 Ley Cody Smith
Ley Cody Smith Mohankumar Ramar
Mohankumar Ramar Gregory L. Riley III1
Gregory L. Riley III1 Clinton B. Mathias
Clinton B. Mathias Ji-Young Lee
Ji-Young Lee- 1Department of Pharmaceutical Sciences, University of Connecticut School of Pharmacy, Storrs, CT, United States
- 2Department of Nutritional Sciences, University of Connecticut, Storrs, CT, United States
The metabolism of immune cells adapts to support the energy demands for their activation, differentiation, and effector functions through a process known as metabolic reprogramming. This metabolic plasticity is influenced by both extrinsic and intrinsic factors, including steroid hormones such as glucocorticoids, androgens, progestogens, and estrogens. These critical mediators modulate immune function and inflammatory responses through genomic and non-genomic regulation of intracellular metabolic pathways, including glycolysis, the tricarboxylic acid cycle, and oxidative phosphorylation. Interestingly, these effects appear to be dependent on cell type, hormonal concentration, and microenvironmental context. Herein, we discuss how steroid hormones regulate inflammation and immunometabolism and summarize recent studies highlighting immunometabolic regulation by steroid hormones as the key driver of their immunomodulatory effects. We also address potential mechanisms contributing to their seemingly dichotomous and context-specific regulation. Understanding the link between steroid hormone signaling, immunometabolism, host defense, chronic inflammation, and immunity will expand our understanding about how biological sex and stress influence the immune system and facilitate more precise therapeutic targeting of immune cell activity to mitigate inflammation- and immune-mediated diseases.
1 Introduction
Immune cells modify the metabolic pathways by which they generate energy to appropriately respond to sterile and infectious stimuli (1). This metabolic reprogramming is triggered by the activation of pattern recognition receptors and cytokine receptors that initiate signaling cascades and activate diverse transcriptional regulators that induce or inhibit the expression of substrate uptake transporters, enzymes, and mitochondrial proteins. In turn, this shifts energy production between aerobic glycolysis, the TCA cycle, and oxidative phosphorylation (2). Changes in metabolism can also modulate immune cell activity, as highlighted by numerous studies reporting that modifying substrate availability or utilization can influence immune cell proliferation, differentiation, activation, and effector functions (3). Thus, understanding the intricate and bidirectional relationship between intracellular metabolism and immune cell activation and function is essential to develop targeted therapeutic strategies to modulate immune and inflammatory responses and mitigate disease.
Immune cell activation and function are regulated by sex- and environmental-based factors. For example, males and females respond to foreign and self-antigens differently, and these responses may change over the course of the lifespan, highlighting gonadal sex hormones as major immunoregulatory agents. In this regard, females exhibit increased susceptibility to autoimmune disease development whereas males show increased susceptibility to non-reproductive malignant cancers (4). Environmental factors such as stress are also known to affect susceptibility to illness by modulating the immune system, sometimes in a sexually dimorphic manner (reviewed in 5 and 6). Underpinning these sex- and environmental-based differences are the actions of steroid hormones, including estrogens, progestogens, androgens, and glucocorticoids. The steroid hormones are derived from cholesterol and regulate reproduction, stress responses, metabolism, and immunity (7). Their levels fluctuate following circadian and ultradian rhythms throughout the estrus (in rodents) and menstrual (in humans) cycle, as well as throughout the lifespan (8–12).
Steroid hormones signal through their cognate steroid hormone receptors, NR3 class nuclear receptors that share similar structures, including an N-terminal transactivation domain, a highly conserved central DNA binding domain, and a C-terminal ligand-binding domain (13). Upon ligand binding, the receptors undergo conformational changes that facilitate the recruitment of co-regulatory proteins and their translocation to the nucleus, where they interact with hormone response elements (HREs) to regulate gene transcription (13). In addition to these direct genomic mechanisms, some steroid hormone receptors (e.g., estrogen receptors and the glucocorticoid receptor) can indirectly regulate gene expression at distinct promoters by tethering to other transcription factors (e.g., AP-1 and NF-κB) (14, 15). The steroid hormones also signal through non-genomic mechanisms initiated at the cell membrane by palmitoylated steroid hormone receptor variants or GPCRs (e.g., G-protein coupled estrogen receptor 1, GPER1); these receptors activate kinase pathways (e.g., MAPK/ERK and phosphoinositide 3-kinase [PI3K]/protein kinase B [Akt]) and modulate gene expression by regulating the activity of the cytoplasmic nuclear receptors and growth factor receptors (16).
Several reviews have thoroughly described the roles of steroid hormones in regulating intracellular metabolic pathways and immune cell development, activation, and function separately, but they rarely connect the two (17–21). Interestingly, these effects appear to vary depending on the cellular microenvironment, cell type, disease or life stage, and concentration of ligand. Parallel to these discordant responses on immune cells are corresponding changes in intracellular metabolism. Thus, the purpose of this review is to explore the interaction between metabolism and immune regulation with a focus on steroid hormone modulation as a key mediator of these seemingly dichotomous effects. This review is focused on immune cells which have been observed to exhibit metabolic reprogramming and be regulated by steroid hormones; these include T cells, macrophages, myeloid-derived suppressor cells (MDSCs), and dendritic cells.
2 Metabolic reprogramming in immune cells
Immune cells play a pivotal role in protecting us from disease and in maintaining tissue integrity during conditions of homeostasis. The rapid activation and resolution of immune and inflammatory responses, accompanied by the secretion of numerous proteins, lipids, and other mediators, incur a significant bioenergetic cost requiring the increased uptake of glucose, fatty acids, and amino acids. This often requires a shift in the metabolism of immune cells corresponding to the state they are in, i.e., quiescent, effector, memory, or regulatory. Each of these states can present with a different metabolic phenotype.
2.1 T cells
T cells are the major cellular mediators of the adaptive immune system and comprise diverse subpopulations with unique effector functions (22). Both T cell development and function are marked by distinct stages that are associated with changes in morphology and phenotype. Furthermore, the induction of an immune response is accompanied by rapid shifts in T cell status including naïve, activated, effector, and memory phenotypes that come at a significant bioenergetic cost to the cell. Metabolic reprogramming fuels the various stages allowing T cells to maintain optimal metabolic fitness and prime for maximum responsiveness during states of heightened immune activity (23). In T-cell development, elevated expression of hypoxia-inducible factor 1-alpha (HIF-1α) leads to increased induction of glucose transporter type 1 (GLUT1) and enhanced glycolysis and flux through the pentose phosphate pathway to support β-selection (24–26). Then, CD4 or CD8 selection results in a sharp reduction in glucose metabolism and shift to mitochondrial oxidative phosphorylation through down-regulation of HIF-1α (26). This shift is essential for the maturation of memory-phenotype T cells (27).
Naïve or quiescent T cells have minimal metabolic needs and maximize the glycolysis and oxidative phosphorylation pathways by fully oxidizing glucose and generating ATP. The predomination of these pathways provides these cells with the energy required to conduct immune surveillance (1). In contrast, the processes of T cell activation, clonal expansion, differentiation into various effector T cell subsets, and subsequent effector function are characterized by increased energetic demands, resulting in the successive generation and replenishment of biosynthetic precursors that power rapid cell growth and proliferation (23, 28). In CD4+ cells, Th1 and Th17 activation is associated with increased expression of glutamine transporters, which provides more substrates for the TCA cycle as well as enhanced fatty acid biosynthesis (29–31). On the other hand, Th2 and T regulatory cell (Treg) differentiation and activation involve transitions to fatty acid oxidation for energy acquisition (30, 32). In CD8+ T cells, increased expression of GLUT1, lactate dehydrogenase, and hexokinase and a concomitant reduction in carnitine palmitoyltransferase 1A (CPT1a) expression shifts metabolism from fatty acid oxidation to glycolysis, supporting the synthesis of nucleotides and serine for activation (33).
At the end of an immune response, the cells that survive to become memory T cells revert back to lipid oxidation with an increased capacity for efficient energy generation. Because memory T cells do not proliferate or produce cytokines, they have a similar metabolic profile as naïve T cells, wherein fatty acid oxidation and oxidative phosphorylation are sufficient to sustain T cell survival and to meet their energetic requirements. However, unlike naïve T cells, they have increased spare respiratory and glycolytic capacities, with elongated mitochondria and increased expression of electron transport chain proteins. As such, memory T cells exist in a state of improved metabolic fitness where they are ready to engage pathogens upon antigen stimulation (34).
2.2 Macrophages
Macrophages are innate immune cells that play important regulatory roles in various tissues and are essential in host defense. They comprise diverse subpopulations characterized by their ontogeny and polarization state (35). The subpopulations are broadly classified into two phenotypes: pro-inflammatory/M1 that promotes Th1-mediated responses against intracellular bacteria with increased phagocytic activity, and an anti-inflammatory/M2 phenotype that promotes type 2 inflammation (36). Macrophages undergo metabolic reprogramming to efficiently perform their major functions of immunomodulation, phagocytosis, and antigen presentation (35, 37). Tissue resident macrophages are long-lived cells and thus rely on fatty acid oxidation and oxidative phosphorylation for both redox balance and prolonged survival (38–40). Conversely, proinflammatory macrophages upregulate glycolysis and exhibit two breaks in the TCA cycle at succinate dehydrogenase (SDH) and isocitrate dehydrogenase (IDH), leading to the accumulation of citrate and succinate (41–43). These metabolites increase flux through the pentose phosphate pathway and upregulate fatty acid synthesis to promote the synthesis of ROS and intermediates for proinflammatory cytokine production and cell proliferation (39, 44–46).
During the resolution of inflammation, macrophages accumulate glutamine from the extracellular environment or through the engulfment of apoptotic cells and break it down into the TCA cycle intermediate α-ketoglutarate (47, 48). The resulting upregulation of oxidative phosphorylation facilitates enhanced efferocytosis and production of the anti-inflammatory and pro-resolving cytokine, IL-10 (47, 49). Enhanced IL-10 signaling potentiates oxidative phosphorylation and inhibits glucose uptake, further suppressing glycolysis (50, 51). It should be noted that there is some controversy regarding the requirement of fatty acid oxidation for anti-inflammatory activation (52–58). Nonetheless, the clear reliance of pro-inflammatory macrophages on aerobic glycolysis and that of anti-inflammatory and tissue-resident cells on oxidative phosphorylation suggests that targeting metabolic reprogramming may be an efficacious strategy to modulate macrophage activity.
Recent studies suggest that crosstalk between macrophages and other immune cells such as mast cells may affect their metabolic fates and enhance functional outcomes during immune activity. Holter et al. demonstrated that IgE-activated mast cells can drive macrophage polarization, phagocytosis, and antibacterial activity by influencing metabolic and epigenetic processes in macrophages (59). Macrophages exposed to supernatants from IgE-activated mast cells exhibited increased basal oxygen consumption rates (representative of mitochondrial respiration), ATP production, maximal respiration, and spare respiratory capacity compared to cells exposed to supernatants from non-sensitized mast cells. This suggests that during inflammatory states such as allergic responses, crosstalk between immune cells may have the potential to regulate metabolic plasticity and function.
2.3 Myeloid-derived suppressor cells
Myeloid-derived suppressor cells (MDSCs) comprise pathologically activated neutrophils and monocytes that exhibit potent immunosuppressive activity (60). In cancer, MDSCs adapt to the anoxic tumor microenvironment by upregulating mechanistic target of rapamycin (mTOR) and HIF-1α signaling, as well as expression of lipid uptake scavenger receptors (e.g., CD36 and macrophage scavenger receptor 1 [MSR1]) and key fatty acid oxidation enzymes (e.g., CPT1A, acyl-CoA dehydrogenase medium chain [ACADM], hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit alpha [HADHA], and peroxisome proliferator-activated receptor coactivator 1 beta [PGC-1β]) (61–64). This leads to a simultaneous increase in glucose uptake and glycolysis, as well as lipid uptake and oxidative phosphorylation that have both been shown to enhance the immunosuppressive activity of these cells (65–67).
2.4 Dendritic cells
Dendritic cells are professional antigen-presenting cells that link the innate to the adaptive immune system. They detect and proteolytically degrade pathogen-derived components, which are in turn loaded onto MHC class I or class II molecules to activate cytotoxic CD8+ T cells or helper CD4+ T cells, respectively (68). Dendritic cells encompass diverse subpopulations, including conventional cDC1s that are efficient at cross-presenting antigens to CD8+ T cells and cDC2s that specialize in CD4+ T cell activation (69). Plasmacytoid dendritic cells (pDCs) are critical in viral defenses by producing Type-I interferon, whereas monocyte-derived, inflammatory DCs (infDCs) regulate inflammation and infection (70). The differentiation and activation of dendritic cells are regulated by intracellular metabolism (71). At baseline, cDC1s are more heavily reliant on oxidative phosphorylation compared to cDC2s or pDCs, likely due to their larger mitochondrial mass and membrane potential (72–74). Treatment with LPS and poly(I:C) was shown to induce PI3K/AKT, nitric oxide (NO), and BNIP33 signaling that resulted in a rapid increase in glucose uptake and glycolytic flux, as well as a decrease in oxidative phosphorylation in cDC1s, cDC2s, pDCs, and infDCs (75–81). The requirement for this metabolic reprogramming in dendritic cell activity is evidenced by studies showing that inhibition of glycolysis with 2-deoxyglucose (2-DG) or knockdown of glycolytic inducers [e.g., HIF-1α and alpha enolase (ENO1)], impaired dendritic cell maturation, and subsequent T cell activation (78, 80, 82, 83). Similarly, in a model of allergic asthma, intranasal treatment with D-2-hydroxyglutarate suppressed allergic sensitization mediated by dendritic cells and promoted the activity of follicular regulatory T cells (84).
3 Steroid hormone regulation of inflammation
3.1 Glucocorticoids
The production of endogenous glucocorticoids is rapidly induced in response to inflammation and other stressors, but the secretion patterns are also affected by circadian and ultradian rhythms (85). Glucocorticoid receptors are expressed in essentially all immune cells in mice and humans where they exhibit time-, concentration-, cell type-, and in some cases, species-specific effects (86). Glucocorticoids bind to cytosolic glucocorticoid receptors and regulate gene expression by interacting with glucocorticoid response elements (GREs) in gene promoters. In addition, they induce transcription through protein-protein interactions in the cytosol and by composite binding to other transcription factors at DNA sequences containing GREs and a response element of a distinct transcription factor (85, 87). Glucocorticoids may also signal through membrane-bound glucocorticoid receptors to mediate rapid signaling (88). High concentrations or chronic administration of glucocorticoids promote apoptosis of immune cells, including T cells, macrophages, dendritic cells, and eosinophils in mice and humans (18, 89), whereas lower concentrations and shorter exposure windows exhibit more nuanced effects on inflammation and immunity.
In murine macrophages, glucocorticoids generally suppress production of pro-inflammatory eicosanoids (e.g., prostaglandins and leukotrienes), and pro-inflammatory cytokines (e.g., IL-1β, IL-6, TNFα, and IL-12), and they inhibit leukocyte migration (85, 90–94). Interestingly, expression of pro-inflammatory cytokines (e.g., IL-6 and TNFα) was significantly reduced in male rat livers compared to females after LPS administration, a response that correlated with enhanced survival (95). Conversely, the estrogen receptor antagonist ICI 182,780 was found to inhibit dexamethasone-induced reductions in TNFα, IL-1β, and iNOS in the lungs of rats treated with carrageenan (96).
Glucocorticoids also promote the resolution of inflammation by inducing expression of scavenger receptors (e.g., CD153, CD206, and MERTK), anti-inflammatory cytokines (e.g., TGF-β and IL-10), and the receptor for the pro-resolving lipid mediator, lipoxin A4, in human and murine macrophages, all of which potentiate macrophage phagocytosis and efferocytosis (97–101). Dichotomous effects have been observed in human T cells in which glucocorticoids suppress pro-inflammatory responses of Th1 and Th17 cells but promote the function of anti-inflammatory and immunosuppressive Th2 and Treg cells (85, 89, 102). Likewise, the expression of pro-inflammatory cytokines (e.g., IL-12 and TNFα) is reduced while that of anti-inflammatory cytokines (e.g., IL-10) is increased in dendritic cells treated with glucocorticoids (103). These effects on pro- and anti-inflammatory signaling and immune responses have resulted in their widespread use in asthma therapy and are associated with distinct changes in intracellular metabolism and metabolic reprogramming as described below (104, 105).
3.2 Androgens
Androgens are commonly known as male sex hormones but are produced by both sexes (106). The main androgen, testosterone, along with other androgens such as androstenedione, are produced by testicular Leydig cells, ovarian theca cells, and adrenal zona reticularis cells (107). Testosterone is locally converted into the more potent dihydrotestosterone (DHT) by 5α-reductase (108). Androgens signal by binding to the androgen receptor (AR), which interacts with androgen response elements (AREs) and recruits co-activators and co-repressors to regulate transcription of AR target genes. While no membrane-bound AR has been reported, androgens may mediate rapid, non-genomic effects by interacting with kinases such as SRC and PI3K in the cytoplasm (109). Androgen receptors are expressed by murine and human macrophages, monocytes, neutrophils, T cells, and B cells, in which they exhibit predominantly anti-inflammatory and immunosuppressive effects (17).
In human monocyte-derived macrophages, murine peritoneal macrophages, and human and murine macrophage cell lines (e.g., RAW 264.7 and J774A.1), testosterone dose-dependently inhibited LPS-induced production of nitric oxide (NO) and TNFα, as well as expression of inducible nitric oxide synthase (iNOS/Nos2) and TLR4 (110–115). Likewise, testosterone treatment increased production of anti-inflammatory IL-10 after LPS stimulation in a human macrophage cell line (111). Similar findings have been reported in T cells where androgens decreased murine and human T cell numbers and dampened the production of type I (T1) and type II (T2) inflammatory mediators (e.g., IFNγ, IL-4, IL-5) while enhancing production of immunosuppressive IL-10 (17, 116–121).
3.3 Progestogens
The main natural progestogen, progesterone (P4), is synthesized from cholesterol by the cholesterol side-chain cleavage enzyme and 3β-hydroxysteroid dehydrogenase (122). Progesterone signals by binding to progesterone receptors, which have two isoforms (A and B) that exhibit tissue-specific expression and functions (123). They exert agonistic and antagonistic effects on other steroid hormone pathways by interacting with other receptors, including estrogen receptors, and androgen receptor, glucocorticoid receptor, and mineralocorticoid receptor (123–126). Progesterone receptors are expressed by murine and human T cells, murine dendritic cells, human natural killer cells, and murine and human macrophages, where they generally suppress pro-inflammatory and enhance immunosuppressive activity (123, 127, 128).
For example, progesterone inhibited NF-κB signaling and reduced NO and IL-12 production in LPS-stimulated murine bone marrow-derived and alveolar macrophages, respectively (129–132). In this regard, the synthetic progestin, medroxyprogesterone acetate, triggered the differentiation of the human monocyte cell line, THP-1, into a more anti-inflammatory phenotype (e.g., upregulated CD163 and IL-10 expression) (133). In human, murine, and bovine CD4+ T cells, progesterone promotes Th2 and Treg expansion and activation (increased IL-4 and IL-10) and inhibits Th17 differentiation (reduced IL-2 and IL-12) (127, 134–138). Likewise, progesterone inhibited LPS and TLR-driven responses in human dendritic cells (e.g., reduced IL-1β, IL-6, and IFN-β); these immunosuppressive effects are important in ensuring successful pregnancies by preventing fetal rejection (128, 139–142).
3.4 Estrogens
Endogenous estrogens are steroid hormones derived from cholesterol (e.g., estrone [E1], 17β-estradiol [E2], estriol, estetrol [E4], and 27-hydroxycholesterol), which exhibit tissue- and life stage-specific production (143, 144). The different estrogens exhibit distinct pharmacology, with E2 exhibiting the greatest affinity for the estrogen receptors (ERs) (143, 144). Estrogens signal through myriad pathways, including genomic mechanisms mediated by two nuclear receptors (ERα and ERβ) that recruit various co-regulatory proteins and induce transcription at estrogen response elements (EREs), in addition to other promoters by tethering to transcription factors (e.g., AP-1 and NF-κB). Estrogens also signal through membrane-bound receptors (e.g., GPER1 and palmitoylated-ERα variants) to initiate non-genomic signaling, and these pathways often intersect and cross-regulate each other (145–148). ERα is the most widely expressed receptor isoform in immune cells, with positive expression in human and murine natural killer cells, T cells, neutrophils, eosinophils, dendritic cells, monocytes, and macrophages (113), while ERβ expression is less widespread and heavily debated (113, 149). GPER1 has been detected in human neutrophils and eosinophils, and palmitoylated ERα variants in natural killer cells, monocytes, and macrophages (113).
The effects of estrogens on immunity have been thoroughly reviewed by multiple groups who report cell- and disease-specific effects (19, 113). In acute inflammation models, E2 appears to enhance TLR-mediated pro-inflammatory responses in human and murine macrophages while also promoting T2 and anti-inflammatory activity in allergic asthma and cutaneous wound healing (110, 115, 150–157). The positive regulation of pro-inflammatory macrophage activity appears to be dependent on activation of STAT1, whereas the increased anti-inflammatory activation is driven by ERα-mediated phosphorylation and activation of STAT3 and interference with p65 subunit binding to the NF-κB complex (158–161). Estrogens modulate T cell activity throughout their entire life cycle, and similar discordant results have been reported in diverse T cell subpopulations. For example, E2 increased human and murine Th1 differentiation by inducing T-bet and enhancing IFNγ production while also promoting IL-4 production and favoring Treg expansion (162–169). These disparate results are likely due to the differences in the cellular microenvironment, cell type-specific utilization of coregulatory proteins, and concentration-dependent effects of estrogens (170–172). Several studies also suggest that estrogen may modulate human and murine mast cell behavior and activity during allergic diseases and other conditions such as endometriosis (173–175).
4 Immunometabolism as the link between steroid hormones and inflammation and immunity
4.1 Glucocorticoids
As discussed, glucocorticoid receptor signaling promotes anti-inflammatory and immunosuppressive activities of innate immune cells, including macrophages and myeloid-derived suppressor cells. Emerging evidence indicates that this is driven by metabolic changes. In a zebrafish model of hair cell ablation injury, activation of glucocorticoid signaling initiated a linear sequence of IL-10, polyamine, and IL-4 signaling, culminating in an increase in the expression of genes involved in oxidative phosphorylation facilitating wound repair (176). Other studies have shown that cortisol increased the expression of carnitine palmitoyltransferase II (CPT2) and glutaminase (GLS) in human monocyte-derived macrophages; this upregulated fatty acid oxidation and glutaminolysis, respectively, resulting in TCA cycle anaplerosis (177). In this regard, dexamethasone increased the expression of carnitine palmitoyltransferase I (CPT1) in human myeloid-derived suppressor cells, leading to an increase in fatty acid oxidation, mitochondrial respiration, and enhanced immunosuppressive activity (178). Collectively, these studies indicate that glucocorticoid receptor signaling promotes immunosuppression and the resolution of inflammation by enhancing oxidative phosphorylation in innate immune cells.
In addition, glucocorticoids have been found to indirectly increase mitochondrial respiration by inhibiting glycolytic activity. For example, treatment of macrophages (e.g., murine bone marrow-derived macrophages [BMDMs] and human THP-1 cells) and myeloid-derived suppressor cells with dexamethasone after exposure to E. coli, LPS, and ovalbumin (OVA) inhibited glycolysis and led to a compensatory increase in glutamine metabolism, anaplerosis, TCA cycle flux, and a restoration of basal and maximal oxidative phosphorylation (179–181). These studies highlighted reduced expression and inhibition of HIF-1α activity and subsequent reductions in expression of key glycolytic enzymes (e.g., Glut1, PDK, pyruvate kinase M2 [PKM2], lactate dehydrogenase a [LDH], and hexokinases) as major drivers of this metabolic plasticity (179, 180, 182). Of note, the shift toward oxidative phosphorylation was associated with reduced expression of T1 and T2 cytokines (e.g., IL-4, -5, -13, -1β), iNOS, and ROS production in these cells (179–181, 183). Thus, in addition to direct effects on oxidative phosphorylation, GR signaling downregulates HIF-1α, leading to a suppression of glycolysis and impaired pro-inflammatory activation; this results in a compensatory shift toward oxidative phosphorylation, enhancing anti-inflammatory and immunosuppressive activity.
The well-established immunosuppressive effects of glucocorticoids on T cells appear to also be mediated, in part, by changes in intracellular metabolism. Dexamethasone inhibited mRNA expression of glycolytic proteins [e.g., Glut1, Glut3, Ldha) in mouse and human T cells and leukemic cells (184, 185)]. This was associated with reduced pro-inflammatory gene expression (e.g., Il2, Ifng) (185). In Tregs, dexamethasone enhanced immunosuppressive activity by reducing glycolysis and increasing oxidative phosphorylation through miR-342-dependent inhibition of rapamycin-insensitive companion of mTOR (RICTOR) (186). It should be noted that the immunosuppressive effects of glucocorticoids are not always desirable. For example, the glucocorticoid, prednisolone, impaired the differentiation and effector function of memory CD8+ T cells by reducing glycolysis; this resulted in impairments in memory formation, recall antigen responses, and anti-tumor activity (187).
Conversely, glucocorticoids have also been shown to suppress oxidative phosphorylation; however, this appears to be a dose-dependent effect. For example, high-dose dexamethasone reduced mRNA and protein expression of mitochondrially encoded cytochrome c oxidase subunit I (MT-CO1) in murine macrophages and PKM2 expression in human chronic lymphocytic leukemia cells (188, 189). This was associated with reduced mitochondrial respiratory chain complex IV activity and loss of mitochondrial membrane potential, respectively, and increased apoptosis (188, 189). Thus, selectively regulating pro- or anti-inflammatory activity of immune cells may require targeting distinct metabolic pathways by careful titration of glucocorticoids.
4.2 Androgens
Although androgens do not appear to directly modulate glycolysis, they have been shown to regulate the expression of genes involved in oxidative phosphorylation. An analysis of previously published single-cell RNA-sequencing data collected from the prostates of castrated male mice identified reduced expression of complex (C) I, CIV, and CV subunits of the mitochondrial oxidative phosphorylation system in macrophage and T cell clusters. Of note, this response was reversed after administration of exogenous testosterone (190). In murine myeloid-derived suppressor cells, androgen receptor up-regulated mitochondrial pyruvate carrier 2 (MPC-2) expression to enhance TCA cycle flux, mitochondrial respiration, and immunosuppressive activities (191). Inhibition of the androgen receptor with enzalutamide reduced oxidative phosphorylation, which in turn resulted in a compensatory increase in GLUT1 expression, glucose uptake, and glycolytic flux (191). The authors suggested this metabolic plasticity led to persistent immunosuppressive activity and limited the effectiveness of anti-androgen therapy in prostate cancer (191). In a mouse model of allergic asthma, androgen receptor signaling was found to restrict allergen-induced airway inflammation and airway hyperresponsiveness by reducing glutamine uptake and glutaminolysis in CD4+ T cells by reducing the expression of glutamine uptake transporters (e.g., Slc1a5 and Slc38a1) (192). The reduction in glutamine shunted metabolism away from the TCA cycle and decreased glutathione levels; this resulted in increased ROS production, which prevented Th17 differentiation and effector function by promoting a regulatory phenotype (192). Thus, the effects of testosterone on bioenergetics and inflammatory activity appear to depend on the cell type.
4.3 Progestogens
In general, progesterone seems to enhance the metabolic activity and function of myeloid cells. Gonadectomy of female rats reduced the production of hydrogen peroxide and phagocytic capacity of both unstimulated macrophages and those stimulated by phorbol myristate acetate (PMA). Progesterone supplementation reversed these effects, and this was associated with an increase in glutaminase activity (193). In immature human dendritic cells, progesterone increased expression of mitochondrial complex II-V genes (succinate dehydrogenase complex iron sulfur subunit B, [SDHB], succinate dehydrogenase complex subunit C [SDHC], ubiquinol-cytochrome C reductase complex III subunit VII [UQCRQ], cytochrome c oxidase subunit II [COX II], and ATP synthase F1 subunit alpha [ATP5A]), and mitochondrial pyruvate carrier 1 (MPC1) (194). In addition, genes involved in fatty acid oxidation (e.g., hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit beta [HADHB], CPT1, and CPT2), as well as genes involved in fatty acid synthesis (e.g., acetyl-CoA carboxylase alpha [ACACA] and fatty acid synthase [FASN]) were upregulated (194). Another study by the same group found that progesterone also increased mRNA expression of glycolytic enzymes [e.g., HK2, LDHA, and pyruvate dehydrogenase (PDH)] in immature dendritic cells (194). This was associated with enhancements in parameters of oxidative phosphorylation (e.g., basal and maximal oxidative phosphorylation and spare respiratory capacity), as well as glycolytic capacity, and increased cell-surface expression of immunosuppressive inhibitor receptor Ig-like transcript 4 (ILT4) (194). The authors concluded that the progesterone-mediated enhancement of metabolic activity in immature dendritic cells helped maintain immune tolerance in the decidual microenvironment to prevent fetal rejection and ensure reproductive success.
4.4 Estrogens
Estrogens appear to differentially regulate intracellular bioenergetics, inflammatory, and immune responses in a cell type- and microenvironment-dependent manner. Spare respiratory capacity, differentiation, and activation were reduced in murine Th17 cells lacking ERα (195). Similarly, treatment of murine RAW 264.7 cells with E2 blunted LPS/IFNγ-induced reductions in basal oxidative phosphorylation, and this was associated with reduced TNF-α and increased IL-10 secretion compared to cells treated with LPS/IFNγ; however, no response was observed in murine BMDMs (196). It appears that the effects on mitochondrial function and inflammatory activity were mediated by E2-dependent increases in Sirt3 expression and mitochondrial protein acetylation (196). A key role for ERα was established in one study showing that murine BMDMs deficient in ERα exhibited reduced fatty acid oxidation compared to wild types (155). In contrast, other studies have found that E2 inhibited receptor activator of nuclear factor-κB ligand (RANKL)-induced upregulation of oxidative phosphorylation (197–199). The authors of the latter studies concluded that the reduced mitochondrial respiration led to enhanced apoptosis and postulated that this immunosuppressive effect of E2 explained why post-menopausal women exhibit increased osteoclast activity and bone resorption (197–199). Interestingly, these seemingly dichotomous effects of E2 on intracellular bioenergetics could be a result of differential receptor activation, as E2 was found to inhibit F0F1-ATP synthase activity via non-genomic mechanisms in a human osteoclastic cell line (FLG 29.1 cells) (200). In this regard, E2 rapidly increased lactate production in human MCF-7 breast cancer cells in a PI3K/AKT-dependent mechanism that was speculated to be initiated by a membrane-bound pool of ERα (201).
In murine alveolar macrophages, genetic deletion of ERα resulted in increased glycolytic capacity and glycolytic reserve with no effect on mitochondrial oxidative phosphorylation; these data suggest that ERα functions to suppress glycolysis (202). It appears this may be a result of the alveolar microenvironment, which is characterized by low glucose levels. One study found that in high glucose conditions, E2 dose-dependently increased lactate and glucose-6-phosphate (G6P) production in human MCF-7 breast cancer cells, whereas in low glucose conditions, E2 dose-dependently reduced lactate production and enhanced TCA cycle flux (201). These results indicate that the local fuel supply can dictate the effect of E2 on metabolic plasticity and may explain the observations of increased glycolysis in ERα-deficient murine alveolar macrophages. While estrogen also regulates mitochondrial DNA transcription, mitochondrial morphology, and mitochondrial fission and fusion in breast cancer cells (reviewed in 203), these mechanisms are understudied in the context of immune cells.
Estrogenic endocrine-disrupting chemicals also influence macrophage activity by modulating intracellular bioenergetics. Exposure of murine J774A.1 cells to the weakly estrogenic plasticizer, bisphenol S (BPS), increased levels of glycolytic metabolites (e.g., lactate, pyruvate, and glucose-6-phosphate) and increased mRNA and protein expression of glycolytic enzymes (e.g., PKM2, LDHA, hexokinase 1 [HK1], and HK2). The BPS-induced enhancements in glycolytic activity were associated with increased expression of pro-inflammatory (e.g., TNF-α, IL-1β, and IL-6) and reduced expression of anti-inflammatory (e.g., TGFβ and IL-10) cytokines and growth factors (204). Another study found that the analogous plasticizer, bisphenol F (BPF), dose-dependently increased expression of glycolytic enzymes (e.g., HIF-1α, GLUT1, PKM2), lactate production, and glucose consumption, and increased the secretion of pro-inflammatory cytokines (e.g., TNF-α, IL-1β, and IL-6) in murine RAW 264.7 cells; this was confirmed to be through an ERα and PI3K/AKT-dependent mechanism (205). These studies highlight environmental estrogens as immunomodulatory agents and demonstrate the critical role of metabolic plasticity in pro- and anti-inflammatory macrophage activation.
5 Discussion
Immune cells undergo metabolic reprogramming to meet the energetic and biosynthetic demands of their activation, differentiation, and effector functions. This reprogramming involves dynamic shifts between glycolysis, the TCA cycle, and oxidative phosphorylation and is influenced by both intrinsic signaling pathways and extrinsic factors such as steroid hormones. Steroid hormones—including glucocorticoids, androgens, progestogens, and estrogens—regulate immune responses by modulating intracellular metabolism in rodents and humans, although species-specific effects have been reported. The effects of steroid hormones can also vary depending on the microenvironment and disease-state. Nonetheless, these findings underscore the intricate and bidirectional relationship between metabolism and immune regulation and highlight steroid hormones as key modulators of immunometabolic activity across multiple immune cell types (Figure 1).
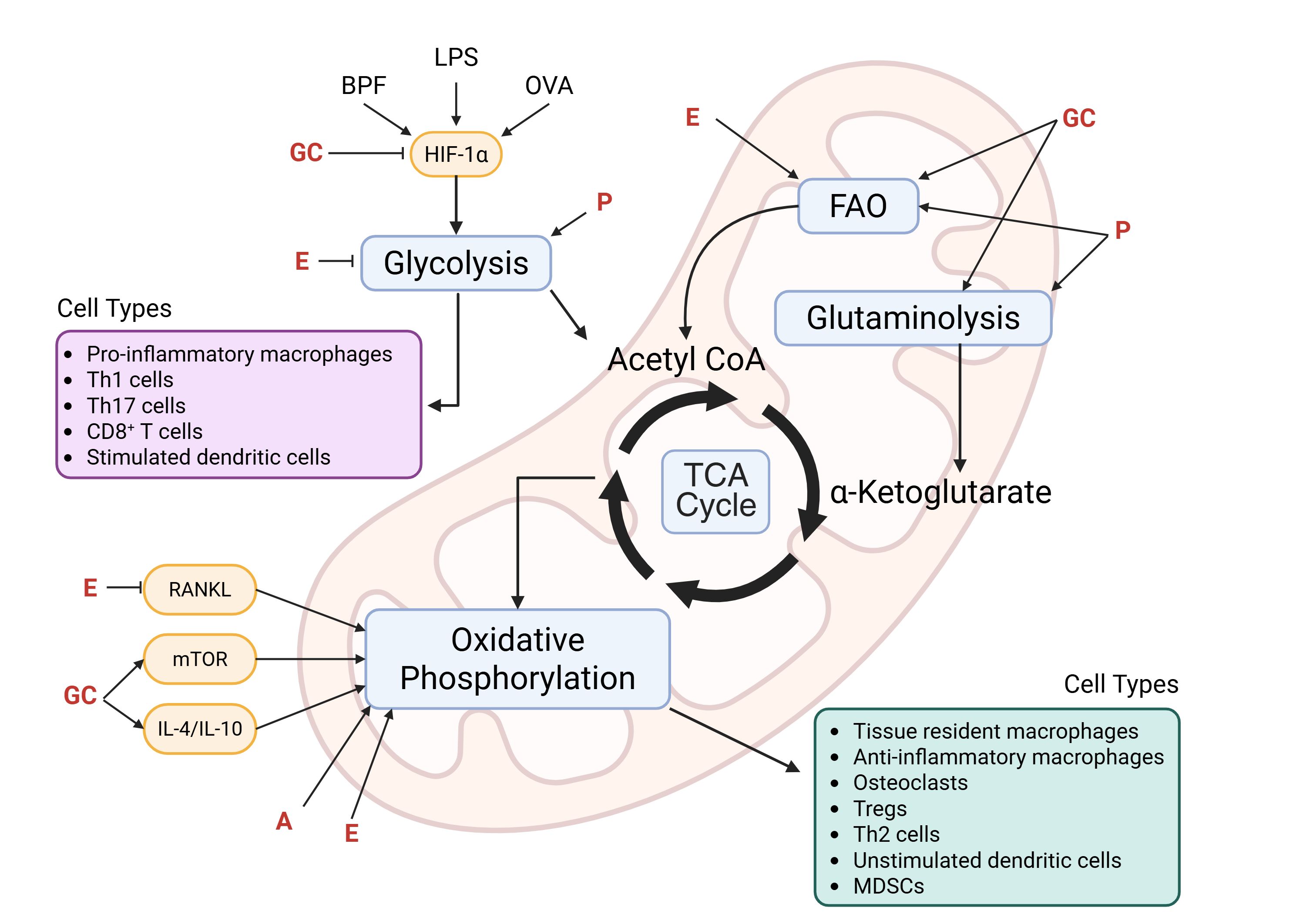
Figure 1. Immunometabolic targets of steroid hormones. Activated cells (e.g., pro-inflammatory macrophages, Th1, Th17, CD8+ T cells, and stimulated dendritic cells) preferentially rely on glycolysis and exhibit dysregulation of the TCA cycle. This rapidly generates glycolytic intermediates that can be used for cell proliferation and production of ROS and inflammatory mediators. On the other hand, immunomodulatory cells (e.g., anti-inflammatory macrophages, Tregs, Th2 cells, and myeloid-derived suppressor cells [MDSCs]), tissue resident cells (e.g., macrophages and osteoclasts), and unstimulated dendritic cells, are long-lived and rely on the more efficient oxidative phosphorylation pathway to satisfy their energy requirements. Key mediators (e.g., hypoxia-inducible factor 1-alpha [HIF-1α], receptor activator of nuclear factor-κB ligand [RANKL], mechanistic target of rapamycin [mTOR], interleukin [IL]-4, and IL-10) that are known to be activated by inflammatory stimuli (e.g., bisphenol-F [BPF], lipopolysaccharide [LPS], and ovalbumin [OVA]) and/or modulated by steroid hormones are depicted in yellow, and the immunometabolic pathways they target are shown in blue. Of note, some steroid hormones exhibit dichotomous regulation driven by cell type-, microenvironment-, and concentration-dependent mechanisms as described in the review. GC, glucocorticoids; E, estrogens; A, androgens; P, progestogens; FAO, fatty acid oxidation. Created in BioRender. Smith, C. (2025) https://BioRender.com/su4w0te.
While not the focus of this review, it should be noted that the adrenal-derived steroid hormone and androgen and estrogen precursor, dehydroepiandrosterone (DHEA), regulates intracellular metabolism and inflammatory activity. It has been found to inhibit glucose-6-phosphate dehydrogenase and the pentose phosphate pathway in human and mouse endometrial stromal cells and human breast cancer cells (206, 207), and to increase oxidative phosphorylation in cumulus cells of aged infertile patients and in liver mitochondria of developing rats (208, 209). Although it is not clear if DHEA modulates bioenergetic pathways in immune cells, it has been shown to attenuate LPS-induced production of pro-inflammatory mediators in murine RAW 264.7 macrophages (210); thus, future investigators into a role for this hormone in regulating immunometabolism are warranted. In conclusion, defining the cell type-, microenvironment-, disease-state, and concentration-dependent effects of steroid hormones on metabolic reprogramming in immune cells will facilitate more precise therapeutic targeting of immune cell activity to mitigate inflammatory- and immune-mediated diseases.
Author contributions
LS: Conceptualization, Writing – original draft, Funding acquisition, Project administration, Visualization, Writing – review & editing. MR: Writing – original draft, Writing – review & editing. GR: Writing – original draft, Writing – review & editing. CM: Writing – original draft, Writing – review & editing. JL: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the National Institutes of Health (ES032473 and AI167884).
Acknowledgments
Figure 1 was created in BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Pearce EL and Pearce EJ. Metabolic pathways in immune cell activation and quiescence. Immunity. (2013) 38:633–43. doi: 10.1016/j.immuni.2013.04.005
2. Hu C, Xuan Y, Zhang X, Liu Y, Yang S, and Yang K. Immune cell metabolism and metabolic reprogramming. Mol Biol Rep. (2022) 49:9783–95. doi: 10.1007/s11033-022-07474-2
3. Conner KR, Duberstein PR, and Conwell Y. The validity of proxy-based data in suicide research: A study of patients 50 years of age and older who attempted suicide. I. Psychiatric diagnoses. Acta Psychiatrica Scandinavica. (2001) 104:204–9. doi: 10.1034/j.1600-0447.2001.00405.x
4. Klein SL and Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. (2016) 16(10):626–38. doi: 10.1038/nri.2016.9
5. Kroon J, Pereira AM, and Meijer O. Glucocorticoids sexual dimorphism in metabolism: Dissecting the role of sex hormones. Trends Endocrinol Metab. (2021) 31:357–67. doi: 10.1016/j.tem.2020.01.010
6. Alotiby A. Immunology of Stress: A Review Article. J Clin Med. (2024) 13(21):6394. doi: 10.3390/jcm13216394
7. Moon J-Y, Choi MH, and Kim J. Metabolic profiling of cholesterol and sex steroid hormones to monitor urological diseases. Endocrine-Related Cancer. (2016) 23:R455–67. doi: 10.1530/ERC-16-0285
8. Grundy SM. Metabolic complications of obesity. Endocrine. (2000) 13:155–65. doi: 10.1385/ENDO:13:2:155
9. Lee JH and Lee SW. Monthly variations in serum testosterone levels: results from testosterone screening of 8,367 middle-aged men. J Urol. (2021) 205:1438–43. doi: 10.1097/JU.0000000000001546
10. Shahid W, Noor R, and Bashir MS. Effects of exercise on sex steroid hormones (estrogen, progesterone, testosterone) in eumenorrheic females: A systematic to review and meta-analysis. BMC Women’s Health. (2024) 24:354. doi: 10.1186/s12905-024-03203-y
11. Spiga F, Walker JJ, Terry JR, and Lightman SL. HPA axis-rhythms. Compr Physiol. (2014) 4:1273–98. doi: 10.1002/cphy.c140003
12. Wang C, Catlin DH, Starcevic B, Leung A, DiStefano E, Lucas G, et al. Testosterone metabolic clearance and production rates determined by stable isotope dilution/tandem mass spectrometry in normal men: Influence of ethnicity and age. J Clin Endocrinol Metab. (2004) 89:2936–41. doi: 10.1210/jc.2003-031802
13. Geserick C, Meyer H-A, and Haendler B. The role of DNA response elements as allosteric modulators of steroid receptor function. Mol Cell Endocrinol. (2005) 236:1–7. doi: 10.1016/j.mce.2005.03.007
14. Webb P, Nguyen P, Valentine C, Lopez GN, Kwok GR, McInerney E, et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol Endocrinol (Baltimore Md.). (1999) 13:1672–85. doi: 10.1210/mend.13.10.0357
15. Xavier AM, Anunciato AKO, Rosenstock TR, and Glezer I. Gene expression control by glucocorticoid receptors during innate immune responses. Front Endocrinol. (2016) 7:31. doi: 10.3389/fendo.2016.00031
16. Wilkenfeld SR, Lin C, and Frigo DE. Communication between genomic and non-genomic signaling events coordinate steroid hormone actions. Steroids. (2018) 133:2–7. doi: 10.1016/j.steroids.2017.11.005
17. Ainslie RJ, Simitsidellis I, Kirkwood PM, and Gibson DA. RISING STARS: Androgens and immune cell function. J Endocrinol. (2024) 261:e230398. doi: 10.1530/JOE-23-0398
18. Bereshchenko O, Bruscoli S, and Riccardi C. Glucocorticoids, sex hormones, and immunity. Front Immunol. (2018) 9:1332. doi: 10.3389/fimmu.2018.01332
19. Chakraborty B, Byemerwa J, Krebs T, Lim F, Chang C-Y, and McDonnell DP. Estrogen receptor signaling in the immune system. Endocrine Rev. (2023) 44:117–41. doi: 10.1210/endrev/bnac017
20. Kobayashi A, Azuma K, Ikeda K, and Inoue S. Mechanisms underlying the regulation of mitochondrial respiratory chain complexes by nuclear steroid receptors. Int J Mol Sci. (2020) 21:6683. doi: 10.3390/ijms21186683
21. Vancolen S, Sébire G, and Robaire B. Influence of androgens on the innate immune system. Andrology. (2023) 11:1237–44. doi: 10.1111/andr.13416
22. Sun L, Su Y, Jiao A, Wang X, and Zhang B. T cells in health and disease. Signal Transduction Targeted Ther. (2023) 8:235. doi: 10.1038/s41392-023-01471-y
23. MacIver NJ, Michalek RD, and Rathmell JC. Metabolic regulation of T lymphocytes. Annu Rev Immunol. (2013) 31:259–83. doi: 10.1146/annurev-immunol-032712-095956
24. Balyan R, Gautam N, and Gascoigne NRJ. The ups and downs of metabolism during the lifespan of a T cell. Int J Mol Sci. (2020) 21:7972. doi: 10.3390/ijms21217972
25. Werlen G, Jain R, and Jacinto E. MTOR signaling and metabolism in early T cell development. Genes. (2021) 12:728. doi: 10.3390/genes12050728
26. Zhang M, Lin X, Yang Z, Li X, Zhou Z, Love PE, et al. Metabolic regulation of T cell development. Front Immunol. (2022) 13:946119. doi: 10.3389/fimmu.2022.946119
27. Jacobs SR, Herman CE, MacIver NJ, Wofford JA, Wieman HL, Hammen JJ, et al. Glucose uptake is limiting in T cell activation and requires CD28-mediated akt-dependent and independent pathways. J Immunol (Baltimore Md. : 1950). (2008) 180:4476–86. doi: 10.4049/jimmunol.180.7.4476
28. Verbist KC, Wang R, and Green DR. T cell metabolism and the immune response. Semin Immunol. (2012) 24:399–404. doi: 10.1016/j.smim.2012.12.006
29. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of th17 and th1 cell differentiation by glutaminase-dependent metabolism. Cell. (2018) 175:1780–1795.e19. doi: 10.1016/j.cell.2018.10.001
30. Ma S, Ming Y, Wu J, and Cui G. Cellular metabolism regulates the differentiation and function of T-cell subsets. Cell Mol Immunol. (2024) 21:419–35. doi: 10.1038/s41423-024-01148-8
31. Yu Q, Tu H, Yin X, Peng C, Dou C, Yang W, et al. Targeting glutamine metabolism ameliorates autoimmune hepatitis via inhibiting T cell activation and differentiation. Front Immunol. (2022) 13:880262. doi: 10.3389/fimmu.2022.880262
32. Stark JM, Tibbitt CA, and Coquet JM. The metabolic requirements of th2 cell differentiation. Front Immunol. (2019) 10:2318. doi: 10.3389/fimmu.2019.02318
33. Cao J, Liao S, Zeng F, Liao Q, Luo G, and Zhou Y. Effects of altered glycolysis levels on CD8+ T cell activation and function. Cell Death Dis. (2023) 14:407. doi: 10.1038/s41419-023-05937-3
34. Corrado M and Pearce EL. Targeting memory T cell metabolism to improve immunity. J Clin Invest. (2022) 132:e148546. doi: 10.1172/JCI148546
35. Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Signal Transduction Targeted Ther. (2023) 8:207. doi: 10.1038/s41392-023-01452-1
36. Locati M, Curtale G, and Mantovani A. Diversity, mechanisms, and significance of macrophage plasticity. Annu Rev Pathol. (2020) 15:123–47. doi: 10.1146/annurev-pathmechdis-012418-012718
37. Galván-Peña S and O’Neill LAJ. Metabolic reprograming in macrophage polarization. Front Immunol. (2014) 5:420. doi: 10.3389/fimmu.2014.00420
38. Bennett CL and Perona-Wright G. Metabolic adaption of mucosal macrophages: Is metabolism a driver of persistence across tissues? Mucosal Immunol. (2023) 16:753–63. doi: 10.1016/j.mucimm.2023.06.006
39. Michaeloudes C, Bhavsar PK, Mumby S, Xu B, Hui CKM, Chung KF, et al. Role of metabolic reprogramming in pulmonary innate immunity and its impact on lung diseases. J Innate Immun. (2020) 12:31–46. doi: 10.1159/000504344
40. Stevenson ER, Smith LC, Wilkinson ML, Lee SJ, and Gow AJ. Etiology of lipid-laden macrophages in the lung. Int Immunopharmacol. (2023) 123:110719. doi: 10.1016/j.intimp.2023.110719
41. Bailey JD, Diotallevi M, Nicol T, McNeill E, Shaw A, Chuaiphichai S, et al. Nitric oxide modulates metabolic remodeling in inflammatory macrophages through TCA cycle regulation and itaconate accumulation. Cell Rep. (2019) 28:218–230.e7. doi: 10.1016/j.celrep.2019.06.018
42. Jha AK, Huang SC-C, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. (2015) 42:419–30. doi: 10.1016/j.immuni.2015.02.005
43. Liu Y, Xu R, Gu H, Zhang E, Qu J, Cao W, et al. Metabolic reprogramming in macrophage responses. biomark Res. (2021) 9:1. doi: 10.1186/s40364-020-00251-y
44. Van den Bossche J and Saraber DL. Metabolic regulation of macrophages in tissues. Cell Immunol. (2018) 330:54–9. doi: 10.1016/j.cellimm.2018.01.009
45. Xu R, He X, Xu J, Yu G, and Wu Y. Immunometabolism: Signaling pathways, homeostasis, and therapeutic targets. MedComm. (2024) 5:e789. doi: 10.1002/mco2.789
46. Zhang H, Cao N, Yang Z, Fang X, Yang X, Li H, et al. Bilobalide alleviated dextran sulfate sodium-induced experimental colitis by inhibiting M1 macrophage polarization through the NF-κB signaling pathway. Front Pharmacol. (2020) 11:718. doi: 10.3389/fphar.2020.00718
47. Merlin J, Ivanov S, Dumont A, Sergushichev A, Gall J, Stunault M, et al. Non-canonical glutamine transamination sustains efferocytosis by coupling redox buffering to oxidative phosphorylation. Nat Metab. (2021) 3:1313–26. doi: 10.1038/s42255-021-00471-y
48. Schilperoort M, Ngai D, Sukka SR, Avrampou K, Shi H, and Tabas I. The role of efferocytosis-fueled macrophage metabolism in the resolution of inflammation. Immunol Rev. (2023) 319:65–80. doi: 10.1111/imr.13214
49. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. (2019) 29:443–456.e5. doi: 10.1016/j.cmet.2018.12.004
50. Batista-Gonzalez A, Vidal R, Criollo A, and Carreño LJ. New insights on the role of lipid metabolism in the metabolic reprogramming of macrophages. Front Immunol. (2019) 10:2993. doi: 10.3389/fimmu.2019.02993
51. Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Sci (New York N.Y.). (2017) 356:513–9. doi: 10.1126/science.aal3535
52. Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, et al. Etomoxir inhibits macrophage polarization by disrupting coA homeostasis. Cell Metab. (2018) 28:490–503.e7. doi: 10.1016/j.cmet.2018.06.001
53. Huang SC-C, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. (2014) 15:846–55. doi: 10.1038/ni.2956
54. Huang SC-C, Smith AM, Everts B, Colonna M, Pearce EL, Schilling JD, et al. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity. (2016) 45:817–30. doi: 10.1016/j.immuni.2016.09.016
55. Malandrino MI, Fucho R, Weber M, Calderon-Dominguez M, Mir JF, Valcarcel L, et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab. (2015) 308:E756–769. doi: 10.1152/ajpendo.00362.2014
56. Namgaladze D and Brüne B. Fatty acid oxidation is dispensable for human macrophage IL-4-induced polarization. Biochim Et Biophys Acta. (2014) 1841:1329–35. doi: 10.1016/j.bbalip.2014.06.007
57. Reales-Calderón JA, Aguilera-Montilla N, Corbí Á.L, Molero G, and Gil C. Proteomic characterization of human proinflammatory M1 and anti-inflammatory M2 macrophages and their response to Candida albicans. Proteomics. (2014) 14:1503–18. doi: 10.1002/pmic.201300508
58. Van den Bossche J, Baardman J, Otto NA, van der Velden S, Neele AE, van den Berg SM, et al. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. (2016) 17:684–96. doi: 10.1016/j.celrep.2016.09.008
59. Holter DB, Zahalka S, Brösamlen J, Radhouani M, Watzenboeck ML, Artner TJ, et al. Mast cells activated in vitro can modulate macrophage polarization and antibacterial responses. J Allergy Clin Immunol. (2025) 156(3):754–73. doi: 10.1016/j.jaci.2025.02.040
60. Veglia F, Sanseviero E, and Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
61. Fleming V, Hu X, Weber R, Nagibin V, Groth C, Altevogt P, et al. Targeting myeloid-derived suppressor cells to bypass tumor-induced immunosuppression. Front Immunol. (2018) 9:398. doi: 10.3389/fimmu.2018.00398
62. Hossain F, Al-Khami AA, Wyczechowska D, Hernandez C, Zheng L, Reiss K, et al. Inhibition of fatty acid oxidation modulates immunosuppressive functions of myeloid-derived suppressor cells and enhances cancer therapies. Cancer Immunol Res. (2015) 3:1236–47. doi: 10.1158/2326-6066.CIR-15-0036
63. Renner K, Singer K, Koehl GE, Geissler EK, Peter K, Siska PJ, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol. (2017) 8:248. doi: 10.3389/fimmu.2017.00248
64. Russell DG, Cardona P-J, Kim M-J, Allain S, and Altare F. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. (2009) 10:943–8. doi: 10.1038/ni.1781
65. Al-Khami AA, Zheng L, Del Valle L, Hossain F, Wyczechowska D, Zabaleta J, et al. Exogenous lipid uptake induces metabolic and functional reprogramming of tumor-associated myeloid-derived suppressor cells. Oncoimmunology. (2017) 6:e1344804. doi: 10.1080/2162402X.2017.1344804
66. Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PloS Pathog. (2008) 4:e1000204. doi: 10.1371/journal.ppat.1000204
67. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. (2013) 496:238–42. doi: 10.1038/nature11986
68. Mellman I. Dendritic cells: Master regulators of the immune response. Cancer Immunol Res. (2013) 1:145–9. doi: 10.1158/2326-6066.CIR-13-0102
69. Amon L, Lehmann CHK, Baranska A, Schoen J, Heger L, and Dudziak D. Transcriptional control of dendritic cell development and functions. Int Rev Cell Mol Biol. (2019) 349:55–151. doi: 10.1016/bs.ircmb.2019.10.001
70. Anderson DA, Dutertre C-A, Ginhoux F, and Murphy KM. Genetic models of human and mouse dendritic cell development and function. Nat Rev Immunol. (2021) 21:101–15. doi: 10.1038/s41577-020-00413-x
71. Wu L, Yan Z, Jiang Y, Chen Y, Du J, Guo L, et al. Metabolic regulation of dendritic cell activation and immune function during inflammation. Front Immunol. (2023) 14:1140749. doi: 10.3389/fimmu.2023.1140749
72. Chougnet CA, Thacker RI, Shehata HM, Hennies CM, Lehn MA, Lages CS, et al. Loss of phagocytic and antigen cross-presenting capacity in aging dendritic cells is associated with mitochondrial dysfunction. J Immunol (Baltimore Md.: 1950). (2015) 195:2624–32. doi: 10.4049/jimmunol.1501006
73. Du X, Wen J, Wang Y, Karmaus PWF, Khatamian A, Tan H, et al. Hippo/Mst signaling couples metabolic state and immune function of CD8α+ dendritic cells. Nature. (2018) 558:141–5. doi: 10.1038/s41586-018-0177-0
74. Kratchmarov R, Viragova S, Kim MJ, Rothman NJ, Liu K, Reizis B, et al. Metabolic control of cell fate bifurcations in a hematopoietic progenitor population. Immunol Cell Biol. (2018) 96:863–71. doi: 10.1111/imcb.12040
75. Bajwa G, DeBerardinis RJ, Shao B, Hall B, Farrar JD, and Gill MA. Cutting edge: critical role of glycolysis in human plasmacytoid dendritic cell antiviral responses. J Immunol (Baltimore Md.: 1950). (2016) 196:2004–9. doi: 10.4049/jimmunol.1501557
76. Basit F, Mathan T, Sancho D, and de Vries IJM. Human dendritic cell subsets undergo distinct metabolic reprogramming for immune response. Front Immunol. (2018) 9:2489. doi: 10.3389/fimmu.2018.02489
77. Everts B, Amiel E, van der Windt GJW, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood. (2012) 120:1422–31. doi: 10.1182/blood-2012-03-419747
78. Everts B, Amiel E, Huang SC-C, Smith AM, Chang C-H, Lam WY, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol. (2014) 15:323–32. doi: 10.1038/ni.2833
79. Grzes KM, Sanin DE, Kabat AM, Stanczak MA, Edwards-Hicks J, Matsushita M, et al. Plasmacytoid dendritic cell activation is dependent on coordinated expression of distinct amino acid transporters. Immunity. (2021) 54:2514–2530.e7. doi: 10.1016/j.immuni.2021.10.009
80. Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. (2010) 115:4742–9. doi: 10.1182/blood-2009-10-249540
81. Malinarich F, Duan K, Hamid RA, Bijin A, Lin WX, Poidinger M, et al. High mitochondrial respiration and glycolytic capacity represent a metabolic phenotype of human tolerogenic dendritic cells. J Immunol (Baltimore Md.: 1950). (2015) 194:5174–86. doi: 10.4049/jimmunol.1303316
82. Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, et al. Hypoxia and hypoxia-inducible factor-1 alpha modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol (Baltimore Md.: 1950). (2008) 180:4697–705. doi: 10.4049/jimmunol.180.7.4697
83. Ryans K, Omosun Y, McKeithen DN, Simoneaux T, Mills CC, Bowen N, et al. The immunoregulatory role of alpha enolase in dendritic cell function during Chlamydia infection. BMC Immunol. (2017) 18:27. doi: 10.1186/s12865-017-0212-1
84. Tharakan A, Kumar A, Allegood J, Cowart LA, Conrad DH, and Martin RK. D-2-hydroxyglutarate suppresses allergic sensitization in a murine model of experimental asthma. Allergy. (2023) 78:3014–6. doi: 10.1111/all.15809
85. Cain DW and Cidlowski JA. Immune regulation by glucocorticoids. Nat Rev Immunol. (2017) 17:233–47. doi: 10.1038/nri.2017.1
86. Rocamora-Reverte L, Villunger A, and Wiegers GJ. Cell-specific immune regulation by glucocorticoids in murine models of infection and inflammation. Cells. (2022) 11:2126. doi: 10.3390/cells11142126
87. Diamond MI, Miner JN, Yoshinaga SK, and Yamamoto KR. Transcription factor interactions: Selectors of positive or negative regulation from a single DNA element. Sci (New York N.Y.). (1990) 249:1266–72. doi: 10.1126/science.2119054
88. Harrison LM and Tasker JG. Multiplexed membrane signaling by glucocorticoids. Curr Opin Endocrine Metab Res. (2022) 26:100390. doi: 10.1016/j.coemr.2022.100390
89. Karagiannidis C, Akdis M, Holopainen P, Woolley NJ, Hense G, Rückert B, et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol. (2004) 114:1425–33. doi: 10.1016/j.jaci.2004.07.014
90. Bhattacharyya S, Brown DE, Brewer JA, Vogt SK, and Muglia LJ. Macrophage glucocorticoid receptors regulate Toll-like receptor 4-mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood. (2007) 109:4313–9. doi: 10.1182/blood-2006-10-048215
91. Kleiman A, Hübner S, Rodriguez Parkitna JM, Neumann A, Hofer S, Weigand MA, et al. Glucocorticoid receptor dimerization is required for survival in septic shock via suppression of interleukin-1 in macrophages. FASEB Journal: Off Publ Fed Am Societies Exp Biol. (2012) 26:722–9. doi: 10.1096/fj.11-192112
92. Li CC, Munitic I, Mittelstadt PR, Castro E, and Ashwell JD. Suppression of dendritic cell-derived IL-12 by endogenous glucocorticoids is protective in LPS-induced sepsis. PloS Biol. (2015) 13:e1002269. doi: 10.1371/journal.pbio.1002269
93. Wüst S, van den Brandt J, Tischner D, Kleiman A, Tuckermann JP, Gold R, et al. Peripheral T cells are the therapeutic targets of glucocorticoids in experimental autoimmune encephalomyelitis. J Immunol (Baltimore Md.: 1950). (2008) 180:8434–43. doi: 10.4049/jimmunol.180.12.8434
94. Yang N, Zhang W, and Shi X-M. Glucocorticoid-induced leucine zipper (GILZ) mediates glucocorticoid action and inhibits inflammatory cytokine-induced COX-2 expression. J Cell Biochem. (2008) 103:1760–71. doi: 10.1002/jcb.21562
95. Duma D, Collins JB, Chou JW, and Cidlowski JA. Sexually dimorphic actions of glucocorticoids provide a link to inflammatory diseases with gender differences in prevalence. Sci Signaling. (2010) 3:ra74. doi: 10.1126/scisignal.2001077
96. Cuzzocrea S, Bruscoli S, Crisafulli C, Mazzon E, Agostini M, Muia C, et al. Estrogen receptor antagonist fulvestrant (ICI 182,780) inhibits the anti-inflammatory effect of glucocorticoids. Mol Pharmacol. (2007) 71:132–44. doi: 10.1124/mol.106.029629
97. Ehrchen J, Steinmüller L, Barczyk K, Tenbrock K, Nacken W, Eisenacher M, et al. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood. (2007) 109:1265–74. doi: 10.1182/blood-2006-02-001115
98. Giles KM, Ross K, Rossi AG, Hotchin NA, Haslett C, and Dransfield I. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol (Baltimore Md.: 1950). (2001) 167:976–86. doi: 10.4049/jimmunol.167.2.976
99. Liu Y, Cousin JM, Hughes J, Van Damme J, Seckl JR, Haslett C, et al. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J Immunol (Baltimore Md.: 1950). (1999) 162:3639–46. doi: 10.4049/jimmunol.162.6.3639
100. Martinez FO, Sica A, Mantovani A, and Locati M. Macrophage activation and polarization. Front Bioscience: A J Virtual Library. (2008) 13:453–61. doi: 10.2741/2692
101. Perretti M, Chiang N, La M, Fierro IM, Marullo S, Getting SJ, et al. Endogenous lipid- and peptide-derived anti-inflammatory pathways generated with glucocorticoid and aspirin treatment activate the lipoxin A4 receptor. Nat Med. (2002) 8:1296–302. doi: 10.1038/nm786
102. Gillis S, Crabtree GR, and Smith KA. Glucocorticoid-induced inhibition of T cell growth factor production. I. The effect on mitogen-induced lymphocyte proliferation. J Immunol (Baltimore Md.: 1950). (1979) 123:1624–31. doi: 10.4049/jimmunol.123.4.1624
103. Szatmari I and Nagy L. Nuclear receptor signaling in dendritic cells connects lipids, the genome and immune function. EMBO J. (2008) 27:2353–62. doi: 10.1038/emboj.2008.160
104. Oppong E, Flink N, and Cato ACB. Molecular mechanisms of glucocorticoid action in mast cells. Mol Cell Endocrinol. (2013) 380:119–26. doi: 10.1016/j.mce.2013.05.014
105. Umland SP, Schleimer RP, and Johnston SL. Review of the molecular and cellular mechanisms of action of glucocorticoids for use in asthma. Pulmonary Pharmacol Ther. (2002) 15:35–50. doi: 10.1006/pupt.2001.0312
106. Dotto G-P. Gender and sex-time to bridge the gap. EMBO Mol Med. (2019) 11:e10668. doi: 10.15252/emmm.201910668
107. Turcu AF, Rege J, Auchus RJ, and Rainey WE. 11-Oxygenated androgens in health and disease. Nat Rev Endocrinol. (2020) 16:284–96. doi: 10.1038/s41574-020-0336-x
108. Marchetti PM and Barth JH. Clinical biochemistry of dihydrotestosterone. Ann Clin Biochem. (2013) 50:95–107. doi: 10.1258/acb.2012.012159
109. Dotto GP, Buckinx A, Özdemir BC, and Simon C. Androgen receptor signaling in non-prostatic Malignancies: Challenges and opportunities. Nat Rev Cancer. (2025) 25:93–108. doi: 10.1038/s41568-024-00772-w
110. Corcoran MP, Meydani M, Lichtenstein AH, Schaefer EJ, Dillard A, and Lamon-Fava S. Sex hormone modulation of proinflammatory cytokine and C-reactive protein expression in macrophages from older men and postmenopausal women. J Endocrinol. (2010) 206:217–24. doi: 10.1677/JOE-10-0057
111. D’Agostino P, Milano S, Barbera C, Di Bella G, La Rosa M, Ferlazzo V, et al. Sex hormones modulate inflammatory mediators produced by macrophages. Ann New York Acad Sci. (1999) 876:426–9. doi: 10.1111/j.1749-6632.1999.tb07667.x
112. Friedl R, Brunner M, Moeslinger T, and Spieckermann PG. Testosterone inhibits expression of inducible nitric oxide synthase in murine macrophages. Life Sci. (2000) 68:417–29. doi: 10.1016/s0024-3205(00)00953-x
113. Kadel S and Kovats S. Sex hormones regulate innate immune cells and promote sex differences in respiratory virus infection. Front Immunol. (2018) 9:1653. doi: 10.3389/fimmu.2018.01653
114. Lai J-J, Lai K-P, Chuang K-H, Chang P, Yu I-C, Lin W-J, et al. Monocyte/macrophage androgen receptor suppresses cutaneous wound healing in mice by enhancing local TNF-alpha expression. J Clin Invest. (2009) 119:3739–51. doi: 10.1172/JCI39335
115. Rettew JA, Huet-Hudson YM, and Marriott I. Testosterone reduces macrophage expression in the mouse of toll-like receptor 4, a trigger for inflammation and innate immunity. Biol Reprod. (2008) 78:432–7. doi: 10.1095/biolreprod.107.063545
116. Araneo BA, Dowell T, Diegel M, and Daynes RA. Dihydrotestosterone exerts a depressive influence on the production of interleukin-4 (IL-4), IL-5, and gamma-interferon, but not IL-2 by activated murine T cells. Blood. (1991) 78:688–99. doi: 10.1182/blood.V78.3.688.688
117. Dulos GJ and Bagchus WM. Androgens indirectly accelerate thymocyte apoptosis. Int Immunopharmacol. (2001) 1:321–8. doi: 10.1016/s1567-5769(00)00029-1
118. Fijak M, Damm L-J, Wenzel J-P, Aslani F, Walecki M, Wahle E, et al. Influence of testosterone on inflammatory response in testicular cells and expression of transcription factor foxp3 in T cells. Am J Reprod Immunol (New York N.Y.: 1989). (2015) 74:12–25. doi: 10.1111/aji.12363
119. Kissick HT, Sanda MG, Dunn LK, Pellegrini KL, On ST, Noel JK, et al. Androgens alter T-cell immunity by inhibiting T-helper 1 differentiation. Proc Natl Acad Sci United States America. (2014) 111:9887–92. doi: 10.1073/pnas.1402468111
120. Olsen NJ, Watson MB, Henderson GS, and Kovacs WJ. Androgen deprivation induces phenotypic and functional changes in the thymus of adult male mice. Endocrinology. (1991) 129:2471–6. doi: 10.1210/endo-129-5-2471
121. Olsen NJ, Viselli SM, Fan J, and Kovacs WJ. Androgens accelerate thymocyte apoptosis. Endocrinology. (1998) 139:748–52. doi: 10.1210/endo.139.2.5729
122. Barros LA, Tufik S, and Andersen ML. The role of progesterone in memory: An overview of three decades. Neurosci Biobehav Rev. (2015) 49:193–204. doi: 10.1016/j.neubiorev.2014.11.015
123. Jacobsen BM and Horwitz KB. Progesterone receptors, their isoforms and progesterone regulated transcription. Mol Cell Endocrinol. (2012) 357:18–29. doi: 10.1016/j.mce.2011.09.016
124. Giangrande PH and McDonnell DP. The A and B isoforms of the human progesterone receptor: Two functionally different transcription factors encoded by a single gene. Recent Prog Hormone Res. (1999) 54:291–313; discussion 313-314.
125. Sitruk-Ware R and El-Etr M. Progesterone and related progestins: Potential new health benefits. Climacteric: J Int Menopause Soc. (2013) 16 Suppl 1:69–78. doi: 10.3109/13697137.2013.802556
126. Tung L, Mohamed MK, Hoeffler JP, Takimoto GS, and Horwitz KB. Antagonist-occupied human progesterone B-receptors activate transcription without binding to progesterone response elements and are dominantly inhibited by A-receptors. Mol Endocrinol (Baltimore Md.). (1993) 7:1256–65. doi: 10.1210/mend.7.10.8123133
127. Collins MK, McCutcheon CR, and Petroff MG. Impact of estrogen and progesterone on immune cells and host-pathogen interactions in the lower female reproductive tract. J Immunol (Baltimore Md.: 1950). (2022) 209:1437–49. doi: 10.4049/jimmunol.2200454
128. Motomura K, Miller D, Galaz J, Liu TN, Romero R, and Gomez-Lopez N. The effects of progesterone on immune cellular function at the maternal-fetal interface and in maternal circulation. J Steroid Biochem Mol Biol. (2023) 229:106254. doi: 10.1016/j.jsbmb.2023.106254
129. Jones LA, Anthony J-P, Henriquez FL, Lyons RE, Nickdel MB, Carter KC, et al. Toll-like receptor-4-mediated macrophage activation is differentially regulated by progesterone via the glucocorticoid and progesterone receptors. Immunology. (2008) 125:59–69. doi: 10.1111/j.1365-2567.2008.02820.x
130. Menzies FM, Henriquez FL, Alexander J, and Roberts CW. Selective inhibition and augmentation of alternative macrophage activation by progesterone. Immunology. (2011) 134:281–91. doi: 10.1111/j.1365-2567.2011.03488.x
131. Miller L and Hunt JS. Sex steroid hormones and macrophage function. Life Sci. (1996) 59:1–14. doi: 10.1016/0024-3205(96)00122-1
132. Robert R and Spitzer JA. Effects of female hormones (17beta-estradiol and progesterone) on nitric oxide production by alveolar macrophages in rats. Nitric Oxide: Biol Chem. (1997) 1:453–62. doi: 10.1006/niox.1997.0157
133. Tsai Y-C, Tseng JT, Wang C-Y, Su M-T, Huang J-Y, and Kuo P-L. Medroxyprogesterone acetate drives M2 macrophage differentiation toward a phenotype of decidual macrophage. Mol Cell Endocrinol. (2017) 452:74–83. doi: 10.1016/j.mce.2017.05.015
134. AbdulHussain G, Azizieh F, Makhseed M, and Raghupathy R. Effects of progesterone, dydrogesterone and estrogen on the production of th1/th2/th17 cytokines by lymphocytes from women with recurrent spontaneous miscarriage. J Reprod Immunol. (2020) 140:103132. doi: 10.1016/j.jri.2020.103132
135. Hellberg S, Raffetseder J, Rundquist O, Magnusson R, Papapavlou G, Jenmalm MC, et al. Progesterone dampens immune responses in in vitro activated CD4+ T cells and affects genes associated with autoimmune diseases that improve during pregnancy. Front Immunol. (2021) 12:672168. doi: 10.3389/fimmu.2021.672168
136. Lee JH, Ulrich B, Cho J, Park J, and Kim CH. Progesterone promotes differentiation of human cord blood fetal T cells into T regulatory cells but suppresses their differentiation into Th17 cells. J Immunol (Baltimore Md.: 1950). (2011) 187:1778–87. doi: 10.4049/jimmunol.1003919
137. Maeda Y, Ohtsuka H, Tomioka M, and Oikawa M. Effect of progesterone on Th1/Th2/Th17 and regulatory T cell-related genes in peripheral blood mononuclear cells during pregnancy in cows. Veterinary Res Commun. (2013) 37:43–9. doi: 10.1007/s11259-012-9545-7
138. Mao G, Wang J, Kang Y, Tai P, Wen J, Zou Q, et al. Progesterone increases systemic and local uterine proportions of CD4+CD25+ Treg cells during midterm pregnancy in mice. Endocrinology. (2010) 151:5477–88. doi: 10.1210/en.2010-0426
139. Ivanova E, Kyurkchiev D, Altankova I, Dimitrov J, Binakova E, and Kyurkchiev S. CD83 monocyte-derived dendritic cells are present in human decidua and progesterone induces their differentiation in vitro. Am J Reprod Immunol (New York N.Y.: 1989). (2005) 53:199–205. doi: 10.1111/j.1600-0897.2005.00266.x
140. Jitprasertwong P, Charadram N, Kumphune S, Pongcharoen S, and Sirisinha S. Female sex hormones modulate Porphyromonas gingivalis lipopolysaccharide-induced Toll-like receptor signaling in primary human monocytes. J Periodontal Res. (2016) 51:395–406. doi: 10.1111/jre.12320
141. Polan ML, Loukides J, Nelson P, Carding S, Diamond M, Walsh A, et al. Progesterone and estradiol modulate interleukin-1 beta messenger ribonucleic acid levels in cultured human peripheral monocytes. J Clin Endocrinol Metab. (1989) 69:1200–6. doi: 10.1210/jcem-69-6-1200
142. Sun Y, Cai J, Ma F, Lü P, Huang H, and Zhou J. miR-155 mediates suppressive effect of progesterone on TLR3, TLR4-triggered immune response. Immunol Lett. (2012) 146:25–30. doi: 10.1016/j.imlet.2012.04.007
143. Holinka CF, Diczfalusy E, and Coelingh Bennink HJT. Estetrol: A unique steroid in human pregnancy. J Steroid Biochem Mol Biol. (2008) 110:138–43. doi: 10.1016/j.jsbmb.2008.03.027
144. Qureshi R, Picon-Ruiz M, Aurrekoetxea-Rodriguez I, Nunes de Paiva V, D’Amico M, Yoon H, et al. The major pre- and postmenopausal estrogens play opposing roles in obesity-driven mammary inflammation and breast cancer development. Cell Metab. (2020) 31:1154–1172.e9. doi: 10.1016/j.cmet.2020.05.008
145. Díaz-Chico BN, Ogasawara Y, Chamness GC, Salman M, and McGuire WL. A 46-kDa antigen associated with estrogen receptor in human breast cancer. J Steroid Biochem. (1988) 30:315–20. doi: 10.1016/0022-4731(88)90114-8
146. Fuentes N and Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. (2019) 116:135–70. doi: 10.1016/bs.apcsb.2019.01.001
147. Hall JM and McDonnell DP. The estrogen receptor beta-isoform (ERbeta) of the human estrogen receptor modulates ERalpha transcriptional activity and is a key regulator of the cellular response to estrogens and antiestrogens. Endocrinology. (1999) 140:5566–78. doi: 10.1210/endo.140.12.7179
148. Stender JD, Kim K, Charn TH, Komm B, Chang KCN, Kraus WL, et al. Genome-wide analysis of estrogen receptor alpha DNA binding and tethering mechanisms identifies Runx1 as a novel tethering factor in receptor-mediated transcriptional activation. Mol Cell Biol. (2010) 30:3943–55. doi: 10.1128/MCB.00118-10
149. Nelson AW, Groen AJ, Miller JL, Warren AY, Holmes KA, Tarulli GA, et al. Comprehensive assessment of estrogen receptor beta antibodies in cancer cell line models and tissue reveals critical limitations in reagent specificity. Mol Cell Endocrinol. (2017) 440:138–50. doi: 10.1016/j.mce.2016.11.016
150. Calippe B, Douin-EChinard V, Laffargue M, Laurell H, Rana-Poussine V, Pipy B, et al. Chronic estradiol administration in vivo promotes the proinflammatory response of macrophages to TLR4 activation: Involvement of the phosphatidylinositol 3-kinase pathway. J Immunol (Baltimore Md.: 1950). (2008) 180:7980–8. doi: 10.4049/jimmunol.180.12.7980
151. Campbell L, Emmerson E, Williams H, Saville CR, Krust A, Chambon P, et al. Estrogen receptor-alpha promotes alternative macrophage activation during cutaneous repair. J Invest Dermatol. (2014) 134:2447–57. doi: 10.1038/jid.2014.175
152. Chao TC, Chao HH, Chen MF, Greager JA, and Walter RJ. Female sex hormones modulate the function of LPS-treated macrophages. Am J Reprod Immunol (New York N.Y.: 1989). (2000) 44:310–8. doi: 10.1111/j.8755-8920.2000.440511.x
153. Melgert BN, Oriss TB, Qi Z, Dixon-McCarthy B, Geerlings M, Hylkema MN, et al. Macrophages: Regulators of sex differences in asthma? Am J Respir Cell Mol Biol. (2010) 42:595–603. doi: 10.1165/rcmb.2009-0016OC
154. Pepe G, Braga D, Renzi TA, Villa A, Bolego C, D’Avila F, et al. Self-renewal and phenotypic conversion are the main physiological responses of macrophages to the endogenous estrogen surge. Sci Rep. (2017) 7:44270. doi: 10.1038/srep44270
155. Ribas V, Drew BG, Le JA, Soleymani T, Daraei P, Sitz D, et al. Myeloid-specific estrogen receptor alpha deficiency impairs metabolic homeostasis and accelerates atherosclerotic lesion development. Proc Natl Acad Sci United States America. (2011) 108:16457–62. doi: 10.1073/pnas.1104533108
156. Routley CE and Ashcroft GS. Effect of estrogen and progesterone on macrophage activation during wound healing. Wound Repair Regeneration. (2009) 17:42–50. doi: 10.1111/j.1524-475X.2008.00440.x
157. Scotland RS, Stables MJ, Madalli S, Watson P, and Gilroy DW. Sex differences in resident immune cell phenotype underlie more efficient acute inflammatory responses in female mice. Blood. (2011) 118:5918–27. doi: 10.1182/blood-2011-03-340281
158. Dai R, Phillips RA, Karpuzoglu E, Khan D, and Ahmed SA. Estrogen regulates transcription factors STAT-1 and NF-kappB to promote inducible nitric oxide synthase and inflammatory responses. J Immunol (Baltimore Md:1950). (2009) 183:6998–7005. doi: 10.4049/jimmunol.0901737
159. Ghisletti S, Meda C, Maggi A, and Vegeto E. 17beta-estradiol inhibits inflammatory gene expression by controlling NF-kappaB intracellular localization. Mol Cell Biol. (2005) 25:2957–68. doi: 10.1128/mcb.25.8.2957-2968.2005
160. Dziennis S, Jia T, Rønnekleiv OK, Hurn PD, and Alkayed NJ. Role of signal transducer and activator of transcription-3 in estradiol-mediated neuroprotection. J Neurosci. (2007) 27:7268–74. doi: 10.1523/jneurosci.1558-07.2007
161. Bjornstrom L and Sjoberg M. Signal transducers and activators of transcription as downstream targets of nongenomic estrogen receptor actions. Mol Endocrinol (Baltimore Md). (2002) 16:2202–14. doi: 10.1210/me.2002-0072
162. Faas M, Bouman A, Moesa H, Heineman MJ, de Leij L, and Schuiling G. The immune response during the luteal phase of the ovarian cycle: A Th2-type response? Fertility Sterility. (2000) 74:1008–13. doi: 10.1016/s0015-0282(00)01553-3
163. Fish EN. The X-files in immunity: Sex-based differences predispose immune responses. Nat Rev Immunol. (2008) 8:737–44. doi: 10.1038/nri2394
164. Fox HS, Bond BL, and Parslow TG. Estrogen regulates the IFN-gamma promoter. J Immunol (Baltimore Md.: 1950). (1991) 146:4362–7. doi: 10.4049/jimmunol.146.12.4362
165. Grasso G and Muscettola M. The influence of beta-estradiol and progesterone on interferon gamma production in vitro. Int J Neurosci. (1990) 51:315–7. doi: 10.3109/00207459008999730
166. Karpuzoglu E, Phillips RA, Gogal RM, and Ansar Ahmed S. IFN-gamma-inducing transcription factor, T-bet is upregulated by estrogen in murine splenocytes: Role of IL-27 but not IL-12. Mol Immunol. (2007) 44:1808–14. doi: 10.1016/j.molimm.2006.08.005
167. Karpuzoglu-Sahin E, Hissong BD, and Ansar Ahmed S. Interferon-gamma levels are upregulated by 17-beta-estradiol and diethylstilbestrol. J Reprod Immunol. (2001) 52:113–27. doi: 10.1016/s0165-0378(01)00117-6
168. Lengi AJ, Phillips RA, Karpuzoglu E, and Ahmed SA. 17beta-estradiol downregulates interferon regulatory factor-1 in murine splenocytes. J Mol Endocrinol. (2006) 37:421–32. doi: 10.1677/jme.1.02122
169. Ziegler Y, Laws MJ, Sanabria Guillen V, Kim SH, Dey P, Smith BP, et al. Suppression of FOXM1 activities and breast cancer growth in vitro and in vivo by a new class of compounds. NPJ Breast Cancer. (2019) 5:45. doi: 10.1038/s41523-019-0141-7
170. McDonnell DP and Norris JD. Connections and regulation of the human estrogen receptor. Sci (New York N.Y.). (2002) 296:1642–4. doi: 10.1126/science.1071884
171. Straub RH. The complex role of estrogens in inflammation. Endocrine Rev. (2007) 28:521–74. doi: 10.1210/er.2007-0001
172. Vrtačnik P, Ostanek B, Mencej-Bedrač S, and Marc J. The many faces of estrogen signaling. Biochemia Med. (2014) 24:329–42. doi: 10.11613/BM.2014.035
173. McCallion A, Nasirzadeh Y, Lingegowda H, Miller JE, Khalaj K, Ahn S, et al. Estrogen mediates inflammatory role of mast cells in endometriosis pathophysiology. Front Immunol. (2022) 13:961599. doi: 10.3389/fimmu.2022.961599
174. Narita S, Goldblum RM, Watson CS, Brooks EG, Estes DM, Curran EM, et al. Environmental estrogens induce mast cell degranulation and enhance IgE-mediated release of allergic mediators. Environ Health Perspect. (2007) 115:48–52. doi: 10.1289/ehp.9378
175. Zierau O, Zenclussen AC, and Jensen F. Role of female sex hormones, estradiol and progesterone, in mast cell behavior. Front Immunol. (2012) 3:169. doi: 10.3389/fimmu.2012.00169
176. Denans N, Tran NTT, Swall ME, Diaz DC, Blanck J, and Piotrowski T. An anti-inflammatory activation sequence governs macrophage transcriptional dynamics during tissue injury in zebrafish. Nat Commun. (2022) 13:5356. doi: 10.1038/s41467-022-33015-3
177. Sharma A, Vikramdeo KS, Sudan SK, Anand S, Deshmukh SK, Singh AP, et al. Cortisol affects macrophage polarization by inducing miR-143/145 cluster to reprogram glucose metabolism and by promoting TCA cycle anaplerosis. J Biol Chem. (2024) 300:107753. doi: 10.1016/j.jbc.2024.107753
178. Hou Y, Xie J, Wang S, Li D, Wang L, Wang H, et al. Glucocorticoid receptor modulates myeloid-derived suppressor cell function via mitochondrial metabolism in immune thrombocytopenia. Cell Mol Immunol. (2022) 19:764–76. doi: 10.1038/s41423-022-00859-0
179. Chen N, Xie Q-M, Song S-M, Guo S-N, Fang Y, Fei G-H, et al. Dexamethasone protects against asthma via regulating Hif-1α-glycolysis-lactate axis and protein lactylation. Int Immunopharmacol. (2024) 131:111791. doi: 10.1016/j.intimp.2024.111791
180. Clayton SA, Lockwood C, O’Neil JD, Daley KK, Hain S, Abdelmottaleb D, et al. The glucocorticoid dexamethasone inhibits HIF-1α stabilization and metabolic reprogramming in lipopolysaccharide-stimulated primary macrophages. Discov Immunol. (2023) 2:kyad027. doi: 10.1093/discim/kyad027
181. Stifel U, Wolfschmitt E-M, Vogt J, Wachter U, Vettorazzi S, Tews D, et al. Glucocorticoids coordinate macrophage metabolism through the regulation of the tricarboxylic acid cycle. Mol Metab. (2022) 57:101424. doi: 10.1016/j.molmet.2021.101424
182. Lu Y, Liu H, Bi Y, Yang H, Li Y, Wang J, et al. Glucocorticoid receptor promotes the function of myeloid-derived suppressor cells by suppressing HIF1α-dependent glycolysis. Cell Mol Immunol. (2018) 15:618–29. doi: 10.1038/cmi.2017.5
183. Auger J-P, Zimmermann M, Faas M, Stifel U, Chambers D, Krishnacoumar B, et al. Metabolic rewiring promotes anti-inflammatory effects of glucocorticoids. Nature. (2024) 629:184–92. doi: 10.1038/s41586-024-07282-7
184. Buentke E, Nordström A, Lin H, Björklund A-C, Laane E, Harada M, et al. Glucocorticoid-induced cell death is mediated through reduced glucose metabolism in lymphoid leukemia cells. Blood Cancer J. (2011) 1:e31. doi: 10.1038/bcj.2011.27
185. Meyer-Heemsoth L, Mitschke K, Bier J, Schütz K, Villunger A, Legler TJ, et al. T cell energy metabolism is a target of glucocorticoids in mice, healthy humans, and MS patients. Cells. (2023) 12:450. doi: 10.3390/cells12030450
186. Kim D, Nguyen QT, Lee J, Lee SH, Janocha A, Kim S, et al. Anti-inflammatory Roles of Glucocorticoids Are Mediated by Foxp3+ Regulatory T Cells via a miR-342-Dependent Mechanism. Immunity. (2020) 53:581–596.e5. doi: 10.1016/j.immuni.2020.07.002
187. Konishi A, Suzuki J, Kuwahara M, Matsumoto A, Nomura S, Soga T, et al. Glucocorticoid imprints a low glucose metabolism onto CD8 T cells and induces the persistent suppression of the immune response. Biochem Biophys Res Commun. (2022) 588:34–40. doi: 10.1016/j.bbrc.2021.12.050
188. Tung S, Shi Y, Wong K, Zhu F, Gorczynski R, Laister RC, et al. PPARα and fatty acid oxidation mediate glucocorticoid resistance in chronic lymphocytic leukemia. Blood. (2013) 122:969–80. doi: 10.1182/blood-2013-03-489468
189. Zhao X, Huang X, Huang C, Wang X, Yang Y, Dang R, et al. Study on the mechanism of glucocorticoid receptor mitochondrial translocation and glucocorticoid-induced apoptosis in macrophages. Immunopharmacol Immunotoxicol. (2024) 46:482–95. doi: 10.1080/08923973.2024.2366867
190. Li Y, Li L, Wu G, Xie G, Yi L, Zhu J, et al. The unique interplay of mitochondrial oxidative phosphorylation (OXIDATIVE PHOSPHORYLATION) and immunity and its potential implication for the sex- and age-related morbidity of severe COVID-19 patients. MedComm. (2023) 4:e371. doi: 10.1002/mco2.371
191. Consiglio CR, Udartseva O, Ramsey KD, Bush C, and Gollnick SO. Enzalutamide, an androgen receptor antagonist, enhances myeloid cell-mediated immune suppression and tumor progression. Cancer Immunol Res. (2020) 8:1215–27. doi: 10.1158/2326-6066.CIR-19-0371
192. Chowdhury NU, Cephus J-Y, Henriquez Pilier E, Wolf MM, Madden MZ, Kuehnle SN, et al. Androgen signaling restricts glutaminolysis to drive sex-specific Th17 metabolism in allergic airway inflammation. J Clin Invest. (2024) 134:e177242. doi: 10.1172/JCI177242
193. De Azevedo RB, Rosa LF, Lacava ZG, and Curi R. Gonadectomy impairs lymphocyte proliferation and macrophage function in male and female rats. Correlation with key enzyme activities of glucose and glutamine metabolism. Cell Biochem Funct. (1997) 15:293–8. doi: 10.1002/(SICI)1099-0844(199712)15:4<293::AID-CBF755>3.0.CO;2-1
194. Liu S, Zhang S, Hong L, Diao L, Cai S, Yin T, et al. Characterization of progesterone-induced dendritic cells in metabolic and immunologic reprogramming. J Reprod Immunol. (2023) 159:104128. doi: 10.1016/j.jri.2023.104128
195. Fuseini H, Cephus J-Y, Wu P, Davis JB, Contreras DC, Gandhi VD, et al. ERα Signaling increased IL-17A production in th17 cells by upregulating IL-23R expression, mitochondrial respiration, and proliferation. Front Immunol. (2019) 10:2740. doi: 10.3389/fimmu.2019.02740
196. Barcena ML, Christiansen-Mensch C, Aslam M, Haritonow N, Ladilov Y, and Regitz-Zagrosek V. Upregulation of mitochondrial sirt3 and alleviation of the inflammatory phenotype in macrophages by estrogen. Cells. (2024) 13:1420. doi: 10.3390/cells13171420
197. Kim H-N, Ponte F, Nookaew I, Ucer Ozgurel S, Marques-Carvalho A, Iyer S, et al. Estrogens decrease osteoclast number by attenuating mitochondria oxidative phosphorylation and ATP production in early osteoclast precursors. Sci Rep. (2020) 10:11933. doi: 10.1038/s41598-020-68890-7
198. Marques-Carvalho A, Sardão VA, Kim H-N, and Almeida M. ECSIT is essential for RANKL-induced stimulation of mitochondria in osteoclasts and a target for the anti-osteoclastogenic effects of estrogens. Front Endocrinol. (2023) 14:1110369. doi: 10.3389/fendo.2023.1110369
199. Marques-Carvalho A, Silva B, Pereira FB, Kim H-N, Almeida M, and Sardão VA. Oestradiol and osteoclast differentiation: Effects on p53 and mitochondrial metabolism. Eur J Clin Invest. (2024) 54:e14195. doi: 10.1111/eci.14195
200. Massart F, Paolini S, Piscitelli E, Brandi ML, and Solaini G. Dose-dependent inhibition of mitochondrial ATP synthase by 17 beta-estradiol. Gynecological Endocrinol. (2002) 16:373–7. doi: 10.1080/gye.16.5.373.377
201. O’Mahony F, Razandi M, Pedram A, Harvey BJ, and Levin ER. Estrogen modulates metabolic pathway adaptation to available glucose in breast cancer cells. Mol Endocrinol (Baltimore Md.). (2012) 26:2058–70. doi: 10.1210/me.2012-1191
202. Smith LC, Abramova E, Vayas K, Rodriguez J, Gelfand-Titiyevksiy B, Roepke TA, et al. Transcriptional profiling of lung macrophages following ozone exposure in mice identifies signaling pathways regulating immunometabolic activation. Toxicological Sci. (2024) 201:103–17. doi: 10.1093/toxsci/kfae081
203. Klinge C. Estrogenic control of mitochondrial function. Redox Biol. (2020) 31:101435. doi: 10.1016/j.redox.2020.101435
204. Zhao C, Tang Z, Yan J, Fang J, Wang H, and Cai Z. Bisphenol S exposure modulate macrophage phenotype as defined by cytokines profiling, global metabolomics and lipidomics analysis. Sci Total Environ. (2017) 592:357–65. doi: 10.1016/j.scitotenv.2017.03.035
205. Zhang W, Li L, Chen H, Zhang Y, Zhang Z, Lin Z, et al. Bisphenol F promotes the secretion of pro-inflammatory cytokines in macrophages by enhanced glycolysis through PI3K-AKT signaling pathway. Toxicol Lett. (2021) 350:30–9. doi: 10.1016/j.toxlet.2021.06.011
206. Liu H-F, Chou S-C, Huang S-C, Huang T-Y, Hsiao P-Y, Chou F-P, et al. Dehydroepiandrosterone-α-2-deoxyglucoside exhibits enhanced anticancer effects in MCF-7 breast cancer cells and inhibits glucose-6-phosphate dehydrogenase activity. Chem Biol Drug Design. (2024) 104:e14624. doi: 10.1111/cbdd.14624
207. Frolova AI, O’Neill K, and Moley KH. Dehydroepiandrosterone inhibits glucose flux through the pentose phosphate pathway in human and mouse endometrial stromal cells, preventing decidualization and implantation. Mol Endocrinol. (2011) 25:1444–55. doi: 10.1210/me.2011-0026
208. Patel MA and Katyare SS. Treatment with dehydroepiandrosterone (DHEA) stimulates oxidative energy metabolism in the liver mitochondria from developing rats. Mol Cell Biochem. (2006) 293:193–201. doi: 10.1007/s11010-006-9242-3
209. Li C-J, Lin L-T, and Tsui K-H. Dehydroepiandrosterone shifts energy metabolism to increase mitochondrial biogenesis in female fertility with advancing age. Nutrients. (2021) 13:2449. doi: 10.3390/nu13072449
Keywords: steroid hormones, immunometabolism, immune, inflammation, glycolysis, oxidative phosphorylation
Citation: Smith LC, Ramar M, Riley III GL, Mathias CB and Lee J-Y (2025) Steroid hormone regulation of immunometabolism and inflammation. Front. Immunol. 16:1654034. doi: 10.3389/fimmu.2025.1654034
Received: 25 June 2025; Accepted: 31 August 2025;
Published: 17 September 2025.
Edited by:
Luis Eduardo Alves Damasceno, University of São Paulo, BrazilReviewed by:
Maria Luisa Barcena, Tübingen University Hospital, GermanyNatalia Santucci, CONICET Rosario, Argentina
Copyright © 2025 Smith, Ramar, Riley, Mathias and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ley Cody Smith, bC5jb2R5LnNtaXRoQHVjb25uLmVkdQ==
†These authors have contributed equally to this work and share first authorship
‡ORCID: Ley Cody Smith, orcid.org/0000-0003-2388-4032