 Hui Li1,2
Hui Li1,2 Yingqi Wang1,2
Yingqi Wang1,2 Hongxia Duan1,2
Hongxia Duan1,2 Yidie Bao1
Yidie Bao1 Xinliao Deng1,3
Xinliao Deng1,3 Yucheng He1
Yucheng He1 Qian Gao4
Qian Gao4 Peijun Li1,2*
Peijun Li1,2* Xiaodan Liu1,2,3,5*
Xiaodan Liu1,2,3,5*- 1Shanghai University of Traditional Chinese Medicine, School of Rehabilitation Medicine, Shanghai, China
- 2The Second Rehabilitation Hospital of Shanghai, Shanghai, China
- 3Engineering Research Center of Intelligent Rehabilitation of Traditional Chinese Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai, China
- 4Shanghai Sports University (SSU), Shanghai, China
- 5School of Sports and Health Sciences, Shanghai University of Sport, Rehabilitation Institute of Shanghai University of Traditional Chinese Medicine, Shanghai, China
Background: Chronic Obstructive Pulmonary Disease (COPD) is a leading cause of global mortality, characterized by chronic inflammation and abnormal immune responses in the lower airways. Recent studies have highlighted the critical role of immune function in the pathogenesis and progression of COPD. The disease is characterized by abnormal immune responses in the lower respiratory tract, with its progression associated with the infiltration of innate and adaptive inflammatory immune cells into the lungs and the formation of lymphoid follicles, mediated by cytokines and inflammasomes. Increasing evidence suggests that cell-mediated immunity has an important role in the pathogenesis of COPD, which is characterized by immune senescence leading to decreased resistance to infection, enhanced neutrophil and macrophage activation, T-cell infiltration, and aberrant B-cell activity, all of which combine to contribute to airway inflammation and lung injury in patients with COPD.
Objective: This review aimed to explore the pivotal role of the immune system in COPD and its therapeutic potential.
Methods: We reviewed, categorized, and summarized literature on immunity and COPD published in the last five years from Web of Science and PubMed databases.
Results: This study elucidates the pivotal role of immune dysregulation in COPD pathogenesis, particularly the dysfunctional transition from innate to adaptive immunity. We delineate how specific immune cell populations—including macrophages, neutrophils, and T-lymphocytes—contribute to sustained airway inflammation and lung injury in COPD through aberrant activation, infiltration, and impaired function. Mechanistically, key features of this dysregulation involve aberrant cytokine signaling pathways and defective resolution of inflammation. These insights reveal potential therapeutic targets for immunomodulatory strategies aimed at interrupting the chronic inflammatory cascade, restoring immune homeostasis, and mitigating infection susceptibility in COPD. Promising approaches highlighted include targeting specific cytokines, modulating macrophage polarization states, and enhancing mucosal immune defenses.
1 Introduction
Chronic Obstructive Pulmonary Disease (COPD) represents a prevalent respiratory disorder characterized by persistent airflow limitation, manifesting clinically as progressive worsening of dyspnea, chronic cough, sputum production, and ultimately predisposing to life-threatening complications including respiratory and cardiac failure when left uncontrolled. The disease course typically alternates between stable phases and acute exacerbations (AECOPD), with the latter significantly accelerating pulmonary function decline. Current epidemiological projections indicate COPD will emerge as the third leading global cause of mortality by 2030 (1), constituting a substantial public health challenge due to its escalating prevalence, debilitating morbidity, and substantial socioeconomic burden.
The human immune system orchestrates a sophisticated defense network against pathogenic invasion through complementary innate and adaptive mechanisms. Innate immunity provides immediate, non-specific protection via physical barriers (e.g., epithelial linings), phagocytic cells (neutrophils, macrophages), and humoral components (complement proteins, antimicrobial peptides) (2). In contrast, adaptive immunity mediates antigen-specific responses through lymphocyte-mediated memory formation and targeted effector functions. While these subsystems employ distinct cellular players (dendritic cells, T/B lymphocytes) and molecular mediators (cytokines, antibodies), they synergistically integrate through both humoral (complement, antibodies) and cell-mediated (phagocytosis, cytotoxic killing) mechanisms (Figure 1). Notably, specialized neurovascular barriers (blood-brain, blood-cerebrospinal fluid) compartmentalize central nervous system immunity from peripheral immune activity (3).
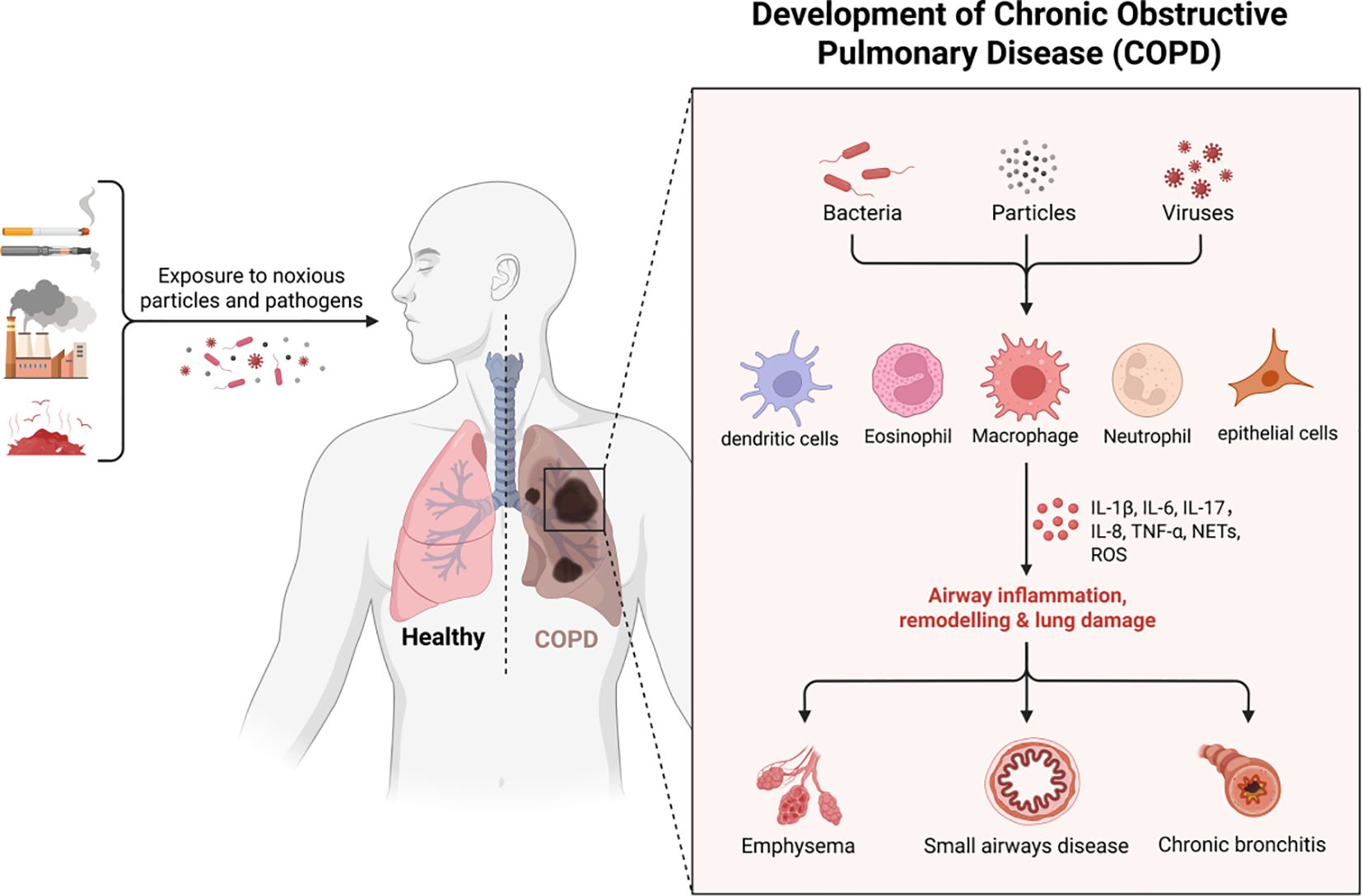
Figure 1. The pathogenesis of COPD.
Pathogen evolution drives continuous immune adaptation through conserved defense strategies across phylogeny. Prokaryotic organisms employ restriction enzymes as primitive antiviral defenses, while ancestral eukaryotes developed foundational mechanisms including phagocytosis, defensin production, and complement-like systems preserved in modern plants and invertebrates. Jawed vertebrates evolved sophisticated adaptive immunity featuring somatic recombination of antigen receptors, enabling immunological memory that enhances secondary responses (4). This evolutionary arms race has shaped a multi-layered defense paradigm: 1) Immediate innate detection via pattern recognition receptors (PRRs) 2) Coordinated inflammatory recruitment 3) Antigen-specific adaptive targeting 4) Long-term immunological memory - all critical for maintaining host-pathogen equilibrium. This study categorized and summarized literature related to immunity and COPD published in the Web of Science and PubMed databases over the past five years (Tables 1, 2).
Table 1. Articles related to immunization and COPD.
Table 2. Research articles related to immunization and COPD.
2 Innate immunity and COPD: cells and related mechanisms
Innate immunity refers to the defense mechanisms that the body is born with, including the body’s physical barriers, nonspecific immune cells and serum proteins, and other nonspecific defense factors, which in the progression of COPD include cellular pattern recognition, surface barriers, cellular components, inflammation, and the complement system. The innate immune system employs specialized germline-encoded detectors called pattern recognition receptors (PRRs) to identify pathogenic signatures. These transmembrane or cytoplasmic proteins function as molecular surveillance systems, primarily expressed on sentinel immune cells including dendritic cells, macrophages, monocytes, neutrophils, and epithelial barriers (17). PRRs demonstrate dual specificity by recognizing: 1) Pathogen-associated molecular patterns (PAMPs) - conserved microbial components essential for pathogen survival, and 2) Damage-associated molecular patterns (DAMPs) - endogenous molecules released during tissue injury or necrotic cell death (124).
Among the cellular pattern recognition covered, surface barrier studies focus on COPD with gut flora, lung-gut axis, and cellular components including macrophages, neutrophils, dendritic cells and innate lymphocytes. Macrophages are one of the major sources of cytokines and inflammatory mediators in the airways, and in addition to phagocytosis of particles, bacteria, and apoptotic cells, the production and secretion of inflammatory mediators promotes the accumulation of other inflammatory cells, such as neutrophils. Neutrophils migrate from the pulmonary capillaries to the airways and kill pathogenic microorganisms (fungi, bacteria, viruses, etc.) by means of reactive oxygen species, antimicrobial proteins, and degradative enzymes (e.g., elastase). Overproduction, release, and inadequate neutralization of these potentially tissue-damaging molecules have been shown to contribute to tissue destruction in COPD. In addition, these infiltrating inflammatory immune cells can activate adaptive immune responses by modulating antigen presentation by antigen-presenting cells in lung tissue, including dendritic cells and macrophages (Figure 2).
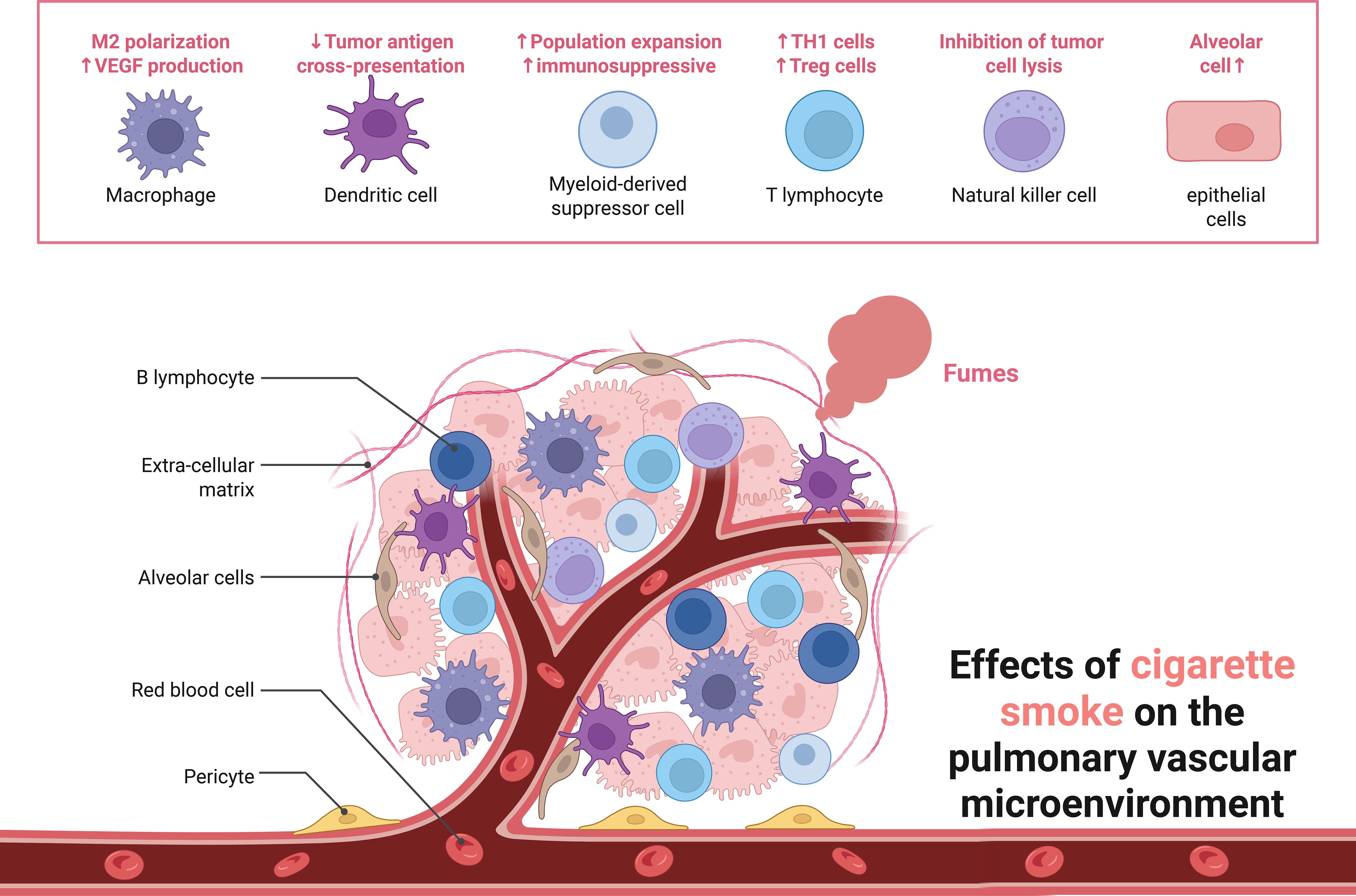
Figure 2. Effects of cigarette smoke on the pulmonary vascular micro environment.
2.1 Macrophage
AMs play a central role in processing and clearing lung particles in COPD patients. Increased recruitment of monocytes in response to monocyte-selective CCL2 and CXC chemokine ligand 1 (CXCL1) further leads to increased levels of macrophages in sputum and bronchoalveolar lavage fluid (BALF) of COPD patients. And sustained exposure to cigarette smoke or biomass significantly depleted intracellular antioxidants, such as glutathione, leading to excessive oxidative stress, which inhibited bacterial phagocytosis and cytophagocytosis in AMs. As a result, AMs in COPD produce more pro-inflammatory factors, leading to tissue damage, as well as defective phagocytosis of AMs, which together influence the progression of COPD (125). Macrophages are highly plastic and can differentiate into distinct phenotypes based on microenvironmental signals, primarily including pro-inflammatory M1 and anti-inflammatory M2 phenotypes. In COPD, macrophage plasticity is regulated by multiple factors, such as transcription factors, signaling pathways, and epigenetic modifications. Studies demonstrate that the polarization state of alveolar macrophages directly influences both the inflammatory response and tissue remodeling in COPD (126). Additionally, macrophages contribute to COPD-associated airway inflammation and fibrosis through regulation of inflammatory mediator release and extracellular matrix metabolism (127).
Macrophage plasticity and polarization status constitute pivotal determinants of COPD pathogenesis. Classically activated M1 macrophages, typically induced by pro-inflammatory stimuli (e.g., LPS and IFN-γ), perpetuate airway inflammation through robust secretion of inflammatory mediators including TNF-α, IL-1β, and IL-6. Conversely, alternatively activated M2 macrophages, polarized by anti-inflammatory cytokines (IL-4/IL-13), actively contribute to tissue remodeling processes by promoting fibrosis and repair mechanisms. Critically, the imbalance between pro-inflammatory M1 and pro-fibrotic M2 phenotypes drives progressive pulmonary destruction in COPD. Polarized M1 and M2 macrophages are also involved in the pathogenesis of COPD: the phagocytic capacity of M1 macrophages and the antigen-presenting properties of Th1 produce a variety of Th1 cytokines (128), e.g.IL-1β, IL-6, IL-12, and TNF-α. It has been found that the activation of the Notch signaling pathway in the cigarette-induced model of COPD and the further polarization of macrophages shifted toward the M1 phenotype with increased levels of IL-6, TNF-α and reactive oxygen species. In contrast, in the face of foreign particulate matter and microbial exposure, M2 macrophages induced by Th2 cytokines overexpressed arginase-1 (Arg-1), IRF4, and mannose receptor CD206 and released TGF-β, leading to myofibroblast activation, smooth muscle proliferation, and aberrant tissue repair. Another study found that e-cigarette exposure enhanced airway dilation, mucus secretion and fibrogenesis in COPD mice, which was associated with an increased M2 macrophage phenotype (79).
2.2 Congenital intrinsic lymphoid cells
Innate lymphoid cells (ILC) are a group of innate immune cells that are derived from common lymphoid progenitor cells (CLP) and belong to the lymphoid lineage. There are no antigen-specific B or T cell receptors to define these cells due to the lack of recombination activation genes (RAG). ILCs do not express myeloid or dendritic cell markers (129).
Natural killer cells, one of the member ILCs (130), are lymphocytes that are part of the innate immune system and do not directly attack invading microorganisms. Instead, NK cells destroy damaged host cells, such as tumor cells or virus-infected cells, and recognize such cells through a condition known as the “missing self”. The term describes low levels of the cell surface marker MHC I (major histocompatibility complex), which can occur in viral infections of host cells. They were called “natural killers” because the original concept was that they did not need to be activated to kill cells that “lacked self”. For many years, it was not clear how NK cells recognized tumor cells and infected cells. It is now known that the MHC composition on the surface of those cells is altered and NK cells are activated by recognizing the “missing self”. Normal human cells are not recognized and attacked by NK cells because they express their own MHC antigens intact. Those MHC antigens are recognized by the killer cell immunoglobulin receptor (KIR), which actually puts the brakes on NK cells (13, 131).
In a study on the phenotyping and quantification of human lung ILC subpopulations (132), the investigators set up control and COPD groups with the presence of COPD as a variable, and identified ILC subpopulations in digested human lungs by flow cytometry, further defining subsets based on specific surface markers, and found that in the control group, both ILC2 and NCR-ILC3 were the most abundant ILC subpopulations; whereas in the COPD group, the lung ILC subpopulation was more heavily weighted toward NCR- ILC3.Although all 3 ILC subpopulations have been identified in the airways, most studies to date have focused on ILC2, emphasizing their importance in tissue repair, protection against helminthic infections, and involvement in a wide range of allergic diseases. However, ILC3 is actually the most common ILC subpopulation in human lung tissue.ILC3 and LTi cells rapidly secrete IL-17 and IL-22, driving the development of COPD (80).
In mice, cigarette smoke exposure leads to an increase in all ILC subpopulations in bronchoalveolar lavage fluid, particularly IFN γ+ and IL-17+ ILC.IL-17 is considered a key driver of neutrophil inflammation in COPD (133), inducing neutrophil maturation and recruitment, and through the release of its proteolytic enzymes (elastase, histone G, and protease-3), facilitating Sputum IL-17 is higher in COPD patients than in non-smoking controls, and bronchial biopsies from COPD patients also show increased expression of IL-17, IL-22 and IL-23. ILC3-secreted GM-CSF is critical in allergic airway disease, human lung anti-microbial defense, and homeostasis of alveolar surfactant. ILC1, Th1 cells and CD8+ cells are essential in the development of allergic airway disease. ILC1, Th1 cells and CD8+ T cells can produce IFN γ, which is involved in the pathogenesis of COPD by inducing the production of elastase and nitric oxide by alveolar macrophages, leading to emphysema (134).
2.3 Dendritic cells
Dendritic cells (DCs) are phagocytes in tissues that come into contact with the external environment. In the lungs, dendritic cells exist in multiple subpopulations, including myeloid dendritic cells (mDCs) and plasmacytoid dendritic cells (pDCs). mDCs primarily mediate T cell-mediated immunity and antibody production, while pDCs play a crucial role in antiviral immunity and immune tolerance (114).In COPD patients, the numbers and functions of these subpopulations undergo significant changes. Studies have shown that in the small airways and alveoli of COPD patients, the number of immature dendritic cells significantly increases, while the number of mature dendritic cells decreases, particularly in advanced COPD patients, where alveolar CD1a+langerin-, BDCA-2+CD11c+dendritic cell subpopulations significantly increase (135).These changes are closely associated with the immunopathology of COPD. Plasmacytoid dendritic cells (pDCs) accumulate significantly in pulmonary lymphoid follicles of COPD patients, especially in those with mild to moderate COPD (136). This accumulation may be associated with viral infections and autoimmune responses in COPD. Additionally, pDCs exhibit enhanced secretion of TNF-α and IL-8 in COPD, further exacerbating inflammatory responses (137).
Dendritic cells serve as a critical bridge between tissues and the immune system, coordinating immune responses by presenting antigens to T cells. Studies have shown that cigarette smoke-induced dendritic cell activation in chronic obstructive pulmonary disease (COPD) promotes the differentiation of ILCs into NCR-ILC3, thereby influencing disease severity (26). These cells initiate immune responses by recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through Toll-like receptors (TLRs) (2).In the progression of COPD, dendritic cells exhibit four major functional dysregulations: First, their antigen-presenting function is inhibited, and exposure to cigarette smoke impairs cell maturation and cytokine secretion, weakening immune defense against bacteria such as Streptococcus pneumoniae (13); second, TLR2-activated dendritic cells drive Th17 cell differentiation, promoting emphysema formation and chronic inflammation; third, the immune tolerance mechanism mediated by dendritic cells is impaired, leading to reduced regulatory T cell (Treg) responses and triggering excessive inflammation and autoimmune reactions; additionally, oxidative stress further disrupts dendritic cell function, accelerating the pathological process (11).These functional dysregulations collectively cause T cell response imbalance, closely associated with airway remodeling and persistent inflammation in COPD (Figure 3).
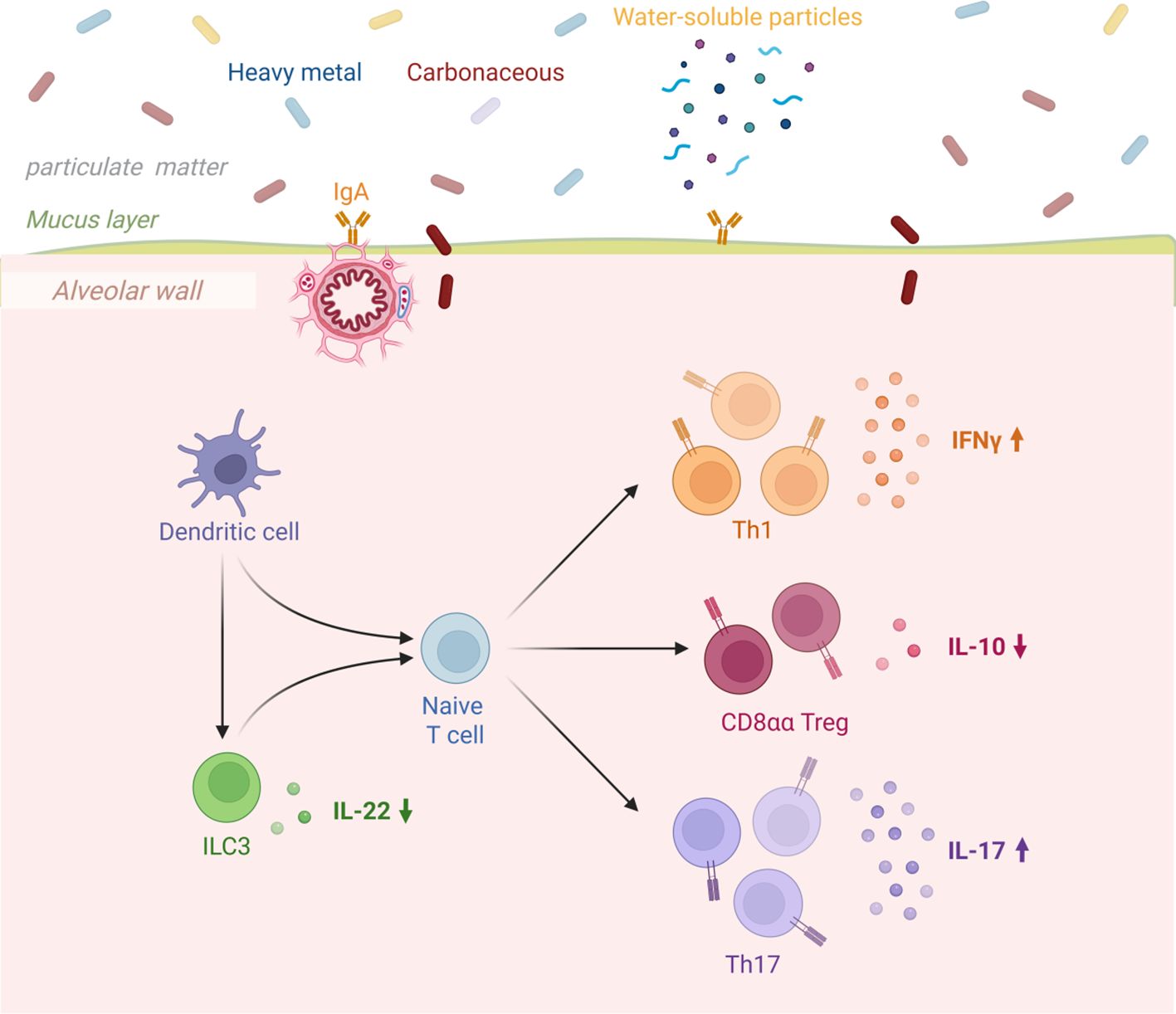
Figure 3. Congenital intrinsic lymphocytes in relation to COPD.
Dendritic cells recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through Toll-like receptors (TLRs), thereby initiating an immune response (17).In COPD, the antigen-presenting function of dendritic cells is impaired, leading to an imbalance in T cell responses (138), in COPD, dendritic cells are activated via TLR2, promoting the differentiation of Th17 cells and contributing to the development of emphysema. This Th17 response is closely associated with the chronic inflammation and airway remodeling characteristic of COPD. Additionally, oxidative stress further exacerbates the pathological process of COPD by impairing dendritic cell function. Impaired tolerance function of dendritic cells leads to excessive inflammatory and autoimmune responses, characterized by reduced regulatory T cell (Treg) responses, further exacerbating immune imbalance (139).
2.4 Granulocyte
Granulocytes are white blood cells that have granules in their cytoplasm. In this category are neutrophils, mast cells, basophils and eosinophils. Mast cells reside in connective tissue and mucous membranes and regulate inflammatory responses. They are most often associated with allergies and allergic reactions. Basophils and eosinophils are related to neutrophils. They secrete chemical mediators that defend against parasites and play a role in allergic reactions such as asthma (140). Eosinophils and basophils have been studied more in COPD related studies (38), one study on the anatomical distribution pattern of infiltrating eosinophils in COPD patients found a novel heterogeneity in the immunopathology of COPD patients, which manifested itself in the identification of a spatially restricted eosinophil-rich type 2 microenvironment, and another study focusing on eosinophils demonstrated that patients with COPD Tissue-infiltrating eosinophils, basophils, and eosinophil-promoting immune mechanisms in the lungs (141).
It is worth noting that under the regulation of multiple inflammatory factors, oxidative stress, and hypoxic environments, neutrophils play a key role in the pathogenesis of COPD. Abnormalities in neutrophil recruitment, neutrophil extracellular trap (NET) formation, and protease activity are closely associated with disease progression (142) (Figure 4). First, inflammatory mediators such as IL-8, LTB4, and CXCL1 activate endothelial cells, mediating neutrophil migration to the airways. This process involves the release of chemokines by lung epithelial cells and macrophages, prompting neutrophils to interact with endothelial cells via adhesion molecules such as CD11b/CD18, ultimately crossing the vascular wall to infiltrate lung tissue (143).Infiltrating neutrophils release reactive oxygen species (ROS) and proteases, directly damaging lung structures and exacerbating inflammatory responses. Neutrophil extracellular traps (NETs) exhibit dual effects in COPD: on one hand, their DNA-histone-antimicrobial protein complexes can capture pathogens, providing protective effects against infections during acute exacerbations; on the other hand, excessive NET formation releases histones and proteases that damage alveolar epithelial and endothelial cells and exacerbate airway inflammation and fibrosis by activating inflammasomes and promoting cytokine release, ultimately leading to lung tissue damage (144). Proteases released by neutrophils (including elastase, matrix metalloproteinases, and cathepsins) are core pathological mediators, elastase directly degrades elastic and collagen fibers in alveolar walls, promoting the formation of pulmonary emphysema; these proteases amplify the inflammatory cascade by activating inflammatory mediators such as IL-8 and TNF-αand disrupting theα1-antitrypsin barrier; simultaneously, they induce airway smooth muscle proliferation and fibrosis, leading to airway remodeling and progressive airflow limitation (145).
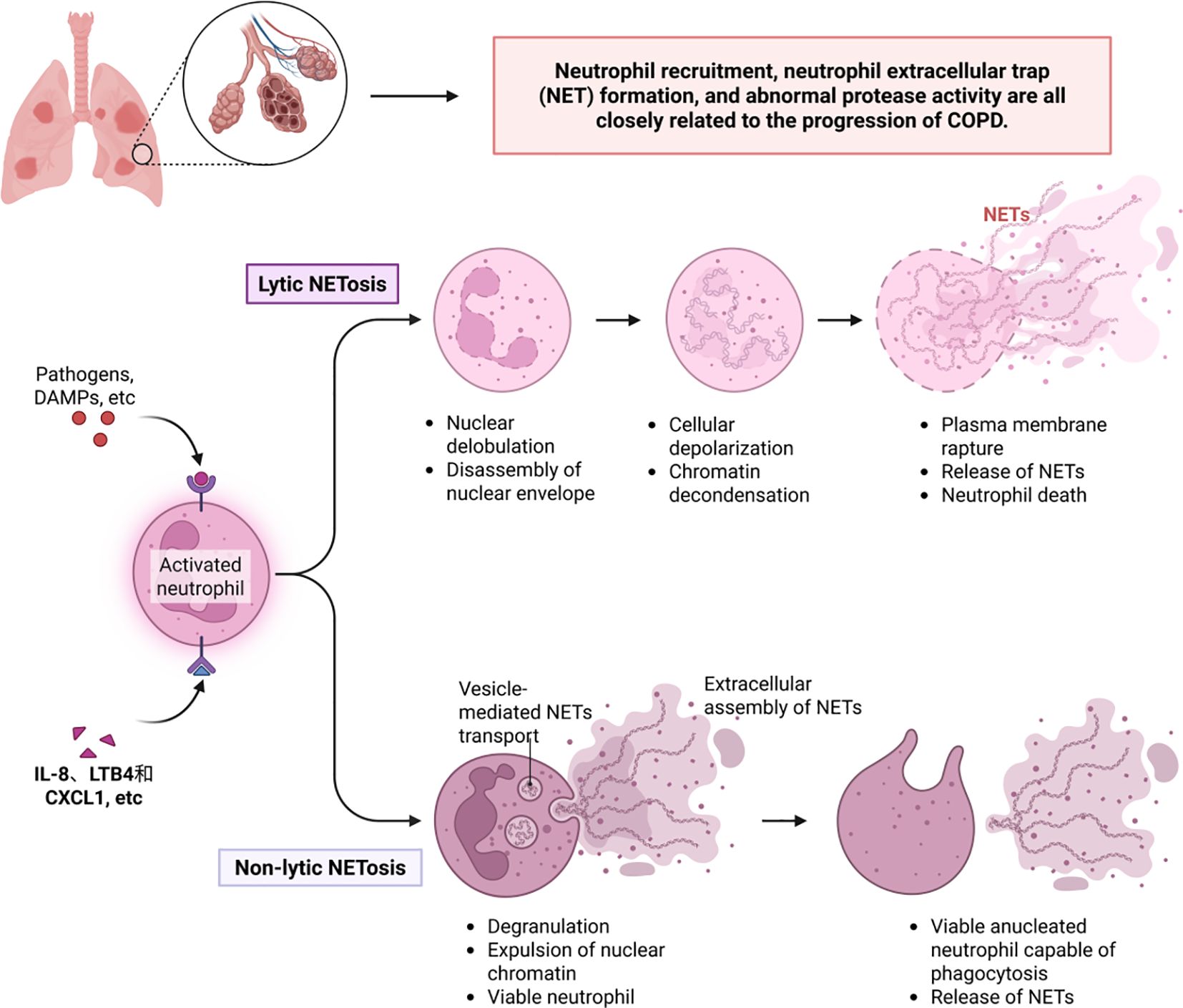
Figure 4. Neutrophil extracellular traps and COPD.
Neutrophilic COPD is the primary phenotype of COPD and differs fundamentally from eosinophilic COPD (accounting for 20–40%) (146). The eosinophilic subtype is characterized by a Th2 immune response, releasing IL-5/IL-13 and eosinophil cationic protein. Clinically, it presents with mild airflow limitation but responds well to glucocorticoid therapy. Elevated eosinophil counts in blood or sputum indicate a risk of acute exacerbation and a favorable prognosis; the neutrophilic type mediates tissue destruction through elastase, MMP-9, and IL-8/IL-17, presenting with severe airflow obstruction, high sputum volume, and hypoxemia. Patients are resistant to corticosteroids and require bronchodilators/antibiotics. Elevated neutrophil/lymphocyte ratios and C-reactive protein (CRP) levels predict high hospitalization rates and mortality (141, 147). Microbiome analysis shows that the eosinophil-dominant phenotype is associated with an enrichment of the genus Campylobacter, whereas the neutrophil-dominant phenotype is dominated by the genus Haemophilus and accompanied by elevated IL-1β/TNF-α levels (148).
2.5 Epithelial cells
Respiratory epithelial cells play a dual barrier function in the pathogenesis of COPD, and the disruption of their structural integrity is a central component of disease progression. Long-term exposure to cigarette smoke and other irritants directly damages epithelial cells, triggering a three-part cascade of reactions: increased susceptibility to pathogens, persistent worsening of chronic inflammation, and reduced tissue repair capacity, all of which collectively drive the pathological progression of COPD. Epithelial cells dynamically regulate the immune microenvironment by releasing mediators such as TSLP (149). In this process, activated immune cells (such as mast cells) release substances like tryptase, which specifically degrade epithelial junction proteins (e.g., E-cadherin, zonula occludens-1). This degradation establishes a vicious cycle of “epithelial damage–immune activation” (150). Simultaneously, epithelial signals may activate the Th2/IL-13 pathway, promoting excessive mucus secretion and airway remodeling (Figure 5). Targeted therapy focuses on two main directions: at the molecular level, blocking TSLP signaling or inhibiting mast cell protease activity aims to reduce inflammation and restore the epithelial barrier; at the environmental level, reducing exposure to pollutants from the source (151). Existing research on the mechanisms of respiratory diseases provides important insights for COPD pathogenesis. Future studies should directly validate the specific role of the epithelial-immune axis in COPD to explore new therapeutic targets.
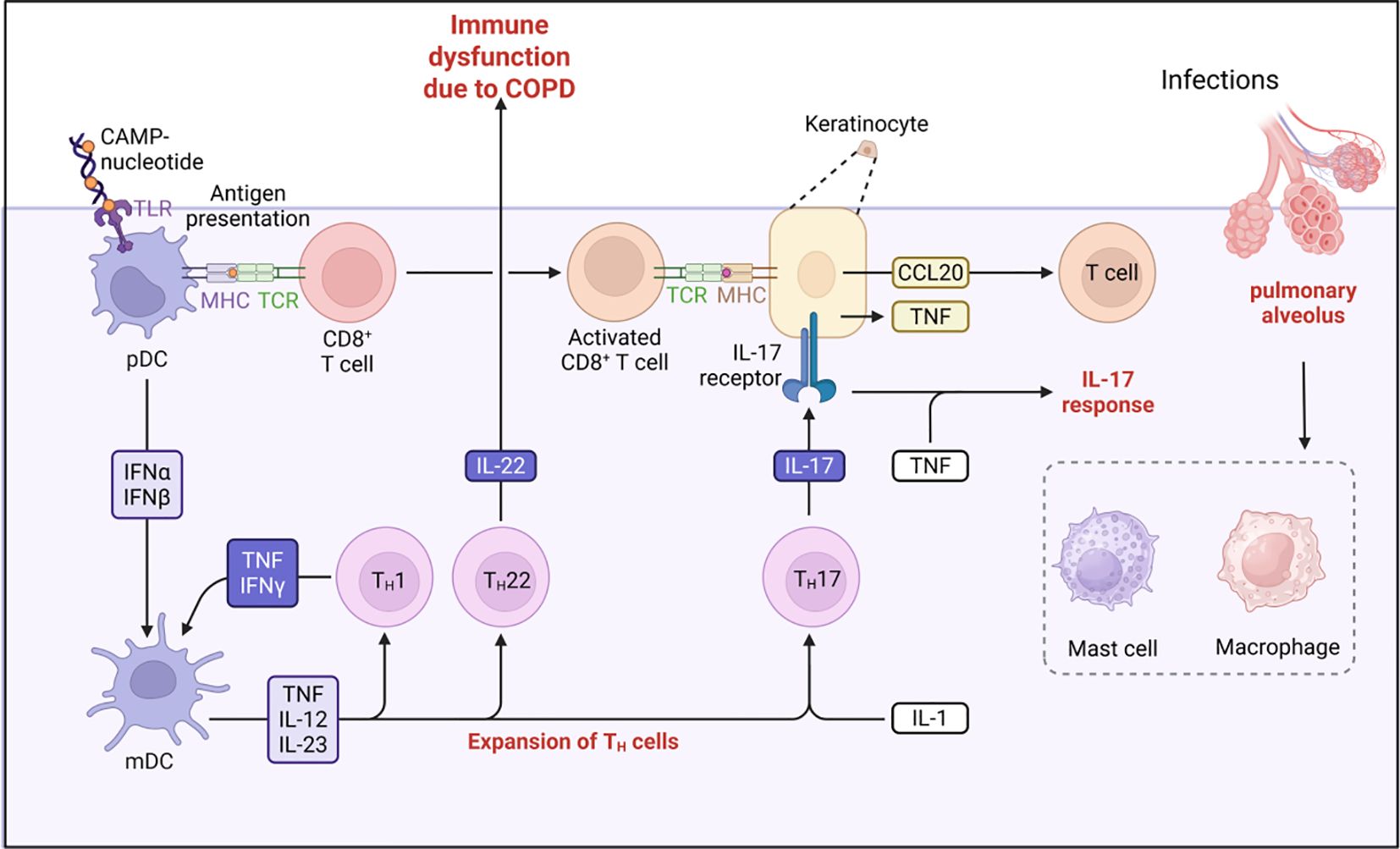
Figure 5. The immune response in relation to chronic obstructive pulmonary disease.
Epithelial-immune interactions and barrier dysfunction play a central role in COPD. Prolonged exposure to noxious gases (such as cigarette smoke) and particulate matter induces epithelial cell damage, triggering subsequent barrier dysfunction. Epithelial cells regulate local immune responses by releasing cytokines (e.g., TSLP) and chemokines. T cells and mast cells are pivotal in mediating epithelial barrier disruption and inflammatory responses. Mast cells release proteases (such as tryptase and chymotrypsin) that disrupt intercellular junctions of epithelial cells (including E-cadherin and zonula occludens-1), compromising barrier function. During acute exacerbations, mast cell activation exacerbates inflammatory responses and epithelial damage (152). Therapeutic strategies targeting epithelial-immune interactions and barrier dysfunction hold significant potential for COPD management. For example, agents targeting TSLP or inhibiting mast cell protease activity may reduce inflammation and restore barrier function (153). Additionally, mitigating environmental factors (e.g., reducing airborne pollutant exposure) and enhancing epithelial barrier function represent promising directions for COPD prevention and treatment.
2.6 Complement system
The complement system is a biochemical cascade that attacks the surface of foreign cells. It contains more than 20 different proteins and is named for its ability to “complement” antibodies in killing pathogens. Complement is a major humoral component of the innate immune response. In humans, the response is activated by binding of complement to antibodies already attached to these microorganisms or by binding of complement proteins to carbohydrates on the microbial surface. This recognition signal triggers a rapid killing response. The speed of the response is the result of signal amplification, which occurs after the activation of sequential protein hydrolysis of complement molecules (also proteases) (4). After the complement proteins initially bind to the microbe, they activate its protease activity, which in turn activates other complement proteases, and so on. This produces a catalytic cascade that is controlled by positive feedback. The cascade leads to the production of peptides that attract immune cells, increase vascular permeability, and condition (cover) the surface of the pathogen so that it is destroyed. This deposition of complement can also kill cells directly by disrupting the cytoplasmic membrane (154). In chronic lung disease, actin-related proteins are more commonly studied, and a study of AM phagocytosis in COPD mice demonstrated that PM2.5 exacerbates AM phagocytosis in COPD mice by inhibiting the Arp2/3 complex and F-actin, and exacerbating AM phagocytosis in COPD mice by the (Arp) 2/3 complex (4).
2.7 Surface barrier particulate matter
COPD is the most common chronic respiratory disease, a respiratory disorder characterized by permanent and progressive loss of lung function associated with smoking and exposure to harmful stimuli. Long-term inhalation of harmful gases and particulate matter are the main etiologic factors in the development and progression of COPD (155), and they are capable of inducing inflammation in the large and small airways and spreading it to the lung parenchyma, causing destruction of the alveolar walls, which in turn leads to emphysema. Studies have shown that a large number of inflammatory mediators are involved in inducing the infiltration of immune inflammatory cells and the production and release of their destructive enzymes, and that these inflammatory mediators are associated with the progressive destruction of the lungs in patients with COPD and trigger the remodeling of the central airways, distal airways, and lung parenchyma. In addition, oxidative stress from inhalation of noxious gases and particulate matter triggers a complex inflammatory response in the airways and lung tissues, ultimately leading to pathological changes in COPD.
3 The adaptive immune system and COPD: humoral and cellular immune responses
The adaptive immune system evolved in early vertebrates and allows for a stronger immune response as well as immune memory, in which each pathogen is “remembered” by a characteristic antigen. The adaptive immune response is antigen-specific and requires the recognition of specific “non-self” antigens in a process called antigen presentation. Antigen specificity produces a response against a specific pathogen or pathogen-infected cell. The ability to carry out these customized responses is maintained in the body by “memory cells”. If a pathogen infects the body more than once, these specific memory cells are used to quickly eliminate it. Adaptive immunity consists of antigen recognition, antigen presentation to T-lymphocytes, cell-mediated immunity, humoral immune responses, and immune memory. Among the most significant correlations between COPD and adaptive immunity are lymphocytes, helper T cells, and other.
3.1 Lymphocyte and antigen recognition
The cells of the adaptive immune system are specialized types of white blood cells called lymphocytes. B cells and T cells are the major types of lymphocytes and are derived from hematopoietic stem cells in the bone marrow. B cells are involved in humoral immune responses while T cells are involved in cell-mediated immune responses. Killer T cells recognize only antigens coupled to class I MHC molecules, whereas helper and regulatory T cells recognize only antigens coupled to class II MHC molecules (156). These two mechanisms of antigen presentation reflect the different roles of the two types of T cells. In contrast, B-cell antigen-specific receptors are antibody molecules on the surface of B cells that recognize whole pathogens without any antigen processing. Each B-cell lineage expresses a different antibody, so the complete B-cell antigen receptor represents all the antibodies that the body can make. Both B cells and T cells carry receptor molecules that recognize specific targets (157).
T cells can recognize a pathogen only after the antigen (a small fragment of the pathogen) has been processed and bound to the “self” receptor called a major histocompatibility complex (MHC) molecule. T cells can only recognize a pathogen after the antigen has been processed and bound to the “self” receptor called a major histocompatibility complex (MHC) molecule (26). T cells recognize “non-self” targets, such as pathogens, only after the antigen (small fragments of the pathogen) has been processed and bound to “self” receptors called major histocompatibility complex (MHC) molecules T and B lymphocytes provide specific immune protection by recognizing and eliminating pathogens and other harmful substances. These two types of adaptive immune cells are critical to the immune response in COPD (158).
3.2 Th immune response
T cells are an important component of the adaptive immune system and can be divided into CD4 helper T cells and CD8 helper T cells. They are involved in immunomodulation and inflammatory regulation in COPD.CD4 helper T cells can differentiate into different subpopulations such as Th1, Th2, Th17 and regulatory T cells (Treg cells). In COPD, the increased activity of Th1 and Th17 cells leads to the production of cytokines, such as interferon-gamma (IFN-γ), by Th1 cells, which stimulate inflammatory responses and cellular damage, as detailed in the Figure 6 (159).
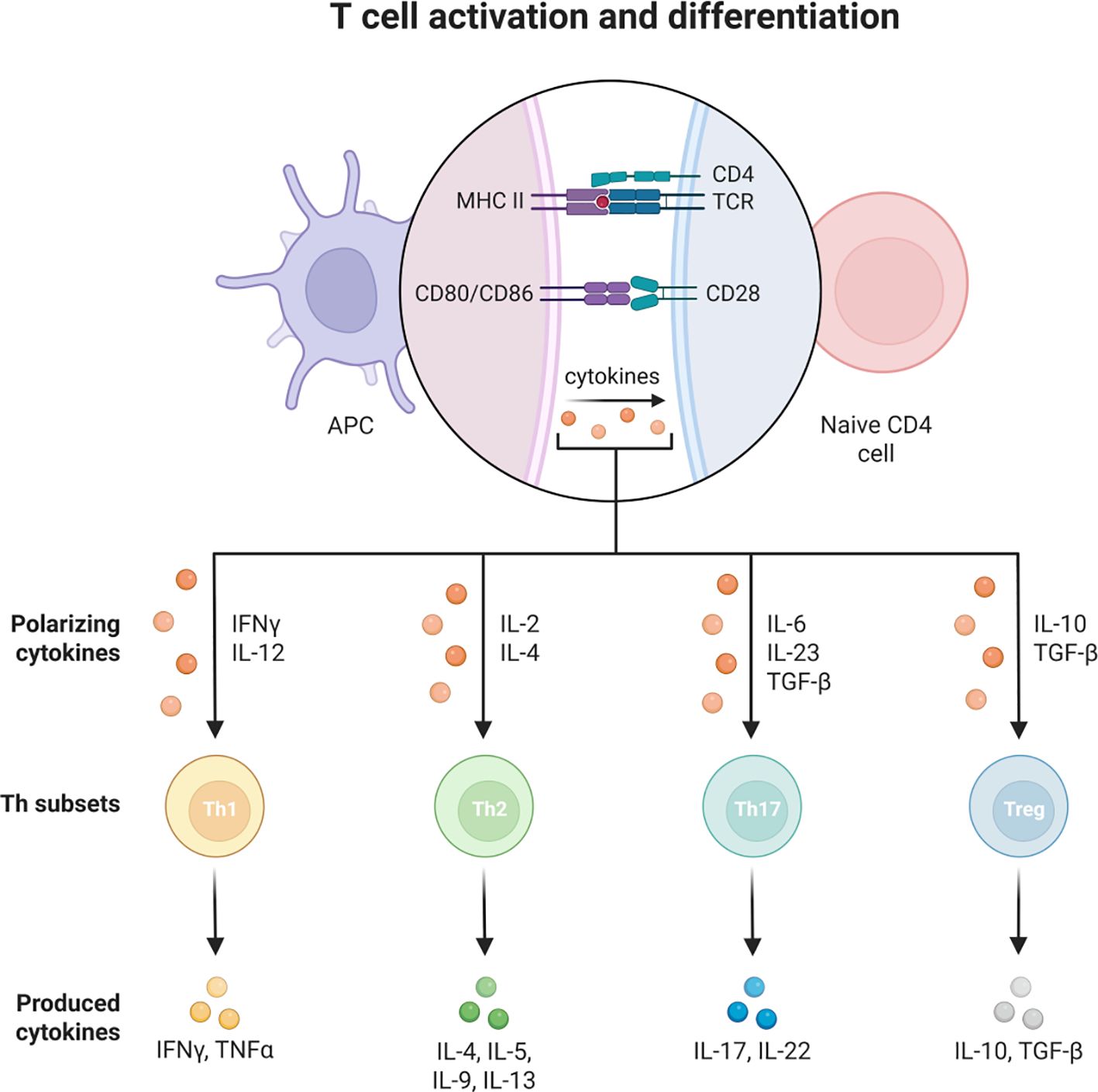
Figure 6. The role of T cells in COPD.
CD8+ T cells play a key role in the inflammatory response and tissue damage associated with COPD. In the lung tissue of patients with mild to moderate COPD, there is a significant increase in CD8+ T cell subsets, particularly KLRG1+TIGIT+CX3CR1+ TEMRA (effector memory CD45RA+ T cells) and DNAM-1+CCR5+ TRM cells. These cells interact with myeloid cells and alveolar type II cells by secreting interferon-γ (IFN-γ) and exhibit highly expanded T cell receptor clonality (134). Additionally, CD8+ TEMRA cells play an important role in early-stage inflammation of the disease (160). In patients with severe emphysema, the number of CD8+ T cells is significantly increased and positively correlated with the degree of airway obstruction and alveolar wall damage (160). Interestingly, smoking and HIV-1 infection have also been found to promote the retention of CD8+ T cells in the airway mucosa, further exacerbating inflammation and tissue remodeling (161).
It is worth noting that T cells playing a crucial role in the chronic inflammation of COPD also include memory T cells (TRM) that persistently reside in tissues. COPD patients exhibit a significant increase in CD8+ TRM cells in lung tissue, which participate in local immune responses through the expression of CCR5 and DNAM-1 (156). In systemic sclerosis-associated interstitial lung disease (SSc-ILD), an increase in CD8+ and TRM cells has also been observed, suggesting their widespread role in chronic lung diseases (162). Interestingly, the function of TRM cells in COPD is not limited to immune surveillance but may also drive inflammation and tissue damage through interactions with other immune cells, such as NK cells and macrophages (162).
In COPD, there are complex interactions between cytotoxic CD8+ T cells and TRM cells. The high-frequency clonal expansion of CD8+ TEMRA cells and the local residency of TRM cells jointly promote the persistence of chronic inflammation (156). Additionally, the differentiation of CD8+ T cells into memory cells depends on the assistance of CD4+ T cells. This process may be further regulated in COPD patients. CD8+ TEMRA cells drive early inflammation by releasing cytotoxic mediators, while TRM cells maintain a crucial role in local immune surveillance and chronic inflammation.
Antigen presenting cells (APC) present antigens on their class II MHC molecules (MHC2). Helper T cells recognize these with the help of their CD4 co-receptor (CD4 +) expression. Activation of resting helper T cells causes them to release cytokines and other stimulatory signals that stimulate the activity of macrophages, killer T cells, and B cells (the latter of which produce antibodies.) (163). In COPD patients, the number of DCs in alveoli and distal airways is significantly increased, especially in the late stages of the disease. These DCs not only express mature markers (such as MHC II and B7) but also exhibit characteristics of both innate and adaptive immune phenotypes, indicating their important role in the immunopathology of COPD. Additionally, alveolar macrophages in the early stages of COPD exhibit impaired antigen-presenting function, which may lead to defects in immune activation and further exacerbate disease progression (136).
3.3 Cytokines
In the pathophysiological network of COPD, IL-17 (produced by Th17/γδ T/NKT cells) drives neutrophil recruitment and activation by inducing epithelial secretion of CXCL8 and IL-6, thereby exacerbating pulmonary inflammation and emphysema formation. IL-17 expression levels in bronchial mucosa are positively correlated with disease severity; whereas this factor enhances antimicrobial defense during acute infections, its dysregulation in COPD patients contributes to immune deficiency and increased infection risk. IL-17’s positive correlation with FEV1 suggests a potential role in lung function regulation (mechanism unknown) (164). In stable COPD patients, serum and sputum IL-17 levels are significantly higher than in healthy controls and rise further during acute exacerbations (AECOPD), indicating that IL-17 is not only associated with persistent inflammation in COPD but may also serve as a biomarker for disease progression (165). Additionally, IL-17 upregulation is closely associated with neutrophil-mediated inflammatory responses, providing a theoretical basis for IL-17-targeted inhibitor development.
IL-6 is a key inflammatory mediator in COPD, operating through the IL-6 receptor (IL-6R)-mediated signaling pathway. Studies demonstrate significant activation of the IL-6 trans-signaling pathway (IL-6TS) in COPD inflammation, particularly in neutrophil-dominant subtypes. IL-6TS activation correlates closely with neutrophil extracellular trap (NET) formation and bacterial infections (e.g., Haemophilus influenzae) (166). Moreover, high IL-6TS activity links to persistent neutrophilic inflammation, reduced quality of life, and elevated inflammatory mediators (e.g., IL-8, MMPs) in COPD.
IL-22 (from Th22/ILC3) maintains barrier function by promoting epithelial repair; its deficiency impairs tissue repair and delays bacterial clearance. Although inducing antimicrobial proteins to enhance defense, IL-22 suppression in COPD compromises immune responses and increases acute exacerbation risk (97). Critically, IL-22 and IL-17 form a synergistic axis: IL-22 deficiency amplifies IL-17-driven inflammation, whereas IL-22 supplementation restores bacterial clearance and attenuates inflammation (167)―This axis is vital during acute exacerbations, where bacterial infections (e.g., Streptococcus pneumoniae) cause delayed clearance due to IL-17/IL-22 defects, while viral infections (e.g., influenza) suppress their secretion by inhibiting IL-1β/IL-23, raising secondary infection risks (168).
Among other cytokines, IL-33 is markedly elevated in COPD patients with lung cancer, showing negative correlation with FEV1/FVC and increasing with GOLD staging (150), Conversely, IL-6/IL-1β rise in tuberculosis patients, positively correlating with total lung capacity (TLC) and RV/TLC, respectively—collectively serving as disease severity indicators.
3.4 Biomarker
Regarding biomarkers, low plasma levels of CPa9-HNE (a calmodulin fragment) indicate mild COPD with air trapping and elevated MMP-2; elevated suPAR correlates with GOLD staging (stage IV > stage I) and serves as a progression marker (169);dysregulated extracellular miRNAs (e.g., miR-374b-5p/miR-223-3p) during acute exacerbations aid staging differentiation (124). For treatment response prediction, patients with high eosinophil counts exhibit lower mortality and shorter hospitalizations. Endocan elevation during acute exacerbations shows negative correlation with FEV1/FVC, establishing it as an independent predictive factor (170), High NLR (neutrophil-to-lymphocyte ratio) predicts mechanical ventilation need and poor prognosis. Altered ABA (acid desquamation) levels—decreased in early-stage COPD but elevated in advanced disease—may indicate disease stage (171).
Studies on COPD biomarker-treatment response correlations reveal that Endocan, a marker of endothelial dysfunction, shows significantly higher levels during acute exacerbations versus stable periods and exhibits negative correlation with lung function (e.g., FEV1/FVC), establishing it as an independent acute exacerbation predictor; Abscisic acid (ABA) displays unique dynamics in COPD patients—reduced overall yet positively correlated with immune regulators, but elevated in advanced-stage disease, suggesting potential as a staging biomarker; The neutrophil-to-lymphocyte ratio (NLR) rises significantly during acute exacerbations, correlating directly with disease severity and poor outcomes. Elevated NLR further predicts mechanical ventilation need, establishing it as a key outcome assessment indicator for acute exacerbations.
3.5 BAL and sputum immunological profile
Bronchoalveolar lavage fluid (BAL) provides important insights into airway inflammation and immune responses in COPD. Immunological profiling of BAL and sputum delivers crucial information on COPD pathophysiology, disease classification, and therapeutic targets. Studies demonstrate abnormal expression of immune/inflammatory markers in COPD BAL. Extracellular vesicle (EV) protein and miRNA profiles in COPD BAL show significant alterations, particularly increased expression of inflammation-associated proteins (e.g., neutrophil degranulation markers) (172),Notably, BAL from COPD patients induces stronger neutrophil chemotaxis, potentially linked to disease severity. Moreover, secretory immunoglobulin A (SIgA) concentration is markedly reduced in COPD BAL and positively correlates with FEV1 (173). Protease-antiprotease imbalance contributes to COPD pathogenesis, evidenced by elevated matrix metalloproteinases (MMP-9, MMP-12) and tissue inhibitors (TIMP-1) in BAL (174).
Sputum analysis serves as a key tool for assessing airway inflammation and microbiome in COPD. Elevated mucin MUC5AC concentration in sputum (when measured with MUC5B) directly associates with disease severity, exacerbation frequency, and lung function decline; microbiome analysis reveals increased abundance of Proteobacteria and Haemophilus genera, closely linked to mucus plug formation and severity (175); inflammatory marker testing shows upregulated MMP-9, LTB4R, and A1AR mRNA, while CC16 mRNA is significantly reduced—the latter particularly associating with impaired lung function and eosinophilic inflammation.
Clinically, BAL and sputum profiling demonstrates utility in COPD management: First, MUC5AC, CC16, and EV-related proteins serve as diagnostic/prognostic biomarkers; second, sputum microbiome and inflammatory markers (e.g., eosinophil levels) guide personalized therapies (e.g., targeted biologics); lastly, longitudinal monitoring enables precise assessment of disease progression and treatment response. These approaches establish a clinical management loop from diagnosis to intervention and outcome evaluation.
4 COPD management strategies—drug treatment strategies
4.1 Innate immunity and the treatment of COPD
Chronic Obstructive Pulmonary Disease (COPD) is a common respiratory disease characterized by persistent airflow limitation, which has a significant impact on patients’ quality of life. As the disease progresses, patients experience symptoms such as dyspnea and decreased activity tolerance, which can even be life-threatening in severe cases. Scientific and rational drug therapy is essential to improve patients’ symptoms, enhance quality of life, and reduce acute exacerbation events.
4.1.1 Bronchodilators
Bronchodilators are the core drugs used in the treatment of COPD to dilate narrowed airways by relaxing bronchial smooth muscle, relieve dyspnea, and improve the patient’s respiratory function. This class of drugs includes three groups: beta2-agonists, anticholinergics, and methylxanthines. beta2-agonists Beta2-agonists, such as salbutamol, formoterol, and salbutamol, are fast-acting bronchodilators that rapidly relieve shortness of breath and wheezing symptoms in COPD patients (176). These drugs increase the concentration of cyclic adenosine monophosphate in the lung smooth muscle by activating β2- receptors in the lungs, leading to the diastole of bronchial smooth muscle. They are suitable for the rapid relief of acute exacerbations of COPD as well as for long-term maintenance therapy, and can help to improve the lung function and quality of life of patients. However, long-term overdose of β2-agonists may lead to side effects such as palpitations, tremors or hypokalemia, and need to be used properly under medical supervision (177).
4.1.2 Anticholinergics
Anticholinergics, such as tiotropium bromide and ipratropium bromide, reduce airway narrowing by blocking the action of parasympathetic nerves. These drugs help COPD patients reduce symptoms and improve quality of life by steadily dilating the airways over time and reducing sputum production (178). They are particularly beneficial in reducing the frequency of acute exacerbations and improving respiratory symptoms in COPD patients, but regular monitoring is needed to avoid potential side effects such as dry mouth and urinary retention. Methylxanthines Methylxanthines, such as theophylline, are less commonly used in current COPD treatment but still have value in some patients. These drugs can increase cAMP levels by inhibiting phosphodiesterase, which induces relaxation of bronchial smooth muscle and helps relieve airway narrowing (179). Theophylline also has a mild anti-inflammatory effect. Because of the narrow window between its efficacy and side effects, blood levels need to be closely monitored to ensure safe and effective treatment (180).
4.1.3 ICS
Glucocorticoid selection Glucocorticoids are primarily used to control airway inflammation in COPD. Inhaled glucocorticoids (ICS) are effective in reducing airway inflammation, improving respiratory function, and reducing the risk of acute exacerbations. The use of ICS is of critical importance, especially in patients who experience frequent acute exacerbations or COPD combined with asthma. Although ICS (181) is extremely effective in controlling COPD, patients should not use it arbitrarily and must follow strict physician’s instructions. Inappropriate use (e.g., too high a dose or too long a duration of use) may result in a range of side effects, such as oral candida infections, hoarseness, osteoporosis, and an increased risk of cataracts. Clinicians will adjust the dose of ICS based on the patient’s symptom severity, history of acute exacerbations, and other co-morbidities to ensure that treatment is both effective and safe (182). Patients with COPD receiving ICS should be evaluated regularly for response to therapy, such as monitoring improvement in symptoms, changes in lung function, and frequency of acute exacerbations, to help the physician determine if adjustments to the treatment plan are needed, including changes in ICS dosage or consideration of the addition of other therapies, such as a long-acting bronchodilator or a phosphodiesterase-4 (PDE4) inhibitor, for further symptom control and improvement in quality of life (183).
4.2 Adaptive Immunity and COPD treatment
4.2.1 Immunosuppressant
In the treatment of COPD, immunomodulatory strategies that focus on regulating T-cell and B-cell activity are expected to reduce the inflammatory response and oxidative stress in COPD, thereby alleviating symptoms and slowing disease progression. Immunosuppressive agents, including those that inhibit T-cell activity, may help reduce inflammatory lesions caused by over-activation of the immune response. Modulation of Th1/Th2/Th17/Treg balance is expected to reduce inflammation in COPD (184). Antibody drugs may also selectively interfere with B-cell activity, reducing antibody production and the release of inflammatory mediators. Overall, T cells and B cells play an important role in the pathogenesis and inflammatory process of COPD.123 Therefore, immunomodulatory and antioxidant therapies may be critical in the treatment of COPD. Balancing the immune response, suppressing hyperactivation of inflammation, and mitigating oxidative stress are expected to improve symptoms and quality of life in patients with COPD while slowing disease progression. This approach provides new ideas for personalized treatment in this field.
4.2.2 IcnRNA
Dexmedetomidine, an alpha2-adrenoceptor agonist, is mainly used for sedation during perioperative procedures, endotracheal intubation and mechanical ventilation, and has a protective effect against inflammatory lung injury. It was shown that lncRNA PACER was highly expressed in the serum of COPD patients (185). Subsequently, it was found that overexpression of PACER in rat alveolar epithelial cells enhanced cell proliferation and migration through activation of protein phosphatase 2 by establishing a COPD rat model. After treatment with dexmedetomidine, it reduced PACER expression and proliferation and migration ability of alveolar epithelial cells in COPD rats, which could help COPD treatment. Andrographolide is a common anti-inflammatory agent that reduces the expression of inflammatory cytokines induced by cigarette smoke as well as infections. A study found that elevated inflammatory factors such as IL-6 and IL-8 in human bronchial epithelial cells exposed to CSE activated downstream signal transducer and activator of transcription 3 (STAT3) (186), which up-regulated the expression of lncRNA HOTAIR and enhancer of zeste homologue 2 (EZH2) (187). Andrographolide reversed CSE-induced HB inflammation and epithelial-mesenchymal transition by lowering IL-6 levels, and it prevented CSE-induced lung inflammation and small airway remodeling in an animal model, suggesting that andrographolide has potential clinical applications for cigarette-induced pulmonary dysfunction and COPD.
4.2.3 Antimicrobial
Antimicrobial studies have found a strong association between acute exacerbations in COPD patients and bacterial infections (188), with common causative organisms including Catamoeba and Pneumococcus. Many drug studies are now beginning to consider the possibility of long-term application of low-dose macrolide antibiotics in the treatment of patients with chronic stable COPD. Studies have shown that this class of drugs is effective in anti-inflammatory and enhanced immunomodulation, which can greatly reduce the inflammatory mediators in the patient’s body and improve the patient’s exercise tolerance (189). However, even with the use of small doses of macrolide antibiotics, in the process of long-term medication, it is possible to develop drug resistance or cause an imbalance in the bacterial flora, and if the patient subsequently develops an acute attack, the use of antimicrobial drugs will be affected to a certain extent. For this reason, the use of this class of drugs in clinical treatment must be cautious. And at present, the time of clinical intervention is not yet specific, and its use time needs to be studied in depth.
4.3 Cytokine-targeted treatment strategies
In mixed-type COPD, concurrent neutrophilic and eosinophilic inflammation requires treatment strategies targeting multiple inflammatory pathways. IL-1β is believed to play a key role in the inflammatory response of COPD (1). In recent years, IL-1β inhibitors such as canakinumab and MEDI8968 have become a hot topic in COPD treatment research. Canakinumab is a humanized anti-IL-1β monoclonal antibody that specifically neutralizes IL-1β signaling, thereby inhibiting the inflammatory response. Preliminary studies indicate that it can improve lung function and pathological damage in COPD patients by reducing pulmonary inflammatory cell infiltration and inflammatory cytokine release, and it also demonstrates anti-inflammatory potential in autoimmune diseases (190). However, there is currently a lack of large-scale clinical trial data to fully validate its efficacy and safety in COPD. MEDI8968 is a fully humanized monoclonal antibody that blocks the activation of IL-1α and IL-1β by binding to the IL-1 receptor 1 (IL-1R1). A Phase II randomized, double-blind, placebo-controlled trial indicated that while the drug did not significantly reduce the rate of acute exacerbations or improve quality of life in moderate-to-severe COPD patients, it demonstrated good tolerability with no serious adverse events, suggesting potential for further investigation (191) (Figure 7).
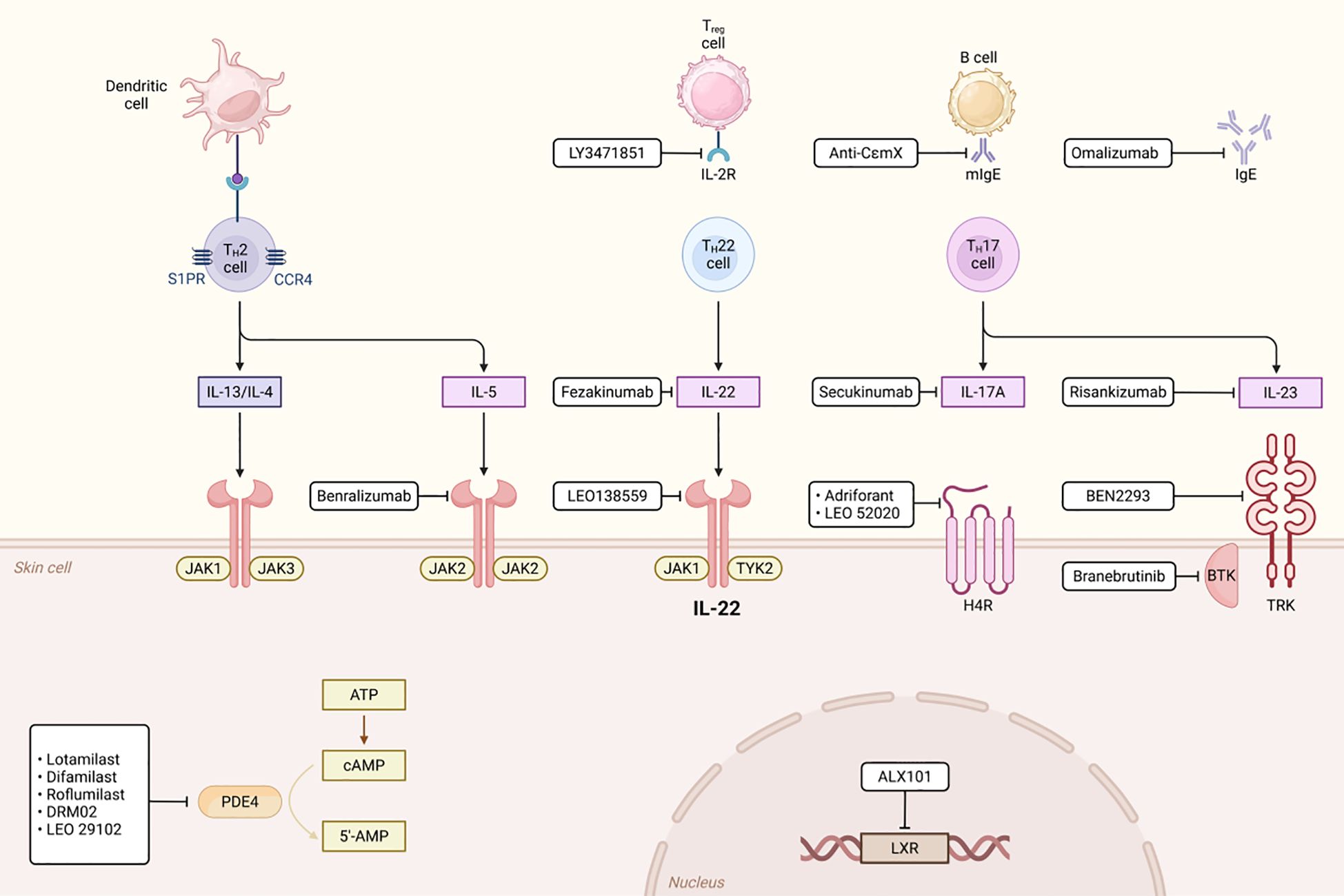
Figure 7. The Role of PDE4 in Immunity.
IL-17 and IL-6 inhibitors demonstrate therapeutic potential, offering new options for mixed-type patients. Specifically, IL-17 inhibitors may benefit neutrophil-dominant subtypes, whereas IL-6 inhibitors could target patients with activated IL-6TS pathways. Combination therapy may enhance anti-inflammatory effects, though further research is needed to evaluate synergy and safety (192). Blocking the IL-17 pathway effectively inhibits neutrophil-mediated inflammation, thereby alleviating chronic inflammation and acute exacerbations. Although preliminary studies support IL-17 inhibitors in COPD, larger clinical trials must confirm safety and efficacy (193). Suppressing the IL-6TS pathway may reduce neutrophilic inflammation and NET formation, potentially improving disease progression. For instance, tocilizumab (an IL-6R inhibitor) has proven effective in other inflammatory diseases, justifying further COPD investigation (166). However, current evidence remains exploratory, necessitating robust clinical validation. Future research should prioritize stratified treatment by inflammatory subtype to optimize individualized therapy.
Itepekimab is a human monoclonal antibody targeting IL-33. In a Phase 2 trial, it demonstrated potential in reducing exacerbations and improving lung function, particularly in former smokers with chronic obstructive pulmonary disease (COPD). For example, a Phase 2a trial showed that itepekimab reduced the annualized rate of exacerbations and improved FEV1 compared to placebo in former smokers. However, no significant benefits were observed in current smokers, highlighting the importance of patient stratification. An ongoing Phase 3 trial aims to confirm these findings in former smokers (2).
TSLP and IL-33 are key alarm molecules released by epithelial cells in response to environmental stimuli (such as allergens, cigarette smoke). They activate immune cells (such as dendritic cells, ILC2s) to promote type 2 (T2) and non-type 2 inflammatory responses, particularly in the eosinophilic COPD (eCOPD) subgroup, where TSLP and IL-33 expression are significantly elevated. ST2 is a functional receptor for IL-33, and its signaling pathway plays a crucial role in inflammation and fibrosis in COPD (25). Studies have shown that ST2+ILC2s are significantly increased in sputum from eCOPD patients and are positively correlated with elevated eosinophil percentages. Additionally, ST2 signaling is involved in modulating the production of T2 cytokines, such as IL-5 and IL-13, which play a key role in the pathophysiology of COPD (8). Therefore, blocking ST2 signaling may help inhibit T2 inflammation and fibrosis, thereby improving clinical symptoms in COPD patients.
Tezepelumab is a monoclonal antibody targeting TSLP, approved for the treatment of severe asthma and is being investigated in COPD. The TSLP and IL-33/ST2 pathways have partially overlapping yet independent regulatory roles in the inflammation and fibrosis of COPD. Studies have shown that simultaneously blocking TSLP and IL-33/ST2 signaling can more effectively suppress chronic T2 inflammation and fibrosis (51). In mouse models, combined blockade of TSLP, IL-25, and IL-33 significantly reduced pulmonary eosinophil infiltration and fibrosis. Therefore, a combined strategy of anti-ST2 and anti-TSLP blockade may provide a more comprehensive therapeutic effect for COPD patients (58).
Biological agents targeting interleukin-5 (IL-5), such as mepolizumab and benalizumab, have demonstrated significant clinical potential, but their application still faces numerous challenges. These drugs are primarily effective for COPD patients with eosinophilic inflammation, with efficacy concentrated in subgroups with higher baseline peripheral blood eosinophil counts (133). They only provide approximately 9%-12% improvement in reducing moderate-to-severe exacerbations, resulting in limited overall efficacy. Additionally, IL-5 inhibitors have minimal impact on lung function metrics such as FEV1, and some studies have even observed no improvement or a decline in lung function, suggesting they may struggle to reverse the core pathological change of airflow limitation. Another significant limitation lies in their single-target mechanism of action: the inflammatory network in COPD involves multiple cells and mediators, and blocking IL-5 alone may not fully suppress disease progression driven by non-eosinophilic inflammation (134). Furthermore, there is currently insufficient evidence regarding long-term safety and pharmacoeconomics, limiting their widespread application. Future research should focus on advancing precision medicine strategies, optimizing patient stratification based on biomarkers such as eosinophil counts to enhance treatment response rates. Concurrently, multi-target combination therapies should be explored, such as combining IL-5 inhibitors with anti-IL-4/IL-13 or anti-TSLP agents to synergistically regulate inflammatory pathways. Dosing regimens also require further optimization to balance efficacy and safety. Conducting long-term follow-up studies to assess sustained efficacy and adverse reactions, while identifying new biomarkers and targets, will be key to achieving personalized, efficient treatment.
4.4 NLRP3 inflammasome inhibitor
In recent years, the role of NLRP3 inflammasomes in the pathogenesis of COPD has been increasingly elucidated, emerging as a potential therapeutic target. NLRP3 inflammasome inhibitors, such as Dapantusin (MCC950), may offer new treatment options for COPD patients by inhibiting inflammasome activation. The NLRP3 inflammasome is a key component of the innate immune system, mediating inflammatory responses and pyroptosis. In COPD, excessive activation of the NLRP3 inflammasome triggers the release of inflammatory cytokines such as IL-1β and IL-18, exacerbating airway inflammation and oxidative stress (194). Studies have shown that NLRP3 expression is significantly elevated in the lung tissue and serum of COPD patients, confirming its key role in disease progression. MCC950, a highly selective NLRP3 inflammasome inhibitor, suppresses inflammasome assembly and activation. A study demonstrated that MCC950 significantly reduces the release of inflammatory cytokines by inhibiting NLRP3 inflammasome activation, thereby alleviating airway inflammation and oxidative stress (194), MCC950 also improves lung function in COPD model mice, reduces inflammatory cell infiltration, and mitigates airway wall damage. In an LPS-induced acute lung injury model, MCC950 significantly inhibited the accumulation of neutrophils and macrophages and reduced the levels of IL-1β and IL-18. These results suggest that MCC950 may provide a new therapeutic strategy for COPD patients by inhibiting NLRP3 inflammasome activation (128). Although MCC950 demonstrated promising effects in preclinical studies, its safety and efficacy in COPD patients require further validation. Evaluating the efficacy and safety of MCC950 in COPD patients, identifying biomarkers that can predict treatment response to MCC950, and exploring the combination of MCC950 with other anti-inflammatory drugs or antioxidants are priorities for future research.
4.5 scRNA-seq and CyTOF
Single-cell RNA sequencing (scRNA-seq) and mass cytometry (CyTOF) provide breakthrough tools for analyzing airway heterogeneity in COPD. scRNA-seq studies have revealed that airway basal cells in COPD patients exhibit a continuous molecular spectrum from health to disease, with stress genes (such as GADD45B/CITED2) significantly upregulated, potentially related to hypoxic adaptation; in the microenvironment of COPD combined with lung squamous cell carcinoma (LSCC), where tumor-associated macrophages (TAMs) and CD8+ T cell exhaustion markers are increased, while CD74+ tumor cells exhibit strong tumorigenicity and poor prognosis (195). High-dimensional protein analysis using CyTOF further revealed that in asthma-COPD overlap (ACO), AMs regulate the aging process via PPARγ; T cell subsets show Th1 cell imbalance, with inflammatory responses regulated by the NFIL3/Tim3 axis. The value of technological integration is highlighted in mechanism analysis: the combination of scRNA-seq and CyTOF in a mouse model confirmed that the HCK-mediated IL-17A/G-CSF/granulocyte axis is the core driver pathway of COPD inflammation and emphysema (175). Future efforts should focus on deepening target identification through single-cell multi-omics integration (transcriptome-proteome-epigenome), developing targeted therapies based on specific cell subpopulations (e.g., CD74+ tumor cells) and pathways (e.g., PPARγ), and ultimately achieving personalized clinical decision-making.
5 Discussion
Both innate and adaptive immune mechanisms underpin the chronic inflammatory characteristics of chronic obstructive pulmonary disease (COPD), and disruptions in these mechanisms play a key role in the pathogenesis of the disease. This complex immunopathological process involves coordinated interactions between macrophages, neutrophils, dendritic cells, and lymphocytes. Within the adaptive immune system, T lymphocytes (divided into CD4+ and CD8+ subpopulations) and B lymphocytes are the primary effector cells. Additionally, this process involves the participation of cytokines and signaling pathways. The redundancy of cytokine pathways constitutes the core bottleneck of monotherapy. Multiple factors (IL-17/TNF-α/IL-6/IL-8) amplify inflammatory responses through independent or overlapping signaling axes such as NF-κB, MAPK, and PI3K/AKT. For example, IL-17 mediates airway remodeling via the β-catenin/ACT1 pathway (175), while TNF-α drives tissue damage via NF-κB/MAPK. This complexity is further exacerbated by pathway interactions: NF-κB and MAPK interact at multiple nodes, and PI3K/AKT cross-talks with IL-17 signaling, leading to compensatory activation of other pathways when inhibiting a single pathway (e.g., using anti-IL-17 antibodies or NF-κB inhibitors), ultimately manifesting as effective animal models but limited clinical efficacy (196). Overcoming this challenge requires a multi-target strategy: combined targeting (such as dual inhibition of IL-17 and NF-κB) or using natural compounds (flavonoids/terpenoids) to simultaneously regulate the JAK3/STAT3/NF-κB and MAPK pathways. Future directions focus on three levels: elucidating pathway interaction networks (e.g., NF-κB-MAPK cross-nodal points); developing multi-target drugs based on network pharmacology-molecular docking; and customizing individualized treatment plans based on patient-specific pathway activity (e.g., IL-6/IL-17-dominant types).
IL-17 and IL-22 play a complex dual role in COPD, participating in both inflammatory responses and immune defense but are also closely associated with disease progression and acute exacerbations. During COPD progression, during bacterial and viral infections, IL-17 and IL-22 have distinct clinical significance. Therapeutic strategies targeting the IL-17/IL-22 pathway (such as IL-22 supplementation therapy) show promise in controlling acute exacerbations of COPD. Therefore, further exploration of therapeutic strategies targeting the IL-17/IL-22 pathway to improve clinical outcomes in COPD patients represents a promising future research direction. Cytokines and biomarkers in COPD play a significant role in assessing disease severity, predicting treatment response, and monitoring disease progression. For example, markers such as IL-33, suPAR, and miRNA can help distinguish between different stages and severities of the disease, while eosinophils and NLR can be used to predict treatment response and the risk of acute exacerbations. Future research should further validate the clinical utility of these biomarkers and explore their potential in personalized treatment. By integrating multiple biomarkers, it may be possible to develop more precise strategies for the diagnosis and treatment of COPD.
COPD is a highly heterogeneous disease, and its complex pathophysiological mechanisms and clinical manifestations pose significant challenges for clinical trial design. To improve trial efficiency and achieve personalized treatment, biomarkers and patient stratification strategies have become key tools. Among inflammation-related biomarkers, elevated levels of C-reactive protein (CRP) and fibrinogen are significantly associated with increased risk of acute exacerbations, making them useful for identifying high-risk patients and adjusting treatment; elevated white blood cell counts also indicate increased risk, particularly in patients with multiple coexisting inflammatory markers; and blood eosinophil counts are important biomarkers for predicting inhaled corticosteroids’ efficacy, with patients having higher levels potentially benefiting more significantly (197). Oxidative stress-related markers such as fibronectin 1 (FN1) are highly expressed in the serum and lung tissue of COPD patients and are associated with impaired lung function, potentially indicating disease progression (197); markers of imbalance between reactive oxygen species (ROS) and the antioxidant defense system can help assess disease progression and treatment response. Other prognostic markers include D-dimer (elevated levels associate with all-cause mortality) and red blood cell distribution width (RDW, associated with increased exacerbation risk, particularly useful for stratifying hypertensive patients) (132).
Patient stratification strategies should integrate clinical characteristics and biological mechanisms. Stratification based on clinical phenotypes (e.g., frequent exacerbation type, emphysema type) is limited due to phenotype overlap; whereas genotype-based stratification enables more precise targeted therapy. Combinations of multiple biomarkers (e.g., C-reactive protein, fibrinogen, and white blood cell count) significantly enhance stratification accuracy; specific proteins identified through plasma proteomics aid in early identification of rapidly progressive COPD. Functional tests such as the 6-minute walk distance (6MWD) are core stratification tools for predicting mortality, hospitalization rates, and acute exacerbation risk (198).
In trial design practice, biomarkers should optimize stratification aligned with individualized treatment goals in the latest GOLD guidelines (improving symptoms and reducing future risks). Targeted therapy trials should incorporate phenotype-specific biomarkers (e.g., for anti-inflammatory drugs) to precisely screen patients; eosinophil-directed ICS treatment strategies can be adjusted based on count levels to enhance efficacy while reducing side effects. Data-driven technologies such as machine learning models (SuStaIn) and network analysis can identify disease subtypes and progression trajectories, providing trial design support (175). Overall, the synergistic application of biomarkers and stratification strategies will drive precision development of COPD clinical trials. Future efforts should further validate biomarkers’ clinical utility and explore new technologies to advance precision medicine practices.
Clinical translation in COPD research faces multiple challenges, with the core issue being animal-human model mismatch. First, model design overlooks key clinical variables; despite COPD primarily affecting the elderly, studies predominantly use young animals (e.g., mice) (126). Gender factors are also neglected, with male animals dominating experiments, failing to reflect female patients’ unique pathophysiological mechanisms. Second, species differences exist in pathophysiological mechanisms. While cigarette smoke exposure models can simulate some human COPD features (e.g., inflammation, epithelial-mesenchymal transition), inflammatory biomarker expression (e.g., IL-6, IL-8) and regulatory mechanisms differ significantly between species, and human-specific molecular pathways remain poorly understood. Therapeutic translation bottlenecks are prominent: compounds effective in preclinical studies (e.g., andrographolide) often fail in human trials, as animal models cannot replicate patients’ individual differences and complex pathology (124). Technical limitations also apply. Diagnostic tools successful in animal studies (e.g., PET imaging for CCR2 assessment) have questionable human sensitivity/applicability. Differences in experimental parameters (e.g., drug dosage, exposure time) versus clinical practice further undermine result reliability. Gender differences in chemical reflex sensitivity during acute lung injury recovery are documented, but insufficiently explored in COPD models. Young animal models struggle to simulate elderly patients’ physiological decline and comorbidities. Optimizing model clinical relevance by incorporating elderly animals and balancing gender ratios; advancing technical clinical adaptability; and establishing humanized detection standards to bridge preclinical-clinical parameters represent critical future efforts.
Author contributions
HL: Conceptualization, Visualization, Writing – original draft. YW: Methodology, Writing – review & editing. HD: Data curation, Investigation, Writing – review & editing. YB: Investigation, Project administration, Writing – review & editing. XD: Writing – review & editing. YH: Formal Analysis, Writing – review & editing. QG: Validation, Writing – review & editing. PL: Resources, Supervision, Writing – review & editing. XL: Funding acquisition, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This research was supported by the National Natural Science Foundation of China (82372573, 82072551), the Shanghai Municipal Health Commission (2022XD044).
Acknowledgments
This study was supported by the Shanghai University of Traditional Chinese Medicine and the School of Rehabilitation Medicine, Shanghai University of Traditional Chinese Medicine.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Venkatesan P. GOLD COPD report: 2024 update [J. Lancet Respir Med. (2024) 12:15–6. doi: 10.1016/S2213-2600(23)00461-7
2. De Cunto G, Cavarra E, Bartalesi B, Lucattelli M, and Lungarella G. Innate immunity and cell surface receptors in the pathogenesis of COPD: insights from mouse smoking models [J. Int J Chronic Obstruct Pulm Dis. (2020) 15:1143–54. doi: 10.2147/COPD.S246219
3. Polverino F. Adaptive immune responses and protein homeostasis in COPD: the immunoproteasome [J. Eur Respir J. (2022) 59. doi: 10.1183/13993003.02557-2021
4. Detsika MG, Palamaris K, Dimopoulou I, Kotanidou A, and Orfanos SE. The complement cascade in lung injury and disease [J. Respir Res. (2024) 25:20. doi: 10.1186/s12931-023-02657-2
5. Arimura K, Sekine Y, Hiroshima K, Shimizu S, Shibata N, Kondo M, et al. PD-L1, FGFR1, PIK3CA, PTEN, and p16 expression in pulmonary emphysema and chronic obstructive pulmonary disease with resected lung squamous cell carcinoma [J. BMC Pulm Med. (2019) 19:169. doi: 10.1186/s12890-019-0913-8
6. Arora A and Singh A. Exploring the role of neutrophils in infectious and noninfectious pulmonary disorders [J. Int Rev Immunol. (2024) 43:41–61. doi: 10.1080/08830185.2023.2222769
7. Asare PF, Tran HB, Hurtado PR, Perkins GB, Nguyen P, Jersmann H, et al. Inhibition of LC3-associated phagocytosis in COPD and in response to cigarette smoke [J. Ther Adv Respir Dis. (2021) 15:17534666211039769. doi: 10.1177/17534666211039769
8. Blackburn JB, Li NF, Bartlett NW, and Richmond BW. An update in club cell biology and its potential relevance to chronic obstructive pulmonary disease [J. Am J Physiol Lung Cell Mol Physiol. (2023) 324:L652–l65. doi: 10.1152/ajplung.00192.2022
9. Bu T, Wang LF, and Yin YQ. How do innate immune cells contribute to airway remodeling in COPD progression? [J. Int J Chronic Obstruct Pulm Dis. (2020) 15:107–16. doi: 10.2147/COPD.S235054
10. Budde J and Skloot G. Aging and susceptibility to pulmonary disease [J. Compr Physiol. (2022) 12:3509–22. doi: 10.1002/j.2040-4603.2022.tb00221.x
11. Bulanda E and Wypych TP. Bypassing the gut-lung axis via microbial metabolites: implications for chronic respiratory diseases [J. Front Microbiol. (2022) 13:857418. doi: 10.3389/fmicb.2022.857418
12. Canè L, Poto R, Palestra F, Pirozzi M, Parashuraman S, Iacobucci I, et al. TSLP is localized in and released from human lung macrophages activated by T2-high and T2-low stimuli: relevance in asthma and COPD [J. Eur J Internal Med. (2024) 124:89–98. doi: 10.1016/j.ejim.2024.02.020
13. Castro ÍA, Yang Y, Gnazzo V, Kim DH, Van Dyken SJ, and López CB. Murine parainfluenza virus persists in lung innate immune cells sustaining chronic lung pathology [J. Nat Microbiol. (2024) 9:2803–16. doi: 10.1038/s41564-024-01805-8
14. Chen G, Mu Q, and Meng ZJ. Cigarette smoking contributes to th1/th2 cell dysfunction via the cytokine milieu in chronic obstructive pulmonary disease [J. Int J Chronic Obstruct Pulm Dis. (2023) 18:2027–38. doi: 10.2147/COPD.S426215
15. Chen J, Wang T, Li X, Gao L, Wang K, Cheng M, et al. DNA of neutrophil extracellular traps promote NF-κB-dependent autoimmunity via cGAS/TLR9 in chronic obstructive pulmonary disease [J. Signal Transduct Target Ther. (2024) 9:163. doi: 10.1038/s41392-024-01881-6
16. Cho WK, Lee CG, and Kim LK. Copd as a disease of immunosenescence [J. Yonsei Med J. (2019) 60:407–13. doi: 10.3349/ymj.2019.60.5.407
17. De Nuccio F, Piscitelli P, and Toraldo DM. Gut-lung microbiota interactions in chronic obstructive pulmonary disease (COPD): potential mechanisms driving progression to COPD and epidemiological data [J. Lung. (2022) 200:773–81. doi: 10.1007/s00408-022-00581-8
18. Devulder JV. Unveiling mechanisms of lung aging in COPD: A promising target for therapeutics development [J. Chin Med J Pulm Crit Care Med. (2024) 2:133–41. doi: 10.1016/j.pccm.2024.08.007
19. Du B, Fu Y, Han Y, Sun Q, Xu J, Yang Y, et al. The lung-gut crosstalk in respiratory and inflammatory bowel disease [J. Front Cell Infect Microbiol. (2023) 13:1218565. doi: 10.3389/fcimb.2023.1218565
20. Flayer CH, Linderholm AL, Ge MQ, Juarez M, Franzi L, Tham T, et al. COPD with elevated sputum group 2 innate lymphoid cells is characterized by severe disease [J. medRxiv: preprint Server Health Sci. (2024). doi: 10.1101/2023.11.21.23298837
21. Gajanayaka N, Dong SXM, Ali H, Iqbal S, Mookerjee A, Lawton DA, et al. TLR-4 Agonist Induces IFN-γ Production Selectively in Proinflammatory Human M1 Macrophages through the PI3K-mTOR- and JNK-MAPK-Activated p70S6K Pathway [J. J Immunol (Baltimore Md: 1950). (2021) 207:2310–24. doi: 10.4049/jimmunol.2001191
22. Gałecka E, Kumor-Kisielewska A, and Górski P. Association of serum deiodinase type 2 level with chronic obstructive pulmonary disease in the Polish population [J. Acta Biochim Polonica. (2019), 66(2). doi: 10.18388/abp.2018_2761
23. George L, Taylor AR, Esteve-Codina A, Soler Artigas M, Thun GA, Bates S, et al. Blood eosinophil count and airway epithelial transcriptome relationships in COPD versus asthma [J. Allergy. (2020) 75:370–80. doi: 10.1111/all.14016
24. Guan X, Yuan Y, Wang G, Zheng R, Zhang J, Dong B, et al. Ginsenoside Rg3 ameliorates acute exacerbation of COPD by suppressing neutrophil migration [J. Int Immunopharmacol. (2020) 83:106449. doi: 10.1016/j.intimp.2020.106449
25. Guo-Parke H, Linden D, Weldon S, Kidney JC, and Taggart CC. Mechanisms of virus-induced airway immunity dysfunction in the pathogenesis of COPD disease, progression, and exacerbation [J. Front Immunol. (2020) 11:1205. doi: 10.3389/fimmu.2020.01205
26. He F, Wang N, Yu X, Zheng Y, Liu Q, Chen Q, et al. GATA3/long noncoding RNA MHC-R regulates the immune activity of dendritic cells in chronic obstructive pulmonary disease induced by air pollution particulate matter [J. J Hazard Mater. (2022) 438:129459. doi: 10.1016/j.jhazmat.2022.129459
27. He Q, Li P, Han L, Yang C, Jiang M, Wang Y, et al. Revisiting airway epithelial dysfunction and mechanisms in chronic obstructive pulmonary disease: the role of mitochondrial damage [J. Am J Physiol Lung Cell Mol Physiol. (2024) 326:L754–l69. doi: 10.1152/ajplung.00362.2023
28. Higham A, Scott T, Li J, Gaskell R, Dikwa AB, Shah R, et al. Effects of corticosteroids on COPD lung macrophage phenotype and function [J. Clin Sci (London England: 1979). (2020) 134:751–63. doi: 10.1042/CS20191202
29. Ho T, Nichols M, Nair G, Radford K, Kjarsgaard M, Huang C, et al. Iron in airway macrophages and infective exacerbations of chronic obstructive pulmonary disease [J. Respir Res. (2022) 23:8. doi: 10.1186/s12931-022-01929-7
30. Hu T, Xu L, Jiang M, Zhang F, Li Q, Li Z, et al. N6-methyladenosine-methylomic landscape of lung tissues of mice with chronic obstructive pulmonary disease [J. Front Immunol. (2023) 14:1137195. doi: 10.3389/fimmu.2023.1137195
31. Huang X, Li Y, Guo X, Zhu Z, Kong X, Yu F, et al. Identification of differentially expressed genes and signaling pathways in chronic obstructive pulmonary disease via bioinformatic analysis [J. FEBS Open Bio. (2019) 9:1880–99. doi: 10.1002/2211-5463.12719
32. Huang X, Zhu Z, Guo X, and Kong X. The roles of microRNAs in the pathogenesis of chronic obstructive pulmonary disease [J. Int Immunopharmacol. (2019) 67:335–47. doi: 10.1016/j.intimp.2018.12.013
33. Huo SJ, Hu Y, Zeng XL, Bao HR, and Liu XJ. The effect of fine particles on the phagocytosis of alveolar macrophages potentially by Arp2/3 complex in a mouse model of chronic obstructive pulmonary disease] [J. Zhonghua jie he he hu xi za zhi = Zhonghua jiehe he huxi zazhi = Chin J Tuberc Respir Dis. (2019) 42:907–15.
34. Hwang JY, Silva-Sanchez A, Carragher DM, Garcia-Hernandez ML, Rangel-Moreno J, and Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) attenuates pulmonary pathology in a mouse model of allergic airway disease [J. Front Immunol. (2020) 11:570661. doi: 10.3389/fimmu.2020.570661
35. Ito JT, Cervilha DAB, Lourenço JD, Gonçalves NG, Volpini RA, Caldini EG, et al. Th17/Treg imbalance in COPD progression: A temporal analysis using a CS-induced model [J. PloS One. (2019) 14:e0209351. doi: 10.1371/journal.pone.0209351
36. Jairaman A and Prakriya M. Calcium signaling in airway epithelial cells: current understanding and implications for inflammatory airway disease [J. Arteriosc Thromb Vasc Biol. (2024) 44:772–83. doi: 10.1161/ATVBAHA.123.318339
37. Jia Y, He T, Wu D, Tong J, Zhu J, Li Z, et al. The treatment of Qibai Pingfei Capsule on chronic obstructive pulmonary disease may be mediated by Th17/Treg balance and gut-lung axis microbiota [J. J Trans Med. (2022) 20:281. doi: 10.1186/s12967-022-03481-w
38. Jogdand P, Siddhuraj P, Mori M, Sanden C, Jönsson J, Walls AF, et al. Eosinophils, basophils and type 2 immune microenvironments in COPD-affected lung tissue [J. Eur Respir J. (2020) 55. doi: 10.1183/13993003.00110-2019
39. Kavianfar A, Taherkhani H, Ahmadi A, Salimi M, Lanjanian H, and Masoudi-Nejad A. Restoring the epigenetic landscape of lung microbiome: potential therapeutic approach for chronic respiratory diseases [J. BMC Pulm Med. (2024) 24:2. doi: 10.1186/s12890-023-02789-7
40. Kayongo A, Nyiro B, Siddharthan T, Kirenga B, Checkley W, Lutaakome Joloba M, et al. Mechanisms of lung damage in tuberculosis: implications for chronic obstructive pulmonary disease [J. Front Cell Infect Microbiol. (2023) 13:1146571. doi: 10.3389/fcimb.2023.1146571
41. Kayongo A, Robertson NM, Siddharthan T, Ntayi ML, Ndawula JC, Sande OJ, et al. Airway microbiome-immune crosstalk in chronic obstructive pulmonary disease [J. Front Immunol. (2022) 13:1085551. doi: 10.3389/fimmu.2022.1085551
42. Kheradmand F, Zhang Y, and Corry DB. Contribution of adaptive immunity to human COPD and experimental models of emphysema [J. Physiol Rev. (2023) 103:1059–93. doi: 10.1152/physrev.00036.2021
43. Koné B, Pérez-Cruz M, Porte R, Hennegrave F, Carnoy C, Gosset P, et al. Boosting the IL-22 response using flagellin prevents bacterial infection in cigarette smoke-exposed mice [J. Clin Exp Immunol. (2020) 201:171–86. doi: 10.1111/cei.13445
44. Kotlyarov S. Involvement of the innate immune system in the pathogenesis of chronic obstructive pulmonary disease [J. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms23020985
45. Krotova K, Khodayari N, Oshins R, Aslanidi G, and Brantly ML. Neutrophil elastase promotes macrophage cell adhesion and cytokine production through the integrin-Src kinases pathway [J. Sci Rep. (2020) 10:15874. doi: 10.1038/s41598-020-72667-3
46. Leahy D, Grant C, Jackson A, Duff A, Tardiota N, Van Haeften J, et al. Discrimination of methionine sulfoxide and sulfone by human neutrophil elastase [J. Mol (Basel Switzerland). (2021) 26. doi: 10.3390/molecules26175344
47. Lee JW, Chun W, Lee HJ, Min JH, Kim SM, Seo JY, et al. The role of macrophages in the development of acute and chronic inflammatory lung diseases [J. Cells. (2021) 10. doi: 10.3390/cells10040897
48. Lee Y, Song J, Jeong Y, Choi E, Ahn C, and Jang W. Meta-analysis of single-cell RNA-sequencing data for depicting the transcriptomic landscape of chronic obstructive pulmonary disease [J. Comput Biol Med. (2023) 167:107685. doi: 10.1016/j.compbiomed.2023.107685
49. Li L, Li Z, Peng Y, Fu Y, Zhang R, Wen J, et al. Bletilla striata polysaccharide alleviates chronic obstructive pulmonary disease via modulating gut microbiota and NR1H4 expression in mice [J. Microb Pathogen. (2024) 193:106767. doi: 10.1016/j.micpath.2024.106767
50. Li Q, Hu Y, Chen Y, Lv Z, Wang J, An G, et al. IL-33 induces production of autoantibody against autologous respiratory epithelial cells: a potential mechanism for the pathogenesis of COPD [J. Immunology. (2019) 157:137–50. doi: 10.1111/imm.13054
51. Li R, Li J, and Zhou X. Lung microbiome: new insights into the pathogenesis of respiratory diseases [J. Signal Transduct Target Ther. (2024) 9:19. doi: 10.1038/s41392-023-01722-y
52. Li Y, Wang Y, Wu R, Li P, and Cheng Z. HTR2B as a novel biomarker of chronic obstructive pulmonary disease with lung squamous cell carcinoma [J. Sci Rep. (2024) 14:13206. doi: 10.1038/s41598-024-63896-x
53. Liang C, Shen Y, Xu Y, Liang Y, Qiu S, Tang H, et al. Dendritic cells promote the differentiation of ILCs into NCR(-)ILC3s in the lungs of mice exposed to cigarette smoke [J. Copd. (2024) 21:2389909. doi: 10.1080/15412555.2024.2389909
54. Liang Z, Long F, Deng K, Wang F, Xiao J, Yang Y, et al. Dissociation between airway and systemic autoantibody responses in chronic obstructive pulmonary disease [J. Ann Trans Med. (2020) 8:918. doi: 10.21037/atm-20-944
55. Liao K, Wang F, Xia C, Xu Z, Zhong S, Bi W, et al. The cGAS-STING pathway in COPD: targeting its role and therapeutic potential [J. Respir Res. (2024) 25:302. doi: 10.1186/s12931-024-02915-x
56. Lin J, Xia H, Yu J, Wang Y, Wang H, Xie D, et al. circADAMTS6 via stabilizing CAMK2A is involved in smoking-induced emphysema through driving M2 macrophage polarization [J. Environ Int. (2024) 190:108832. doi: 10.1016/j.envint.2024.108832
57. Lin J, Xue Y, Su W, Zhang Z, Wei Q, Huang T, et al. Identification of dysregulated mechanisms and candidate gene markers in chronic obstructive pulmonary disease [J. Int J Chronic Obstruct Pulm Dis. (2022) 17:475–87. doi: 10.2147/COPD.S349694
58. Lin WC and Fessler MB. Regulatory mechanisms of neutrophil migration from the circulation to the airspace [J. Cell Mol Life Sci: CMLS. (2021) 78:4095–124. doi: 10.1007/s00018-021-03768-z
59. Lu Y, Deng M, Yin Y, Hou G, and Zhou X. Global trends in research regarding macrophages associated with chronic obstructive pulmonary disease: A bibliometric analysis from 2011 to 2022 [J. Int J Chronic Obstruct Pulm Dis. (2023) 18:2163–77. doi: 10.2147/COPD.S419634
60. Luo X, Zeng W, Tang J, Liu W, Yang J, Chen H, et al. Multi-modal transcriptomic analysis reveals metabolic dysregulation and immune responses in chronic obstructive pulmonary disease [J. Sci Rep. (2024) 14:22699. doi: 10.1038/s41598-024-71773-w
61. Matarazzo L, Hernandez Santana YE, Walsh PT, and Fallon PG. The IL-1 cytokine family as custodians of barrier immunity [J. Cytokine. (2022) 154:155890. doi: 10.1016/j.cyto.2022.155890
62. Mei D, Liao W, Gan PXL, Tran QTN, Chan C, Heng CKM, et al. Angiotensin II type-2 receptor activation in alveolar macrophages mediates protection against cigarette smoke-induced chronic obstructive pulmonary disease [J. Pharmacol Res. (2022) 184:106469. doi: 10.1016/j.phrs.2022.106469
63. Meng H, Long Q, Wang R, Zhou X, Su H, Wang T, et al. Identification of the key immune-related genes in chronic obstructive pulmonary disease based on immune infiltration analysis [J. Int J Chronic Obstruct Pulm Dis. (2022) 17:13–24. doi: 10.2147/COPD.S333251
64. Moradi S, Jarrahi E, Ahmadi A, Salimian J, Karimi M, Zarei A, et al. PI3K signalling in chronic obstructive pulmonary disease and opportunities for therapy [J. J Pathol. (2021) 254:505–18. doi: 10.1002/path.5696
65. Morán G, Uberti B, and Quiroga J. Role of cellular metabolism in the formation of neutrophil extracellular traps in airway diseases [J. Front Immunol. (2022) 13:850416. doi: 10.3389/fimmu.2022.850416
66. Naessens T, Morias Y, Hamrud E, Gehrmann U, Budida R, Mattsson J, et al. Human lung conventional dendritic cells orchestrate lymphoid neogenesis during chronic obstructive pulmonary disease [J. Am J Respir Crit Care Med. (2020) 202:535–48. doi: 10.1164/rccm.201906-1123OC
67. Nedeva D, Kowal K, Mihaicuta S, Guidos Fogelbach G, Steiropoulos P, Jose Chong-Neto H, et al. Epithelial alarmins: a new target to treat chronic respiratory diseases [J. Expert Rev Respir Med. (2023) 17:773–86. doi: 10.1080/17476348.2023.2262920
68. Norman KC, Freeman CM, Bidthanapally NS, Han MK, Martinez FJ, Curtis JL, et al. Inference of cellular immune environments in sputum and peripheral blood associated with acute exacerbations of COPD [J. Cell Mol Bioeng. (2019) 12:165–77. doi: 10.1007/s12195-019-00567-2
69. Odajiu I, Covantsev S, Sivapalan P, Mathioudakis AG, Jensen JS, Davidescu EI, et al. Peripheral neuropathy: A neglected cause of disability in COPD - A narrative review [J. Respir Med. (2022) 201:106952. doi: 10.1016/j.rmed.2022.106952
70. Oliveira FMS, Kraemer L, Vieira-Santos F, Leal-Silva T, Gazzinelli-Guimarães AC, Lopes CA, et al. The long-lasting Ascaris suum antigens in the lungs shapes the tissue adaptation modifying the pulmonary architecture and immune response after infection in mice [J. Microb Pathogen. (2024) 186:106483. doi: 10.1016/j.micpath.2023.106483
71. Ouyang Y, Liu J, Wen S, Xu Y, Zhang Z, Pi Y, et al. Association between chronic obstructive pulmonary disease and periodontitis: The common role of innate immune cells? [J. Cytokine. (2022) 158:155982. doi: 10.1016/j.cyto.2022.155982
72. Panda K, Chinnapaiyan S, Rahman MS, Santiago MJ, Black SM, and Unwalla HJ. Circadian-coupled genes expression and regulation in HIV-associated chronic obstructive pulmonary disease (COPD) and lung comorbidities [J. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24119140
73. Pilaczyńska-Cemel M, Gołda R, Dąbrowska A, and Przybylski G. Analysis of the level of selected parameters of inflammation, circulating immune complexes, and related indicators (neutrophil/lymphocyte, platelet/lymphocyte, CRP/CIC) in patients with obstructive diseases [J. Central European J Immunol. (2019) 44:292–8. doi: 10.5114/ceji.2019.87498
74. Prasad C, Dasgupta D, Tripathi A, Steele N, Pyaram K, Sundar IK, et al. Cadmium-induced lung injury disrupts immune cell homeostasis in the secondary lymphoid organs in mice [J. Toxicology. (2024) 509:153971. doi: 10.1016/j.tox.2024.153971
75. Puttur F, Gregory LG, and Lloyd CM. Airway macrophages as the guardians of tissue repair in the lung [J. Immunol Cell Biol. (2019) 97:246–57. doi: 10.1111/imcb.12235
76. Qi Y, Yan Y, Tang D, Han J, Zhu X, Cui M, et al. Inflammatory and immune mechanisms in COPD: current status and therapeutic prospects [J. J Inflammation Res. (2024) 17:6603–18. doi: 10.2147/JIR.S478568
77. Raby KL, Michaeloudes C, Tonkin J, Chung KF, and Bhavsar PK. Mechanisms of airway epithelial injury and abnormal repair in asthma and COPD [J. Front Immunol. (2023) 14:1201658. doi: 10.3389/fimmu.2023.1201658
78. Raftery AL, Tsantikos E, Harris NL, and Hibbs ML. Links between inflammatory bowel disease and chronic obstructive pulmonary disease [J. Front Immunol. (2020) 11:2144. doi: 10.3389/fimmu.2020.02144
79. Ran B, Qin J, Wu Y, and Wen F. Causal role of immune cells in chronic obstructive pulmonary disease: Mendelian randomization study [J. Expert Rev Clin Immunol. (2024) 20:413–21. doi: 10.1080/1744666X.2023.2295987
80. Rao Y, Le Y, Xiong J, Pei Y, and Sun Y. NK cells in the pathogenesis of chronic obstructive pulmonary disease [J. Front Immunol. (2021) 12:666045. doi: 10.3389/fimmu.2021.666045
81. Richmond BW, Mansouri S, Serezani A, Novitskiy S, Blackburn JB, Du RH, et al. Monocyte-derived dendritic cells link localized secretory IgA deficiency to adaptive immune activation in COPD [J. Mucosal Immunol. (2021) 14:431–42. doi: 10.1038/s41385-020-00344-9
82. Riera-Martínez L, Cànaves-Gómez L, Iglesias A, Martin-Medina A, and Cosío BG. The role of IL-33/ST2 in COPD and its future as an antibody therapy [J. Int J Mol Sci. (2023) 24. doi: 10.3390/ijms24108702
83. Salih MM, Robinson EK, Malekos E, Perez E, Capili A, Kim K, et al. LincRNA-Cox2 regulates smoke-induced inflammation in murine macrophages [J. Am J Respir Cell Mol Biol. (2023) 68:511–22. doi: 10.1165/rcmb.2022-0413OC
84. Shah BK, Singh B, Wang Y, Xie S, and Wang C. Mucus hypersecretion in chronic obstructive pulmonary disease and its treatment [J. Mediators Inflammation. (2023) 2023:8840594. doi: 10.1155/2023/8840594
85. Shaikh SB, Goracci C, Tjitropranoto A, and Rahman I. Impact of aging on immune function in the pathogenesis of pulmonary diseases: potential for therapeutic targets [J. Expert Rev Respir Med. (2023) 17:351–64. doi: 10.1080/17476348.2023.2205127
86. Shanmugam G, Rakshit S, and Sarkar K. Hdac inhibitors: Targets for tumor therapy, immune modulation and lung diseases [J. Trans Oncol. (2022) 16:101312. doi: 10.1016/j.tranon.2021.101312
87. Sidletskaya K, Vitkina T, and Denisenko Y. The role of toll-like receptors 2 and 4 in the pathogenesis of chronic obstructive pulmonary disease [J. Int J Chronic Obstruct Pulm Dis. (2020) 15:1481–93. doi: 10.2147/COPD.S249131
88. Sidletskaya KA, Vitkina TI, Denisenko YK, and Mineeva EE. Role of toll-like receptor 2 in regulation of T-helper immune response in chronic obstructive pulmonary disease [J. Can Respir J. (2021) 2021:5596095. doi: 10.1155/2021/5596095
89. Singanayagam A, Footitt J, Marczynski M, Radicioni G, Cross MT, and Finney LJ. Airway mucins promote immunopathology in virus-exacerbated chronic obstructive pulmonary disease [J. J Clin Invest. (2022) 132. doi: 10.1172/JCI120901
90. Singh D, Bassi M, Balzano D, Lucci G, Emirova A, Anna Nandeuil M, et al. COPD patients with chronic bronchitis and higher sputum eosinophil counts show increased type-2 and PDE4 gene expression in sputum [J. J Cell Mol Med. (2021) 25:905–18. doi: 10.1111/jcmm.16146
91. Song Z, Meng Y, Fricker M, Li X, Tian H, Tan Y, et al. The role of gut-lung axis in COPD: Pathogenesis, immune response, and prospective treatment [J. Heliyon. (2024) 10:e30612. doi: 10.1016/j.heliyon.2024.e30612
92. Stoleriu MG, Ansari M, Strunz M, Schamberger A, Heydarian M, Ding Y, et al. COPD basal cells are primed towards secretory to multiciliated cell imbalance driving increased resilience to environmental stressors [J. Thorax. (2024) 79:524–37. doi: 10.1136/thorax-2022-219958
93. Tanno A, Fujino N, Yamada M, Sugiura H, Hirano T, Tanaka R, et al. Decreased expression of a phagocytic receptor Siglec-1 on alveolar macrophages in chronic obstructive pulmonary disease [J. Respir Res. (2020) 21:30. doi: 10.1186/s12931-020-1297-2
94. Trivedi A, Khan MA, Bade G, and Talwar A. Orchestration of neutrophil extracellular traps (Nets), a unique innate immune function during chronic obstructive pulmonary disease (COPD) development [J. Biomedicines. (2021) 9. doi: 10.3390/biomedicines9010053
95. Unninayar D, Abdallah SJ, Cameron DW, and Cowan J. Polyvalent immunoglobulin as a potential treatment option for patients with recurrent COPD exacerbations [J. Int J Chronic Obstruct Pulm Dis. (2021) 16:545–52. doi: 10.2147/COPD.S283832
96. Vasudevan S, Vásquez JJ, Chen W, Aguilar-Rodriguez B, Niemi EC, Zeng S, et al. Lower PDL1, PDL2, and AXL expression on lung myeloid cells suggests inflammatory bias in smoking and chronic obstructive pulmonary disease [J. Am J Respir Cell Mol Biol. (2020) 63:780–93. doi: 10.1165/rcmb.2020-0085OC
97. Wan R, Srikaram P, Xie S, Chen Q, Hu C, Wan M, et al. PPARγ attenuates cellular senescence of alveolar macrophages in asthma-COPD overlap [J. Respir Res. (2024) 25:174. doi: 10.1186/s12931-024-02790-6
98. Wang C, Wei Z, Han Z, Wang J, Zhang X, Wang Y, et al. Neutrophil extracellular traps promote cadmium chloride-induced lung injury in mice [J. Environ pollut (Barking Essex: 1987). (2019) 254:113021. doi: 10.1016/j.envpol.2019.113021
99. Wang J, Xia B, Ma R, and Ye Q. Comprehensive analysis of a competing endogenous RNA co-expression network in chronic obstructive pulmonary disease [J. Int J Chronic Obstruct Pulm Dis. (2023) 18:2417–29. doi: 10.2147/COPD.S431041
100. Wang L, Yu Q, Xiao J, Chen Q, Fang M, and Zhao H. Cigarette Smoke Extract-Treated Mouse Airway Epithelial Cells-Derived Exosomal LncRNA MEG3 Promotes M1 Macrophage Polarization and Pyroptosis in Chronic Obstructive Pulmonary Disease by Upregulating TREM-1 via m(6)A Methylation [J. Immune Netw. (2024) 24:e3. doi: 10.4110/in.2024.24.e3
101. Wang X, Gao Y, Zhu J, Zhang X, Wu F, Yang Q, et al. Bioinformatics analysis of the molecular mechanism of Qibaipingfei Capsule in the regulation of chronic obstructive pulmonary disease related immune cells] [J. Xi bao yu fen zi mian yi xue za zhi = Chin J Cell Mol Immunol. (2022) 38:961–6.
102. Wang X, Zhu J, Gao Y, Wu F, Yang Q, Tong J, et al. Liuwei Buqi capsule modulates immune function by targeting multiple immune cell subsets in lung tissue of patients with COPD] [J. Nan fang yi ke da xue xue bao = J South Med Univ. (2021) 41:1492–500. doi: 10.12122/j.issn.1673-4254.2021.10.07
103. Wang X, Zhu J, Tong J, Wu F, Gao Y, Wang X, et al. Bioinformatical analysis of differentially expressed genes in alveolar macrophages of chronic obstructive pulmonary disease(COPD)] [J. Xi bao yu fen zi mian yi xue za zhi = Chin J Cell Mol Immunol. (2020) 36:961–6.
104. Wang YQ, Liao Q, Tang SH, Cai Z, and Zhang HC. Gubenzhike recipe ameliorates respiratory mucosal immunity in mice with chronic obstructive pulmonary disease through upregulation of the γδT lymphocytes and KGF levels [J. Evidence-Based Complement Altern Med: eCAM. (2020) 2020:3056797. doi: 10.1155/2020/3056797
105. Wang Z, Liang W, Ma C, Wang J, Gao X, and Wei L. Macrophages inhibit ciliary protein levels by secreting BMP-2 leading to airway epithelial remodeling under cigarette smoke exposure [J. Front Mol Biosci. (2021) 8:663987. doi: 10.3389/fmolb.2021.663987
106. Wen H, Luo Y, Ge P, Lan B, Dong X, Zhang G, et al. Research progress on molecular mechanism of transcription factor C/EBPβ in lung diseases] [J. Zhonghua wei zhong bing ji jiu yi xue. (2022) 34:875–80. doi: 10.3760/cma.j.cn121430-20220809-00731
107. Wu TJ, Wang SH, Chen ES, Tsai HH, Chang YC, Tseng YH, et al. Loss of core-fucosylation of SPARC impairs collagen binding and contributes to COPD [J. Cell Mol Life Sci: CMLS. (2022) 79:348. doi: 10.1007/s00018-022-04381-4
108. Wu XC, Liu Y, and Bai ZP. Compound Tinglizi Decoction intervenes COPD-associated pulmonary hypertension through regulation of HMGB1-mediated pyroptosis and immune imbalance] [J. Zhongguo Zhong yao za zhi = Zhongguo zhongyao zazhi = China J Chin Mater Med. (2023) 48:3055–65. doi: 10.19540/j.cnki.cjcmm.20221014.702
109. Wu Y, Ma M, Choi W, Xu W, and Dong J. Identification of immune-related gene signatures for chronic obstructive pulmonary disease with metabolic syndrome: evidence from integrated bulk and single-cell RNA sequencing data [J. Int Immunol. (2024) 36:17–32. doi: 10.1093/intimm/dxad043
110. Xiao X, Ding Z, Shi Y, and Zhang Q. Causal role of immune cells in chronic obstructive pulmonary disease: A two-sample Mendelian randomization study [J. Copd. (2024) 21:2327352. doi: 10.1080/15412555.2024.2327352
111. Xie B, Dai Z, Jiang C, Gao X, Yang S, Peng M, et al. ZC3H13 promotes ITGA6 m(6)A modification for chronic obstructive pulmonary disease progression [J. Cell Signal. (2024) 120:111190. doi: 10.1016/j.cellsig.2024.111190
112. Xie S, Yan P, Yao C, Yan X, Huo Y, Zhang J, et al. Efficacy and safety of Xuebijing injection and its influence on immunomodulation in acute exacerbations of chronic obstructive pulmonary disease: study protocol for a randomized controlled trial [J. Trials. (2019) 20:136. doi: 10.1186/s13063-019-3204-z
113. Yang D, Guo X, Huang T, and Liu C. The role of group 3 innate lymphoid cells in lung infection and immunity [J. Front Cell Infect Microbiol. (2021) 11:586471. doi: 10.3389/fcimb.2021.586471
114. Yu H, Guo W, Liu Y, and Wang Y. Immune characteristics analysis and transcriptional regulation prediction based on gene signatures of chronic obstructive pulmonary disease [J. Int J Chronic Obstruct Pulm Dis. (2021) 16:3027–39. doi: 10.2147/COPD.S325328
115. Yuan R, Yu J, Jiao Z, Li J, Wu F, Yan R, et al. The roles of tissue-resident memory T cells in lung diseases [J. Front Immunol. (2021) 12:710375. doi: 10.3389/fimmu.2021.710375
116. Zhang F, Cui Y, Zhang T, and Yin W. Epigenetic regulation of macrophage activation in chronic obstructive pulmonary disease [J. Front Immunol. (2024) 15:1445372. doi: 10.3389/fimmu.2024.1445372
117. Zhang J, Liu J, Xu S, Yu X, Zhang Y, Li X, et al. Bioinformatics analyses of the pathogenesis and new biomarkers of chronic obstructive pulmonary disease [J. Medicine. (2021) 100:e27737. doi: 10.1097/MD.0000000000027737
118. Zhang X, Li X, Ma W, Liu F, Huang P, Wei L, et al. Astragaloside IV restores Th17/Treg balance via inhibiting CXCR4 to improve chronic obstructive pulmonary disease [J. Immunopharmacol Immunotoxicol. (2023) 45:682–91. doi: 10.1080/08923973.2023.2228479
119. Zhao C, Pu W, Niu M, Wazir J, Song S, Wei L, et al. Respiratory exposure to PM2.5 soluble extract induced chronic lung injury by disturbing the phagocytosis function of macrophage [J. Environ Sci pollut Res Int. (2022) 29:13983–97. doi: 10.1007/s11356-021-16797-9
120. Zhao S, Lin C, Yang T, Qian X, Lu J, and Cheng J. Expression of long non-coding RNA LUCAT1 in patients with chronic obstructive pulmonary disease and its potential functions in regulating cigarette smoke extract-induced 16HBE cell proliferation and apoptosis [J. J Clin Lab Anal. (2021) 35:e23823. doi: 10.1002/jcla.23823
121. Zhao Y, Li M, Yang Y, Wu T, Huang Q, Wu Q, et al. Identification of macrophage polarization-related genes as biomarkers of chronic obstructive pulmonary disease based on bioinformatics analyses [J. BioMed Res Int. (2021) 2021:9921012. doi: 10.1155/2021/9921012
122. Zhu Y and Chang D. Interactions between the lung microbiome and host immunity in chronic obstructive pulmonary disease [J. Chronic Dis Trans Med. (2023) 9:104–21. doi: 10.1002/cdt3.66
123. Zou H, Zhu N, and Li S. The emerging role of dipeptidyl-peptidase-4 as a therapeutic target in lung disease [J. Expert Opin Ther Targets. (2020) 24:147–53. doi: 10.1080/14728222.2020.1721468
124. Zoulikha M, Xiao Q, Boafo GF, Sallam MA, Chen Z, and He W. Pulmonary delivery of siRNA against acute lung injury/acute respiratory distress syndrome [J. Acta Pharm Sin B. (2022) 12:600–20. doi: 10.1016/j.apsb.2021.08.009
125. Liu J, Zhang Z, Yang Y, Di T, Wu Y, and BIan T. NCOA4-mediated ferroptosis in bronchial epithelial cells promotes macrophage M2 polarization in COPD emphysema [J. Int J Chronic Obstruct Pulm Dis. (2022) 17:667–81. doi: 10.2147/COPD.S354896
126. Kim GD, Lim EY, and Shin HS. Macrophage polarization and functions in pathogenesis of chronic obstructive pulmonary disease [J. Int J Mol Sci. (2024), 25(11). doi: 10.3390/ijms25115631
127. Li Y, Xiao G, Fu X, Luo X, Yang F, Li Y, et al. CH25H/25-HC promotes pulmonary fibrosis via AMPK/STAT6 pathway-dependent M2 macrophage polarization in COPD [J. Immunobiology. (2025) 230:152908. doi: 10.1016/j.imbio.2025.152908
128. Ryan EM, Sadiku P, Coelho P, Watts ER, Zhang A, Howden AJM, et al. NRF2 activation reprograms defects in oxidative metabolism to restore macrophage function in chronic obstructive pulmonary disease [J. Am J Respir Crit Care Med. (2023) 207:998–1011. doi: 10.1164/rccm.202203-0482OC
129. Ritzmann F, Lunding LP, Bals R, Wegmann M, and Beisswenger C. IL-17 cytokines and chronic lung diseases [J. Cells. (2022) 11. doi: 10.3390/cells11142132
130. De Pasquale C, Campana S, Bonaccorsi I, Carrega P, and Ferlazzo G. ILC in chronic inflammation, cancer and targeting with biologicals [J. Mol Aspects Med. (2021) 80:100963. doi: 10.1016/j.mam.2021.100963
131. Chen J, Wang X, Schmalen A, Haines S, Wolff M, Ma H, et al. Antiviral CD8+ T-cell immune responses are impaired by cigarette smoke and in COPD [J. Eur Respir J. (2023) 62. doi: 10.1183/13993003.01374-2022
132. Blomme EE, Provoost S, De Smet EG, De Grove KC, Van Eeckhoutte HP, De Volder J, et al. Quantification and role of innate lymphoid cell subsets in Chronic Obstructive Pulmonary Disease [J. Clin Transl Immunol. (2021) 10:e1287. doi: 10.1002/cti2.1287
133. Gutiérrez-Romero KJ, Falfán-Valencia R, Ramírez-Venegas A, Hernández-Zenteno RDJ, Flores-Trujillo F, Sansores-Martínez R, et al. Altered levels of IFN-γ, IL-4, and IL-5 depend on the TLR4 rs4986790 genotype in COPD smokers but not those exposed to biomass-burning smoke [J. Front Immunol. (2024) 15:1411408. doi: 10.3389/fimmu.2024.1411408
134. Cazzola M, Hanania NA, Page CP, and Matera MG. Novel anti-inflammatory approaches to COPD [J. Int J Chronic Obstruct Pulm Dis. (2023) 18:1333–52. doi: 10.2147/COPD.S419056
135. Mori M, Clausson CM, Sanden C, Jönsson J, Andersson CK, Siddhuraj P, et al. Expansion of phenotypically altered dendritic cell populations in the small airways and alveolar parenchyma in patients with chronic obstructive pulmonary disease [J. J Innate Immun. (2023) 15:188–203. doi: 10.1159/000526080
136. Colombo SAP, Brown SL, Hepworth MR, Hankinson J, Granato F, Kitchen SJ, et al. Comparative phenotype of circulating versus tissue immune cells in human lung and blood compartments during health and disease [J. Discov Immunol. (2023) 2:kyad009. doi: 10.1093/discim/kyad009
137. Graf J, Trautmann-Rodriguez M, Sabnis S, Kloxin AM, and Fromen CA. On the path to predicting immune responses in the lung: Modeling the pulmonary innate immune system at the air-liquid interface (ALI) [J. Eur J Pharm Sci: Off J Eur Fed Pharm Sci. (2023) 191:106596. doi: 10.1016/j.ejps.2023.106596
138. Beyeler S, Steiner S, Wotzkow C, Tschanz SA, Adhanom Sengal A, Wick P, et al. Multi-walled carbon nanotubes activate and shift polarization of pulmonary macrophages and dendritic cells in an in vivo model of chronic obstructive lung disease [J. Nanotoxicology. (2020) 14:77–96. doi: 10.1080/17435390.2019.1663954
139. Pu J, Xu J, Chen L, Zhou H, Cao W, Hao B, et al. Exposure to biomass smoke induces pulmonary Th17 cell differentiation by activating TLR2 on dendritic cells in a COPD rat model [J. Toxicol Lett. (2021) 348:28–39. doi: 10.1016/j.toxlet.2021.05.010
140. Lan C-C, Su W-L, Yang M-C, et al. Predictive role of neutrophil-percentage-to-albumin, neutrophil-to-lymphocyte and eosinophil-to-lymphocyte ratios for mortality in patients with COPD: Evidence from NHANES 2011–2018 [J. Respirol (Carlton Vic). (2023) 28:1136–46. doi: 10.1111/resp.14589
141. Bhatt SP, Rabe KF, Hanania NA, Vogelmeier CF, Bafadhel M, Christenson SA, et al. Dupilumab for COPD with blood eosinophil evidence of type 2 inflammation [J. New Engl J Med. (2024) 390:2274–83. doi: 10.1056/NEJMoa2401304
142. Metzemaekers M, Gouwy M, and Proost P. Neutrophil chemoattractant receptors in health and disease: double-edged swords [J. Cell Mol Immunol. (2020) 17:433–50. doi: 10.1038/s41423-020-0412-0
143. Cazzola M, Calzetta L, Rogliani P, et al. Emerging anti-inflammatory COPD treatments: potential cardiovascular impacts [J. Int J Chronic Obstruct Pulm Dis. (2024) 19:2481–95. doi: 10.2147/COPD.S498255
144. Katsoulis O, Toussaint M, Jackson MM, Mallia P, Footitt J, Mincham KT, et al. Neutrophil extracellular traps promote immunopathogenesis of virus-induced COPD exacerbations [J. Nat Commun. (2024) 15:5766. doi: 10.1038/s41467-024-50197-0
145. Kelly-Robinson GA, Reihill JA, Lundy FT, McGarvey LP, Lockhart JC, Litherland GJ, et al. The serpin superfamily and their role in the regulation and dysfunction of serine protease activity in COPD and other chronic lung diseases [J. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22126351
146. Gueçamburu M and Zysman M. COPD and eosinophils] [J. Rev Des Maladies Respiratoires. (2022) 39:685–97. doi: 10.2147/COPD.S498255
147. Bhatt SP, Rabe KF, Hanania NA, Vogelmeier CF, Cole J, Bafadhel M, et al. Dupilumab for COPD with type 2 inflammation indicated by eosinophil counts [J. New Engl J Med. (2023) 389:205–14. doi: 10.1056/NEJMoa2303951
148. Lea S, Higham A, Beech A, and Singh D. How inhaled corticosteroids target inflammation in COPD [J. Eur Respir Rev: an Off J Eur Respir Soc. (2023) 32. doi: 10.1183/16000617.0084-2023
149. Caminati M, Vatrella A, Rogliani P, Carpagnano E, Spanevello A, and Senna G. Tezepelumab for severe asthma: elevating current practice to recognize epithelial driven profiles [J. Respir Res. (2024) 25:367. doi: 10.1186/s12931-024-02998-6
150. Strickson S, Houslay KF, Negri VA, Ohne Y, Ottosson T, Dodd RB, et al. Oxidised IL-33 drives COPD epithelial pathogenesis via ST2-independent RAGE/EGFR signalling complex [J. Eur Respir J. (2023) 62. doi: 10.1056/NEJMoa2401304
151. Rabe KF, Rennard S, Martinez FJ, Celli BR, Singh D, Papi A, et al. Targeting type 2 inflammation and epithelial alarmins in chronic obstructive pulmonary disease: A biologics outlook [J. Am J Respir Crit Care Med. (2023) 208:395–405. doi: 10.1164/rccm.202303-0455CI
152. Dwyer DF, Ordovas-Montanes J, Allon SJ, Buchheit KM, Vukovic M, Derakhshan T, et al. Human airway mast cells proliferate and acquire distinct inflammation-driven phenotypes during type 2 inflammation [J. Sci Immunol. (2021) 6. doi: 10.1126/sciimmunol.abb7221
153. Calderon AA, Dimond C, Choy DF, Pappu R, Grimbaldeston MA, Mohan D, et al. Targeting interleukin-33 and thymic stromal lymphopoietin pathways for novel pulmonary therapeutics in asthma and COPD [J. Eur Respir Rev: an Off J Eur Respir Soc. (2023) 32. doi: 10.1183/16000617.0144-2022
154. Pei Y, Zhang J, Qu J, Rao Y, Li D, Gai X, et al. Complement component 3 protects human bronchial epithelial cells from cigarette smoke-induced oxidative stress and prevents incessant apoptosis [J. Front Immunol. (2022) 13:1035930. doi: 10.3389/fimmu.2022.1035930
155. Lee P-H, Park S, Lee Y-G, Choi SM, An MH, and Jang AS. The impact of environmental pollutants on barrier dysfunction in respiratory disease [J. Allergy Asthma Immunol Res. (2021) 13:850–62. doi: 10.4168/aair.2021.13.6.850
156. Villaseñor-Altamirano AB, Jain D, Jeong Y, Menon JA, Kamiya M, Haider H, et al. Activation of CD8+ T cells in chronic obstructive pulmonary disease lung [J. Am J Respir Crit Care Med. (2023) 208:1177–95. doi: 10.1126/sciimmunol.abb7221
157. Lee J, Machin M, Russell KE, Pavlidis S, Zhu J, Barnes PJ, et al. Corticosteroid modulation of immunoglobulin expression and B-cell function in COPD [J. FASEB J. (2016) 30:2014–26. doi: 10.1096/fj.201500135
158. Wang X, Liu C, Liang R, Zhou Y, Kong X, Wang W, et al. Elucidating the beneficial impact of exercise on chronic obstructive pulmonary disease and its comorbidities: Integrating proteomic and immunological insights [J. Br J Pharmacol. (2024) 181:5133–50. doi: 10.1111/bph.17328
159. Ke J, Huang S, He Z, Lei S, Lin S, Li Y, et al. NFIL3/Tim3 axis regulates effector Th1 inflammation in COPD mice [J. Front Immunol. (2024) 15:1482213. doi: 10.3389/fimmu.2024.1482213
160. Liu L, Zhou M, Sun S, Chen L, Li D, Lv J, et al. Developmental immune network of airway lymphocytes and innate immune cells in patients with stable COPD [J. Front Immunol. (2025) 16:1614655. doi: 10.3389/fimmu.2025.1614655
161. Corleis B, Cho JL, Gates SJ, et al. Smoking and human immunodeficiency virus 1 infection promote retention of CD8(+) T cells in the airway mucosa [J. Am J Respir Cell Mol Biol. (2021) 65:513–20. doi: 10.1165/rcmb.2021-0168OC
162. Padilla CM, Valenzi E, Tabib T, Nazari B, Sembrat J, Rojas M, et al. Increased CD8+ tissue resident memory T cells, regulatory T cells and activated natural killer cells in systemic sclerosis lungs [J. Rheumatol (Oxford England). (2024) 63:837–45. doi: 10.1093/rheumatology/kead273
163. Husebø GR, Gabazza EC, D’alessandro Gabazza C, Yasuma T, Toda M, Aanerud M, et al. Coagulation markers as predictors for clinical events in COPD [J. Respirol (Carlton Vic). (2021) 26:342–51. doi: 10.3389/fimmu.2024.1482213
164. Schleich F, Bougard N, Moermans C, Sabbe M, and Louis R. Cytokine-targeted therapies for asthma and COPD [J. Eur Respir Rev: an Off J Eur Respir Soc. (2023) 32. doi: 10.1183/16000617.0193-2022
165. Sulaiman I, Wu BG, Chung M, Isaacs B, Tsay JJ, Holub M, et al. Lower airway dysbiosis augments lung inflammatory injury in mild-to-moderate chronic obstructive pulmonary disease [J. Am J Respir Crit Care Med. (2023) 208:1101–14. doi: 10.1164/rccm.202210-1865OC
166. Keir HR and Chalmers JD. IL-6 trans-signalling: how Haemophilus surfs the NET to amplify inflammation in COPD [J. Eur Respir J. (2021) 58. doi: 10.1165/rcmb.2021-0168OC
167. Aggor FEY, Break TJ, Trevejo-Nuñez G, et al. Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis [J. Sci Immunol. (2020) 5. doi: 10.1126/sciimmunol.aba0570
168. Liu J, Ouyang Y, Zhang Z, Wen S, Pi Y, Chen D, et al. The role of Th17 cells: explanation of relationship between periodontitis and COPD? [J. Inflammation research: Off J Eur Histamine Res Soc [et al]. (2022) 71:1011–24. doi: 10.1007/s00011-022-01602-1
169. Hansen AH, Mortensen JH, Rønnow SR, Karsdal MA, Leeming DJ, and Sand JMB. A serological neoepitope biomarker of neutrophil elastase-degraded calprotectin, associated with neutrophil activity, identifies idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease more effectively than total calprotectin [J. J Clin Med. (2023) 12. doi: 10.3390/jcm12247589
170. In E, Kuluöztürk M, Turgut T, Altıntop Geçkil AG, and İlhan N. Endocan as a potential biomarker of disease severity and exacerbations in COPD [J. Clin Respir J. (2021) 15:445–53. doi: 10.1111/crj.13320
171. Guo P, Li R, Piao TH, et al. Pathological mechanism and targeted drugs of COPD [J. Int J Chronic Obstruct Pulm Dis. (2022) 17:1565–75. doi: 10.2147/COPD.S366126
172. Ditz B, Christenson S, Rossen J, et al. Sputum microbiome profiling in COPD: beyond singular pathogen detection [J. Thorax. (2020) 75:338–44. doi: 10.1136/thoraxjnl-2019-214168
173. Baffetta F, Buonsanti C, Moraschini L, Aprea S, Canè M, Lombardi S, et al. Lung mucosal immunity to NTHi vaccine antigens: Antibodies in sputum of chronic obstructive pulmonary disease patients [J. Hum Vaccines Immunother. (2024) 20:2343544. doi: 10.1080/21645515.2024.2343544
174. Li CL and Liu SF. Exploring molecular mechanisms and biomarkers in COPD: an overview of current advancements and perspectives [J. Int J Mol Sci. (2024), 25(13). doi: 10.3390/ijms25137347
175. Radicioni G, Ceppe A, Ford AA, Alexis NE, Barr RG, Bleecker ER, et al. Airway mucin MUC5AC and MUC5B concentrations and the initiation and progression of chronic obstructive pulmonary disease: an analysis of the SPIROMICS cohort [J. Lancet Respir Med. (2021) 9:1241–54. doi: 10.1016/S2213-2600(21)00079-5
176. Macleod M, Papi A, Contoli M, Beghé B, Celli BR, Wedzicha JA, et al. Chronic obstructive pulmonary disease exacerbation fundamentals: Diagnosis, treatment, prevention and disease impact [J. Respirol (Carlton Vic). (2021) 26:532–51. doi: 10.1111/resp.14041
177. Hsu E and Bajaj T. Beta2-agonists. In: StatPearls. StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC, Treasure Island (FL (2025). Disclosure: Tushar Bajaj declares no relevant financial relationships with ineligible companies.
178. Bourbeau J, Bhutani M, Hernandez P, Aaron SD, Beauchesne MF, Kermelly SB, et al. 2023 Canadian thoracic society guideline on pharmacotherapy in patients with stable COPD [J. Chest. (2023) 164:1159–83. doi: 10.1016/j.chest.2023.08.014
179. Kunisaki KM and Sin DD. Methylxanthines in COPD: yes to caffeine, no to theophylline [J. Eur Respir J. (2021) 57. doi: 10.1183/13993003.04564-2020
180. Journey JD and Bentley TP. Theophylline toxicity. In: StatPearls. StatPearls Publishing Copyright © 2025, StatPearls Publishing LLC, Treasure Island (FL (2025). Disclosure: Thomas Bentley declares no relevant financial relationships with ineligible compani.
181. Mccormack M, Paczkowski R, Gronroos NN, Noorduyn SG, Lee L, Veeranki P, et al. Outcomes of patients with COPD treated with ICS/LABA before and after initiation of single-inhaler triple therapy with fluticasone furoate/umeclidinium/vilanterol (FF/UMEC/VI) [J. Adv Ther. (2024) 41:1245–61. doi: 10.1007/s12325-023-02776-8
182. Choi KY, Kim HI, Rhee CK, et al. Comparing Costs and Healthcare Resource Utilization (HCRU) Using LAMA versus LABA/ICS at Treatment Initiation for COPD: Findings from CITRUS (Comparing the Incidence of Tiotropium and ICS/LABA in Real-World Use in South Korea) Study [J. Int J Chronic Obstruct Pulm Dis. (2024) 19:1661–71. doi: 10.2147/COPD.S448492
183. Calzetta L, Cazzola M, Gholamalishahi S, and Rogliani P. The novel inhaled dual PDE3 and PDE4 inhibitor ensifentrine for the treatment of COPD: A systematic review and meta-analysis protocol on trough FEV1 and exacerbation according to PRISMA statement [J. Curr Res Pharmacol Drug Discov. (2024) 7:100195. doi: 10.1016/j.crphar.2024.100195
184. Martínez-García MÁ, Oscullo G, García-Ortega A, Matera MG, Rogliani P, Cazzola M, et al. Inhaled corticosteroids in adults with non-cystic fibrosis bronchiectasis: from bench to bedside. A narrative review [J. Drugs. (2022) 82:1453–68. doi: 10.1007/s40265-022-01785-1
185. Uwagboe I, Adcock IM, Lo Bello F, et al. New drugs under development for COPD [J. Minerva Med. (2022) 113:471–96. doi: 10.23736/S0026-4806.22.08024-7
186. Huan W, Tianzhu Z, Yu L, and Shumin W. Effects of ergosterol on COPD in mice via JAK3/STAT3/NF-κB pathway [J. Inflammation. (2017) 40:884–93. doi: 10.1007/s10753-017-0533-5
187. Song B and Chen Y. Long non-coding RNA SNHG4 aggravates cigarette smoke-induced COPD by regulating miR-144-3p/EZH2 axis [J. BMC Pulm Med. (2023) 23:513. doi: 10.1186/s12890-023-02818-5
188. Brennan M, Mcdonnell MJ, Harrison MJ, Duignan N, O'Regan A, Murphy DM, et al. Antimicrobial therapies for prevention of recurrent acute exacerbations of COPD (AECOPD): beyond the guidelines [J. Respir Res. (2022) 23:58. doi: 10.1186/s12931-022-01947-5
189. Hoult G, Gillespie D, Wilkinson TMA, Thomas M, and Francis NA. Biomarkers to guide the use of antibiotics for acute exacerbations of COPD (AECOPD): a systematic review and meta-analysis [J. BMC Pulm Med. (2022) 22:194. doi: 10.1186/s12890-022-01958-4
190. Garon EB, Chih-Hsin Yang J, and Dubinett SM. The role of interleukin 1β in the pathogenesis of lung cancer [J. JTO Clin Res Rep. (2020) 1:100001. doi: 10.1016/j.jtocrr.2020.100001
191. Rabe KF, Celli BR, Wechsler ME, et al. Safety and efficacy of itepekimab in patients with moderate-to-severe COPD: a genetic association study and randomised, double-blind, phase 2a trial [J. Lancet Respir Med. (2021) 9:1288–98. doi: 10.1016/S2213-2600(21)00167-3
192. Camargo LDN, Righetti RF, De Almeida FM, et al. Modulating asthma-COPD overlap responses with IL-17 inhibition [J. Front Immunol. (2023) 14:1271342. doi: 10.3389/fimmu.2023.1271342
193. Henen C, Johnson EA, and Wiesel S. Unleashing the power of IL-17: A promising frontier in chronic obstructive pulmonary disease (COPD) treatment [J. Cureus. (2023) 15:e41977. doi: 10.7759/cureus.41977
194. Wang L, Lei W, Zhang S, et al. MCC950, a NLRP3 inhibitor, ameliorates lipopolysaccharide-induced lung inflammation in mice [J. Bioorg Medicinal Chem. (2021) 30:115954. doi: 10.1016/j.bmc.2020.115954
195. Wu H, Ma H, Wang L, et al. Regulation of lung epithelial cell senescence in smoking-induced COPD/emphysema by microR-125a-5p via Sp1 mediation of SIRT1/HIF-1a [J. Int J Biol Sci. (2022) 18:661–74. doi: 10.7150/ijbs.65861
196. Dong Y, Dong Y, Zhu C, et al. Targeting CCL2-CCR2 signaling pathway alleviates macrophage dysfunction in COPD via PI3K-AKT axis [J. Cell Commun Signal: CCS. (2024) 22:364. doi: 10.1186/s12964-024-01746-z
197. Dong LL, Liu ZY, Chen KJ, et al. The persistent inflammation in COPD: is autoimmunity the core mechanism? [J. Eur Respir Rev: an Off J Eur Respir Soc. (2024) 33. doi: 10.1183/16000617.0137-2023
Keywords: COPD, immune system, innate immunity, autoadaptive immunity, immune cell regulatory networks
Citation: Li H, Wang Y, Duan H, Bao Y, Deng X, He Y, Gao Q, Li P and Liu X (2025) Immune cell regulatory networks in chronic obstructive pulmonary disease: mechanistic analysis from innate to adaptive immunity. Front. Immunol. 16:1651808. doi: 10.3389/fimmu.2025.1651808
Received: 22 June 2025; Accepted: 29 September 2025;
Published: 16 October 2025.
Edited by:
Firdos Ahmad, University of Sharjah, United Arab EmiratesReviewed by:
Shahram Salek-Ardakani, Inhibrx, United StatesAruna Pal, West Bengal University of Animal and Fishery Sciences, India
Copyright © 2025 Li, Wang, Duan, Bao, Deng, He, Gao, Li and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peijun Li, bHBqMDIyN0AxNjMuY29t; Xiaodan Liu, aHpocDQwM2FAMTI2LmNvbQ==