 Cheng-chi Chao1,2
Cheng-chi Chao1,2 Chongming Jiang
Chongming Jiang- 1Terasaki Institute for Biomedical Innovation, Los Angeles, CA, United States
- 2ImmuX Consulting, San Jose, CA, United States
- 3Department of Melanoma Medical Oncology, Division of Cancer Medicine, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 4Department of Immunology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
- 5Department of GI Medical Oncology, The University of Texas MD Anderson Cancer Center, Houston, TX, United States
T cells play a dual role in various physiopathological states, capable of eliminating tumors and infected cells, while also playing a pathogenic role when activated by autoantigens, causing self-tissue damage. The regulation of T cell-peptide/major histocompatibility complex (TCR-pMHC) recognition is crucial for maintaining disease balance and treating cancer, infections, and autoimmune diseases. Despite efforts, predictive models of TCR-pMHC specificity are still in the early stages. Inspired by advances in protein structure prediction via deep neural networks, we evaluated AlphaFold 3 (AF3)-based AI computation as a method to predict TCR epitope specificity. We demonstrate that AlphaFold can model TCR-pMHC interactions, distinguishing valid epitopes from invalid ones with increasing accuracy. Immunogenic epitopes can be identified for vaccine development through in silico high-throughput processes. Additionally, higher-affinity and specific T cells can be designed to enhance therapy efficacy and safety. An accurate TCR-pMHC prediction model is expected to greatly benefit T-cell-mediated immunotherapy and aid drug design. Overall, precise prediction of T-cell immunogenicity holds significant therapeutic potential, allowing the identification of peptide epitopes linked to tumors, infections, and autoimmune diseases. Although there is much work to be done before these predictions achieve widespread practical use, we are optimistic that deep learning-based structural modeling is a promising pathway for the generalizable prediction of TCR-pMHC interactions.
In-silico prediction of peptide-MHC binding and TCR recognition
There are many previous interests and attempts about predicting the antigen specificity in the T cell activity (1–19). Models for standalone prediction of peptide and MHC binding have existed for decades, such as NetMHC, NetMHCpan, MHCflurry, IEDB-AR, SYFPEITHI, TEPITOPE, MixMHCpred, DeepHLApan, PickPocket, MARIA, SMM, ARBO-MHC, HBond-MHC, MHCnuggets, PepCNN, BigMHC (20), and so on (21) (5, 21–30). Yet, for an assessment of antigen specificity and immunogenicity, the precise interaction of a given TCR to its corresponding pMHC complex should be further considered (31–33). Although tools, such as NetTCR (34), IMRex (35), ERGO (19), TEINet (36), AEPCAM (37), PanPep (18), pMTnet (38), TEIM-Res (39), PISTE (7), BERTrand (40), BigMHC (20), and HLAIImaster (41, 42), have been available, few models among them are able to accurately predict the recognition of pMHC complexes by T-cell receptors (TCRs) (31–33). The most precise method to dissect TCR-pMHC interactions involve experimentally generating X-ray crystallography structures, which is a time-consuming and technically demanding process. The primary hurdle in accurately predicting T-cell recognition of pMHC complexes lies in the inherent difficulties of protein structure prediction for TCRs and pMHC complexes. While numerous computational models employing various mechanisms have been developed to predict the structure of proteins like antibodies and TCRs, few of them have achieved satisfactory results (31–33). With the advent of the AI/ML era and models, many algorithms, such as AlphaFold 2 and AlphaFold 3 (43, 44), RoseTTAFold/RFdiffusion (45, 46), TrRosetta (47), HADDOCK (48), DeepFRI (49), CANDOCK (50), and Boltz-2 (51), have brought major improvements to predict protein structures and interactions. The accuracy of computational modeling has greatly improved. We have leveraged these advancements to explore AI/ML-driven computational protein structure prediction. Using our current model system, we can exploratively predict T-cell binding to pMHC complexes with significantly higher docking precision, enhancing reliability and biological relevance (14, 14, 31, 43–46, 52–58).
AI/ML-powered computational design for predicting TCR-pMHC recognition
Computationally predicting TCR-pMHC interactions using AI/ML approaches offers a rapid, accurate, and scalable alternative to traditional experimental methods, thereby significantly facilitating antigen discovery and immune response modeling. AF3 is a publicly released model developed by DeepMind, trained extensively on more than 120 million protein sequences from the UniProt database and more than 2.2 million experimentally determined protein structures from the Protein Data Bank (PDB). We utilized the model as-is, without retraining or further fine-tuning. The default hyperparameters provided by AlphaFold 3 were employed, including three cycles of recycling, a multiple sequence alignment (MSA) depth of 256, and a template dropout rate of 15% (43, 44). A comparative analysis of AF3 with other structure prediction tools confirms that AF3 predictions outperform other tools in terms of structural accuracy and reliability (59). The results present a comparative analysis of TCR-pMHC recognition between the experimentally determined x-ray crystallography structure and AF3 computational predictions (Figure 1). The experimentally resolved crystal structure of the TCR-pMHC complex is shown in Figure 1A, serving as a reference for evaluating AF3 prediction accuracy. The crystal structure offers detailed insights into the spatial arrangement and interactions among the T-cell receptor (TCR) and peptide-MHC molecules. AF3’s prediction of TCR binding in the presence of peptide-MHC complex is shown in Figure 1B. This prediction closely mirrors the crystal structure in Figure 1A, demonstrating high accuracy in modeling the ternary complex. This highlights AF3’s ability to effectively predict TCR-pMHC interactions once the peptide is bound to the MHC groove. Conversely, AF3’s prediction of TCR binding to MHC in the absence of the same peptide (SLLMWITQC) is illustrated in Figure 1C. This prediction does not align well with the expected TCR binding conformation shown in Figure 1B. The reduced predictive performance from the AF3-based model highlights the importance of peptide presence for accurate binding predictions. This suggests that the conformation of the peptide-MHC complex is essential for accurate TCR interaction. The presence of the peptide resulted in a higher predicted interface template modeling (ipTM) score compared to its absence in AF3’s TCR-pMHC binding prediction (ipTM = 0.92 vs. 0.54, respectively) in the advanced protein structure prediction analysis, as shown in Figures 1B, D in contrast to Figures 1C, E. These high TM-score values confirm strong agreement between AF3 predictions and the crystal structure for the TCR-pMHC complex. The results demonstrated that the AF3-enabled approach reliably predicts TCR-pMHC interactions, supported by a high correlation with crystal structures and favorable TM-scores (Figures 1B, D). Instead, predictive accuracy decreases notably without peptides (Figures 1C, E). A comparative analysis in more TCR-pMHC binding structures assesses how peptide presence influences TCR-pMHC binding, as reflected in the ipTM values. In this analysis, the same set of 9 TCR-pMHC complexes (60–67) were included under both conditions: one group is the TCR-pMHC binding structure with peptides (+Peptides) and the other without peptides (-Peptides). The results demonstrate that the ipTM scores of TCR-pMHC binding structures with peptides are significantly higher than those without peptides (two-sided Wilcoxon tests, p-value =6e-04), as shown in Figure 1F. These findings highlight the significance of achieving accurate TCR-pMHC binding predictions with AF3.

Figure 1. The x-ray crystallography structure and AF3-based prediction of T cell-pMHC interaction. (A) The x-ray crystallography structure of TCR binding to NY-ESO-1 derived peptide (SLLMWITQC)/HLA-A*02:01complexes (https://www.imgt.org/3Dstructure-DB/cgi/details.cgi?pdbcode=2PYE&Part=JMOL#jmolvisu) (60). (B) AF3 prediction of the interaction between specific TCR and NY-ESO-1 derived peptide (SLLMWITQC)/HLA-A*02:01 complexes. (C) AF3 prediction of NY-ESO-1-specific TCR binding to MHC molecules alone in the absence of its peptide epitope. (D) The predicted aligned error (PAE) of AlphaFold score, ipTM=0.92, from (B). (E) PAE of AlphaFold score, ipTM=0.54, from (C). (F) A total of 9 TCR-pMHC complexes (60–67) were included under both conditions: one group is the TCR-pMHC binding structure with peptides (+Peptides) and the other without peptides (-Peptides). All complexes involved the same class I MHC molecule (HLA-A*02:01) presenting 9–10mer peptides. The ipTM scores of the TCR-pMHC binding structure with peptides (+Peptides) are significantly higher than the TCR-pMHC binding structure without peptides(-Peptides), two-sided Wilcoxon tests, p-value =6e-04.
Future perspectives and challenges
AI-driven T-cell-pMHC modeling holds significant potential for drug discovery and clinical applications. However, a major hurdle in the existing models is their inability to accurately predict T-cell recognition of its cognate antigen (68–70). Developing in-silico tools to assess T-cell immunogenicity is crucial from both biological and therapeutic standpoints. Computational identification and screening of TCR specificity can greatly advance T-cell-related therapeutics, such as T cell therapy and vaccines, enhancing efficacy and safety (31–33, 40, 55, 68, 69, 71). A generalizable model of TCR-pMHC interactions can significantly accelerate the identification of dominant antigenic epitopes with high affinity for their respective MHC. Improving predictions of TCR binding to the pMHC complex can help fine-tune TCR affinity and address a key challenge in the field. Accurate predictions of the T-cell-pMHC complex structure can aid in designing agonistic or antagonistic peptide analogs to stimulate tumor-specific or tolerize (auto)antigen-specific T cells (34, 48, 72–75). For vaccine design, an AI-enabled model can assist in selecting suitable epitopes with strong immunogenicity, helping to accurately identify and validate those capable of activating T cells (76, 77). Moreover, accurate predictions of peptide-MHC interaction are expected to enable more effective assessment of the risk of anti-drug responses in patients, although current models often overestimate the results (34, 72–74). Ultimately, we hope that such a model will be able to predict T-cell functional activity in an exploratory manner, thereby contributing to the development of potential therapeutic strategies (Table 1).
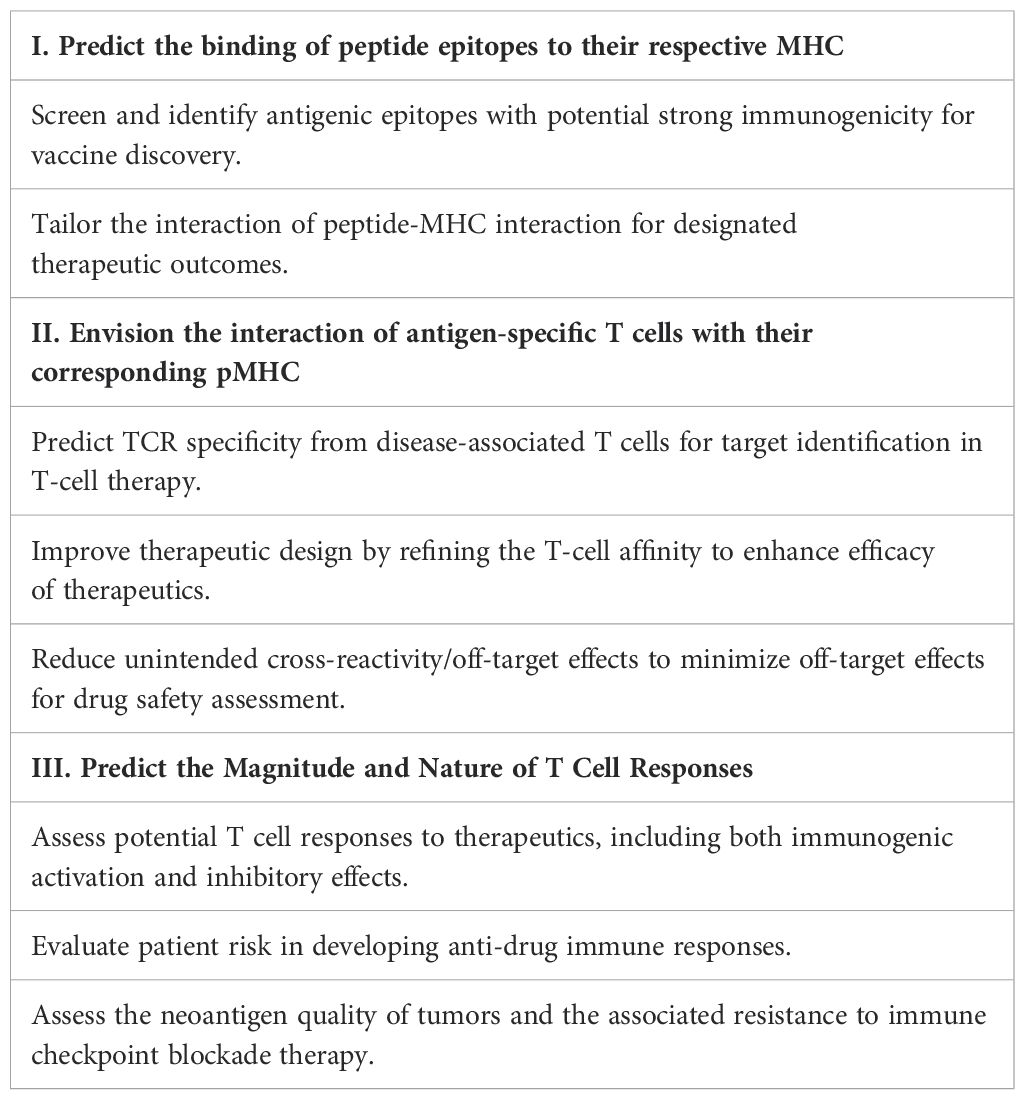
Table 1. Exploring three key facets of therapeutic applications for accurate T cell–pMHC recognition prediction.
While the pharmaceutical and biotech industries have leveraged AI across various stages of drug discovery, primarily focusing on small molecules and antibody drugs, the application of AI/ML and digital biology to T-cell therapeutics remains relatively limited (68, 69, 76). As demonstrated by the results above, our advanced protein prediction modeling approach enables accurate prediction of T-cell-pMHC interactions, offering significant potential to enhance T-cell-mediated therapies. Despite advancements, AI-assisted protein design and protein-protein interactions (PPI) still face significant unresolved challenges (Table 2). Tools like AF3, Rosetta, and Boltz-2 have revolutionized protein engineering, yet several critical obstacles remain, as outlined below. One major challenge is the limited availability of high-quality data, especially for underrepresented antigens, rare HLA alleles, and paired TCR alpha and beta chains (42, 78–80). The lower the quality and quantity of training data available to AI systems, the less reliable their predictions of binding interactions become. Additionally, TCRs naturally exhibit a wide range of binding affinities and can be polyspecific, making it difficult to train models that accurately capture their complex interaction profiles (43, 44, 46, 52, 58, 81). A further complication arises from protein conformational dynamics. Proteins exist in multiple conformations; they open, close, twist, and bend. These conformational changes depend on factors such as temperature, pH, chemical environment, and interactions with other molecules (31, 33, 55). Moreover, TCR binding affinity alone is not sufficient to guarantee a functional immune response. A robust response requires a complex interplay of factors, including antigen processing and presentation, TCR binding,as well as T cell activation, differentiation and the diseased microenvironment (82–85). While AlphaFold can help discriminate between correct and incorrect binding partners, it doesn’t directly predict binding affinity in a quantitative way. Additional modeling approaches, such as Rosetta, are required to calculate binding energy changes, which can then be used to predict the effects of mutations on TCR affinity and correlate them with binding affinity. Ultimately, the development of a reliable and high-throughput screening system is essential for identifying effective therapeutic candidates in drug discovery. While our exploratory model system is an initial step, we aspire for it to drive future advancements in T-cell-based therapeutics.

Table 2. The major challenges in predicting TCR immunogenicity in silico.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Author contributions
C-cC: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. YC: Data curation, Formal analysis, Validation, Writing – review & editing. LY: Writing – review & editing, Data curation, Validation, Investigation, Visualization. CY: Conceptualization, Data curation, Resources, Validation, Writing – review & editing. CJ: Data curation, Formal analysis, Investigation, Methodology, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. XS: Funding acquisition, Resources, Supervision, Writing – original draft, Writing – review & editing.
Funding
The authors declare financial support was received for the research and/or publication of this article. This work is supported by the National Institutes of Health, United States (NIH) R01 DK119795, R35 GM122465, and the Cancer Prevention Research Institute of Texas (CPRIT) (RR240007).
Acknowledgments
This work is supported by the National Institutes of Health, United States (NIH) R01 DK119795, R35 GM122465, and the Cancer Prevention Research Institute of Texas (CPRIT) (RR240007). Thank Xiuying Li for useful suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The authors declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, et al. Identifying specificity groups in the T cell receptor repertoire. Nat Publishing Group. (2017) 547:94–8. doi: 10.1038/nature22976
2. George JT, Kessler DA, and Levine H. Effects of thymic selection on T cell recognition of foreign and tumor antigenic peptides. Proc Natl Acad Sci. (2017) 114:E7875–81. doi: 10.1073/pnas.1708573114
3. Calis J, Maybeno M, Greenbaum J a., Weiskopf D, De Silva AD, Sette A, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PloS Comput Biol. (2013) 9:e1003266. doi: 10.1371/journal.pcbi.1003266
4. Dash P, Fiore-gartland AJ, Hertz T, Wang GC, Sharma S, Souquette A, et al. Quantifiable predictive features define epitope- specific T cell receptor repertoires. Nat Publishing Group. (2017) 547:89–93. doi: 10.1038/nature22383
5. Zhao W and Sher X. Systematically benchmarking peptide-MHC binding predictors: From synthetic to naturally processed epitopes. PloS Comput Biol. (2018) 14:1–28. doi: 10.1371/journal.pcbi.1006457
6. Parkhurst M, Goff SL, Lowery FJ, Beyer RK, Halas H, Robbins PF, et al. Adoptive transfer of personalized neoantigen-reactive TCR-transduced T cells in metastatic colorectal cancer: phase 2 trial interim results. Nat Med. (2024) 30:2586–95. doi: 10.1038/s41591-024-03109-0
7. Feng Z, Chen J, Hai Y, Pang X, Zheng K, Xie C, et al. Sliding-attention transformer neural architecture for predicting T cell receptor–antigen–human leucocyte antigen binding. Nat Mach Intell. (2024) 6:19–21. doi: 10.1038/s42256-024-00901-y
8. Karnaukhov VK, Shcherbinin DS, Chugunov AO, Chudakov DM, Efremov RG, and Zvyagin IV. Structure-based prediction of T cell receptor recognition of unseen epitopes using TCRen. Nat Comput Sci. (2024) 4:510–21. doi: 10.1038/s43588-024-00653-0
9. Saethang T, Hirose O, Kimkong I, Tran VA, Dang XT, Nguyen LAT, et al. PAAQD: Predicting immunogenicity of MHC class I binding peptides using amino acid pairwise contact potentials and quantum topological molecular similarity descriptors. J Immunol Methods. (2013) 387:293–302. doi: 10.1016/j.jim.2012.09.016
10. Tickotsky N, Sagiv T, Prilusky J, Shifrut E, and Friedman N. McPAS-TCR: A manually curated catalogue of pathology-associated T cell receptor sequences. Bioinformatics. (2017) 33:2924–9. doi: 10.1093/bioinformatics/btx286
11. Rasmussen M, Fenoy E, Harndahl M, Kristensen AB, Nielsen IK, Nielsen M, et al. Pan-specific prediction of peptide-MHC class I complex stability, a correlate of T cell immunogenicity. J Immunol. (2016) 197:1517–24. doi: 10.4049/jimmunol.1600582
12. Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, et al. Deconstructing the peptide-MHC specificity of T cell recognition. Cell. (2014) 157(5):1073–87. doi: 10.1016/j.cell.2014.03.047
13. Huang J, Zeng X, Sigal N, Lund PJ, Su LF, Huang H, et al. Detection, phenotyping, and quantification of antigen-specific T cells using a peptide-MHC dodecamer. (2016) 113(13):E1890–7. doi: 10.1073/pnas.1602488113
14. Mikhaylov V, Brambley CA, Keller GLJ, Arbuiso AG, Weiss LI, Baker BM, et al. Accurate modeling of peptide-MHC structures with AlphaFold. Structure. (2024) 32:228–241.e4. doi: 10.1016/j.str.2023.11.011
15. Wu J, Wang W, Zhang J, Zhou B, Zhao W, Su Z, et al. DeepHLApan: A deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity. Front Immunol. (2019) 10:2559. doi: 10.3389/fimmu.2019.02559
16. Huppa JB, Axmann M, Mo MA, Davis MM, Brameshuber M, Klein LO, et al. TCR–peptide–MHC interactions in situ show accelerated kinetics and increased affinity. Nature (2010) 463:963–7. doi: 10.1038/nature08746
17. Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, et al. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. (1999) 286:1913–21. doi: 10.1126/science.286.5446.1913
18. Gao Y, Gao Y, Fan Y, Zhu C, Wei Z, Zhou C, et al. Pan-Peptide Meta Learning for T-cell receptor–antigen binding recognition. Nat Mach Intell. (2023) 5:236–49. doi: 10.1038/s42256-023-00619-3
19. Springer I, Besser H, Tickotsky-Moskovitz N, Dvorkin S, and Louzoun Y. Prediction of specific TCR-peptide binding from large dictionaries of TCR-peptide pairs. Front Immunol. (2020) 11:1803. doi: 10.3389/fimmu.2020.01803
20. Albert BA, Yang Y, Shao XM, Singh D, Smit KN, Anagnostou V, et al. Deep neural networks predict class I major histocompatibility complex epitope presentation and transfer learn neoepitope immunogenicity. Nat Mach Intell. (2023) 5:861–72. doi: 10.1038/s42256-023-00694-6
21. Vita R, Mahajan S, Overton JA, Dhanda SK, Martini S, Cantrell JR, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. (2019) 47:D339–43. doi: 10.1093/nar/gky1006
22. O’Donnell TJ, Rubinsteyn A, Bonsack M, Riemer AB, Laserson U, and Hammerbacher J. MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst. (2018) 7:129–132.e4. doi: 10.1016/j.cels.2018.05.014
23. Rammensee H, Bachmann J, Emmerich NP, Bachor OA, and Stevanović S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. (1999) 50:213–9. doi: 10.1007/s002510050595
24. Wu J, Li J, Chen S, and Zhou Z. DeepHLApan: A deep learning approach for the prediction of peptide-HLA binding and immunogenicity. Methods Mol Biol. (2024) 2809:237–44. doi: 10.1007/978-1-0716-3874-3_15
25. Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology. (2018) 154:394–406. doi: 10.1111/imm.12889
26. Dönnes P and Elofsson A. Prediction of MHC class I binding peptides, using SVMHC. BMC Bioinf. (2002) 3:25. doi: 10.1186/1471-2105-3-25
27. Zhang H, Lund O, and Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics. (2009) 25:1293–9. doi: 10.1093/bioinformatics/btp137
28. Wieczorek M, Abualrous ET, Sticht J, Álvaro-Benito M, Stolzenberg S, Noé F, et al. Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front Immunol. (2017) 8:292. doi: 10.3389/fimmu.2017.00292
29. Chandra A, Sharma A, Dehzangi I, Tsunoda T, and Sattar A. PepCNN deep learning tool for predicting peptide binding residues in proteins using sequence, structural, and language model features. Sci Rep. (2023) 13:20882. doi: 10.1038/s41598-023-47624-5
30. Sales AP, Tomaras GD, and Kepler TB. Improving peptide-MHC class I binding prediction for unbalanced datasets. BMC Bioinf. (2008) 9:385. doi: 10.1186/1471-2105-9-385
31. Hudson D, Fernandes RA, Basham M, Ogg G, and Koohy H. Can we predict T cell specificity with digital biology and machine learning? Nat Rev Immunol. (2023) 23:511–21. doi: 10.1038/s41577-023-00835-3
32. Croce G, Bobisse S, Moreno DL, Schmidt J, Guillame P, Harari A, et al. Deep learning predictions of TCR-epitope interactions reveal epitope-specific chains in dual alpha T cells. Nat Commun. (2024) 15:3211. doi: 10.1038/s41467-024-47461-8
33. Kohlgruber AC, Dezfulian MH, Sie BM, Wang CI, Kula T, Laserson U, et al. High-throughput discovery of MHC class I- and II-restricted T cell epitopes using synthetic cellular circuits. Nat Biotechnol. (2024) 43:623–34. doi: 10.1038/s41587-024-02248-6
34. Montemurro A, Schuster V, Povlsen HR, Bentzen AK, Jurtz V, Chronister WD, et al. NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data. Commun Biol. (2021) 4:1–13. doi: 10.1038/s42003-021-02610-3
35. Moris P, De Pauw J, Postovskaya A, Gielis S, De Neuter N, Bittremieux W, et al. Current challenges for unseen-epitope TCR interaction prediction and a new perspective derived from image classification. Brief Bioinform. (2021) 22:1–12. doi: 10.1093/bib/bbaa318
36. Jiang Y, Huo M, and Cheng Li S. TEINet: a deep learning framework for prediction of TCR-epitope binding specificity. Brief Bioinform. (2023) 24:bbad086. doi: 10.1093/bib/bbad086
37. Chen J, Zhao B, Lin S, Sun H, Mao X, Wang M, et al. TEPCAM: Prediction of T-cell receptor-epitope binding specificity via interpretable deep learning. Protein Sci. (2024) 33:e4841. doi: 10.1002/pro.4841
38. Lu T, Zhang Z, Zhu J, Wang Y, Jiang P, Xiao X, et al. Deep learning-based prediction of the T cell receptor-antigen binding specificity. Nat Mach Intell. (2021) 3:864–75. doi: 10.1038/s42256-021-00383-2
39. Peng X, Lei Y, Feng P, Jia L, Ma J, Zhao D, et al. Characterizing the interaction conformation between T-cell receptors and epitopes with deep learning. Nat Mach Intell. (2023) 5:395–407. doi: 10.1038/s42256-023-00634-4
40. Myronov A, Mazzocco G, Król P, and Plewczynski D. BERTrand-peptide:TCR binding prediction using Bidirectional Encoder Representations from Transformers augmented with random TCR pairing. Bioinformatics. (2023) 39:1–9. doi: 10.1093/bioinformatics/btad468
41. Yang Q, Xu L, Dong W, Li X, Wang K, Dong S, et al. HLAIImaster: a deep learning method with adaptive domain knowledge predicts HLA II neoepitope immunogenic responses. Brief Bioinform. (2024) 25:bbae302. doi: 10.1093/bib/bbae302
42. Xu L, Yang Q, Dong W, Li X, Wang K, Dong S, et al. Meta learning for mutant HLA class I epitope immunogenicity prediction to accelerate cancer clinical immunotherapy. Brief Bioinform. (2024) 26:bbae625. doi: 10.1093/bib/bbae625
43. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. (2021) 596:583–89. doi: 10.1038/s41586-021-03819-2
44. Guan X, Tang Q, Ren W, Chen M, Wang W, Wolynes PG, et al. Predicting protein conformational motions using energetic frustration analysis and AlphaFold2. Proc Natl Acad Sci U S A. (2024) 121(35):1–11. doi: 10.1073/pnas.2410662121/-/DCSupplemental.Published
45. Vázquez Torres S, Leung PJY, Venkatesh P, Lutz ID, Hink F, Huynh HH, et al. De novo design of high-affinity binders of bioactive helical peptides. Nature. (2024) 626:435–42. doi: 10.1038/s41586-023-06953-1
46. Watson JL, Juergens D, Bennett NR, Trippe BL, Yim J, Eisenach HE, et al. De novo design of protein structure and function with RFdiffusion. Nature. (2023) 620:1089–100. doi: 10.1038/s41586-023-06415-8
47. Adelborg K, Szentkúti P, Henriksen JE, Thomsen RW, Pedersen L, Sundbøll J, et al. Cohort profile: the Funen Diabetes Database-a population-based cohort of patients with diabetes in Denmark. BMJ Open. (2020) 10:e035492. doi: 10.1136/bmjopen-2019-035492
48. Peacock T and Chain B. Information-driven docking for TCR-pMHC complex prediction. Front Immunol. (2021) 12:686127. doi: 10.3389/fimmu.2021.686127
49. Pagiatakis C, Musolino E, Gornati R, Bernardini G, and Papait R. Epigenetics of aging and disease: a brief overview. Aging Clin Exp Res. (2021) 33:737–45. doi: 10.1007/s40520-019-01430-0
50. Feghali J, Kim J, Gami A, Rapaport S, Caplan JM, McDougall CG, et al. Monocyte-based inflammatory indices predict outcomes following aneurysmal subarachnoid hemorrhage. Neurosurg Rev. (2021) 44:3499–507. doi: 10.1007/s10143-021-01525-1
51. Passaro S, Corso G, Wohlwend J, Reveiz M, Thaler S, Somnath VR, et al. Boltz-2: Towards Accurate and Efficient Binding Affinity Prediction. (2025) 2025.06.14.659707. doi: 10.1101/2025.06.14.659707
52. McMaster B, Thorpe C, Ogg G, Deane CM, and Koohy H. Can AlphaFold’s breakthrough in protein structure help decode the fundamental principles of adaptive cellular immunity? Nat Methods. (2024) 21:766–76. doi: 10.1038/s41592-024-02240-7
53. Bradley P. Structure-based prediction of T cell receptor:peptide-MHC interactions. Elife. (2023) 12:e82813. doi: 10.7554/eLife.82813
54. Cheng J, Novati G, Pan J, Bycroft C, Žemgulyte A, Applebaum T, et al. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Sci (1979). (2023) 381:eadg7492. doi: 10.1126/science.adg7492
55. Marzella DF, Parizi FM, van Tilborg D, Renaud N, Sybrandi D, Buzatu R, et al. PANDORA: A fast, anchor-restrained modelling protocol for peptide: MHC complexes. Front Immunol. (2022) 13:878762. doi: 10.3389/fimmu.2022.878762
56. Qiao Z, Nie W, Vahdat A, Miller TF, and Anandkumar A. State-specific protein–ligand complex structure prediction with a multiscale deep generative model. Nat Mach Intell. (2024) 6:195–208. doi: 10.1038/s42256-024-00792-z
57. Agarwal V and McShan AC. The power and pitfalls of AlphaFold2 for structure prediction beyond rigid globular proteins. Nat Chem Biol. (2024) 20:950–9. doi: 10.1038/s41589-024-01638-w
58. Wu D, Yin R, Chen G, Ribeiro-Filho HV, Cheung M, Robbins PF, et al. Structural characterization and AlphaFold modeling of human T cell receptor recognition of NRAS cancer neoantigens. bioRxiv. (2024) 2024.05.21.595215:2024.05.21.595215. doi: 10.1126/sciadv.adq6150
59. Xu S, Feng Q, Qiao L, Wu H, Shen T, Cheng Y, et al. FoldBench: An All-atom Benchmark for Biomolecular Structure Prediction. (2025) 2025.05.22.655600. doi: 10.1101/2025.05.22.655600
60. Sami M, Rizkallah PJ, Dunn S, Molloy P, Moysey R, Vuidepot A, et al. Crystal structures of high affinity human T-cell receptors bound to peptide major histocompatibility complex reveal native diagonal binding geometry. Protein Eng Des Sel. (2007) 20:397–403. doi: 10.1093/protein/gzm033
61. Yang X, Gao M, Chen G, Pierce BG, Lu J, Weng N-P, et al. Structural basis for clonal diversity of the public T cell response to a dominant human cytomegalovirus epitope. J Biol Chem. (2015) 290:29106–19. doi: 10.1074/jbc.M115.691311
62. Song I, Gil A, Mishra R, Ghersi D, Selin LK, and Stern LJ. Broad TCR repertoire and diverse structural solutions for recognition of an immunodominant CD8+ T cell epitope. Nat Struct Mol Biol. (2017) 24:395–406. doi: 10.1038/nsmb.3383
63. Ishizuka J, Stewart-Jones GBE, van der Merwe A, Bell JI, McMichael AJ, and Jones EY. The structural dynamics and energetics of an immunodominant T cell receptor are programmed by its Vbeta domain. Immunity. (2008) 28:171–82. doi: 10.1016/j.immuni.2007.12.018
64. Ding YH, Smith KJ, Garboczi DN, Utz U, Biddison WE, and Wiley DC. Two human T cell receptors bind in a similar diagonal mode to the HLA-A2/Tax peptide complex using different TCR amino acids. Immunity. (1998) 8:403–11. doi: 10.1016/s1074-7613(00)80546-4
65. Bulek AM, Cole DK, Skowera A, Dolton G, Gras S, Madura F, et al. Structural basis for the killing of human beta cells by CD8(+) T cells in type 1 diabetes. Nat Immunol. (2012) 13:283–9. doi: 10.1038/ni.2206
66. Madura F, Rizkallah PJ, Holland CJ, Fuller A, Bulek A, Godkin AJ, et al. Structural basis for ineffective T-cell responses to MHC anchor residue-improved “heteroclitic” peptides. Eur J Immunol. (2015) 45:584–91. doi: 10.1002/eji.201445114
67. Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem. (2012) 287:1168–77. doi: 10.1074/jbc.M111.289488
68. Pun FW, Ozerov IV, and Zhavoronkov A. AI-powered therapeutic target discovery. Trends Pharmacol Sci. (2023) 44:561–72. doi: 10.1016/j.tips.2023.06.010
69. Qureshi R, Irfan M, Gondal TM, Khan S, Wu J, Hadi MU, et al. AI in drug discovery and its clinical relevance. Heliyon. (2023) 9:e17575. doi: 10.1016/j.heliyon.2023.e17575
70. Rollins ZA, Curtis MB, George SC, and Faller R. A computational strategy for the rapid identification and ranking of patient-specific T cell receptors bound to neoantigens. Macromol Rapid Commun. (2024) 45:e2400225. doi: 10.1002/marc.202400225
71. Pham MN, Nguyen T-N, Tran LS, Nguyen Q-TB, Nguyen T-PH, Pham TMQ, et al. epiTCR: a highly sensitive predictor for TCR-peptide binding. Bioinformatics. (2023) 39:btad284. doi: 10.1093/bioinformatics/btad284
72. Deng L, Ly C, Abdollahi S, Zhao Y, Prinz I, and Bonn S. Performance comparison of TCR-pMHC prediction tools reveals a strong data dependency. Front Immunol. (2023) 14:1128326. doi: 10.3389/fimmu.2023.1128326
73. Kim SH, Lee BR, Kim S-M, Kim S, Kim M-S, Kim J, et al. The identification of effective tumor-suppressing neoantigens using a tumor-reactive TIL TCR-pMHC ternary complex. Exp Mol Med. (2024) 56:1461–71. doi: 10.1038/s12276-024-01259-2
74. Yadav S, Vora DS, Sundar D, and Dhanjal JK. TCR-ESM: Employing protein language embeddings to predict TCR-peptide-MHC binding. Comput Struct Biotechnol J. (2024) 23:165–73. doi: 10.1016/j.csbj.2023.11.037
75. Zhang J, Ma W, and Yao H. Accurate TCR-pMHC interaction prediction using a BERT-based transfer learning method. Brief Bioinform. (2023) 25:bbad436. doi: 10.1093/bib/bbad436
76. Kumar A, Dixit S, Srinivasan K, M D, and Vincent PMDR. Personalized cancer vaccine design using AI-powered technologies. Front Immunol. (2024) 15:1357217. doi: 10.3389/fimmu.2024.1357217
77. Bhattacharya M, Alshammari A, Alharbi M, Dhama K, Lee S-S, and Chakraborty C. A novel mutation-proof, next-generation vaccine to fight against upcoming SARS-CoV-2 variants and subvariants, designed through AI enabled approaches and tools, along with the machine learning based immune simulation: A vaccine breakthrough. Int J Biol Macromol. (2023) 242:124893. doi: 10.1016/j.ijbiomac.2023.124893
78. Sarkizova S, Klaeger S, Le PM, Li LW, Oliveira G, Keshishian H, et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat Biotechnol. (2020) 38:199–209. doi: 10.1038/s41587-019-0322-9
79. Jiang C, Li J, Zhang W, Zhuang Z, Liu G, Hong W, et al. Potential association factors for developing effective peptide-based cancer vaccines. Front Immunol. (2022) 13:931612. doi: 10.3389/fimmu.2022.931612
80. Kawakita S, Shen A, Chao C-C, Wang Z, Cheng S, Li B, et al. An integrated database of experimentally validated major histocompatibility complex epitopes for antigen-specific cancer therapy. Antib Ther. (2024) 7:177–86. doi: 10.1093/abt/tbae011
81. Riley TP, Keller GLJ, Smith AR, Davancaze LM, Arbuiso AG, Devlin JR, et al. Structure based prediction of neoantigen immunogenicity. Front Immunol. (2019) 10:2047. doi: 10.3389/fimmu.2019.02047
82. Gálvez J, Gálvez JJ, and García-Peñarrubia P. Is TCR/pMHC affinity a good estimate of the T-cell response? An answer based on predictions from 12 phenotypic models. Front Immunol. (2019) 10:349. doi: 10.3389/fimmu.2019.00349
83. al-Ramadi BK, Jelonek MT, Boyd LF, Margulies DH, and Bothwell AL. Lack of strict correlation of functional sensitization with the apparent affinity of MHC/peptide complexes for the TCR. J Immunol. (1995) 155:662–73. doi: 10.4049/jimmunol.155.2.662
84. Bhattacharyya ND, Counoupas C, Daniel L, Zhang G, Cook SJ, Cootes TA, et al. TCR affinity controls the dynamics but not the functional specification of the antimycobacterial CD4+ T cell response. J Immunol. (2021) 206:2875–87. doi: 10.4049/jimmunol.2001271
Keywords: TCR-pMHC recognition, AI/ML-driven structure prediction, immunogenicity modeling, T-cell therapy design, protein-protein interactions
Citation: Chao C-c, Chiu Y, Yeung L, Yee C, Jiang C and Shen X (2025) AI/ML-empowered approaches for predicting T Cell-mediated immunity and beyond. Front. Immunol. 16:1651533. doi: 10.3389/fimmu.2025.1651533
Received: 21 June 2025; Accepted: 04 August 2025;
Published: 29 August 2025.
Edited by:
Hyejin Choi, Johnson & Johnson, United StatesReviewed by:
Qiang Yang, Harbin Institute of Technology, ChinaCopyright © 2025 Chao, Chiu, Yeung, Yee, Jiang and Shen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiling Shen, eHNoZW4zQG1kYW5kZXJzb24ub3Jn; Chongming Jiang, Y2hvbmdtaW5nLmppYW5nQHRlcmFzYWtpLm9yZw==