 Sumon Mukherjee1
Sumon Mukherjee1 Ahana Ghosh1
Ahana Ghosh1 Sourio Chakraborty1
Sourio Chakraborty1 Udit Basak1
Udit Basak1 Sumoyee Mukherjee2Tanya Das1
Sumoyee Mukherjee2Tanya Das1 Gaurisankar Sa1*
Gaurisankar Sa1*- 1Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata, India
- 2Department of Biochemistry and Medical Biotechnology, Calcutta School of Tropical Medicine, Kolkata, India
Immunotherapy has transformed the landscape of cancer treatment, offering hope to patients who were once considered beyond the reach of effective care. However, its success is restricted to a limited fraction of patients. This discrepancy in response is largely due to the complex and dynamic nature of the tumor immune-microenvironment. At the heart of this complexity is the concept of cancer immunoediting—a dynamic process through which the immune system both sculpts and is shaped by the tumor. This process unfolds in three key stages: Elimination, Equilibrium, and Escape, each representing a shifting balance between immune defenses and tumor adaptation. Central to this interaction are tumor-infiltrating lymphocytes (TILs) and tumor-associated macrophages (TAMs). TILs are frontline defenders in targeting tumor cells, while TAMs can either hinder or facilitate tumor growth based on their polarization. As cancer progresses, immune selection pressure induces phenotypic alterations that promote immune evasion, fostering an environment detrimental to effective immune response. This review explores the role of these immunological components in each phase of immunoediting and their impact on the efficacy or failure of immunotherapy. Gaining deeper insight into these interactions is crucial for developing advanced immunotherapies that reshape tumor microenvironment and expand the reach of immunotherapy to more patients.
Introduction to cancer-immunoediting: overview and phases
Tumor suppression has traditionally been thought of as a cell-intrinsic function driven by pathways involving proteins such as p53. When these signaling pathways fail, it leads to malignant transformations. On the other hand, the cancer immunosurveillance concept suggests that external factors, such as the immune system, also inhibit tumor growth (1). The crosstalk between immune cells and tumor cells is becoming an emerging subject in tumor immunology. Cancer immunoediting focuses on the immune system’s combined host-protective and tumor-sculpting functions. The immune system can simultaneously inhibit as well as promote tumor growth (2, 3). Robert Schreiber and his colleagues, using mouse tumor models, hypothesized that cancer immunosurveillance remains active in immunocompetent hosts during cancer progression (4). The authors further showed that by attempting to control cancer proliferation, the immune system is pushing the cancer cells to evolve into more resistant variants. This dual role of immunity to control cancer (elimination, equilibrium) and thereby promote it (escape) served as the basis for the theory of cancer immunoediting (3). The three E’s, elimination, equilibrium, and escape, are the three crucial stages that have been suggested as the major features of cancer immunoediting that progress from immune surveillance to immunological escape (Figure 1) (2, 3).
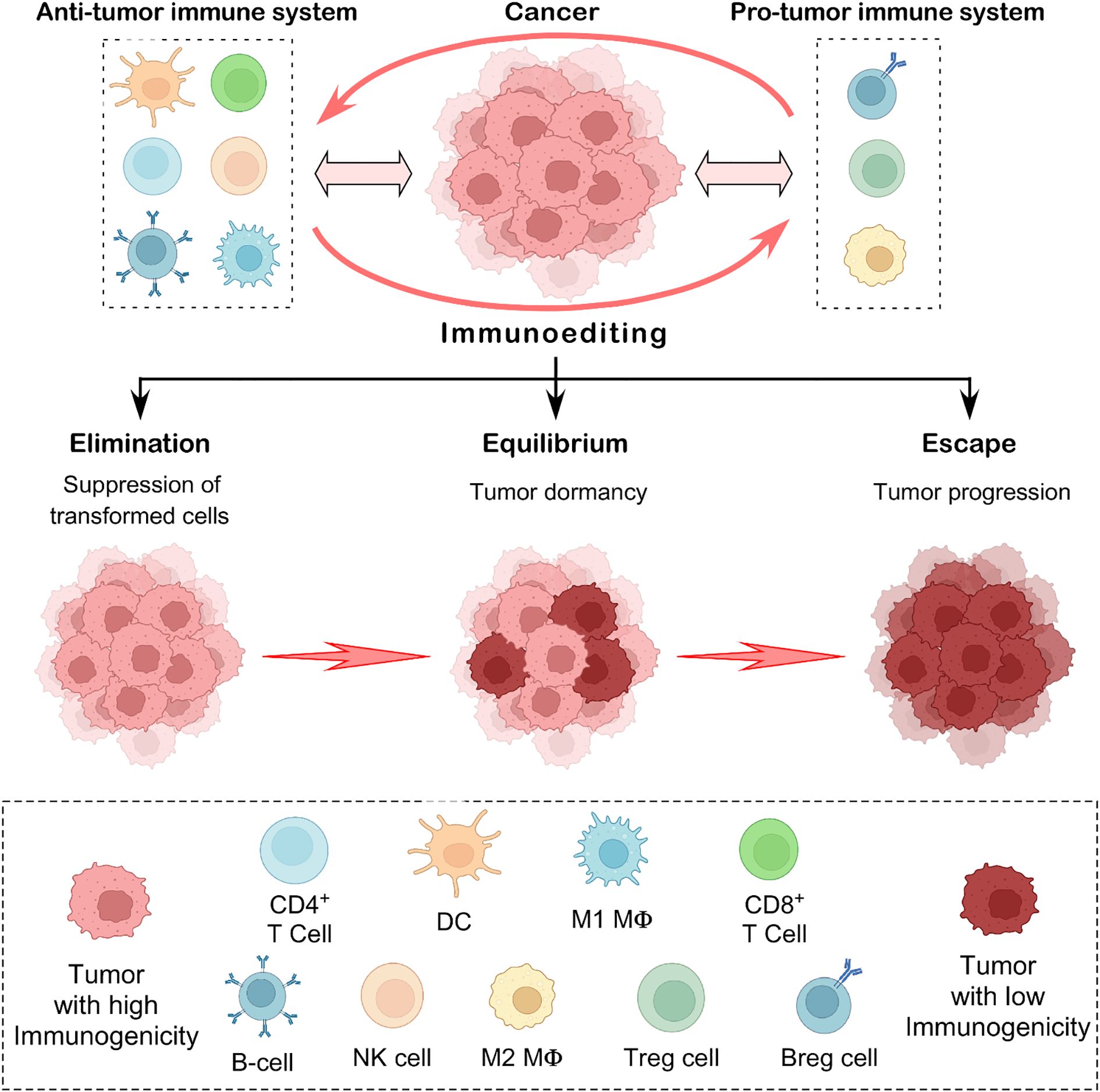
Figure 1. The concept of cancer immunoediting. This illustration depicts the three key phases of cancer immunoediting: elimination, equilibrium, and escape. In the elimination stage, anti-tumor immune effector cells act as defenders, identifying and destroying emerging transformed tumor cells. When some cancer cells evade this response, the process enters the equilibrium phase, where the immune system keeps the tumor in a dormant state but cannot eliminate it. Eventually, in the escape phase, resistant tumor cells adapt and grow unchecked, often spreading to other parts of the body. This concept captures the complex and evolving relationship between the immune system and cancer, reflecting its ability to both protect against and support tumor progression.
Under the fundamental principles of cancer immunoediting, it is hypothesized that in the elimination phase, the immune effector cells remove the newly transformed cells. If they are unable to completely eradicate the tumor, then the tumor cells enter the equilibrium phase (5). The tumor variants entering the equilibrium phase are characterized by reduced immunogenicity and higher resistance to immune response. They arise from immune selection and immune sculpting that they adopt to survive the elimination phase. Tumors eventually enlarge and employ several defense mechanisms to evade immune response within the tumor-microenvironment (3, 5). Studies in the mouse MCA sarcoma model provided the experimental evidence in favor of the cancer immunoediting theory. These investigations revealed the significance of several immune cell populations including T cells and natural killer (NK) cells and molecules (such as FASL, TRAIL, Perforin, and type-I and type-II IFNs) in the immunoediting of cancer (2, 6, 7). The idea of cancer immunoediting has evolved, changing our understanding of the interactions between the immune system and tumors. However, much more study is required to clarify the molecular and cellular dynamics of cancer immunoediting. This review discusses the fundamentals of immunoediting, specifically immune surveillance and escape, as well as the crucial role of immune effector cells, particularly tumor-infiltrating lymphocytes (TILs) and tumor-associated macrophages (TAMs), in this process. Gaining more knowledge about the processes by which immunoediting occurs during tumor growth could lead to new developments in cancer immunotherapy.
Cancer-immunity cycle meets cancer immunoediting
An efficient anti-cancer immune response requires a sequence of events called the cancer-immunity cycle (Figure 2) (8, 9). To begin the process, the secreted tumor neoantigens are first taken up by antigen-presenting cells such as dendritic cells (DCs). DCs present the collected antigens to T cells on major histocompatibility complex (MHC)-I and MHC-II molecules. The next step involves priming and activating effector T cell responses against the specific antigens. At this point, the immune response strategy is clear, and the ratio of T-effector cells to T-regulatory cells, a crucial balance, determines the outcome. Following their trafficking to the tumor bed, the activated effector T cells kill their target cancer cell after selectively identifying and binding to cancer cells through the interaction of their T cell receptor (TCR) to the corresponding antigen coupled to MHC-I. Additional tumor-associated antigens are released when the tumor cell is killed, expanding the scope and depth of the response in later cycle revolutions (9). If this cycle becomes ineffective in the elimination stage of cancer immunoediting, it leads to immunosurveillance evasion and poor patient survival. Tumor-derived factors in the tumor microenvironment (TME) may suppress those effector cells by generating immunosuppressive immune cell types, hiding tumor antigens, influencing DCs and T cells to perceive antigens as self rather than foreign, thereby preventing effector responses; or preventing T cells from entering the tumor (9, 10). Thus, the cancer-immunity cycle is especially important at the initial stage of cancer immunoediting and entails the immune system’s identification and destruction of cancer cells. Here, we’ve covered the role that TILs and TAMs play in each stage of cancer immunoediting and tried to provide insight into how to apply this understanding to the development of future immunotherapies.
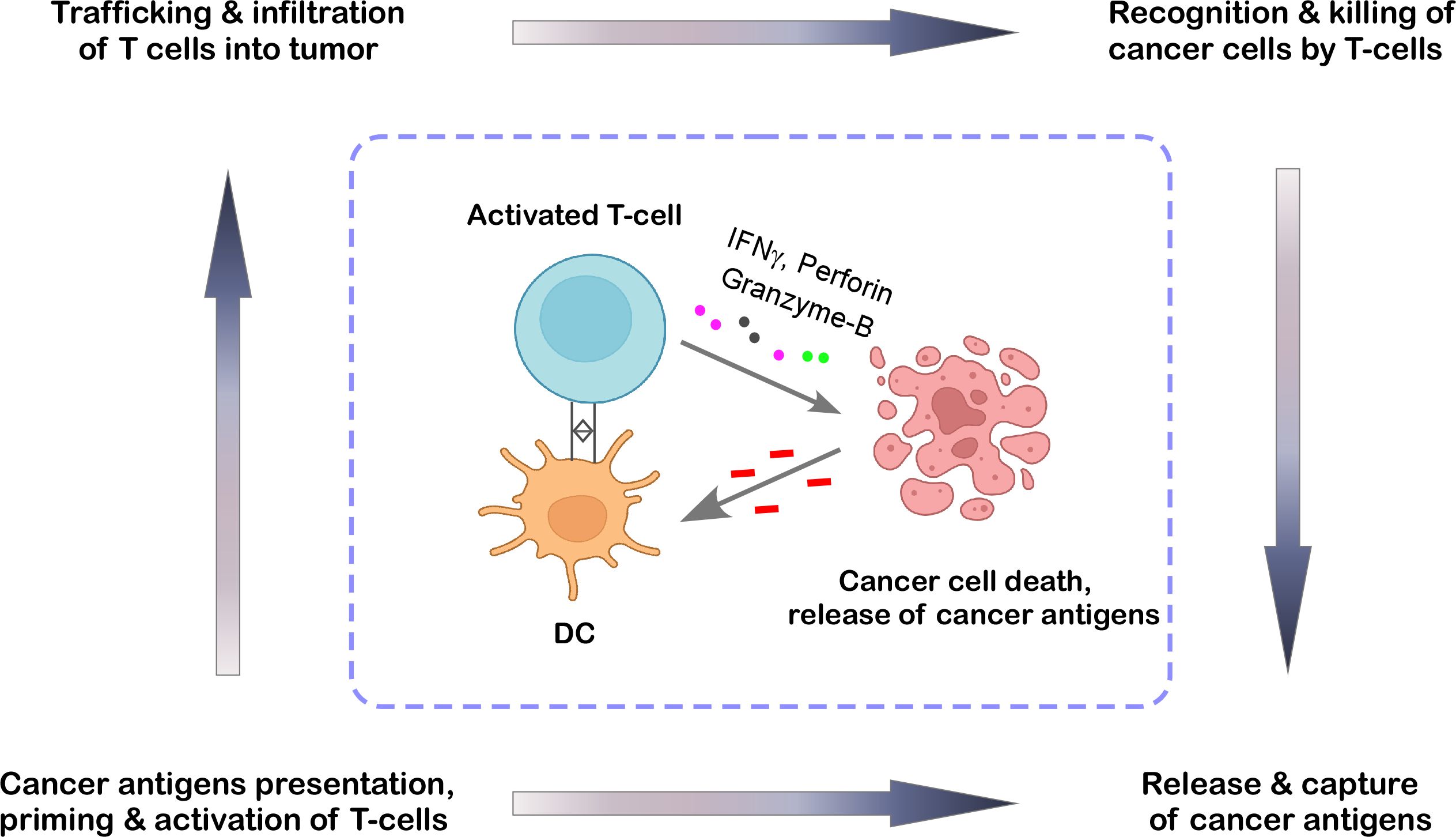
Figure 2. The cancer-immunity cycle. This cycle represents the stepwise process through which the immune system detects and restricts tumor growth. It begins with dendritic cells (DCs) capturing antigens released by cancer cells. These antigens are then presented to T cells, which become activated and proliferate. Once primed, the T cells travel to the tumor site, where they recognize and destroy cancer cells by releasing cytotoxic molecules, thereby reinforcing the immune response in a continuous loop. The illustration was created from Biorender.com.
Roles of immune cells in the phases of cancer immunoediting
Elimination phase
In this initial stage of cancer immunoediting, many anti-tumor immune subsets, including T cells, B cells, innate lymphoid cells (ILCs), such as NK cells and NKT cells, and M1-type macrophages, destroy cancer cells by invading the tumor sites in response to molecular signals (11, 12).
T cells
Since T cells are one of the largest subsets in the TME and primarily contribute to TILs, they have been the subject of extensive research (13). Both the antitumor response and the invasion of the TME depend on them. Numerous signaling molecules, including chemokines (e.g., CCL3, CCL4, CCL5, CCL20, CXCL9, CXCL10, CXCL11, and CXCL16) and their receptors, cause infiltration into the TME (14–17). In head and neck cancer, MHC-I molecules display antigens to TCRs, which in turn activate cytotoxic T lymphocytes to specifically attack tumor cells and trigger their apoptosis (18, 19).
Death receptor ligation, FASL, TRAIL, and the release of granzyme B and perforin can all trigger apoptosis (Figure 3) (20). Naïve CD4+ T cells, when activated by tumor antigen-primed APC and exposed to various cytokines, can differentiate into different subsets, such as T-helper cells: Th1, Th2, Th9, Th17, Th22, and T-regulatory (Treg) cells (21, 22). Th1 cells secrete interleukin-2 (IL2) and interferon-γ (IFNγ) to aid in CTL activation. IFNγ plays a crucial role in inhibiting tumor growth, enhancing MHC expression, and limiting angiogenesis in cancers such as brain tumors, melanoma, and colon cancer (23–25). Th2 cells release cytokines like IL4 and IL13, which can induce eosinophil recruitment into the TME (26). CD4+ T cells, together with immune partners like DCs, B cells, and NK cells support cytotoxic T cells in carrying out their effector functions; (27–29). Evidence indicates that Th1 cell differentiation and function are closely related to those of other immune cells in the TME. For example, it has been demonstrated that DCs, M1 macrophages, and B cells promote Th1 cell development by generating cytokines such as IL12 and IFNγ (30–32).
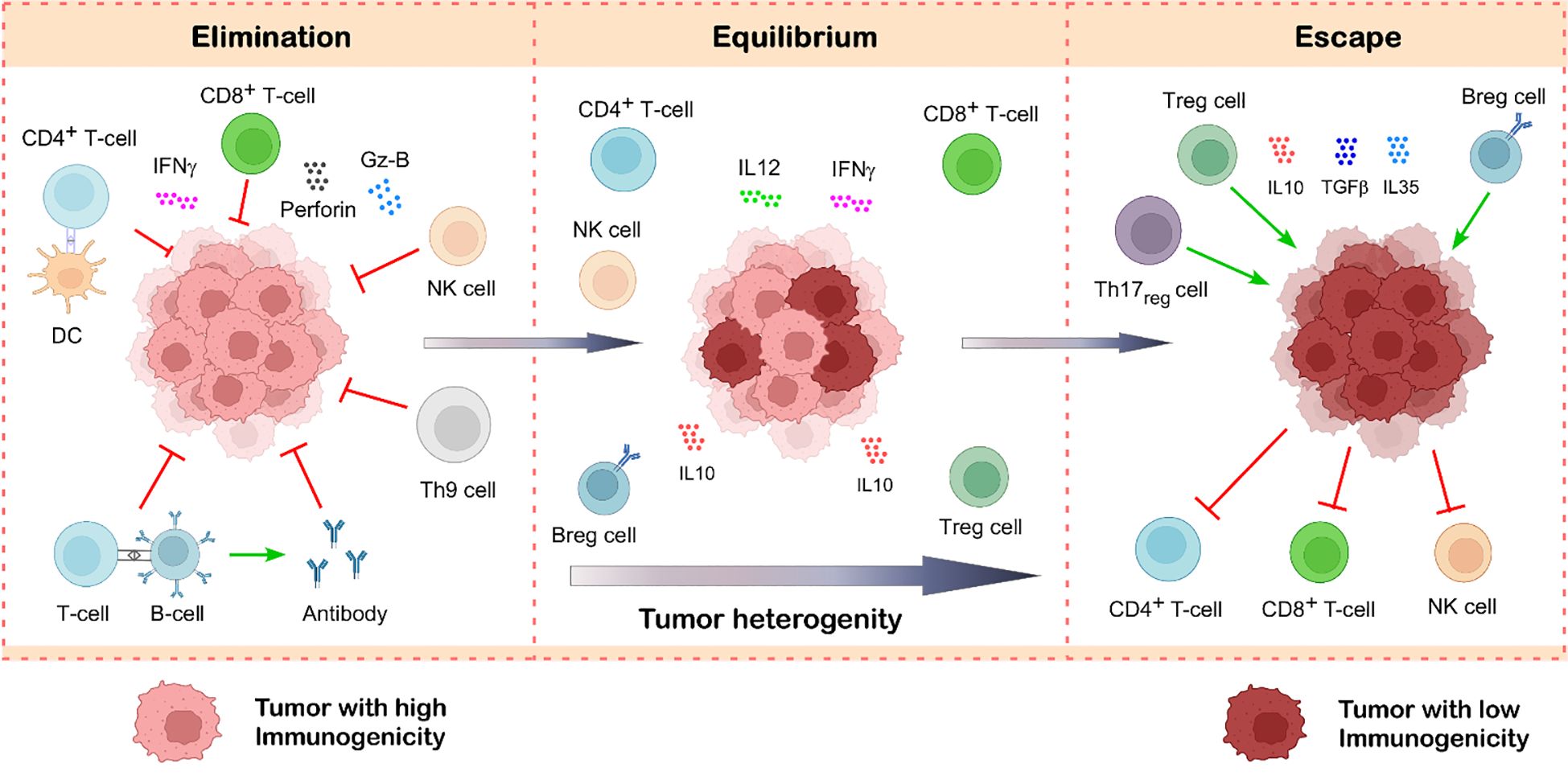
Figure 3. Tumor-infiltrating lymphocytes in cancer immunoediting. Cancer immunoediting unfolds in three interconnected stages: elimination, equilibrium, and escape. During the elimination phase, immune cells such as CD8+ T cells, NK cells, CD4+ T cells, B cells, and Th9 cells actively detect and destroy newly transformed tumor cells through various effector mechanisms. If a subset of tumor cells survives this immune attack, the process transitions into the equilibrium phase, where immune surveillance limits tumor expansion without fully eliminating it. Over time, immune pressure leads to the selection of tumor variants with reduced immunogenicity. Simultaneously, the tumor promotes the accumulation of immunosuppressive cells like Treg cells and Breg cells, which makes the tumor microenvironment immunosuppressive by suppressing the function of anti-tumor immune cells. This shift creates an immunosuppressive tumor microenvironment, marking the escape phase, wherein the tumor evades immune control, resumes growth, and metastasize. Illustration created with BioRender.com.
According to a recent analysis, Th9 cells in the TME of solid tumors are linked to a strong anti-tumor immune response via both innate and adaptive immunological pathways (33). Th9 cells primarily rely on IL9 and IL21 to perform their anti-tumor function. CCR6+ DC and CCR6+ CD8+ T cells are drawn into the tumor bed and have a higher chance of surviving when IL9 stimulates epithelial lung cells to generate CCL20 (34). IL21 from Th9 cells stimulates CD8+ T cell proliferation and boosts NK cytolytic activity and IFNγ production (34). Recombinant IL9 activation increased the cytotoxicity of tumor-specific mice CD8+ T cells, whereas blocking IL9-signalling decreased the production of granzyme B and perforin in human CD8+ T cells (35). In a murine model, studies showed that a reduction in tumor-specific Th17 cells facilitated the advancement of B16 melanoma, with this effect directly associated with the production of IFNγ. Martin-Orozco et al. provide solid evidence that Th17 facilitates the activation of tumor-specific CD8+ T cells, hence indirectly inhibiting tumor growth (36, 37). Th17 cells possess the ability to acquire Th1-like characteristics and secrete substantial amounts of IFNγ, likely enhancing anticancer immune responses (38, 39). DCs release IL12, which promotes CD4+ T cells to develop into Th1 cells and triggers a strong Th1 immune response (40). Memory T cells may stay in pathogenic tissue, as in the case of resident memory T (TRM) cells, or they may circulate in the immune system to react quickly and strongly to subsequent exposure to antigens (41).
Natural killer and natural killer T cells
Similar to CTLs, NK cells induce apoptosis in tumor cells by releasing cytolytic granules that include granzymes and perforins (Figure 3) (20). Table 1 lists examples of activating and inhibitory receptors present on NK cells and NKT cells. Natural killer group-2 member-D (NKG2D), DNAX accessory molecule-1 (DNAM-1), NKp30, NKp44, and NKp46 are some of these activating receptors. Activating receptors use an immunoreceptor tyrosine-based activation motif (ITAM) located on their cytoplasmic tail to transduce signals. NKG2D is also expressed by NKT cells, CD8+ αβT cells, and γδ T cells (20, 54). As a result of signal transduction triggered by NKG2D receptor-ligand interaction, the tumor cells are degranulated by NK cells. While activating receptors initiate cytolytic activity, inhibiting receptors, such as those that detect MHC-I, limit their ability to kill normal, healthy cells (54, 55).
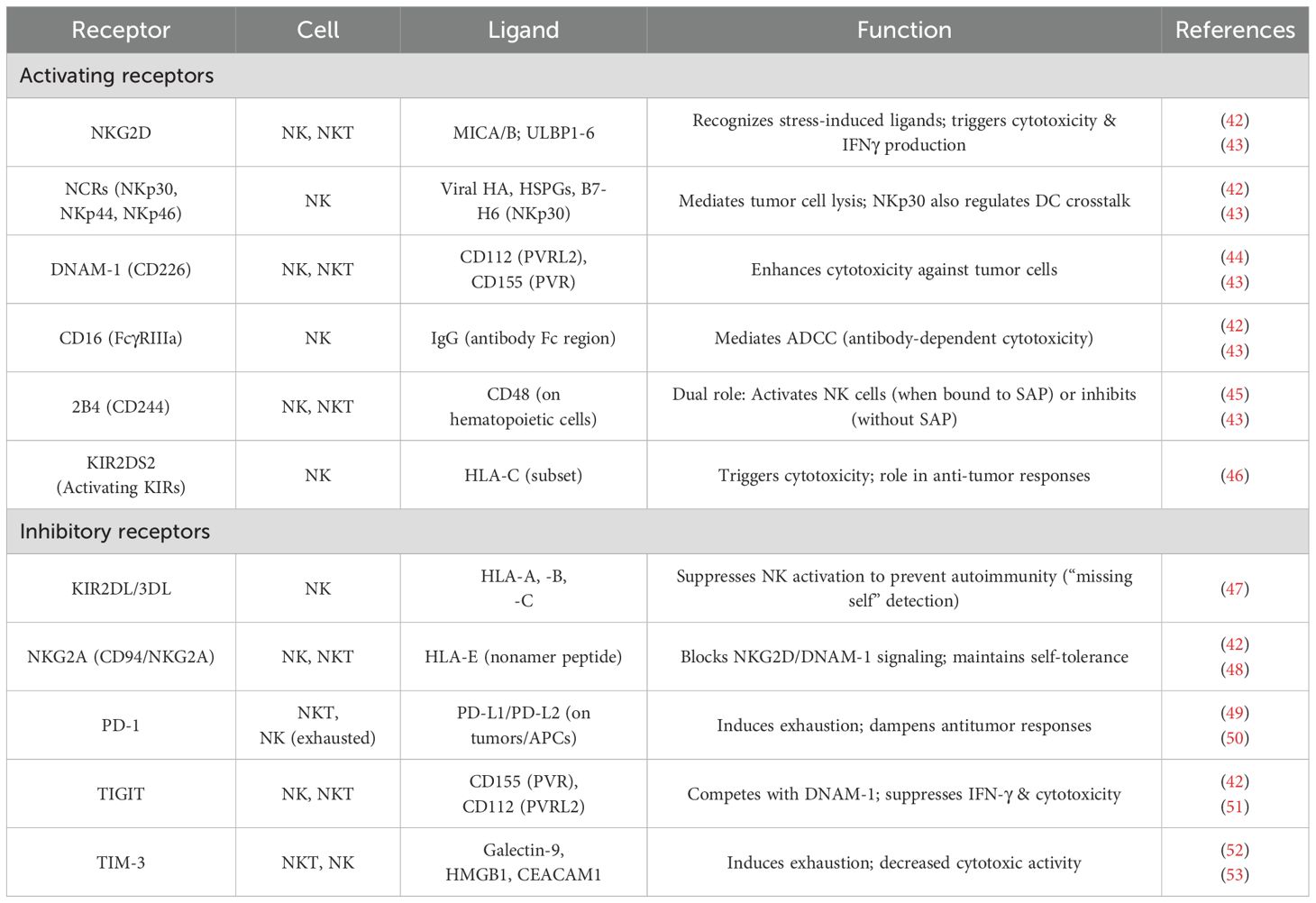
Table 1. Activating and inhibitory receptors expressed by NK and NKT cells.
NKT cells are a subset that is characterized by their dual roles. They express αβ-TCR to identify endogenous and foreign lipid antigens presented on CD1d, a non-classical antigen-presenting protein resembling MHC class-I. On the other hand, they show NK cell characteristics due to the expression of CD56, CD16, and Fc receptor on their surface and granzyme production (15, 56). Leukocyte function-associated antigen-1 (LFA1) expression, CCR2, and CXCR6 can all mediate the recruitment of NKT cells (20, 56). NKT cells release Th1 and Th2 cytokines, including IFNγ, tumor necrosis factor-α (TNFα), IL2, IL4, IL5, IL6, IL10, IL13, IL17, IL21, transforming growth factor-β (TGFβ), and granulocyte monocyte-colony stimulating factor (GM-CSF), upon activation via αβ-TCR and CD1d interaction (57).
B cells
B cells are vital for their antigen-presenting capability since B cell receptor (BCR) is far more specific to antigen recognition than the other APCs (58). In the TME, B cells act in multiple ways. They increase the density of T cells in the tumor by activating CD4+ T cells. They also mediate the conversion of CD4+ T cells and CD8+ T cells into distinct functional subsets (59). B cells can modulate the activity of other APCs, such as DCs and macrophages, enhancing their ability to present antigens effectively (58, 60, 61). A “helper” function for B cells in the tumor immune response is that activated CTLs can interact with soluble CD27 produced by CD19+ B cells, which promotes their survival and proliferation (62). The discovery that IgG2b-mediated activated B cells were highly lethal to tumor cells raised the possibility that B cells also contributed to the inhibition of tumor growth (63). Additionally, pulmonary host B cells were found to enhance IFNγ production and facilitate NK cell killing of tumor cells, suggesting that effector B cells may have a protective role against cancer (64, 65). CD20+ B cells recruit CD8+ T cells by the production of chemokines such as CCL3, CCL4, CCL5, CXCL10, and CXCL13. CXCL13 is a major chemokine that recruits B and T cells to malignancies (66–68). By stimulating T-cells, particularly CD4+ T cells, B cells improve the density and responsiveness of T cells in the TME (59).
M1-tumor-associated macrophages
Type-1 macrophages (M1) have anti-tumor characteristics that allow them to distinguish between transformed and healthy cells. The way by which M1-type macrophages eliminate tumor cells after identifying them has been demonstrated to be impacted by two different factors (69). Using a variety of mechanisms, M1-type macrophages directly mediate cytotoxicity to kill tumor cells (70). One of the ways they achieve this is by generating molecules that kill tumors, such as ROS and NO, which have a cytotoxic effect on tumor cells (71) (Figure 4). The second pathway, known as antibody-dependent cell-mediated cytotoxicity (ADCC), kills tumor cells by selectively targeting them using anti-tumor antibodies (72). The destruction of tumor cells by NK cells is improved when Dectin1 is expressed on M1 macrophages (69). By secreting large amounts of pro-inflammatory cytokines IFNγ and IL12, which have anti-tumor activity, M1 macrophages incite NK cell and cytotoxic T cell infiltration and activation in the tumor site, indicating an indirect mechanism of stopping the spread of cancer (69, 73) (Figure 4).
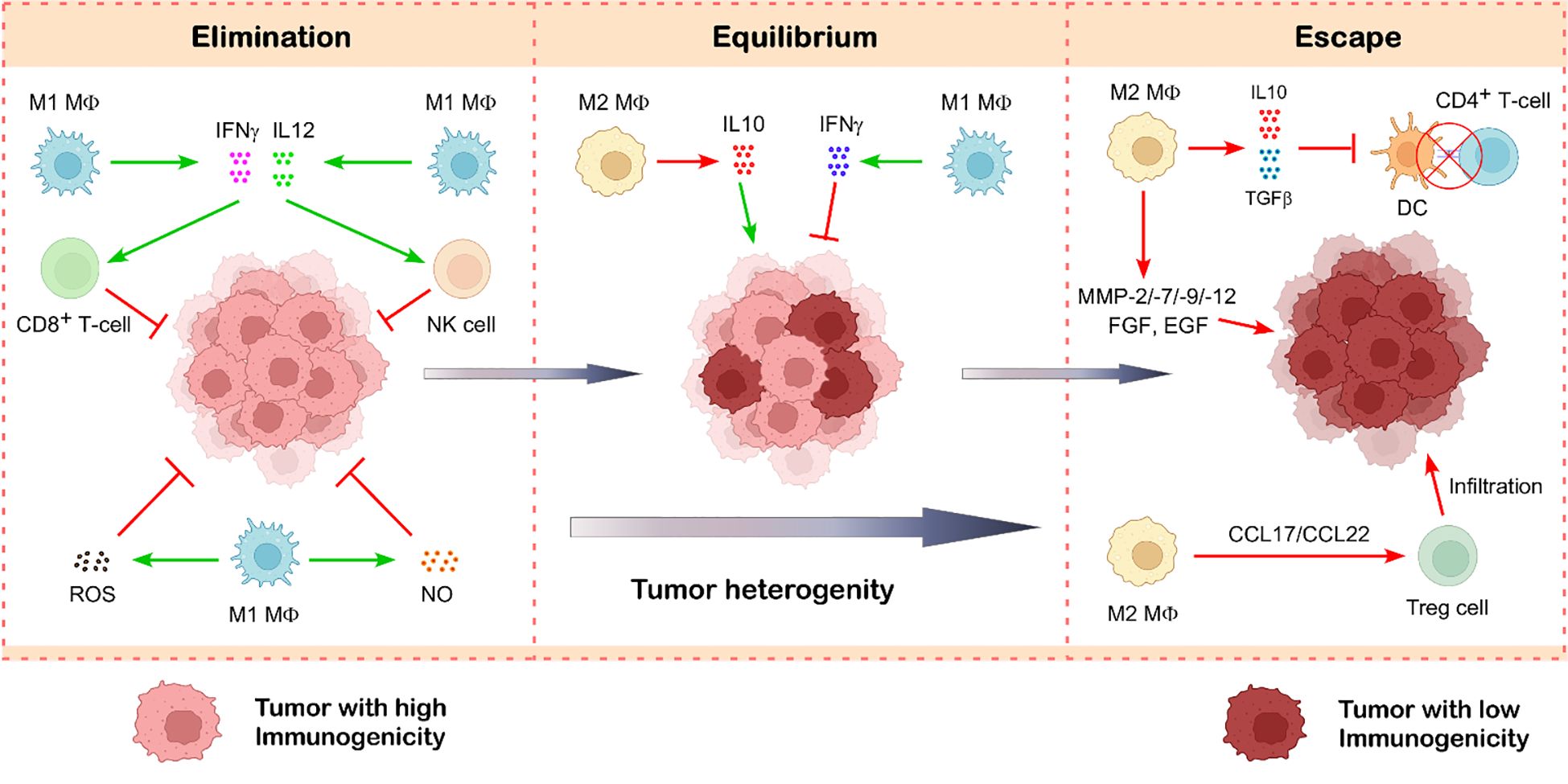
Figure 4. Role of tumor-associated macrophages in cancer immunoediting. Tumor-associated macrophages exist in two main functional states—M1 and M2—that play opposing roles in tumor progression. M1-like TAMs are typically anti-tumorigenic and contribute to tumor suppression by releasing cytotoxic molecules such as ROS, NO, IFNγ, and IL12, which help destroy cancer cells. In contrast, M2-like TAMs support tumor growth and progression. They secrete immunosuppressive cytokines like TGFβ and IL10, which dampen the anti-tumor immunity. Additionally, M2 macrophages produce growth factors and matrix-remodeling enzymes such as metalloproteinases, facilitating tumor growth and metastasis. These macrophages also promote the infiltration of Treg cells, further reinforcing an immunosuppressive tumor microenvironment. Illustration created with BioRender.com.
Equilibrium phase
The subsequent phase of cancer immunoediting, referred to as the equilibrium phase, occurs when tumor cells undergo continuous reshaping to increase their resistance to immune effector cells (74). Immune selection promotes tumor cells with lower immunogenicity as a result of this strategy. The paradox of tumor growth in an immunocompetent individual may be answered with the emergence of highly evolved resistant cancer cells (1, 2). Although these tumor cell variations are less immunogenic, random genetic changes inside tumor cells may result in more unstable malignancies. Nucleotide-excision repair instability (NIN), microsatellite instability (MIN), and chromosomal instability (CIN) are the three forms of genetic instability linked to cancer that have been suggested to be the cause of the “mutator phenotype” of tumor cells (74). Moreover, immune selection pressure increases the likelihood of tumor cell clones with a non-immunogenic phenotype (1, 3).
Of the three stages of cancer immunoediting, the equilibrium phase is probably the longest and may continue for many years (1, 74). It entails the constant removal of tumor cells and the emergence of resistant tumor variants as a result of immune selection pressure. It is unclear how exactly TILs and TAMs contribute to preserving equilibrium, even though their function in the elimination and escape phase has been thoroughly investigated. Research has shown that adaptive immunity components CD4+ T cells, CD8+ T cells, IL12, and IFNγ are in charge of keeping tumor cells in balance by exerting immune selection pressure (Figure 3) (1, 2). This Darwinian selection phase destroys many of the original tumor variants while tumor variants with distinct mutations that boost resistance to immune attack survive and proliferate to reach the next phase (1, 74). Moreover, the relative balance between effector and regulatory immune cells is also considered to be the most important factor in this phase (75). Additionally, studies showed that tumors remain in an equilibrium condition when IL12, which promotes elimination, and IL23, which promotes persistence, are balanced (76). Besides this, Th1 to Th2 cytokine bias also plays a very important role in maintaining the tumor in this phase (77, 78). By decreasing the activity of anti-tumor immune cells, anti-inflammatory cytokines such as IL10 and its expression level play a significant role in the equilibrium phase and cause the tumor to enter the escape phase (79).
Escape phase
In the escape process, the surviving tumor variants that bypass the immune detection and elimination start to proliferate in an uncontrolled manner and employ various mechanisms to create an environment for their survival (80). In this phase, tumor-promoting immune cells like Treg cells, Breg cells, and M2-type TAMs infiltrate the tumor and promote tumor growth by suppressing the function of anti-tumor immune subsets (69, 81). In addition, Th1 to Th2 cytokine bias is also considered an important factor that maintains the tumor in an equilibrium phase. Immunosuppressive cytokines like IL10 and its expression level determine the shift from equilibrium to the escape phase.
T cells
Tumor cells downregulate MHC class-I to evade CTL-mediated death by impairing their ability to process and deliver antigens (19). Additionally, tumor cells frequently exhibit dysregulated expression of death receptors like FAS and/or upregulate anti-apoptotic molecules like BCL2. TH2 cells secrete IL4, IL5, IL10, IL13, and IL17 - all of which have been shown to contribute to the tumor-promoting role of this subtype, even though IL4, IL5, and IL13 have been shown to contribute to the growth and metastasis of cancer (82, 83). CD4+ Th2 cells encourage the growth of tumors by suppressing Th1 cell-mediated immunity and boosting angiogenesis (84). However, recent literature has reported that IL10 has a dual pro- and anti-tumorigenic role (85, 86). IL10 elicits an anti-inflammatory immune response, downregulates Th1 cytokine function, and MHC class-II antigen presentation (87). At the same time, binding of IL10 with its cognate receptor activates signal-transducer and activator of transcription-3 (STAT3)-signaling and transcription of anti-apoptosis and cell cycle progression genes, further strengthening the pro-tumorigenic effect (88). Greater IL9+ cell infiltration in the tumor tissue was associated with worse prognosis, greater frequencies of Treg cells, and decreased cytotoxic capability of CD8+ T cells and NK cells in patients with muscle-invasive bladder cancer (89). The main issue with the Th9 function in the TME is that different tumor types exhibit inconsistent behavior.
One essential cytokine that controls the activity of mast cells and Treg cells is IL9. Feng et al. found that in B-cell non-Hodgkin’s lymphoma (NHL), IL9 was associated with immunosuppression mediated by CD117+ mast cells and FOXP3+ T-regulatory cells (90). Additionally, studies showed that Th9 cells enhanced the phosphorylation of STAT3, which has been linked to a poor prognosis for patients with HCC, hence increasing the production of CCL20. CCL20 is known to stimulate the migration and proliferation of tumor cells (34, 91). Th17 cells have two functions in the TME. This cell subgroup inhibits anti-cancer responses by producing immunosuppressive adenosine, and promotes angiogenesis as well as tumor development by releasing pro-inflammatory IL17A (92). The TME may encourage Th17 cells to differentiate into suppressive Treg cells, which would aid in immunosuppression (37, 93). Compared to the density of Th17 cells in the patient’s surrounding, non-tumor tissue, a noticeably higher number of Th17 cells infiltrate malignancies. This increased Th17 cell presence in tumor tissue is consistent across a wide spectrum of cancers, suggesting that tumors themselves generate components that facilitate Th17 cell trafficking to the site of illness (94). Th17, along with Th2, are involved in chronic inflammation, which encourages the growth and development of tumors (37, 84).
Natural killer cells
To avoid immune surveillance by NK cells, tumor cells downregulate their surface ligands, thereby obstructing anti-tumor recognition. TGFβ, IFNγ, STAT3, hypoxia, proteolytic shedding, the generation of soluble ligands, and certain microRNAs collectively facilitate the downregulation of ligands (95–97). Cancer cells release immunosuppressive microvesicles, such as exosomes, that display NKG2D ligands (NKG2DLs) on their surface. These ligands bind to NKG2D receptors on NK cells and CD8+ T cells, leading to receptor downregulation, internalization, or degradation. This decoy interaction prevents NKG2D from recognizing tumor-expressed ligands, reducing tumor detection and impairing the anti-tumor activity of NK cells and CD8+ T cells (95, 98, 99).
T-regulatory cells
T-regulatory cells, characterized by their immunosuppressive properties, infiltrate the TME via four distinct mechanisms. Initially, by transitioning to the TME from the circulatory or lymphatic systems. Signaling molecules such as chemokines and their receptors (e.g., CCL1-CCR8, CCL5-CCR5, CCL22-CCR4, CCL28-CCR10, and CXCL12-CXCR4) facilitate the ingress of Tregs into the TME (17, 100, 101). Secondly, immunosuppressive chemokines and cytokines can enhance their development in the TME. Third, by the expansion mediated by DC activation. Ultimately, effector T cell differentiation into Treg cells by TGFβ (102). CD25+ and FOXP3lo Treg cell progenitors are the origins of thymically mature Tregs (101, 103). Tregs facilitate tumor growth and metastasis when they are recruited to the TME. Various suppressive functions of Treg cells are facilitated by immunosuppressive cytokines secreted by Tregs, such as TGFβ, IL10, and IL35 (Figure 3) (103). Treg cells can restrict the activity of antigen-presenting cells by downregulating the expression of CD80 and CD86 in a CTLA4-dependent way, therefore preventing the presentation of tumor antigens and the activation of tumor-specific T cells (104). Since an almost total suppression of CD8-mediated cytolytic activity is primarily reliant on TGFβ-signaling, and CD8+ T cells with a dominant negative TGFβ receptor were resistant to this suppression, and CD8+ T cells with a dominant negative TGFβ receptor were resistant to this suppression, TGFβ secretion from Treg cells can regulate CTL function and reduce anti-tumor immunity (105).
Previous findings indicate that Treg cells inhibit the proliferation and functionality of CD4+ and CD8+ T cells by depleting available IL2 and activating IL2/IL2-receptor signaling (101, 106). A recent study investigating the antigen specificity of Treg cells across several malignancies revealed that intra-tumoral Treg cells selectively responded to tumor antigens, leading to the activation and clonal expansion of Treg cells (107). Treg cells inhibit the interactions between antigen-presenting cells and T cells, hence obstructing the maturation and functionality of APCs, which subsequently impedes the activity of effector T cells (108, 109). Furthermore, Treg cells diminish the efficacy of NK cells. Treg cells can suppress anti-tumor immunity by diminishing responses from both the innate and adaptive immune systems (110, 111). T-regulatory cells are associated with a low survival probability in cancer patients (112, 113).
B-regulatory cells
Numerous studies indicate that Breg cells infiltrate human malignancies and suppress anti-tumor immune responses through the expression of immune checkpoint molecules (PDL1) and immunosuppressive cytokines (IL10, IL35, and TGFβ) (Figure 3) (114, 115). Additionally, a significant presence of Breg cells was observed to diminish antibody-mediated humoral immunity in cancer patients (115). The effector molecules of Breg cells, such as TGFβ, IL10, and IL35, can diminish effector T cell responses and/or promote Treg differentiation, thereby resulting in enhanced tumor progression (116). Breg cells impede the development of naïve T cells into Th1 and Th17 (117). They also exert analogous effects on dendritic cells and macrophages (117). Advanced hepatocellular carcinoma, gastric carcinoma, and prostate carcinoma often exhibit elevated levels of Breg cells, indicating that Breg cells may influence tumor development and metastasis (118–120). Breg cells secrete cytokines such as TGFβ, which can convert M1 macrophages into an M2 immunosuppressive phenotype and induce the differentiation of naive CD4+ T cells into Treg cells, leading to remodeling of the TME (121, 122).
M2-tumor-associated macrophages
The growth of the tumor is closely associated with macrophage infiltration. Multiple studies have shown that tumor-associated macrophages can secrete various cytokines, such as platelet-derived growth factor (PDGF), TGFβ1, hepatocyte growth factor (HGF), epidermal growth factor (EGF) family, and basic fibroblast growth factor (BFGF), that facilitate tumor cell proliferation and survival (123). Another study indicates that the malignant invasion of phyllodes tumors is facilitated by a positive feedback loop of CCL5 and CCL18 between TAM and myofibroblasts. CCL5 triggers the AKT-signaling upon binding to its receptor to attract and repolarize tumor-associated macrophages. Consequently, TAMs secrete CCL18, which promotes the invasion of malignant tumors by converting mesenchymal fibroblasts into myofibroblasts (124). M2 macrophages can enhance the migration of tumor and stromal cells by secreting matrix metalloproteinases (MMPs), cathepsins, serine proteases, and various collagen types, along with other extracellular matrix components, which degrade the endothelial cell matrix membrane to facilitate neo-angiogenesis (70). The secreted factors promote epithelial-mesenchymal transition (EMT), resulting in tumor spread (125). In addition to synthesizing angiogenic factors, macrophages can express many enzymes that regulate angiogenesis, including MMP-2, cyclooxygenase-2, MMP-7, MMP-9, and MMP-12 (Figure 4) (70, 126). M2 macrophages secrete immunosuppressive cytokines that promote the Th2 response, whereas M1 macrophages elicit the Th1 response through the production of pro-inflammatory cytokines (127). TAM-derived TGFβ has been shown to induce HIF1/TRIB3-signaling, hence facilitating the advancement of cancer (128).
Tumor-associated macrophages produce large amounts of the immunosuppressive cytokine IL10, which inhibits the cytotoxic activity of Th1 cells, NK cells, and CD8+ T cells against tumor cells (129). This constrains the TME’s capacity to inhibit tumor growth. The recruitment of Tregs to the TME through the chemokine receptor CCR4 is significantly enhanced by tumor-associated macrophage-derived CCL17/CCL22 (17) (Figure 4). Reports indicate TAM-mediated Treg recruitment in liver, nasopharyngeal, and ovarian malignancies. These Tregs suppress anti-cancer CTL activity (130). M2-TAMs preserve their immunosuppressive characteristics and effectively deplete the anti-tumoral immune response by exhibiting elevated synthesis of immune checkpoint inhibitor (ICI) PDL1 and cytotoxic T-lymphocyte antigen-4 (CTLA4) ligand (69). The TGFβ and IL10 generated by TAM hinder the maturation and proliferation of DCs. Consequently, antigen presentation diminishes, leading to a compromised adaptive immune response (69, 131) (Figure 4).
Interaction between tumor-infiltrating lymphocytes and tumor-associated macrophages
The crosstalk between tumor-infiltrating lymphocytes and tumor-associated macrophages plays a pivotal role in shaping the TME and influencing cancer progression. IFNγ secreted by Th1 cells can reprogram TAMs toward an M1 phenotype by activating the JAK–STAT1 signaling pathway, thereby enhancing anti-tumor immune responses (132, 133). M1 macrophages, in turn, produce cytokines and chemokines such as IL12, CXCL9, and CXCL10, which promote the polarization and recruitment of Th1 cells, strengthening type-1 immune responses (31) (Figure 5). In contrast, Th2-derived cytokines, including IL4 and IL13, drive M2 macrophage polarization. IL4, upon receptor binding, activates JAK, leading to STAT6 activation, a key regulator of M2 differentiation, which also induces peroxisome proliferator-activated receptor γ (PPARγ) to control the transcription of M2-specific genes (31, 132). In glioblastoma, the TME-derived glycoprotein chitinase-3-like protein 1 (CHI3L1), in association with Gal3, promotes M2 polarization, resulting in reduced CD4+ and CD8+ T cell populations (134). M2-TAM–secreted factors such as IL10, TGFβ, and CCL22 facilitate Treg cell expansion and recruitment into tumors (31, 135) (Figure 5). Additionally, MARCO+ macrophages suppress CD8+ T cell activity by promoting Treg proliferation (136), while M2-TAM–expressed ARG1 depletes L-arginine from the microenvironment, limiting T cell proliferation (137). B cell-derived IL10 has also been shown to contribute to M2 polarization in a melanoma model (31, 138), and in colon cancer, B cell–derived γ-aminobutyric acid (GABA) enhances IL10+ macrophage populations, which in turn inhibit CD8+ T cell function (139).
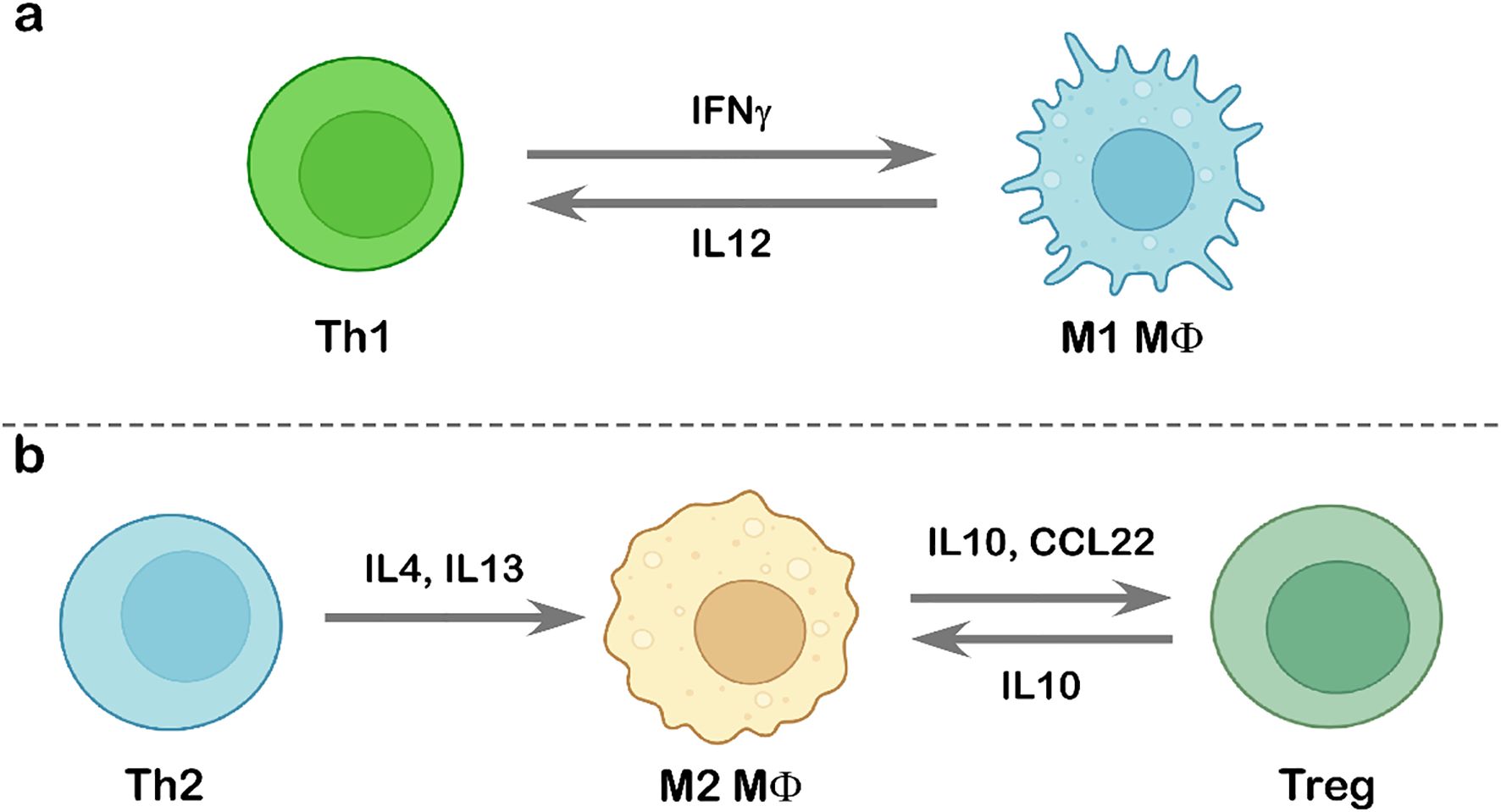
Figure 5. Interaction between tumor-infiltrating lymphocytes and tumor-associated macrophages. Schematic representation of the interplay between TILs and TAMs. (a) Interaction between Th1 cells and M1-polarized macrophages, highlighting the cytokine-mediated enhancement of anti-tumor immunity. (b) Th2 cell-induced M2 polarization through IL4 and IL13 signaling (left) and the subsequent interaction between M2-like macrophages and Treg cells (right). Illustration created using BioRender.com.
Immunotherapy to counter immunoescape mechanisms
In the escape phase, tumor and stromal cells proliferate in a manner that evades the immune system. This generates a tumor-microenvironment that makes the immune system less effective. The objectives of immunotherapies are to restore the balance between the immune system and tumor cells, inhibit tumor growth, and eradicate tumor cells (140).
Therapeutic approaches for tumor-infiltrating lymphocytes
Therapeutic strategies targeting TILs have evolved with the development of a novel adoptive cell therapy (ACT) approach known as TIL therapy, which harnesses the patient’s own immune system to fight cancer. In this method, immune cells, primarily T cells, are extracted from a patient’s tumor, expanded in the laboratory, and reinfused to boost the body’s ability to recognize and eliminate cancer cells. In February 2024, the U.S. Food and Drug Administration (FDA) approved Lifileucel, an autologous TIL-based therapy (141). A phase-II clinical trial (NCT03645928) is currently evaluating Lifileucel in combination with immune checkpoint inhibitors in patients with solid tumors (142). Recent advances have also focused on generating PD1–deficient TILs using CRISPR-Cas9 gene editing, which has shown enhanced anti-tumor efficacy. For instance, IOV-4001, a genetically engineered PD1 knockout TIL, is being investigated in a Phase I/II trial (IOV-GM1-201) for advanced non-small-cell lung cancer and unresectable or metastatic melanoma (141). Multiple TIL-based ACT trials (NCT01174121, NCT05417750, and NCT06488950) are actively recruiting for solid tumor studies (ClinicalTrials.gov). Another candidate, AGX148, a CD8+ TIL product expressing CD39 and CD103, is under Phase-I evaluation (NCT05902520) for advanced solid tumors, both as monotherapy and in combination with siRNA-mediated PD1 silencing (PH-762) (143). TBio-4101, a neoantigen-targeted TIL therapy designed using peptides with tumor-specific mutations to enrich tumor-reactive TILs, is being tested as a monotherapy in melanoma (NCT05628883) and alongside pembrolizumab in solid tumors (NCT05576077) (143).
Targeting inhibitory immune receptors may serve as a therapeutic strategy for restoring immune normalcy. To develop new therapeutic strategies, numerous innovative immune checkpoint inhibitors, such as Lymphocyte Activation Gene 3 (LAG3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-domain immunoglobulin suppressor of T cell activation (VISTA), and T cell immunoglobulin and mucin-domain containing-3 (TIM3), are undergoing extensive studies alongside PD1, PDL1, and CTLA4. Enhancing the proliferative and anti-tumor capabilities of effector T cells can be accomplished by targeting the elevated expression of LAG3 on CD8+ and CD4+ T cells in the bone marrow and blood of patients with multiple myeloma (144). Moreover, it has been shown that the combination of anti-LAG3 and anti-PD1 monoclonal antibodies enhances IFNγ production and T cell cytotoxicity, leading to more effective tumor growth inhibition (Figure 6) (145). Blocking TIGIT has been demonstrated to reverse NK cell depletion and enhance CD8+ T cell activity (146, 147). The co-expression of TIM3 and PD1 significantly affects T cell exhaustion and the loss of stemness (148). Unlike single-agent therapy, the simultaneous inhibition of TIM3 and PD1 has been shown to reinstate effector T cell functionality, induce more significant tumor shrinkage, and amplify the anti-tumor immune response (Figure 6) (149). In addition, CAR-T therapy has demonstrated considerable advantages in cancer treatment. It entails the genetic modification of T lymphocytes to express chimeric antigen receptors, so allowing them to specifically target tumor cells. Clinical trials indicated that CAR-T therapy exhibits a 45% efficacy rate in the treatment of multiple myeloma (150).
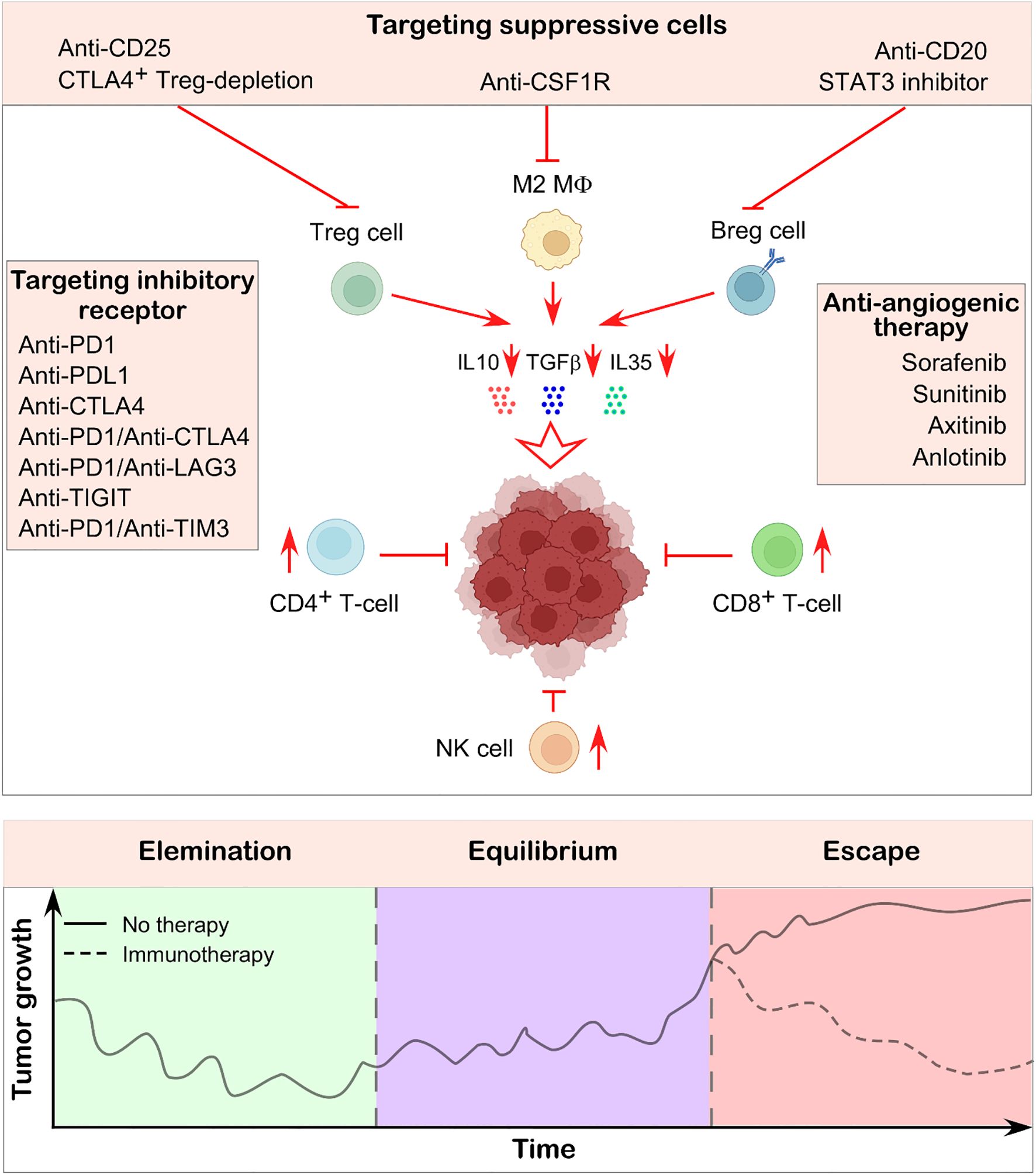
Figure 6. Immunotherapeutic strategy to suppress tumor growth. Immunotherapy aims to reinvigorate the body’s naturally hard-wired immune system to recognize and eliminate cancer cells. One approach involves using specific antibodies to target immunosuppressive cells within the tumor-microenvironment, thereby restoring the activity of effector immune cells and promoting anti-tumor responses. Another widely used strategy targets inhibitory immune checkpoints such as PD1, CTLA4, LAG3, PDL1 etc., which are often expressed on both immune cells and tumor cells. Blocking these molecules has been shown to enhance immune-mediated tumor destruction. Additionally, anti-angiogenic therapies contribute by normalizing the tumor microenvironment, improving immune cell infiltration, and further restricting tumor progression. The illustration in the lower panel shows how tumor growth rates alter depending on whether immunotherapy was used to counteract the immunosuppressive environment and restore immune balance. Illustration created using BioRender.com.
In a phase I/II clinical trial (NCT03056339), patients with recurrent or refractory B-cell lymphoma received cord blood-derived, CD19-targeted CAR-NK therapy, resulting in an overall response rate in 21 patients, 13 achieving complete responses and 8 partial responses. Among 49 treated patients, 18 demonstrated objective responses, including 14 complete and 4 partial responses (151). Another phase-I trial (NCT03383978) is evaluating the safety and tolerability of HER2-targeted NK-92 cells in combination with the immune checkpoint inhibitor ezabenlimab in patients with recurrent HER2-positive glioblastoma. In the United States, a phase-II study (NCT04847466) is underway to assess the efficacy of irradiated PDL1 CAR-NK cells combined with pembrolizumab and N-803 for recurrent or metastatic head and neck and gastric cancers. Collectively, these studies highlight the potential of CAR-NK cell therapies, particularly when paired with anticancer drugs, as a promising strategy for future cancer treatment.
It is crucial to target several immunosuppressive cells present in the TME to reshape the TME. Neutralizing antibodies against CD25 effectively eradicated CD25+ Treg cells in tumor-bearing mice, resulting in an enhanced infiltration of CD8+ T cells in the tumor (Figure 6) (152, 153). Furthermore, a mouse model has shown that the targeted elimination of Ctla4+ Treg cells results in complete tumor remission (154, 155). In contrast, the application of anti-CD20 monoclonal antibodies to eradicate Breg cells has yielded unsatisfactory outcomes for solid tumors, although it has demonstrated significant therapeutic efficacy in hematologic malignancies (156). Conversely, STAT3 inhibitors have been shown to reduce the synthesis of immunosuppressive cytokines and the proliferation of Breg cells (Figure 6) (157, 158). B-cell epitope–based vaccines represent an innovative direction in B-cell immunotherapy. For instance, a vaccine targeting the HER-2/neu peptide, an established tumor antigen, is being used in breast and ovarian cancers (159). Additionally, a phase I/II clinical trial (NCT04416984) is currently evaluating the safety and effectiveness of ALLO-501A, an allogeneic anti-CD19 CAR T cell therapy, alongside ALLO-647, an anti-CD52 monoclonal antibody, in patients with relapsed or refractory large B-cell lymphoma. Aside from this, current studies reveal that anti-angiogenic therapy enhances the immune-microenvironment and tumor vasculature. Anti-angiogenic therapy has led to the development of several strategies that target multi-targeted tyrosine kinases, including sorafenib, sunitinib, axitinib, and anlotinib (Figure 6) (160). Research has shown that neutralizing antibodies targeting VEGFA can effectively enhance CD8+ T cell activity and inhibit endothelial cell expression of FAS ligand, hence promoting effector T cell infiltration (161, 162). Anlotinib, a small-molecule tyrosine kinase inhibitor, was demonstrated to reduce PDL1 expression on endothelial cells, hence enhancing the equilibrium of immune cells within the tumor (163). Table 2 also summarizes TIL-targeting therapies currently listed on ClinicalTrials.gov.
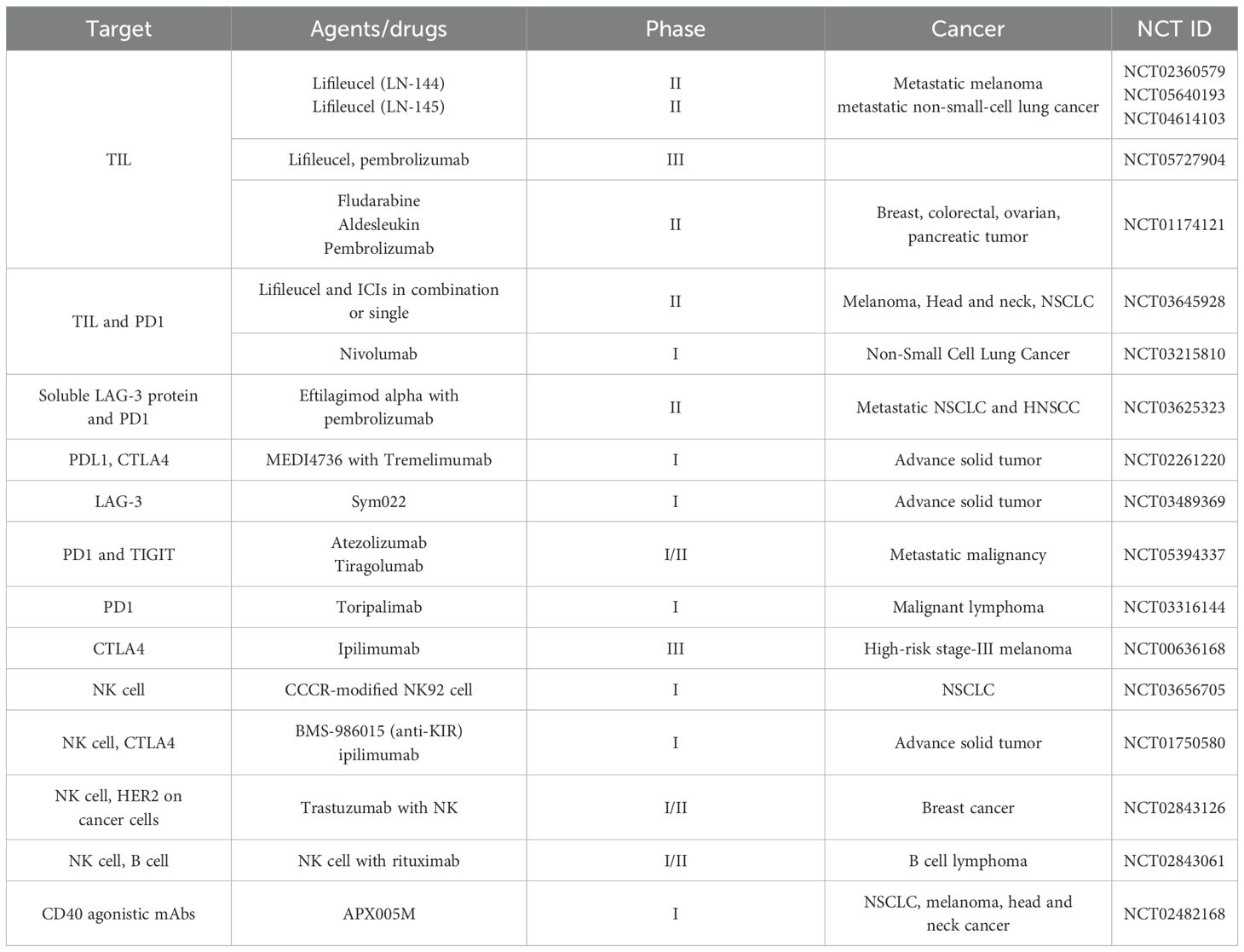
Table 2. Therapeutic approaches for tumor-infiltrating lymphocytes.
Therapeutic approaches for tumor-associated macrophages
One promising strategy to combat cancer involves reprogramming M2-type TAMs into the pro-inflammatory, anti-tumor M1 subtype. This repolarization can be stimulated through activation of toll-like receptors (TLR) on macrophages. Preclinical studies have shown that TLR7 agonists like imiquimod and 852A exhibit notable anticancer effects with durable therapeutic benefits (164, 165). Similarly, the TLR9 agonist lefitolimod has been evaluated in multiple clinical trials, including NCT02668770, where it promotes M1 polarization of TAMs and enhances anti-tumor immune responses (69, 166).
Targeting signal regulatory protein-α (SIRPα) or CD47 to restore the phagocytosis activity of TAM is another immunotherapeutic approach being used to treat different cancers. It is now feasible to successfully suppress tumor cell growth and metastasis by inhibiting CD47-SIRPα signaling, restoring macrophage phagocytosis of tumor cells, and enhancing the effector CD8+ T cell response (167). The versatile drug RRx-001, currently being investigated in clinical trials (NCT02518958), has shown the ability to promote the M1 phenotype in TAM. It also blocks the interaction between SIRPα on macrophages and CD47 on cancer cells, enhancing immune response against tumors (168, 169). Additionally, molecules like microRNA miR-340 and the polypeptide PEP-20 have been found to target CD47, boosting macrophage phagocytosis of cancer cells (170, 171). Macrophages expressing the scavenger receptor MARCO, which contributes to their transformation into immunosuppressive TAMs, display a pro-tumor, anti-inflammatory phenotype. Studies have demonstrated that targeting MARCO with specific antibodies can suppress TAM activation, shift macrophages toward the M1 phenotype, and enhance the effectiveness of anti-tumor immunotherapies in breast, colon cancer, and melanoma models (172, 173).
Macrophage colony stimulating factor-1 (CSF1)-CSF1R plays a critical role in TAM recruitment and survival, hence blocking this route could be a possible TAM treatment method. Study shown that the specific targeting of the CSF1R significantly inhibits tumor growth in mice with EL4 tumor and mouse mammary tumor virus transgenic mammary tumor model by depleting M2-type macrophages, indicating a promising target for cancer immunotherapy (Figure 6) (174). A randomized phase-II clinical trial involving patients with advanced triple-negative breast cancer found that combining the CSF1-targeting monoclonal antibody lacnotuzumab (NCT02435680) with chemotherapy led to better outcomes compared to chemotherapy alone (175). Bisphosphonates have also been reported to reduce the recruitment and infiltration of TAMs while promoting their apoptosis (69). Emactuzumab, another monoclonal antibody against CSF1, enhances anti-tumor immune responses by decreasing the number of F4/80+ TAMs within tumors and increasing the CD8+/CD4+ T cell ratio through inhibition of the CSF1/CSF1R-signaling pathway (164). When combined with paclitaxel, emactuzumab effectively suppresses the growth of advanced solid tumors and significantly reduces CSF1R+ TAM populations (176). Additionally, blocking the CCL2/CCR2 axis lowers invasive TAM numbers in tumors and enhances the effectiveness of combined immunotherapy and chemoradiotherapy (164, 177).
In glioblastoma animal models, inhibitors targeting the CCR2/CCL2 axis have been shown to significantly reduce M2-type TAM infiltration at tumor sites, leading to improved survival outcomes (69). In a clinical trial (NCT01413022) involving patients with locally advanced pancreatic cancer, the CCR2 inhibitor PF-04136309 was found to be safe and well tolerated when combined with chemotherapy (178). Suppressing TAM activity by blocking the CCL2/CCR2–STAT3 pathway using siRNA or pharmacological agents enhances macrophage phagocytosis and inhibits tumor growth and metastasis (164, 179). The CXCL12/CXCR4-signaling pathway also plays a key role in macrophage recruitment, and its inhibition alongside standard treatments has demonstrated anti-tumor effects (180). For example, a phase-I clinical trial (NCT02737072) combining durvalumab with the CXCR4 antagonist LY2510924 showed a favorable safety profile and promising responses in patients with advanced, treatment-resistant solid tumors (181). Additionally, in preclinical studies, macrophages engineered to express chimeric antigen receptors (CAR-M) markedly slowed tumor progression and extended survival (182, 183). Building on this, a clinical trial (NCT04660929) is investigating CAR-M therapies targeting HER2 for tumors that overexpress this receptor (184). Table 3 provides a comprehensive overview of therapies currently approved or under active investigation that aim to target TAMs, based on information from ClinicalTrials.gov.
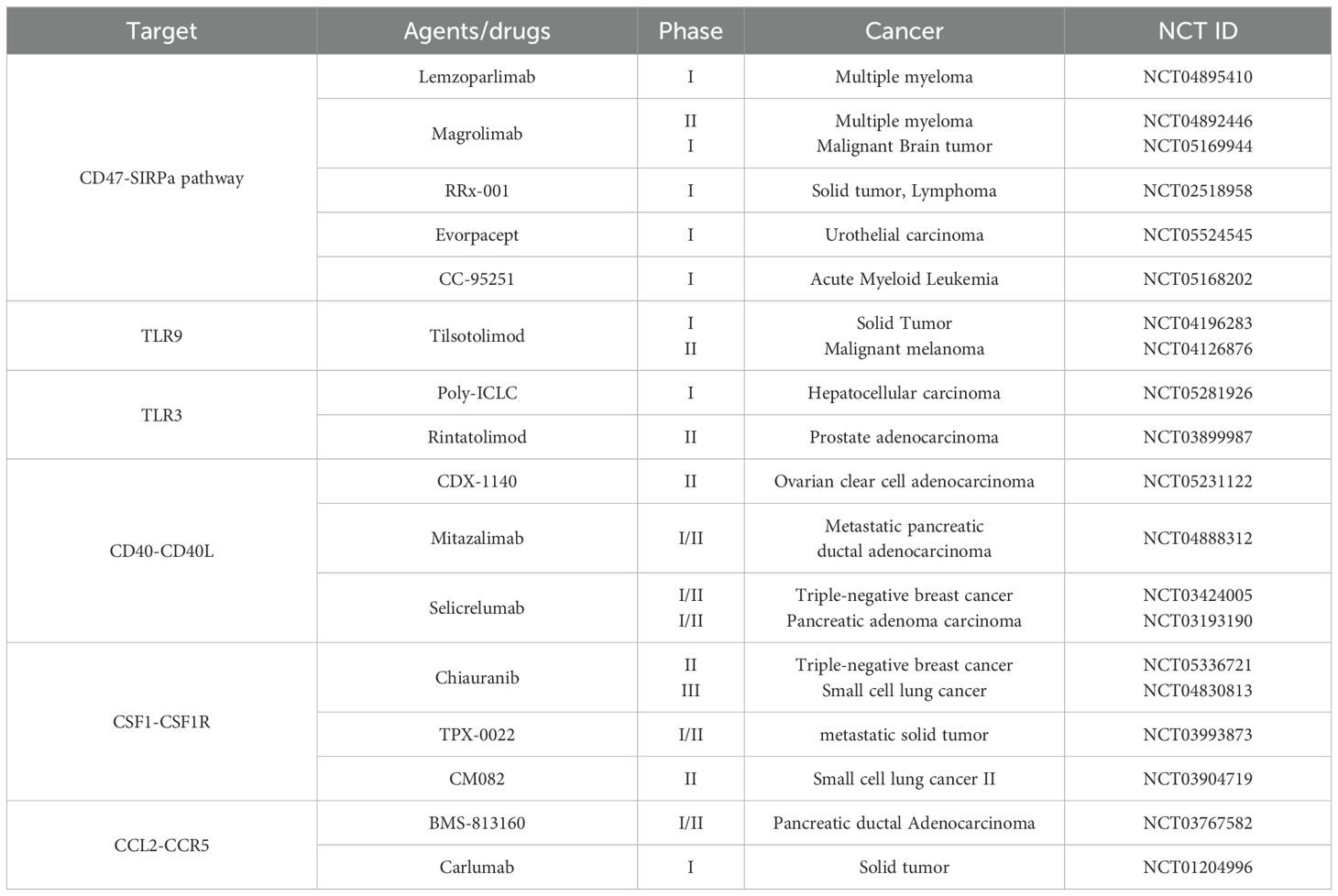
Table 3. Therapeutic approaches for tumor-associated macrophages.
Therapeutic approaches targeting tumor-infiltrating lymphocytes and tumor-associated macrophages
Emerging therapeutic strategies increasingly focus on simultaneously targeting tumor-infiltrating lymphocytes and tumor-associated macrophages to reshape the tumor immune microenvironment and boost treatment efficacy. In lung cancer, tumor-derived IL37 promotes the generation of MARCO-positive TAMs, which contribute to an immunosuppressive environment. Targeting MARCO or the IL37 receptor on macrophages has been shown to repolarize TAMs toward a more pro-inflammatory state, enhancing the cytolytic function of both natural killer cells and T cells while reducing Treg cell activity (136). Inhibition of the PI3Kγ pathway represents another promising avenue, as it leads to TAM repolarization with increased production of IL12 and IFNγ, alongside greater recruitment and maturation of CD8+ T cells within tumors (185, 186). Building on these findings, a phase-II randomized clinical trial (NCT03980041) is evaluating the combination of the PI3Kγ inhibitor IPI-549 with anti-PD1 immune checkpoint inhibitors in patients with advanced urothelial cancer.
Preclinical studies in mouse models of hepatocellular and liver cancer demonstrate that CCR2 antagonists reduce TAM populations and increase infiltration of CD8+ T cells, further promoting anti-tumor immunity (187, 188). Likewise, stimulator of interferon genes (STING) agonists have been shown to reprogram TAMs into the pro-inflammatory M1 phenotype and enhance CD8+ T cell infiltration in murine colorectal cancer models (189). A first-in-human phase-I trial (NCT03843359) is currently investigating GSK3745417, a STING agonist, combined with dostarlimab (anti-PD1) to assess safety, tolerability, and preliminary efficacy in patients with advanced solid tumors. Targeting molecular interactions within the tumor-microenvironment also shows promise. For example, disrupting the interaction between CHI3L1 and Galectin-3 (Gal3) using GMP, a Gal3-binding peptide mimetic, inhibits M2-TAM polarization and supports T cell proliferation in brain tumors (134). Similarly, combining anti-MS4A4A antibodies with PD1 blockade improves immune checkpoint inhibition by preventing macrophage M2 polarization and enhancing CD8+ T cell infiltration in advanced colorectal cancer (190).
In colon cancer, the GABA-A receptor antagonist picrotoxin significantly reduces IL10–secreting macrophages and restores effector T cell activity, further illustrating the potential of modulating TAMs to boost anti-tumor immunity (139). Ongoing clinical research includes a phase-I trial (NCT06637306) testing dupilumab, an IL4 receptor blocker, in combination with ICIs to repolarize M2 TAMs in breast cancer patients (191). Together, these approaches demonstrate the growing recognition that coordinated targeting of both TILs and TAMs holds substantial potential to remodel the immune landscape within tumors and improve therapeutic outcomes across a range of cancers.
Factors determining immunotherapy outcomes
Biomarkers
A key element influencing the success of immunotherapy lies within the TME, particularly the presence and characteristics of tumor-infiltrating lymphocytes and tumor-associated macrophages. Biomarkers associated with these immune cells serve as important predictors of treatment response. For instance, a high density of TILs, especially CD8+ T cells, has consistently been linked to improved clinical outcomes across multiple cancer types (192). Recent clinical analyses reveal that elevated levels of intratumoral tissue-resident memory T cells (TRMs), characterized by CD103+ and CD69+ expression, correlate with better responses to immune checkpoint inhibitor therapies in non-small cell lung cancer and oral cancer (193).
Markers indicative of T cell exhaustion, including TIM3, CTLA4, and PD1, reflect impaired T cell function and significantly impact the effectiveness of checkpoint blockade treatments (193). In melanoma patients treated with anti-PD1 therapy, the proportion of precursor exhausted T cells (TPEX cells) has been associated with longer-lasting responses, underscoring their potential as predictive biomarkers (193, 194). Furthermore, the presence of tertiary lymphoid structures (TLS), organized aggregates of B cells, DCs, and T cells, has been linked to enhanced therapeutic responses in ICIs-treated soft tissue sarcoma and melanoma patients (195, 196). Conversely, elevated FOXP3 expression, a marker for Treg cells, is often associated with immunotherapy resistance, immunosuppression, and poorer prognoses (197).
On the macrophage front, molecules expressed by different TAM subsets also play a critical role in shaping immunotherapy outcomes. M1 macrophages, known for their pro-inflammatory and anti-tumor functions, typically express markers such as CD80, CD86, and IL12. In contrast, M2 macrophages, which tend to support tumor progression and immune suppression, are characterized by markers including CD163, CD204, CD206, and IL10 (198). The ratio of M1 to M2 macrophages within the tumor has been linked to both patient survival and response to immunotherapy in cancers such as breast cancer (199). Additionally, high expression levels of TAM infiltration markers like CD68 and CD163 have been correlated with poor prognosis and treatment resistance in cervical and oral squamous cell carcinomas (200, 201). Together, these biomarkers provide valuable insights into the immune landscape of tumors and help guide personalized immunotherapy approaches for improved patient outcomes.
In the foreseeable future, immune checkpoint inhibitor therapy will predominate in cancer treatment (202–204). Neglecting immunophenotyping prior to the initiation of checkpoint inhibitor therapy may accelerate tumor progression (205). As a result, immunophenotyping contributes significantly to cancer research by revealing the immune landscape of tumors and supporting the design of targeted and personalized treatment strategies for diverse immunophenotypes, ultimately enhancing the efficacy of checkpoint inhibitor treatments.
Tumor-infiltrating lymphocytes and tumor-associated macrophages heterogeneity
Immunotherapy outcomes can vary widely even among patients diagnosed with the same type of cancer, and one of the key factors influencing this variability is the composition of tumor-infiltrating immune cells within the TME. Tumors are often classified as “hot” or “cold” based on their immune cell infiltration profiles. “Hot tumors” are characterized by a TME rich in tumor-infiltrating lymphocytes, particularly CD8+ T cells, while “cold tumors” display lower levels of these cytotoxic immune cells (206). It is generally accepted that immunotherapies, such as immune checkpoint inhibitors, tend to be more effective in “hot tumors,” where CD8+ T cells play a crucial role in mounting an anti-tumor immune response (206, 207).
This section highlights how the heterogeneity of TILs and TAMs correlates with disease progression across various cancers. A recent study found that tumors with higher infiltration of CD8+ T cells and NK cells, combined with lower levels of M2-polarized TAMs, were significantly associated with improved progression free survival (PFS), indicated by hazard ratios (HR) less than 1, which reflects better PFS (208). For example, patients with adrenocortical carcinoma (ACC), liver hepatocellular carcinoma (LIHC), cervical squamous cell carcinoma and endocervical adenocarcinoma (CESC) exhibited lower M2 macrophage abundance and correspondingly better PFS (HR < 1). In contrast, cancers such as lower-grade glioma (LGG) and glioblastoma (GBM) demonstrated reduced median levels of CD8+ T cells and NK cells, alongside elevated M2 macrophage presence, correlating with poorer PFS (HR > 1) (208). The study also revealed notable differences within breast cancer subtypes: basal-like and HER2-enriched tumors showed significantly higher numbers of CTLs and a substantially lower likelihood of disease progression compared to luminal-A and luminal-B subtypes. Additionally, increased Treg cell infiltration was specifically associated with worse outcomes in renal clear cell carcinoma (208).
Together, these findings emphasize the complexity and heterogeneity of immune cell populations within the TME and underscore their critical influence on immunotherapy effectiveness and cancer progression. Understanding these nuances is essential for tailoring personalized treatment strategies and improving clinical outcomes.
Heterogeneity of immune infiltration
Within a single tumor, the immune landscape can vary significantly across different regions, which greatly influences how the tumor responds to immunotherapy. Some areas may be rich in immune infiltration, characterized by the presence of activated T cells and dendritic cells, making these regions more susceptible to treatments like immune checkpoint blockade. Conversely, other parts of the TME may exhibit immunosuppressive features, marked by increased levels of myeloid-derived suppressor cells (MDSCs), Treg cells, Breg cells, and TAMs. These immunosuppressive cells dampen the activation and function of effector immune cells, leading to a diminished therapeutic response (209).
Immune-related adverse events
Immune-related adverse events (irAEs) are commonly observed in patients undergoing treatment with immune checkpoint inhibitors and can significantly impact patient outcomes and the overall success of immunotherapy. ICIs work by blocking inhibitory immune checkpoints, thereby enhancing T cell–mediated immune responses against tumors. However, this disruption of immune regulation can lead to a loss of immune tolerance, causing autoreactive T cells to attack healthy tissues, a process that results in irAEs resembling autoimmune diseases (210). Additionally, ICIs may increase levels of pre-existing autoantibodies, such as antithyroid antibodies, which contribute to these immune complications (211). Overproduction of inflammatory cytokines in patients receiving ICIs is also associated with systemic toxicities (212).
Among the most common irAEs are gastrointestinal issues, with symptoms generally more frequent and severe in patients treated with anti-CTLA4 therapies compared to those receiving PD1 or PDL1 inhibitors (213). Clinical trials report that ICI-associated hepatitis occurs in approximately 2% to 15% of patients (214). Inflammatory arthritis affects about 1% of patients, while arthralgia is reported in 3% to 7%. Unlike dermatologic toxicities, which often appear early, other irAEs, such as arthritis, tend to develop later, typically around two months after starting PD1/PDL1 inhibitors and about one month following initiation of anti-CTLA4 therapy (212). Understanding and managing these immune-related side effects are critical to optimizing immunotherapy safety and effectiveness.
Acquired resistance to therapy
Acquired resistance to immunotherapy often arises from disruptions in the tumor’s ability to present antigens effectively to T cells. Mutations affecting MHC molecules or components of the antigen-processing machinery can impair antigen presentation, preventing T cells from recognizing and attacking cancer cells. Such mechanisms have been documented in cancers like metastatic melanoma and prostate cancer (215).
Tumor metabolic changes also contribute to resistance. Cancer cells frequently undergo metabolic reprogramming, producing high levels of lactic acid through glycolysis. This acidifies the TME, which in turn suppresses T cell metabolism and impairs their function (215). Moreover, the tumor’s consumption of glucose can limit its availability to T cells, reducing mTOR signaling, IFNγ production, and the glycolytic capacity of T cells, further dampening their anti-tumor activity (216).
Another factor involved in acquired resistance is the increased production of adenosine by tumor cells, often driven by elevated expression of CD38. Adenosine inhibits T cell proliferation and cytotoxic functions, contributing to immune evasion (217). Genetic alterations can also play a role; for example, melanoma patients who develop resistance to PD1 blockade have been found to carry loss-of-function mutations in JAK1 or JAK2 genes. These mutations render tumor cells less responsive to the antiproliferative effects of T cell–derived IFNγ, allowing cancer cells to escape immune attack (218).
Lastly, tumor cells may undergo “antigenic drift,” where changes in tumor epitopes alter their antigenicity. This shift enables cancer cells to evade recognition by T cells, facilitating immune escape and therapy resistance (219). Together, these mechanisms highlight the complexity of acquired resistance and underscore the need for strategies that can overcome or prevent these barriers to effective immunotherapy.
Concluding remarks
In conclusion, comprehending cancer through the lens of immunoediting provides valuable insight into the dynamic and often paradoxical relationship between the immune system and tumor cells. By focusing on the regulatory roles of tumor-infiltrating lymphocytes and tumor-associated macrophages, we highlight the importance of decoding the immune landscape within the tumor-microenvironment. Restoring and enhancing the immune system’s inherent defenses requires a deep grasp of how these cellular components interact under both healthy and pathological conditions. As immunotherapy continues to evolve, identifying the reasons for therapeutic resistance remains a critical step toward improving outcomes.
Resistance remains a significant challenge in cancer treatment, as tumor cells employ diverse strategies to escape immune detection. These include modifying immune evasion pathways, increasing the expression of alternative immune checkpoints, and losing tumor antigens that are targets for immune cells. However, several promising research directions are emerging. Advances in personalized medicine now allow for the development of tailored therapies that consider each patient’s unique immune landscape and tumor characteristics, potentially improving treatment effectiveness. Additionally, growing evidence highlights the important role of the gut microbiota in shaping responses to cancer immunotherapy, making modulation of the gut microbiome a compelling approach to enhance therapeutic outcomes.
With growing access to advanced immune profiling and the integration of artificial intelligence, we are poised to develop more precise and personalized immunotherapeutic strategies. Ultimately, these advancements possess the capacity to transform immunotherapy into a widely effective and enduring remedy in the battle against cancer.
Author contributions
SumonM: Methodology, Formal analysis, Investigation, Visualization, Data curation, Writing – original draft. AG: Formal analysis, Writing – original draft, Data curation, Validation, Investigation. SC: Writing – original draft, Formal analysis, Resources, Methodology, Data curation, Validation. UB: Writing – original draft, Data curation, Validation, Methodology. SumoyM: Formal analysis, Writing – original draft, Data curation. TD: Validation, Writing – review & editing, Resources, Funding acquisition, Supervision. GS: Conceptualization, Project administration, Supervision, Writing – review & editing, Resources, Visualization, Funding acquisition.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. GS (HRD/IES/2023(01)) and TD (74/1/2020-Pers) are ICMR Emeritus Scientists. The study was funded by the Indian Council for Medical Research, Government of India. We acknowledge the BioRender platform (https://www.biorender.com) for the creation of the illustrations.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
APC: Antigen Presenting Cell
Breg: B-regulatory cell
CCL: Chemokine (C-C motif) ligand
CAR-T: Chimeric Antigen Receptor – T cell
CD: Cluster of Differentiation
CSF1: Colony stimulating factor-1
CTL: Cytotoxic T lymphocyte
CTLA4: Cytotoxic T Lymphocyte Associated Protein – 4
CXCL: Chemokine (C-X-C motif) ligand
CXCR: Chemokine (C-X-C motif) receptor
DC: Dendritic cell
FASL: FAS Ligand
ICI: Immune Checkpoint Inhibitor
IFN: Interferon
IL: Interleukin
LAG-3: Lymphocyte Activation Gene 3
MHC: Major Histocompatibility Complex
MMP: Matrix Metalloproteinase
NK: Natural Killer
NKG2D: Natural Killer Group 2 Member D
NKT: Natural Killer T cells
NO: Nitric Oxide
PD1: Programmed Death receptor 1
PDL-1: Programmed Death Ligand 1
STAT3: Signal Transducer and Activator of Transcription 3
TAM: tumor-associated macrophage
TCR: T cell receptor
TGFβ: Transforming Growth Factor β
Th: T helper cell
TIGIT: T cell immunoreceptor with Ig and ITIM domains
TIL: Tumor Infiltrating Lymphocyte
Tim-3: T cell immunoglobulin and mucin-domain containing – 3
TLR: Toll-like receptor
TME: Tumor microenvironment
TRAIL: TNF-related apoptosis-inducing ligand
Treg: T-regulatory cell
References
1. Kim R, Emi M, and Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. (2007) 121:1–14. doi: 10.1111/j.1365-2567.2007.02587.x
2. Dunn GP, Bruce AT, Ikeda H, Old LJ, and Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. (2002) 3:991–8. doi: 10.1038/ni1102-991
3. Gubin MM and Vesely MD. Cancer immunoediting in the era of immuno-oncology. Clin Cancer Res. (2022) 28(18):3917–28. doi: 10.1158/1078-0432.CCR-21-1804
4. Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. (1998) 95:7556–61. doi: 10.1073/pnas.95.13.7556
5. Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. (2007) 450:903–7. doi: 10.1038/nature06309
6. Vesely MD, Kershaw MH, Schreiber RD, and Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. (2011) 29:235–71. doi: 10.1146/annurev-immunol-031210-101324
7. Fenton SE, Saleiro D, and Platanias LC. Role of T cells in cancer immunotherapy: Opportunities and challenges. Cancers (Basel). (2021) 13:1037. doi: 10.3390/cancers13051037
8. Mellman I, Chen DS, Powles T, and Turley SJ. The cancer-immunity cycle: Indication, genotype, and immunotype. Immunity. (2023) 56:2188–205. doi: 10.1016/j.immuni.2023.09.011
9. Chen DS and Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
10. Motz GT and Coukos G. Deciphering and reversing tumor immune suppression. Immunity. (2013) 39:61–73. doi: 10.1016/j.immuni.2013.07.005
11. Gonzalez H, Hagerling C, and Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. (2018) 32:1267–84. doi: 10.1101/gad.314617.118
12. Swann JB and Smyth MJ. Immune surveillance of tumors. J Clin Invest. (2007) 117:1137–46. doi: 10.1172/JCI31405
13. Oliveira G and Wu CJ. Dynamics and specificities of T cells in cancer immunotherapy. Nat Rev Cancer. (2023) 23:295–316. doi: 10.1038/s41568-023-00560-y
14. Jung H and Paust S. Chemokines in the tumor microenvironment: implications for lung cancer and immunotherapy. Front Immunol. (2024) 15:1443366. doi: 10.3389/fimmu.2024.1443366
15. Tiwary S, Berzofsky JA, and Terabe M. Altered lipid tumor environment and its potential effects on NKT cell function in tumor immunity. Front Immunol. (2019) 10:2187. doi: 10.3389/fimmu.2019.02187
16. Maimela NR, Liu S, and Zhang Y. Fates of CD8+ T cells in tumor microenvironment. Comput Struct Biotechnol J. (2019) 17:1–13. doi: 10.1016/j.csbj.2018.11.004
17. Sarkar T, Dhar S, Chakraborty D, Pati S, Bose S, Panda AK, et al. FOXP3/HAT1 axis controls Treg infiltration in the tumor microenvironment by inducing CCR4 expression in breast cancer. Front Immunol. (2022) 13:740588. doi: 10.3389/fimmu.2022.740588
18. Xiang Y, Gong M, Deng Y, Wang H, and Ye D. T cell effects and mechanisms in immunotherapy of head and neck tumors. Cell Communication Signaling. (2023) 21:49. doi: 10.1186/s12964-023-01070-y
19. Cornel AM, Mimpen IL, and Nierkens S. MHC class I downregulation in cancer: underlying mechanisms and potential targets for cancer immunotherapy. Cancers (Basel). (2020) 12:1760. doi: 10.3390/cancers12071760
20. Labani-Motlagh A, Ashja-Mahdavi M, and Loskog A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune responses. Front Immunol. (2020) 11:940. doi: 10.3389/fimmu.2020.00940
21. Ahmed H, Mahmud AR, Siddiquee Mohd F-R, Shahriar A, Biswas P, Shimul Md EK, et al. Role of T cells in cancer immunotherapy: Opportunities and challenges.Cancer Pathog Ther.(2022) 1:116–26. doi: 10.1073/pnas.0408197102
22. Yang W, Chen X, and Hu H. CD4+ T-Cell Differentiation In Vitro. Methods Mol Biol. (2020) 21111:91–9. doi: 10.1007/978-1-0716-0266-9_8
23. Beatty G and Paterson Y. IFN-gamma-dependent inhibition of tumor angiogenesis by tumor-infiltrating CD4+ T cells requires tumor responsiveness to IFN-gamma. J Immunol. (2001) 166:2276–82. doi: 10.4049/jimmunol.166.4.2276
24. Propper DJ, Chao D, Braybrooke JP, Bahl P, Thavasu P, Balkwill F, et al. Low-dose IFN-γ Induces tumor MHC expression in metastatic Malignant melanoma. Clin Cancer Res. (2003) 9:84–92.
25. Hoepner S, Loh JMS, Riccadonna C, Derouazi M, Maroun CY, Dietrich P-Y, et al. Synergy between CD8 T cells and Th1 or Th2 polarised CD4 T cells for adoptive immunotherapy of brain tumours. PLoS One. (2013) 8:e63933. doi: 10.1371/journal.pone.0063933
26. Ellyard JI, Simson L, and Parish CR. Th2-mediated anti-tumour immunity: friend or foe? Tissue Antigens. (2007) 70:1–11. doi: 10.1111/j.1399-0039.2007.00869.x
27. Bevan MJ. Helping the CD8(+) T-cell response. Nat Rev Immunol. (2004) 4:595–602. doi: 10.1038/nri1413
28. DiLillo DJ, Yanaba K, and Tedder TF. B cells are required for optimal CD4+ and CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma growth in mice. J Immunol. (2010) 184:4006–16. doi: 10.4049/jimmunol.0903009
29. Uzhachenko RV and Shanker A. CD8+ T lymphocyte and NK cell network: circuitry in the cytotoxic domain of immunity. Front Immunol. (2019) 10:1906. doi: 10.3389/fimmu.2019.01906
30. Wang Z-T, Deng Z-M, Dai F-F, Yuan M-Q, Liu S-Y, Li B-S, et al. Tumor immunity: A brief overview of tumor−infiltrating immune cells and research advances into tumor−infiltrating lymphocytes in gynecological Malignancies (Review). Exp Ther Med. (2024) 27:166. doi: 10.3892/etm.2024.12453
31. Biswas SK and Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. (2010) 11:889–96. doi: 10.1038/ni.1937
32. Zhu S, Wang X, Wang J, Lin J, Cong Y, and Qiao G. CD21lo/medCD27+ proinflammatory B cells are enriched in breast cancer patients and promote antitumor T cell responses. Exp Cell Res. (2017) 361:149–54. doi: 10.1016/j.yexcr.2017.10.013
33. Zheng N and Lu Y. Targeting the IL-9 pathway in cancer immunotherapy. Hum Vaccin Immunother. (2020) 16:2333–40. doi: 10.1080/21645515.2019.1710413
34. Chen T, Guo J, Cai Z, Li B, Sun L, Shen Y, et al. Th9 cell differentiation and its dual effects in tumor development. Front Immunol. (2020) 11:1026. doi: 10.3389/fimmu.2020.01026
35. Nonomura Y, Otsuka A, Nakashima C, Seidel JA, Kitoh A, Dainichi T, et al. Peripheral blood Th9 cells are a possible pharmacodynamic biomarker of nivolumab treatment efficacy in metastatic melanoma patients. Oncoimmunology. (2016) 5:e1248327. doi: 10.1080/2162402X.2016.1248327
36. Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. (2009) 31:787–98. doi: 10.1016/j.immuni.2009.09.014
37. Ye J, Livergood RS, and Peng G. The role and regulation of human Th17 cells in tumor immunity. Am J Pathol. (2013) 182:10–20. doi: 10.1016/j.ajpath.2012.08.041
38. Zhou L, Chong MMW, and Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. (2009) 30:646–55. doi: 10.1016/j.immuni.2009.05.001
39. Basdeo SA, Cluxton D, Sulaimani J, Moran B, Canavan M, Orr C, et al. Ex-Th17 (Nonclassical Th1) cells are functionally distinct from classical Th1 and Th17 cells and are not constrained by regulatory T cells. J Immunol. (2017) 198:2249–59. doi: 10.4049/jimmunol.1600737
40. Terhune J, Berk E, and Czerniecki BJ. Dendritic cell-induced Th1 and Th17 cell differentiation for cancer therapy. Vaccines (Basel). (2013) 1:527–49. doi: 10.3390/vaccines1040527
41. Gavil NV, Cheng K, and Masopust D. Resident memory T cells and cancer. Immunity. (2024) 57:1734–51. doi: 10.1016/j.immuni.2024.06.017
42. Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, et al. Innate or adaptive immunity? The example of natural killer cells. Science. (2011) 331:44–9. doi: 10.1126/science.1198687
43. Shimizu K, Iyoda T, Yamasaki S, Kadowaki N, Tojo A, and Fujii S. NK and NKT cell-mediated immune surveillance against hematological Malignancies. Cancers. (2020) 12:817. doi: 10.3390/cancers12040817
44. Tahara-Hanaoka S, Shibuya K, Onoda Y, Zhang H, Yamazaki S, Miyamoto A, et al. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int Immunol. (2004) 16:533–8. doi: 10.1093/intimm/dxh059
45. Mathew SO, Rao KK, Kim JR, Bambard ND, and Mathew PA. Functional role of human NK cell receptor 2B4 (CD244) isoforms. Eur J Immunol. (2009) 39:1632–41. doi: 10.1002/eji.200838733
46. Blunt MD, Pulido AV, Fisher JG, Graham LV, Doyle A, Fulton R, et al. KIR2DS2 expression identifies NK cells with enhanced anti-cancer activity. J Immunol. (2022) 209:379–90. doi: 10.4049/jimmunol.2101139
47. Dębska-Zielkowska J, Moszkowska G, Zieliński M, Zielińska H, Dukat-Mazurek A, Trzonkowski P, et al. KIR receptors as key regulators of NK cells activity in health and disease. Cells. (2021) 10:1777. doi: 10.3390/cells10071777
48. Kawamura T, Takeda K, Kaneda H, Matsumoto H, Hayakawa Y, Raulet DH, et al. NKG2A inhibits iNKT cell activation in hepatic injury. J Immunol. (2009) 182:250–8. doi: 10.4049/jimmunol.182.1.250
49. Beldi-Ferchiou A, Lambert M, Dogniaux S, Vély F, Vivier E, Olive D, et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget. (2016) 7:72961–77. doi: 10.18632/oncotarget.12150
50. Al-Mterin MA, Murshed K, and Elkord E. PD-1 expression, among other immune checkpoints, on tumor-infiltrating NK and NKT cells is associated with longer disease-free survival in treatment-naïve CRC patients. Cancer Immunol Immunother. (2023) 72:1933–9. doi: 10.1007/s00262-022-03337-8
51. Tang W, Chen J, Ji T, and Cong X. TIGIT, a novel immune checkpoint therapy for melanoma. Cell Death Dis. (2023) 14:466. doi: 10.1038/s41419-023-05961-3
52. Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, et al. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. (2015) 29:635–41. doi: 10.1016/j.intimp.2015.09.017
53. Wu H, Tang T, Deng H, Chen D, Zhang C, Luo J, et al. Immune checkpoint molecule Tim-3 promotes NKT cell apoptosis and predicts poorer prognosis in Sepsis. Clin Immunol. (2023) 254:109249. doi: 10.1016/j.clim.2023.109249
54. Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, et al. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. (1999) 285:727–9. doi: 10.1126/science.285.5428.727
55. Tan G, Spillane KM, and Maher J. The role and regulation of the NKG2D/NKG2D ligand system in cancer. Biol (Basel). (2023) 12:1079. doi: 10.3390/biology12081079
56. Kumar V and Delovitch TL. Different subsets of natural killer T cells may vary in their roles in health and disease. Immunology. (2014) 142:321–36. doi: 10.1111/imm.12247
57. Wong CHY and Kubes P. Imaging natural killer T cells in action. Immunol Cell Biol. (2013) 91:304–10. doi: 10.1038/icb.2013.6
58. Rastogi I, Jeon D, Moseman JE, Muralidhar A, Potluri HK, and McNeel DG. Role of B cells as antigen presenting cells. Front Immunol. (2022) 13:954936. doi: 10.3389/fimmu.2022.954936
59. Zhang E, Ding C, Li S, Zhou X, Aikemu B, Fan X, et al. Roles and mechanisms of tumour-infiltrating B cells in human cancer: a new force in immunotherapy. biomark Res. (2023) 11:28. doi: 10.1186/s40364-023-00460-1
60. Maddur MS, Sharma M, Hegde P, Stephen-Victor E, Pulendran B, Kaveri SV, et al. Human B cells induce dendritic cell maturation and favour Th2 polarization by inducing OX-40 ligand. Nat Commun. (2014) 5:4092. doi: 10.1038/ncomms5092
61. Harvey BP, Gee RJ, Haberman AM, Shlomchik MJ, and Mamula MJ. Antigen presentation and transfer between B cells and macrophages. Eur J Immunol. (2007) 37:1739–51. doi: 10.1002/eji.200636452
62. Deola S, Panelli MC, Maric D, Selleri S, Dmitrieva NI, Voss CY, et al. Helper B cells promote cytotoxic T cell survival and proliferation independently of antigen presentation through CD27/CD70 interactions1. J Immunol. (2008) 180:1362–72. doi: 10.4049/jimmunol.180.3.1362
63. Li Q, Teitz-Tennenbaum S, Donald EJ, Li M, and Chang AE. In vivo sensitized and in vitro activated B cells mediate tumor regression in cancer adoptive immunotherapy. J Immunol. (2009) 183:3195–203. doi: 10.4049/jimmunol.0803773
64. Jones HP, Wang Y-C, Aldridge B, and Weiss JM. Lung and splenic B cells facilitate diverse effects on in vitro measures of antitumor immune responses. Cancer Immun. (2008) 8:4.
65. Sarvaria A, Madrigal JA, and Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol Immunol. (2017) 14:662–74. doi: 10.1038/cmi.2017.35
66. Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun. (2019) 10:4186. doi: 10.1038/s41467-019-12160-2
67. Workel HH, Lubbers JM, Arnold R, Prins TM, van der Vlies P, de Lange K, et al. A transcriptionally distinct CXCL13+CD103+CD8+ T-cell population is associated with B-cell recruitment and neoantigen load in human cancer. Cancer Immunol Res. (2019) 7:784–96. doi: 10.1158/2326-6066.CIR-18-0517
68. Zhang Q-F, Li J, Jiang K, Wang R, Ge J-L, Yang H, et al. CDK4/6 inhibition promotes immune infiltration in ovarian cancer and synergizes with PD-1 blockade in a B cell-dependent manner. Theranostics. (2020) 10:10619–33. doi: 10.7150/thno.44871
69. Basak U, Sarkar T, Mukherjee S, Chakraborty S, Dutta A, Dutta S, et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front Immunol. (2023) 14:1295257. doi: 10.3389/fimmu.2023.1295257
70. Pan Y, Yu Y, Wang X, and Zhang T. Tumor-associated macrophages in tumor immunity. Front Immunol. (2020) 11:583084. doi: 10.3389/fimmu.2020.583084
71. Aminin D and Wang Y-M. Macrophages as a “weapon” in anticancer cellular immunotherapy. Kaohsiung J Med Sci. (2021) 37:749–58. doi: 10.1002/kjm2.12405
72. Bruns H, Büttner M, Fabri M, Mougiakakos D, Bittenbring JT, Hoffmann MH, et al. Vitamin D-dependent induction of cathelicidin in human macrophages results in cytotoxicity against high-grade B cell lymphoma. Sci Transl Med. (2015) 7:282ra47. doi: 10.1126/scitranslmed.aaa3230
73. Duluc D, Corvaisier M, Blanchard S, Catala L, Descamps P, Gamelin E, et al. Interferon-gamma reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int J Cancer. (2009) 125:367–73. doi: 10.1002/ijc.24401
74. Dunn GP, Old LJ, and Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
75. Quezada SA, Peggs KS, Simpson TR, and Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. (2011) 241:104–18. doi: 10.1111/j.1600-065X.2011.01007.x
76. Teng MWL, Vesely MD, Duret H, McLaughlin N, Towne JE, Schreiber RD, et al. Opposing roles for IL-23 and IL-12 in maintaining occult cancer in an equilibrium state. Cancer Res. (2012) 72:3987–96. doi: 10.1158/0008-5472.CAN-12-1337
77. Shimato S, Maier LM, Maier R, Bruce JN, Anderson RCE, and Anderson DE. Profound tumor-specific Th2 bias in patients with Malignant glioma. BMC Cancer. (2012) 12:561. doi: 10.1186/1471-2407-12-561
78. Ziegler A, Heidenreich R, Braumüller H, Wolburg H, Weidemann S, Mocikat R, et al. EpCAM, a human tumor-associated antigen promotes Th2 development and tumor immune evasion. Blood. (2009) 113:3494–502. doi: 10.1182/blood-2008-08-175109
79. Mittal D, Gubin MM, Schreiber RD, and Smyth MJ. New insights into cancer immunoediting and its three component phases–elimination, equilibrium and escape. Curr Opin Immunol. (2014) 27:16–25. doi: 10.1016/j.coi.2014.01.004
80. Drake CG, Jaffee E, and Pardoll DM. Mechanisms of immune evasion by tumors. Adv Immunol. (2006) 90:51–81. doi: 10.1016/S0065-2776(06)90002-9
81. Pati S, Chowdhury A, Mukherjee S, Guin A, Mukherjee S, and Sa G. Regulatory lymphocytes: the dice that resolve the tumor endgame. Appl Cancer Res. (2020) 40:7. doi: 10.1186/s41241-020-00091-0
82. Basu A, Ramamoorthi G, Albert G, Gallen C, Beyer A, Snyder C, et al. Differentiation and regulation of TH cells: A balancing act for cancer immunotherapy. Front Immunol. (2021) 12:669474. doi: 10.3389/fimmu.2021.669474
83. Bankaitis KV and Fingleton B. Targeting IL4/IL4R for the treatment of epithelial cancer metastasis. Clin Exp Metastasis. (2015) 32:847–56. doi: 10.1007/s10585-015-9747-9
84. Jou E. Type 1 and type 2 cytokine-mediated immune orchestration in the tumour microenvironment and their therapeutic potential. Explor Target Antitumor Ther. (2023) 4:474–97. doi: 10.37349/etat.2023.00146
85. Mannino MH, Zhu Z, Xiao H, Bai Q, Wakefield MR, and Fang Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. (2015) 367:103–7. doi: 10.1016/j.canlet.2015.07.009
86. Oft M. IL-10: master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol Res. (2014) 2:194–9. doi: 10.1158/2326-6066.CIR-13-0214
87. Kidd P. Th1/Th2 balance: the hypothesis, its limitations, and implications for health and disease. Altern Med Rev. (2003) 8:223–46.
88. Acuner-Ozbabacan ES, Engin BH, Guven-Maiorov E, Kuzu G, Muratcioglu S, Baspinar A, et al. The structural network of Interleukin-10 and its implications in inflammation and cancer. BMC Genomics. (2014) 15 Suppl 4:S2. doi: 10.1186/1471-2164-15-S4-S2
89. Zhou Q, Zhang H, Wang Z, Zeng H, Liu Z, Huang Q, et al. Poor clinical outcomes and immunoevasive contexture in interleukin-9 abundant muscle-invasive bladder cancer. Int J Cancer. (2020) 147:3539–49. doi: 10.1002/ijc.33237
90. Feng L-L, Gao J-M, Li P-P, and Wang X. IL-9 contributes to immunosuppression mediated by regulatory T cells and mast cells in B-cell non-hodgkin’s lymphoma. J Clin Immunol. (2011) 31:1084–94. doi: 10.1007/s10875-011-9584-9
91. Tan H, Wang S, and Zhao L. A tumour-promoting role of Th9 cells in hepatocellular carcinoma through CCL20 and STAT3 pathways. Clin Exp Pharmacol Physiol. (2017) 44:213–21. doi: 10.1111/1440-1681.12689
92. Qi W, Huang X, and Wang J. Correlation between Th17 cells and tumor microenvironment. Cell Immunol. (2013) 285:18–22. doi: 10.1016/j.cellimm.2013.06.001
93. Roy D, Bose S, Pati S, Guin A, Banerjee K, Saha S, et al. GFI1/HDAC1-axis differentially regulates immunosuppressive CD73 in human tumor-associated FOXP3+ Th17 and inflammation-linked Th17 cells. Eur J Immunol. (2021) 51:1206–17. doi: 10.1002/eji.202048892
94. He S, Fei M, Wu Y, Zheng D, Wan D, Wang L, et al. Distribution and clinical significance of Th17 cells in the tumor microenvironment and peripheral blood of pancreatic cancer patients. Int J Mol Sci. (2011) 12:7424–37. doi: 10.3390/ijms12117424
95. Papak I, Chruściel E, Dziubek K, Kurkowiak M, Urban-Wójciuk Z, Marjański T, et al. What inhibits natural killers’ Performance in tumour. Int J Mol Sci. (2022) 23:7030. doi: 10.3390/ijms23137030
96. Cantoni C, Wurzer H, Thomas C, and Vitale M. Escape of tumor cells from the NK cell cytotoxic activity. J Leukoc Biol. (2020) 108:1339–60. doi: 10.1002/JLB.2MR0820-652R
97. Pesce S, Greppi M, Ferretti E, Obino V, Carlomagno S, Rutigliani M, et al. miRNAs in NK cell-based immune responses and cancer immunotherapy. Front Cell Dev Biol. (2020) 8:119. doi: 10.3389/fcell.2020.00119
98. Dhar P and Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol. (2018) 51:55–61. doi: 10.1016/j.coi.2018.02.004
99. Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, and Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol. (2008) 180:7249–58. doi: 10.4049/jimmunol.180.11.7249
100. Tie Y, Tang F, Wei Y-Q, and Wei X-W. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J Hematol Oncol. (2022) 15:61. doi: 10.1186/s13045-022-01282-8
101. Li C, Jiang P, Wei S, Xu X, and Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. (2020) 19:116. doi: 10.1186/s12943-020-01234-1
102. Mukherjee S, Chakraborty S, Basak U, Pati S, Dutta A, Dutta S, et al. Breast cancer stem cells generate immune-suppressive T regulatory cells by secreting TGFβ to evade immune-elimination. Discov Onc. (2023) 14:220. doi: 10.1007/s12672-023-00787-z
103. Attias M, Al-Aubodah T, and Piccirillo CA. Mechanisms of human FoxP3+ Treg cell development and function in health and disease. Clin Exp Immunol. (2019) 197:36–51. doi: 10.1111/cei.13290
104. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science. (2011) 332:600–3. doi: 10.1126/science.1202947
105. Chen ML, Pittet MJ, Gorelik L, Richard A, Flavell RA, et al. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc Natl Acad Sci U S A. (2005) 102:419–24. doi: 10.1073/pnas.0408197102
106. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. (2016) 17:1322–33. doi: 10.1038/ni.3540
107. Ahmadzadeh M, Pasetto A, Jia L, Deniger DC, Stevanović S, Robbins PF, et al. Tumor-infiltrating human CD4+ regulatory T cells display a distinct TCR repertoire and exhibit tumor and neoantigen reactivity. Sci Immunol. (2019) 4:eaao4310. doi: 10.1126/sciimmunol.aao4310
108. Schmidt A, Oberle N, and Krammer PH. Molecular mechanisms of Treg-mediated T cell suppression. Front Immunol. (2012) 3:51. doi: 10.3389/fimmu.2012.00051
109. Dhar S, Sarkar T, Bose S, Pati S, Chakraborty D, Roy D, et al. FOXP3 transcriptionally activates fatty acid scavenger receptor CD36 in tumour-induced Treg cells. Immunology. (2025) 174:296–309. doi: 10.1111/imm.13887
110. O’Garra A and Vieira P. Regulatory T cells and mechanisms of immune system control. Nat Med. (2004) 10:801–5. doi: 10.1038/nm0804-801
111. Kajal K, Bose S, Panda AK, Chakraborty D, Chakraborty S, Pati S, et al. Transcriptional regulation of VEGFA expression in T-regulatory cells from breast cancer patients. Cancer Immunol Immunother. (2021) 70:1877–91. doi: 10.1007/s00262-020-02808-0
112. Ohue Y and Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. (2019) 110:2080–9. doi: 10.1111/cas.14069
113. Mukherjee S, Dutta S, Pati S, Sarkar T, Paul S, Chakraborty S, et al. Nuclear factor kappa-B: The El Dorado of inflammatory immune response. J Cell Mol Immunol Volume. (2024) 3:6–19. doi: 10.46439/immunol.3.025
114. He Y, Qian H, Liu Y, Duan L, Li Y, and Shi G. The roles of regulatory B cells in cancer. J Immunol Res. (2014) 2014:215471. doi: 10.1155/2014/215471
115. Pati S, Mukherjee S, Dutta S, Guin A, Roy D, Bose S, et al. Tumor-associated CD19+CD39- B regulatory cells deregulate class-switch recombination to suppress antibody responses. Cancer Immunol Res. (2023) 11:364–80. doi: 10.1158/2326-6066.CIR-21-1073
116. Berthelot J-M, Jamin C, Amrouche K, Le Goff B, Maugars Y, and Youinou P. Regulatory B cells play a key role in immune system balance. Joint Bone Spine. (2013) 80:18–22. doi: 10.1016/j.jbspin.2012.04.010
117. Rosser EC and Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. (2015) 42:607–12. doi: 10.1016/j.immuni.2015.04.005
118. Shao Y, Lo CM, Ling CC, Liu XB, Ng KT-P, Chu ACY, et al. Regulatory B cells accelerate hepatocellular carcinoma progression via CD40/CD154 signaling pathway. Cancer Lett. (2014) 355:264–72. doi: 10.1016/j.canlet.2014.09.026
119. Moreira H, Dobosz A, Cwynar-Zając Ł., Nowak P, Czyżewski M, Barg M, et al. Unraveling the role of Breg cells in digestive tract cancer and infectious immunity. Front Immunol. (2022) 13:981847. doi: 10.3389/fimmu.2022.981847
120. Roya N, Fatemeh T, Faramarz M-A, Milad S-G, Mohammad-Javad S, Najmeh S-V, et al. Frequency of IL-10+CD19+ B cells in patients with prostate cancer compared to patients with benign prostatic hyperplasia. Afr Health Sci. (2020) 20:1264–72. doi: 10.4314/ahs.v20i3.31
121. Sezginer O and Unver N. Dissection of pro-tumoral macrophage subtypes and immunosuppressive cells participating in M2 polarization. Inflammation Res. (2024) 73:1411–23. doi: 10.1007/s00011-024-01907-3
122. Lee KM, Stott RT, Zhao G, SooHoo J, Xiong W, Lian MM, et al. TGF-β-producing regulatory B cells induce regulatory T cells and promote transplantation tolerance. Eur J Immunol. (2014) 44:1728–36. doi: 10.1002/eji.201344062
123. Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J Clin Invest. (2016) 126:4157–73. doi: 10.1172/JCI87252
124. Nie Y, Huang H, Guo M, Chen J, Wu W, Li W, et al. Breast phyllodes tumors recruit and repolarize tumor-associated macrophages via secreting CCL5 to promote Malignant progression, which can be inhibited by CCR5 inhibition therapy. Clin Cancer Res. (2019) 25:3873–86. doi: 10.1158/1078-0432.CCR-18-3421
125. Dongre A and Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. (2019) 20:69–84. doi: 10.1038/s41580-018-0080-4
126. Egawa M, Mukai K, Yoshikawa S, Iki M, Mukaida N, Kawano Y, et al. Inflammatory monocytes recruited to allergic skin acquire an anti-inflammatory M2 phenotype via basophil-derived interleukin-4. Immunity. (2013) 38:570–80. doi: 10.1016/j.immuni.2012.11.014
127. Wang N, Liang H, and Zen K. Molecular mechanisms that influence the macrophage M1–M2 polarization balance. Front Immunol. (2014) 5:614. doi: 10.3389/fimmu.2014.00614
128. Liu C, Zhang W, Wang J, Si T, and Xing W. Tumor-associated macrophage-derived transforming growth factor-β promotes colorectal cancer progression through HIF1-TRIB3 signaling. Cancer Sci. (2021) 112:4198–207. doi: 10.1111/cas.15101
129. Guruvayoorappan C. Tumor versus tumor-associated macrophages: how hot is the link? Integr Cancer Ther. (2008) 7:90–5. doi: 10.1177/1534735408319060
130. Bied M, Ho WW, Ginhoux F, and Blériot C. Roles of macrophages in tumor development: a spatiotemporal perspective. Cell Mol Immunol. (2023) 20:983–92. doi: 10.1038/s41423-023-01061-6
131. Ito M, Minamiya Y, Kawai H, Saito S, Saito H, Nakagawa T, et al. Tumor-derived TGFbeta-1 induces dendritic cell apoptosis in the sentinel lymph node. J Immunol. (2006) 176:5637–43. doi: 10.4049/jimmunol.176.9.5637
132. Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H, Xiao GG, et al. Macrophages in immunoregulation and therapeutics. Sig Transduct Target Ther. (2023) 8:207. doi: 10.1038/s41392-023-01452-1
133. Shaheryar ZA, Khan MA, Adnan C, Zaidi AA, Hänggi D, and Muhammad S. Neuroinflammatory triangle presenting novel pharmacological targets for ischemic brain injury. Front Immunol. (2021) 12:748663. doi: 10.3389/fimmu.2021.748663
134. Chen A, Jiang Y, Li Z, Wu L, Santiago U, Zou H, et al. Chitinase-3-like 1 protein complexes modulate macrophage-mediated immune suppression in glioblastoma. J Clin Invest. (2021) 131:e147552. doi: 10.1172/JCI147552
135. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. (2004) 10:942–9. doi: 10.1038/nm1093
136. La Fleur L, Botling J, He F, Pelicano C, Zhou C, He C, et al. Targeting MARCO and IL37R on immunosuppressive macrophages in lung cancer blocks regulatory T cells and supports cytotoxic lymphocyte function. Cancer Res. (2021) 81:956–67. doi: 10.1158/0008-5472.CAN-20-1885
137. Karadima E, Chavakis T, and Alexaki VI. Arginine metabolism in myeloid cells in health and disease. Semin Immunopathol. (2025) 47:11. doi: 10.1007/s00281-025-01038-9
138. Wong S-C, Puaux A-L, Chittezhath M, Shalova I, Kajiji TS, Wang X, et al. Macrophage polarization to a unique phenotype driven by B cells. Eur J Immunol. (2010) 40:2296–307. doi: 10.1002/eji.200940288
139. Zhang B, Vogelzang A, Miyajima M, Sugiura Y, Wu Y, Chamoto K, et al. B cell-derived GABA elicits IL-10+ macrophages to limit anti-tumour immunity. Nature. (2021) 599:471–6. doi: 10.1038/s41586-021-04082-1
140. Liu S, Sun Q, and Ren X. Novel strategies for cancer immunotherapy: counter-immunoediting therapy. J Hematol Oncol. (2023) 16:38. doi: 10.1186/s13045-023-01430-8
141. Shaik R, Royyala SA, Inapanuri B, Durgam A, Khan H, and Unnisa A. Tumor infiltration therapy: from FDA approval to next-generation approaches. Clin Exp Med. (2025) 25:254. doi: 10.1007/s10238-025-01574-6
142. Hensel J, Metts J, Gupta A, Ladle BH, Pilon-Thomas S, and Mullinax J. Adoptive cellular therapy for pediatric solid tumors: beyond chimeric antigen receptor-T cell therapy. Cancer J. (2022) 28:322–7. doi: 10.1097/PPO.0000000000000603
143. Tseng D and Lee S. Tumor-infiltrating lymphocyte therapy: A new frontier. Transplant Cell Ther. (2025) 31:S599–609. doi: 10.1016/j.jtct.2024.11.014
144. Bae J, Accardi F, Hideshima T, Tai Y-T, Prabhala R, Shambley A, et al. Targeting LAG3/GAL-3 to overcome immunosuppression and enhance anti-tumor immune responses in multiple myeloma. Leukemia. (2022) 36:138–54. doi: 10.1038/s41375-021-01301-6
145. Gestermann N, Saugy D, Martignier C, Tillé L, Fuertes Marraco SA, Zettl M, et al. LAG-3 and PD-1+LAG-3 inhibition promote anti-tumor immune responses in human autologous melanoma/T cell co-cultures. Oncoimmunology. (2020) 9:1736792. doi: 10.1080/2162402X.2020.1736792
146. Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. (2018) 19:723–32. doi: 10.1038/s41590-018-0132-0
147. Guillerey C, Harjunpää H, Carrié N, Kassem S, Teo T, Miles K, et al. TIGIT immune checkpoint blockade restores CD8+ T-cell immunity against multiple myeloma. Blood. (2018) 132:1689–94. doi: 10.1182/blood-2018-01-825265
148. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J Exp Med. (2010) 207:2175–86. doi: 10.1084/jem.20100637
149. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, and Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. (2010) 207:2187–94. doi: 10.1084/jem.20100643
150. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. New Engl J Med. (2019) 380:1726–37. doi: 10.1056/NEJMoa1817226
151. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N Engl J Med. (2020) 382:545–53. doi: 10.1056/NEJMoa1910607
152. Shimizu J, Yamazaki S, and Sakaguchi S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J Immunol. (1999) 163:5211–8. doi: 10.4049/jimmunol.163.10.5211
153. Arce Vargas F, Furness AJS, Solomon I, Joshi K, Mekkaoui L, Lesko MH, et al. Fc-optimized anti-CD25 depletes tumor-infiltrating regulatory T cells and synergizes with PD-1 blockade to eradicate established tumors. Immunity. (2017) 46:577–86. doi: 10.1016/j.immuni.2017.03.013
154. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. (2008) 322:271–5. doi: 10.1126/science.1160062
155. Togashi Y and Nishikawa H. Regulatory T cells: molecular and cellular basis for immunoregulation. Curr Top Microbiol Immunol. (2017) 410:3–27. doi: 10.1007/82_2017_58
156. Bodogai M, Lee Chang C, Wejksza K, Lai J, Merino M, Wersto RP, et al. Anti-CD20 antibody promotes cancer escape via enrichment of tumor-evoked regulatory B cells expressing low levels of CD20 and CD137L. Cancer Res. (2013) 73:2127–38. doi: 10.1158/0008-5472.CAN-12-4184
157. Lee-Chang C, Bodogai M, Martin-Montalvo A, Wejksza K, Sanghvi M, Moaddel R, et al. Inhibition of breast cancer metastasis by resveratrol-mediated inactivation of tumor-evoked regulatory B cells. J Immunol. (2013) 191:4141–51. doi: 10.4049/jimmunol.1300606
158. Wang R-X, Yu C-R, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. (2014) 20:633–41. doi: 10.1038/nm.3554
159. Disis ML, Goodell V, Schiffman K, and Knutson KL. Humoral epitope-spreading following immunization with a HER-2/neu peptide based vaccine in cancer patients. J Clin Immunol. (2004) 24:571–8. doi: 10.1023/B:JOCI.0000040928.67495.52
160. Cook KM and Figg WD. Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin. (2010) 60:222–43. doi: 10.3322/caac.20075
161. de Almeida PE, Mak J, Hernandez G, Jesudason R, Herault A, Javinal V, et al. Anti-VEGF treatment enhances CD8+ T-cell antitumor activity by amplifying hypoxia. Cancer Immunol Res. (2020) 8:806–18. doi: 10.1158/2326-6066.CIR-19-0360
162. Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med. (2014) 20:607–15. doi: 10.1038/nm.3541
163. Liu S, Qin T, Liu Z, Wang J, Jia Y, Feng Y, et al. anlotinib alters tumor immune microenvironment by downregulating PD-L1 expression on vascular endothelial cells. Cell Death Dis. (2020) 11:1–16. doi: 10.1038/s41419-020-2511-3
164. Luo S, Yang G, Ye P, Cao N, Chi X, Yang W-H, et al. Macrophages are a double-edged sword: molecular crosstalk between tumor-associated macrophages and cancer stem cells. Biomolecules. (2022) 12:850. doi: 10.3390/biom12060850
165. Adams S, Kozhaya L, Martiniuk F, Meng T-C, Chiriboga L, Liebes L, et al. Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin Cancer Res. (2012) 18:6748–57. doi: 10.1158/1078-0432.CCR-12-1149
166. Kapp K, Volz B, Oswald D, Wittig B, Baumann M, and Schmidt M. Beneficial modulation of the tumor microenvironment and generation of anti-tumor responses by TLR9 agonist lefitolimod alone and in combination with checkpoint inhibitors. Oncoimmunology. (2019) 8:e1659096. doi: 10.1080/2162402X.2019.1659096
167. Willingham SB, Volkmer J-P, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U.S.A. (2012) 109:6662–7. doi: 10.1073/pnas.1121623109
168. Cabrales P. RRx-001 acts as a dual small molecule checkpoint inhibitor by downregulating CD47 on cancer cells and SIRP-α on monocytes/macrophages. Transl Oncol. (2019) 12:626–32. doi: 10.1016/j.tranon.2018.12.001
169. Oronsky B, Paulmurugan R, Foygel K, Scicinski J, Knox SJ, Peehl D, et al. RRx-001: a systemically non-toxic M2-to-M1 macrophage stimulating and prosensitizing agent in Phase II clinical trials. Expert Opin Investig Drugs. (2017) 26:109–19. doi: 10.1080/13543784.2017.1268600
170. Wang H, Sun Y, Zhou X, Chen C, Jiao L, Li W, et al. CD47/SIRPα blocking peptide identification and synergistic effect with irradiation for cancer immunotherapy. J Immunother Cancer. (2020) 8:e000905. doi: 10.1136/jitc-2020-000905
171. Xi Q, Zhang J, Yang G, Zhang L, Chen Y, Wang C, et al. Restoration of miR-340 controls pancreatic cancer cell CD47 expression to promote macrophage phagocytosis and enhance antitumor immunity. J Immunother Cancer. (2020) 8:e000253. doi: 10.1136/jitc-2019-000253
172. Georgoudaki A-M, Prokopec KE, Boura VF, Hellqvist E, Sohn S, Östling J, et al. Reprogramming tumor-associated macrophages by antibody targeting inhibits cancer progression and metastasis. Cell Rep. (2016) 15:2000–11. doi: 10.1016/j.celrep.2016.04.084
173. La Fleur L, Boura VF, Alexeyenko A, Berglund A, Pontén V, Mattsson JSM, et al. Expression of scavenger receptor MARCO defines a targetable tumor-associated macrophage subset in non-small cell lung cancer. Int J Cancer. (2018) 143:1741–52. doi: 10.1002/ijc.31545
174. Fend L, Accart N, Kintz J, Cochin S, Reymann C, Le Pogam F, et al. Therapeutic effects of anti-CD115 monoclonal antibody in mouse cancer models through dual inhibition of tumor-associated macrophages and osteoclasts. PLoS One. (2013) 8:e73310. doi: 10.1371/journal.pone.0073310
175. Kuemmel S, Campone M, Loirat D, Lopez RL, Beck JT, De Laurentiis M, et al. A randomized phase II study of anti-CSF1 monoclonal antibody lacnotuzumab (MCS110) combined with gemcitabine and carboplatin in advanced triple-negative breast cancer. Clin Cancer Res. (2022) 28:106–15. doi: 10.1158/1078-0432.CCR-20-3955
176. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
177. Kalbasi A, Komar C, Tooker GM, Liu M, Lee JW, Gladney WL, et al. Tumor-derived CCL2 mediates resistance to radiotherapy in pancreatic ductal adenocarcinoma. Clin Cancer Res. (2017) 23:137–48. doi: 10.1158/1078-0432.CCR-16-0870
178. Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, et al. Targeting tumour-associated macrophages with CCR2 inhibition in combination with FOLFIRINOX in patients with borderline resectable and locally advanced pancreatic cancer: a single-centre, open-label, dose-finding, non-randomised, phase 1b trial. Lancet Oncol. (2016) 17:651–62. doi: 10.1016/S1470-2045(16)00078-4
179. Izumi K, Fang L-Y, Mizokami A, Namiki M, Li L, Lin W-J, et al. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol Med. (2013) 5:1383–401. doi: 10.1002/emmm.201202367
180. Eckert F, Schilbach K, Klumpp L, Bardoscia L, Sezgin EC, Schwab M, et al. Potential role of CXCR4 targeting in the context of radiotherapy and immunotherapy of cancer. Front Immunol. (2018) 9:3018. doi: 10.3389/fimmu.2018.03018
181. O’Hara MH, Messersmith W, Kindler H, Zhang W, Pitou C, Szpurka AM, et al. Safety and pharmacokinetics of CXCR4 peptide antagonist, LY2510924, in combination with durvalumab in advanced refractory solid tumors. J Pancreat Cancer. (2020) 6:21–31. doi: 10.1089/pancan.2019.0018
182. Anderson NR, Minutolo NG, Gill S, and Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. (2021) 81:1201–8. doi: 10.1158/0008-5472.CAN-20-2990
183. Morrissey MA, Williamson AP, Steinbach AM, Roberts EW, Kern N, Headley MB, et al. Chimeric antigen receptors that trigger phagocytosis. Elife. (2018) 7:e36688. doi: 10.7554/eLife.36688
184. Klichinsky M, Ruella M, Shestova O, Lu XM, Best A, Zeeman M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat Biotechnol. (2020) 38:947–53. doi: 10.1038/s41587-020-0462-y
185. Foubert P, Kaneda MM, and Varner JA. PI3Kγ Activates integrin α4 and promotes immune suppressive myeloid cell polarization during tumor progression. Cancer Immunol Res. (2017) 5:957–68. doi: 10.1158/2326-6066.CIR-17-0143
186. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kγ Drives pancreatic ductal adenocarcinoma progression. Cancer Discov. (2016) 6:870–85. doi: 10.1158/2159-8290.CD-15-1346
187. Li X, Yao W, Yuan Y, Chen P, Li B, Li J, et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. (2017) 66:157–67. doi: 10.1136/gutjnl-2015-310514
188. Yao W, Ba Q, Li X, Li H, Zhang S, Yuan Y, et al. A natural CCR2 antagonist relieves tumor-associated macrophage-mediated immunosuppression to produce a therapeutic effect for liver cancer. EBioMedicine. (2017) 22:58–67. doi: 10.1016/j.ebiom.2017.07.014
189. Deng A, Fan R, Hai Y, Zhuang J, Zhang B, Lu X, et al. A STING agonist prodrug reprograms tumor-associated macrophage to boost colorectal cancer immunotherapy. Theranostics. (2025) 15:277–99. doi: 10.7150/thno.101001
190. Li Y, Shen Z, Chai Z, Zhan Y, Zhang Y, Liu Z, et al. Targeting MS4A4A on tumour-associated macrophages restores CD8+ T-cell-mediated antitumour immunity. Gut. (2023) 72:2307–20. doi: 10.1136/gutjnl-2022-329147
191. de Groot AE, Myers KV, Krueger TEG, Brennen WN, Amend SR, and Pienta KJ. Targeting interleukin 4 receptor alpha on tumor-associated macrophages reduces the pro-tumor macrophage phenotype. Neoplasia. (2022) 32:100830. doi: 10.1016/j.neo.2022.100830
192. Barnes TA and Amir E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. Br J Cancer. (2017) 117:451–60. doi: 10.1038/bjc.2017.220
193. Yamaguchi H, Hsu J-M, Sun L, Wang S-C, and Hung M-C. Advances and prospects of biomarkers for immune checkpoint inhibitors. Cell Rep Med. (2024) 5:101621. doi: 10.1016/j.xcrm.2024.101621
194. Miller BC, Sen DR, Al Abosy R, Bi K, Virkud YV, LaFleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
195. Italiano A, Bessede A, Pulido M, Bompas E, Piperno-Neumann S, Chevreau C, et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: a phase 2 PEMBROSARC trial cohort. Nat Med. (2022) 28:1199–206. doi: 10.1038/s41591-022-01821-3
196. Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. (2020) 577:561–5. doi: 10.1038/s41586-019-1914-8
197. Saleh R and Elkord E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. (2020) 490:174–85. doi: 10.1016/j.canlet.2020.07.022
198. Rakina M, Larionova I, and Kzhyshkowska J. Macrophage diversity in human cancers: New insight provided by single-cell resolution and spatial context. Heliyon. (2024) 10:e28332. doi: 10.1016/j.heliyon.2024.e28332
199. Oshi M, Tokumaru Y, Asaoka M, Yan L, Satyananda V, Matsuyama R, et al. M1 Macrophage and M1/M2 ratio defined by transcriptomic signatures resemble only part of their conventional clinical characteristics in breast cancer. Sci Rep. (2020) 10:16554. doi: 10.1038/s41598-020-73624-w
200. Guo F, Kong W, Zhao G, Cheng Z, Ai L, Lv J, et al. The correlation between tumor-associated macrophage infiltration and progression in cervical carcinoma. Biosci Rep. (2021) 41:BSR20203145. doi: 10.1042/BSR20203145
201. Chohan MH, Perry M, Laurance-Young P, Salih VM, and Foey AD. Prognostic role of CD68+ and CD163+ Tumour-associated macrophages and PD-L1 expression in oral squamous cell carcinoma: A meta-analysis. Br J BioMed Sci. (2023) 80:11065. doi: 10.3389/bjbs.2023.11065
202. Leach DR, Krummel MF, and Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. (1996) 271:1734–6. doi: 10.1126/science.271.5256.1734
203. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. (2002) 8:793–800. doi: 10.1038/nm730
204. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. (2000) 192:1027–34. doi: 10.1084/jem.192.7.1027
205. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. (2014) 515:568–71. doi: 10.1038/nature13954
206. Wang L, Geng H, Liu Y, Liu L, Chen Y, Wu F, et al. Hot and cold tumors: Immunological features and the therapeutic strategies. MedComm (2020). (2023) 4:e343. doi: 10.1002/mco2.343
207. Raskov H, Orhan A, Christensen JP, and Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer. (2021) 124:359–67. doi: 10.1038/s41416-020-01048-4
208. Bhinder B, Friedl V, Sethuraman S, Risso D, Chiotti KE, Mashl RJ, et al. Pan-cancer immune and stromal deconvolution predicts clinical outcomes and mutation profiles. Sci Rep. (2025) 15:23921. doi: 10.1038/s41598-025-09075-y
209. Zhang M, Liu C, Tu J, Tang M, Ashrafizadeh M, Nabavi N, et al. Advances in cancer immunotherapy: historical perspectives, current developments, and future directions. Mol Cancer. (2025) 24:136. doi: 10.1186/s12943-025-02305-x
210. Postow MA, Sidlow R, and Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. (2018) 378:158–68. doi: 10.1056/NEJMra1703481
211. Kimbara S, Fujiwara Y, Iwama S, Ohashi K, Kuchiba A, Arima H, et al. Association of antithyroglobulin antibodies with the development of thyroid dysfunction induced by nivolumab. Cancer Sci. (2018) 109:3583–90. doi: 10.1111/cas.13800
212. Choi J and Lee SY. Clinical characteristics and treatment of immune-related adverse events of immune checkpoint inhibitors. Immune Netw. (2020) 20:e9. doi: 10.4110/in.2020.20.e9
213. Schachter J, Ribas A, Long GV, Arance A, Grob J-J, Mortier L, et al. Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet. (2017) 390:1853–62. doi: 10.1016/S0140-6736(17)31601-X
214. Wang W, Lie P, Guo M, and He J. Risk of hepatotoxicity in cancer patients treated with immune checkpoint inhibitors: A systematic review and meta-analysis of published data. Int J Cancer. (2017) 141:1018–28. doi: 10.1002/ijc.30678
215. Bai R, Chen N, Li L, Du N, Bai L, Lv Z, et al. Mechanisms of cancer resistance to immunotherapy. Front Oncol. (2020) 10:1290. doi: 10.3389/fonc.2020.01290
216. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
217. Gao L, Liu Y, Du X, Ma S, Ge M, Tang H, et al. The intrinsic role and mechanism of tumor expressed-CD38 on lung adenocarcinoma progression. Cell Death Dis. (2021) 12:680. doi: 10.1038/s41419-021-03968-2
218. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. New Engl J Med. (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
Keywords: immunoediting, immunotherapy, TIL, TAM, tumor microenvironment
Citation: Mukherjee S, Ghosh A, Chakraborty S, Basak U, Mukherjee S, Das T and Sa G (2025) Tumor-infiltrating lymphocytes and tumor-associated macrophages in cancer immunoediting: a targetable therapeutic axis. Front. Immunol. 16:1655176. doi: 10.3389/fimmu.2025.1655176
Received: 27 June 2025; Accepted: 22 August 2025;
Published: 11 September 2025.
Edited by:
Sonam Mittal, Medical College of Wisconsin, United StatesReviewed by:
Sarika Gunjan, Medical College of Wisconsin, United StatesSudhir Kumar, Emory University, United States
Priyanga Jayakrishnan, Medical College of Wisconsin, United States
Bishnu Joshi, Gel4Med, Inc. (A Harvard Innovation Labs Company), United States
Copyright © 2025 Mukherjee, Ghosh, Chakraborty, Basak, Mukherjee, Das and Sa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gaurisankar Sa, Z2F1cmlAamNib3NlLmFjLmlu