 Ran Chen
Ran Chen Yan Sun
Yan Sun Ying Hu1
Ying Hu1 Wenlin Tai
Wenlin Tai- 1Clinical Laboratory, The Second Affiliated Hospital of Kunming Medical University, Kunming, Yunnan, China
- 2Pharmaceutical College and Key Laboratory of Pharmacology for Natural Products of Yunnan Province, Kunming Medical University, Kunming, Yunnan, China
- 3College of Modern Biomedical Industry, Kunming Medical University, Kunming, Yunnan, China
Primary Biliary Cholangitis (PBC) is a chronic autoimmune liver disease characterized by immune-mediated destruction of intrahepatic bile ducts. This review synthesizes current knowledge on the critical role of innate immunity, specifically involving cholangiocytes, bile components, and associated immune cells. Cholangiocytes function not only as passive targets but also as active immunomodulators through mechanisms including Toll-like receptor (TLR) signaling, antigen presentation, and immune cell recruitment. Dysregulated bile acid signaling via receptors like TGR5 disrupts immune homeostasis, while apoptosis of biliary epithelial cells releases antigens (e.g., PDC-E2), triggering aberrant innate and adaptive immune responses. Innate lymphoid cells (ILCs), natural killer (NK) cells, and macrophages exhibit altered frequencies and functions in PBC, driving chronic inflammation and fibrosis through cytokine cascades (e.g., IL-17, IFNγ) and interactions within the gut-liver axis. Furthermore, biliary microbiota dysbiosis exacerbates disease by promoting bacterial translocation, modifying bile acid metabolism, and activating innate immune pathways. Current clinical management with ursodeoxycholic acid (UDCA) and obeticholic acid (OCA) primarily addresses cholestasis. However, the immunomodulatory effects of these agents remain constrained. Targeted therapeutic strategies addressing innate immune pathways—exemplified by RIPK2 (Receptor Interacting Serine/Threonine Kinase 2) inhibition, IL-1 blockade(Canakinumab), and T cell immunoglobulin mucin domain-containing protein 3 (TIM-3) modulation—alongside cell-based interventions such as mesenchymal stem cell therapy, demonstrate considerable therapeutic potential. Advancing these modalities necessitates multidisciplinary integration to facilitate clinical translation. Additionally, Prognostic indices like the neutrophil-to-lymphocyte ratio (NLR) and monocyte-to-lymphocyte ratio (MLR) reflect systemic inflammation and correlate with disease progression. Achieving therapeutic precision requires deeper elucidation of the gut-biliary-immune axis, trained immunity mechanisms, and cholangiocyte senescence, paving the way for targeted interventions in PBC. Establishing a comprehensive treatment burden assessment system is imperative to facilitate the transition from investigational platforms to clinical care.
1 Introduction
Primary Biliary Cholangitis (PBC) is a chronic autoimmune disorder characterized by immune-mediated destruction of interlobular and septal bile ducts. Left untreated, this pathological trajectory evolves through progressive cholestasis and chronic inflammation, culminating in irreversible hepatic fibrosis and cirrhosis via sustained fibrogenesis (1, 2). The disease pathogenesis entails multifactorial interactions among genetic predisposition (notably HLA class II alleles), epigenetic alterations, and environmental triggers, collectively initiating autoimmune targeting of biliary epithelial cells (1, 2). Diagnostic classification for PBC includes three key parameters: AMA status, histopathological staging (Ludwig or Nakanishi), and clinical phenotype (3–5). AMA sensitivity is 90-95%, but AMA-negative cases require autotaxin levels and cholangiographic imaging for confirmation (5–7). First-line treatment is UDCA at 13–15 mg/kg/day, achieving biochemical response in 60-70% of patients via bile acid modulation and anti-apoptotic mechanisms (8, 9). For UDCA non-responders (40% of cases), second-line agents like obeticholic acid and PPAR agonists are implemented to impede progression (9). Post-liver transplant patients with relapse and poor UDCA response exhibit elevated mortality risk (10). Clinical trials should identify and validate surrogate markers as endpoints for evaluating second-line therapies (11, 12). The Ursodeoxycholic Acid Response Score (URS) may serve as a predictive tool for long-term clinical outcomes—such as liver transplantation or death—following 12 months of pre-therapeutic UDCA administration (13, 14). Specifically, the GLOBE and UK-PBC Risk Scores guide second-line therapy allocation in practice (15). High-risk patients can be prioritized for prompt second-line treatments such as obeticholic acid (16), while Paris II criteria offer standardized patient stratification for trials (17). With individualized medicine advancing, models integrating multifactorial data are essential for optimizing PBC management (18). Although well-validated prognostic models are applicable to all patients with PBC, their clinical utility in guiding therapy is most pronounced in individuals responsive to UDCA. For up to 40% of patients who do not respond to UDCA as first - line therapy, and considering that a substantial proportion may also experience failure of second - line treatment options, the development of therapeutic strategies remains an unmet clinical requirement.
Autoimmune bile duct lesions may arise following disruption of tolerance mechanisms mediated by bile duct epithelial cells (19). Biliary epithelial cells establish a sophisticated interactive network with diverse hepatic immune cell populations, facilitating leukocyte recruitment to specific anatomical sites through cytokine and chemokine expression (20). In cholangiopathies, immune cells—including monocytes, lymphocytes, neutrophils, and mast cells—are recruited to the liver. Within this microenvironment, they interact with biliary epithelial cells and resident Kupffer cells, collectively influencing disease progression (21).
Crucially, interactions between biliary epithelial cells and immune cells are pivotal for maintaining homeostasis. Additionally, biliary epithelial cells can mitigate cholangitis development through upregulation of PD-L1 expression, thereby conferring protection against CD8+T cell-mediated cytotoxicity (22). During PBC pathogenesis, Damage-associated Molecular Patterns (DAMPs) released during biliary epithelial cell apoptosis trigger innate immune responses via activation of Pattern Recognition Receptors (PRRs), including TLRs and NOD-like receptors (NLRs) (23). These DAMPs encompass autoantigens such as the mitochondrial Antigen Pyruvate Dehydrogenase Complex- E2 (PDC-E2), whose aberrant exposure disrupts normal immune tolerance (24). Disordered bile acid metabolism potentiates DAMP release, establishing a pathological positive feedback loop (25). Activated PRRs induce chemokine secretion through MyD88-dependent signaling pathways, promoting infiltration of innate immune cells—including Ly6C+monocytes, macrophages, and Dendritic Cells (DCs) (26). These cells subsequently establish the foundation for adaptive immune responses by presenting PDC-E2 antigen and secreting pro-inflammatory cytokines (such as IL-12, IL-23) (24). Metabolites generated during intestinal dysbiosis, such as short-chain fatty acids, can further perturb Myeloid-derived Suppressor Cell (MDSC) homeostasis, exacerbating innate immune dysregulation (27). The innate-adaptive immune crosstalk constitutes a core immunological principle, facilitating synergistic amplification of the immune response. Autoantigens presented by innate immune cells activate CD4+T cells, specifically the CCR5+CD4+T cell subset (28). IL-15Rα+B cells promote activation and expansion of these T cells via IL-15 signaling, orchestrating targeted immune attacks against the biliary epithelium (28).This paradigm emphasizes that innate immunity functions not merely as an initiator but also as a critical modulator of adaptive immunity, thereby mediating dynamic cross-regulation between these two systems.
AMA is the most classic hallmark, but other antibodies such as elevated serum IgM and specific antinuclear antibodies also play critical roles in diagnosis and prognosis. Non-AMA autoantibodies, particularly specific subtypes of antinuclear antibodies (ANA), including anti-gp210, anti-sp100, and anti-centromere, constitute an essential component of PBC diagnosis. They are especially important in cases where AMA is negative (3–5). Immunomodulation constitutes a systemic therapeutic approach. Despite the presence of characteristic AMA-M2, current immunomodulatory strategies still lack cellular specificity and precise targeting. Furthermore, the translational relevance of animal models remains debatable, as they fail to accurately identify therapeutic windows or adequately replicate the spatiotemporal heterogeneity of the immune microenvironment. Achieving therapeutic efficacy necessitates maintaining a delicate balance—a critical consideration warranting in-depth investigation.
Therefore, this review provides an overview of biliary tract innate immunity, focusing on cholangiocytes, bile constituents, and immune cells in PBC from an immunological perspective (as illustrated in Figure 1).
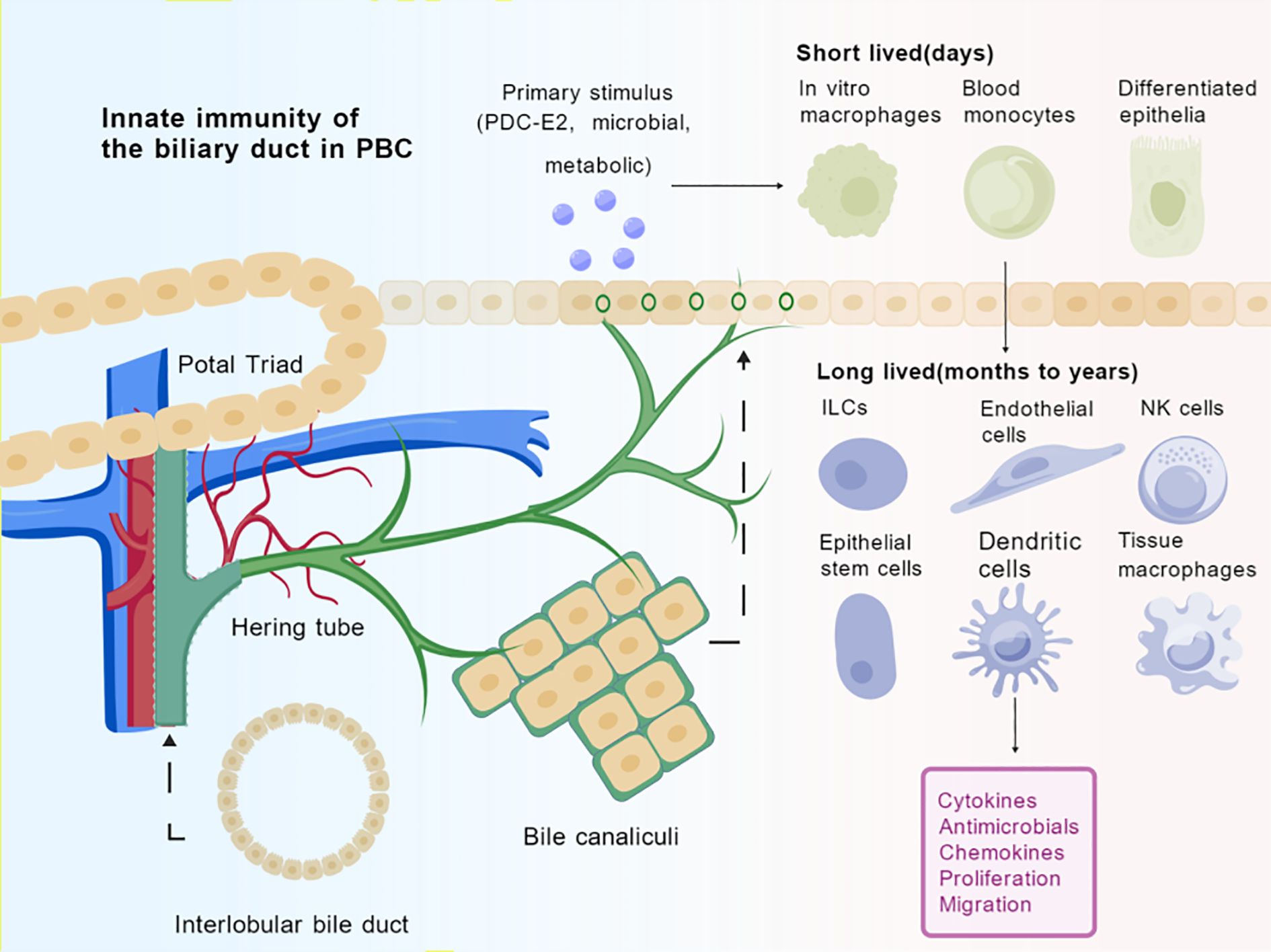
Figure 1. Innate immune cells in the bile duct environment of PBC. Spatial arrangements and dynamic transformations of cells in biliary duct transport compartments, organized around Hering tube and bile canaliculi to form a conduit for immune cell trafficking. The initial immunogenic insult (e.g., biliary epithelial exposure to PDC-E2, microbial products, or metabolic stressors) induces the recruitment of short-lived innate immune cells (e.g., monocytes, macrophages) from peripheral circulation. Within the tissue microenvironment, a complex interplay takes place among long-lived tissue-resident cells (e.g., macrophages, NK cells, ILCs, dendritic cells), cholangiocytes, and endothelial cells. The sustained release of pro-inflammatory cytokines, chemokines, antimicrobial peptides, aberrant proliferation, and migratory responses, culminating in the progressive autoimmune targeting of interlobular bile ducts. Created by BioGDP.com (29). PDC-E2, Pyruvate Dehydrogenase E2; ILCs, Innate lymphoid cells, NK cells, Natural Killer cells.
We aim to concentrate on the role of innate immune imbalance in PBC pathogenesis and how addressing these mechanisms might offer new therapeutic possibilities. The first-line therapy for PBC remains UDCA. Based on validated prognostic models such as the URS, GLOBE, and UK-PBC scores, patients with high-risk PBC are prioritized candidates for second-line therapies, these established prognostic markers and treatment strategies are supported by robust clinical evidence and recommended for use in international guidelines. However, it is worth noting that emerging therapeutic approaches including RIPK2 inhibition, IL-1 blockade, TIM-3 modulation, and MSCs therapy are currently given the involvement of innate immunity in PBC pathogenesis. Prognostic indices such as NLR and MLR reflect systemic inflammation and correlate with disease progression. Moving forward, achieving therapeutic precision will require deeper mechanistic insights into the gut-biliary-immune axis, trained immunity, and cholangiocyte senescence to enable targeted interventions. Nevertheless, it must be mentioned that these approaches are currently primarily at the experimental stage, and their clinical translation necessitates rigorous validation through further clinical trials to ensure both safety and efficacy as potential innate immunomodulatory interventions.
2 The innate immune system of the biliary tract
2.1 Cell composition, function and pathways
The biliary tract orchestrates innate immunity through coordinated interactions between its cellular components, biochemical mediators, and microbial residents(as shown in Figure 2).
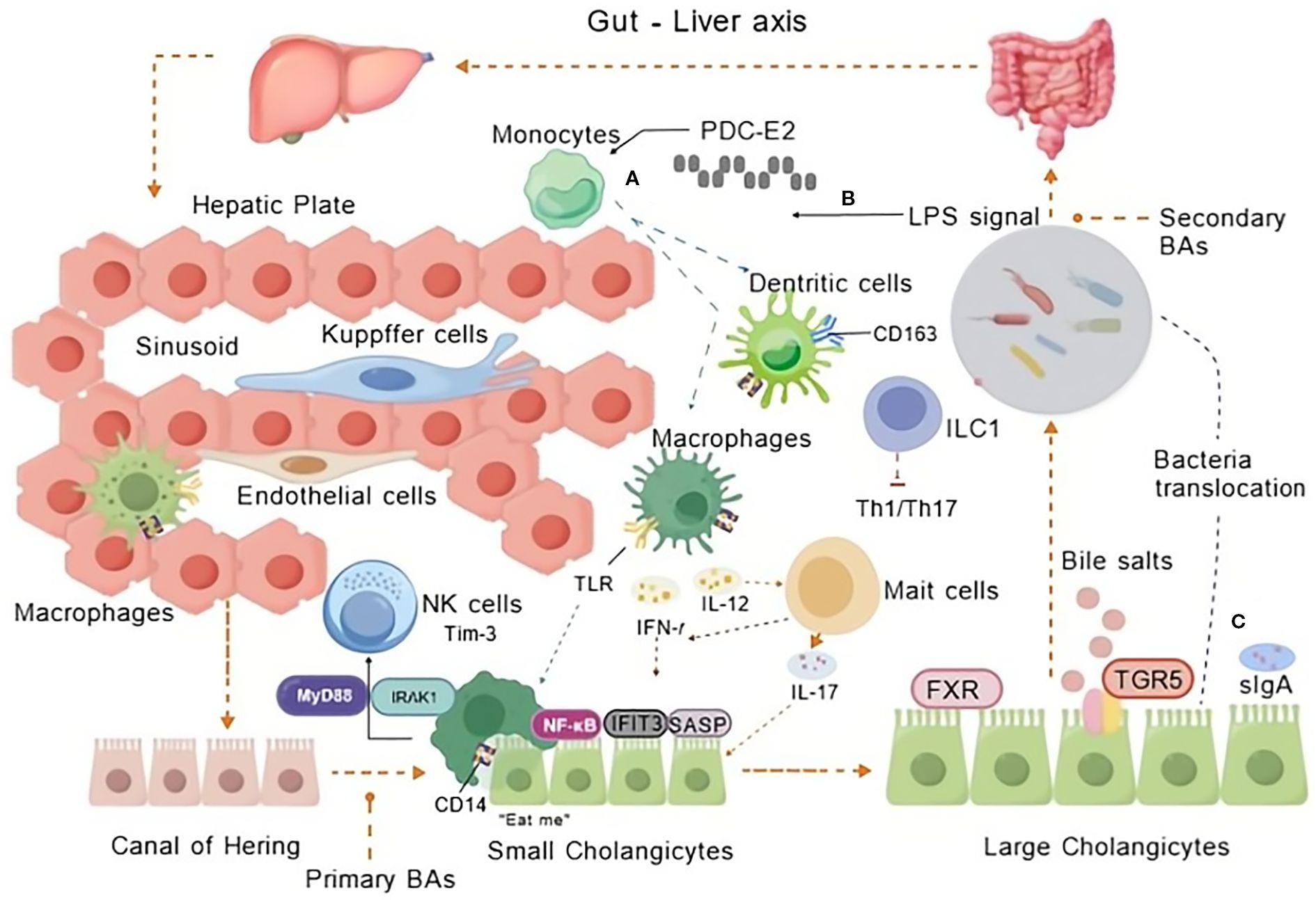
Figure 2. Innate immune cells in PBC. The interplay of innate immune cells in PBC’s hepatic sinusoids and biliary system with micro-organisms traversing hepatoenteric circulation during BA metabolism. Innate immune mechanisms via the gut-liver axis contribute to PBC pathogenesis through gut-derived signals and hepatic inflammation. (A) Monocyte recruitment and hepatic activation: Monocytes infiltrate the liver sinusoids and interact with endothelial cells, Kupffer cells, and macrophages. Mitochondrial antigen PDC-E2 triggers monocyte recruitment and activation via NF-κB signaling (through CD14/TLR4), alongside regulatory interactions with NK cells and immune checkpoint regulator TIM-3 modulate cytotoxicity. (B) LPS-induced inflammation: LPS translocated from the gut activates TLR signaling on innate immune cells (e.g., macrophages, dendritic cells), promoting pro-inflammatory cytokine production (e.g., IL-12, IL-23, IL-17, IFN-γ). (C) Bacterial translocation and bile acid dysregulation: sIgA function and gut barrier failure lead to bacterial translocation. Secondary BAs activate TGR5 receptors, disrupting BA homeostasis and amplifying inflammatory and cholestatic injury in small and large cholangiocytes. Created by BioGDP.com (29). BAs, Bile Acids; TLR, Toll-like receptor; LPS, Lipopolysaccharide; MyD88, Myeloid differentiation factor 88; IRAK1, Immune Recognition of Alphavirus Capsid Protein; IFIT3, Interferon-induced protein with tetratricopeptide repeats 3; SASP, Senescence-Associated Secretory Phenotype; TIM-3, an immune checkpoint molecule; FXR, Farnesoid X Receptor; TGR5, Takeda G protein-coupled receptor 5; ILC1, Innate Lymphoid Cells; CD163, a marker for macrophages; sIgA, Secretory Immunoglobulin A.
As the primary defense mechanism, macrophages, dendritic cells, and innate lymphoid cells strategically localize within portal tracts and bile ducts, establishing surveillance networks that detect gastrointestinal-derived pathogens via pattern recognition receptors (30). These immune effectors collaborate with cholangiocytes—transcending their structural role—which actively modulate immune responses through cytokine secretion, antigen presentation, and secretory IgA production to neutralize invading microorganisms (31). Significantly, this cellular interplay is biochemically regulated by Bile Acids(BAs) via TGR5 receptor signaling on cholangiocytes. BA-mediated induction of anti-inflammatory mediators counterbalances pro-inflammatory signals to maintain mucosal homeostasis. The system’s resilience further relies on bidirectional crosstalk with the biliary microbiota: commensal organisms metabolize BAs into immunoregulatory derivatives, while dysbiosis disrupts this equilibrium, predisposing to cholangiopathies such as primary biliary cholangitis through aberrant immune activation (32, 33). Notably, cholangiocyte viability constitutes a critical determinant—their senescence impairs antimicrobial defense, whereas apoptosis initiates fibrotic cascades, thereby delineating how epithelial-immune coordination governs disease progression (18). Consequently, emerging therapeutic strategies target this multifaceted network, aiming to recalibrate BA signaling, restore microbial symbiosis, and preserve cholangiocyte function to intercept pathological cascades.
The innate immune signaling pathways within the biliary tract encompass complex molecular mechanisms regulating host responses to microbial stimuli. While direct investigations of the biliary tract remain limited, insights derived from related tissues and cell types provide valuable insights into these pathways. NF-κB signaling constitutes a pivotal component of innate immune responses across diverse epithelial tissues. NF-κB activation is essential for epithelial defense, with cortactin playing a critical role in facilitating NF-κB-mediated cytokine production, such as IL-8, during bacterial infection (32). This underscores the significance of NF-κB in mediating inflammatory responses, likely relevant to biliary epithelial cells due to their epithelial characteristics. Similarly, microbial components modulate innate immune signaling, as demonstrated in studies of pathogen recognition. Chlamydia trachomatis can induce TLR3 expression while concurrently downregulating the NF-κB and IRF3 pathways in Sertoli cells, resulting in the suppression of pro-inflammatory cytokine production (33). This indicates that certain pathogens can manipulate innate signaling pathways, potentially influencing immune responses in tissues such as the biliary tract that are exposed to microbial stimuli.
Type I and III interferons play critical roles in innate antiviral immunity. Studies demonstrate differential interferon responses in human nasal versus lung tissues following SARS-CoV-2 infection, revealing restricted interferon induction in lung tissue despite productive viral infection (34, 35). These findings indicate tissue-specific regulation of interferon signaling pathways, suggesting parallels to the response mechanisms observed in biliary epithelium upon exposure to pathogens. The role of cytokines, such as Interferon Gamma (IFNγ), in linking immune responses to tissue pathology is underscored (36), who found that chronic IFNγ expression alters hepatic immune microenvironments, potentially contributing to autoimmune conditions like primary biliary cholangitis.
This connection underscores the critical role of cytokine signaling pathways in biliary immune regulation. Furthermore, the involvement of nuclear receptors in modulating immune responses is evident within the context of liver and biliary diseases. Studies indicate that PPARα and FXR may alter the hepatic immune microenvironment in biliary atresia (37, 38), suggesting that nuclear receptor pathways are integral to immune regulation in biliary tissues. Additional insights into innate immune regulation derive from investigations of tissue-specific responses. Bacterial outer membrane proteins activate innate immunity via neural-immune communication pathways, demonstrating that microbial components may influence immune signaling beyond classical receptor pathways (39). Moreover, the multifunctional role of CD14 in innate immunity and tissue homeostasis has highlighted its potential regulatory functions in barrier tissues characterized by rapid cell turnover, such as the biliary epithelium (40).
2.2 The relationship between innate biliary immunity and PBC
Typically, Biliary epithelial cells (BECs) are integral to immune response, functioning not only as a physical barrier but also actively participating in immune signaling. The apoptosis of BECs induces the release of auto-antigenic epitopes, which subsequently activate the immune system, driving dysregulation of both innate and adaptive immunity in PBC (41). Mass cytometry analyses demonstrate quantitative alterations in peripheral immune cell subsets in patients, including decreased γδ T cells and memory B cells, alongside increased monocytes and naïve B cells (42). This cellular imbalance underscores the critical role of innate immunity in disease progression (43).
Cytokines and immune mediators orchestrate PBC pathogenesis. Elevated expression of interferon-induced proteins, such as IFIT3, within senescent BECs indicates their involvement in inflammatory processes (44). Furthermore, TIM-3-mediated modulation of chemokine receptors on NK cells contributes to immune dysregulation, suggesting therapeutic potential through targeting this pathway (45). The gut microbiome exhibits significant interaction with innate immunity in PBC. Disease-associated dysbiosis, characterized by reduced microbial diversity and overgrowth of specific bacterial taxa, may influence immune responses via the gut-liver axis (46). Microbial metabolites can modulate both innate and adaptive immunity, thereby linking intestinal homeostasis to biliary inflammation (47).
Identification of disease-associated immune cell subsets and cytokine profiles holds promise for yielding novel biomarkers and therapeutic targets. Promising strategies include modulating the TIM-3 pathway and restoring gut microbiome balance (45, 46). Concurrently, genome-wide association studies identify risk loci predominantly related to immune function, highlighting the contribution of innate immunity to genetic susceptibility in PBC (43).
3 The mechanism of onset and progression
3.1 The autoimmune response
The innate immune system plays a critical role in the pathogenesis of PBC. NK cells mediate the destruction of BECs through both direct and indirect mechanisms. Specifically, the enhanced frequency and cytotoxicity of NK cells observed in the peripheral blood and liver tissues of PBC patients amplify autoimmune responses via the activation of autoreactive CD4+ T cells and the secretion of inflammatory cytokines (48). Innate Lymphoid Cells (ILCs) exhibit imbalances in subtype distribution among PBC patients, with alterations in cytokine production patterns correlating with disease severity. These ILC dysregulations may promote inflammatory and autoimmune pathways during PBC progression (49). Emerging evidence highlights trained immunity—a persistent functional reprogramming of innate immune cells following initial stimuli—as a contributor to exaggerated inflammatory responses upon secondary challenges (50). This innate immune hyper-reactivity to self-antigens may perpetuate chronic inflammation and tissue injury. The metabolic and epigenetic remodeling underlying trained immunity represents potential therapeutic targets. Reversing these adaptations may attenuate PBC-associated chronic inflammation (51). Pro-inflammatory cytokines secreted by activated innate immune cells establish a destructive microenvironment for BECs. Notably, the Th17/Treg imbalance, characterized by elevated Th17 cell levels and reduced Treg cell levels, reflects the inflammatory shift characteristic of PBC (52).
Gut microbiome dysbiosis in PBC patients correlates with altered microbial diversity and metabolite profiles. Microbiota-derived metabolites (e.g., short-chain fatty acids) modulate innate immunity, suggesting microbiome-targeted therapies could influence disease progression (46). Furthermore, metabolites derived from the gut microbiota reach the liver via the portal venous system, providing persistent low-grade immune stimulation. This continuous gut-derived immunogenic signal is hypothesized to disrupt immune tolerance towards mitochondrial antigens (53). Consequently, this ongoing microbial-driven immune activation provides a mechanistic explanation for the suboptimal efficacy and lack of durability observed with therapeutic strategies solely targeting systemic immune responses (53). Concurrently, the pivotal role of Peroxisome Proliferator-Activated Receptor (PPAR) in regulating the gut-liver immune axis has emerged as a significant finding (23). PPAR agonists to modulate immune responses, Low-dose IL-2 to restore immune balance (52), Trained immunity pathway inhibitors (50, 54). Their involvement in modulating immune and metabolic pathways offers promising novel targets for therapeutic intervention in PBC.
3.2 Injury of bile duct epithelial cells
Biliary epithelial cells are primary targets in PBC, where apoptosis releases autoantigen epitopes that trigger immune activation. This process is modulated by genetic predisposition and environmental factors that disrupt immune tolerance, driving disease progression (41). Bile acid accumulation within BECs induces cellular damage through membrane-disrupting detergent effects (55). Concurrently, diminished bicarbonate production compromises the protective “bicarbonate umbrella,” worsening BEC injury and cholestasis (56). Innate immunity critically influences PBC pathogenesis. Inflammatory cytokines released during BEC injury recruit and activate macrophages and NK cells, amplifying bile duct damage. Invariant NK T cells exacerbate liver fibrosis via Interleukin-17A (IL-17A) production, correlating with disease severity in PBC patients (56). Mast cells further contribute by interacting with innate immune cells to promote inflammation and fibrosis in cholestatic liver diseases (57).
Cholangiocytes exhibit injury responses including proliferation, differentiation, and senescence, collectively termed the Ductular Reaction (DR) to repair bile ducts (58). However, maladaptive responses exacerbate fibrosis. Senescent cholangiocytes adopt a Senescence-associated Secretory Phenotype (SASP), releasing pro-inflammatory mediators that sustain fibrogenesis and perpetuate injury cycles (44). Advancements in organoid technology have enabled the modeling of BEC injury, providing insights into cholangiocyte pathophysiology. These model systems facilitate the investigation of cholangiocyte apoptosis and fibrogenic responses critical to the progression of cholangiopathies, including biliary atresia and PBC (59). Such models hold promise for identifying therapeutic targets to mitigate BEC damage in PBC.
UDCA targets enhanced bile flow and reduced cholestatic injury but frequently fails to halt disease progression (60). Emerging therapeutic strategies focus on immunomodulatory agents and anti-fibrotic therapies to improve BEC survival and restore biliary function (55). Targeting cholangiokines—cytokines secreted by cholangiocytes—may also modulate the hepatic microenvironment for therapeutic benefit (61). The complex interplay between BEC injury, immune activation, and cholangiocyte adaptive responses underscores the disease pathogenesis.
3.3 Inflammatory factors
Inflammatory factors—ranging from molecular signaling pathways like TGF-β1/Smad and IRF3 phosphorylation, to immune cell subsets such as ILCs, and systemic cytokines—play crucial roles in the innate immune response of the biliary tract in PBC. These factors contribute to inflammation, tissue injury, and fibrosis, shaping the disease course and offering potential targets for therapeutic intervention.
MicroRNA-34a has been identified as a potential marker and regulator of fibrogenesis in PBC which promotes Epithelial-Mesenchymal Transition (EMT) and liver fibrosis by modulating the TGF-β1/Smad pathway, suggesting its pivotal role in inflammatory and fibrotic processes within the biliary system (62). However, the precise role of EMT in the pathogenesis of PBC remains controversial. Although in vitro studies demonstrate that TGF-β1 can induce EMT in cholangiocytes (63, 64), lineage-tracing animal models have failed to provide definitive evidence for EMT occurrence in vivo (65, 66). Cholangiocyte senescence (67, 68), autophagy dysfunction (69), and inflammatory cytokines such as IL-17A (70) are recognized as significant pathogenic mechanisms in PBC. As a matter of fact, whether EMT acts as a primary driver of bile duct injury or a secondary consequence requires further investigation. The extent of concordance between EMT manifestations observed in human PBC and those in experimental animal models also warrants critical evaluation.
Moreover, substantial evidence suggests that interferon regulatory factor 3 (IRF3) phosphorylation serves as a critical mediator of inflammation and tissue injury. Elevated IRF3 phosphorylation levels observed in the livers of patients with PBC and Primary Sclerosing Cholangitis (PSC) reveal that bile acid–induced IRF3 activation mediates cell death, inflammatory responses, and fibrosis, highlighting the pivotal role of innate immune signaling in disease pathology (71). The immune cell landscape also undergoes alterations in PBC. ILC subsets, specifically ILC1s and ILC3s in both patients and murine models, are implicated in the disease process, potentially contributing to inflammatory responses and biliary tract fibrosis (72).
Bile acids themselves function as modulators of inflammation. The roles of bile acids and their cognate receptors underscore their influence on immune responses in autoimmune liver diseases (73). This connection implies that bile acid–mediated signaling pathways may intersect with inflammatory cascades, further modulating innate immunity in PBC. The microbial milieu within the biliary system likewise influences inflammatory responses. Altered biliary microbial patterns correlate with disease progression and reduced transplant-free survival in PSC, suggesting microbial factors modulate innate immune activation and inflammation in cholangiopathies (74).
Additionally, systemic inflammatory cytokines and their mediators have been investigated for causal roles (Table 1). Mendelian randomization analyses demonstrate that circulating inflammatory cytokines mediate the relationship between plasma metabolites and bile duct or gallbladder calculus formation (91), collectively illustrating the integral contribution of inflammatory mediators to disease pathogenesis and progression.

Table 1. Primary cytokines associated with cholangiocytes and their functional roles in PBC.
3.4 Biliary microbiota
The biliary microbiome constitutes a complex microbial community inhabiting the biliary tract. Dysbiosis—an imbalance within this ecosystem. Specifically, patients with PBC exhibit compositional alterations in their biliary microbiota, which may drive immune dysregulation and disease progression (92, 93). Certain bacterial genera, including Enterococcus and Fusobacteria, demonstrate significant correlations with PBC severity. The detection of Enterococcus in bile samples is associated with heightened risks of disease advancement, suggesting that specific microbial populations exacerbate inflammatory cascades within the biliary tract (41, 94).
Biliary epithelial cells regulate local immune responses through secretory IgA expression and chemokine receptor modulation, while bile acids confer cytoprotection via TGR5 receptor activation (95). Conversely, secondary bile acids generated by microbial metabolism exhibit concentration-dependent and microbiota-contextual effects on cholangiocytes, ranging from protective to detrimental outcomes (41, 96). The interplay between biliary microbiota and inflammation involves multifactorial mechanisms. Dysbiosis may promote intestinal barrier dysfunction, facilitating bacterial translocation and metabolite influx into the biliary system. This process can incite aberrant immune activation and cholangiocyte injury (93, 97). Distinct microbial signatures within the bile of PBC patients correlate with disease duration and severity. Elevated microbial richness and enrichment of specific taxa are linked to advanced fibrotic stages (98).
Given the emerging role of biliary dysbiosis in PBC, microbiome-targeted interventions represent a promising therapeutic strategy. Approaches including probiotics, prebiotics, and fecal microbiota transplantation aim to restore microbial homeostasis and enhance biliary immune function (99, 100). Pharmacological agents modulating bile acid metabolism and cholangiocyte signaling pathways also hold therapeutic potential as PPAR agonists (23, 32).
4 Diagnosis and treatment strategies for innate biliary immunity
4.1 The standard of care
UDCA persists as the cornerstone of PBC management, attenuating cholestatic injury through pleiotropic mechanisms (32, 100). Although its cytoprotective effects on cholangiocytes against bile acid toxicity are well-established, the precise molecular pathways—particularly its interactions with bile acid transporters and nuclear receptors—require further elucidation. This knowledge gap holds clinical significance: approximately 40% of patients exhibit suboptimal biochemical responses to UDCA, necessitating second-line therapies (41). OCA, a potent FXR agonist, targets this therapeutic gap by reprogramming bile acid homeostasis. Its efficacy derives from the transcriptional upregulation of efflux transporters (BSEP, MRP2/3, MDR3), thereby reducing hepatocellular bile acid retention and apoptosis (43, 101). However, the dose-dependent pruritus of OCA and heterogeneous treatment responses underscore the necessity for personalized FXR agonist selection based on patient genetics and disease phenotype. The autoimmune pathogenesis of PBC positions innate immunity regulators as promising therapeutic targets, as illustrated in Figure 3.
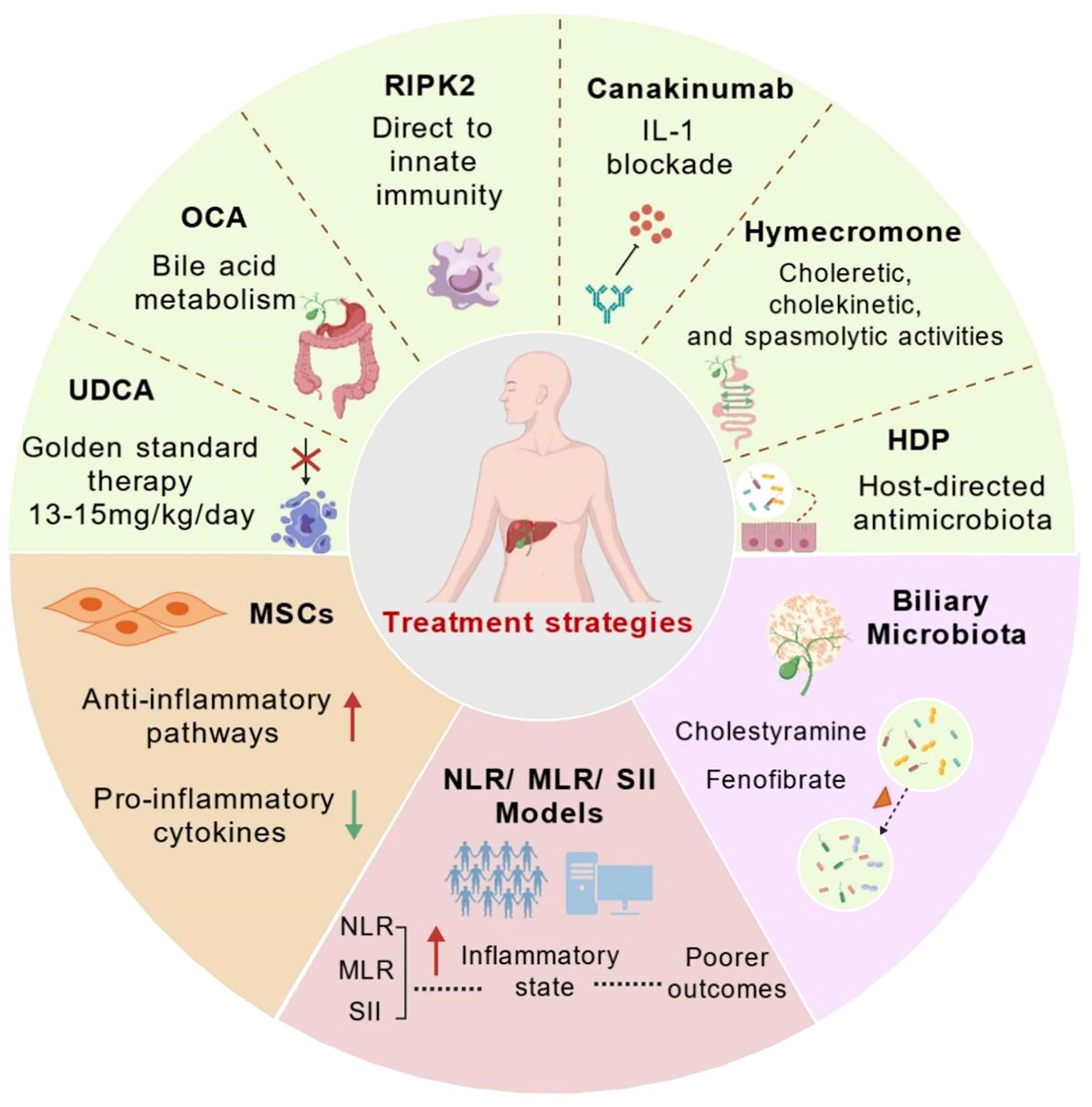
Figure 3. Therapeutic approaches in primary biliary cholangitis. The figure summarizes current and emerging approaches to modulate innate immune responses and inflammation in PBC. Schematic overview of current standard of care (UDCA and OCA) and emerging therapeutic strategies (RIPK2 inhibition, IL-1 blockade, TIM-3 modulation, MSCs, Biliary Microbiota therapy and novel prognostic markers - NLR/MLR/SII), highlighting potential molecular targets and pathways in PBC. Established therapies: UDCA (first-line) and OCA (obeticholic acid, FXR agonist) primarily target bile acid metabolism. Investigational and adjunct therapies: PPAR-α agonists (e.g., fenofibrate) and IL-1 blockade (e.g., canakinumab). Exploratory strategies: RIPK2 inhibition (innate immunity), Hymecromone (choleretic effects), MSCs therapy(anti-inflammatory), and HDPs (biliary microbiota modulation). Systemic inflammation indices NLR, MLR, SII serve as prognostic biomarkers, with elevated levels correlating with a pro-inflammatory state and poorer outcomes, guiding personalized treatment decisions. Created by BioGDP.com (29). UDCA, ursodeoxycholic acid; OCA, Obeticholic Acid; RIPK2 inhibitors, immunomodulation; Canakinumab, IL-1 blockade; MSCs, anti-inflammatory modulation; HDPs, Host-Directed Therapies; Cholestyramine, bile acid sequestrant; Fenofibrate, PPAR-α agonist; NLR, Neutrophil-to-Lymphocyte Ratio; MLR, Monocyte-to-Lymphocyte Ratio; SII, Systemic Immune-Inflammation Index.
4.2 Emerging experimental strategies targeting innate immunity
Receptor Interacting Serine/Threonine Kinase 2 (RIPK2) functions as a critical signaling hub within the NOD-mediated inflammatory cascade (102). Pharmacological inhibition of RIPK2 may disrupt the self-perpetuating cycle of biliary epithelial cell damage and cytokine release- a strategy potentially synergistic with bile acid-directed therapies. Similarly, interleukin-1 blockade (Canakinumab) represents a rational therapeutic approach to attenuate inflammasome activation in patients refractory to UDCA/OCA (103). Its established efficacy in autoinflammatory disorders such as Still’s disease suggests applicability in PBC patients exhibiting prominent serological inflammation (elevated CRP/SAA). Nevertheless, strategic stratification of patients is essential: IL-1 antagonists may benefit individuals with dominant innate immune activation, whereas RIPK2 inhibitors might target those with NOD pathway dysregulation. Non-immunosuppressive adjuncts play vital roles in symptom control and mucosal protection. Hymecromone exemplifies this approach—its triple mechanisms of action (choleretic, cholekinetic, spasmolytic) alleviate biliary pain without inducing gallbladder contraction, rendering it uniquely suited for biliary dyskinesia (104, 105). Concurrently, the gut-liver axis emerges as a therapeutic target. Host defense peptides (HDPs), induced by dietary components, enhance mucosal barrier integrity and exert selective antimicrobial effects without exacerbating inflammation (105). This host-directed strategy could counteract dysbiosis-associated disease progression, particularly given evidence that UDCA modulates gut microbiota composition- a potential biomarker for treatment response (106).
The therapeutic landscape of PBC is poised for transformation through innovative strategies extending beyond conventional approaches. Drug repurposing spearheads this evolution, with network pharmacology identifying promising candidates—including IL-1, EGFR, and TNF-α inhibitors, branched-chain amino acids, and curcumin—tailored to distinct PBC endotypes (107). These agents offer accelerated translational potential due to established safety profiles, enabling rapid deployment for phenotype-specific interventions. Building on cross-disciplinary insights, oncology-inspired therapeutic synergies present compelling paradigms. The efficacy of CDK4/6 inhibitors combined with cytotoxic chemotherapy in biliary tract cancers demonstrates how autophagy blockade overcomes treatment resistance (108). While direct applicability to PBC requires validation, this mechanistic approach—particularly triple therapy regimens targeting autophagic flux—could inform combinational strategies for advanced, treatment-refractory PBC.
Furthermore, advanced delivery systems are redefining precision targeting. Engineered exosomes exemplify this frontier, functioning as modular platforms for cholangiocyte-directed immunomodulation (109). Their capacity to deliver bespoke cargo (e.g., miRNA silencing RIPK2 or anti-inflammatory cytokines) could overcome limitations inherent to systemic drug administration, enabling site-specific pathway modulation with reduced off-target effects. Additionally, immunogenic microenvironment reprogramming—inspired by oncology’s “cold-to-hot” tumor conversion strategies—holds untapped potential. Activating Double-stranded RNA (dsRNA) sensors within fibrotic hepatic niches may reverse immunological anergy, priming PBC microenvironments for enhanced responsiveness to checkpoint inhibitors (110). This approach could synergize with existing immunomodulators to interrupt cycles of autoimmune-driven fibrosis.
4.3 Stem cell therapy drugs
Mesenchymal stem cells (MSCs) represent a promising therapeutic strategy for PBC owing to their immunomodulatory properties, capacity for multilineage differentiation, and potential to promote tissue repair (111, 112). MSCs modulate immune responses by suppressing pro-inflammatory cytokine production and augmenting anti-inflammatory pathways, thereby potentially restoring immune homeostasis in PBC patients (41, 111). MSC therapy may mitigate the effects of trained immunity—a phenomenon wherein innate immune cells exhibit hyperresponsiveness to secondary stimuli that may otherwise exacerbate pathologies such as stroke (41, 112).
However, the clinical translation of MSC therapy for PBC remains nascent, with ongoing research focused on optimizing therapeutic protocols. Clinical trials have demonstrated promising efficacy, indicating MSC administration improves hepatic function and attenuates inflammation in autoimmune liver diseases (112–114). Nevertheless, clinical trials in autoimmune liver diseases are ongoing, with results awaited (NCT02997878). The challenges persist, including the necessity for standardized methodologies for MSC isolation, expansion, and delivery, alongside concerns regarding long-term therapeutic safety and efficacy (113, 114).
Furthermore, integrating MSC therapy with established treatments may enhance clinical outcomes. Combining MSC therapy with immunomodulatory agents could yield synergistic effects, advancing overall PBC management (115, 116). Recent advances in stem cell membrane-camouflaged nanoparticles demonstrate potential for targeted delivery to inflamed tissues, offering a particularly advantageous approach for PBC treatment (117).
4.4 Prognostic model of NLR, MLR, and SII
Neutrophil-to-Lymphocyte Ratio (NLR) and Monocyte-to-Lymphocyte Ratio (MLR) are readily available and cost-effective markers of systemic inflammation (118–120). The potential utility of NLR and MLR as predictors of treatment response to novel therapies for PBC. However, these biomarkers have not yet been validated for routine clinical application and are not incorporated into current international management guidelines. NLR reflects the balance between innate (neutrophils) and adaptive (lymphocytes) immune responses (121). Elevated NLR indicates a heightened inflammatory state, often associated with poorer outcomes in various diseases (120). Similarly, MLR reflects the proportion of monocytes relative to lymphocytes, providing another dimension of immune system activity (121, 122).
Several studies have demonstrated the prognostic value of NLR and MLR in various diseases, including autoimmune conditions and cancers (119–130). In seropositive autoimmune encephalitis, a high NLR was associated with a higher likelihood of first-line treatment failure (129). In advanced gastric and colorectal cancers, lower MLR was associated with prolonged progression-free survival and overall survival (120). Higher NLR has been correlated with poor prognosis in several cancers, as well as being a reliable marker of inflammation, infection and sepsis (121, 129). High NLR levels independently associated with poor prognosis in heart failure (129). Therefore, NLR and MLR can serve as indicators of disease severity and predictors of treatment response and survival. A study found the maximal NLR had the best predictive value for in-hospital and 30-day mortality in ICU patients with CAD and CKD (128). Meanwhile, the total cholesterol, ALP, and NLR were the three independent risk factors associated with early biochemical nonresponse to UDCA treatment (129).
While specific studies focusing on NLR and MLR as prognostic markers in PBC are still emerging, the existing literature provides a strong rationale for their potential utility. Combining NLR and MLR with other inflammatory markers, such as the Systemic Immune-inflammation Index (SII) (122, 123, 125, 126), to improve prognostic accuracy (38).
4.5 Biliary microbiota therapy
Dysbiosis, or an imbalance in the gut microbiota, has been implicated in the pathogenesis of PBC. For instance, alterations in the gut microbiome have been associated with the severity of liver disease and the response to treatment. Research indicates that specific microbial taxa may influence the immune response and contribute to the autoimmune processes observed in PBC patients (131). Furthermore, the salivary microbiota of PBC patients exhibited significant differences, suggesting that oral microbiota may play a role in the disease’s pathogenesis (132).
Consequently, given the emerging evidence of the gut-liver axis’s role in PBC, several therapeutic strategies are being explored to manipulate the biliary microbiota. One promising approach involves the use of bile acid sequestrants, such as cholestyramine, which have been shown to alter the gut microbiome and improve cholestatic symptoms in PBC patients (133). Additionally, the use of fenofibrate as a second-line therapy for patients with inadequate responses to UDCA has shown promise. Studies have demonstrated that fenofibrate can improve liver biochemistry and histological features in PBC patients, suggesting that it may also exert beneficial effects on the gut microbiota (134). The combination of UDCA and fenofibrate has been associated with improved biochemical responses, indicating a synergistic effect that may be mediated by changes in the gut microbiome (135). The exploration of biliary microbiota therapy in PBC is still in its infancy. Moreover, clinical trials investigating the efficacy of probiotics, prebiotics, and other microbiota-targeted therapies in PBC patients are warranted (136). Beyond cholestatic pathologies, Hypoxia-inducible Factors (HIFs) exhibit immunosuppressive properties in hepatic malignancies by fostering pro-tumorigenic microenvironments, positioning them as therapeutic targets in liver cancer management (137).
4.6 CAR-T therapy
Originally developed for hematological malignancies, Chimeric Antigen Receptor (CAR)-T cell therapy has recently demonstrated sustained, profound depletion of autoreactive B cells in autoimmune diseases, exhibiting promising safety and efficacy profiles (138). This transition from oncology to autoimmunity represents a transformative advancement in therapeutic strategy (139). Currently, CAR-T therapy is under investigation across 372 institutions in 40 countries/regions for various autoimmune conditions, including Systemic Lupus Erythematosus (SLE) and multiple sclerosis (140). The development of novel CAR architectures, such as fourth-generation constructs, continues to enhance therapeutic potential (141, 142).
However, CAR-T therapy entails significant risks, including Cytokine Release Syndrome (CRS), neurotoxicity, and organ-specific toxicities. These adverse events are influenced by inflammatory microenvironments, limitations inherent in CAR design, and systemic immune disruption (143). Safety optimization may be achievable through innovative strategies—e.g., logic-gated CAR systems or alternative cellular carriers—coupled with rigorous clinical monitoring. Future efforts should prioritize generating disease-specific clinical evidence and developing adaptable CAR designs to facilitate broader, safer application.
Although CAR-T expansion into autoimmunity offers new therapeutic potential for refractory cases (144–147), its adverse effects present distinctive challenges within autoimmune pathological contexts (138, 139). For instance, inflammatory microenvironments can impair CAR-T persistence and exacerbate toxicity (140). Disease-specific toxicities, such as renal dysfunction or cutaneous manifestations, have also been reported, particularly with CD19-directed CAR-T therapy in SLE (146). Research characterizing toxicity profiles is evolving rapidly. Critical future directions include developing allogeneic CAR-T products to circumvent limitations associated with autologous approaches and extending CAR technology to alternative effector cells (e.g., CAR-macrophages or CAR-Natural Killer (NK) cells) to improve safety and efficacy (148, 149). Integration of precision medicine methodologies may facilitate enhanced risk stratification and personalized monitoring protocols (150). Therefore, Refined CAR designs and systematic real-world evidence are essential for safe, effective translation from oncology to autoimmunity. It must be emphasized that the application of CAR-T therapy in PBC remains speculative, there is no clinical evidence currently supports its potential efficacy in this condition.
5 Conclusions and future perspectives
Innate immune pathways—including TIM3/Gal9, NF-κB, and cGAS-STING—emerge as promising intervention points for biliary tract disorders, given their central role in stone-associated inflammatory cascades (151–154).
The innate immune system constitutes a pivotal pathogenic mechanism in PBC. Dysregulation of innate immune responses can activate autoreactive T and B lymphocytes, contributing to autoimmunity (155). Recent scientific advances demonstrate that apoptosis of biliary epithelial cells releases auto-antigens, which subsequently activate the immune system and disrupt immune tolerance (41, 156). Furthermore, the gut-liver axis is implicated in PBC pathogenesis, with gut microbiota and bile acids influencing immune responses and disease progression (75).
Although strategies targeting innate immunity—such as RIPK2 inhibition, IL-1 blockade, TIM-3 modulation, and MSCs therapy—hold therapeutic promise, they remain largely at the experimental stage and require further validation before clinical translation. Furthermore, novel prognostic inflammatory markers including NLR and MLR have been shown to reflect systemic inflammation and correlate with disease progression. In parallel, deeper investigation into mechanisms such as the gut–biliary–immune axis and trained immunity is ongoing. However, these approaches currently lack sufficient clinical evaluation, and their application remains speculative. Firstly, the complexity of the immune response in PBC complicates the identification of specific intervention targets. For instance, while Tregs are crucial for maintaining immune tolerance, their therapeutic application in autoimmune liver diseases has yielded inconsistent outcomes (156). While the role of autoimmunity in the pathogenesis of PBC is well established, immunomodulatory therapies (including biologics) effective in other autoimmune diseases may be ineffective in PBC (157). Furthermore, existing experimental models of PBC inadequately recapitulate the key immunopathological features, progression kinetics, and heterogeneity observed in human (158, 159). Consequently, immunomodulatory strategies developed based on model data frequently fail to achieve anticipated efficacy in the clinical trials of human. Moreover, the protracted progression characteristic of PBC presents substantial challenges for clinical trial design. Studies often rely on surrogate endpoints, and the implementation of long-term placebo-controlled trials is problematic. These limitations impede the definitive confirmation of benefits on hard clinical endpoints. Additionally, trial durations may be insufficient to demonstrate meaningful therapeutic effects (28, 160).
Critically, a fundamental limitation of failed therapies lies in their inability to effectively target core liver/bile duct-specific immunopathological mechanisms. Specifically, these include pathogenic CD8+T cells, dysregulated IL-15 signaling, and tissue-resident memory T cells. Many therapeutic approaches, however, induce generalized immunosuppression without effectively intervening in the early stages of the autoimmune cascade (28).
Additionally, precise modulation of the immune response is critical, as overactivation can exacerbate hepatic damage. Advancements in immunotherapy offer promising avenues for PBC treatment. The therapeutic application of MSCs has emerged as a potential strategy due to their immunomodulatory properties and capacity to facilitate tissue regeneration (114). Concurrently, biopolymer immune implants designed to sequentially activate innate and adaptive immunity show promise in other contexts, suggesting potential applicability in PBC (161). Moreover, exploration of the gut microbiota-bile acid-immunity network provides a novel perspective for therapeutic strategies. Targeting immune factors associated with gut microbiota dysbiosis and bile acid imbalance may yield breakthroughs in PBC management (75, 120). Converging research underscores the critical regulatory role of the STING pathway in innate immunity, revealing novel therapeutic opportunities (162). Genetic associations between PBC and extrahepatic autoimmune disorders (e.g., inflammatory bowel disease) may indicate shared pathogenic pathways, potentially informing the development of targeted interventions (163, 164).
Collectively, the biliary innate immune system in PBC critically regulates disease progression through intricate cellular and molecular mechanisms. Damage to bile duct epithelial cells, release of inflammatory mediators, and immune cell imbalance collectively drive autoimmune responses and fibrotic processes. Technological innovations, including T Cell Receptor (TCR) sequencing and Chimeric Antigen Receptor (CAR) platforms, demonstrate potential for engineering personalized immunotherapies addressing PBC-specific immune dysfunction (141, 145).
Emerging therapeutic strategies—including targeting innate immune signaling pathways, modulating the gut microbiota, and applying stem cell therapy—provide novel approaches for PBC management. These findings elucidate key immunological nodes in PBC pathogenesis, thereby establishing a foundation for developing precise therapeutic strategies. Nevertheless, these methods presently do not have adequate clinical assessment, and their use is still hypothetical. Consequently, thorough mechanistic studies and stringent treatment burden evaluation are crucial to promote these strategies for clinical application. Future research should prioritize elucidating the gut-liver axis, bile acid metabolism and CAR-T cells, which are anticipated to yield more effective PBC treatment options.
Author contributions
RC: Writing – review & editing, Writing – original draft. YS: Writing – original draft, Writing – review & editing. YH: Writing – review & editing, Writing – original draft. WT: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This study was supported by the National Natural Science Foundation of China 82560413, the Joint Foundation of Department of Science and Technology of Yunnan Province 202501AY070001-248/202401AY070001-091/202201AY070001-008, the Yunnan Provincial Department of Education Science Research Fund Project 2023J0256, the Investigator Initiated Trail Projects of the Second Affiliated Hospital of Kunming Medical University Project ynIIT2023018, University Students Innovative Experimental Program Fund Project 202210678036, and The Applied Basic Research Foundation of Yunnan Province 202501AT070594. The above institutions did not participate in the design of the study, collection, analysis, interpretation of data, or in writing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Park JW, Kim JH, Kim SE, Jung JH, Jang MK, Park SH, et al. Primary biliary cholangitis and primary sclerosing cholangitis: current knowledge of pathogenesis and therapeutics. Biomedicines. (2022) 10:1288. doi: 10.3390/biomedicines10061288
2. Vögelin M and Kremer AE. Advancing care in primary biliary cholangitis: emerging insights and novel therapies. Expert Opin Pharmacother. (2025) 16:1–14. doi: 10.1080/14656566.2025.2516622
3. Jiang Y, Wang J, Wu H, Zhou W, Li S, and Jin M. Diagnostic and prognostic value of monocyte-to-lymphocyte ratio and red cell distribution width to lymphocyte ratio in primary biliary cholangitis. Turk J Gastroenterol. (2023) 34:170–6. doi: 10.5152/tjg.2023.21768
4. You H, Duan W, Li S, Lv T, Chen S, Lu L, et al. Chinese society of hepatology, chinese medical association. Guidelines on the diagnosis and management of primary biliary cholangitis (2021). J Clin Transl Hepatol. (2023) 11:736–46. doi: 10.14218/JCTH.2022.00347
5. Yang Y, Liu B, Zang B, Liu Q, Zhao C, Yao Y, et al. Autotaxin: A Potential biomarker for primary biliary cholangitis. Heliyon. (2023) 10:e23438. doi: 10.1016/j.heliyon.2023.e23438
6. Cristoferi L, Calvaruso V, Overi D, Viganò M, Rigamonti C, Degasperi E, et al. Accuracy of transient elastography in assessing fibrosis at diagnosis in naïve patients with primary biliary cholangitis: A dual cut-off approach. Hepatology. (2021) 74:1496–508. doi: 10.1002/hep.31810
7. Lopes Vendrami C, Thorson DL, Borhani AA, Mittal PK, Hammond NA, Escobar DJ, et al. Imaging of biliary tree abnormalities. Radiographics. (2024) 44:e230174. doi: 10.1148/rg.230174
8. John BV, Aitcheson G, Schwartz KB, Khakoo NS, Dahman B, Deng Y, et al. Male sex is associated with higher rates of liver-related mortality in primary biliary cholangitis and cirrhosis. Hepatology. (2021) 74:879–91. doi: 10.1002/hep.31776
9. Barba Bernal R, Ferrigno B, Medina Morales E, Castro CM, Goyes D, Trivedi H, et al. Management of primary biliary cholangitis: current treatment and future perspectives. Turk J Gastroenterol. (2023) 34:89–100. doi: 10.5152/tjg.2023.22239
10. Montano-Loza AJ, Lytvyak E, Hirschfield G, Hansen BE, Ebadi M, Berney T, et al. Prognostic scores for ursodeoxycholic acid-treated patients predict graft loss and mortality in recurrent primary biliary cholangitis after liver transplantation. J Hepatol. (2024) 81:679–89. doi: 10.1016/j.jhep.2024.05.010
11. Kawata K, Joshita S, Shimoda S, Yamashita Y, Yamashita M, Kitsugi K, et al. The ursodeoxycholic acid response score predicts pathological features in primary biliary cholangitis. Hepatol Res. (2021) 51:80–9. doi: 10.1111/hepr.13584
12. Roberts SB, Choi WJ, Worobetz L, Vincent C, Flemming JA, Cheung A, et al. Loss of biochemical response at any time worsens outcomes in UDCA-treated patients with primary biliary cholangitis. JHEP Rep. (2024) 6:101168. doi: 10.1016/j.jhepr.2024.101168
13. Chang JI, Kim JH, Sinn DH, Cho JY, Kim KM, Oh JH, et al. Clinical outcomes and validation of ursodeoxycholic acid response scores in patients with korean primary biliary cholangitis: A multicenter cohort study. Gut Liver. (2023) 17:620–8. doi: 10.5009/gnl220420
14. Gazda J, Drazilova S, Gazda M, Janicko M, Koky T, Macej M, et al. Treatment response to ursodeoxycholic acid in primary biliary cholangitis: A systematic review and meta-analysis. Dig Liver Dis. (2023) 55:1318–27. doi: 10.1016/j.dld.2022.12.010
15. Pinyopornpanish K, Chadalavada P, Talal Sarmini M, Khoudari G, Alomari M, Padbidri V, et al. Simplified 6-month prediction scores for primary biliary cholangitis patients treated with ursodeoxycholic acid. Eur J Gastroenterol Hepatol. (2022) 34:411–6. doi: 10.1097/MEG.0000000000002216
16. Scaravaglio M and Carbone M. Prognostic scoring systems in primary biliary cholangitis: an update. Clin Liver Dis. (2022) 26:629–42. doi: 10.1016/j.cld.2022.06.005
17. European Association for the Study of the Liver. EASL Clinical Practice Guidelines on non-invasive tests for evaluation of liver disease severity and prognosis - 2021 update. J Hepatol. (2021) 75:659–89. doi: 10.1016/j.jhep.2021.05.025
18. Warnes TW, Roberts SA, Smith A, Cope VM, Vales P, and McMahon R. Portal pressure is of significant prognostic value in primary biliary cholangitis. Liver Int. (2023) 43:139–46. doi: 10.1111/liv.15289
19. Horst AK, Kumashie KG, Neumann K, Diehl L, and Tiegs G. Antigen presentation, autoantibody production, and therapeutic targets in autoimmune liver disease. Cell Mol Immunol. (2021) 18:92–111. doi: 10.1038/s41423-020-00568-6
20. Zhang H, Leung PSC, Gershwin ME, and Ma X. How the biliary tree maintains immune tolerance? Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1367–73. doi: 10.1016/j.bbadis.2017.08.019
21. Lan T, Qian S, Tang C, and Gao J. Role of immune cells in biliary repair. Front Immunol. (2022) 13:866040. doi: 10.3389/fimmu.2022.866040
22. Stein S, Henze L, Poch T, Carambia A, Krech T, Preti M, et al. IL-17A/F enable cholangiocytes to restrict T cell-driven experimental cholangitis by upregulating PD-L1 expression. J Hepatol. (2021) 74:919–30. doi: 10.1016/j.jhep.2020.10.035
23. Wang C, Shi Y, Wang X, Ma H, Liu Q, Gao Y, et al. Peroxisome proliferator-activated receptors regulate hepatic immunity and assist in the treatment of primary biliary cholangitis. Front Immunol. (2022) 13:940688. doi: 10.3389/fimmu.2022.940688
24. Hou X, Yang Y, Chen J, Jia H, Zeng P, Lv L, et al. TCRβ repertoire of memory T cell reveals potential role for Escherichia coli in the pathogenesis of primary biliary cholangitis. Liver Int. (2019) 39:956–66. doi: 10.1111/liv.14066
25. Yang H and Duan Z. Bile acids and the potential role in primary biliary cirrhosis. Digestion. (2016) 94:145–53. doi: 10.1159/000452300
26. Reuveni D, Gore Y, Leung PSC, Lichter Y, Moshkovits I, Kaminitz A, et al. The critical role of chemokine (C-C motif) receptor 2-positive monocytes in autoimmune cholangitis. Front Immunol. (2018) 9:1852. doi: 10.3389/fimmu.2018.01852
27. Wang R, Li B, Huang B, Li Y, Liu Q, Lyu Z, et al. Gut microbiota-derived butyrate induces epigenetic and metabolic reprogramming in myeloid-derived suppressor cells to alleviate primary biliary cholangitis. Gastroenterology. (2024) 167:733–49. doi: 10.1053/j.gastro.2024.05.014
28. Pu X, Liu Y, Lyu Z, Zhou Y, Zhao Y, Huang B, et al. B cells drive CCR5+CD4+ tissue-resident memory T cell cytotoxicity via IL-15Rα-IL-15 signaling in primary biliary cholangitis. J Hepatol. (2025). doi: 10.1016/j.jhep.2025.06.037
29. Jiang S, Li H, Zhang L, Mu W, Zhang Y, Chen T, et al. Generic Diagramming Platform(GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. (2025) 53:D1670–6. doi: 10.1093/nar/gkae973
30. Reuveni D, Assi S, Gore Y, Brazowski E, Leung PSC, Shalit T, et al. Conventional type 1 dendritic cells are essential for the development of primary biliary cholangitis. Liver Int. (2024) 44:2063–74. doi: 10.1111/liv.15961
31. Deutschmann K, Reich M, Klindt C, Dröge C, Spomer L, Häussinger D, et al. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1319–25. doi: 10.1016/j.bbadis.2017.08.021
32. Tegtmeyer N, Soltan Esmaeili D, Sharafutdinov I, Knorr J, Naumann M, Alter T, et al. Importance of cortactin for efficient epithelial NF-ĸB activation by Helicobacter pylori, Salmonella enterica and Pseudomonas aeruginosa, but not Campylobacter spp. Eur J Microbiol Immunol (Bp). (2022) 11:95–103. doi: 10.1556/1886.2021.00023
33. Di Pietro M, Filardo S, Alfano V, Pelloni M, Splendiani E, Po A, et al. Chlamydia trachomatis elicits TLR3 expression but disrupts the inflammatory signaling down-modulating NFκB and IRF3 transcription factors in human Sertoli cells. J Biol Regul Homeost Agents. (2020) 34:977–86. doi: 10.23812/20-80-A-29
34. Alfi O, Yakirevitch A, Wald O, Wandel O, Izhar U, Oiknine-Djian E, et al. Human nasal and lung tissues infected ex vivo with SARS-coV-2 provide insights into differential tissue-specific and virus-specific innate immune responses in the upper and lower respiratory tract. J Virol. (2021) 95:e0013021. doi: 10.1128/JVI.00130-21
35. Li Y, Renner DM, Comar CE, Whelan JN, Reyes HM, Cardenas-Diaz FL, et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc Natl Acad Sci U S A. (2021) 118:e2022643118. doi: 10.1073/pnas.2022643118
36. Bae HR, Choi M-S, Kim S, Young HA, Gershwin ME, Jeon S-M, et al. IFNγ is A key link between obesity and th1-mediated autoimmune diseases. Int J OF Mol Sci. (2020) 22(1):208. doi: 10.3390/ijms22010208
37. Ma Y, Lu L, Tan K, Li Z, Guo T, Wu Y, et al. Reduced peroxisome proliferator-activated receptor-α and bile acid nuclear receptor NR1H4/FXR may affect the hepatic immune microenvironment of biliary atresia. Front Immunol. (2022) 13:875593. doi: 10.3389/fimmu.2022.875593
38. Tran DN, Go SM, Park SM, Jung EM, and Jeung EB. Loss of Nckx3 Exacerbates Experimental DSS-Induced Colitis in Mice through p53/NF-κB Pathway. Int J Mol Sci. (2021) 22:2645. doi: 10.3390/ijms22052645
39. Geng S, Li Q, Zhou X, Zheng J, Liu H, Zeng J, et al. Gut commensal E. coli outer membrane proteins activate the host food digestive system through neural-immune communication. Cell Host Microbe. (2022) 30:1401–1416.e8. doi: 10.1016/j.chom.2022.08.004
40. Na K, Oh BC, and Jung Y. Multifaceted role of CD14 in innate immunity and tissue homeostasis. Cytokine Growth Factor Rev. (2023) 74:100–7. doi: 10.1016/j.cytogfr.2023.08.008
41. Li H, Guan Y, Han C, Zhang Y, Liu Q, Wei W, et al. The pathogenesis, models and therapeutic advances of primary biliary cholangitis. BioMed Pharmacother. (2021) 140:111754. doi: 10.1016/j.biopha.2021.111754
42. Jang JS, Juran BD, Cunningham KY, Gupta VK, Son YM, Yang JD, et al. Single-cell mass cytometry on peripheral blood identifies immune cell subsets associated with primary biliary cholangitis. Sci Rep. (2020) 10:12584. doi: 10.1038/s41598-020-69358-4
43. Cordell HJ, Fryett JJ, Ueno K, Darlay R, Aiba Y, Hitomi Y, et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J Hepatol. (2021) 75:572–81. doi: 10.1016/j.jhep.2021.04.055
44. Sasaki M, Sato Y, and Nakanuma Y. Interferon-induced protein with tetratricopeptide repeats 3 may be a key factor in primary biliary cholangitis. Sci Rep. (2021) 11:11413. doi: 10.1038/s41598-021-91016-6
45. Xu J, Fu H, Yang Y, Yu H, Ai X, Lei Y, et al. Modulation of CXCR1 and CXCR3 expression on NK cells via TIM-3 in a murine model of primary biliary cholangitis. Mol Immunol. (2021) 135:342–50. doi: 10.1016/j.molimm.2021.04.014
46. Kitahata S, Yamamoto Y, Yoshida O, Tokumoto Y, Kawamura T, Furukawa S, et al. Ileal mucosa-associated microbiota overgrowth associated with pathogenesis of primary biliary cholangitis. Sci Rep. (2021) 11:19705. doi: 10.1038/s41598-021-99314-9
47. Zheng Y, Ran Y, Zhang H, Wang B, and Zhou L. The microbiome in autoimmune liver diseases: metagenomic and metabolomic changes. Front Physiol. (2021) 12:715852. doi: 10.3389/fphys.2021.715852
48. Gianchecchi E, Delfino DV, and Fierabracci A. Natural killer cells: potential biomarkers and therapeutic target in autoimmune diseases? Front Immunol. (2021) 12:616853. doi: 10.3389/fimmu.2021.616853
49. Li H, Zhan H, Cheng L, Yan S, Wang L, and Li Y. Imbalanced distribution of group 2 innate lymphoid cells (ILCs) and ILC precursors in peripheral blood of patients with primary biliary cholangitis. Scand J Immunol. (2022) 96:e13166. doi: 10.1111/sji.13166
50. Municio C and Criado G. Therapies targeting trained immune cells in inflammatory and autoimmune diseases. Front Immunol. (2021) 11:631743. doi: 10.3389/fimmu.2020.631743
51. Mora VP, Loaiza RA, Soto JA, Bohmwald K, and Kalergis AM. Involvement of trained immunity during autoimmune responses. J Autoimmun. (2023) 137:102956. doi: 10.1016/j.jaut.2022.102956
52. Wang Z, Liu Z, Zheng J, Huang L, Jin R, Wang X, et al. The effects of low-dose IL-2 on Th17/Treg cell imbalance in primary biliary cholangitis mouse models. BMC Gastroenterol. (2024) 24:87. doi: 10.1186/s12876-024-03176-0
53. Colapietro F, Gershwin ME, and Lleo A. PPAR agonists for the treatment of primary biliary cholangitis: Old and new tales. J Transl Autoimmun. (2023) 6:100188. doi: 10.1016/j.jtauto.2023.100188
54. Fiorucci S, Urbani G, Di Giorgio C, Biagioli M, and Distrutti E. Current landscape and evolving therapies for primary biliary cholangitis. Cells. (2024) 13:1580. doi: 10.3390/cells13181580
55. Pham HN, Pham L, and Sato K. Deconvolution analysis identified altered hepatic cell landscape in primary sclerosing cholangitis and primary biliary cholangitis. Front Med (Lausanne). (2024) 11:1327973. doi: 10.3389/fmed.2024.1327973
56. Jia H, Chen J, Zhang X, Bi K, Zhou H, Liu T, et al. IL-17A produced by invariant natural killer T cells and CD3+ CD56+ αGalcer-CD1d tetramer- T cells promote liver fibrosis in patients with primary biliary cholangitis. J Leukoc Biol. (2022) 112:1079–87. doi: 10.1002/JLB.2A0622-586RRRR
57. Bernard JK, Marakovits C, Smith LG, and Francis H. Mast cell and innate immune cell communication in cholestatic liver disease. Semin Liver Dis. (2023) 43:226–33. doi: 10.1055/a-2104-9034
58. Hrncir HR, Hantelys F, and Gracz AD. Panic at the bile duct: how intrahepatic cholangiocytes respond to stress and injury. Am J Pathol. (2023) 193:1440–54. doi: 10.1016/j.ajpath.2023.02.012
59. Chusilp S, Lee C, Li B, Lee D, Yamoto M, Ganji N, et al. A novel model of injured liver ductal organoids to investigate cholangiocyte apoptosis with relevance to biliary atresia. Pediatr Surg Int. (2020) 36:1471–9. doi: 10.1007/s00383-020-04765-2
60. Mayo MJ. Mechanisms and molecules: What are the treatment targets for primary biliary cholangitis? Hepatology. (2022) 76:518–31. doi: 10.1002/hep.32405
61. Cai X, Tacke F, Guillot A, and Liu H. Cholangiokines: undervalued modulators in the hepatic microenvironment. Front Immunol. (2023) 14:1192840. doi: 10.3389/fimmu.2023.1192840
62. Pan Y, Wang J, He L, and Zhang F. MicroRNA-34a promotes EMT and liver fibrosis in primary biliary cholangitis by regulating TGF-β1/smad pathway. J Immunol Res. (2021) 2021:6890423. doi: 10.1155/2021/6890423
63. Fan J, Wang Q, Zhang Z, and Sun L. Curcumin mitigates the epithelial-to-mesenchymal transition in biliary epithelial cells through upregulating CD109 expression. Drug Dev Res. (2019) 80:992–9. doi: 10.1002/ddr.21580
64. Kim Y, Lee EJ, Jang HK, Kim CH, Kim DG, Han JH, et al. Statin pretreatment inhibits the lipopolysaccharide-induced epithelial-mesenchymal transition via the downregulation of toll-like receptor 4 and nuclear factor-κB in human biliary epithelial cells. J Gastroenterol Hepatol. (2016) 31:1220–8. doi: 10.1111/jgh.13230
65. Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat Commun. (2013) 4:2823. doi: 10.1038/ncomms3823
66. Chu AS, Diaz R, Hui JJ, Yanger K, Zong Y, Alpini G, et al. Lineage tracing demonstrates no evidence of cholangiocyte epithelial-to-mesenchymal transition in murine models of hepatic fibrosis. Hepatology. (2011) 53:1685–95. doi: 10.1002/hep.24206
67. Sasaki M, Sato Y, and Nakanuma Y. A heterogeneous subtype of biliary epithelial senescence may be involved in the pathogenesis of primary biliary cholangitis. Clin Res Hepatol Gastroenterol. (2025) 49:102512. doi: 10.1016/j.clinre.2024.102512
68. Sasaki M, Sato Y, and Nakanuma Y. An involvement of Hippo-yes-associated protein pathway in biliary epithelial senescence in primary biliary cholangitis. Clin Res Hepatol Gastroenterol. (2023) 47:102106. doi: 10.1016/j.clinre.2023.102106
69. Zhu Y, Wang Q, Tang X, Yao G, and Sun L. Mesenchymal stem cells enhance autophagy of human intrahepatic biliary epithelial cells in vitro. Cell Biochem Funct. (2018) 36:280–7. doi: 10.1002/cbf.3340
70. Huang Q, Chu S, Yin X, Yu X, Kang C, Li X, et al. Interleukin-17A-induced epithelial-mesenchymal transition of human intrahepatic biliary epithelial cells: implications for primary biliary cirrhosis. Tohoku J Exp Med. (2016) 240:269–75. doi: 10.1620/tjem.240.269
71. Zhuang Y, Ortega-Ribera M, Thevkar Nagesh P, Joshi R, Huang H, Wang Y, et al. Bile acid-induced IRF3 phosphorylation mediates cell death, inflammatory responses, and fibrosis in cholestasis-induced liver and kidney injury via regulation of ZBP1. Hepatology. (2024) 79:752–67. doi: 10.1097/HEP.0000000000000611
72. Liu Y, Hu Y, Li B, Su R, Han Z, Jin B, et al. Innate lymphoid cell subsets in the pathogenesis of primary biliary cholangitis. J Gastroenterol Hepatol. (2024) 39:1431–41. doi: 10.1111/jgh.16547
73. Zhou T, Ismail A, and Francis H. Bile acids in autoimmune liver disease: unveiling the nexus of inflammation, inflammatory cells, and treatment strategies. Cells. (2023) 12:2725. doi: 10.3390/cells12232725
74. Özdirik B, Scherf M, Brumercek A, Nicklaus JM, Kruis T, Haber PK, et al. Biliary microbial patterns in primary sclerosing cholangitis are linked to poorer transplant-free survival. Hepatol Commun. (2023) 7:e0156. doi: 10.1097/HC9.0000000000000156
75. Nagano T, Yamamoto K, Matsumoto S, Okamoto R, Tagashira M, Ibuki N, et al. Cytokine profile in the liver of primary biliary cirrhosis. J Clin Immunol. (1999) 19:422–7. doi: 10.1023/a:1020511002025
76. Wang CB, Wang Y, Yao Y, Wang JJ, Tsuneyama K, Yang Q, et al. The gut microbiome contributes to splenomegaly and tissue inflammation in a murine model of primary biliary cholangitis. Ann Transl Med. (2022) 10:507. doi: 10.21037/atm-21-5448
77. Rogalska M, Błachnio-Zabielska A, Zabielski P, Janica JR, Roszczyc-Owsiejczuk K, Pogodzińska K, et al. Acylcarnitine and free fatty acid profiles in primary biliary cholangitis: associations with fibrosis and inflammation. Nutrients. (2025) 17:1097. doi: 10.3390/nu17071097
78. Park J, Gores GJ, and Patel T. Lipopolysaccharide induces cholangiocyte proliferation via an interleukin-6-mediated activation of p44/p42 mitogen-activated protein kinase. Hepatology. (1999) 29:1037–43. doi: 10.1002/hep.510290423
79. Zhang S, Tao X, Wang L, Chen H, Zhao L, Sun J, et al. Downregulation of programmed death-1 pathway promoting CD8 + T cell cytotoxicity in primary biliary cholangitis. Dig Dis Sci. (2022) 67:2981–93. doi: 10.1007/s10620-021-07165-1
80. Gallucci GM, Alsuwayt B, Auclair AM, Boyer JL, Assis DN, and Ghonem NS. Fenofibrate downregulates NF-κB signaling to inhibit pro-inflammatory cytokine secretion in human THP-1 macrophages and during primary biliary cholangitis. Inflammation. (2022) 45:2570–81. doi: 10.1007/s10753-022-01713-1
81. Sun Q, Wang Q, Feng N, Meng Y, Li B, Luo D, et al. The expression and clinical significance of serum IL-17 in patients with primary biliary cirrhosis. Ann Transl Med. (2019) 7:389. doi: 10.21037/atm.2019.07.100
82. Yang CY, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasani NP, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology. (2014) 59:1944–53. doi: 10.1002/hep.26979
83. Lan RY, Salunga TL, Tsuneyama K, Lian ZX, Yang GX, Hsu W, et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J Autoimmun. (2009) 32:43–51. doi: 10.1016/j.jaut.2008.11.001
84. Chan CW, Chen HW, Wang YW, Lin CI, and Chuang YH. IL-21, not IL-17A, exacerbates murine primary biliary cholangitis. Clin Exp Immunol. (2024) 215:137–47. doi: 10.1093/cei/uxad107
85. Liaskou E and Hirschfield GM. Genetic association studies and the risk factors for developing the “Immuno-bile-logic” disease primary biliary cholangitis. Hepatology. (2018) 67:1620–2. doi: 10.1002/hep.29603
86. Yang F, Zhou L, Shen Y, Wang X, Fan X, and Yang L. Multi-omics approaches for drug-response characterization in primary biliary cholangitis and autoimmune hepatitis variant syndrome. J Transl Med. (2024) 22:214. doi: 10.1186/s12967-024-05029-6
87. Chung Y, Tsou HLP, Heneghan MA, Chokshi S, and Riva A. Soluble herpes virus entry mediator and type II/III interferons are upregulated in primary biliary cholangitis. Int J Mol Sci. (2025) 26:605. doi: 10.3390/ijms26020605
88. Huang B, Lyu Z, Qian Q, Chen Y, Zhang J, Li B, et al. NUDT1 promotes the accumulation and longevity of CD103+ TRM cells in primary biliary cholangitis. J Hepatol. (2022) 77:1311–24. doi: 10.1016/j.jhep.2022.06.014
89. Bauer A and Habior A. Concentration of serum matrix metalloproteinase-3 in patients with primary biliary cholangitis. Front Immunol. (2022) 13:885229. doi: 10.3389/fimmu.2022.885229
90. Liang M, Ye S, Jing R, Zhu B, Yuan W, Chu X, et al. Estrogen receptor alpha-mediated mitochondrial damage in intrahepatic bile duct epithelial cells leading to the pathogenesis of primary biliary cholangitis. Environ Toxicol. (2023) 38:2803–18. doi: 10.1002/tox.23906
91. Peng S, Wei Y, Huang H, Lan C, Zeng Z, Zhu G, et al. The mediating role of circulating inflammatory cytokines in causal associations between plasma metabolites and asymptomatic bile duct and cholecyst calculus: A Mendelian randomization study. Med (Baltimore). (2025) 104:e41745. doi: 10.1097/MD.0000000000041745
92. Saab M, Mestivier D, Sohrabi M, Rodriguez C, Khonsari MR, Faraji A, et al. Characterization of biliary microbiota dysbiosis in extrahepatic cholangiocarcinoma. PloS One. (2021) 16:e0247798. doi: 10.1371/journal.pone.0247798
93. Xia Q, Liu Q, and Ma X. Intestinal microbiota in biliary diseases. Curr Opin Gastroenterol. (2023) 39:95–102. doi: 10.1097/MOG.0000000000000910
94. Zigmond E, Zecher BF, Bartels AL, Ziv-Baran T, Rösch T, Schachschal G, et al. Bile duct colonization with enterococcus sp. Associates with disease progression in primary sclerosing cholangitis. Clin Gastroenterol Hepatol. (2023) 21:1223–1232.e3. doi: 10.1016/j.cgh.2022.09.006
95. Farina A, Delhaye M, Lescuyer P, and Dumonceau JM. Bile proteome in health and disease. Compr Physiol. (2014) 4:91–108. doi: 10.1002/cphy.c130016
96. Lenci I, Milana M, Signorello A, Grassi G, and Baiocchi L. Secondary bile acids and the biliary epithelia: The good and the bad. World J Gastroenterol. (2023) 29:357–66. doi: 10.3748/wjg.v29.i2.357
97. Zhang L, Yang L, and Chu H. Targeting gut microbiota for the treatment of primary biliary cholangitis: from bench to bedside. J Clin Transl Hepatol. (2023) 11:958–66. doi: 10.14218/JCTH.2022.00408
98. Lammert C, Shin A, Xu H, Hemmerich C, O’Connell TM, and Chalasani N. Short-chain fatty acid and fecal microbiota profiles are linked to fibrosis in primary biliary cholangitis. FEMS Microbiol Lett. (2021) 368:fnab038. doi: 10.1093/femsle/fnab038
99. Wang H, Gong J, Chen J, Zhang W, Sun Y, and Sun D. Intestinal microbiota and biliary system diseases. Front Cell Infect Microbiol. (2024) 14:1362933. doi: 10.3389/fcimb.2024.1362933
100. Abenavoli L, Scarlata GG, Scarpellini E, Procopio AC, Ponziani FR, Boccuto L, et al. Therapeutic success in primary biliary cholangitis and gut microbiota: a safe highway? Minerva Gastroenterol (Torino). (2024) 70:430–41. doi: 10.23736/S2724-5985.23.03590-8
101. Lu Q, Zhu Y, Wang C, Zhang R, Miao Y, Chai Y, et al. Obeticholic acid protects against lithocholic acid-induced exogenous cell apoptosis during cholestatic liver injury. Life Sci. (2024) 337:122355. doi: 10.1016/j.lfs.2023.122355
102. Zhao W, Leng RX, and Ye DQ. RIPK2 as a promising druggable target for autoimmune diseases. Int Immunopharmacol. (2023) 118:110128. doi: 10.1016/j.intimp.2023.110128
103. Segú-Vergés C, Coma M, Kessel C, Smeets S, Foell D, and Aldea A. Application of systems biology-based in silico tools to optimize treatment strategy identification in Still’s disease. Arthritis Res Ther. (2021) 23:126. doi: 10.1186/s13075-021-02507-w
104. Elaskandrany MA, Ismail M, Liu Y, and Wang WW. Biliary dyskinesia with reduced gallbladder ejection fraction: A diagnostic and therapeutic shift in management. J Brown Hosp Med. (2025) 4:6–9. doi: 10.56305/001c.127836
105. Whitmore M, Tobin I, Burkardt A, and Zhang G. Nutritional modulation of host defense peptide synthesis: A novel host-directed antimicrobial therapeutic strategy? Adv Nutr. (2024) 15:100277. doi: 10.1016/j.advnut.2024.100277
106. Ma PJ, Wang MM, and Wang Y. Gut microbiota: A new insight into lung diseases. BioMed Pharmacother. (2022) 155:113810. doi: 10.1016/j.biopha.2022.113810
107. Shahini E, Pasculli G, Mastropietro A, Stolfi P, Tieri P, Vergni D, et al. Network proximity-based drug repurposing strategy for early and late stages of primary biliary cholangitis. Biomedicines. (2022) 10:1694. doi: 10.3390/biomedicines10071694
108. Arora M, Bogenberger JM, Abdelrahman AM, Yonkus J, Alva-Ruiz R, Leiting JL, et al. Synergistic combination of cytotoxic chemotherapy and cyclin-dependent kinase 4/6 inhibitors in biliary tract cancers. Hepatology. (2022) 75:43–58. doi: 10.1002/hep.32102
109. Abedi A, Moosazadeh Moghaddam M, Kachuei R, and Imani Fooladi AA. Exosomes as a therapeutic strategy in cancer: potential roles as drug carriers and immune modulators. Biochim Biophys Acta Rev Cancer. (2025) 1880:189238. doi: 10.1016/j.bbcan.2024.189238
110. Young AA, Bohlin HE, Pierce JR, and Cottrell KA. Suppression of double-stranded RNA sensing in cancer: molecular mechanisms and therapeutic potential. Biochem Soc Trans. (2024) 52:2035–45. doi: 10.1042/BST20230727
111. He C, Yang Y, Zheng K, Chen Y, Liu S, Li Y, et al. Mesenchymal stem cell-based treatment in autoimmune liver diseases: underlying roles, advantages and challenges. Ther Adv Chronic Dis. (2021) 12:2040622321993442. doi: 10.1177/2040622321993442
112. Yang Y, Zhao RC, and Zhang F. Potential mesenchymal stem cell therapeutics for treating primary biliary cholangitis: advances, challenges, and perspectives. Front Cell Dev Biol. (2022) 10:933565. doi: 10.3389/fcell.2022.933565
113. Feng YW, Wu C, Liang FY, Lin T, Li WQ, Jing YH, et al. hUCMSCs mitigate LPS-induced trained immunity in ischemic stroke. Front Immunol. (2020) 11:1746. doi: 10.3389/fimmu.2020.01746
114. Feng Y, Guo M, Zhao H, Han S, Dong Q, and Cui M. Mesenchymal-stem-cell-derived extracellular vesicles mitigate trained immunity in the brain. Front Bioeng Biotechnol. (2020) 8:599058. doi: 10.3389/fbioe.2020.599058
115. Floreani A, Gabbia D, and De Martin S. Update on the pharmacological treatment of primary biliary cholangitis. Biomedicines. (2022) 10:2033. doi: 10.3390/biomedicines10082033
116. Jallouli I, Doulberis M, and Kountouras J. Primary biliary cholangitis: a summary of pathogenesis and therapies. Ann Gastroenterol. (2025) 38:121–32. doi: 10.20524/aog.2025.0953
117. Zhang W and Huang X. Stem cell membrane-camouflaged targeted delivery system in tumor. Mater Today Bio. (2022) 16:100377. doi: 10.1016/j.mtbio.2022.100377
118. Broadley J, Wesselingh R, Seneviratne U, Kyndt C, Beech P, Buzzard K, et al. Peripheral immune cell ratios and clinical outcomes in seropositive autoimmune encephalitis: A study by the Australian autoimmune encephalitis consortium. Front Immunol. (2021) 11:597858. doi: 10.3389/fimmu.2020.597858
119. Fan X, Wang D, Zhang W, Liu J, Liu C, Li Q, et al. Inflammatory markers predict survival in patients with advanced gastric and colorectal cancers receiving anti-PD-1 therapy. Front Cell Dev Biol. (2021) 9:638312. doi: 10.3389/fcell.2021.638312
120. Zahorec R. Neutrophil-to-lymphocyte ratio, past, present and future perspectives. Bratisl Lek Listy. (2021) 122:474–88. doi: 10.4149/BLL_2021_078
121. Zhu S, Cheng Z, Hu Y, Chen Z, Zhang J, Ke C, et al. Prognostic value of the systemic immune-inflammation index and prognostic nutritional index in patients with medulloblastoma undergoing surgical resection. Front Nutr. (2021) 8:754958. doi: 10.3389/fnut.2021.754958
122. Mai RY, Bai T, Luo XL, and Wu GB. Preoperative fibrinogen-to-albumin ratio predicts the prognosis of patients with hepatocellular carcinoma subjected to hepatectomy. BMC Gastroenterol. (2022) 22:261. doi: 10.1186/s12876-022-02328-4
123. Liao J, Wei D, Sun C, Yang Y, Wei Y, and Liu X. Prognostic value of the combination of neutrophil-to-lymphocyte ratio, monocyte-to-lymphocyte ratio and platelet-to-lymphocyte ratio on mortality in patients on maintenance hemodialysis. BMC Nephrol. (2022) 23:393. doi: 10.1186/s12882-022-03020-1
124. Guo J, Lv W, Wang Z, Shang Y, Yang F, Zhang X, et al. Prognostic value of inflammatory and nutritional markers for patients with early-stage poorly-to moderately-differentiated cervical squamous cell carcinoma. Cancer Control. (2023) 30:10732748221148913. doi: 10.1177/10732748221148913
125. Duan X, Yang B, Zhao C, Tie B, Cao L, and Gao Y. Prognostic value of preoperative hematological markers in patients with glioblastoma multiforme and construction of random survival forest model. BMC Cancer. (2023) 23:432. doi: 10.1186/s12885-023-10889-0
126. Hoppe M, Gersey ZC, Muthiah N, Abdallah HM, Plute T, Abou-Al-Shaar H, et al. The utility of inflammatory biomarkers in predicting overall survival and recurrence in skull base chordoma. Neurosurg Focus. (2024) 56:E16. doi: 10.3171/2024.2.FOCUS2421
127. Li X, Zhang J, and Fu Z. Development and validation of an inflammation-combined prognostic index (ICPI)-based nomogram for predicting overall survival in gastric cancer. J Inflammation Res. (2024) 17:5439–52. doi: 10.2147/JIR.S476346
128. An S, Che W, Gao Y, Duo X, Li X, and Li J. Predictive value of complete blood count indicators for short-term mortality in patients with combined coronary artery disease and chronic kidney disease. Int J Nephrol Renovasc Dis. (2025) 18:113–22. doi: 10.2147/IJNRD.S508019
129. García-Escobar A, Vera-Vera S, Tébar-Márquez D, Rivero-Santana B, Jurado-Román A, Jiménez-Valero S, et al. Neutrophil-to-lymphocyte ratio an inflammatory biomarker, and prognostic marker in heart failure, cardiovascular disease and chronic inflammatory diseases: New insights for a potential predictor of anti-cytokine therapy responsiveness. Microvasc Res. (2023) 150:104598. doi: 10.1016/j.mvr.2023.104598
130. Zhu H, Zheng M, He H, Lei H, Tai W, Yang J, et al. Development and external validation of an early prediction model to identify irresponsive patients and prognosis of UDCA treatment in primary biliary cholangitis. Sci Rep. (2024) 14:31369. doi: 10.1038/s41598-024-82854-1
131. Guo Z, He K, Pang K, Yang D, Lyu C, Xu H, et al. Exploring advanced therapies for primary biliary cholangitis: insights from the gut microbiota-bile acid-immunity network. Int J Mol Sci. (2024) 25:4321. doi: 10.3390/ijms25084321
132. Lv L, Jiang H, Chen X, Wang Q, Wang K, Ye J, et al. The salivary microbiota of patients with primary biliary cholangitis is distinctive and pathogenic. Front Immunol. (2021) 12:713647. doi: 10.3389/fimmu.2021.713647
133. Li B, Zhang J, Chen Y, Wang Q, Yan L, Wang R, et al. Alterations in microbiota and their metabolites are associated with beneficial effects of bile acid sequestrant on icteric primary biliary Cholangitis. Gut Microbes. (2021) 13:1946366. doi: 10.1080/19490976.2021.1946366
134. Wang L, Sun K, Tian A, Liu Y, Zhang M, Zhou X, et al. Fenofibrate improves GLOBE and UK-PBC scores and histological features in primary biliary cholangitis. Minerva Med. (2022) 113:974–82. doi: 10.23736/S0026-4806.21.07316-X
135. Ding D, Guo G, Liu Y, Zheng L, Jia G, Deng J, et al. Efficacy and safety of fenofibrate addition therapy in patients with cirrhotic primary biliary cholangitis with incomplete response to ursodeoxycholic acid. Hepatol Commun. (2022) 6:3487–95. doi: 10.1002/hep4.2103
136. Yang J, Ma G, Wang K, Yang H, Jiang S, Fan Q, et al. Causal associations between gut microbiota and Cholestatic liver diseases: a Mendelian randomization study. Front Med (Lausanne). (2024) 11:1342119. doi: 10.3389/fmed.2024.1342119
137. Yuen VW and Wong CC. Hypoxia-inducible factors and innate immunity in liver cancer. J Clin Invest. (2020) 130:5052–62. doi: 10.1172/JCI137553
138. Hagen M, Müller F, Wirsching A, Kharboutli S, Spoerl S, Düsing C, et al. Local immune effector cell-associated toxicity syndrome in CAR T-cell treated patients with autoimmune disease: an observational study. Lancet Rheumatol. (2025) 7:e424–33. doi: 10.1016/S2665-9913(25)00091-8
139. Yu J, Yang Y, Gu Z, Shi M, La Cava A, and Liu A. CAR immunotherapy in autoimmune diseases: promises and challenges. Front Immunol. (2024) 15:1461102. doi: 10.3389/fimmu.2024.1461102
140. Zeng L, Li Y, Xiang W, Xiao W, Long Z, and Sun L. Advances in chimeric antigen receptor T cell therapy for autoimmune and autoinflammatory diseases and their complications. J Autoimmun. (2025) 150:103350. doi: 10.1016/j.jaut.2024.103350
141. Lungova K and Putman M. Barriers to CAR T-cell therapy in rheumatology. Lancet Rheumatol. (2025) 7:e212–6. doi: 10.1016/S2665-9913(24)00240-6
142. Caël B, Bôle-Richard E, Garnache Ottou F, and Aubin F. Chimeric antigen receptor-modified T-cell therapy: Recent updates and challenges in autoimmune diseases. J Allergy Clin Immunol. (2025) 155:688–700. doi: 10.1016/j.jaci.2024.12.1066
143. Ji X, Sun Y, Xie Y, Gao J, and Zhang J. Advance in chimeric antigen receptor T therapy in autoimmune diseases. Front Immunol. (2025) 16:1533254. doi: 10.3389/fimmu.2025.1533254
144. Cheever A, Kang CC, O’Neill KL, and Weber KS. Application of novel CAR technologies to improve treatment of autoimmune disease. Front Immunol. (2024) 15:1465191. doi: 10.3389/fimmu.2024.1465191
145. Avouac J, Scherlinger M, and Club for Innovative Immunotherapies in Immune-Mediated Inflammatory Diseases (C3I). CAR T-cell therapy for rheumatic diseases: what does the future hold? BioDrugs. (2025) 39:5–19. doi: 10.1007/s40259-024-00692-z
146. Nie W, Yang X, Ma M, Xu X, and Hou J. Chimeric antigen receptor T cell therapy for autoimmune diseases. Curr Opin Immunol. (2025) 95:102596. doi: 10.1016/j.coi.2025.102596
147. Khalifeh M and Salman H. Engineering resilient CAR T cells for immunosuppressive environment. Mol Ther. (2025) 33(6):2391–2405. doi: 10.1016/j.ymthe.2025.01.035
148. Sun Y, Yuan Y, Zhang B, and Zhang X. CARs: a new approach for the treatment of autoimmune diseases. Sci China Life Sci. (2023) 66:711–28. doi: 10.1007/s11427-022-2212-5
149. Li YR, Lyu Z, Chen Y, Fang Y, and Yang L. Frontiers in CAR-T cell therapy for autoimmune diseases. Trends Pharmacol Sci. (2024) 45:839–57. doi: 10.1016/j.tips.2024.07.005
150. Schett G, Müller F, Taubmann J, Mackensen A, Wang W, Furie RA, et al. Advancements and challenges in CAR T cell therapy in autoimmune diseases. Nat Rev Rheumatol. (2024) 20:531–44. doi: 10.1038/s41584-024-01139-z
151. Kandel S, Adhikary P, Li G, and Cheng K. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett. (2021) 510:67–78. doi: 10.1016/j.canlet.2021.04.011
152. Gehrke N, Nagel M, Straub BK, Wörns MA, Schuchmann M, Galle PR, et al. Loss of cellular FLICE-inhibitory protein promotes acute cholestatic liver injury and inflammation from bile duct ligation. Am J Physiol Gastrointest Liver Physiol. (2018) 314:G319–33. doi: 10.1152/ajpgi.00097.2017
153. Jain M, Singh MK, Shyam H, Mishra A, Kumar S, Kumar A, et al. Role of JAK/STAT in the neuroinflammation and its association with neurological disorders. Ann Neurosci. (2021) 28:191–200. doi: 10.1177/09727531211070532
154. Wang J, Yao N, Chen Y, Li X, and Jiang Z. Research progress of cGAS-STING signaling pathway in intestinal diseases. Int Immunopharmacol. (2024) 135:112271. doi: 10.1016/j.intimp.2024.112271
155. Yang Y, He X, Rojas M, Leung PSC, and Gao L. Mechanism-based target therapy in primary biliary cholangitis: opportunities before liver cirrhosis? Front Immunol. (2023) 14:1184252. doi: 10.3389/fimmu.2023.1184252
156. Richardson N, Wootton GE, Bozward AG, and Oo YH. Challenges and opportunities in achieving effective regulatory T cell therapy in autoimmune liver disease. Semin Immunopathol. (2022) 44:461–74. doi: 10.1007/s00281-022-00940-w
157. Itoh A, Adams D, Huang W, Wu Y, Kachapati K, Bednar KJ, et al. Enoxacin up-regulates microRNA biogenesis and down-regulates cytotoxic CD8 T-cell function in autoimmune cholangitis. Hepatology. (2021) 74:835–46. doi: 10.1002/hep.31724
158. Hebbandi Nanjundappa R, Kolla HB, Edgar A, Leung PS, Chauhan DS, Ridgway WM, et al. Immunological and fibrotic profiling in the NOD.c3c4 murine model of autoimmune cholangitis. J Autoimmun. (2025) 156:103473. doi: 10.1016/j.jaut.2025.103473
159. Yamashita M, Honda A, Shimoyama S, Umemura M, Ohta K, Chida T, et al. Breach of tolerance versus burden of bile acids: Resolving the conundrum in the immunopathogenesis and natural history of primary biliary cholangitis. J Autoimmun. (2023) 136:103027. doi: 10.1016/j.jaut.2023.103027
160. Cançado GGL, Lleo A, Levy C, Trauner M, and Hirschfield GM. Primary biliary cholangitis and the narrowing gap towards optimal disease control. Lancet Gastroenterol Hepatol. (2025) 10:855–70. doi: 10.1016/S2468-1253(25)00025-1
161. Ji G, Zhang Y, Si X, Yao H, Ma S, Xu Y, et al. Biopolymer immune implants’ Sequential activation of innate and adaptive immunity for colorectal cancer postoperative immunotherapy. Adv Mater. (2021) 33:e2004559. doi: 10.1002/adma.202004559
162. He X, Wedn A, Wang J, Gu Y, Liu H, Zhang J, et al. IUPHAR ECR review: The cGAS-STING pathway: Novel functions beyond innate immune and emerging therapeutic opportunities. Pharmacol Res. (2024) 201:107063. doi: 10.1016/j.phrs.2024.107063
163. Huang W, Jiang R, Li S, Zeng R, Li Y, Zhang Y, et al. Investigating shared genetic architecture between inflammatory bowel diseases and primary biliary cholangitis. JHEP Rep. (2024) 6:101037. doi: 10.1016/j.jhepr.2024.101037
Keywords: primary biliary cholangitis (PBC), innate immunity, biliary tract, cholangiocyte, therapeutic target, prognostic assessment
Citation: Chen R, Sun Y, Hu Y and Tai W (2025) Innate immunity of bile and cholangiocytes in primary biliary cholangitis. Front. Immunol. 16:1655287. doi: 10.3389/fimmu.2025.1655287
Received: 27 June 2025; Accepted: 05 September 2025;
Published: 02 October 2025.
Edited by:
Francesca Granucci, University of Milano-Bicocca, ItalyReviewed by:
Debby Reuveni, The Center for Liver Diseases, Sheba Tel-Hashomer, IsraelMiki Scaravaglio, University of Milano-Bicocca, Italy
Copyright © 2025 Chen, Sun, Hu and Tai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenlin Tai, dGFpd2VubGluQGttbXUuZWR1LmNu