 Jia-Wen Wang†
Jia-Wen Wang† Qi Feng
Qi Feng Jian-Jun Xun
Jian-Jun Xun- Department of Orthopedics, The Fourth Hospital of Hebei Medical University, Shijiazhuang, Hebei, China
Malignant melanoma is characterized by high heterogeneity, aggressive metastatic potential, and a profoundly immunosuppressive “cold” tumor microenvironment, contributing to broad therapeutic resistance and suboptimal responses to immunotherapy. Conventional PD-1 inhibitors yield an ORR of only 38%. As an emerging class of immunotherapeutic agents, oncolytic viruses (OV) induce ICD, promoting the release of DAMPs and activating innate immune pathways such as cGAS-STING, thereby transforming “cold” tumors into “hot” phenotypes and eliciting robust anti-tumor responses. Mechanistically, OV therapy increases the proportion of CD103+ dendritic cells (DCs) in lymph nodes from 5% to 25% and enhances DC–tumor synapse formation by 300%, facilitating efficient cross-presentation of tumor antigens and T-cell priming. Clinically, T-VEC combined with pembrolizumab achieves a 48.6% ORR with grade ≥3 AEs occurring in <20% of patients—superior to either monotherapy or conventional chemoradiotherapy. Nonetheless, OV therapy faces challenges including tumor heterogeneity, core mechanistic limitations, viral shedding risks, and regulatory hurdles. Over the next 5–10 years, single-cell RNA sequencing is expected to unravel molecular heterogeneity in melanoma, while CRISPR/Cas systems may enable the design of tailored OV to overcome resistance. Additional strategies such as serotype switching, JAK/STAT inhibition, and arming OV with hyaluronidase or STING agonists are under investigation to overcome immune and stromal barriers. Integration of artificial intelligence with biomarkers—such as neutralizing antibody titers, ISG expression, and STING methylation—may further enable personalized OV-based therapies. This review discusses OV therapy’s mechanisms, clinical impact, and future prospects in melanoma treatment.
1 Introduction
Malignant melanoma is notoriously difficult to treat due to its pronounced heterogeneity, aggressive metastatic behavior, and extensive drug resistance (1, 2). Under hypoxic microenvironmental pressures, melanoma cells can undergo phenotype switching between MITF-high and MITF-low states, contributing to a population of slow-cycling, therapy-resistant cells (1). Even with PD-1 inhibitor therapy, the objective response rate (ORR) remains limited at 38% (2), with a significant proportion of patients developing resistance, primarily attributed to the tumor’s immunologically “cold” microenvironment (3, 4). Histologically, these tumors typically exhibit low tumor mutation burden, scarce T-cell infiltration, impaired antigen presentation, and enrichment of immunosuppressive signaling molecules (4, 5)—collectively presenting major obstacles to effective immune reactivation. As a result, strategies to awaken or reprogram suppressed anti-tumor immunity have become a central focus of melanoma immunotherapy research (6, 7).
Oncolytic viruses (OV), a novel class of immunotherapeutic agents, exert effects that extend far beyond direct tumor cell lysis (8, 9). Upon infecting tumor cells, OV trigger immunogenic cell death (ICD), leading to the release of key damage-associated molecular patterns (DAMPs)—including high-mobility group box 1 (HMGB1), ATP, and calreticulin (CRT) (10, 11). These molecules activate innate immune sensors such as the STING pathway and Toll-like receptors (TLRs), initiating a robust immune cascade (10–13). Concurrently, OV remodel the tumor microenvironment, promoting M1 polarization of macrophages, enhancing natural killer (NK) cell activity, and activating dendritic cells (DCs). Notably, OV treatment elevates CD103+ DC levels in lymph nodes from 5% to 25% and increases the formation of DC–tumor synapses by 300%, thereby enabling efficient cross-presentation of tumor antigens and activation of cytotoxic T cells (14, 15). This sequential activation of innate and adaptive immunity positions OV as “in situ vaccines,” offering a promising avenue to break through the immunotherapy plateau in melanoma (8, 16).
Despite the clinical success of OV such as talimogene laherparepvec (T-VEC), which is FDA-approved for advanced melanoma (12, 17), substantial challenges remain. These include extracellular matrix (ECM) barriers limiting viral penetration, highly immunosuppressive tumor microenvironments, difficulties in accurately evaluating treatment response (e.g., >40% false positives in PET-CT), and systemic delivery inefficiencies (12, 17). These limitations must be addressed for broader and more effective application of OV therapy.
This review aims to explore four key questions:
1. Mechanistic Axis: How do OV activate innate immunity via the ICD–DAMP–STING axis? How does this pathway convert melanoma from a “cold” to “hot” phenotype, and how do tertiary lymphoid structures (TLSs) and ferroptosis contribute to sustained anti-tumor immunity?
2. Vector Comparison and Clinical Outcomes: What are the mechanistic advantages of different viral vectors (e.g., HSV-1, adenovirus, VSV)? Why does T-VEC combined with pembrolizumab achieve a 48.6% ORR, and how is its safety reflected in a <20% incidence of grade ≥3 AEs?
3. Resistance and Regulatory Challenges: How does tumor heterogeneity impact treatment response (e.g., differential progression-free survival in BRAF-mutant vs. wild-type patients)? How do neutralizing antibody clearance, type I IFN/ISG overexpression, and stromal barriers cooperatively limit OV efficacy? What are the clinical safety and regulatory concerns associated with viral shedding?
4. Future Innovations: How can single-cell RNA sequencing and CRISPR/Cas gene editing technologies address melanoma heterogeneity? How might serotype switching, JAK/STAT inhibitors, and “armed” OV (e.g., hyaluronidase, STING agonists) overcome core mechanistic limitations? How can artificial intelligence integrate biomarkers such as NAb titers, ISG expression, and STING methylation to enable personalized therapeutic strategies and predictive modeling?
Figure 1 provides a schematic overview of the central theme: OV-mediated reprogramming of the tumor microenvironment from “cold” to “hot.”

Figure 1. Oncolytic virus therapy transforms “cold” melanoma into “hot” tumors.
2 Oncolytic virus therapy for malignant melanoma: mechanisms and opportunities
2.1 Molecular immunological mechanisms of oncolytic virus therapy
Immunogenic cell death (ICD) represents a critical mechanism by which oncolytic viruses (OV) exert antitumor effects in malignant melanoma. Upon infecting tumor cells, OV induce ICD, leading to the release of key danger signals such as high mobility group box 1 (HMGB1), adenosine triphosphate (ATP), and calreticulin (CRT). These molecules act as damage-associated molecular patterns (DAMPs), which activate the host innate immune system and initiate a cascade of immune responses (18–20).
Specifically, HMGB1, normally confined to the nucleus, is released extracellularly during ICD and binds to pattern recognition receptors such as Toll-like receptors (TLRs), thereby activating downstream pathways like NF-κB and promoting proinflammatory cytokine secretion and immune cell activation (19, 20). ATP engages the P2X7 purinergic receptor, which in turn activates inflammasomes, resulting in the maturation and release of interleukin-1β (IL-1β) and recruitment of innate immune cells (19, 20). CRT translocates to the cell surface, serving as an “eat-me” signal that enhances recognition and uptake by antigen-presenting cells (APCs), particularly dendritic cells (DCs), facilitating efficient cross-presentation of tumor antigens to CD4+ helper T cells and CD8+ cytotoxic T lymphocytes (CTLs) (18).
During this process, CD4+ T cells secrete cytokines such as IL-2 and interferon-gamma (IFN-γ) to promote CD8+ T cell activation, proliferation, and differentiation, ultimately enhancing their cytolytic capacity. Upon antigen presentation, T cell receptors (TCRs) recognize the antigen–major histocompatibility complex (MHC) complex, and co-stimulatory signals such as B7-CD28 further amplify T cell activation. This results in the secretion of IFN-γ, tumor necrosis factor-alpha (TNF-α), and other cytotoxic mediators, leading to tumor cell death and the generation of long-lived central memory T cells (Tcm) and effector memory T cells (Tem), which provide durable antitumor immunity (18–20).
Moreover, the ICD process activates intracellular cytosolic DNA sensors, including the cGAS–STING pathway, which stimulates interferon regulatory factor 3 (IRF3) and NF-κB, thereby inducing type I interferons (IFNs). These cytokines boost the activity of natural killer (NK) cells and macrophages, enhancing innate immune responses and contributing to overall antitumor efficacy (19, 20).
In addition to directly killing tumor cells, OV remodel tumor immunogenicity by altering surface markers, increasing susceptibility to NK cell-mediated lysis, and recruiting macrophages. OV infection promotes the polarization of macrophages from an M2 immunosuppressive phenotype to an M1 immunostimulatory phenotype, enhancing the production of IFN-α and IFN-γ and further augmenting NK cell cytotoxicity (18, 19, 21).
Within the tumor microenvironment (TME), OV-infected tumor cells secrete immunomodulatory cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-α, and chemokines that facilitate immune cell recruitment while downregulating immunosuppressive cells and molecules, thereby orchestrating a comprehensive reprogramming of the TME (18, 22, 23). Specifically: GM-CSF promotes the proliferation, differentiation, and activation of granulocytes and macrophages, enhancing their phagocytic and cytotoxic functions. It also supports DC maturation, thereby improving antigen presentation and activating adaptive immune responses (18, 22, 23); IFN-α induces tumor cell apoptosis and boosts the cytotoxic functions of NK and T cells, suppressing tumor growth and proliferation (18, 22, 23); CXC chemokine ligand 10 (CXCL10) and other chemokines guide OV-activated effector T cells to the tumor site, enhancing immune infiltration and antitumor activity (18).
Additionally, OV reduce the presence and function of immunosuppressive cell types such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), while also inhibiting the expression of programmed death-ligand 1 (PD-L1) and other immunosuppressive molecules. This release from immunosuppression restores immune cell functionality and amplifies antitumor immunity (18, 19, 22, 24, 25).
Figure 2 visually summarizes this molecular network, integrating the two core mechanisms of OV therapy: ICD–DAMP axis activation and tumor microenvironment reprogramming, and elucidating their synergistic roles in initiating and sustaining robust antitumor immune cascades.
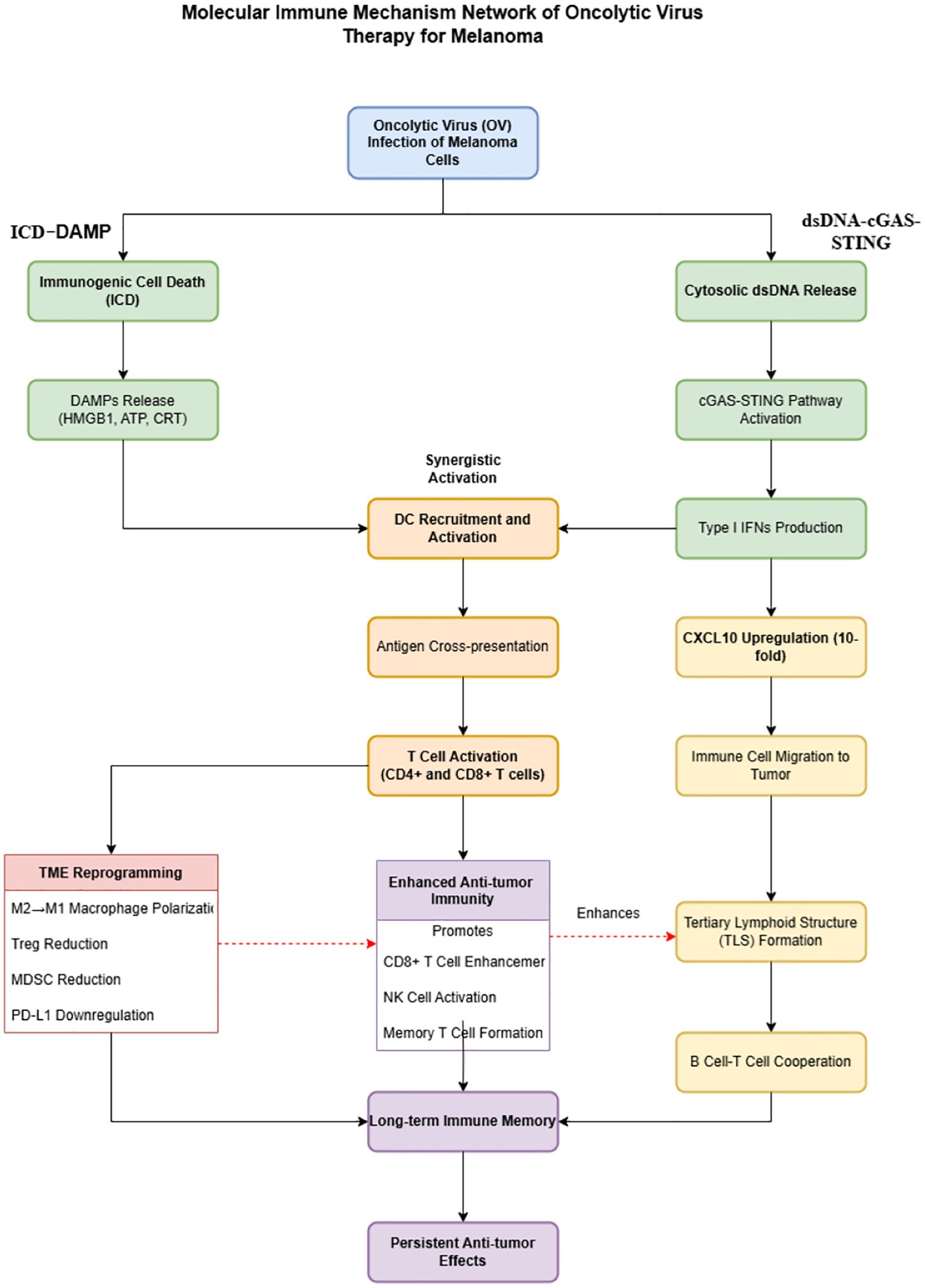
Figure 2. Molecular immune mechanisms network of oncolytic virus therapy for melanoma.
Beyond the classical ICD–DAMP axis and TME remodeling (summarized in Table 1), emerging molecular immune mechanisms further enhance the efficacy of OV therapy against malignant melanoma (Table 2): the cGAS–STING innate immune amplification mechanism is triggered by cytosolic dsDNA released during OV infection, leading to the production of chemokines such as CXCL10 and CCL5, forming an “interferon–chemokine” cascade. This process converts “cold” tumors into “hot” immunogenic phenotypes, enhancing immune infiltration and tumor clearance (26); tertiary lymphoid structure (TLS) induction remodels the tumor stroma and facilitates coordinated B/T cell responses. Notably, patients with high TLS density exhibit significantly prolonged 2-year recurrence-free survival, reaching up to 81.5% (27); ferroptosis synergy: OV efficacy is augmented by ferroptosis inducers such as Erastin, achieving tumor inhibition rates as high as 72% (28); antigen presentation and epigenetic activation: OV therapy upregulates MHC-I expression and, in combination with EZH2 inhibition, significantly enhances CD8+ T cell tumor recognition, thereby potentiating cytotoxic immune responses (29).
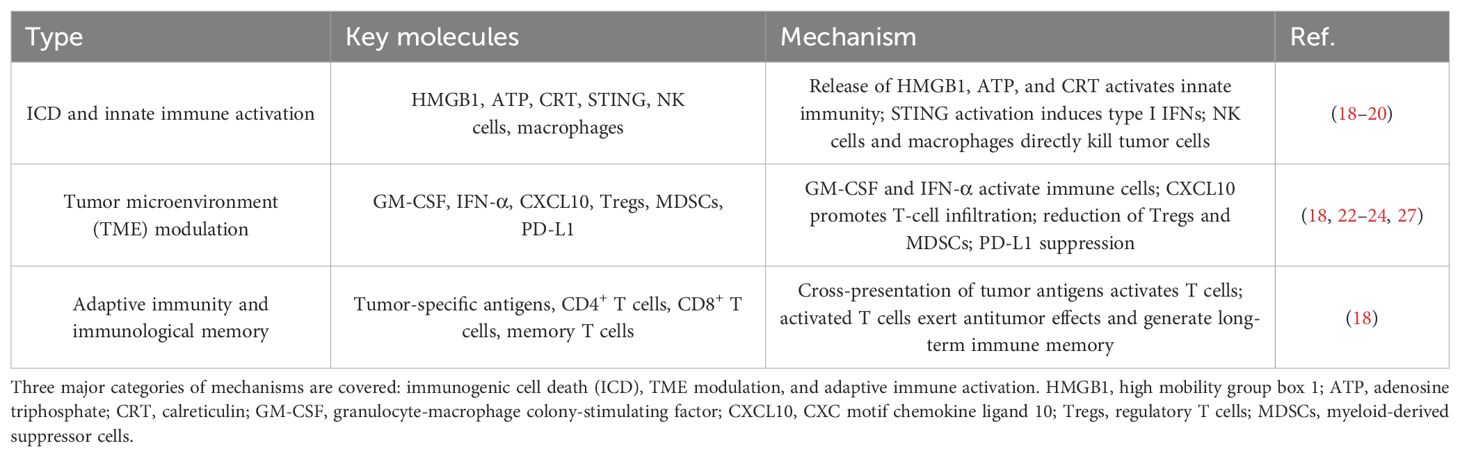
Table 1. Canonical mechanisms of oncolytic virus therapy in malignant melanoma.

Table 2. Emerging molecular-immune mechanisms of OV therapy in malignant melanoma.
2.2 Efficacy and safety of oncolytic virus therapy
Different oncolytic virus (OV) vectors demonstrate distinct mechanistic advantages and significant variability in efficacy and safety profiles in the treatment of malignant melanoma.
Mechanistically (Table 3), HSV-1–based vectors exert immune-activating effects through STING pathway activation (e.g., T-VEC) and enhanced cell-to-cell fusion (e.g., RP1). Adenoviral vectors primarily induce anti-tumor responses via cGAS–STING pathway activation and the induction of immunogenic cell death (e.g., ONCOS-102, which achieved a 70% tumor shrinkage rate), as well as through dual co-stimulatory mechanisms (e.g., LOAd703). In contrast, vesicular stomatitis virus (VSV) vectors activate innate immune signaling via the RIG-I/MDA5 axis, achieving up to a 60% tumor clearance rate (30–34).
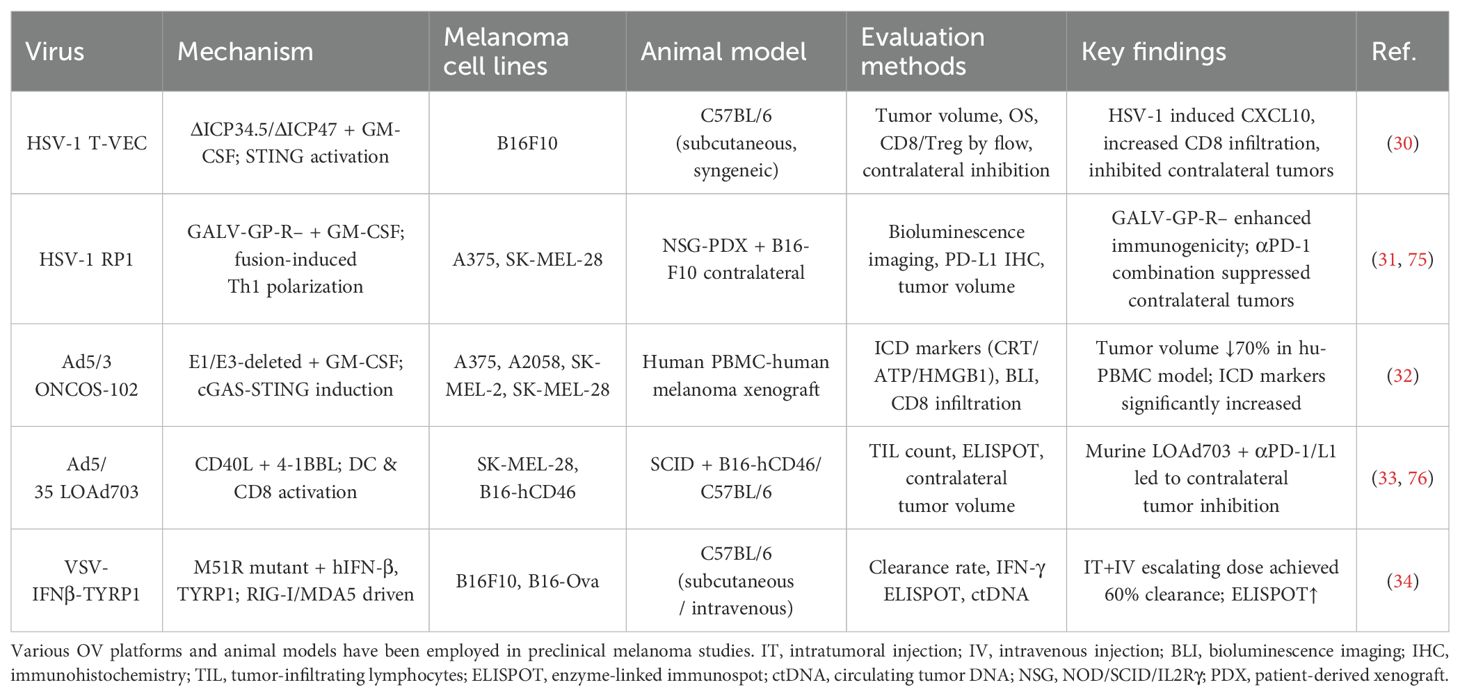
Table 3. Preclinical efficacy of OV therapies in malignant melanoma.
Clinically (Table 4), the therapeutic efficacy of OV varies by vector and treatment strategy:T-VEC monotherapy has demonstrated an objective response rate (ORR) of up to 31.5% and a median overall survival (mOS) of 23.3 months. Notably, it led to a >50% reduction in lesion size in 34% of non-visceral and 15% of visceral uninjected tumors (35); RP1 combined with nivolumab in PD-1–refractory patients achieved an ORR of 32.9%, a median duration of response (mDoR) of 33.7 months, and a 1-year survival rate of 75.3%. Impressively, 96.6% of patients experienced regression in uninjected lesions, and 39.5% achieved complete lesion clearance (36); OH2 monotherapy, in patients previously treated with PD-1 inhibitors, resulted in an ORR of 58.3% and a 1-year survival rate of 94.3%, with 40% experiencing reductions in uninjected lesions (19);ONCOS-102 combined with pembrolizumab achieved a 35% ORR in PD-1–resistant patients, with 53% showing regression of uninjected lesions (37);T-VEC combined with pembrolizumab reached an ORR of 48.6%, along with a 33% reduction rate in non-injected visceral lesions (38).
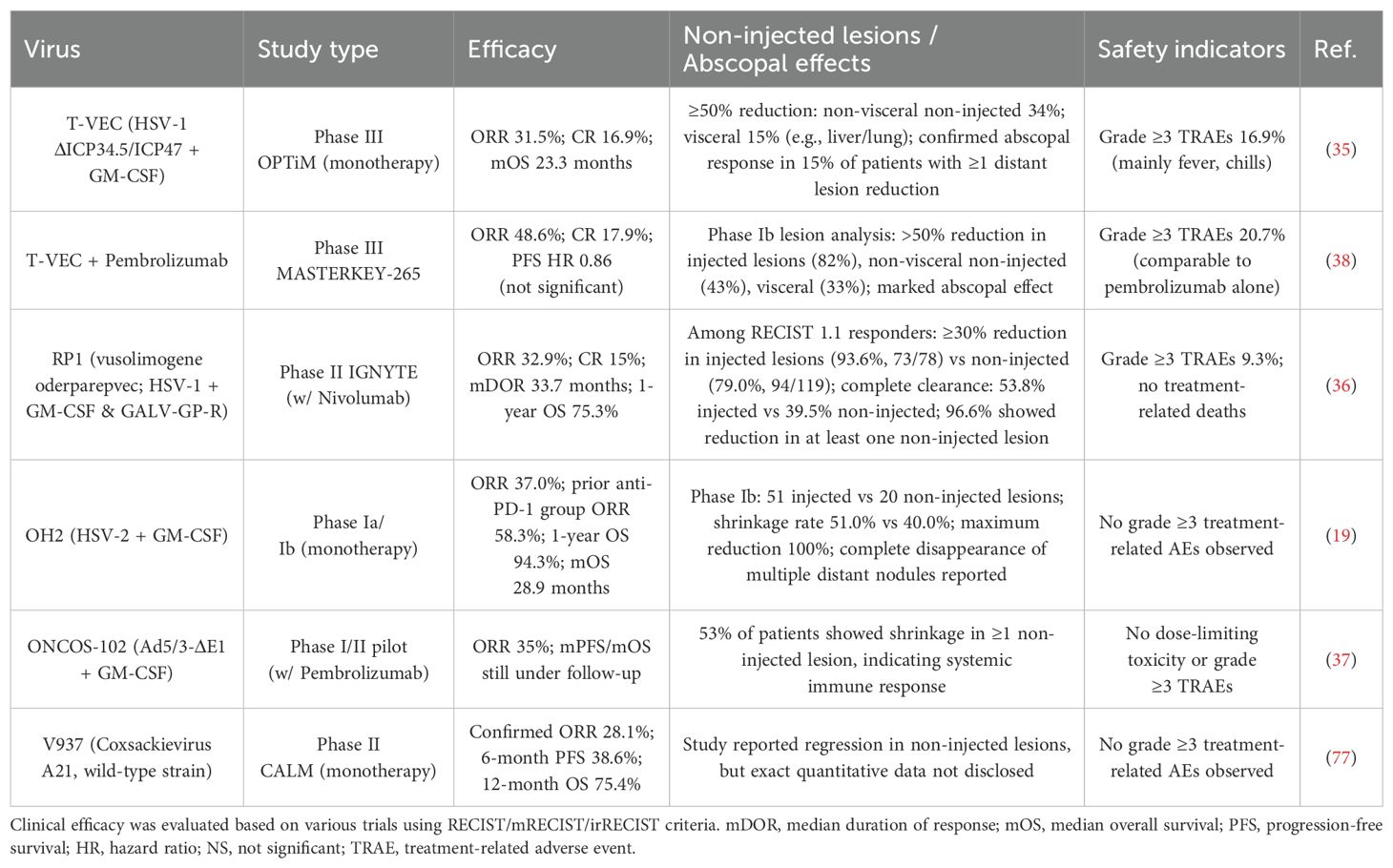
Table 4. Clinical efficacy, systemic (abscopal) responses, and safety profile of OV therapies in melanoma.
In terms of safety (Table 4), most OV-based regimens were well tolerated, with the incidence of grade ≥3 treatment-related adverse events (TRAEs) generally remaining below 20%. Notably, several trials reported no grade ≥3 TRAEs at all. This is in stark contrast to traditional chemotherapy and many targeted therapies, which typically report severe adverse events in 30% to 60% of cases (39–42).
3 Key bottlenecks and challenges in OV-based immunotherapy
In clinical practice, patient responses to oncolytic virus (OV) therapy vary significantly (23, 43, 44), primarily due to the high heterogeneity of immune status, tumor microenvironment, and tumor cell gene expression profiles (23, 43, 44). Among these, tumor heterogeneity plays a critical role in determining therapeutic outcomes (Table 5):Patients with BRAF wild-type tumors exhibit significantly longer disease-free survival than those with BRAF mutations (P = 0.04), with the former achieving a local objective response rate (ORR) of 80%, compared to 65% in the latter (45). Similarly, patients with high MITF expression show an ORR of 49%, whereas those with AXL-high tumors demonstrate a much lower ORR of only 15% (46). Notably, STING-low patients (~25%) can achieve 100% regression of uninjected lesions and reversal of PD-1 resistance (47), whereas in STING-high patients, the regression rate of uninjected tumors is <10% (48).
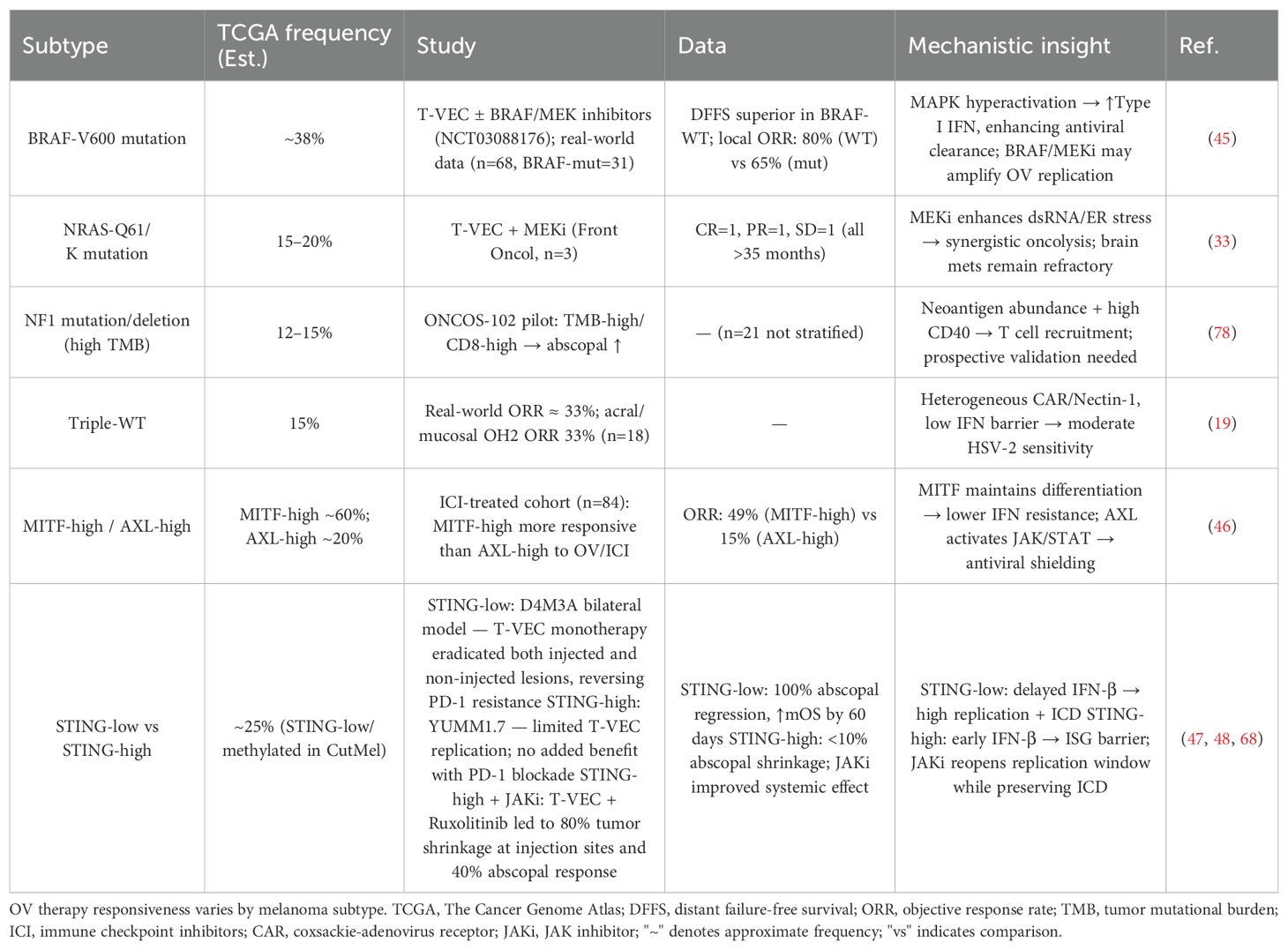
Table 5. Efficacy of OV therapy across distinct melanoma subtypes.
Beyond tumor heterogeneity, fundamental mechanistic barriers also constrain OV efficacy (Table 6): the clearance of OV by pre-existing neutralizing antibodies (NAbs) significantly impairs therapeutic effectiveness. Patients with high baseline NAbs exhibit a median overall survival (mOS) of only 12.5 months, whereas those with low titers reach 21.2 months (HR ≈ 2.0) (49); intrinsic antiviral pathways limit viral propagation. High expression of type I interferons (IFNs) and interferon-stimulated genes (ISGs) in melanoma cells activates the IFNAR–JAK/STAT pathway, upregulating PD-1 and enhancing endogenous antiviral defenses. Consequently, ISG-high patients show a much lower response rate (15%) compared to ISG-low patients (45%) (50, 51); the tumor microenvironment (TME) also suppresses OV efficacy. Specifically, PD-L1 in the TME induces an immunosuppressive milieu via the glycolysis–lactate axis, inhibiting IFN-β production and dampening immune activation (50, 51).
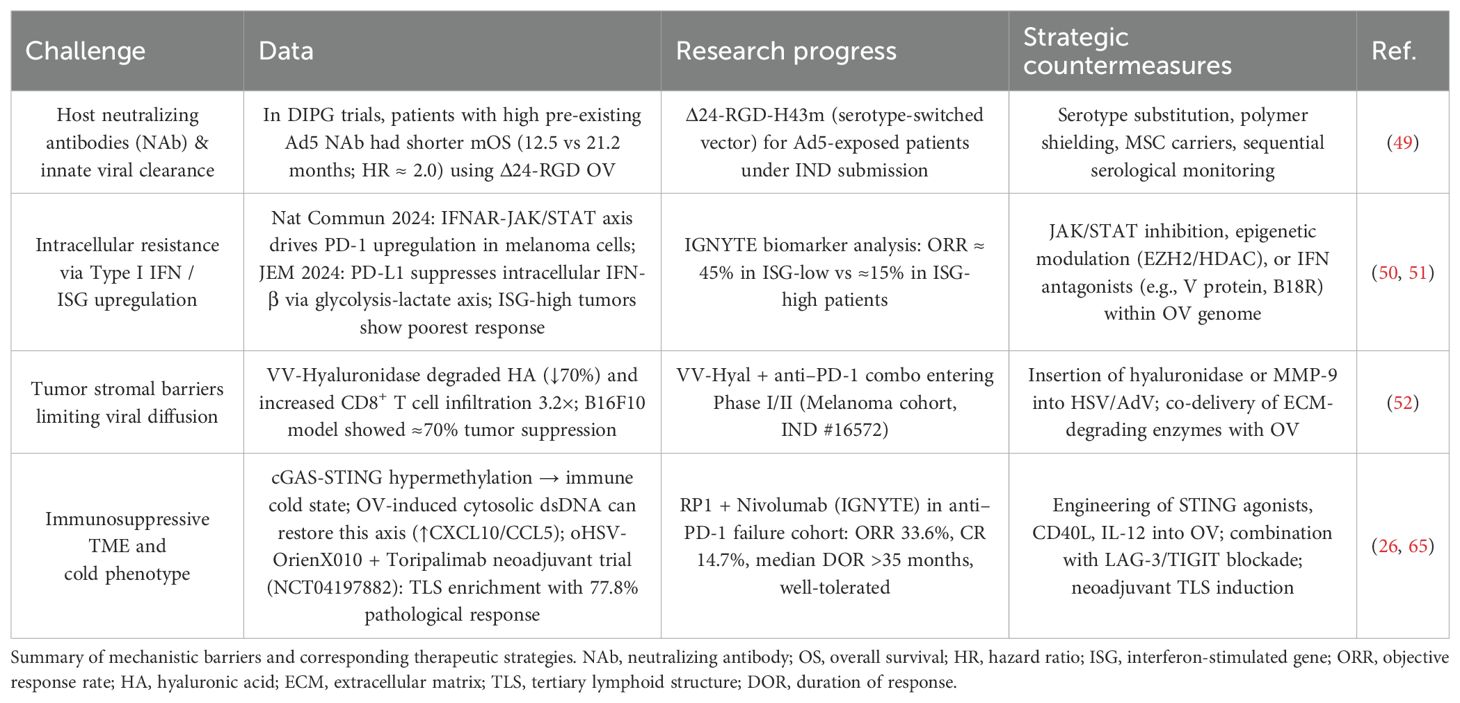
Table 6. Key mechanistic barriers to OV therapy and corresponding strategies.
In addition, the stromal barrier contributes to immune exclusion. For instance, VV-Hyal—a hyaluronidase-expressing vaccinia virus—can degrade up to 70% of intratumoral hyaluronic acid (HA), resulting in a 3.2-fold increase in T cell infiltration and enhanced antitumor immunity (52). Despite these findings, the molecular mechanisms underlying these resistance factors remain incompletely understood (23, 43, 44, 53, 54).
Moreover, although OV therapies have generally demonstrated favorable safety profiles, off-target risks and viral shedding remain significant safety and regulatory concerns. For example, HSV-DNA has been detected in wound exudate dressings in up to 37% of cases, and rare instances of disseminated HSV infection have been reported even in immunocompetent patients (55, 56), indicating the potential for viral shedding and transmission. Regulatory agencies including the FDA (57), EMA (58), and PMDA (59) have acknowledged these concerns. However, >60% of OV clinical trials monitor only blood samples (60), suggesting an underestimation or neglect of shedding risk. This limited sampling approach hinders the implementation of crucial regulatory requirements, such as assessing multi-route shedding and defining sustained shedding as >3 log10 copies in urine/saliva/feces, which mandates isolation measures (59, 61) (Table 7). Therefore, the safety risks and regulatory oversight in OV therapy demand renewed scrutiny and should not be underestimated.
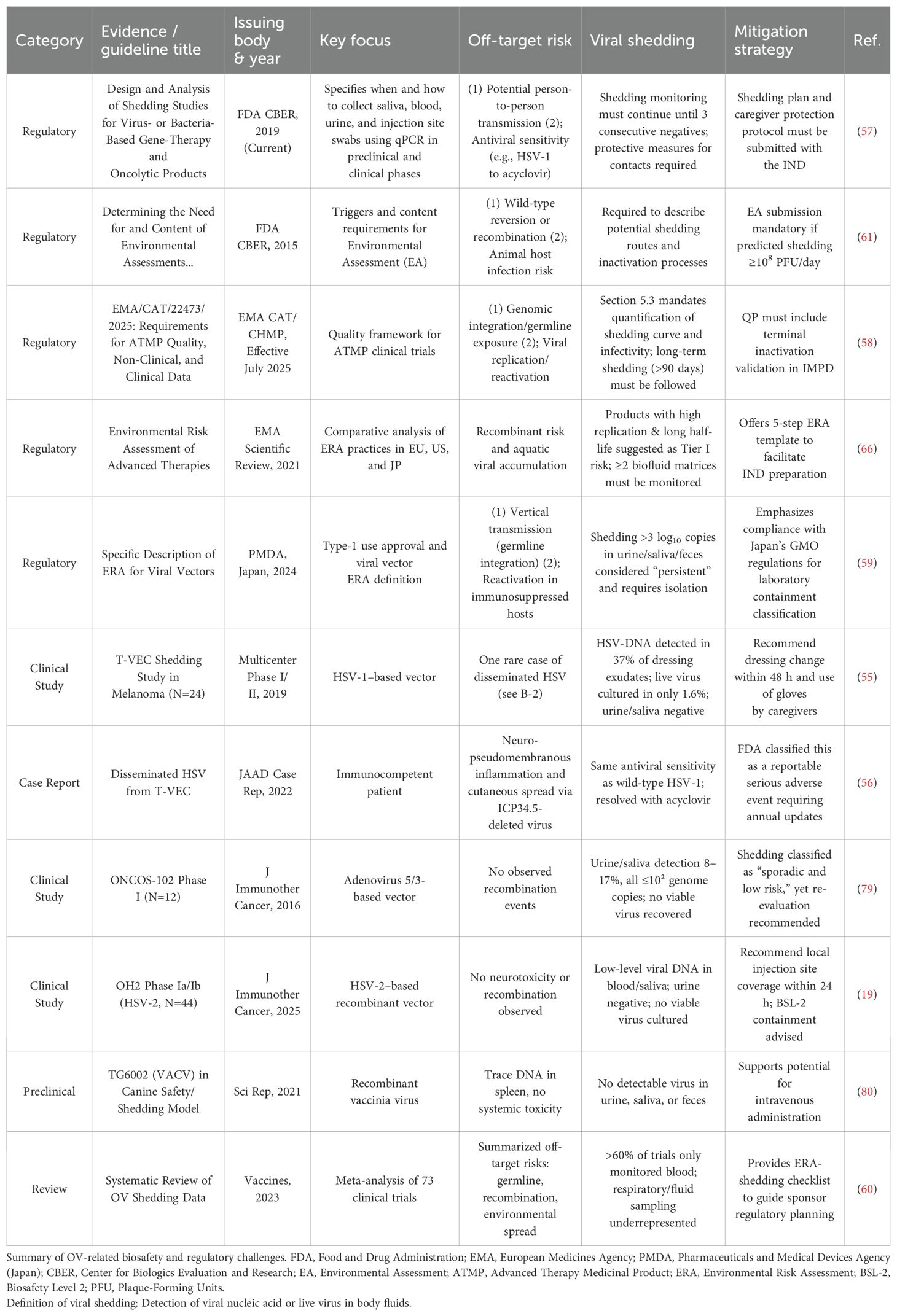
Table 7. Safety concerns (off-target effects and viral shedding) and regulatory challenges of oncolytic virus (OV) therapy, along with corresponding mitigation strategies.
4 Discussion
In the next 5–10 years, technologies such as single-cell RNA sequencing (scRNA-seq) are expected to enable a more refined understanding of gene expression profiles, cellular subtypes, and functional states of both tumor and immune cells at single-cell resolution. This will facilitate the resolution of tumor microenvironment (TME) complexity and intratumoral heterogeneity (62–64). Furthermore, molecular profiling will enable the design of OV with selective infectivity and cytotoxicity toward specific tumor cell subpopulations, thereby improving treatment precision and efficacy (62–64). Among these approaches, genome editing technologies such as CRISPR/Cas systems stand out as promising tools for enhancing the immunostimulatory potency of OV and addressing tumor heterogeneity (20, 22, 24, 63, 64).
To overcome key mechanistic barriers (Table 6), multiple strategies are being developed. For instance, host-mediated viral clearance due to pre-existing neutralizing antibodies (NAbs) can be mitigated by employing rare serotype recombinants such as Δ24-RGD-H43m (in Ad5-preexposed populations), polymer shielding, or mesenchymal stem cell (MSC)-based delivery platforms (49). Intracellular resistance resulting from elevated type I interferon (IFN) or interferon-stimulated gene (ISG) expression may be alleviated through combination therapies involving JAK/STAT inhibitors, EZH2/HDAC epigenetic modulators, or the insertion of IFN antagonistic genes (e.g., V protein, B18R) into the OV genome (50, 51). Dense stromal barriers may be addressed by engineering OV (e.g., HSV/AdV) to express hyaluronidase or MMP-9, co-administering ECM-degrading enzymes, or using combination therapies such as VV-Hyal with anti-PD-1 antibodies (52). To remodel immunosuppressive TMEs and convert “cold” tumors into “hot” phenotypes, arming OV with STING agonists, CD40L, IL-12, or combining them with LAG-3/TIGIT inhibitors has demonstrated therapeutic potential (26, 65).
From a safety and regulatory standpoint (Table 7), the next 5–10 years will see the progressive implementation of policies and clinical guidelines. Regulatory authorities such as the FDA, PMDA, and EMA are enforcing standards including the requirement for “three consecutive negative tests before sampling cessation” (57) and “isolation for patients with viral shedding >3 log10 copies” (59, 66). In clinical settings, protocols such as “dressing changes within 48 hours” (55) and “monitoring across ≥2 fluid matrices” (66) will help enhance biosafety awareness and improve safety evaluation frameworks. Collectively, these advances are expected to contribute to the establishment of standardized international regulations and risk-stratified safety management systems, addressing both off-target risks and viral shedding concerns associated with OV therapy.
In parallel, research into predictive biomarkers for OV therapy will continue to expand and shape clinical decision-making, especially in balancing antiviral and antitumor immune responses (Table 8). Predictive biomarkers such as baseline NAb titers and ISG signatures will become more widely used to anticipate treatment responses and therapeutic efficacy (67, 68). Functional biomarkers, including STING promoter methylation and MITF/AXL expression ratios, will inform virus vector selection and combination strategies (26, 69). Dynamic response biomarkers—such as post-treatment increases in CD8+ tumor-infiltrating lymphocytes (TILs) and PD-L1 expression, as well as the rate of circulating tumor DNA (ctDNA) clearance—will facilitate real-time efficacy assessment and dose adjustment (36). In addition, tumor mutational burden (TMB), a known double-edged biomarker, will be integrated into refined prognostic models (37).
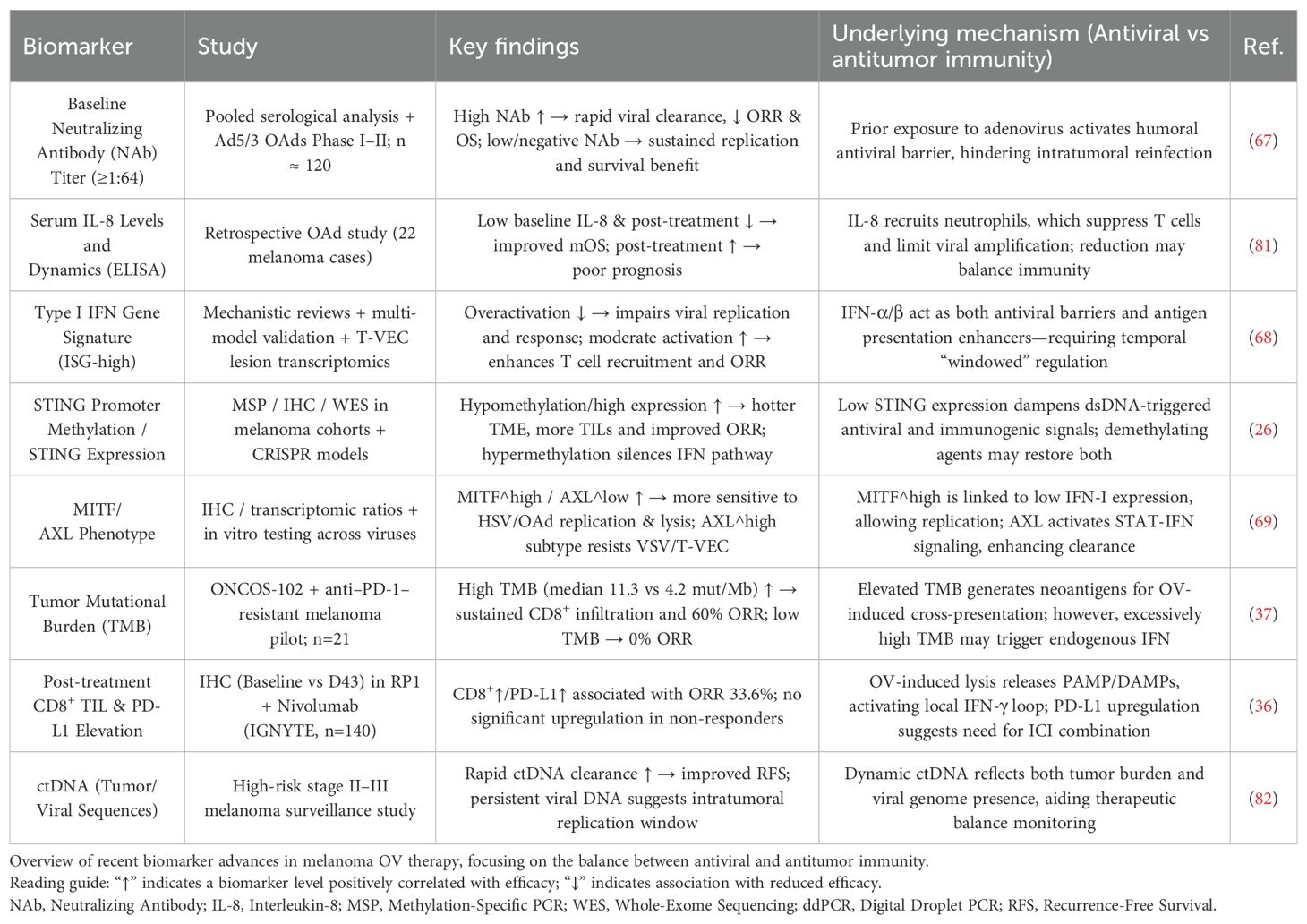
Table 8. Emerging biomarkers for Oncolytic Virus (OV) therapy in malignant melanoma.
The rapid advancement of artificial intelligence (AI) technologies is poised to revolutionize OV-based cancer therapy. Machine learning algorithms will integrate clinical, transcriptomic, radiologic, and biomarker datasets to generate individualized predictive models, guiding vector selection, dose optimization, and combinatorial design (62–64). When coupled with single-cell omics, AI will aid in addressing tumor heterogeneity at the cellular level (62–64). Integration of AI with CRISPR/Cas-based editing will further accelerate the development of OV with enhanced immunogenicity and safety profiles (20, 22, 24, 63, 64). Nanotechnology platforms will facilitate the co-delivery of OV with immunomodulators or chemotherapeutics, promoting synergistic therapeutic effects (62–64). The emergence of multi-targeting viral vectors will allow simultaneous infection of diverse tumor cell subsets and sustained release of immunoregulatory molecules, thereby amplifying antitumor immunity (20, 22, 24, 63, 64).
Finally, as large-scale randomized clinical trial data accumulates and AI algorithms continue to evolve, key prognostic factors—such as the optimal timing, dosage, and sequencing of OV combination regimens—will be more accurately defined (23, 53, 54, 70–74), ultimately enabling a broader population of patients with malignant melanoma to benefit from OV therapy.
Oncolytic virus (OV) therapy induces immunogenic cell death (ICD), which releases damage-associated molecular patterns (DAMPs) and activates the cGAS-STING pathway, thereby transforming immunologically “cold” tumors (with a PD-1 inhibitor objective response rate [ORR] of only 38%) into “hot” tumors. This process increases the proportion of CD103+ dendritic cells (DCs) from 5% to 25%. Clinically, T-VEC combined with pembrolizumab has demonstrated an ORR of 48.6% with a treatment-related serious adverse event rate below 20%. In the future, artificial intelligence (AI)-guided approaches may facilitate individualized and precise OV-based therapies.
This diagram illustrates the immunoactivation network triggered by OV therapy in melanoma. Upon infection, OV activates two parallel pathways: the left axis represents the ICD pathway, leading to the release of DAMPs, while the right axis shows the cGAS-STING pathway, upregulating type I interferons and CXCL10. Together, these pathways enhance dendritic cell activation, tumor antigen cross-presentation, and T-cell responses, and promote tumor microenvironment (TME) reprogramming and tertiary lymphoid structure (TLS) formation, establishing durable antitumor immunity. Color-coded stages include: initiation events (blue), early molecular signals (green), immune cell activation (orange), TME remodeling (red), and long-term immune memory (purple). Solid arrows indicate direct effects, and dashed arrows indicate promoting relationships.
4.1 Permission to reuse and copyright
Permission must be obtained for use of copyrighted material from other sources (including the web). Please note that it is compulsory to follow figure instructions.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author contributions
J-WW: Conceptualization, Investigation, Visualization, Writing – original draft. QF: Investigation, Project administration, Resources, Writing – review & editing. J-HL: Supervision, Writing – review & editing. J-JX: Project administration, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ahmed F and Haass NK. Microenvironment-driven dynamic heterogeneity and phenotypic plasticity as a mechanism of melanoma therapy resistance. Front Oncol. (2018) 8:173. doi: 10.3389/fonc.2018.00173
2. Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, et al. Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat Med. (2019) 25:1916–27. doi: 10.1038/s41591-019-0654-5
3. Ding L, Sun L, Bu M-T, Zhang Y, Scott LN, Prins RM, et al. Antigen presentation by clonally diverse CXCR5+ B cells to CD4 and CD8 T cells is associated with durable response to immune checkpoint inhibitors. Front Immunol. (2023) 14:1176994. doi: 10.3389/fimmu.2023.1176994
4. Lim S-Y, Shklovskaya E, Lee J-H, Pedersen B, Stewart A, Ming Z, et al. The molecular and functional landscape of resistance to immune checkpoint blockade in melanoma. Nat Commun. (2023) 14:1516. doi: 10.1038/s41467-023-36979-y
5. Tang T, Huang X, Zhang G, Hong Z, Bai X, and Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal transduction targeted Ther. (2021) 6:72. doi: 10.1038/s41392-020-00449-4
6. Cui Y, Miao Y, Cao L, Guo L, Cui Y, Yan C, et al. Activation of melanocortin-1 receptor signaling in melanoma cells impairs T cell infiltration to dampen antitumor immunity. Nat Commun. (2023) 14:5740. doi: 10.1038/s41467-023-41101-3
7. Gruber T, Kremenovic M, Sadozai H, Rombini N, Baeriswyl L, Maibach F, et al. IL-32γ potentiates tumor immunity in melanoma. JCI Insight. (2020) 5:e138772. doi: 10.1172/jci.insight.138772
8. Kaufman HL, Kohlhapp FJ, and Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. (2015) 14:642–62. doi: 10.1038/nrd4663
9. Vonderheide RH. The immune revolution: A case for priming, not checkpoint. Cancer Cell. (2018) 33:563–9. doi: 10.1016/j.ccell.2018.03.008
10. Ahmed A and Tait S. Targeting immunogenic cell death in cancer. Mol Oncol. (2020) 14:2994–3006. doi: 10.1002/1878-0261.12851
11. Shao X, Wang X, Guo X, Jiang K, Ye T, Chen J, et al. STAT3 contributes to oncolytic newcastle disease virus-induced immunogenic cell death in melanoma cells. Front Oncol. (2019) 9:436. doi: 10.3389/fonc.2019.00436
12. Takasu A, Masui A, Hamada M, Imai T, Iwai S, and Yura Y. Immunogenic cell death by oncolytic herpes simplex virus type 1 in squamous cell carcinoma cells. Cancer Gene Ther. (2016) 23:107–13. doi: 10.1038/cgt.2016.8
13. Yun CO, Hong J, and Yoon AR. Current clinical landscape of oncolytic viruses as novel cancer immunotherapeutic and recent preclinical advancements. Front Immunol. (2022) 13:953410. doi: 10.3389/fimmu.2022.953410
14. Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, and Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. (2017) 16:829–42. doi: 10.1038/nrd.2017.178
15. Shi T, Song X, Wang Y, Liu F, and Wei J. Combining oncolytic viruses with cancer immunotherapy: establishing a new generation of cancer treatment. Front Immunol. (2020) 11:683. doi: 10.3389/fimmu.2020.00683
16. Romero D. HDAC inhibitors tested in phase III trial. Nat Rev Clin Oncol. (2019) 16:465. doi: 10.1038/s41571-019-0224-2
17. Kong D, Yang Z, Li G, Wu Q, Gu Z, Wan D, et al. SIRPα antibody combined with oncolytic virus OH2 protects against tumours by activating innate immunity and reprogramming the tumour immune microenvironment. BMC Med. (2022) 20:376. doi: 10.1186/s12916-022-02574-z
18. Stull CM, Clark D, Parker T, Idriss MH, Patel VA, and Migden MR. Current and emerging intralesional immunotherapies in cutaneous oncology. J Am Acad Dermatol. (2024) 91:910–21. doi: 10.1016/j.jaad.2024.05.095
19. Wang X, Tian H, Chi Z, Si L, Sheng X, Hu H, et al. Oncolytic virus OH2 extends survival in patients with PD-1 pretreated melanoma: phase Ia/Ib trial results and biomarker insights[J. J Immunother Cancer. (2025) 13:e010662. doi: 10.1136/jitc-2024-010662
20. Wang Z-M, Li M-K, Yang Q-L, Duan S-X, Lou X-Y, Yang X-Y, et al. Recombinant human adenovirus type 5 promotes anti-tumor immunity via inducing pyroptosis in tumor endothelial cells. Acta pharmacologica Sin. (2024) 45:2646–56. doi: 10.1038/s41401-024-01349-x
21. Mohite P, Yadav V, Pandhare R, Maitra S, Saleh FM, Saleem RM, et al. Revolutionizing cancer treatment: unleashing the power of viral vaccines, monoclonal antibodies, and proteolysis-targeting chimeras in the new era of immunotherapy. ACS omega. (2024) 9:7277–95. doi: 10.1021/acsomega.3c06501
22. Dugan MM, Shannon AB, DePalo DK, Perez MC, and Zager JS. Intralesional and infusional updates for metastatic melanoma. Cancers. (2024) 16:1957. doi: 10.3390/cancers16111957
23. Wang H, Borlongan M, Kaufman HL, Le U, Nauwynck HJ, Rabkin SD, et al. Cytokine-armed oncolytic herpes simplex viruses: a game-changer in cancer immunotherapy. J immunotherapy Cancer. (2024) 12:e008025. doi: 10.1136/jitc-2023-008025
24. Ammour Y, Nikolaeva E, Sagimbaeva O, Shestakov A, Ivanov A, Krivoruchko A, et al. Human melanoma and glioblastoma cells express cathepsins supporting reovirus moscow strain infection. Viruses. (2024) 16:1944. doi: 10.3390/v16121944
25. Bhatt DK, Boerma A, Bustos SO, de Gruijl TD, van den Bos H, Hoeben RC, et al. Oncolytic alphavirus-induced extracellular vesicles counteract the immunosuppressive effect of melanoma-derived extracellular vesicles. Sci Rep. (2025) 15:803. doi: 10.1038/s41598-024-82331-9
26. Mahin J, Xu X, Li L, Chen Q, Wang Y, Huang T, et al. cGAS/STING in skin melanoma: from molecular mechanisms to therapeutics[J. Cell Commun Signal. (2024) 22:553. doi: 10.1186/s12964-024-01860-y
27. Liu J, Wang X, Li Z, Chen H, Li Y, Yang R, et al. Neoadjuvant oncolytic virus orienx010 and toripalimab in resectable acral melanoma: a phase Ib trial. Signal transduction targeted Ther. (2024) 9:318. doi: 10.1038/s41392-024-02029-2
28. Li W, Ji T, Ye J, Liu Y, Wu J, Chen W, et al. Ferroptosis enhances the therapeutic potential of oncolytic adenoviruses KD01 against cancer. Cancer Gene Ther. (2025) 32:403–17. doi: 10.1038/s41417-025-00882-z
29. Yan Z, Zhang Z, Chen Y, Xu J, Wang J, and Wang Z. Enhancing cancer therapy: the integration of oncolytic virus therapy with diverse treatments. Cancer Cell Int. (2024) 24:242. doi: 10.1186/s12935-024-03424-z
30. Uche IK, Kousoulas KG, and Rider P. The effect of herpes simplex virus-type-1 (HSV-1) oncolytic immunotherapy on the tumor microenvironment[J. Viruses. (2021) 13:1200. doi: 10.3390/v13071200
31. Thomas S, Kuncheria L, Roulstone V, Mansfield D, Coffey M, Harrington KJ, et al. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1[J. J Immunother Cancer. (2019) 7:214. doi: 10.1186/s40425-019-0682-1
32. Kuryk L, Møller AW, and Jaderberg M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model[J. Oncoimmunology. (2019) 8:e1532763. doi: 10.1080/2162402X.2018.1532763
33. Simon S, Müller V, and Utikal JS. Case report: Therapeutic potential of T-VEC in combination with MEK inhibitors in melanoma patients with NRAS mutation[J. Front Oncol. (2023) 13:1111119. doi: 10.3389/fonc.2023.1111119
34. AuYeung A, Mould RC, Stegelmeier AA, Sharma H, Rowe MC, Stojdl DF, et al. Mechanisms that allow vaccination against an oncolytic vesicular stomatitis virus-encoded transgene to enhance safety without abrogating oncolysis[J. Sci Rep. (2021) 11:15290. doi: 10.1038/s41598-021-94483-z
35. Andtbacka R, Collichio F, Harrington KJ, Puzanov I, Hodi FS, Sznol M, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma[J. J Immunother Cancer. (2019) 7:145. doi: 10.1186/s40425-019-0623-z
36. Wong MK, Milhem MM, Sacco JJ, Hodi FS, Long GV, Chesney JA, et al. RP1 combined with nivolumab in advanced anti-PD-1-failed melanoma (IGNYTE)[J. J Clin Oncol. (2025) 43:101200JCO2501346. doi: 10.1200/JCO-25-01346
37. Shoushtari AN, Olszanski AJ, Nyakas M, Aksamitiene E, Grivas P, Postow MA, et al. Pilot study of ONCOS-102 and pembrolizumab: remodeling of the tumor microenvironment and clinical outcomes in anti-PD-1-resistant advanced melanoma[J. Clin Cancer Res. (2023) 29:100–9. doi: 10.1158/1078-0432.CCR-22-2046
38. Chesney JA, Ribas A, Long GV, Robert C, Hamid O, Daud AI, et al. Randomized, double-blind, placebo-controlled, global phase III trial of talimogene laherparepvec combined with pembrolizumab for advanced melanoma[J. J Clin Oncol. (2023) 41:528–40. doi: 10.1200/JCO.22.00343
39. Zhao X, Gao F, Yang J, Wang T, Zhang Y, Wang X, et al. Risk of adverse events in cancer patients receiving nivolumab with ipilimumab: A meta-analysis[J. Front Oncol. (2022) 12:877434. doi: 10.3389/fonc.2022.877434
40. Dülgar Ö, Saha A, Elleson KM, Ekiz HA, Grotz TE, Cheng Y, et al. Successful treatment with carboplatin and paclitaxel in melanoma progression after immune-related adverse events[J. Immunotherapy. (2023) 15:993–9. doi: 10.2217/imt-2022-0213
41. Huang Y-F, Xie W-J, Fan H-Y, Li S-S, Zheng J-C, Xu X, et al. Comparative risks of high-grade adverse events among FDA-approved systemic therapies in advanced melanoma: systematic review and network meta-analysis[J. Front Oncol. (2020) 10:571135. doi: 10.3389/fonc.2020.571135
42. Moreira A, Heinzerling L, Bhardwaj N, Friedlander P, Blank C, Westphal G, et al. Current melanoma treatments: where do we stand?[J. Cancers (Basel). (2021) 13:221. doi: 10.3390/cancers13020221
43. Monberg TJ, Pakola SA, Albieri B, Dominguez C, Hyvönen M, Ogawa Y, et al. Safety and efficacy of combined treatment with tumor-infiltrating lymphocytes and oncolytic adenovirus TILT-123 in metastatic melanoma. Cell Rep Med. (2025) 6:102016. doi: 10.1016/j.xcrm.2025.102016
44. Wang J, Chen Q, Shan Q, Liang T, Forde P, and Zheng L. Clinical development of immuno-oncology therapeutics. Cancer Lett. (2025) 617:217616. doi: 10.1016/j.canlet.2025.217616
45. Balasubramanian A, Salusti-Simpson M, Callas P, O’Neill C, Rehman H, Ahmed S, et al. Factors associated with response to talimogene laherparepvec in the treatment of advanced melanoma[J. MI. (2024) 1:95–105. doi: 10.36922/mi.3445
46. Willemsen M, Bulgarelli J, Chauhan SK, Miao Y, Mahoney KM, Rizos H, et al. Changes in AXL and/or MITF melanoma subpopulations in patients receiving immunotherapy[J. Immunooncol Technol. (2024) 24:101009. doi: 10.1016/j.iotech.2024.101009
47. Bommareddy PK, Zloza A, Rabkin SD, Kaufman HL, Martuza RL, Saha D, et al. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma[J. Oncoimmunology. (2019) 8:1591875. doi: 10.1080/2162402X.2019.1591875
48. Kaufman HL, Shalhout SZ, and Iodice G. Talimogene laherparepvec: moving from first-in-class to best-in-class[J. Front Mol Biosci. (2022) 9:834841. doi: 10.3389/fmolb.2022.834841
49. Sanchez Gil J, Fudaba H, and Wakimoto H. Chimeric oncolytic adenovirus to break away from neutralizing antibodies. Mol therapy: J Am Soc Gene Ther. (2024) 32:875–7. doi: 10.1016/j.ymthe.2024.03.017
50. Hodgins JJ, Abou-Hamad J, O’Dwyer CE, Donnelly CR, Ramos I, Cook L, et al. PD-L1 promotes oncolytic virus infection via a metabolic shift that inhibits the type I IFN pathway. J Exp Med. (2024) 221:e20221721. doi: 10.1084/jem.20221721
51. Holzgruber J, Martins C, Kulcsar Z, Praznik A, Krimbacher M, Leitner J, et al. Type I interferon signaling induces melanoma cell-intrinsic PD-1 and its inhibition antagonizes immune checkpoint blockade. Nat Commun. (2024) 15:7165. doi: 10.1038/s41467-024-51496-2
52. Wang S, Li Y, Xu C, Dong J, and Wei J. An oncolytic vaccinia virus encoding hyaluronidase reshapes the extracellular matrix to enhance cancer chemotherapy and immunotherapy. J immunotherapy Cancer. (2024) 12:e008431. doi: 10.1136/jitc-2023-008431
53. Bommareddy PK, Wakimoto H, Martuza RL, Kaufman HL, Rabkin SD, and Saha D. Oncolytic herpes simplex virus expressing IL-2 controls glioblastoma growth and improves survival. J immunotherapy Cancer. (2024) 12:e008880. doi: 10.1136/jitc-2024-008880
54. Jiang H, Nace R, Carrasco TF, Zhang L, Whye Peng K, and Russell SJ. Oncolytic varicella-zoster virus engineered with ORF8 deletion and armed with drug-controllable interleukin-12. J immunotherapy Cancer. (2024) 12:e008307. doi: 10.1136/jitc-2023-008307
55. Andtbacka R, Amatruda T, Nemunaitis J, Zager JS, Desai J, Bhanja P, et al. Biodistribution, shedding, and transmissibility of the oncolytic virus talimogene laherparepvec in patients with melanoma[J. EBioMedicine. (2019) 47:89–97. doi: 10.1016/j.ebiom.2019.07.066
56. Shmuylovich L, McEvoy AM, Fields RC, Clark LN, Le BH, Babar N, et al. Durable melanoma control following disseminated talimogene laherparepvec herpetic infection[J. JAAD Case Rep. (2022) 29:131–3. doi: 10.1016/j.jdcr.2022.09.012
57. U.S. Food and Drug Administration, Center for Biologics Evaluation and Research. Design and analysis of shedding studies for virus or bacteria-based gene therapy and oncolytic products: guidance for industry. (MD, USA: Silver Spring) (2015)
58. (EMA) EMA. Guideline on quality, non-clinical and clinical requirements for investigational advanced therapy medicinal products in clinical trials. Amsterdam, The Netherlands: European Medicines Agency (EMA). Vol. 20. (2025).
59. Maruyama Y. Regulatory update of cell and gene therapy products in Japan. Japan: Mesa, AZ, USA, other: Pharmaceuticals and Medical Devices Agency (PMDA). (2024).
60. Onnockx S, Baldo A, and Pauwels K. Oncolytic viruses: an inventory of shedding data from clinical trials and elements for the environmental risk assessment[J. Vaccines (Basel). (2023) 11:1448. doi: 10.3390/vaccines11091448
61. Administration USFaD. Determining the need for and content of environmental assessments for gene therapies, vectored vaccines, and related recombinant viral or microbial products: guidance for industry. FDA guidance document (2015).
62. Mortezaee K and Majidpoor J. Immunotherapy of human melanoma: past, present, future. Curr medicinal Chem. (2024) 32:3548–70. doi: 10.2174/0109298673283943240227104122
63. Santry LA, van Vloten JP, AuYeung A, Wilson MA, Wootton SK, Bell JC, et al. Recombinant Newcastle disease viruses expressing immunological checkpoint inhibitors induce a pro-inflammatory state and enhance tumor-specific immune responses in two murine models of cancer. Front Microbiol. (2024) 15:1325558. doi: 10.3389/fmicb.2024.1325558
64. Sierra-Davidson K, Dedeilia A, Lawless A, Liang J, Wong RW, Longino C, et al. Genetic factors associated with clinical response in melanoma patients treated with talimogene laherparapvec: A single-institution retrospective analysis. Ann Surg Oncol. (2025) 32:482–94. doi: 10.1245/s10434-024-16346-x
65. Doherty K. New data with RP1 plus nivolumab in PD-1–refractory melanoma build on positive findings. OncLive (June. (2024).
66. Whomsley R, Palmi Reig V, and Hidalgo-Simon A. Environmental risk assessment of advanced therapies containing genetically modified organisms in the EU[J. Br J Clin Pharmacol. (2021) 87:2450–8. doi: 10.1111/bcp.14781
67. Ono R, Nishimae F, Wakida T, Takehara M, Ito F, Nakayama T, et al. Effects of pre-existing anti-adenovirus antibodies on transgene expression levels and therapeutic efficacies of arming oncolytic adenovirus[J. Sci Rep. (2022) 12:21560. doi: 10.1038/s41598-022-26030-3
68. Li Q, Tan F, Wang Y, Gong J, Zhang S, Zhao X, et al. The gamble between oncolytic virus therapy and IFN[J. Front Immunol. (2022) 13:971674. doi: 10.3389/fimmu.2022.971674
69. Ballotti R, Cheli Y, and Bertolotto C. The complex relationship between MITF and the immune system: a Melanoma ImmunoTherapy (response) Factor?[J. Mol Cancer. (2020) 19:170. doi: 10.1186/s12943-020-01290-7
70. Chen Y, Chen X, Bao W, Liu G, Wei W, and Ping Y. An oncolytic virus-T cell chimera for cancer immunotherapy. Nat Biotechnol. (2024) 42:1876–87. doi: 10.1038/s41587-023-02118-7
71. Dummer R, Robert C, Scolyer RA, Long GV, Blank CU, Larkin J, et al. Neoadjuvant anti-PD-1 alone or in combination with anti-TIGIT or an oncolytic virus in resectable stage IIIB-D melanoma: a phase 1/2 trial. Nat Med. (2025) 31:144–51. doi: 10.1038/s41591-024-03411-x
72. Haugh A and Daud AI. Therapeutic strategies in BRAF V600 wild-type cutaneous melanoma. Am J Clin Dermatol. (2024) 25:407–19. doi: 10.1007/s40257-023-00841-0
73. Kulbay M, Tuli N, Mazza M, Hernandez F, Uden Y, Anastasopoulos I, et al. Oncolytic viruses and immunotherapy for the treatment of uveal melanoma and retinoblastoma: the current landscape and novel advances. Biomedicines. (2025) 13:108. doi: 10.3390/biomedicines13010108
74. Natarelli N, Aleman SJ, Mark IM, Walters S, Xie Q, Shirin T, et al. A review of current and pipeline drugs for treatment of melanoma. Pharm (Basel Switzerland). (2024) 17:214. doi: 10.3390/ph17020214
75. Izumikawa M, Minoda R, Kawamoto K, Tanaka K, Kojima H, Kuriyama H, et al. Auditory hair cell replacement and hearing improvement by Atoh1 gene therapy in deaf mammals[J. Nat Med. (2005) 11:271–6. doi: 10.1038/nm1193
76. Shirazi M, Saedi TA, Moghaddam Z, Hashemi M, Jafari S, Keshavarz M, et al. Nanotechnology and nano-sized tools: Newer approaches to circumvent oncolytic adenovirus limitations[J. Pharmacol Ther. (2024) 256:108611. doi: 10.1016/j.pharmthera.2024.108611
77. Andtbacka R, Curti B, Daniels GA, Hallmeyer S, Diab A, LaRocca R, et al. Clinical responses of oncolytic coxsackievirus A21 (V937) in patients with unresectable melanoma[J. J Clin Oncol. (2021) 39:3829–38. doi: 10.1200/JCO.20.03246
78. Cerqueira O, Antunes F, Assis NG, Luz A, Pina-Cabral S, Gonçalves R, et al. Perspectives for combining viral oncolysis with additional immunotherapies for the treatment of melanoma[J. Front Mol Biosci. (2022) 9:777775. doi: 10.3389/fmolb.2022.777775
79. Ranki T, Pesonen S, Hemminki A, Kipar A, Kairemo K, Guse K, et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers[J. J Immunother Cancer. (2016) 4:17. doi: 10.1186/s40425-016-0121-5
80. Béguin J, Gantzer M, Farine I, Beiner O, Zurbriggen A, Segura MM, et al. Safety, biodistribution and viral shedding of oncolytic vaccinia virus TG6002 administered intravenously in healthy beagle dogs[J. Sci Rep. (2021) 11:2209. doi: 10.1038/s41598-021-81831-2
81. Taipale K, Tähtinen S, Havunen R, Grundy Z, Guse K, Kipar A, et al. Interleukin 8 activity influences the efficacy of adenoviral oncolytic immunotherapy in cancer patients[J. Oncotarget. (2018) 9:6320–35. doi: 10.18632/oncotarget.23967
Keywords: oncolytic viruses, malignant melanoma, immunogenic cell death, tumor microenvironment, innate immunity, STING pathway, dendritic cells, precision immunotherapy
Citation: Wang J-W, Feng Q, Liu J-H and Xun J-J (2025) Opportunities, challenges, and future perspectives of oncolytic virus therapy for malignant melanoma. Front. Immunol. 16:1653683. doi: 10.3389/fimmu.2025.1653683
Received: 25 June 2025; Accepted: 20 August 2025;
Published: 04 September 2025.
Edited by:
Max Levin, University of Gothenburg, SwedenReviewed by:
Wang Yang, Hubei University of Technology, ChinaCopyright © 2025 Wang, Feng, Liu and Xun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-Jun Xun, NDY3MDA3MDRAaGVibXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship