroblast transition via inhibiting fatty acid oxidation. Inflammation Res. (2023) 72(12):2111–26. doi: 10.1007/s00011-023-01803-2
64. Ye ZH, Yang SY, Chen LJ, Yu WM, Xia YQ, Li BJ, et al. Luteolin alleviated calcium oxalate crystal induced kidney injury by inhibiting Nr4a1-mediated ferroptosis. Phytomedicine. (2025) 136. doi: 10.1016/j.phymed.2024.156302
65. Xu LY, Sharkey D, and Cantley LG. Tubular GM-CSF promotes late MCP-1/CCR2-mediated fibrosis and inflammation after ischemia/reperfusion injury. J Am Soc Nephrology. (2019) 30:1825–40. doi: 10.1681/asn.2019010068
66. Abu-Serie MM, Habashy NH, and Maher AM. In vitro anti-nephrotoxic potential of Ammi visnaga, Petroselinum crispum, Hordeum vulgare, and Cymbopogon schoenanthus seed or leaf extracts by suppressing the necrotic mediators, oxidative stress and inflammation. BMC Complementary Altern Med. (2019) 19. doi: 10.1186/s12906-019-2559-8
67. Xia YQ, Zhou XJ, Ye ZH, Yu WM, Ning JZ, Ruan Y, et al. Construction and analysis of immune infiltration-related ceRNA network for kidney stones. Front Genet. (2021) 12:774155. doi: 10.3389/fgene.2021.774155
68. Zhang QK, Wei HL, Huang G, and Jin L. CCL7 and olfactory transduction pathway activation play an important role in the formation of CaOx and CaP kidney stones. Front Genet. (2024) 14:1267545. doi: 10.3389/fgene.2023.1267545
69. Huang HX, Chen BH, Feng C, Chen W, and Wu DP. Genetic insights into kidney stone formation: a Mendelian randomization study of protein quantitative trait loci. Urolithiasis. (2024) 53. doi: 10.1007/s00240-024-01667-z
70. Singhto N and Thongboonkerd V. Exosomes derived from calcium oxalate-exposed macrophages enhance IL-8 production from renal cells, neutrophil migration and crystal invasion through extracellular matrix. J Proteomics. (2018) 185:64–76. doi: 10.1016/j.jprot.2018.06.015
71. Song QL, Song C, Chen X, Xiong YH, Li LJ, Liao WB, et al. FKBP5 deficiency attenuates calcium oxalate kidney stone formation by suppressing cell-crystal adhesion, apoptosis and macrophage M1 polarization via inhibition of NF-κB signaling. Cell Mol Life Sci. (2023) 80. doi: 10.1007/s00018-023-04958-7
72. Chen HL, Hu KQ, Liang YR, Gao YQ, Zeng CY, Xu K, et al. Ample dietary fat reduced the risk of primary vesical calculi by inducing macrophages to engulf budding crystals in mice. Acta Pharm Sin B. (2022) 12:747–58. doi: 10.1016/j.apsb.2021.08.001
73. Ye ZH, Xia YQ, Zhou XJ, Li BJ, Yu WM, Ruan Y, et al. CXCR4 inhibition attenuates calcium oxalate crystal deposition-induced renal fibrosis. Int Immunopharmacol. (2022) 107. doi: 10.1016/j.intimp.2022.108677
74. Jin X, Jian ZY, Chen XT, Ma YC, Ma HW, Liu Y, et al. Short chain fatty acids prevent glyoxylate-induced calcium oxalate stones by GPR43-dependent immunomodulatory mechanism. Front Immunol. (2021) 12:729382. doi: 10.3389/fimmu.2021.729382
75. Fang YD and Hong XQ. miR-124-3p inhibits microglial secondary inflammation after basal ganglia hemorrhage by targeting TRAF6 and repressing the activation of NLRP3 inflammasome. Front Neurol. (2021) 12:653321. doi: 10.3389/fneur.2021.653321
76. Liu FH, Qiu FY, and Chen HY. miR-124-3p ameliorates isoflurane-induced learning and memory impairment via targeting STAT3 and inhibiting neuroinflammation. Neuroimmunomodulation. (2021) 28:248–54. doi: 10.1159/000515661
77. Zhai CN, Cong HL, Hou K, Hu YC, Zhang JX, Zhang YY, et al. Effects of miR-124-3p regulation of the p38MAPK signaling pathway via MEKK3 on apoptosis and proliferation of macrophages in mice with coronary atherosclerosis. Adv Clin Exp Med. (2020) 29:803–12. doi: 10.17219/acem/121926
78. Zhou Q, He DX, Deng YL, Wang CL, Zhang LL, Jiang FM, et al. MiR-124-3p targeting PDE4B attenuates LPS-induced ALI through the TLR4/NF-κB signaling pathway. Int Immunopharmacol. (2022) 105. doi: 10.1016/j.intimp.2022.108540
79. Liu Q, Shen Y, Xiao YF, Xiang H, Chu L, Wang TS, et al. Increased miR-124-3p alleviates type 2 inflammatory response in allergic rhinitis via IL-4Rα. Inflammation Res. (2022) 71:1271–82. doi: 10.1007/s00011-022-01614-x
80. Chen ZY, Wang XY, Yang YM, Wu MH, Yang L, Jiang DT, et al. LncRNA SNHG16 promotes colorectal cancer cell proliferation, migration, and epithelial-mesenchymal transition through miR-124-3p/MCP-1. Gene Ther. (2022) 29:193–205. doi: 10.1038/s41434-020-0176-2
81. Li YH, Yan GL, Zhang J, Chen W, Ding T, Yin YP, et al. LncRNA HOXA11-AS regulates calcium oxalate crystal-induced renal inflammation via miR-124-3p/MCP-1. J Cell Mol Med. (2020) 24:238–49. doi: 10.1111/jcmm.14706
82. Nakamachi Y, Uto K, Hayashi S, Okano T, Morinobu A, Kuroda R, et al. Exosomes derived from synovial fibroblasts from patients with rheumatoid arthritis promote macrophage migration that can be suppressed by miR-124-3p. Heliyon. (2023) 9. doi: 10.1016/j.heliyon.2023.e14986
83. Yin DJ and Shen GL. Exosomes from adipose-derived stem cells regulate macrophage polarization and accelerate diabetic wound healing via the circ-Rps5/miR-124-3p axis. Immun Inflammation Disease. (2024) 12. doi: 10.1002/iid3.1274
84. Guan HT, Zhang GH, Li SJ, Chen WZ, Zhang JP, Duan XL, et al. Chloroquine-loaded selenium nanoparticles alleviated nephrolithiasis by inhibiting NLRP3 inflammasome activation. Chem Eng J. (2025) 519. doi: 10.1016/j.cej.2025.164680
85. Qiu MQ, Li SH, Zhang Y, Yan JX, Qin ST, Yin XJ, et al. Multi - dimensional mechanism analysis of Choerospondias axillaris (Roxb.) Burtt et Hill in treating kidney stones: network pharmacology, molecular docking and in vitro experimental verification. Front Pharmacol. (2025) 16:1501386. doi: 10.3389/fphar.2025.1501386
86. Kawase K, Hamamoto S, Unno R, Taguchi K, Okada A, and Yasui T. Prolyl hydroxylase domain inhibitors prevent kidney crystal formation by suppressing inflammation. Urolithiasis. (2024) 53. doi: 10.1007/s00240-024-01677-x
87. Washino S, Hosohata K, and Miyagawa T. Roles played by biomarkers of kidney injury in patients with upper urinary tract obstruction. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21155490
88. Khan SR and Canales BK. Proposal for pathogenesis-based treatment options to reduce calcium oxalate stone recurrence. Asian J urology. (2023) 10:246–57. doi: 10.1016/j.ajur.2023.01.008
89. Sun Y, Sun H, Zhang Z, Tan F, Qu Y, Lei X, et al. New insight into oxidative stress and inflammatory responses to kidney stones: Potential therapeutic strategies with natural active ingredients. Biomedicine pharmacotherapy. (2024) 179:117333. doi: 10.1016/j.biopha.2024.117333
90. Williams JC Jr. and El-Achkar TM. Recent developments in the study of cellular inflammation in the papillae of stone formers. Urolithiasis. (2025) 53:34. doi: 10.1007/s00240-025-01707-2
Keywords: kidney stone, interleukin, chemokines, inflammation, renal fibrosis
Citation: Liang F, Guo F, Liu R and Wang X (2025) Research progress on the mechanisms of interleukin and chemokine families in driving calcium oxalate nephrolithiasis formation. Front. Immunol. 16:1651003. doi: 10.3389/fimmu.2025.1651003
Received: 23 June 2025; Accepted: 29 August 2025;
Published: 12 September 2025.
Edited by:
Remo Castro Russo, Federal University of Minas Gerais, BrazilReviewed by:
Diogo B Peruchetti, Federal University of Minas Gerais, BrazilXudong Shen, First Affiliated Hospital of Anhui Medical University, China
Copyright © 2025 Liang, Guo, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiang Wang, d2FuZ194aWFuZzEyMEAxNjMuY29t
†ORCID: Xiang Wang, orcid.org/0000-0002-8267-4644